
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Accelerated screening of gas diffusion electrodes for carbon dioxide reduction†
Ryan J. R.
Jones
 ,
Yungchieh
Lai
,
Dan
Guevarra
,
Kevin
Kan
,
Joel A.
Haber
and
John M.
Gregoire
*
,
Yungchieh
Lai
,
Dan
Guevarra
,
Kevin
Kan
,
Joel A.
Haber
and
John M.
Gregoire
*
Division of Engineering and Applied Science, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: gregoire@caltech.edu
First published on 30th April 2024
Abstract
The electrochemical conversion of carbon dioxide to chemicals and fuels is expected to be a key sustainability technology. Electrochemical carbon dioxide reduction technologies are challenged by several factors, including the limited solubility of carbon dioxide in aqueous electrolyte as well as the difficulty in utilizing polymer electrolytes. These considerations have driven system designs to incorporate gas diffusion electrodes (GDEs) to bring the electrocatalyst in contact with both a gaseous reactant/product stream as well as a liquid electrolyte. GDE optimization typically results from manual tuning by select experts. Automated preparation and operation of GDE cells could be a watershed for the systematic study of, and ultimately the development of a materials acceleration platform (MAP) for, catalyst discovery and system optimization. Toward this end, we present the automated GDE (AutoGDE) testing system. Given a catalyst-coated GDE, AutoGDE automates the insertion of the GDE into an electrochemical cell, the liquid and gas handling, the quantification of gaseous reaction products via online mass spectroscopy, and the archiving of the liquid electrolyte for subsequent analysis.
Introduction
The development of gas diffusion electrodes (GDEs) has enabled electrochemical carbon dioxide reduction to achieve industrially-relevant current densities while avoiding both parasitic hydrogen evolution, which is often observed when using proton-conducting polymer electrolytes, and parasitic sorption of the carbon dioxide, which is often observed when using hydroxide-conducting polymer electrolytes.1 A GDE mimics the triple phase boundary of membrane electrode assemblies (MEAs) used for electrochemical hydrogen technologies,2 with the polymer electrolyte replaced by a liquid, often aqueous, electrolyte. While strategies for managing the gas-liquid-solid boundary of the reactant stream, electrolyte, and catalyst are less mature than those of MEAs, considerable progress has been made in GDE systems by employing a hydrophobic gas diffusion layer (GDL), such as polytetrafluoroethylene (PTFE), with a metal catalyst coating on the electrolyte-facing side.3The manual assembly and operation of GDEs into electrochemical cells present an opportunity to accelerate experimentation via automation. Materials acceleration platforms (MAPs)4 comprise strategies for integrating experiment automation and data science, which hold great promise for developing electrocatalysts in general5 and CO2 reduction systems in particular.6 In the ecosystem of MAPs for electrocatalyst discovery, a host of techniques have been developed for bulk liquid electrolytes,7–9 including for CO2 reduction with analytical quantification of product distribution.10–14 Driven mostly by fuel cell technologies, high throughput screening has also been demonstrated for MEAs.15,16 Building upon these concepts, high throughput screening of GDEs for the electroreduction of dinitrogen was demonstrated with a 16-channel parallel testing system.17
In the present work, we demonstrate an instrument that automates GDE cell assembly and screening in a manner suitable for future incorporation in a MAP via integration with robotic synthesis and GDE sample exchange. We are not aware of comparable hardware for automating the operation of a GDE electrochemical cell for incorporation into a MAP. The description of the instrument and its validation using a PTFE-supported Cu GDE are provided below. The ESI† and associated data repository include pseudo-code for operation, Python code for incorporation into the HELAO-async18 instrument control platform, a fabrication guide, machining instructions for custom parts, computer drawings, and a bill of materials with estimated pricing.
