
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SMARTpy: a Python package for the generation of cavity steric molecular descriptors and applications to diverse systems†
Beck R.
Miller
 ,
Ryan C.
Cammarota
and
Matthew S.
Sigman
*
,
Ryan C.
Cammarota
and
Matthew S.
Sigman
*
Department of Chemistry, University of Utah, 315 South 1400 East, Salt Lake City, UT 84112, USA. E-mail: matt.sigman@utah.edu
First published on 3rd January 2025
Abstract
Steric molecular descriptors designed for machine learning (ML) applications are critical for connecting structure–function relationships to mechanistic insight. However, many of these descriptors are not suitable for application to complex systems, such as catalyst reactive site pockets. In this context, we recently disclosed a new set of 3D steric molecular descriptors that were originally designed for dirhodium(II) tetra-carboxylate catalysts. Herein, we expand the spatial molding for rigid targets (SMART) descriptor toolkit by releasing SMARTpy; an automated, open-source Python API package for computational workflow integration of SMART descriptors. The impact of the structure of the molecular probe for generation of SMART descriptors was analyzed. Resultant SMART descriptors and pocket features were found to be highly dependent upon probe selection, and do not scale linearly. Flexible probes with smaller substituents can explore narrow pocket regions resulting in a higher resolution pocket imprint. Macrocyclic probes with larger substituents are more applicable to larger cavities with smooth boundaries, such as dirhodium paddlewheel complexes. In these cases, SMARTpy provides comparable descriptors to the original calculation method using UCSF Chimera. Finally, we analyzed a series of case studies demonstrating how SMART descriptors can impact other areas of catalysis, such as organocatalysis, biocatalysis, and protein pocket analysis.
Introduction
Structure–function relationships are leveraged to provide mechanistic insight into the connections between catalyst structural features and observed experimental outcome. A diverse array of steric molecular descriptors has historically captured structural features of 3D-representations for application to statistical modeling and machine learning (ML) prediction of reaction performance. Traditional steric descriptors, including Sterimol1,2 (L, B1, B5) and buried volume3,4 (VBur), are successfully applied to diverse areas of catalysis and provide unique insight into structure–function relationships from resultant ML models. However, limitations of many steric molecular descriptors prevent their application to certain complex systems.We recently developed a set of steric molecular descriptors tailored for dirhodium paddlewheel catalysts.5–7 These are privileged catalyst scaffolds with large, conical reactive pockets, that provide a confined environment conducive to selective transformations.8–10 As a result of these complex 3D-conformations, steric features of these catalysts cannot be adequately parametrized using traditional molecular descriptors.5 For instance, Sterimol descriptors are highly dependent upon the selection of the L-axis. This can be difficult to apply to systems with multiple bridging ligands and distinct axial binding sites (Fig. 1a). Similarly, VBur assumes a spherical binding environment around the metal center of interest, and the radius of search space is often too small to encompass the distal ligand environment (Fig. 1b). As a result, these steric descriptors that were designed for small molecule catalysts were found to be insufficient to describe the complex cavity environments in dirhodium catalysts that contain subunits beyond the scope of amino acids or DNA-bases, and thus cannot be applied to many small molecule transition metal catalysts. Second, these programs are designed to analyze pockets encompassed within or between larger molecules and can struggle to provide a reasonable cut off for pockets with a wide entry. Finally, most methods interpret pocket accessibility based on solvent access.11–13 This method typically relies on generating a “space filling”14 model of points with assigned van der Waals radii to parametrize an active cavity through its interactions with solvent models.
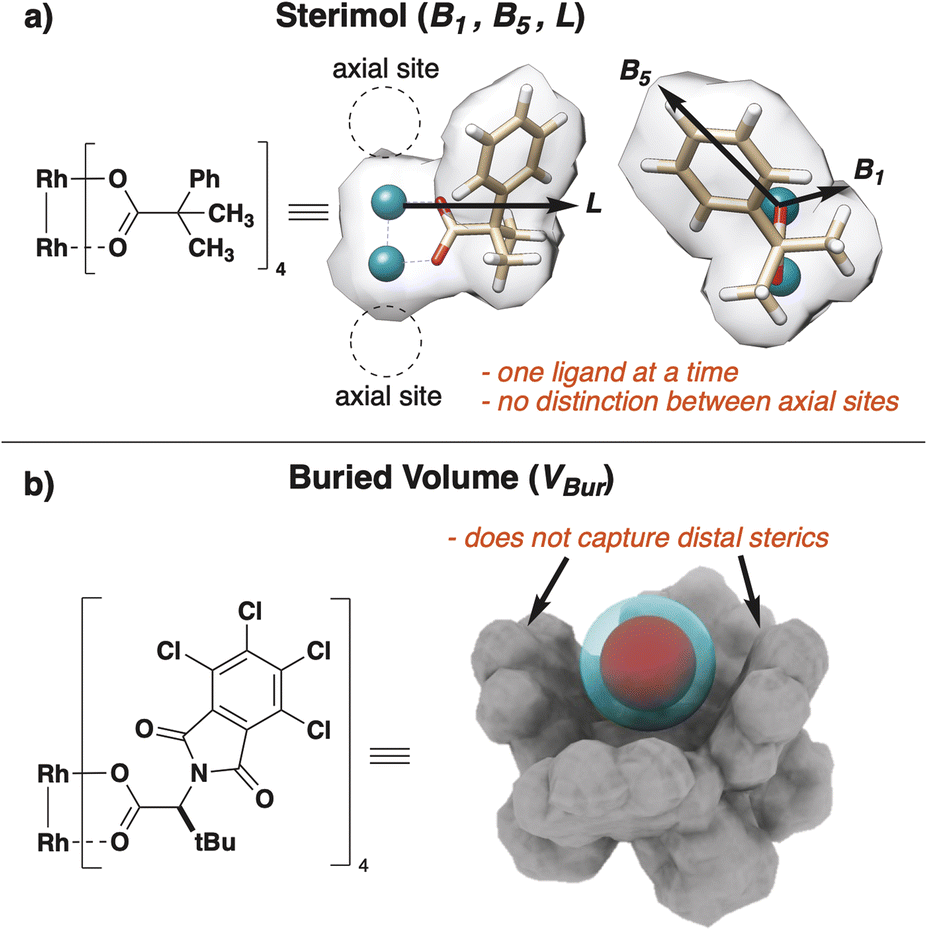 | ||
| Fig. 1 (a) Challenges of dirhodium catalysts for Sterimol descriptors. (b) Challenges of dirhodium catalysts for VBur descriptors. | ||
Other approaches for pocket description have been explored, including generating representations based on ligand docking, electron density maps, grid-based approaches,15 and machine learning algorithms.16–18 These established methods can still overestimate the size and accessibility of specific regions within a pocket from the perspective of a bound molecule. Approaches based on experimental assessment of a series of docked molecules can inherently limit the domain of applicability of the pocket information to structurally similar molecules.19 Thus, a general method to generate quantitative pocket representations remain of interest.
Spatial molding for approachable rigid targets (SMART) descriptors quantify structural features at the reactive pockets of catalysts, such as cavity volume (VCAVITY), entry surface area (ESA), and contact surface area (CSA) with the surrounding ligands. These descriptors are obtained through conformational sampling of reactive site space using a generalized molecular probe. SMART descriptors were initially applied to quantify the origins of regioselectivity in dirhodium C–H functionalization of donor/acceptor carbenes5 and diastereoselectivity in dirhodium C–H insertion of donor/donor carbenes.7 Although we envisioned broader applicability to diverse areas of catalysis, the original implementation of SMART was challenging for widespread adoption, including a significant reliance on user input and the necessity for commercial software. These two factors have prevented the rapid analysis of larger data sets and limited the accessibility of the tool to a broader community of potential users.
