
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computation of 31P NMR chemical shifts in Keggin−based lacunary polyoxotungstates†
Jake A.
Thompson
and
Laia
Vilà-Nadal
 *
*
School of Chemistry, University of Glasgow, UK. E-mail: laia.vila-nadal@chem.gla.ac.uk
First published on 23rd November 2023
Abstract
Density Functional Theory (DFT) calculations were employed to systematically study the accuracy of various exchange-correlation functionals in reproducing experimental 31P NMR chemical shifts, δExp(31P) for Keggin, [PW12O40]3− and corresponding lacunary clusters: [PW11O39]7−, [A-PW9O34]9−, and [B-PW9O34]9−. Initially, computed chemical shifts, δCalc(31P) were obtained with without neutralising their charge in which associated error, δError(31P), decreased as a function of Hartree–Fock (HF) exchange, attributed to constriction of the P–O tetrahedron. By comparison, δCalc(31P) performed with explicitly located counterions to render the system charge neutral, reduced discrepancies, δError(31P) by 1–2 ppm. However, uncertainties in δCalc(31P) remain, particularly for [B-PW9O34]9− anions attributed to direct electrostatic interactions between the counterions and the central tetrahedron. Optimal results were achieved using the PBE/TZP//PBE0/TZP method, achieving a mean absolute error (MAE) and a mean squared error (MSE) of 4.03 ppm. Our results emphasize that understanding the nature of the electrolyte and solvent environment is essential to obtaining reasonable agreement between theoretical and experimental results.
Introduction
Polyoxometalates (POMs) are a large group of discrete, polynuclear metal–oxo clusters comprised of early transition-metal (addenda) and oxide atoms.1–6 Addenda atoms are fully oxidized to d0 electron configurations capable of forming various topologies employing {MOx} as the principal building block.1–6 Polyoxo clusters are formed from the one-pot acidification of aqueous molybdate or tungstate oxoanions.6–8 Self-assembly can be controlled by several synthetic variables including but not limited to: (i) concentration/type of metal oxide anion; (ii) pH and type of acid; (iii) presence of reducing agent; (iv) heteroatom concentration; (v) and solvent.6–10 The synthesis and self-assembly mechanism of POMs requires a complex network of interdependent reactions, in which any slight variation to the listed five experimental variables can change the dominant species of the combinatorial library. Hence the outcome is often hard to predict.11 Recent work exploiting the self-assembly properties of POMs with non-ionic organic matter was employed to enhance the structural and functional diversity of cyclodextrin-based open frameworks.12Partial hydrolysis of poly-oxo clusters is achieved through the controlled addition of base produces lacunary clusters9,10 Lacunary clusters formally ‘lose’ one or several M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vertices and possess reactive cavities with high charge density around the defective region due to the negatively charged oxygen ligands.13–17 The defect can react with transition-metal cations forming a new class of compounds, an example shown are mono-transition-metal-substituted polyoxotungstates posessing the general formula [XW11M(L)O39]q− (X = e.g. P(V), Si(IV), L = H2O, DMSO, etc.).13–18
O vertices and possess reactive cavities with high charge density around the defective region due to the negatively charged oxygen ligands.13–17 The defect can react with transition-metal cations forming a new class of compounds, an example shown are mono-transition-metal-substituted polyoxotungstates posessing the general formula [XW11M(L)O39]q− (X = e.g. P(V), Si(IV), L = H2O, DMSO, etc.).13–18
Nuclear magnetic resonance (NMR) of active nuclei has been routinely employed for characterising molecular structures in solution and solid state.19 The employment of 1H, 29Si, and 31P nuclei are common, producing resonance signals across a wide range of chemical shifts providing direct information of the chemical environment. For some cases, poorly sensitive nuclei (17O, 183W) require high concentrations (≥ 0.1 M) coupled with long acquisition times.19 By contrast, 31P is a highly sensitive nucleus with 100% abundance, requiring significantly lower acquisition time and concentrations (< 0.01 M).19 This spectroscopy provides structural information reflecting in the range of −350 to 300 ppm, relative to 85% H3PO4. A fundamental variable of POM chemistry is controlling solubility achieved through the incorporation of inorganic and organic counterions.9 Their tuneable solubility in aqueous and organic media makes them suitable precursors for 31P NMR investigations.9 For decades, 31P NMR has been used to identify intermediates to understand self-assembly mechanisms in POMs,20 for example, the base degradation of H3PW12O40 and the formation of high-nuclearity clusters such as [P8W48O184]40− wheels.20,21
