
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical space analysis and property prediction for carbon capture solvent molecules†
James L.
McDonagh‡
*ab,
Stamatia
Zavitsanou
 c,
Alexander
Harrison
a,
Dimitry
Zubarev
d,
Theordore
van Kessel
e,
Benjamin H.
Wunsch
e and
Flaviu
Cipcigan
*a
c,
Alexander
Harrison
a,
Dimitry
Zubarev
d,
Theordore
van Kessel
e,
Benjamin H.
Wunsch
e and
Flaviu
Cipcigan
*a
aIBM Research Europe, Hartree Centre, SciTech Daresbury, Warrington, Cheshire WA4 4AD, UK. E-mail: flaviu.cipcigan@ibm.com; james.mcdonagh@serna.bio
bUniversity of Edinburgh, School of Mathematics, Bayes Centre, 47 Potterrow, Edinburgh EH8 9BT, UK
cUniversity of Oxford, Physical and Theoretical Chemistry Laboratory, Oxford, UK
dIBM Research, IBM Almaden Research Center, San Jose, CA 95120, USA
eIBM Research, IBM T. J. Watson Research Center, Yorktown Heights, New York 10598, USA
First published on 21st February 2024
Abstract
We present a new chemical representation (the CCS fingerprint) and data set (ccs-98) for carbon capture solvents. We then assess the chemical space, data availability and utility of common machine learning algorithms for high throughput virtual screening in the carbon capture solvents field. This is an area of growing importance, as carbon capture and storage is part of the road map towards net zero for many countries around the world. A major class of commercial carbon capture technology involves using solvents, which are commonly blends of amines and N-heterocyclic molecules in water. Whilst these blends have proved valuable, there is an increasing need to identify new candidate molecules which are more efficient and improve performance. We found that the CCS fingerprint can out-perform other common chemical representations when combined with standard machine learning approaches for classifying molecules based on absorption capacity. We demonstrate models achieving classification accuracy for absorption capacity of over 80%.
1 Introduction
Climate change driven by emissions from human activities now poses the greatest environmental concern of this century.1 Emissions of greenhouse gases such as CO2, methane and nitrous oxides (NOx) are the primary drivers of global warming. CO2 is the largest fraction of greenhouse gases emitted.2 Electricity generation from fossil fuel burning is the largest point source of CO2 emissions around the world. Yet, fossil fuel burning infrastructure is still being built.1 Due to this trend, committed emissions from existing energy generation infrastructure jeopardise climate targets.3Modelling suggestes that Carbon Capture, Utilization and Storage (CCUS) for CO2 emissions is a necessary part of the technological solutions required to meet the Paris climate accord.1,4 CCUS is the only technology that can be used to help decarbonise existing energy infrastructure without decommissioning. CCUS is also important for hard-to-abate emissions, such as those in heavy industries.5 There are a growing number of planned CCUS plants. A recent survey suggested that there are at least 87 planned CCUS plants between 2020–2030 according to the map of global CCUS projects by the International Association of Oil and Gas producers.6
Of the currently available CCUS technologies, absorption using carbon capture solvents is the most mature, seeing commercial usage with further plans for new developments.7,8 The technology is dominated by the use of amine and N-heterocyclic based solvents such as Monoethanolamine (MEA) or proprietary formulations of blends of amines and N-heterocyclic molecules. MEA has become a defacto standard, as it has shown good performance in terms of capture capability as well as being relatively cheap. However, it has several drawbacks: high-energy penalty on regeneration, thermal degradation and corrosion.7 As a result, new solvent candidates and new solvent mixtures are being investigated in both academic and industrial research laboratories.9
In this context, computational techniques can be used to screen, rank and predict new carbon capture solvents.10–14 These computational techniques hold promise to improve the speed of discovery and innovation if paired with suitable data sets of solvent performance. In particular, the field of Chemical Informatics has developed a multitude of methods and practices, which can be used to address problems in the field of carbon capture.15 Access to good quality research data and methods is critical to the fast progress of a field, as demonstrated by examples such as those in solid state materials design16 that have benefited from open innovation and widely shared data sets.
To inform this study and demonstrate the usefulness of computational approaches to this field, we have identified 167 unique amine and N-heterocyclic molecules which have been reported in the literature17–25 in relation to a range of carbon capture performance metrics. We have extracted string representations for these molecules from PubChem26 and ChemSpider,27 using chemical name and/or SMILES searches through the web portals and APIs, in order to perform an analysis of the chemical space of carbon capture amines and N-heterocyclic molecules. In addition, we have created a new data set of 98 amine and N-heterocyclic molecules. For this set we have performed new experiment to determine the molecules absorption capacity as an aqueous solution of 30% w/w (g solvent per g solution). We have used a consistent set of experimental measures, making the new data set highly valuable for training Machine Learning (ML) models upon. All data sets generated in this work can be found in ESI† and on GitHub at https://github.com/Jammyzx1/Carbon-capture-fingerprint-generation.
In this work, we therefore make the following contributions:
(1) A new data set of 167 molecular structures from pre-existing literature, which identifies small molecules that have been experimentally tested for carbon capture capability. We name this data set ccs-lit-167.
(2) We use ccs-lit-167, coupled with a set of commercially available amine and N-heterocyclic molecules from ZINC (zinc-20938), in Section 3.1 to analyse the chemical space of amines and generate a new molecular representation named the CCS fingerprint.
(3) We measure an own experimental data set, which to the best of our knowledge, is the largest data set of single experimental source measurements for absorption capacity in the carbon capture solvents literature. This set contains 98 molecules. We name this dataset ccs-98. It is applied in Section 3.2 to build and test high throughput screening models for carbon capture molecules.
2 Methods
There are a variety of methods and data sets deployed and created in this work. To clarify where data is input, data sets are created and how these data sets are used Fig. 1 provides a high level view.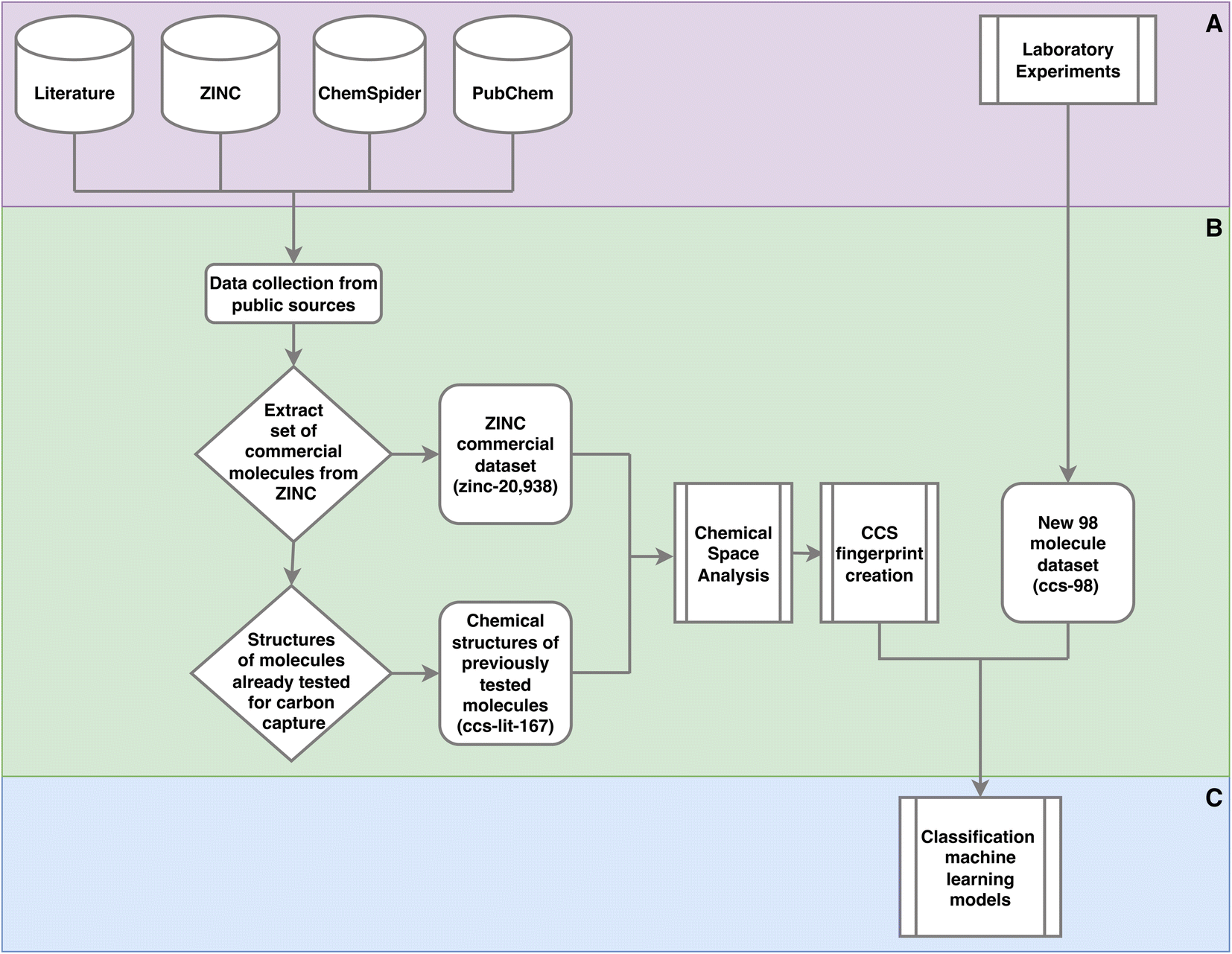 | ||
| Fig. 1 Outline of the methods used in this work. This is a high level outline detailing: (A) the input data and experiments; (B) processing of the data leading the generation of three data sets and their usage; (C) machine learning. The online databases in section A, were accessed via there web interfaces and APIs to extract molecular data. This work was performed throughout 2021. Section C is given in more detail in Fig. 2. | ||
2.1 Data collection and curation
Initially, we reviewed the literature searching for experimental absorption capacity measurements. It became clear that there were potentially issues comparing data over multiple experimental techniques and conditions and that the field lacked common data standards for carbon capture solvents research. Unlike counterparts in the solid state, such as Metal Organic Frameworks (MOFs), for which extensive crystal structure databases have been provided,28–30 carbon capture solvents is a relatively data poor field. This in many ways is likely related to the field's success in being one of the first commercially applied carbon capture technologies. As a result, data may often be considered too sensitive to be released. This is especially true of formulations and blended solvents.This situation is historically reminiscent of fields such as pharmaceuticals, which, in some cases, have seen benefits from opening larger internal data sets from commercial organizations in recent years.31 These benefits are both scientific (faster development of new ideas)32 and also economic.33 Woelfle et al.34 provides an example case study on how a community accelerated the development of a route to enanitopure Praziquantel. The authors of this manuscript have demonstrated the use of open data sets towards predicting molecular and material properties such as water solubility and partition coefficients previously.35–37
Opening data in the carbon capture solvents field could enable a proliferation of data driven modelling. The establishment of common standards upon which to relay data and fairly compare methods is however an important prerequisite. In this regard, a conversation should be encouraged across the community to aid in establishing such standards. In this work we provide:
(1) IUPAC chemical names
(2) SMILES strings
(3) InChI strings
(4) Experimental values and units
(5) Solution concentrations
(6) Software version numbers
(7) Description of the experimental procedures
We suggest these items for inclusion as a minimum starting point for data sets in the carbon capture solvents field. As the data here is still small we share data as csv files, however, a more robust online repository would be more suitable in the longer term.
