
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgress and future of the computational design of antimicrobial peptides (AMPs): bio-inspired functional molecules
Miroslava
Nedyalkova
 *ac,
Andrew S.
Paluch
b,
Diana Potes
Vecini
ac and
Marco
Lattuada
*a
*ac,
Andrew S.
Paluch
b,
Diana Potes
Vecini
ac and
Marco
Lattuada
*a
aDepartment of Chemistry, Fribourg University, Chemin Du Musée 9, 1700 Fribourg, Switzerland. E-mail: miroslava.nedyalkova@unifr.ch
bDepartment of Chemical, Paper, and Biomedical Engineering, Miami University, Oxford, Ohio 45056, USA
cSwiss National Center for Competence in Research (NCCR) Bio-inspired Materials, University of Fribourg, Chemin des Verdiers 4, CH-1700 Fribourg, Switzerland
First published on 29th November 2023
Abstract
The effectiveness of antibiotics is greatly enhanced by their ability to target invasive organisms involved in the ancient evolutionary battle between hosts and pathogens. Conventional antibiotics no longer offer adequate protection due to the evolution of strategies to evade them. As a result, efforts are needed to design novel replacement antibiotics, making them unique from most other forms of drug development. As drug discovery costs have steadily increased along with the need for novel antibiotics, the interest in antimicrobial peptides (AMPs) as alternative antimicrobial treatments has grown in recent years. As a complement to experimental high-throughput screening, computational methods have become essential in hit and lead discovery in pharmaceutical research. It may be possible to access unexplored chemical space with customized virtual compound libraries. It has been questioned whether screening billions of molecules virtually with the risk of false positives is practical despite their unlimited potential size. In terms of finding novel chemical compounds capable of solving many global problems, machine learning, deep learning, and generative models hold significant promise. It is anticipated that the current challenges and limitations about the applicability of the stated approaches will be overcome in the coming years. However, plenty of advances are still required to achieve their full potential. In this perspective, we review the previous and ongoing work based on the latest scientific breakthroughs and technologies that could offer new opportunities and alternative strategies for developing novel AMPs.
Introduction
Motivation
Pathogens resistant to conventional antibiotics have emerged and spread rapidly, increasing difficult-to-treat infections that threaten global health. By 2050, drug-resistant pathogens are predicted to account for the highest number of deaths worldwide due to infections.1 Due to poor economic incentives and market failure, discovery and development efforts have gradually declined, and few new antibiotics have been commercialized in recent decades.2 It is imperative that this trend be reversed with new strategies to develop novel antimicrobial treatments.In terms of antimicrobial potential, metal complexes are an unexploded source. For instance, Rhenium complexes are particularly attractive given their low in vivo toxicity and high antimicrobial activity. However, their targets and mechanism of action need further research.3–5 Microbial metabolites are the source of many existing antibiotics and other medicines. For their high diversity and broad bioactivity spectra, short peptides are among the most widely studied secondary metabolites, and the large group of antimicrobial peptides (AMPs) produced by bacteria has been used to treat bacterial, fungal, and viral infections and even cancer.6 Previously, antibiotics were developed from bacterial antimicrobial peptides (AMPs), primarily non-ribosomally synthesized peptides and ribosomally synthesized and post-translationally modified peptides. Furthermore, class II and class III bacteriocins can be synthesized ribosomally and function unmodified.7 As a result, they can be directly identified from microbial genomes, like AMPs found in eukaryotic genomes, such as human LL37 (cathelicidin).8 In contrast to conventional small-molecule antibiotics, AMPs exhibit lower susceptibility to developing resistance in pathogens and encounter stronger phylogenetic barriers inside bacteria against horizontal transmission of developed resistance. While AMPs are diverse in amino acid sequence, they share several similarities, such as cationic charge (+1 to +7) generated by the presence of arginine/lysine/histidine residues, short amino acid sequences (frequently up to 50 residues in length), and are usually amphiphilic.9,10 With contribution from both hydrophobic and hydrophilic regions in these peptides, AMPs can interact with bacterial membranes to gain entry into cells. This characteristic enables them to attach to anionic (negatively charged) bacterial membranes and exert antibacterial activity. The interaction between AMPs and bacterial membranes primarily determines their antimicrobial function.11,12 Amino acid diversity at the N-terminus of AMPs directly influences which AMPs can disrupt the membranes of specific bacteria while not being active against others.13 The observations that cationic AMPs cause increased membrane disruption and permeabilization in bacterial membranes agree with this conclusion.14
The structural attributes of AMPs (including α-helices, β-sheets, combined α-helix and β-sheets, and extended structures; see Fig. 1)15–18 give rise to distinct mechanisms of action. As the peptide interacts with the target membrane, it folds into an amphipathic α-helix.19 The class of AMPs based on β-sheets is characterized by a strand of antiparallel peptide chains stabilized by two or more disulfide bonds.15 It includes, for example, the defense peptides of vertebrates, insects, and plants.20 There are high proportions of specific amino acids in extended peptides such as indolicidin, including tryptophan, histidine, and proline.21 Most of these peptides adopt extended configurations upon interaction with the membrane. They are stabilized by hydrogen bonds and van der Waals forces with lipids rather than inter-residue hydrogen bonds. As the name suggests, loop peptides possess a loop structure imparted by the presence of a single bond, such as disulfide or amide.
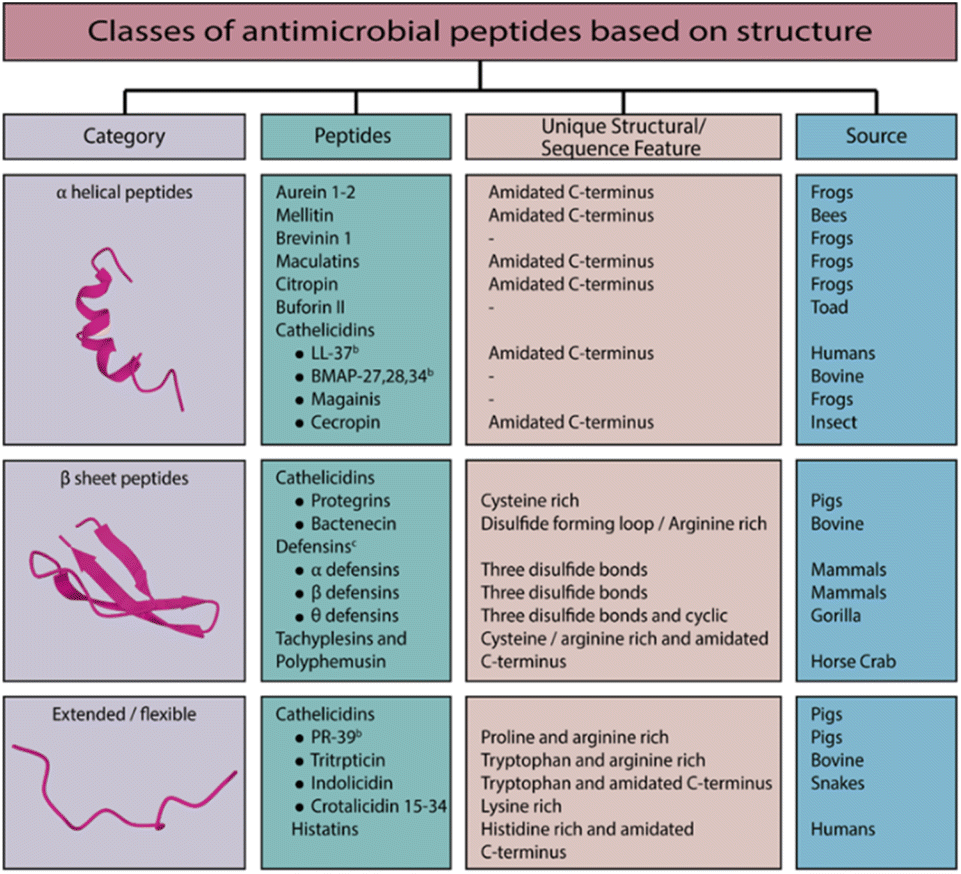 | ||
| Fig. 1 Classes of antimicrobial peptides based on structure. | ||
Based on the structural features, the most important characteristic of all AMPs is their solubility in aqueous environments and their ability to partition into lipid environments.22 As AMPs are generally cationic, they preferentially target bacterial membranes over zwitterionic eukaryotic membranes due to their anionic nature.17,23–25 The hydrophobic amino acid content of AMPs facilitates the interaction of AMPs with cell membranes. AMPs that are non-ribosomal can also contain features such as lipids that may facilitate their interaction with membranes. As different organisms have different membrane compositions, AMPs have different selectivities.
Sometimes, AMPs inhibit biofilm production, cross the cell membrane, and inhibit cellular functions. However, AMPs generally cause cell death through membrane disruption and eventual cell lysis. Despite this, there are a variety of mechanisms of action among membranolytic AMPs have been proposed. A three-pronged mechanism of membrane disruption has been identified for ribosomally synthesized AMPs, particularly those that are helical or tetrahelical. “Barrel stave”, “toroidal pore”, and “carpet” are all terms describing these mechanisms.18,23,26,27 (see Fig. 2).
 | ||
| Fig. 2 The interaction between peptide and bacterial cellular membrane. This figure is based on the work of Saeed et al.28 | ||
AMPs of other types are less well-known regarding their mechanisms of action. In Fig. 3,29–39 we summarize the most important AMPs and their relation to the source and mechanism of action.
