
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Machine learning interatomic potentials for amorphous zeolitic imidazolate frameworks†
Nicolas
Castel
ab,
Dune
André
a,
Connor
Edwards
c,
Jack D.
Evans
 *c and
François-Xavier
Coudert
*a
*c and
François-Xavier
Coudert
*a
aChimie ParisTech, CNRS, Institut de Recherche de Chimie Paris, PSL University, 75005, Paris, France. E-mail: j.evans@adelaide.edu.au; fx.coudert@chimieparistech.psl.eu
bÉcole des Ponts, 77420, Marne-la-Vallée, France
cSchool of Physics, Chemistry and Earth Sciences, The University of Adelaide, Adelaide, SA 5005, Australia
First published on 8th January 2024
Abstract
The detailed understanding of the microscopic structure of amorphous phases of metal–organic frameworks (MOFs) remains a widely open question: characterization of these systems is very difficult, both from the experimental and computational point of view. In molecular simulations, approaches have been proposed that rely either on reactive force field, that lack chemical accuracy, or first-principles calculations, that are too computationally expensive. Here, we have found an innovative solution to these problems by training a machine learning potential for the description of disordered phases of a zeolitic imidazolate framework (ZIF). We then used it to produce high-quality atomistic models of ZIF glasses, with accuracy close to density functional theory (DFT) but at far lower computational cost in production runs.
Introduction
Metal–organic frameworks (MOFs) are a widely-studied class of nanoporous materials composed of metal nodes connected by organic linkers, exhibiting a wide range of applications including gas storage, catalysis, and drug delivery. While crystalline MOFs have been extensively studied and characterized, their amorphous counterparts have gained interest in the past decade.1 However, the fundamental understanding of their structure at the microscopic level remains limited, and their physical and chemical properties are not well known. One key reason for the challenge lies in the inherent disorder and lack of long-range order in amorphous MOFs, which results in a complex and heterogeneous local environment.2 Diffraction methods, which are widely used for the structural resolution of the crystalline phases, only offer indirect information about the structure of amorphous MOFs.3,4Another factor is the intrinsically dynamic nature of amorphous MOFs, involving fluctuations in the coordination environment of metal nodes, conformational changes of organic linkers, and even guest molecule interactions. The lack of a static, “perfect” structure makes it difficult to capture using simple models the fundamental nature of amorphous MOFs, and their structure–property relationships.5 Overcoming these challenges requires the development of new experimental techniques and theoretical models to better probe the local atomic arrangements and dynamic processes within amorphous MOFs. Advances in this area are needed not only to deepen our understanding of these promising materials, but also pave the way for their rational design and optimization for applications.
Here, we turn our attention specifically to computational methods for the description of amorphous MOFs. While we refer the reader to ref. 6 for a full review, we can summarize the current state of the art by stating that four classes of methods have been used to build atomistic models of amorphous MOFs: (i) the application of inverse problem methods using constraints from available experimental data, in particular from X-ray or neutron-scattering experiments, in methods like Reverse Monte Carlo;7,8 (ii) solving the same inverse problem with a physics-informed description of the interactions, for example in the polymatic approach;9,10 (iii) mimicking the experimental melt-quench process in silico, through the use of ab initio molecular dynamics;11,12 (iv) following the same process with reactive force fields, such as ReaxFF.13,14
Most of these methods rely on a description of the molecular interactions in the system. While the quality of this level of description is crucial to the validity of the models derived, none of the currently used options are truly satisfying. Classical force fields do not directly allow the description of bond breaking and formation. Reactive force fields are typically limited in the accuracy of their description of the chemical environment around metals,15 leading to issues in the structures obtained and their properties.16Ab initio methods have high chemical accuracy, but their expensive computational cost limits their use to small time and length scales, which in turn limits the statistical accuracy of the atomistic models obtained. In this work, we decided to apply a radically new approach to solve this issue by training a machine learning potential for the description of disordered phases of a zeolitic imidazolate framework (ZIF), and using it to produce high-quality atomistic models of ZIF glasses.
The use of machine learning potentials (MLPs) has emerged as a promising approach for describing interatomic interactions in both molecular systems and condensed matter.17–19 MLPs leverage the power of machine learning algorithms to learn complex potential energy surfaces from a large amount of training data, enabling accurate and efficient modeling of interatomic forces. The use of MLPs bypasses the need for explicit functional forms or empirical parameterization typically used in traditional force fields, leading to more universal and adaptable models. A large diversity of MLPs are currently available and being developed, with contrasting features when it comes to their data requirements, computational cost in training and production, generalization ability, etc. In their most common use, MLPs are typically trained from large databases of atomic configurations and their associated energies and atomic forces, allowing the MLPs to encode complex relationships between atomic positions and the potential energy surface. This enables MLPs to accurately describe a wide range of phenomena, including bond breaking and formation, chemical reactions, and phase transitions – as long as the potential has been trained on representative data for these phenomena. Several studies have shown that MLPs are, in particular, well adapted for the description of amorphous phases of condensed matter – including multiple glass-forming materials – such as oxide glasses,20 chalcogenide compounds,21 silicates22 or amorphous carbons.23
MLPs are trained on high-accuracy data that has been produced with computational chemistry methods of high computational cost, such as density functional theory (DFT) calculations, subsequently the trained MLP potential can rapidly evaluate interatomic forces and energies for arbitrary atomic configurations. There is therefore a large advantage in computational efficiency (several orders of magnitude) compared to the use of ab initio methods, while retaining an accuracy close to the reference level. This speed-up can allow for efficient exploration of complex systems, such as large-scale molecular dynamics simulations or high-throughput materials screening. In our case, it enables the simulations of amorphous ZIFs at time and length scales previously considered unreachable through first-principle calculations.
Despite the dramatic increase in the use of MLPs for both molecules and materials, only seven articles proposed MLPs for MOFs at the time of writing, all of them consisting of neural network potentials (NNP). MOFs are particularly challenging for multiple reasons, in particular because of the different interactions that need to be correctly modeled – from strong covalent bonds and ionic interactions to weak van der Waals interactions, along with their chemically complex composition. In addition, the (often) large unit cell size impedes ab initio simulations from generating training data.24,25 In 2019, Eckhoff and Behler24 proposed the first MLP for a MOF system (MOF-5) and circumvented the need for large ab initio calculations by using DFT calculations of small molecular fragments to train their MLP for the periodic bulk framework. This method was applied on different systems in three later works in different groups.26–28 However, the molecular fragments approach suffers from a lack of universal method to choose the fragments working on all MOFs, and from the sensitivity of its predicted energies to the choice of fragments. To overcome its shortcomings, four other works relied on periodic DFT calculations on the primitive cell of the framework to train their NNP.25,29–31 An option proposed by Vandenhaute et al.29 to solve the high computational cost of running ab initio simulations with large unit cells, was to use a data-efficient NNP (NequIP32) to reduce the number of ab initio calculations. Interestingly, this work also demonstrated that it was possible to simulate a phase transition for MOFs with MLPs, although the transition was non-displacive and did not involve any bond breaking. During the writing of this work, Goeminne et al. applied MLPs to describe the adsorption of CO2 in two MOFs (ZIF-8 and Mg-MOF-74) to derive adsorption isotherms from first principles.33
Here, we use the case of the ZIF family of materials to show that machine learning potentials offer an attractive atomistic description of disordered phases of MOFs. Based on first-principles molecular dynamics, we trained neural network-based MLPs and used them to produce high-quality atomistic models of ZIF glasses, with accuracy close to density functional theory (DFT) but at much lower computational cost.
