
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResolving a structural issue in cerium-nickel-based oxide: a single compound or a two-phase system?†
Jelena
Kojčinović
 a,
Dalibor
Tatar
a,
Stjepan
Šarić
a,
Cora
Bartus Pravda
b,
Andraž
Mavrič
c,
Iztok
Arčon
cd,
Zvonko
Jagličić
ef,
Maximilian
Mellin
g,
Marcus
Einert
g,
Angela
Altomare
h,
Rocco
Caliandro
h,
Ákos
Kukovecz
b,
Jan Philipp
Hofmann
g and
Igor
Djerdj
*a
a,
Dalibor
Tatar
a,
Stjepan
Šarić
a,
Cora
Bartus Pravda
b,
Andraž
Mavrič
c,
Iztok
Arčon
cd,
Zvonko
Jagličić
ef,
Maximilian
Mellin
g,
Marcus
Einert
g,
Angela
Altomare
h,
Rocco
Caliandro
h,
Ákos
Kukovecz
b,
Jan Philipp
Hofmann
g and
Igor
Djerdj
*a
aDepartment of Chemistry, Josip Juraj Strossmayer University of Osijek, Cara Hadrijana 8/A, 31000 Osijek, Croatia. E-mail: igor.djerdj@kemija.unios.hr
bDepartment of Applied and Environmental Chemistry, University of Szeged, 6720 Szeged, Hungary
cUniversity of Nova Gorica, Vipavska 13, 5000 Nova Gorica, Slovenia
dInstitute Jožef Stefan, Jamova 39, 1000 Ljubljana, Slovenia
eInstitute of Mathematics, Physics, and Mechanics, University of Ljubljana, Jamova 2, 1000 Ljubljana, Slovenia
fFaculty of Civil & Geodetic Engineering, University of Ljubljana, Jadranska 19, 1000 Ljubljana, Slovenia
gSurface Science Laboratory, Department of Materials and Earth Sciences, Technical University of Darmstadt, Otto-Berndt-Strasse 3, 64287 Darmstadt, Germany
hInstitute of Crystallography, CNR, via Amendola 122/o, Bari 70126, Italy
First published on 22nd December 2023
Abstract
CeNiO3 has been reported in the literature in the last few years as a novel LnNiO3 compound with promising applications in different catalytic fields, but its structure has not been correctly reported so far. In this research, CeNiO3 (RB1), CeO2 and NiO have been synthesized in a nanocrystalline form using a modified citrate aqueous sol–gel route. A direct comparison between the equimolar physical mixture (n(CeO2)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(NiO) = 1:1) and compound RB1 was made. Their structural differences were investigated by laboratory powder X-ray diffraction (PXRD), selected area electron diffraction (SAED), transmission electron microscopy (TEM) with an energy-dispersive X-ray spectroscopy (EDS) detector, and Raman spectroscopy. The surface of the compounds was analyzed by X-ray photoelectron spectroscopy (XPS), while the thermal behaviour was explored by thermogravimetric analysis (TGA). Their magnetic properties were also investigated with the aim of exploring the differences between these two compounds. There were clear differences between the physical mixture of CeO2 + NiO and RB1 presented by all of these employed methods. Synchrotron methods, such as atomic pair distribution function analysis (PDF), X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), were used to explore the structure of RB1 in more detail. Three different models for the structural solution of RB1 were proposed. One structural solution proposes that RB1 is a single-phase pyrochlore compound (Ce2Ni2O7) while the other two solutions suggest that RB1 is a two-phase system of either CeO2 + NiO or Ce1−xNixO2 and NiO.
:n(NiO) = 1:1) and compound RB1 was made. Their structural differences were investigated by laboratory powder X-ray diffraction (PXRD), selected area electron diffraction (SAED), transmission electron microscopy (TEM) with an energy-dispersive X-ray spectroscopy (EDS) detector, and Raman spectroscopy. The surface of the compounds was analyzed by X-ray photoelectron spectroscopy (XPS), while the thermal behaviour was explored by thermogravimetric analysis (TGA). Their magnetic properties were also investigated with the aim of exploring the differences between these two compounds. There were clear differences between the physical mixture of CeO2 + NiO and RB1 presented by all of these employed methods. Synchrotron methods, such as atomic pair distribution function analysis (PDF), X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), were used to explore the structure of RB1 in more detail. Three different models for the structural solution of RB1 were proposed. One structural solution proposes that RB1 is a single-phase pyrochlore compound (Ce2Ni2O7) while the other two solutions suggest that RB1 is a two-phase system of either CeO2 + NiO or Ce1−xNixO2 and NiO.
Introduction
LnNiO3 compounds (Ln – lanthanide cation) have already been investigated for different purposes, including electrocatalysis,1 energy storage,2,3 heterogeneous catalysis,4,5 artificial intelligence,6 gas sensing,2etc. Their metal-to-insulator transition has also been widely inspected.7–17 In the previous literature,9,18,19 it has been reported that LnNiO3 compounds are distorted perovskites with defined crystallographic parameters. However, there are a lot of discrepancies in the literature concerning Ce-based compounds. Most of the recently published articles claim that they obtained phase pure CeNiO3 compound which crystallizes as a perovskite-type oxide.20–27 Nonetheless, the studies suffer from a lack of thorough physicochemical characterization, typically only showing powder X-ray analysis without providing any crystallographic information. Some authors tried to index the X-ray diffraction (XRD) pattern to the orthorhombic crystal system, space group Pnma with no detailed structural characterization.20,21,26,27 Moreover, when comparing the XRD patterns given for CeNiO3 with those of PrNiO328,29 or other lanthanide nickelates,30 clear differences in Bragg positions are observed, so there is no isostructurality.In one study, it was claimed that CeNiO3 was obtained using a citrate–nitrate combustion technique and provided Rietveld refinement data clearly showing a mixture of CeO2 and NiO.31 Detailed literature studies were conducted to find a crystallographic information file (.cif) to directly compare our experimentally obtained XRD pattern and to obtain crystallographic information but without success. Therefore, there is still no crystallographic proof of the existence of CeNiO3 crystallizing in the perovskite structure.
Importantly, we noticed that what the literature claims to be the XRD pattern of the “CeNiO3 perovskite” suspiciously has the same Bragg positions as the mixture of constituent oxides, fluorite CeO2, and cubic NiO. There could be two explanations: either this is just a coincidence or CeNiO3 is a mixture of CeO2 and NiO at the nanoscale level.
Another possibility is that this “CeNiO3” perovskite is a two-phase system containing solid solution Ce1−xNixO2 and NiO. Various authors have investigated the Ni2+ doping effect of the CeO2 structure.32–39 These studies were mostly used for hydrogen production via water splitting35 and as catalysts for the reduction of NO40 or unsaturated organic compounds.34,36,41 A similar application has also been reported for the “CeNiO3” perovskite.20,22–24,26
Therefore, it is important to elucidate the actual structure of what the literature claims to be CeNiO3 so that the structure–property relationship is established. If this is resolved, its properties could be improved by further research and consequently, its application. In this research, a compound that corresponds to CeNiO3 (RB1) and the substituent oxides, CeO2 and NiO, were synthesized in a nanocrystalline form by means of the previously developed modified aqueous sol–gel method.42–48 Ceria and nickel oxide were mixed in an equimolar molar ratio (n(CeO2):n(NiO) = 1:1) after synthesis to obtain the same amounts of constituent oxides as there are in RB1, so the direct comparison between the physical mixture (CeO2 + NiO) and a “compound” (RB1) can be made. Their structural features were thoroughly investigated using laboratory X-ray and electron diffraction, synchrotron, microscopic and spectroscopic methods, with the aim of solving the actual structure of RB1 (CeNiO3). Their magnetic properties have also been explored to point out the differences in the synthesized compounds.
Experimental
Materials and methods
For the synthesis of materials, the following chemicals were used as presented in Table 1.| Chemical name | Manufacturer |
|---|---|
| Cerium(III) nitrate hexahydrate, 99.5% | Acros Organics, USA |
| Nickel nitrate hexahydrate, p.a. | T.T.T., Croatia |
| Citric acid monohydrate, Ph Eur | T.T.T., Croatia |
| Concentrated ammonia solution (w = 25%), p.a. | Gram-Mol, Croatia |
Chemicals were used as purchased, without further purification. MilliQ ultrapure water was obtained using a PURELAB Flex device for ultrapure water preparation. For pH value adjustment, a pH-meter HANNA pH 211 was used. The reaction solution was heated on a magnetic hotplate stirrer DLAB MS-H-S and dried in a drying oven Instrumentaria ST-01/02 afterwards. Calcination was performed in a muffle furnace Nabertherm LT5/11/B410.
Synthesis procedure
Stoichiometric amounts of metal cation precursors (see Table S1†) were dissolved in a 10% solution of citric acid in MilliQ water. Then, the pH value was adjusted to 5 using a concentrated ammonia solution. The as-prepared reaction solution was heated on a hotplate with constant stirring until a black resin was formed. The black resin was further dried at 120 °C overnight until it was completely solid. Finally, it was ground in a mortar and calcined in a furnace at 600 °C, heating rate 2 °C min−1, and held at this temperature for 8 hours (stabilization time).Characterization techniques
000 ms each measurement.
