
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic dehalogenation with activated borane, a porous borane cluster polymer†
Abhishek
Udnoor
ab,
Béla
Urbán
c,
Karel
Škoch
a,
Jan
Hynek
a,
Michal
Horáček
 c,
Martin
Lamač
*c and
Jan
Demel
*a
c,
Martin
Lamač
*c and
Jan
Demel
*a
aInstitute of Inorganic Chemistry of the Czech Academy of Sciences, Husinec-Řež 1001, 250 68, Řež, Czech Republic. E-mail: demel@iic.cas.cz
bDepartment of Inorganic Chemistry, University of Chemistry and Technology Prague, Technická 5, 166 28 Praha 6, Czech Republic
cJ. Heyrovsky Institute of Physical Chemistry of the Czech Academy of Sciences, Dolejškova 2155, 182 00, Prague 8, Czech Republic. E-mail: lamac@jh-inst.cas.cz
First published on 22nd July 2024
Abstract
Activated borane (ActB), a metal- and halogen-free porous borane cluster polymer with a significant Lewis acidity, has been successfully used as a heterogeneous catalyst for hydrodehalogenation reactions of aliphatic fluorides and chlorides using triethylsilane as a reductant. In analogy to known homogeneous systems, full dehalogenation of organohalides is achieved with a predominant formation of Friedel–Crafts alkylation products in aromatic reaction media. Importantly, the herein described material is robust, tolerates elevated reaction temperatures and can be re-used, while reaching a turnover number (TON) of up to 5190. These features make it an attractive candidate for a sustainable disposal of halogenated pollutants by heterogeneous catalysis.
Introduction
Organic halides are widely used in many applications of modern industry. Unfortunately, many of them are harmful to human health as well as the environment and because of the strong C–Cl and C–F bonds many organic fluorides and chlorides are persistent pollutants.1 For these reasons, new strategies for dechlorination and defluorination are highly sought after. Several approaches have been developed for the disposal of organic halides, such as incineration, enzymatic or microbial breakdown,2 chemical oxidation,3 photochemical decomposition,4 ultrasonic treatment,5 electrolysis,6 and catalytic dehalogenation.7Catalytic dehalogenation is recognized as a cost-effective and ecologically benign technology for dealing with pollutants that can be transformed into significantly less dangerous species or potentially valuable chemicals. Traditionally, C–X bonds were cleaved using low valent transition metal catalysts.8 This is done through oxidative addition of the C–X bond to the metal centre. However, the reactivity decreases in the order C–I > C–Br > C–Cl > C–F, which makes the most persistent pollutants, organofluorides, least reactive. Additionally, in order to increase the activity of transition metal-based catalysts various phosphine or carbene ligands are used to stabilize the low valent active species. This is rather challenging when it comes to the design of heterogeneous catalysts that could be used in flow reactors or otherwise easily separated from the reaction mixture.9
In recent years, transition metal free catalysis has gained significant interest. The impetus being not only the price of transition metals, but also the environmental aspects related with their recovery and refining. Main group element based strong Lewis acids are perceived as an alternative,10 while various approaches to their heterogenization have been developed.11 Strong molecular boron Lewis acids, such as B(C6F5)3 (commonly abbreviated as BCF), have been shown to readily hydrodefluorinate primary, secondary and tertiary alkyl fluorides in the presence of triethylsilane.12 More recently, the BCF-catalysis has been applied to selective C–F bond functionalisations.10,13 C–F bond cleavage in gem-difluorocyclopropenes enabled an elegant preparation of substituted cyclopropane derivatives by using silylated nucleophiles. It has been demonstrated that the fluoroborate anion [FB(C6F5)3]− is a plausible intermediate in these reactions.14 In a related work, Paradies et al. have utilised benzylic fluorides as substrates for C–C bond forming reactions under BCF catalysis.15