Systems and methods
We provide here a brief summary of the system studied, and describe in detail the different computational methods used in this work. In order to make our work fully reproducible by others, representative input files for each type of simulation are available online in our data repository at https://github.com/fxcoudert/citable-data and the data set of configurations generated through ab initio MD and used for training and testing is made available on Zenodo at https://doi.org/10.5281/zenodo.10015594.ZIF-4
Our systems throughout this study belong to multiple phases of ZIF-4: the crystal, liquids, and multiple melt-quenched glasses.34 The first amorphous MOF discovered and studied in detail,7 ZIF-4 has since been the subject of numerous works and can be seen as a prototypical amorphous ZIF system.6 It is built up from Zn2+ metal nodes and imidazolate (Im) organic linkers, which are organized in the crystalline state as Zn(Im)4 tetrahedra linked by Zn–N coordinative bonds as depicted in Fig. 1. There are several amorphous phases of this system,5 which can be formed from the parent crystal by a variety of experimental methods, including ball milling, melt-quenching, or pressure-induced amorphization.2,35 A representative model of a melt-quenched glass of ZIF-4, gained from modelling synchrotron and neutron total scattering data, is presented in Fig. 2. | ||
| Fig. 1 Representation of the assembly of ZIF-4 as a three-dimensional network of Zn(Im)4 tetrahedra. Reproduced from ref. 6. Copyright 2022 American Chemical Society. | ||
 | ||
| Fig. 2 Atomic configuration of the melt-quenched glass of ZIF-4. Reproduced from ref. 34. | ||
Ab initio molecular dynamics
DFT-based MD simulations were performed following the protocol established in earlier work,16 using the Quickstep module36 of the CP2K software (version 6.1).37 The exchange–correlation energy was evaluated in the PBE approximation,38 and the dispersion interactions were treated at the DFT-D3 level.39 Valence electrons were described by double-ζ valence polarized basis sets and norm-conserving Goedecker–Teter–Hutter pseudopotentials.40The simulations were performed in the constant–volume (N, V, T) ensemble with a fixed size and shape of the unit cell. A time step of 0.5 fs was used in the MD runs; the temperature was controlled by velocity rescaling41 with a time constant of 1000 fs.
Reference data consisted in four trajectories of ZIF-4 liquids at four different volumes, and at temperatures of either 1500 K or 1750 K, all run for more than 50 ps. The preparation of these systems is detailed in the ESI.† We also included a simulation of the ZIF-4 crystal at 300 K, for purposes of comparison.
Flat histogram sampling
Flat histogram sampling is a powerful Monte Carlo approach used to overcome the limitations of traditional MC simulations, particularly when treating complex energy landscapes that feature rare events. Unlike standard simulations that rely on random sampling, flat histogram methods aim to ensure an equal exploration of all possible states or configurations within a system's phase space. For example, the Wang–Landau algorithm performs a non-Markovian random walk to build the density of states by quickly visiting all the available energy spectrum.42 This resulting sampling distribution leads to a simulation where the energy barriers are invisible, meaning that the algorithm visits all accessible states (favorable and less favorable) much faster than a Metropolis algorithm. Flat histogram sampling has been applied to drive simulations to calculate density of states, explore the flexibility of nanoporous framework, and explore water adsorption.43–45 We took inspiration from these approaches to develop sampling strategies to extract frames from the large amount of AIMD trajectories that represent the reference data.The complete data set produced from AIMD trajectories features a total of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 189836 frames. We compared two approaches: random sampling and the flat-histogram approach. In the random sampling approach, configurations are taken at random from our very long AIMD trajectories, and then split randomly between training and test data. Due to the very large nature of our AIMD trajectories (four trajectories of more than 50 ps), the configurations available are widely spaced in time and there is no need to use specific methods to avoid correlation between configurations. In the flat-histogram approach, we first bin with respect to energy (with the number of bins corresponding to the requested number of samples), and then a random sample is taken from each bin. The resulting difference in energy distributions for the two different approaches is demonstrated in Fig. 3a. This process is completed without replacement, and repeated, to give a ‘training’ data set and the leftover frames were subsequently sampled in the same way to give a ‘test’ set (data binning is repeated and unique for each data set). The unsampled frames were saved as ‘validation’. An initial train-test data set was produced for both the random sampling approach for 1000 training samples and 100 test samples for hyperparameter screening, and a final train-test data set produced with the flat-histogram approach for 2500 training samples and 200 test samples. It is worth noting that, because of empty bins the flat-histogram approach at times produces less samples than bins; for examples the initial train-test data set was 964 and 96 samples and a final data set of 2357 and 190 samples.
189836 frames. We compared two approaches: random sampling and the flat-histogram approach. In the random sampling approach, configurations are taken at random from our very long AIMD trajectories, and then split randomly between training and test data. Due to the very large nature of our AIMD trajectories (four trajectories of more than 50 ps), the configurations available are widely spaced in time and there is no need to use specific methods to avoid correlation between configurations. In the flat-histogram approach, we first bin with respect to energy (with the number of bins corresponding to the requested number of samples), and then a random sample is taken from each bin. The resulting difference in energy distributions for the two different approaches is demonstrated in Fig. 3a. This process is completed without replacement, and repeated, to give a ‘training’ data set and the leftover frames were subsequently sampled in the same way to give a ‘test’ set (data binning is repeated and unique for each data set). The unsampled frames were saved as ‘validation’. An initial train-test data set was produced for both the random sampling approach for 1000 training samples and 100 test samples for hyperparameter screening, and a final train-test data set produced with the flat-histogram approach for 2500 training samples and 200 test samples. It is worth noting that, because of empty bins the flat-histogram approach at times produces less samples than bins; for examples the initial train-test data set was 964 and 96 samples and a final data set of 2357 and 190 samples.
