
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficiently solving the curse of feature-space dimensionality for improved peptide classification
Mario
Negovetić
a,
Erik
Otović
 a,
Daniela
Kalafatovic
*bc and
Goran
Mauša
*ac
a,
Daniela
Kalafatovic
*bc and
Goran
Mauša
*ac
aUniversity of Rijeka, Faculty of Engineering, Vukovarska 58, 51000 Rijeka, Croatia. E-mail: goran.mausa@uniri.hr
bUniversity of Rijeka, Faculty of Biotechnology and Drug Development, R. Matejčić 2, 51000 Rijeka, Croatia. E-mail: daniela.kalafatovic@uniri.hr
cUniversity of Rijeka, Center for Artificial Intelligence and Cybersecurity, R. Matejčić 2, 51000 Rijeka, Croatia
First published on 23rd May 2024
Abstract
Machine learning is becoming an important tool for predicting peptide function that holds promise for accelerating their discovery. In this paper, we explore feature selection techniques to improve data mining of antimicrobial and catalytic peptides, boost predictive performance and model explainability. SMILES is a widely employed software-readable format for the chemical structures of peptides, and it allows for extraction of numerous molecular descriptors. To reduce the high number of features therein, we conduct a systematic data preprocessing procedure including the widespread wrapper techniques and a computationally better solution provided by the filter technique to build a classification model and make the search for relevant numerical descriptors more efficient without reducing its effectiveness. Comparison of the outcomes of four model implementations in terms of execution time and classification performance together with Shapley-based model explainability method provide valuable insight into the impact of feature selection and suitability of the models with SMILE-derived molecular descriptors. The best results were achieved using the filter method with a ROC-AUC score of 0.954 for catalytic and 0.977 for antimicrobial peptides, with the execution time of feature selection lower by 2 or 3 orders of magnitude. The proposed models were also validated by comparison with established models used for the prediction of antimicrobial and catalytic functions.
1 Introduction
The use of machine learning (ML) predictions can speed up the process of identifying and optimizing peptides for therapeutic applications.1–3 To develop effective predictive models, it is necessary to have access to extensive data that include both positive (e.g. active) and negative (e.g. inactive) instances. An example is the collection of numerous antimicrobial peptides (AMP) in publicly available databases.4–7 Due to the increasing global health risk posed by antimicrobial resistance, which jeopardizes the efficacy of current treatment options,8 AMPs are considered a promising alternative to conventional antibiotics, and consequently their investigation must be urgently accelerated.9,10 However, it can take decades and considerable resources to discover new preclinical candidates for peptide-based therapies, and despite the considerable effort invested in developing AMPs, the availability of peptide-based drugs on the market remains relatively low and presents a continuous challenge.11,12 Hence, the application of efficient machine intelligence that enables searching through a large combinatorial space of peptide sequences and pinpointing promising candidates is essential.The choice of ML algorithm and representation scheme can significantly affect the predictive performance of the model.13 Derived from the FASTA annotation, various representation schemes have been developed to transform peptide sequences into machine-interpretable formats, such as physico-chemical properties,3,13,14 graph-based chemical structures,3,15,16 or sequences of amino acids.3,13,14,17 In this paper, we challenge existing models by extracting molecular descriptors from the more information-rich SMILES format that encodes the chemical structure. However, extracting a comprehensive range of features from SMILES and identifying the most informative is a time-consuming process.18,19 The high feature-to-instance ratio, known as the curse of dimensionality, influences the performance of classifiers, as it can indicate that the model is learning noise in the dataset, which negatively affects its accuracy.20,21 This risk can be mitigated by eliminating redundant or irrelevant features, thereby improving the generalizability of the model.22 Preserving only the most important features leads to the development of a simpler model and typically results in faster convergence.
The choice of feature selection plays a significant role in ML, as there are methods of various computational complexities that operate under different assumptions about the model and data.23,24 Modern feature selection techniques are designed to avoid exhaustive search, whose complexity is O(2n) where n denotes the number of features. Wrapper methods evaluate feature subsets using the performance of a specific ML model as a search criterion. The model is repeatedly trained and evaluated on different subsets of features, and the one that yields the best results is selected as the optimal set of features for the model at hand. Although effective in finding the most relevant features, wrapper methods can be computationally expensive and may lead to overfitting if the dataset is not sufficiently large.25 On the other hand, filter methods assess feature relevance based on their intrinsic characteristics, independent of any specific ML algorithm. These methods use statistical techniques or correlation measures to rank features according to their individual importance or relevance to the target variable. By filtering out less informative features during preprocessing, filter methods significantly reduce computational costs and improve model generalization.26 Little emphasis has been placed on feature selection methods in ML-based classifiers for active peptides that would improve not only the model's performance, but also our understanding of the underlying sequence-to-activity relationship.
The contributions of this paper are treefold: (i) suitability of the SMILES-based feature extraction method for peptide activity prediction, (ii) green data mining strategy for data preprocessing, and (iii) high level of performance for a catalytic peptides dataset composed of less than 100 instances. This paper explores an array of 1613 features derived from the SMILES format, suggests a thorough data cleaning process, and provides the cost–benefit analysis of feature selection techniques to develop ML-based models for prediction of peptide activity. For this purpose, three distinct methods for feature selection are utilized to reduce the large number of features extracted from the SMILES format. In addition, two baseline cases are also taken into account, where no feature selection is performed and with a FASTA representation that contains no descriptors. Comparison of the outcomes of all four cases that use molecular descriptors in terms of execution time and predictive performance provides valuable insight into the impact of feature selection and suitability of the models with SMILES-derived peptide features. Two datasets of different peptide activities were explored, a large one for AMP and a small one for catalytic peptides,27,28 which contains a considerably smaller number of experimentally validated examples.29 The potential of ML models for the prediction of catalytic peptides has not yet been fully explored. Therefore, we also assessed the suitability of ML models and SMILES-derived molecular descriptors for learning from a small catalytic dataset.
2 Background
Various ML algorithms have been used to establish the relationship between the properties of peptides and their function. While the support vector machine model was the preferred choice in the last decade,30 recent attention has shifted toward modern ML models, particularly to deep neural networks.31 Universal Language Model Fine-Tuning is one of the transfer learning techniques that yields high performance in the domain of chemical records.32 However, a popular choice in various classification problems is the Random Forest (RF) algorithm due to (i) a good overall performance attributed to an improved estimate of the average training error,33 (ii) fast training due to parallelization, which is particularly important in high-dimensional problems,34 and (iii) excellent adaptability to data imbalance.35AMPs have been extensively investigated in ML studies, mainly due to the abundance of available data.36,37 In our previous research, we investigated the potential of various string-based representations for this purpose. We found that a model employing one-hot encoding in conjunction with the theoretical peptide properties enhanced the predictive performance of the support vector machine models. This approach increased precision and reduced the number of false positives for the prediction of AMPs.30 Based on these observations, a hybrid sequential representation scheme was developed for the recurrent neural network (RNN) model to increase its predictive power, resulting in a high ROC-AUC score for both AMP and antiviral peptides.13 On the other hand, catalytic peptides have received less attention because of limited data availability. To overcome this challenge and facilitate the exploration of ML approaches for this category of peptides, we have collected and made available the manually curated dataset of peptides that catalyze ester hydrolysis.29 The dataset was used to develop an RNN-based classifier, which was combined with a genetic algorithm for a computer-driven search of undiscovered catalytic peptides.1 Such a computer-aided approach mitigates rational design limitations and expert bias, often rooted in prejudice, assumptions, and other human restrictions.
