
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergism between B–N atomic pair for promoting the catalytic cracking of 1,2-dichloroethane†
Yaqi
Yao
ab,
Hongying
Zhuo
*a,
Fanan
Wang
 c,
Guiyue
Bi
ad,
Jinming
Xu
*a,
Xiaofeng
Yang
a and
Yanqiang
Huang
a
c,
Guiyue
Bi
ad,
Jinming
Xu
*a,
Xiaofeng
Yang
a and
Yanqiang
Huang
a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, No, 457 Zhongshan Road, Dalian 116023, China. E-mail: zhuohongying@dicp.ac.cn; xujm@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cFujian University of Technology, Fuzhou 350118, China
dUniversity of Science and Technology of China, Hefei 230026, China
First published on 9th August 2024
Abstract
The catalytic cracking of 1,2-dichloroethane (EDC) to obtain vinyl chloride (VCM) monomer is a crucial step in the production of polyvinyl chloride (PVC). The heteroatom-doped carbon catalysts have exhibited desired performance; however, the underlying mechanism is still not fully understood. Herein, a series of B–N co-doped carbon (BNC), N-doped carbon (NC), B-doped carbon (BC) and pure carbon (C) catalysts were prepared for EDC catalytic cracking, and the synergistic mechanism between B and N was carefully investigated. The BNC catalyst exhibits prominently higher activities with an EDC conversion of 53.9% at 250 °C. Through a combination of experimental and theoretical analyses, it is rationalized that the formation of the B–N atomic pair contributes to the enhanced performance and the electronic interaction between the B–N atomic pair imparts greater basicity to the N sites, which reduces the activation energy barrier for C–H bond cleavage by 0.34 eV. The present results provide a theoretical foundation for the precise design of highly efficient non-metallic carbon-based catalysts for EDC cracking.
1. Introduction
Cracking of EDC to obtain VCM, the key monomer for the production of PVC, plays a significant role in the plastic manufacturing industry.1–3 Currently, the primary industrial route is thermal pyrolysis, which requires a high reaction temperature (500–550 °C) and pressure (25–35 bar) and suffers from low efficiency and severe coking in the device.4,5 It is thus a highly energy-consuming as well as environmentally unfriendly process. To this end, the catalytic cracking of EDC at a mild reaction condition with high efficiency has been considered the most promising replacement. Considerable research has been devoted to various catalyst systems, such as activated carbon (AC),6–8 metal oxides,9,10 zeolites,11,12 and ionic liquids.13,14 Among them, AC exhibited promising stability and selectivity, as well as considerably high conversion toward VCM within the temperature range of 210–300 °C and ambient pressure.15–17Activated carbon, which solely contains carbon atoms, does not demonstrate catalytic activity in the catalytic cracking reaction of EDC; it requires heteroatom doping to generate active sites. The most common approach is the introduction of nitrogen species,6,8,17–19 which can act as basic sites that can effectively activate the C–H bond in EDC,8,19 thereby enhancing the catalytic performance. Hence, nitrogen-doped carbon is the most extensively studied material, and improving the catalytic activity based on nitrogen doping has become a central focus of various studies.7,15 Recently, Yu et al.20 successfully obtained N, P co-doped graphene using a two-step pyrolysis method and demonstrated that the introduction of acidic P species promoted EDC cracking by altering the distribution of nitrogen species. Chen et al.21 found that B3N3-doped activated carbon, synthesized from arylacetylene compounds containing B3N3, exhibited significantly enhanced catalytic performance compared to the previously reported N-doped activated carbon. Compared to carbon's electronegativity (2.55), boron (2.04) is less electronegative and nitrogen (3.04) is more electronegative.22–24 These properties would confer on boron a significantly different catalytic profile compared to that of nitrogen. Although Chen21 has introduced B into the EDC cracking reaction, demonstrating the efficacy of B, N co-doping, the fundamental reasons for the enhanced activity were not elucidated and it remains unclear whether boron influences the electronic structure of nitrogen or participates as an acidic site in the reaction. Understanding the structure–activity relationship at the atomic/molecular level continues to be a complex and enigmatic task. Elucidating the synergistic effects between different dopants to enhance their inherent catalytic activity is of significant importance.
To this end, B, N co-doped carbon-based catalysts were synthesized using a simple hydrothermal method. Through a combination of experimental analysis and DFT calculations, we theoretically elucidate for the first time, how doped boron modulates the electronic structure of active nitrogen, thereby lowering the activation barrier for C–H bond cleavage. This study reveals an atomic-level understanding of how the second heteroatom dopant influences the catalytic activity of nitrogen-doped carbon materials. It provides a strategy for the rational development of highly active non-metallic carbon-based catalysts for EDC catalytic cracking.
2. Experimental
2.1 Synthesis
BNC was prepared by a hydrothermal method. Monohydrate glucose (6 g, 30.27 mmol), urea (1.8178 g, 30.27 mmol) and boric acid (5.616 g, 90.83 mmol) were dissolved in deionized water (130 ml). Then, the mixture was transferred into a Teflon autoclave and heated at 200 °C for 12 h. After cooling to room temperature, the solid in the mixture was separated by centrifugation, washed with deionized water and dried at 80 °C for 12 h. The obtained powder was further calcined in a tube furnace under N2 at a fixed temperature (500–1000 °C) for 3 h, donated as BNC-T (T = 500–1000 °C).For comparison, NC, BC and pure C were also prepared by the same method without boric acid, urea or both of boric acid and urea.
