DOI:
10.1039/D3DT03193D
(Paper)
Dalton Trans., 2024,
53, 1817-1832
Coordination chemistry effects of the space-demanding solvent molecule N,N′-dimethylpropyleneurea†
Received
28th September 2023
, Accepted 12th December 2023
First published on 13th December 2023
Abstract
Crystallographic investigations of eight homoleptic N,N′-dimethylpropyleneurea (dmpu) coordinated metal ions in the solid state, [Mg(dmpu)5]I2 (1), [Ca(dmpu)6]I2 (2), [Ca(dmpu)6](ClO4)2 (3), [Ca(dmpu)6](CF3SO3)2 (4), [Sr(dmpu)6](CF3SO3)2 (5), [Ba(dmpu)6](CF3SO3)2 (6), [Sc(dmpu)6]I3 (7), and [Pr(dmpu)6]I(I3)2 (8), and the complex [CoBr2(dmpu)2] (9) as well as the structures of the dmpu coordinated calcium, strontium, barium, scandium(III) and cobalt(II) ions and the cobalt(II) bromide complex in dmpu solution as determined by EXAFS are reported. The methyl groups in the dmpu molecule are close to the oxygen donor atom, causing steric restrictions, and making dmpu space-demanding at coordination to metal ions. The large volume required by the dmpu ligand at coordination contributes to crowdedness around the metal ion with often lower coordination numbers than for oxygen donor ligands without such steric restrictions. The crowdedness is seen in M⋯H distances equal to or close to the sum of the van der Waals radii. To counteract the space-demand at coordination, the dmpu molecule has an unusual ability to increase the M–O–C bond angle to facilitate as large coordination numbers as possible. M–O–C bond angles in the range of 125–170° are reported depending on the crowdedness caused by the coordination figure and the M–O bond distance. All reported structures of dmpu coordinated metal ions in both the solid state and dmpu solution are summarized to study the relationship between the M–O–C bond angle and the crowdedness around the metal ion. However, highly symmetric complexes seem to be favoured in the solid state due to favourable lattice energies. As a result, the dmpu coordinated lanthanoid(III) ions are octahedral in the solid state, while they, except lutetium, are seven-coordinate in the dmpu solution.
Introduction
1,3-Dimethyl-3,4,5,6-tetrahydro-2(H)pyrimidone, known as N,N′-dimethylpropyleneurea (dmpu), CAS number: 7226-23-5, is a commonly used aprotic organic solvent with high relative permittivity (εr = 36.1),1 large dipole moment (μ = 4.23 D),2 high thermal and chemical stability, wide liquid range (mp −23.8 °C, bp 246.5 °C), low vapor pressure (flash point 125 °C), low toxicity and limited corrosive properties. It is used as a versatile solvent for the synthesis of pharmaceuticals and agrochemicals, dyestuffs and polymers, epoxides, polyimides, and other engineering resins, and for BTX extraction, spinning, dyeing of fibers, etc.3–7 The high relative permittivity is the reason for its excellent dissolving properties, not only for organic compounds but also for inorganic compounds due to its ability to form stable metal dmpu complexes and compounds.8 Furthermore, the closeness of the two methyl groups to the oxygen donor atom leads to the dmpu molecule demanding a lot of space at coordination to metal ions, often forcing them to adopt lower coordination numbers than in most other oxygen donor solvents without such steric restrictions, Table S1 (ESI).† A molecule becomes space-demanding at coordination when the sizes of other groups in the molecule, except the size of the coordinating atom, have an impact on the volume it requires. This leads to the crowdedness around the coordinating metal ion to increase, e.g. as seen in short M⋯H distances equal to or close to the sum of the van der Waals radii, and the coordination number to decrease. A decrease of the coordination number and symmetry of a dmpu coordinated metal ion may change its physico-chemical properties, e.g. its ability to form complexes with halide ions increases significantly in solvents which are space-demanding at coordination, such as dmpu9,10 and hexamethylphosphoramide (hmpa).11
In this paper, we report the structures of eight homoleptic dmpu coordinated metal ions and the [CoBr2(dmpu)2] complex in the solid state and dmpu solution, and provide an overview of the structures of dmpu coordinated metal ions and the complexes reported so far. It is observed in the reported crystal structures that the coordinated dmpu molecule can reduce the required volume at coordination by straightening out the M–O–C bond angle to almost linearity. The ideal bond angle seems to be ca. 125°, as seen in complexes without steric hindrance as in the square-planar tetrakis(dmpu)copper(II) complex,12 and in tetrahedral dmpu coordinated bisbromometal complexes.9,10,13,14
The aim of the present study is to obtain a deeper insight into the coordination chemistry of dmpu coordinated metal ions and of space-demanding ligands in general. Dmpu has been chosen for this purpose as many structures of dmpu coordinated metal ions and complexes are reported in both the solid state and solution, Table S1 (ESI),† and some physico-chemical studies have also been conducted.9,10 The structures of dmpu coordinated metal ions and complexes in both the solid state and solution are summarized and analysed with the aim to obtain an overview of preferred coordination numbers and geometries, as well as the flexibility in the M–O–C bond angle.
Experimental
Solvent and salts
N,N′-Dimethylpropyleneurea (BASF) was distilled under reduced pressure over calcium hydride, CaH2 (Sigma-Aldrich), and stored in dark bottles over 3 Å molecular sieves. Anhydrous magnesium, calcium, scandium(III) and praseodymium(III) iodide, and cobalt(II) bromide (Sigma-Aldrich, 99.9% purity) were received in glass ampoules and used as purchased. Anhydrous calcium trifluoromethanesulfonate, Ca(CF3SO3)2, was prepared by dissolving calcium oxide (Merck, 99% purity) in dilute trifluoromethanesulfonic acid (Fluka), followed by filtration, boiling off excess water and acid, and finally drying and storing in an oven at 470 K. Anhydrous strontium, barium and scandium(III) trifluoromethanesulfonate were prepared accordingly using strontium, barium and scandium oxide (Merck, 99.9% purity). Hexakis(N,N′-dimethylpropyleneurea)calcium perchlorate, [Ca(dmpu)6](ClO4)2, was prepared by dissolving calcium perchlorate tetrahydrate, Ca(ClO4)2·4H2O (99%, Merck), in a minimum amount of dry acetone and a four-fold excess of 2,2-dimethoxymethane, (CH3)2C(OCH3)2 (Merck), was added and the mixture was stirred vigorously for two hours.15 A six-fold excess of dmpu was added and the resulting mixture was stirred for another 30 min. White crystals were obtained on cooling.
Warning
Metal perchlorates are powerful explosives, and under certain circumstances, violent reactions are easily triggered. To minimize the risk of accidents, the handling of metal perchlorates and perchloric acid should be performed in solution under a hood without heating, with physical protection around the experiment setup, and using as small amounts of chemicals as possible. Efficient safety glasses and gloves should always be used when handling metal perchlorates and perchloric acid.
Sample preparation
Single crystals of [Mg(dmpu)5]I2 (1), [Ca(dmpu)6]I2 (2), [Ca(dmpu)6](ClO4)2 (3), [Ca(dmpu)6](CF3SO3)2 (4), [Sr(dmpu)6](CF3SO3)2 (5), [Ba(dmpu)6](CF3SO3)2 (6), [Sc(dmpu)6]I3 (7), [Pr(dmpu)6]I(I3)2 (8) and [CoBr2(dmpu)2] (9) of X-ray quality were obtained after dissolving the anhydrous salt in freshly distilled dmpu, gently heating to ca. 310 K and allowing to slowly cool to room temperature. If no crystals were formed, the solutions were cooled in a refrigerator. 3 was recrystallized in dmpu to obtain crystals of sufficient quality for X-ray analysis.
Crystals of compound 8 were formed during the mounting of [Pr(dmpu)6]I3·3dmpu crystals, reported elsewhere,16 which included non-coordinated dmpu molecules in the unit cell. To facilitate the selection of a good crystal for X-ray diffraction from a larger amount of these crystals, a few drops of Merck immersion oil (Merck) resulted in the formation of a compound with different unit cell parameters and without non-coordinated dmpu molecules and partially oxidized iodide ions with the formation of triiodide; for details see the ESI.†
Crystallography
Single crystal diffraction data were collected at room temperature or 100 K using Mo-Kα or Cu-Kα radiation on either a Bruker SMART platform equipped with a CCD area detector and a graphite monochromator (Bruker, 1998) for compounds 2, 3, 4, 5, 6, 7 and 9; a Rigaku/Oxford diffraction Supernova equipped with an EOS CCD detector for compound 1; or a Rigaku/Oxford diffraction Excalibur III equipped with a Sapphire detector for compound 8. The software used to process the data was SMART, ver. 5.046 (area detector control), SAINT, ver. 5.01 (integration software), SDABS (program for empirical absorption correction), and SHELXTL, ver. 5.1 (Bruker Analytical X-ray Systems, Madison, Wisconsin, USA) for the Bruker data sets for 2, 3, 4, 5, 6, 7 and 9, while it was CrysAlisPRO (Rigaku/Oxford diffraction) for 1 and 8.
The details about data collection and the refined cell parameters for samples 1–9 are summarized in Table 1. Intensity decay was negligible for all samples and any needed correction was taken care of by respective diffractometer control software. Data reduction and empirical absorption correction were performed using the program packages Bruker SAINT and SADABS, for the Bruker data. The structures were solved using direct methods and refined using full-matrix least-squares on F2 with various programs in the SHELX system.17 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were placed in geometrically calculated positions and refined as riding atoms with common fixed isotropic thermal factors. The Diamond 2.1e program18 was used for crystal graphics.