Despite commonly used in bio-informatics, linear textual chemical representations such as SMILES, SELFIES and DeepSMILES are still rarely employed for ML. Several case studies led to the conclusion that the SMILES notation enables good activity prediction and computer-driven design of molecules.38–40 The SMILES representation is also important because it embodies valid chemical structures,41 allows for the representation of the molecular structure in a textual format, and maintains information on spatial relationships between atoms in molecules.42 The fundamental rules for SMILES notation can be summarized as follows:42 (i) organic atoms are indicated with capital letters, while in the case of inorganic atoms, the charge and number of hydrogen atoms must be indicated; (ii) bonds between atoms can go from single to quadruple; (iii) parentheses are used to preserve spatial notation; (iv) ring structures are broken so that the first letter indicates the beginning of an open ring, and the last letter indicates the atom that closes the ring. By following these rules, for some molecules, it is possible to derive different but equally valid SMILES strings. This is known as the randomized SMILES issue, and it is a consequence of traversing the graph where there are multiple starting points.43 With this approach, it is possible to get n! different records, where n represents heavy atoms.44 Generating unique canonical records is still a problem in this annotation that can potentially be solved with the InChI45 or SELFIES46 notations. The development of canonicalization algorithms allowed the comparison of chemical sequences in the SMILES format.47
3 Methods
The project was carried out using the Python 3.7 programming language with the Scikit-learn ML library. The supercomputer “Bura” from the University of Rijeka was used to achieve parallelization and speed up the experiment. A single node with two Intel® Xeon® E5-2690 v3 processors was used for feature selection. Each processor has 12 physical cores and 24 threads with a maximum frequency of 3.5 GHz.3.1 Data collection
The antimicrobial (AMP) and catalytic (CAT) datasets were obtained from publicly available sources.13,29 The number of instances in each dataset can be seen in Table 1, alongside the numbers of positive and negative instances for each peptide category. While the AMP dataset has an approximately even distribution of positive and negative instances, the CAT datasets show imbalance towards the positive class. In this paper, the FASTA format for all peptides contained in both datasets were converted to the SMILES representation that maintains the data on chemical structure. Subsequently, the Mordred software library version 1.2.0 (ref. 48) was used to calculate 1826 possible features, of which 1613 were 2D and 213 were 3D. In this study, we only used the set of 2D features derived from SMILES.| Antimicrobial peptides | Catalytic peptides | |
|---|---|---|
| Positive | 4640 (44.87%) | 58 (68.24%) |
| Negative | 5701 (53.13%) | 27 (31.76%) |
| Total | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 341 341 |
85 |
3.2 Data analysis and preprocessing
Prior to any data preprocessing or feature selection, statistical analysis of all features was performed using measures of skewness and kurtosis, widely applied in descriptive statistics to summarize the shape of a distribution. CAT follows a symmetrical distribution of data with 53% of the asymmetry coefficients in the [−1, 1] range together with AMP which has 83% of the coefficients in the [−1, 1] range. On the other hand, in the case of catalytic peptides, a platykurtic distribution is visible since 59% coefficients are lower than 3, with the maximum coefficient being 85.0 and the minimum −1.9690. AMP also has a platykurtic data distribution because 82% of the coefficients are less than 3 with the minimum value being −1.9 and the maximum value being 3443.6.In the first step of data preprocessing, features that contain (i) outliers in the form of positive or negative infinity, (ii) only constant values, or (iii) NULL values were removed because they lack critical information for decision making. Features that contain (iv) less than 10% unique values were also considered non-informative and removed. In addition, features reported as (v) overflow by the Mordred library were discarded. The second step was to impute the missing values by using the k-nearest neighbors (k-NN) algorithm. The algorithm identified the five closest samples and used their Euclidean distance to estimate and fill in the missing value. The third step was to normalize the data into the [0, 1] range to alleviate the scale problem that arises from a wide range of non-standardized features. This method is known to lead to better convergence during training.
3.3 Sampling
The sampling was carried out in two different ways because the AMP and CAT datasets drastically differ in the number of instances, as shown in Table 1. In the case of a small CAT dataset containing less than 100 instances, a leave-One-out cross-validation was utilized. It uses all peptides to train the model, except one that is left to test its performance, and this process is repeated until each peptide is used once to test the model. In the case of large AMP dataset containing more than 10000 instances, this approach is inefficient and therefore a stratified K-fold cross-validation with 10 folds was applied. This procedure randomly divides the dataset into folds of the same size preserving the ratio between the positive and negative classes. In each iteration, one fold is used for testing, while other folds are used for training.
3.4 Feature selection
Feature selection is a dimensionality reduction method that selects relevant features with the aim to speed up the training phase and increase the predictive performance of the model. In this paper, we employ and compare filter and wrapper feature selection methods.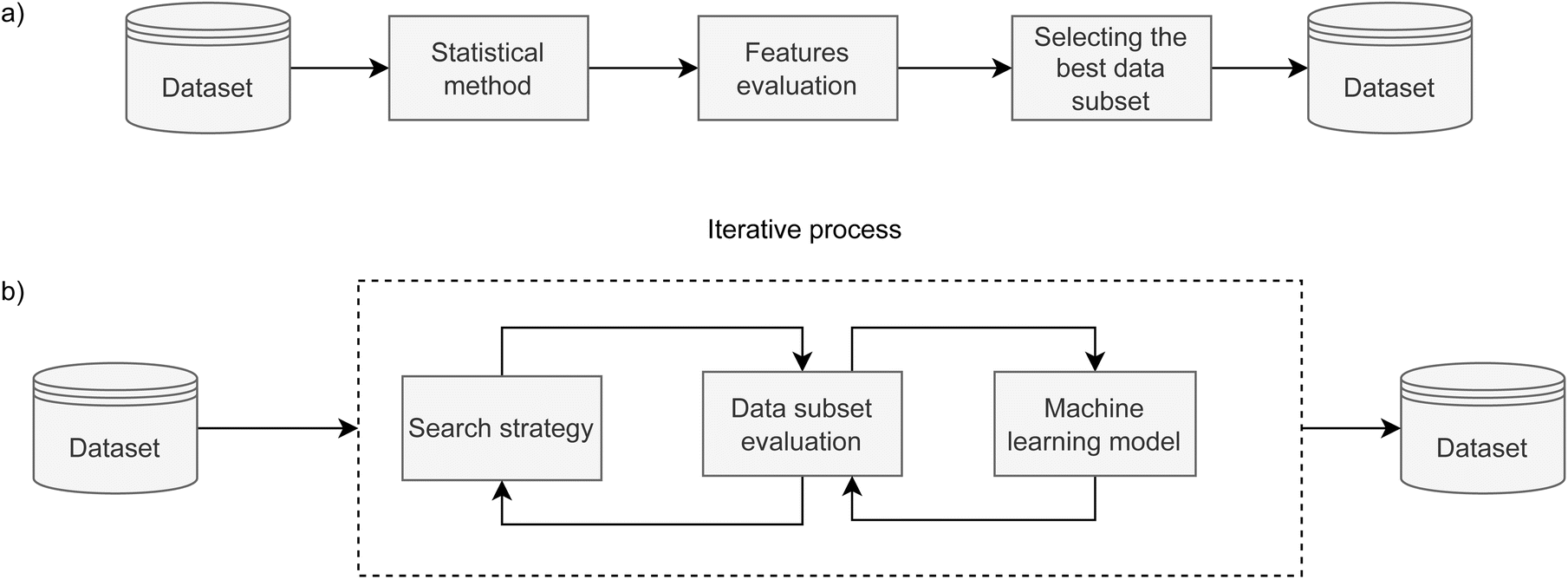 | ||
| Fig. 1 Schematic representation of: (a) filter methods and (b) wrapper methods for feature selection. While the filter method (a) is a straightforward algorithm, the wrapper method is computationally expansive as it contains an iterative section where sequential feature selection can have the backward and forward search direction. | ||
To get a broader picture of the complexity of finding the best set of features, we used two ML algorithms to navigate the iterative search, as presented in Fig. 1b. In the case of forward search, Gaussian Naive Bayes was used, where all parameters were kept at their default values. The backward search used decision tree, and all parameters were kept at their default values, except the tree depth. For the AMP search, the depth is limited to 5 levels, while it is limited to 8 levels for catalytic peptides. Stratified K-fold cross-validation was used in both wrapper methods to further split the training set into 4 folds and to reliably estimate model's performance for each feature set. Forward and backward approaches were run until all features were consumed to obtain their relative ranking. In the case of forward search, the ranking is equal to the order in which they were selected, while in the case of backward search, their ranking is inverse to the order they were discarded. Subsequently, the top-ranked features that yielded the highest F1 score in inner 4-fold cross-validation were selected as the result of feature selection.