Herein, we release SMARTpy; a Python suite uniting open-source computational packages in a fully automated workflow for the generation of SMART descriptors. In addition to description of the construction of the SMART cavities, we evaluated the impact of probe design on resultant descriptors. Finally, we demonstrate the applicability of SMART descriptors through a series of case studies. This code is open-source and available on GitHub (https://github.com/SigmanGroup/SMART-molecular-descriptors.git). A detailed description of the API is supplied in the ESI,† and all structures analyzed are available in the Git repository.
Workflow
Original workflow for SMART descriptor calculation
The workflow for generating SMART descriptors has been partially disclosed by Davies and Sigman.5 In this workflow, molecular probes were added to catalysts, checked for atomic overlap with the structure, then conformer searched, all requiring manual user input for every step. This implementation was time consuming and limited the possibility for high throughput catalyst parametrization. The most significant limitation of the original workflow is that probe conformer ensembles were generated using the OPLS3e forcefield20 and a torsional Monte Carlo (MC) algorithm implemented in MacroModel, a commercial software distributed by Schrödinger. Molecular descriptors were then calculated using the free program UCSF Chimera.21 SMARTpy employs exclusively free and open-source Python modules to generate conformer ensembles. Additionally, the package employs multiple methods for computing an array of steric descriptors.Molecular probe conformational generation in SMARTpy
The initial method for conformer searching implemented in the SMART package was a Monte Carlo inspired torsional search algorithm that rejected moves based on van der Waals overlaps with the structure. This method performed well for acyclic probes with freely rotatable bonds, but conformational searching for macrocycles was not possible using this method. Macrocyclic structures are a known limitation of torsional algorithms as rotating one bond along a macrocycle causes multiple other bonds to simultaneously rotate on the structure in different directions to maintain atomic geometry. This makes the search space difficult to explore by simple torsional methods. MacroModel conformational searching employs a version of the ConfGen algorithm disclosed by Watts et al.11 to expand applicability and speed up conformer searching. ConfGen employs a template-based method where substructures of the molecule of interest are matched to precomputed templates of conformer ensembles. Inspired by the format of the ConfGen algorithm, a similar approach was employed in SMARTpy.Using the RDKit function EmbedMultipleMolecules command, a conformer ensemble template is first generated for the free probe using the MMFF forcefield. This represents the accessibility of space to the probe unhindered by a catalyst structure (Scheme 1). This template is then fit into the pocket of interest aligned to a defined binding axis vector, and conformers are saved or rejected based on van der Waals overlap with the structure. The orientation of the probe template is rotated about the binding axis by a randomly generated displacement from a uniform distribution, and the fitting and assessing process is repeated for a user-defined number of steps. To provide a stable conformational ensemble, several methods are recommended for application of SMARTpy. First, the default number of fitting iterations is set to be 50 which was found to provide descriptor stability for the dirhodium case study structures (Fig. S4†). Second, it is recommended that the results of multiple SMARTpy runs are averaged to reduce artifacts of the fitting algorithm. The saved conformers from each fitting iteration are compiled into a single ensemble and returned as an object or optionally saved to an SDF file for later analysis.
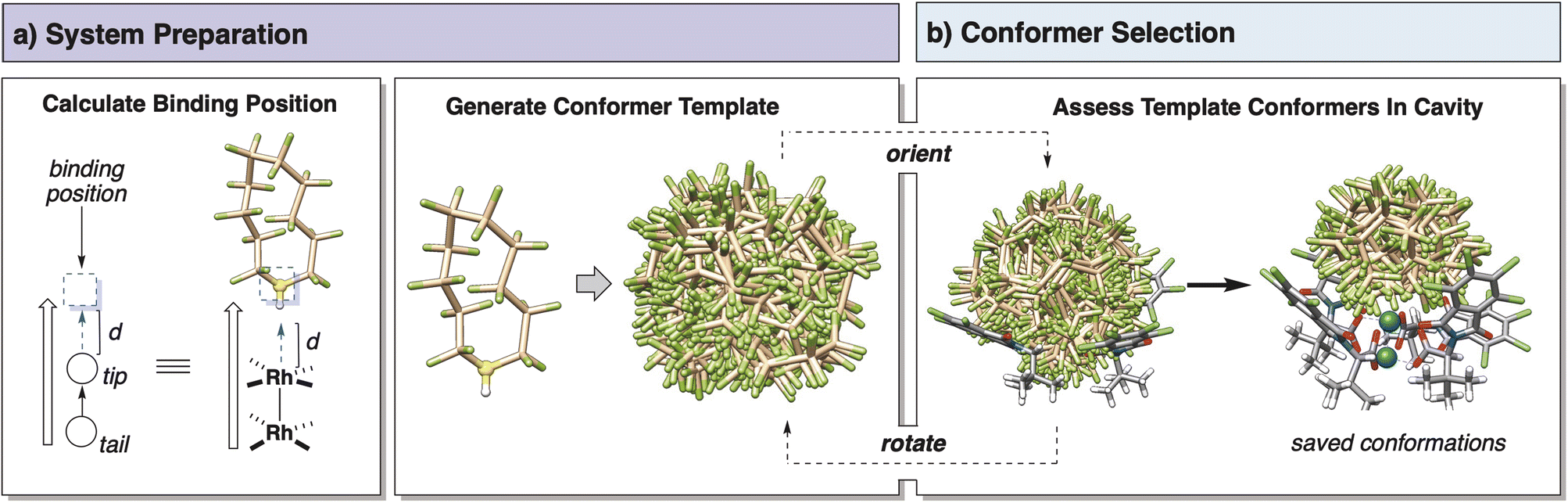 | ||
| Scheme 1 SMART template conformational search protocol. | ||
Molecular descriptor computational methods available in SMARTpy
In UCSF Chimera, the command molmap was used to enclose the probe conformers in a molecular surface from which VCAVITY and ACAVITY were computed. The molmap function in UCSF Chimera is a density-based computation that computes a surface around select atoms in a manner proportional to the atomic numbers. Open-source Python packages were implemented instead for either speed or expanded functionality to compute SMART descriptors from probe ensembles.Volume descriptors, such as VCAVITY, can be computed through two different methods. In the first method, algebraic triangulation and the alpha method12 are used to compute a surface encompassing all atoms of the probe ensemble using PyVista13 (Fig. 2b). Proximal (proxVCAVITY) and distal (distVCAVITY) volume can be computed by defining a radius for spherical intersection with the ensemble and computing the space taken up by separate portions of the cavity (Fig. 2a). This first method was implemented for speed of descriptor calculation, as assessment of the probe ensembles proved to be the fastest (Table S1†).
 | ||
| Fig. 2 (a) VCAVITY and proxVCAVITY measured using VBur computed at different radii (12 Å and 5 Å). (b) VCAVITY and ACAVITY measured by triangulation of the probe ensemble point cloud. (c) Sterimol values (L, B1, B5) measured for the cavity ensemble. | ||
In the second method, VBur is first calculated for the total probe ensemble using Morfeus. To accomplish this, the ensemble is enclosed within a large sphere and the percentage of sphere volume occupied by the conformers is computed (Fig. 2a). This method is significantly slower than the first, but is implemented for11 the opportunity to compute an extended array of SMART descriptors. The cavity space can be further subdivided into quadrants (VQUADRANT) and octants (VOCTANT). Sterimol descriptors can also be computed for the conformational ensemble, as an interpretable method to parametrize the shape of the cavity by the maximal (B5) and minimal (B1) widths perpendicular to the structure binding axis (L) (Fig. 2c).