Early computational work in POMs has primarily focused on charged systems without implicit solvation models; see Fig. 1, in which we summarise the insights gained over the past 20-years regarding computed chemical shifts in POMs. In 2002, Bagno and co-workers calculated 99Ru chemical shifts in [PW11Ru(DMSO)O39]5− without accounting for counterions, solvation, relativistic corrections.22 The authors shown that shortening of d(Ru–S) by 0.045 Å induced shielding of the 99Ru nucleus by 235 ppm.22 These approximations produced results that were significantly underestimated with respect to experimental 99Ru chemical shifts, obtained at 7737 ppm.23 Later, Bagno and co-workers computed 183W NMR signals in [W5O18]6−, [Ru(DMSO)PW11O39]5−, and [PW11O39]7− clusters accounting for relativistic effects using zero-order regular scalar approximations.23 The authors reported the computed W1–W5 signals were deshielded relative to W6; only W2 and W3 reproduced trends shown by experimental work.23 Counterion dependence of approx. 53 ppm was accounted for modelling with the [LiPW11O39]6− adduct.23 The adduct demonstrated improved ordering of the signals, however, such chemical shifts did not accurately reproduce experimental work. Later, Bagno reported that incorporation of spin–orbit (SO) corrections, coupled with implicit solvation, provided optimal (average) accuracy for 183W chemical shifts, determined at 35 ppm.23 In 2009, Kortz and co-workers performed relativistic 183W NMR calculations for the mono-lacunary clusters, [GeW10X(H2O)O36]7− (X+ = Li, Na, K) which incorporated counterions with explicitly located water.24 The ordering of three resonances in [GeW10K(H2O)O36]7− was successfully reproduced; although slightly shielded, replicated the experimental distance between W(B) and W(C) signals (Δδ = 26 ppm vs. 29 ppm).24 The influence of counterions was shown by Δδ which was reduced to 19 ppm for [GeW10O36]8− systems.24 Vilà-Nadal and co-workers rationalised these observations in [XW12O40]q− (X = B, Al, Si, P, Ga, Ge, As, Zn) by showing 183W chemical shifts were linearly correlated with the tungsten-bridging oxygen (Ob and Oc) bond distances.25 The authors proposed that the contraction of the WO6 polyhedral increased the energy gap between the occupied and unoccupied orbitals involved in electronic transition, hence, the principal contribution to paramagnetic shielding, Uai, becomes less negative, deshielding the nucleus.25
 | ||
| Fig. 1 Timeline showing the progression of NMR calculations in polyoxometalates.22–29 | ||
Linear scaling has been employed to reduce mean absolute error (MAE) of computed chemical shifts. In 2014, Pascual-Borràs and co-workers calculated 17O chemical shifts for [W6O19]2− including spin–orbit corrections and implicit solvation providing an average mean absolute error of 39 ppm.26 By incorporating scaling corrections, MAE was reduced to approx. 26 ppm across 75 signals.26 Later, Pascual-Borràs and co-workers calculated 31P chemical shifts in Keggin, [PW12O40]3−, and Wells–Dawson, [P2W18O62]6− anions reporting MAE values of 2.62 ppm. The authors performed a systematic benchmark employing several exchange−correaltion (x-c) functionals and concluded that the optimal methodology was TZP/PBE//TZ2P/OPBE (NMR//optimization step)coupled with spin–orbit (SO) corrections and implicit solvation. Linear scaling corrections were applied which further reduced MAE to 0.57 ppm.27 The authors rationalised chemical shifts with orbital gaps as an approximation to the probability of electronic transition. For example, chemical shifts became more positive as a function of metal oxidation state (X = W(VI) > V(V) > Sn(IV) > Ru(II)) in [P2W17M(H2O)O61]q− anions.27
Contemporary quantum chemical calculations have focused on charged systems with spin–orbit (SO) corrections and implicit solvation models. Obtaining accurate models for computing 31P chemical shifts is crucial for spectral assignments of experimental data. The role of counterions in computed 31P chemical shifts have not been throughly investigated. DFT calculations currently rely on linear scaling to obtain sufficiently accurate results requiring a significant number of calculations to generate these regression models. In this work, we have employed density functional theory (DFT) calculations to reproduce experimental 31P NMR shifts in Keggin and corresponding mono- and tri-lacunary clusters, see Fig. 2. The present work will assess the accuracy of various exchange-correlation functionals and applied basis sets in reproducing experimental 31P NMR chemical shifts and provide a detailed analysis on the effect of the geomtric factors controlling it.
 | ||
| Fig. 2 Polyhedral representations of the hydrolytic conversions of [PW12O40]3− to lacunary: [PW11O39]7−, [A-PW9O34]9−, and [B-PW9O34]9− clusters. Colours corresponding to W = cyan, O = red, and P = orange. | ||
Experimental
Materials and methods
All reagents and solvents were supplied by Sigma Aldrich Chemical Company Ltd, Thermo Fisher Scientific, and Cambridge Isotope Laboratories. All materials were used without further purification.FT-IR spectra were recorded on a Nicolet 170SX-FT/IR spectrometer in the range of 400–4000 cm−1. UV-Vis spectra of the prepared compounds were measured in the region (200–400) nm for 30 μM solutions in distilled water at 25 °C using a Shimadzu 1800 spectrophotometer matched quartz cell. All 31P NMR spectroscopy was performed using a Bruker DPX 400 spectrometer with 85% phosphoric acid as an external standard. All spectra were recorded using 50 mg samples dissolved in D2O. All tetrabutylammonium (TBA) salts were recorded using 50 mg samples dissolved in CD3CN. Electrospray ionization mass spectrometry (ESI-MS) was obtained in negative-ion mode on the Agilent Q-TOF 6520 LC/MS mass spectrometer. The electrospray ionization source conditions: Vcap, 3500 V; skimmer, 65 V; nebulizer, 30 psi; drying and nebulizer gas, N2; drying gas flow, 10 L min−1; drying gas temperature, 300 °C; fragmentor, 80 V; scan range 100–2000 m/z. The sample solutions with the concentration of approximately 1000 ppm were made and analysed by direct injection using an automatic sampler with a flow rate of 0.2 mL min−1.
