
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvothermal synthesis of VO2 nanoparticles with locally patched V2O5 surface layer and their morphology-dependent catalytic properties for the oxidation of alcohols†
Dorothea
Gömpel
a,
Muhammad Nawaz
Tahir
 *bc,
Mujeeb
Khan
d,
Syed Farooq
Adil
d,
Mohammed Rafi
Shaik
d,
Mufsir
Kuniyil
d,
Abdulrahman
Al-Warthan
d and
Wolfgang
Tremel
*a
*bc,
Mujeeb
Khan
d,
Syed Farooq
Adil
d,
Mohammed Rafi
Shaik
d,
Mufsir
Kuniyil
d,
Abdulrahman
Al-Warthan
d and
Wolfgang
Tremel
*a
aChemistry Department, Johannes Gutenberg-Universität Mainz, Duesbergweg 10-14, D-55128 Mainz, Germany. E-mail: tremel@uni-mainz.de
bInterdisciplinary Research Center for Hydrogen Technologies and Carbon Management (IRC-HTCM), King Fahd University of Petroleum & Minerals KFUPM, Dahran 31261, Saudi Arabia. E-mail: muhammad.tahir@kfupm.edu.sa
cInterdisciplinary Research Center for Hydrogen and Energy Storage (IRC-HES), King Fahd University of Petroleum and & Minerals, Dahran 31261, Saudi Arabia
dDepartment of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Kingdom of Saudi Arabia
First published on 8th January 2024
Abstract
Vanadium oxides are promising oxidation catalysts because of their rich redox chemistry. We report the synthesis of VO2 nanocrystals with VO2(B) crystal structure. By varying the mixing ratio of the components of a binary ethanol/water mixture, different VO2 nanocrystal morphologies (nanorods, -urchins, and -sheets) could be made selectively in pure form. Polydisperse VO2(B) nanorods with lengths between 150 nm and a few micrometers were formed at large water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol ratios between 4:1 and 3:2. At a water:ethanol ratio of 1:9 VO2 nanosheets with diameters of ∼50–70 nm were formed, which aggregated to nano-urchins with diameters of ∼200 nm in pure ethanol. The catalytic activity of VO2 nanocrystals for the oxidation of alcohols was studied as a function of nanocrystal morphology. VO2 nanocrystals with all morphologies were catalytically active. The activity for the oxidation of benzyl alcohol to benzaldehyde was about 30% higher than that for the oxidation of furfuryl alcohol to furfural. This is due to the substrate structure. The oxidation activity of VO2 nanostructures decreases in the order of nanourchins > nanosheets > nanorods.
:ethanol ratios between 4:1 and 3:2. At a water:ethanol ratio of 1:9 VO2 nanosheets with diameters of ∼50–70 nm were formed, which aggregated to nano-urchins with diameters of ∼200 nm in pure ethanol. The catalytic activity of VO2 nanocrystals for the oxidation of alcohols was studied as a function of nanocrystal morphology. VO2 nanocrystals with all morphologies were catalytically active. The activity for the oxidation of benzyl alcohol to benzaldehyde was about 30% higher than that for the oxidation of furfuryl alcohol to furfural. This is due to the substrate structure. The oxidation activity of VO2 nanostructures decreases in the order of nanourchins > nanosheets > nanorods.
Introduction
Multivalent transition metal oxides (TMOs) display a wide range of intriguing and useful properties.1–4 The ground state in TMOs is dictated by the valence state of the transition metal cations. In particular, the interplay between spin, orbital, charge, and lattice degrees of freedom has been studied in this category of solids with partially filled d-band.5,6 Vanadium oxides are prototypical examples of multivalent TMOs with strongly correlated electrons.7,8 They can undergo reversible metal–insulator phase transitions accompanied by changes in their crystallographic, magnetic, optical, and electrical properties.7–10 Electron–lattice interactions and electron–electron correlation modify the properties of vanadium oxides for a variety of applications as chemical sensors,11 electrode materials for lithium batteries,12–14 capacitors and supercapacitors,15 for electronic and optical2 devices, and in particular in heterogeneous catalysis.5,16–18 They are used for the production of important chemicals (e.g., sulfuric acid, phthalic anhydride) and in the alleviation of environmental pollution (e.g., nitrogen oxides from power plant flue gas).19–21The high catalytic activity of V-oxides can be attributed to the ability of vanadium to easily switch between its V3+, V4+ and V5+ oxidation states under the respective reaction conditions22 and to the presence of unsaturated surface coordination sites, which represent oxygen-deficient defects that allow reactions involving bulk oxygen atoms.9,23–26 Oxide surfaces undergo restructuring and can exchange oxygen atoms with the environment in ways that are difficult to predict.27,28 VO2 in particular is a catalyst for the desulfurization of dibenzothiophene,29 the oxidative dehydrogenation of propane30 or the electrochemical reduction of trinitrotoluene.31
Nanocrystals can serve as model catalysts with defined surface structure to study the structure–property relationships of powder catalysts. Apart from the oxidation state and crystallinity of vanadium oxide catalysts there are a variety of variables such as structure, surface area, morphology, surface wetting and interface interactions.32 Colloidal synthesis allows the controlled preparation of metal oxide nanocrystals with defined and uniform morphology. Different facets of a nanocrystal have different surface energies, surface structures, and chemical reactivity, which can significantly determine catalytic performance. By controlling the morphology and size of the nanocrystals, it is possible to tune the exposed facets and enhance the catalytic activity and selectivity for a given reaction.22,33,34
The activities of nanostructured VOx catalysts have been reported as a function of crystal morphology, crystal size and surface area.35,36 Most of these studies, however, have been carried out with Magneli-type vanadium oxides such as VnO2n+1 (V3O7, V4O9, and V6O13),9 whereas morphology-dependent catalytic properties of VO2 NPs have rarely been reported.29 Several routes have been reported for the synthesis of VO2 and other vanadium oxide nanocrystals.37–40 The most common ones are based on hydro- or solvothermal techniques.41 The choice of the solvent controls the reactivity, solubility and the diffusion of the precursors.42 In binary solvent mixtures these factors can be tuned accurately and independently. This allows the preparation of nanocrystals with different morphologies, e.g., spheres,43–45 rods,46–49 sheets,50–53 and urchins54,55 only by varying the volume ratio of the solvents.56
Sheet- and urchin-like morphologies are the most promising options for large surface areas, but the morphological effects can be offset by the particle size. Often the crystallites are several hundred nanometers in diameter. This leads in total to a significant reduction of the surface area. Therefore, a synthesis of nanometer-sized urchins and sheets is highly desirable. This requires the reactivity during the reaction to be fine-tuned to prevent uncontrolled aggregation into larger particles.