Table 1 Crystallographic measurement data for [Mg(dmpu)5]I2 (1), [Ca(dmpu)6]I2 (2), [Ca(dmpu)6](ClO4)2 (3), [Ca(dmpu)6](CF3SO3)2 (4) [Sr(dmpu)6](CF3SO3)2 (5), [Ba(dmpu)6](CF3SO3)2 (6), [Sc(dmpu)6]I3 (7), [Pr(dmpu)6]I(I3)2 (8) and [CoBr2(dmpu)2] (9)
| Complex |
1
|
2
|
3
|
4
|
5
|
| CCDC |
2206123
|
2206128
|
2297623
|
2297621
|
2249334
|
| Chemical formula |
C30H60I2N10O2Mg |
C36H72I2N12O6Ca |
C36H72Cl2N12O14Ca |
C38H72F6N12O12Ca |
C38H72F6N12O12Sr |
| Formula weight |
919.008 |
1062.960 |
1008.052 |
1107.2888 |
1154.83 |
| Temperature (K) |
100(2) |
296(2) |
295(2) |
293(2) |
293(2) |
|
λ (Å) |
1.54184 (CuKα) |
0.71073 (MoKα) |
0.71073 (MoKα) |
0.71073 (MoKα) |
0.71073 (MoKα) |
| Crystal system |
Monoclinic |
Trigonal |
Trigonal |
Trigonal |
Trigonal |
| Space group |
P21/n (no. 14) |
P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (no. 147) (no. 147) |
P (no. 147) |
P (no. 147) |
P (no. 147) |
|
a (Å) |
15.0348(2) |
12.9549(18) |
13.083(3) |
13.532(2) |
13.7677(7) |
|
b (Å) |
20.2696(2) |
12.9549(18) |
13.083(3) |
13.532(2) |
13.7677(7) |
|
c (Å) |
14.7659(2) |
8.5602(12) |
8.553(3) |
8.7287(14) |
8.5413(5) |
|
α (°) |
90 |
90 |
90 |
90 |
90 |
|
β (°) |
116.7976(17) |
90 |
90 |
90 |
90 |
|
γ (°) |
90 |
120 |
120 |
120 |
120 |
| Volume (Å3) |
4016.62(10) |
1244.2(4) |
1267.8(6) |
1384.3(5) |
1402.09(16) |
|
Z
|
4 |
1 |
1 |
1 |
1 |
|
D
calc. (g cm−3) |
1.520 |
1.419 |
1.320 |
1.328 |
1.368 |
|
μ (mm−1) |
12.846 |
1.418 |
0.299 |
0.272 |
1.118 |
|
F (000) |
1872 |
546 |
538 |
586 |
604 |
| Crystal size (mm) |
0.20 × 0.15 × 0.10 |
0.15 × 0.12 × 0.10 |
0.40 × 0.25 × 0.20 |
0.35 × 0.20 × 0.17 |
0.17 × 0.15 × 0.10 |
| Index ranges |
−18 ≤ h ≤ 15 |
−14 ≤ h ≤ 14 |
−16 ≤ h ≤ 16 |
−9 ≤ h ≤ 17 |
−14 ≤ h ≤ 15 |
| −24 ≤ k ≤ 24 |
−14 ≤ k ≤ 14 |
−16 ≤ k ≤ 14 |
−18 ≤ k ≤ 13 |
−15 ≤ k ≤ 8 |
| −17 ≤ l ≤ 16 |
−9 ≤ l ≤ 9 |
−11 ≤ l ≤ 10 |
−11 ≤ l ≤ 11 |
−10 ≤ l ≤ 10 |
|
θ range (°) |
3.293–69.454 |
2.993–23.536 |
1.80–27.47 |
2.33–28.55 |
3.417–25.645 |
| Reflections collected |
42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 715 715 |
7894 |
9888 |
7993 |
4522 |
| Independent reflections |
7489 |
1246 |
1270 |
1768 |
1734 |
|
R
int
|
0.0393 |
0.0164 |
0.0368 |
0.1043 |
0.0346 |
| Data/restraints/parameters |
7489/0/443 |
1246/0/90 |
1914/0/116 |
1768/27/105 |
1734/89/110 |
| GOF (F2) |
1.038 |
1.069 |
0.916 |
0.845 |
0.928 |
| Final R1, wR2 [I > 2σ(I)] |
0.0224, 0.0556 |
0.0494, 0.1299 |
0.0450, 0.1269 |
0.0651, 0.1402 |
0.1017, 0.2620 |
| Final R1, wR2 [I > 2σ(I)] |
0.0244, 0.0566 |
0.0547, 0.1338 |
0.0654, 0.1348 |
0.2134, 0.2620 |
0.1150, 0.2769 |
| Max. peak/hole (e Å−3) |
1.227/−0.590 |
0.714/−0.557 |
0.459/−0.254 |
0.848/−0.598 |
1.454/−0.991 |
| Complex |
6
|
7
|
8
|
9
|
| CCDC |
2249335
|
2209379
|
2209377
|
2209424
|
| Chemical formula |
C38H72F6N12O12Ba |
C36H72I3N12O6Sc |
C36H72I7N12O6Pr |
C12H24Br2N4O2Co |
| Formula weight |
1204.538 |
|
1798.3120 |
475.099 |
| Temperature (K) |
293(2) |
|
100(2) |
296(2) |
|
λ (Å) |
0.71073 (MoKα) |
0.71073 (MoKα) |
0.71073 (MoKα) |
0.71073 (MoKα) |
| Crystal system |
Trigonal |
Monoclinic |
Trigonal |
Monoclinic |
| Space group |
P (no. 147) |
P21/n (no. 14) |
P (no. 147) |
P21/c (no. 14) |
|
a (Å) |
13.9909(3) |
11.9413(10) |
11.6236(18) |
8.0226(15) |
|
b (Å) |
13.9909(3) |
12.0708(10) |
11.6236(18) |
8.4660(16) |
|
c (Å) |
8.4390(3) |
17.9935(16) |
12.2134(12) |
26.796(5) |
|
α (°) |
90 |
90 |
90 |
90 |
|
β (°) |
90 |
91.590(2) |
90 |
92.364(2) |
|
γ (°) |
120 |
90 |
120 |
90 |
| Volume (Å3) |
1430.58(8) |
2592.6(4) |
1429.1(5) |
1818.4(6) |
|
Z
|
1 |
2 |
1 |
4 |
|
D
calc. (g cm−3) |
1.510 |
1.530 |
2.090 |
1.735 |
|
μ (mm−1) |
0.854 |
1.975 |
1.978 |
5.347 |
|
F (000) |
670 |
1188 |
1200 |
948 |
| Crystal size (mm) |
0.10 × 0.08 × 0.07 |
0.45 × 0.40 × 0.35 |
0.20 × 0.15 × 0.12 |
0.21 × 0.17 × 0.15 |
| Index ranges |
−16 ≤ h ≤ 17 |
−14 ≤ h ≤ 14 |
−15 ≤ h ≤ 7 |
−9 ≤ h ≤ 9 |
| −15 ≤ k ≤ 17 |
−14 ≤ k ≤ 12 |
−15 ≤ k ≤ 15 |
−9 ≤ k ≤ 8 |
| −10 ≤ l ≤ 10 |
−20 ≤ l ≤ 21 |
−16 ≤ l ≤ 16 |
−31 ≤ l ≤ 27 |
|
θ range (°) |
2.413–25.674 |
2.021–25.681 |
3.883–28.269 |
2.523–24.812 |
| Reflections collected |
9903 |
17098 |
11630 |
8656 |
| Independent reflections |
1811 |
4932 |
1328 |
2471 |
|
R
int
|
0.0233 |
0.0330 |
0.561 |
0.325 |
| Data/restraints/parameters |
1811/117/122 |
4932/0/271 |
2370/0/98 |
3117/0/205 |
| GOF(F2) |
1.206 |
0.902 |
0.703 |
1.042 |
| Final R1, wR2 [I > 2σ(I)] |
0.0534, 0.1424 |
0.0337, 0.0880 |
0.0241, 0.0398 |
0.0311, 0.0696 |
| Final R1, wR2 [I > 2σ(I)] |
0.0537, 0.1429 |
0.0585, 0.0925 |
0.0412, 0.0513 |
0.0450, 0.0739 |
| Max. peak/hole(e Å−3) |
0.958/−0.441 |
0.988/−0.580 |
0.616/−0.647 |
0.461/−0.531 |
EXAFS
The details of EXAFS measurements and data treatment are given in the ESI.†
Results and discussion
Crystal structure of pentakis(N,N′-dimethylpropyleneurea)magnesium iodide, 1
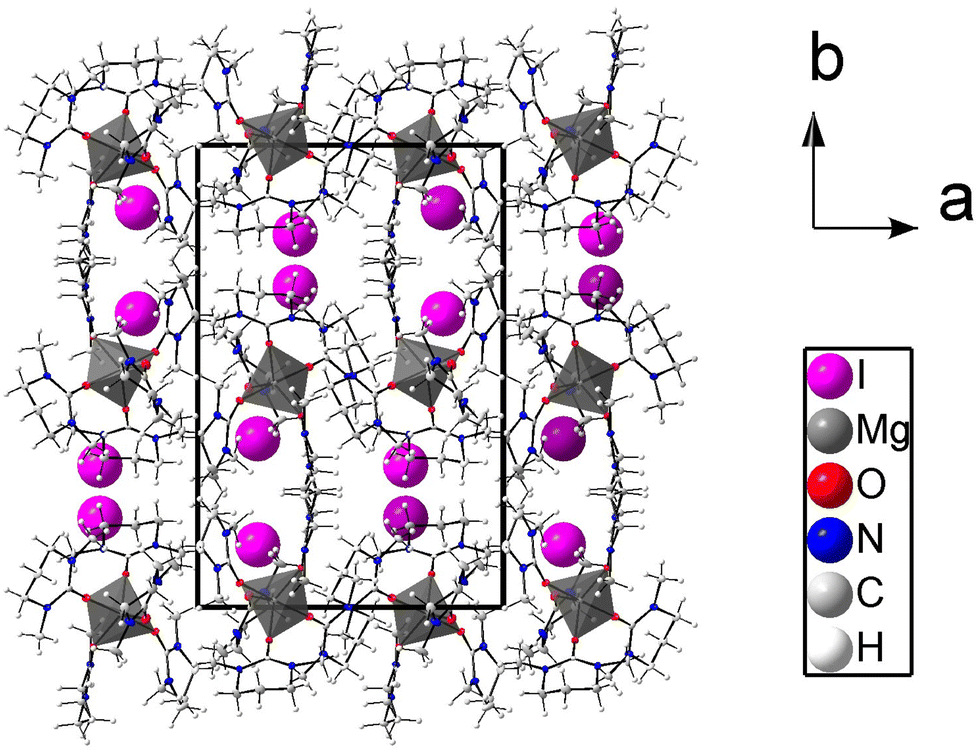
The crystal structure of 1 consists of discrete pentakis(dmpu)magnesium cations and iodide anions. The magnesium ion binds five dmpu oxygen atoms in a slightly distorted trigonal bipyramidal geometry as shown in Fig. 1. The packing of 1 is shown in Fig. 2. The three oxygens forming the base of the bipyramid have Mg–O bond lengths of 1.972(2), 1.987(1) and 1.991(2) Å which are only slightly shorter than the axial bond distances, 2.023(2) and 2.030(2) Å. The mean Mg–O bond distance is 2.000(2) Å. The Mg–O–C bond angles are in the range of 133.4(2)–165.7(2)°, mean 150.7°, Table 2. The O, N and C atoms, except the mid-carbon in the propylene group, are almost planar. Two of the dmpu molecules in the trigonal positions and those in the axial position are pair-wise stacked, which is an unusual orientation. Two pairs of hydrogens in the neighbouring dmpu rings are close to the sum of the van der Waals radii of hydrogen, 2.4 Å, Fig. 1.19 In one of these pairs of stacked dmpu rings, the mid-propylene carbons point towards each other with an H⋯H distance of 2.414 Å (H15B and H21A). In the same pair of rings, two methyl hydrogens have an H⋯H distance of 2.527 Å (H24B and H18A). In the other pair of neighbouring dmpu rings, the mid-propylene carbons are oriented in the same direction, and the shortest H⋯H distance is between the mid-propylene methylene and a methylene group binding to nitrogen, 2.374 Å (H3B and H8A), and a methylene group binding to nitrogen and methyl hydrogen, 2.515 Å (H6A⋯H10B). The crowdedness of the dmpu molecules at coordination to magnesium is seen in short Mg⋯H distances, 2.87–3.59 Å, where the shortest distances are equal to the sum of the van der Waals radii of hydrogen and magnesium, Table S2 (ESI).†19,20 The crowdedness in octahedral dmpu complexes is further discussed below. Selected bond distances and angles are presented in Table 2.