Methods
Structures for analysis
Computed dirhodium(II) catalyst structures from a study by Shaw and Sigman7 were used to assess the impact of probe features on SMART descriptors. All conformers with symmetrical, asymmetrical, and chiral ligands were analyzed with the intent to maximize representative ligand feature diversity. All molecular probes (Table S2†) used in analysis have tetrahedral Si core atoms functionalized with either H or F. The tether atom that binds to the structure is S with a dummy H atom that is removed after initial docking. The choice of Si was initially practical for ease of pocket manipulation in UCSF Chimera with the legacy method, but many molecular units can now be used as a molecular probe core using SMARTpy.Computation of case study structures
Each case study is adapted from a literature data set or series of literature data sets. Protein and enzyme structures were obtained from the RCSB Protein Data Bank (PDB). A subset of 1,1′-bi-2-napthol (BINOL) and 1,1′-spirobiindane-7,7′-diol (SPINOL) catalysts were selected from a published computational study on BINOL catalysts to represent a diverse set of substituent steric environments.22 Initial structures of all chiral phosphoric acid (CPA) catalysts were optimized by xTB-GFN2 using the ALPB solvation method in dichloromethane. All 3D images are generated in UCSF Chimera.General utility guide
The case for general cavity descriptors
The first application of SMART descriptors aided in mechanistic understanding and modeling for dirhodium(II) catalyzed site-selective C–H functionalization of 1-bromo-4-pethylbenzene via donor/acceptor carbenes (Fig. 3a).5 This initial study explicitly quantified that more confined and rigid catalysts allowed for functionalization at the less hindered C2 site. The authors noted direct comparisons showing that traditional Sterimol and Vbur steric descriptors were unable to capture peripheral steric hindrance, the flexibility of catalyst shape, and the resulting variable accessibility of the bound carbene to the approaching substrate C–H bonds. | ||
| Fig. 3 Dirhodium(II) catalyzed reactions for SMART descriptor application. (a) First disclosure of SMART descriptors in C–H functionalization of 1-bromo-4-pethylbenzene. (b) Subsequent application and expansion of SMART descriptors in diastereoselective C–H insertion. Subsequent application of SMART descriptors. | ||
SMART descriptors were subsequently used to model diastereoselectivity in the C–H insertion of donor/donor carbenes for the cyclization of benzodihydrofurans (Fig. 3b).7 However, due to the steric demands of the intramolecular cyclization transition state, this system required a different molecular probe and a set of proximal and distal SMART descriptors. In this general utility guide, we present a mechanistic analysis of the different SMART methods utilized in these two applications to contextualize the practical considerations analyzed.
Parametrizing dirhodium(II) cavity subspace
In the initial SMART application, the full cavity space was parametrized. This proved to be advantageous for an intermolecular C–H insertion as the second substrate enters the catalyst cavity and is directed towards the rhodium carbene (Fig. 4, top). In the intramolecular cyclization, the site for C–H insertion is already within the pocket upon carbene formation, thus the space proximal to the rhodium is likely to be most influential to selectivity (Fig. 4, bottom). | ||
| Fig. 4 Ligands can direct intermolecular regioselectivity during entrance of a substrate into the pocket (top). On the other hand, ligands can direct intramolecular diastereoselectivity within the pocket after binding (bottom). | ||
This analysis prompted the division of space within the SMART cavity into proximal vs. distal with respect to the rhodium. Excluding the large, distal portion of the pocket allows for focused parametrization of the proposed active space of the cavity for the diastereoselectivity determining step. To accomplish this, a sphere was centered 2.0 Å from the rhodium (along the Rh–Rh vector) to simulate the position of a bound donor/donor carbene. The proximal cavity space was then separately parametrized from the full space.
It is generally recommended that the position of the probe be determined using information about the structure via computational or experimental methods. If a mechanistically guided “docking point” is not available, then consistency of the positioning and distance between the structure binding point and the molecular probe should be conserved across a data set.
Discussion
Analyzing molecular probe design
Users may wish to carefully tailor the probe structure to a specific system of interest, thus the careful design of a molecular probe is essential. The general SMART molecular probe is a feature with two main modes of modularity: shape and substituent radius (Fig. 5a). Probe shape can significantly influence the determination of accessible pocket space. Acyclic probes allow for exploration of smaller areas with more hindrance, such as between dirhodium ligands, resulting in a more irregular pocket than macrocyclic probes (Fig. 5b). Though both studies using SMART utilize macrocyclic probes acyclic probes are noteworthy variants that may be preferred in certain applications where high flexibility is essential, such as shape-dependent analysis.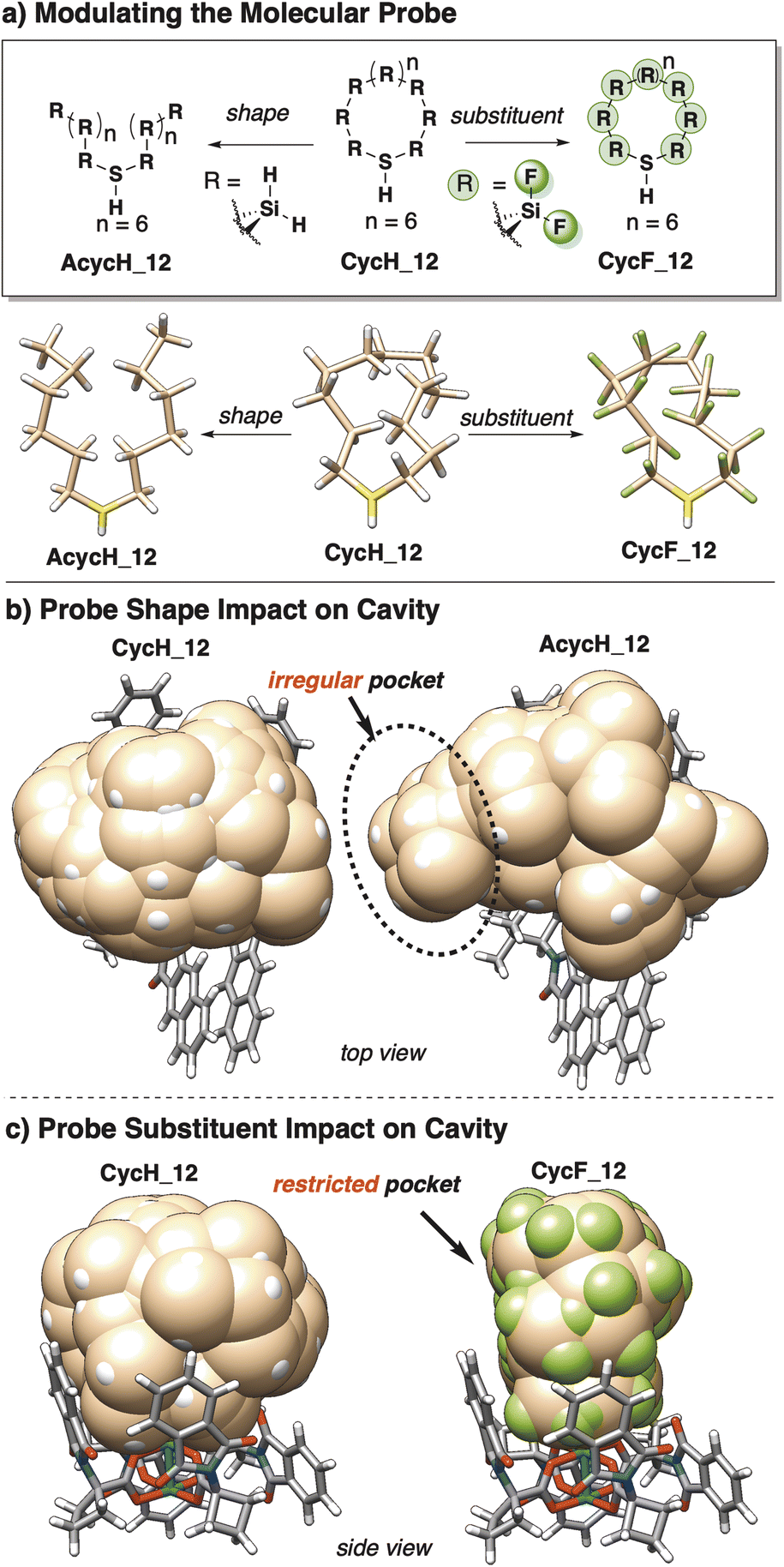 | ||
| Fig. 5 (a) Opportunities for modulating the generalized molecular probe; the core shape (left) and substituents (right). (b) The probe shape greatly impacts the shape of the cavity. Flexible probes (AcycH_12) can explore more hindered regions of a cavity than rigid probes (CycH_12). (c) Probe substituents also impact the size of the cavity. Small substituents (CycH_12) can parametrize more space than larger substituents (CycF_12). | ||
Cavities generated using acyclic probes generally result in larger values for SMART descriptors due to their increased flexibility and therefore larger search space compared to macrocyclic probes. This is shown to impact VCAVITY when varying the conformational search energy window (Fig. 6).
