
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1D Zn(II)/2D Cu(I) halogen pyridyl coordination polymers. Band gap engineering by DFT for predicting more efficient photocatalysts in water treatment†
Andrea
García-Hernán
a,
Fernando
Aguilar-Galindo
 bc,
Oscar
Castillo
d and
Pilar
Amo-Ochoa
*ac
bc,
Oscar
Castillo
d and
Pilar
Amo-Ochoa
*ac
aDpto. de Química Inorgánica, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: pilar.amo@uam.es
bDpto. Química, Universidad Autónoma de Madrid, 28049 Madrid, Spain
cInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
dDepartment of Organic and Inorganic Chemistry, University of the Basque Country UPV/EHU, 48080 Bilbao, Spain
First published on 18th September 2024
Abstract
One of the key factors determining the photocatalytic capacity of a compound is its optical band gap, which, within an optimal range, maximizes the absorption and utilization of solar light. Recent research shows that modifying organic ligands and metal centers in metal–organic frameworks (MOFs) plays a crucial role in tailoring specific band gap characteristics. Many research articles use different structural MOF motives for photocatalysis, but few delve into band gap engineering using quantum chemistry calculations. Density functional theory (DFT) has been widely used to understand the possible mechanisms of action of some photocatalysts, but few studies directly relate the band gap to the ligands present in the compounds. In our study, DFT calculations are used to get a deeper understanding of the relationship between crystal and band structures aimed at predicting and enhancing the photocatalytic properties of new isostructural coordination polymers (CPs) with the general formula [ZnX2(L)2]n or [Cu2X2(L)]n from X = Cl and Br to X = I, and from 1,2-bis(4-pyridyl) ethane (BPE) to 1,2-bis(4-pyridyl)ethylene (BPEE) offering significant time and cost savings by enabling predictions. Despite the good band gap value of [ZnI2(BPEE)2]n, all members of the Zn/BPEE family show partial hydrolysis in water, which limits their use as photocatalysts. However, the 2D [Cu2X2(BPEE)]n (X= Cl, Br, and I) CPs are more insoluble and stable in water. Following the DFT results, the study of [Cu2I2(BPEE)]n as photocatalysts with water-persistent organic dyes (methylene blue (MB), methyl orange (MO), and tartrazine (Trz)) has been done. This approach allows us to assess whether the experimental synthesis of novel compounds with improved optical properties is worthwhile for their potential use as heterogeneous photocatalysts for water remediation.
Introduction
Among the key factors determining the capacity of a compound to act as a photocatalyst, the optical band gap stands out.1 The semiconducting character of materials used in photocatalysis is crucial, as it involves the absorption of light and the generation of electron–hole pairs, which is a fundamental process for promoting chemical reactions. The band gap of the semiconductor directly influences its ability to absorb light efficiently. Consequently, the generation of charge carriers, including reactive oxygen species (ROS), plays an essential role in photocatalytic reactions, particularly in the degradation of organic pollutants.2 High-performance photocatalysts typically exhibit an appropriate band gap ranging from 1.23 eV to 3.0 eV.3 This optimal range maximizes the absorption and utilization of solar light, whose maximum is located at 2.4 eV approximately. The positions of the valence band maximum (VBM) and conduction band minimum (CBM) are also critical, as they must cover the redox potentials of water.4The pursuit of improved photocatalytic activity remains a constant goal in the search for more efficient photocatalysts than those commonly used in industrial applications (such as titanium dioxide, TiO2).5–8 Scientists seek other alternatives, such as using different compounds with better properties. In this search, MOFs (metal–organic frameworks), a sub-family of coordination polymers (CPs), appear.9 These MOFs have undergone extensive experimental study. Researchers have diligently focused on controlling the structure and size of MOFs to achieve the desired physical properties. Indeed, there are many research articles using MOFs for photocatalysis,10–17 and although is clear that the modification of the organic linkers and metal centers plays a crucial role in tailoring specific band gap characteristics within MOFs, only a few of them delve into band gap engineering.11–14 These types of investigations generally use the principles of quantum chemistry calculations in predicting the electronic structure of materials, including their electronically excited states, and mainly focus on density functional theory (DFT) due to its relatively low computational cost (especially when compared to ab initio methods like Möller–Plesset or coupled cluster), its ability to predict material properties with good accuracy, and the ability to impose periodic boundary conditions (PBC). These conditions allow to simulate “infinite” systems, effectively avoiding spurious boundary effects. While generalized gradient approximation (GGA) functionals are computationally efficient and provide excellent agreement with experimental geometry values, more expensive hybrid functionals have been successfully employed to study band gaps. Hybrid functionals are particularly useful because the HOMO–LUMO energy differences are typically close to experimentally measured values. Through computational modeling, we not only replicate experimental observations but also gain insights into various factors that can modify electron density. These factors include the electronegativity of link atoms, ligand flexibility, and the crystal structure of the materials, among others. Although DFT has been used to understand the possible mechanism of action of some photocatalysts,15 or to corroborate the results of the theoretical band gap with the experimental one,16,17 there are very few studies that relate the band-gap with the ligands that are part of the compound.18,19 Indeed, recently Lu et al. proposed that band gaps in MOFs for electronic applications can be decreased by increasing the degree of conjugation of linkers, or by choosing electron-rich metal nodes and organic molecules.20 In this study, we employ DFT calculations as a tool to predict the band gap of new compounds. This approach allows us to assess whether the experimental synthesis of novel compounds with improved optical properties is worthwhile for their potential use as heterogeneous photocatalysts in treating persistent water pollutants. The use of DFT calculations offers significant time and cost savings by enabling predictions. Researchers can then focus their efforts on compounds that are genuinely of experimental interest.
In this work, we focus on CPs because they can present some advantages over MOFs. Indeed, the practical implementation of MOFs faces challenges. One significant hurdle is the complexity of their synthesis, often requiring solvothermal conditions. These conditions lead to substantial costs and limit scalability. CPs offer other compelling compounds with similar chemical stability, but they stand out due to their more direct and economical synthesis process.21–24 Many CPs can be synthesized under mild, single-step, and even solvent-free conditions.25 These accessible and scalable synthesis capabilities position CPs as a promising option for heterogeneous photocatalytic applications.26–28 Our CP selection criteria prioritize compounds with potential semiconducting properties, which are easily obtainable using commercial and non-toxic ligands. We follow the proposal of Lu et al.,20 trying to control the band gap using isostructural CPs in which the electronegativity of the halogen will be decreased (increase in size) (1) and the degree of linker conjugation will be increased (2). Additionally, we study the band gap behavior of similar tetrahedral coordination environments by comparing 1D Zn(II) and 2D Cu(I) CPs (3).
To do this we start with two 1D [ZnX2(BPE)]n CPs (X = Cl, Br, BPE = 1,2-bis(4-pyridyl)ethane) where the Cl and Br halides have already been synthesized.29,30 Building on theoretical and experimental results, we extend our study to the iodine analog for the first time, and explore a new family of 1D [ZnX2(BPEE)]n compounds (X = Cl, Br, and BPEE = 1,2-bis(4-pyridyl)ethylene), where the BPEE has a higher degree of conjugation. Once again, theoretical data, supported by experimental evidence, guide our synthesis efforts. As a result, we successfully synthesized the new iodine [ZnI2(BPEE)]n, validating the theoretical calculations. Despite the good band gap value of [ZnI2(BPEE)2]n, all this family shows partial hydrolysis in water, which limits their use as photocatalysts. For this reason, the study is extended with new 2D CPs [Cu2X2(BPEE)]n (X = Cl, Br, and I) that are more insoluble and stable in water than the Zn(II) analogs. Following the DFT results, and considering that currently, up to 80% of industrial wastewater containing dyes generated in low- and middle-income countries is dumped untreated into waterways or used directly for irrigation, which represents a wide range of direct and indirect threats to human, animal and plant health,31 the study of [Cu2I2(BPEE)]n as photocatalysts of water-persistent organic dyes (methylene blue (MB), methyl orange (MO) and tartrazine (Trz)) has been done.