 |
| | Fig. 1 The molecular structure of the [Mg(dmpu)5]2+ ion in 1 viewed along two directions to show how two pairs of bound dmpu molecules face each other. The atoms are shown with 50% probability ellipsoids. | |
 |
| | Fig. 2 Packing structure of regular Mg[(dmpu)5]2+ with iodide ions seen along the c axis. The atoms are shown with 50% probability ellipsoids, except iodides that are shown as spheres. | |
Table 2 Selected bond distances and angles in the crystal structures of 1–9
| Bond distance |
(Å) |
Bond angle |
(°) |
Bond angle |
(°) |
| [Mg(dmpu)5]I2 (1) |
| Mg–O1 |
2.0227(16) |
O1–Mg–O2 |
90.25(7) |
Mg–O1–C1 |
165.65(16) |
| Mg–O2 |
1.9907(17) |
O1–Mg–O3 |
88.23(7) |
Mg–O2–C7 |
133.41(14) |
| Mg–O3 |
1.9865(16) |
O1–Mg–O4 |
177.77(7) |
Mg–O3–C13 |
146.91(13) |
| Mg–O4 |
2.0302(15) |
O1–Mg–O5 |
90.47(7) |
Mg–O4–C19 |
152.08(14) |
| Mg–O5 |
1.9711(16) |
O2–Mg–O3 |
122.41(7) |
Mg–O5–C25 |
155.66(15) |
| O1–C1 |
1.254(3) |
O2–Mg–O4 |
91.12(7) |
|
|
| O2–C7 |
1.269(3) |
O2–Mg–O5 |
114.02(7) |
|
|
| O3–C13 |
1.258(3) |
O3–Mg–O4 |
89.55(7) |
|
|
| O4–C19 |
1.254(3) |
O3–Mg–O5 |
123.55(7) |
|
|
| O5–C25 |
1.257(3) |
O4–Mg–O5 |
90.58(6) |
|
|
| Mean d(Mg–O) = 2.0002 Å, d(O–C) = 1.258 Å, ∠Mg–O–C = 150.74° |
| |
| [Ca(dmpu)6]I2 (2) |
| Bond distance |
(Å) |
Bond angle |
(°) |
Bond angle |
(°) |
| Ca–O |
2.319(2) |
O–Ca–O |
180.00(11) |
Ca–O–C |
162.4(2) |
| O–C |
1.246(4) |
O–Ca–O |
89.73(9) |
|
|
| |
| [Ca(dmpu)6](ClO4)2 (3) |
| Bond distance |
(Å) |
Bond angle |
(°) |
Bond angle |
(°) |
| Ca–O |
2.3282(12) |
O–Ca–O |
180.00(7) |
Ca–O–C |
161.97(12) |
| O–C |
1.242(2) |
O–Ca–O |
89.77(5) |
|
|
| [Ca(dmpu)6](CF3SO3)2 (4) |
| Bond distance |
(Å) |
Bond angle |
(°) |
Bond angle |
(°) |
| Ca–O |
2.333(4) |
O–Ca–O |
180.0(2) |
Ca–O–C |
163.9(4) |
| O–C |
1.240(7) |
O–Ca–O |
89.68(14) |
|
|
| [Sr(dmpu)6](CF3SO3)2 (5) |
| Bond distance |
(Å) |
Bond angle |
(°) |
Bond angle |
(°) |
| Sr–O |
2.470(5) |
O–Sr–O |
180.0(4) |
Sr–O–C |
161.4(5) |
| O–C |
1.232(8) |
O–Sr–O |
89.06(18) |
|
|
| [Ba(dmpu)6](CF3SO3)2 (6) |
| Bond distance |
(Å) |
Bond angle |
(°) |
Bond angle |
(°) |
| Ba–O |
2.616(3) |
O–Ba–O |
180.00(11) |
Ba–O–C |
159.5(3) |
| O–C |
1.235(5) |
O–Ba–O |
88.79(11) |
|
|
| [Sc(dmpu)6]I3 (7) |
| Bond distance |
(Å) |
Bond angle |
(°) |
Bond angle |
(°) |
| Sc–O1 |
2.068(2) |
O1–Sc–O1 |
180.00(12) |
Sc–O1–C1 |
175.4(2) |
| Sc–O11 |
2.0735(19) |
O11–Sc–O11 |
180.00 |
Sc–O11–C11 |
168.9(2) |
| Sc–O21 |
2.0801(18) |
O21–Sc–O21 |
180.00 |
Sc–O21–C21 |
172.0(2) |
| O1–C1 |
1.265(4) |
O1–Sc–O11 |
89.89(8) |
|
|
| O11–C11 |
1.265(3) |
O1–Sc–O21 |
89.26(8) |
|
|
| O21–C21 |
1.270(3) |
O11–Sc–O21 |
88.65(7) |
|
|
| Mean d(Sc–O) = 2.074 Å, d(O–C) = 1.267 Å, ∠Sc–O–C = 172.1°, ∠O–Sc–O = 180.00, 89.65° |
| |
| [Pr(dmpu)6]I(I3)2 (8) |
| Bond distance |
(Å) |
Bond angle |
(°) |
Bond angle |
(°) |
| Pr–O |
2.346(2) |
O–Pr–O |
180.0(2) |
Pr–O–C |
159.2(2) |
| O–C |
1.266(4) |
O–Pr–O |
89.56(8) |
|
|
| I21–I23 |
2.9202(7) |
I21–I23–I22 |
180.00 |
|
|
| I22–I23 |
2.8961(7) |
|
|
|
|
| |
| [Co(dmpu)2Br2] (9) |
| Bond distance |
(Å) |
Bond angle |
(°) |
Bond angle |
(°) |
| Co–O1 |
1.969(2) |
O1–Co–O2 |
106.67(10) |
Co–O1–C1 |
124.9(2) |
| Co–O2 |
1.953(2) |
O1–Co–Br1 |
103.49(7) |
Co–O2–C2 |
131.0(2) |
| Co–Br1 |
2.3809(7) |
O1–Co–Br2 |
112.51(8) |
|
|
| Co–Br2 |
2.3773(7) |
O2–Co–Br1 |
110.99(8) |
|
|
| O1–C1 |
1.267(4) |
O2–Co–Br2 |
105.12(7) |
|
|
| O1–C2 |
1.266(4) |
Br1–Co–Br2 |
117.70(3) |
|
|
| Mean d(Co–O) = 1.961 Å, d(Co–Br) = 2.3791 Å, d(C–O) = 1.267 Å, ∠Co–O–C = 128.0°, ∠O–Co–Br = 108.0° |
Mononuclear five-coordinate magnesium complexes with oxygen donor ligands are unusual with only a couple of reported structures in the solid state. Magnesium forms a square-pyramidal complex with trimethylarsine-oxide, [Mg(OAs(C6H5)3)5](ClO4)2, with a mean Mg–O bond distance of 2.028 Å in the equatorial positions, but the bond distance in the axial position is significantly shorter, 1.918 Å, mean 2.006 Å.21 Magnesium forms a five-coordinate complex with tetrahydofuran, thf, in solid [Mg(thf)5][U7C96H108Cl6N12O6], where the thf molecules form a regular trigonal bipyramid around magnesium.22 The Mg–O bond distances in the trigonal base are 1.773 Å, which is surprisingly short, and the axial Mg–O bonds are 2.154 Å. The mean Mg–O bond distance, 1.925 Å, is significantly shorter than that of magnesium in five-coordination, vide infra. A third structure containing a homoleptic five-coordinate magnesium ion is [Mg(H2O)5]2(C20H22N10O12P2)·16H2O.23 However, the structural details of [Mg(H2O)5]2+ ions have not been reported. Moreover, four complexes with the composition [Mg(thf)3(phenolate/benzamide)2] have been reported. These structures have a slightly distorted square-pyramidal configuration with short Mg–O bond distances to the phenolate or benzamide group, ca. 1.90 Å, and much longer Mg–O bond distances to the thf molecules completing the square-plane, ca. 2.10 Å, while the axially bound thf molecule is at a slightly shorter Mg–O bond distance, ca. 2.07 Å.24–26 The mean Mg–O bond distance is 2.02 Å. The mean Mg–O bond distance, 2.00 Å, is independent of the trigonal bipyramidal or square-pyramidal configuration, giving an ionic radius of 0.66 Å for magnesium in five-coordination, assuming an oxygen radius of 1.34 Å. This is in perfect agreement with the ionic radius proposed by Shannon.27
Crystal structure of hexakis(N,N′-dimethylpropyleneurea)calcium iodide, 2
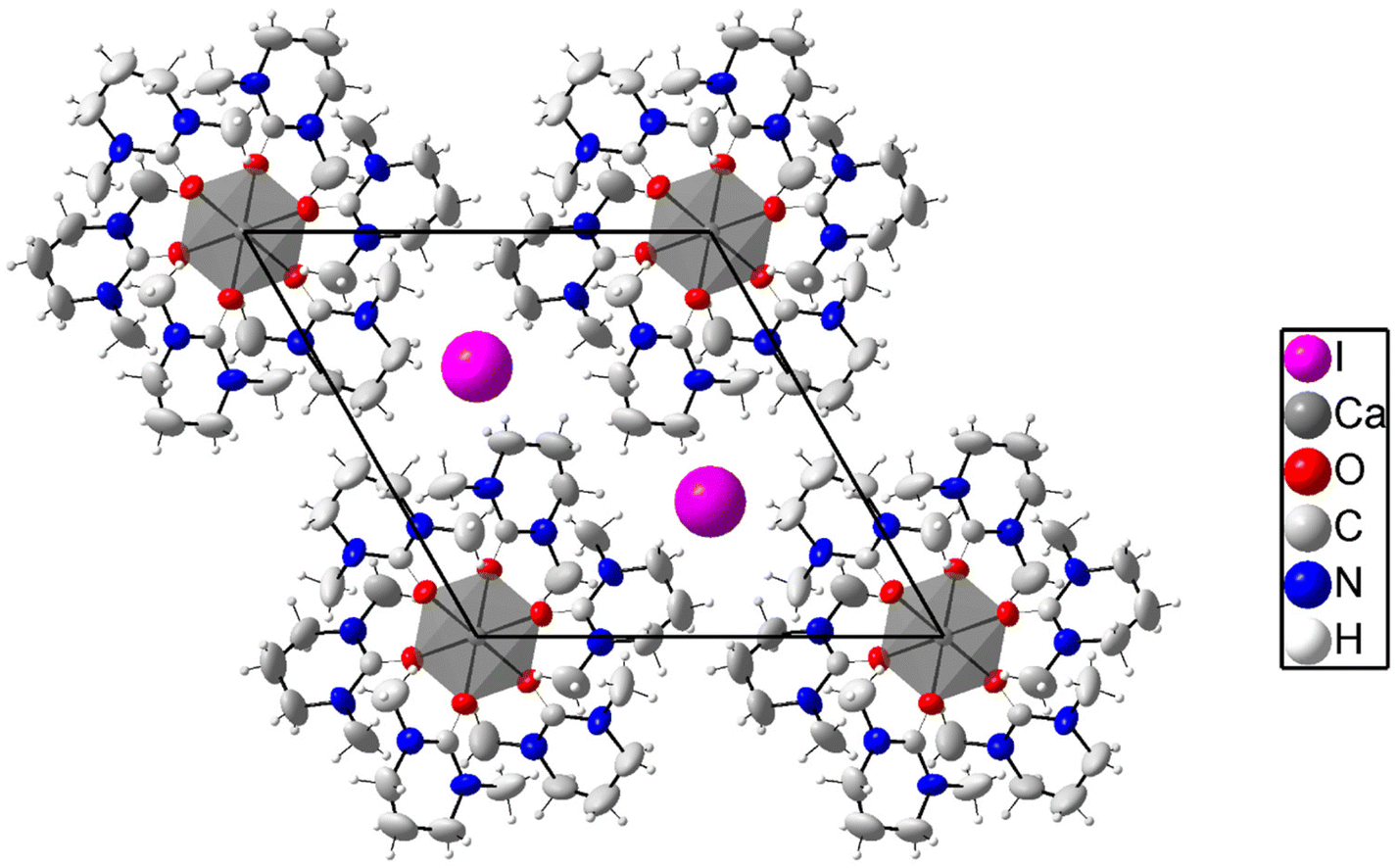
The calcium ion binds six oxygen atoms of dmpu in an octahedral geometry with all Ca–O bond distances being equal, 2.319(2) Å, and the O–Ca–O angles are 89.73(9) and 180.00(11)°. The Ca–O–C bond angle is 162.4(2)°. The [Ca(dmpu)6]2+ complex and the iodide ions are packed in a regular trigonal lattice without any signs of disorder. The packing of 2 is shown in Fig. 3. Selected bond distances and angles are listed in Table 2.