Results and discussion
AutoGDE mechanical design
The objectives for the AutoGDE design include automation of gas and liquid handling, coupling to analytical detection of reaction products, and simplifying the exchange of working electrodes to enable future automation of sample loading and unloading. This last aspect required the greatest departure from traditional GDE experimentation, which relies on manual assembly of fasteners and gaskets to maintain hermetic seals during operation. While modern and future robotics may rival the dexterity of human GDE assemblers, we aimed to simplify the mechanical insertion of a GDE sample into the electrochemical cell as well as the establishment of an operational gas-electrolyte-catalyst boundary. Toward this end, we commenced with a cell design composed of 3 custom fixtures corresponding to the core components of a GDE electrochemical cell: the counter electrode (CE), working electrode (WE), and gas handling (GH) fixtures.Each fixture is machine from polyether ether ketone (PEEK) and contains liquid or gas handling ports. The bill of materials for building the AutoGDE system, not including analytical detection of reaction products, is summarized in Table S1.† The CE and WE fixtures contain electrical contacts for interfacing to the power supply or potentiostat. The CE and WE fixtures are separated using a polymer membrane, which is Selemion anion-exchange membrane in the present work.
A cross-section drawing of an assembly of these fixtures is shown in Fig. 1. Atop the mounting platform, which is mechanically coupled to the lab bench via rigid framing, the GH fixture is affixed and is intended to remain stationary. The CE fixture on top can be retracted via a spring-loaded translation stage and motor. When in the “engaged” position, the spring-loaded stage and motor enable controlled application of ca. 200 N of downward force to maintain the o-ring seals between the CE fixture and membrane, the membrane and WE fixture, and the WE and GH fixtures. With the CE fixture in the “disengaged” positions, the WE fixture is removable along with the GDE sample, which is held in place with a friction fit of the GDL paper with a regular octagon profile whose ca. 8 mm outer diameter is ca. 5% larger than the bored hole in the WE fixture.
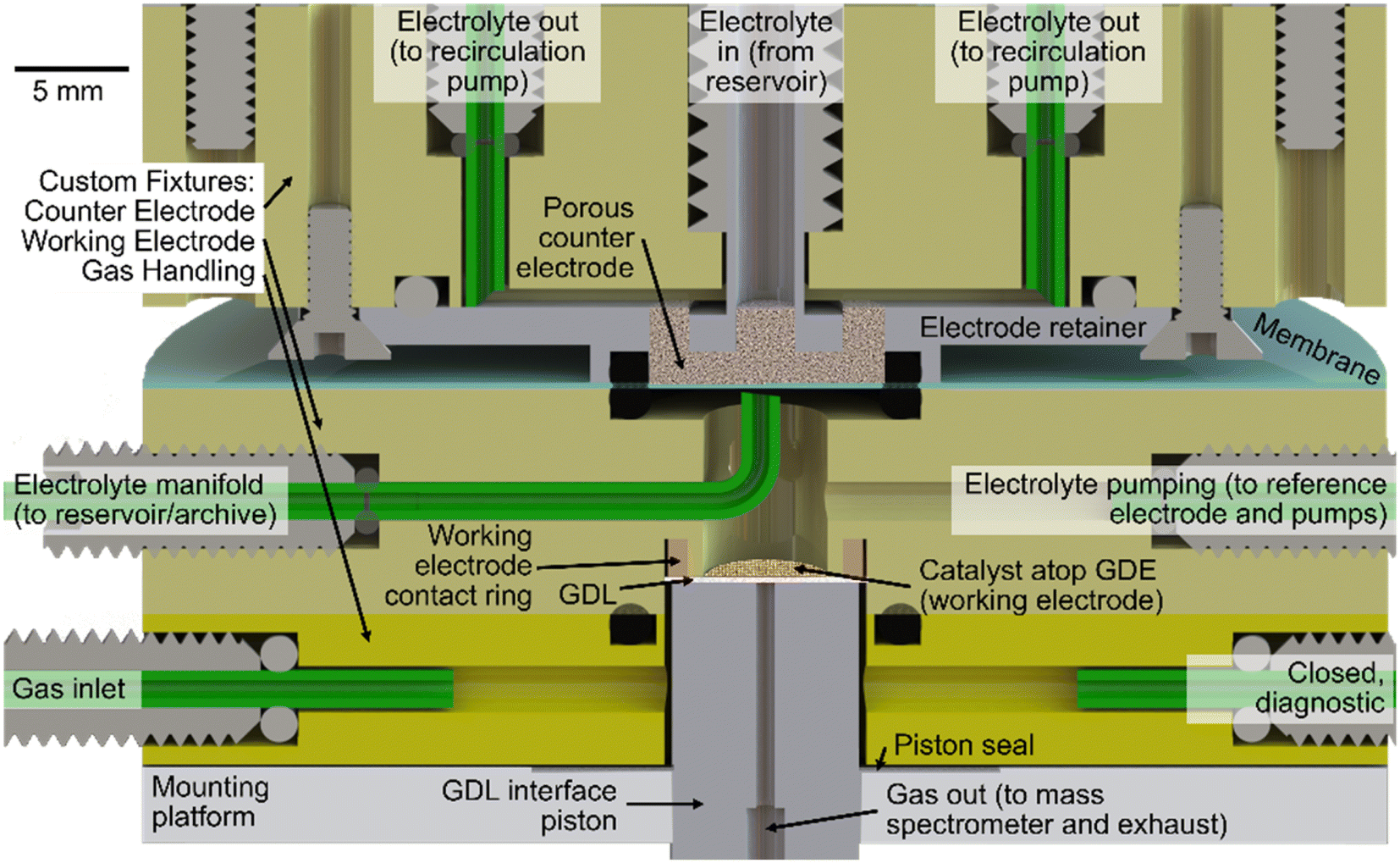 | ||
| Fig. 1 The AutoGDE electrochemical cell. The cross-section view shows the cell's three custom PEEK fixtures (shaded yellow) atop the mounting platform. The ports in each fixture are labelled by their purpose and/or ancillary pump and valve components to which they are connected. The GDL interface piston and gas handling fixture (bottom) manage the delivery of gas to the catalyst and to subsequent analytical characterization of reaction products. The working electrode fixture (middle) manages the electrical contact to the catalyst as well as the working electrolyte. Separated by a membrane, the counter electrode fixture (top) features a porous counter electrode and recirculating electrolyte. | ||