The samples were prepared in the form of homogeneous pellets, pressed from micronized powder mixed with micronized BN, with a total absorption thickness of about 1.5 above the Ni K-edge or Ce L3-edge, and inserted in the monochromatic beam between the first two ionization detectors. The absorption spectra were measured in the energy region from −150 eV to +1000 eV relative to the Ni K-edge (8333 eV), while for the Ce L3-edge (5724 eV) EXAFS scans were stopped at the Ce L2-edge (6165 eV). Equidistant energy steps of 0.3 eV were used in the XANES region, while for the EXAFS region, equidistant k steps of 0.03 Å−1 were adopted, with an integration time of 2 s per step. The exact energy calibration was established with simultaneous absorption measurement on 5 μm thick Ni metal foil for the Ni K-edge, or the CeO2 reference sample (calibrated with vanadium metal foil) for the Ce L3-edge, placed between the second and third ionization chambers. The energy reproducibility of the measured spectra was ±0.03 eV. The quantitative analysis of XANES and EXAFS spectra was performed using the Demeter (IFEFFIT) program package,50 in combination with the FEFF6 program code for ab initio calculations of photoelectron scattering paths in quantitative EXAFS analysis.51
sinϑ/λ by using the DIOPTAS program.52 PDF profiles were calculated from the background-subtracted Q profiles by using the PDFGetX3 program.53 The parameters for PDF calculations (background subtraction, scale factor, minimum and maximum values of Q, degree of data-correction polynomial) were chosen to avoid large termination effects and preserve the signal-to-noise ratio. The Qmax value was 28.5 Å−1. The PDF profile has been fitted with structural models by using the PDFGUI program.54
Results and discussion
Structural and microstructural analysis
The studied literature20–24,26,27 shows powder X-ray patterns that are similar to experimental XRD patterns shown in Fig. 1. However, a combination of XRD patterns of CeO2 and NiO also gives the same XRD pattern that the literature20–24,26,27 claims to be the CeNiO3 perovskite. For this purpose, CeO2 and NiO were synthesized separately by the same synthesis procedure as that used for RB1 (CeNiO3) and were mixed in a mortar in an equimolar ratio (n(CeO2):n(NiO) = 1:1). The results of the Rietveld refinement of RB1 and the CeO2 + NiO mixture are given in Table 2.
 | ||
| Fig. 1 Rietveld plots of (a) a structural solution of RB1 as a two-phase system: tetragonal solid solution Ce1−xNixO2 + cubic NiO; (b) a refinement of the physical mixture of CeO2 + NiO (n:n = 1:1) (c) a structural solution of RB1 as a single-phase pyrochlore Ce2Ni2O7; (d) a structural solution of RB1 as a two-phase system: cubic CeO2 + NiO. Red curves represent experimental patterns versus calculated patterns as black curves. | ||
| Compound | RB1 (CeNiO3) | CeO2 + NiO (n:n = 1:1) |
|||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | |||||
| Chemical formula | CeO2 | NiO | Ce0.97Ni0.03O2 | NiO | Ce2Ni2O7 | CeO2 | NiO |
| Crystal system | Cubic | Tetragonal | Cubic | Cubic | Cubic | ||
| Space group |
Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
P42/nmc |
Fmm |
Fdm |
Fmm |
||
| Z | 8 | 2 | 8 | 8 | 8 | ||
| Calculated density (g cm−3) | 7.20 | 6.76 | 7.07 | 6.74 | 7.83 | 7.19 | 6.80 |
| Unit cell parameters (Å) | 5.4165(2) | 4.1785(2) | a = 3.8208(3); c = 5.4492(2) | 4.1903(2) | 10.8406(9) | 5.4162(2) | 4.1861(2) |
| Unit cell volume (Å3) | 158.88(6) | 73.36(1) | 79.55(1) | 73.57(1) | 1273.98(8) | 158.91(6) | 72.96(1) |
| Phase content (wt%) | 71.0(2) | 29.0(3) | 71.34(1) | 28.66(4) | 100(4) | 66.73(2) | 33.27(2) |
| Average crystallite size (nm) | 5.1 | 4.6 | 5.2 | 4.7 | 5.4 | 9.6 | 9.8 |
| Average apparent microstrain (×10−4) | 0.95 | 0.95 | 4.47 | 0.95 | |||
| R B | 0.91 | 1.02 | 0.88 | 0.75 | 4.40 | 1.45 | 0.68 |
| R p, Rwp, Re | 10.9, 9.19, 8.08 | 11.0, 9.21, 8.13 | 19.9, 17.9, 8.59 | 9.87, 8.44, 6.56 | |||
| χ 2 | 1.29 | 1.28 | 4.32 | 1.66 | |||
Based on the literature, the actual structure of RB1 (CeNiO3) is unknown, so we proposed three different possibilities:
(1) RB1 is an equimolar mixture of both cubic Fmm CeO2 and NiO.
(2) RB1 is a mixture of tetragonal P42/nmc Ni-doped CeO2 and cubic Fmm NiO.
(3) RB1 is a novel cubic Fdm compound with a pyrochlore structure and the chemical formula Ce2Ni2O7.
The main challenge in a direct comparison of RB1 and previously reported CeNiO3 is the lack of crystallographic data obtained by the Rietveld refinement in those research studies. The results presented in this study unequivocally indicate that our RB1 sample corresponds to the previously reported CeNiO3. However, variations in synthesis procedures undoubtedly generate visible differences in structural features such as the degree of crystallinity. For instance, Harikrishnan et al.21 employed a co-precipitation method using metal nitrates, NaOH, and potassium carbonate. After prolonged stirring of the solution followed by calcination at 650 °C, the targeted CeNiO3 has been obtained. In another research conducted by the same author,27 CeNiO3 synthesis involved Ni-foam for active material growth, followed by autoclaving at 180 °C for 12 hours and then calcination at 600 °C under an inert (argon) atmosphere. These differences in synthesis approaches affected only the degree of crystallinity, whilst the positions of Bragg reflections of the claimed CeNiO3 and their relative intensities are almost intact. Our initial goal was to directly compare the equimolar physical mixture of CeO2 + NiO (n(CeO2):n(NiO) = 1:1) and RB1 prepared under the same synthesis conditions to identify potential differences in the crystallographic structure. Here, we assumed that RB1 consists of both cubic CeO2 and NiO, so the physical mixture of these two constituent oxides should not differ much from RB1. As for the unit cell parameters and crystal structure, these two samples appear to be the same. However, the phase content slightly differs since there is around 4 wt% more of CeO2 in RB1. Also, there is a large difference in average crystallite size values. The refinement of the physical mixture of CeO2 + NiO reveals crystallite sizes of constituent oxides that are two times larger than those found in RB1. This can be directly seen from the broadened Bragg peaks of RB1 compared to the physical mixture.
Another possibility was that cerium and nickel form a unique single-phase compound. For the proposal of an actual crystal structure of RB1, we have tried to solve it ab initio from the powder XRD pattern. However, this is a complex task since there are only a few broad peaks present, so our attempt was unsuccessful. Therefore, we have tried to find XRD patterns of different compounds in the literature that are similar to the experimental XRD pattern of RB1. The most suitable XRD pattern that was found was that of pyrochlore Ce2Zr2O755 which was used as a starting point. However, the content of the unit cell was changed to refine it with the obtained XRD pattern of RB1. Although this refinement appears to be correct, the plot given in Fig. 1c demonstrates that there are slight differences in the peak positions between the calculated and experimental patterns.
Mahammadunnisa et al.32 investigated the impact of doping CeO2 with 5–30 wt% of Ni2+. In their synthesis procedure, specific amounts of metal nitrates and citric acid were dissolved in distilled water, sonicated, and placed in a preheated furnace at around 450 °C, causing a spark, leading to the formation of a solid product. The samples further underwent calcination at 600 °C for 4 hours to eliminate the carbon content. They have noticed that there is a slight decrease in unit cell parameters due to doping CeO2 with a smaller cation Ni2+ (C.N. = 6, r = 0.69 Å (ref. 56)) in NiO, compared to Ce4+ (C.N. = 8, r = 0.97 Å (ref. 56)) in CeO2, which is also visible in RB1 compared to the physical mixture of constituent oxides (CeO2 + NiO). Two additional peaks in the XRD pattern of Ce–Ni–O systems32 at 2θ = 37° and 2θ = 43° correspond to the NiO phase and only appear when doping of CeO2 is beyond 15 wt%. Zhou et al.57 used three types of synthesis procedures, all including metal nitrates as the starting material: (i) the citrate acid method by adding citric acid into the nitrate aqueous solution, heating until gel was formed and then calcination at 450 °C; (ii) the coprecipitation method with addition of potassium carbonate, followed by the adjustment of the pH value and then calcination at 450 °C, and (iii) the ammonia evaporation method, which included addition of NH3 to aqueous solution of metal nitrates until a specific pH value, followed by calcination at 450 °C. They have shown that Ni-loading in ceria must be beyond 20 mol% for these additional peaks to appear. Therefore, the NiO phase detected by XRD in RB1 might be the NiO that is dispersed on the surface of ceria, while the rest of it could be dissolved in the cerium-nickel-based tetragonal solid solution Ce1−xNixO2−δ.57 Another difference between the physical mixture of CeO2 + NiO and RB1 is in average crystallite size values which can also be estimated from the Bragg peak widths on X-ray diffraction patterns (Fig. 1a compared to Fig. 1b). In the physical mixture CeO2 + NiO, the average crystallite sizes of the constituent oxides are twice as large as those in RB1 (Table 2). Mahammadunisa et al.32 also reported this behaviour as an effect of doping CeO2 with NiO. They attributed it to the incorporation of nickel cations in the initial structure of ceria.58 Even though these authors did not refine the XRD patterns they obtained, it was probably assumed that both Ce1−xNixO2−δ and NiO are cubic, at least according to the given results. Small dopant concentrations can still preserve the cubic crystal structure of ceria.59 However, doping ceria with other elements can also result in symmetry breaking from the cubic to the tetragonal crystal structure, which cannot be detected by XRD.5,60,61
Another method that is sensitive to phase formation and therefore useful for distinction between cubic and tetragonal crystal structures is Raman spectroscopy. Therefore, the Raman spectra of RB1 and a physical mixture of CeO2 + NiO were recorded in the range from 1200 cm−1 to 80 cm−1 at an excitation wavelength of 532 nm and are shown in Fig. 2. According to Kroumova et al.,62 CeO2 with a fluorite structure should possess only one Raman active mode, T2g, also known in the available literature as the F2g vibration.43 However, cubic NiO has no Raman active modes.62 Therefore, the Raman spectrum of the physical mixture of CeO2 and NiO should contain only one Raman active mode corresponding to fluorite ceria, as shown in Fig. 2 (red). Sole NiO would not be visible in the Raman spectrum. A strong F2g vibration of CeO2 in the Raman spectrum of the physical mixture CeO2 + NiO is found at 450 cm−1. The Raman spectrum of RB1 shows a strong peak slightly shifted to higher wavenumbers, at 455 cm−1. Also, an additional defect peak at 576 cm−1 coincides with the vibrations that are often assigned to oxygen vacancies in CeO2-based compounds.48,59,63–67
 | ||
| Fig. 2 Raman spectra of the RB1 sample and physical mixture of cerium(IV) and nickel(II) oxide (1:1). | ||
Cop et al.59 have shown that doping ceria with different cations results in the appearance of additional defect bands that are a result of increased concentration of oxygen vacancies. Various other authors have shown that these bands are expected when CeO2 is doped with other cations,32,59,68 especially when these cations are aliovalent,59 such as Ni2+. Also, a peak at 220 cm−1 is not active in the fluorite-structured ceria. This phonon mode is often activated in doped ceria, especially if there is a mismatch in ionic radii and ionic charge of dopants.61,69 Therefore, a tetragonal solid solution would explain the symmetry breaking observed in the Raman spectrum of RB1. Popović et al.38 have investigated charge delocalization in ceria upon doping with Fe2+/3+. They observed that the doping of ceria with smaller cations has an impact on the position of F2g mode because of the shrinkage of the unit cell. Also, charge mismatch and the small crystallite size impact oxygen vacancy concentration and the intensity of the vacancy mode.38 Atzori et al.70 have also investigated CeO2–NiO systems synthesized using the surface-templated method with direct synthesis of metal oxides from nitrates, dissolved in water with the template and NaOH, then filtered and calcined to obtain the targeted compound. Another used route was the synthesis of CeO2 while Ni was deposited through incipient wetness impregnation technique. They have shown that this large defect band increases with increased Ni loading. Several other authors have studied Ce1−xNixO2−δ for various catalytic applications.33,41,71,72 Barrio et al.33 even reported that the limit of the solubility of Ni in ceria ranges from 10 to 12 mol%, even though they did not give information on the Ce1−xNixO2−δ unit cell content. These values were calculated from the difference in the NiO content; they assumed that the remaining Ni was dissolved in CeO2. The total Ni-content in RB1 is 30 wt%, which is 50 mol%, but there is only 3 mol% of total Ni-content dissolved in the tetragonal solid solution in our case according to the Rietveld refinement results. It seems that in our case, there is much less Ni that is dissolved in CeO2, but none of the mentioned research papers calculated the actual amount of Ni dissolved in the solid solution directly from the unit cell content. Therefore, we must consider their results carefully.
