
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced catalytic performance of single-atom Cu on Mo2C toward CO2/CO hydrogenation to methanol: a first-principles study†
Anna
Vidal-López
 a,
Estefanía
Díaz López
a and
Aleix
Comas-Vives
*ab
a,
Estefanía
Díaz López
a and
Aleix
Comas-Vives
*ab
aDepartment of Chemistry, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Catalonia, Spain. E-mail: Aleix.Comas@uab.cat
bInstitute of Materials Chemistry, Technische Universität Wien, 1060 Vienna, Austria. E-mail: aleix.comas@tuwien.ac.at
First published on 8th October 2024
Abstract
CO2 emissions harm the environment due to their pivotal role in fostering climate change and ocean acidification. One way to take advantage of CO2 is to use it as a precursor to chemical materials to enable energy transition. The CO2 to methanol conversion from green H2 is a promising option. The silica-supported Cu/Mo2CTx (MXene) catalyst displayed higher activity than the industrial reference system Cu/ZnO/Al2O3. To better understand CO2 hydrogenation in Cu/Mo2CTx and related processes under reaction conditions (CO hydrogenation and reverse water gas shift reaction), we performed periodic DFT calculations to evaluate the methanol synthesis reaction mechanism using our previously calibrated theoretical model against experiment characterization. Our results show the crucial role played by the Cu/Mo2CTx interface in providing low-energy pathways to facilitate the hydrogenation of CO2 to methanol, where both the Cu atom and the Mo2CTx support participate in the reaction mechanism. The findings showcase the unique pathways provided by this supported single-atom catalyst, allowing the successive heterolytic cleavages of molecular hydrogen (H2) to form HCOO*, HCOOH*, and H2COOH* species with co-adsorbed hydrogen in contrast with classical heterogeneous catalysts based on Cu NPs supported on oxides. Thus, CH3OH is readily formed under reaction conditions. CO also forms via the reverse water-gas shift (RWGS) reaction, which can be hydrogenated to methanol. These findings open new avenues to understanding CO2 and CO hydrogenation by exploiting single-atom catalysts and metal–support interfaces.
1. Introduction
Climate and energy crises are critical challenges we must face to keep life as we know it. CO2 is pivotal in this problem,1 being a primary target to mitigate. To that aim, one strategy is to close the carbon cycle by converting CO2 to methanol. Methanol is a crucial organic feedstock in the chemical industry as it is a precursor of several valuable chemicals like formaldehyde, acetic acid, and methyl-tert butyl ether.2 Moreover, methanol is an excellent transportation and industrial fuel, able to replace gasoline, diesel fuel, and natural gas. Among different approaches to CO2 conversion, hydrogenation using “green” H2 from renewable energy sources is an emergent strategy in the so-called methanol economy framework.3 The methanol economy is partly based on its production by chemical recycling of CO2 from the gases of fossil fuel-burning power plants as well as other industrial and natural sources or even atmospheric CO2. The methanol economy represents a chemical regenerative carbon cycle alternative to natural photosynthesis. Selecting the appropriate catalyst is crucial to driving the use of hydrogen and avoiding competitive reactions such as the reverse water gas shift (RWGS) reaction. Many heterogeneous catalysts, typically Cu-based, have recently been reported for this purpose.4,5 The reported Cu-based industrial catalyst (Cu–Zn–Al2O3), which displays a highly dynamic nature,6–10 promotes methanol synthesis11 from a synthesis gas mixture (CO2/CO/H2) at elevated pressures P (50 to 100 bar) and temperatures T (200° to 300 °C).MXenes12,13 arise as an attractive family of materials with suitable characteristics such as stability and physical and mechanical properties. They have also displayed attractive electronic, optical, plasmonic, and thermoelectric properties13 or are emerging materials in thermocatalytic applications.14 These 2D-materials are a family of transition metal carbides, carbonitrides, and nitrides15 with the general formula of Mn+1XnTx (where M is an early transition metal, n = 1, 2, 3, X is C and/or N and T is surface –O–, –OH and/or –F groups). MXenes without surface termination groups are oxophilic; thus, they can bind and activate CO2.16 Another class of emerging catalysts is single-atom catalysts (SACs).17 They are based on an isolated metallic atom stabilized by the support or embedded into another metal. They have shown exceptional performance, drastic cost reduction, high activity, selectivity, metal atom utilization, and stability.18–20 SACs can incorporate a range of metals, heteroatoms, and supports.17 Several studies have shown that SACs exhibit superior performance in reactions such as hydrogenation,21 CO oxidation,22,23 water-gas shift (RWGS),24 and CO2 conversion.25,26 Moreover, the Cu-SAC family has gained interest due to their ability to convert CO2 into C2+ products with high selectivity.18 Hence, the combination of 2D materials (MXene) and single-site systems (SACs) has allowed the development of a new framework for hydrogenating CO2.27–29
In a recent study, we reported30 the catalytic activity of a single Cu atom supported on a molybdenum MXene, Mo2CTx. The resulting material, Cu/Mo2COx, had a fixed coverage of 0.67 O* ML during the reaction, and the Cu atom's movement revealed two adsorption sites: one on Mo-hollow sites30 and another in a bridge position between two Mo atoms. The latter, more stable by 17 kJ mol−1, was used as the reference for energy profile construction in this work. Cu/Mo2COx provides an interface with methanol's increased intrinsic formation rate by hydrogenating CO2. We benchmarked the theoretical model against the experimental characterization. We found out via theoretical models that those with a high oxygen surface lead to a stronger CO adsorption and a higher blueshift in the resulting IR stretching CO frequency, having a high cationic character on the Cu atom, in agreement with IR spectroscopical measurements. Moreover, the H2 adsorption was more stable on the Cu/Mo2C interface, which aligned with H2 thermal programmed desorption (TPD) experiments. These results suggested that the Cu/2D-Mo2C 0.67 O* ML is a catalytic model in agreement with the experimental characterization. The evaluation of the catalytic activity of the Cu/Mo2COx system by joint experimental and computational work suggested that CO2 hydrogenation occurred via the formate species, which was also detected experimentally. Still, the proposed mechanism of CO2 hydrogenation over Cu/Mo2COx based on theoretical calculations showed that the most energy-demanding step of the pathway corresponded to the formation of dioxymethylene, having an energy barrier of ca. 140 kJ mol−1. Hence, since this barrier is rather energy-demanding, the question of whether alternative pathways can catalyze the methanol formation in an even more feasible way remains open.
Further, the involvement of various reactivity sites of the system (Cu atom, Cu–Mo2C interface, and support) in a reaction mechanism remains an open question. Besides how the MXene–SAC (Cu/Mo2COx–SiO2, Fig. 1a) catalyzes the CO/CO2 hydrogenation, it can provide relevant insights into the catalysis of SACs and the role played by the metal support interface toward the CO2 hydrogenation in contrast to the mechanisms for typical catalytic/industrial systems based on supported copper nanoparticles such as Cu/ZnO–Al2O3 (Fig. 1b).31
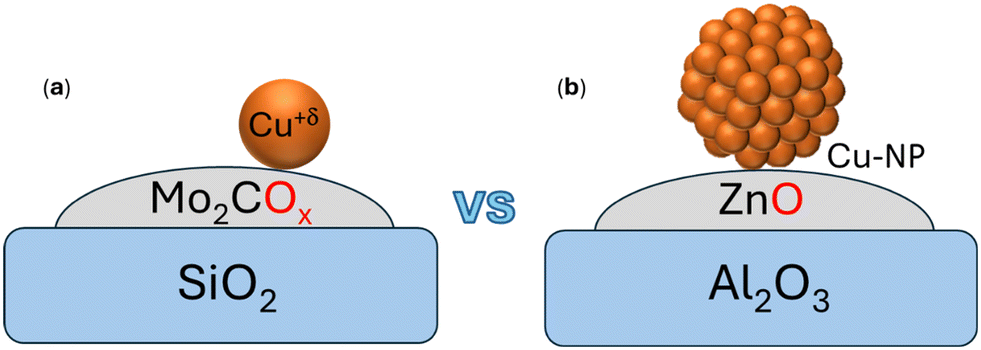 | ||
| Fig. 1 (a) Representation of the catalytic system (Cu/Mo2COx–SiO2) used in the study vs. (b) the Cu-NP based catalyst (Cu/ZnOx–Al2O3) typically used. | ||
Moreover, recently, attention has been refocused on the hydrogenation of pure CO2 or CO2-rich gas feeds containing carbon monoxide (CO).32–36 CO is present in the reaction mix, provided that there is activity in the RWGS reaction. Besides, a small fraction of CO2 in the mixture significantly affects the oxygen chemical potential of the mix.37–40 One of the most important open questions for Cu-based materials and the Cu/Mo2CTx system is the nature of the preferred carbon source to produce methanol: CO or CO2.41,42 Also, evaluating the hydrogenation of CO and CO2 separately is desirable to avoid conflicting views on methanol synthesis, as in the same way as CO2, CO has many possible reaction pathways our former work did not consider that should be deeply explored. Another critical aspect of direct CO2 hydrogenation is the competitive RWGS reaction, which decreases the selectivity towards methanol by forming CO, especially in the low-pressure range.43 The production of large quantities of water may also decrease the catalytic activity and reduce the long-term catalyst stability.44 Besides, the RWGS reaction is also a highly relevant process.45–47 In this context, computational chemistry arises as a powerful technique for understanding heterogeneous catalysts38,48–50 and unraveling the role played by the catalyst at the atomic level in methanol synthesis.39,40
For all the noted above, in the present work, we use DFT calculations to answer the critical mechanistic aspects of the CO2/CO hydrogenation and the RWGS reaction catalyzed by the recently reported Cu/Mo2COX catalyst via an extensive mechanistic analysis. The evaluation of the resulting energy profiles provides crucial aspects of the MXene–Cu-based catalyst, such as the role of each catalytic component, the Cu atom, the interface, and the support, besides revealing critical chemical aspects of this material concerning its reactivity and selectivity in the methanol synthesis by CO/CO2 hydrogenation.