Upon engaging the CE fixture to establish a sealed cell, the final mechanical preparation of the cell involves compression of the GDL interface piston to a force of ca. 100 N using a linear actuator (Firgelli Automation). This piston resides within the GH fixture, where a gasket creates an air-tight seal around its perimeter while providing flexibility in its vertical position so that in the “engaged” (raised) position, the piston presses the catalyst layer atop the GDE against the working electrode contact ring. This positioning also establishes a gas flow path from the perimeter of the piston, laterally through the GDL, to the center bore of the piston. A photograph of the AutoGDE system and the fixtures in engaged and disengaged states are shown in Fig. 2.
 | ||
| Fig. 2 The AutoGDE electrochemical cell and mounting hardware. The realized version of the electrochemical cell in Fig. 1 is shown with its mounting platform, as well as the linear actuators for engaging the GDL interface piston (bottom) and compressing the counter electrode fixture to the stack of cell components (top). | ||
AutoGDE incorporation in autonomous workflows
We pause the description of the AutoGDE instrument to highlight the future action items for incorporation of the instrument in a fully-automated MAP: (1) the preparation of the GDE sample, which involves profiling of the GDL to the appropriate size and application of a catalyst on one side of the GDL; (2) the insertion of the GDL in the WE fixture, or one of several duplicate WE fixtures; (3) the placement of the WE fixture atop the GH fixture; (4) the connection of tubing to the 2 liquid handling ports on the WE fixture; and (5) the connection of the potentiostat to the wire that is attached to the working electrode contact ring, which is embedded within the WE fixture. We assert that automation of these steps is achievable with standard lab automation practices and focus the present work on the description of AutoGDE operation that is agnostic to how the WE fixture that was positioned and connected. To be clear, these steps were performed manually to generate the data of the present work.AutoGDE liquid and gas handling
As shown in Fig. 1, the CE fixture contains a central inlet port for electrolyte, which passes through a porous stainless-steel frit, which serves as the counter electrode. The electrolyte is returned to its reservoir via a peristaltic recirculation pump attached to four symmetric “electrolyte out” ports. The flat lower surface of the porous counter electrode provides mechanical stability for the membrane. The counter electrode, electrolyte, and membrane are intended to be used for testing any number of GDE electrodes.The GH fixture contains a single gas inlet port for flowing gas around the perimeter of the piston, through the GDL, and into the central exhaust port, which interfaces to analytical detection of gaseous reaction products. The present work employs a quadrupole mass spectrometer (MS, Hiden HPR20) for quasi-real-time product analysis.