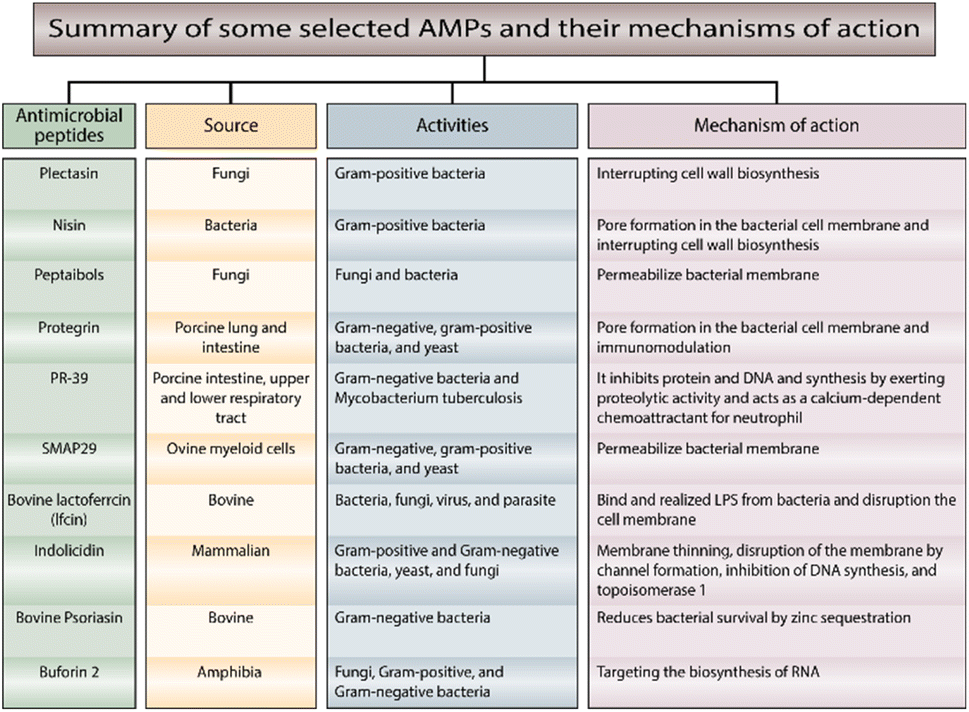 | ||
| Fig. 3 Summary of selected AMPs and their mechanisms of action. | ||
As well as damaging the membranes, the peptide can kill bacteria by inhibiting the biosynthesis of nucleic acids, proteins, and some essential enzymes involved in synthesizing cell walls. The mechanisms for intracellular AMPs are summarized in Fig. 4.
 | ||
| Fig. 4 Mechanism for intracellular AMP activity. This figure is based on the work of Saeed et al.28 | ||
The unique characteristic of the interactions involving AMPs is to inhibit a specific bacterium. It is believed that the molecular net charge of the peptide is the most important characteristic that makes it effective against bacteria, as predicted based on AMP features contributing to antimicrobial activity.40 Recent studies have also demonstrated that some other characteristics of AMPs play an important role in their antimicrobial activity, and these features may vary according to the bacterial species they target.41,42 It is, therefore, possible to discover new characteristics of AMPs important for specific bacteria through a machine learning (ML) analysis of AMP characteristics associated with bacterium-specific efficacy. Doing so makes it possible to develop new antimicrobial drugs and better understand AMP's microscopic mechanisms.
AMPs constitute a promising class of compounds with a wide range of applications. This, in part, is a result of the diverse molecular chemistries of AMPs. However, herein lies the challenge of selecting and designing AMPs for novel applications. The number of potential AMP candidates for a specific application is enormous. Developing computational methods for selecting and designing AMPs is of utmost importance.
Faccone et al.50 have demonstrated different strategies for generating effective drug candidates based on de novo algorithms. By combining computer-assisted approaches with omiganan (MBI-226) peptides, these authors have engineered an AMP. They identified functionally relevant natural or synthetic peptide motifs at specific amino acid positions. Database filtering technology (DFT) has also been proposed as a promising approach for the de novo design of improved AMPs. Mishra and Wang51 first began exploring this concept. The features identified as filters for designing novel peptides were peptide activity, length, amino acid frequency, charge, hydrophobicity, and structure profile. It was shown that the obtained peptide efficiently inhibited a Methicillin-resistant Staphylococcus aureus (MRSA).
As a key tool for combating multidrug-resistant bacteria, quantitative structure–activity relationship (QSAR) methods provide useful information for the rational design of new active molecules at a minimal cost. Much progress has been made in QSAR methods, from traditional 2D-QSAR methods to 3D-QSAR methods, incorporating parameters such as molecular and spatial variety and protein flexibility. In QSAR, two of the most commonly used methods are comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA), both of which are linear. QSAR methods have been widely used in discovering and growing different libraries for new antibacterial agents.52–55
Various linear and nonlinear statistical methods are used to develop these models based on the 2D or 3D representations of molecules. As a result of its simplicity, transparency, reproducibility, and ease of interpretation, multiple linear regression (MLR) is often used to obtain QSAR models. Due to the direct correlation between each descriptor's coefficient and its algebraic sign, it is easy to interpret its contribution to the model.
In pattern recognition, linear discriminant analysis (LDA) separates two or more class objects based on a linear combination of variables and can be applied to classification problems. The differences among data classes are explicitly modeled as part of the LDA method.
Nonlinear techniques (machine learning) are becoming more influential. Among the machine learning methods used in QSAR are artificial neural networks (ANNs), random forests (RFs), and support vector machines (SVMs).
In traditional QSAR models, the relationship between activities and the variables of the descriptors is identified. Additionally, RF (random forest) and DNN (deep neural network) methods from the machine learning approach were used to develop the prediction model. A decision tree (DT) is a classification method using ensemble learning. The final model was based on the highest score from individually developed trees in the forest. The DNN algorithm is a mathematical method designed to mimic the neurons (nodes) of the human brain to recognize objects and analyze them progressively, improving previous neural network algorithms. Thus, more features are identified as more executed nodes are added to each layer.
where f is our function that maps from X (vector of descriptors) to y (scalar physical property of interest). The difference between traditional and ML-based QSAR is in the functional mapping, f. In both cases, we can use a similar set of descriptors, X, and we will train on similar data to predict a desired physical property by minimizing the squared difference between the reference and predicted data. Using traditional QSAR, we are typically limited in our functional map and, as mentioned earlier, usually employ MLR, e.g.:
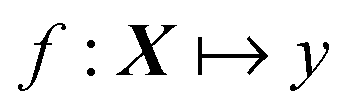
| y = a0 + a1x1 + a2x2 + … |
Using traditional models (i.e., MLR), we end up with an analytic equation that can readily be presented and shared. On the other hand, with machine learning we do not have an analytical model that we can share and summarize, but instead some computer code that we can share.
This Perspective article summarizes strategies for the design of the new AMPs. As part of our review, we addressed the challenges that de novo AI strategies for new AMPs must overcome. For example, proper molecular representation is a key point for de novo molecule generation. Model benchmark and how and which metrics should be used to evaluate the obtained models is the other Achille's heel of de novo design. A de novo molecule generation model benchmarking and validation can be challenging. To validate newly generated molecules, it is best to synthesize them and then test their predicted properties experimentally. This review aims to provide readers with the information and context needed to utilize generative modeling effectively.
Training database
The overarching goal of computational strategies for identifying and designing AMPs is two-fold. First, we seek methods to identify promising AMP candidates. Second, we strive to understand the underlying molecular-level properties of AMP candidates that lead to a specific activity. This second point is crucial for optimization and intuitive design applications—additionally, our computational strategies generally fall into two camps. First, molecular dynamic (MD) simulations (structural and mechanistic analyses) may be used to study AMP-target interactions in atomistic detail. Theoretically, this presents a way to identify promising AMP candidates and gain insight into the underlying intermolecular interactions. The MD simulations provide mechanistic insights into the different modes of action during the early stages of interaction and the time-dependent information about the structure of AMPs and the nature of the interactions. We have summarized the MD methods in Table 1. Simulating AMP-lipid interactions using MD simulations has been common practice for many years.56–59 Almost all simulation studies, however, have been of AMPs that form stable α-helical or β-hairpin structures upon binding to and/or insertion into the membrane. These AMPs are generally more likely to act via a pore-forming mechanism. Less is known, however, about the mechanism of action of unstructured AMPs such as the linear battacin analogs. Unfortunately, such strategies are inefficient and impractical for early-stage design applications and the identification of candidates.| Process/property of interest | Simulation technique | Considerations for AMPs |
|---|---|---|
| Peptide secondary structure | Conventional atomistic60–66 | * Can be used to monitor conformational stability |
| Molecular dynamics (MD) | ||
| Replica exchange approaches67–71 | * Can capture slower peptide conformational changes than conventional MD | |
| * Fewer simulation repeats are needed, but it is still advisable to check that simulations starting from different conformations converge to the same point | ||
| Accelerated (a) MD72,73 | * Can capture slower peptide conformational changes than conventional MD | |
| High-temperature (HT) MD69,74 | * Elevated temperatures can speed up the kinetics associated with peptide insertion and folding in the membrane | |
| Peptide aggregation | Conventional atomistic MD62,75–77 | * Can be used to monitor the stability of experimentally determined |
| Coarse-grained (CG) MD64,78,79 | * Larger systems and longer simulation times can be achieved | |
| * Atomic detail is lost | ||
| Metadynamics76,80,81 | * The technique can be used to enhance the sampling of slow aggregation event | |
| Umbrella sampling (US)82,83 | * The technique can be used to enhance the sampling of slow aggregation | |
| Peptide–membrane interactions | Conventional atomistic MD62,84,85 | * Can be used to investigate the surface interactions of AMPs |
| * Multiple simulation repeats may be required to achieve statistical significance | ||
| CG MD86–88 | * Larger systems and longer simulation times can be achieved atomic detail is lost | |
| US85,89,90 | * US can be used with the reaction coordinate set as the center of mass (COM) distance between a peptide and the membrane | |
| Metadynamics81,91 | * Metadynamics can be used to enhance the sampling | |
| HT-MD74,92 | * Temperatures can speed up the kinetics involved in AMP insertion into the membrane and folding | |
| Electroporation93,94 | * The technique forces the poration of bilayers and can, therefore, be used to increase the sampling of AMPs entering pores | |
| aMD72,73 | * Increasing the sampling of peptide conformations should be able to escape energy minima quicker and access different metastable states |
Second, machine learning-based quantitative structure–activity relation (ML-QSAR) methods can be used. The database used to train the model is central to successfully identifying AMP candidates. Within our earlier discussion, the role of the database is the development of the functional mapping from descriptor to property space. AMPs are short peptides containing up to 100 amino acids, with most AMPs containing less than 50 amino acids.16,95 In the work of Sharma et al., the authors considered AMPs contain 10 to 200 amino acids. If one were to try to enumerate all possible unique structures, the problem quickly becomes intractable.96
The database used to train the model must cover the important range of phase space, as the promising AMP candidates will be a subset of this. Put differently, we seek to interpolate within the dataset, if possible, compared to extrapolate. Data availability continues to expand, as does the computational ability to process massive amounts of information and readily available packages to train ML-QSAR methods. The dataset used to train ML-QSAR methods comprises AMPs (positives) and non-AMPs (negatives). In 2018, Bhadra et al.97 constructed a positive database of naturally occurring and experimentally validated AMPs from APD3,97 CAMPR3,98 and LAMP99 databases.