2. Computational details
All DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP)51–53 with a cut-off energy of 500 eV. The structures were optimized using the projector augmented wave (PAW) method (plane-wave basis set with pseudopotentials)54,55 using the official VASP pseudopotentials56 and the Bayesian error estimation functional with van der Waals corrections (BEEF-vdW).57 In the present work context, other research groups have also used it in previous mechanistic studies on CO2 hydrogenation. Studt and co-workers compared the performance of the BEEF-vdW with the RPBE functional for the reaction catalyzed by a stepped copper surface,11 showing that the selectivity with respect to CO and CO2 in methanol formation is only described correctly with BEEF-vdW in contrast to other commonly used functionals such as RPBE. The electronic SCF-loop (EDIFF) convergence criterion was set to 10−5 eV. The convergence criteria for the ionic relaxation were set to forces' norms less than 0.01 eV Å−1. We use the conjugate gradient algorithm (IBRION = 2) to relax the ions. The Monkhorst–Pack with a 3 × 3 × 1 k-point mesh was used to sample the Brillouin zone. The catalyst models were illustrated using Atomic Simulation Environment (ASE), a 3D visualization program for structural models.58 The energy of isolated molecules was determined by a single-point calculation placing each species in a box with dimensions of 15 × 15 × 15 Å. The nudged elastic band method (NEB)59 with intermediate images was used to locate all the transition states (TS). Then, all the TS structures were refined using a Newton-based algorithm with the same convergence criteria as for the minima. All reported energy values in the text correspond to electronic energies. In all energy profiles, the values were calculated and referenced against the energy of the initial reactants (in kJ mol−1). Thermodynamic corrections were calculated using statistical thermodynamics at 503.15 K and pressure values of 5 bar (CO2, H2O), 15 bar (H2), and 2.5 bar (CO, CH3OH) to build the Gibbs energy profile (see the ESI†). The Gibbs energy of the gas phase species was calculated by considering translational, rotational, and vibrational contributions. In contrast, only the vibrational contributions of the chemisorbed species were considered. Finally, an algorithm60–63 was employed to perform Bader's analysis on a charge density grid, determining the total charge associated with each atom and defining the zero flux surfaces delineating the Bader volumes.3. Results
3.1 CO2 and CO hydrogenation and RWGS on the Cu/2D-Mo2COx system
In former work,30 we evaluated the CO2 hydrogenation to methanol catalyzed by the Cu/2D-Mo2C 0.67 O ML surface via the BEEF-vdW functional, using a model benchmarked against experimental characterization (Fig. 2a). This work evaluates, using the same model and methodology, how Cu/Mo2CTx catalyzes methanol, CO, and water formation from CO2 and H2, as summarized in Fig. 2b. We describe each section as the most feasible path based on the calculated energy barriers. By building up on our previous localized intermediates and transition states for this reaction/catalyst (A1), we explored additional CO2 hydrogenation mechanisms (A2–A3). Moreover, since Cu/Mo2COx was also found to experimentally form CO under CO2 hydrogenation reaction conditions via the RWGS reaction, we also checked the feasibility of the CO hydrogenation to methanol (B), as it is another potential operating route for ultimately catalyzing CO2 to methanol via CO as a reaction intermediate. Moreover, we considered alternative pathways for the competitive reverse water-gas shift (RWGS, C) reaction where successive hydrogen transfers to O* to produce water (H2O*).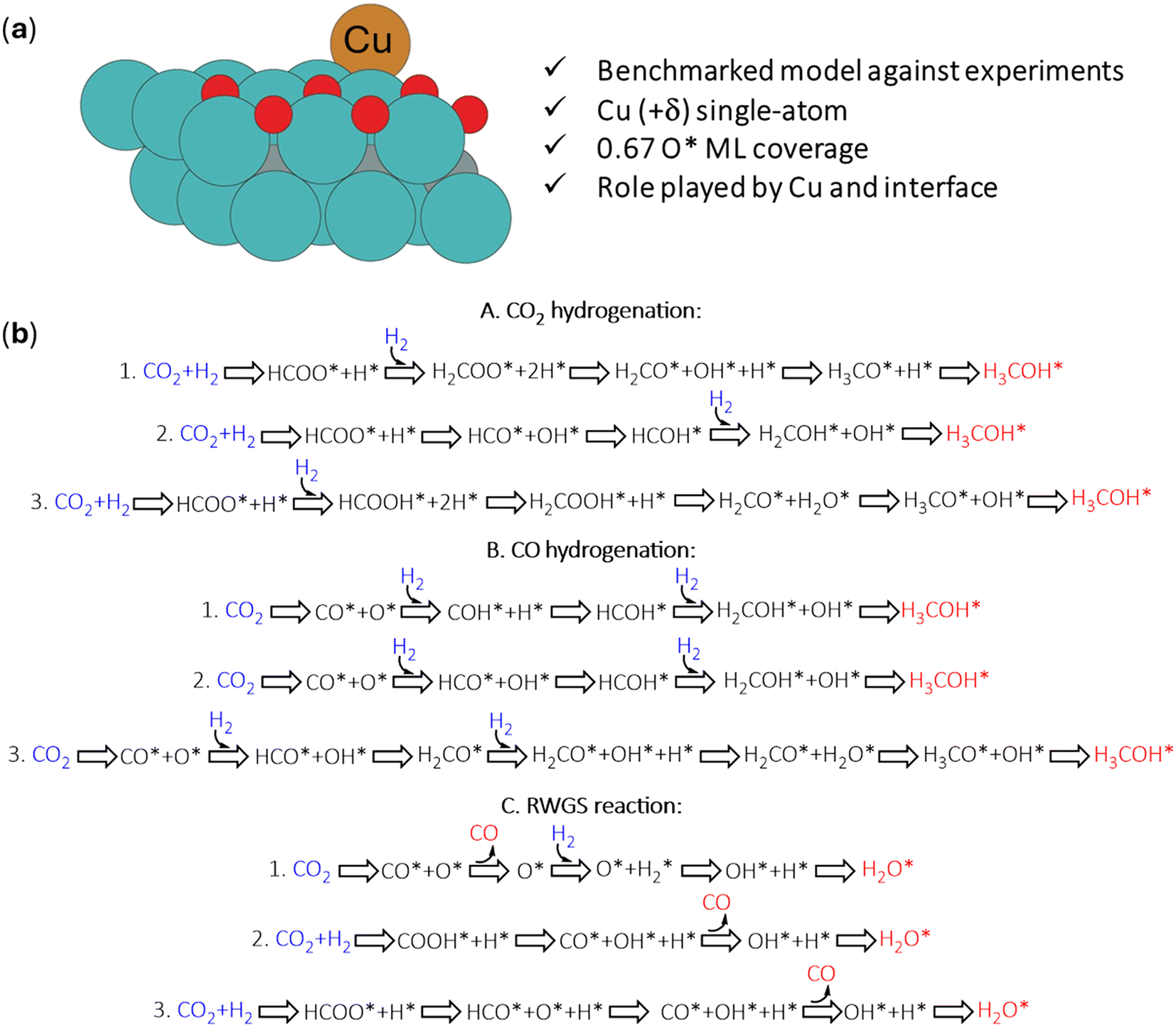 | ||
| Fig. 2 (a) Optimized structure of the catalytic system (Cu/Mo2COx) used in that study, where the atoms are represented by colors (Cu-ochre, Mo-blue, O-red, H-white, C-gray). (b) Summary of the reactants (blue), intermediates (black), and products (red) involved in each of the different reaction mechanisms reported previously and, in this study, corresponding to the hydrogenation of CO2 (A) and CO (B) and the competitive RWGS reaction (C). | ||
 | ||
| Fig. 3 Energy profile (A2) for the reaction of CO2 and 2H2 to form methanol from HCOO* species. Energies of minima and transition states (framed) are referenced against the sum of the initial reactants and catalyst energies in kJ mol−1 (Erel). | ||
Once HCOO* is obtained, the cleavage of one of the C–O bonds forms HCO* and O* via an energy barrier of 92 kJ mol−1 (A2-TS2). The resulting HCO* is bonded to the Cu center via oxygen atoms with an H* (metal hydride) adsorbed at the Cu/Mo2COx interface (HCO-s1) that was produced during the H2(g) heterolytic cleavage. Afterward, OH* forms by combining O* and H*. The formation of the OH* group is required because it participates in subsequent hydrogenation steps, described later. This step must overcome an energy barrier of 70 kJ mol−1 (A2-TS3). The HCO* species rotation makes the subsequent migration next to the Cu center more feasible. It requires an energy of 11 kJ mol−1, being a shallow and affordable energy barrier (A2-TS4). At this point, the HCO-s2* species rotated geometry is obtained. The profile proceeds via its migration over the Cu center through a formerly reported transition state (A2-TS5), forming the HCO–Cu* species. This step has an energy barrier equal to 85 kJ mol−1. This migration causes a slight rotation of the HCO* compound around the Cu center to facilitate its subsequent hydrogenation (A2-TS6) of about 0.3 kJ mol−1, forming the HCO–Cu-rot* species. Afterward, the new configuration promotes the proton transfer from the surface OH* to the HCO* group (A2-TS7) subjected to 2 kJ mol−1 as an energy barrier, forming O* and HCOH*. Then, the H2(g) molecule splits on O* and HCOH* via a heterolytic transition state (A2-TS8) as an Eley–Rideal step with an energy barrier equal to 87 kJ mol−1, forming OH* and H2COH*, respectively. This step is the second most energy-demanding step of this energy profile. The OH* group bonds the Mo2COx surface on the resulting structure, while the H2COH* species binds the Cu center. Finally, methanol can be readily obtained by proton transfer from the OH* group to H2COH (A2-TS9), with 61 kJ mol−1 as an energy barrier. This final state corresponds to the final CH3OH on the Cu center. Finally, CH3OH* can be desorbed, having an energetic cost of 99 kJ mol−1, and water can be easily formed with the oxygen remaining on the surface with two main energy barriers of 73 and 100 kJ mol−1 and an endo-energetic desorption by 65 kJ mol−1, as shown in Fig. S1 from the ESI.† Overall, this reaction mechanism provides very feasible energy barriers. The HCOO* formation, the most energy-demanding step of the whole route, has an energy barrier of 103 kJ mol−1. Route A3 goes through the formation of formic acid (HCOOH*), followed by its hydrogenation (H2COOH*) and its subsequent cleavage to formaldehyde (H2CO). Therefore, these steps present higher energy barriers than the previous A2 route; thus, this pathway (A3) is described in the ESI† (Fig. S2).