The GH fixture also plays a pivotal role in the preparation of the WE electrolyte, which commences by closing the gas inlet port and applying a ca. 100 Torr vacuum on the center port of the GDL interface piston, which also evacuates the WE fixture through the porous GDE. By closing the electrolyte pumping port of the WE fixture and connecting the electrolyte manifold pump to an electrolyte reservoir open to atmosphere, the vacuum within the WE fixture pulls electrolyte into the fixture and against the GDE. While any trapped gas between the electrolyte and GDE is readily evacuated, the electrolyte does not flow through the GDE due to the hydrophobicity of the GDL. This initial wetting of the GDE automates one of the trickiest parts of traditional GDE cell preparation. We note that the tubing for the electrolyte manifold port approaches but not touches the membrane. The proximity helps ensure that no bubbles get caught on the membrane surface, which could cause an open circuit condition, and the small displacement and angled cut of the tube ending helps ensure that the membrane does not seal the tube end upon commencement of the vacuum-fill procedure.
The final preparation of the cell for electrochemical testing involves connecting the WE electrolyte manifold port to waste and pumping electrolyte into the WE fixture through the electrolyte pumping port, which removes the air gap in the tubing and flushes electrolyte through the WE chamber, ensuring electrolyte contact to the membrane. A reference electrode, in this case Ag/AgCl (Innovative Instrument Inc. LF-2-24, 2 mm diameter, 24 mm barrel leak-free reference electrode), can be inserted via a tee adjacent to the electrolyte pumping port, in which case this electrolyte flow step contacts the reference electrode to the working electrode chamber near the working electrode surface. With the electrolyte management complete, the reactant gas flow commences, which in the present work is 3 sccm of CO2, followed by electrochemical experimentation. The electrolyte used in the present work is aqueous 1 M potassium bicarbonate.
Given the ca. 2 mL of internal volume of tubing between the GDL and the inlet port to the mass spectrometer, the 3 sccm flow rate corresponds to a ca. 40 s delay between the formation of gaseous products at the working electrode and their detection in the mass spectrometer. This delay time is measured as the difference between the start of a CA measurement and the first detection of products in the MS and is used to shift the MS time axis to better match that of the electrochemistry data.
To enable off-line quantification of reaction products dissolved in the WE electrolyte, the electrolyte can be archived upon completion of the electrochemistry. By connecting the electrolyte manifold port to the desired vial, air (or another gas) is pumped into the electrolyte pumping port to push practically all electrolyte from the WE fixture, including its attached tubing, to the vial. In the present work, ca. 15 sccm of air via a peristaltic pump provides near complete removal of the electrolyte from the WE fixture. The volume of electrolyte within the tubing, which remains from the electrolyte filling procedure, is comparable to the volume of the WE chamber, which results in dilution of the reaction products by approximately a factor of 2. When using analytical methods with excellent levels of detection, for example liquid-injection gas chromatography (GC) and high-performance liquid chromatography (HPLC) in the present work (Thermo Trace 1310 and Ultimate 3000), the dilution is inconsequential. The total amount of liquid product in the vial is the lower limit of that synthesized during the electrochemical experiment due to possible loss of product, for example via incomplete electrolyte extraction from the cell, product evaporation during electrochemistry or during electrolyte extraction, and product crossover through the membrane.
To rinse the cell and/or load fresh electrolyte for further electrochemical interrogation of the GDE sample, the electrolyte manifold port is connected to waste and ca. 2.1 mL of electrolyte is pumped through the electrolyte pumping port, which flushes the WE fixture. When interrogation of the loaded sample is completed, the above air-pumping procedure is performed to extract all remaining liquid from the working electrode fixture, effectively preparing the working electrode chamber for exchange with a new GDE sample.
To facilitate quantitative analysis of the MS data, a calibration experiment can also be performed by switching the gas stream to a calibration gas with known concentrations of products of interest. In the present work, the calibration gas is 1% C2H4, 5% H2, and 5% CH4 with balance CO2 and is diluted by CO2 to obtained different levels of concentrations.