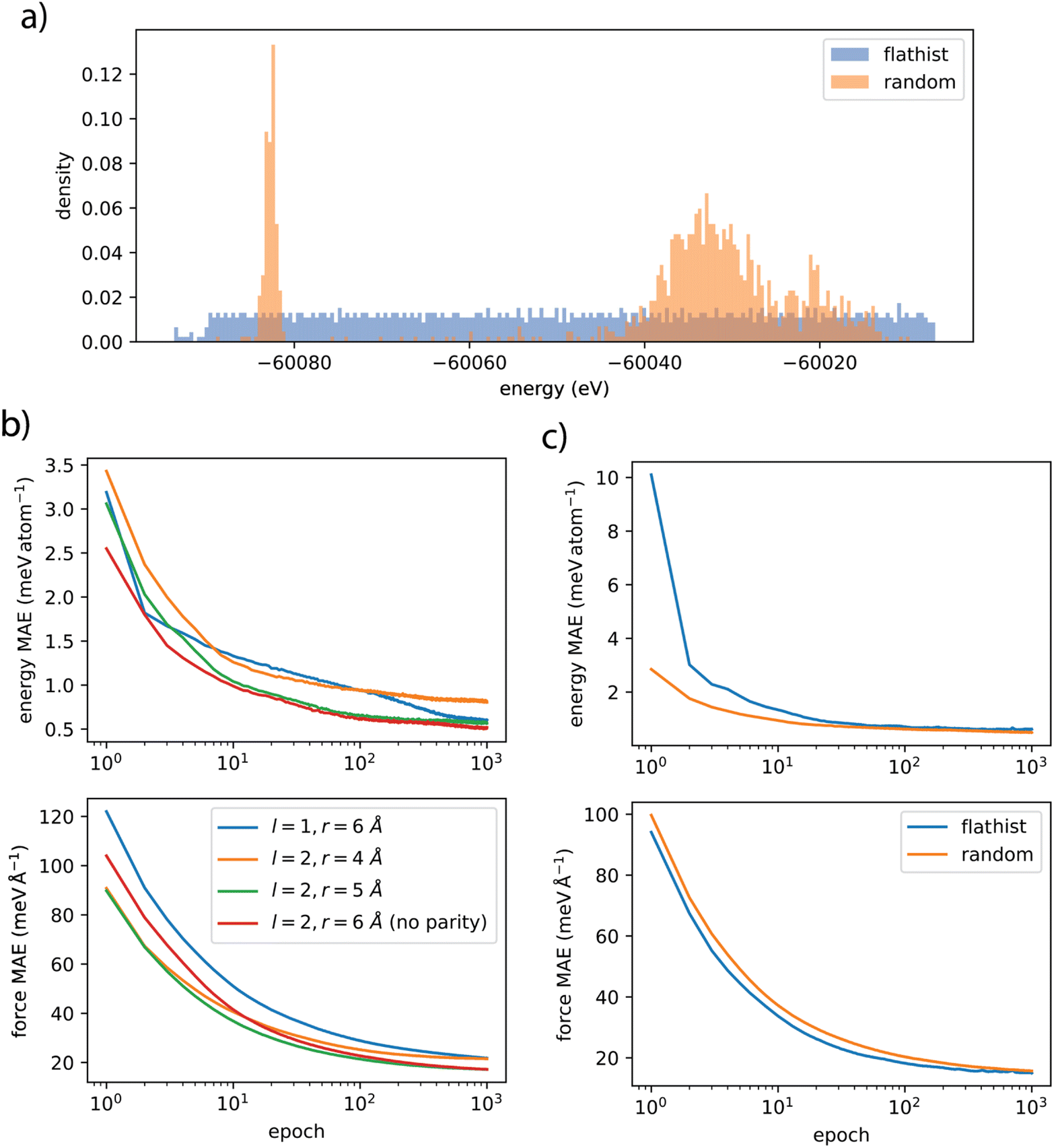 | ||
| Fig. 3 (a) Comparison of the energy distributions of the initial data set with either random or flat-histogram (flathist) sampling. (b) Testing accuracy curves for energies and forces as a function of epochs, for difference values of hyperparameters on a random train-test data set. (c) Accuracy curves comparing the performance of random and flat-histogram sampling. | ||
MLP architectures
NequIP (Neural Equivariant Interatomic Potential) is a recently published open-source code for building E(3)-equivariant interatomic potentials,32 building on the e3nn library.46 It is a state-of-the-art message-passing neural network (MPNN)47 framework that has been shown to produce high accuracy potentials with good training data efficiency,48 due to its equivariance and because it directly operates on relative interatomic positions. It has demonstrated high accuracy across a wide variety of systems, including MOFs29 and amorphous solids.32As our goal is to produce large-scale models of amorphous systems, we also compared NequiP with Allegro, another approach of the same family and developed by the same group. The Allegro model is implemented as an extension of NequIP. It is a strictly local equivariant deep learning interatomic potential. Because it does not rely on atom-centered message passing, Allegro achieves much higher scalability in parallel (multi-GPU) computations, allowing the description of larger systems – up to 100 million atoms were demonstrated by its authors.49
Choice of ML hyperparameters
The MLP models in this study were constructed using NequIP version 0.5.6 and Allegro (which does not have released version numbers). The models employed a cutoff radius chosen between 4 Å and 6 Å for the atomic environments and used five interaction layers. Feature representations were restricted to maximal rotation orders of either l = 1 or l = 2, and for the largest cutoff radius (6 Å) features with odd mirror parity were neglected. The loss function consists of a weighted average of potential energy and force errors, and was optimized using the Adam algorithm and a learning rate of 0.005. We tested the choice of hyperparameters on the 1000–100 train-test data set with random sampling to determine the influence of rotation orders and cutoff radius. We also compared the choice of random or flat-histogram. This lead us to choose a production model that used l = 2 and a cut-off radius of 6 Å without parity as this enabled the highest accuracy with good efficiency. The production model was trained on the final flat-histogram data-set featuring 2357–190 samples.For the Allegro model, we chose the same hyperparameters as for NequIP, in order to compare the accuracy and efficiency of the two models in a fair manner.
MLP-based molecular dynamics
MD simulations with NequIP were performed using LAMMPS32,50 with the pair_nequip patch (available at https://github.com/mir-group/pair_nequip). By default, a time step of 0.5 fs was used in the MD runs, and was occasionally changed to 0.25 fs to simulate very high temperatures (above 1500 K). The temperature and pressure (when applicable) were controlled using a Nosé–Hoover thermostat and barostat. Temperature and pressure damping parameters were fixed at 100 fs and 1000 fs respectively. Several values around the chosen damping parameters were tested to check their relevance. Simulations were typically performed on a single unit cell for NequIP, and on a 2 × 2 × 2 supercell for Allegro.A typical (N, V, T) simulation of a ZIF-4 unit cell (272 atoms) for 100 ps, required around 5 hours of computation on a single NVIDIA V100 GPU. It is a significant speedup compared to AIMD, for which 4 days and 160 CPUs (4 nodes of Intel Cascade Lake 6248) are required for the same system and duration.
Structural properties
Structural analyses were performed using the Python library aMOF and the parameters detailed in a previous study.15 aMOF is available online at https://github.com/coudertlab/amof, and the version used in this work is v1.1.0.Unless explicitly stated in the figure caption, properties were averaged over an (N, V, T) trajectory of 100 ps for MLP simulations. Frames were taken every 100 fs for the radial distribution function (RDF), bond angle distribution and coordination number, every 500 fs for the mean square displacement (MSD), every 1 ps for ring statistics and every 10 ps for pore statistics. Reference ab initio properties were computed as in ref. 15.
The potential of mean force (PMF) was computed from the RDF g(r) through the relationship F(r) = −kBT(lng(r) − lngmax), where gmax is the maximum of g(r) over r. The N–Zn–N angle distribution and Zn–N coordination numbers are computed by taking a cutoff radius of 2.5 Å for Zn–N distances, a value determined based on the Zn–N PMF, and validated in previous ab initio11,12,34 and ReaxFF15 studies. The total pore volume is computed on individual frames using the Zeo++ software,51–53 as the sum of accessible and non-accessible volume with a helium probe of radius 1.2 Å.