3.5 Machine learning model
Once feature selection and data preprocessing are performed, the final ML model is trained. For our experiment, we used RF and a trial-and-error approach to determine the hyperparameters of the model, which optimized the accuracy. Finally, we opt for 600 decision trees with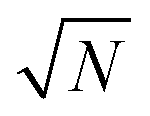 features (N being the total number of features that enter the model), setting the minimum number of features in a node to 6, and ensuring that the dataset has been shuffled. These settings provide an adequate execution time, but also avoid overfitting. For a fair comparison, the same hyperparameters values were used for all RF configurations.
features (N being the total number of features that enter the model), setting the minimum number of features in a node to 6, and ensuring that the dataset has been shuffled. These settings provide an adequate execution time, but also avoid overfitting. For a fair comparison, the same hyperparameters values were used for all RF configurations.
As a quality indicator for the proposed models, we used a RF model trained with peptides in the FASTA format. This type of format is often represented by one-hot encoding that encodes each position in sequence with a binary vector having zero values in all positions except one, which contains the value of one, indicating the presence of a specific amino acid. As RF expects the number of input features to be constant, all sequences were padded to the length of the longest sequence with binary vectors containing only zero values. This approach serves as a baseline for comparison and is widely used in related studies.13,14
3.6 Evaluation metrics
The confusion matrix is used to count the number of correct and incorrect predictions after translating the output probability into classification using a cut-off probability. In the case of binary classification, it is a 2 × 2 matrix and consists of cells representing the number of True Negative (TN), False Positive (FP), False Negative (FN), and True Positive (TP) predictions. The binary classification metrics, which are derived from the confusion matrix, are used to evaluate the performance of prediction models.49Table 2 shows the list of metrics that are used in this paper and their mathematical definitions. The area under the Receiver Operating Characteristic curve (ROC-AUC) is also used as an evaluation metric, and presents the only metric which does not depend on the cut-off probability of a classifier. It is a more general metric because it measures how well a binary classifier can distinguish between two classes, based on the true positive rate versus the false positive rate at different thresholds.| Metric | Calculation expression |
|---|---|
| Accuracy |
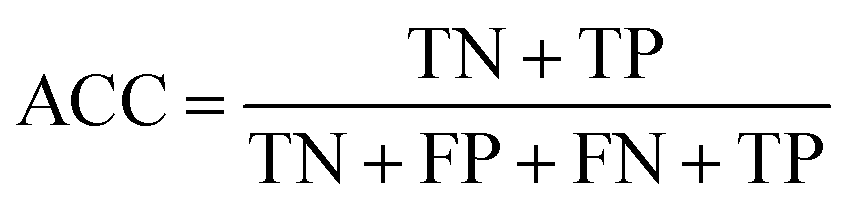
|
| Precision |
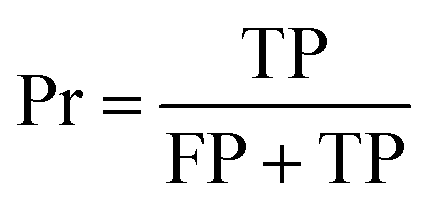
|
| Recall (true positive rate) |
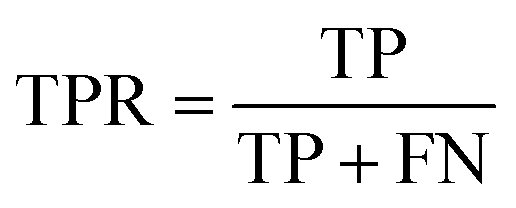
|
| Specificity (true negative rate) |

|
| F1 score (TPR – Pr harmonic mean) |
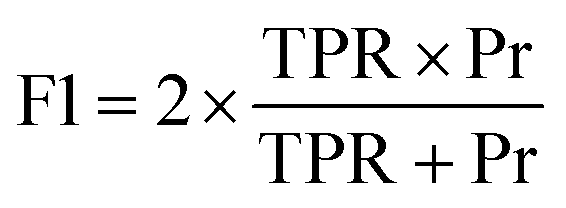
|
| Geometric mean accuracy |
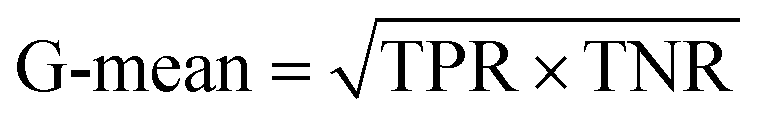
|
3.7 SHAP
SHapley Additive exPlanations (SHAP) is used to bridge the gap between accuracy and interpretability of complex ML models.50 SHAP assigns each feature an importance value by computing its contribution to the prediction of a model. After computing SHAP values for all instances in the dataset, a beeswarm plot of SHAP values is used to provide an understanding of the relationship between feature intensity and output probability.4 Results
In this paper, antimicrobial (AMP)13 and catalytic (CAT)29 datasets were used and the respective number of instances for each dataset is shown in Table 1. The distribution of data affects the feature selection process and plays an important role in a proper understanding of the results obtained. The AMP dataset shows an approximately even distribution of positive and negative records, whereas the CAT dataset contains more positive than negative instances.The same data preprocessing and prediction model training methodology was carried out for both datasets. Fig. 2 shows a representation of an example catalytic peptide annotated in the FASTA format as IHIHIQI and its equivalent record in the SMILES format, which is much longer and represents the complete chemical structure. FASTA format represents a peptide as a string of letters, where each letter corresponds to the one-letter amino acid code. This format leads to a loss of information about the spatial structure. Therefore, all FASTA strings were converted into SMILES representation, which allowed us to retain all the spatial features of chemical structures.
 | ||
| Fig. 2 An example of one short (catalytic) peptide in FASTA and SMILES formats with representation of chemical and spatial 3D structure. | ||
The methodology overview along with the breakdown of the features removed in each step of data cleaning is depicted in Fig. 3 for both CAT and AMP datasets. The first step of data preprocessing led to a reduction from 1613 of 2D features calculated by Mordred to 1151 features for the CAT dataset and 1087 features for the AMP dataset. Furthermore, the stage of data cleaning found and replaced missing values in 30 instances in CAT and 35 instances in AMP dataset by k-NN algorithm. In the experimental phase, we tested the effect of using three feature selection methods with RF classifier for a substantial dataset of 10341 AMPs and a small dataset of 85 catalytic peptides. In the following subsections we highlight their advantages and disadvantages, discuss the selection time as well as the consumption of computer resources, which differed drastically and their cost–benefit on classification performance. Furthermore, we also determined the relative significance of features by considering the frequency with which they were selected by various feature selection techniques.