The ability to fairly perform comparisons can drive rapid advancement of computational screening. This will help to bring research in this area in line with solid state carbon capture which sees wide spread computational modelling.38,39 Similar arguments have been proposed and discussed in other related fields, for example environmental toxicity and formulation chemistry.40,41 In order to demonstrate the value of consistent data, we have gathered our own data using a single experimental method. We gathered 98 data points in total. These molecules were chosen as they represent a sub-set of previously explored molecules and unexplored molecules to the best of our knowledge. The unexplored molecules were chosen based upon expert input and computational similarity screening. The similarity screening was carried out against the zinc-20938 data set of 20938 purchasable amines and N-heterocyclic molecules from the ZINC database. We used the ccs-lit-167 chemical structures as queries to search for similar molecules within a sub-set of purchasable small molecules from the ZINC database.42 Similarity molecules were identified by extended Murko scaffold matching and Tanimoto similarity searching applying a 0.7 similarity threshold. A final set of 98 purchasable molecules was then selected from the similarity screening based on purchasableity constraints and expert input. We refered to this final data set as ccs-98.
For each of the ccs-98 molecules we extracted the identifiers and 2D structures of the molecules from ZINC. We proceeded to search the PubChem26 and ChemSpider27 databases for entries of these molecules and extracting further identifiers such that all molecules were specified by: IUPAC name, InChI, InChIKey and SMILES. In some cases an entry could not be found and we manually determined the name and generated the SMILES string, from which, we generated the InChI and InChIKey using RDKit43 (version 2022.03.2). These representations are the most commonly used and are easily parsed by standard chemical informatics tool kits such as RDKit and OpenBabel.44 This information is provided in the ESI.†
Monoethanolamine (MEA), 30% w/w aqueous, was used as a calibrant as it has a well-established capture capacity of α = 0.50 mol CO2 per mol N.17 The estimated apparatus delay time is 0.16 min, and control experiments with pure water show a background absorption of ∼20 μmol CO2.
A range of capacity units are used in the literature. The most common are:  . Another unit which we encountered several times was
. Another unit which we encountered several times was  . The latter unit requires knowledge of density to accurately convert, as the solution includes the solvent volume as well as the active capture molecule(s) volume. We have used the unit
. The latter unit requires knowledge of density to accurately convert, as the solution includes the solvent volume as well as the active capture molecule(s) volume. We have used the unit 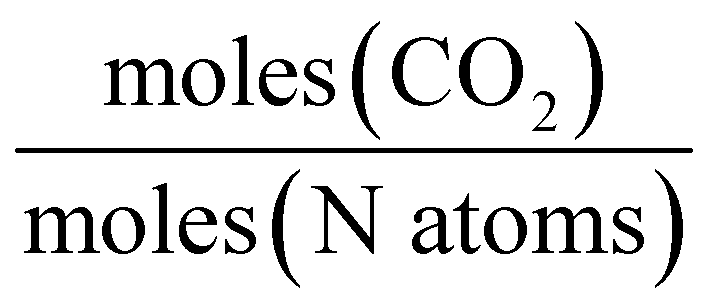 for our absorption capacities and provide conversion factors in the ESI eqn (1).†
for our absorption capacities and provide conversion factors in the ESI eqn (1).†
2.2 Infrastructure
In this work we used cloud based computing as this offers us flexibility to scale the resources to our needs. This cluster consisted of eight nodes, each with 8 virtual CPUs and 32 GB of RAM. This allowed us to quickly provision infrastructure to run our modeling.41,46,47 The computational time for each step of this work varies with the features and the ML algorithms used. The majority of time is typically spent on training over the cross validation steps. The feature generation steps must be computationally efficient to enable high throughput screening. The features used here are faster to generate than the time taken to train a model.2.3 Computational modelling
In this work we have applied a range of methods to explore the properties of the proposed solvents. These methods broadly fall into the category of data driven chemical informatics, including chemical graph analysis, sub-structure searching and machine learning.15 To our knowledge, the application of chemical space analysis and the subsequent bespoke fingerprinting is a novel contribution to this field and present a new analysis of the molecules most commonly used for carbon capture solvents.We begin by analyzing the molecular graphs of the molecules. Firstly, we provide a summary of common sub-structures found in the zinc-20938 and ccs-lit-167 data sets and compare the relative abundance of some of these common sub-structures. The relative abundance is plotted in Fig. 6. The sub-structures displayed in Fig. 6 make up the new CCS fingerprint representation.
A molecular similarity analysis follows the sub-structure analysis. Molecular similarity is commonly applied in chemical informatics typically applying a distance metrics to a vector representation of two molecules. A graph representing the entire chemical similarity space formed by zinc-20938 and ccs-lit-167 data sets is presented in Fig. 5.
Further, analyze the chemical space with Topological Data Analysis (TDA) to produce a skeletonized representation of the chemical space via Mapper TDA, which is displayed in Fig. 7.48–59 Mapper TDA is a technique to visualise the topology of high-dimensional data, such as point clouds. The construction is related to the concepts of a Reeb graph and pullback covers.50,57 Mapper TDA tracks the evolution of the level sets of a real-valued function associated with the data points, known as the filter function. The filter function can be selected to reflect some geometric properties of the points in the data set, such as eccentricity (position relative to the center of the data) or local density. The range of filter function values is split into overlapping intervals, also referred to as level sets. Mapper TDA tracks evolution of these level sets. For each interval, the corresponding subset of the data points is clustered. Finally, a graph is constructed where each node represents a cluster and two nodes are linked if the corresponding clusters overlap. Two Mapper TDA clusters can overlap because the filter function intervals are allowed to overlap. Further, it is customary to associate some attributes, such as filter function values or some scalar properties, with the nodes and visualize them as colors. The number of data points in the cluster is often visualized as the node size. The output of Mapper TDA is highly dependent on the choice of hyper-parameters. A comprehensive analysis of Mapper TDA parameters can be quite involved and equivalent to a standalone computational task.53
Gaussian processes have been used in chemical modelling in many instances.67,71–73 These are a stochastic process, which perform Bayesian inference over a space of functions that map a representation to a probability space, for the class of a molecule. A prior is used to define a probability distribution over functions. As data is provided to train the model, the distribution of functions, which most suitably represent the data, is updated leading to the posterior probability distribution. For classification, a logit function is used to output class probabilities. More details are give in chapter 3 of Williams et al.65
Ada Boost, as implied by the name, is a boosting algorithm that combines multiple weak classifiers to increase the accuracy. In our case we use decision trees as our weak learners. The Ada Boost method works by initializing all training data with equal weights. After the first classifier is trained, examples which are incorrectly classified by the first classifier are given a higher weighting. The process is repeated for N weak learners.
Finally, Logistic Regression in its most basic form uses a logistic function to model a binary dependent variable. This is done using a standard linear regression model which is mapped through a logistic function to give probabilities. Each molecule is assigned a probability for class 0 and 1 with a sum of one.
All models are assessed in terms of multiple performance metrics: accuracy, sensitivity, specificity, Receiver Operating Characteristics (ROC) curves and74 Matthews Correlation Coefficient (MCC).75,76 These metrics can all be formulated mathematically from a confusion matrix, which identifies the correct predictions, True Positives (TP) and True Negatives (TN), along its main diagonal and the two types of error associated with binary classification (classification where the model chooses between two possible outcomes), False Positive (FP) and False Negative (FN), in the off diagonal elements. The equations used for these metrics are given in the ESI eqn (2)–(7).†
Briefly, these metrics comprise the most commonly applied metrics for classification problems and well characterise the performance of our methods. Accuracy is likely the most common classification metric.76 It is a ratio of the number of correct predictions over the total number of predictions. This leads to a ratio describing the fraction of predictions which are correctly classified in the set. This simple metric is a valuable high level overview of the performance of a classifier. The sensitivity and specificity each focus on the models ability to correctly predict the positive or negative class respectively. These metrics provide a greater insight into the potential errors and biases of the models. The ROC curves describe the model performance over decision thresholds with a FN rate on the x axis and TP rate on the y axis. These thresholds can be considered as balancing the positive and negative predictions, i.e. lowering the threshold will increase the number of positive predictions, which is the sum of true positive and false positive predictions. The Area Under the Curve (AUC) for a ROC curve is the integral of the area under the ROC curve and provides a single value metric for this trade off. The MCC metric is a powerful summary metric which ranges from −1 to +1 describing the skill of the classifier to predict positive cases as positive and negative cases as negative even when the classes are imbalanced.76
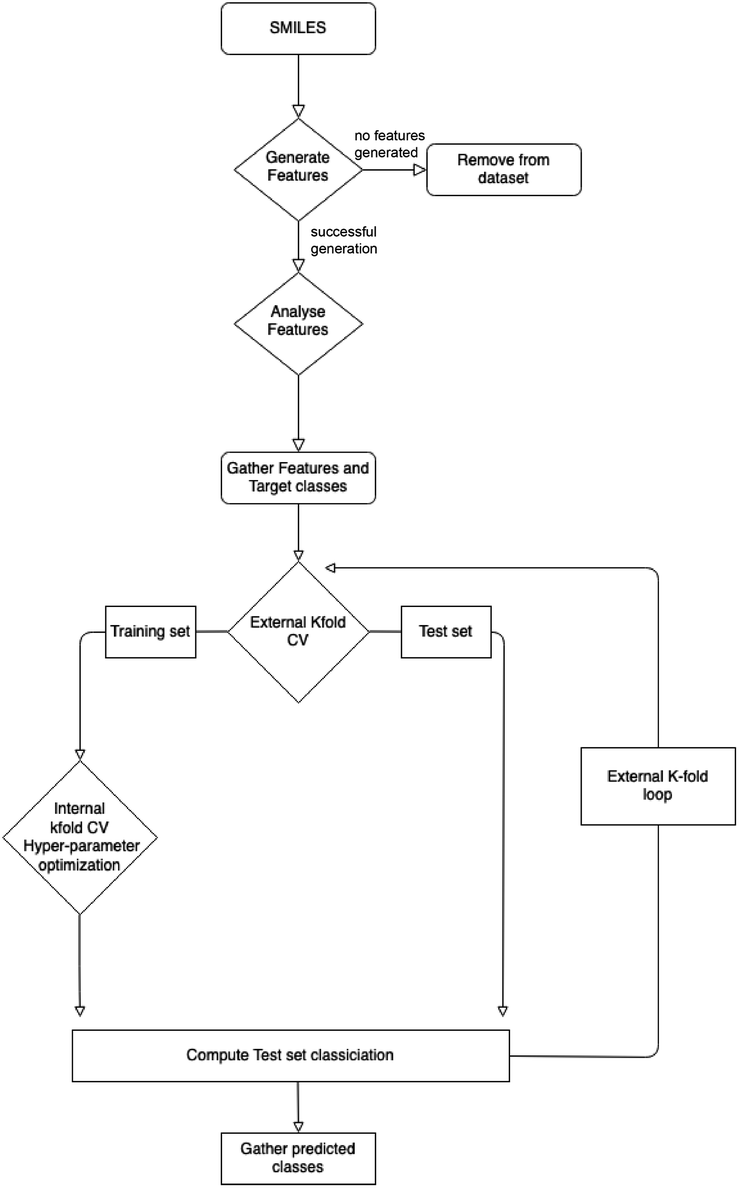 | ||
| Fig. 2 Workflow to make classification predictions of each molecule in the ccs-98 data set. This workflow generates random splits of the data for training and testing building a model for each of the external k-folds. | ||
To describe these molecules, we used three methods. The first are standard chemical informatics descriptors, generated through the Mordred descriptor calculator,78 which produces over 1800 features of molecular characteristics. From the 1800 descriptors calculated, we identified the ones that correlate significantly with the properties of interest using the Spearman correlation coefficient between each Mordred descriptor and the respective property of interest.