Preparation
Computational details
All computational results were obtained using the ARCHIE-WeSt High-Performance Computer based at the University of Strathclyde. Density Functional Theory (DFT) calculations were performed using the Amsterdam Modelling Suite (AMS 2020.1) package.32 In this work, two classes of exchange–correlation (x–c) functionals were employed, which include: (i) generalized gradient approximation (GGA); (ii) and hybrid methodologies. GGA functionals considered were Perdew–Burke–Ernzerhof (PBE).33 The hybrid x–c functionals considered were: (i) Becke, 3-parameter, Lee–Yang–Parr (B3LYP*,34 B3LYP35); (ii) Perdew–Burke–Ernzerhof (PBE0);36 (iii) and Becke's half-and-half (BH&H).37 Hybrid functionals were selected on their contributions of Hartree–Fock (HF) exchange (15% B3LYP*, 20% B3LYP, 25% PBE0, and 50% BH&H). We employed Slater basis sets comprising: (i) triple-ζ polarization (TZP); (ii) triple-ζ plus polarization (TZ2P); (iii) and quadruple-ζ plus polarization (QZ4P).38,39 Relativistic corrections were included by means of the zeroth order regular approximation (ZORA) formalism.40 The effects of aqueous solvent were approximated by using the conductor-like screening model (COSMO), as implemented by AMS.4131P-NMR signals were computed using several x–c functionals with varying degrees of HF exchange (0% PBE, 15% B3LYP*, 20% B3LYP, 25% PBE0, 50% BH&H). Also, the effect of larger basis sets (TZ2P & QZ4P) was investigated, restricted to the GGA–PBE level of theory. Relativistic corrections were included by the zeroth order regular approximation (ZORA) formalism and the aqueous solution was approximated by using the conductor-like screening model (COSMO), as implemented by AMS. Thereafter, single-point calculations were employed to calculate 31P-NMR parameters (hyperfine couplings and isotropic shielding constants) with spin–orbit (SO) corrections. The notation for this procedure is expressed throughout the text as FunctionalNMR/BasisNMR//FunctionalOPT/BasisOPT. Herein, all 31P NMR calculations were performed using PBE/TZP. The chemical shifts were referenced to 85% H3PO4 using PH3 as a secondary standard following the method suggested by van Wüllen:42
| δ(XCalc) = σ(PH3Calc) − σ(XCalc) − 266.1 | (1) |
The chemical shift of a nucleus is dependent on the magnetic shielding tensor, σ. The shielding tensor can be rewritten in terms of the diamagnetic, paramagnetic, and spin–orbit contributions, see below:
| σ = σd + σp + σSO | (2) |
 | (3) |
Finally, to evaluate the discrepancy of the calculated vs. experimental chemical shifts, we employed the mean unassigned error (MUE), the mean signed error (MSE) obtained as:
 | (4) |
 | (5) |
 | (6) |
Results and discussion
Anionic model
In this section, we seek to establish an optimal computational practise for calculating 31P NMR signals by using several x–c functionals with varying contributions of exact Hartree–Fock (HF) exchange (0% PBE, 15% B3LYP*, 20% B3LYP, 25% PBE0, 50% BH&H). The influence of applied basis set (TZ2P & QZ4P) was restricted to the GGA–PBE functional. Our initial work focussed on anionic systems, shown in Fig. 3. | ||
| Fig. 3 Computed chemical shift, δCalc(31P), plotted as a function of P–O distance for the hydrolytic conversions of (a) [PW12O40]3− to lacunary: (b) [PW11O39]7−, (c) [A-PW9O34]9−, and (d) [B-PW9O34]9− clusters. Optimisations were performed x–c functionals with varying degrees of Hartree–Fock (HF) exchange (0% PBE–50% BH&H). All NMR calculations were performed using PBE/TZP. | ||