After eliminating duplicates and removing unnatural amino acids, they obtained a training database of 3268 AMPs. Their negative database was sourced from the UniProt database limited to proteins containing 5 to 255 amino acids.100 After eliminating sequences labeled as AMP (and similar labels) and unnatural amino acids, they were left with 166![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 791 non-AMPs.
791 non-AMPs.
Subsequently, in 2021, Sharma et al. constructed a positive database using the protein database of NCBI (US National Center for Biotechnology Information)101 and the StarPepDB database.102–104 After eliminating duplicates, removing unnatural amino acids, and restricting the set to AMPs containing 10 to 200 amino acids, they obtained a training database of 10187 AMPs. We note that by restricting the AMPs to 10 to 200 amino acids in length, only 576 AMPs were eliminated. The authors sourced their negative database from UniProt, again restricting the results to proteins containing 10 to 200 amino acids. After removing sequences labeled as AMP (and similar labels) and unnatural amino acids, they were left with 10422 non-AMPs.
The works of Bhadra et al. and Sharma et al. highlight important issues concerning the database used to train. First, they found that there is no universal AMP database. Moreover, overlaps between the databases exist. In Table 2 below, we summarize the most important general AMP databases. Table 2 follows Ramazi et al.'s recent review, providing additional details on each database.105
| Database name | Number of covered classes and AMPs | Size | Type of database | Type of data | Years | URL |
|---|---|---|---|---|---|---|
| dbAMP 3.0 | 52 biological activities in 3044 organisms | ∼57304 |
Exp. and pred. | Natural and synthetic | 2023 | http://awi.cuhk.edu.cn/dbAMP |
| dbAMP 2.0 (ref. 106) | 52 biological activities in 3044 organisms | ∼28709 |
Exp. and pred. secondary | Natural and synthetic | 2021 | http://awi.cuhk.edu.cn/dbAMP |
| dbAMP107 | 26 biological activities in 2048 organisms | ∼12389 |
Exp. and pred. secondary | Natural and synthetic | 2018 | http://csb.cse.yzu.edu.tw/dAMP/ |
| DBAASP108 | Antibacterial, antifungal, antiviral, anticancer, and antitumor in seven organisms and cancer cells and mammalian cells | ∼15700 |
Exp. and pred. secondary | Natural, synthetic, and patent | 2021 | http://dbaasp.org/home |
| LAMP109 | 8 major functional classes and 38 functional activities | ∼23250 |
Exp. and pred. secondary | Natural, synthetic, and patent | 2020 | http://biotechlab.fudan.edu.cn/database/lamp/index.php |
| DRAMP110 | Antibacterial, antifungal, antiviral, anticancer, antitumor, antiprotozoal, and insecticidal | ∼22250 |
Exp. and pred. secondary | Natural, synthetic, patent, and AMPs in drug development | 2019 | http://dramp.cpu-bioinfor.org/ |
| InverPep111 | Invertebrates phyla Arthropoda, Mollusca, Nematoda, Annelida, Echinodermata, Platyheminthes, Placozoa, the Hydridae family (Cnidaria) and the subphylum Tunicata (Chordate) | ∼770 | Exp. primary | Natural | 2017 | http://ciencias.medellin.unal.edu.co/prospeccionydisenobiomoleculas/InverPep/public/home_en |
| CAMPR3 (ref. 112) | Antibacterial, antifungal and/or antiviral | ∼8160 sequences and 757 structures | Exp. and pred. | Natural, Predicted and patented | 2016 | http://www.camp3.bicnirrh.res.in/ |
| MEGARes113 | Antimicrobial compounds, e.g., drugs, biocides, multi-compound, and metals | ∼9000 | Exp. primary | Natural | 2022 | http://megares.meglab.org/ |
| ADAM114 | Archaea, bacteria, plants, and animals | ∼7000 | Exp. primary | Natural | 2015 | http://bioinformatics.cs.ntou.edu.tw/adam/index.html |
| APD115 | Antibacterial | ∼1230 | Exp. and pred. primary | Natural and patent | 2008 | https://webs.iiitd.edu.in/raghava/satpdb/catalogs/apd2/ |
| Defensins knowledge-base115 | Defensin, antimicrobial | ∼360 | Exp. primary | Natural | 2007 | http://defensins.bii.a-star.edu.sg/ |
| YADAMP116 | Antibacterial | ∼2525 | Exp. and predicted | Natural | 2018 | http://www.yadamp.unisa.it/ |
| MLAMP (unbalanced dataset)117 | Antibacterial, anticancer, antifungal, anti-HIV and antiviral | ∼879 AMP | Predicted | Predicted | 2016 | http://www.jci-bioinfo.cn/MLAMP |
| ∼2405 non-AMP | ||||||
| DADP118 | Antimicrobial, antibacterial, anticancer | ∼2571 | Prediction | Prediction | 2012 | http://split4.pmfst.hr/dadp/ |
| Bactibase119 | Bacteriocin | ∼230 | Prediction | Predicted | 2010 | http://bactibase.hammamilab.org/main.php |
| BAGEL4 (ref. 120) | Bacteriocin | ∼814 | Prediction | Predicted | 2018 | http://bagel4.molgenrug.nl/index.php |
| ADAPTABLE121 | Antimicrobial, antibacterial, antifungal, anticancer | >40000 |
Prediction | Predicted | 2019 | http://gec.u-picardie.fr/adaptable |
Additionally, care needs to be taken concerning the data coverage; the ML-QSAR-identified candidates depend on the database's quality.122 Sharma et al. only considered AMPs containing 10 to 200 amino acids. While only a small number of AMPs were eliminated, AMPs are generally short in length to prevent issues related to folding in large AMPs.123 Both Bhadra et al. and Sharma et al. also eliminated unnatural amino acids. While this is a common practice, doing so also removes the possibility of the ML-QSAR method to identify AMP candidates containing unnatural amino acids. However, the recent work of Murakami et al. demonstrates that including unnatural amino acids in the design of anti-bacterial AMPs may be advantageous.124
One needs to be careful concerning the source of the data. From Table 2, we consider the most recent available version (2.0) of dbAMP available online.125 The database currently contains 28709 AMPs. Of these, 18345 (63.9%) are validated, and the remaining 10364 (36.1%) are predicted. Fortunately, in dbAMP, the predicted AMPs are labeled and can readily be filtered out. This is indicated in Table 2 in that the primary database contains experimentally validated AMPs, with a secondary database containing predicted AMPs. This split is an improvement over earlier releases of the database. As posted on the previous dbAMP website, in the dbAMP 2.0 database released on 30 June 2021, only 31.6% (9062 of 28709) of the AMPs were validated. Similarly, in the dbAMP database released on 15 June 2018, only 34.5% (4271 of 12389) of the AMPs were validated.
Further, using the available online filters within the 18345 validated AMPs contained in the most recent available version (2.0) of dbAMP available online, we find that the majority (11431; 62.3%) contain fewer than 20 amino acids, 16599 (90.5%) contain fewer than 40 amino acids, and 17495 (95.4%) contain fewer than 60 amino acids, further emphasizing that AMPs are commonly short peptides. Furthermore, within dbAMP, it is noted that 2262 of the compounds are considered antimicrobial proteins, which are longer.126
The diversity of the database is also of the utmost importance. It has previously been found that many of the common databases are unbalanced concerning AMP activity, which presents challenges for ML-QSAR and classification methods.127 Within the most recent available version (2.0) of dbAMP available online, let us consider the biological function of the AMP. Of the 18345 validated AMPs, 13538 (73.8%) are classified as anti-bacterial, the largest group within the database. For comparison, only 1592 (8.7%) validated AMPs are classified as anti-viral. The major ratio of the predicted AMPs within dbAMP (58.7%) is anti-bacterial. We note that the anti-bacterial class is further broken down into eight types.
The mode of action and the AMP molecular structure are dependent on the biological function of the AMP. For example, anti-bacterial AMPs generally contain hydrophobic cationic amino acids, which interact via electrostatic interactions with the negatively charged bacteria surface, leading to membrane disorder.128 Conversely, anti-viral AMPs may bind to the target (DNA or RNA) to prevent virus replication.129,130 Having found that most of the verified AMPs in the dbAMP database are anti-bacterial, ensuring the employed database is suitable for the desired application must be taken. In the same vein, the recent work of Murakami et al. demonstrates that the inclusion of unnatural amino acids, which are likely not well represented in the database, can affect the α-helical structure of the AMP and increase its anti-bacterial activity while reducing the net charge.124
In addition to the need for an AMP (positive) database, it is also essential to have a non-AMP (negative) database, which is a database of peptides validated not to exhibit antimicrobial behavior. In comparing the work of Bhadra et al. and Sharma et al., the size of the negative database sourced from UniProt is relatively unchanged. On the other hand, the positive AMP database's size change is significant. Moreover, a major effect of this is that the positive to the negative ratio (P: N) is close to 1:1. When developing classification methods, the conventional goal is to obtain a P:N ratio as close to 1:1 as possible. In the work of Bhadra et al., their P:N ratio was not close to 1:1. Interestingly, using their positive database and varying their negative database, they investigated the sensitivity of their model on the P:N ratio used. The authors suggest that a P:N ratio of 1:3 was best, in line with the databases they used. However, further studies are needed. The conventional practice of a P:N close to 1:1 is analogous to the desire for an AMP database equally distributed with respect to activity.