Another way to describe molecules is via molecular fingerprints. Molecular fingerprints are vectors that encode structural information about a molecule. Commonly, this information is stored as binary digits representing presence and absence of a structural feature. There are different types of fingerprints available such as Morgan fingerprints,79 MACCS fingerprints80 or MinHashed Atom Pair (MAP) fingerprints.81 In this work we have used the commonly applied MACCS fingerprints.
Additionally, we have defined our own structure based fingerprint (CCS fingerprint) following consideration of the literature and our own chemical space analysis. The latest version of the source code for generating these fingerprints and the ccs-98 data set can be found https://github.com/Jammyzx1/Carbon-capture-fingerprint-generation and archived under https://zenodo.org/record/8304466. Documentation for the code can be found at https://jammyzx1.github.io/Carbon-capture-fingerprint-generation/. This fingerprint is a fixed length (72 elements) with each element representing a chemical group or groups. These chemical groups comprise those commonly seen in carbon capture solvents and those found more broadly across amine and N-heterocyclic chemical space. We discuss the details of this in the Section 3.1.1.
3 Results and discussion
3.1 Chemical space analysis of carbon capture amines and N-heterocyclic molecules
First, we explore and compare the structures of the amines and N-heterocyclic molecules in the ccs-lit-167 data set and zinc-20938 data sets.Several authors have reported chemical sub-structures which influence carbon capture capabilities.10,17–19 In particular, Singh et al.18,19 developed structure activity relationships based on chemical functionalities. Their work studies the effects of many chemical functionalities on carbon capture loading and develops design considerations for carbon capture molecules. These included alkyl chain lengths and functional group separation, measured in number of carbon atoms. Additionally, consideration of ring substituent and their positions was provided in a later publication.19 Work by Papadopoulos et al.10 provided a computational design system. This work also identified a small number of chemical structures which were useful as descriptors for their models. Work by Puxty et al.17 reports the position of OH moieties relative to the amine nitrogen to be important. Steric hindrance§ (presence of physically voluminous moieties in close proximity to a site of interest) around the amine nitrogen is another chemical feature reported to be of importance. It has been shown for example, that steric hindrance can change the reaction route of primary and secondary amines towards that of tertiary amines. This is an important observation owing to the differing atom efficiency between the two routes. Primary and secondary amines have been shown to react with CO2 through a pathway requiring a second molecule to complete the reaction, see Fig. 3. The second molecule may be water in some cases or a second primary or secondary amine. Tertiary amines have been shown to react in a one to one fashion with CO2 effectively acting as a catalyst see Fig. 4.12,17,83,84
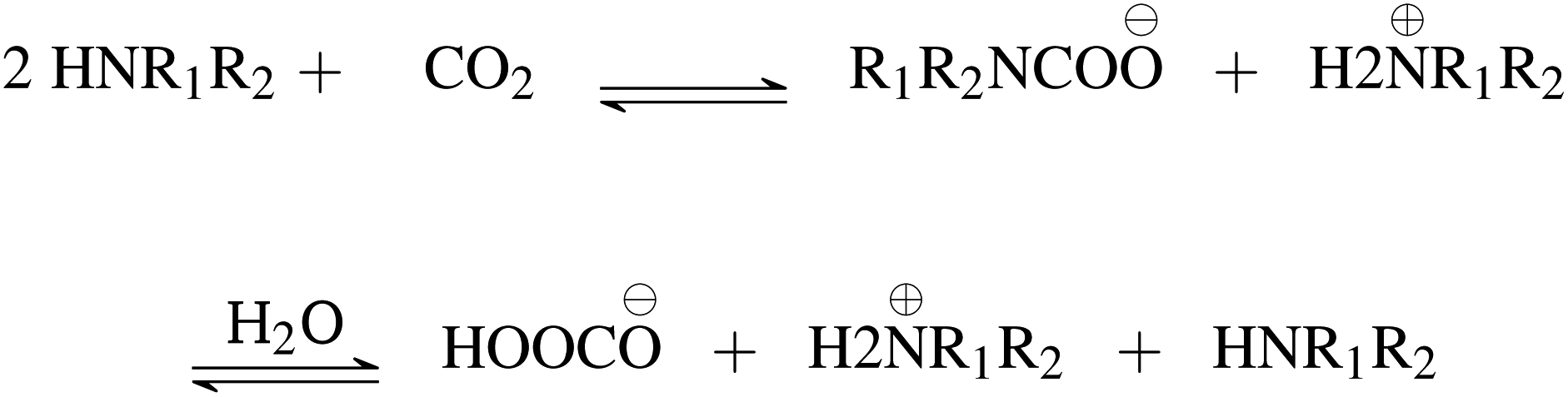 | ||
| Fig. 3 Primary and secondary amine general reaction scheme. | ||
 | ||
| Fig. 4 Tertiary amine general reaction scheme. | ||
Each bit in the CCS fingerprint is defined by a SMARTS string. Substructure searching for these SMARTS patterns over a molecule is carried out in parallel (over molecules) using DASK85 (version 2022.02.0) and RDKit43 (version 2022.03.2) to generate the fingerprint vector(s). The source code also enables others to define there own structure based binary fingerprint using SMARTS86 for any application. As a result others can easily build on this initial version.
The inclusion, of chemical functionalities more prominent in the ccs-lit-167 set compared to the zinc-20938 set and vice versa, was done to enable the fingerprint to capture the differentiation between the two groups. The sub-structure searching is done in a fixed order defined by the order of the SMARTS strings, in order to give a consistent signal from the CCS fingerprint. The fingerprint definition in terms of the order and SMARTS patterns used for substructure matching are included in the ESI.† Each of the SMARTS patterns defines one bit in our fingerprint. In total there are 72 elements and hence 72 sub-structure searches per molecule. In order to make this computationally reasonable in terms of cost we have found parallelizing over batches of 1000 molecules to be effective.
The list of carbon capture molecules collected in this work is not exhaustive, but is a representative sample of the published carbon capture solvent molecules which have been openly reported. As a result the aim is to provide an analysis which highlights the most explored regions of the carbon capture solvent chemical space and point out synthetically accessible areas which may be under explored. Fig. 6 displays a histogram with the normalized count of occurrences of the given sub-structures across molecules in the ccs-lit-167 (blue) and zinc-20938 (red) sets. Clearly there is a substantial difference in the size of these data sets, hence the normalization allows one to consider relative abundance rather than absolute counts. The figure demonstrates that the CCS fingerprint captures several chemical sub-structures which are proportionately over and under expressed in the ccs-lit-167 set compared with the background zinc-20938 commercial set, suggesting these functionalities presence or absence are important when considering carbon capture applications.
From Fig. 6 it is clear that the ccs-lit-167 data set includes molecules which contain a sub-set of chemical moieties from the CCS fingerprint at a proportionately high rate than the zinc-20938 data set. For example, in the alkanolamines substructures in the centre of the y-axis. This subset may be somewhat expected given the wide spread use of MEA and related molecules. It is also clear that structures such as carbonyls, halo-carbons and aromatic groups are found at a proportionately lower rate in the ccs-lit-167 data set compared with the zinc-20938 data set. We note that substances such as benzylamine have been used as promoters within formulated blends rather than capture solvents themselves. Such molecules are not captured in this analysis.87,88 This analysis suggests there is likely a defined chemical sub-space of amine and N-heterocyclic molecules which is more likely to be associated with molecules suitable for carbon capture.