Additionally, it was necessary to record Raman spectra of CeO2 and NiO separately, to further inspect their structure. Fig. 3a and b shows individual Raman spectra of CeO2 and NiO. The Raman spectrum of pure CeO2 shows typical strong F2g vibration at 462 cm−1 and a wide defect peak from 550–650 cm−1 that occurs due to the presence of oxygen vacancies.73 Now, it can be observed that F2g vibration is red-shifted in both RB1 and the CeO2 + NiO mixture. Also, the NiO spectrum reveals a low-intensity wide peak at 506 cm−1 that cannot be classified as Raman-active mode since its intensity is 150 times lower than the intensity of F2g vibration in CeO2. Also, there are two additional broad peaks at around 190 cm−1 and 1075 cm−1, with even lower intensity. The inset in Fig. 3b shows the Raman spectrum of NiO on the same scale as the CeO2 spectrum in Fig. 3a to visualize the difference in intensities and thus to explain the absence of these peaks in the Raman spectrum of the CeO2 + NiO mixture. According to George et al.74 these peaks appear due to the first- and second-order Raman scattering in NiO-nanostructures because of the structural defects. To conclude, there is a clear difference between RB1 and the CeO2 + NiO mixture in terms of Raman spectroscopy.
 | ||
| Fig. 3 Raman spectrum of (a) CeO2 and (b) NiO. | ||
Nevertheless, additional analyses are necessary to resolve this structural issue. One of them is transmission electron microscopy. Fig. 4 and 5 show STEM, TEM, and SAED images of RB1. STEM-EDS mapping in Fig. 4 shows a uniform distribution of involved cations in RB1. The physical mixture of CeO2 + NiO does not have a uniform distribution of cerium and nickel, as shown in Fig. S1 and S2 in the ESI.† This is a difference that can have an impact on the difference in catalytic activity or some other physical properties.
 | ||
| Fig. 4 STEM-EDS mapping of RB1 (CeNiO3) nanoparticles agglomerate. | ||
 | ||
| Fig. 5 (a) TEM image of RB1 (CeNiO3) nanoparticle agglomerate and (b) a corresponding SAED with assigned d-values in Å. (c) HRTEM of RB1 (CeNiO3) nanoparticles, d-values in Å. | ||
TEM, HRTEM and SAED images confirm the nanocrystalline nature of synthesized RB1. The crystallite size values nearly correspond to the ones calculated by the Rietveld refinement of the XRD pattern of RB1. Measured interplanar distances (d values) were compared to values obtained from the Rietveld refinement results of RB1 for three different cases. A comparison is given in Table 3.
| SAED pattern | Rietveld refinement | |||||
|---|---|---|---|---|---|---|
| Cubic Fmm |
Tetragonal P42/nmc | Cubic pyrochlore Fdm |
||||
| d (Å) | hkl | d (Å) | hkl | d (Å) | hkl | |
| 3.17 | 3.13 | 111 | 3.13 | 101 | 3.13 | 222 |
| 2.79 | 2.71 | 200 | 2.72 | 002 | 2.71 | 400 |
| 2.48 | 2.41 | 111 | 2.42 | 111 | 2.49 | 331 |
| 2.13 | 2.09 | 200 | 2.10 | 200 | 2.09 | 511 |
| 1.96 | 1.92 | 220 | 1.92 | 112 | 1.92 | 440 |
| 1.67 | — | — | 1.64 | 103 | 1.65 | 533 |
| 1.61 | 1.63 | 311 | 1.63 | 211 | 1.63 | 622 |
| 1.52 | 1.56 | 222 | 1.56 | 202 | 1.52 | 551 |
| 1.39 | 1.35 | 400 | 1.36 | 004 | 1.36 | 800 |
As seen in Table 3, the cubic Fmm crystal structure can be ruled out due to the missing interplanar distance. Therefore, the actual compound present in RB1 could be in the form of a solid solution Ce1−xNixO2, or it could crystalize as a pyrochlore Ce2Ni2O7.
Normalized Ni K-edge XANES spectra recorded for nanocrystalline NiO and RB1 are shown in Fig. 6, together with the spectra of the corresponding crystalline NiO reference compound. The energy position of the Ni K-edge and the edge profile in NiO and RB1 coincide with the energy position and edge profile of crystalline NiO with Ni in the 2+ state. This shows that the valence state and local symmetry of Ni cations is the same as in the crystalline NiO. Ni K-edge EXAFS analysis is used to detect the average local environment of Ni cations in the nanocrystalline NiO and RB1 samples. In the Fourier transform (FT), the magnitude of the k3-weighted EXAFS spectra are plotted in Fig. 7. The contributions of photoelectron scattering on the nearest shells of neighbours around the Ni atoms are observed in the R range of up to about 6 Å. Already by qualitative comparison of the FT EXAFS spectra, it is evident that both samples exhibit the same Ni local structure characteristic of crystalline NiO with an FCC crystal structure, as in the crystalline NiO reference.75 Quantitative EXAFS analysis is used to determine the structural parameters of the average local Ni neighbourhood (the type and average number of neighbours; the radii and Debye–Waller factor of neighbouring shells) in the samples. The structural parameters are quantitatively resolved from the EXAFS spectra by comparing the measured EXAFS signal with the model signal. The FEFF model for the crystalline NiO nanoparticles is based on the cubic crystal structure of NiO with the space group Fmm with the lattice constant a = 4.177 Å,76 where Ni is coordinated with 6 oxygen atoms at a distance of 2.09 Å, 12 Ni atoms at 2.95 Å and 8 oxygen atoms at 3.62 Å.
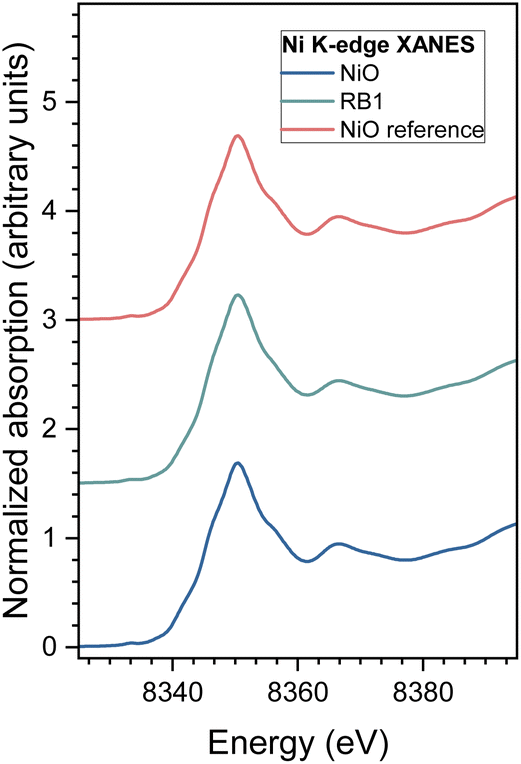 | ||
| Fig. 6 Ni K-edge XANES spectrum of nanocrystalline NiO and RB1 samples with the crystalline NiO reference compound. Spectra are shifted vertically for clarity. | ||
 | ||
| Fig. 7 Fourier transform magnitude of the k3-weighted Ni K-edge EXAFS spectra of the nanocrystalline NiO and RB1 samples, calculated in the k range of 3–12 Å. Experiment – (solid line); the best fit EXAFS model calculated in the R-range of 1.2 to 3.3 Å – (red dashed line). Graph curves are shifted vertically for clarity. | ||
The FEFF model comprised three single scattering and two significant multiple scattering paths up to 3.6 Å, with 8 variable parameters: coordination shell distance (R) and Debye–Waller factors (σ2) of all single scattering paths, and the amplitude reduction factor (S02) and shift of the energy origin of the photoelectron (ΔEo), common to all scattering paths, are introduced. The structural parameters of multiple scattering paths are constrained to those of the corresponding single scattering paths. The model was tested on the EXAFS spectrum measured for the crystalline NiO.75 The same FEFF model is used in the fit of the EXAFS spectra of the nanocrystalline NiO and RB1 samples, but here, the amplitude reduction factor (S02) was fixed to a value of 0.90 obtained for the crystalline NiO, and the shell coordination numbers were used as variable parameters. A very good EXAFS fit (Fig. 7) is obtained in the k range of 3–12 Å and the R-range of 1.2–3.3 Å. The best-fit structural parameters are given in Table 4. The results show that the local structure around Ni cations in the nanocrystalline RB1 sample is the same as that in the nanocrystalline NiO compound. The structural parameters of the RB1 sample are the same as those of the nanocrystalline NiO sample, except that the Debye–Waller factors are slightly larger in RB1, indicating a larger structural disorder in the crystalline structure.
| Neighbour | N | R (Å) | σ 2 (Å2) | R-factor |
|---|---|---|---|---|
| NiO | ||||
| O | 6.0(5) | 2.079(4) | 0.0052(7) | 0.00086 |
| Ni | 12.0(5) | 2.953(2) | 0.0058(2) | |
| O | 8.0(5) | 3.46(3) | 0.010(7) | |
| RB1 | ||||
| O | 6.1(5) | 2.074(4) | 0.0061(8) | 0.0019 |
| Ni | 11.0(7) | 2.957(3) | 0.0067(3) | |
| O | 7.3(4) | 3.46(2) | 0.010(3) | |
To test for the eventual presence of the RB1 nanocrystalline structure one single scattering path from Ce neighbours is added to the FEFF model at the distance characteristic for Ni–Ce in the RB1 crystal structure. The presence of Ce neighbour in the local neighbourhood around Ni cations is excluded by the fit, showing there is no presence of Ce in Ni neighbourhood or it is below the detection limit.