2.2 Characterization
X-ray diffraction (XRD) patterns were taken using a Malvern Panalytical Empyrean diffractometer operated at 40 kV, 40 mA with Cu Kα radiation source. The elemental state analysis was obtained on the ThermoFisher Escalab 250Xi instrument using the X-ray photoelectron spectroscopy (XPS) method with C 1s (284.8 eV) of the external polluting carbon as the reference peak for correction. N2 physical adsorption was conducted on Quantachrome Quadrasorb evo to evaluate the textual structure of the catalyst. Thermogravimetric and mass spectrum (TG-MS) was tested on STA449F5-Thermostar. The catalyst morphology characterization was obtained by scanning electron microscopy (SEM) and transmission electron microscopy (TEM), conducted on the JSM-7800F and HEM-2100F instruments, respectively, from JEOL Ltd.2.3 Activity evaluation
The EDC catalytic cracking performance was evaluated in a continuous-flow fixed-bed reactor under ambient pressure in a temperature range of 210–280 °C. 50 mg of the catalyst was packed into a U-shaped quartz reactor and then the reactor was purged with N2 at a rate of 5 ml min−1 to remove the air from the system. When the temperature was ramped up to the target reaction temperature, the EDC gas was introduced into the reactor in a bubbling manner by N2. Product analysis was performed at the reactor outlet with an Agilent 8890 gas chromatograph with a DB-624 column and a flame ionization detector (FID).2.4 Theoretical calculations
Spin-polarized calculations were performance-based on density functional theory (DFT) by using the Vienna ab initio simulation package (VASP5.3.5).25–27 The generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional was utilized to describe the interactions of electrons.28 The projector augmented-wave (PAW) potentials with scalar relativistic effects were used for taking into account the interaction between the valence electrons and ionic core with the nucleus. The valence electrons were designated by 2s22p2 for C, 2s22p3 for N, 2s22p1 for B, and 1s1 for H. 400 eV of energy cut-off was used for the plane wave expansion. Ground-state geometries and energies were obtained by minimizing the forces on the atoms below 0.02 eV Å−1. The transition states were acquired using the climbing image nudged elastic band (CI-NEB) method by relaxing the force below 0.05 eV Å−1, which was further confirmed by the analysis of only one imaginary frequency.A hydrogen-terminated graphene nanoribbon with an armchair edge was used as the initial model of the catalyst. It is periodic along the a-axis, as shown in Fig. S1.† To avoid the interactions between different images, a vacuum layer of 20 Å along the z direction was placed between the graphene sheets. Brillouin zone (BZ) integration was sampled over a 2 × 1 × 1 Monkhorst–Pack (MP) k-point grid. The key parameters, such as the energy cut-off, vacuum layer along the z-direction as well as the k-point grid are tested, as shown in Tables S1–S3.† Bader charge analysis was used to evaluate the electron transfer.
3. Results and discussion
3.1 Textural features
In this work, the BNC was synthesized by a simple hydrothermal method with glucose, urea, and boric acid as precursors. For comparison, three different C samples, the corresponding NC, BC and pure C, were also prepared through the same method without boric acid, urea or both of urea and boric acid, respectively. The morphology of the obtained samples is shown in Fig. 1. All the obtained materials presented a spherical morphology. Different from the particle fusion for pure C (C-700), the regularity and the surface smoothness of the spheres are significantly enhanced by the addition of boric acid (BC-700), while the surface roughness is increased in N-doped C (NC-700). Accordingly, the B and N co-doped C particles (BNC-700) reveal a much higher dispersion with smaller sizes and rough texture on the surface. It is indicated that, during the hydrothermal process, boric acid promotes the conversion of glucose to fructose, which is conducive to further hydrolysis into furan.29–31 The hydrophobicity of the polymerized product is enhanced, improving the dispersibility of the sphere. Urea is hydrolyzed into ammonia and grafted onto the glucose hydrolysis polymer, increasing the solubility of the product, which is not conducive to re-precipitation from the solution and forming a rough surface.32,33 The elemental distribution mapping of BNC (Fig. 1c and S6†) shows notably the overlapping and uniform distribution of B, N, and C, confirming the effective co-doping of B and N elements with carbon. The SEM images of the samples calcined at different temperatures (Fig. S2–S5†) further illustrate that temperature has no significant effect on the macroscopic morphology of the catalyst, but has a more pronounced impact on its microstructure and performance. | ||
| Fig. 1 (a) SEM images (b) TEM images of BNC-700, NC-700, BC-700, C-700, and (c) the elemental distribution mapping of C, B, N in BNC-700. | ||
The XRD patterns of BNC under different calcination temperatures are shown in Fig. 2a. The two broad and weak peaks at ∼26.6° and ∼44.3° correspond to (002) and (100) crystal planes of graphite, respectively, indicating that all the BNC samples are partially graphitized.34,35 With the increase of the calcination temperature, a peak shift at ∼26.6° is observed. The refined XRD with slow scan in the 15–30° range was conducted by adding silicon powder as a standard. As shown in Fig. S7,† with the increase of the calcination temperatures, a clear position shift towards the high angle direction exists for BNC compared with NC. Such lattice contraction indicates the successful introduction of B in NC,22,24 and higher calcination temperature is more conducive to the B doping.36 In the Raman spectrum (Fig. S8a†), the wide bands at around ∼1320 and ∼1580 cm−1 correspond to the D and G bands of typical carbons, respectively, representing the vibrations of C atoms within disordered graphite and the vibration of sp2-bonded carbon atoms within aromatic carbon rings.37,38 Therefore, the intensity ratio (ID/IG) is commonly used to indicate the degree of defects in carbon materials.39,40 As listed in Table 1 and Fig. S8,† increasing the calcination temperature results in a certain degree of defect increase, but there is no significant difference between BNC, NC, BC and C at the same temperature.