Care must be taken when assembling reference AMP and non-AMP data to train an ML-QSAR or classification method. Further studies are needed to understand the effect of the dataset on the resulting predictions. As raised by Elliott et al.,123 we also question whether conventional design schemes and rules for small molecules apply to the design of AMPs. We imagine, for example, Lapinski's Rule of 5.131 Could we learn from our conventional strategies and leverage them (i.e., as filters) in assembling a database to train our models?
The database is crucial for developing the functional map from the descriptor to the physical property space. Next, we will discuss suitable descriptors required to develop the desired quantitative structure–activity (or property) relationship (QSAR) to make the most of the AMP and non-AMP databases. In chemical and materials informatics, descriptor and fingerprint are terms used interchangeably to describe heuristically determined molecular properties that are easier to calculate than the quantities one wishes to predict. During the development of quantitative structure–property (or activity) relationship (QSPR or QSAR) techniques, one uses the database (reference chemical property space) with the descriptors along with a suitable cheminformatics approach (functional map132) to make predictions (explore the desired chemical property space).
Molecular features
Generally, the descriptors used to model peptides are the same as those used in conventional small molecule drug design.133 The detailed representation of larger molecules, such as peptides or polymers, might be more closely related to features' overall distribution and spatial arrangement. Therefore, the same methodology used to model small molecules may not be used directly. In this regard, it was proposed that the representations of peptides be simplified into amino acid (AA) scales, where each AA side chain is assigned a value for its characteristic value representing the whole molecule.134,135There are different ways in which peptide sequences can be processed based on the AA scales. In either case, a global average value is calculated for all side chains in the sequence or the values are computed based on the position of the corresponding AA in the sequence. There are several ways to retain such positional information.136 The most widely used method involves autocorrelation and cross-correlation measures on discrete descriptor scales. To serve this need, extensive research has been devoted to developing freely available and commercial packages of molecular and quantum mechanical-based descriptors, with several excellent reviews and comparisons available.137–140 The descriptor packages can be facilitated using freely available online servers.141–143 Nonetheless, their application is not without challenge. The issue is twofold. First, it is desirable to not only use QSPR (or QSAR) to predict suitable AMPs for a particular application, but it is of great value to gain insight into the underlying molecular-level driving forces for intuitive (early stage) design processes. Too often, insight is clouded by the employed descriptors. Consider that the commonly used and freely available PADEL package contains 1875 descriptors, while the popular commercial DRAGON package contains 4885 descriptors. This leads to the second challenge of overfitting.138
Within QSPR (or QSAR), the primary technique to reduce the number of descriptors by identifying interrelated descriptors is by a principal component analysis (PCA).144 This reduces the number of parameters to prevent overfitting and can help highlight the essential molecular features. Related to this, Bhadra et al.97 developed an ML-based AMP classification method. Using the Distribution (DF) descriptor set from the Global Protein Sequence Descriptors, they could reduce the number of descriptors used from 80 to 23 while maintaining high accuracy.145 Kleandrova et al.142 have developed a multitask computational model utilizing Moreau–Broto autocorrelation descriptors to predict the activity and cytotoxicity of AMPs. ModlAMP is a software package that includes functions for calculating correlated descriptors for various AA scales. Furthermore, peptide descriptors can be classified according to different AA scales: one-dimensional or global descriptors, which average over the whole sequence, and multi-dimensional descriptors, partly keeping positional information.
Future efforts on the development of descriptors for use with AMPs will be of great value. Existing property–activity relationships can be leveraged, such as AMPs with anti-bacterial activity commonly have a significant net positive charge. Moreover, one should consider key structural features that distinguish peptides from small molecules, such as their large size and flexibility and their make-up from a series of amino acids. If descriptor sets can be constructed containing only the most important features of AMPs, it can help provide insight into the underlying structural property relationships. The molecular structures can be used to calculate all the previously mentioned representations.
In addition to activity, AMPs must exhibit various properties to be effective for a specific application, which can be used as descriptors. A fundamental property of interest is the octanol/water partition coefficient, log P, measured as the equilibrium distribution of a dilute peptide (solute) between water and octanol-saturated phases. The octanol/water partition coefficient is commonly used to characterize the lipophilic/hydrophilic balance of the peptide. It is an important parameter to determine the fate of the peptide in the body for pharmaceutical applications. Leo et al.143 provide a comprehensive review of partition coefficient theory.
Nonetheless, log P is limited in only considering the partitioning of a single peptide form. To overcome this limitation, the octanol–water distribution coefficient, log D, considers protonation, deprotonation, and tautomerization.146 The task of measuring log P and log D for small molecules can be difficult, cumbersome, and imprecise, which we expect to be even worse for the case of peptides.147
Given the difficulty in measuring log P and log D, there is an excellent opportunity to use structural-based descriptors and computational methods to make predictions. We see three reasons for this. First, as already described, log P and log D are great physical values. Second, they can provide insight into the important AMP characteristics for a particular activity. Log P and log D may offer insight during classification processes and ML-QSAR methods to identify and design AMPs for a specific application. Moreover, log P and log D may be used as descriptors themselves. Third, previous work has demonstrated that conventional QSAR tools trained on small molecules are not suitable for predicting log P of peptides.148 This is not unexpected, given the complex chemistry of peptides. This, therefore, presents an opportunity for transfer learning, wherein one could first develop an ML-QSPR method to predict the log P of AMPs. This would allow one to identify the most important peptide descriptors, which could be used to predict additional properties and activities.
Similarly, in the work of Zhou et al.,147 HPLC retention times have been used to study amphipathic helical peptides. They found that the retention time of the peptides correlates with their antibacterial properties, as demonstrated in the case of amphipathic helices, which have been found to have antibacterial properties. Using a reverse approach, Meek et al. were able to predict retention times for peptides up to 20 amino acids in length.148 As measured by CD spectroscopy, other empirical properties used as peptide descriptors are aqueous solubility, refractive index, and helicity. Strøm et al. performed a multivariate analysis of several empirical properties for modeling variants of murine amplactoferricin.149 According to the authors, helicity and global charge were the most critical factors determining the activity of peptides.
Leveraging AI for expanding the AMP space
There is an urgent need for computational methods that promise to expedite the discovery of new drugs because of the proliferation of drug-resistant pathogens and the slow and costly development of antibiotics. In this part of the review, we describe advances in discovering AMPs (antimicrobial peptides) facilitated by AI (artificial intelligence). Given the antimicrobial resistance crisis, we analyze best practices in AI-driven antibiotic discovery and advocate for openness and reproducibility to accelerate preclinical research. As a final point, the literature trends and areas for future research are discussed, as AI enhancements to drug discovery at large provide many opportunities for future applications in antibiotic development.Available improvements in ML and deep learning technologies, particularly deep learning techniques, have demonstrated their favorable impact on generative chemistry and computer-aided drug discovery.150,151 The application of ML to drug discovery, and antibiotic discovery specifically, has been greatly facilitated by the public availability of empirical datasets (Table 2), advances in computer engineering, and the proliferation of free and open-source ML libraries.
Several deep learning techniques, including generative adversarial networks (GANs),152–154 have been used to develop novel peptides and proteins for drug targets in generative chemistry during the drug screening and discovery stages of the drug development process. In light of the above, it has been indicated that deep learning techniques such as GAN algorithms will be essential to the future of generative chemistry as well as computer-aided drug discovery and design, as these advantageous approaches can be applied to numerous aspects of generative chemistry and drug discovery by computer. Several drawbacks of the ML can be attributed not only to the selection of an appropriate model and/or use parameters, but the question is how to scale the selected descriptor features and handle unbalanced data classes.155 We are dealing with a data-rich library based on a sequence representation for the peptides (and/or small molecules), which can be represented by a set of features (numbers) describing the molecule's characteristics in a machine-understandable way. The question of which representation to choose or drop for a given problem is feature selection.156 There is a problem with this inequality in ML models, which must be eliminated to prevent overestimating single features by their magnitude rather than their real underlying issue. However, there was a lack of consistency and reproducibility in the application and robustness of AI-based antibiotic discovery models. It will be necessary for antibiotic discovery to be accelerated using computer-aided approaches for new drugs with novel mechanisms of action (MOAs157), for example, with the application of ML techniques such as support vector machines (SVMs40,158).
A wide range of chemical space is available to design computational antibiotics. In recent years, the developments in the gut microbiome and progress in sequencing analysis opened a new avenue for harvesting antibiotic-resistant genes, and the human gut is an alternative reservoir for AMP structures.159 According to bioinformatics analysis, many potential AMP families in the human gut microbiome remain to be studied. The large number of potential AMPs derived from the human gut microbiome, thus, theoretically, could serve as a source of candidates against infectious bacteria.160
Using artificial intelligence approaches, such as natural language processing (NLP), it is possible to identify candidate AMPs by identifying sequence features from genome sequences, even short sequences with low homology, and features from DNA sequences. Ma et al.161 demonstrate that combining neural network models (NNMs) for autonomous learning of AMP sequence features and large-scale human microbiome data resources can discover AMPs with high antibacterial potency.