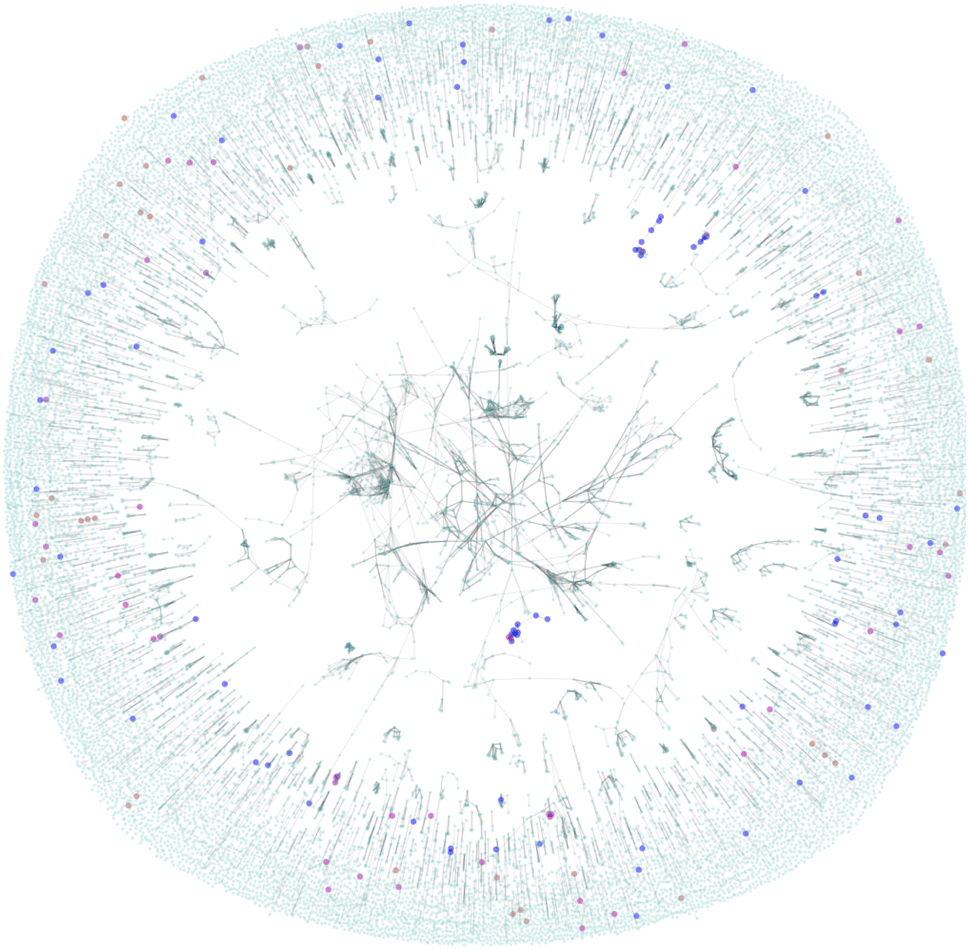 | ||
| Fig. 5 Force directed graph of the amine chemical space. The highlighted nodes are molecules which have been reported in the literature as trialled for carbon capture capability previously. The cyan nodes are commercially available amines which to the best of our knowledge have not been tested for carbon capture capability. | ||
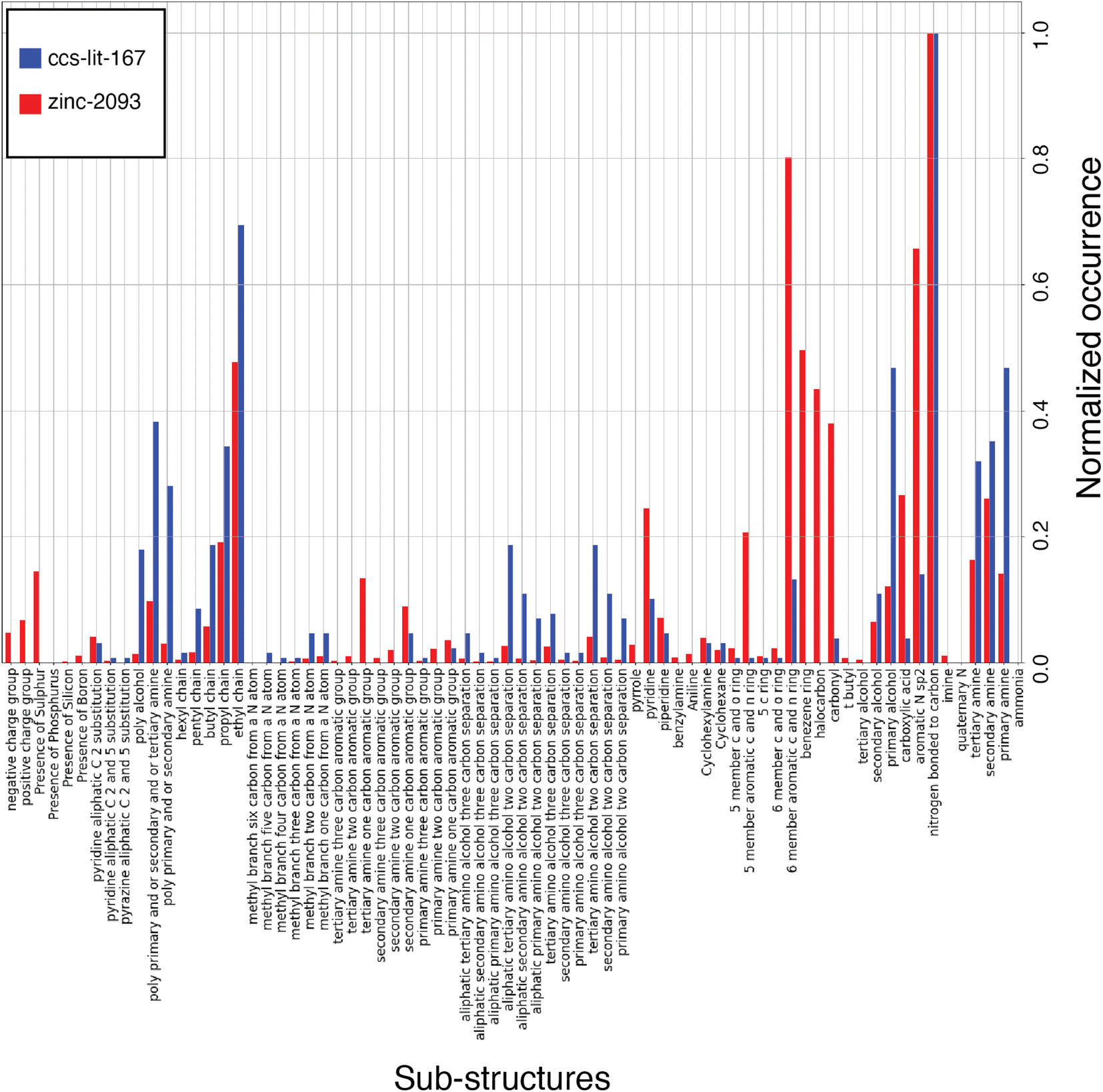 | ||
| Fig. 6 Fingerprint comparison over two data sets ccs-lit-167 and zinc-20938. All bits are found in the larger data set at least once except ammonia, however their occurrence may be rare enough that it is not clearly visible on the normalized x-axis. Where this occurs we have decided to include the bit as it has been noted in other literature sources as potentially important. | ||
The figure is displaying a 2D representation of the chemical space based on commonly applied molecular similarity (Tanimoto similarity ≥0.7). We interpret the figure as follows:
• The highly connected core region contains molecules over the zinc-20938 and ccs-lit-167 data sets with highly conserved structural features defining them as highly similar.
• We note that there are almost no carbon capture molecules highlighted in this core region. This suggests the most common core structural motifs in the zinc-20938 set are rare in the ccs-lit-167 set.
• The ccs-lit-167 molecules do tend to have connections showing that they are typically not isolated in this chemical space the highly connected core demonstrating the molecules.
Taken together this analysis demonstrates that the ccs-lit-167 set are not evenly distributed in the chemical similarity space displayed in Fig. 5. As the carbon capture molecules tend to exist outside of the highly connected core region they can be considered relatively dissimilar to many of the commercial amines within the zinc-20938 set but not totally isolated. Generally the reported carbon capture molecules appear to inhabit sub-sections of the chemical space, this may suggest there is room for innovation in some of the unreported/unexplored regions. Additional related analysis for carbon capture solvent molecules is provided in Elmegreen et al. 2023.91
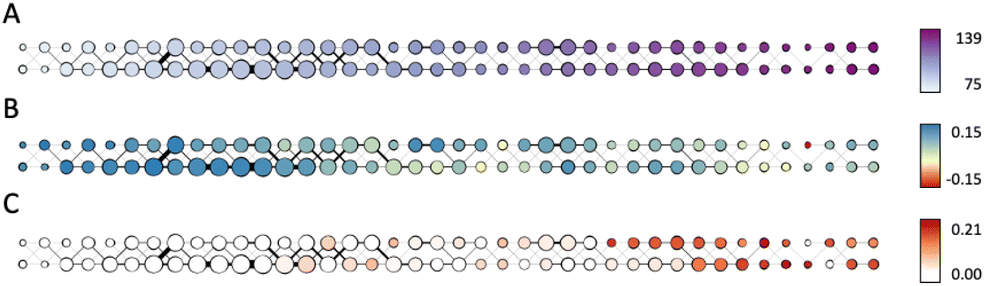 | ||
| Fig. 7 Mapper graph of the combined data set of zinc-20938 and ccs-lit-167. Eccentricity of amines and N-heterocyclic molecules in the combined data set is used as the filter during Mapper construction. Node size is proportional to the number of molecules associated with the node. Thickness of a link between two nodes is proportional to the number of molecules that are associated with both nodes. Panel (A): color encodes mean eccentricity (distance from the centre) of the molecules associated with the node. Panel (B): color encodes mean anomaly score (Isolation Forest – how structurally dissimilar the molecules of the node are compared to the data set) of the molecules associated with the node. Panel (C): fraction of molecules from the ccs-lit-167 data set among the molecules associated with the node in total. | ||
Fig. 7A shows the produced Mapper graph where nodes represent clusters within level sets, nodes are linked if respective clusters have common members, color encodes the filter function (eccentricity), and the node size encodes the number of amines in the respective cluster. Fig. 7B and C maintain the layout of the graph in Fig. 7A and the encoding of the number of amines in a cluster by the node size. Fig. 7B shows the anomaly scores of the molecules in the data set evaluated using the Isolation Forest algorithm, averaged over clusters, and encoded as the node color. High positive values of the anomaly score indicate inliers, decreasing values indicate higher level of abnormality, and negative values indicate outliers. Fig. 7C uses color to encode the fraction of the carbon capture amines in each cluster. We note that the highest content of carbon capture amines in the Mapper clusters does not exceed 20%.
Comparison of Fig. 7A and C suggests that carbon capture molecules are not present in the left most (most central) nodes. This finding can be interpreted as a sign of under-utilization of the space. One possible reason could be a bias of the majority of amines and N-heterocyclic molecules towards biochemical/medicinal applications leading to specificity in the structures towards such applications. Comparison of Fig. 7B and C shows that carbon capture amines and N-heterocyclic molecules are not outliers, as the only cluster with the average anomaly score characteristic of outliers has zero fraction of carbon capture amines. Carbon capture amines are not the most “normal” amines either as the average anomaly scores of the clusters rich in carbon capture amines are shifted towards zero.
Considering all aspects of this analysis it appears that the carbon capture amines considered here are representatives of a sub-space in amine and N-heterocyclic molecule chemistry. Many of the zinc-20938 molecules are likely to have been developed for diverse industrial applications and as such many will be unsuitable, in terms of cost, quantity and structure, for carbon capture. The analysis does suggest though that there is considerable unexplored, or at least unreported, areas of amine and N-heterocyclic molecule chemical space which may hold novel candidates for carbon capture.
3.2 Carbon capture absorption capacity classification
In this section we outline our absorption capacity classifications. We begin generating QSAR models for the classification of molecules based on absorption capacity. We complete this work by evaluating our models and considering the impact of our predictions.We report the results for the classification models generated with MACCS fingerprints, CCS fingerprints and Mordred descriptors against absorption capacity in units of (molCO2 molN−1).
There are 98 molecules in our absorption capacity data set denoted as ccs-98, classified to binary groups. Class 1 represents higher values and class 0 represents lower values of absorption capacity. The molecules are classified based upon the nitrogen centric organic functionalities they contain. Both primary and secondary amines are thought to react with CO2 through a mechanism requiring two amine molecules to complete the reaction. Therefore, a primary or secondary amine has a theoretical absorption capacity of 0.5 per primary or secondary amine group. Tertiary amines are thought to react in a one to one mechanism therefore have a theoretical absorption capacity of 1.0 per tertiary amine group. We classify molecules by summing up these expected contributions per amine group. For sp2 nitrogens in rings the pKa tends to be lower than for amines therefore it is likely a much less active functionality. sp2 nitrogens in rings are the only containing functionality in the molecules the molecule is assigned a theoretical capacity of 0.5, however, if the molecule contains one or more amines then the theoretical capacity is set to the values associated with the amines. Functions to generate these classes are provided in the CCS fingerprint library https://github.com/Jammyzx1/Carbon-capture-fingerprint-generation. Where mixtures of primary or secondary with tertiary amines arise we apply a weighting based upon the number of tertiary amine groups, as both of the proposed amine reaction routes are possible and can be competitive in terms of the kinetics. We therefore down scale the tertiary contributions to 0.5. If the approximate expected value for absorption capacity is below the experimental absorption capacity then class 0 is assigned to the molecule. If the experimental absorption capacity is greater than or equal to the approximate expected value then class 1 is assigned to the molecule. From the ccs-98 data set 71 molecules are class 0, and 27 molecules are class 1.