However, –CF3 groups proved to be challenging substrates for BCF.16 To overcome this limitation, BCF needs to be combined with various bases, thus employing the frustrated Lewis pairs (FLP) mechanism.17 These systems, however, require a proper selection of a matching Lewis base.18 In parallel, Ozerov et al. showed that triethylsilylium (Et3Si+) ions generated in situ from Et3SiH and Ph3C[B(C6F5)4] can defluorinate even –CF3 groups using triethylsilane as the hydride source.19 The borate anion proved to be unstable over several catalytic cycles, for this reason systems employing closo-borane anions such as halogenated CB11H12− and B12H122− were developed.20 Analogously, silylium ions can be used for catalytic dechlorination,20a or utilized in tandem hydrodechlorination/Friedel–Crafts alkylation reaction,21 however, silylium ions are very unstable and commonly are generated in situ.22 Alternatively 4-coordinate phosphonium salts or 3-coordinate dications can act as potent Lewis acids inducing defluorination of fluoroalkanes.17b–d,18,23
Strong heterogeneous Lewis acids were also tested for dehalogenation reactions. For example, amorphous aluminium chlorofluoride in the presence of Et3SiH was shown to form a silylium-ion-like surface species capable of defluorination.24 In a similar way, silylium species generated on the surface of sulphated zirconia were active defluorination catalysts.25 Moreover, materials such as aluminium chlorofluoride can be also used for transhalogenation reactions,26 and related zirconium chlorofluoride can be used for HF elimination and subsequent HF addition to a triple bond.27
Unfortunately, the majority of the abovementioned molecular as well as heterogeneous Lewis acids contain fluorine and/or chlorine atoms as electron withdrawing groups making those systems controversial for the use of dehalogenation. For this reason, we turned our attention to halogen-free boron-based solid Lewis acids.
An example of such materials is activated borane (hereafter abbreviated as ActB), which is a porous polymer formed by co-thermolysis of decaborane(14) (nido-B10H14) and an organic solvent.28 In this report, we describe the ability of ActB to promote dehalogenation reactions (Scheme 1).
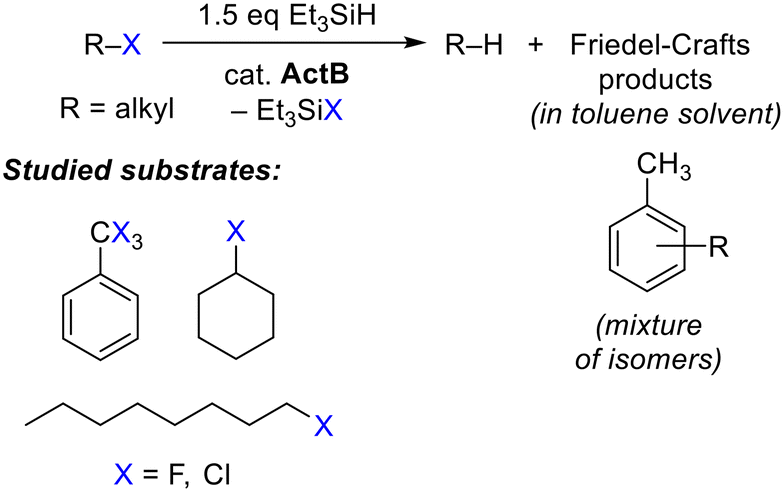 | ||
| Scheme 1 Hydrodehalogenation (HDH) of organohalides with triethylsilane in the presence of ActB – a general reaction scheme and substrate scope. | ||
Results and discussion
ActB was prepared analogously as described earlier,28b in short, a mixture of 2 g of nido-B10H14 and 50 mL of dry toluene was heated at 250 °C in Ar-filled autoclave for 24 h. In our recent study, we showed that ActB contains strong Lewis acid sites capable of activation of Si–H bond in triethylsilane. Preliminary experiments supported our hypothesis that ActB can be used for hydrodefluorination of α,α,α,-trifluorotoluene (PhCF3) at elevated temperatures. For this reason, we employed a microwave reactor as a convenient tool for performing reactions at elevated pressures and temperatures up to 200 °C.Initial screening was performed using PhCF3 at 140 °C with a reaction time of 1 h. We varied the amount of catalyst, silane, and the solvent, see Table 1. As expected, increasing the amount of ActB and excess of silane resulted in higher conversion. The use of (EtO)3SiH or PMHS (polymethylhydrosiloxane) as a substitute for Et3SiH did not yield satisfactory results. It shall be noted that in the complete absence of the catalyst and/or silane, no conversion of the substrate was observed. Toluene was found to be the best solvent for the reaction, while the solvent molecules were involved in scavenging the carbocations produced by the fluoride abstraction. Such Friedel–Crafts (FC) reactivity involving the aromatic solvents leads to a mixture of isomeric arene alkylation products (Scheme 1). This, together with the identification of Et3SiF and, to a small extent also Et2SiF2, (by 19F and 1H NMR) as the fluorine-containing product, is an additional confirmation that the mechanism (Scheme 2) proceeds similarly to homogeneous Lewis acid catalysts, like BCF.19 The use of aliphatic chlorinated solvent, 1,2-dichloroethane, resulted in comparable reaction rates to toluene, while benzylated toluenes originating solely from the PhCF3 substrate were detected as the major reaction products.