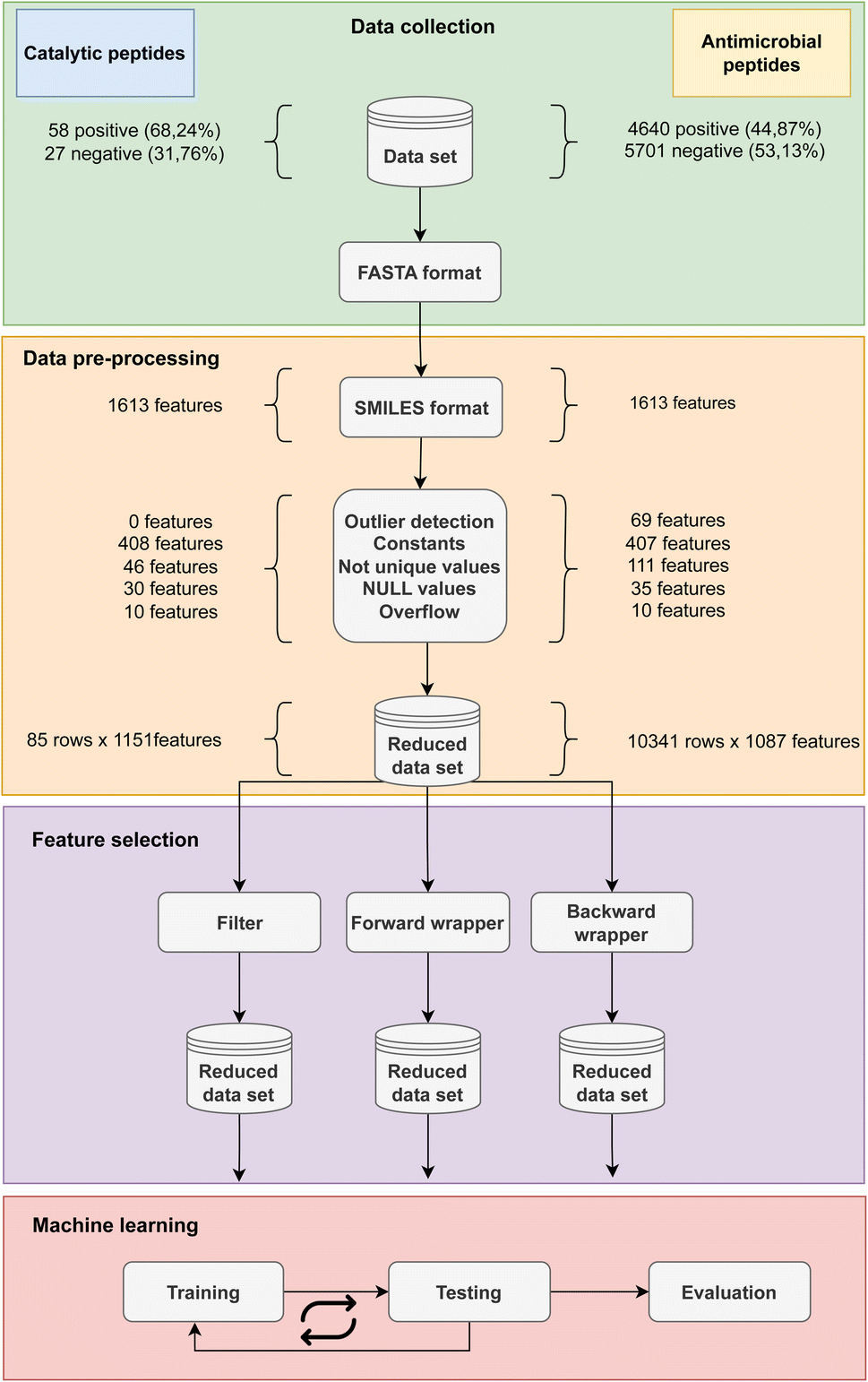 | ||
| Fig. 3 Schematic representation from data collection based on available catalytic and AMP dataset through data pre-processing including the FASTA to SMILES conversion and computing of features using Mordred library to the reduction of important features. Next, the features selection was performed using three different techniques: filter, forward wrapper and backward wrapper. The final step was machine learning which included cross-validation and classification testing. | ||
4.1 Performance of catalytic peptide models
In the case of CAT dataset, Kendall's Tau filter method had the best execution time of 00:01:01.59 hours and resulted in the selection of 263 features. The forward search was running for 00:18:02.07 hours and 46 features were selected, while the backward search resulted in a larger feature set of 477 features and took 00:36:04.34 hours to complete.Once feature selection was completed, the RF models were trained using the reduced feature set and the prediction results are shown in Table 3 and together with the baseline FASTA model in (Fig. 5a). Every RF model that used molecular descriptors from the SMILES format outperformed the baseline model in terms of AUC. The best results (ACC = 95.3%, F1 = 96.7% and ROC-AUC = 95.4%) were achieved using the filter method, which was also the fastest. It is worth noting that the filter method not only runs significantly faster, but also gives the highest ROC-AUC score, the only metric that takes into account how well the positive and negative classes are separated in terms of predicted probability, and not the final classification label. Moreover, when filter, backward wrapper or no selection were used, the RF classifier yielded only 4 false positive and 0 false negative predictions, which explains why their accuracy, precision and recall, F1 and G-mean are the same. The lowest values of these metrics are achieved by the forward wrapper, which yielded 4 false positives and 1 false negative. Although the backward search resulted in a larger feature set compared to the other methods, the model performed worse on the ROC-AUC metric by 6% compared to the filter method, showing that less complex models are generally a better choice.
| Feature selection | Without | Filter | Forward wrapper | Backward wrapper |
|---|---|---|---|---|
| Number of features | 1151 | 263 | 46 | 477 |
| Selection time [h] | 00:00:00 | 00:01:01.59 | 00:18:02.07 | 00:36:04.34 |
| Validation time [h] | 00:01:01.99 | 00:00:59.52 | 00:00:54.26 | 00:01:00.43 |
| Accuracy | 0.953 | 0.953 | 0.941 | 0.953 |
| Precision | 0.935 | 0.935 | 0.934 | 0.935 |
| Recall | 1.0 | 1.0 | 0.983 | 1.0 |
| F1 measure | 0.967 | 0.967 | 0.958 | 0.967 |
| G-mean | 0.967 | 0.967 | 0.958 | 0.967 |
| ROC-AUC | 0.931 | 0.954 | 0.923 | 0.896 |
In the next step, we analyzed which features were found to be more frequent among the 10 most important with respect to the Gini importance of the RF classifier. As shown by the schematic representation of their importance in Fig. 4, two features stood out, in particular F1 and F2. F1 corresponds to ATSC4i (autocorrelation of lag 4 weighted by ionization potential) and appears in the 10 most important features for three models (3×): the one without feature selection and in both models that use wrapper feature selections. F2 is GATS7s (Geary coefficient of lag 7 weighted by intrinsic state) and also appears in three models (3×): the one that is trained without a feature selection, the one with Wrapper forward selection and the one that uses the filter method. Furthermore, F3 and F4 were among the most important features for two (2×) out of four models tested. The AMID_N (averaged molecular ID on nitrogen atoms) feature is important in models using forward and backward feature selection methods, while JGI5 (5-ordered mean topological charge) is employed by models using filter and backward selection methods. There are 26 other features (F5⋯F30) among the 10 most important ones, but they appeared only in one of the models under consideration (1×).
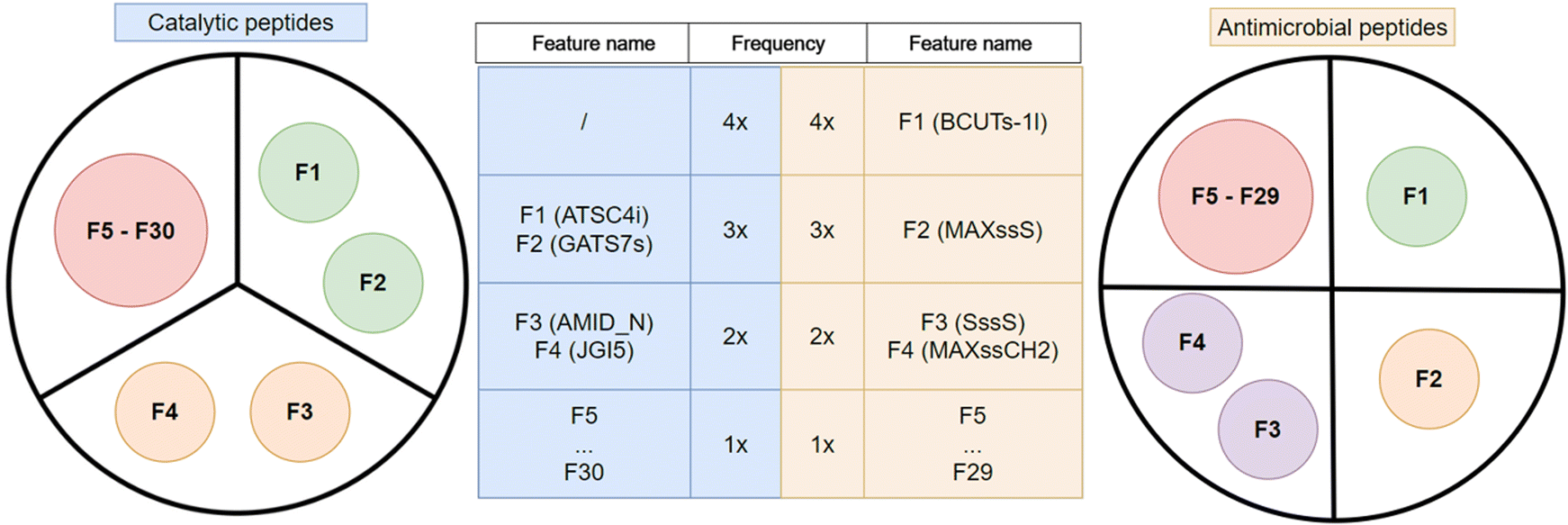 | ||
| Fig. 4 Schematic representation of the most important features in the CAT and AMP datasets. The frequency shown in the table refers to the number of RF models that contain the corresponding feature among the 10 most important ones. For example, BCUTs-1l was among the 10 most important features after (1) no feature selection, (2) filter, (3) forward wrapper, and (4) backward wrapper for AMP, and no feature was among top 10 in all four cases for CAT. | ||
4.2 Performance of antimicrobial peptide models
In the case of the AMP dataset, the filter method was the fastest by completing in 00:04:15.87 hours and selecting 291 features. The forward feature selection lasted 04:42:17.79 hours and selected 126 features, while the backward feature selection completed in 2 days and 10:46:08.57 hours and selected 45 features.The feature selection and classification results are shown in (Table 4 and Fig. 5b). Similarly to the CAT dataset, the use of SMILES-based molecular descriptors gave better performance than the baseline FASTA model in terms of AUC. The highest level of performance was achieved after using the filter method, which is confirmed by the metrics F1 = 91.0%, ACC = 91.9% and ROC-AUC = 97.7%. Although the classification performance was similar to the results achieved by forward selection, the execution time of the filter method was two orders of magnitude lower. On the other hand, backward feature selection resulted in the smallest feature set of 45 features and the performance was only marginally lower than other models, however, its execution time was three orders of magnitude higher and therefore it exhibited the worst cost–benefit ratio.