 |
| | Fig. 3 Packing structure of the regular [Ca(dmpu)6]2+ complex with iodide ions. The atoms are shown with 50% probability ellipsoids, except iodides that are shown as spheres. | |
In the dmpu molecules in 2, non-hydrogen atoms except the middle methylene carbon (C14) form a virtual plane with an average deviation of about 0.04 Å, Fig. 3. In turn, for these methylene carbons, the deviation from the virtual planes is approximately 0.60 Å, Fig. 3. Furthermore, these virtual planes are parallel to each other. The planes formed by three Ca(dmpu)2 fragments are almost perpendicular to each other, approximately 76°. The crowdedness in 2 is illustrated in Fig. 4 (left) where green dotted lines show the closest hydrogen in the twelve methyl groups. These Ca⋯H distances are in the range of 3.51–3.69 Å, with the sum of the van der Waals radii being 3.51 Å,19,20 Table S2 (ESI).† In Fig. 4 (right), only these hydrogens are shown for clarity. These hydrogens form a cage in the form of an icosahedron.
 |
| | Fig. 4 Left: the molecular structure of 2 with the twelve shortest Ca⋯H distances marked with dashed green lines, right: only the Ca⋯H distances are shown for clarity to highlight the high symmetry of the cage in the form of an icosahedron of these methyl hydrogens around the calcium ion. | |
The molecular structures of the [Ca(dmpu)6]2+ ion in [Ca(dmpu)6](ClO4)2 (3) and [Ca(dmpu)6](CF3SO3)2 (4) are very similar to that of 2. However, the perchlorate ions in 3 and the trifluoromethanesulfonate ions in 4 are severely distorted. As the structure of the [Ca(dmpu)6]2+ ion is almost identical without distortion in 2, 3 and 4, the description of the structures of 3 and 4 is only briefly reported in the ESI.† The packing of 3 and 4 is shown in Fig. S1 and S2 (ESI),† respectively.
Structure of the dmpu coordinated calcium ion in solution
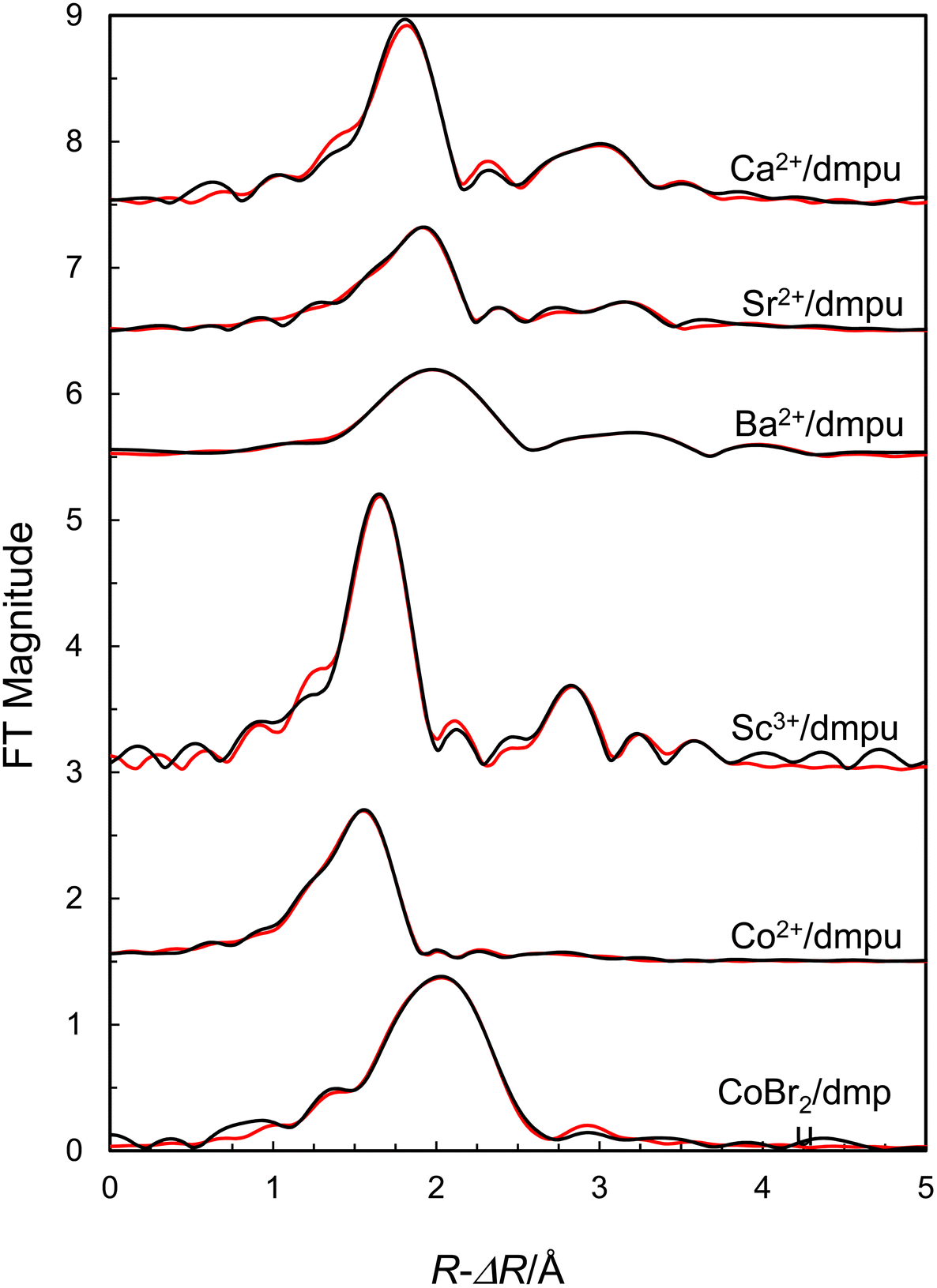
The EXAFS data of a 0.3 mol dm−3 dmpu solution of Ca(CF3SO3)2 reveal the same structure as in the solid state with six dmpu molecules being bound to calcium at 2.319(5) Å in an octahedral fashion, Table 3. Significant contributions from linear Ca–O–O and Ca–O–Ca–O multiple scattering further support a regular octahedral configuration around calcium. The Ca⋯C distance is 3.43 Å giving a mean Ca–O–C bond angle of 144(3)° assuming a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distance of 1.27 Å. The refined model parameters are summarized in Table 3, and the fits of the EXAFS raw data and the Fourier transform are shown in Fig. 5 and 6, respectively.
O bond distance of 1.27 Å. The refined model parameters are summarized in Table 3, and the fits of the EXAFS raw data and the Fourier transform are shown in Fig. 5 and 6, respectively.
 |
| | Fig. 5 Fit of raw EXAFS data of dmpu solutions of calcium trifluoromethanesulfonate (offset: 48), strontium trifluoromethanesulfonate (offset: 41), barium trifluoromethanesulfonate (offset: 34), scandium(III) trifluoromethanesulfonate (offset: 22), cobalt(II) trifluoromethanesulfonate (offset: 10) and cobalt(II) bromide (no offset). Black line – experimental data, red line – modelled function using the parameters in Table 3. | |
 |
| | Fig. 6 Fit of Fourier transforms of EXAFS data of dmpu solutions of calcium trifluoromethanesulfonate (offset: 7.5), strontium(II) trifluoromethanesulfonate (offset: 6.5), barium trifluoromethanesulfonate (offset: 5.5), scandium(III) trifluoromethanesulfonate (offset: 3.0), cobalt(II) trifluoromethanesulfonate (offset: 1.5) and cobalt(II) bromide (no offset). Black line – experimental data, red line – the modelled function using the parameters in Table 3. | |
Table 3 Mean bond distances, d/Å, Debye–Waller factors, σ2, number of distances, N, the threshold energy, E0/eV, and the amplitude reduction factor, S02, from EXAFS data of the studied dmpu solutions of scandium(III), calcium, strontium, barium and cobalt(II) trifluoromethanesulfonate and cobalt(II) bromide in the k range 2–13 Å−1 at ambient room temperature
| Solvent interaction |
N
|
D
|
σ
2
|
E
0
|
S
0
2
|
| Ca(CF3SO3)2 in dmpu solution |
| Ca–O |
6 |
2.319(2) |
0.0049(2) |
4058.4(3) |
0.88(2) |
| Ca⋯C |
6 |
3.43(2) |
0.008(2) |
|
|
| Ca–O–C |
12 |
3.510(8) |
0.009(1) |
|
|
| MS (CaO6) |
3 × 6 |
4.62(3) |
0.021(4) |
|
|
| Ca–O–C bond angle 144(3)° |
| |
| Sr(CF3SO3)2 in dmpu solution |
| Sr–O |
6 |
2.467(2) |
0.0069(2) |
16128.7(3) |
0.89(2) |
| Sr⋯C |
6 |
3.524(9) |
0.0087(9) |
|
|
| Sr–O–C |
12 |
3.615(10) |
0.0078(11) |
|
|
| MS (SrO6) |
3 × 6 |
4.94(2) |
0.018(2) |
|
|
| Sr–O–C bond angle 139(2)° |
| |
| Ba(CF3SO3)2 in dmpu solution |
| Ba–O |
6 |
2.628(4) |
0.0167(6) |
5270.5(4) |
1.23(5) |
| Ba⋯C |
6 |
3.646(4) |
0.0083(8) |
|
|
| Ba–O–C |
12 |
3.818(9) |
0.016(2) |
|
|
| MS (BaO6) |
3 × 6 |
5.12(3) |
0.037(6) |
|
|
| Ba–O–C bond angle 136(1)° |
| |
| Sc(CF3SO3)3 in dmpu solution |
| Sc–O |
6 |
2.091(2) |
0.0031(2) |
4520.5(3) |
0.87(2) |
| Sc⋯C |
6 |
3.335(5) |
0.0040(4) |
|
|
| Sc–O–C |
12 |
3.346(6) |
0.012(1) |
|
|
| MS (SrO6) |
3 × 6 |
4.182 |
0.0091(8) |
|
|
| Sc–O–C bond angle 165(3)° |
| |
| Co(CF3SO3)2 in dmpu solution |
| Co–O |
5 |
1.996(1) |
0.0072(1) |
7123.7(3) |
0.94(1) |
| Co⋯C |
5 |
2.993(4) |
0.0142(6) |
|
|
| Co–O–C |
10 |
3.158(8) |
0.020(2) |
|
|
| Co–O–C bond angle 131.5(5)° |
| |
| CoBr2 in dmpu solution |
| Co–O |
2 |
1.993(3) |
0.0025(1) |
7123.7(3) |
0.87(1) |
| Co–Br |
2 |
2.394(1) |
0.0035(1) |
|
|
| Co⋯C |
2 |
2.980(7) |
0.0038(9) |
|
|
| Co–O–C |
4 |
3.113(9) |
0.011(2) |
|
|
| Co–O–C bond angle 130.6(1.0)° |
Crystal structure of hexakis(N,N′-dimethylpropyleneurea)strontium trifluoromethanesulfonate, 5
The strontium ion binds six oxygen atoms of dmpu in a regular octahedral fashion with a Sr–O bond distance of 2.470(5) Å, and the O–Sr–O angles are 89.06(18) and 180.0(4)°. The Sr–O–C bond angle is 161.4(5)°, Table 2. The packing of 5 is shown in Fig. S3 (ESI).† The [Sr(dmpu)6]2+ complex is well ordered with regular octahedral coordination without any signs of disorder. The dmpu molecules are arranged in the very same way as in 2, with the slightly dented Sr(dmpu)2 entities perpendicular to each other. However, the large disorder of trifluoromethanesulfonate ions gives some hints of weak interactions between the “outside” of the dmpu ligands and the CF3SO3− ions. The Sr⋯H distances are in the range of 3.64–3.96 Å, Table S2 (ESI).†
EXAFS data of the dmpu coordinated strontium ion in solution
The EXAFS data of a 0.2 mol dm−3 dmpu solution of Sr(CF3SO3)2 reveal the same structure as that in the solid state with six dmpu molecules being bound to strontium at a mean Sr–O bond distance of 2.467(4) Å, cf. Table 3. Significant contributions from linear Sr–O–O and Sr–O–Sr–O multiple scattering further support a regular octahedral configuration around strontium. The Sr⋯C distance was refined to 3.524(9) Å, corresponding to a Sr–O–C bond angle of 139(2)° assuming a CO bond distance of 1.27 Å. The refined model parameters are summarized in Table 3, and the fit of the EXAFS raw data and the corresponding Fourier transform are given in Fig. 5 and 6, respectively.