Experimental details
Materials
All reagents were used as obtained from the suppliers. Ethanol (≥99.9%) and acetonitrile (99.93%) were purchased from Scharlau. The supplier for 1,2-di(4-pyridyl)ethylene (≥97%), 1,2-bis(4-pyridyl)ethane (≥99%), zinc iodide (≥99.95%), zinc chloride (≥99.95%), copper(I) chloride (≥99%), copper(I) bromide (≥98%), copper(I) iodide (≥99%), methyl orange, methyl blue and tartrazine is Sigma Aldrich. Zinc bromide (≥98%) and was supplied by TCI.Synthesis
(1a) Elemental analysis (%) calculated for [ZnCl2(C12H12N2)]n: C, 44.97; H, 3.77; N, 8.74. Experimental: C, 44.85; H,3.70; N, 8.69. (2a) Elemental analysis (%) calculated for [ZnBr2(C12H12N2)]n: C, 35.2; H, 2.95; N, 6.84. Experimental: C, 35.1; H, 2.90; N, 6.86. (3a) Elemental analysis (%) calculated for [ZnI2(C12H12N2)]n: C, 28.63; H,2.40; N, 5.56. Experimental: C, 28.58; H, 2.60; N, 5.53.
IR (1a–3a): 3050 (w), 2958 (w), 2930 (w), 1937 (w), 1842 (w), 1616 (s), 1561 (m), 1509 (m), 1451 (m), 1430 (s), 1225 (m), 1219 (m), 1072 (m), 1075 (m), 1027 (s), 827 (s). P-XRD and IR (Fig. S1 and S2†).
(1b) Elemental analysis (%) calculated for [ZnCl2(C12H10N2)]n: C, 45.25; H, 3.16; N, 8.88. Experimental: C, 45.15; H,3.21; N, 8.82. (2b) Elemental analysis (%) calculated for [ZnBr2(C12H10N2)]n: C, 35.4; H, 2.47; N, 6.88. Experimental: C, 35.1; H, 2.53; N, 6.91. (3b) Elemental analysis (%) calculated for [ZnI2(C12H10N2)]n: C, 29.75; H, 2.23; N, 5.66. Experimental: C, 29.82; H, 2.32; N, 5.78.
IR (1b–3b): 2969 (w), 1611 (s), 1556 (w), 1505 (m), 1451 (m), 1428 (s), 1350 (w), 1298 (w), 1202 (m), 1067 (w), 1023 (m), 967 (m), 954 (m), 827 (s). P-XRD and IR (Fig. S3 and S4†).
Solutions of CuX (X = Cl, Br, and I) 0.1 mmol (9 mg, 14.3 mg, and 19.0 mg respectively) in CH3CN (2 mL), and BPEE (22.0 mg, 0.1 mmol) in CH3CN (2 mL) are mixed for 5 minutes at room temperature, under magnetic stirring (700 rpm). Reddish-colored precipitates are formed immediately. The obtained precipitates are filtered off under vacuum and washed with CH3CN (3 mL × 2). Finally, the precipitates are dried under vacuum for 24 h with yields of 93% (47 mg, 4b), 97% (53 mg, 5b), and 98% (55 mg, 6b), based on Cu.
(4b) Elemental analysis (%) calculated for [Cu2Cl2(C12H10N2)]n: C, 33.25; H,3.16; N, 7.33. Experimental: C, 33.02; H, 3.22; N, 7.27. (5b) Elemental analysis (%) calculated for [Cu2Br2(C12H10N2)]n: C, 30.72; H, 2.15; N, 5.97. Experimental: C, 31.02; H, 2.30; N, 6.06. (6b) Elemental analysis (%) calculated for [Cu2I2(C12H10N2)]n: C, 25.59; H, 1.79; N, 4.97. Experimental: C, 25.65; H, 1.85; N, 5.01.
IR (4–6b): 3048 (w), 1605 (s), 1545 (m), 1502 (m), 1425 (m), 1352 (m), 1303 (w), 1207 (w), 1016 (m), 974 (s), 828 (s). P-XRD, and IR (Fig. S7 and S8†).
Methods and equipment's
Infrared (IR). The infrared spectra were obtained using a PerkinElmer spectrophotometer with the MIRacle Universal Attenuated Total Reflectance (ATR). Elemental analysis (EA) is performed using an elemental analyzer LECO CHNS-93217. The Powder X-ray diffraction (P-XRD) data were colleted using a PANalytical X'Pert PRO diffractometer equipped with a primary θ/2θ monochromator and a X'Celerator fast detector and monochromator with 1° for Kα1. Samples were analyzed with a θ/2θ sweep from 5° to 35° with an angular increase of 0.0167 and a time of 1 s per increment. Single-crystal X-ray diffraction (SC-XRD) single-crystal diffraction X-ray diffraction data for structure determination were collected on Bruker Kappa Apex II diffractometers (λ CuKα = 1.54184 Å at 200 K for 3a and 3b; λ MoKα = 0.71073 Å at 298 K under inert atmosphere for 3a and 3b). Crystal structures were solved by direct methods using SUPERFLIP37 and refined by full-matrix least-squares on F2 including all reflections (SHELXL).38 All calculations for these structures were performed using the OLEX2 crystallographic software package programs.39 Details of the structure determination and refinement of all compounds are summarized in Table S5 of the ESI.†Diffuse reflectance (DRS), by using a VARIAN model CARY 500 spectrophotometer equipped with an integrating sphere accessory, the Kubelka–Munk function can be plotted versus wavelength. This function depends on diffuse reflectance and relates to the transformation of the diffuse reflectance spectrum to its equivalent in absorbance units. Efficiency studies of the CPs in the degradation of the different organic dyes were carried out by measuring the loss of absorbance using an Ultraviolet-visible (UV-vis) Agilent 8453 ultraviolet-visible spectrophotometer in the range of 300 to 800 nm (λ).
The photocatalytic studies were carried out with light irradiation using a photoreactor with a 15 W purple LED at 20 °C temperature. To measure the emission of the LED (range λ = 300–600 nm, integration time CR2-AP + 200 ms, and intensity 20.5674 W m−2), a Stellarnet spectroradiometer model Blue-Wave UVNB50 was used. To conduct the study, 2 mg of each compound is placed in a vial along with 2 ml of a 10−5 M dilution of the dye. A blank vial containing 2 mL of the same 10−5 M dye is also prepared for comparison. These vials are then introduced into a white light photoreactor equipped with a stirring plate. After allowing a moderate amount of time to elapse, any changes in the blank are observed. The CPs are subsequently removed from the photoreactor, and the degradation of the dye is measured using UV-vis equipment.
Thermogravimetric analysis coupled with differential thermal analysis (TGA–DTA) was carried out on a TA Instruments Q500 thermobalance oven with a Pt sample holder. The particle size is measured using the scanning electron microscopy (SEM) Philips XL 30 S-FEG electron microscope, applying an electron beam of 10.0 kV of potential and 300 μA of intensity, at a pressure of 10−7 Pa. Compounds were metalized with a 10 nm thick Cr layer, at a pressure of 10−3 Pa. Theoretical calculations have been performed using density functional theory (DFT), a fundamental tool for understanding and predicting molecular and material properties, focusing on the application of periodic boundary conditions and full structure optimization using the Vienna Ab initio Simulation Package (VASP).40 In this process, a meticulous methodology is employed, expanding the electron density on a plane-wave basis up to a cutoff value of 420 eV, setting a threshold for the electronic energies of 10−5 eV, and using the projected augmented wave (PAW) pseudopotentials, from the VASP database. The complete optimization of the structures, encompassing both atomic positions and lattice vectors, has been carried out with precision, ensuring that all Hellmann–Feynman forces were below 0.01 eV Å−1. The OPTPBE functional41 has been used, which allows us to include weak interactions, such as van der Waals forces, in our calculations. On the top of the optimized structures, single point calculations with the HSE06 functional42 were performed, since the use of hybrid functionals allows for a reduction of the self-interaction error, thus leading to more accurate band gap energies. Reciprocal spaces were sampled using the Monkhorst–Pack scheme, assuring convergence with respect of the number of K-points. Atomic charges were obtained with the quantum theory of atoms in molecules (QTAIM)43 using the code developed by Henkelman et al.44 Proton nuclear magnetic resonance (1H-NMR) spectra were recorded, at room temperature on two-channel 300 MHz spectrometer Bruker Avance III-HD Nanobay 300 MHz equipped with 5 mm BBO 1H/X probe, equipped with Z gradients. A D2O 1H-NMR spectrums are studied with 2 mg of the CPs in 10−3 M of organic dyes. The 1H-NMR tubes of CPs and dyes are compared at the initial time (t0) and complete degradation time (tf) in the photoreactor. Finally, the CPs are isolated and dried for 24 h and dissolved in DMSO-d6 and two drops of DCl3 in the Cu(I) CPs. The 1H-NMR spectra were compared with the 1H-NMR of CPs before the photocatalytic process.