| Solvent | Conversionb (%) |
|---|---|
| Toluene | 26 |
| Benzene | 13 |
| Hexane | 5 |
| Dichloromethane | 2 |
| 1,2-Dichloroethane | 25 |
| 1,4-Difluorobenzene | 9 |
| Et3SiH (mmol) | Conversionc (%) |
|---|---|
| None | 0 |
| 1.5 | 9 |
| 4.5 | 26 |
| 9.0 | 38 |
| Neat Et3SiH | 58 |
| 4.5 eq. (EtO)3SiH instead of Et3SiH | 0 |
| 4.5 eq. PMHS instead of Et3SiH | 0 |
| Catalyst (mg) | Conversiond (%) |
|---|---|
| a Total conversion of C–F bonds determined by 19F NMR; conditions as described below. b PhCF3 (1 mmol), Et3SiH (4.5 mmol), ActB (20 mg), indicated solvent (2 mL), 140 °C, 1 h. c PhCF3 (1 mmol), Et3SiH (indicated amount), ActB (20 mg), toluene (2 mL), 140 °C, 1 h. d PhCF3 (1 mmol), Et3SiH (4.5 mmol), ActB (indicated amount), toluene (2 mL), 140 °C, 1 h. | |
| None | 0 |
| 20 | 26 |
| 40 | 53 |
| 60 | 78 |
 | ||
| Scheme 2 Proposed reaction mechanism of hydrodehalogenation (HDH) using ActB. | ||
In order to demonstrate the synthetic utility of the observed defluorination/arylation reaction sequence, the main monobenzylated FC product, benzyltoluene 1 (Scheme 3), was isolated in a 67% yield as a mixture of 2- and 4-isomers (ratio ca. 2/3). Toluene was also replaced by other aromatic substrates serving as solvents for the reaction and the corresponding monobenzylated products 2–4 were isolated as summarized in Scheme 3. As evident, for benzene, p-xylene, and mesitylene, respectively, a single-isomer product was formed. The low isolated yield of diphenylmethane 2 could be explained by a more pronounced concurrent formation of multiply alkylated or oligomeric products.
 | ||
| Scheme 3 Reaction of PhCF3 with Et3SiH and selected aromatic substrates. Reaction conditions: PhCF3 (1 mmol), Et3SiH (4.5 mmol), ActB (20 mg), indicated substrate (2 mL), 140 °C, 16 h. Yields are of isolated products after column chromatography. | ||
For further testing we defined uniform conditions using 1 mmol of a halogenated substrate, 20 mg of ActB, 50% excess of Et3SiH and toluene as a solvent. In order to assess the catalytic activity of ActB, we screened aliphatic (1-fluorooctane), cyclic aliphatic (fluorocyclohexane) and benzylic (PhCF3) substrates (Scheme 1, Fig. 1–4).