Here we describe the solvo-/hydrothermal synthesis of VO2(B) nanorods and VO2 nanosheets and -urchins. Therefore, the effect of solvent, especially the impact of chain length of aliphatic alcohols and the composition of binary mixtures on morphology and phase of VO2 nanoparticles was explored systematically. The catalytic activity of VO2 nanoparticles with different morphology and active surface area was tested for the oxidation of benzyl alcohol to benzaldehyde and furfuryl alcohol to furfural as a model reaction (Scheme 1). To the best of our knowledge, this is the first investigation of the catalytic properties of VO2 nanoparticles in oxidation reactions as a function of particle size and morphology.
 | ||
| Scheme 1 Preparation of VO2 nanoparticles with different morphologies and their catalytic applications in the oxidation of furfuryl alcohol. | ||
Results and discussion
The morphology of the nanoparticles was controlled by the reaction conditions, i.e., by the solvent composition in the solvothermal reaction.57,58 For this purpose, the synthesis of vanadium oxide nanoparticles was carried out in water/ethanol mixtures with different mixing ratios. The other reaction variables (reaction time, temperature, amount of pluronic capping agent and the concentration of the output compound) were kept constant.The electron micrographs in Fig. 1 show that the morphology of the VO2 nanoparticles is determined by the composition of the solvent mixture. The ratio of water to ethanol was systematically varied between 0% (pure water) and 100% (pure ethanol). Nanorods with very different aspect ratios formed in water (Fig. 1a). The length of the rods varied between 150 nm and several micrometers, while the width was between 20 and 200 nm. At water to ethanol ratios of 4:1 (20 mL:5 mL, Fig. 1b) and 3:2 (15:10 mL, Fig. 1c), the morphology was similar (nanorods), but the rods were significantly smaller and less polydisperse than when synthesized in pure water. Their width was in the range between 20 and 80 nm and their length between 100 and 220 nm. There was no visible difference in the morphology of the nanorods from water:ethanol mixtures between water:ethanol ratios of 4:1 and 3:2. The particles remained rod-like for higher ethanol concentrations. However, their polydispersity increased again (Fig. 1d, water:ethanol ratio 2:3). At a water:ethanol ratio of 1:4, the rods agglomerated into disordered bundles (Fig. 1e), and at a ratio of 1:9, nanorods no longer formed. The particles became increasingly isotropic but without defined morphology (Fig. 1f).
 | ||
| Fig. 1 TEM micrographs of the reaction products with different water:ethanol ratios. (a) pure water (25 mL:0 mL), (b) 4:1 (20 mL:5 mL), (c) 3:2 (15 mL:10 mL), (d) 2:3 (10 mL:15 mL), (e) 1:4 (5 mL:20 mL), (f) 1:9 (2.5 mL:22.5 mL), (g) 1:49 (0.5 mL:24.5 mL), (h) 1:82.3 (0.3 mL:24.7 mL) and (i) pure ethanol (0 mL:25 mL). | ||
At high ethanol concentrations (water:ethanol < 1:49), a significant change in morphology occurred, and sheet-like nanocrystals with a narrow size distribution were formed (Fig. 1g and h).
The morphology of some nanoparticles in Fig. 1g is reminiscent of structures obtained at a water–ethanol ratio of 1:9 (Fig. 1f). In both cases, the diameter of the sheets was about 50–70 nm. The thickness of about 5 nm can be deduced from some sheets that were oriented vertically on the grid accidentally. Nanoparticles synthesized in pure ethanol have urchin-like morphologies with a total diameter of about 200 nm (Fig. 1i), as illustrated in Fig. 2. The wet chemistry methods used for the synthesis of nanoparticles are ideal to control the morphology, size and composition. In particular, hydrothermal/solvothermal methods59,60 where the solubility and re-precipitation of the precursors define the number of nuclei and the growth (size) of particles, can be utilized to control size and morphology. We used pure water (high dielectric constant) as solvent, resulting in a medium of low supersaturation, leading to a small number of nuclei and the formation of larger particles (nanorods). However, a gradual increase of the amount of ethanol (low dielectric constant that provides a medium with high supersaturation), the number of nuclei increases and ultimately results in small size particles sizes. Using pure ethanol, where the number of nuclei is highest, these nuclei combine to form nano-urchins to compensate their surface energy. Basically, a single urchin consists of several sheets that agglomerate in a random manner. Therefore, the crystallinity of the resulting particles is very low as indicated by the very broad diffraction intensities (Fig. 3, vide infra). The driving force for this agglomeration could be the different colloidal stability of the sheets depending on the solvent. Moreover, the sheets that compose the nano-urchins were smaller than free-standing sheets, thereby favoring aggregation. Thus, the composition of the solvent mixture is an effective means of controlling the morphology of vanadium oxide nanoparticles. By varying the water:ethanol ratio, three different morphologies – rods, sheets, and urchins (resulting from sheet agglomeration) – could be made in a reproducible fashion. We note that the surfactant F-127 helps to obtain well dispersed (but not porous) nanoparticles as indicated by the TEM images in Fig. S1.† The morphology of the nanoparticles was primarily controlled by using different ethanol:water ratios.
 | ||
| Fig. 2 Schematic representation of the morphological relation between the sheets and urchins. | ||
 | ||
| Fig. 3 Powder X-ray diffraction patterns of the reaction products prepared with different water:ethanol ratios. (a) Pure water (25 mL:0 mL, VO2 nanorods), (b) 4:1 (20 mL:5 mL, nanorods, less polydisperse), (c) 3:2 (15 mL:10 mL, nanorods, less polydisperse), (d) 2:3 (10 mL:15 mL, nanorods, more polydisperse), (e) 1:4 (5 mL:20 mL, nanorods agglomerated into disordered bundles), (f) 1:9 (2.5 mL:22.5 mL, isotropic nanoparticles, no distinct morphology), (g) 1:49 (0.5 mL:24.5 mL, sheet-like nanoparticles with narrow size distribution), (h) 1:82.3 (0.3 mL:24.7 mL) and (i) pure ethanol (0 mL:25 mL, sheets agglomerating to urchin-like morphologies). | ||
Fig. 3 shows the PXRD patterns of the products prepared at different water:ethanol ratios. The diffractograms of the products prepared with water and water:ethanol ratios between 5:1 and 1:4 show the reflections of VO2(B) (JCPDS 812392, Fig. 3a–e). Some reflections display sharper profiles than others. This is indicative of strongly anisotropic crystallites, as the crystallite size is inversely proportional to the full width at half maximum (fwhm).61,62 Anisotropic crystallite sizes are in good agreement with the rod-like morphology of the nanoparticles (Fig. 3a–e), where the reflections should be sharper along the lattice direction associated with the long axis of the rods. For the rods obtained in pure water, the 00l reflections are weak. For the product obtained at a water:ethanol ratio of 1:9 (Fig. 3f), there or virtually no resolved reflections i.e., the intensities of the X-ray diffraction patterns are extremely broad (compared to the diffractograms in Fig. 3a–e). The principal reflection is roughly at the same 2θ value as that for VO2(B), indicating that the main product is also VO2(B), albeit with very low crystallinity, i.e., no long range order.