Crystal structure of hexakis(N,N′-dimethylpropyleneurea)barium trifluoromethanesulfonate, 6
The barium ion in the homoleptic dmpu complex binds six oxygen atoms of dmpu in a regular octahedral fashion with a Ba–O bond distance of 2.616(3) Å, and the O–Ba–O angles are 88.79(11) and 180.00(11)°, and the mean Ba–O–C bond angle is 159.5(3)°, Table 2. The [Ba(dmpu)6]2+ complex is well ordered with regular octahedral coordination without disorder. The dmpu molecules are arranged in the very same way as in 2 and 5 with the slightly dented Ba(dmpu)2 entities perpendicular to each other. Ba⋯H distances are in the range of 3.78–4.02 Å, Table S2 (ESI).† As in 4 and 5, the trifluoromethanesulfonate ion is heavily disordered. The crystal structure of 6 showing the packing of molecules as well as the molecular structure of the [Ba(dmpu)6]2+ complex is shown in Fig. S4 (ESI).†
Structure of the dmpu coordinated barium ion in solution
The EXAFS data of a 0.2 mol dm−3 dmpu solution of Ba(CF3SO3)2 reveal the same structure as that in the solid state with six dmpu molecules being bound to barium at 2.628(8) Å, cf. Table 3. Significant contributions from Ba–O–O and Ba–O–Ba–O multiple scattering further support a regular octahedral configuration around the metal center. The Ba⋯C distance was refined to 3.646(4) Å, corresponding to a Ba–O–C bond angle of 136(1)° assuming a CO bond length of 1.27 Å. The refined model parameters are summarized in Table 3, and the fit of the EXAFS raw data and the corresponding Fourier transform are given in Fig. 5 and 6, respectively.
Crystal structure of hexakis(N,N′-dimethylpropyleneurea)scandium(III) iodide, 7
The crystal structure of 7 consists of discrete hexakis(N,N′-dimethylpropyleneurea)scandium(III) and iodide ions, Fig. S5 (ESI).† The scandium ion coordinates six oxygen atoms in octahedral configuration with Sc–O bond distances in the range of 2.068(2)–2.081(2) Å, O–Sc–O bond angles in the range of 88.67(7)–89.88(8)° and 180.00(12)°, and Sc–O–C angles in the range of 168.8(2)–175.5(2)°, Table 2. The Sc–O bond length is the shortest one observed in an octahedrally dmpu coordinated metal ion, Table 4, accompanied by the largest M–O–C angle observed in any metal dmpu complex reported so far. The dmpu molecules are arranged in the very same way as in 2, 5 and 6 with the slightly dented Sc(dmpu)2 entities perpendicular to each other.
Table 4 Overview of mean M–O bond distances (Å) and mean M–O–C bond angles (degrees) in N,N′-dimethylpropyleneurea coordinated metal ions in the solid state
| Metal dmpu complex |
d(M–O)dmpu |
d(CO) |
M–O–C bond angle |
Coordination figure |
Ref. |
| [Mg(dmpu)5]I2 |
2.000 |
1.258 |
150.8 |
Trigonal bipyramid |
This work |
| [Ca(dmpu)6]I2 |
2.319(2) |
1.246(4) |
162.4(2) |
Octahedron |
This work |
| [Ca(dmpu)6](ClO4)2 |
2.328(1) |
1.242(2) |
162.0(2) |
Octahedron |
This work |
| [Ca(dmpu)6](CF3SO3)2 |
2.333(4) |
1.240(7) |
163.7(4) |
Octahedron |
This work |
| [Sr(dmpu)6](CF3SO3)2 |
2.470(5) |
1.232(8) |
161.4(5) |
Octahedron |
This work |
| [Ba(dmpu)6](CF3SO3)2 |
2.617(4) |
1.234(7) |
160.1(4) |
Octahedron |
This work |
| [Sc(dmpu)6]I3 |
2.074 |
1.267 |
172.1 |
Octahedron |
This work |
| [Y(dmpu)6]I3 |
2.219 |
1.268 |
169.18 |
Octahedron |
29
|
| [Sm(dmpu)6]I2 |
2.472 |
2.472 |
151.41 |
Octahedron |
30
|
| [Sm(dmpu)6](CF3SO3)3 |
|
|
|
Octahedron |
31
|
| [La(dmpu)6]I3 |
2.368 |
1.288 |
160.25 |
Octahedron |
16
|
| [Ce(dmpu)6]I3 |
2.440 |
2.44 |
|
Octahedron |
16
|
| [Pr(dmpu)6]I3 |
2.343 |
1.260 |
160.35 |
Octahedron |
16
|
| [Pr(dmpu)6]I(I3)2 |
2.346(2) |
1.266(4) |
159.2(2) |
Octahedron |
This work |
| [Nd(dmpu)6]I3 |
2.319 |
1.281 |
1.281 |
Octahedron |
16
|
| [Sm(dmpu)6]I3 |
|
|
|
Octahedron |
16
|
| [Gd(dmpu)6]I3 |
2.259 |
1.269 |
165.21 |
Octahedron |
16
|
| [Tb(dmpu)6]I3 |
2.247 |
1.268 |
164.87 |
Octahedron |
16
|
| [Er(dmpu)6]I3 |
2.216 |
1.254 |
165.36 |
Octahedron |
16
|
| [Yb(dmpu)6]I3 |
2.191 |
1.268 |
164.82 |
Octahedron |
16
|
| [Yb(dmpu)6](N(SO2CF3)2)3 |
2.202 |
|
165.5 |
Octahedron |
32
|
| [Lu(dmpu)6]I3 |
2.181 |
1.275 |
169.4 |
Octahedron |
16
|
| [Cu(dmpu)4](CF3SO3)2 |
1.901 |
1.256 |
128.4 |
Square-planar |
12
|
| [Zn(dmpu)4](CF3SO3)2 |
1.912 |
1.271 |
136.5 |
Tetrahedron |
33
|
| [Cd(dmpu)6](CF3SO3)2 |
2.260 |
1.266 |
158.0 |
Octahedron |
33
|
| [Pb(dmpu)6](CF3SO3)2 |
2.520 |
1.248 |
154.93 |
Octahedron |
34
|
| [Bi(dmpu)6](CF3SO3)2 |
2.323 |
1.259 |
148.55 |
Octahedron |
35
|
| [Bi(dmpu)6][Bi3I12] |
2.311 |
1.279 |
155.76 |
Octahedron |
36
|
| [MnBr2(dmpu)2] |
2.025 |
1.252 |
157.7 |
Distorted tetrahedron |
9
|
| [FeBr3(dmpu)2] |
1.981 |
1.294 |
137.00 |
Trigonal bipyramid |
13
|
| [FeBr3(dmpu)2] |
1.981 |
1.294 |
136.98 |
Trigonal bipyramid |
37
|
| [CoBr2(dmpu)2] |
1.938(6) |
1.261(10) |
125.8 |
Distorted tetrahedron |
This work |
| [CdI2(dmpu)2] |
2.208 |
1.293 |
125.39 |
Distorted tetrahedron |
14
|
| [NiBr2(dmpu)2] |
1.948 |
1.277 |
125.90 |
Distorted tetrahedron |
10
|
| [InBr3(dmpu)2] |
2.202 |
1.293 |
133.4 |
Trigonal bipyramid |
38
|
| [TlBr3(dmpu)2] |
2.352 |
1.256 |
129.7 |
Trigonal bipyramid |
39
|
| [VO(dmpu)4](CF3SO3)2 |
1.967 |
1.277 |
135.81 |
Distorted pyramid |
40
|
| [UO2(dmpu)2(NO2)2] |
2.363 |
1.271 |
140.0 |
Hexagonal bipyramid |
41
|
| [UO2(dmpu)5]I2·dmpu |
2.356 |
1.270 |
148.44 |
Pentagonal bipyramid |
41
|
| [UO2Cl2(dmpu)2] |
2.287 |
1.275 |
153.1 |
Distorted octahedron |
42
|
| [Al(OH)2(dmpu)2] |
1.809 |
1.282 |
148.0 |
Distorted square pyramid |
43
|
Structure of the dmpu coordinated scandium ion in solution
The EXAFS data of a 0.3 mol dm−3 dmpu solution of Sc(CF3SO3)3 reveal the same structure as in the solid state with six dmpu molecules being bound to scandium at 2.091(4) Å, cf. Table 3. Significant contributions from Sc–O–O and Sc–O–Sc–O multiple scattering further support a regular octahedral configuration around the scandium ion. The Sc⋯C distance was refined to 3.335(5) Å, corresponding to a nearly linear Sc–O–C configuration, 167(3)°, which is in full agreement with the observation in the solid state, vide supra. The refined model parameters are summarized in Table 3, the fit of the raw data is shown in Fig. 5, and the Fourier transform is shown in Fig. 6.