Ring statistics were computed using the R.I.N.G.S. code54 on the Zn–Im periodic graph obtained after identification of the building units. The definition of rings used in this work corresponds to the definition of “primitive rings” in the R.I.N.G.S. code (see ref. 15 for an extended definition). We used a maximum search depth of 32, safely above the largest rings of the ZIF-4 crystal.16
Mechanical properties
Computation of finite temperature mechanical properties were performed as detailed in our recent study.16 Two approaches were used in this work: the finite strain difference approach and the strain-fluctuation method. We focused on the example of the bulk modulus K, defined as K = −V(∂P/∂V)T. Values of K reported in this work correspond to the equilibrium volume.The finite strain difference approach consists in first establishing the P–V relationship, and then fitting it with an equation of state (EoS). P–V data is generated here by running multiple MD simulations in the (N, V, T) ensemble by enforcing the volume V, with the resulting pressure P(V) of the equilibrated system being measured. The well-behaved region of the P–V data was fitted with the second order Birch–Murnaghan EoS.55
Mechanical properties at a given temperature T can also be evaluated from the fluctuations of a system at equilibrium, simulated under a constant stress in the (N, σ, T) ensemble. The bulk modulus can be obtained directly from the fluctuations of the volume, and multiple mechanical properties – bulk modulus K, Young's modulus E, shear modulus G, and Poisson's ratio ν – can be computed from the elastic stiffness tensor C obtained from the fluctuations of the unit cell matrix.56 Their computation was performed using the ELATE code57 interfaced with the Python library aMOF. Values reported here were obtained with the Hill averaging scheme.
Results and discussion
Training and accuracy
We find that each of the models rapidly learns the energy and force of these complicated molecular systems, with the learning curves reaching a plateau in most cases before or near 1000 epochs, as seen on Fig. 3. Using the smaller initial data set we find the best accuracy is afforded by the l = 2, r = 6 Å model which is unsurprising as this is the model with the most features. The l = 1, r = 6 Å model provides a greater error in forces similar to l = 2, r = 4 Å but at very high efficiency. Models with l = 2, r = 5 Å or r = 6 Å without parity provide similar accuracy to r = 6 Å with parity, but again with much better efficiency.Furthermore, we see that sampling the data set with the flat histogram method does not appear to provide more accuracy when tested against a large random sample. However, as observed in the training curves (Fig. 3c), it does appear to learn marginally faster for forces, in our specific case. As a result of the initial training on the smaller data set, we trained a model with l = 2, r = 6 Å without parity (denoted here as “nequip deployed”) on a large data set created by flat histogram sampling. This model achieves near DFT accuracy on energy and forces, as summarized in Table 1. We also trained a model with Allegro, for the purpose of comparison, and in order to be able to perform molecular simulations on much larger systems. As expected, we found out that the strictly local Allegro model has lower accuracy, with a mean average error (MAE) of 0.70 meV on energy per atom (compared to 0.60 for NequIP), and 30.1 meV Å−1 on forces (compared to 15.0 for nequip). On the other hand, we will show later than it has great scalability, and it uses only a fraction of the memory that NequIP requires.
000 frames of held-out data and estimated efficiency (using a NVIDIA Tesla V100) of the models based on 300 calls with 6 used for warm-up. Models are trained on the smaller random data set unless stated, while the deployed models are trained on the larger flat histogram sampled data set
| Energy MAE (meV) | Force MAE (meV Å−1) | Stress MAE (MPa) | Efficiency (μs per atom per call) | |
|---|---|---|---|---|
| l = 1, r = 6 Å | 0.70 | 20.5 | 352 | 240 |
| l = 2, r = 4 Å | 0.90 | 20.7 | 304 | 371 |
| l = 2, r = 5 Å | 0.70 | 16.3 | 320 | 369 |
| l = 2, r = 6 Å (no parity) | 0.60 | 16.4 | 352 | 280 |
| l = 2, r = 6 Å | 0.58 | 15.2 | 361 | 452 |
| l = 2, r = 6 Å (flat histogram) | 0.61 | 17.8 | 360 | 453 |
| NequIP deployed | 0.60 | 15.0 | 352 | 282 |
| Allegro deployed | 0.70 | 30.1 | 352 | 347 |
During our study, we tested several different loss functions to simultaneously minimize the errors on the energy (E), forces ({Fi}) and stress (σ) of the system. We found that training MLPs on energy and forces only is sufficient to reproduce the mechanical properties with reasonable accuracy (see ESI†) as expected from the errors in stress computed, and that including stress in the optimization of the MLP typically does not improve the quality of the model (and sometimes degrades it with respect to errors on energy and force). While this is counter-intuitive, it may come from the fact that instantaneous stress tensors computed during the ab initio dynamics suffer from a higher relative noise than the energy and forces.
Reproducing structural properties
Fig. 4 shows three local properties: the partial Zn–N radial distribution functions (RDF) and potential of mean force (PMF), along with the N–Zn–N bond angle distribution. In every case, we note an excellent reproduction of the ab initio properties. The MLP accurately emulates the crystal properties, thus demonstrating excellent transferability on this system, although it was trained on a different state (the liquid). Additionally, no change in coordination nor ring statistics is observed for the crystal (see Table S5†), and the computed porous volumes are fairly similar for every phase (see Table S6†). These data point to an excellent reproduction of the structural properties of both the system they were trained on, and the crystal.
 | ||
| Fig. 4 (a) Radial distribution functions (RDF), (b) potentials of mean force (PMF) for the Zn–N atom pairs, and (c) distribution of the N–Zn–N angle for the ZIF-4 crystal (left) and liquid (right) with the NequIP MLP compared to AIMD. | ||
Their local properties are reported on Fig. S3,† and accurately reproduce every local ab initio property (RDF, PMF and angle distribution). Additionally, no change in coordination between every MLP and AIMD data was reported, with coordination numbers and ring statistics being identical. It provides strong evidence that our NequIP MLP can accurately model a variety of ZIF-4 phases.
 | ||
| Fig. 5 Temperature as a function of time during the glass formation procedure, consisting of preparation (green), melting (red and orange), quenching (grey) and equilibration (blue). For clarity, a moving average over 25 fs is used. | ||
Melt-quenching is mainly parameterized by the maximal temperature Tmax and the heating/cooling rate r. Consistently with the previous ab initio work,12 we aimed for a maximal temperature of 1500 K, and chose a rate of 20 K ps−1.
Contrary to ReaxFF simulations which led to excessive decoordination, we find that ZIF-4 in simulations with the MLP does not melt that easily. After a procedure consisting of only two ramps (one for heating, one for cooling), as with ReaxFF, the final system was still crystalline for Tmax = 1500 K and r in the 50–2.5 K ps−1 region. Either lowering the rate to r = 1 K ps−1, or increasing the temperature to Tmax = 1900 K, both led to the successful formation of glasses. However, the first option requires rather long simulations, while the second leads to an increased risk of loss of physical integrity.