 | ||
| Fig. 6 Comparison of VCAVITY for molecular probes with an increasing energy window. Conformational sampling was performed using MacroModel. | ||
Macrocyclic probes (CycH_8, CycH_10, CycH_12) reach maximum VCAVITY quickly, and higher energy conformers are unable to continue to parametrize additional space by further window increases beyond 5.0 kcal mol−1. These probes are more constrained in shape, generally resulting in more regular, spherical pockets. Acyclic probes (ACycH_8, AcycH_10, AcycH_12) explore more space (larger VCAVITY) with higher conformer energy windows. The flexibility and narrow side arms of acyclic probes can access smaller cavities within a pocket of interest, such as gaps and channels between ligands, parametrizing unique cavity space compared to macrocyclic probes.
Substituents bound to probes can also determine how small of space is accessible to the probe, and consequently the amount of detail in the resultant pocket information. Probes with H and F substituents from literature probes were compared as test cases. Smaller substituents (H) allow for exploration of space closer to the surrounding structure, resulting in a larger pocket on average. VCAVITY computed by probes CycH_12 and CycF_12 show poor correlation at low VCAVITY, indicating that they are disparately parametrizing highly confined cavities (Fig. 7). The smaller CycH_12 substituents increase flexibility of the probe, allowing it to explore tighter spaces more completely.
 | ||
| Fig. 7 Comparison of H and F probe substituents. Descriptors do not correlate as well at low values of VCAVITY. The lowest volumes are more limited using CycF_12 instead of CycH_12 due to less flexibility. | ||
SMARTpy computed VCAVITY and proxVCAVITY are found to correlate well to Chimera-computed descriptors (Fig. 8a). A few interesting outliers are observed in these correlations (Fig. 8a, dashed line). These structures were visually assessed and found to have highly hindered pockets, resulting in only a single probe conformer fit (Fig. S1†). Such small probe ensembles are hypothesized to give disparate VCAVITY due to the significant dependence upon the exact probe conformation, which are fit into the pocket using a stochastic algorithm.
 | ||
| Fig. 8 (a) VCAVITY (blue, R2 = 0.80) and proxVCAVITY (red, R2 = 0.82) computed using SMARTpy correlate well to the UCSF Chimera volume descriptors. The two outliers observed are thought to be an artifact of sparse conformational ensembles where the final VCAVITY is more dependent on individual conformations than with larger ensembles. (b) ACAVITY (blue) and ESA (red) computed using SMARTpy correlate well to the UCSF Chimera area descriptors above ∼200 Å2 (blue R2 = 0.66, red R2 = 0.67). Below ∼200 Å2 (grey area), descriptors are uncorrelated (blue R2 = 0.20, red R2 = 0.10). | ||
V AREA and ESA are also shown to correlate well to UCSF Chimera descriptors (Fig. 8b). This correlation does not hold for smaller areas (Fig. 8b, gray region), attributed again to the high variability of SMART descriptors for sparse probe ensembles. We again attribute this to the area of the conformer ensemble being highly variable for sparse ensembles. VAREA is thus found to be less stable than VCAVITY, suggesting that area descriptors should only be used for dense ensembles.
Applications
SMART molecular descriptors are envisioned with broad applicability to the study and design of catalysts with irregular shapes. In this section, we demonstrate the utility of SMART for describing chiral phosphoric acids, enantioselective metalloenzyme catalysis, and protein side pockets by analyzing mechanistic implications of computed descriptors.Chiral phosphoric acid scaffolds
Chiral phosphoric acid (CPA) catalysts mediate a vast array of enantioselective transformations.23 The axially chiral scaffold asymmetrically hinders the binding site around the phosphoric acid moiety, encouraging selectivity. Diverse CPA backbones and scaffolds have been designed to sterically modulate the phosphoric acid site. Some of the most employed scaffolds include BINOL and SPINOL backbones (Fig. 9a). Variants of these scaffolds were considered to assess the ability of SMART to parametrize the steric hindrance of the reactive sites of CPAs (Fig. 9c). | ||
| Fig. 9 (a) Structure of SPINOL and BINOL backbones. (b) Probe positioning for CPA catalysts. The phosphoric acid was replaced by the molecular probe. (c) Scope of substituents analysed for both BINOL and SPINOL. | ||
One design feature commonly leveraged is the confinement and rigidity of the binding pocket.24 Similar to the dirhodium(II) catalysts, a more hindered CPA binding site is often connected to higher enantioselectivity. The dependence of CPA performance on 3,3′ substitution was assessed by Goodman, showing that the positioning of steric bulk around the phosphoric acid controls reactivity by directing substrate orientation.25,26 From this model of reactivity it was hypothesized that SMART descriptors could aid in the comparison and selection of sterically hindered CPA structures.
Due to the proposed proximal influence of the steric environment around the phosphoric acid moiety on selectivity, the probe was docked taking the place of the P atom in the BINOL and SPINOL backbones (Fig. 9b). The original CycH_12 molecular probe was implemented in the SMART workflow for these structures. Upon visual inspection of the docked scaffolds the probe was determined to be too long and would likely parametrize redundant space far from the binding site. While this could be resolved during the descriptor computation step by only considering proxVCAVITY, we employed a shorter probe (CycH_10) to increase the speed of conformer generation.