 | ||
| Fig. 2 (a) XRD patterns of BNC calcinated at different temperature and (b) N2 adsorption and desorption isotherm (for the convenience of observation, the offset values of the isothermal adsorption are 0 cm3 g−1 (BNC-500), 80 cm3 g−1 (BNC-600), 170 cm3 g−1 (BNC-700), 270 cm3 g−1 (BNC-800), 360 cm3 g−1 (BNC-900) and 440 cm3 g−1 (BNC-1000), respectively). | ||
| Sample | S BET (m2 g−1) | V pore (cm3 g−1) | V meso (cm3 g−1) | V micro (cm3 g−1) | I D/IG |
|---|---|---|---|---|---|
| BNC-500 | 326 | 0.40 | 0.28 | 0.12 | 0.91 |
| BNC-600 | 278 | 0.37 | 0.24 | 0.13 | 0.95 |
| BNC-700 | 190 | 0.37 | 0.29 | 0.08 | 0.97 |
| BNC-800 | 142 | 0.33 | 0.28 | 0.05 | 0.98 |
| BNC-900 | 90 | 0.31 | 0.28 | 0.03 | 0.99 |
| BNC-1000 | 94 | 0.44 | 0.42 | 0.02 | 0.97 |
The N2 adsorption–desorption isothermal analysis was also performed, as shown in Fig. 2(b). BNCs calcined under different temperatures exhibited similar H4-type isotherms41 without adsorption saturation in the high-pressure region, indicating narrow slit-like pores with no irregular structure. On the other hand, the pore size distribution listed in Table 1 (also shown in Fig. S9†) displays a remarkable decrease in the micropore volume for BNC with the increase in the calcination temperature, as well as the specific surface area. It is suggested that, during the calcination and carbonization of the polymer precursor, the carbon framework contracts and micropores expand, resulting in a decrease of the micropores and specific surface area. In comparison, the specific surface area value for the samples calcined at 700 °C decreased in the following sequence: BC > C > BNC > NC (Tables S4–S6†).
3.2 Catalytic performances
The catalytic performance of EDC catalytic cracking on the as-prepared BNC, NC, BC and C catalysts was evaluated under the reaction temperature of 250 °C at the atmospheric pressure condition, which is presented in Fig. 3 and Table S7.† All the tested catalysts exhibit high selectivity (∼99.5%) toward VCM. However, distinguished from NC, BC and C which displayed the highest EDC conversion of 16.3%, 0.369% and 0.420%, respectively (Table S7†), BNC showed considerably high catalytic performance with the highest EDC conversion reaching ∼53.9% for BNC-700, and the lowest of ∼20.8% for BNC-1000. As illustrated above, there is no significant difference in the ID/IG ratios among C, BC, NC and BNC, indicating that the defect density is not the critical factor that determines the EDC cracking activity. Also, there is no obvious link between the catalyst activity and specific surface area, as BNC-700 with a moderate specific surface area (190 m2 g−1) exhibits the highest EDC conversion. Thus, it is cautiously speculated that the co-introduction of B and N primarily accounts for the high EDC catalytic activity in our system.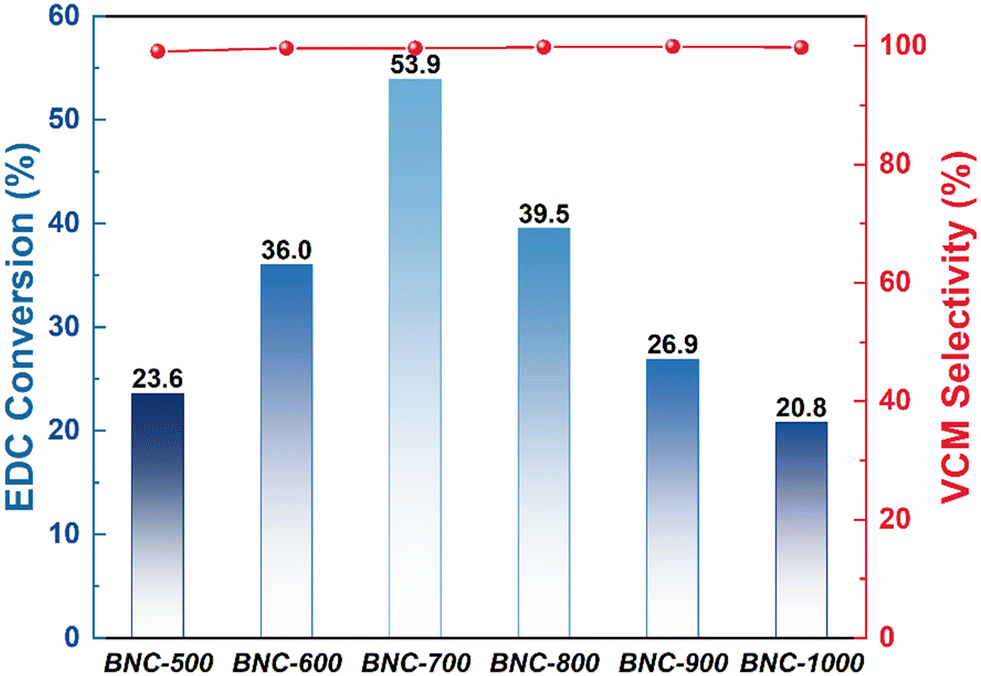 | ||
| Fig. 3 Catalytic performance of BNC for EDC cracking. Reaction conditions: T = 250 °C, P = 0.1 MPa, mcat = 50 mg. | ||
In order to confirm the significant contribution of B and N dopants in the enhancement of EDC conversion, XPS characterization was conducted. The high-resolution N 1s and B 1s spectra of the samples are shown in Fig. 4 and S10,† with the N 1s deconvolution results listed in Tables S8 and S9.† Compared with NC (Fig. S10a, Table S9†), besides the conventional N species in the high-resolution N 1s spectrum, which are pyridinic N (∼398.5 eV), pyrrolic N (∼400 eV), graphitic N (∼401 eV), and nitrogen oxide species (∼402 eV), a new species assigned to B–N (∼398 eV) for BNC was observed (Fig. 4a, Table S8†).42–45 The lowest binding energy could be attributed to the largest electron density in N for B–N species. In addition, in comparison with BC (Fig. S10b†), the additional signal peak appeared around 191 eV for BNC (Fig. 4b) further confirmed the formation of B–N bond.34,45,46 The content of pyridinic and pyrrolic N species gradually decrease while the graphitic N presents a growth trend with the increase of calcination temperature, which is consistent with the fact that graphitic nitrogen is the most thermally stable N species47 and the pyridinic and pyrrolic N may transfer to graphitic nitrogen under high temperature.48,49 At the same time, the content of B–N increases from 6.09% to 15.36% when the calcination temperature increases from 500 °C to 700 °C but decreases to 11.08% when the temperature further rises to 1000 °C. This trend matches that of their EDC conversion well. On the other hand, the EDC cracking process is accompanied by carbon deposition on the active sites. For the used catalysts, the relative content of pyridinic N and B–N decreases (Table S8†). On the other hand, the NC with N species dominated by pyridinic N displays notably higher EDC conversion (16.3%), compared to pure C (0.369%) and BC (0.420%) without N species. As previously reported, the pyridinic N might provide the basicity for the EDC cracking. It could be inferred that the basicity of the N sites in the B–N bond with enriched electrons might be strengthened, promoting the cleavage of the C–H bond. Therefore, the high conversion rate of EDC over BNC-700 could be rationally attached to the large content of B–N bonds.