AutoGDE demonstration and validation
To prepare nominally duplicate electrodes, two GDE samples were extracted from a PTFE GDL sheet (0.45 μm Aspire Laminated membrane from lot# 685459, Sterlitech corporation) coated with ca. 300 nm of evaporated Cu. Each GDE was operated using a sequence of cyclic voltammetry (CV) and chronoamperometry (CA) experiments, with the CVs assessing the time-resolved product analysis and the CAs used for product quantification and evaluation of reproducibility between comparable experiments with the nominally duplicate electrodes.CVs acquired with 3 voltage cycles at 50 mV s−1 and 20 mV s−1 scan rates from −0.33 to −1.33 V vs. RHE (without resistance compensation) are shown in Fig. 3. The working electrode current density is calculated on a geometric area basis corresponding to the 0.155 cm2 aperture of the WE fixture. The MS signal (pressure) for H2, CH4, and C2H4 are shown in subsequent panels, along with the ratio of the MS signal from CO2 to the total for C-containing gases, which approximates the pressure-based, single-pass conversion efficiency of the CO2 to hydrocarbon products. While CO is a known prominent reaction product, its MS quantification is hampered by overlap with a CO2 fragment, and the liquid products are not quantified for the CV experiments. We note that additional calibrations are required to fully interpret the MS signal from a time-varying product stream.13
 | ||
| Fig. 3 Cyclic voltammograms and associated gaseous products for a Cu GDE operated in the AutoGDE system with online mass spectroscopy. The 3 voltage cycles are show with 2 different scan rates. The gaseous products are shown as the MS partial pressure for mass-to-atomic number (m/z) values of 2, 15, and 26 for H2, CH4, and C2H4, respectively. The CO2 signal (m/z = 22) is normalized by the sum of partial pressures for C-containing products. | ||
To quantitatively validate the AutoGDE for CO2 reduction on a Cu GDE, a series of CA experiments were performed, each with a fresh working electrode electrolyte, which was archived for subsequent liquid product analysis. The time series data for the CA experiments is shown in Fig. S1.† The time integral over each 10 min CA experiment was used to quantify the total electrochemical charge and partial current densities for each quantified product, as shown in Fig. 4 and Table S2,† which shows the approximately reproduced values for the electrochemical and partial current densities at 3 CA potentials. We note that in some experiments at applied bias below −1.33 V vs. RHE, the CA experiment appears unstable, which we attribute to degradation of the GDE via extended operation at current densities in excess of 50 mA cm−2.
 | ||
| Fig. 4 Chronoamperometry results for a sequence of 10 min experiments with the same GDE sample and fresh electrolyte. The time series data in Fig. S1† is averaged to provide the total and current partial densities for the electrochemistry and gaseous products (quantified using the same MS channels as Fig. 3). The liquid products are analyzed with off-line GC and HPLC characterization of the electrolyte from each CA measurement. A second GDE sample was measured on a different day at 3 of the CA potentials, showing reproduction of the potential-dependent product distribution. | ||
Although CO2RR performance on GDE could vary among electrochemical cell designs due to variation in, for example, mass transport and current distribution,19 our AutoGDE produces CO2RR product distributions commensurate with traditional Cu-based GDEs. To quantitatively compare faradaic efficiencies (FE), we consider the results acquired near −1.1 V vs. RHE, a potential commonly used to evaluate Cu catalyst in bicarbonate electrolytes due to the prevalence of C2+ products over H2. Our results of 48% FE toward C2+ products and 14% for H2 are commensurate with the literature values of 40% and 15%, respectively (see Table S2†).20
Regarding the acceleration in experiment throughput, the time spent acquiring electrochemical data can be considered to be independent of the level of experiment automation. Automation shortens the time spent on other experiment steps, with ancillary benefits including increased reproducibility and robust data tracking. For the AutoGDE demonstration reported herein, there are 2 primary experiment preparation steps. The first step is the assembly of the electrochemical reactor with a new GDE sample, which for a traditional GDE experiment comparable to those presented herein takes 15–25 min due to the multi-step manual cell assembly. The equivalent time on the AutoGDE is less than 1 min, providing an approximate 20-fold acceleration. Second, electrolyte handling involves cell cleaning and (re)filling electrolyte, which occurs prior each electrochemical experiment for which independent liquid product characterization is desired, as well as archiving of the electrolytes for subsequent liquid product detection. These liquid handling tasks require at least 10 minutes of human researcher time with traditional methods, while AutoGDE reduces the time to 3 min and does not require human intervention to manage the electrolyte (or to subsequently start an electrochemical experiment). The overall increase of experiment throughput must be ascertained for a given electrochemical testing protocol and the availability of human researchers for conducting multiple experiments. More detailed comparison between conventional- and Auto-GDE can be found at the ESI.†