Computed δ(31P) signals for Keggin, [PW12O40]3− and their corresponding lacunary: [PW11O39]7−, [A-PW9O34]9−, and [B-PW9O34]9− clusters are presented in Fig. 3. δCalc(31P) for Keggin, [PW12O40]3−, were reproduced within ca. 8 ppm across all methods with respect to the experimental, δexp(31P) – see Table 1. δCalc(31P) for anionic Keggin systems, [PW12O40]3− were in closer agreement with δexp(31P) of Na3[PW12O40]. The discrepancy between δCalc(31P) and δExp(31P) was represented by δError(31P). Herein, δError(31P) decreased as a function of exact Hartree–Fock (HF) exchange which was attributed shortening of P–O distances. For example, as exchange was increased from 15% (B3LYP*) to 50% (BH&H), P–O in [PW12O40]3− decreased by 0.026 Å, reflecting in the range of 7.87 ppm. All computed entries for [PW12O40]3− produced P–O geometries in the region of 1.519–1.545 Å, closely replicating the crystallographic structure of 1.530 Å.45
| PBENMR/TZPNMR//FunctionalOPT/BasisOPT | δ Calculated(31P) | δ Experimental(31P) | ||||||
|---|---|---|---|---|---|---|---|---|
| PBE/TZ2P | PBE/QZ4P | B3LYP*/TZP | B3LYP/TZP | PBE0/TZP | BH&H/TZP | Our work | Lit.19,46 | |
| a Protonated species. (Value) Discrepancy values in parentheses calculated using: δExp(31P) minus δCalc(31P). | ||||||||
| [PW12O40]3−/H2O | −7.56 | −9.66 | −9.63 | −10.43 | −11.30 | −17.50 | −14.60a | |
| Li3[PW12O40] | −7.90 (7.5) | −10.22 (5.2) | −10.27 (5.1) | −11.03 (4.4) | −12.84 (2.6) | −18.55 (3.1) | −15.41 | |
| Na3[PW12O40] | −8.28 (7.1) | −10.80 (4.6) | −10.65 (4.7) | −11.39 (4.0) | −13.04 (2.3) | −19.04 (3.7) | −15.36 | |
| [PW12O40]3−/MeCN | −7.55 | −9.62 | −9.27 | −9.97 | −11.38 | −17.50 | ||
| TBA3[PW12O40] | −8.25 (7.1) | −11.40 (4.0) | −9.75 (5.6) | −10.68 (4.7) | −12.33 (3.0) | −19.65 (4.3) | −15.35 | |
| [PW11O39]7−/H2O | −2.29 | −4.43 | −4.21 | −5.00 | −6.59 | −14.35 | ||
| Li7[PW11O39] | −3.47 (8.2) | −5.38 (6.3) | −5.35 (6.3) | −6.16 (5.5) | −7.27 (4.4) | −14.92 (3.5) | −11.65 | −11.20 |
| Na7[PW11O39] | −2.33 (8.5) | −4.65 (6.2) | −4.58 (6.2) | −5.37 (5.5) | −7.35 (3.5) | −14.28 (3.5) | −10.82 | −10.65 |
| K7[PW11O39] | −3.40 (7.7) | −6.06 (5.1) | −6.05 (5.1) | −6.82 (4.3) | −8.5 (2.6) | −16.28 (5.2) | −11.13 | −10.80 |
| [A-PW9O34]9−/H2O | 0.55 | −1.00 | −0.86 | −1.61 | −3.87 | −13.86 | ||
| Na8H[A-PW9O34] | −0.99 (6.8) | −2.34 (5.4) | −2.39 (5.4) | −3.24 (4.5) | −5.10 (2.7) | −12.83 (5.1) | −7.76 | −7.10 |
| [B-PW9O34]9−/H2O | 3.41 | 1.89 | 2.33 | 1.60 | −1.96 | −11.64 | ||
| Na8H[B-PW9O34] | 11.05 (14.8) | 9.23 (13.0) | 9.17 (12.9) | 8.15 (11.9) | 6.28 (2.5) | −2.96 (0.8) | −3.76 | −3.00 |
| MAE | 8.65 | 6.37 | 6.61 | 5.79 | 4.03 | 3.74 | ||
| MSE | 8.65 | 6.37 | 6.61 | 5.79 | 4.03 | −3.54 | ||
| STD | 2.54 | 2.83 | 2.66 | 2.58 | 2.53 | 1.57 | ||
Computed chemical shifts, δCalc(31P), will be rationalised as a function of structural geometry. The magnetic shielding tensor, σ, is comprised of the sum of three contributions: (i) diamagnetic; (ii) paramagnetic; (iii) and the spin–orbit (SO) correction – see eqn (2). The diamagnetic contribution to shielding is dominated by core orbitals contributions which are largely unaffected by the chemical environment. Hence, δCalc(31P) can be attributed to the paramagnetic term, σp. eqn (3) shows the principal contributor to the paramagnetic term is uai, which depends on the reciprocal of the energy gap between the occupied and virtual orbitals involved in transition. HOMO LUMO gaps are not the same as transitions atttributed to the P nucleus!
Generally, employment of hybrid x–c functionals proved superior and were in closer agreement with δexp(31P) with respect to GGA-PBE coupled with triple-ζ plus polarization (TZ2P) or quadruple-ζ plus polarization (QZ4P) basis sets. This observation was attributed to an over-expansion of the PO4 tetrahedron. Our computed signals shown an inverse correlation with Hartree–Fock (HF) exchange, shown in Fig. 3. The contraction of the PO4 tetrahedron induced shielding of the nucleus. Consequently, the energy gap between the occupied and virtual orbitals involved in transition have increased with contraction of the P–O distances. One would expect that for small energy gaps, this will reinforce σp (more negative), and the shielding tensor, σ, becomes less positive, displacing δCalc(31P) to more positive regions. Hence, the energy gap between the occupied and virtual orbitals increases and uai becomes less negative.