The Gibbs energy profile (Fig. S3†) maintains the trend of the electronic profiles. The most notable difference between the two profiles lies in the energy barriers, especially those involving the addition or removal of a gas-phase species, due to the entropic contributions in the energy correction of these species. This occurs when gas-phase incoming molecules react on the surface, such as CO2 or H2, increasing the energy barrier due to the entropic penalty and for the desorption of products, such as CO or methanol and H2O, which are favored entropically.
In summary, we provided more feasible CO2 hydrogenation pathways than the previously reported one.30 The most feasible mechanism avoids forming dioxymethylene, previously reported as the most energetically demanding step of the CO2 hydrogenation pathway.30 The most preferred path (A2) forms methanol via formate (HCOO*), its subsequent cleavage to HCO* and O*, followed by the hydrogenation of HCO* to HCOOH*, H2COH* to form methanol. The evaluated pathways share critical steps and similar energy barriers with the CO hydrogenation mechanism to methanol (vide infra).
 bending and C–O bond breaking. CO2 pre-activation leads to a structure where the ∠O–C–O angle is ca. 136°, and subsequent low energy barriers for the bending and activation equal 35 and 3 kJ mol−1, respectively. Thus, since CO(g) is an experimental product and its formation is highly feasible, the mechanism to hydrogenate CO* to methanol is a route deserving further investigation.
bending and C–O bond breaking. CO2 pre-activation leads to a structure where the ∠O–C–O angle is ca. 136°, and subsequent low energy barriers for the bending and activation equal 35 and 3 kJ mol−1, respectively. Thus, since CO(g) is an experimental product and its formation is highly feasible, the mechanism to hydrogenate CO* to methanol is a route deserving further investigation.
Once CO* is formed, we considered two pathways for its subsequent hydrogenation to methanol: via the COH* intermediate or the HCO* one. The formation of COH* species occurs on the Cu atom by hydrogenating the CO* molecule. The COH* pathway (Fig. 2b, B1) proceeds via subsequent hydrogenations to HCOH*, H2COH*, and methanol species by heterolytic H2 cleavage or transfers of remaining H* at the interface and of protons from OH* groups on the surface. Since already at the thermodynamic level do the mechanisms and intermediates have a low stability compared to the others (by ca. 60–80 kJ mol−1), we did not perform additional transition-state calculations for this pathway.
Conversely, when the HCO* species formation occurs on the support, it is significantly more stable than the COH* one by ca. 175 kJ mol−1. Thus, we evaluated three possible mechanisms going through the HCO* intermediate (Fig. 2b, B2-B2sup-B3). These mechanisms differ in the reaction intermediates' stabilization energy and the participation of the support in the reaction mechanism. The corresponding three energy profiles, only considering the energetics of the reaction intermediates, are depicted in Fig. S4 in the ESI.† The formed HCO* species is bonded to the Cu center, and one OH* group is also obtained at the Cu/2D-Mo2COx surface due to the CO2 cleavage. This process involved the same transition states in the evaluated routes to form the subsequent HCOH and H2CO intermediates.
HCOH path. Fig. 4 presents methanol formation from HCO* species (B2) through different intermediates and located transition states (TS), the most favorable pathway among all the evaluated ones. It should be mentioned that the last part of the profile is shared with the route described before for the CO2 hydrogenation described in the former section to generate the HCO* species (A2). The first step of this mechanism is the heterolytic cleavage of H2(g) through an Eley–Rideal mechanism that allows obtaining OH* and one H* atom adsorbed on the Cu/Mo2COx interface with an energy barrier of 96 kJ mol−1 (B-TS3). A subsequent transition state to form HCO* by reaction of H* and CO* for HCO* (B-TS4) is 102 kJ mol−1. This transition state is the mechanism's most energy-demanding step (RDS). Upon forming the HCO* and OH* species, the mechanism bifurcates into two pathways (B2, B2-sup) on the Cu/2D-Mo2COx surface. In the first route (B2), through the HCOH* species, the reactivity takes place on the Cu center. This pathway exhibits the most feasible energies in terms of transition state energies and reaction energies. Thus, it is described and depicted in Fig. 4. The other path (B2sup) on the support is reported in the ESI† (Fig. S5).
 | ||
| Fig. 4 Energy profile for the reaction of CO2 and 2H2 to form methanol from the HCOH* species. Dashed traces are steps common for all profiles. Energies of minima and transition states (framed) are referenced against the sum of the initial reactants and catalyst energies in kJ mol−1 (Erel). | ||
Once the HCO* species and the OH* group are obtained, the mechanism forms the HCOH* species through a proton transfer from OH* to HCO*. However, a slight rotation of the HCO* species around the Cu center is first required to enable the subsequent proton transfer. The rotation is about 0.3 kJ mol−1 (B2-TS5). Afterward, the new configuration allows the proton transfer from the OH* adsorbed on Mo2C to the HCO* group (B2-TS6), subjected to 2 kJ mol−1 as an energy barrier. Then, when a second H2(g) molecule enters the reactive path, the system slightly stabilizes, releasing 2 kJ mol−1. Subsequently, the H2(g) molecule splits through a heterolytic transition state (B2-TS7) as an Eley–Rideal step again with an energy barrier equal to 87 kJ mol−1, forming the H2COH* species coordinated to Cu and an OH* group bonded to the support. This step is the second most energy-demanding for this energy profile. Finally, adsorbed methanol can be readily obtained by proton transfer from the OH* group to H2COH* with 61 kJ mol−1 as an energy barrier (B2-TS8). Methanol is obtained and adsorbed on the Cu center. Finally, CH3OH* can be desorbed as described above on the A2 profile. The resulting energy profile confirms the feasibility of this reaction mechanism from CO hydrogenation.