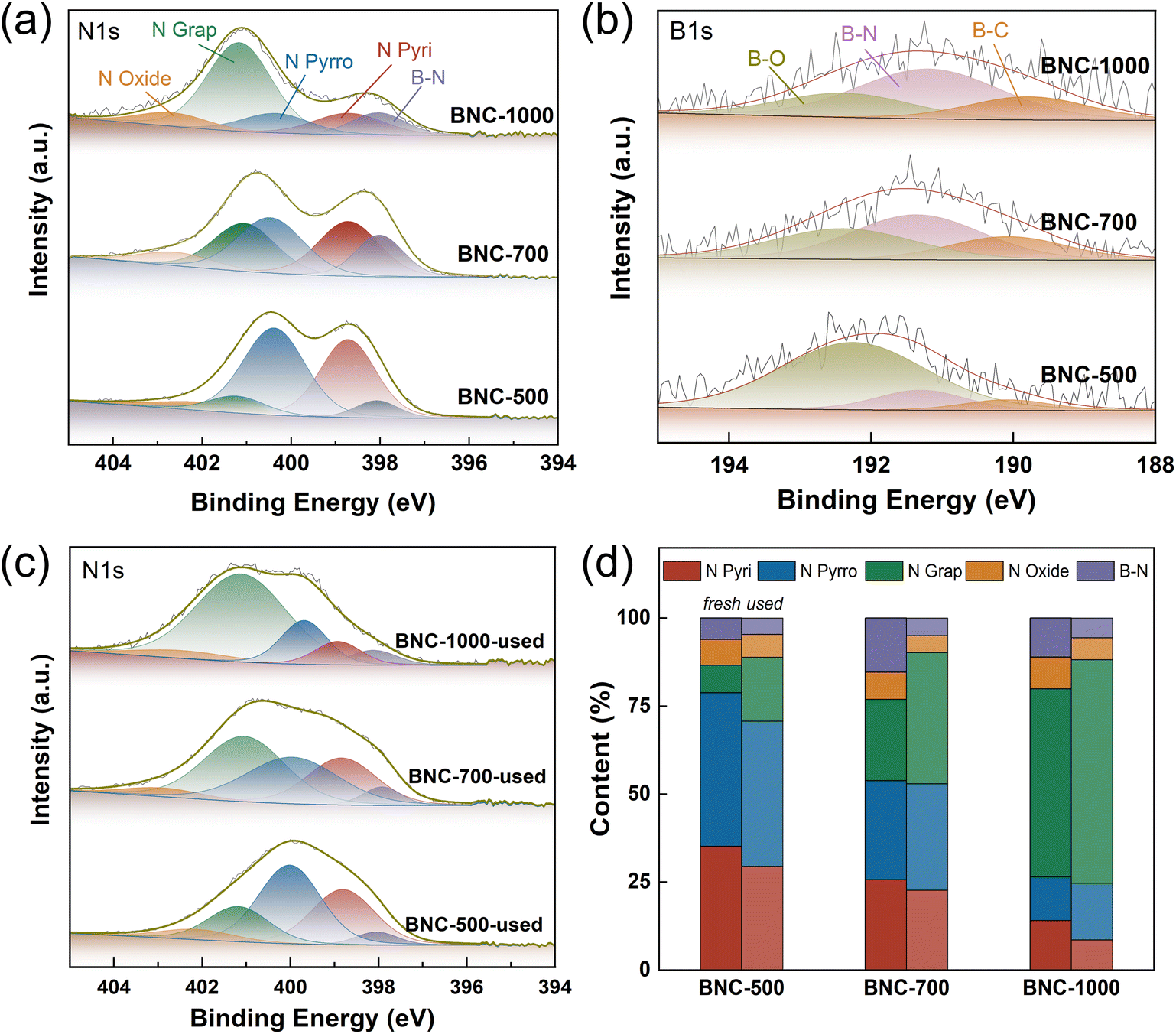 | ||
| Fig. 4 (a) XPS of N 1s and (b) B 1s of fresh BNC, and (c) XPS of N 1s of spent BNC after reaction for 20 h and (d) quantitative analysis of different N species before and after the reaction. | ||
DFT calculations were further performed to investigate the significant role of the B–N atomic pair on the enhancement of EDC conversion. Only the pyridinic N and B–N active sites are considered. The pyridinic N and B–N in the C framework were modeled by replacing the C–H atomic pair in the pristine graphene nanoribbon, as shown in the inset Fig. 5. Specifically, different doping sites of B were considered, as shown in Fig. S11,† and the formation of the B–N bond is determined with the lowest total energy, which consists of the experimental results. The EDC cracking process on pyridinic N and B–N site were calculated. The EDC molecules are firstly adsorbed on the active N site, followed by the activation of the C–H bond, where the EDC molecules are directly decomposed into VCM and HCl via an E2-like mechanism, with the cleaved H atom adsorbed on the N site and release of VCM and HCl. As can be seen, the breaking of the C–H bond accounts for the rate-determining step (RDS) for the cracking of EDC. The activation barrier (Ea) of this step on pyridinic N is 1.53 eV, which is reduced by 0.34 eV to 1.19 eV on the B–N atomic pair, highlighting the higher activity on the latter. Bader charge analysis further substantiates larger electron density (0.86 e for pyridinic N and 0.52 e for N paired with B), i.e., stronger basicity, of N paired with B, with intimate electronic interaction between B and N, contributing to the decreased energy barrier. It was reported that desorption barriers were, in general, lower than the absolute value of the adsorption energies of the adsorbents and the absolute value of adsorption energy can be used as a measure for the desorption barrier.50 Thus, according to our results, the calculated desorption barrier of VCM is 0.24 and 0.18 eV for the pyridinic N and B–N atomic pair, respectively. While, the desorption energy of HCl is 0.61 and 1.15 eV for the pyridinic N and B–N atomic pair, indicating the removal of HCl is more difficult and maybe the main reason for the deactivation of our catalysts, as discussed in the following part.