Results and discussion
DFT calculations to explore [ZnX2(BPE)]n theoretical band gaps and correlation with the experimental ones
In our quest for potential CPs, we focused on candidates that are easily synthesizable and feature accessible, with non-toxic metallic centers capable of exhibiting semiconductor behavior.45 With the idea of seeing how electronegativity and the size of the halogen affect the band gap we investigated two isostructural 1D-CPs previously synthethized29,30 with the formula [ZnX2(BPE)]n, where X represents either chloride (1a) or bromide (2a), and BPE stands for 1,2-bis(4-pyridyl)ethane (Fig. 1a).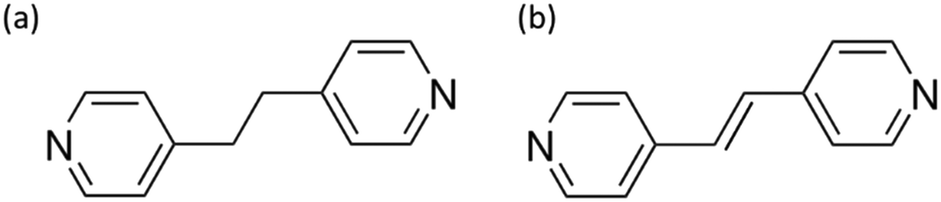 | ||
| Fig. 1 Employed ligands: a) 1,2-bis(4-pyridyl)ethane (BPE) and b) 1,2-bis(4-pyridyl)ethylene (BPEE). | ||
By employing DFT calculations to explore their theoretical band gaps, we observed an intriguing correlation: as the halogen size increases (and electronegativity decreases), the optical bandgap decreases. Specifically, the bandgap decreased from 4.95 eV (1a) to 4.66 eV (2a) (as indicated in Table 1) and there is a good correlation with the experimental band-gap (Table 1 and Fig. S9†). The character of the band gap is mainly “ZnX2 → ligands”, as shown by the projected density of states (PDOS, Fig. S10–S12†). This phenomenon can be explained by the different energies of the p orbital of the halogen: along the series Cl–Br–I, this orbital has less negative energy, reducing the gap with the first empty orbital of the Zn (4s orbital). Also, due to the lower gap, both 4s and np orbitals can hybridize, increasing the covalent character of the interaction, as corroborated by the reduction of the absolute values of the atomic charges (Table S1†).
| Theoretical band gap (eV) | Experimental band gap (eV) | |
|---|---|---|
| (1a) [ZnCl2(BPE)]n | 4.95 | 4.50 |
| (1b) [ZnCl2(BPEE)]n | 3.36 | 3.15 |
| (2a) [ZnBr2(BPE)]n | 4.66 | 4.40 |
| (2b) [ZnBr2(BPEE)]n | 3.43 | 3.40 |
| (3a) [ZnI2(BPE)]n | 4.08 | 4.12 |
| (3b) [ZnI2(BPEE)]n | 3.08 | 2.90 |
Consistent with these findings, we anticipated that the isostructural [ZnI2(BPE)]n CP would exhibit an even lower band gap, following the rules of Lu et al.,20 and our theoretical reasoning. To verify this trend, we successfully synthesized it using a rapid and easily scalable chimie douce approach (Table S2†). The synthesis involved mixing ZnI2 and BPE ligand at room temperature in a water/CH3CN solvent mixture. We applied the same synthetic procedure to the chloride and bromide analogs, obtaining sufficient samples for comparative studies on their photocatalytic behavior.
Remarkably, both the experimentally measured optical band-gap (4.12 eV) and the DFT calculated band-gap (4.08 eV) values align with this trend (Table 1).
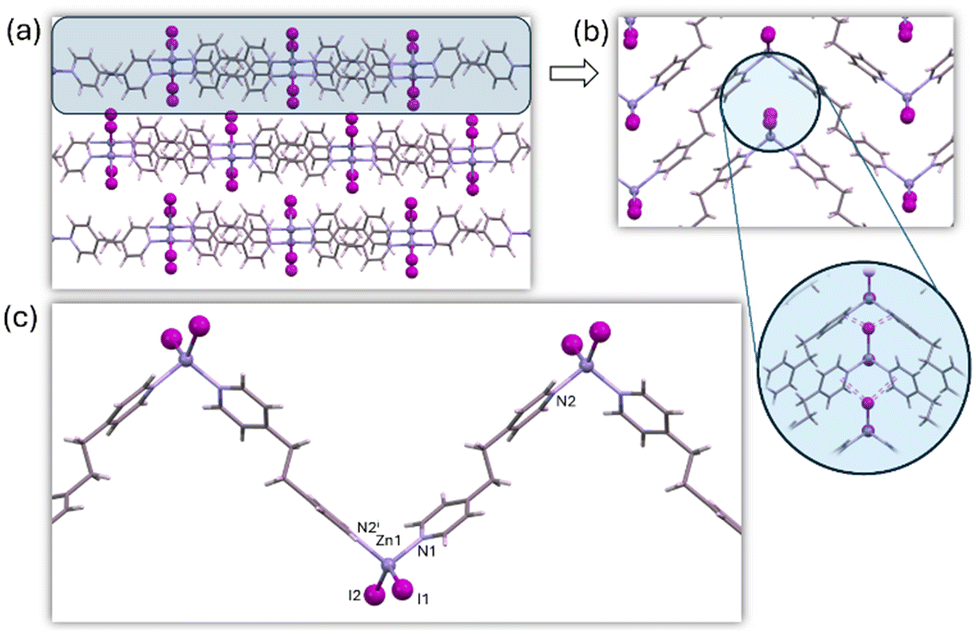
The crystal structure of 3a was solved by employing SC-XRD on single crystals obtained through diffusion techniques (Fig. S5†). 3a shows the same structural features within the 1D chain observed for the rest of the members of this family of compounds: tetrahedral ZnX2N2 coordination geometry (4-coordinate geometry index τ4: 0.89 (Cl), 0.90 (Br), 0.93 (I)), the halides acting as terminal ligands and BPE ligands bridging adjacent metal centers through its two pyridinic nitrogen atoms (Fig. 2). Interestingly, 3a shows a different arrangement of the chains because of the presence of different supramolecular interactions. In the chloride and bromide analogs C–H⋯X hydrogen bonding interactions took a predominant role in directing the arrangement of the chains, but in the iodide-based compound the dispersive forces take this role, and iodide⋯π-cloud interactions are observable (Fig. 2b).46–48 In this sense, one of the iodide anions is involved in an intrachain hydrogen bonding with an aromatic C–H donor group of the BPE bridging ligand. The second iodide provides these I⋯π-cloud interactions connecting the chains, by locating between two BPE ligands coordinated to the same metal center establishing I⋯N/C distances in the range 3.9–4.2 Å. The conformation of the BPE also differs being planar for the chloride and bromide analogs, but for the iodide compound the two pyridinic rings are no longer coplanar (dihedral angle: 40°). The angle between three consecutive copper atoms within the chain changes from 116° (Cl) and 114° (Br) to 99° (I), in the search for a better conformation to maximize the I⋯π-cloud interactions. This different supramolecular arrangement of the otherwise quite similar chains leads to completely different unit cell parameters and space groups (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) for chloride and bromide analogs and C2/c for the iodide one).
for chloride and bromide analogs and C2/c for the iodide one).
 | ||
| Fig. 2 Crystal packing (a) of compound 3a showing the supramolecular I⋯π-cloud interactions (b) holding the 1D coordination polymer chains (c) together. | ||
This structural change could be attributed to the flexibility of the BPE ligand, which can adopt different conformations to accommodate the structural requirements of both a crystal structure directed by C–H⋯X hydrogen bonding or X⋯π-cloud interactions.