Again, the Gibbs energy profile (Fig. S6†) maintains the trend of the electronic energy profile. They showed the same differences described above due to the entropic contributions in the energy correction of these species. Frequency analysis confirmed the nature of all transition states shown in the electron profile, except CO2 dissociation (TS2), where no saddle point was found, i.e., the transition was lower in Gibbs energy than the respective reactants according to the Gibbs energy. This suggests that the dissociation of CO2 is practically spontaneous after the formation of bent CO2, with a CO bond elongated by 1.466 Å.
B2+B2sup path. Fig. 5 presents the last path of this section, namely B2+B2sup. This route comprises previous HCOH routes (B2, B2sup), connected via a transition state (TS6). Thus, this pathway goes through the formation of HCO* on the support (Fig. S5†), followed by its hydrogenation to HCOH* on Cu (Fig. 4).
 | ||
| Fig. 5 Energy profile for the reaction of CO2 and 2H2 to form methanol from HCOH* species on the support (purple labels) to HCOH* species on the Cu center (orange labels). The dashed line shows steps that are common for all profiles. Energies of minima and transition states (framed) are referenced against the sum of the initial reactants and catalyst energies in kJ mol−1 (Erel). | ||
This mechanism starts with the CO* migration from Cu to the Mo2C support, where its stabilization increases considerably, releasing 72 kJ mol−1. In this case, the C atom of CO* is directly bonded to Mo hollow sites, and its desorption has an energy cost of 152 kJ mol−1, much higher than the CO desorption energy from the Cu atom. After that, a new molecule of H2(g) participates in the pathway. In contrast to the previously evaluated paths, its activation proceeds via a homolytic transition state (B2sup-TS3) via an Eley–Rideal step, having an energy barrier of 107 kJ mol−1 and forming a couple of H–Cu bonds. The preference for the homolytic cleavage of H2 is probably due to the distance between the CO* group and the Cu interface (3.165 Å), which does not allow a heterolytic cleavage. Once this activation occurs, the mechanism forms HCO* species by transferring the H* from the Cu/Mo2COx interface to CO*, with an energy barrier of 122 kJ mol−1 (B2sup-TS4). Afterward, we considered the OH* formation by hydrogen transfer from the second H*, having an energy barrier of 40 kJ mol−1 (B2sup-TS5). Thus, the step sequence ends when both HCO* and co-adsorbed OH* species are obtained at the Mo-hollow sites of the support. Overall, this route allows the formation of the HCO* intermediate via one of the lowest energy barriers obtained. The next step (TS6) connects the HCOH-support pathway (B2-sup) with the HCOH one (B2). This transition state corresponds to the simultaneous migration of the HCO* species to Cu and OH* near the Cu/Mo2COx interface. This step requires an energy barrier of 85 kJ mol−1. Therefore, at this point, the subsequent reactions leading to methanol directly involve the Cu center (orange labels), as described in the main text in the HCOH-path section (Fig. 3, B2). This mixed route is possibly another feasible mechanism for obtaining methanol despite the higher values of some energy barriers, namely 107 and 122 kJ mol−1 for the hydrogen activation and HCO formation steps, respectively. The final Gibbs energy profile (Fig. S7†) was also analyzed, maintaining the trends of the electronic energy profile.
The other energy profiles via the HCO* species are described in the ESI† due to their higher energy barriers. The first one (B2sup) considers the formation and hydrogenations of HCO* on the support because it considers the prior migration of CO* from the Cu atom to the 2D-Mo2COx support, increasing its stability (Fig. S5†). The second one forms formaldehyde (B3, H2CO*), where the chemistry moves from the Cu atom to the support and its subsequent hydrogenations to methoxy and methanol species (Fig. S8†).
In summary, DFT calculations suggest a feasible energy profile for the CO hydrogenation pathway to methanol catalyzed by the Cu/2D-Mo2C 0.67 O ML system. This occurs via the HCOH intermediate on the Cu atom through the HCO species (B2 pathway). The Cu atom, the Cu/Mo2COx interface, and the support provide active sites and play a crucial role in the CO hydrogenation reaction. Among all the profiles, we can see their active participation in the reaction mechanism by simultaneously lowering the energy barriers for successive heterolytic H2(g) cleavages required to form HCO*, HCOH*, and H2COH* species, respectively, besides H* adsorbed at the interface. The most energetically demanding step of the most affordable energy path leading to methanol is the formation of the HCO* species (102 kJ mol−1) with an energy barrier like the reported one30 for HCOO* formation.
Competition and selectivity. After the electronic and Gibbs energies for the evaluated pathways have been obtained, it is necessary to discuss their competition and how it may affect the selectivity toward CO or CH3OH. We need to consider the energy values comprising barriers and desorption.
Concerning CO2, the most plausible route to obtain methanol (A2 path) is through the HCO* species obtained by the previous dissociation of HCOO*. The energy barrier associated with the formation of HCOO* is about 103 kJ mol−1, and its dissociation to HCO* and O* of 92 kJ mol−1. The HCO* is subsequently hydrogenated to methanol or can be dissociated into CO* and H* with an energy barrier of 100 kJ mol−1, suggesting CO* as a feasible side product of the pathway.
Concerning CO as a reactant, it has three significant barriers that might affect the selectivity of the reaction. CO* formation is less energy-demanding than the HCOO* one since its cleavage into CO* and O* is ca. 35 kJ mol−1. Its subsequent hydrogenation to HCO* (B2 path) is a two-step process involving energy barriers of 96 kJ mol−1 and 102 kJ mol−1 for the H2(g) heterolytic activation and the subsequent HCO* formation, respectively. On the other hand, the CO desorption costs 80 kJ mol−1. CO can also migrate from the Cu atom to the support, i.e., towards a hollow-Mo site of Mo2C (B2sup). This step is favorable by 72 kJ mol−1 upon adsorption. Thus, the CO* desorption from the support has an energy cost of 152 kJ mol−1, much higher than the CO desorption energy from the Cu atom. Hence, this route will likely increase selectivity toward methanol since CO desorption is difficult. At the same time, the hydrogenation of CO* (CO-s) is more feasible energetically, i.e., the formation of the HCO* species is the highest energy barrier toward methanol, with an energy barrier of 122 kJ mol−1. These results suggest the preference for CO-s to be hydrogenated rather than desorbed.
Evaluating the Gibbs profiles and the computed energies about barriers, we observed the same tendency as in the electronic scenario described above, considering the thermodynamic corrections. The critical difference in Gibbs energies compared to the electronic energies is that CO desorption becomes easier than the respective transition state of hydrogenation, both from Cu and the support (by 107 kJ mol−1 and by 48 kJ mol−1, respectively). Nevertheless, since the gas-phase CO and CH3OH have similar energy (CO is more stable in Gibbs energy than methanol by only 16 kJ mol−1) and the Gibbs energy barriers for hydrogenation are affordable under the evaluated conditions, one can expect forming both products, without having a high selectivity for either of them.
All pathways start with the bending of CO2, which is pre-activated via its bending (∠O–C–O is ca. 136°), having an energetic cost of 35 kJ mol−1. Then, the mechanism (Fig. 2, C1) can proceed through the C–O cleavage with the lowest energy barrier, about 3 kJ mol−1. Thus, after CO desorption, which required 80 kJ mol−1, H2(g) was energetically favorably adsorbed on the Cu atom by 13 kJ mol−1.  was coordinated to the Cu atom in a η2 mode with an H–H distance of 0.759 Å and −0.44 eV as adsorption energy. Thus, in the next step, through a Langmuir–Hinshelwood path, the heterolytic cleavage of
was coordinated to the Cu atom in a η2 mode with an H–H distance of 0.759 Å and −0.44 eV as adsorption energy. Thus, in the next step, through a Langmuir–Hinshelwood path, the heterolytic cleavage of  simultaneously produced O–H and Cu–H bonds. This step was endo-energetic by 29 kJ mol−1 with an energy barrier of 73 kJ mol−1. Finally, H2O* was formed by the subsequent reaction of OH* with H* at the interface, with an energy barrier of 100 kJ mol−1, i.e., the rate-limiting step of the RWGS pathway. The resulting intermediate was 3 kJ mol−1 higher than the initial reactants. The final step, i.e., water desorption, was endo-energetic and required 65 kJ mol−1 (see below, Fig. 6a).
simultaneously produced O–H and Cu–H bonds. This step was endo-energetic by 29 kJ mol−1 with an energy barrier of 73 kJ mol−1. Finally, H2O* was formed by the subsequent reaction of OH* with H* at the interface, with an energy barrier of 100 kJ mol−1, i.e., the rate-limiting step of the RWGS pathway. The resulting intermediate was 3 kJ mol−1 higher than the initial reactants. The final step, i.e., water desorption, was endo-energetic and required 65 kJ mol−1 (see below, Fig. 6a).