 | ||
| Fig. 1 Dehalogenation of PhCX3 (conversions determined by 19F NMR for PhCF3 and by GC for PhCCl3); (a) temperature dependence of the C–X bond cleavage conversion (reaction time 1 h), (b) kinetic profile of the reactions performed at 140 °C, (c) monitoring of the reaction progress by 19F NMR spectroscopy. | ||
 | ||
| Fig. 2 Dehalogenation of 1-halooctanes (conversions determined by 19F NMR or GC); (a) temperature dependence of the C–X bond cleavage conversion (reaction time 1 h), (b) kinetic profile of the reactions performed at 140 °C. | ||
 | ||
| Fig. 3 Dehalogenation of halocyclohexanes (conversions determined by GC); (a) temperature dependence of the C–X bond cleavage conversion (reaction time 1 h), (b) kinetic profile of the reactions performed at the indicated temperatures. | ||
 | ||
| Fig. 4 Dehalogenation of 1-fluorocyclohexane in the absence of Et3SiH (conversion determined by GC); (a) temperature dependence of the C–F bond cleavage conversion (reaction time 1 h), (b) kinetic profile of the reaction performed at 60 °C. | ||
From the temperature optimization (Fig. 1a) and kinetic profiles (Fig. 1b) of dehalogenation of PhCF3 and PhCCl3 it is clear that the reaction proceeds very slowly below 120 °C and dechlorination proceeds faster than defluorination, which is in line with previous observations done by Ozerov et al.20a For both PhCF3 and PhCCl3, the reaction is complete after 1 h at 180 and 200 °C, respectively, or after 6–8 hours at 140 °C. Interestingly, for the defluorination of PhCF3, we did not observe mono- or difluorotoluene as intermediates during the dehalogenation, however, in the case of PhCCl3 after 1 h reaction at 30% overall conversion ca. 7% of dichlorotoluene was detected, which was later fully converted to the dehalogenated products. This is also in accordance with previous studies suggesting that once the first halogen atom is removed the substrate becomes much more reactive.19 The main products of the dehalogenation reactions were fluoro- and chloro-silanes along with the FC products. When the reaction of PhCF3 was performed in benzene as described in Scheme 3, the GC yield of toluene as a simple product of reductive dehalogenation was only around 7% (see the ESI† for details).
The rate of dehalogenation for 1-fluorooctane was faster than for 1-chlorooctane (Fig. 2), which is likely due to the different affinity of the Lewis acid to fluoride and chloride anion, i.e., a hard acid prefers a lighter halogen. This is consistent with the observations made for homogeneous silylium-based systems.20a The conversion of halocyclohexane (Fig. 3) was significantly faster, with fluorocyclohexane the reaction was by far the fastest of all the studied ones, being complete after only 1 h at 100 °C. In this case, however, we observed an almost exclusive formation of cyclohexene (92% cyclohexene and 8% of FC reaction products; determined by GC) and etching of the glass reaction vessel. Formation of the unsaturated product suggested that in this case the formed carbocation follows a dehydrofluorination pathway eliminating HF, which is then responsible for the glass etching. To understand the role of Et3SiH in the reaction mixture we performed the reaction without silane, heating only fluorocyclohexane with ActB in toluene. Surprisingly, the reaction also led to the full conversion after 1 h at 100 °C (Fig. 4) and exhibited the same distribution of reaction products (i.e., >90% of cyclohexene). Motivated by those results we tested also chlorocyclohexane, 1-fluorooctane, and 1-chlorooctane for the reaction without silane, however, in all cases the reaction did not proceed even at temperatures as high as 200 °C.
Analogous HF elimination from alkyl fluorides was previously reported by Braun et al. using heterogeneous Lewis acid catalysts based on aluminium or zirconium chlorofluoride.24b,27,29 Furthermore, they used this reactivity to perform hydrofluorination reactions of various alkyne substrates by the in situ formed HF.27
To establish the limitations of the studied catalytic system, we also tested chloro- and fluoro-benzene as substrates, in both cases, however, the reaction did not proceed even at 200 °C, confirming that aromatic C–halogen bonds are inert under these conditions. Similarly, we tested perfluorinated substrates that are considered to be persistent pollutants. In the case of perfluorooctanoic acid at 200 °C we observed the formation of Et3SiF, however, the catalyst was deactivated rapidly and the overall conversion was below 1%. In the case of perfluorooctane no reaction was observed.