Incorporation of alkali metal cations
In this work, we have explicitly located alkali metal (X = Li+, Na+, K+) cations to render our systems charge neutral. On the surface of the molecule, the counterion can interact with 4-fold (pocket A–F) or 3-fold pockets – see Fig. S9.†14 The 4-fold pockets feature four oxygen atoms capable of interacting with counterions, whilst 3-fold pockets are comprised of three adjacent oxygen atoms which assume a triangular shape. In our previous work, we established alkal-.metal counterions preferentially interact with the 4-fold pockets because of improved Coulomb interactions.14 Herein, we have explicitly located alkali metal (X = Li+, Na+, K+) cations into 4-fold hollow sites, unless specified otherwise, to render the system neutral.Computed δ(31P) signals for Keggin, X3[PW12O40] and their corresponding lacunary: X7[PW11O39], X9[A-PW9O34], and X9[B-PW9O34] (X = Li+, Na+, K+, TBA+) clusters are presented in Table 1. Incorporation of counterions to [PW12O40]3− induced (minimal) deshielding by approx. 0.5–1.5 ppm. The active nuclei in X3[PW12O40] (X = Li+, Na+) became more shielded (0.3 – 0.5 ppm) as the ionic radii of alkali–metal cation was increased from ca. 2.125 to 2.250 Å (Li+to Na+). δCalc(31P) calculated using PBE0/TZP produced the strongest agreement with δExp(31P) shifted upfield by 2.5 ppm, relative to experimental data.
The accuracy of x–c functionals was assessed, in which, increasing HF exchange progressively shifted signals upfield from −10.27 (15% B3LYP*) to −18.55 ppm (50% BH&H) in Li3[PW12O40]3− salts and −10.80 (15% B3LYP*) to −19.04 ppm (50% BH&H) in Na3[PW12O40], salts. Negligible effects to δ(31P) were induced by changing the implicit solvent (H2O → MeCN) shown for [PW12O40]3− in which Δδ(31P) did not exceed 0.5 ppm.
Assessment of the lacunary, X7[PW11O39] (X = Li+, Na+) clusters were shifted downfield by 4.0–5.0 ppm with respect to Keggin, X3[PW12O40]. δ(31P) signal in Na7[PW11O39] clusters were shifted upfield by 1.0–1.5 ppm, with respect to Li7[PW11O39] clusters. Conversely, δ(31P) signals in K7[PW11O39] clusters were shifted downfield by ca. 1.0 ppm relative to Na7[PW11O39] revealing the overall trend: K+ (upfield) > Li+ > Na+ (downfield). Unfortunately, our computational model is unable to replicate the order of δExp(31P) values in X7[PW11O39]: Li+ (upfield) > K+ > Na+ (downfield). This is probably since our model overlooks the ion pairing effect in aqueous solutions of polyoxometalate anions with different cations. In 2008 Poblet and co-workers studied the delicate interplay between electrostatic interactions and the proximity effect in the aqueous solutions of three different POMs, [PW12O40]3−, [SiW12O40]4−, and [AlW12O40]5−, with respect to three different monovalent cations Li+, Na+, and K+.47 Later, Nyman and co-workers successfully investigated the ion-pairing effect in Mo− based Lindqvist([(CH3)4N]+,Cs)8[M6O19] (M = Nb, Ta), combining experiments and theory. Their system is particularly challenging as the clusters water solubility increases with stronger ion-pairing, contrary to most ionic salts.48 Although, the best overall computational methodology was not consistent across the lacunary clusters. The influence of exact exchange progressively shielded δ(31P) values, for example: from −5.35 (15% B3LYP*) to −14.92 ppm (50% BH&H) in Li7[PW11O39], salts. Generally, optimisations using PBE0/TZP (20% exchange) accurately replicated the experimental, reporting a mean absolute error (MAE) of 4.03 ppm. Computation with BH&H/TZP demonstrated the lowest value of MAE (3.74 ppm), attributed to an excellent replication of δ(31P) in Na8H[B-PW9O34].
Incorporation of alkylammonium cations
To further this work, we have explicitly incorporated alkylammonium cations, neutralising our systems to: [(n-CxH2x+1)4N]3[PW12O40]; where x = 1–4 (Table 2). The alkylammonium cations adopted a triangular arrangement around the Keggin, [PW12O40]3− in which the central heteroatom is co-planar with the cations, see Fig. S10.†| [(n-CxH2x+1)4N]+ | δ Calc(31P) | ΔδCalc(31P) |
|---|---|---|
| [NH4]+ | −13.00 | 0.67 |
| [(n-CH3)4N]+ | −12.68 | 0.35 |
| [(n-C2H5)4N]+ | −12.70 | 0.37 |
| [(n-C3H7)4N]+ | −12.83 | 0.50 |
| [(n-C4H9)4N]+ | −12.33 | 0.00 |
The arrangement of [(n-CxH2x+1)4N]3 can be assessed by the average central heteroatom (P) – nitrogen (ammonium) distance calculated at 5.601, 7.227, 8.003, 7.919, and 8.043 Å for NH4+, n-(CH3)4+, n-(C2H5)4+, n-(C3H7)4+, and n-(C4H9)4+, respectively. Optimizations using PBE0/TZP for [(n-C4H9)4N]3[PW12O40] produced δ(31P) with reasonable agreement with the experimental (ca. 3.0 ppm). However, optimizations of [(n-CxH2x+1)4N]3[PW12O40] (x = 3 or 4) with hybrid x–c functionals is CPU expensive, hence, we explored the sensitivity of δ(31P) of alkyl chain length in [(n-CxH2x+1)4N]3. Computed δCalc(31P) was shielded by 0.67 ppm from −12.33 ([(n-C4H9)4N]+) to −13.00 ([NH4]+). Interestingly, optimisations using [NH4]+ cations yielded the closest agreement with the experimental values.