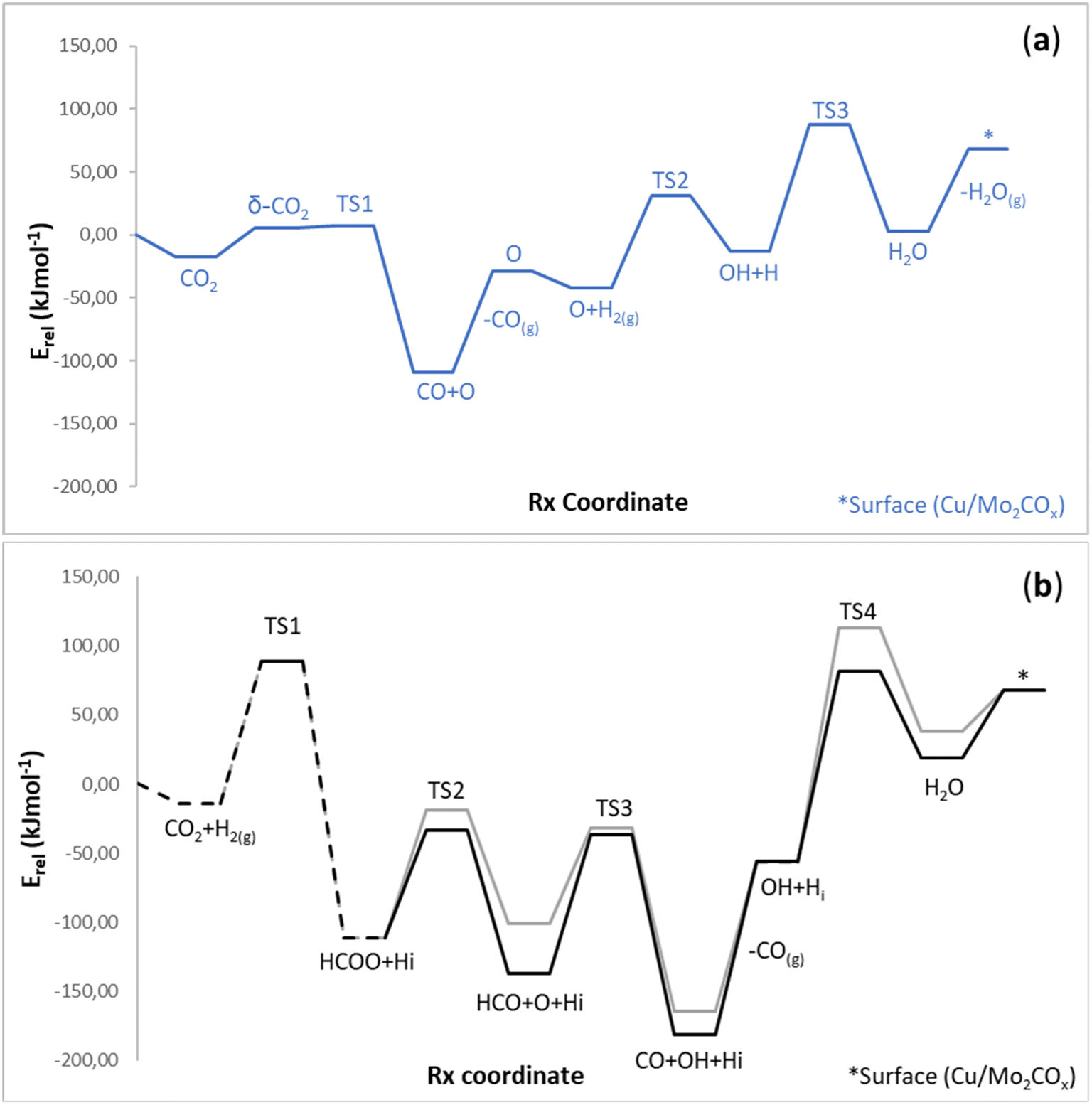 | ||
| Fig. 6 Energy profiles for the RWGS reaction of CO and H2 to form water through two different pathways: (a) CO* + O* (blue line) or (b) HCOO* (C21-gray and C22-black lines), respectively. Intermediates in the dashed line are common in more than evaluated pathways. Energies are referenced against the initial reactants' energy in kJ mol−1 (Erel). | ||
Next, we describe the most feasible pathways from the δ-CO2 species forming the HCOO* species (C3). Here, we discuss the transition states of the most viable routes, taking those with the most stable reaction intermediates. It is written with more details in the ESI, where the corresponding energies of each intermediate and transition state are also provided. The COOH* species (C2) formation from the δ-CO2 and subsequent water formation are described in the ESI† (Fig. S10).
HCOO routes. The HCOO route begins with the direct hydrogenation of CO2 to obtain HCOO* on the support. This heterolytic transition state (A2-TS1) formally produces a metal hydride and a positive H* attached to a carbon atom from the bent
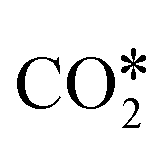 . This step required an energy barrier of 103 kJ mol−1 (TS1, Fig. 3). As mentioned above, the HCOO* intermediate on RWGS can take different pathways to obtain water from HCOO* cleavage. We subsequently describe the two most feasible energy routes (Fig. 6b): C3-1 (gray) and C3-2 (black).
. This step required an energy barrier of 103 kJ mol−1 (TS1, Fig. 3). As mentioned above, the HCOO* intermediate on RWGS can take different pathways to obtain water from HCOO* cleavage. We subsequently describe the two most feasible energy routes (Fig. 6b): C3-1 (gray) and C3-2 (black).
The C3-1 route from HCOO* starts with its cleavage into HCO* and O* species in the Mo-hollow site with an energy barrier of 92 kJ mol−1 (C3-1 TS2). In this step, the C–O cleavage occurs on the oxygen far from the copper atom. The HCO* and H* atoms are then adsorbed at the Cu/2D-Mo2C 0.67 O ML interface. After that, HCO* promotes proton transfer to obtain the OH* group via an energy barrier of 69 kJ mol−1 (C3-1 TS3). Thus, in this third transition state, CO* forms on the Cu atom while the OH* group remains on the surface and the H* atom on the interface. The CO* can then be desorbed in a step endo-energetic by 109 kJ mol−1. The final step for water formation is the transfer of the proton at the interface to the OH* with an energy barrier of 170 kJ mol−1 (C3-1 TS4), the most energy-demanding step of the mechanism. Finally, water can be desorbed by 30 kJ mol−1. The ESI† (Fig. S11) shows the resulting energy profile.
The C3-2 route from HCOO* started with its cleavage into HCO* and O* species in Mo-hollow with an energy barrier of 77 kJ mol−1 (C3-2 TS2). The C–O bond split through the oxygen next to the copper atom. HCO* is adsorbed in Mo-hollow, while H* is at the metal–surface interface. After that, HCO* promotes the transfer of the proton to the adsorbed oxygen atom (O*) through an energy barrier of 100 kJ mol−1 (C3-2 TS3). In this transition state, the CO* and OH* groups remain on the surface at the Mo-hollow sites. The CO* can then be desorbed at an energy cost of ca. 125 kJ mol−1 due to its higher stability when adsorbed on the support compared to when adsorbed on the Cu center (CO + OH + Hi: Fig. S11,† −164 kJ mol−1vs. Fig. S12,† −181 kJ mol−1). The final step for water formation is the transfer of the proton from the H* atom at the interface to OH* with an energy barrier of 138 kJ mol−1 (C3-2 TS4). Again, obtaining H2O* is the most energy-demanding step of this second mechanism on the HCOO* intermediate. Finally, water desorption costs 49 kJ mol−1. The ESI† (Fig. S12) shows the resulting energy profile.