| Feature selection | Without | Filter | Forward wrapper | Backward wrapper |
|---|---|---|---|---|
| Number of features | 1087 | 291 | 126 | 45 |
| Selection time [h] | 00:00:00 | 00:04:15.87 | 04:42:17.79 | 2 days, 10:46:08.57 |
| Validation time [h] | 00:17:34.82 | 00:09:39.78 | 00:05:35.28 | 00:03:07.53 |
| Accuracy | 0.918 | 0.919 | 0.918 | 0.912 |
| Precision | 0.912 | 0.908 | 0.908 | 0.900 |
| Recall | 0.903 | 0.912 | 0.910 | 0.904 |
| F1 measure | 0.908 | 0.910 | 0.909 | 0.902 |
| G-mean | 0.908 | 0.910 | 0.909 | 0.902 |
| ROC-AUC | 0.975 | 0.977 | 0.975 | 0.974 |
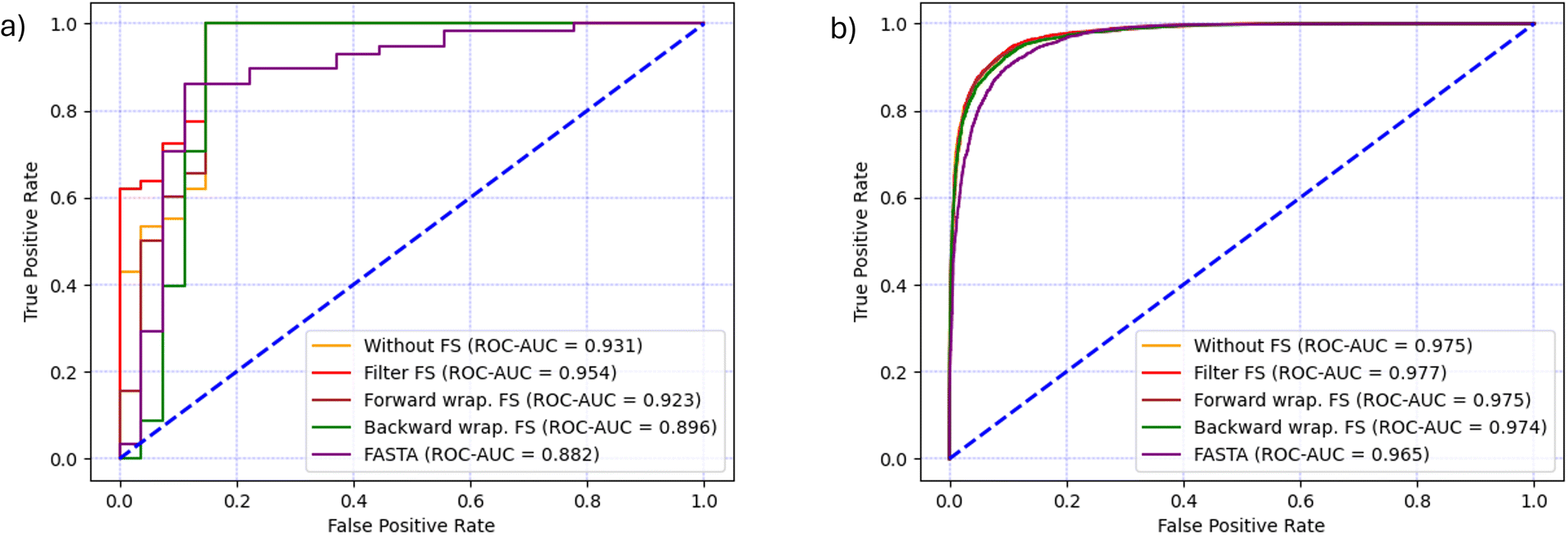 | ||
| Fig. 5 Comparison of classification performance in terms of ROC-AUC curve and AUC for each feature selection technique and the baseline model (FASTA) for (a) CAT dataset and (b) AMP dataset. | ||
All models achieved a high level of performance, as shown in Table 4, and the confusion matrix was analyzed to corroborate the differences in the predictions. It is important to point out that less than 10% of the predictions were false for all models. Thus, after using the filter technique, the model had an incorrect prediction for 835 peptides, while the correct prediction was made for 9506 peptides. On the other hand, after applying the forward wrapper feature selection, the model differs for an additional 10 false negative predictions. The use of backward wrapper feature selection further deteriorated the results, with 909 misclassified and 9432 correctly classified peptides.
The feature importance was analyzed in the same way as for the CAT dataset and is represented schematically on the right side of Fig. 4. The BCUTs − 1l (first lowest eigenvalue of Burden matrix weighted by intrinsic state) feature (F1) appears in all four models (4×) and appears to be essential for the classification of AMPs. The second most important feature (F2) is MAXssS (maximum E-state index for Sulfur with two single bonds –S–), which was found to be important in three models (3×), those using filter and both wrapper methods. The SssS (sum of E-state indices for Sulfur with two single bonds –S–) and MAXssCH2 (maximum E-state index for Methylene with two single bonds –CH2–) features (F3 and F4, respectively) are of importance only for the model that uses filter method and the model trained without feature selection (2×). The remaining 25 highlighted features (F5⋯F29) appear in the top 30 most important ones only for one model (1×).
5 Discussion
Identifying the relevant peptide features during data preprocessing and training the classifier can help us gain insight into the functioning of ML models. This can improve our understanding of peptide sequence-to-activity relationship and facilitate the discovery of novel peptides with desired functions. However, the choice of feature selection method depends on the trade-off between execution time and ML performance. Therefore, in this study, we compared the filter and wrapper methods in terms of these criteria. The results demonstrated the superior efficiency of the filter technique and the suitability of the SMILES-based representation for building a reliable peptide prediction model. RF-based antimicrobial and catalytic prediction models are compared in terms of average score to current state-of-the-art models that use sequential properties and recurrent neural networks1,13 in Table 5. The high performance of the developed models indicates that this procedure is on par with the existing models. To the best of our knowledge, the results presented in this paper represent the first comprehensive study on the relevance of features for the prediction of peptide catalytic activity.| Dataset | AMP | CAT | ||
|---|---|---|---|---|
| Model | Filter + RF | RNN13 | Filter + RF | RNN1 |
| F1 | 0.910 | 0.901 | 0.967 | 0.844 |
| ROC-AUC | 0.977 | 0.977 | 0.954 | 0.713 |
Overall, this study confirmed that SMILES-based features extracted for the prediction of peptide activity by ML are a valid alternative to existing representation schemes for both large datasets containing approximately 10000 peptides and small datasets containing approximately 100 peptides. Regardless of feature selection, every trained model reached a ROC-AUC greater than 0.97 in the AMP dataset and greater than 0.89 in the CAT dataset, which is considered excellent performance for prediction models. The advantage of using the SMILES annotation is the preservation of the chemical and spatial information of the peptide molecules, as shown in Fig. 2, which allows the derivation of informative features for ML. Although data cleaning ensured that each feature provided a comprehensive perspective on the peptides, our results revealed that numerous atom-count features derived from SMILES are irrelevant for the prediction of antimicrobial and catalytic activities of peptides. Interestingly, most of them are discarded by all feature selection methods we employed; however, the three feature selection techniques rarely selected the same ones. Among the selected features, we identified several that are also considered important by the RF classifier and in descending order of importance these are ATSC4 (F1), GATS7s (F2), AMID_N (F3), JGI5 (F4) for CAT, while for the AMP dataset they are BCUTs − 1l (F1), MAXssS (F2), SssS (F3) and MAXssCH2 (F4).