A closed-loop approach combining experimental and machine learning techniques requires a template with known antimicrobial activity and a series of homologous sequences. Using a generalized linear model, new AMPs with 160-fold higher antimicrobial activity against Escherichia coli could be created by training a generalized linear model.162 Most machine learning-based antibiotic development approaches utilize molecular descriptors space exploring as the basis of new representations for drug candidates and new models to predict their activity. In contrast, phenotypic drug discovery emphasizes the molecule's effects on target organisms rather than the molecule itself. Using cell imaging, for example, a recent study used a random forest model to predict antimicrobial activity without describing each molecule individually.163 A focus on the effects of drugs on pathogens, rather than comparing molecular descriptors directly, can expand the search space for new medicines.
In recent years, deep learning techniques have made it possible to model generative adversarial networks (GANs) that can be used to design new peptides and proteins. As opposed to artificial neural networks consisting of only one layer, deep learning uses artificial neural networks that consist of multiple layers.164
Generative modeling reframes molecule design as an inverse design problem, which provides an alternative method of discovering new molecules.165 Generative models offer a promising solution. By leveraging recent advances in deep learning, generative models help to solve the inverse molecular design problem: what set of molecules will satisfy a given set of properties? Generic models enable rapid identification of diverse sets of molecules highly optimized for specific applications by identifying a function that maps properties to structures.
Deep neural networks are highly dependent on their architecture, which consists of the types of layers and how they are arranged. The classification of deep generative models for molecular discovery can be divided into three classes: variational autoencoders (VAEs), generative adversarial networks (GANs), and normalizing flow models. A VAE is a generative model consisting of an encoder, which maps molecules into continuous embeddings, followed by a decoder, which reconstructs molecules based on the learned embeddings.166 VAEs are directed probabilistic models, learning continuous latent variables through a variational Bayesian approach to generative DL.
The loss function of VAEs consists of two terms: (1) a reconstruction loss which forces the decoder to recover the correct molecule from the embedded structure, and (2) a Kullback–Leibler divergence term that regularizes the distribution of learned molecular embeddings so that the distribution of generated molecules closely resembles the distribution of training molecules. In molecule generation, VAEs have been used to generate SMILES strings and molecular graphs.167–170
As a first step, it is necessary to formulate these distinct applications as concrete problem statements; for example, we seek to discover molecules with X properties subject to Y constraints. Broadly, molecular generation problem statements fall into three classes: (1) unconstrained molecular generation, (2) property-constrained molecular generation, and (3) structure-constrained molecular generation.
By appropriately tuning models and their associated latent spaces, targeted sampling of new antimicrobial peptides with ideal characteristics can be achieved. For the relevant test case of generating novel antimicrobial peptide sequences, Renaud and Mansbach170 focus on the question of the quality of -latent spaces and their interpretability. To evaluate and compare the different behaviors of deep generative models with VAE-like latent spaces in terms of reconstruction accuracy, generative capability, and interpretability, we will use deep generative models with VAE-like latent spaces. In specific regions of the latent space, the obtained models can generate unique and diverse sequences and grow more AMP-like.
An overview of deep generative models for peptides was presented in a recent review.171 Several challenges still need to be addressed. For example, no single deep generative model framework consistently produces superior results compared to other deep generative models. Due to this, selecting an appropriate model from various deep generative frameworks can be challenging given a peptide dataset of interest. Additionally, benchmarking datasets and metrics in peptide generation evaluation are lacking, further complicating comparing and selecting models. There have been several benchmarking platforms developed in the field of molecular generation, including GuacaMol172 and MOSES,173 that use a variety of criteria to assess the quality of the generated data, such as novelty, uniqueness, validity, and Fréchet ChemNet distance. A similar benchmarking platform for peptide generation models is urgently needed.
Since generative modeling criteria may vary from application to application, developing a set of benchmarks is difficult. In an ideal benchmarking set, metrics relevant to a wide range of applications would be included, and solutions to most of the obstacles associated with using generative models for molecular discovery. We anticipate this set of benchmarks to include synthetic feasibility, safety and handling, uncertainty quantification, and other relevant factors relevant to deploying generative models in real-world applications.
GAN-based algorithms can go beyond other models only when the generative network module can generate continuous output values, such as a vector of numbers, as in the image generation task. Using a vector of numbers, we can train the generative network module and adjust its weights based on the gradient of the loss function from the discriminative network module. Nevertheless, peptide/protein structures are represented in text strings, not continuous numbers. This is one fundamental trick of GAN-based algorithms in de novo peptide and protein design. Thus, we must design an approach to facilitate gradients through peptide/protein structures. The existing solutions in the literature are as follows. First, a pairwise distance matrix between α-carbons on the protein backbone represents protein structures. Second, a four-dimensional (4D) tensor is employed to describe the positions of active atoms in proteins. Third, DNA/gene sequences are used and converted into protein sequences. Moreover, Ramachandran angles, the main chain torsion angles (i.e., phi and psi) in each amino acid, are used to represent a protein structure. In addition, the generative and discriminative network modules directly deal with a latent vector encoded by 20 canonical amino acids.
For the deep generative models, we should consider the limitations arising from the dynamic and conformational states of the peptides. Input PDB structures of peptides may not contain sufficient information for computational modeling, for example, due to the static condition. It won't be possible to capture enough data for computational modeling. An alternative to using a single PDB structure for the generative models is to take a set of peptide conformers or a trajectory of structure changes (computed using molecular dynamics) as inputs. We anticipate that deep generative models will play a significant role in drug discovery in the future as we become more adept at generating structural and functional data about peptides and deep learning advances. Then, in our opinion, coupling algorithms should be proposed and developed in the future to overcome these problems with the flexibility of the AMPs, but which is the main property in the other way. Degiacomi174 presents a usage of generative neural networks for the characterization of the conformational space of proteins featuring domain-level dynamics. The generated protein-like structures can be sampled with a protein–protein docking algorithm to score the conformations (poses) close to the bound state. In addition to pre-existing MD simulation data, the autoencoders can generate new, realistic AMP conformation space. Suppose there is a sufficiently large dataset available. In that case, it may be possible to train a general neural network suitable for molecular modeling that can be rapidly trained using transfer learning to tackle a specific conformational space sampling problem.
Outlook and future perspective
Antimicrobial peptides hold great promise in addressing a wide range of pressing issues. They are natural-based compounds with unique chemistries. As the availability of reference data continues to grow, computational methods are poised to make significant contributions to the selection and development of antimicrobial peptides for specific applications. This is aided by the increased availability of computational tools (machine learning and deep learning) and readily available, user-friendly software. As the field advances, we must establish best practices. We also believe that rather than thinking of antimicrobial peptides in a vacuum, we must recall what has been successfully done in small molecule drug discovery and leverage their known physical properties.It is anticipated that the current challenges in generative modeling will be overcome in the coming years, even though many advances are still required for generative modeling to achieve its full potential.
Data availability
As this is a Perspective article, no primary research results, data, software or code have been included.Author contributions
Conceptualization: M. N. and M. L.; investigation: M. N., A. S. P., D. P. V., M. L.; writing – original draft: M. N., A. S. P., D. P. V., M. L.; writing – review & editing: M. N., A. S. P., M. L.; visualization: M. N.; project administration: M. N.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NCCR Bioinspired Materials financially supported the authors (MN and DPV).References
-
J. O'Neill, Tackling Drug-Resistant Infections Globally: Final Report and Recommendations, 2016 Search PubMed
.