At this point, it is necessary to compare the new RWGS pathways, forming CO and H2O via the HCOO* intermediate with the previous results30 of the direct CO2 activation to CO* and O*, CO desorption, and the water formation from O* (described above). Fig. 6a and b show the energy profiles corresponding to the cleavage of the formate (gray, black) with the first step (TS1) in common (dashed) and from the activation of CO2 (blue). The first difference between the two reaction mechanisms (HCOO vs. CO) is the first transition state. Formate formation shows a higher energy barrier than for cleavage of the previously activated C–O bond, ca. 102 kJ mol−1, as described in the previous profiles in this manuscript. From here, the hydrogenation of the oxygen cleaved from CO2 presents two more TS with energy barriers of 73 and 100 kJ mol−1, respectively, already reported.30 In the case of TS on the HCOO route, the following are mentioned: it must be said that the barrier to obtaining formate is feasible, being a species detected experimentally.30 Therefore, in this case, the formation of H2O marks the viability of one or another mechanism. In this case, the CO2 cleavage pathway (blue) is the one with the lowest barrier, approximately 38 kJ mol−1 compared to HCOO (Fig. 6, a-blue line: TS3 – 100 kJ mol−1vs. b-black line: C3-2 TS4 – 138 kJ mol−1).
In summary, the DFT calculations allowed studying alternative pathways for obtaining water in the RWGS compared to the previous route explored through CO* and O* species on the Cu/2D-Mo2C 0.67 O ML model.30 Also, they highlight the possible intermediates and transition states. The copper atom, the support, and the interface between them participate in the reaction mechanism by reducing the energy barriers heterolytic cleavages of H2 required to form HCOO* and COOH* species, simultaneously with H* adsorbed or cleaved bonds. The most energy-demanding step of the COOH pathway is the formation of CO* and OH* species (C2-TS2, 139 kJ mol−1 in Fig. S10†). In contrast, for HCOO pathways, the most energy-demanding step is forming water (C3-1 TS4, 170 kJ mol−1 in Fig. S11† and C3-2 TS4, 138 kJ mol−1, in Fig. S12†). Based on the energy barriers, the formation of the HCOO* intermediate is more likely than the COOH* one. Moreover, the water formation step determines the preferred RWGS mechanism, corresponding to the direct cleavage of CO2 to CO* and O*, followed by CO* desorption and the subsequent hydrogenation of O* to form H2O.
3.2 Bader charge analysis
The nature of the observed heterolytic cleavage was of interest to understanding the mechanism of successive hydrogenations. In contrast, the activation of CO2 and H2 was also analyzed. We used the Bader charge analysis for this approach.60–63 This computational tool allows us to obtain valuable information about the charge change along the evaluated reaction paths, highlighting the importance of the Cu/Mo2COx interface in the reaction mechanism.According to the previous results (see Fig. 3 and 4), we found several steps for which the direct cleavage for the H2(g) molecule showed charge polarization between the hydrogen atoms on the transition states, suggesting the heterolytic cleavage of H2(g). This scenario is present for structures along all the reaction profiles for CO2 hydrogenation (a), CO hydrogenation (b), and RWGS (c). This cleavage also allowed the production of formate and hydroxymethylene (HCOH) hydrogenation on the CO2 hydrogenation profile before obtaining methanol. On the CO hydrogenation, a similar H2(g) heterolytic cleavage is found for the previous step of HCO formation and the HCOH hydrogenation, as mentioned before. On the RWGS, this transition state was observed before the water formation step through an adsorbed  , as reported in previous studies.30
, as reported in previous studies.30
Bader charge analysis shows a negative charge (red) acquired by the carbon or oxygen atom as they hydrogenate. In contrast, the Cu atom had a positive charge value (blue) due to hydride formation, as depicted in Fig. 7. The results suggest a heterolytic cleavage of H2(g), as reported in the abovementioned energy barriers. This type of cleavage is associated with relatively energy-demanding energy barriers (kJ mol−1) because it involves two different bonding processes of breaking and forming in a single-step transition state. However, the reported energy barriers are affordable, in a range of 73–102 kJ mol−1 in different scenarios (Fig. 7) and provide unique paths characteristic of single-atom catalysts, differing from those on extended Cu metal surfaces, present on Cu NPs supported on oxides.
 | ||
| Fig. 7 Bader charges aquired by the key atoms involved in the transition states (positive in blue, negative in red) corresponding to HCOO formation on CO2 hydrogenation (a), in CO hydrogenation (b), and the key step in the water formation path (c). | ||
On the other hand, we also analyzed the Bader charges for the CO2 activation to CO* and O*. Fig. 8a shows the trend along the CO2 activation path to CO* and O*. The first transition state presented the bending from its linear equilibrium geometry for the uncharged state, which required a slight energy barrier. This bending (180 to 157°) induces changes in the shape and energy level of the molecular orbitals, possibly stabilizing the LUMO on the carbon and making it more electrophilic, as suggested in the literature.64 Bader values on the TS and δ-CO2 indicated the negative character added (+0.19) on the carbon and oxygen atoms by increased reactivity due to the bending step. The C atom bonded with Cu on the interface while remaining bonded to the oxygen atoms. The bond length is elongated on the resulting bent configuration, indicating the weakening of the C–O bond. The second transition state shows CO2 polarization between the C–O group and oxygen near Mo sites. Then, it takes the formation of two new bonds: OC–Cu and O–Mo. This cleavage is favored by the electron-transfer process between C (negative character) and O (positive character), which is facilitated when CO2 is bent as the LUMO level lowers its energy.64 Next, Bader values indicated carbon monoxide formation where C localized negative charge due to being bonded to O and Cu while these last two atoms remain positive. Finally, the activation of the H2 molecule was also evaluated along the CO hydrogenation pathway (Fig. 8b). Conversely to the former cases, this activation goes through a homolytic cleavage on the Cu atom, which has an energy barrier of 106 kJ mol−1. The Bader charge analysis showed that both H atoms acquire a negative charge when bonded to the Cu atom. Thus, there was no difference in charge polarization between the hydrogen atoms for this cleavage, while Cu also acquired a positive charge (Fig. 8c).
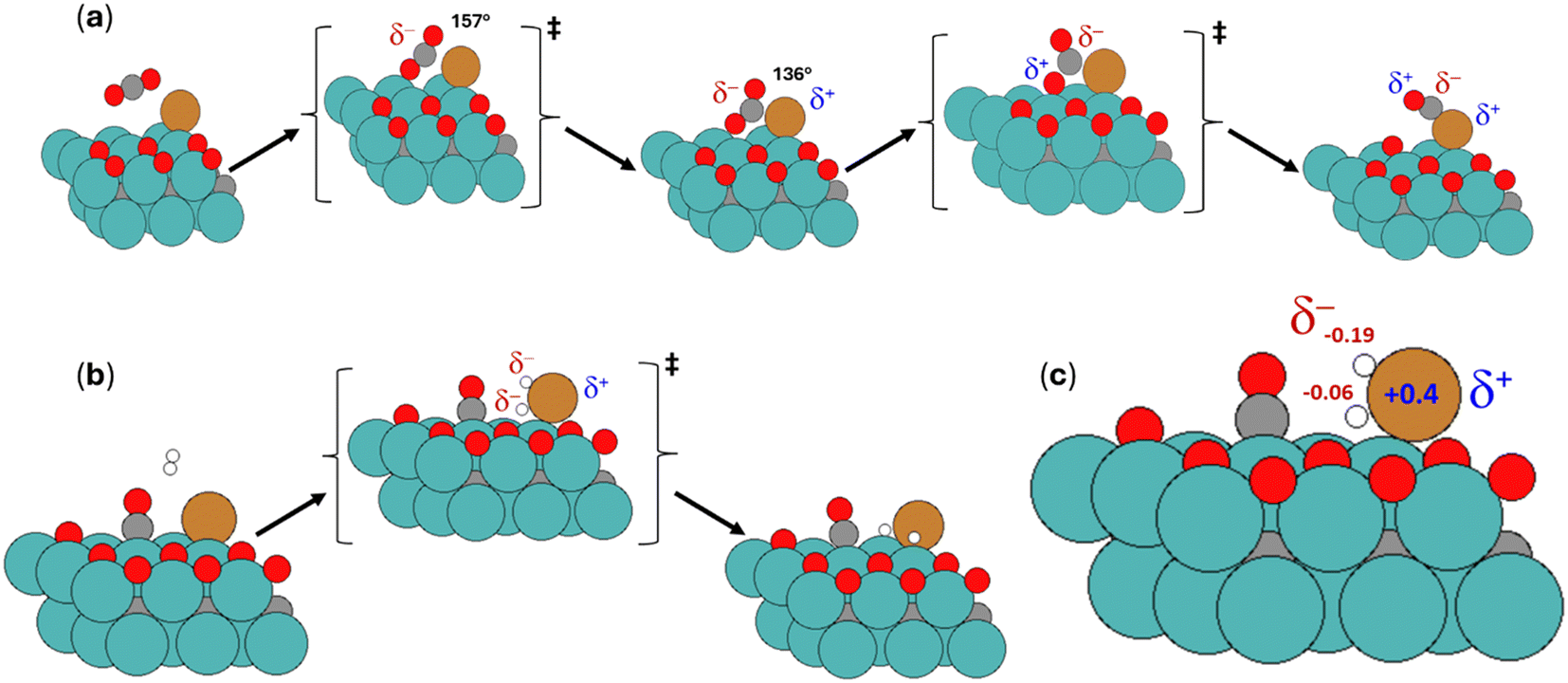 | ||
| Fig. 8 (a) Optimized geometries of minima and transition states obtained for the CO2 activation into CO* and O*. It also depicts the degree of bending of CO2 and the character acquired by the atoms involved. (b) Optimized geometries of minima and transition states were obtained for the H2 activation. (c) Bader charges aquired by the key atoms involved in the transition states (positive in blue, negative in red). | ||
Thus, via Bader charge analysis, we further characterize the nature of the bond cleavages along the critical reaction intermediates along the methanol path. Via this analysis, we can also confirm the crucial role of the interface and the Cu atom in driving methanol synthesis, where the latter actively participates in the cleavages; the interface allows the resulting species to be firmly bonded to the system, avoiding their desorption. All the calculated Bader values in this section are summarized in the ESI† (Tables S13–S15).