Crystal structure of hexakis(N,N′-dimethylpropyleneurea)praseodymium iodide di(triodide), 8
The crystal structure of 8 consists of isolated hexakis(N,N′-dimethylpropyleneurea)praseodymium, iodide, and triiodide ions, as shown in Fig. 7, S6a and S6b (different view angles) (ESI).† The praseodymium ion binds six oxygen atoms of dmpu in an octahedral geometry with a mean Pr–O bond distance of 2.346(2) Å, and the O–Pr–O angles are 89.56(8) and 180.0(2) °, and the Pr–O–C angle is 159.2(2)°. The triiodide ions are linear with I–I bond distances of 2.8961(7) and 2.9202(7) Å, mean 2.9082 Å, which is in close agreement with the reported structures containing triiodide ions. The orientation of the dmpu molecules is the same as in the other octahedral dmpu complexes reported in this study. The packing of 8 can be described as chains of [Pr(dmpu)6]3+ complexes with iodide ions in between, both centered on the three-fold axis passing through the origin. The triiodide ions are situated between these chains on pairs of the other three-fold axis. Selected bond distances and angles are presented in Table 2, and the packing and the molecular structure of 8 are shown in Fig. 7, S6a, and S6b (ESI).†
 |
| | Fig. 7 Packing structure of [Pr(dmpu)6]3+ with a mix of iodide and triiodide ions viewed along the a axis in a hexagonal representation of the structure in the space group P (No. 147). The atoms are shown with 50% probability ellipsoids except for the iodine atoms that are shown as spheres. | |
The structure of 8 is in very close agreement with the corresponding iodide salt, [Pr(dmpu)6]I3·dmpu, in which the mean Pr–O bond distance is 2.343 Å, and the Pr–O–C angle is 159.3°.16 The Pr⋯H distances are in the range of 3.81–3.83 Å, Table S2 (ESI),† and they are arranged in the same way as in 2 and 5–7. However, it has been shown that in solution the dmpu-coordinated praseodymium ion is seven-coordinate, with a mean Pr–O bond distance of 2.420 Å, as well as all the other dmpu coordinated lanthanoid(III) ions in dmpu solution, except lutetium(III) which is six-coordinate.16
Structure of the dmpu coordinated cobalt(II) ion in solution
It has not been possible to crystallize any dmpu coordinated cobalt(II) trifluoromethanesulfonate salt as only very viscous solutions are obtained on cooling saturated dmpu solutions. The EXAFS data of the dmpu coordinated cobalt(II) ion in dmpu solution reveal a mean Co–O distance of 1.996 Å, Table 3. The expected Co–O bond distances in 4-, 5- and 6-coordination from the ionic radii given by Shannon27 and the oxygen radius in dmpu, 1.34 Å,16 are 1.92, 2.01, and 2.085 Å, respectively. The obtained Co–O bond distance in the dmpu coordinated cobalt(II) ion clearly indicates that the cobalt(II) ion is 5-coordinate. The Co⋯C single scattering and Co–O–C three-leg scattering pathways of 2.993 and 3.158 Å, respectively, give a Co–O–C bond angle of 131.5(1.0)° assuming a CO bond distance of 1.27 Å in dmpu. The structural parameters used in the modelling are summarized in Table 3. The fit of the raw EXAFS data and the Fourier transforms are shown in Fig. 5 and 6, respectively.
Crystal structure of bisbromobis(N,N′-dimethylpropyleneurea)cobalt(II), 9
The crystal structure of bis(N,N′-dimethylpropyleneurea)dibromocobalt(II) is shown in Fig. 8, S7a and S7b (ESI).† The cobalt(II) ion, binding two dmpu molecules and two bromide ions, has an approximately tetrahedral geometry with the Br1–Co–Br2 and O1–Co–O2 angles being 117.68(5) and 106.60(10)°, respectively. The Co–Br distances are 2.3776(7) and 2.3817(6) Å, whereas the Co–O distances are 1.954(2) and 1.968(2) Å. The Co–O–C angles are 125.0(7) and 131.1(7)°, which are in good agreement with the angles in the bis(N,N′-dimethylpropyleneurea)dibromonickel(II) complex.10 Selected bond distances and angles are listed in Table 2.
 |
| | Fig. 8 Molecular structure of the [CoBr2(dmpu)2] complex in 9 with atoms shown as 50% probability ellipsoids. | |
One of the dmpu rings in the [Co(dmpu)2Br2] complex is slightly disordered, as can be seen in Fig. 8. This is related only to one of the rings, which means that most probably it is not related to extra symmetry elements. The packing of the structure of 9 is shown along the a-axis in Fig. S7a† and along the b-axis in Fig. S7b (ESI).† It can be clearly seen in Figs. S7a and S7b (ESI)† that the packing is governed by interactions between the parallel dmpu rings. Most probably the flipping of the envelope carbon, the disordered one, has small effects on the actual packing.
Structure of the dmpu coordinated cobalt(II) bromide complex in solution
The EXAFS data of a 0.2 mol dm−3 dmpu solution of CoBr2 reveal similar structural parameters as those in solid 9. Co–Br and Co–O bond distances were refined to 2.394(2) and 1.993(6) Å, respectively, which are reasonably close to the mean bond distances in 9, that are 2.379 and 1.961 Å, respectively, vide supra. The slightly longer Co–O and Co–Br bond distances may indicate a 5-coordinated complex, [CoBr2(dmpu)3], or a mixture of [Co(dmpu)5]2+ and [CoBr4]2− complexes. However, a quite intense pre-edge on the absorption edge strongly indicates the presence of a tetrahedral complex,28 Fig. S8 (ESI).† By introducing multiple scattering in the model where a regular [CoBr4]2− complex is present, the fit becomes significantly worse. Therefore, the dominating complex is a slightly distorted tetrahedral [CoBr2(dmpu)2] complex similar to the one in the solid state. The Co⋯C distance was refined to 2.980(15) Å, corresponding to a Co–O–C bond angle of 131(2)° assuming a CO bond length of 1.27 Å. The refined model parameters are summarized in Table 3, and the fit of the EXAFS raw data and the corresponding Fourier transform are given in Fig. 5 and 6.
Coordination chemistry effects of the steric demand of dmpu
The smallest metal ion able to coordinate six dmpu molecules is scandium(III), with a mean Sc–O bond distance of 2.074 and 2.091 Å in the solid state and dmpu solution, respectively. Metal ions with the same or a slightly larger ionic radius than scandium(III), but with lower charge, as the divalent transition metal ions and magnesium(II), form five- or four-coordinate dmpu complexes, Table 4 (solids) and Table 5 (solutions). Of these, we have only been able to crystallize the homoleptic magnesium dmpu complex. The dmpu coordinated lanthanoid(III) ions are seven-coordinate in dmpu solution, except the smallest one, lutetium, but only the regular octahedral dmpu complexes are formed in the solid state. The structure of the square-planar dmpu coordinated copper(II) complex shows clearly that one methyl group of two coordinated dmpu molecules prevent coordination in the axial positions, Fig. S9,† and a combined longer M–O bond distance and a larger M–O–C bond angle are required for the formation of a six-coordinate dmpu complex.
Table 5 Overview of M–O bond distances (Å) and M–O–C bond angles (degrees) in N,N′-dimethylpropyleneurea coordinated metal ions in dmpu solution
| Metal dmpu complex |
d(M–O) |
M–O–C |
Ref. |
| [Ca(dmpu)6]2+/dmpu |
2.319 |
160 |
This work |
| [Sr(dmpu)6]2+/dmpu |
2.467 |
pu154 |
This work |
| [Ba(dmpu)6]2+/dmpu |
2.626 |
136 |
This work |
| [Sc(dmpu)6]3+/dmpu |
2.090 |
170 |
This work |
| [Y(dmpu)6]3+/dmpu |
2.242 |
133 |
29
|
| [La(dmpu)7]3+/dmpu |
2.454 |
160 |
16
|
| [La(dmpu)7]3+/dmpu |
2.454 |
160 |
44
|
| [Ce(dmpu)7]3+/dmpu |
2.438 |
160 |
16
|
| [Pr(dmpu)7]3+/dmpu |
2.420 |
160 |
16
|
| [Nd(dmpu)7]3+/dmpu |
2.408 |
160 |
16
|
| [Sm(dmpu)7]3+/dmpu |
2.380 |
160 |
16
|
| [Gd(dmpu)7]3+/dmpu |
2.345 |
160 |
16
|
| [Tb(dmpu)7]3+/dmpu |
2.332 |
160 |
16
|
| [Dy(dmpu)7]3+/dmpu |
2.324 |
159 |
16
|
| [Er(dmpu)7]3+/dmpu |
2.300 |
160 |
16
|
| [Ho(dmpu)7]3+/dmpu |
2.311 |
158 |
16
|
| [Tm(dmpu)7]3+/dmpu |
2.284 |
160 |
16
|
| [Yb(dmpu)7]3+/dmpu |
2.278 |
159 |
16
|
| [Lu(dmpu)6]3+/dmpu |
2.178 |
165 |
16
|
| [Th(dmpu)8]4+/dmpu |
2.404 |
157 |
45
|
| [Mn(dmpu)5]2+/dmpu |
2.087 |
137 |
9
|
| [Fe(dmpu)5]2+/dmpu |
2.048 |
132 |
13
|
| [Fe(dmpu)5]3+/dmpu |
2.001 |
125 |
13
|
| [Co(dmpu)5]2+/dmpu |
1.996 |
132 |
This work |
| [Ni(dmpu)5]2+/dmpu |
2.004 |
128 |
10
|
| [Cu(dmpu)4]2+/dmpu |
1.939 |
124 |
12
|
| [Ag(dmpu)2+2]+/dmpu |
2.313 + 2.537 |
133 + 128 |
46
|
| [Zn(dmpu)5]2+/dmpu |
2.01 |
137 |
34
|
| [Cd(dmpu)6]2+/dmpu |
2.259 |
159 |
34
|
| [Cd(dmpu)6]2+/dmpu |
2.26 |
146 |
47
|
| [Ga(dmpu)5]3+/dmpu |
1.924 |
146 |
38
|
| [In(dmpu)6]3+/dmpu |
2.26 |
134 |
38
|
| [Pb(dmpu)6]2+/dmpu |
2.488 |
137 |
34
|
| [Bi(dmpu)6]2+/dmpu |
2.322 |
150 |
35
|
| [MnBr2(dmpu)3]/dmpu |
2.076 |
158 |
9
|
| [FeBr3(dmpu)2]/dmpu |
1.986 |
131 |
13
|
| [CoBr2(dmpu)2]/dmpu |
1.993 |
131 |
This work |