DFT calculations to explore [ZnX2(BPEE)]n theoretical band gaps and correlation with the experimental ones
In addition, the use of BPEE ligand, with a higher degree of conjugation, may potentially reduce the CPs band-gap making them more optically interesting for their potential application as photocatalysts, due to the greater electronic delocalization of their π-systems.49 Consequently, we decided to conduct a similar study using the related but more rigid BPEE ligand (Fig. 1b).We found that a higher degree of conjugation of the organic ligand (BPE→BPEE) drastically decreases the band-gap value, making the compounds more interesting as semiconductors with possible photocatalytic activity (Table 1). This is explained based on the electronic structure of the ligand and the chemical nature of the fundamental (HOMO–LUMO) transition, thanks to the analysis of the PDOS (Fig. S13–S15†). For BPEE, the presence of the double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond allows for greater electronic delocalization, which lowers the energy of the LUMO orbital, thus reducing the band-gap of the material. This higher conjugation also generates an occupied state with less negative energy, which is above in energy than the orbitals of the ZnX2 for X = Cl, Br (Table S3†). In the case of X = I, due to the lower electronegativity of I, the states located around the ZnI2 are less stabilized and their energies become like the HOMO of BPEE.
C bond allows for greater electronic delocalization, which lowers the energy of the LUMO orbital, thus reducing the band-gap of the material. This higher conjugation also generates an occupied state with less negative energy, which is above in energy than the orbitals of the ZnX2 for X = Cl, Br (Table S3†). In the case of X = I, due to the lower electronegativity of I, the states located around the ZnI2 are less stabilized and their energies become like the HOMO of BPEE.
Based on their possible interest and future applications, we have increased the efficiency and sustainability of the previously reported synthesis (Table S2†),32,33 developing and optimizing the methodology employing a less toxic solvent and environmentally friendly procedures. In this improved approach, acetonitrile serves as the sole solvent, completely replacing the use of dimethylformamide (DMF). Additionally, we have adjusted the reaction conditions, resulting in a one-step synthesis with instantaneous precipitation at ambient temperature. This modification aims to minimize energy consumption, in contrast to the previously published method that required a reaction at 100 °C for 96 hours. Under these synthesis conditions, the particle size varies between 0.8 and 2.5 microns in length and 0.4 and 0.9 microns in width (Fig. S16 and Table S4†).
Remarkably, all the structures of both the previously published chloride and bromide compounds29,30 and the newly reported iodide-based compound are isostructural. These structures rely on underlying supramolecular interactions that connect the chains, with X⋯π-cloud interactions playing a significant role (Fig. 3). The 1D CP shares features with BPE-based compounds: ZnX2N2 tetrahedral coordination geometry (τ4: 0.88 for Cl and Br, 0.89 for I) involving terminal halides and bridging BPEE ligands. Interestingly, in these compounds, while the planarity of the BPEE ligands is maintained, the angle between three consecutive zinc atoms within the chain is approximately 90° (94° for Cl, 93° for Br, and 91° for I) this phenomenon is also observed in [ZnI2(BPE)]n (91°). Additionally, the unit cell parameters of [ZnX2(BPEE)]n and [ZnI2(BPE)]n exhibit a striking similarity, suggesting an isostructural relationship (Tables S5†). Notably, the more rigid [ZnX2(BPEE)]n compounds are seen to be less soluble in water than their BPE analogs, which opens up opportunities for water-based applications.
 | ||
| Fig. 3 Crystal packing (a) of 3b showing the supramolecular I⋯π-cloud interactions (b) holding the 1D CP chains (c) together. | ||
However, these supramolecular arrangements should not have directly a significant impact on the band-gap value of the material as it is most dependent on the direct bonds established between the zinc(II), iodide, and BPE/BPEE ligands. In this sense, Table 2 shows how the coordination bond distances and angles evolve along the halide series for each family of compounds. These experimental data agree with the theoretical calculations (Table 1). However, it is known that strong supramolecular interactions can modify to some extent the coordination bond features (distance and angles). Among these compounds, the strongest supramolecular interactions are expected to be those between the chloride anions and the aromatic C–H groups of the BPEE ligand. Additionally, when analyzing the theoretical and experimental gap values along the BPE and BPEE-based compounds there is a change in the trend for 1b (3.36 eV theoretical/3.15 eV experimental), 2b (3.43 eV theoretical, 3.40 eV experimental), and 3b (3.08 eV theoretical, 2.9 eV experimental) (Table 1 and Fig. S17†). Usually for full shell cations, the weaker the metal-to-ligand interaction, as happens along the Cl, Br, and I series, the smaller the band gap. Simultaneously, the steric hindrance of the big iodide anions leads to longer Zn–N coordination bond distances which reinforces this trend. Therefore, BPE-based compounds show band-gap values that completely agree with this tendency. However, this is not true for the BPEE-based compounds as the Zn–N bond distances are unusually longer in the case of 1b probably because the strength of its supramolecular interactions is forcing them to adopt slightly unusual coordination bond distances. Fig. 4 shows a distribution of the Zn–N coordination bond distances in the context of similar ZnX2N2 compounds found in the CSD database (Fig. S18–S20†).50 These longer coordination bond distances create such a reduction of the band-gap value that 1b provides a smaller band-gap value than its analogous 2b (Table 2).