AutoGDE safety and build validation
In addition to standard chemical and electrical safety practices, the specific safety concerns include a mechanical pinch hazard due to the motorized pressurization of the cell components, as well as a tubing rupture hazard if the inlet gas pressure is not limited and gas flow becomes obstructed. As with all GDE experiments, the catalyst may suffer from delamination from the GDL, resulting in a potential exposure to unsupported nanomaterials.The assembly instructions provided in the instrument design repository are intended to produce a fully functional AutoGDE system, assuming manual insertion of the GDE into the WE fixture and incorporation of that fixture into the system. The diagnostic port noted in Fig. 1 can be attached to a pressure gauge to leak-check the system. The calibration gas can be used to validate the product detection. Upon these checks, the electrochemical operation of the cell, for example via reproduction of the data in Fig. 4 or S1,† is the ultimate validation of the AutoGDE build.
The most common anticipated issue is an open circuit condition. Diagnostic steps for identifying the cause of the open circuit include checking the electrical contact between the catalyst and the WE contact ring, which can be done with the WE fixture removed from the cell, or by removing the CE fixture and membrane to enable line of site access to the catalyst surface so that it can be electrically contacted to check connectivity with the WE contact wire. The cause of an electrolyte-based open circuit conditions can be harder to identify. One strategy is to test the electrolyte fill of each chamber. For the WE chamber, replace the CE fixture and membrane with a transparent plate so that the presence of bubbles on the top of the cell can be visually observed. Similarly, operating the CE fixture with a transparent plate can be used to confirm electrolyte penetration through the porous counter electrode to the plate.
The AutoGDE build has been replicated at Caltech, and given that the design is being shared with the community via this manuscript, we are not aware of any other replication at the time of manuscript preparation. The computer drawings and machining instructions are provided under an open hardware license to facilitate such replication.
Conclusions
We present the AutoGDE instrument, which automates the preparation and operation of a gas diffusion electrode sample, demonstrated herein for electroreduction of CO2 with a polycrystalline copper catalyst supported on a PTFE GDL. While automation of sample handling has been well demonstrated in the field, the automation of liquid and gas handling in gas diffusion electrochemical cells, as well as integration with analytical detection of reaction products, is uniquely enabled by AutoGDE. We present this open hardware design to enable future development of MAPs for GDE-based electrochemical devices.Data and code availability
The instrument control software is available at https://github.com/High-Throughput-Experimentation/helao-async, with dependencies in https://github.com/High-Throughput-Experimentation/helao-core. These repositories contain the MIT License. The hardware design for AutoGDE is comprised of drawings and machining instructions, assembly instructions, pseudo-code for instrument operation, python code for the HELAO sequence for collection of demonstration data, data files acquired for the present work, and the source code for the analysis and plotting of those data, which are provided under the CERN Open Hardware License, CERN-OHL-P, via CaltechData at https://data.caltech.edu/records/f40n8-cv274 (doi: https://doi.org/10.22002/f40n8-cv274).Author contributions
R. J. J. and J. M. G. designed AutoGDE with input from all co-authors. R. J. J. and Y. L. assembled AutoGDE. D. G., K. K., and Y. L. developed the instrument control software. Y. L. procured and analyzed the demonstration data. J. M. G. was the primary author of the manuscript with contributions from all authors. R. J. R and Y. L. were the primary authors of the assembly and operation instructions. J. A. H. and J. M. G. supervised the project.Conflicts of interest
J. M. G. is an industrial consultant for experiment automation.Acknowledgements
This material is based on work performed by the Liquid Sunlight Alliance, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Fuels from Sunlight Hub under Award Number DE-SC0021266. The Resnick Sustainability Institute at Caltech is acknowledged for its support of enabling infrastructure and facilities. The authors thank Aidan Fenwick, Gavin Heim, and Theodor Agapie for helpful discussion, and Aidan Fenwick for deposition of the Cu on the GDE.Notes and references
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- Y. Wang, Y. Pang, H. Xu, A. Martinez and K. S. Chen, Energy Environ. Sci., 2022, 15, 2288–2328 RSC.