Normalized Ce L3-edge XANES spectra measured on CeO2 and RB1 are shown in Fig. 8, together with the spectra of the corresponding Ce3+ reference compounds (crystalline CeVO4).77
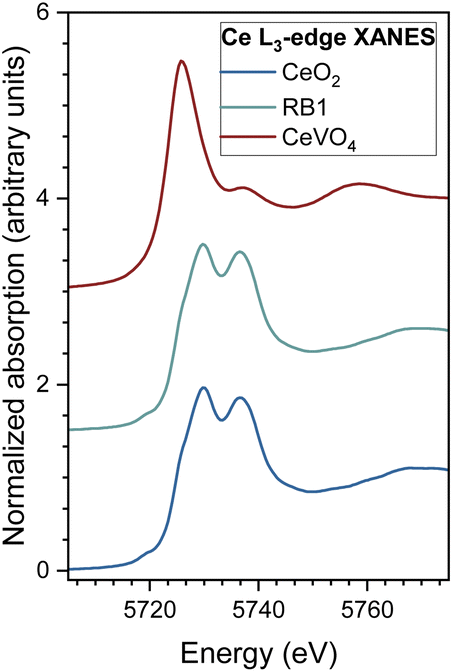 | ||
| Fig. 8 Ce L3-edge XANES spectra of CeO2 and RB1 samples, and CeVO4 as a reference for Ce3+. | ||
The energy position of the Ce L3-edge and the edge profile in RB1 completely coincide with the energy position and edge profile of CeO2, indicating that all Ce cations in the sample are in the Ce4+ valence state. If the sample contains a mixture of two or more compounds of the same cation with different local structures and valence states, the measured XANES spectrum is a linear combination of individual XANES spectra of different cation sites. In such cases, the relative amounts of the cation at each site and the average valence state of the cation in the sample can be determined by the linear combination fit (LCF) with XANES spectra. LCF analysis of the XANES spectra on RB1 with the XANES spectra of the reference compounds (CeO2 as reference for Ce4+, and CeVO4 as reference for Ce3+) shows that there is no significant presence of Ce3+ species. Quantitative EXAFS analysis to determine the structural parameters of the average local Ce neighbourhood was carried out for CeO2 and RB1 samples (see ESI, Fig. S3 and Table S2†). The structural parameters for RB1 are quantitatively and equally well resolved from the EXAFS spectra using the FEFF models for CeO2 with a cubic (Fmm) and tetragonal (P42/nmc) structure. This shows that EXAFS cannot resolve between cubic and tetragonal structures for the RB1 sample. The obtained structural parameters for RB1 are in agreement with those obtained for pure CeO2 (Table S2†).
The atomic pair distribution function (PDF) profile of RB1 has been fitted to test the three modelling hypotheses. The crystal cell parameters and atomic displacement parameters and positions not constrained by symmetry have been refined for all the crystal phases. Results are shown in Fig. 9, while the main fitting parameters are reported in Table 5. It can be noted that the hypothesized pyrochlore structure is ruled out by the poorer PDF fitting, as the corresponding calculated profile shows significant differences compared to the observed one even at small interatomic distances (Fig. 9c). For the remaining structural hypotheses (1) and (2) there is an overall agreement with reciprocal space determinations (Rietveld analysis) for crystal cell and weight fraction parameters, and the average crystallite size is in good agreement with the particle diameter estimated by PDF (SPdiameter). However, PDF data definitively resolves the question of which structural description is the best among the two linear combinations of crystal phases. A better fit is achieved when the cubic NiO crystal phase is coupled with the tetragonal Ni-doped CeO2 rather than the cubic CeO2 one. In the latter case, a weight agreement factor Rw = 0.163 is obtained and the observed-calculated difference profile in Fig. 9b only contains only high-frequency noise throughout the profile.
 | ||
| Fig. 9 Fit of the PDF profile of RB1 (experimental data = blue circles, fit curves = red line, residuals = green line) by using a structural model obtained by combining (a) cubic Fmm CeO2 and NiO, (b) tetragonal P42/nmc Ni-doped CeO2 and cubic Fmm NiO and (c) a hypothetical cubic Fdm compound with a pyrochlore structure and the chemical formula Ce2Ni2O7. | ||
m CeO2 and NiO (1), tetragonal P42/nmc Ni-doped CeO2 and cubic Fmm NiO (2) and a hypothetical cubic Fdm compound with a pyrochlore structure and chemical formula Ce2Ni2O7 (3). Rw is the weighted agreement factor between observed and calculated PDF, Qbroad describes the peak broadening from increased intensity noise at high Q, scale is an overall scale factor, δ1 is the coefficient for 1/r contribution to the peak sharpening, SPdiameter is the particle diameter for the PDF shape damping function
| 1 | 2 | 3 | |||
|---|---|---|---|---|---|
| Chemical formula | CeO2 | NiO | Ce0.97Ni0.03O2 | NiO | Ce2Ni2O7 |
| Crystal system | Cubic | Tetragonal | Cubic | Cubic | |
| Space group |
Fmm |
P42/nmc |
Fmm |
Fdm |
|
| Fit range | 1.4–40.0 Å | 1.4–40.0 Å | 1.4–40.0 Å | ||
| R w | 0.193 | 0.163 | 0.261 | ||
| Q broad | 0.040 | 0.028 | 0.040 | ||
| Scale | 0.136 | 0.141 | 0.08 | ||
| Phase content (wt%) | 76.9 | 23.1 | 77.0 | 23.0 | 100 |
| δ 1 | 2.07 | 1.50 | 2.10 | 1.54 | 2.44 |
| Unit cell parameters (Å) | 5.402 | 4.175 | a = 3.812; c = 5.417 | 4.175 | 10.804 |
| SPdiameter (nm) | 5.5 | 5.2 | 5.4 | 5.0 | 5.7 |
Additionally, X-ray photoelectron spectroscopy (XPS) was conducted to investigate the surface of the materials. XPS spectra were recorded for NiO and CeO2 separately and for all synthesized compounds. Fig. 10 shows high-resolution Ce 3d spectra for CeO2 and RB1, respectively. The Ce 3d spectrum of CeO2 shows the typical 3d5/2 at 883.1 eV and the 3d3/2 at 901.7 eV with a spin–orbit splitting of 18.6 eV. The spectra exhibit strong charge-transfer satellites78,79 and additional overlapping peaks from multiplet effects.80 The position and intensity ratio between those peaks depends on the oxidation state, the ligand type, and the next-nearest neighbour.80 This makes analysing core spectra of mixed transition metal and lanthanide oxides difficult. It was pointed out81 that satellites 3 and 4 can be directly related to cerium 4+ (CeO2), which shows that both materials in Fig. 10 mainly consist of cerium 4+. However, the CeO2 deviates from PLD-deposited films shown in ref. 81 indicating that cerium 3+ is present on the surface, which could be due to the formation of cerium hydroxide, clearly seen in the O 1s spectra (Fig. S4b†).
 | ||
| Fig. 10 High-resolution Ce 3d spectra of CeO2 and RB1. | ||
In the Ce 3d spectra of RB1, the left shoulder of the 3d5/2 main peak broadens while the 3d3/2 main peak increases in intensity, which indicates the higher cerium 3+ content in RB1 compared to the CeO2 sample. This could hint at a mixed oxidation state of cerium and nickel or an oxygen-deficient structure. However, in the O 1s spectra of RB1 (Fig. S4c†), the cerium hydroxide content on the surface is high, which would result in cerium in the 3+ state. Reference spectra without the influence of hydroxide could be used in the fitting procedure to analyze the content of different cerium components. Peak position analysis is summarized in Table S3.†
Similar analysis was performed for the high-resolution Ni 2p spectra, in order to investigate the stoichiometry of the compounds and the oxidation state of nickel cations in both samples. The Ni 2p spectra are shown in Fig. 11 for NiO and RB1 and the peak position analysis is summarized in Table S4.†
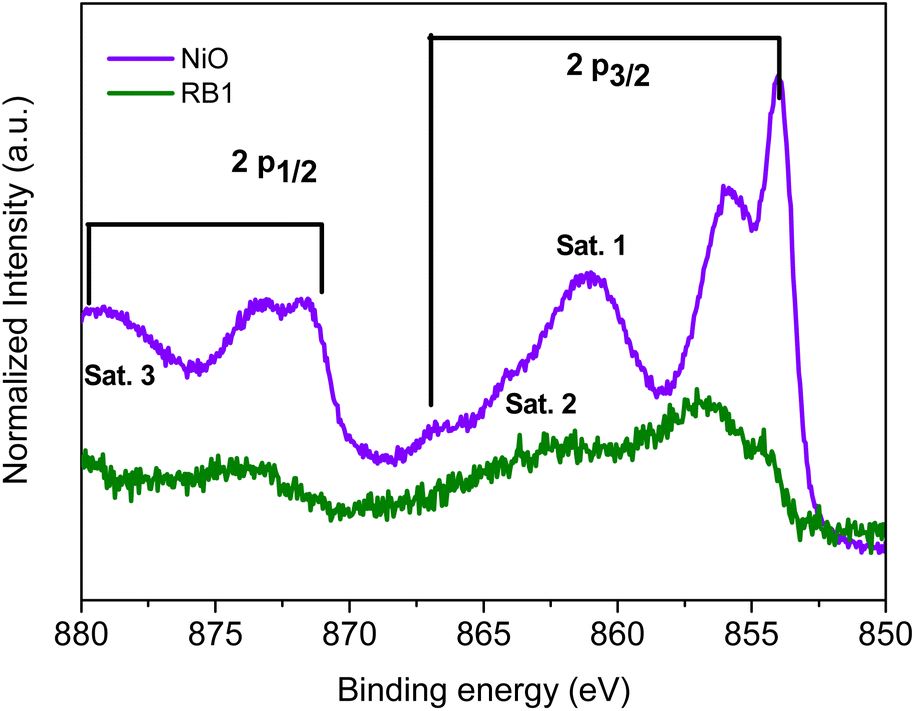 | ||
| Fig. 11 High-resolution Ni 2p XPS spectra of NiO and RB1. | ||
The Ni 2p spectrum of NiO shows 2p3/2 (853.0 eV) and 2p1/2 (871.5 eV) doublets with a spin–orbit splitting of 17.5 eV.82 The core level peaks at 856.1 and 873.2 eV are a sign of the presence of non-stoichiometry in NiO and the existence of Ni3+, respectively.82,83 It has been previously reported that satellite peaks 2 and 3 also point out towards the existence of Ni3+.83,84 Nickel oxide prepared at lower temperatures (>700 °C) has a higher concentration of Ni3+ than Ni2+.82,85 Interestingly, a fully stoichiometric NiO compound is formed after calcination at 1100 °C.