| 1a | 2a | 3a | |
|---|---|---|---|
| Zn–X (Å) | 2.2400(7); 2.2244(7) | 2.3634(7); 2.3779(7) | 2.5527(7); 2.5883(6) |
| Zn–N (Å) | 2.043(2); 2.047(2) | 2.040(5); 2.044(4) | 2.049(3); 2.056(3) |
| X–Zn–X (°) | 123.60 | 122.99 | 121.62 |
| N–Zn–N (°) | 110.64 | 109.85 | 103.46 |
| Zn geometry, τ4 | 0.89 | 0.90 | 0.93 |
| Zn⋯Zn⋯Zn (°) | 115.88 | 113.77 | 90.96 |
| BPE torsion (°) | 0 | 0 | 37.99 |
| 1b | 2b | 3b | |
| Zn–X (Å) | 2.216(2); 2.245(2) | 2.3501(5); 2.3886(5) | 2.5430(5); 2.5890(5) |
| Zn–N (Å) | 2.062(4); 2.064(4) | 2.047(3); 2.054(3) | 2.064(3); 2.064(4) |
| X–Zn–X (°) | 128.98 | 127.39 | 124.69 |
| N–Zn–N (°) | 106.87 | 106.32 | 105.11 |
| Zn geometry, τ4 | 0.88 | 0.88 | 0.89 |
| Zn⋯Zn⋯Zn (°) | 93.74 | 92.92 | 91.38 |
| BPEE torsion (°) | 0 | 0 | 0 |
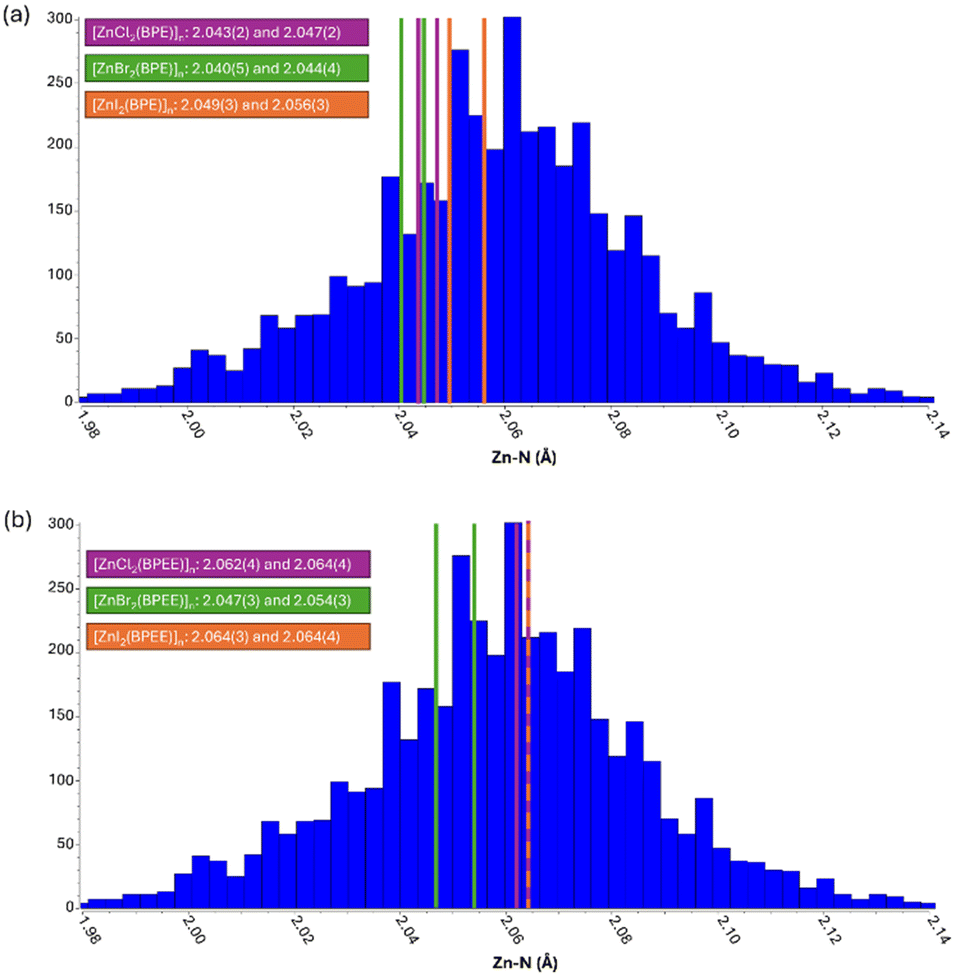 | ||
| Fig. 4 Zn–N coordination bond distances found in compounds: (a) [ZnX2(BPE)]n and (b) [ZnX2(BPEE)]n placed within the distribution found in the CSD database for analogous ZnX2N2 coordination environments. | ||
Water and thermal stability of [ZnX2(BPEE)]n
The studied compounds show reasonable thermal stability up to 250 °C as can be seen in the TGA studies (Fig. S21†). Additionally, it is well known that Zn(II) in water undergoes significant hydrolysis forming several mononuclear and polynuclear species.51,52 Although Zn(II) CPs with BPEE are much more insoluble in water than the BPE analogs, we studied their behavior in water and at different pHs.53 For this purpose, [ZnX2(BPEE)]n (1b, 2b, and 3b) are suspended in water at room temperature (pH = 6.58). Small amounts of diluted 0.01 M of HCl and NaOH are added to the suspensions to assess their stability. At pH < 4.2 [ZnX2(BPEE)]n solubilize in water. P-XRD and IR analysis of the solids after base treatment reveal that 1b, 2b, and 3b remain stable within the pH range of 4 to 8.4 (Fig. S22 and S23†). Moreover, the IR, P-XRD, and 1H-NMR studies of 3b in D2O at 25 °C and pH = 6.6, conducted at different times after irradiation in the photoreactor (from t0 = 0 minutes to tf = 20 minutes) indicates that the compound partially dissolves (Fig. S24†). The observed signals correspond to BPEE ligand shifting relative to the initial ligand position, suggesting the presence of an alternative coordination environment for the Zn(II) compound with BPEE. This phenomenon may be attributed to the ingress of water molecules into the Zn(II) coordination sphere, leading to the displacement of the coordinated ligand and the formation of other species (Table S8 and Fig. S25†).DFT calculations to explore 2D [Cu2X2(BPEE)]n (X = Cl, Br, I) theoretical band gaps and correlation with the experimental ones
The partial hydrolysis of Zn(II) compounds with BPEE in water, leads us to focus the study using CPs that can be chemically more inert but that may have an appropriate band gap. Cu(I) bipyridine-type ligands CPs have been extensively studied for their optical properties, with many of them exhibiting semiconductor characteristics.54 Additionally, this broad family of CPs tends to be stable and insoluble in water.22 Moreover, Cu(I) typically adopts a tetrahedral coordination environment analogous to Zn(II) compounds. Based on these principles, we have selected an isostructural family of 2D CPs with the formula [Cu2X2(BPEE)]n (where X = Cl (4b), Br (5b), I (6b)). These related [Cu2(μ-I)2(μ-BPEE)]n compounds, which have been previously reported,34,55 differ from the previously described zinc analogous in many different aspects: (i) the amount of BPEE ligand is reduced to half; (ii) the copper(I) coordination environment is also tetrahedral but with a NI3 donor set instead of N2I2; (iii) the halides behave as bridging ligands leading to ladder like [CuI]n chains which are connected by bridging BPEE ligands to generate 2D planar sheets (Fig. 5); (iv) there is no polymorphism within this family. The coordination bond distances show relatively similar values for chloride and bromide analogs (Cu–Cl: 2.338(1), 2.410(1), 2.557(1) Å and Cu–N: 2.004(3) Å vs. Cu–Br: 2.472(1), 2.520(1), 2.564(1) Å and Cu–N: 2.009 Å) but significantly longer for the iodide analog (Cu–I: 2.6199(3), 2.6563(4), 2.6582(4) Å and Cu–N: 2.040(2) Å). Another distinctive feature of these compounds is the presence of relatively short Cu⋯Cu distances within the ladder-like chain (2.985(3) Å for chloride, 2.885(2) Å for bromide, and 2.7851(5) Å for iodide). The latter value implies some kind of copper⋯copper contact as this distance is just below double the copper van der Waals radii. These 2D sheets are held together employing weak hydrogen bonding interactions in which every halide receives a double C–H⋯X from a BPEE ligand from the adjacent sheet.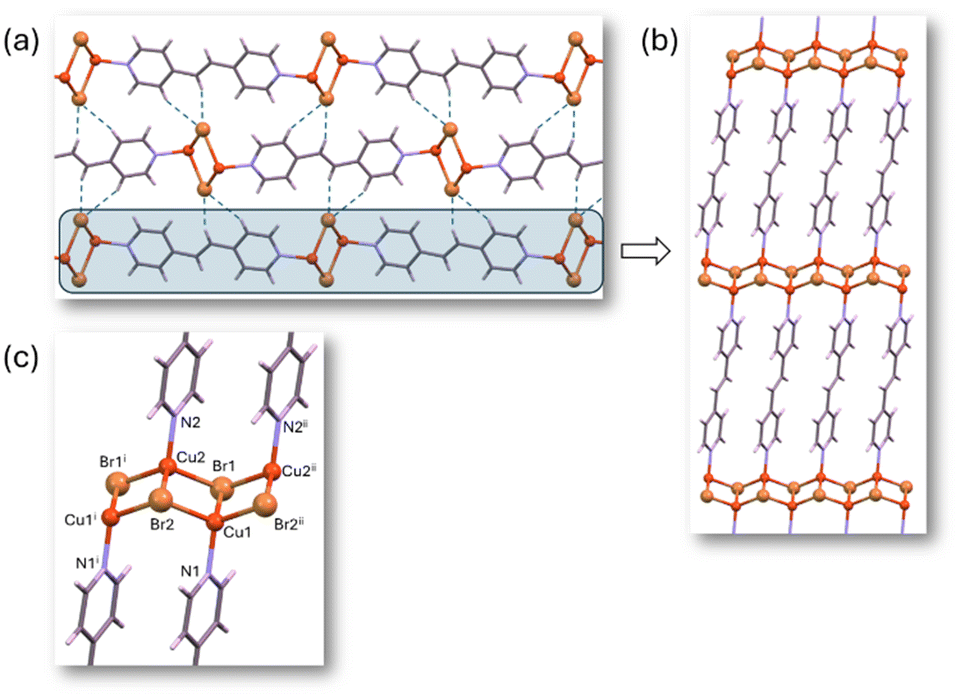 | ||
| Fig. 5 (a) Crystal packing of compound 5b showing the hydrogen bonding interactions holding together the 2D planar sheets. (b and c) Detail of the 2D planar sheet showing the CuBr ladder-like chain connected by bridging BPEE ligands. | ||
The employed syntheses have been sustainably modified compared to the initially published ones (Table S2†). They now occur in a single step, using water, and with high yields. Their particle size varies between 0.9 and 1.1 microns in length and 0.2 and 0.5 microns in width (Fig. S26 and Table S9†).