SMARTpy descriptor analysis for phosphoric acid catalysts
SPINOL catalysts were initially designed to provide more constrained reactive cavities than their BINOL analogs. Analysis of the VCAVITY for various substituted SPINOL and BINOL catalysts shows a correlation between backbones (Fig. S2†) supporting linear scaling of substituent bulk between backbone scaffolds. SPINOL catalysts have a smaller VCAVITY than BINOL analogs (Fig. 10a), supporting the initial design impetus for SPINOL scaffolds. | ||
| Fig. 10 (a) Comparison of VCAVITY for BINOL (solid line) and SPINOL (dashed line) computed using SMARTpy. BINOL catalysts have consistently larger pockets than SPINOL analogs. (b) Comparison of SMARTpy to VBur calculation of BINOL CPAs. Both proxVCAVITY and VBur were computed using a radius of 5.0 Å. VBur (red line) correlates well to the SMART descriptor distVCAVITY (grey line), indicating that VBur is being heavily influenced by the size of substituents farther from the phosphoric acid site. VBur volume does not correlate to proxVCAVITY (black, dashed line). | ||
Steric influence from substituents both proximal and distal to the active site has been shown to be influential to reactivity in different modes.25 It was hypothesized that proximal steric features dictate the orientation a bound substrate can adopt, while distal steric features can interact with a bound substrate. As a result, it is necessary to capture both proximal and distal steric effects in CPA catalysts. The values of distVCAVITY and proxVCAVITY computed at a radius of 5.0 Å are found to be uncorrelated (Fig. S3†), supporting the analysis that the two regions can impart independent influences.
Comparing these SMART descriptors directly to VBur computed analogously at a radius of 5.0 Å shows a correlation with distVCAVITY, but not with proxVCAVITY (Fig. 10b). This was an unexpected result, as the VBur computed within 5.0 Å of P could be directly analogous to proxVCAVITY. However, this implies that VBur descriptors are more dependent on the substituent identity rather than the proximal pocket environment experienced by a bound substrate. Thus, SMART descriptors provide valuable information upon decomposition of cavity space that traditional steric parameters cannot capture.
Further decomposition of the cavity into quadrants, analogous to the original CPA quadrant model (Fig. 11a and b), and octants also shows a stark difference in featurization between SMARTpy and VBur. When subtracting the maximal octant volume (MAX VOCTANT) for a SPINOL catalyst from the MAX VOCTANT for the BINOL analog, SMARTpy descriptors show that all studied BINOL catalysts have a larger MAX VOCTANT than SPINOL (Fig. 11c). This trend supports the analysis that BINOL catalysts have larger pockets than their SPINOL analogs. However, performing the same analysis with the maximal octant volume computed with Morfeus (MAX VBur,Octant) indicated that catalysts h and l have smaller octant volumes with the SPINOL scaffold than BINOL (Fig. 11d). As SPINOL was designed to afford more enclosed pockets, an effect observed in VCAVITY, this indicates that SMART octant analysis captures different information about cavity environment from VBur calculations.
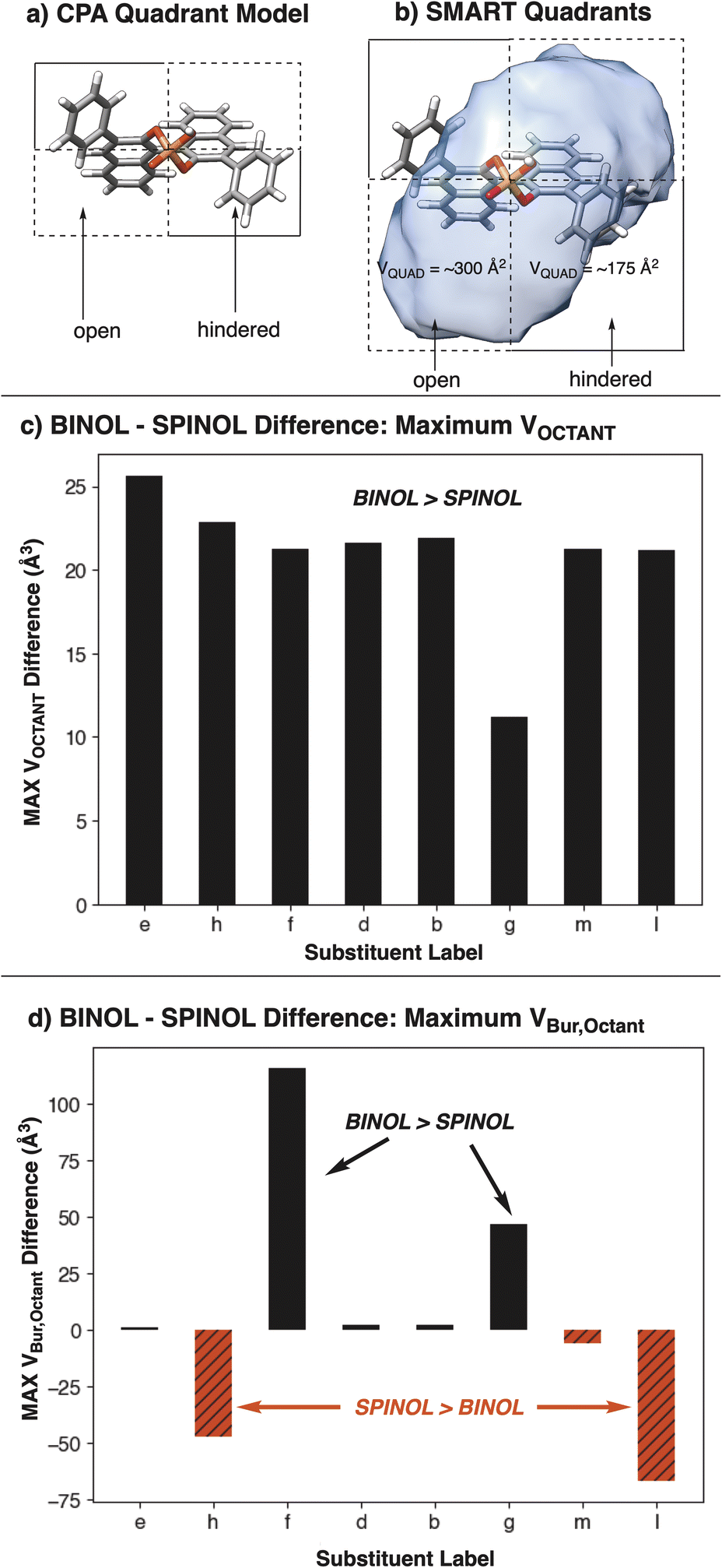 | ||
| Fig. 11 (a) Quadrant model for CPA catalysts. (b) Analogous quadrant model for SMART cavities. (c) Difference between the maximal octant VOCTANT between BINOL and SPINOL CPAs computed using SMARTpy. All BINOL catalysts have larger maximal octant volumes than the SPINOL analogs. (d) Difference between VBur,Octant of BINOL and SPINOL CPAs computed using Morfeus. Black bars represent catalysts where the maximal BINOL VOCTANT is larger than the maximal SPINOL VOCTANT. Red bars represent catalysts where the maximal SPINOL VOCTANT is larger than the maximal BINOL VOCTANT. | ||
The utility of SMARTpy is highlighted in an application to MLR modeling of asymmetric acylation catalyzed by a set of BINOL scaffold CPAs.22 Previously, Sterimol descriptors were used to model the site selectivity of primary versus secondary alcohols on 19-hydroxydehydroepiandro-sterone. Analysis of this SMARTpy MLR model indicates that more hindrance in both proximal and distal pocket regions encourages higher selectivity for the secondary alcohol position (Fig. 12). This is a similar conclusion to the steric implications of the original model.
 | ||
| Fig. 12 SMARTpy used to make an MLR model for a BINOL CPA-catalyzed transformation previously modeled by Toste and Sigman.22 The SMART descriptor MLR model shows comparable R2 to the original model. The descriptors proxVCAVITY and distVCAVITY enhance the mechanistic understanding gained from the original MLR model, where proximal and distal steric effects both impact selectivity. | ||
Protein binding pockets
The binding of small molecules to protein receptors is fundamental to many biological processes. Assessment of the binding environment in protein active sites is crucial to the design of small molecule ligands and pharmaceuticals. Important features to assess in docking studies include the size and shape of the active cavity as shape matching influences binding. Multiple free methods for quantifying cavity environments in biological ensembles, but these are generally computed using solvent accessibility and cannot subdivide cavity space according to specific binding positions. To illustrate the utility of SMART for quantifying protein binding pockets, the structure of the G-coupled protein receptor (GPR) was selected for analysis.GPRs are responsible for a variety of biological functions, and design of small molecule antagonists for GPRs is of interest in the field of computational drug design.27 The structure and dynamics of the side binding pocket of the GPR101-Gs complex (PDB: 8W8R) have been shown to influence binding in computational antagonist design.28 The structure of the GPR101-Gs protein was obtained from the PDB (PDB: 8W8R) and truncated to the side binding pocket of GPR101. Water molecules and ions were removed from the structure to allow space for the molecular probe. Multiple conformations of these proteins were not considered to reduce computational cost, but in principle this workflow could be applied to analyze pockets changes across conformational ensembles.