Even if no breaking of the imidazolate rings was observed for 1500 K ≤ T ≤ 1900 K, we chose a safer third option which consisted in keeping Tmax = 1500 K and spending more time at the maximal temperature. A plateau of 50 ps at Tmax was enough to ensure melting, and therefore obtain a glass as highlighted on Fig. S4.†
 | ||
| Fig. 6 Radial distribution function (RDF, top panel) and potential of mean force (PMF, bottom panel) for the Zn–N atom pairs of the MLP glass (blue), ab initio glass (orange) and ab initio crystal (red). | ||
We investigated the N–Zn–N angle distribution (plotted on Fig. 7a): the MLP glass demonstrates an excellent reproduction of the ab initio data, which is crucial because that property is key to the features of ZIFs, and is not well reproduced by other levels of description (such as ReaxFF). The angular PMF shown on Fig. 7b further confirms this accuracy, showing minimal deviations for angles away from the 109.5° of the Zn(Im)4 tetrahedral structure. We then computed the porosity of the glasses (see Fig. S6†) and found a total porous volume of 61 cm3 kg−1 for the MLP glass, comprised between the 54 cm3 kg−1 of the crystal and 68 cm3 kg−1 of the ab initio glass. This is in good agreement with the experimental measurements of the porosity of a ZIF-4 glass made by positron annihilation lifetime spectroscopy (PALS).58
 | ||
| Fig. 7 Angular distribution (top panel) and potential of mean force (PMF, bottom panel) of the N–Zn–N angle for the MLP glass (blue), ab initio glass (orange) and ab initio crystal (red). | ||
Finally, we investigated the differences in the medium-range order of the glasses, by examining the coordination network built from the alternating Zn–Im units. We first calculated the average Zn–N coordination numbers and found a coordination of 3.93 for the MLP glass, the same value as the ab initio glass. We also computed Zn–Im ring statistics, to characterize the topology at a larger scale. Fig. 8 evidences that both glasses have topologies that deviate from the crystal's perfectly defined 8, 12 and 16-membered rings. While not identical, the two glass models display rather similar topologies.
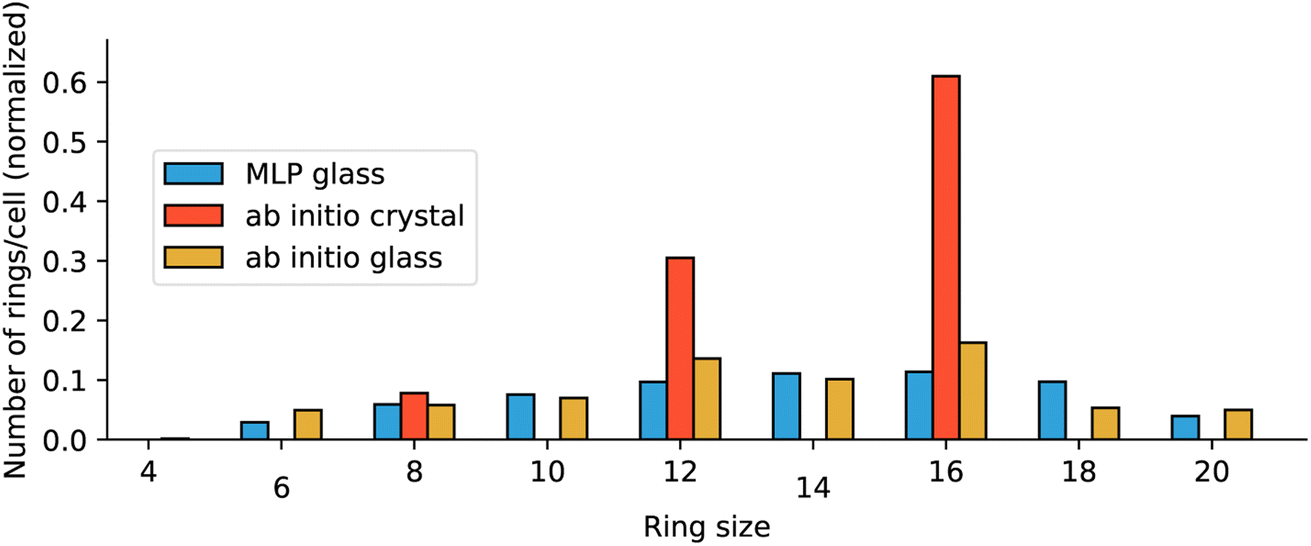 | ||
| Fig. 8 Distributions of size of zinc–imidazolate alternate rings for the MLP glass (blue), ab initio glass (orange) and ab initio crystal (red). | ||
Overall, these elements show that the glass model generated with our MLP reproduces well many properties of the ab initio glasses, all the more when compared to how ReaxFF fares. Moreover, the MLP glass has characteristics in line with known experimental data,34 providing strong evidence that aZIF models can be generated by MD simulations of melt-quenching with chemically accurate force fields.
Mechanical properties
While the mechanical stability of MOFs is also essential to fully achieve their potential in industrial-scale processes, the study of how these materials respond to mechanical stress is comparatively still emerging.59,60 While a series of experimental techniques have successfully been employed to determine the mechanical properties of crystalline MOFs, there is a lack of studies on the amorphous phases and very little data is available for now.35 Computational methods offer an alternative method, but are rather expensive at the ab initio level,16 which is why MLPs are of high interest in this context, being able to explore larger spatial and time scales. Several studies demonstrated how MLPs can be used to study various mechanical properties, including four applied to MOF materials.24,27,29,31Here we focus on the bulk modulus (K), a property that provides key information for applications, being related to MOF stability during the shaping process61 or pressure swing adsorption (PSA) cycles.59
In order to offer a meaningful comparison, we considered several glass models: the same ab initio glass models studied with AIMD, and MLP glasses obtained by melt-quenching in the previous section. For consistency between ab initio and MLP glasses, we used for both 10 single cell models and averaged results. These models are thoroughly presented in the ESI.† We checked that the use of a supercell did not seem to significantly impact the resulting bulk moduli K and density at zero pressure ρ0, as exemplified on Table S7† in the case of the ZIF-4 crystal. The P–V plot with the standard deviations as well as the fitted EoS is shown on Fig. S8† in the case of the crystal.
As shown on Table 2 and Fig. 9, we obtain a bulk modulus of Kcrystal = 1.69 GPa, in reasonable agreement with AIMD (1.39 GPa16) and previous experimental works – which found a value of 2.01 GPa with high-pressure crystallography62 and 1.42 GPa with mercury intrusion.63 Further interpretation is limited, as the precise value determined depends on the computational methodology and – for experiments – also on subtle differences between MOFs from different batches or on the experimental setup.62,64 However, the comparison between two different systems within the same methodology is meaningful, and calculations with the NequIP MLP reproduced the trend previously identified with other MD schemes, where glasses have larger K than the crystalline phase.
| Crystal | Ab initio glass | MLP glass | |
|---|---|---|---|
| K (GPa) | 1.69 | 3.43 ± 1.23 | 3.24 ± 0.72 |
| ρ 0 (g cm−3) | 1.16 | 1.25 ± 0.06 | 1.29 ± 0.04 |
 | ||
| Fig. 9 Bulk modulus K for the ZIF-4 crystal and two glasses with the finite strain difference method with the NequIP MLP compared with AIMD. | ||
Interestingly, this finite strain difference approach also yields the density at zero pressure ρ0 which is of 1.16 g cm−3 for the crystal and of in the 1.21 to 1.31 g cm−3 range for the glasses. It is consistent with previous AIMD results16 and experimental works65,66 which showed that glasses have larger ρ0 than the crystalline phase. However, it should be noted that all densities are smaller than the experimental values of ρglass = 1.38 g cm−3 (CO2 physisorption study66) and ρcrystal = 1.22 g cm−3 (crystallographic density67).