DFT calculations have allowed us to understand the influence of the halogen on the band gap value. In the case of the Cu-based, the observed trends in the calculations are consistent with the Zn analogs: a reduction of the band gap from Cl and Br (which DFT predicts to be almost degenerate) to I, showing the effect of the electronegativity (and energy of the p orbital) of the halogen atom (Fig. S27–S29†). The covalent character of the Cu–X bond is also affected by the chemical nature of the halogen, in a similar way to the Zn-based compounds, as pointed out by the atomic charges (Table S10†). Additionally, the experimental band-gap study reveals a strong correlation with the theoretical values (Fig. S30†). These results enable us to select the potentially most efficient compound as a heterogeneous photocatalyst, namely 6b (Table 1).
Water and thermal stability of [Cu2X2(BPEE)]n
TGA studies show that this family of compounds is stable up to values of 200 °C (Fig. S31†). Compound 4b shows a slight increase in mass at this temperature which may be due to a possible partial oxidation of CuCl since it is quite unstable.The stability study in water at different pHs has been conducted for the three compounds (4–6b) by creating dispersions of each one in water within the pH range of 2 to 11. IR spectra of the solids separated from the suspensions indicate that the original structure remains unchanged within this pH range (Fig. S32†). Additionally, compound 6b was suspended in D2O. After 24 hours its 1H-NMR spectrum was obtained (Fig. S33 up†), and it was compared with the BPEE spectrum in D2O (Fig. S33 down†). The absence of signals corresponding to the ligand confirms the compound's insolubility under these conditions.
Study of the photocatalytic efficiency of compounds [Cu2X2(BPEE)]n (X = Cl, Br, and I) in the degradation of organic dyes methyl blue (MB), methyl orange (MO) and tetrazine (Trz)
The band gap values, and stability in water, along with their resistance to change in pH, enable the study of this family of CPs as photocatalysts for water-persistent organic dyes. The experiments were performed mixeding 2 mg of CPs with 10−5 M aqueous solutions of MB, MO, and Trz inside a white light photoreactor (Fig. S34†). After the degradation experiment, the dye is separated from the catalyst, and its absorbance loss is analyzed using UV-vis spectroscopy (Fig. 6, S35 and S37†). As anticipated, the 6b compound exhibits greater efficiency in degrading these dyes (Table 3).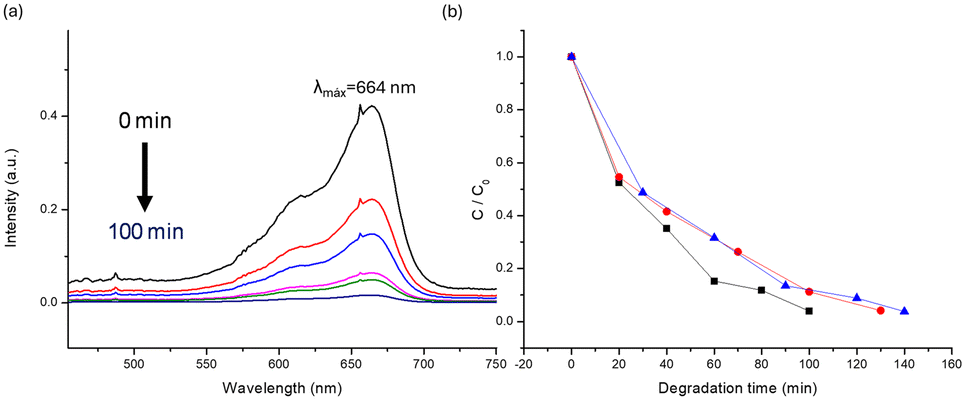 | ||
| Fig. 6 MB dye degradation: a) UV-vis degradation 0 to 100 min for [Cu2I2(BPEE)]n and b) plots of MB absorbance intensity loss as a function of exposure time under photoreactor 6b (black), 5b (red), and 4b (blue). | ||
| 2D [Cu2X2(BPEE)]n | Theoretical band-gap (eV) | Experimental band-gap (eV) | MO min (n° cycles) | MB | Trz (min) (n° cyles) |
|---|---|---|---|---|---|
| 4b | 1.89 | 2.45 | 90 | 130 | 120 |
| 5b | 1.89 | 2.38 | 80 | 120 | 110 |
| 6b | 1.66 | 2.04 | 60 (5) | 110 | 90 (3) |
The 1H NMR spectra of compound 6b, along with MB, MO, and Trz under photoirradiation, reveal that at time 0 minutes, free MB, MO, and Trz signals in the solution can be observed (Fig. 7d, S36b and S38b†). However, after the final photodegradation time (tf) for each dye (Fig. 7a, S36a and S38a†) there are no signals from either MB/MO/Trz or BPEE in the solutions. These results confirm the effectiveness of compound 6b as a heterogeneous photocatalyst. Furthermore, the solid separated from the solution after the final photodegradation time was dissolved in DMSO-d6 with two drops of DCl and its 1H-NMR spectrum exclusively shows signals from BPEE (Fig. S39†). This finding rules out MB/MO/Trz adsorption processes and supports their photodegradation.
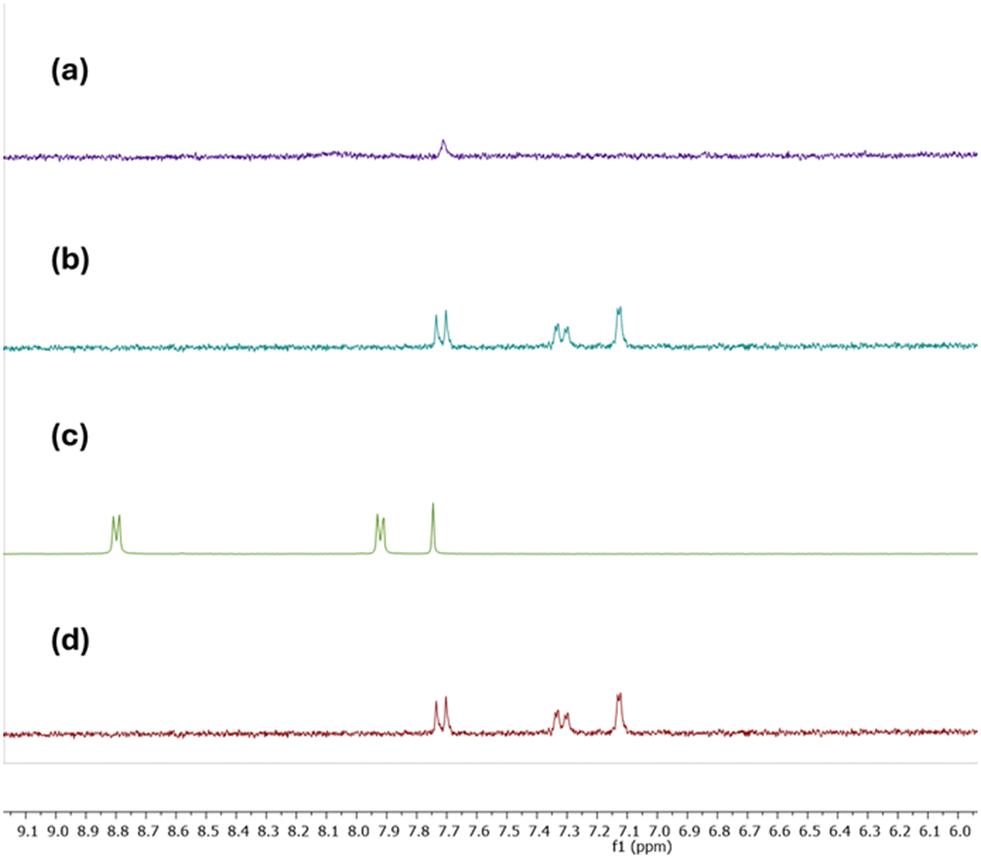 | ||
| Fig. 7 1H-NMR study with 6b and MB 10−3 M in D2O: a) 6b + MB (t = 100 min.), b) 6b + MB (t = 0 min) and c) BPEE and d) MB 60 min in D2O respectively. | ||
Finally, the kinetics of the reaction have been studied in the case of compound 6b, being of order 1 for each of the dyes (Fig. S40†). Likewise, the degradation mechanisms of dyes in the presence of compounds 4–6b have been studied, using trappers such as benzoquinone (BQ), tert-butanol (t-but), and ammonium oxalate (AO) (Fig. S41†). The degradation mechanism was found to occur via the formation of OH˙ type radicals (Fig. S42†). Hydroxyl radicals are generated when holes in the valence band interact with adsorbed OH ions or water molecules. These radicals can react with molecules on the surface of the photocatalyst or move into solution. Since t-but reacts with hydroxyl radicals at a rate almost equal to the diffusion limit, it acts as an excellent trapping agent for these radicals. On the other hand, the superoxide anion radical is formed when oxygen molecules on the catalyst surface capture electrons from the CBM.56
Conclusions
This work represents a detailed study that demonstrates the advantages of using DFT calculations for selecting and designing new coordination compounds with optimal band-gaps for use as heterogeneous photocatalysts. The study highlights how employing these calculations can save time, resources, and energy by allowing researchers to focus solely on compounds whose theoretical calculations exhibit optimal band gaps.Our theoretical calculations allow us to correlate how the increase of size and decrease of electronegativity of the halogens affects the energies of their p orbitals concerning the 4s Zn(II)/Cu(I) orbital, increasing the covalent character and the M–X bond distances, weakening its strength by hybridizing, favoring a reduction in the gap. Notably, strong supramolecular interactions can alter the coordination bond distances (M–X), thereby modifying the band-gap value. The decrease in band-gap values due to halogen size ranges from 0.6 to 0.3 eV. However, the energies of the p orbitals concerning the 4s Zn(II)/Cu(I) orbital, and the M–N distances do not significantly affect the band-gap reduction; rather, it is the enhanced conjugation of the organic ligand resulting from structural rigidity and facilitated electronic delocalization along the chain (in this case, the band gap reduction is approximately 1.6–1.0 eV).