One of the main advantages of heterogeneous catalysts is the possibility of catalyst recycling. To test the reusability of ActB for defluorination of PhCF3, we consecutively added portions of the substrate and silane (all under Ar atmosphere) after the complete conversion was reached (8 h reaction time) at 140 °C (Fig. 5). During six consecutive reactions, a full conversion was achieved, only in the seventh run we observed a slight decrease in the activity. This reveals a significant robustness of the catalyst that can convert over 20 mmol of C–F bonds using only 20 mg of the catalyst, reaching overall turnover number (TON) of 5190 (based on Si–F bond formation, see below for the assessment of the concentration of active centres). It should be noted that when ActB recycling was studied in hydrosilylation and deoxygenation reaction, the catalytic activity was gradually declining.28b We believe that this was caused by the presence of oxygen-containing species that over the course of the reaction poisoned the Lewis acid centres while fluorosilanes and aromatic hydrocarbon products are inert and therefore are not interfering with the catalytic centres.
 | ||
| Fig. 5 Catalyst feeding experiment – defluorination of PhCF3: 1 mmol of PhCF3 and 1.5 eq. of Et3SiH were added in each consecutive run and the conversion of the substrate was determined after 8 h at 140 °C. | ||
In order to assess the concentration of active sites in ActB catalyst we adapted a procedure described by M. Conley et al. which consists in measuring the catalytic activity in the presence of titrated amounts of triethylphosphine oxide (TEPO). Because TEPO forms a strong adduct with strong Lewis acids its presence decreases the amount of available catalytic centres. We selected defluorination of PhCF3 done at 140 °C for 2 h in the presence of increasing amounts of TEPO and the C–F bond cleavage conversion was determined. As can be seen from Fig. 6, the decrease in activity is not linear. We fitted two lines which suggest the presence of two types of catalytic centres. This analysis indicates that the concentration of catalytic centres is approximately 0.2 mmol g−1.
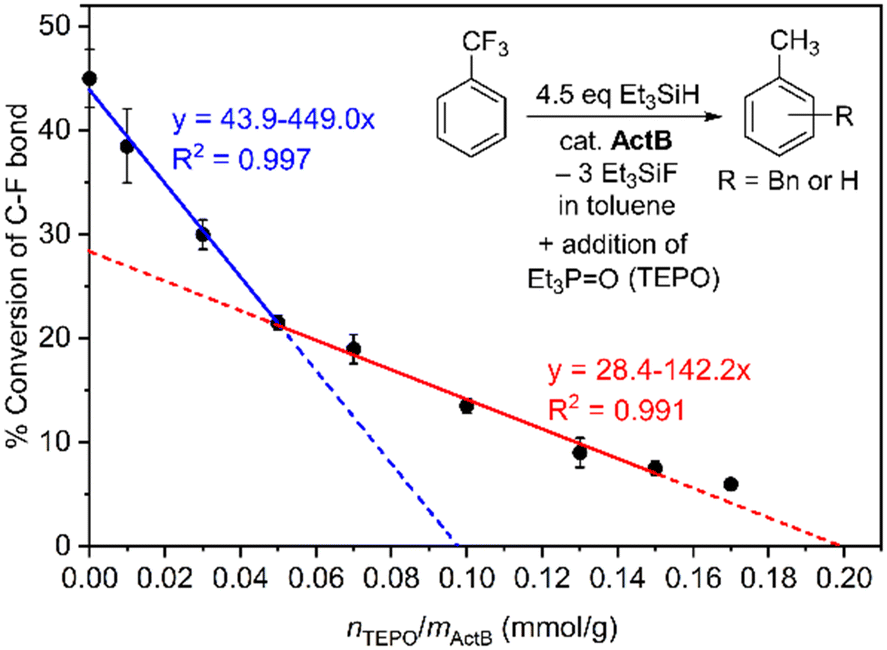 | ||
| Fig. 6 C–F bond conversion in the PhCF3 dehalogenation catalysed by ActB in the presence of increasing amounts of TEPO. Conditions: 140 °C, 2 h, toluene solvent. | ||
Characterisation of the ActB after catalytic reaction
FT-IR of the ActB catalyst recovered after the reaction of PhCF3 at 140 °C and 8 h revealed that the main structural features of the material were retained (Fig. 7a), including the B–H vibration band at 2549 cm−1. Presence of some additional aromatic organic material in the catalyst was also detected (aromatic C–H stretching at 3026 cm−1 and C–C vibration band at 1494 cm−1). | ||
| Fig. 7 (a) FT-IR spectra of ActB before (segmented line) and after (solid line) the catalytic reaction, the spectra are shifted along the y axes to avoid overlaps; (b) adsorption isotherms of Ar measured at 87 K before and after the catalytic reaction; (c) SEM images of ActB before (top) and after (bottom) the catalytic reaction. | ||
The adsorption isotherm of Ar at 87 K before and after the reaction (Fig. 7b) revealed that ActB lost nearly all porosity during the catalytic reaction. We believe that blocking of the pores is a result of the additional organic matter detected by FT-IR and EDX (see below). We would like to point out that even though the catalyst after reaction is non-porous it retained its catalytic activity which suggests that the catalytic reaction is taking place only at the surface of the catalyst.