The flexibility of the M–O–C bond angle to compensate for the steric demand
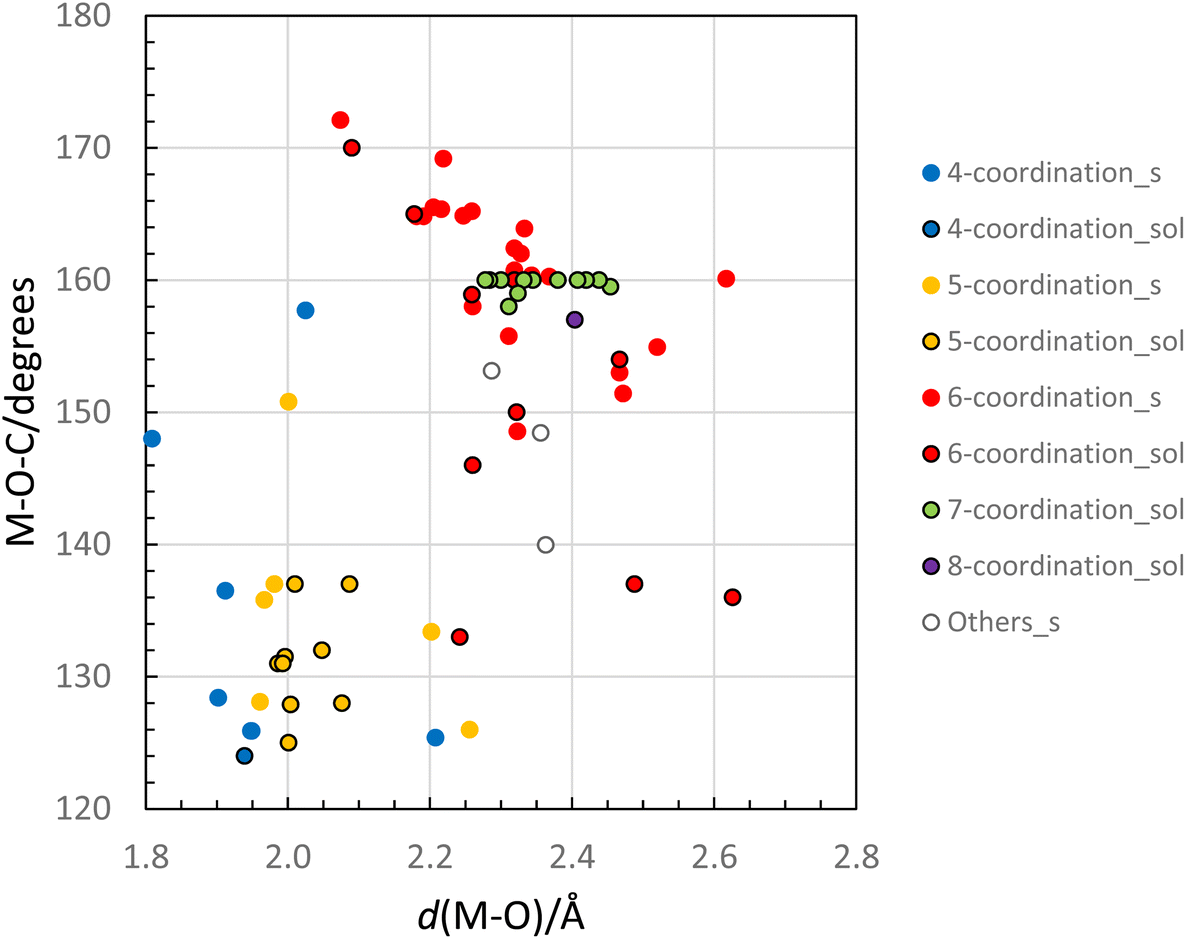
The M–O–C bond angle in homoleptic metal dmpu complexes is very flexible with the ability to decrease the steric demand during coordination, thereby increasing the possibility for higher coordination numbers. The M–O–C bond angle without any steric constraints is ca. 125° as observed in dmpu coordinated metal and mixed halide/dmpu complexes, where the coordination number is four or five allowing the dmpu molecules to be sufficiently far from each other and not interfere, Tables 4 and 5, and Fig. 9. However, in complexes with the coordination number six, the dmpu molecules will come sufficiently close to interfere and to minimize the interference the M–O–C angle becomes increasingly larger with decreasing M–O bond distance, Table 4 and Fig. 9. The smallest M–O–C angles in hexakis(dmpu)metal ion complexes are close to 140° in e.g. the dmpu coordinated barium ion, while the smallest metal ion able to form an octahedral metal dmpu complex, scandium(III), has a mean Sc–O–C bond angle of 172°. There is a correlation between the M–O bond distance and the M–O–C bond angle for octahedral dmpu coordinated metal ions, Fig. 9. A similar correlation is also present for six- and seven-coordinate dmpu coordinated metal ions in dmpu solution, Fig. 9. There is also a weak correlation for four- and five-coordinate complexes and only the smallest dmpu coordinated metal ions, aluminum ([Al(dmpu)2(OH)2]) and magnesium ([Mg(dmpu)5]I2) exhibit M–O–C bond angles significantly larger than 135°. An exception from this pattern is solid [MnBr2(dmpu)2] with an Mn–O–C angle of 158° in spite of no steric constraints.9 The M–O–C bond angle seems to be a compromise of a coordination number as large as possible and an M–O–C bond angle where the bond angle at the same time strives to be as small as possible seen in the diagonal pattern in Fig. 9.
 |
| | Fig. 9 Relationship between the M–O bond distance in dmpu coordinated metal complexes and the corresponding M–O–C bond angle. Data from the solid state are denoted with a filled circle, and data from the solution with a circle with a black border. Blue, yellow, red, green, and purple-filled circles represent complexes with the 4-, 5-, 6-, 7- and 8-coordinate metal ion, respectively. The numerical values in this figure are presented in Tables 4 and 5. | |
Physico-chemical properties of metal ions in space-demanding solvents
Complex formation studies with halide ions of the manganese(II)- and nickel(II)-bromide systems in dmpu,9,10 and the manganese(II)-chloride system in hmpa,11 where dmpu coordinated metal ions are five-coordinate, show a significantly stronger complex formation ability than in water and other oxygen-donor solvents where the metal ion is six-coordinate in an octahedral fashion, Table 6. The solvation thermodynamics of metal and halide ions are similar in dimethylsulfoxide and dmpu,52 and can hardly be the reason for the significantly stronger complex formation in dmpu. High symmetry of solvated metal ions in solution seems to be a stabilizing factor. However, if the metal ion has a lower symmetry, as e.g. in five-coordinated complexes, it seems to become significantly more reactive as can be seen in the complex formation ability.
Table 6 Stability constants (logK1 and logK2) of the manganese(II) and nickel(II)-bromide systems in dmpu, hmpa, water and dimethylsulfoxide
| Solvent |
Dmpu |
Hmpa |
Dmso |
Water |
| LogK1 |
LogK2 |
LogK1 |
LogK2 |
LogK1 |
LogK1 |
LogK2 |
|
Ref. 9.
Ref. 11.
Ref. 48.
Ref. 49.
Ref. 50.
Ref. 10.
Ref. 51.
|
| Mn2+–Br− |
3.36a |
2.63a |
4.1b |
2.4a |
|
0.27c |
0.26c |
| Mn2+–Cl− |
|
|
5.8b |
4.4b |
2.20d |
0.14e |
|
| Ni2+–Br− |
3.31f |
2.55f |
|
|
|
−0.3g |
|
Conclusions
The crystal structures of eight homoleptic dmpu metal complexes and the [CoBr2(dmpu)2] complex have been described. All reported structures of metal ions and metal halide complexes coordinating dmpu in both the solid state and dmpu solution are summarized with an emphasis to study the relationship between the M–O–C bond angle and crowdedness around the metal ion. The crowdedness can be seen in M⋯H distances that are close to the sum of the van der Waals radii of the metal and hydrogen, Fig. 4 and Table S2 (ESI).† The M–O–C bond angle is unusually flexible with angles in the range of 125–170° depending on the coordination figure and M–O bond distance. In complexes without any steric crowdedness, the M–O–C bond angle is ca. 125°, Tables 4 and 5, as also found in other urea and amide complexes.53 In octahedral complexes in the solid state, also for the largest metal ions such as barium, bond angles of least 140° are observed, while in solution the M–O–C bond angles tend to be slightly smaller. For scandium(III), the smallest ion binding six dmpu molecules in an octahedral fashion, the Sc–O–C bond angle is ca. 170°. This shows that the dmpu molecule has an unusual ability to increase the M–O–C bond angle to counteract the crowdedness and facilitate as large coordination numbers as possible. However, in the solid state, the highly symmetric complexes seem to be favored due to more favorable lattice constants as seen for the dmpu coordinated lanthanoid(III) ions, which are six-coordinate octahedra in the solid state, while they are seven-coordinate in dmpu solution, except for the smallest one, lutetium.16
Author contributions
Lundberg prepared compounds 1, 2, 3, 5, 6, 8 and 9 and solved their crystal structures together with Eriksson. Lindqvist-Reis prepared compounds 4 and 7 and determined the preliminary crystal structures of these compounds. Łyczko solved the crystal structure of 1. Lars Eriksson determined the final structures of compounds 2–9 and the plotted all reported crystal structures. Persson was responsible for project administration, conceptualization, EXAFS data collection and analysis, and for the final draft and revision of the manuscript. All authors have contributed to the writing of the first drafts and contributed to the revision of the manuscript.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
Financial support from the Swedish Research Council is gratefully acknowledged. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. Parts of this research were carried out at beamline I811, MAX-Lab synchrotron radiation source, Lund University, Sweden. Funding for the beamline I811 project was kindly provided by the Swedish Research Council and Knut och Alice Wallenbergs Stiftelse.
References
-
J. A. Riddick and W. B. Bunger, Techniques of Chemistry, Organic Solvents, Wiley-Interscience, New York, 1970, vol. II Search PubMed.
-
Y. Marcus, The Properties of Solvents, John Wiley & Sons, Chichester, 1998, ISBN: 978-0-471-98369-9 Search PubMed.
- T. Mukhopadhyay and D. Seebach, Substitution of HMPT by the cyclic urea DMPU as a cosolvent for highly reactive nucleophiles and bases, Helv. Chim. Acta, 1982, 65, 385–391 CrossRef CAS.
- H. Sukari and F. Kondo, Chemistry of organosilicon compounds: LXXX. Useful modifications in the preparation of trimethylsilylsodium and trimethylsilyl-potassium, J. Organomet. Chem., 1975, 92, C46–C48 CrossRef.
- B. J. Barker, J. Rosenfarb and J. A. Caruso, Harnstoffe als Lösungsmittel in der chemischen Forschung, Angew. Chem., 1979, 91, 560–564 CrossRef CAS.
- E. J. C. Lien and W. D. Kumler, Dipole moments and pharmacological activity of cyclic ureas, cyclic thioureas, and the N,N’-dimethylated compounds, J. Med. Chem., 1968, 11, 214–219 CrossRef CAS PubMed.
-
A. K. Beck, D. Seebach and G. Nikonov, Encyclopedia of Reagents for Organic Synthesis, Chap, in: N,N′-Dimethylpropyleneurea, 2014. ISBN: 0-471-93623-5, 9780470842898 Search PubMed.
-
C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003. ISBN: 3-527-30618-8 Search PubMed.
- H. Konieczna, D. Lundberg and I. Persson, Solvation and Coordination Chemistry of Manganese(II) Ion in Solvents with Solvation Properties. A Transfer Thermodynamic, Complex Formation, EXAFS Spectroscopic and Crystallographic Study, Polyhedron, 2021, 195, 114961 CrossRef CAS.
- D. Bobicz, O. Kristiansson and I. Persson, Reactivity of Five- and Six-Coordinated Solvates. A Complex Formation and Crystallographic Study of the Nickel(II) Bromide and Iodide Systems in Dimethylsulfoxide and N,N′-Dimethylpropyleneurea, J. Chem. Soc., Dalton Trans., 2002, 4201–4205 RSC.
- K. Ozutsumi, Y. Abe, R. Takahashi and S.-I. Ishiguro, Chloro and Bromo Complexation of the Manganese(II) Ion and Solvation Structure of the Manganese(II), Iron(II), Cobalt(II), Nickel(II), Copper(II), and Zinc(II) Ions in Hexamethylphosphoric Triamide, J. Phys. Chem., 1994, 98, 9894–9899 CrossRef CAS.