Empirical scaling was performed to correct the discrepancies between the computed and experimental work. The present work employed data points obtained using the PBE/TZP//PBE0/TZP methodology, thereby computed isotropic shielding (σ) was plotted taking the form δ = bσ + a, in which the slope, b, is the scaling factor, see Fig. 4. Table S2† reports the comparison between experimental, δExp, computed, δComp, and fitted, δFitted, chemical shifts (Fig. 5).
 | ||
| Fig. 4 Computed isotropic shielding, σCalc using the PBE/TZP//PBE0/TZP methodology, plotted as a function of experimental δExp(31P) values. | ||
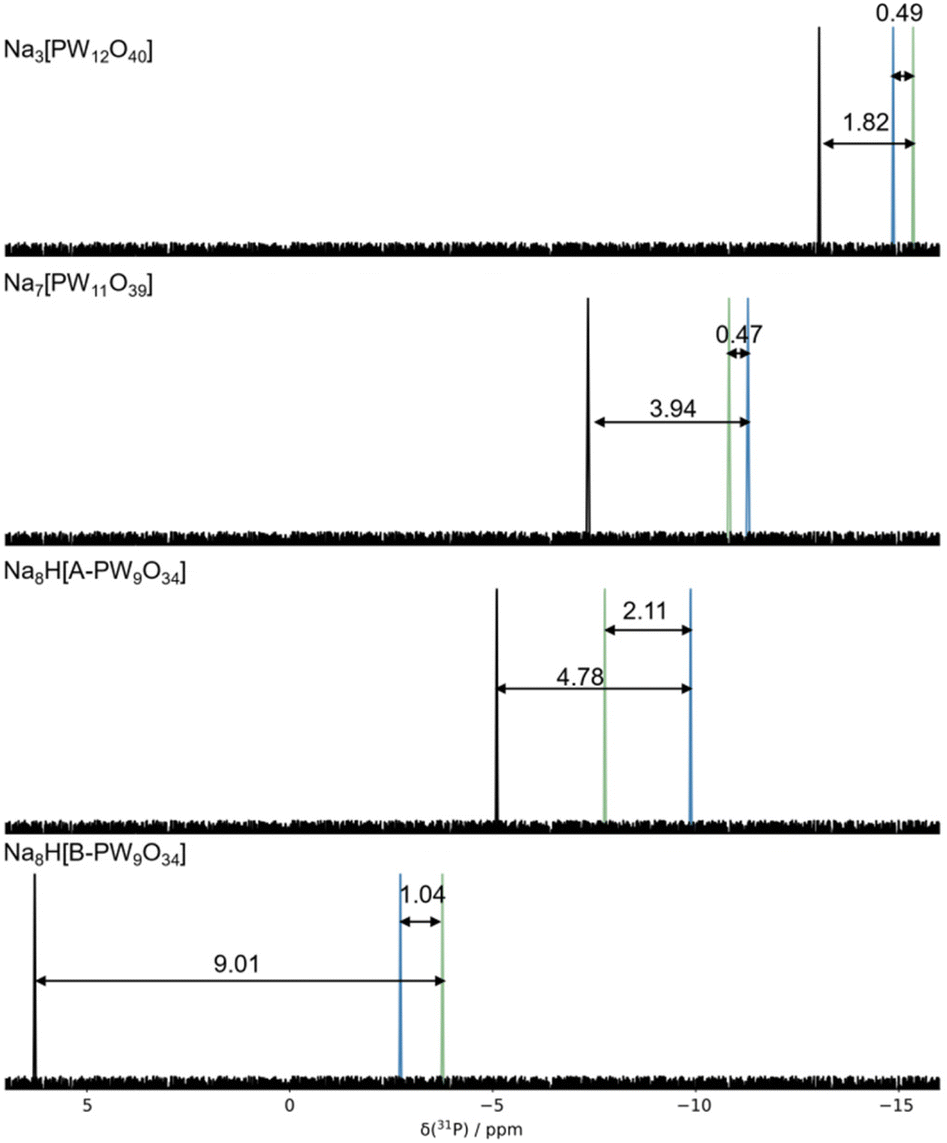 | ||
| Fig. 5 Computed (black), δExp(31P), fitted (blue), δFitted(31P), and experimental (green), δExp(31P), signals for Keggin, Na3[PW12O40] and their corresponding lacunary: Na7[PW11O39], Na9[A-PW9O34], and Na9[B-PW9O34] clusters. All NMR calculations were performed using PBE/TZP. | ||
The fitted values, δFitted(31P), revealed significantly lower MAE values of 0.86 and were not systematically underestimated shown by MSE of −0.02 ppm. δFitted(31P) replicated the order of δ(31P) values in X7[PW11O39] (X = Li+, Na+, K+) replicating the experimental within ca. 0.9 ppm, see Fig. 4 and Table S2.† A significant improvement for the tri-lacunary: Na8H[A-PW9O34] and Na8H[B-PW9O34] clusters were observed, in which δError(31P) was reduced from 2.66 and 10.04 using PBE/TZP//PBE0/TZP to 2.11 and 1.04 using fitted values, δFitted(31P). Computed δ(31P) signals for Na8H[B-PW9O34] were challenging to accurately compute without correctional fitting. No correlation between ion-pair proximity and chemical shift was observed, contrasted to redox potentials shown in our previous work. The effect of the functional outweighs the effect of the counterions.