4. Conclusions
We evaluated via first-principles calculations the mechanism of methanol and water formation from CO2/CO and H2 catalyzed by the Cu/2D-Mo2C 0.67 O ML model, finding additional feasible routes for CO2 hydrogenation to those previously reported.30 The energy barriers confirm that the route via HCOO*, HCO*, HCOH*, and H2COH* as reaction intermediates allows methanol production via lower energy barriers than those previously reported (A2 path). Formate formation is the most energy-demanding step for these alternative pathways. On the other hand, the best and easiest way to obtain CO is by directly cleaving CO2, forming CO* and O*. Subsequently, the most feasible path for the CO hydrogenation to methanol occurs via the HCOH* intermediate (B2 path) from HCO* species adsorbed on Cu. Also, the migration of CO to the support and its subsequent hydrogenation, as described on the B2+B2sup path, is another feasible mechanism for obtaining methanol. Based on the electronic and free energies evaluated, we can conclude that CH3OH and CO formation are affordable under reaction conditions. Therefore, the catalytic system allows both, and it seems reasonable that there is no high selectivity toward either of the two products.Our study revealed that the Cu/Mo2COx interface is crucial in providing low-energy pathways for CO2/CO hydrogenation to methanol. Both the Cu atom and the Mo2COx support are actively involved in the reaction mechanism, facilitating successive heterolytic cleavages of molecular hydrogen (H2) to form critical intermediates such as HCOO*, HCOOH*, H2COOH*, and H3CO*. This contrasts with classical heterogeneous catalysts based on Cu nanoparticles supported on oxides. We found that the most energy-demanding step in these pathways is the formation of HCOO* or HCO* species, with energy barriers around 103–102 kJ mol−1, respectively. Our results also identified feasible energy profiles for the RWGS reaction, with the HCOO* pathways (C3 path) more likely than via the COOH* species (C2). DFT calculations highlight the most likely intermediates and transition states and allow us to propose a feasible energy profile on the Cu/2D-Mo2C 0.67 O ML. The most energy-demanding step of the HCOO pathways is forming water. Nevertheless, the most viable paths to obtain CO* and H2O* are those starting with the direct cleavage of one C–O bond of CO2.
Using Bader charge analysis, we confirmed the nature of the heterolytic cleavages that enable successive hydrogenations, highlighting the crucial role of charge polarization. These findings provide insights into CO2 and CO hydrogenation, demonstrating the unique advantages of single-atom catalysts and metal–support interfaces in enhancing catalytic performance.
Data availability
Data for this article, including input and output files will be available at Zenodo at [https://doi.org/10.5281/zenodo.13950412].Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the Spanish “Ministerio de Ciencia e Innovación” for funding the “I + D Generación del Conocimiento” project (PID 2021-128416NB-I00) and acknowledge the grant to AV-L (PRE 2019-089647).References
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Irreversible climate change due to carbon dioxide emissions, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(6), 1704–1709 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Recycling of carbon dioxide to methanol and derived products—closing the loop, Chem. Soc. Rev., 2014, 43(23), 7995–8048 RSC.
- G. A. Olah, Beyond oil and gas: the methanol economy, Angew. Chem., Int. Ed., 2005, 44(18), 2636–2639 CrossRef CAS PubMed.
- R. Guil-López, N. Mota, J. Llorente, E. Millán, B. Pawelec, J. L. G. Fierro and R. M. Navarro, Methanol Synthesis from CO2: A Review of the Latest Developments in Heterogeneous Catalysis, Materials, 2019, 12(23), 3902–3926 CrossRef PubMed.
- S. T. Yong, C. W. Ooi, S. P. Chai and X. S. Wu, Review of methanol reforming-Cu-based catalysts, surface reaction mechanisms, and reaction schemes, Int. J. Hydrogen Energy, 2013, 38(22), 9541–9552 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker and F. Abild-Pedersen, et al., The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts, Science, 2012, 336(6083), 893–897 CrossRef CAS PubMed.
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlögl and M. G. Willinger, Formation of a ZnO overlayer in industrial Cu/ZnO/Al2O3 catalysts induced by strong metal-support interactions, Angew. Chem., Int. Ed., 2015, 54(15), 4544–4548 CrossRef CAS PubMed.
- R. Schlögl, Chemical batteries with CO2, Angew. Chem., Int. Ed., 2022, 61(7), e202007397 CrossRef PubMed.
- G. Pacchioni, From CO2 to Methanol on Cu/ZnO/Al2O3 Industrial Catalyst. What Do We Know about the Active Phase and the Reaction Mechanism?, ACS Catal., 2024, 14(4), 2730–2745 CrossRef CAS.
- A. Beck, M. A. Newton, L. G. van de Water and J. A. van Bokhoven, The Enigma of Methanol Synthesis by Cu/ZnO/Al2O3-Based Catalysts, Chem. Rev., 2024, 124(8), 4543–4678 CrossRef CAS PubMed.
- F. Studt, F. Abild-Pedersen and J. B. Varley, et al., CO and CO2 Hydrogenation to Methanol Calculated Using the BEEF-vdW Functional, Catal. Lett., 2013, 143, 71–73 CrossRef CAS.
- S. Posada-Pérez, P. J. Ramírez, R. A. Gutiérrez, D. J. Stacchiola, F. Viñes and P. Liu, et al., The conversion of CO2 to methanol on orthorhombic β-Mo2C and Cu/β-Mo2C catalysts: mechanism for admetal induced change in the selectivity and activity, Catal. Sci. Technol., 2016, 6, 6766–6777 RSC.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, 2D metal carbides and nitrides (MXenes) for energy storage, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- A. Kurlov, E. B. Deeva and P. M. Abdala, et al., Exploiting two-dimensional morphology of molybdenum oxycarbide to enable efficient catalytic dry reforming of methane, Nat. Commun., 2020, 11, 4920 CrossRef CAS PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu and M. Heon, et al., Two-dimensional nanocrystals produced by exfoliation of Ti3AlC2, Adv. Mater., 2011, 23(37), 4248–4253 CrossRef CAS PubMed.
- Á. Morales-García, A. Fernández-Fernández, F. Viñes and F. Illas, CO2 abatement using two-dimensional MXene carbides, J. Mater. Chem. A, 2018, 6, 3381–3385 RSC.
- C. E. Creissen and M. Fontecave, Keeping sight of copper in single-atom catalysts for electrochemical carbon dioxide reduction, Nat. Commun., 2022, 13, 2280 CrossRef CAS PubMed.
- N. Cheng, L. Zhang and K. Doyle-Davis, et al., Single-Atom Catalysts: From Design to Application, Electrochem. Energy Rev., 2019, 2, 539–573 CrossRef.
- H. Wei, X. Liu and A. Wang, et al., FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes, Nat. Commun., 2014, 5(1), 1–8 Search PubMed.
- B. Qiao, A. Wang and X. Yang, et al., Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3(8), 634–641 CrossRef CAS PubMed.
- Y. Lu, J. Wang and L. Yu, et al., Identification of the active complex for CO oxidation over single-atom Ir-on-MgAl2O4 catalysts, Nat. Catal., 2019, 2(2), 149–156 CrossRef CAS.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang and X. Wang, et al., Remarkable performance of Ir1/FeO x single-atom catalyst in water gas shift reaction, J. Am. Chem. Soc., 2013, 135(41), 15314–15317 CrossRef CAS PubMed.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, et al., Heterogeneous single-atom catalysts for electrochemical CO2 reduction reaction, Adv. Mater., 2020, 32(34), 2001848 CrossRef CAS PubMed.