-
PEW, Analysis shows continued deficiencies in antibiotic developments since 2014, 2021 Search PubMed
- K. Schindler, Y. Cortat, M. Nedyalkova, A. Crochet, M. Lattuada, A. Pavic and F. Zobi, Pharmaceuticals, 2022, 15, 1107 CrossRef CAS PubMed
- Y. Cortat, M. Nedyalkova, K. Schindler, P. Kadakia, G. Demirci, S. Nasiri Sovari, A. Crochet, S. Salentinig, M. Lattuada, O. M. Steiner and F. Zobi, Antibiotics, 2023, 12, 619 CrossRef CAS PubMed
- S. N. Sovari, N. Radakovic, P. Roch, A. Crochet, A. Pavic and F. Zobi, Eur. J. Med. Chem., 2021, 226, 113858 CrossRef CAS
- B. P. Lazzaro, M. Zasloff and J. Rolff, Science, 2020, 368(6490), eaau5480 CrossRef CAS
- N. C. K. Heng and J. R. Tagg, Nat. Rev. Microbiol., 2006, 4, 160 CrossRef CAS
- X. Chen, X. Zou, G. Qi, Y. Tang, Y. Guo, J. Si and L. Liang, Cell. Physiol. Biochem., 2018, 47, 1060–1073 CrossRef CAS
- C. de la Fuente-Núñez, O. N. Silva, T. K. Lu and O. L. Franco, Pharmacol. Ther., 2017, 178, 132–140 CrossRef
- S. Clark, T. A. Jowitt, L. K. Harris, C. G. Knight and C. B. Dobson, Commun. Biol., 2021, 4, 605 CrossRef CAS PubMed
- T. N. Nguyen, H. Teimouri, A. Medvedeva and A. B. Kolomeisky, J. Phys. Chem. B, 2022, 126, 7365–7372 CrossRef CAS
- L. Otvos, J. Pept. Sci., 2005, 11, 697–706 CrossRef CAS PubMed
- N. Papo and Y. Shai, Peptides, 2003, 24, 1693–1703 CrossRef CAS PubMed
- V. Teixeira, M. J. Feio and M. Bastos, Prog. Lipid Res., 2012, 51, 149–177 CrossRef CAS
- J. P. S. Powers and R. E. W. Hancock, Peptides, 2003, 24, 1681–1691 CrossRef CAS PubMed
- M. Pushpanathan, P. Gunasekaran and J. Rajendhran, Int. J. Pept., 2013, 675391 Search PubMed
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2006, 6, 1–10 Search PubMed
- P. Kumar, J. Kizhakkedathu and S. Straus, Biomolecules, 2018, 8, 4 CrossRef
- Y. Sang and F. Blecha, Anim. Health Res. Rev., 2008, 9, 227–235 CrossRef
- P. H. Mygind, R. L. Fischer, K. M. Schnorr, M. T. Hansen, C. P. Sönksen, S. Ludvigsen, D. Raventós, S. Buskov, B. Christensen, L. De Maria, O. Taboureau, D. Yaver, S. G. Elvig-Jørgensen, M. V. Sørensen, B. E. Christensen, S. Kjærulff, N. Frimodt-Moller, R. I. Lehrer, M. Zasloff and H.-H. Kristensen, Nature, 2005, 437, 975–980 CrossRef
- R. Hancock and A. Patrzykat, Curr. Drug Targets Infect. Disord., 2002, 2, 79–83 CrossRef PubMed
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS
- L. Zhang and R. L. Gallo, Curr. Biol., 2016, 26, R14–R19 CrossRef CAS
- R. E. W. Hancock and G. Diamond, Trends Microbiol., 2000, 8, 402–410 CrossRef CAS
- T.-H. Lee, K. N. Hall and M.-I. Aguilar, Curr. Top. Med. Chem., 2015, 16, 25–39 CrossRef
- Y. Shai, Biochim. Biophys. Acta, 1999, 1462, 55–70 CrossRef CAS PubMed
- N. Raheem and S. K. Straus, Front. Microbiol., 2019, 10 Search PubMed
- S. I. Saeed, A. Mergani, E. Aklilu and N. F. Kamaruzzaman, Front. vet. sci., 2022, 9, 851052 CrossRef PubMed
- L. Li, L. Wang, Y. Gao, J. Wang and X. Zhao, Front. Microbiol., 2017, 8, 2386 CrossRef PubMed
- J. Li, S. Hu, W. Jian, C. Xie and X. Yang, Bot. Stud., 2021, 62, 5 CrossRef CAS
- A. R. M. Ribeiro, H. P. Felgueiras, S. P. G. Costa and S. M. M. A. Pereira-Lima, Proceedings, 2021, 78, 47 Search PubMed
- C. Guo, P. Cong, Z. He, D. Mo, W. Zhang, Y. Chen and X. Liu, Antivir. Ther., 2014, 20, 573–582 CrossRef PubMed
- C. Subbalakshmi and N. Sitaram, FEMS Microbiol. Lett., 1998, 160, 91–96 CrossRef CAS
- C. M. A. Linde, S. E. Hoffner, E. Refai and M. Andersson, J. Antimicrob. Chemother., 2001, 47, 575–580 CrossRef CAS
- M. R. Ackermann, J. M. Gallup, J. Zabner, R. B. Evans, C. W. Brockus, D. K. Meyerholz, B. Grubor and K. A. Brogden, Microb. Pathog., 2004, 37, 21–27 CrossRef CAS
- N. Bruni, M. Capucchio, E. Biasibetti, E. Pessione, S. Cirrincione, L. Giraudo, A. Corona and F. Dosio, Molecules, 2016, 21, 752 CrossRef
- C. H. Hsu, C. Chen, M. L. Jou, A. Y. Lee, Y. C. Lin, Y. P. Yu, W. T. Huang and S. H. Wu, Nucleic Acids Res., 2005, 33, 4053–4064 CrossRef CAS PubMed
- K. V. R. Reddy, R. D. Yedery and C. Aranha, Int. J. Antimicrob. Agents, 2004, 24, 536–547 CrossRef CAS PubMed
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed
- E. Y. Lee, B. M. Fulan, G. C. L. Wong and A. L. Ferguson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13588–13593 CrossRef CAS PubMed
- E. Strandberg, J. Zerweck, D. Horn, G. Pritz, M. Berditsch, J. Bürck, P. Wadhwani and A. S. Ulrich, J. Pept. Sci., 2015, 21, 436–445 CrossRef CAS
- Z. Liu, A. Brady, A. Young, B. Rasimick, K. Chen, C. Zhou and N. R. Kallenbach, Antimicrob. Agents Chemother., 2007, 51, 597–603 CrossRef CAS PubMed
-
W. Yu and A. D. MacKerell, Antibiotics: Methods and Protocols, ed. P. Sass, Springer New York, New York, NY, 2017, pp. 85–106 Search PubMed
-
W. Yu and A. D. MacKerell, Antibiotics: Methods and Protocols, ed. P. Sass, Springer New York, New York, NY, 2023, pp. 123–152 Search PubMed
- D. J. Danziger and P. M. Dean, Proc. R. Soc. London, Ser. B, 1989, 236, 101–113 CAS
- A. Miranker and M. Karplus, Proteins, 1991, 23, 472–490 CrossRef
- D. A. Pearlman and M. A. Murcko, J. Med. Chem., 1996, 39, 1651–1663 CrossRef CAS
- A. C. Pierce, G. Rao and G. W. Bemis, J. Med. Chem., 2004, 47, 2768–2775 CrossRef CAS PubMed
- D. N. Woolfson, G. J. Bartlett, A. J. Burton, J. W. Heal, A. Niitsu, A. R. Thomson and C. W. Wood, Curr. Opin. Struct. Biol., 2015, 33, 16–26 CrossRef CAS PubMed
- D. Faccone, O. Veliz, A. Corso, M. Noguera, M. Martinez, C. Payes, L. Semorile and P. C. Maffía, Eur. J. Med. Chem., 2014, 71, 31–35 CrossRef CAS PubMed
- B. Mishra and G. Wang, J. Am. Chem. Soc., 2012, 134, 12426–12429 CrossRef CAS PubMed
- B. Suay-Garcia, J. I. Bueso-Bordils, A. Falcó, M. T. Pérez-Gracia, G. Antón-Fos and P. Alemán-López, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1472 CAS
- J. I. Bueso-Bordils, P. A. Aleman, L. L. Zamora, R. Martin-Algarra, M. J. Duart and G. M. Anton-Fos, Curr. Comput.-Aided Drug Des., 2015, 11, 336–345 CrossRef CAS PubMed
- D. Hodyna, V. Kovalishyn, S. Rogalsky, V. Blagodatnyi, K. Petko and L. Metelytsia, Chem. Biol. Drug Des., 2016, 88, 422–433 CrossRef
- M. A. Abdelrahman, I. Salama, M. S. Gomaa, M. M. Elaasser, M. M. Abdel-Aziz and D. H. Soliman, Eur. J. Med. Chem., 2017, 138, 698–714 CrossRef
- P. La Rocca, P. C. Biggin, D. P. Tieleman and M. S. P. Sansom, Biochim. Biophys. Acta, 1999, 1462, 185–200 CrossRef
- D. P. Tieleman and M. S. P. Sansom, Int. J. Quantum Chem., 2001, 83, 166–179 CrossRef
- E. Matyus, C. Kandt and D. Tieleman, Curr. Med. Chem., 2007, 14, 2789–2798 CrossRef PubMed
- J. Mondal, Drug Dev. Res., 2019, 80, 28–32 CrossRef CAS PubMed
- S. Baeriswyl, B. H. Gan, T. N. Siriwardena, R. Visini, M. Robadey, S. Javor, A. Stocker, T. Darbre and J. L Reymond, ACS Chem. Biol., 2019, 14, 758–766 CrossRef CAS PubMed
- S. Benetti, P. B. Timmons and C. M. Hewage, Eur. Biophys. J., 2019, 48, 203–212 CrossRef CAS
-
A. R. Leach, Molecular Modelling: Principles and Applications, Prentice Hall, Harlow, 2nd edn, 2001 Search PubMed
- L. Zhao, Z. Cao, Y. Bian, G. Hu, J. Wang and Y. Zhou, Int. J. Mol. Sci., 2018, 19, 1186 CrossRef PubMed
- G. Manzo, P. M. Ferguson, V. B. Gustilo, C. K. Hind, M. Clifford, T. T. Bui, A. F. Drake, R. A. Atkinson, J. M. Sutton, G. Batoni, C. D. Lorenz, D. A. Phoenix and A. J. Mason, Sci. Rep., 2019, 9, 1–16 CrossRef
- P. K. Hazam, R. Akhil, G. Jerath, J. Saikia and V. Ramakrishnan, Biophys. Chem., 2019, 248, 1–8 CrossRef CAS