SMARTpy descriptor analysis for protein pocket regions
The significance of residues around the binding site can be difficult to discern, as dynamic, noncovalent interactions between the substrate and protein are influential to docking. Three residues were selected along the binding pocket to capture the local environments at different depths, represented by noncovalent attachment to different types of residues (Fig. 13a). The centroid of P30 was used as the binding reference to assess environment around the N-terminus. The C2 of the W441 residue was selected to parametrize the transmembrane domain between the N-terminus and the deeper region of the pocket. Finally, T111 was selected to probe the deeper region of the larger binding cavity. The default probe CycH_12 was unable to dock in the side pocket without overlapping with protein residues. Due to the narrow shape of the binding pocket, a linear probe (LinH_6) was utilized for protein descriptor calculation. | ||
| Fig. 13 (a) Select GPR101 (PDB: 8W8R) residues along the transmembrane domain and binding pocket. Depiction of probe positioning for each residue (center). (b) SMART cavities for GPR101 (PDB: 8W8R) side biding pocket at different residues. The N-terminus is very hindered resulting in a small cavity (purple). The deepest region of the pocket (blue) is larger and likely has more flexibility in binding molecule features. The transmembrane between the two pockets is too hindered for the probe to enter. Either conformational dynamics or favourable electrostatic interactions are hypothesized to dictate binding in this domain. | ||
The N-terminus is shown to be significantly hindered in GPR101 (Fig. 13b, purple). Additionally, the environment around residue P30 is too hindered to fit a general molecular probe (Fig. 13b, red). Based on this analysis, entrance of a small molecule into the side pocket is likely dictated by either protein flexibility to open the entrance to the binding pocket, or by favorable electrostatic interactions with neighboring residues. Further investigation into the dynamics of GPRs is outside the scope of this study but is envisioned to be achieved through SMART analysis of representative frames from molecular dynamics simulations. The deepest part of the pocket is shown to be large and irregular in shape (Fig. 13b, blue), which could allow for multiple antagonist binding orientations within the cavity.
Selectivity in Fe-porphyrin enzymes
Enzymes with engineered reactive sites can induce highly selective transformations.29–32 One well established transformation is intermolecular carbene insertion bio-catalyzed by Fe-porphyrin residues.33–35 The orientation and approach of the substrate to the Fe-carbene intermediate influences the observed selectivity, thus the residues around the porphyrin site are often specifically targeted for mutations.In 2022, Arnold disclosed a site selective C–H functionalization using engineered enzyme catalysts derived from the P411-PFA variant.36 Three variants were assessed to provide insight into the structural relationship between active site residues and observed reactivity. We reasoned that SMART descriptors could provide additional insight into the porphyrin site proximal to Fe, representative of the approach of N-phenyl-morpholine to a Fe-carbene intermediate.
Enzyme structures for the two most investigated structures were obtained from the PDB (IDs: 5UCW, 8DSG) and truncated to a single chain for analysis using SMARTpy. Due to the structural significance of the bridging water in P411-PFA (8DSG), this molecule was retained in the truncated structure.36 Remaining water, ion, and non-covalently bound residues were removed from each enzyme to allow for assessment of the empty cavity. The linear probe, LinH_6, was docked at the axial position of the Fe site to represent a bound carbene intermediate.
The major structural difference between P411-PFA and the P-4 (5UCW) variant used previously for selective amination is the perturbation of the helix directly over the binding site. In P4110PFA, a residue mutation induces a flip in orientation resulting in a site of increased steric hindrance. This artifact hinders the distal portion of the binding cavity, shown by a decrease in distVCAVITY from 260 Å3 to 237 Å3 (Fig. 14a and b). This distal hindrance around the porphyrin likely influences observed selectivity by restricting degrees of freedom during the approach of the N-phenyl-morpholine substrate.
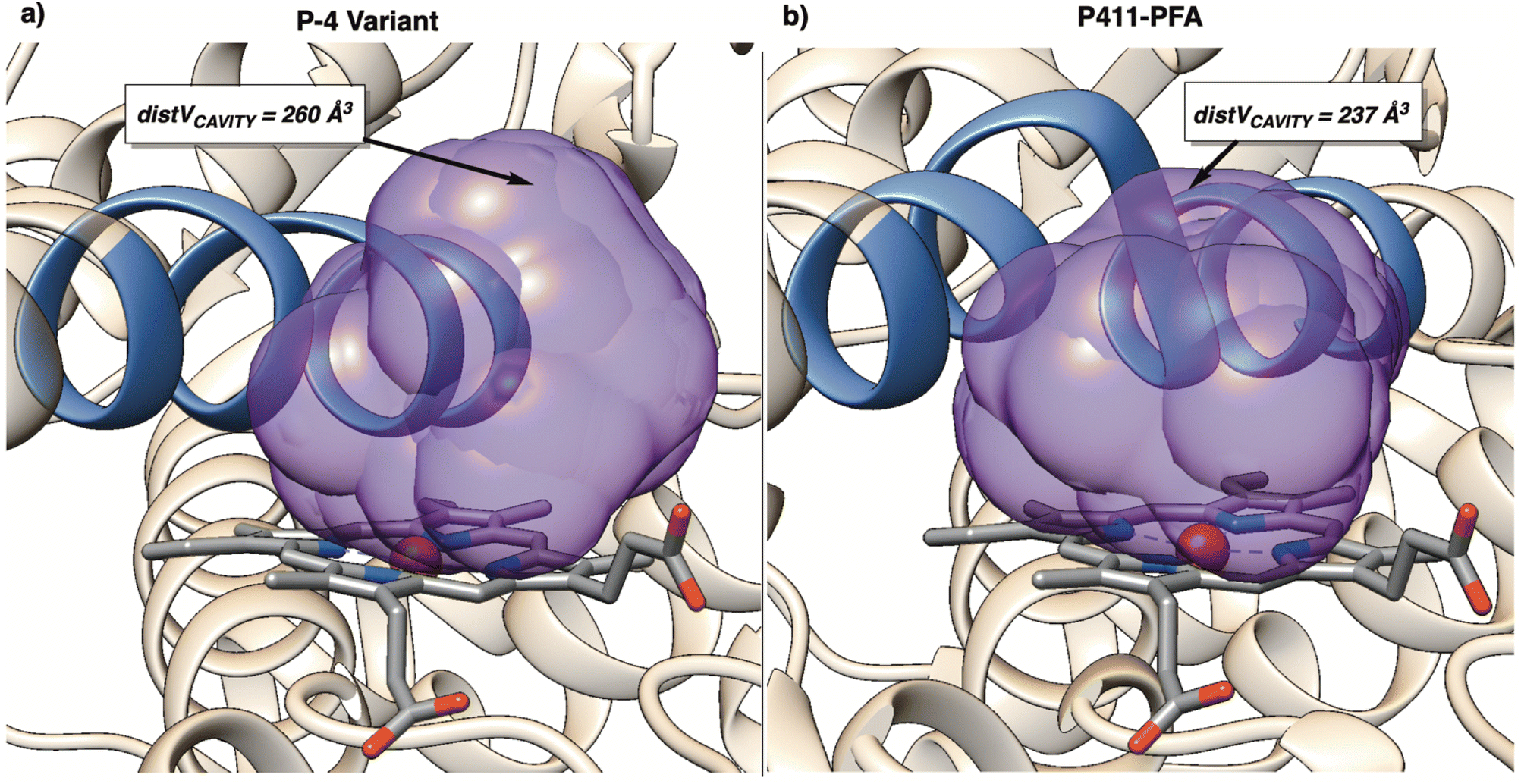 | ||
| Fig. 14 (a) SMART cavity (purple) for P-4 variant (PDB: 5UCW). (b) SMART cavity (purple) for P411-PFA enzyme (PDB: 8DSG). The bulge in the helix proximal to the porphyrin site exerts more hindrance on the cavity than in 5UCW. | ||
Conclusions
Reactive cavities are difficult to sterically parametrize in mechanistically meaningful ways using traditional molecular descriptors. A free, open-source Python package, SMARTpy, is released to compute SMART molecular descriptors that have only been disclosed in application to modeling dirhodium(II) selectivity. SMART descriptors provide information about the steric environment within a reactive cavity from the perspective of a bound or docked substrate. Though originally designed for dirhodium catalysts, we envision a broad scope of applicability to diverse systems.SMARTpy performs a template-based conformational search that generates an ensemble representative of the topology of the cavity. The choice of molecular probe is shown to influence the information obtained from SMART parameters. Acyclic probes are shown to generate highly irregular cavities, parametrizing the space between ligands. Macrocyclic probes generate regular, more spherical pockets due to rotational barriers. The flexibility of macrocyclic probes can be increased by the selection of small substituents bound to the core, such as H. This allows the probe to explore space closer to the ligands, resulting in a “high definition” representation of the cavity. Depending on the flexibility of the substrates coming together within the pocket in the transformation of interest, smaller or larger probes may be more suitable for generating SMART descriptors.