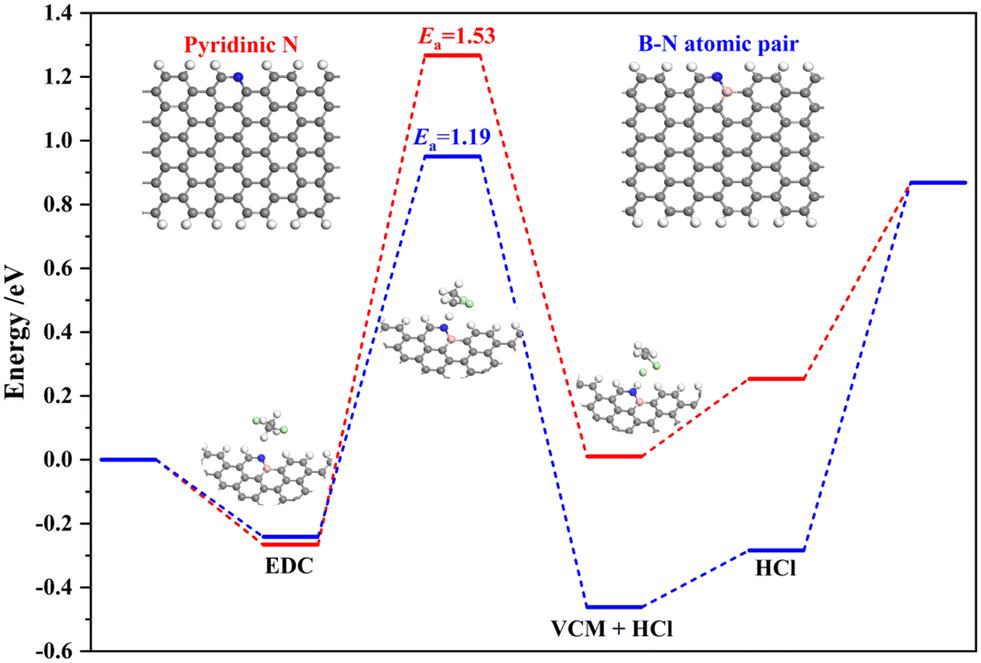 | ||
| Fig. 5 Energy profiles of EDC dehydrogenation on pyridinic N (red line) and B–N atomic pair (blue line). The insets are the models of the pyridinic N and B–N in the C framework and the EDC dehydrogenation on B–N in the C framework. The C, N, B, Cl and H atoms are depicted in gray, blue, pink, green and white, respectively. | ||
3.3 Catalyst deactivation analysis
The stability of different catalysts calcined at different temperatures is shown in Fig. 6(a). It can be seen that except for the 900 and 1000 °C calcinated catalysts, almost all the tested catalysts suffer a decreasing EDC conversion within 20 hours. BNC-700, which initially exhibits the highest activity, experiences the fastest deactivation. To analyze the reason for catalyst deactivation, N2 physisorption characterization was first performed (Fig. S12†). As can be read from Table 1 and Table S10,† the used catalysts display a decrease in the specific surface area and the total pore volume compared to the fresh ones, which could be indicative of the deposition of carbon on the catalyst surface during EDC cracking. TG-MS analysis further reveals that, within the temperature range of 150–500 °C, the used catalysts show higher weight loss compared to the fresh ones, verifying the coke deposition.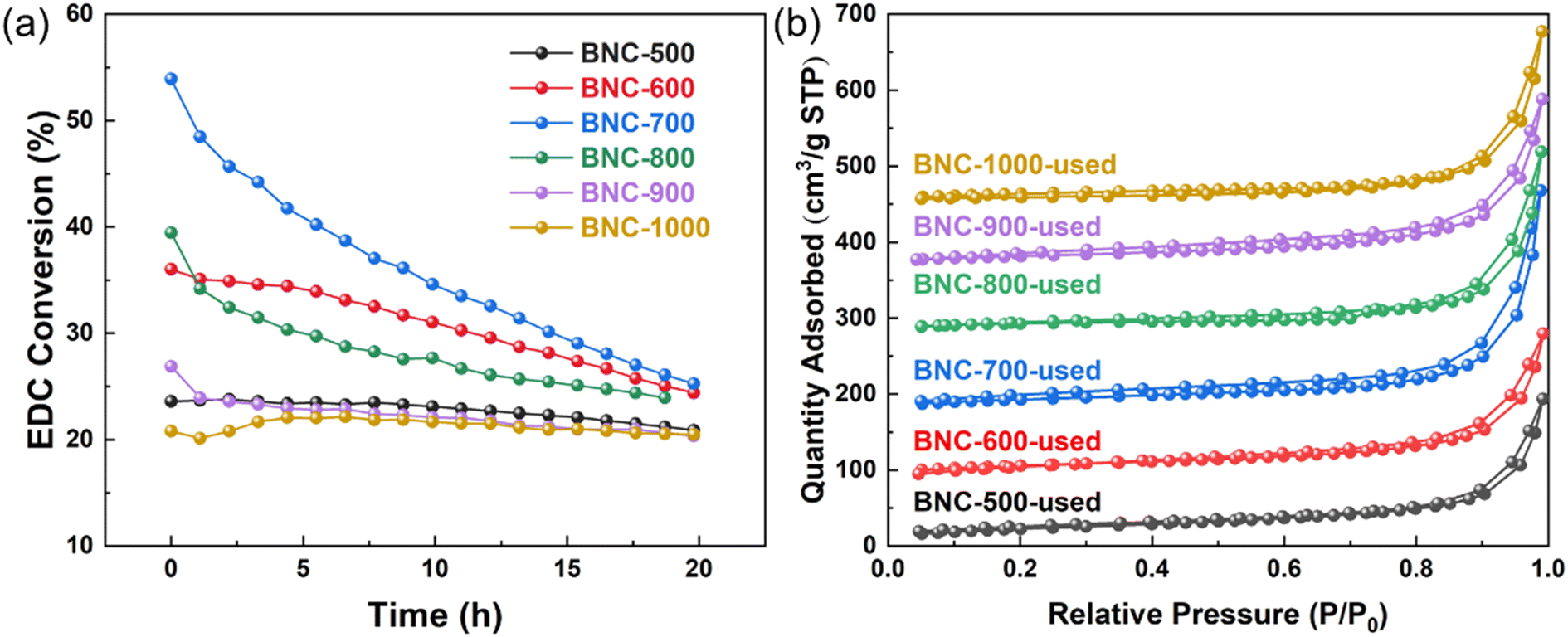 | ||
| Fig. 6 (a) Stability of BNC (b) N2 adsorption and desorption isotherm of spent BNC (for the convenience of observation, the offset values of the isothermal adsorption are 0 cm3 g−1 (BNC-500), 80 cm3 g−1 (BNC-600), 170 cm3 g−1 (BNC-700), 270 cm3 g−1 (BNC-800), 360 cm3 g−1 (BNC-900) and 440 cm3 g−1 (BNC-1000), respectively). | ||
In addition, HCl generated during the EDC dehydrochlorination could be another toxicant that is prone to adsorb over the active sites and accounts for deactivation. Given that the carbon deposition could be accelerated with the reaction temperature increasing, while the HCl-adsorption could be mitigated at high temperatures,51,52 the activity of BNC-700 was evaluated at different reaction temperatures. The variation of the activity in the range of 210–280 °C is shown in Fig. 7(a). The derivative of conversion with respect to time was plotted to obtain the deactivation rate, as shown in Fig. 7(b). Within the first 4 hours of the reaction, a noticeable trend of accelerated deactivation with increasing reaction temperature is observed, where the reaction at 280 °C exhibits the highest initial activity with the fastest deactivation rate. It can be confirmed that the carbon deposition is primarily ascribed to the deactivation in this reaction.
 | ||
| Fig. 7 (a) Stability and (b) deactivation rate of BNC-700 at different reaction temperatures. | ||
4. Conclusions