- N. Agadi, S. Vasudevan and A. Kumar, J. Struct. Biol., 2018, 204, 435–448 CrossRef CAS PubMed
- J. Li, Z. Hu, R. Beuerman and C. Verma, J. Phys. Chem. B, 2017, 121, 2739–2747 CrossRef CAS
- S. J. Fox, R. Lakshminarayanan, R. W. Beuerman, J. Li and C. S. Verma, J. Phys. Chem. B, 2018, 122, 8698–8705 CrossRef CAS PubMed
- C. H. Chen, G. Wiedman, A. Khan and M. B. Ulmschneider, Biochim. Biophys. Acta, 2014, 1838, 2243–2249 CrossRef CAS PubMed
- G. Bussi, Mol. Phys., 2014, 112, 379–384 CrossRef CAS
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS
- S. Mukherjee, R. K. Kar, R. P. R. Nanga, K. H. Mroue, A. Ramamoorthy and A. Bhunia, Phys. Chem. Chem. Phys., 2017, 19, 19289–19299 RSC
- D. Hamelberg, J. Mongan and J. A. McCammon, J. Chem. Phys., 2004, 120, 11919–11929 CrossRef CAS PubMed
- J. P. Ulmschneider and M. B. Ulmschneider, Acc. Chem. Res., 2018, 51, 1106–1116 CrossRef CAS
- D. Sengupta, H. Leontiadou, A. E. Mark and S. J. Marrink, Biochim. Biophys. Acta, 2008, 1778, 2308–2317 CrossRef CAS PubMed
- C. Liao, M. Esai Selvan, J. Zhao, J. L. Slimovitch, S. T. Schneebeli, M. Shelley, J. C. Shelley and J. Li, J. Phys. Chem. B, 2015, 119, 10390–10398 CrossRef CAS
- A. Iscen, C. R. Brue, K. F. Roberts, J. Kim, J. G. C. Schatz and T. J. Meade, J. Am. Chem. Soc., 2019, 141, 16685–16695 CrossRef CAS
- E. Han and H. Lee, RSC Adv., 2015, 5, 2047–2055 RSC
- R. Marinova, P. Petkov, N. Ilieva, E. Lilkova and L. Litov, Stud. Comput. Intell., 2019, 793, 257–265 CrossRef PubMed
- R. C. Bernardi, M. C. R. Melo and K. Schulten, Biochim. Biophys. Acta, 2015, 1850, 872–877 CrossRef PubMed
- A. Laio and F. L. Gervasio, Rep. Prog. Phys., 2008, 71, 126601 CrossRef
- R. Zou, X. Zhu, Y. Tu, J. Wu and M. P. Landry, Biochemistry, 2018, 57, 2606–2610 CrossRef PubMed
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 Search PubMed
- R. Lipkin, A. Pino-Angeles and T. Lazaridis, J. Phys. Chem. B, 2017, 121, 9126–9140 CrossRef PubMed
- B. Liu and M. Karttunen, Biochim. Biophys. Acta, 2018, 1860, 1949–1954 CrossRef PubMed
- S. J. Marrink and D. P. Tieleman, Chem. Soc. Rev., 2013, 42, 6801–6822 RSC
- Z. X. Deng, J. L. Li, B. Yuan and K. Yang, Commun. Theor. Phys., 2019, 71, 887–902 CrossRef
- Y. Shi, M. Wan, L. Fu, S. Zhang, S. Wang, L. Gao and W. Fang, Biophys. J., 2018, 115, 1518–1529 CrossRef PubMed
- R. Yeasmin, M. Buck, A. Weinberg and L. Zhang, J. Phys. Chem. B, 2018, 122, 11883–11894 CrossRef CAS PubMed
- W. F. D. Bennett, C. K. Hong, Y. Wang and D. P. Tieleman, J. Chem. Theory Comput., 2016, 12, 4524–4533 CrossRef CAS PubMed
- J. Liu, S. Xiao, J. Li, B. Yuan, K. Yang and Y. Ma, Biochim. Biophys. Acta, 2018, 1860, 2234–2241 CrossRef CAS PubMed
- Y. Wang, C. H. Chen, D. Hu, M. B. Ulmschneider and J. P. Ulmschneider, Nat. Commun., 2016, 7, 1–9 CAS
- P. K. Lai and Y. N. Kaznessis, ACS Omega, 2018, 3, 6056–6065 CrossRef CAS PubMed
- T. J. Piggot, D. A. Holdbrook and S. Khalid, J. Phys. Chem. B, 2011, 115, 13381–13388 CrossRef CAS
- E. Y. Lee, M. W. Lee, B. M. Fulan, A. L. Ferguson and G. C. L. Wong, Interface Focus, 2017, 38(7), 20160153 CrossRef
- R. Sharma, S. Shrivastava, S. Kumar Singh, A. Kumar, S. Saxena and R. Kumar Singh, Briefings Bioinf., 2021, 22, bbab242 CrossRef
- P. Bhadra, J. Yan, J. Li, S. Fong and S. W. I. Siu, Sci. Rep., 2018, 8, 1697 CrossRef
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS PubMed
- F. H. Waghu, R. S. Barai, P. Gurung and S. Idicula-Thomas, Nucleic Acids Res., 2016, 44, D1094–D1097 CrossRef CAS PubMed
- X. Zhao, H. Wu, H. Lu, G. Li and Q. Huang, PLoS One, 2013, 8, e66557 CrossRef CAS
- A. Bateman, M. J. Martin, S. Orchard, M. Magrane, S. Ahmad, E. Alpi, E. H. Bowler-Barnett, R. Britto, H. Bye-A-Jee, A. Cukura, P. Denny, T. Dogan, T. G. Ebenezer, J. Fan, P. Garmiri, L. J. da Costa Gonzales, E. Hatton-Ellis, A. Hussein, A. Ignatchenko, G. Insana, R. Ishtiaq, V. Joshi, D. Jyothi, S. Kandasaamy, A. Lock, A. Luciani, M. Lugaric, J. Luo, Y. Lussi, A. MacDougall, F. Madeira, M. Mahmoudy, A. Mishra, K. Moulang, A. Nightingale, S. Pundir, G. Qi, S. Raj, P. Raposo, D. L. Rice, R. Saidi, R. Santos, E. Speretta, J. Stephenson, P. Totoo, E. Turner, N. Tyagi, P. Vasudev, K. Warner, X. Watkins, R. Zaru, H. Zellner, A. J. Bridge, L. Aimo, G. Argoud-Puy, A. H. Auchincloss, K. B. Axelsen, P. Bansal, D. Baratin, T. M. Batista Neto, M. C. Blatter, J. T. Bolleman, E. Boutet, L. Breuza, B. C. Gil, C. Casals-Casas, K. C. Echioukh, E. Coudert, B. Cuche, E. de Castro, A. Estreicher, M. L. Famiglietti, M. Feuermann, E. Gasteiger, P. Gaudet, S. Gehant, V. Gerritsen, A. Gos, N. Gruaz, C. Hulo, N. Hyka-Nouspikel, F. Jungo, A. Kerhornou, P. Le Mercier, D. Lieberherr, P. Masson, A. Morgat, V. Muthukrishnan, S. Paesano, I. Pedruzzi, S. Pilbout, L. Pourcel, S. Poux, M. Pozzato, M. Pruess, N. Redaschi, C. Rivoire, C. J. A. Sigrist, K. Sonesson, S. Sundaram, C. H. Wu, C. N. Arighi, L. Arminski, C. Chen, Y. Chen, H. Huang, K. Laiho, P. McGarvey, D. A. Natale, K. Ross, C. R. Vinayaka, Q. Wang, Y. Wang and J. Zhang, Nucleic Acids Res., 2023, 51, D523–D531 CrossRef CAS PubMed
- E. W. Sayers, E. E. Bolton, J. R. Brister, K. Canese, J. Chan, D. C. Comeau, R. Connor, K. Funk, C. Kelly, S. Kim, T. Madej, A. Marchler-Bauer, C. Lanczycki, S. Lathrop, Z. Lu, F. Thibaud-Nissen, T. Murphy, L. Phan, Y. Skripchenko, T. Tse, J. Wang, R. Williams, B. W. Trawick, K. D. Pruitt and S. T. Sherry, Nucleic Acids Res., 2022, 50, D20–D26 CrossRef PubMed
- L. Aguilera-Mendoza, Y. Marrero-Ponce, J. A. Beltran, R. Tellez Ibarra, H. A. Guillen-Ramirez and C. A. Brizuela, Bioinformatics, 2019, 35, 4739–4747 CrossRef
- L. Aguilera-Mendoza, Y. Marrero-Ponce, C. R. García-Jacas, E. Chavez, J. A. Beltran, H. A. Guillen-Ramirez and C. A. Brizuela, Sci. Rep., 2020, 10, 18074 CrossRef
- S. Ramazi, N. Mohammadi, A. Allahverdi, E. Khalili and P. Abdolmaleki, Database, 2022, 2022, baac011 CrossRef PubMed
- J. H. Jhong, L. Yao, Y. Pang, Z. Li, C. R. Chung, R. Wang, S. Li, W. Li, M. Luo, R. Ma, Y. Huang, X. Zhu, J. Zhang, H. Feng, Q. Cheng, C. Wang, K. Xi, L. C. Wu, T. H. Chang, J. T. Horng, L. Zhu, Y. C. Chiang, Z. Wang and T. Y. Lee, Nucleic Acids Res., 2022, 50, D460–D470 CrossRef PubMed
- J. H. Jhong, Y. H. Chi, W. C. Li, T. H. Lin, K. Y. Huang and T. Y. Lee, Nucleic Acids Res., 2019, 47, D285–D297 CrossRef PubMed
- M. Pirtskhalava, A. A. Amstrong, M. Grigolava, M. Chubinidze, E. Alimbarashvili, B. Vishnepolsky, A. Gabrielian, A. Rosenthal, D. E. Hurt and M. Tartakovsky, Nucleic Acids Res., 2021, 49, D288–D297 CrossRef PubMed
- G. Ye, H. Wu, J. Huang, W. Wang, K. Ge, G. Li, J. Zhong and Q. Huang, Database, 2020, 2020, baaa061 CrossRef PubMed
- X. Kang, F. Dong, C. Shi, S. Liu, J. Sun, J. Chen, H. Li, H. Xu, X. Lao and H. Zheng, Sci. Data, 2019, 6, 148 CrossRef PubMed
- E. A. Gomez, P. Giraldo and S. Orduz, J. Global Antimicrob. Resist., 2017, 8, 13–17 CrossRef PubMed
- F. H. Waghu, R. S. Barai, P. Gurung and S. Idicula-Thomas, Nucleic Acids Res., 2015, 44, gkv1051 Search PubMed
- N. Bonin, E. Doster, H. Worley, L. J. Pinnell, J. E. Bravo, P. Ferm, S. Marini, M. Prosperi, N. Noyes, P. S. Morley and C. Boucher, Nucleic Acids Res., 2022, 51, D744–D752 CrossRef PubMed
- H. T. Lee, C. C. Lee, J. R. Yang, J. Z. Lai and K. Y. Chang, BioMed Res. Int., 2015, 2015, 475062 Search PubMed