The PXRD patterns of the sheet-like products prepared with an ethanol excess (water–ethanol ratio < 1:49, Fig. 3g–i) also show broad reflections, indicating a low crystallinity. However, the weak reflection at 2θ = 15.7° in Fig. 3g and h, which is not present in the sample prepared from pure ethanol, does not match the diffraction pattern of VO2(B). Its position roughly corresponds to the stacking separation of layers in the structure of layered vanadium oxides (like V2O5), although no other reflections of V2O5 are present. This might be compatible with the formation of a second phase which may be present as a surface (mono)layer on the crystallites of VO2(B), but might also be due to the sheet-like structure. The remaining broad intensities match in essence the diffraction pattern of VO2(B), indicating that it is still the principal phase, although the reflections are also very broad and weak. The diffractogram in Fig. 3i is again not well defined, and the broad reflections are in harmony with the morphology of the particles seen in the TEM images (Fig. 3g–i). These products are made up of nanostructures with thin layers forming sheets or urchins. Typical for layered structures (stacked in a random manner) are broad reflections with partial overlap. The overall crystallinity of the samples is very low, resulting in weak scattering (and broad/weak reflections). Due to the poor crystallinity and the lamellar morphology it was difficult to clearly assign individual reflections and to assign the crystalline phase unambiguously. The change in morphology from rod- to sheet-like structures observed in the TEM would be compatible with a change in crystal structure of VO2(B) to a sheet-like structure and possibly the formation of a surface layer containing fragments of the V2O5 structure. This assumption would also be in harmony with a Raman band at ∼990 cm−1 (Fig. 5, vide infra) that indicates the presence of polymeric V2O563,64 containing distorted VO5 pyramids sharing edges and corners as structural motif, while VO2 (whose structure is based on edge-sharing VO6 octahedra) is responsible for the remaining bands.65
This might arise from a “monolayer” of V2O5 on the surface of the VO2 nanocrystals (that does not show up in the X-ray diffractograms due to the absence of long-range order). Although the XPS spectra (Fig. 7, vide infra) could indicate that vanadium remains in oxidation state +4 (V 2p at 516.9 eV) consistent with a VO2 composition the V 2p signals of V4+ and V5+ are to close (516.3, 517.3 eV)66 to allow for a clear distinction.
The VO2 nanorods, -sheets and -urchins were characterized by Raman spectroscopy (Fig. 5). The Raman spectrum of the nanorods shows bands at 283 and 406 cm−1 that can be assigned to V–O bending vibrations. The band at 477 cm−1 belongs to the V–O–V bending vibration, and the broad peak at 526 cm−1 corresponds to the stretching mode of triply coordinated V3–O. The V2–O band is located at 695 cm−1 whereas the V–O stretch appears at 878 cm−1. Additionally, there is another band at 929 cm−1 which is attributed to a (local, i.e., defect-related) V4+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety.67
O moiety.67
The strong Raman peak at 995 cm−1 might be caused by the V–O stretching mode of a V2O5 surface layer.63,64,68,69 Still, the Raman and X-ray data corroborate that the principle phase of the rods is VO2. The different signal-to-noise ratios between sheets and urchins on one hand and rods on the other hand are due to different filters. The nanosheets and -urchins decompose under the measurement conditions for the rods. Therefore, the radiation intensity is lower for the sheets and urchins, while no peaks were observed for the rods. Since the local structure of the phases crystallizing in sheet and urchin form is not complete from the PXRD and the vibrational spectra, it remains difficult to assign the bands and phase identity unambiguously. Both, urchins and sheets, exhibit their strongest band at the same wavenumber (855 cm−1). This position is typical of a V–O stretch. Additionally, a weak band appears at 366 cm−1, which is due to V–O bending modes.70 The Raman spectra suggest in agreement with the PXRD and IR data that the sheets and urchins have the same structure, because they show the same characteristic bands. The difference in fwhm of the VO2 urchins and sheets is due to their respective morphology. The urchins show a higher degree of disorder as the petals are randomly oriented which in turn leads to a strong peak broadening in the Raman spectrum.