SMARTpy descriptors were found to capture salient trends across BINOL and SPINOL CPAs. Lower VCAVITY in SPINOL catalysts supports the prevalence of higher selectivity compared to BINOL catalysts. SMARTpy was also demonstrated to outperform VBur in the analysis of subdivided pocket space. SMARTpy was also demonstrated with a GPR101-Gs side binding pocket. The hindrance of the N-terminus and transmembrane domain are emphasized, suggesting that protein dynamics, or favorable non-covalent interactions are likely responsible for the initial procedure of small molecule binding. Finally, two enzymes used for different selective transformations are shown to differ in the distal region of the porphyrin cavity. The more hindered distal cavity observed in P411-PFA is hypothesized to constrain the approach of substrates to the Fe-carbene, directing selectivity for C–H functionalization.
In summary, SMARTpy provides a convenient tool for the precise quantification of steric environments for complex, irregularly shaped 3D cavities, which are critical for controlling reactivity in disparate chemical and biochemical systems. Although non-covalent interactions are not currently supported for the generation of molecular probe ensembles, this is an area of current development.
Data availability
ESI Figures† and an overview of SMARTpy usage is provided in the ESI Materials.† Biological structures were acquired from the PDB with accession codes 8W8R28 for the GPR101-Gs complex, and 5UCW31 and 8DSG36 for the porphyrin enzymes. The truncated structures and SMARTpy data supporting this article have been included as part of the GitHub repository (https://github.com/SigmanGroup/SMART-molecular-descriptors.git). The data analysis scripts used in these case studies and other examples of SMARTpy usage are also available on the GitHub repository.Author contributions
B. Miller contributed to the original implementation, designed and wrote the SMARTpy architecture, wrote the manuscript, and performed case study analysis. R. Cammarota formulated the initial idea for SMART descriptors, the original implementation of the workflow, and edited the manuscript. M. Sigman acquired funding, managed the project, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the insight from our beta-testers, especially Dr James Howard. Support for this project was provided by the National Science Foundation (CHE-2154502). The support and resources from the Center for High-Performance Computing at the University of Utah are gratefully acknowledged.Notes and references
- A. Verloop, The Sterimol Approach: Further Development of the Method and New Applications, International Union of Pure and Applied Chemistry, 1983, DOI:10.1016/b978-0-08-029222-9.50051-2.
- A. V. Brethomé, S. P. Fletcher and R. S. Paton, Conformational effects on physical-organic descriptors: the case of sterimol steric parameters, ACS Catal., 2019, 9(3), 2313–2323, DOI:10.1021/acscatal.8b04043.
- H. Clavier and S. P. Nolan, Percent buried volume for phosphine and n-heterocyclic carbene ligands: steric properties in organometallic chemistry, Chem. Commun., 2010, 46(6), 841–861, 10.1039/b922984a.
- K. Wu and A. G. Doyle, Parameterization of phosphine ligands demonstrates enhancement of nickel catalysis via remote steric effects, Nat. Chem., 2017, 9(8), 779–784, DOI:10.1038/NCHEM.2741.
- R. C. Cammarota, W. Liu, J. Bacsa, H. M. L. Davies and M. S. Sigman, Mechanistically guided workflow for relating complex reactive site topologies to catalyst performance in C−H functionalization reactions, J. Am. Chem. Soc., 2022, 144(4), 1881–1898, DOI:10.1021/jacs.1c12198.
- Y. T. Boni, R. C. Cammarota, K. Liao, M. S. Sigman and H. M. L. Davies, Leveraging regio- and stereoselective C(Sp3)–H functionalization of silyl ethers to train a logistic regression classification model for predicting site-selectivity bias, J. Am. Chem. Soc., 2022, 144(34), 15549–15561, DOI:10.1021/jacs.2c04383.
- L. W. Souza, B. R. Miller, R. C. Cammarota, A. Lo, I. Lopez, Y.-S. Shiue, B. D. Bergstrom, S. N. Dishman, J. C. Fettinger, M. S. Sigman and J. T. Shaw, Deconvoluting nonlinear catalyst–substrate effects in the intramolecular dirhodium-catalyzed C–H insertion of donor/donor carbenes using data science tools, ACS Catal., 2023, 104–115, DOI:10.1021/acscatal.3c04256.
- C. Qin and H. M. L. Davies, Role of sterically demanding chiral dirhodium catalysts in site-selective C–H functionalization of activated primary C–H bonds, J. Am. Chem. Soc., 2014, 136(27), 9792–9796, DOI:10.1021/ja504797x.
- J. Hansen and H. M. L. Davies, High symmetry dirhodium(II) paddlewheel complexes as chiral catalysts, Coord. Chem. Rev., 2008, 252(5–7), 545–555, DOI:10.1016/j.ccr.2007.08.019.
- H. M. L. Davies and D. Morton, Guiding principles for site selective and stereoselective intermolecular C–H functionalization by donor/acceptor rhodium carbenes, Chem. Soc. Rev., 2011, 40(4), 1857–1869, 10.1039/c0cs00217h.