Theoretical data is compared with experimental results, allowing us to predict which compound will have a lower band-gap value and, consequently, initially exhibit better photocatalytic properties within these CPs families.
Moreover, while the study of band-gap value is essential, the stability of compounds in water is paramount. This study demonstrates that such stability is enhanced by an increase in structural rigidity facilitated by the presence of conjugated ligands. The increase in conjugation of the organic ligand (from BPE to BPEE) not only contributes to a drastic reduction in the band gap value but also allows the creation of more water-stable compounds. However, although the stability of Zn(II) CPs has increased with the use of BPEE, partial hydrolysis of these compounds continues to hinder their use as heterogeneous photocatalysts. This leads us to replace the metal center with Cu(I), which is richer in electrons and has the same tetrahedral environment. The theoretical calculations for the new CPs family ([Cu2X2(L)]n) have corroborated the results from the Zn(II) family, further improving the compounds' stability in water. This work is complemented by an investigation into the photocatalytic efficiency of the studied compounds against persistent dyes in water. This subject can be of great international interest considering that currently, up to 80% of industrial wastewater containing dyes generated in low- and middle-income countries is dumped untreated into waterways or used directly for irrigation, which represents a wide range of direct and indirect threats to human, animal and plant health.
[Cu2I2(BPEE)]n, the compound with the lowest band-gap and highest stability, exhibits the shortest dye degradation times and a greater number of useful cycles without generated byproducts. Additionally, it shows a comparable and/or superior degradation efficiency with respect of well-studied semiconductors such as TiO2. (Table 4). Moreover, the study modifies the synthetic conditions of the compounds to make them more sustainable, achieving one-step synthesis at room temperature with reduced reaction times.
| Compounds | MO [mg L−1] | mg photocat/ml dye | MO degradation time (cycles) | Band gap (eV) |
|---|---|---|---|---|
| a Solvothermal. b Magnetic stirring. | ||||
| Cu(I) MOF,57,a | 10 | 50/250 | 120 (1) | 2.32 |
| Cu(I) MOF,58,a | 20 | 30/10 | 45 (3) | 2.49 |
| Cu(I/II) MOF,59,a | 20 | 10/10 | 90 (3) | 1.07 |
| TiO2,60,a | 2000 | 2000/80 | 240 (8) | 3.20 |
| [Cu2I2(BPEE)]nb | 3 | 2/2 | 60 (6) | 2.04 |
Data availability
The data supporting this article have been included as part of the ESI.† Experimental details. DFT calculations to explore theoretical band gaps and correlation with the experimental ones. Calculation of atomic economy (AE) and E-factor in compounds using the optimizing methodologies employing environmentally friendly procedures. Studies of the particle size of the compounds. Crystallographic data and structure analysis. Analysis of the coordination bonds distances distribution. Water and thermal stability of the study compounds. Study of the photocatalytic efficiency of [Cu2X2(BPEE)]n in the degradation of methyl blue (MB), methyl orange (MO), and tartrazine (Trz). Study of the mechanism of photocatalysis via ROS species traps in CPs (PDF). CIFs and checkcifs (PDF). CCDC 2350813-2350819 contain the ESI† crystallographic data for this paper https://www.ccdc.cam.ac.uk/deposit.Author contributions
The manuscript was written through the contributions of all authors. Andrea García-Hernán: investigation, visualization, writing review and editing. Fernando Aguilar-Galindo: visualization, investigation, formal analysis, software, writing, review and editing. Oscar Castillo: visualization, investigation, formal analysis, writing, review and editing. Pilar Amo-Ochoa: visualization, conceptualization, funding acquisition, investigation, project administration, resources, supervision, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by MCINN/AEI/10.13039/5011000011033 under the National Program of Sciences and Technological Materials (PID2022-138968NB-C21, PID2022-138968NB-C22, PID2022-138470NB-I00, TED2021-129810B-C22, and TED2021-131132B-C22) and by the Basque Government (T1722-22). The authors thank to the Single Crystal XRD laboratory, to the Universidad Autónoma de Madrid (UAM), and to the Servicio Interdepartamental de Investigación (SIdI), for the use of their scientific equipment.References
- D.-P. Bui, M.-T. Pham, H.-H. Tran, T.-D. Nguyen, T. M. Cao and V. V. Pham, ACS Omega, 2021, 6, 27379–27386 CrossRef CAS PubMed.
- A. Pavanello, A. Blasco, P. F. Johnston, M. A. Miranda and M. L. Marin, Catalysts, 2020, 10, 774 CrossRef CAS.
- K. Kočí, L. Obalová and Z. Lacný, Chem. Pap., 2008, 62, 1–9 Search PubMed.
- M. Murillo, A. García-Hernan, J. López, J. Perles, I. Brito and P. Amo-Ochoa, Catal. Today, 2023, 418, 114072 CrossRef CAS.
- S. J. Armaković, M. M. Savanović and S. Armaković, Catalysis, 2023, 13, 26–55 Search PubMed.
- C. B. Anucha, I. Altin, E. Bacaksiz and V. N. Stathopoulos, Chem. Eng. J. Adv., 2022, 10, 100262 CrossRef CAS.
- H. Atta ul, S. Muhammad, K. Samreen Gul and I. Muhammad, Adv. App, in Titanium Dioxide, ed. A. Hafiz Muhammad, ch. 4, 2022 Search PubMed.
- Y. Zhang and X. Xu, ACS Omega, 2020, 5, 15344–15352 CrossRef CAS PubMed.
- Y. Li, H. Xu, S. Ouyang and J. Ye, Phys. Chem. Chem. Phys., 2016, 18, 7563–7572 RSC.
- E. Flage-Larsen, A. Røyset, J. H. Cavka and K. Thorshaug, J. Phys. Chem. C, 2013, 117, 20610–20616 CrossRef CAS.
- S. O. Odoh, C. J. Cramer, D. G. Truhlar and L. Gagliardi, Chem. Rev., 2015, 115, 6051–6111 CrossRef CAS PubMed.
- X. P. Wu, I. Choudhuri and D. G. Truhlar, Energy Environ. Mater., 2019, 2, 251–263 CrossRef CAS.
- A. Aziz, A. R. Ruiz-Salvador, N. C. Hernández, S. Calero, S. Hamad and R. Grau-Crespo, J. Mater. Chem. A, 2017, 5, 11894–11904 RSC.