Conclusions
In this work, we have employed Density Functional Theory (DFT) calculations to systematically study the accuracy of exchange– correlation functionals and applied basis sets for reproducing experimental chemical shift, δExp(31P) values. Our objective has been to establish a methodology for obtaining accurate δCalc(31P) signals in Keggin-based clusters and to provide an insight into the geometric factors controlling it.Computation of δExp(31P) for Keggin, [PW12O40]3−, were in closer agreement to δCalc(31P) obtained for Na3[PW12O40]. Generally, discrepancies between δCalc(31P) and δExp(31P) represented by δError(31P) decreased as a function of Hartree–Fock (HF) exchange, attributed to shortening of P–O. Explicitly located counterions reduced discrepancies by 1.0–2.0 ppm across all Keggin and corresponding lacunary compounds which was attributed to their deshielding effect. However, obtaining accurate approximations for for [B-PW9O34]9− remains a challenge due to direct electrostatic interactions between the located counterions and central tetrahedron. Optimal results were achieved using the PBE/TZP//PBE0/TZP method, achieving an MAE and MSE of 4.03 ppm. Linear scaling was performed to reduce discrepancies between the computed and experimental work,in which δFitted(31P), revealed lower MAE values of 0.86 ppm and were not systematically underestimated shown by MSE of −0.02 ppm.
Data availability
The datasets generated using DFT calculations are available in the ioChem-BD database: https://iochem-bd.bsc.es/browse/handle/100/312985Author contributions
L.V.-N. conceived the idea together with J. A. T. who designed the modelling approach, performed all calculations, synthesised, and fully characterised all the compounds and analysed all the data. J. A. T. wrote the paper and made all tables and figures in the manuscript with input from L.V.-N.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by University of Glasgow and the Engineering and Physical Sciences Research Council Grants (EP/S030603/1; EP/V048341/1; EP/S031170/1), Royal Society of Chemistry RSC Hardship Grant (COVID-19). We thank the EPSRC Doctoral Training Partnership (DTP) funding received by the University of Glasgow for Jake A. Thompson PhD studentship ‘Exploring N2 fixation with metal oxides’ project 2442596 (EP/R513222/1; EP/T517896/1). We also thank the University of Glasgow (UofGla) Early Career Development Programme (ECDP) 2021, the UofGla Reinvigorating Research Scheme 2022, and the School of Chemistry for long-lasting support. We thank ScotCHEM for 2022 ECR equipment funding and we also thank the Cronin Group for upcylced glassware and equipment. Finally, we thank Mr Alec Mungall, for help collecting NMR samples, and all the technical staff in the School of Chemistry for support during this work. Results were obtained using the ARCHIE-WeSt High-Performance Computer (https://www.archie-west.ac.uk) based at the University of Strathclyde.References
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- N. I. Gumerova and A. Rompel, Chem. Soc. Rev., 2020, 49, 7568–7601 RSC.
- N. V. Izarova, M. T. Pope and U. Kortz, Angew. Chem., Int. Ed., 2012, 51, 9492–9510 CrossRef CAS PubMed.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- A. S. Cherevan, S. P. Nandan, I. Roger, R. Liu, C. Streb and D. Eder, Adv. Sci., 2020, 7, 1903511 CrossRef CAS PubMed.
- S. M. Wilson, B. E. Petel, E. Schreiber, M. L. Maiola, P. Su, E. M. Matson and J. Laskin, Chem. – Eur. J., 2023, 29, 1–10 CrossRef.
- C. P. Pradeep, D. L. Long and L. Cronin, J. Chem. Soc., Dalton Trans., 2010, 39, 9443–9457 RSC.
- M. S. Balulla, J. A. Gamelas, H. M. Carapuça, A. M. V. Cavaleiro and W. Schlindwein, Eur. J. Inorg. Chem., 2004, 619–628 CrossRef.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596–612 CrossRef CAS.
- D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC.
- H. N. Miras, L. Vilà-Nadal and L. Cronin, Chem. Soc. Rev., 2014, 43, 5679–5699 RSC.
- S. Khlifi, J. Marrot, M. Haouas, W. E. Shepard, C. Falaise and E. Cadot, J. Am. Chem. Soc., 2022, 144, 4469–4477 CrossRef CAS.
- S. Azuma, T. Kadoguchi, Y. Eguchi, H. Hirabaru, H. Ota, M. Sadakane, K. Yanagisawa, T. Hasegawa and T. Ueda, Dalton Trans., 2020, 49, 2766–2770 RSC.
- J. A. Thompson, R. González-Cabaleiro and L. Vilà-Nadal, Inorg. Chem., 2023, 62, 12260–12271 CrossRef CAS PubMed.