- K. S. Watts, P. Dalal, R. B. Murphy, W. Sherman, R. A. Friesner and J. C. Shelley, ConfGen: a conformational search method for efficient generation of bioactive conformers, J. Chem. Inf. Model., 2010, 50(4), 534–546, DOI:10.1021/ci100015j.
- H. Edelsbrunner and E. P. Mücke, Three-dimensional alpha shapes, ACM Trans. Graphics, 1994, 13(1), 43–72, DOI:10.1145/174462.156635.
- C. Sullivan and A. Kaszynski, PyVista: 3D plotting and mesh analysis through a streamlined interface for the visualization toolkit (VTK), J. Open Source Softw., 2019, 4(37), 1450, DOI:10.21105/joss.01450.
- F. M. Richards, Areas, volumes, packing, and protein structure, Ann. Rev. Biphys. Bioeng., 1977, 6, 151–176 CrossRef CAS.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning, Science, 1979, 2019(6424), 363, DOI:10.1126/science.aau5631.
- A. Meller, M. Ward, J. Borowsky, M. Kshirsagar, J. M. Lotthammer, F. Oviedo, J. L. Ferres and G. R. Bowman, Predicting locations of cryptic pockets from single protein structures using the pocketminer graph neural network, Nat. Commun., 2023, 14(1), 1–15, DOI:10.1038/s41467-023-36699-3.
- L. Shen, J. Fang, L. Liu, F. Yang, J. L. Jenkins, P. S. Kutchukian and H. Wang, Pocket crafter: a 3D generative modeling based workflow for the rapid generation of hit molecules in drug discovery, J. Cheminform., 2024, 16(1), 1–17, DOI:10.1186/s13321-024-00829-w.
- W. Feng, L. Wang, Z. Lin, Y. Zhu, H. Wang, J. Dong, R. Bai, H. Wang, J. Zhou, W. Peng, B. Huang and W. Zhou, Generation of 3D molecules in pockets via a language model, Nat. Mach. Intell., 2024, 6(1), 62–73, DOI:10.1038/s42256-023-00775-6.
- G. Kudo, T. Hirao, R. Yoshino, Y. Shigeta and T. Hirokawa, Pocket to concavity: a tool for the refinement of protein-ligand binding site shape from alpha spheres, Bioinformatics, 2023, 39(4), 1–3, DOI:10.1093/bioinformatics/btad212.
- K. Roos, C. Wu, W. Damm, M. Reboul, J. M. Stevenson, C. Lu, M. K. Dahlgren, S. Mondal, W. Chen, L. Wang, R. Abel, R. A. Friesner and E. D. Harder, OPLS3e: extending force field coverage for drug-like small molecules, J. Chem. Theory Comput., 2019, 15(3), 1863–1874, DOI:10.1021/acs.jctc.8b01026.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera – a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25(13), 1605–1612, DOI:10.1002/jcc.20084.
- J. Li, S. Grosslight, S. J. Miller, M. S. Sigman and F. D. Toste, Site-selective acylation of natural products with binol-derived phosphoric acids, ACS Catal., 2019, 9(11), 9794–9799, DOI:10.1021/acscatal.9b03535.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Complete field guide to asymmetric BINOL-phosphate derived brønsted acid and metal catalysis: history and classification by mode of activation; brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates, Chem. Rev., 2014, 114(18), 9047–9153, DOI:10.1021/cr5001496.
- L. Schreyer, R. Properzi and B. List, IDPi catalysis, Angew. Chem., Int. Ed., 2019, 58(37), 12761–12777, DOI:10.1002/anie.201900932.
- J. P. Reid and J. M. Goodman, Goldilocks catalysts: computational insights into the role of the 3,3′ substituents on the selectivity of BINOL-derived phosphoric acid catalysts, J. Am. Chem. Soc., 2016, 138(25), 7910–7917, DOI:10.1021/jacs.6b02825.
- J. P. Reid, L. Simón and J. M. Goodman, A practical guide for predicting the stereochemistry of bifunctional phosphoric acid catalyzed reactions of imines, Acc. Chem. Res., 2016, 49(5), 1029–1041, DOI:10.1021/acs.accounts.6b00052.
- M. Zhang, T. Chen, X. Lu, X. Lan, Z. Chen and S. Lu, G protein-coupled receptors (GPCRs): advances in structures, mechanisms, and drug discovery, Signal Transduction Targeted Ther., 2024 DOI:10.1038/s41392-024-01803-6.
- Z. Yang, J. Y. Wang, F. Yang, K. K. Zhu, G. P. Wang, Y. Guan, S. L. Ning, Y. Lu, Y. Li, C. Zhang, Y. Zheng, S. H. Zhou, X. W. Wang, M. W. Wang, P. Xiao, F. Yi, C. Zhang, P. J. Zhang, F. Xu, B. H. Liu, H. Zhang, X. Yu, N. Gao and J. P. Sun, Structure of GPR101–Gs enables identification of ligands with rejuvenating potential, Nat. Chem. Biol., 2024, 20(4), 484–492, DOI:10.1038/s41589-023-01456-6.
- S. V. Athavale, S. Gao, A. Das, S. C. Mallojjala, E. Alfonzo, Y. Long, J. S. Hirschi and F. H. Arnold, Enzymatic nitrogen insertion into unactivated C–H bonds, J. Am. Chem. Soc., 2022, 144(41), 19097–19105, DOI:10.1021/jacs.2c08285.
- K. Chen, X. Huang, S. B. Jennifer Kan, R. K. Zhang and F. H. Arnold, Enzymatic construction of highly strained carbocycles, Science, 2018, 360(6384), 71–75, DOI:10.1126/science.aar4239.
- C. K. Prier, R. K. Zhang, A. R. Buller, S. Brinkmann-Chen and F. H. E. Arnold, Intermolecular benzylic C–H amination catalysed by an engineered iron-haem enzyme, Nat. Chem., 2017, 9(7), 629–634, DOI:10.1038/nchem.2783.
- Z. Liu, C. Calvó-Tusell, A. Z. Zhou, K. Chen, M. Garcia-Borràs and F. H. Arnold, Dual-function enzyme catalysis for enantioselective carbon–nitrogen bond formation, Nat. Chem., 2021, 13(12), 1166–1172, DOI:10.1038/s41557-021-00794-z.
- T. Rogge, Q. Zhou, N. J. Porter, F. H. Arnold and K. N. Houk, Iron heme enzyme-catalyzed cyclopropanations with diazirines as carbene precursors: computational explorations of diazirine activation and cyclopropanation mechanism, J. Am. Chem. Soc., 2024, 146(5), 2959–2966, DOI:10.1021/jacs.3c06030.
- Y. Yang and F. H. Arnold, Navigating the unnatural reaction space: directed evolution of heme proteins for selective carbene and nitrene transfer, Acc. Chem. Res., 2021, 54(5), 1209–1225, DOI:10.1021/acs.accounts.0c00591.
- K. Chen and F. H. Arnold, Engineering cytochrome P450s for enantioselective cyclopropenation of internal alkynes, J. Am. Chem. Soc., 2020, 142(15), 6891–6895, DOI:10.1021/jacs.0c01313.
- J. Zhang, A. O. Maggiolo, E. Alfonzo, R. Mao, N. J. Porter, N. M. Abney and F. H. Arnold, Chemodivergent C(Sp 3)–H and C(Sp 2)–H cyanomethylation using engineered carbene transferases, Nat. Catal., 2023, 6(2), 152–160, DOI:10.1038/s41929-022-00908-x.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dd00329b |
| This journal is © The Royal Society of Chemistry 2025 |