The explanation of the nonstoichiometry in nanocrystalline nickel oxide lies in the formation of Ni2+ vacancies85,86 because of excess oxygen. Positively charged nickel-vacant holes (h++) are typically neutralized by receiving two electrons from the neighbouring Ni2+ species, causing them to increase their oxidation state to Ni3+. In addition, there is another possible way of neutralizing the charge mismatch by neighbouring O2− species, thus creating reactive electrophilic O− species.85,87 These reactions are summarized in eqn (1) and (2):
| 2Ni2+ + h++ → 2Ni3+ | (1) |
| 2O2− + h++ → 2O−. | (2) |
The electrophilic oxygen species are ideal candidates for nucleophilic attack by hydroxide or water thus making them highly active in the formation of O–O bonds.87 Therefore, they are very important for the oxygen evolution reaction (OER).88–90 However, electrophilic oxygen can also be used for the reduction of unsaturated organic compounds.34,36,41
When comparing the Ni 2p spectrum of NiO and RB1, one can see that the RB1 spectrum is very noisy and cannot be interpreted easily. This is probably due to the interference of the charge-transfer satellite peaks in the Ni 2p region with the Ce 3d region in RB1.91 However, there is a sign of core level peaks at 854 and 856 eV, where the peak at 856 is higher in intensity and broadened compared to the peak at 854 which could indicate a higher content of Ni3+ in RB1 than in pure NiO. This could be due to the higher hydroxide content on the surface of RB1 and the formation of NiOOH, as seen in the high-resolution O 1s spectra shown in Fig. S4.†
All O 1 s spectra in Fig. S4† show a lattice oxygen (OL) peak at 530 eV. While the NiO and CeO2 spectra show two additional peaks at 531 eV and 533 eV. The peak at 531 eV can be related to loosely bound/adsorbed surface oxygen species (Oads), while the peak at around 533 eV is ascribed to adsorbed OH-groups or water molecules (OOH), in good agreement with the literature.85,88,92 The deconvolution parameters of O 1s spectra are summarized in Table S5 (ESI).† NiO and CeO2 have a similar concentration of adsorbed oxygen species (around 30%), while RB1 contains almost 67% of adsorbed hydroxide groups. This could be attributed to the almost two times smaller crystallite size of RB1, providing substantially more surface area for oxygen species to adsorb at.
The results of thermogravimetric analysis (Fig. S5†) can also confirm the presence of higher content of Ni3+ and Ce3+ in RB1 than in a physical mixture of constituent oxides since there is a higher oxygen uptake in RB1. During heating in an oxidative atmosphere, Ce3+ is oxidized to Ce4+, while the surface hydroxides are removed. However, heating non-stoichiometric Ni-based oxides increases stoichiometry and thus decreases the concentration of nickel vacancies.82
To further corroborate our structural findings, aiming to clarify the earlier assumption that RB1 is CeNiO3 or not, we have synthesized an additional compound Ce0.97Ni0.03O2 and conducted several additional measurements (PXRD and Raman spectroscopy). These additional measurements prove that, indeed, the equimolar introduction of Ni into the CeO2 lattice does not yield the single CeNiO3 compound. Instead, it likely forms a mixture with two distinct phases: Ce1−xNixO2 and NiO. The synthesis of 3% Ni-doped CeO2 yielded phase-pure, tetragonal Ce0.97Ni0.03O2, which agrees well with crystallographic data reported for RB1. Rietveld refinement results presented in Fig. 12a and Table S6† confirmed almost identical values for this compound compared to the phases reported for RB1 in Table 2.
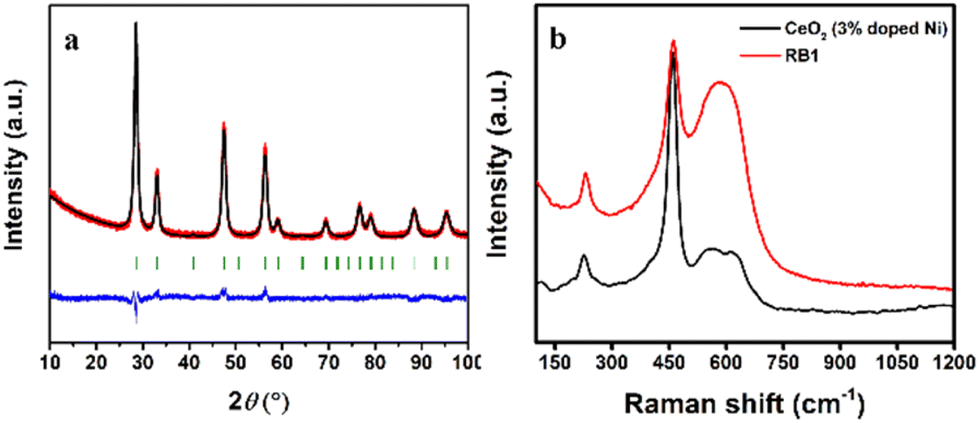 | ||
| Fig. 12 Rietveld plot (a) and Raman spectroscopy (b) for 3% Ni-doped CeO2. | ||
In addition to that, Raman spectroscopy measurements were employed to provide a further proof (Fig. 12b). The defect peak at 576 cm−1 decreased proportionally with the reduction in Ni-dopant concentration, aligning with the 50% dopant level in RB1 and the 3% Ni-doped sample. Moreover, a consistent increase in the CeO2 Raman active mode intensity (F2g at 450 cm−1) occurred with decreasing dopant content. Additionally, the intensity evident at 220 cm−1 suggests Ni integration into CeO2 as in the RB1 compound.
Magnetic properties
Since nickel is one of the elements of the iron triad group, nickel-based compounds are known as magnetically active compounds. Therefore, magnetic measurements of RB1 (CeNiO3) and a physical mixture of CeO2 + NiO should also give an insight into a possible difference between the properties of these compounds. Fig. 13a shows a temperature-dependent susceptibility of two input compounds and RB1. The measured negative susceptibility of CeO2 above 50 K means that the compound is a diamagnet at room temperature. The result is consistent with the expected nonmagnetic Ce4+ ion in CeO2. Only below 50 K a week paramagnetic signal appears (1/T dependence of the susceptibility), which can be attributed to lattice defects or oxygen vacancies.93,94 The isothermal magnetization at 2 K (Fig. 13b) can be described as a sum of paramagnetic and diamagnetic contributions. The maximal value at 50 kOe is as small as 0.033 emu g−1. We can conclude that the magnetic signal of CeO2 is much smaller than the magnetic signals of NiO and RB1. Bulk NiO is an antiferromagnet with the Néel temperature of 523 K.95 The almost temperature-constant susceptibility and linear magnetization curve at 2 K of NiO nanoparticles that were used as the input material in the synthesis, are consistent with the antiferromagnetic order of NiO. Weak splitting of zero-field cooled (ZFC) and field cooled (FC) susceptibilities and tiny hysteresis in M(H) are due to uncompensated magnetic moments on the surface of the nanoparticles.96 | ||
| Fig. 13 Temperature dependent susceptibility (a) and isothermal magnetization (b) of CeO2, NiO, RB1 and 3% Ni doped CeO2. | ||
The magnetic behaviour of the newly synthesised compounds RB1 and CeO2 (3% doped Ni) differ significantly from the magnetic properties of CeO2 and NiO nanoparticles. A much larger splitting between ZFC and FC susceptibility and hysteresis with a coercive magnetic field of 600 Oe and a remanent magnetization of 0.02 emu g−1, are clear evidence of a ferromagnetic order in CeNiO3 (RB1). The local maximum of ZFC susceptibility at about 100 K and the temperature at which the ZFC and FC curves diverge (about 220 K) are quite different, indicating a rather broad size distribution of RB1 nanoparticles. The temperature where ZFC and FC curves diverge shifts to lower temperatures in CeO2 (3% doped Ni) while the isothermal magnetization at 2 K of only 3% doped CeO2 is already quite similar to the M(H) curve of RB1. These results support the finding that RB1 is the mixture of Ce0.97Ni0.03O2 and NiO. Several publications have reported on the ferromagnetism of Ce1−xNixO2 nanoparticles.97–101 In our final product (RB1) there is a lot of NiO and only a small portion of Ce1−xNixO2, which makes the ferromagnet even more interesting as this small portion of Ce1−xNixO2 makes a huge difference in the magnetic properties of the system.
Conclusions
The compound that corresponds to CeNiO3 (RB1) was synthesized in a nanocrystalline form along with constituent oxides CeO2 and NiO in order to resolve the structural issue regarding this compound. The recent literature claims that CeNiO3 is a perovskite-type compound. However, XRD patterns from the CeNiO3 perovskite suspiciously resemble the phase mixture of CeO2 and NiO in a 1:1 molar ratio. Thus, we have tried to investigate this issue by proposing three different structural solutions. The first structural solution is that CeNiO3 (RB1) is a two-phase system containing both cubic CeO2 and NiO, the second one is that this two-phase system contains tetragonal solid solution Ce1−xNixO2 and cubic NiO, while the final proposed solution is that CeNiO3 (RB1) is actually a single-phase compound with a pyrochlore structure Ce2Ni2O7, rather than a perovskite. According to the detailed structural and microstructural analysis, the most probable solution is that RB1 is a two-phase system comprising Ce1−xNixO2 and NiO. The average crystallite size of RB1 is twice as small (around 5 nm) compared to the physical mixture of the constituent oxides (around 10 nm) in a molar ratio of 1:1. The involved elements in RB1 are homogeneously distributed along the surface. Raman spectroscopy reveals defects in RB1 that are probably related to symmetry breaking from the cubic to the tetragonal phase, which is the case when cubic CeO2 is doped with Ni2+. The magnetic properties also differ significantly: CeO2 is diamagnetic, NiO is antiferromagnetic, and RB1 shows ferromagnetic behaviour.