In summary, a simple hydrothermal method was employed to successfully prepare B–N co-doped C catalysts for EDC catalytic cracking. The BNC catalyst calcinated at 700 °C, achieves a maximum conversion rate of ∼53.9% for EDC conversion, significantly higher than the NC and BC counterparts prepared through the same method. Structural characterizations and DFT calculations elucidate that the B–N atomic pair and pyridinic N are both active sites for EDC cracking. The electronic interaction between B and N is identified as the key factor enhancing the activity of N species, leading to a reduction in the reaction barrier by 0.34 eV. The primary cause of catalyst deactivation is the carbon deposition covering active sites during the reaction, and higher initial activity results in faster deactivation. The B–N co-doping method proposed in this study offers a new perspective for the design of catalysts in EDC cracking, but the instability of catalysts should be carefully considered.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yaqi Yao: writing – original draft, data curation, software, writing – review and editing. Hongying Zhuo: writing – review and editing, investigation. Fanan Wang: writing – review and editing. Guiyue Bi: data curation. Jinming Xu: conceptualization, methodology, writing – review and editing. Xiaofeng Yang: writing – review and editing, funding acquisition. Yanqiang Huang: supervision, resource, funding acquisition, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the financial support from the CAS Project for Young Scientists in Basic Research (YSBR-022), the National Key Research and Development Program of China (No. 2021YFB4000700), National Natural Science Foundation of China (22008230), the Doctoral Scientific Research Foundation of Liaoning Province (2022-BS-014), DICP I202224.References
- D. Braun, Recycling of PVC, Prog. Polym. Sci., 2002, 27, 2171–2195 CrossRef CAS.
- L. Montero de Espinosa, A. Gevers, B. Woldt, M. Graß and M. A. R. Meier, Sulfur-containing fatty acid-based plasticizers via thiol–ene addition and oxidation: synthesis and evaluation in PVC formulations, Green Chem., 2014, 16, 1883–1896 RSC.
- R. H. Lin, A. P. Amrute and J. Pérez-Ramírez, Halogen-mediated conversion of hydrocarbons to commodities, Chem. Rev., 2017, 117, 4182–4247 CrossRef CAS PubMed.
- I. Mochida, T. Tsunawaki, C. Sotowa, Y. Korai and K. Higuchi, Coke produced in the commercial pyrolysis of ethylene dichloride into vinyl chloride, CHEMTECH, 1996, 35, 3803–3807 CAS.
- Q. Zhang, W. Huang, W. H. Cui, X. Z. Dong, G. Y. Liu, Y. P. Xu and Z. M. Liu, Optimization and modification of ZSM-5 zeolite for efficient catalytic cracking of 1,2-dichloroethane, Mol. Catal., 2023, 545, 113189 CrossRef CAS.
- I. Mochida, Y. Yasumoto, Y. Watanabe, H. Fujitsu, Y. Kojima and M. Morita, Catalytic dehydrochlorination of 1,2-dichloroethane into vinyl chloride over polyacrylonitrile-based active carbon fiber (PAN-ACF), Chem. Lett., 1994, 23, 197–200 CrossRef.