In terms of interpretability of the identified features for CAT, F1 and F2 are autocorrelation-based descriptors which encode the atomic properties related to ionization potential and intrinsic state, respectively. By calculating the separation between atom pairs, they allow for finding repeating patterns in the topological structure.51 F3 is a molecular ID descriptor of the nitrogen atom and F4 is a descriptor indicating topological charge. These features might suggest the importance of specific intrinsic atomic properties and the presence of nitrogen to guide the ML decision process toward the identification of catalytic peptides. For example, nitrogen atoms are present in the main chain of all peptides (every amino acid has at least one nitrogen atom) and are found in side chains of arginine, histidine, and lysine, as well as asparagine, glutamine, and tryptophan. Recently, lysine was identified as the amino acid that promotes the catalytic activity of short peptide sequences, through its side chain amino group,52,53 which points in the same direction as F3. The importance of F3 could be related to its specific position within the molecule, as the topological features F1, F2, and F4 might suggest; however, it remains inconclusive as its relation to a specific amino acid is unknown. Therefore, identification of important features alone might not lead to conclusive information about a specific design strategy or chemical detail applicable to catalytic propensity improvement, nor could the correlation of the identified feature with a specific molecular design be established.
Similarly, in terms of interpretability of the identified features for AMPs, F1 is linked to Burden matrix descriptors that relate to relevant aspects of molecular structure, often used for structural similarity search. The F2, F3 and F4 features are related to the electrotopological state (e-state) indices for atom types for sulfur (MAXssS, SssS) and methylene (MAXssCH2) groups calculated based on electronic, topological and valence state information.54 These main features indicate that sulfur atoms, found in cysteine and methionine side chains, and methylene groups present in many side chains, together with other molecular structure aspects, play an important role in AMP activity prediction. Although many reported AMPs contain sulfur atoms,55–57 their specific positions within the peptide and a specific chemical microenvironment probably underline their importance. Consequently, as in the case of CAT, they remain inconclusive about specific design strategies and to what extent they should be applied to increase the antimicrobial activity of peptides.
As it is challenging to rationalize the molecular descriptors and directly link them to specific peptide designs, we applied the SHAP method for the explainability of machine learning models to determine the impact of the most frequent features on the predictions. The SHAP values of the top 10 features according to the Gini importance of the most successful classifier with the filter method were calculated and plotted in the beeswarm plot. The favorable distribution of SHAP values, from the explainability point of view, is when the blue and red dots in the beeswarm plot do not overlap and appear only on one side of the zero-impact vertical line. The beeswarm plot for CAT in (Fig. 6a) indicates that higher values of F2 always increase the probability of catalytic function, while lower values in some cases may strongly decrease it. The opposite behavior is evident for F4, where lower values always increase the probability of catalytic function, and higher values mostly decrease it. The most important feature for AMP (F1) discriminates between high values that increase the probability of output and low values that decrease it, as indicated by the clear separation of blue and red dots with respect to the vertical axis at value 0 in (Fig. 6b). The other three most important AMP features (F2–F4) exhibit the opposite effect on the output probability, but with a less decisive discrimination between high and low values. This is evident from the blurred red and blue colors in the beeswarm plot, which occur because there are examples of similar feature values on both the positive and negative end of the SHAP values. With this insight, which was beyond our reach when using FASTA-derived features, we gained a deeper understanding of the relationship between ML-based decision making and specific features from a biological perspective. The methodology proposed in this paper may also allow further investigation by experts with domain-specific experience and knowledge to design better peptide descriptors and further improve the performance of predictive models.
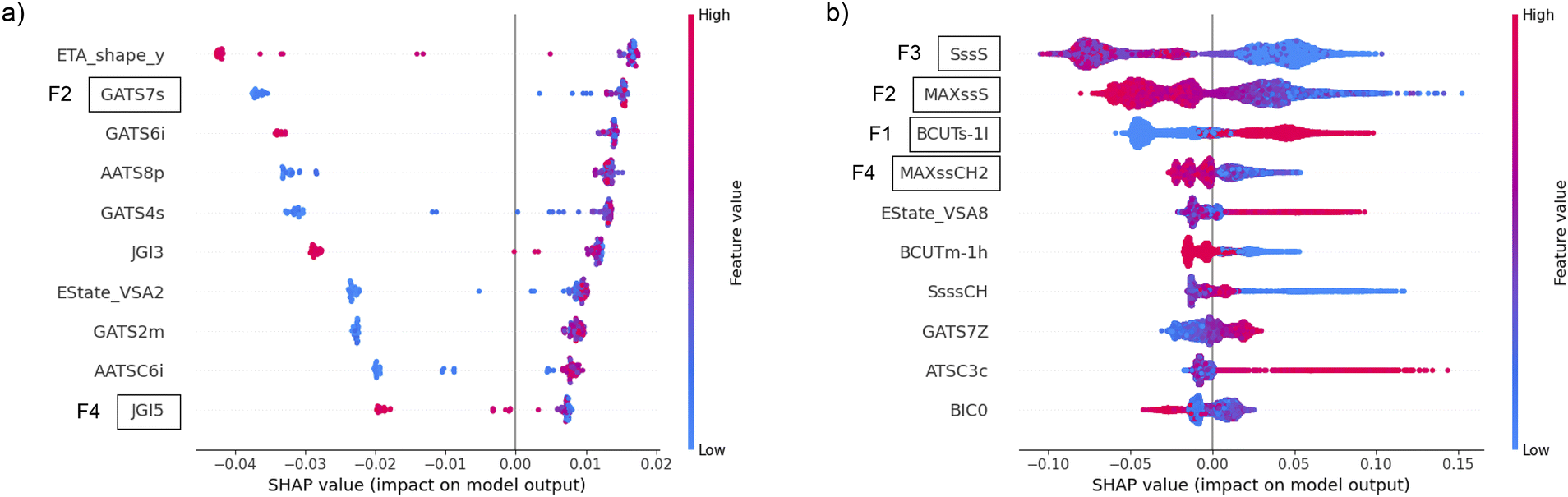 | ||
| Fig. 6 The beeswarm plot of SHAP values using all instances in the (a) CAT and (b) AMP datasets for 10 most important features after using filter method. With higher values of features colored in red and lower values colored in blue, their impact on models output probability is quantified on horizontal axis. Positive SHAP values contribute to higher probability of a peptide to be functional according to the classifier, while negative values decrease that output probability. | ||
The choice of feature selection method significantly affects the execution time of the preprocessing phase and the ML setup, as well as the final results. The measurements confirmed that wrapper methods are computationally expensive for data preprocessing, despite being the dominant method of choice for feature selection. The biggest obstacle to using wrapper methods is their execution time to select the best feature subset, especially in the case of high-dimensional data like we had in this case study with more than 1600 features. In the case of a small CAT dataset, the filter method was 17 times faster than forward feature selection and 35 times faster than backward feature selection. In the case of a relatively large AMP dataset, the filter method was 66 times faster than forward feature selection and 829 times faster than backward feature selection. This demonstrates that filter methods scale better with a dataset size than wrapper methods. When comparing the two wrapper methods in the case of CAT, forward feature selection was twice as fast as backward feature selection, while in the case of AMP, it was 12.5 times faster. Although they are based on the same principles and their algorithmic complexity is the same, the drastic difference in execution time can be attributed to the search direction. If the optimal feature set that the algorithms try to identify is relatively small, then forward feature selection will perform faster since it only needs a few iterations to arrive at the optimal feature set. On the contrary, backward feature selection would require many iterations to discard most of the features from a dataset before arriving at the same optimal feature set. However, if the optimal feature set contains nearly all features and only a small number of features need to be discarded, backward feature selection will outperform a forward feature selection timewise because it will require a smaller number of iterations to arrive at the optimal set. In addition, to speed up the search with a simpler classifier, we have also tried Naive Bayes instead of decision tree algorithm in backward feature selection. In the case of the CAT dataset, the number of selected features was reduced by a factor of 12.5, but the selection time was of the same order of magnitude. On the other hand, 15 fewer features were selected in the case of the AMP dataset and the selection time was reduced by 6.5 times. However, the performance of the final RF classifier was unaffected by the choice of classifier within the backward wrapper.