The two classes are highly imbalanced. To achieve better performance in the models, we generate additional sampling points for the minority class using the Synthetic Minority Over-Sampling Technique (SMOTE)93 for non-categorical features and Synthetic Minority Over-sampling Technique for Nominal (SMOTEN)93 for categorical features. This is implemented in the imbalanced learn Python package (version 0.9.0). In both cases, these methods select the five nearest minority class neighbours in feature space to the kth example minority point, choose at random one of the five and generate a synthetic sample point along the connecting line between the example point and the random neighbour. Note that the methods have no information about the majority class.
SMOTE provide a better balance between the classes, hence improving the learning of a decision boundary. We apply the SMOTE algorithms to each training set in the k-fold cross validation independently to avoid data leakage from the test sets. We note that pre-computing the SMOTE synthetic points prior to train test splits in the k-fold cross validation can lead to notable data leakage and over optimistic metrics for the model performance. We explored the impact of this in our work and found that on the headline accuracy metrics data leakage could provide approximately an 7–8% over estimate in a models predictive accuracy. This experiment was performed by generating the SMOTE examples prior to running the 10 fold-CV and calculating the equivalent metrics to those reported later in the manuscript. Here we present how Gaussian Process, Logistic Regression and Ada Boost methods perform on the SMOTE balanced ccs-98 data set.
Additionally, we compared our CCS fingerprint with the well established MACCS keys.97,98 The MACCS keys are composed of 166 binary bits which also represent the presence and absence of chemical features. MACCS keys have been widely used, especially in the pharmaceutical industry. The bits represent a wide sub-set of chemical space.
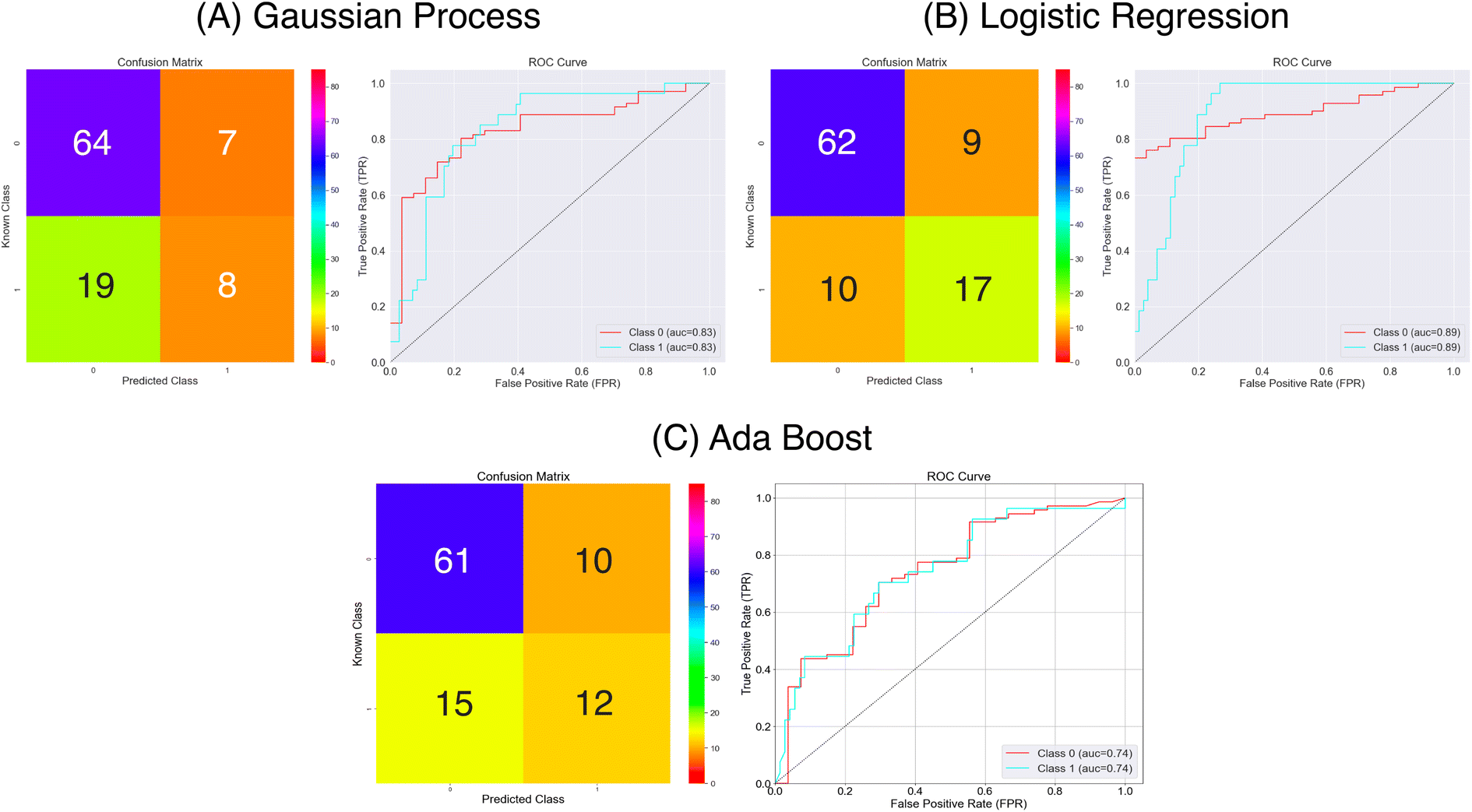 | ||
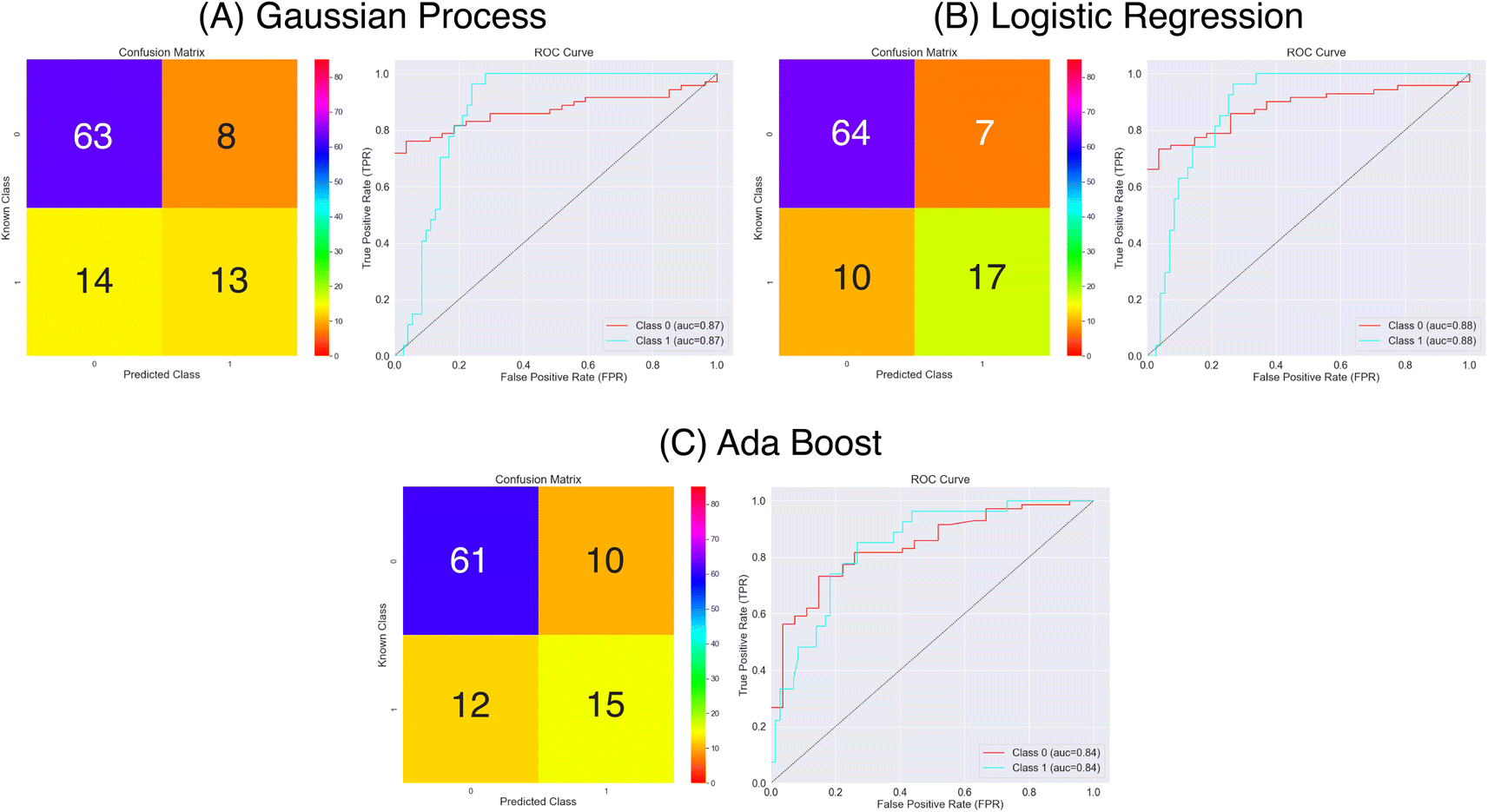
| Fig. 8 Confusion matrices and ROC curves for the balanced data against absorption capacity classification using the Mordred chemical features. Confusion matrices calculated over all external folds. | ||
| Algorithm | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|
| Gaussian process | 0.73 | 0.30 | 0.90 | 0.25 |
| Logistic regression | 0.81 | 0.63 | 0.87 | 0.51 |
| Adaboost | 0.74 | 0.48 | 0.85 | 0.34 |
From the results in Fig. 8 and Table 1 the models have a fair predictive accuracy between 0.73 and 0.81. The Gaussian Process and Ada Boost methods have broadly performed similarly in terms of accuracy, but the Logistic Regression method has a notable improvement with an accuracy over 0.80. However, for all three model there are notable differences in the sensitivity and specificity. The Gaussian Process and Ada Boost models both struggle similarly in terms of sensitivity. This is demonstrated clearly in Fig. 8A and C. Plot A shows roughly the same number of true positives and false positives predictions coupled with a larger number of false negatives predictions whilst plot C shows a near even spread over true positives, false positives and false negatives. This suggests the models are very poor in terms of predicting the positive class. The Logistic Regression model shows improvement beyond Gaussian Process and Ada Boost with respect to sensitivity, with notably higher true positives prediction proportion. All models show much better performance in terms of predicting true negatives. The MCC values highlight this imbalanced predictive accuracy with fairly low values; noting that values of 0.0 for MCC correspond to random, these predictions are showing limited improvement above this.
 | ||
| Fig. 9 Confusion matrices and ROC curves for the balanced data against absorption capacity classification using the MACCS keys as features. Confusion matrices calculated over all external folds. | ||
| Algorithm | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|
| Gaussian process | 0.78 | 0.48 | 0.89 | 0.40 |
| Logistic regression | 0.83 | 0.63 | 0.90 | 0.55 |
| Adaboost | 0.78 | 0.56 | 0.86 | 0.43 |
Using the MACCS fingerprints, and considering the metrics in Fig. 9 and Table 2 all three models again make a reasonable prediction of the molecules class considering the accuracy metric that ranges between 0.78 and 0.83. As for the Mordred descriptors, delving a bit deeper using the sensitivity and specificity metrics we find that predictions of the positive class are poorer that for the negative class. Again we the Logistic Regression model out performing the other two, however, there is a notable improvement in the prediction of the positive class for the Gaussian Process and Ada Boost models. The specificity has remained at a similar level of accuracy compared to the Mordred models. We note that the MCC scores have improved overall representing the better balance over the three model in predicting both classes.