The scanning electron microscopy (SEM, Fig. 7c) showed that the morphology of ActB did not change during the reaction and EDX measurement did not reveal the presence of fluorine in the material suggesting that all fluorine was effectively scavenged by triethylsilane. In line with the data obtained from FTIR, the amount of carbon increased significantly, from C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B ratio close to 1:1 for as prepared ActB to 14:5 for ActB after the reaction.
:B ratio close to 1:1 for as prepared ActB to 14:5 for ActB after the reaction.
Conclusions
We have demonstrated that the strength of Lewis acidity of ActB is sufficient to promote reductive defluorination and dechlorination reactions on several substrates that are relevant models of numerous halogenated organic pollutants. Due to the high thermal stability of ActB, elevated reaction temperatures can be applied which clearly enhances the reaction rates. ActB was also proved to be highly robust in the catalytic testing being active even after 6 consecutive runs and achieving an overall TON of 5190.The fact that ActB does not contain any halogen atom or transition metal to induce a strong catalytic activity for hydrodefluorination and hydrodechlorination reactions makes ActB a rare example among heterogeneous catalysts.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Czech Science Foundation (No. GA 23-05818S), Research Infrastructure NanoEnviCz by the Ministry of Education, Youth and Sports of the Czech Republic under Project no. LM2023066, and the Ministry of Education, Youth and Sports of the Czech Republic and The European Union – European Structural and Investments Funds within the project Pro-NanoEnviCz II (No. CZ.02.1.01/0.0/0.0/18_046/0015586) and the JHIPC team by the Advanced Multiscale Materials for Key Enabling Technologies project (No. CZ.02.01.01/00/22_008/0004558).References
- (a) R. A. Dickman and D. S. Aga, J. Hazard. Mater., 2022, 436, 129120 CrossRef CAS PubMed; (b) J. Snow, J. Lederer, P. Kuráň and P. Koutník, Fuel Process. Technol., 2023, 248, 107823 CrossRef CAS.
- (a) A. Berhanu, I. Mutanda, J. Taolin, M. A. Qaria, B. Yang and D. Zhu, Sci. Total Environ., 2023, 859, 160010 CrossRef CAS PubMed; (b) P. Ebrahimbabaie and J. Pichtel, Environ. Sci. Pollut. Res., 2021, 28, 7710 CrossRef CAS PubMed; (c) P. Pimviriyakul, T. Wongnate, R. Tinikul and P. Chaiyen, Microb. Biotechnol., 2020, 13, 67 CrossRef PubMed; (d) V. Agarwal, Z. D. Miles, J. M. Winter, A. S. Eustáquio, A. A. El Gamal and B. S. Moore, Chem. Rev., 2017, 117, 5619 CrossRef CAS PubMed; (e) I. Nijenhuis and K. Kuntze, Curr. Opin. Biotechnol., 2016, 38, 33 CrossRef CAS PubMed.