- G. Capano, F. Ambrosio, S. Kampouri, K. C. Stylianou, A. Pasquarello and B. Smit, J. Phys. Chem. C, 2020, 124, 4065–4072 CrossRef CAS.
- Z.-L. Wu, C.-H. Wang, B. Zhao, J. Dong, F. Lu, W.-H. Wang, W.-C. Wang, G.-J. Wu, J.-Z. Cui and P. Cheng, Angew. Chem., Int. Ed., 2016, 55, 4938–4942 CrossRef CAS PubMed.
- M. Hu, J. Zhang, F. Tian, W. Yan, J. Tang, Z. Chen, W. Liang, D. Shi and D. Chen, J. Mol. Struct., 2024, 1301, 137332–137338 CrossRef CAS.
- L. H. da Silveira Lacerda, E. Longo, J. Andres and M. A. San-Miguel, J. Solid State Chem., 2022, 305, 122670–122681 CrossRef.
- K. Uemura, D. Ito, J. Pirillo, Y. Hijikata and A. Saeki, ACS Omega, 2020, 5, 30502–30518 CrossRef CAS PubMed.
- J. Songkerdthong, T. Thanasarnsurapong, A. Boonchun, D. J. Harding and P. Pattanasattayavong, Mol. Syst. Des. Eng., 2024, 9, 814–825 RSC.
- M. Usman, S. Mendiratta and K. L. Lu, Adv. Mater., 2017, 29, 1605071–1605076 CrossRef PubMed.
- H. Zhang, G. Liu, L. Shi, H. Liu, T. Wang and J. Ye, Nano Energy, 2016, 22, 149–168 Search PubMed.
- J. Troyano, J. Perles, P. Amo-Ochoa, J. I. Martínez, F. Zamora and S. Delgado, CrystEngComm, 2014, 16, 8224–8231 RSC.
- K. Hassanein, O. Castillo, C. J. Gómez-García, F. Zamora and P. Amo-Ochoa, Cryst. Growth Des., 2015, 15, 5485–5494 CrossRef CAS.
- V. G. Vegas, R. Lorca, A. Latorre, K. Hassanein, C. J. Gomez-Garcia, O. Castillo, A. Somoza, F. Zamora and P. Amo-Ochoa, Angew. Chem., Int. Ed., 2017, 56, 987–991 CrossRef CAS PubMed.
- M. Murillo, R. Wannemacher, J. Cabanillas-González, U. R. Rodríguez-Mendoza, J. Gonzalez-Platas, A. Liang, R. Turnbull, D. Errandonea, G. Lifante-Pedrola, A. García-Hernán, J. I. Martínez and P. Amo-Ochoa, Inorg. Chem., 2023, 62, 10928–10939 CrossRef CAS PubMed.
- Q.-Q. Liu, M. K. Ghosh, S.-L. Cai, X.-H. Liu, L. Lu, M. Muddassir, T. K. Ghorai and J. Wang, Polyhedron, 2024, 247, 116731 CrossRef CAS.
- J. Zhao, Z. Dang, M. Muddassir, S. Raza, A. Zhong, X. Wang and J. Jin, Molecules, 2023, 28, 6848 CrossRef CAS PubMed.
- S. Wang, J. Chen, W. Kou, L. Qiu, S. Luo, X. Wang, L. Fu and T. Yang, Cryst. Growth Des., 2022, 22, 2935–2945 CrossRef CAS.
- S. J. Hong, J. Y. Lee, C. Kim, D.-H. Lee, S.-J. Kim and Y. Kim, Anal. Sci.: X-Ray Struct. Anal. Online, 2005, 21, x65–x66 CAS.
- S. J. Hong, J. H. Lee, E. Y. Lee, C. Kim, Y. Kim and S.-J. Kim, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1561–m1562 CrossRef CAS.
- J. Lin, W. Ye, M. Xie, D. H. Seo, J. Luo, Y. Wan and B. Van der Bruggen, Nat. Rev. Earth Environ., 2023, 4, 785–803 CrossRef CAS.
- C.-Y. Qin and J.-W. Xu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m717–m718 CrossRef CAS.
- M. Nagarathinam, A. Chanthapally, S. H. Lapidus, P. W. Stephens and J. J. Vittal, Chem. Commun., 2012, 48, 2585–2587 RSC.
- A. J. Blake, N. R. Brooks, N. R. Champness, P. A. Cooke, M. Crew, A. M. Deveson, L. R. Hanton, P. Hubberstey, D. Fenske and M. Schröder, CrystEngComm, 1999, 2, 181–195 CAS.
- Z.-G. Li, J.-W. Xu, H.-Q. Jia and N.-H. Hu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m205–m207 CrossRef PubMed.
- F. M. Shen and S. F. Lush, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m1071 CrossRef CAS PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- R. Pawar and A. A. Sangolkar, Comput. Theor. Chem., 2021, 1205, 113445 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- A. García-Hernán, G. Brito-Santos, E. de la Rubia, F. Aguilar-Galindo, O. Castillo, G. Lifante-Pedrola, J. Sanchiz, R. Guerrero-Lemus and P. Amo-Ochoa, Inorg. Chem., 2024, 63, 4646–4656 CrossRef PubMed.
- V. Milašinović, V. Vuković, A. Krawczuk, K. Molčanov, C. Hennig and M. Bodensteiner, IUCrJ, 2023, 10, 156–163 CrossRef PubMed.
- O. B. Berryman and D. W. Johnson, Chem. Commun., 2009, 3143–3153 RSC.
- O. B. Berryman, F. Hof, M. J. Hynes and D. W. Johnson, Chem. Commun., 2006, 506–508 RSC.
- L. Fotović and V. Stilinović, Crystals, 2021, 11, 1240 CrossRef.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- J. K. Conrad and P. R. Tremaine, Chem. Eng. Sci., 2022, 254, 117596 CrossRef CAS.
- M. Zhu and G. Pan, J. Phys. Chem. A, 2005, 109, 7648–7652 CrossRef CAS PubMed.
- H. Kumari, Sonia, Suman, R. Ranga, S. Chahal, S. Devi, S. Sharma, S. Kumar, P. Kumar, S. Kumar, A. Kumar and R. Parmar, Water, Air, Soil Pollut., 2023, 234, 349–395 CrossRef CAS PubMed.
- J. Conesa-Egea, C. D. Redondo, J. I. Martínez, C. J. Gomez-Garcia, O. Castillo, F. Zamora and P. Amo-Ochoa, Inorg. Chem., 2018, 57, 7568–7577 CrossRef CAS PubMed.
- Z. G. Li, J. W. Xu, H. Q. Jia and N. H. Hu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m205–m207 CrossRef PubMed.
- L. G. Devi, M. L. ArunaKumari, B. G. Anitha, R. Shyamala and G. Poornima, Surf. Interfaces, 2016, 1-3, 52–58 CrossRef CAS.
- L. Liu, Y. Wu, L. Ma, G. Fan, W. Gao, W. Wang and X. Ma, J. Struct. Chem., 2022, 63, 302–309 CrossRef CAS.
- T. Wen, D.-X. Zhang, J. Liu, R. Lin and J. Zhang, Chem. Commun., 2013, 49, 5660–5662 RSC.
- L. Li, L. Huang, Z.-Y. Liu, X.-J. Zhao and E.-C. Yang, Z. Anorg. Allg. Chem., 2019, 645, 623–630 CrossRef CAS.
- P. Jantawasu, T. Sreethawong and S. Chavadej, Chem. Eng. J., 2009, 155, 223–233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. DFT calculations to explore theoretical band gaps and correlation with the experimental ones. Calculation of atomic economy (AE) and E-factor in compounds using the optimizing methodologies employing environmentally friendly procedures. Studies of the particle size of the compounds. Crystallographic data and structure analysis. Analysis of the coordination bonds distances distribution. Water and thermal stability of the study compounds. Study of the photocatalytic efficiency of [Cu2X2(BPEE)]n in the degradation of methyl blue (MB), methyl orange (MO), and tartrazine (Trz). Study of the mechanism of photocatalysis via ROS species traps in CPs (PDF). CIFs and checkcifs (PDF). CCDC 2350813–2350819. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cy00969j |
| This journal is © The Royal Society of Chemistry 2024 |