Starting from the initial systems for the finite strain difference method, we performed further (N, P, T) simulations with a flexible cell (LAMMPS keyword tri), corresponding to the (N, σ, T) ensemble required by the method, until convergence of the volume (see Fig. S9†) and elastic constants (see Fig. S10†). From the results shown in Table S8,† we see that the values of K obtained from the elastic constants are consistent with the more robust volume fluctuations, as are the K and ρ0 values.
We summarize in Table 3 the mechanical properties we have calculated – Young's modulus E, shear modulus G, and Poisson's ratio ν – some of which have never been reported for ZIF-4 glasses. In particular we reproduce the trend found in nanoindentation studies, with E being significantly larger in the glass than in the crystal (respectively 8.2 GPa65 and 4.6 GPa7), although the values computed with this method are significantly smaller.
| Crystal | Ab initio glass | |
|---|---|---|
| Bulk modulus K (GPa) | 1.58 | 2.72 ± 0.97 |
| Young's modulus E (GPa) | 2.49 | 2.93 ± 0.78 |
| Shear modulus G (GPa) | 1.01 | 1.12 ± 0.29 |
| Poisson's ratio ν (dimensionless) | 0.24 | 0.31 ± 0.04 |
We conclude that MLPs are very powerful in the calculation of physical properties, such as mechanical properties, that are typically very hard or impossible to compute at the DFT level of accuracy. Other physical quantities of interest, such as thermal properties, could be determined in the future from similar simulations.
Producing larger glass models
The training of our Allegro MLP has been described above already, as well as its accuracy on energies and atomic forces (see Table 1). The learning curves, and their comparison with NequIP, are presented in Fig. S11.† The main conclusion is that the MAE on forces for that Allegro model is twice as large as for NequIP, and we investigated the impact of this slightly lower accuracy on the physical properties of our systems. We found that the Allegro MLP reproduces very well the properties of low temperature systems (300 K): the ZIF-4 crystal and the ab initio glasses. As shown on Fig. 10, structural properties are reproduced as well as with NequIP when evaluated on 100 ps (N, V, T) trajectories.
 | ||
| Fig. 10 (a) Radial distribution functions (RDF), (b) potentials of mean force (PMF) for the Zn–N atom pairs, and (c) distribution of the N–Zn–N angle for the ZIF-4 crystal (left) and ab initio glasses (right) with Allegro MLP compared to NequIP MLP and AIMD. | ||
We also verified that Allegro can very efficiently simulate larger length scales, and found that that system sizes as large as a 5 × 5 × 5 supercell (containing ∼34 × 103 atoms) can be computed easily. The local structural properties are – as expected – identical for different system sizes for a number of systems (see Fig. S17†) and therefore we report here results for (2 × 2 × 2) supercells.
We therefore adapted our procedure to produce glass models, by running a simulation at a sufficiently high temperature to observe structural rearrangement while keeping the simulation duration short enough to avoid the unphysical events. We carried an MD melt-quenching simulation with a maximal temperature (Tmax) of 1400 K, maintaining the simulation at this temperature for 100 ps: we observed melting while witnessing no imidazolate breaking during the process.
The final properties of the melt-quenched glass obtained with the Allegro model closely resemble those generated by NequIP, as depicted in Fig. S15.† The generated glass structures are however not identical, possessing distinct ring statistics (see Fig. S16†), as is expected because of the larger length scales accessible to the system. This shows the power of Allegro MLPs for the description of amorphous phases, as well as the creation of large-scale atomistic models of these phases.
Conclusions and perspectives
In this work, we provide strong evidence that the development of machine learning potentials can pave the way towards the generation of multiple amorphous MOF models and the study of their properties. Using a dataset consisting of ab initio trajectories of liquid ZIF-4, we trained MLPs that led to an exceptional reproduction of the structural properties of multiple phases – crystal, liquid and several glasses – of ZIF-4. By generating new aZIF-4 models by melt-quenching, we demonstrated the potential of these MLPs to generate disordered models of MOFs at a fraction of the computational cost of AIMD. These models reproduce very well the local structure, framework topology, and macroscopic physical properties (such as mechanical behaviour) of the amorphous MOFs. Modern parallelized MLPs can even allow us to reach model sizes up ten of thousands of atoms at a modest computational cost.We think this development is a great perspective in the field of computational studies of disordered and amorphous framework materials. Previously, the choice of approaches required us to choose between the chemical accuracy of the description of intermolecular interactions, and the time and length scales achievable. On the one hand, first-principle methods were accurate but very expensive, and therefore limited by the lack of sampling of the phase space explored, as well as restricted to relatively short dynamics. On the other hand, reactive force field-based approaches were often inaccurate in their depiction of the chemistry at play. The use of MLPs based on ab initio data offer us a new way to approach the problem, and accelerate our quest of understanding the physical and chemical properties of these systems, and their relationship to local structure.
Data availability
Representative input files for each type of simulation are available online in our data repository at https://github.com/fxcoudert/citable-data and the data set of configurations generated through ab initio MD and used for training and testing is made available on Zenodo at https://doi.org/10.5281/zenodo.10015594.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Kim Jelfs and Tom Bennett for many inspiring discussions on this topic. Access to high-performance computing platforms was provided by GENCI grant A0150807069. This work was funded under the Imperial College London – CNRS PhD joint programme. JDE is the recipient of an Australian Research Council Discovery Early Career Award (project number DE220100163) funded by the Australian Government. Phoenix HPC service at the University of Adelaide are thanked for providing high-performance computing resources. This research was supported by the Australian Government's National Collaborative Research Infrastructure Strategy (NCRIS), with access to computational resources provided by Pawsey Supercomputing Research Centre through the National Computational Merit Allocation Scheme.References
- T. D. Bennett and A. K. Cheetham, Amorphous Metal–Organic Frameworks, Acc. Chem. Res., 2014, 47, 1555–1562 CrossRef PubMed.
- T. D. Bennett and S. Horike, Liquid, glass and amorphous solid states of coordination polymers and metal–organic frameworks, Nat. Rev. Mater., 2018, 3, 431–440 CrossRef.
- D. A. Keen and T. D. Bennett, Structural investigations of amorphous metal–organic frameworks formed via different routes, Phys. Chem. Chem. Phys., 2018, 20, 7857–7861 RSC.
- A. F. Sapnik, C. Sun, J. E. M. Laulainen, D. N. Johnstone, R. Brydson, T. Johnson, P. A. Midgley, T. D. Bennett and S. M. Collins, Mapping nanocrystalline disorder within an amorphous metal–organic framework, Commun. Chem., 2023, 6, 1555 Search PubMed.
- R. N. Widmer, G. I. Lampronti, S. Chibani, C. W. Wilson, S. Anzellini, S. Farsang, A. K. Kleppe, N. P. M. Casati, S. G. MacLeod, S. A. T. Redfern, F.-X. Coudert and T. D. Bennett, Rich Polymorphism of a Metal–Organic Framework in Pressure–Temperature Space, J. Am. Chem. Soc., 2019, 141, 9330–9337 CrossRef.