- I. Persson, D. Lundberg, É. G. Bajnoczi, K. Klementiev, J. Just and K. G. V. Sigfridsson Clauss, EXAFS study on the coordination chemistry of the solvated copper(II) ion in a series of oxygen donor solvents, Inorg. Chem., 2020, 59, 9538–9550 CrossRef CAS PubMed.
- D. Lundberg, A.-S. Ullström, P. D'Angelo, D. Warminska and I. Persson, On the Complex Formation of Iron(III) Bromide in the Space-Demanding Solvent N,N′-dimethylpropyleneurea and the Structure of Trisbromoiron(III) Complex in Solution and Crystalline State, Inorg. Chim. Acta, 2007, 360, 2744–2750 CrossRef CAS.
- D. Lundberg and L. Eriksson, Bis[1,3-dimethyl-3,4,5,6-tetrahydropyrimidin-2(1H)-one-κO]diiodocadmium(II), Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m400–m401 CrossRef CAS.
- K. Starke, The preparation of anhydrous metal compounds by dehydration with 2,2-dimethoxypropane, J. Inorg. Nucl. Chem., 1959, 11, 77–78 CrossRef CAS.
- D. Lundberg, I. Persson, L. Eriksson, P. D'Angelo and S. De Panfilis, Structural Study of the N,N′-Dimethylpropyleneurea Solvated Lanthanoid(III) Ions in Solution and Solid State with an Analysis of the Ionic Radii of Lanthanoid(III) Ions, Inorg. Chem., 2010, 49, 4420–4432 CrossRef CAS PubMed . and references therein.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
-
K. Brandenburg, Diamond ver. 2.1e, Crystal Impact GbR, Bonn, Germany, 2001 Search PubMed.
- R. S. Rowland and R. Taylor, Intermolecular Nonbonded Contact Distances in Organic Crystal Structures: Comparisonwith Distances Expected from van der Waals Radii, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, Consistent van der Waals Radii for the Whole Main Group, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- Y. S. Ng, G. A. Rodley and W. T. Robinson, Crystal structures of the Pentakis(trimethylarsine oxide)metal(II) perchlorates [Ni(Me3AsO)5](ClO4)2 and [Mg(Me3AsO)5](ClO4)2, Inorg. Chem., 1976, 15, 303–309 CrossRef CAS.
- J.-C. Berthet, P. Thuery and M. Ephritikhine, Polyimido Uranium(IV) Clusters: Imidometalates with an M7(μ3-N)6(μ2-N)6 Core Analogous to the Anderson-Type Polyoxometalate Motif, Angew. Chem., Int. Ed., 2008, 47, 5586–5589 CrossRef CAS PubMed.
- C. A. Frederick, M. Coll, G. A. van der Marel, J. H. van Boom and A. H.-J. Wang, Molecular structure of cyclic deoxydiadenylic acid at atomic resolution, Biochemistry, 1988, 27, 8350–8361 CrossRef CAS PubMed.
- C. A. Zechmann, T. J. Boyle, M. A. Rodriguez and R. A. Kemp, Solvent influences on the molecular aggregation of magnesium aryloxides: synthesis, crystal structure, and solution characterization of Mg(OAr)2(L)3 (OAr=DMP, DIP, TCP) and [Mg(DIP)2]3, Polyhedron, 2000, 19, 2557–2564 CrossRef CAS.
- H. W. Roesky, M. Scholz and M. Noltemeyer, Über Reaktionen des 2,4,6-Tris(trifluormethyl)phenols mit Verbindungen von Hauptgruppen- und Nebengruppen-Elementen (Li, Na, Mg, Ca, Ba, Ge, Sn und Ti, W, Mn, Cd), Chem. Ber., 1990, 123, 2303–2309 CrossRef CAS.
- T. E. Boddie, S. H. Carpenter, T. M. Baker, J. C. DeMuth, G. Cera, W. W. Brennessel, L. Ackermann and M. L. Neidig, Identification and Reactivity of Cyclometalated Iron(II) Intermediates in Triazole-Directed Iron-Catalyzed C–H Activation, J. Am. Chem. Soc., 2019, 141, 12338–12345 CrossRef CAS PubMed.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalocgenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- M. Uchikoshi and K. Shinoda, Determination of structures of cobalt(II)-chloro complexes in hydrochloric acid solutions by X-ray absorption spectroscopy at 298 K, Struct. Chem., 2019, 30, 945–954 CrossRef CAS.
- P. Lindqvist-Reis, J. Näslund, I. Persson and M. Sandström, Structure of the solvated yttrium(III) ion in the oxygen donor solvents dimethyl sulfoxide, N,N-dimethylformamide and N,N’-dimethylpropyleneurea and crystal structures of Y(OSMe2)8I3 and Y(OCN2Me2(CH2)3)6I3, J. Chem. Soc., Dalton Trans., 2000, 2703–2710 RSC.
- M. Nishiura, K. Katagiri and T. Imamoto, Isolation, X-ray Structural Characterization, and Reactivities of Diiodosamarium(II) Complexes Bearing Amide Compounds as Ligands, Bull. Chem. Soc. Jpn., 2001, 74, 1417–1424 CrossRef CAS.
- M. Nishiura, K. Katagiri and T. Imamoto, Isolation, X-Ray Structural Analyses of Rare Earth Trifluoromethanesulfonate Complexes Bearing Urea Derivatives, Bull. Chem. Soc. Jpn., 1999, 72, 1793–1801 CrossRef CAS.
- K. Mikami, O. Kotera, Y. Motoyama and M. Tanaka, Synthesis, structure and high catalytic activity in the Diels–Alder reaction of ytterbium(III) and yttrium(III) bis(trifluoromethanesulfonyl)amide complexes, Inorg. Chem. Commun., 1998, 1, 10–11 CrossRef CAS.
- D. Lundberg, L. Eriksson, P. D'Angelo and I. Persson, A Structural Study of the N,N′-Dimethylpropyleneurea Solvated Zinc(II) and Cadmium(II) in Solution and Crystalline State, J. Mol. Liq., 2007, 131–132, 105–112 CrossRef CAS.
- I. Persson, K. Łyczko, D. Lundberg, L. Eriksson and A. Płaczek, A Coordination Chemistry Study of Hydrated and Solvated Lead(II) Ions in Solution and Solid State, Inorg. Chem., 2011, 50, 1058–1072 CrossRef CAS PubMed.
- J. Näslund, I. Persson and M. Sandström, Solvation of the Bismuth(III) Ion by Water, Dimethyl Sulfoxide, N,N’-Dimethylpropyleneurea, and N,N-Dimethylthioformamide. An EXAFS, Large-Angle X-ray Scattering, and Crystallographic Structural Study, Inorg. Chem., 2000, 39, 4012–4021 CrossRef PubMed.
- C. J. Carmalt, L. J. Farrugia and N. C. Norman, Structural Studies on some Iodoantimonate and Iodobismuthate Anions, Z. Anorg. Allg. Chem., 1995, 621, 47–56 CrossRef CAS.
- R. E. Marsh, Space groups P1 and Cc: how are they doing?, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 782–783 CrossRef CAS PubMed.
- Ö. Topel, I. Persson, D. Lundberg and A.-S. Ullström, On the Structure of the N,N-Dimethylpropyleneurea and Dimethylsulfoxide Solvated Gallium(III) and Indium(III) Ions and Bromide Complexes in Solution and Solid State, and the Complex Formation of the Gallium(III) and Indium(III) Bromide Systems in N,N-Dimethylpropyleneurea, Inorg. Chim. Acta, 2010, 363, 988–994 CrossRef.
- C. J. Carmalt, L. J. Farrugia and N. C. Norman, Coordination Complexes of Thallium(III) Chloride, Main Group Chem., 1996, 1, 339–344 CrossRef.
- J. Krakowiak, D. Lundberg and I. Persson, A Coordination Chemistry Study of Hydrated and Solvated Cationic Vanadium Ions in Oxidation States +III, +IV, and +V in Solution and Solid State, Inorg. Chem., 2012, 51, 9598–9609 CrossRef CAS PubMed.
- T. Suzuki, T. Kawasaki and Y. Ikeda, Bis(1,3-dimethyl-1,3-diazinan-2-one)dinitratodioxidouranium(VI), Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m18–m18 CrossRef CAS PubMed.
- E. M. Weis, P. B. Duval and S. S. Jurisson, Syntheses and structures of two uranyl complexes with DMPU, Radiochim. Acta, 2012, 100, 237–241 CrossRef CAS.
- D. Lundberg and K. Łyczko, Crystal structure of hexakis(dmpu)-di-μ2-hydroxido-dialuminium tetraiodide dmpu tetrasolvate [dmpu is 1,3-dimethyltetrahydropyrimidin-2(1H)-one]: a centrosymmetric dinuclear aluminium complex containing AlO5 polyhedra, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, 895–898 CrossRef CAS PubMed.
- J. Näslund, P. Lindqvist-Reis, I. Persson and M. Sandström, Steric Effects Control the Structure of the Solvated Lanthanum(III) Ion in Aqueous, Dimethyl Sulfoxide, and N,N’-Dimethylpropyleneurea Solution. An EXAFS and Large-Angle X-ray Scattering Study, Inorg. Chem., 2000, 39, 4006–4011 CrossRef PubMed.
- N. Torapava, D. Lundberg and I. Persson, A Coordination Chemistry Study of Solvated Thorium(IV) Ions in Two Oxygen-Donor Solvents, Eur. J. Inorg. Chem., 2011, 5273–5278 CrossRef CAS.
- I. Persson and K. B. Nilsson, Coordination Chemistry of the Solvated Silver(I) Ion in Oxygen Donor Solvents Water, Dimethyl Sulfoxide and N,N′-Dimethylpropyleneurea, Inorg. Chem., 2006, 45, 7428–7434 CrossRef CAS PubMed.
- P. D'Angelo, G. Chillemi, V. Barone, G. Mancini, N. Sanna and I. Persson, Experimental Evidence for a Variable First Coordination Shell of the Cadmium(II) Ion in Aqueous, Dimethyl Sulfoxide and N,N′-Dimethylpropyleneurea Solution, J. Phys. Chem. B, 2005, 109, 9178–9185 CrossRef PubMed.
- J. Fryer and D. Morris, Stbility constants of manganese(II) bromide complexes, Talanta, 1968, 15, 1309–1312 CrossRef CAS PubMed.
- H. Suzuki, S.-I. Ishiguro and H. Ohtaki, Formation of chloro complexes of manganese(II), cobalt(II), nickel(II) and zinc(II) in dimethyl sulphoxide, J. Chem. Soc., Faraday Trans., 1990, 86, 2179–2185 RSC.
- Z. Libus and H. Tralowska, Stability and nature of complexes of the type MCl+ in aqueous solution (M = Mn, Co, Ni and Zn), J. Solution Chem., 1975, 4, 1011–1022 CrossRef CAS.
- D. A. Netzel and H. A. Droll, Chloride and Bromide Complexes of Nickel(II) in Aqueous Solution, Inorg. Chem., 1963, 2, 412–413 CrossRef CAS.
- P. Smirnov, L. Weng and I. Persson, Determination of the Transfer Thermodynamic Functions for Some Monovalent Ions from Water to N,N′-Dimethylpropyleneurea, Phys. Chem. Chem. Phys., 2001, 3, 5248–5254 RSC.
- F. H. Allen, The Cambridge Structural Database: a quarter of a million crystal structures and rising, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed . ConQuest 2022.2.0 (September 2022).
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Patric
Lindqvist-Reis
a,
Patric
Lindqvist-Reis