- T. N. Nguyen and C.-T. Dinh, Chem. Soc. Rev., 2020, 49, 7488–7504 RSC.
- M. M. Flores-Leonar, L. M. Mejía-Mendoza, A. Aguilar-Granda, B. Sanchez-Lengeling, H. Tribukait, C. Amador-Bedolla and A. Aspuru-Guzik, Curr. Opin. Green Sustainable Chem., 2020, 25, 100370 CrossRef.
- H. S. Stein, A. Sanin, F. Rahmanian, B. Zhang, M. Vogler, J. K. Flowers, L. Fischer, S. Fuchs, N. Choudhary and L. Schroeder, Curr. Opin. Electrochem., 2022, 35, 101053 CrossRef CAS.
- A. Wang, C. Bozal-Ginesta, S. G. H. Kumar, A. Aspuru-Guzik and G. A. Ozin, Matter, 2023, 6, 1334–1347 CrossRef CAS.
- T. H. Muster, A. Trinchi, T. A. Markley, D. Lau, P. Martin, A. Bradbury, A. Bendavid and S. Dligatch, Electrochim. Acta, 2011, 56, 9679–9699 CrossRef CAS.
- A. Kormányos, K. J. Jenewein and S. Cherevko, Trends Chem., 2022, 4, 475–478 CrossRef.
- P. J. McGinn, Mater. Discovery, 2015, 1, 38–53 CrossRef.
- J.-P. Grote, A. R. Zeradjanin, S. Cherevko and K. J. J. Mayrhofer, Rev. Sci. Instrum., 2014, 85, 104101 CrossRef PubMed.
- J. P. Grote, A. R. Zeradjanin, S. Cherevko, A. Savan, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, J. Catal., 2016, 343, 248–256 CrossRef CAS.
- R. J. R. Jones, Y. Wang, Y. Lai, A. Shinde and J. M. Gregoire, Rev. Sci. Instrum., 2018, 89, 124102 CrossRef PubMed.
- Y. Lai, R. J. R. Jones, Y. Wang, L. Zhou and J. M. Gregoire, ACS Comb. Sci., 2019, 21, 692–704 CrossRef CAS PubMed.
- P. Khanipour, M. Lçffler, A. M. Reichert, F. T. Haase, K. J. J. Mayrhofer and I. Katsounaros, Angew. Chem., Int. Ed., 2019, 131, 7273–7277 CrossRef PubMed.
- D. A. Stevens, J. M. Rouleau, R. E. Mar, A. Bonakdarpour, R. T. Atanasoski, A. K. Schmoeckel, M. K. Debe and J. R. Dahn, J. Electrochem. Soc., 2007, 154, B566–B576 CrossRef CAS.
- R. Liu and E. S. Smotkin, J. Electroanal. Chem., 2002, 535, 49–55 CrossRef CAS.
- M. Kolen, G. Antoniadis, H. Schreuders, B. Boshuizen, D. D. van Noordenne, D. Ripepi, W. A. Smith and F. M. Mulder, J. Electrochem. Soc., 2022, 169, 124506 CrossRef CAS.
- D. Guevarra, K. Kan, Y. Lai, R. J. R. Jones, L. Zhou, P. Donnelly, M. Richter, H. S. Stein and J. M. Gregoire, Digital Discovery, 2023, 2(6), 1806–1812 RSC.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave and E. B. Duoss, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- G. P. Heim, M. A. Bruening, C. B. Musgrave, W. A. Goddard, J. C. Peters and T. Agapie, Joule, 2024, 8, 1–10 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Chronoamperometry data as well as the detailed description, pseudocode for operation, and bill of materials for AutoGDE. See DOI: https://doi.org/10.1039/d4dd00061g |
| This journal is © The Royal Society of Chemistry 2024 |