- N. Castel and F.-X. Coudert, Atomistic Models of Amorphous Metal–Organic Frameworks, J. Phys. Chem. C, 2022, 126, 6905–6914 CrossRef CAS.
- T. D. Bennett, A. L. Goodwin, M. T. Dove, D. A. Keen, M. G. Tucker, E. R. Barney, A. K. Soper, E. G. Bithell, J.-C. Tan and A. K. Cheetham, Structure and Properties of an Amorphous Metal-Organic Framework, Phys. Rev. Lett., 2010, 104, 2272 Search PubMed.
- S. Horike, S. S. Nagarkar, T. Ogawa and S. Kitagawa, A New Dimension for Coordination Polymers and Metal–Organic Frameworks: Towards Functional Glasses and Liquids, Angew. Chem., Int. Ed., 2020, 59, 6652–6664 CrossRef CAS.
- A. F. Sapnik, I. Bechis, S. M. Collins, D. N. Johnstone, G. Divitini, A. J. Smith, P. A. Chater, M. A. Addicoat, T. Johnson, D. A. Keen, K. E. Jelfs and T. D. Bennett, Mixed hierarchical local structure in a disordered metal–organic framework, Nat. Commun., 2021, 12, 1213 CrossRef.
- I. Bechis, A. F. Sapnik, A. Tarzia, E. H. Wolpert, M. A. Addicoat, D. A. Keen, T. D. Bennett and K. E. Jelfs, Modeling the Effect of Defects and Disorder in Amorphous Metal–Organic Frameworks, Chem. Mater., 2022, 34, 9042–9054 CrossRef CAS PubMed.
- R. Gaillac, P. Pullumbi and F.-X. Coudert, Melting of Zeolitic Imidazolate Frameworks with Different Topologies: Insight from First-Principles Molecular Dynamics, J. Phys. Chem. C, 2018, 122, 6730–6736 CrossRef CAS.
- R. Gaillac, P. Pullumbi, T. D. Bennett and F.-X. Coudert, Structure of Metal–Organic Framework Glasses by Ab Initio Molecular Dynamics, Chem. Mater., 2020, 32, 8004–8011 CrossRef CAS.
- Y. Yang, Y. K. Shin, S. Li, T. D. Bennett, A. C. T. van Duin and J. C. Mauro, Enabling Computational Design of ZIFs Using ReaxFF, J. Phys. Chem. B, 2018, 122, 9616–9624 CrossRef CAS PubMed.
- S. A. Mohamed and J. Kim, Gas Adsorption Enhancement in Partially Amorphized Metal–Organic Frameworks, J. Phys. Chem. C, 2021, 125, 4509–4518 CrossRef CAS.
- N. Castel and F.-X. Coudert, Challenges in Molecular Dynamics of Amorphous ZIFs Using Reactive Force Fields, J. Phys. Chem. C, 2022, 126, 19532–19541 CrossRef CAS.
- N. Castel and F.-X. Coudert, Computation of Finite Temperature Mechanical Properties of Zeolitic Imidazolate Framework Glasses by Molecular Dynamics, Chem. Mater., 2023, 35, 4038–4047 CrossRef.
- P. Friederich, F. Häse, J. Proppe and A. Aspuru-Guzik, Machine-learned potentials for next-generation matter simulations, Nat. Mater., 2021, 20, 750–761 CrossRef.
- J. Behler and G. Csányi, Machine learning potentials for extended systems: a perspective, Eur. Phys. J. B, 2021, 94, 457 CrossRef.
- A. M. Miksch, T. Morawietz, J. Kästner, A. Urban and N. Artrith, Strategies for the construction of machine-learning potentials for accurate and efficient atomic-scale simulations, Mach. Learn.: Sci. Technol., 2021, 2, 031001 Search PubMed.
- A. Pedone, M. Bertani, L. Brugnoli and A. Pallini, Interatomic potentials for oxide glasses: Past, present, and future, J. Non-Cryst. Solids: X, 2022, 15, 100115 Search PubMed.
- F. C. Mocanu, K. Konstantinou, T. H. Lee, N. Bernstein, V. L. Deringer, G. Csányi and S. R. Elliott, Modeling the Phase-Change Memory Material, Ge2Sb2Te5, with a Machine-Learned Interatomic Potential, J. Phys. Chem. B, 2018, 122, 8998–9006 CrossRef.
- V. L. Deringer, N. Bernstein, G. Csányi, C. Ben Mahmoud, M. Ceriotti, M. Wilson, D. A. Drabold and S. R. Elliott, Origins of structural and electronic transitions in disordered silicon, Nature, 2021, 589, 59–64 CrossRef CAS.
- J. Wang, H. Shen, R. Yang, K. Xie, C. Zhang, L. Chen, K.-M. Ho, C.-Z. Wang and S. Wang, A deep learning interatomic potential developed for atomistic simulation of carbon materials, Carbon, 2022, 186, 1–8 CrossRef.
- M. Eckhoff and J. Behler, From Molecular Fragments to the Bulk: Development of a Neural Network Potential for MOF-5, J. Chem. Theory Comput., 2019, 15, 3793–3809 CrossRef PubMed.
- S. K. Achar, J. J. Wardzala, L. Bernasconi, L. Zhang and J. K. Johnson, Combined Deep Learning and Classical Potential Approach for Modeling Diffusion in UiO-66, J. Chem. Theory Comput., 2022, 18, 3593–3606 CrossRef PubMed.
- Y. Yu, W. Zhang and D. Mei, Artificial Neural Network Potential for Encapsulated Platinum Clusters in MOF-808, J. Phys. Chem. C, 2022, 126, 1204–1214 CrossRef.
- O. Tayfuroglu, A. Kocak and Y. Zorlu, A neural network potential for the IRMOF series and its application for thermal and mechanical behaviors, Phys. Chem. Chem. Phys., 2022, 24, 11882–11897 RSC.
- M. Herbold and J. Behler, Machine learning transferable atomic forces for large systems from underconverged molecular fragments, Phys. Chem. Chem. Phys., 2023, 25, 12979–12989 RSC.
- S. Vandenhaute, M. Cools-Ceuppens, S. DeKeyser, T. Verstraelen and V. Van Speybroeck, Machine learning potentials for metal-organic frameworks using an incremental learning approach, npj Comput. Mater., 2023, 9, 10575 Search PubMed.
- P. Ying, T. Liang, K. Xu, J. Zhang, J. Xu, Z. Zhong and Z. Fan, Sub-Micrometer Phonon Mean Free Paths in Metal–Organic Frameworks Revealed by Machine Learning Molecular Dynamics Simulations, ACS Appl. Mater. Interfaces, 2023, 15, 36412–36422 CrossRef.
- S. Wieser and E. Zojer, Machine learned Force-Fields for an ab-initio Quality Description of Metal-Organic Frameworks, arXiv, 2023, prepint, arXiv:2308.01278, DOI:10.48550/arXiv.2308.01278.
- S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt and B. Kozinsky, E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials, Nat. Commun., 2022, 13, 1 Search PubMed.