Author contributions
J. K. conceptualization, formal analysis, and manuscript preparation; D. T. and S. Š. synthesis, thermogravimetric and X-ray diffraction measurements, and analysis; C. B. P. Raman spectra measurements and analysis; Z. J. magnetic property measurements and analysis; A. M. electron microscopy and diffraction; A. M. and I. A. extended X-ray absorption fine structure and X-ray absorption near edge structure measurements and analysis; M. M. and M. E. X-ray photoelectron spectroscopy measurements and analysis; A. A. powder XRD pattern analysis and structure solution; R. C. atomic pair distribution function measurements and analysis; I. Dj. Rietveld refinement, funding acquisition, project management; I. Dj., Á. K. and J. P. H. review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Croatian Science Foundation project “The study of the role of rare earth metal promoters and ordering on the redox properties of CeO2–ZrO2 system” (PZS-2019-02-2467) for full financial support. This work was partially supported financially by the Slovenian Research Agency (Grant No. P2-0348 and Grants No. P2-0412, P1-0112, J2-2498, J2-50058). The authors acknowledge access to the SR facilities of ELETTRA (beamline XAFS, proposal 20215267). The authors would like to thank Giuliana Aquilanti and Luca Olivi of ELETTRA for expert advice on beamline operation and for assistance during the experiment. The authors also thank Sanjit Ghose, Brookhaven National Laboratory and Rocco Lassando, Istituto di Cristallografia, for their support in collecting PDF data. The access to the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-SC0012704 (NSLS-II Proposal Number 312211).References
- L. Wang, K. A. Stoerzinger, L. Chang, J. Zhao, Y. Li, C. S. Tang, X. Yin, M. E. Bowden, Z. Yang, H. Guo, L. You, R. Guo, J. Wang, K. Ibrahim, J. Chen, A. Rusydi, J. Wang, S. A. Chambers and Y. Du, Tuning Bifunctional Oxygen Electrocatalysts by Changing the A–Site Rare–Earth Element in Perovskite Nickelates, Adv. Funct. Mater., 2018, 28, 1803712 CrossRef.
- D. K. Hwang, S. Kim, J.-H. Lee, I.-S. Hwang and I.-D. Kim, Phase evolution of perovskite LaNiO3 nanofibers for supercapacitor application and p-type gas sensing properties of LaOCl–NiO composite nanofibers, J. Mater. Chem., 2011, 21, 1959–1965 RSC.
- A. A. Yaremchenko, B. I. Arias-Serrano, K. Zakharchuk and J. R. Frade, Perovskite-like LaNiO3−δ as Oxygen Electrode Material for Solid Oxide Electrolysis Cells, ECS Trans., 2019, 91, 2399–2408 CrossRef CAS.
- P. D. K. Nezhad, M. F. Bekheet, N. Bonmassar, A. Gili, F. Kamutzki, A. Gurlo, A. Doran, S. Schwarz, J. Bernardi, S. Praetz, A. Niaei, A. Farzi and S. Penner, Elucidating the role of earth alkaline doping in perovskite-based methane dry reforming catalysts, Catal. Sci. Technol., 2022, 12, 1229–1244 RSC.
- F. J. Trindade, S. Damasceno, L. Otubo, M. R. Felez, D. Z. de Florio, F. C. Fonseca and A. S. Ferlauto, Tuning of Shape, Defects, and Disorder in Lanthanum-Doped Ceria Nanoparticles: Implications for High-Temperature Catalysis, ACS Appl. Nano Mater., 2022, 5, 8859–8867 CrossRef CAS.
- Z. Zhang, Y. Yu, X. Qiao, J. Sun, Y. Ni and J. Chen, Strongly correlated nickelate: Recent progress of synthesis and applications in artificial intelligence, Mater. Sci. Semicond. Process., 2023, 166, 107735 CrossRef CAS.
- G. Catalan, Progress in perovskite nickelate research, Phase Transitions, 2008, 81, 729–749 CrossRef CAS.
- J. Chen, A. Bird, F. Yan, W. Wu, X. Ke, Y. Jiang and N. Chen, Mechanical and correlated electronic transport properties of preferentially orientated SmNiO3 films, Ceram. Int., 2020, 46, 6693–6697 CrossRef CAS.
- D. D. Sarma, N. Shanthi and P. Mahadevan, Electronic structure and the metal-insulator transition in LnNiO3 (Ln=La, Pr, Nd, Sm and Ho): bandstructure results, J. Phys.: Condens.Matter, 1994, 6, 10467–10474 CrossRef CAS.
- A. Ambrosini and J.-F. Hamet, SmxNd1−xNiO3 thin-film solid solutions with tunable metal–insulator transition synthesized by alternate-target pulsed-laser deposition, Appl. Phys. Lett., 2003, 82, 727–729 CrossRef CAS.
- P. Laffez, M. Zaghrioui, I. Monot, T. Brousse and P. Lacorre, Microstructure and metal–insulator transition of NdNiO3 thin films on various substrates, Thin Solid Films, 1999, 354, 50–54 CrossRef CAS.
- F. Yan, Z. Mi, J. Chen, H. Hu, L. Gao, J. Wang, N. Chen, Y. Jiang, L. Qiao and J. Chen, Revealing the role of interfacial heterogeneous nucleation in the metastable thin film growth of rare-earth nickelate electronic transition materials, Phys. Chem. Chem. Phys., 2022, 24, 9333–9344 RSC.
- X. Li, T. Zhang, Z. Li, F. Yan, H. Li, Y. Cui, N. Chen and J. Chen, Nonlinearity in regulating the metal to insulator transition of ReNiO3 towards low temperature range, Ceram. Int., 2022, 48, 31995–32000 CrossRef CAS.
- Y. Bian, H. Li, F. Yan, H. Li, J. Wang, H. Zhang, Y. Jiang, N. Chen and J. Chen, Hydrogen induced electronic transition within correlated perovskite nickelates with heavy rare-earth composition, Appl. Phys. Lett., 2022, 120, 092103 CrossRef CAS.
- J. Chen, H. Hu, J. Wang, T. Yajima, B. Ge, X. Ke, H. Dong, Y. Jiang and N. Chen, Overcoming synthetic metastabilities and revealing metal-to-insulator transition & thermistor bi-functionalities for d-band correlation perovskite nickelates, Mater. Horiz., 2019, 6, 788–795 RSC.
- N. Shukla, T. Joshi, S. Dasgupta, P. Borisov, D. Lederman and S. Datta, Electrically induced insulator to metal transition in epitaxial SmNiO3 thin films, Appl. Phys. Lett., 2014, 105, 012108 CrossRef.
- F. Y. Bruno, S. Valencia, R. Abrudan, Y. Dumont, C. Carrétéro, M. Bibes and A. Barthélémy, Probing the metal-insulator transition in nickelates using soft x-ray absorption spectroscopy, Appl. Phys. Lett., 2014, 104, 021920 CrossRef.
- M. T. Escote, A. M. L. da Silva, J. R. Matos and R. F. Jardim, General Properties of Polycrystalline LnNiO3 (Ln=Pr, Nd, Sm) Compounds Prepared through Different Precursors, J. Solid State Chem., 2000, 151, 298–307 CrossRef CAS.
- T. Moriga, O. Usaka, I. Nakabayashi, Y. Hirashima, T. Kohno, S. Kikkawa and F. Kanamaru, Reduction of the perovskite-type LnNiO3 (Ln=Pr, Nd) to Ln3Ni3O7 with monovalent nickel ions, Solid State Ionics, 1994, 74, 211–217 CrossRef CAS.
- M. P. Harikrishnan and A. C. Bose, Porous CeNiO3 with an enhanced electrochemical performance and prolonged cycle life (>50 000 cycles) via a lemon-assisted sol–gel autocombustion method, New J. Chem., 2022, 46, 15118–15129 RSC.
- M. P. Harikrishnan, A. J. C. Mary and A. C. Bose, Electrochemical performance of ANiO3 (A= La, Ce) perovskite oxide material and its device performance for supercapattery application, Electrochim. Acta, 2020, 362, 137095 CrossRef CAS.
- N. Ahmad, F. Alharthi, M. Alam, R. Wahab, S. Manoharadas and B. Alrayes, Syngas Production via CO2 Reforming of Methane over SrNiO3 and CeNiO3 Perovskites, Energies, 2021, 14, 2928 CrossRef CAS.
- H.-N. Barad, D. A. Keller, K. J. Rietwyk, A. Ginsburg, S. Tirosh, S. Meir, A. Y. Anderson and A. Zaban, How Transparent Oxides Gain Some Color: Discovery of a CeNiO3 Reduced Bandgap Phase as an Absorber for Photovoltaics, ACS Comb. Sci., 2018, 20, 366–376 CrossRef CAS PubMed.
- N. Ahmad, R. Wahab, S. Manoharadas, B. F. Alrayes, M. Alam and F. A. Alharthi, The Role of Strontium in CeNiO3 Nano-Crystalline Perovskites for Greenhouse Gas Mitigation to Produce Syngas, Molecules, 2022, 27, 356 CrossRef CAS PubMed.
- N. Tri, N. P. Anh, T. D. Huy, D. B. Long, H. C. Anh, P. H. Phuong, N. T. T. Van, T.-T. Nguyen and L. C. Loc, In situ synthesis of highly effective nickel nanocatalyst for methane bireforming, J. Sci.: Adv. Mater. Devices, 2023, 8, 100529 CAS.
- M. P. Harikrishnan and A. C. Bose, Co-precipitation route for synthesizing CeNiO3 and its application as excellent pseudocapacitor, AIP Conf. Proc., 2020, 2265, 030631 CrossRef CAS.
- M. P. Harikrishnan and A. C. Bose, Binder–free synthesis of cerium nickel oxide for supercapattery devices, Int. J. Energy Res., 2022, 46, 21826–21840 CrossRef CAS.
- T. C. Huang, W. Parrish, H. Toraya, P. Lacorre and J. B. Torrance, High-temperature crystal structures of orthorhombic and rhombohedral PrNiO3, Mater. Res. Bull., 1990, 25, 1091–1098 CrossRef CAS.
- V. Vibhu, A. Flura, C. Nicollet, S. Fourcade, N. Penin, J.-M. Bassat, J.-C. Grenier, A. Rougier and M. Pouchard, Characterization of PrNiO3−δ as oxygen electrode for SOFCs, Solid State Sci., 2018, 81, 26–31 CrossRef CAS.
- J. A. Alonso, M. J. Martínez-Lope, M. T. Casais, M. A. G. Aranda and M. T. Fernández-Díaz, Metal−Insulator Transitions, Structural and Microstructural Evolution of RNiO3 (R = Sm, Eu, Gd, Dy, Ho, Y) Perovskites: Evidence for Room-Temperature Charge Disproportionation in Monoclinic HoNiO3 and YNiO3, J. Am. Chem. Soc., 1999, 121, 4754–4762 CrossRef CAS.