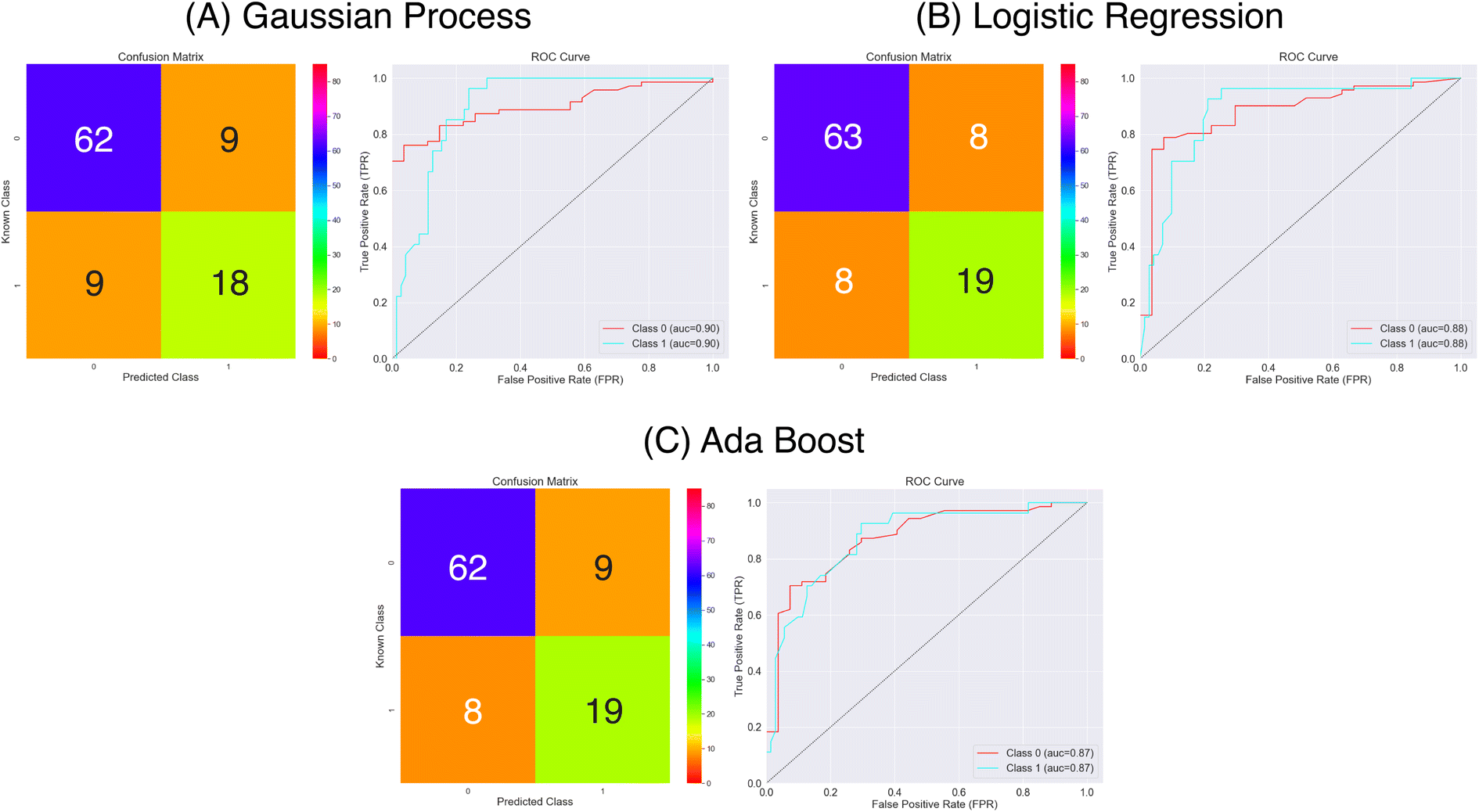 | ||
| Fig. 10 Confusion matrices and ROC curves for the balanced data against absorption capacity using CCS fingerprints as features. Confusion matrices calculated over all external folds. | ||
| Algorithm | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|
| Gaussian process | 0.82 | 0.67 | 0.87 | 0.54 |
| Logistic regression | 0.84 | 0.70 | 0.89 | 0.59 |
| Adaboost | 0.83 | 0.70 | 0.87 | 0.57 |
From Fig. 10 and Table 3 it appears that all three models make good predictions of the molecules classes. The accuracy of all models is greater than 0.8, with the accuracy range of 0.82–0.84. In the Logistic Regression and Ada Boost models we note a much improved sensitivity of 0.70 shown diagrammatically in Fig. 10 where we can now see the majority of positive class molecules are predicted correctly by all three models. There is a slight improvement in the specificity also over the three models compared to the models using Mordred or MACCS representations. Overall the MCC scores are now all over 0.5 showing the more balanced predictive accuracy.
Comparing the models on their summary metrics we see that in general Fig. 8–10 and Tables 1–3 suggest that classification of molecules using shallow learning algorithms for absorption capacity is a difficult task. Across the models presented we have used several molecular representations. The Mordred descriptors are composed of a range of well known 2D molecular descriptors encoding information of electronic state, graph topologies and molecular properties. We found 35 had a notable correlation with absorption capacity but this vector extended to 84 when one-hot encoding was applied. This means a notable part of the representation contains a null representation. It is possible that with a larger data set the most explanatory features could be more readily identified and the models improved. The current models struggle particularly to correctly separate molecules into the promising class, with a fairly balanced error rate across false positives and false negatives predictions.
The MACCS fingerprints are a standard fingerprint representation which has been employed many times in materials modelling. To our knowledge, it has not been applied previously to predicting absorption capacity. In this work we see that the MACCS fingerprint performs reasonably as a representation but struggles with the classification of molecules in the promising class. This is clearly shown in the sensitivity and specificity values. The MACCS fingerprints are the largest representation used in this work at 164 elements each, with every element requiring a sub-structure match to build the representation. This can be a relatively computationally expensive task.
Having considered these two standard representation methods, we developed our own fingerprint, inspired by the MACCS scheme, which encoded the sub-structures noted by the carbon capture community to correlate with carbon capture performance. We also wished to generate a more condensed representation which with equivalent software implementation could reasonably be expected to be generated with fewer sub-structure matches. From this we developed the CCS fingerprint. The models generated above show the result is promising. All of the models built using the CCS fingerprint perform with an accuracy higher than the standard features together with much improved predictive accuracy for the positive class, of approximately 70%. The models using the CCS fingerprint maintain high predictive accuracy for the negative class inline with the values seen from the standard features of 0.85–0.90. Owing to the improved predictive performance of the positive class these models also achieve the highest MCC scores demonstrating a more balanced predictive capability over the classes.
The best overall positive class predictor comes from the use of the CCS fingerprint features using the Logistic Regression classifier with 0.89 promising class correctly predicted 0.89 negative class correctly predicted and an overall accuracy of 0.84. The Logistic Regression models across all feature sets have tended to provide the most promising predictive accuracy over the classes. All models show a reasonable capability to predict the molecules which are unlikely to be promising in terms of capacity, which for HTVS may still be a useful and computationally inexpensive filter. The use of the CCS fingerprint provides improved predictions of the positive class suggesting it could be useful in HTVS in terms of prioritisation of laboratory testing.
3.3 Feature importance
We have performed feature importance analysis using the Logistic Regression classifiers over the difference feature sets. The importance of a feature is reflected by the magnitude of the linear regression coefficients in the models. We show in Fig. 11 the mean feature importance over the 10 cross validation.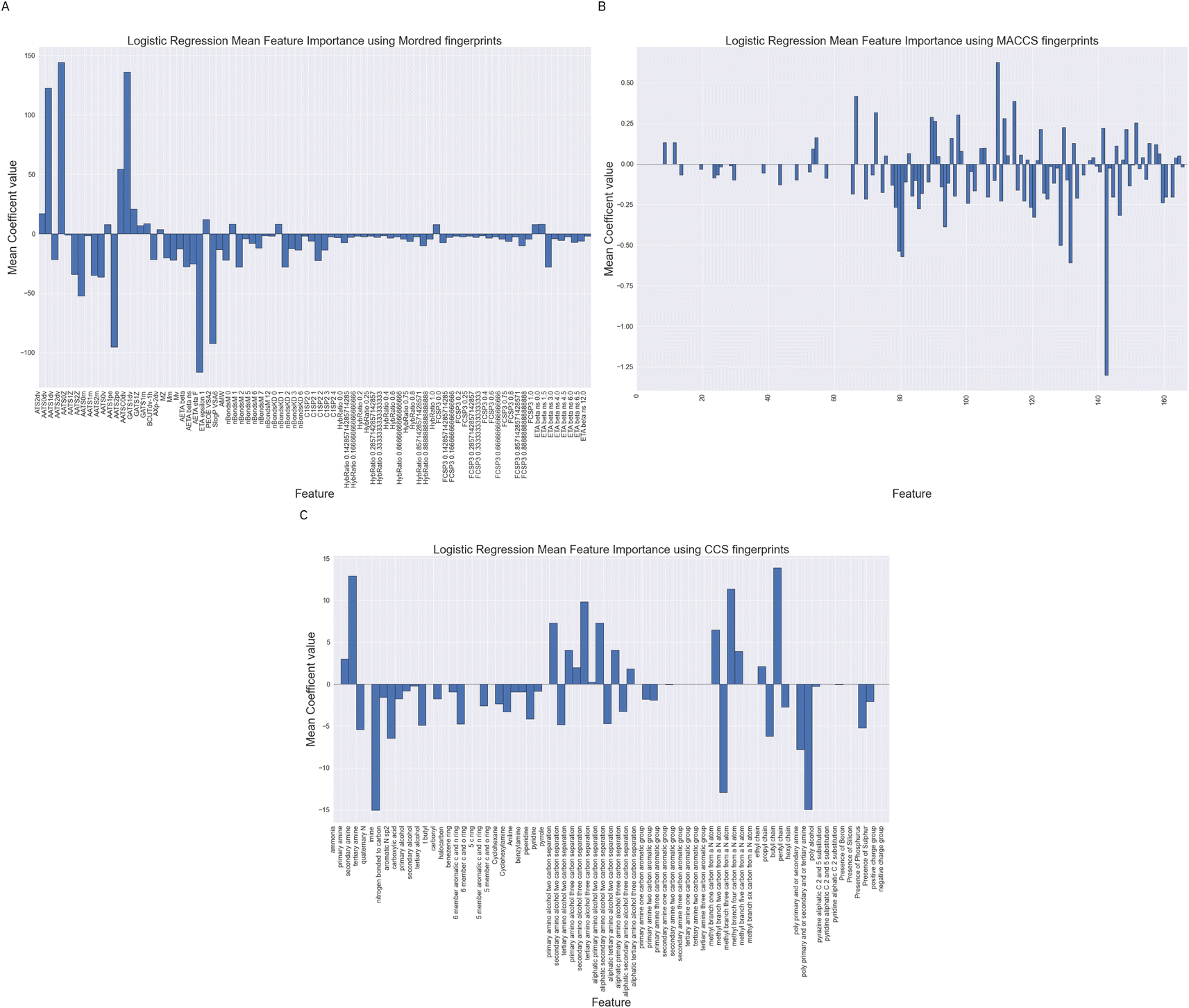 | ||
| Fig. 11 Feature importance metrics using Logistic Regression over all feature sets. The mean regression coefficients are plotted as measures of importance. Sub-figure (A) is for Logistic Regression using the Mordred feature set, sub-figure (B) is for Logistic Regression using the MACCS fingerprints and sub-figure (C) is for Logistic Regression using the CCS fingerprints. | ||
Whilst being careful not to over interpret Fig. 11, as they are based on no underlying fundamental physics or chemical theory, we can see some trends in the feature which are important. Looking at sub-Fig. 11A, using Mordred descriptors we note number of auto-correlation feature have large magnitude coefficients. These auto-correlation coefficients relate to valence electrons and charges suggesting the model is largely relying on fairly simplistic representations of the electronic structure of the molecule. These models may be improved with a better description of the electronic structure.