The predictive efficacy of each model was closely monitored because the reduction of features should not come at the cost of reduced performance. The filter method also stood out in this aspect and allowed the RF model to obtain the best scores, as presented in Fig. 5. The lower performance of the baseline FASTA model in terms of ROC-AUC can be attributed to the inability of RF to process sequential information and interaction between amino acids. Despite being the appropriate format for categorical variables, i.e. amino acids, the one-hot encoding results in a sparse matrix with the majority of the bits set to zero, which increases the complexity of the model. Although the combination of RF and one-hot encoding is widely used, our results underscore the need to use a more appropriate combination of representations and models that capture relevant information. The dataset size played an important role, because a higher number of AMPs allowed each model to perform equally well, but for the smaller number of CAT peptides, the difference in performance was more pronounced. The filter method selected a consistent number of features, and 291 were chosen for the AMP dataset and 263 for the CAT dataset. A sufficient number of features, together with a thorough data cleaning, allowed the correct prediction of 95.3% catalytic and 91.9% antimicrobial peptides. These are excellent performance results for predictive models in peptide chemistry, especially when the size of the datasets is taken into account, and suggest that appropriate data preprocessing is essential. This was particularly important for the CAT dataset, which also has a higher level of class imbalance (ratio 68:32) and a high feature-to-instance ratio. Certainly, the choice of the appropriate ML algorithm played an important role in this case study. The random forest, used to perform the predictions on both balanced and unbalanced datasets, proved to be a robust and suitable classifier, regardless of the input dataset size in the training phase.
6 Conclusions
The SMILES format, already applied to the prediction of molecular binding and for the construction of molecular generative models, can also offer a viable approach for calculating a large number of numerical features for antimicrobial and catalytic peptides. In this study, we examine the potential of peptide features derived from the atomic level of granularity to train a RF classification model that could pave the way for more accurate and efficient prediction of peptide function in general. For the proof of concept, we targeted two categories of peptides: a widely investigated one represented by a large AMP dataset and an underinvestigated CAT dataset containing less than 100 peptides. However, this approach is applicable to any peptide activity or function.Importantly, we compared wrapper and filter methods for selecting representative numerical features, with a focus on selection and validation time, accuracy, F1-score and ROC-AUC. With the goal of achieving a favorable feature-to-instance ratio, we demonstrate that the filter technique is the most efficient approach that reduces the complexity of the model and improves its predictive performance. The filter technique proposed in this study is based on the combination of non-parametric Kendall Tau and parametric Pearson correlation coefficients and provides a complementary set of features that enables the model to predict the peptide function effectively. Kendall Tau is the method of choice for datasets with outliers and non-linear relationships, as it uses ranks within the dataset, while Pearson correlation is more suitable for capturing relationships in continuous feature space. Our results demonstrate that the outcomes of the ML model are significantly influenced by data preprocessing and that a careful implementation of the feature selection method is essential. Utilizing an over-limited number of features, such as the 45 selected by backward wrapper for the CAT dataset, could lead to an increased number of incorrect predictions. In contrast, employing an excessive number of features, such as the complete set of 1151 features in the AMP dataset, could lead to an inadequate environment that hinders predictive accuracy and obstructs the interpretability of the model, demonstrating the principle that, indeed, less proves to be more. Based on our results, the most suitable number of features for the selected datasets falls between 250 and 300.
In addition to preprocessing optimization, it is also crucial to select an appropriate classifier according to the characteristics of the dataset and the desired prediction target. Our analyses have shown that the RF model achieves a high level of performance, reaching a ROC-AUC of 0.967 with the catalytic dataset, and 0.977 with the antimicrobial dataset. The significance of these results is even greater considering that the algorithm performs well with both balanced AMP data (44.87% positive and 53.13% negative) and unbalanced CAT data (68.24% positive and 31.76% negative). The results indicate that the features computed from the SMILES representation, in combination with the RF model, present an ML framework suitable for predicting peptide activity. In general, simpler models, such as RF, are preferred over those based on neural networks due to their faster training and better interpretability. However, it is worth noting that features computed from peptide sequences or SMILES inherently lead to the loss of information on the amino acid order within the sequence which may be important in certain applications. A situation where a dataset includes peptides with high similarity, but with permuted sequences showing opposite activity levels may require the use of recurrent neural networks and related methodologies that are able to process sequential or time series data.
In the future, it would be beneficial to extend our understanding of the peptide activity prediction by placing greater emphasis on analyzing and interpreting the features used for a specific peptide activity prediction. Our findings indicate that specific features are consistently found in multiple models, highlighting the importance of investigating their actual significance and role from a chemical and biological viewpoint.
Data availability
The code supporting this article have been uploaded at https://github.com/mario11596/peptides-project. This study was carried out using publicly available data. Active antimicrobial peptides were collected from the DRAMP 2.0 repository (https://dramp.cpu-bioinfor.org/), while inactive peptides were collected from the Uniprot (https://www.uniprot.org/). Publicly available dataset of manually curated catalytic peptides was used (https://doi.org/10.17632/6s9kxj2ndr.2; corresponding paper https://doi.org/10.1016/j.dib.2023.109290).Author contributions
Conceptualization: DK, GM; data curation: MN, EO, GM; funding acquisition: DK, GM; investigation: MN, EO; methodology: GM; DK software: MN, EO; supervision: DK, GM; validation: EO, GM; visualization: MN, GM; writing – original draft: MN, DK, GM; writing – review & editing: MN, EO, DK, GM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Croatian Science Foundation/Hrvatska zaklada za znanost (grant no: UIP-2019-04-7999 and DOK-2020-01-4659), University of Rijeka (UNIRI-INOVA-3-23-2, UNIRI-23-78, UNIRI-23-16) and ERASMUS+ project “promoting sustainability as a fundamental driver in training and education for software development” with the label 2020-1-PT01-KA203-078646. The information and views set out in this paper are those of the authors and do not necessarily reflect the official opinion of the European Union. Neither the European Union institutions and bodies nor any person acting on their behalf may be held responsible for the use which may be made of the information contained therein.Notes and references
- G. Mauša, M. Njirjak, E. Otović and D. Kalafatovic, MRS Adv., 2023, 1–7 Search PubMed.
- F. Wan, F. Wong, J. J. Collins and C. de la Fuente-Nunez, Nat. Rev. Bioeng., 2024, 1–16 Search PubMed.
- M. C. Melo, J. R. Maasch and C. de la Fuente-Nunez, Commun. Biol., 2021, 4, 1050 CrossRef CAS PubMed.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS PubMed.
- X. Kang, F. Dong, C. Shi, S. Liu, J. Sun, J. Chen, H. Li, H. Xu, X. Lao and H. Zheng, Sci. Data, 2019, 6, 148 CrossRef PubMed.
- M. Pirtskhalava, A. A. Amstrong, M. Grigolava, M. Chubinidze, E. Alimbarashvili, B. Vishnepolsky, A. Gabrielian, A. Rosenthal, D. E. Hurt and M. Tartakovsky, Nucleic Acids Res., 2021, 49, D288–D297 CrossRef CAS PubMed.
- S. Ramazi, N. Mohammadi, A. Allahverdi, E. Khalili and P. Abdolmaleki, Database, 2022, 2022, baac011 CrossRef CAS PubMed.