To further characterize the nanosheets, their composition was examined by energy dispersive X-ray spectroscopy (EDX, Fig. 6a). EDX confirms the presence of vanadium, oxygen, carbon and copper in the sample. The copper signal is from the copper grid on which the sample was measured. In addition, the carbon signal is likely caused in part by (i) the carbon coating on the grid. The carbon signal further originates (ii) from the organic components that serve as capping agents for the nanoparticles. The FT-IR spectrum of the sheets shows the presence of alkyl groups (Fig. 4). Therefore, the layers are composed of vanadium and oxygen, while part of the carbon signal likely originates from the ligands covering the nanoparticles. Fig. 6b shows a dark field image of the nanosheets with a size between 50 and 70 nm. Here, upright sheets with a thickness of 5 nm can be seen. Some electron diffraction patterns of the sheets are shown in Fig. 6c and d. Fig. 6c shows the patterns within the sheet layer. The sheet spacings are 6.6 Å and 23.8 Å (compatible with the VO2(B) structure10) with systematic extinctions along the short axis. A diffraction pattern from the side of a platelet is shown in Fig. 6d, which shows spacings of 14.7 Å. Overall, the reflections are very broad, and the crystallinity of the associated phase is very low. Therefore, it was not possible to determine the cell parameters of the VO2 sheets by electron diffraction, especially since the material is radiation sensitive. The sheets (with the organic surface ligands) decompose under the beam. Either bubble formation or hole etching occurs as in Fig. 6e (red circle), which prevents a structure determination by automated diffraction tomography (ADT).71–73
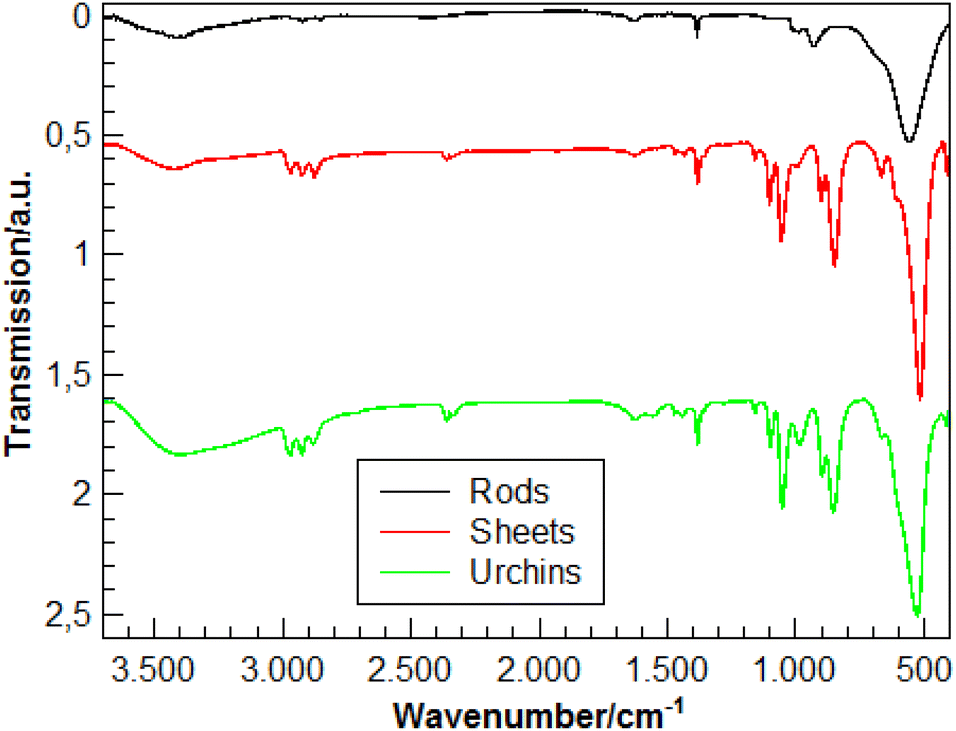 | ||
| Fig. 4 IR spectra of the VO2 nanorods (black), VOx nanosheets (red) and VOx nano-urchins. | ||
Finally, the oxidation state of the vanadium atoms in the vanadium oxide nanoparticles was determined by XPS spectroscopy. In the XPS spectra (Fig. 7), the oxidation state of the respective atoms determines the peak position via the electron binding energies (or electron work functions). The nanorods, -urchins, and -sheets show the vanadium 2p peaks at 516.4 eV. The assignment of oxidation states (V4+vs. V5+) based on XPS data is not unambiguous, because the V 2p signals of the different oxides may appear at similar energies (516.3, 517.3 eV) and the value for the nanorods, -urchins, and -sheets (516.4 eV) is in between. Moreover, the oxygen 1s peak (530 eV) which could not be deconvoluted agrees for the three particle types, i.e., nanorods, -urchins, and -sheets contain vanadium in the same oxidation states. Since the phase identity of the crystalline fraction of the nanorods is assured from the PXRD diffractograms and the Raman spectra, (Fig. 3 and 5) the urchins and -sheets also contain V4+, i.e., VO2. The positions of the vanadium 2p (516.9 eV) and oxygen 1s orbitals (530 eV) agree with literature data for the respective bands.74,75
 | ||
| Fig. 5 Normalized Raman spectra of the VO2 nanorods (black), nano-sheets (red) and nano-urchins (green). | ||
 | ||
| Fig. 6 (a) TEM EDX, (b) darkfield image, (c and d) electron diffraction patterns and (e) darkfield image showing radiation damage of the VO2 sheets. | ||
 | ||
| Fig. 7 XPS data of VO2 nanorods (black), VO2 nanosheets (red) and -urchins (green). | ||
Catalytic evaluation
Many partial oxidation catalysts in industrial applications are based on vanadium and molybdenum oxides.5,19–21,32 The partial oxidation of alcohols is particularly well studied because the resulting aldehydes and ketones are important and reactive intermediates in industrial chemistry. Highly active catalysts have been developed and optimized mostly empirically by high-throughput methods. The structure of these catalysts often consists of several cooperating phases. Moreover, the structure of the surface, where the catalytic reaction proceeds, is rich in structural defects and structurally different from the bulk. Therefore, nanoparticles with a large variety of possible active surface sites are interesting targets for experimentation. They are also difficult to prepare in pure form by high-throughput reactions. In this context, VO2 nanoparticles with different morphologies are interesting model systems.Here we investigate the effect of particle morphology on the catalytic performance of the VO2 nanoparticles. All three morphologies, i.e., VO2 nanorods, -sheets, and -urchins were used as catalysts for the oxidation of two model compounds, benzyl alcohol and furfuryl alcohol. The experimental procedure for the oxidation of alcohols and GC chromatogram showing product purity of furfuryl alcohol and benzyl alcohol oxidation using VO2 nano-urchins is shown in the ESI Fig. S2 and S3.† Oxidation reactions were performed under identical conditions reaction temperature 150 °C with O2 gas as oxygen source for comparison. All three VO2 particle morphologies were active for the oxidation of benzyl alcohol to benzyl aldehyde. The results in Table 1 show that the catalytic efficiency is morphology dependent. VO2 nano-urchins showed highest yield (∼100%) and a specific activity of 3.01 mmol g−1 h−1. VO2 nanorods showed the lowest yield of ∼71% with a specific activity of 2.13 mmol g−1 h−1. VO2 nanosheets yielded a slightly lower conversion (87%) than urchins. For the oxidation of furfuryl alcohol to furfural the catalytic activity was significantly lower (Table 2) with a maximum conversion of ∼67% for the urchin morphology. Still, the trend in terms of morphology dependence remained. VO2 urchins showed the highest conversion (67%), VO2 nanosheets a slightly lower conversion of ∼60%. while the lowest conversion (∼33%) was achieved with VO2 nanorods. The lower product conversion during the oxidation of furfuryl alcohol compared to that of benzyl alcohol can may be attributed to the O heteroatom in the furfuryl ring system.
| Morphology (VO2 NPs) | Benzyl alcohol → Benzaldehyde |
|---|---|
| Conversion (%) | |
| Nano-urchins | 100 |
| Nanosheets | 87 |
| Nanorods | 71 |
| Morphology (VO2 NPs) | Furfuryl alcohol → Furfural |
|---|---|
| Conversion (%) | |
| Nanourchins | 67 |
| Nanosheets | 60 |
| Nanorods | 33 |
Surface area analysis (using multipoint BET measurement) shows the correlation between the catalytic activity of the VO2 nanocatalysts and their surface area. VO2 nanorods have a surface area of 21.7 m2 g−1 measured, which is within the typical size range for nanorods.76 The surface area of the urchins and sheets is significantly higher (124.6 and 73.9 m2 g−1, respectively) due to their higher surface-to-volume ratio. Specifically, the surface area of the VO2 urchins is remarkably high compared to the reported surface areas of sheet- and urchin-like particles. Xu et al.77 synthesized urchin-like V2O3 particles with a surface area of 48.57 m2 g−1, while Pan et al.78 obtained flower-like V2O5 with a surface area of 33.64 m2 g−1. The differences in surface area are due to the difference in the overall size of the particles, which was in the nanometer range for the VO2 urchin-type particles, while the diameters of the particles reported in the literature are in the micrometer range. Another factor is the thickness of the “petals”. The thinner the petals, the larger the surface area. Pang et al.79 obtained flower-like V4O9 microparticles with a specific surface area of 107.9 m2 g−1, which can be attributed to the thin layers of 2 nm. The surface areas of nanoparticles are usually much larger than those of bulk vanadium oxides (e.g., V2O5 with 8.4 m2 g−1). When correlating the surface areas with the catalytic performance, the catalytic performance is high with increasing surface area. The highest conversion of alcohol to aldehyde is obtained for urchins and sheets, while the lower active surface area of nanorods entails a lower conversion. For all morphologies there are indications for surface layers with local V2O5-type characteristics (without long-range order). Since these surface layers with local V2O5-type moieties but without translational symmetry are present on all surfaces, they affect the catalytic activity of the VO2 particles in a comparable way. To confirm that enhanced catalytic activity is related to VO2 nanomaterials with traces of V2O5 surface patches (VO2/V2O5 hybrid) and not just due to the surface V2O5 layer, we provide a comparison based on previous reports.81–86 (Table S1†). The results in Table S1† show that, depending on their morphology, VO2 nanomaterials with patches of a V2O5 surface layer exhibit better catalytic activity than pure V2O5 nanoparticles as well as V2O5 nanoparticles on different support surfaces.
The turnover number (TON) and turnover frequency (TOF) for the three different morphologies of VO2 nanocatalysts are also compiled in Tables 1 and 2. For the oxidation of benzyl alcohol to benzaldehyde, the TON and TOF values range from 9.97 to 7.08 and from 2.49 to 1.77 h−1, respectively. For the oxidation of furfuryl alcohol to furfural, the values are 8.00–3.94 and 2.00–0.99 h−1, respectively. Urchin-like VO2 nanoparticles had the highest, VO2 nanorods the lowest TON and TOF values. The results are illustrated in Fig. 8 and 9. The reusability of the VO2 nano-urchins, the best catalyst, was investigated in five cycles. The catalyst was essentially stable (activity decrease from 100% to ∼96%, Fig. 9). The VO2 materials are structurally stable as shown by the X-ray diffractogram (Fig. S4†) of the VOx nanourchins measured after the catalytic application.
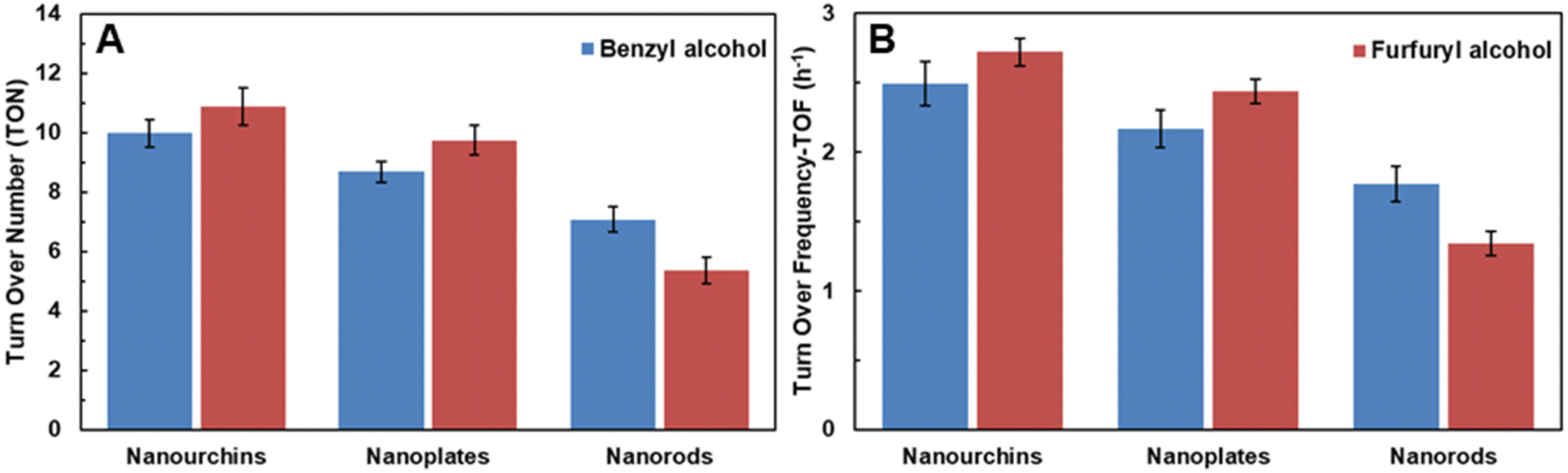 | ||
| Fig. 8 TON and TOF values for as-synthesized VO2 nanocrystals, (A) benzyl alcohol and (B) furfuryl alcohol. | ||
 | ||
| Fig. 9 Catalytic performance of VO2 nano-urchins when reused. | ||
Based on the catalytic results obtained, we propose a possible reaction mechanism for the oxidation of alcohols over the VO2 nanocatalysts (Fig. S5†). The adsorption of alcohols on the surface of nanocatalysts occur in the beginning of the catalytic cycle. Subsequently, breaking of O–H bond and loss of a proton may lead to the formation of VO2-alkoxide on the surface of the cluster, which is suggested by the DFT simulations in an earlier study.80 In the next step, the released proton is captured by adsorbed O2 on the catalyst surface, which lead to the cleavage of the C–H bond of α-C. This may lead to the generation of desired carbonyl compound and regeneration of the VO2 surface (IV).
Conclusion
By simple but very efficient reaction control, VO2 nanocatalysts with different morphologies and activities could be prepared for the selective oxidation of alcohols to aldehydes. We demonstrated this by changing the composition of a binary solvent mixture, which established a facile new hydro-/solvothermal synthesis of VO2 nanoparticles. VO2 nanorods were obtained at water:ethanol ratios between 4:1 and 3:2. Polydisperse VO2(B) nanorods were formed at a large water:ethanol ratio. At a water:ethanol ratio of 1:9 and in pure ethanol, aggregates of VOx nanosheets were formed. The formation of these aggregates may be due to a lack of water leading to aggregation because of insufficient hydration of the products. The VO2 phase identity was determined by TEM and X-ray diffraction on powder samples. XPS spectroscopy clearly showed the presence of V4+, thus supporting the VO2 phase assignment. Weak binding of alcohol surface ligands was demonstrated by IR and Raman spectroscopy. The catalytic activities of VO2 particles in oxidation reactions were investigated using two model reactions, the oxidation of benzyl alcohol to benzaldehyde and the oxidation of furfuryl alcohol to furfural. All VO2 particles were catalytically active. VO2 particles with urchin-like morphology had the larger active BET surface area and showed the highest conversion for the oxidation of benzyl alcohol to benzaldehyde and furfuryl alcohol to furfural with 100% and 67% (spec. activities 3.01 and 2.41 mmol g−1 h−1). VO2 nanorods showed the lowest conversion with 71% and 33% (spec. activities 2.13 and 1.19 mmol g−1 h−1). The conversion for alcohols in the presence of VO2 particles with sheet-like morphology was slightly lower (87% and 60%) than for VO2 with urchin-like morphology (spec. activities 2.61 and 2.16 mmol g−1 h−1). The differences in catalytic activity can be attributed to the active surface area of the catalysts as a result of particle size. The VO2 nanocatalysts are reusable; their activity remained stable even after several catalytic cycles. Our results show that (i) VO2 is a good redox catalyst for the oxidation of alcohols, and (ii) mixed-valent vanadium oxides of the Magneli-type are not needed. (iii) The particle morphology has a strong effect on the catalytic activity of a solid, which for a given compound is (iv) determined to a large extent by the active surface area.
Experimental
Chemicals
All starting materials were used without further purification. Pluronic F-127, vanadyl acetylacetonate (98%, VO(acac)2), and ethanol (p.a.), were purchased from Sigma Aldrich. Benzyl alcohol (BzOH, 99%), furfuryl alcohol (99%), 1-dodecanol (98%) and triethylamine were obtained from Acros Organics.Synthesis of VO2 (B) nanorods
In a typical synthesis, 150 mg of pluronic F-127 was dissolved in 5 mL of ethanol to which 140 mg of VO(acac)2 was added. The resulting solution was stirred for 3 h. Subsequently, it was poured into 20 mL water in a 50 mL Teflon-lined autoclave and kept at 453 K (180 °C) for 24 h. The products were separated by centrifugation (15 min, 9000 rpm) and further purified by washing with 15 mL of ethanol. Finally, the nanoparticles were precipitated by centrifugation and dried in a vacuum oven at 313 K (40 °C) for 12 h. The product was stored at ambient temperature.Synthesis of VO2 sheets
To 150 mg of pluronic F127 dissolved in 24.7 mL of ethanol 140 mg of VO(acac)2 was added, and the resulting solution was stirred for 4 h. Then, 0.3 mL of water was added, and this solution was transferred into a 50 mL Teflon-lined autoclave and kept at 453 K (180 °C) for 24 h. Finally, the products were washed, isolated and dried in a vacuum oven.Synthesis of VO2 urchins
140 mg of VO(acac)2 were added to a 150 mg of pluronic F-127 solution in 25 mL of ethanol, and the solution was stirred for 4 h. Subsequently, the solution was transferred into a 50 mL Teflon-lined autoclave and heated to 453 K (180 °C) for 24 h. The products were washed, isolated and dried in vacuo.Synthesis with dodecanol
Pluronic F127 (150 mg) was dispersed in 25 mL of dodecanol (25 mL) in a water bath at 313 K. 140 mg of VO(acac)2 was added, and the solution was stirred for 4 h. Subsequently the reaction mixture was transferred into a 50 mL Teflon-lined autoclave and heated to 453 K for 24 h. The brown product was precipitated by centrifugation (9000 rpm, 10 min) and washed with 15 mL of ethanol.Heat treatment of VO2 urchins
The dried VO2 was heated to 453 K in a corundum boat with a heating rate of 2 K min−1. This temperature was kept for 4 h before the sample was allowed to cool down at a rate of 2 K min−1.Oxidation catalysis
The catalytic oxidations were performed using benzyl alcohol and furfuryl alcohol as substrates with VO2 nanorods, -sheets and -urchin as catalysts. The reactions were performed in a 100 mL stainless steel autoclave reactor equipped with a mechanical stirrer. A typical procedure for the catalytic oxidation of alcohol was as follows. The autoclave reactor was charged with benzyl alcohol (0.61 mmol), trimethylamine (0.61 mmol), VO2 nanorods (0.050 mmol), toluene (15 mL), and O2 (14 bar) and the reaction solution was heated for 4 h at 150 °C. After completion of the reaction, the catalyst was separated by filtration. The recovered catalyst was washed with ethanol (25 mL), dried at 80 °C and reused. To determine the product selectivity, the liquid products were analyzed by gas chromatography (GC, 7890A) Agilent Technologies Inc., equipped with a flame ionization detector (FID) and a 19019S-001 HP-PONA column.Characterization
Conflicts of interest
There are no conflicts to declare.Author contributions
All authors have given approval to the final version of the manuscript.Acknowledgements
The KSU authors extend their appreciation to the Deputyship for Research and Innovation, “Ministry of Education” in Saudi Arabia for funding this research (IFKSUOR3–103–4).References
- M. Liu, B. Su, Y. Tang, X. Jiang and A. Yu, Adv. Energy Mater., 2017, 7, 1700885 CrossRef.
- J. Meyer, S. Hamwi, M. Kröger, W. Kowalsky, T. Riedl and A. Kahn, Adv. Mater., 2012, 24, 5408–5427 CrossRef CAS PubMed.
- H. Takagi and H. Y. Hwang, Science, 2010, 327, 1601–1602 CrossRef CAS PubMed.
- C. Wu and Y. Xie, Energy Environ. Sci., 2010, 3, 1191–1206 RSC.
- B. M. Weckhuysen and D. E. Keller, Catal. Today, 2003, 78, 25–46 CrossRef CAS.
- C. Wu, F. Feng and Y. Xie, Chem. Soc. Rev., 2013, 42, 5157–5183 RSC.
- J. Jeong, N. Aetukuri, T. Graf, T. D. Schladt, M. G. Samant and S. S. P. Parkin, Science, 2013, 339, 1402–1405 CrossRef CAS PubMed.
- M. Brahlek, L. Zhang, J. Lapano, H.-T. Zhang, R. Engel-Herbert, N. Shukla, S. Datta, H. Paik and D. G. Schlom, MRS Commun., 2017, 7, 27–52 CrossRef CAS.
- G. Kieslich, G. Cerretti, I. Veremchuk, R. P. Hermann, M. Panthöfer, J. Grin and W. Tremel, Phys. Status Solidi A, 2016, 213, 808–823 CrossRef CAS.
- M. Joos, G. Cerretti, I. Veremchuk, P. Hofmann, H. Frerichs, D. H. Anjum, T. Reich, I. Lieberwirth, M. Panthöfer, W. G. Zeier and W. Tremel, Inorg. Chem., 2018, 57, 1259–1268 CrossRef CAS PubMed.
- E. Strelcov, Y. Lilach and A. Kolmakov, Nano Lett., 2009, 9, 2322–2326 CrossRef CAS PubMed.
- Z. Khan, P. Singh, S. A. Ansari, S. R. Manippady, A. Jaiswal and M. Saxena, Small, 2021, 17, 2006651 CrossRef CAS PubMed.
- S. Yang, Y. Gong, Z. Liu, L. Zhan, D. P. Hashim, L. Ma, R. Vajtai and P. M. Ajayan, Nano Lett., 2013, 13, 1596–1601 CrossRef CAS PubMed.
- https://www.swatchgroup.com/en/swatch-group/innovation-powerhouse/industry-40/revolutionary-battery-belenos . Accessed April 12 2023.
- M. Yu, Y. Zang, H. Yan, X. Cheng, W. Zhao, C. Liang, Y. Tong, H. Tang and X. Lu, Adv. Funct. Mater., 2015, 25, 3534–3540 CrossRef CAS.
- I. E. Wachs, Dalton Trans., 2013, 42, 11762–11769 RSC.
- F. Natalio, R. André, A. F. Hartog, B. Stoll, K. P. Jochum, R. Wever and W. Tremel, Nat. Nanotechnol., 2012, 7, 530–535 CrossRef CAS PubMed.
- K. Herget, F. Pfitzner, H. Frerichs, M. N. Tahir and W. Tremel, Adv. Mater., 2018, 30, e1707073 CrossRef PubMed.
- Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, Wiley–VCH, Weinheim, 1997 Search PubMed.
- J. M. Thomas and W. J. Thomas, Principles and Practice of Heterogeneous Catalysis, VCH, Weinheim, 1997 Search PubMed.
- J. Hagen, Industrial Catalysis, A Practical Approach, 3rd edn, Wiley–VCH, Weinheim, 2015 Search PubMed.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS PubMed.
- P. Mars and D. W. van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- D. Matthey, J. G. Wang, S. Wendt, J. Matthiesen, R. Schaub, E. Lægsgaard, B. Hammer and F. Besenbacher, Science, 2007, 315, 1692–1696 CrossRef CAS PubMed.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- B. L. M. Hendriksen, M. D. Ackermann, R. van Rijn, D. Stoltz, I. Popa, O. Balmes, A. Resta, D. Wermeille, R. Felici, S. Ferrer and J. W. M. Frenken, Nat. Chem., 2010, 2, 730–734 CrossRef CAS PubMed.
- C. Wöll, Phys. Chem. Chem. Phys., 2016, 18, 19713–19721 RSC.
- B. Frank, R. Fortrie, C. Hess, R. Schlögl and R. Schomaecker, Appl. Catal., A, 2009, 353, 288–295 CrossRef CAS.
- K. Chen, N. Liu, M. Zhang and D. Wang, Appl. Catal., B, 2017, 212, 32–40 CrossRef CAS.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4(10), 3357–3380 CrossRef CAS.
- M. C. Casey and D. E. Cliffel, Anal. Chem., 2015, 87, 334–337 CrossRef CAS PubMed.
- J. Haber, Catal. Today, 2009, 142, 100–113 CrossRef CAS.
- R. Berenguer, M. O. Guerrero-Perez, I. Guzman, J. Rodríguez-Mirasol and T. Cordero, ACS Omega, 2017, 2, 7739–7745 CrossRef CAS PubMed.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS.
- D. Yun, Y. Song and J. E. Herrea, ChemCatChem, 2017, 9, 3655–3669 CrossRef CAS.
- A. Chieregato, C. Bandinelli, P. Conception, D. Soriano, F. Puzzo, F. Basile, F. Cavani and J. M. Lopez-Nieto, ChemSusChem, 2017, 10, 234–244 CrossRef CAS PubMed.
- A. Pan, H. B. Wu, L. Wu and X. W. Lou, Angew. Chem., 2013, 125, 2282–2286 ( Angew. Chem., Int. Ed. , 2013 , 52 , 2226–2230 ) CrossRef.
- E. Uchaker, N. Zhou, Y. Li and G. Cao, J. Phys. Chem. C, 2013, 117, 1621–1626 CrossRef CAS.
- N. Pinna, M. Willinger, K. Weiss, J. Urban and R. Schlögl, Nano Lett., 2003, 3(8), 1131–1134 CrossRef CAS.
- H. A. Therese, F. Rocker, A. Reiber, J. Li, M. Stepputat, G. Glasser, U. Kolb and W. Tremel, Angew. Chem., 2005, 117, 267–270 ( Angew. Chem., Int. Ed. , 2005 , 44 , 262–265 ) CrossRef.
- Y. Liu, E. Uchaker, N. Zhou, J. Li, Q. Zhang and G. Cao, J. Mater. Chem., 2012, 22, 24439–24445 RSC.
- N. Pinna and M. Niederberger, Angew. Chem., 2008, 120, 5372–5385 ( Angew. Chem. Int. Ed. , 2008 , 47 , 5292–5304 ) CrossRef.
- R. Li and C.-Y. Liu, Mater. Res. Bull., 2010, 45, 688–692 CrossRef CAS.
- W. Li, S. Ji, Y. Li, A. Huang, H. Luo and P. Jin, RSC Adv., 2014, 4, 13026–13033 RSC.
- K. Vikrant, S. Weon, K.-H. Kim and M. Sillanpää, Photonics Nanostruct., 2022, 49, 100993 CrossRef.
- P. Liu, K. Zhu, Y. Gao, Q. Wu, J. Liu, J. Qiu, Q. Gu and H. Zheng, CrystEngComm, 2013, 15, 2753–2760 RSC.
- H. F. Xua, Y. Liu, N. Wei and S. W. Jin, Optik, 2014, 125, 6078–6081 CrossRef.
- K. K. Dey, D. Bhatnagar, A. K. Srivastava, M. Wan, S. Singh, R. R. Yadav and M. Deepa, VO2 nanorods for efficient performance in thermal fluids and sensors, Nanoscale, 2015, 7, 6159–6172 RSC.
- L. Zhang, J. Yao, Y. Guo, F. Xia, F. Cui, B. Liu and Y. Gao, Ceram. Int., 2018, 44, 19301–19306 CrossRef CAS.
- Y. Zhang, M. Fan, X. Liu, G. Xie, H. Li and C. Huang, Solid State Commun., 2007, 144, 259–263 CrossRef.
- P. Liu, Y. Xu, K. Zhu, K. Bian, J. Wang, X. Sun, G. Tai, Y. Gao, H. Luo, L. Lu and J. Liu, J. Mater. Chem. A, 2017, 5, 8307–8316 RSC.
- S. Zhong, Z. Zou, S. Le, C. Shu, S. Zhang and J. Geng, ACS Appl. Nano Mater., 2022, 5, 18023–18034 CrossRef CAS.
- S. Shi, Y. Yu, X. Feng, R. Qi and Y. Zhao, Batteries, 2023, 9, 95 CrossRef CAS.
- A. Kojima, K. Okazaki, S. Ooi and K. Saito, Inorg. Chem., 2009, 48, 1168–1172 CrossRef PubMed.
- O. Karahan, A. Tufani, S. Unal, I. B. Misirlioglu, Y. Z. Menceloglu and K. Serdur, Nanomaterials, 2021, 11, 752 CrossRef CAS PubMed.
- S. Xiong, B. Xi, C. Wang, G. Zou, L. Fei, W. Wang and Y. Qian, Chem. – Eur. J., 2007, 13, 3076–3081 CrossRef CAS PubMed.
- D. M. Minic and V. A. Blagojevic, CrystEngComm, 2013, 15, 6617–6624 RSC.
- M. Niederberger and G. Garnweitner, Chem. – Eur. J., 2006, 12, 7282–7302 CrossRef CAS PubMed.
- H. Weingärtner and E. U. Franck, Angew. Chem., Int. Ed., 2005, 44, 2672–2692 CrossRef PubMed.
- R. I. Walton, Chem. – Eur. J., 2020, 26, 9041–9069 CrossRef CAS PubMed.
- P. Scherrer, Nachr. Ges. Wiss. Göttingen, 1918, 26, 98–100 Search PubMed.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- J. T. Grant, C. A. Carrero, A. M. Love, R. Verel and I. Hermans, ACS Catal., 2015, 5, 5787–5793 CrossRef CAS.
- S. Barman, N. Maity, K. Bhatte, S. Ould-Chikh, O. Dachwald, C. Haeßner, Y. Saih, E. Abou-Hamad, I. Llorens, J.-L. Hazemann, K. Köhler, V. D′ Elia and J.-M. Basset, ACS Catal., 2016, 6, 5908–5921 CrossRef CAS.
- P. Shvets, O. Dikaya, K. Maksimova and A. Goikhman, J. Raman Spectrosc., 2019, 50, 1226–1244 CrossRef CAS.
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. De Gryse, J. Electron Spectrosc. Relat. Phenom., 2004, 135, 167–175 CrossRef CAS.
- S. H. Lee, H. M. Cheong, M. J. Seong, P. Liu, C. E. Tracy, A. Mascarenhas, J. R. Pitts and S. K. Deb, J. Appl. Phys., 2002, 92, 1893 CrossRef CAS.
- M. Pradhan, A. Roy, A. K. Sinha, R. Sahoo, D. Deb and T. Pal, Dalton Trans., 2015, 44, 1889–1899 RSC.
- I. Mjejri, N. Etteyeb and F. Sediri, Mater. Res. Bull., 2013, 48, 3335–3341 CrossRef CAS.
- R. L. Frost, K. L. Erickson, M. L. Weier and O. Carmody, Spectrochim. Acta, Part A, 2005, 61, 829–834 CrossRef PubMed.
- U. Kolb, E. Mugnaioli and T. E. Gorelik, Cryst. Res. Technol., 2011, 46, 542–554 CrossRef CAS.
- E. Mugnaioli, I. Andrusenko, T. Schüler, N. Loges, R. E. Dinnebier, M. Panthöfer, W. Tremel and U. Kolb, Angew. Chem., Int. Ed., 2012, 51, 7041–7045 CrossRef CAS PubMed.
- M. A. Lange, Y. Krysiak, J. Hartmann, M. N. Tahir, M. Panthöfer, T. Reich, M. Mondeshki, U. Kolb and W. Tremel, Adv. Funct. Mater., 2021, 30, 1909051 CrossRef.
- J. Mendialdua, R. Casanova and Y. Barbaux, J. Electron Spectrosc. Relat. Phenom., 1995, 71, 249–261 CrossRef CAS.
- A. Gloskovskii, S. A. Nepijko, G. Schönhense, H. A. Therese, A. Reiber, H. C. Kandpal, G. H. Fecher, C. Felser, W. Tremel and M. Klimenkov, J. Appl. Phys., 2007, 101, 084301 CrossRef.
- A. Birkel, F. Reuter, D. Koll, S. Frank, R. Branscheid, M. Panthöfer, E. Rentschler and W. Tremel, CrystEngComm, 2011, 13, 2487–2493 RSC.
- Y. Xu, L. Zheng, C. Wu, F. Qi and Y. Xie, Chem. – Eur. J., 2011, 17, 384–391 CrossRef CAS PubMed.
- A. Pan, H. B. Wu, L. Yu, T. Zhu and X. W. Lou, ACS Appl. Mater. Interfaces, 2012, 4, 3874–3879 CrossRef CAS PubMed.
- H. Pang, Y. Dong, S. L. Ting, J. Lu, C. Li, D.-H. Kim and P. Chen, Nanoscale, 2013, 5, 7790–7794 RSC.
- S. Zavahir, Q. Xiao, S. Sarina, J. Zhao, S. Bottle, M. Wellard, J. Jia, L. Jing, Y. Huang, J. P. Blinco, H. Wu and H.-Y. Zhu, ACS Catal., 2016, 6, 3580–3588 CrossRef CAS.
- K. Alagiri and K. R. Prabhu, Tetrahedron, 2011, 67, 8544–8551 CrossRef CAS.
- G. K. Kara, J. Rahimi, M. Niksefat, R. Taheri-Ledari, M. Rabbani and A. Maleki, Mater. Chem. Phys., 2020, 250, 122991 CrossRef CAS.
- R. Upadhyay, S. Kumar and S. K. Maurya, ChemCatChem, 2021, 13, 3594–3600 CrossRef CAS.
- N. Anbu, M. B. R. Kamalam, K. Sethuraman and A. Dhakshinamoorthy, ChemistrySelect, 2018, 3, 12725–12733 CrossRef CAS.
- C. Srilakshmi, V. Basava, G. Ramesh and M. Manjunath, ChemistrySelect, 2020, 5, 4500–4508 CrossRef CAS.
- J. Li, B. Ren, X. Yan, P. Li, S. Gao and R. Cao, J. Catal., 2021, 395, 227–235 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt02605a |
| This journal is © The Royal Society of Chemistry 2024 |