- S. J. Kashyap, R. Sankannavar and G. M. Madhu, Synthesis and Characterization of La(Ce, Ba)NiO3 Perovskite-Type Oxides, J. Supercond. Novel Magn., 2022, 35, 2107–2118 CrossRef CAS.
- Sk. Mahammadunnisa, P. M. K. Reddy, N. Lingaiah and Ch. Subrahmanyam, NiO/Ce1−xNixO 2−δ as an alternative to noble metal catalysts for CO oxidation, Catal. Sci. Technol., 2013, 3, 730–736 RSC.
- L. Barrio, A. Kubacka, G. Zhou, M. Estrella, A. Martínez-Arias, J. C. Hanson, M. Fernández-García and J. A. Rodriguez, Unusual Physical and Chemical Properties of Ni in Ce1−xNixO2−y Oxides: Structural Characterization and Catalytic Activity for the Water Gas Shift Reaction, J. Phys. Chem. C, 2010, 114, 12689–12697 CrossRef CAS.
- G. Zhou, L. Barrio, S. Agnoli, S. D. Senanayake, J. Evans, A. Kubacka, M. Estrella, J. C. Hanson, A. Martínez-Arias, M. Fernández-García and J. A. Rodriguez, High Activity of Ce1−xNixO2−y for H2 Production through Ethanol Steam Reforming: Tuning Catalytic Performance through Metal-Oxide Interactions, Angew. Chem., Int. Ed., 2010, 49, 9680–9684 CrossRef CAS PubMed.
- J. Milikić, R. O. Fuentes, J. E. Tasca, D. M. F. Santos, B. Šljukić and F. M. L. Figueiredo, Nickel-Doped Ceria Bifunctional Electrocatalysts for Oxygen Reduction and Evolution in Alkaline Media, Batteries, 2022, 8, 100 CrossRef.
- W. Shan, M. Fleys, F. Lapicque, D. Swierczynski, A. Kiennemann, Y. Simon and P.-M. Marquaire, Syngas production from partial oxidation of methane over Ce1−XNiXOY catalysts prepared by complexation–combustion method, Appl. Catal., A, 2006, 311, 24–33 CrossRef CAS.
- L. Pino, A. Vita, F. Cipitì, M. Laganà and V. Recupero, Catalytic Performance of Ce1−xNixO2 Catalysts for Propane Oxidative Steam Reforming, Catal. Lett., 2008, 122, 121–130 CrossRef CAS.
- Z. V. Popović, Z. D. Dohčević-Mitrović, N. Paunović and M. Radović, Evidence of charge delocalization in Ce1−xFex2+(3+)O2−y nanocrystals (x = 0, 0.06, 0.12), Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 014302 CrossRef.
- S. K. Misra, S. I. Andronenko, M. H. Engelhard, A. Thurber, K. M. Reddy and A. Punnoose, Role of dopant incorporation on the magnetic properties of Ce1−xNixO2 nanoparticles: An electron paramagnetic resonance study, J. Appl. Phys., 2008, 103(7), 07D122 CrossRef.
- Y. Wang, A. Zhu, Y. Zhang, C. T. Au, X. Yang and C. Shi, Catalytic reduction of NO by CO over NiO/CeO2 catalyst in stoichiometric NO/CO and NO/CO/O2 reaction, Appl. Catal., B, 2008, 81, 141–149 CrossRef CAS.
- R. Rameshan, P. Pentyala, S. A. Singh, P. A. Deshpande and S. Roy, Probing the surface active sites of Ce1−xNixO2−δ for catalytic reduction of NO, J. Environ. Chem. Eng., 2022, 10, 108966 CrossRef CAS.
- J. Kojčinović, M. Sahu, S. Hajra, D. Tatar, T. Klaser, Ž. Skoko, Z. Jagličić, E. Sadrollahi, F. J. Litterst, H. J. Kim and I. Djerdj, Nanocrystalline triple perovskite compounds A3Fe2BO9 (A = Sr, Ba; B = W, Te) with ferromagnetic and dielectric properties for triboelectric energy harvesting, Mater. Chem. Front., 2022, 6, 1116–1128 RSC.
- D. Tatar, J. Kojčinović, B. Marković, A. Széchenyi, A. Miletić, S. B. Nagy, S. Ziegenheim, I. Szenti, A. Sapi, Á. Kukovecz, K. Dinjar, Y. Tang, D. Stenzel, G. Varga and I. Djerdj, Sol-Gel Synthesis of Ceria-Zirconia-Based High-Entropy Oxides as High-Promotion Catalysts for the Synthesis of 1,2-Diketones from Aldehyde, Molecules, 2021, 26, 6115 CrossRef CAS PubMed.
- J. Bijelić, A. Stanković, M. Medvidović-Kosanović, B. Marković, P. Cop, Y. Sun, S. Hajra, M. Sahu, J. Vukmirović, D. Marković, Á. Kukovecz, Z. Jagličić, B. M. Smarsly and I. Djerdj, Rational Sol–Gel-Based Synthesis Design and Magnetic, Dielectric, and Optical Properties Study of Nanocrystalline Sr3Co2WO9 Triple Perovskite, J. Phys. Chem. C, 2020, 124, 12794–12807 CrossRef.
- J. Bijelić, D. Tatar, S. Hajra, M. Sahu, S. J. Kim, Z. Jagličić and I. Djerdj, Nanocrystalline Antiferromagnetic High-κ Dielectric Sr2NiMO6 (M = Te, W) with Double Perovskite Structure Type, Molecules, 2020, 25, 3996 CrossRef PubMed.
- J. Bijelić, A. Stanković, B. Matasović, B. Marković, M. Bijelić, Ž. Skoko, J. Popović, G. Štefanić, Z. Jagličić, S. Zellmer, T. Preller, G. Garnweitner, T. Đorđević, P. Cop, B. Smarsly and I. Djerdj, Structural characterization and magnetic property determination of nanocrystalline Ba3Fe2WO9 and Sr3Fe2WO9 perovskites prepared by a modified aqueous sol–gel route, CrystEngComm, 2019, 21, 218–227 RSC.
- J. Bijelić, D. Tatar, M. Sahu, Z. Jagličić and I. Djerdj, Size reduction-induced properties modifications of antiferromagnetic dielectric nanocrystalline Ba2NiMO6 (M = W, Te) double perovskites, Oxford Open Mater. Sci., 2021, 1(1), itaa003 CrossRef.
- S. Nundy, D. Tatar, J. Kojčinović, H. Ullah, A. Ghosh, T. K. Mallick, R. Meinusch, B. M. Smarsly, A. A. Tahir and I. Djerdj, Bandgap Engineering in Novel Fluorite–Type Rare Earth High–Entropy Oxides (RE–HEOs) with Computational and Experimental Validation for Photocatalytic Water Splitting Applications, Adv. Sustainable Syst., 2022, 6, 2200067 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Systematic and collaborative approach to problem solving using X-ray photoelectron spectroscopy, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS : data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. J. Rehr, R. C. Albers and S. I. Zabinsky, High-order multiple-scattering calculations of x-ray-absorption fine structure, Phys. Rev. Lett., 1992, 69, 3397–3400 CrossRef CAS PubMed.
- C. Prescher and V. B. Prakapenka, DIOPTAS : a program for reduction of two-dimensional X-ray diffraction data and data exploration, High Pressure Res., 2015, 35, 223–230 CrossRef CAS.
- P. Juhás, T. Davis, C. L. Farrow and S. J. L. Billinge, PDFgetX3: a rapid and highly automatable program for processing powder diffraction data into total scattering pair distribution functions, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, PDFfit2 and PDFgui: computer programs for studying nanostructure in crystals, J. Phys.: Condens.Matter, 2007, 19, 335219 CrossRef CAS PubMed.
- P. E. Raison, C. C. Pavel, R. Jardin, E. Suard, R. G. Haire and K. Popa, Thermal expansion behavior of Ce2Zr2O7 up to 898 K in conjunction with structural analyses by neutron diffraction, Phys. Chem. Miner., 2010, 37, 555–559 CrossRef CAS.
- Database of Ionic Radii, https://abulafia.mt.ic.ac.uk/shannon/ptable.php.
- H. Zhou, P. Hu, Z. Huang, F. Qin, W. Shen and H. Xu, Preparation of NiCe Mixed Oxides for Catalytic Decomposition of N2O, Ind. Eng. Chem. Res., 2013, 52, 4504–4509 CrossRef CAS.
- R. K. Pati, I. C. Lee, S. Hou, O. Akhuemonkhan, K. J. Gaskell, Q. Wang, A. I. Frenkel, D. Chu, L. G. Salamanca-Riba and S. H. Ehrman, Flame Synthesis of Nanosized Cu−Ce−O, Ni−Ce−O, and Fe−Ce−O Catalysts for the Water-Gas Shift (WGS) Reaction, ACS Appl. Mater. Interfaces, 2009, 1, 2624–2635 CrossRef CAS PubMed.
- P. Cop, R. Maile, Y. Sun, O. Khalid, I. Djerdj, P. Esch, S. Heiles, H. Over and B. M. Smarsly, Impact of Aliovalent/Isovalent Ions (Gd, Zr, Pr, and Tb) on the Catalytic Stability of Mesoporous Ceria in the HCl Oxidation Reaction, ACS Appl. Nano Mater., 2020, 3, 7406–7419 CrossRef CAS.
- M. Varenik, J. C. Nino, E. Wachtel, S. Kim, O. Yeheskel, N. Yavo and I. Lubomirsky, Dopant Concentration Controls Quasi-Static Electrostrictive Strain Response of Ceria Ceramics, ACS Appl. Mater. Interfaces, 2020, 12, 39381–39387 CrossRef CAS PubMed.
- R. Schmitt, A. Nenning, O. Kraynis, R. Korobko, A. I. Frenkel, I. Lubomirsky, S. M. Haile and J. L. M. Rupp, A review of defect structure and chemistry in ceria and its solid solutions, Chem. Soc. Rev., 2020, 49, 554–592 RSC.
- E. Kroumova, M. I. Aroyo, J. M. Perez-Mato, A. Kirov, C. Capillas, S. Ivantchev and H. Wondratschek, Bilbao Crystallographic Server: Useful Databases and Tools for Phase-Transition Studies, Phase Transitions, 2003, 76, 155–170 CrossRef CAS.
- Q. Zhou, C. Zhou, Y. Zhou, W. Hong, S. Zou, X. Q. Gong, J. Liu, L. Xiao and J. Fan, More than oxygen vacancies: A collective crystal-plane effect of CeO2 in gas-phase selective oxidation of benzyl alcohol, Catal. Sci. Technol., 2019, 9, 2960–2967 RSC.