- Y. Zhu, X. Yang, C. Peng, C. Priest, Y. Mei and G. Wu, Carbon-Supported Single Metal Site Catalysts for Electrochemical CO2 Reduction to CO and Beyond, Small, 2021, 17(16), 2005148 CrossRef CAS PubMed.
- H. Xiong, A. K. Datye and Y. Wang, Thermally Stable Single-Atom Heterogeneous Catalysts, Adv. Mater., 2021, 33(50), 2004319 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Single-atom catalysts: emerging multifunctional materials in heterogeneous catalysis, Adv. Energy Mater., 2018, 8(1), 1701343 CrossRef.
- H. Oschinski, Á. Morales-García and F. Illas, Interaction of First Row Transition Metals with M2C (M = Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, and W) MXenes: A Quest for Single-Atom Catalysts, J. Phys. Chem. C, 2021, 125(4), 2477–2484 CrossRef CAS.
- M. Zhang, C. Lai, B. Li, S. Liu, D. Huang and F. Xu, et al., MXenes as Superexcellent Support for Confining Single Atom: Properties, Synthesis, and Electrocatalytic Applications, Small, 2021, 17(29), 2007113 CrossRef CAS PubMed.
- S. Baskaran and J. Jung, Mo2CS2-MXene supported single-atom catalysts for efficient and selective CO2 electrochemical reduction, Appl. Surf. Sci., 2022, 592(1), 153339 CrossRef CAS.
- H. Zhou, Z. Chen and A. V. López, et al., Engineering the Cu/Mo2CTx (MXene) interface to drive CO2 hydrogenation to methanol, Nat. Catal., 2021, 4, 860–871 CrossRef CAS.
- S. Docherty and C. Copéret, J. Am. Chem. Soc., 2021, 143(18), 6767–6780 CrossRef CAS PubMed.
- M. S. Duyar, C. Tsai, J. L. Snider, J. A. Singh, A. Gallo and J. S. Yoo, et al., A Highly Active Molybdenum Phosphide Catalyst for Methanol Synthesis from CO and CO2, Angew. Chem., Int. Ed., 2018, 57, 15045–15050 CrossRef CAS PubMed.
- J. Kim, B. B. Sarma, E. Andrés, N. Pfänder, P. Concepción and G. Prieto, Surface Lewis Acidity of Periphery Oxide Species as a General Kinetic Descriptor for CO2 Hydrogenation to Methanol on Supported Copper Nanoparticles, ACS Catal., 2019, 9(11), 10409–10417 CrossRef CAS.
- M. Zabilskiy, V. L. Sushkevich and D. Palagin, et al., The unique interplay between copper and zinc during catalytic carbon dioxide hydrogenation to methanol, Nat. Commun., 2020, 11, 2409 CrossRef CAS PubMed.
- Z. Zhang, X. Chen and J. Kang, et al., The active sites of Cu–ZnO catalysts for water gas shift and CO hydrogenation reactions, Nat. Commun., 2021, 12, 4331 CrossRef CAS PubMed.
- N. D. Nielsen, J. Thrane and A. D. Jensen, et al., Bifunctional Synergy in CO Hydrogenation to Methanol with Supported Cu, Catal. Lett., 2020, 150, 1427–1433 CrossRef CAS.
- A. Müller, A. Comas-Vives and C. Copéret, Ga and Zn increase the oxygen affinity of Cu-based catalysts for the COx hydrogenation according to ab initio atomistic thermodynamics, Chem. Sci., 2022, 13, 13442–13458 RSC.
- A. Müller, A. Comas-Vives and C. Copéret, Shape and Surface Morphology of Copper Nanoparticles under CO2 Hydrogenation Conditions from First Principles, J. Phys. Chem. C, 2020, 125(1), 396–409 CrossRef.
- E. Lam, J. J. Corral-Pérez, K. Larmier, G. Noh, P. Wolf, A. Comas-Vives, A. Urakawa and C. Copéret, CO2 Hydrogenation on Cu/Al2O3: Role of the Metal/Support Interface in Driving Activity and Selectivity of a Bifunctional Catalyst, Angew. Chem., Int. Ed., 2019, 58(39), 13989–13996 CrossRef CAS PubMed.
- K. Larmier, W. C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Copéret, CO2-to-Methanol Hydrogenation on Zirconia-Supported Copper Nanoparticles: Reaction Intermediates and the Role of the Metal Support Interface, Angew. Chem., Int. Ed., 2017, 56(9), 2318–2323 CrossRef CAS PubMed.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander and A. Tarasov, et al., The mechanism of CO and CO2 hydrogenation to methanol over Cu-based catalysts, ChemCatChem, 2015, 7(7), 1105–1111 CrossRef CAS.
- H. Ruland, H. Song, D. Laudenschleger, S. Stürmer, S. Schmidt and J. He, et al., CO2 hydrogenation with Cu/ZnO/Al2O3: a benchmark study, ChemCatChem, 2020, 12(12), 3216–3222 CrossRef CAS.
- J. Nakamura, Y. Choi and T. Fujitani, On the Issue of the Active Site and the Role of ZnO in Cu/ZnO Methanol Synthesis Catalysts, Top. Catal., 2003, 22, 277–285 CrossRef CAS.
- G. Wang, S. Mine and D. Chen, et al., Accelerated discovery of multi-elemental reverse water-gas shift catalysts using extrapolative machine learning approach, Nat. Commun., 2023, 14, 5861 CrossRef CAS PubMed.
- C. Zhou, J. Zhang, Y. Fu and H. Dai, Recent Advances in the RWG Conversion Reaction, Molecules, 2023, 28, 7657 CrossRef CAS PubMed.
- M. González-Castaño, B. Dorneanu and H. Arellano-García, et al., The reverse water gas shift reaction: a process systems engineering perspective, React. Chem. Eng., 2021, 6, 954–976 RSC.
- M. B. Fichtl, D. Schlereth, N. Jacobsen, I. Kasatkin, J. Schumann and M. Behrens, et al., Kinetics of deactivation on Cu/ZnO/Al2O3 methanol synthesis catalysts, Appl. Catal., A, 2015, 502(5), 262–270 CrossRef CAS.
- L. Grajciar, C. J. Heard, A. A. Bondarenko, M. V. Polynski, J. Meeprasert, E. A. Pidko and P. Nachtigall, Towards operando computational modeling in heterogeneous catalysis, Chem. Soc. Rev., 2018, 47, 8307–8348 RSC.
- M. Figueras, R. A. Gutiérrez, F. Viñes, P. J. Ramírez, J. A. Rodríguez and F. Illas, Supported Molybdenum Carbide Nanoparticles as an Excellent Catalyst for CO2 Hydrogenation, ACS Catal., 2021, 11(15), 9679–9687 CrossRef CAS.
- A. Jurado, Á. Morales-García, F. Viñes and F. Illas, Identifying the Atomic Layer Stacking of Mo2C MXene by Probe Molecule Adsorption, J. Phys. Chem. C, 2021, 125(48), 26808–26813 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(20), 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- G. Kresse and J. Hafner, Norm-conserving and ultrasoft pseudopotentials for first row and transition elements, J. Phys.: Condens. Matter, 1994, 6, 8245–8257 CrossRef CAS.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis and J. K. Nørskov, et al., Density functionals for surface science: exchange-correlation model development with Bayesian error estimation, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85(23), 235149 CrossRef.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen and M. Dułak, et al., The atomic simulation environment—a Python library for working with atoms, J. Phys.: Condens. Matter, 2017, 29(27), 273002 CrossRef PubMed.
- H. Jonsson, G. Mills and K. Jacobsen, Classical and Quantum Dynamics in Condensed Phase Simulations, ed. B. J. Berne, Singapore World Scientific, 1998, pp. 385–404 Search PubMed.
- W. Tang, E. Sanville and G. Henkelman, A grid-based Bader analysis algorithm without lattice bias, J. Phys.: Condens. Matter, 2009, 21(8), 084204 CrossRef CAS PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, Improved grid-based algorithm for Bader charge allocation, J. Comput. Chem., 2007, 28(5), 899–908 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36(3), 354–360 CrossRef.
- M. Yu and D. R. Trinkle, Accurate and efficient algorithm for Bader charge integration, J. Chem. Phys., 2011, 134, 064111 CrossRef PubMed.
- A. Álvarez, M. Borges, J. J. Corral-Pérez, J. Giner Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, CO2 Activation over Catalytic Surfaces, ChemPhysChem, 2017, 18(22), 3135–3141 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00703d |
| This journal is © The Royal Society of Chemistry 2024 |