- J. M. Xu, X. C. Zhao, A. Q. Wang and T. Zhang, Synthesis of nitrogen-doped ordered mesoporous carbons for catalytic dehydrochlorination of 1,2-dichloroethane, Carbon, 2014, 80, 610–616 CrossRef CAS.
- C. Sotowa, Y. Kawabuchi and I. Mochida, Catalytic dehydrochlorination of 1,2-dichloroethane over pyridine deposited pitch-based active carbon fiber, Chem. Lett., 1996, 25, 967–968 CrossRef.
- A. W. A. M. van der Heijden, A. J. M. Mens, R. Bogerd and B. M. Weckhuysen, Dehydrochlorination of intermediates in the production of vinyl chloride over lanthanum oxide-based catalysts, Catal. Lett., 2008, 122, 238–246 CrossRef CAS.
- M. Scharfe, P. A. Lira-Parada, V. Paunović, M. Moser, A. P. Amrute and J. Pérez-Ramírez, Oxychlorination–dehydrochlorination chemistry on bifunctional ceria catalysts for intensified vinyl chloride production, Angew. Chem., Int. Ed., 2016, 55, 3068–3072 CrossRef CAS PubMed.
- R. Lin, A. P. Amrute and J. Perez-Ramirez, Halogen-mediated conversion of hydrocarbons to commodities, Chem. Rev., 2017, 117, 4182–4247 CrossRef CAS PubMed.
- A. S. Shalygin, L. V. Malysheva and E. A. Paukshtis, Mechanism of 1,2-dichloroethane dehydrochlorination on the acid sites of oxide catalysts as studied by IR spectroscopy, Kinet. Catal., 2011, 52, 305–315 CrossRef CAS.
- D. Glas, J. Hulsbosch, P. Dubois, K. Binnemans and D. E. De Vos, End-of-life treatment of poly(vinyl chloride) and chlorinated polyethylene by dehydrochlorination in ionic liquids, ChemSusChem, 2014, 7, 610–617 CrossRef CAS PubMed.
- T. Boudewijns, M. Piccinini, P. Degraeve, A. Liebens and D. De Vos, Pathway to vinyl chloride production via dehydrochlorination of 1,2-dichloroethane in ionic liquid media, ACS Catal., 2015, 5, 4043–4047 CrossRef CAS.
- W. Zhao, M. X. Sun, H. Y. Zhang, Y. Z. Dong, X. Y. Li, W. Li and J. L. Zhang, Catalytic dehydrochlorination of 1,2-dichloroethane to produce vinyl chloride over N-doped coconut activated carbon, RSC Adv., 2015, 5, 104071–104078 RSC.
- H. Zhao, S. Y. Chen, M. T. Guo, D. Zhou, Z. B. Shen, W. J. Wang, B. Feng and B. Jiang, Catalytic Dehydrochlorination of 1,2-Dichloroethane into Vinyl Chloride over Nitrogen-Doped Activated Carbon, ACS Omega, 2019, 4, 2081–2089 CrossRef CAS PubMed.
- Q. Yu, X. L. Mou, L. Y. Guo, Z. P. Chen, R. H. Lin and Y. J. Ding, Triton X-100-directed synthesis of carbon nitride and nitrogen-doped carbon for ethylene dichloride dehydrochlorination, Carbon, 2022, 196, 110–119 CrossRef CAS.
- Z. B. Shen, Y. J. Han, Y. Liu, Y. J. Qin, P. Xing, H. Zhao and B. Jiang, Understanding Surface Basic Sites of Catalysts: Kinetics and Mechanism of Dehydrochlorination of 1,2-Dichloroethane over N-Doped Carbon Catalysts, Catalysts, 2020, 10, 707 CrossRef CAS.
- X. Sun, Y. C. Qin, Q. Li, X. Liu, Z. Liu, L. J. Song and Z. L. Sun, Supported structure-controlled graphitic carbon nitride catalyst for dehydrochlorination of 1,2-dichloroethane, Catal. Sci. Technol., 2018, 8, 5334–5343 RSC.
- H. N. Yu, X. F. Yang, H. B. Yang, J. M. Xu and Y. Q. Huang, Phosphorus and nitrogen co-doped graphene for catalytic dehydrochlorination of 1,2-dichloroethane, Chin. J. Chem. Eng., 2023, 64, 149–155 CrossRef CAS.
- C. Chen, Z. B. Shen, Y. P. Zhu, F. Wang, B. Jiang and H. M. Qi, Construction of activated carbon-supported B3N3 doped carbon as metal-free catalyst for dehydrochlorination of 1,2-dichloroethane to produce vinyl chloride, RSC Adv., 2021, 11, 183–191 RSC.
- B. Chang, L. L. Li, D. Shi, H. H. Jiang, Z. Z. Ai, S. Z. Wang, Y. L. Shao, J. X. Shen, Y. Z. Wu, Y. L. Li and X. P. Hao, Metal-free boron carbonitride with tunable boron Lewis acid sites for enhanced nitrogen electroreduction to ammonia, Appl. Catal., B, 2021, 283, 119622 CrossRef CAS.
- X. Y. Ma, J. J. Du, H. Sun, F. H. Ye, X. Wang, P. F. Xu, C. G. Hu, L. P. Zhang and D. Liu, Boron, nitrogen co-doped carbon with abundant mesopores for efficient CO2 electroreduction, Appl. Catal., B, 2021, 298, 120543 CrossRef CAS.
- C. Van Nguyen, S. Lee, Y. G. Chung, W. H. Chiang and K. C. W. Wu, Synergistic effect of metal-organic framework-derived boron and nitrogen heteroatom-doped three-dimensional porous carbons for precious-metal-free catalytic reduction of nitroarenes, Appl. Catal., B, 2019, 257, 117888 CrossRef CAS.
- G. Kresse and J. Furthmiiller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- T. Ståhlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Metal-free dehydration of glucose to 5-(hydroxymethyl)furfural in ionic liquids with boric acid as a promoter, Chem. – Eur. J., 2011, 17, 1456–1464 CrossRef PubMed.