For the MACCS keys feature importance in Fig. 11B we also see the nitrogen environment as being important. For example bit numbers 70, 80 and 84 all relating to the presence or separation between nitrogen atoms in a molecule. The largest positive Logistic Regression coefficient belongs to bit 109 which represents the presence and absence of a CH2–O which could match to an alcohol functionality.
Fig. 11C displays Logistic Regression coefficients of large magnitudes for the CCS fingerprint on features related to the nitrogen environment, separating distances between amine and alcohol groups and chain lengths together with whether a molecule contains multiple amine functionalities. These are structural features which have been highlight by others as correlating with absorption capacity.
We provide in the ESI† a SHAP99 analysis of each of these models over 10 cross validation for the 20 most important features as determine by SHAP. This analysis was performed on a subset of the each folds test data. This analysis shows similar trends to the feature importance.
4 Conclusions
This work proposes a new molecular representation, CCS fingerprint and data set ccs-98, both of which are available at https://github.com/Jammyzx1/Carbon-capture-fingerprint-generation. An analysis of the chemical space of amines and N-heterocyclic molecules is provided against a background of commercially available amines and N-heterocyclic molecules. This analysis shows that carbon capture solvent molecules inhabit a sub-space, but are not outliers in their structure compared to the wide set of commercially available amines and N-heterocyclic molecules. This is promising as it suggests that there may be other commercially available molecules suitable for carbon capture without expensive new synthesis pathways being required. It also highlights chemical functional groups which in the ccs-lit-167 data set differ in relative abundance compared to commercial amines and N-heterocyclic molecules in the zinc-20938 data set. It remains unclear whether these differences are due to a lack of reporting on carbon capture capabilities for molecules containing these functionalities or due to these chemical functionalities having a consistent detrimental impact on carbon capture performance. This is an area for further exploration which could have a notable impact on the field by improving knowledge, data availability and thus modelling validation capabilities.We used this chemical space analysis to define a novel fingerprint for the modelling of amine molecules used in carbon capture. This fingerprint has been shown to be an effective featurization method for QSAR modelling and a way to analyze the chemical space. We have also tested the use of commonly applied featurization methods through the Mordred engine and MACCS fingerprints. The QSAR models built in this work show that QSAR prediction for absorption capacity is challenging with the limited available data. Some of our model show promise for high throughput virtual screening of carbon capture amines in the future. The use of the CCS fingerprint gave the most accurate classification models for each class. The CCS fingerprint also showed the most balanced model in terms of predictive accuracy for each class.
One of the biggest challenges to this work is relative lack of open available data in this field. This leads to small-data issues and limits the potential use of more complex modelling. Opening data in machine readable formats (such as csv, json, paraquat and HDF5 files for example) will enable computational scientist to better explore this area. A community conversation on data standards is encouraged to enable fair comparisons across data sources and models. As policy shifts towards a net zero carbon world and carbon capture, usage and storage is deployed, the release of more data in the open literature related to these technologies will become more vital. This data can be enhanced with computation to help in the search for more efficient solvents, and carbon capture materials more generally, as we have demonstrated in this work.
The overlap of computational and experimental work is a powerful combination. Computation can rapidly screen and rank materials. Discovering more efficient materials for carbon capture is a goal that is required to avoid the more catastrophic effects of climate change. To mitigate the effects of climate change is likely to require great urgency in collaborating at scale across the world to accelerate the development and understanding of the most promising net zero technologies.
Data availability
The code supporting this paper and datasets (ccs-98, ccs-lit-167, zinc-20938) are on GitHub and archived on Zenodo. https://github.com/Jammyzx1/Carbon-capture-fingerprint-generation for the carbon capture fingerprint generation, archived at https://zenodo.org/record/8304466. https://github.com/flaviucipcigan/ccus_amine_prediction_workflow for the amine prediction workflow, archived at https://zenodo.org/records/10213104.Author contributions
James McDonagh: conceptualization, data curation, formal analysis, methodology, software, project administration, supervision writing – original draft. Stamatia Zavitsanou: data curation, formal analysis, methodology, software, writing – original draft, writing – review & editing. Alexander Harrison: resources, project administration, software. Dimitry Zubarev: formal analysis, methodology, software, writing – original draft. Flaviu Cipcigan: conceptualization, project administration, formal analysis, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Bruce Elmegreen, Mathias Steiner, Stacey Gifford, Binquan Luan, James Hendrick and Nathaniel Park for insightful conversations. Data and required materials for this work can be found in the ESI.† This work was supported by the Hartree National Centre for Digital Innovation, a collaboration between STFC and IBM.References
- X. Wu, M. Wang, P. Liao, J. Shen and Y. Li, Appl. Energy, 2020, 257, 113941 CrossRef CAS.
- J. G. Olivier, K. Schure and J. Peters, PBL Netherlands Environmental Assessment Agency, 2017, vol. 5 Search PubMed.
- D. Tong, Q. Zhang, Y. Zheng, K. Caldeira, C. Shearer, C. Hong, Y. Qin and S. J. Davis, Nature, 2019, 572, 373–377 CrossRef CAS PubMed.
- T. Bruhn, H. Naims and B. Olfe-Kräutlein, Environ. Sci. Policy, 2016, 60, 38–43 CrossRef CAS.
- IEA, CCUS in Clean Energy Transitions, International energy association technical report, 2020 Search PubMed.
- I. A. of Oil and G. Producers, Map of global CCUS projects, 2020, https://web.archive.org/web/20210128061441/https://www.iogp.org/bookstore/product/map-of-global-ccs-projects/ Search PubMed.
- C. Chao, Y. Deng, R. Dewil, J. Baeyens and X. Fan, Renewable Sustainable Energy Rev., 2020, 110490 Search PubMed.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo and L. A. Hackett, et al. , Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- I. M. Bernhardsen and H. K. Knuutila, Int. J. Greenhouse Gas Control, 2017, 61, 27–48 CrossRef CAS.
- A. I. Papadopoulos, S. Badr, A. Chremos, E. Forte, T. Zarogiannis, P. Seferlis, S. Papadokonstantakis, A. Galindo, G. Jackson and C. S. Adjiman, Mol. Syst. Des. Eng., 2016, 1, 313–334 RSC.
- G. Puxty and M. Maeder, J. Chemom., 2020, 34, e3207 CrossRef CAS.
- X. Yang, R. J. Rees, W. Conway, G. Puxty, Q. Yang and D. A. Winkler, Chem. Rev., 2017, 117, 9524–9593 CrossRef CAS PubMed.
- H.-C. Li, J.-D. Chai and M.-K. Tsai, Int. J. Quantum Chem., 2014, 114, 805–812 CrossRef CAS.
- A. A. Orlov, A. Valtz, C. Coquelet, X. Rozanska, E. Wimmer, G. Marcou, D. Horvath, B. Poulain, A. Varnek and F. de Meyer, Commun. Chem., 2022, 5, 1–7 CrossRef PubMed.
- E. N. Muratov, J. Bajorath, R. P. Sheridan, I. V. Tetko, D. Filimonov, V. Poroikov, T. I. Oprea, I. I. Baskin, A. Varnek, A. Roitberg, O. Isayev, S. Curtalolo, D. Fourches, Y. Cohen, A. Aspuru-Guzik, D. A. Winkler, D. Agrafiotis, A. Cherkasov and A. Tropsha, Chem. Soc. Rev., 2020, 49, 3525–3564 RSC.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. a. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- G. Puxty, R. Rowland, A. Allport, Q. Yang, M. Bown, R. Burns, M. Maeder and M. Attalla, Environ. Sci. Technol., 2009, 43, 6427–6433 CrossRef CAS PubMed.
- P. Singh, J. P. Niederer and G. F. Versteeg, Int. J. Greenhouse Gas Control, 2007, 1, 5–10 CrossRef CAS.
- P. Singh, J. P. Niederer and G. F. Versteeg, Chem. Eng. Res. Des., 2009, 87, 135–144 CrossRef CAS.
- Y. E. Kim, S. H. Yun, J. H. Choi, S. C. Nam, S. Y. Park, S. K. Jeong and Y. I. Yoon, Energy Fuels, 2015, 29, 2582–2590 CrossRef CAS.
- F. A. Chowdhury, H. Yamada, T. Higashii, K. Goto and M. Onoda, Ind. Eng. Chem. Res., 2013, 52, 8323–8331 CrossRef CAS.
- S. Evjen, O. S. Løge, A. Fiksdahl and H. K. Knuutila, Energy Fuels, 2019, 33, 10011–10015 CrossRef CAS.
- A. Hartono, S. J. Vevelstad, A. Ciftja and H. K. Knuutila, Int. J. Greenhouse Gas Control, 2017, 58, 201–211 CrossRef CAS.
- B. Rezaei, S. Riahi and A. E. Gorji, Korean J. Chem. Eng., 2020, 37, 72–79 CrossRef CAS.
- Q. Yang, G. Puxty, S. James, M. Bown, P. Feron and W. Conway, Energy Fuels, 2016, 30, 7503–7510 CrossRef CAS.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen and B. Yu, et al. , Nucleic Acids Res., 2021, 49, D1388–D1395 CrossRef CAS PubMed.
- H. E. Pence and A. Williams, ChemSpider: an online chemical information resource, 2010 Search PubMed.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef CAS.
- Y. G. Chung, E. Haldoupis, B. J. Bucior, M. Haranczyk, S. Lee, H. Zhang, K. D. Vogiatzis, M. Milisavljevic, S. Ling and J. S. Camp, et al. , J. Chem. Eng. Data, 2019, 64, 5985–5998 CrossRef CAS.
- F. L. Oliveira, C. Cleeton, R. N. B. Ferreira, B. Luan, A. H. Farmahini, L. Sarkisov and M. Steiner, Sci. Data, 2023, 10, 230 CrossRef CAS PubMed.