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS
- S. Seebah, A. Suresh, S. Zhuo, Y. H. Choong, H. Chua, D. Chuon, R. Beuerman and C. Verma, Nucleic Acids Res., 2007, 35, D265–D268 CrossRef CAS PubMed
- S. Piotto, L. Sessa, S. Concilio and P. Iannelli, Int. J. Antimicrob. Agents, 2012, 39, 346–351 CrossRef CAS
- W. Lin and D. Xu, Bioinformatics, 2016, 32, 3745–3752 CrossRef CAS
- M. Novković, J. Simunić, V. Bojović, A. Tossi and D. Juretić, Bioinformatics, 2012, 28, 1406–1407 CrossRef
- R. Hammami, A. Zouhir, J. Ben Hamida and I. Fliss, BMC Microbiol., 2007, 7, 89 CrossRef PubMed
- A. J. van Heel, A. de Jong, C. Song, J. H. Viel, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2018, 46, W278–W281 CrossRef CAS
- F. Ramos-Martín, T. Annaval, S. Buchoux, C. Sarazin and N. D'Amelio, Life Sci. Alliance, 2019, 2, e201900512 CrossRef
- A. G. Elliott, J. X. Huang, S. Neve, J. Zuegg, I. A. Edwards, A. K. Cain, C. J. Boinett, L. Barquist, C. V. Lundberg, J. Steen, M. S. Butler, M. Mobli, K. M. Porter, M. A. T. Blaskovich, S. Lociuro, M. Strandh and M. A. Cooper, Nat. Commun., 2020, 11, 3184 CrossRef CAS PubMed
- Y. Murakami, S. Ishida, Y. Demizu and K. Terayama, Digital Discovery, 2023, 2, 1347–1353 RSC
- S. Kausar, F. Said Khan, M. Ishaq Mujeeb Ur Rehman, M. Akram, M. Riaz, G. Rasool, A. Hamid Khan, I. Saleem, S. Shamim and A. Malik, Int. J. Immunopathol. Pharmacol., 2021, 35 Search PubMed
- N. Thakur, A. Qureshi and M. Kumar, Nucleic Acids Res., 2012, 40, W199–W204 CrossRef
- I. V. Hartung, B. R. Huck and A. Crespo, Nat. Rev. Chem, 2023, 7, 3–4 CrossRef PubMed
-
M. Karelson, Molecular Descriptors in QSAR/QSPR, Wiley-Interscience, New York, 2000 Search PubMed
- C. D. Fjell, J. A. Hiss, R. E. W. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2012, 11, 37–51 CrossRef
- J. M. Zimmerman, N. Eliezer and R. Simha, J. Theor. Biol., 1968, 21, 170–201 CrossRef PubMed
- H. Jenssen, Expert Opin. Drug Discovery, 2011, 6, 171–184 CrossRef
-
R. Todeschini and V. Consonni, Handbook of Molecular Descriptors, Wiley VCH, Weinheim, 2000 Search PubMed
- L. Wang, J. Ding, L. Pan, D. Cao, H. Jiang and X. Ding, Chemom. Intell. Lab. Syst., 2021, 217, 104384 CrossRef
- Danishuddin and A. U. Khan, Drug Discovery Today, 2016, 21, 1291–1302 CrossRef
- R. Sawada, M. Kotera and Y. Yamanishi, Mol. Inf., 2014, 33, 719–731 CrossRef
-
A. Varnek and A. Tropsha, Chemoinformatics Approaches to Virtual Screening, Royal Society of Chemistry, Cambridge, 2008 Search PubMed
- I. Sushko, S. Novotarskyi, R. Körner, A. K. Pandey, M. Rupp, W. Teetz, S. Brandmaier, A. Abdelaziz, V. V. Prokopenko, V. Y. Tanchuk, R. Todeschini, A. Varnek, G. Marcou, P. Ertl, V. Potemkin, M. Grishina, J. Gasteiger, C. Schwab, I. I. Baskin, V. A. Palyulin, E. V. Radchenko, W. J. Welsh, V. Kholodovych, D. Chekmarev, A. Cherkasov, J. Aires-de-Sousa, Q.-Y. Zhang, A. Bender, F. Nigsch, L. Patiny, A. Williams, V. Tkachenko and I. V. Tetko, J. Comput.-Aided Mol. Des., 2011, 25, 533–554 CrossRef
- S. J. Capuzzi, I. S.-J. Kim, W. I. Lam, T. E. Thornton, E. N. Muratov, D. Pozefsky and A. Tropsha, J. Chem. Inf. Model., 2017, 57, 105–108 CrossRef
- I. V. Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V. Radchenko, N. S. Zefirov, A. S. Makarenko, V. Yu. Tanchuk and V. V. Prokopenko, J. Comput.-Aided Mol. Des., 2005, 19, 453–463 CrossRef
- C. K. Yoo and M. Shahlaei, Chem. Biol. Drug Des., 2018, 91, 137–152 CrossRef PubMed
- I. Dubchak, I. Muchnikt, S. R. Holbrook and S.-H. Kim, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 8700–8704 CrossRef
- V. V. Kleandrova, J. M. Ruso, A. Speck-Planche and M. N. Dias Soeiro Cordeiro, ACS Comb. Sci., 2016, 18, 490–498 CrossRef PubMed
- A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525–616 CrossRef
- S. K. Bhal, K. Kassam, I. G. Peirson and G. M. Pearl, Mol. Pharm., 2007, 4, 556–560 CrossRef
- G. Tse and S. I. Sandler, J. Chem. Eng. Data, 1994, 39, 354–357 CrossRef
- S. J. Thompson, C. K. Hattotuwagama, J. D. Holliday and D. R. Flower, Bioinformation, 2006, 1, 237–241 CrossRef PubMed
- N. E. Zhou, C. T. Mant and R. S. Hodges, Pept. Res., 1990, 3, 8–20 Search PubMed
- J. L. Meek, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 1632–1636 CrossRef
- M. B. Strøm, W. Stensen, J. S. Svendsen and Ø. Rekdal, Pept. Res., 2001, 57, 127–139 CrossRef
- Y. LeCun, Y. Bengio and G. Hinton, Nature, 2015, 521, 436–444 CrossRef
- G. Hinton, JAMA, J. Am. Med. Assoc., 2018, 320, 1101 CrossRef
- D. Xue, Y. Gong, Z. Yang, G. Chuai, S. Qu, A. Shen, J. Yu and Q. Liu, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2019, 9, e1395 CrossRef
- T. Cauchy, J. Leguy and B. Da Mota, Digital Discovery, 2023, 2, 736–747 RSC
- H. Zhang, K. M. Saravanan, Y. Wei, Y. Jiao, Y. Yang, Y. Pan, X. Wu and J. Z. H. Zhang, J. Chem. Inf. Model., 2023, 63, 835–845 CrossRef PubMed
- U. Fayyad, G. Piatetsky-Shapiro and P. Smyth, AI Mag., 1996, 17, 37 Search PubMed
- A. L. Blum and P. Langley, Artif. Intell., 1997, 97, 245–271 CrossRef
- M. D. T. Torres and C. de la Fuente-Nunez, Curr. Opin. Microbiol., 2019, 51, 30–38 CrossRef PubMed
- M. W. Lee, E. Y. Lee, A. L. Ferguson and G. C. L. Wong, Curr. Opin. Colloid Interface Sci., 2018, 38, 204–213 CrossRef PubMed
- M. C. R. Melo, J. R. M. A. Maasch and C. de la Fuente-Nunez, Commun. Biol., 2021, 4 Search PubMed
- C. T. Walsh, Nat. Prod. Rep., 2016, 33, 127–135 RSC
- Y. Ma, Z. Guo, B. Xia, Y. Zhang, X. Liu, Y. Yu, N. Tang, X. Tong, M. Wang, X. Ye, J. Feng, Y. Chen and J. Wang, Nat. Biotechnol., 2022, 40, 921–931 CrossRef CAS
- M. Yoshida, T. Hinkley, S. Tsuda, Y. M. Abul-Haija, R. T. McBurney, V. Kulikov, J. S. Mathieson, S. Galiñanes Reyes, M. D. Castro and L. Cronin, Chem, 2018, 4, 533–543 CAS
- S. Zoffmann, M. Vercruysse, F. Benmansour, A. Maunz, L. Wolf, R. Blum Marti, T. Heckel, H. Ding, H. H. Truong, M. Prummer, R. Schmucki, C. S. Mason, K. Bradley, A. I. Jacob, C. Lerner, A. Araujo del Rosario, M. Burcin, K. E. Amrein and M. Prunotto, Sci. Rep., 2019, 9, 5013 CrossRef PubMed
-
D. P. Kingma and M. Welling, arXiv, 2022, preprint, arXiv:1312.6114, DOI:10.48550/arXiv.1312.6114
- C. Bilodeau, W. Jin, T. Jaakkola, R. Barzilay and K. F. Jensen, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1608 CrossRef
- M. J. Kusner, B. Paige and J. M. Hernández-Lobato, arXiv, 2017, preprint, arXiv:1703.01925, DOI:10.48550/arXiv.1703.01925.
-
H. Dai, Y. Tian, B. Dai, S. Skiena and L. Song, arXiv, 2018, preprint, arXiv:1802.08786, DOI:10.48550/arXiv.1802.08786
- Y. Kwon, J. Yoo, Y.-S. Choi, W.-J. Son, D. Lee and S. Kang, J. Cheminf., 2019, 11, 70 Search PubMed
-
R. Assouel, M. Ahmed, M. H. Segler, A. Saffari and Y. Bengio, arXiv, 2018, preprint, arXiv:1811.09766, DOI:10.48550/arXiv.1811.09766
- S. Renaud and R. A. Mansbach, Digital Discovery, 2023, 2, 441–458 RSC
- F. Wan, D. Kontogiorgos-Heintz and C. de la Fuente-Nunez, Digital Discovery, 2022, 1, 195–208 RSC
- N. Brown, M. Fiscato, M. H. S. Segler and A. C. Vaucher, J. Chem. Inf. Model., 2019, 59, 1096–1108 CrossRef CAS
-
D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy, M. Veselov, A. Kadurin, S. Johansson, H. Chen, S. Nikolenko, A. Aspuru-Guzik and A. Zhavoronkov, arXiv, 2018, preprint, arXiv:1811.12823, DOI:10.48550/arXiv.1811.12823
- M. T. Degiacomi, Structure, 2019, 27, 1034–1040 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2024 |