- K. Kuntaiah, P. Sudarsanam, B. M. Reddy and A. Vinu, Nanocrystalline Ce1-xSmxO2−δ (x = 0.4) solid solutions: Structural characterization versus CO oxidation, RSC Adv., 2013, 3, 7953–7962 RSC.
- O. V. Safonova, A. Guda, Y. Rusalev, R. Kopelent, G. Smolentsev, W. Y. Teoh, J. A. van Bokhoven and M. Nachtegaal, Elucidating the Oxygen Activation Mechanism on Ceria-Supported Copper-Oxo Species Using Time-Resolved X-ray Absorption Spectroscopy, ACS Catal., 2020, 10, 4692–4701 CrossRef CAS.
- N. Shehata, K. Meehan, M. Hudait and N. Jain, Control of oxygen vacancies and Ce+3 concentrations in doped ceria nanoparticles via the selection of lanthanide element, J. Nanopart. Res., 2012, 14, 1173 CrossRef.
- S. Tiwari, G. Rathore, N. Patra, A. K. Yadav, D. Bhattacharya, S. N. Jha, C. M. Tseng, S. W. Liu, S. Biring and S. Sen, Oxygen and cerium defects mediated changes in structural, optical and photoluminescence properties of Ni substituted CeO2, J. Alloys Compd., 2019, 782, 689–698 CrossRef CAS.
- X. Wang, J. C. Hanson, G. Liu, J. A. Rodriguez, A. Iglesias-Juez and M. Fernández-García, The behavior of mixed-metal oxides: Physical and chemical properties of bulk Ce1−xTbxO2 and nanoparticles of Ce1−xTbxOy, J. Chem. Phys., 2004, 121, 5434–5444 CrossRef CAS PubMed.
- M. Yashima, H. Arashi, M. Kakihana and M. Yoshimura, Raman Scattering Study of Cubic-Tetragonal Phase Transition in Zr1-xCexO2 Solid Solution, J. Am. Chem. Soc., 1994, 77, 1067–1071 CAS.
- L. Atzori, M. G. Cutrufello, D. Meloni, C. Cannas, D. Gazzoli, R. Monaci, M. F. Sini and E. Rombi, Highly active NiO-CeO2 catalysts for synthetic natural gas production by CO2 methanation, Catal. Today, 2018, 299, 183–192 CrossRef CAS.
- J. Deng, W. Chu, B. Wang, W. Yang and X. S. Zhao, Mesoporous Ni/Ce1−xNixO2−y heterostructure as an efficient catalyst for converting greenhouse gas to H2 and syngas, Catal. Sci. Technol., 2016, 6, 851–862 RSC.
- W. Shan, Reduction property and catalytic activity of Ce1−XNiXO2 mixed oxide catalysts for CH4 oxidation, Appl. Catal., A, 2003, 246, 1–9 CrossRef CAS.
- C. Schilling, A. Hofmann, C. Hess and M. V. Ganduglia-Pirovano, Raman Spectra of Polycrystalline CeO2 : A Density Functional Theory Study, J. Phys. Chem. C, 2017, 121, 20834–20849 CrossRef CAS.
- G. George and S. Anandhan, Synthesis and characterisation of nickel oxide nanofibre webs with alcohol sensing characteristics, RSC Adv., 2014, 4, 62009–62020 RSC.
- H.-T. Vu, I. Arčon, D. O. de Souza, S. Pollastri, G. Dražić, J. Volavšek, G. Mali, N. Z. Logar and N. N. Tušar, Insight into the interdependence of Ni and Al in bifunctional Ni/ZSM-5 catalysts at the nanoscale, Nanoscale Adv., 2022, 4, 2321–2331 RSC.
- S. Sasaki, K. Fujino and Y. Takéuchi, X-ray determination of electron-density distributions in oxides, MgO, MnO, CoO, and NiO, and atomic scattering factors of their constituent atoms, Proc. Jpn. Acad., Ser. B, 1979, 55, 43–48 CrossRef CAS.
- M. Zabilskiy, I. Arčon, P. Djinović, E. Tchernychova and A. Pintar, In–situ XAS Study of Catalytic N2O Decomposition Over CuO/CeO2 Catalysts, ChemCatChem, 2021, 13, 1814–1823 CrossRef CAS.
- T. Nakano, A. Kotani and J. C. Parlebas, Theory of XPS and BIS Spectra in Ce2O3 and CeO2, J. Phys. Soc. Jpn., 1987, 56, 2201–2210 CrossRef CAS.
- F. de Groot, Multiplet effects in X-ray spectroscopy, Coord. Chem. Rev., 2005, 249, 31–63 CrossRef CAS.
- M. A. van Veenendaal and G. A. Sawatzky, Nonlocal screening effects in 2p x-ray photoemission spectroscopy core-level line shapes of transition metal compounds, Phys. Rev. Lett., 1993, 70, 2459–2462 CrossRef CAS PubMed.
- D. R. Mullins, The surface chemistry of cerium oxide, Surf. Sci. Rep., 2015, 70, 42–85 CrossRef CAS.
- P. Dubey, N. Kaurav, R. S. Devan, G. S. Okram and Y. K. Kuo, The effect of stoichiometry on the structural, thermal and electronic properties of thermally decomposed nickel oxide, RSC Adv., 2018, 8, 5882–5890 RSC.
- U. De Silva, J. See, W. P. R. Liyanage, J. Masud, J. Wu, W. Yang, W.-T. Chen, D. Prendergast and M. Nath, Understanding the Structural Evolution of a Nickel Chalcogenide Electrocatalyst Surface for Water Oxidation, Energy Fuels, 2021, 35, 4387–4403 CrossRef CAS.
- P. S. Bagus, C. J. Nelin, C. R. Brundle, B. V. Crist, E. S. Ilton, N. Lahiri and K. M. Rosso, Main and Satellite Features in the Ni 2p XPS of NiO, Inorg. Chem., 2022, 61, 18077–18094 CrossRef CAS PubMed.
- X. Xu, L. Li, J. Huang, H. Jin, X. Fang, W. Liu, N. Zhang, H. Wang and X. Wang, Engineering Ni3+ Cations in NiO Lattice at the Atomic Level by Li + Doping: The Roles of Ni3+ and Oxygen Species for CO Oxidation, ACS Catal., 2018, 8, 8033–8045 CrossRef CAS.
- B. Savova, S. Loridant, D. Filkova and J. M. M. Millet, Ni–Nb–O catalysts for ethane oxidative dehydrogenation, Appl. Catal., A, 2010, 390, 148–157 CrossRef CAS.
- V. Pfeifer, T. E. Jones, S. Wrabetz, C. Massué, J. J. V. Vélez, R. Arrigo, M. Scherzer, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Reactive oxygen species in iridium-based OER catalysts, Chem. Sci., 2016, 7, 6791–6795 RSC.
- V. Pfeifer, T. E. Jones, J. J. V. Vélez, R. Arrigo, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, In situ observation of reactive oxygen species forming on oxygen-evolving iridium surfaces, Chem. Sci., 2017, 8, 2143–2149 RSC.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, Electrolysis of water on oxide surfaces, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- S. Piccinin, A. Sartorel, G. Aquilanti, A. Goldoni, M. Bonchio and S. Fabris, Water oxidation surface mechanisms replicated by a totally inorganic tetraruthenium–oxo molecular complex, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4917–4922 CrossRef CAS PubMed.
- E. Ioannidou, Ch. Neofytidis, L. Sygellou and D. K. Niakolas, Au-doped Ni/GDC as an Improved Cathode Electrocatalyst for H2O Electrolysis in SOECs, Appl. Catal., B, 2018, 236, 253–264 CrossRef CAS.
- G. J. Kim, S. M. Lee, S. C. Hong and S. S. Kim, Active oxygen species adsorbed on the catalyst surface and its effect on formaldehyde oxidation over Pt/TiO2 catalysts at room temperature; role of the Pt valence state on this reaction?, RSC Adv., 2018, 8, 3626–3636 RSC.
- K. Ackland and J. M. D. Coey, Room temperature magnetism in CeO2 —A review, Phys. Rep., 2018, 746, 1–39 CrossRef CAS.
- L. R. Shah, B. Ali, H. Zhu, W. G. Wang, Y. Q. Song, H. W. Zhang, S. I. Shah and J. Q. Xiao, Detailed study on the role of oxygen vacancies in structural, magnetic and transport behavior of magnetic insulator: Co–CeO2, J. Phys.: Condens.Matter, 2009, 21, 486004 CrossRef PubMed.
- A. Barbier, C. Mocuta, W. Neubeck, M. Mulazzi, F. Yakhou, K. Chesnel, A. Sollier, C. Vettier and F. de Bergevin, Surface and Bulk Spin Ordering of Antiferromagnetic Materials: NiO(111), Phys. Rev. Lett., 2004, 93, 257208 CrossRef PubMed.
- M. Jagodič, Z. Jagličić, A. Jelen, J. B. Lee, Y.-M. Kim, H. J. Kim and J. Dolinšek, Surface-spin magnetism of antiferromagnetic NiO in nanoparticle and bulk morphology, J. Phys.: Condens.Matter, 2009, 21, 215302 CrossRef PubMed.
- P. L. Madhav, K. R. Teja, N. Sreelekha, D. A. Reddy, G. Murali, M. Ramanadha, K. Subramanyam and R. P. Vijayalakshmi, Transition of Magnetic Characteristics from Paramagnetic State to Ferromagnetic Phase in Ce1−xNixO2 Nanoparticles, J. Supercond. Novel Magn., 2018, 31, 1631–1636 CrossRef CAS.
- M. Parveen, V. Asvini, G. Saravanan, K. Ravichandran and D. KalaiSelvi, Investigation of Ni-doped CeO2 nanoparticles–spintronics application, Ionics, 2017, 23, 1285–1291 CrossRef.
- A. Thurber, K. M. Reddy, V. Shutthanandan, M. H. Engelhard, C. Wang, J. Hays and A. Punnoose, Ferromagnetism in chemically synthesized CeO2 nanoparticles by Ni doping, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 165206 CrossRef.
- R. Murugan, G. Vijayaprasath, T. Mahalingam and G. Ravi, Room temperature ferromagnetism of Ni doped cerium oxide single crystalline thin Films deposited by using rf magnetron sputtering, Mater. Lett., 2016, 162, 71–74 CrossRef CAS.
- F. Abbas, T. Jan, J. Iqbal, I. Ahmad, M. S. H. Naqvi and M. Malik, Facile synthesis of ferromagnetic Ni doped CeO2 nanoparticles with enhanced anticancer activity, Appl. Surf. Sci., 2015, 357, 931–936 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt03280a |
| This journal is © The Royal Society of Chemistry 2024 |