- M. Simonovsky and J. Meyers, J. Chem. Inf. Model., 2020, 60, 2356–2366 CrossRef CAS PubMed.
- L. L. Wang and K. Lo, Briefings Bioinf., 2020, 22, 781–799 CrossRef PubMed.
- M. J. Fell, Publications, 2019, 7(3), 46 CrossRef.
- M. Woelfle, P. Olliaro and M. H. Todd, Nat. Chem., 2011, 3, 745–748 CrossRef CAS PubMed.
- J. McDonagh, T. van Mourik and J. B. Mitchell, Mol. Inf., 2015, 34, 715–724 CrossRef CAS PubMed.
- J. L. McDonagh, D. S. Palmer, T. v. Mourik and J. B. Mitchell, J. Chem. Inf. Model., 2016, 56, 2162–2179 CrossRef CAS PubMed.
- P. Das, T. Sercu, K. Wadhawan, I. Padhi, S. Gehrmann, F. Cipcigan, V. Chenthamarakshan, H. Strobelt, C. Dos Santos and P.-Y. Chen, et al. , Nat. Biomed. Eng., 2021, 1–11 Search PubMed.
- G. CONG, A. Gupta, R. N. B. Ferreira, B. O'Conchuir and M. De Bayser, AAAI Conference on Artificial Intelligence, 2022 Search PubMed.
- Y. Luo, S. Bag, O. Zaremba, A. Cierpka, J. Andreo, S. Wuttke, P. Friederich and M. Tsotsalas, Angew. Chem., Int. Ed., 2022, 61, e202200242 CrossRef CAS PubMed.
- L. Molander, M. Agerstrand and C. Ruden, Regul. Toxicol. Pharmacol., 2009, 55, 367–371 CrossRef CAS PubMed.
- J. L. McDonagh, W. C. Swope, R. L. Anderson, M. A. Johnston and D. J. Bray, Polym. Int., 2021, 70, 248–255 CrossRef CAS.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, J. Chem. Inf. Model., 2012, 52, 1757–1768 CrossRef CAS PubMed.
- G. Landrum, RDKit: Open-source cheminformatics, http://www.rdkit.org Search PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 1–14 Search PubMed.
- R. Notz, N. Asprion, I. Clausen and H. Hasse, Chem. Eng. Res. Des., 2007, 85, 510–515 CrossRef CAS.
- IBM Project Photoresist, https://research.ibm.com/interactive/photoresist/, accessed 14 March 2022.
- V. Vassiliadis, M. A. Johnston and J. L. McDonagh, 2022 IEEE International Conference on Services Computing (SCC), 2022, pp. 174–184 Search PubMed.
- L. Wasserman, Annu. Rev. Stat. Appl., 2018, 5, 501–532 CrossRef.
- M. Offroy and L. Duponchel, Anal. Chim. Acta, 2016, 910, 1–11 CrossRef CAS PubMed.
- G. Singh, F. Mémoli and G. E. Carlsson, SPBG, 2007, pp. 91–100 Search PubMed.
- M. Nicolau, A. J. Levine and G. Carlsson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7265–7270 CrossRef CAS PubMed.
- L. Parida, N. Haiminen, D. Haws and J. Suchodolski, Distributed Computing and Internet Technology, Cham, 2015, pp. 134–149 Search PubMed.
- J. L. Nielson, J. Paquette, A. W. Liu, C. F. Guandique, C. A. Tovar, T. Inoue, K.-A. Irvine, J. C. Gensel, J. Kloke, T. C. Petrossian, P. Y. Lum, G. E. Carlsson, G. T. Manley, W. Young, M. S. Beattie, J. C. Bresnahan and A. R. Ferguson, Nat. Commun., 2015, 6, 8581 CrossRef CAS PubMed.
- A. H. Rizvi, P. G. Camara, E. K. Kandror, T. J. Roberts, I. Schieren, T. Maniatis and R. Rabadan, Nat. Biotechnol., 2017, 35, 551–560 CrossRef CAS PubMed.
- W. Guo and A. G. Banerjee, J. Manuf. Syst., 2017, 43, 225–234 CrossRef.
- L. Carlsson, G. Carlsson and M. Vejdemo-Johansson, CoRR, arXiv, 2018, preprint, arXiv:1803.00384, DOI:10.48550/arXiv.1803.00384.
- T. K. Dey, F. Mémoli and Y. Wang, Multiscale Mapper: Topological Summarization via Codomain Covers, Society for Industrial and Applied Mathematics, 2016, pp. 997–1013 Search PubMed.
- Y. Zhou, M. Kamruzzaman, P. Schnable, B. Krishnamoorthy, A. Kalyanaraman and B. Wang, Proceedings of the 12th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics, New York, NY, USA, 2021 Search PubMed.
- Y. Zhou, N. Chalapathi, A. Rathore, Y. Zhao and B. Wang, 2021 IEEE 14th Pacific Visualization Symposium (PacificVis), 2021, pp. 101–110 Search PubMed.
- M. Schmidt, N. L. Roux and F. Bach, Math. Program., 2016, 162, 83–112 CrossRef.
- A. Defazio, F. R. Bach and S. Lacoste-Julien, CoRR, arXiv, 2014, preprint, arXiv:1407.0202, DOI:10.48550/arXiv.1407.0202.
- H.-F. Yu, F.-L. Huang and C.-J. Lin, Mach. Learn., 2010, 85, 41–75 CrossRef.
- Y. Freund and R. E. Schapire, J. Comput. Syst. Sci., 1997, 55, 119–139 CrossRef.
- T. Hastie, S. Rosset, J. Zhu and H. Zou, Stat. Interface, 2009, 2, 349–360 CrossRef.
- C. K. Williams and C. E. Rasmussen, Gaussian processes for machine learning, MIT press Cambridge, MA, 2006, vol. 2 Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss and V. Dubourg, et al. , J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- O. Obrezanova and M. D. Segall, J. Chem. Inf. Model., 2010, 50, 1053–1061 CrossRef CAS PubMed.
- A. J. Sterling, S. Zavitsanou, J. Ford and F. Duarte, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, e1518 CAS.
- L. J. Field, D. D. MacDonald, S. B. Norton, C. G. Ingersoll, C. G. Severn, D. Smorong and R. Lindskoog, Environ. Toxicol. Chem., 2002, 21, 1993–2005 CrossRef CAS PubMed.
- J. Cui, W. Li, C. Fang, S. Su, J. Luan, T. Gao, L. Hu, Y. Lu and G. Chen, IEEE Access, 2019, 7, 38397–38406 Search PubMed.
- V. L. Deringer, A. P. Bartók, N. Bernstein, D. M. Wilkins, M. Ceriotti and G. Csányi, Chem. Rev., 2021, 121, 10073–10141 CrossRef CAS PubMed.
- J. L. McDonagh, A. Shkurti, D. J. Bray, R. L. Anderson and E. O. Pyzer-Knapp, J. Chem. Inf. Model., 2019, 59, 4278–4288 CrossRef CAS PubMed.
- M. J. Burn and P. L. Popelier, J. Chem. Phys., 2020, 153, 054111 CrossRef CAS PubMed.
- T. Fawcett, Pattern Recognit. Lett., 2006, 27, 861–874 CrossRef.
- B. Matthews, Biochim. Biophys. Acta, Protein Struct., 1975, 405, 442–451 CrossRef CAS PubMed.
- D. Chicco and G. Jurman, BMC Genomics, 2020, 21, 1–13 CrossRef PubMed.
- J. L. McDonagh, N. Nath, L. De Ferrari, T. Van Mourik and J. B. Mitchell, J. Chem. Inf. Model., 2014, 54, 844–856 CrossRef CAS PubMed.
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, J. Cheminf., 2018, 10, 4 Search PubMed.
- H. L. Morgan, J. Chem. Doc., 1965, 5, 107–113 CrossRef CAS.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 CrossRef CAS PubMed.
- A. Capecchi, D. Probst and J.-L. Reymond, J. Cheminf., 2020, 12, 43 CAS.
- M. Gallegos, A. Costales and Á. M. Pendás, ChemPhysChem, 2021, 22(8), 775–787 CrossRef CAS PubMed.
- R. B. Said, J. M. Kolle, K. Essalah, B. Tangour and A. Sayari, ACS Omega, 2020, 5, 26125–26133 CrossRef CAS PubMed.
- S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang, A. G. Russell, Q. Wei and M. Fan, RSC Adv., 2013, 3, 22739–22773 RSC.
- Dask Development Team, Dask: Library for dynamic task scheduling, 2016 Search PubMed.
- D. W. C. A. James, Daylight Theory Manual, available at https://web.archive.org/web/20220327064115/https://www.daylight.com/dayhtml/doc/theory/theory.smarts.html, 2022/03/27 Search PubMed.
- G. Richner, Energy Procedia, 2013, 37, 423–430 CrossRef CAS.
- W. Conway, Y. Beyad, G. Richner, G. Puxty and P. Feron, Chem. Eng. J., 2015, 264, 954–961 CrossRef CAS.
- J. G. M. Conn, J. W. Carter, J. J. A. Conn, V. Subramanian, A. Baxter, O. Engkvist, A. Llinas, E. L. Ratkova, S. D. Pickett, J. L. McDonagh and D. S. Palmer, J. Chem. Inf. Model., 2023, 63, 1099–1113 CrossRef CAS PubMed.
- T. M. Fruchterman and E. M. Reingold, Softw. - Pract. Exp., 1991, 21, 1129–1164 CrossRef.
- B. Elmegreen, H. F. Hamann, B. H. Wunsch, T. Van Kessel, B. Luan, T. Elengikal, M. Steiner, R. Neumann, R. Luis Ohta and F. L. Oliveira, et al. , Front. Environ. Sci., 2023, 11, 1204690 CrossRef.
- M. Pirashvili, L. Steinberg, F. Belchi Guillamon, M. Niranjan, J. G. Frey and J. Brodzki, J. Cheminf., 2018, 10, 1–14 Search PubMed.
- N. V. Chawla, K. W. Bowyer, L. O. Hall and W. P. Kegelmeyer, J. Artif. Intell. Res., 2002, 16, 321–357 CrossRef.
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, Descriptor List, accessed: 28/10/ 2021, https://mordred-descriptor.github.io/documentation/master/descriptors.html Search PubMed.
- S. Raschka, J. Open Source Softw., 2018, 3(24), 638 CrossRef.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- MDL Information Systems, Inc., 14600 Catalina Street, San Leandro, CA 94577.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 CrossRef CAS PubMed.
- S. M. Lundberg and S.-I. Lee, Advances in Neural Information Processing Systems 30, Curran Associates, Inc., 2017, pp. 4765–4774 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00073g |
| ‡ Current address: Ladder Therapeutics doing business as Serna Bio, Lab F37, Stevenage Bioscience Catalyst, Gunnels Wood Road, Stevenage, Hertfordshire, SG1 2FX, UK. |
| § Steric hindrance emerged out of chemical intuition. Providing a physical basis for this concept is an important research topic (see for example Gallegos et al.82). Here, chemical intuition is enough to design of the fingerprint reported in Section 3.1.1. |
| This journal is © The Royal Society of Chemistry 2024 |