- (a) M. Trojanowicz, A. Bojanowska-Czajka, I. Bartosiewicz and K. Kulisa, Chem. Eng. J., 2018, 336, 170 CrossRef CAS; (b) E. A. Paukshtis, L. G. Simonova, A. N. Zagoruiko and B. S. Balzhinimaev, Chemosphere, 2010, 79, 199 CrossRef CAS PubMed.
- (a) G. Yan, Chemistry, 2022, 28, e202200231 CrossRef CAS PubMed; (b) J. Cui, P. Gao and Y. Deng, Environ. Sci. Technol., 2020, 54, 3752 CrossRef CAS PubMed; (c) Y. Y. Wang, Y. Wei, W. J. Song, C. C. Chen and J. C. Zhao, ChemCatChem, 2019, 11, 258 CrossRef CAS; (d) S. Wang, Q. Yang, F. Chen, J. Sun, K. Luo, F. Yao, X. Wang, D. Wang, X. Li and G. Zeng, Chem. Eng. J., 2017, 328, 927 CrossRef CAS.
- (a) N. Nzeribe, M. Crimi, S. M. Thagard and T. M. Holsen, Crit. Rev. Environ. Sci. Technol., 2019, 49, 866 CrossRef; (b) M. P. Rayaroth, U. K. Aravind and C. T. Aravindakumar, Environ. Chem. Lett., 2016, 14, 259 CrossRef CAS.
- (a) Y. Guo, Y. Li and Z. Wang, Water Res., 2023, 234, 119810 CrossRef CAS PubMed; (b) H. S. Yin, X. K. Cao, C. Lei, W. Q. Chen and B. B. Huang, ChemElectroChem, 2020, 7, 1825 CrossRef CAS; (c) C. Lei, F. Y. Liang, J. Li, W. Q. Chen and B. B. Huang, Chem. Eng. J., 2019, 358, 1054 CrossRef CAS; (d) Z. J. Chen, Y. W. Liu, W. Wei and B. J. Ni, Environ. Sci.: Nano, 2019, 6, 2332 RSC.
- (a) D. K. Leonard, P. Ryabchuk, M. Anwar, S. Dastgir, K. Junge and M. Beller, ChemSusChem, 2022, 15, e202102315 CrossRef CAS PubMed; (b) D. H. Ge, J. W. Chen, Y. L. Chen, M. T. Ma, Z. L. Shen and X. Q. Chu, Org. Chem. Front., 2023, 10, 3909 RSC; (c) V. S. Saberov, G. F. Rayenko, A. S. Avksentiev, L. M. Vakhitova and N. I. Korotkikh, Theor. Exp. Chem., 2023, 59, 151 CrossRef CAS; (d) Q. Shen, Y. G. Huang, C. Liu, J. C. Xiao, Q. Y. Chen and Y. Guo, J. Fluorine Chem., 2015, 179, 14 CrossRef CAS; (e) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009 CrossRef CAS PubMed.
- S. Sabater, J. A. Mata and E. Peris, Nat. Commun., 2013, 4, 2553 CrossRef PubMed.
- (a) P. Štěpnička, M. Semler, J. Demel, A. Zukal and J. Čejka, J. Mol. Catal. A: Chem., 2011, 341, 97 CrossRef; (b) T. Hara, T. Kaneta, K. Mori, T. Mitsudome, T. Mizugaki, K. Ebitani and K. Kaneda, Green Chem., 2007, 9, 1246 RSC.
- T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578 CrossRef CAS.
- (a) Y. J. Wanglee, J. Hu, R. E. White, M. Y. Lee, S. M. Stewart, P. Perrotin and S. L. Scott, J. Am. Chem. Soc., 2012, 134, 355 CrossRef CAS PubMed; (b) F. Vidal, J. McQuade, R. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2020, 142, 14427 CrossRef CAS PubMed; (c) K. K. Samudrala, M. O. Akram, J. L. Dutton, C. D. Martin and M. P. Conley, Inorg. Chem., 2024, 63, 4939 CrossRef CAS PubMed.
- C. B. Caputo and D. W. Stephan, Organometallics, 2012, 31, 27 CrossRef CAS.
- G. Kumar, S. Roy and I. Chatterjee, Org. Biomol. Chem., 2021, 19, 1230 RSC.