- R. Goeminne, L. Vanduyfhuys, V. V. Speybroeck and T. Verstraelen, DFT-Quality Adsorption Simulations in Metal–Organic Frameworks Enabled by Machine Learning Potentials, J. Chem. Theory Comput., 2023, 19(18), 6313–6325 CrossRef.
- R. Gaillac, P. Pullumbi, K. A. Beyer, K. W. Chapman, D. A. Keen, T. D. Bennett and F.-X. Coudert, Liquid metal–organic frameworks, Nat. Mater., 2017, 16, 1149–1154 CrossRef.
- J. Fonseca, T. Gong, L. Jiao and H.-L. Jiang, Metal–organic frameworks (MOFs) beyond crystallinity: amorphous MOFs, MOF liquids and MOF glasses, J. Mater. Chem. A, 2021, 9, 10562–10611 RSC.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef.
- T. D. Kühne, et al., CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152, 194103 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Separable dual-space Gaussian pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- D. P. Landau, S.-H. Tsai and M. Exler, A new approach to Monte Carlo simulations in statistical physics: Wang-Landau sampling, Am. J. Phys., 2004, 72, 1294–1302 CrossRef.
- M. S. Shell, P. G. Debenedetti and A. Z. Panagiotopoulos, An improved Monte Carlo method for direct calculation of the density of states, J. Chem. Phys., 2003, 119, 9406–9411 CrossRef.
- D. Bousquet, F.-X. Coudert and A. Boutin, Free energy landscapes for the thermodynamic understanding of adsorption-induced deformations and structural transitions in porous materials, J. Chem. Phys., 2012, 137, 044118 CrossRef PubMed.
- A. Datar, M. Witman and L.-C. Lin, Improving Computational Assessment of Porous Materials for Water Adsorption Applications via Flat Histogram Methods, J. Phys. Chem. C, 2021, 125, 4253–4266 CrossRef.
- M. Geiger, et al., e3nn/e3nn: 2022-12-12, 2022, https://zenodo.org/record/3724963.
- E. Kocer, T. W. Ko and J. Behler, Neural Network Potentials: A Concise Overview of Methods, Annu. Rev. Phys. Chem., 2022, 73, 163–186 CrossRef.
- I. Batatia, S. Batzner, D. P. Kovács, A. Musaelian, G. N. C. Simm, R. Drautz, C. Ortner, B. Kozinsky and G. Csányi, The Design Space of E(3)-Equivariant Atom-Centered Interatomic Potentials, arXiv, 2022, preprint, arXiv:2205.06643, DOI: DOI:10.48550/arXiv.2205.06643.
- A. Musaelian, S. Batzner, A. Johansson, L. Sun, C. J. Owen, M. Kornbluth and B. Kozinsky, Learning local equivariant representations for large-scale atomistic dynamics, Nat. Commun., 2023, 14, 1 Search PubMed.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comput. Phys. Commun., 2022, 271, 108171 CrossRef.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef.
- M. Pinheiro, R. L. Martin, C. H. Rycroft, A. Jones, E. Iglesia and M. Haranczyk, Characterization and comparison of pore landscapes in crystalline porous materials, J. Mol. Graphics Modell., 2013, 44, 208–219 CrossRef.
- M. Pinheiro, R. L. Martin, C. H. Rycroft and M. Haranczyk, High accuracy geometric analysis of crystalline porous materials, CrystEngComm, 2013, 15, 7531 RSC.
- S. L. Roux and P. Jund, Ring statistics analysis of topological networks: New approach and application to amorphous GeS2 and SiO2 systems, Comput. Mater. Sci., 2010, 49, 70–83 CrossRef.
- F. Birch, Finite Elastic Strain of Cubic Crystals, Phys. Rev., 1947, 71, 809–824 CrossRef.
- M. Parrinello and A. Rahman, Strain fluctuations and elastic constants, J. Chem. Phys., 1982, 76, 2662–2666 CrossRef.
- R. Gaillac, P. Pullumbi and F.-X. Coudert, ELATE: an open-source online application for analysis and visualization of elastic tensors, J. Phys.: Condens. Matter, 2016, 28, 275201 CrossRef PubMed.
- A. W. Thornton, K. E. Jelfs, K. Konstas, C. M. Doherty, A. J. Hill, A. K. Cheetham and T. D. Bennett, Porosity in metal–organic framework glasses, Chem. Commun., 2016, 52, 3750–3753 RSC.
- K. Yang, G. Zhou and Q. Xu, The elasticity of MOFs under mechanical pressure, RSC Adv., 2016, 6, 37506–37514 RSC.
- L. R. Redfern and O. K. Farha, Mechanical properties of metal–organic frameworks, Chem. Sci., 2019, 10, 10666–10679 RSC.
- P. Vervoorts, J. Stebani, A. S. J. Méndez and G. Kieslich, Structural Chemistry of Metal–Organic Frameworks under Hydrostatic Pressures, ACS Mater. Lett., 2021, 3, 1635–1651 CrossRef.
- P. Vervoorts, C. L. Hobday, M. G. Ehrenreich, D. Daisenberger and G. Kieslich, The Zeolitic Imidazolate Framework ZIF-4 under Low Hydrostatic Pressures, Z. Anorg. Allg. Chem., 2019, 645, 970–974 CrossRef CAS.
- S. Henke, M. T. Wharmby, G. Kieslich, I. Hante, A. Schneemann, Y. Wu, D. Daisenberger and A. K. Cheetham, Pore closure in zeolitic imidazolate frameworks under mechanical pressure, Chem. Sci., 2018, 9, 1654–1660 RSC.
- I. E. Collings and A. L. Goodwin, Metal–organic frameworks under pressure, J. Appl. Phys., 2019, 126, 181101 CrossRef.
- T. D. Bennett, Y. Yue, P. Li, A. Qiao, H. Tao, N. G. Greaves, T. Richards, G. I. Lampronti, S. A. T. Redfern, F. Blanc, O. K. Farha, J. T. Hupp, A. K. Cheetham and D. A. Keen, Melt-Quenched Glasses of Metal–Organic Frameworks, J. Am. Chem. Soc., 2016, 138, 3484–3492 CrossRef CAS PubMed.
- L. Frentzel-Beyme, P. Kolodzeiski, J.-B. Weiß, A. Schneemann and S. Henke, Quantification of gas-accessible microporosity in metal-organic framework glasses, Nat. Commun., 2022, 13, 705 Search PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Exceptional chemical and thermal stability of zeolitic imidazolate frameworks, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- L. Bouëssel du Bourg, A. U. Ortiz, A. Boutin and F.-X. Coudert, Thermal and mechanical stability of zeolitic imidazolate frameworks polymorphs, APL Mater., 2014, 2, 124110 CrossRef.
- S. Stocker, J. Gasteiger, F. Becker, S. Günnemann and J. T. Margraf, How robust are modern graph neural network potentials in long and hot molecular dynamics simulations?, Mach. Learn.: Sci. Technol., 2022, 3, 045010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: System description and methodological details for AIMD simulations, training of the MLPs and computation of mechanical properties. Volume, pressure and elastic constant convergence, P–V data, additional mechanical properties, radial distribution functions, potentials of mean force, angle distributions, porous volumes, ring statistics. See DOI: https://doi.org/10.1039/d3dd00236e |
| This journal is © The Royal Society of Chemistry 2024 |