- D. Laurencin, R. Thouvenot, K. Boubekeur, P. Gouzerh and A. Proust, C. R. Chim., 2012, 15, 135–142 CrossRef CAS.
- M. Sadakane, D. Tsukuma, M. H. Dickman, B. Bassil, U. Kortz, M. Higashijima and W. Ueda, Dalton Trans., 2006, 4271–4276 RSC.
- P. Klonowski, J. C. Goloboy, F. J. Uribe-Romo, F. Sun, L. Zhu, F. Gándara, C. Wills, R. J. Errington, O. M. Yaghi and W. G. Klemperer, Inorg. Chem., 2014, 53, 13239–13246 CrossRef CAS PubMed.
- R. González-Cabaleiro, J. A. Thompson and L. Vilà-Nadal, Front. Chem., 2021, 9, 742565 CrossRef PubMed.
- R. I. Maksimovskaya and G. M. Maksimov, Coord. Chem. Rev., 2019, 385, 81–99 CrossRef CAS.
- L. Vilà-Nadal and L. Cronin, Nat. Rev. Mater., 2017, 2, 17054 CrossRef.
- R. Contant and A. Tézé, Inorg. Chem., 1985, 24, 4610–4614 CrossRef CAS.
- A. Bagno and M. Bonchio, Eur. J. Inorg. Chem., 2002, 1475–1483 CrossRef CAS.
- (a) A. Bagno, M. Bonchio, A. Sartorel and G. Scorrano, Eur. J. Inorg. Chem., 2000, 17–20 CrossRef CAS; (b) A. Bagno, M. Bonchio, A. Sartorel and G. Scorrano, ChemPhysChem, 2003, 4, 517–519 CrossRef CAS; (c) A. Bagno, M. Bonchio and J. Autschbach, Chem. – Eur. J., 2006, 12, 8460–8471 CrossRef CAS PubMed.
- N. Vankova, T. Heine and U. Kortz, Eur. J. Inorg. Chem., 2009, 5102–5108 CrossRef CAS.
- L. Vilà-Nadal, J. P. Sarasa, A. Rodríguez-Fortea, J. Igual, L. P. Kazansky and J. M. Poblet, Chem. – Asian J., 2010, 5, 97–104 CrossRef.
- M. Pascual-Borràs, X. López, A. Rodríguez-Fortea, R. J. Errington and J. M. Poblet, Chem. Sci., 2014, 5, 2031–2042 RSC.
- M. Pascual-Borràs, X. López and J. M. Poblet, Phys. Chem. Chem. Phys., 2015, 17, 8723–8731 RSC.
- J. A. Dias, S. C. L. Dias, E. Caliman, J. Bartis and L. Francesconi, Keggin Structure Polyoxometalates, Inorg. Synth., 2014, 36, 210–217 CrossRef CAS.
- J. A. Gamelas, F. A. S. Couto, M. C. N. Trovão, A. M. V. Cavaleiro, J. A. S. Cavaleiro and J. D. P. de Jesus, Thermochim. Acta, 1999, 326, 165–173 CrossRef CAS.
- N. Haraguchi, Y. Okaue, T. Isobe and Y. Matsuda, Inorg. Chem., 1994, 33, 1015–1020 CrossRef CAS.
- B. Moore and K. Foger, Inorg. Chim. Acta, 1991, 181, 201–205 CrossRef CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 3865–3868 CrossRef.
- M. Reiher, O. Salomon and B. A. Hess, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed.
- C. Lee, E. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785 CrossRef.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- R. Sure, J. G. Brandenburg and S. Grimme, ChemistryOpen, 2016, 5, 94–109 CrossRef CAS.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS.
- E. Van Lenthe, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed.
- C. Van Wüllen, Phys. Chem. Chem. Phys., 2000, 2, 2137–2144 RSC.
- D. B. Chesnut, J. Phys. Chem. A, 2005, 109, 11962–11966 CrossRef CAS PubMed.
- J. Přecechtělová, P. Novák, M. L. Munzarová, M. Kaupp and V. Sklenář, J. Am. Chem. Soc., 2010, 132, 17139–17148 CrossRef.
- G. M. Brown, M.-R. Noe-Spirlet, W. R. Busing and H. A. Levy, Acta Crystallogr., 1977, 1038–1046 CrossRef CAS.
- M. Kozik, C. F. Hammer and L. C. W. Baker, J. Am. Chem. Soc., 1986, 108, 7627–7630 CrossRef CAS.
- F. Leroy, P. Miró, J. M. Poblet, C. Bo and J. Bonet-Ávalos, J. Phys. Chem. B, 2008, 112(29), 8591–8599 CrossRef CAS.
- J. Sures, S. A. Serapian, K. Kozma, P. I. Molina, C. Bo and M. Nyman, Phys. Chem. Chem. Phys., 2017, 19, 8715–8725D RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Selected structural parameters for Keggin, [PW12O40]3− and corresponding lacunary: [PW11O39]7−, [A-PW9O34]9−, and [B-PW9O34]9− clusters. Experimental 31P NMR, IR, UV, and ESI-MS spectra. See DOI: https://doi.org/10.1039/d3dt02694a |
| This journal is © The Royal Society of Chemistry 2024 |