- J. P. Mancinelli, W.-Y. Kong, W. Guo, D. J. Tantillo and S. M. Wilkerson-Hill, J. Am. Chem. Soc., 2023, 145, 17389 CrossRef CAS PubMed.
- A. Hoppe, A. Stepen, L. Köring and J. Paradies, Adv. Synth. Catal., 2024, 366, 2933 CrossRef CAS.
- (a) H. Fang and M. Oestreich, Chem. Sci., 2020, 11, 12604 RSC; (b) D. Dunlop, J. Pinkas, M. Horáček, N. Žilková and M. Lamač, Dalton Trans., 2020, 49, 2771 RSC.
- (a) K. Lye and R. D. Young, Chem. Sci., 2024, 15, 2712 RSC; (b) D. Mandal, R. Gupta, A. K. Jaiswal and R. D. Young, J. Am. Chem. Soc., 2020, 142, 2572 CrossRef CAS PubMed; (c) K. Škoch, C. G. Daniliuc, G. Kehr, S. Ehlert, M. Müller, S. Grimme and G. Erker, Chem. – Eur. J., 2021, 27, 12104 CrossRef PubMed; (d) D. Mandal, R. Gupta and R. D. Young, J. Am. Chem. Soc., 2018, 140, 10682 CrossRef CAS PubMed.
- R. Gupta, D. Csókás, K. Lye and R. D. Young, Chem. Sci., 2023, 14, 1291 RSC.
- V. J. Scott, R. Çelenligil-Çetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852 CrossRef CAS PubMed.
- (a) C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946 CrossRef CAS PubMed; (b) C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188 CrossRef CAS PubMed.
- M. K. Assefa and M. E. Fieser, J. Mater. Chem. A, 2023, 11, 2128 RSC.
- H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176 RSC.
- (a) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374 CrossRef CAS PubMed; (b) J. Zhu, M. Pérez, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2016, 55, 1417 CrossRef CAS PubMed; (c) P. Mehlmann, T. Witteler, L. F. B. Wilm and F. Dielmann, Nat. Chem., 2019, 11, 1139 CrossRef CAS PubMed.
- (a) M. Ahrens, G. Scholz, T. Braun and E. Kemnitz, Angew. Chem., Int. Ed., 2013, 52, 5328 CrossRef CAS PubMed; (b) G. Meißner, D. Dirican, C. Jäger, T. Braun and E. Kemnitz, Catal. Sci. Technol., 2017, 7, 3348 RSC.
- D. B. Culver and M. P. Conley, Angew. Chem., Int. Ed., 2018, 57, 14902 CrossRef CAS PubMed.
- (a) X. Pan, M. Talavera, G. Scholz and T. Braun, ChemCatChem, 2022, 14, e202200029 CrossRef CAS; (b) R. Gupta and R. D. Young, Synthesis, 2022, 54, 1671 CrossRef CAS.
- C. Heinekamp, A. G. Buzanich, M. Ahrens, T. Braun and F. Emmerling, Chem. Commun., 2023, 59, 11224 RSC.
- (a) D. Bůžek, K. Škoch, S. Ondrušová, M. Kloda, D. Bavol, A. Mahun, L. Kobera, K. Lang, M. G. S. Londesborough and J. Demel, Chem. – Eur. J., 2022, 28, e202201885 CrossRef PubMed; (b) M. Lamač, B. Urbán, M. Horáček, D. Bůžek, L. Leonová, A. Stýskalík, A. Vykydalová, K. Škoch, M. Kloda, A. Mahun, L. Kobera, K. Lang, M. G. S. Londesborough and J. Demel, ACS Catal., 2023, 13, 14614 CrossRef PubMed.
- M. Bui, K. F. Hoffmann, T. Braun, S. Riedel, C. Heinekamp, K. Scheurell, G. Scholz, T. M. Stawski and F. Emmerling, ChemCatChem, 2023, 15, e202300350 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details of syntheses and catalytic experiments, additional characterization data for the reaction products and for ActB after use. See DOI: https://doi.org/10.1039/d4cy00732h |
| This journal is © The Royal Society of Chemistry 2024 |