- D. H. Lukamto, P. Wang and T. P. Loh, Catalytic conversion of inert carbohydrates into platform chemical 5-hydroxymethylfurfural using arylboronic acids, Asian J. Org. Chem., 2013, 2, 947–951 CrossRef CAS.
- B. J. Graham and R. T. Raines, Emergent organoboron acid catalysts, J. Org. Chem., 2022, 89, 2069–2089 CrossRef PubMed.
- A. Jung, S. Han, T. Kim, W. J. Cho and K. H. Lee, Synthesis of high carbon content microspheres using 2-step microwave carbonization, and the influence of nitrogen doping on catalytic activity, Carbon, 2013, 60, 307–316 CrossRef CAS.
- S. S. Li, F. Liang, J. K. Wang, H. J. Zhang and S. W. Zhang, Preparation of mono-dispersed carbonaceous spheres via a hydrothermal process, Adv. Powder Technol., 2017, 28, 2648–2657 CrossRef CAS.
- X. H. Chen, P. C. Ye, H. Y. Wang, H. Huang, Y. J. Zhong and Y. Hu, Discriminating active B-N sites in coralloidal B, N dual-doped carbon nano-bundles for boosted Zn-ion storage capability, Adv. Funct. Mater., 2023, 33, 2212915 CrossRef CAS.
- T. K. Zhao, Y. N. Liu and J. W. Zhu, Temperature and catalyst effects on the production of amorphous carbon nanotubes by a modified arc discharge, Carbon, 2005, 43, 2907–2912 CrossRef CAS.
- E. Iyyamperumal, S. Y. Wang and L. M. Dai, Vertically aligned BCN nanotubes with high capacitance, ACS Nano, 2012, 6, 5259–5265 CrossRef CAS PubMed.
- K. Ghosh, M. Kumar, T. Maruyama and Y. Ando, Micro-structural, electron-spectroscopic and field-emission studies of carbon nitride nanotubes grown from cage-like and linear carbon sourc, Carbon, 2009, 47, 1565–1575 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Raman spectra of graphite oxide and functionalized graphene sheets, Nano Lett., 2007, 8, 36–41 CrossRef PubMed.
- S. S. Shang, P. P. Chen, L. Y. Wang, Y. Lv, W. X. Li and S. Gao, Metal-free nitrogen- and boron-codoped mesoporous carbons for primary amides synthesis from primary alcohols via direct oxidative dehydrogenation, ACS Catal., 2018, 8, 9936–9944 CrossRef CAS.
- C. H. Choi, S. H. Park and S. I. Woo, Binary and ternary doping of nitrogen, boron, and phosphorus into carbon for enhancing electrochemical oxygen reduction activity, ACS Nano, 2012, 6, 7084–7091 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- Y. Wang, Y. Y. Shao, D. W. Matson, J. H. Li and Y. H. Lin, Nitrogen-doped graphene and its application in electrochemical biosensing, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- Q. T. Zeng, G. X. Yang, J. H. Chen, Q. Zhang, Z. T. Liu, B. H. Qin and F. Peng, Effects of nitrogen and oxygen on electrochemical reduction of CO2 in nitrogen-doped carbon black, Carbon, 2023, 202, 1–11 CrossRef CAS.
- H. T. Zou and J. Z. Chen, Efficient and selective approach to biomass-based amine by reductive amination of furfural using Ru catalyst, Appl. Catal., B, 2022, 309, 12162 CrossRef.
- H. L. Li, B. H. Ren, W. W. Liu, L. Jing, R. Y. Tay, S. H. Tsang, L. Ricardez-Sandoval, A. P. Yu and E. H. T. Teo, Boron nanosheets induced microstructure and charge transfer tailoring in carbon nanofibrous mats towards highly efficient water splitting, Nano Energy, 2021, 88, 106246 CrossRef CAS.
- T. Yuan, L. Y. Sun, Z. W. Wu, R. Wang, X. Cai, W. Lin, M. F. Zheng and X. C. Wang, Mild and metal-free Birch-type hydrogenation of (hetero)arenes with boron carbonitride in water, Nat. Catal., 2022, 5, 1157–1168 CrossRef CAS.
- Q. Wu, W. Li, J. Tan, Y. J. Wu and S. X. Liu, Hydrothermal carbonization of carboxymethylcellulose: One-pot preparation of conductive carbon microspheres and water-soluble fluorescent carbon nanodots, Chem. Eng. J., 2015, 266, 112–120 CrossRef CAS.
- S. W. Han, J. Bang, S. H. Ko and R. Ryoo, Variation of nitrogen species in zeolite-templated carbon by low-temperature carbonization of pyrrole and the effect on oxygen reduction activity, J. Mater. Chem. A, 2019, 7, 8353–8360 RSC.
- Y. Ito, C. Christodoulou, M. V. Nardi, N. Koch, H. Sachdev and K. Müllen, Chemical vapor deposition of n-doped graphene and carbon films: the role of precursors and gas phase, ACS Nano, 2014, 8, 3337–3346 CrossRef CAS PubMed.
- B. Yang, R. Burch, C. Hardacre, G. Headdock and P. Hu, Origin of the increase of activity and selectivity of nickel doped by Au, Ag, and Cu for acetylene hydrogenation, ACS Catal., 2012, 2, 1027–1032 CrossRef CAS.
- W. Liu, M. H. Qi, X. Y. Chu, S. Y. Peng and D. Y. Han, Investigation of adsorption-diffusion behaviors of elementary O2, CO2, and N2 in coal particles: influence from temperature, Environ. Sci. Pollut. Res., 2023, 30, 78619–78631 CrossRef CAS PubMed.
- C. Guan, S. M. Liu, C. W. Li, Y. Wang and Y. X. Zhao, The temperature effect on the methane and CO2 adsorption capacities of Illinois coal, Fuel, 2018, 211, 241–250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00774c |
| This journal is © The Royal Society of Chemistry 2024 |