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao and E. Wool, et al. , Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- A. Tyagi, A. Tuknait, P. Anand, S. Gupta, M. Sharma, D. Mathur, A. Joshi, S. Singh, A. Gautam and G. P. Raghava, Nucleic Acids Res., 2015, 43, D837–D843 CrossRef CAS PubMed.
- M. D. Torres, S. Sothiselvam, T. K. Lu and C. de la Fuente-Nunez, J. Mol. Biol., 2019, 431, 3547–3567 CrossRef CAS PubMed.
- G. Hummel, U. Reineke and U. Reimer, Mol. BioSyst., 2006, 2, 499–508 RSC.
- N. T. T. Nhàn, T. Yamada and K. H. Yamada, Int. J. Mol. Sci., 2023, 24, 12931 CrossRef PubMed.
- E. Otović, M. Njirjak, D. Kalafatovic and G. Mauša, J. Chem. Inf. Model., 2022, 62, 2961–2972 CrossRef PubMed.
- M. Attique, M. S. Farooq, A. Khelifi and A. Abid, IEEE Access, 2020, 8, 148570–148594 Search PubMed.
- K. Yan, H. Lv, Y. Guo, W. Peng and B. Liu, Bioinformatics, 2023, 39, btac715 CrossRef CAS PubMed.
- T.-J. Sun, H.-L. Bu, X. Yan, Z.-H. Sun, M.-S. Zha and G.-F. Dong, Front. Genet., 2022, 13, 1062576 CrossRef CAS PubMed.
- J. Yan, J. Cai, B. Zhang, Y. Wang, D. F. Wong and S. W. Siu, Antibiotics, 2022, 11, 1451 CrossRef CAS PubMed.
- S. García, S. Ramírez-Gallego, J. Luengo, J. M. Benítez and F. Herrera, Big Data Anal., 2016, 1, 1–22 CrossRef.
- J. Sessa and D. Syed, 2016 5th international conference on electronic devices, systems and applications (ICEDSA), 2016, pp. 1–4 Search PubMed.
- D. Dernoncourt, B. Hanczar and J.-D. Zucker, Proceedings of the 3rd international conference on pattern recognition applications and methods, 2014, pp. 325–330 Search PubMed.
- A. Vabalas, E. Gowen, E. Poliakoff and A. J. Casson, PLoS One, 2019, 14, e0224365 CrossRef CAS PubMed.
- N. Pudjihartono, T. Fadason, A. W. Kempa-Liehr and J. M. O'Sullivan, Front. bioinform., 2022, 2, 927312 CrossRef PubMed.
- U. M. Khaire and R. Dhanalakshmi, J. King Saud Univ., Comp., 2022, 34, 1060–1073 Search PubMed.
- V. Kumar and S. Minz, Smart Comput. Rev., 2014, 4, 211–229 Search PubMed.
- M. Gutlein, E. Frank, M. Hall and A. Karwath, 2009 IEEE symposium on computational intelligence and data mining, 2009, pp. 332–339 Search PubMed.
- M. Cherrington, F. Thabtah, J. Lu and Q. Xu, 2019 International Conference on Computer and Information Sciences (ICCIS), 2019, pp. 1–4 Search PubMed.
- O. Zozulia, M. Dolan and I. Korendovych, Chem. Soc. Rev., 2018, 47, 3621–3639 RSC.
- P. Janković, M. Babić, M. Perčić, A. S. Pina and D. Kalafatovic, Mol. Syst. Des. Eng., 2023, 8, 1371–1380 RSC.
- P. Janković, E. Otović, G. Mauša and D. Kalafatovic, Data Brief, 2023, 109290 CrossRef PubMed.
- I. Erjavac, D. Kalafatovic and G. Mauša, Artif. Intell. Life Sci., 2022, 2, 100034 Search PubMed.
- F. Wan, D. Kontogiorgos-Heintz and C. de la Fuente-Nunez, Digital Discovery, 2022, 1, 195–208 RSC.
- S. Singh and R. B. Sunoj, Digital Discovery, 2022, 1, 303–312 RSC.
- F. Livingston, ECE591Q Machine Learning Journal Paper, 2005, 1–13 Search PubMed.
- R. Genuer, J.-M. Poggi, C. Tuleau-Malot and N. Villa-Vialaneix, Big Data Res., 2017, 9, 28–46 CrossRef.
- C. Zhang and Y. Ma, Ensemble machine learning: methods and applications, Springer, 2012 Search PubMed.
- J. Xu, F. Li, A. Leier, D. Xiang, H.-H. Shen, T. T. Marquez Lago, J. Li, D.-J. Yu and J. Song, Briefings Bioinf., 2021, 22, bbab083 CrossRef PubMed.
- M. Attique, M. S. Farooq, A. Khelifi and A. Abid, IEEE Access, 2020, 8, 148570–148594 Search PubMed.
- S. Lim and Y. O. Lee, 2020 25th International Conference on Pattern Recognition (ICPR), 2021, pp. 3146–3153 Search PubMed.
- S. Hu, P. Chen, P. Gu and B. Wang, IEEE J. Biomed. Health Inform., 2020, 24, 3020–3028 Search PubMed.
- W. Shi, M. Singha, G. Srivastava, L. Pu, J. Ramanujam and M. Brylinski, Front. Pharmacol., 2022, 13, 837715 CrossRef CAS PubMed.
- K. Rajan, C. Steinbeck and A. Zielesny, Digital Discovery, 2022, 1, 84–90 RSC.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- L. David, A. Thakkar, R. Mercado and O. Engkvist, J. Cheminf., 2020, 12, 1–22 Search PubMed.
- J. Arús-Pous, S. V. Johansson, O. Prykhodko, E. J. Bjerrum, C. Tyrchan, J.-L. Reymond, H. Chen and O. Engkvist, J. Cheminf., 2019, 11, 1–13 Search PubMed.
- N. M. O'Boyle, J. Cheminf., 2012, 4, 1–14 Search PubMed.
- M. Krenn, Q. Ai, S. Barthel, N. Carson, A. Frei, N. C. Frey, P. Friederich, T. Gaudin, A. A. Gayle and K. M. Jablonka, et al. , Patterns, 2022, 3, 100588 CrossRef CAS PubMed.
- D. G. Krotko, J. Cheminf., 2020, 12, 48 CAS.
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, J. Cheminf., 2018, 10, 1–14 Search PubMed.
- S. Visa, B. Ramsay, A. L. Ralescu and E. Van Der Knaap, Maics, 2011, 710, 120–127 Search PubMed.
- S. M. Lundberg and S.-I. Lee, A unified approach to interpreting model predictions, NIPS’17: Proceedings of the 31st International Conference on Neural Information Processing Systems, 2017, pp. 4768–4777 Search PubMed.
- G. Sliwoski, J. Mendenhall and J. Meiler, J. Comput. Aided Mol. Des., 2016, 30, 209–217 CrossRef CAS PubMed.
- E. Arad, K. B. Pedersen, O. Malka, S. Mambram Kunnath, N. Golan, P. Aibinder, B. Schiøtt, H. Rapaport, M. Landau and R. Jelinek, Nat. Commun., 2023, 14, 8198 CrossRef CAS PubMed.
- Y. Wang, T. Pan, J. Li, L. Zou, X. Wei, Q. Zhang, T. Wei, L. Xu, R. V. Ulijn and C. Zhang, ACS Appl. Mater. Interfaces, 2024, 16(17), 22369–22378 CrossRef CAS PubMed.
- L. H. Hall and L. B. Kier, J. Chem. Inf. Comput. Sci., 1995, 35, 1039–1045 CrossRef CAS.
- T. Schneider, A. Baldauf, L. A. Ba, V. Jamier, K. Khairan, M.-B. Sarakbi, N. Reum, M. Schneider, A. Röseler and K. Becker, et al. , J. Biomed. Nanotechnol., 2011, 7, 395–405 CrossRef CAS PubMed.
- J. P. Tam, S. Wang, K. H. Wong and W. L. Tan, Pharmaceuticals, 2015, 8, 711–757 CrossRef CAS PubMed.
- J. Koehbach and D. J. Craik, Trends Pharmacol. Sci., 2019, 40, 517–528 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |