
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic N2O decomposition in an electric field at low temperatures†
Ayaka
Shigemoto
 *a,
Takuma
Higo
a,
Chihiro
Ukai
a,
Yuki
Inoda
a,
Kenta
Mitarai
b and
Yasushi
Sekine
*a
*a,
Takuma
Higo
a,
Chihiro
Ukai
a,
Yuki
Inoda
a,
Kenta
Mitarai
b and
Yasushi
Sekine
*a
aApplied Chemistry, Waseda University, 3-4-1, Okubo, Shinjuku, Tokyo, Japan. E-mail: ayaka.shigemoto@nifty.com; ysekine@waseda.jp
bResearch & Development Centre, Yanmar Holdings, 2481, Umegahara, Maibara, Shiga 521-8511, Japan
First published on 8th July 2024
Abstract
Nitrous oxide (N2O) exerts strong effects on global warming and environmental destruction. Various catalytic technologies have been investigated for N2O abatement. We investigated a catalytic system in an electric field, revealing that N2O can be decomposed efficiently, even at low temperatures and in the presence of excess oxygen and water vapour. Reaction mechanisms with and without an electric field have been investigated using kinetics and various operando analyses, which revealed that surface-lattice oxygen on catalyst supports plays a crucially important role in N2O decomposition in an electric field at low temperatures.
Introduction
In recent years, much attention has been devoted to nitrous oxide (N2O) because of its strong environmental effects. In fact, its global warming potential is approximately 300 times greater than that of CO2. It is also a major stratospheric ozone-depleting substance.1–3 Moreover, N2O has a long lifetime of 116 ± 9 years in the atmosphere.1,4 Mainly, N2O emissions are produced by human activities, including nitric acid production, adipic acid synthesis, fossil fuel combustion, waste incineration, and automobile exhaust emissions.1,5–7 Addressing the elimination of N2O at low temperatures has, therefore, become imperative for environmental protection. Various post-treatment technologies have been developed and implemented to address N2O emissions in chemical and energy industries. These are classifiable as thermal decomposition, selective catalytic reduction, direct catalytic decomposition, and plasma and microwave heating.7–11 Among these, catalytic N2O decomposition, as outlined in eqn (1), stands out as a particularly promising approach because of its simplicity, lower energy requirements, and, consequently, lower costs.7,12–14| 2N2O → 2N2 + O2 | (1) |
To date, numerous and diverse catalytic technologies have emerged for N2O abatement, encompassing supported metal oxide/novel metal-supported catalysts,15–18 ion-exchanged zeolites,19–21 and composite oxide catalysts including perovskites,22–24 hydrotalcite,25,26 and spinels.27,28 Most notably, rhodium (Rh) catalysts have emerged as particularly potent catalysts, exhibiting high activity for N2O decomposition at low temperatures.2,7,8,29–31
Regarding the reaction mechanism of direct catalytic N2O decomposition, the widely accepted model comprises the following three fundamental elementary steps:32–35
| N2O + * → N2(g) + O* | (2) |
| O* + O* ↔ O2(g) + 2* | (3) |
| N2O + O* → N2(g) + O2(g) + * | (4) |
The N2O molecule is adsorbed onto active sites (*) of the catalyst surface and is subsequently decomposed into N2(g) and adsorbed oxygen (eqn (2)). The active sites are then regenerated through the Langmuir–Hinshelwood (LH) and/or Eley–Rideal (ER) mechanism, completing the entire catalytic cycle. In other words, the catalyst surface is regenerated through the recombination of adsorbed oxygen species (eqn (3): the LH mechanism) or through a reaction with another gaseous N2O molecule (eqn (4): the ER mechanism). It is noteworthy that the recombination of oxygen atoms is reversible, whereas the regeneration of active sites by N2O is irreversible. The reversibility of the former reaction explains why O2 has an inhibitory effect on most N2O decomposition catalysts, particularly at low temperatures, because N2O and O2 compete for the active sites of the catalyst.36–38 Furthermore, industrial exhaust typically contains excess O2 and H2O, which deactivate active sites and reduce catalytic activity.15,38 Consequently, a catalytic N2O abatement process that is capable of decomposing N2O efficiently, even at low temperatures and even in the presence of an excess of oxygen and water vapour, must be developed.
As a promising method for greenhouse gas removal, an electric-field-applied catalytic system is very suitable for low-temperature catalytic synergistic reactions. In recent years, an increasing number of electric-field-applied catalytic systems has been employed for converting NOx, exhibiting remarkable N2 selectivity.39–41 Within the framework of those earlier studies, results show that the application of a mere few milliamperes of weak direct current to the catalyst bed can enhance the oxygen release capacity of the catalyst at low temperatures, consequently lowering the temperature necessary for gas decomposition and conversion reactions.39–43
For this study, Rh catalysts supported on ceria–zirconia (Ce0.7Zr0.3O2) were prepared for the catalytic decomposition of N2O. The applied electric-field catalytic system operates effectively at temperatures as low as 373 K, even with excess O2 and H2O. A comprehensive understanding of the reaction mechanism was sought through kinetic studies and isotope analysis. The findings in the study have elucidated that the applied electric field mitigates oxygen's inhibition of N2O decomposition, thereby facilitating completion of the catalytic N2O decomposition cycle.
Experimental
Catalyst preparation
We prepared mixed oxides of Ce1−xZrxO2 (x = 0, 0.1, 0.3, 0.5) for the catalyst support. Each is suitable for the catalytic reaction in the electric field. Preparation was conducted using a citric acid complex method with aqueous solutions of Ce(NO3)3·6H2O and ZrO(NO3)2·2H2O (Kanto Chemical Co. Inc.), with excess amounts of citric acid and ethylene glycol (Kanto Chemical Co. Inc.). The molar ratio of metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :citric acid:ethylene glycol was 1:3:3. After the obtained solution was evaporated in a water bath for 17 h at 353 K, the solution was dried on a hot plate with stirring. The obtained powder was pre-calcined and then calcined in air at 1123 K for 10 h.
:citric acid:ethylene glycol was 1:3:3. After the obtained solution was evaporated in a water bath for 17 h at 353 K, the solution was dried on a hot plate with stirring. The obtained powder was pre-calcined and then calcined in air at 1123 K for 10 h.
Next, we supported Rh, Fe, Co, Ni or Cu nanoparticles on Ce1−xZrxO2 (x = 0, 0.1, 0.3, 0.5) using an impregnation method. Table S1† presents the precursors. They were dried at 393 K for 20 h and were calcined in air at 823 K for 3 h. The structural information of the obtained catalysts is presented in Fig. S1 and Table S2 in the ESI.†
Activity tests
Catalytic activity tests were conducted in a fixed-bed flow-type quartz reactor at atmospheric pressure, as shown in Fig. S2 in the ESI.† An electric field was applied using a power supply via stainless steel electrodes, which are in contact with the catalyst bed on the upper and lower sides. Here, 100 or 200 mg of catalyst (sieved to 250–500 μm mesh) was placed in a quartz tube with a 6.0 mm internal diameter. The catalyst was treated in Ar for 20 min at 773 K. Reactant gases were a mixture comprising 1000 ppm of N2O, 10% O2, and 10% H2O (when used). The total gas flow rate was 100 mL min−1. The gas hourly space velocity was approximately 50000 or 100000 h−1.
For activity tests, the catalyst bed temperature was increased stepwise from 373 to 773 K. The steady state activity was evaluated at each temperature. To evaluate the catalytic activity in the electric field, a direct current of 6 mA was applied to the catalyst bed. The applied current and response voltage were measured using a digital phosphor oscilloscope (TDS 2001C with a voltage probe P6015A; Tektronix Inc.). Also, a thermocouple was set at the bottom of the catalyst bed to measure the actual catalyst bed temperature. The outlet gases were detected using a quadrupole mass spectrometer (ThermoStar GSD 350; Pfeiffer Vacuum GmbH). The N2O conversion and N2 selectivity were calculated based on the following:
 | (5) |
 | (6) |
In these equations, [N2O]in and [N2O]out respectively represent the inlet and outlet N2O concentrations. rN2 and rN2O conv. respectively represent the N2 generation rate and N2O conversion rate.
Tests for measuring the dependence on the partial pressure of N2O and O2 were conducted under a N2O–O2–H2O condition at 423 K with the electric field and 638 K without the electric field. The partial reaction orders with respect to N2O and O2 were found with different concentrations of N2O (500–2000 ppm) and O2 (0.05–15%) balanced with Ar.
Transient response tests using isotope-labelled 18O2
Isotope-labelled 18O2 (99.5% 18O2) was obtained from Nippon Sanso Holdings Corp. The 18O tracer-loaded catalyst was prepared as explained hereinafter. The catalyst was treated with 18O2 at 523 K for 10 min after H2 reduction at 773 K for 30 min. After purging the catalyst with Ar to remove physisorbed 18O, activity tests using 1000 ppm of N216O (when used) and 10% 16O2 were conducted at 423 K with the 6 mA electric field. Furthermore, tests were conducted in which the catalyst was treated with 16O2 and was then exposed to 18O2 with and without the electric field. The following AMUs were used for mass spectroscopic identification of the different compounds: 28 (N2), 32 (16O2), 34 (16O18O), 36 (18O2), 40 (Ar), and 44 (N2O).Operando transmission infrared spectroscopy (TIRS)
Operando TIRS measurements were conducted using a Fourier transform infrared spectrometer (FT/IR 4600; Jasco Corp.) with an MCT detector and a CaF2 window. The 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst (50 mg) mixed with KBr was pressed and shaped into a 10 mmϕ disk. All spectra were recorded with 4 cm−1 resolution and 50 scans. Regarding the pretreatment, the catalyst disk was heated at 773 K for 20 min under Ar. The background spectra (denoted as BKG) were measured under inert Ar gas at 373 or 573 K. After the BKG spectra were recorded, 1000 ppm N2O and 10% O2 balanced with Ar were dosed in the IR cell at 373 or 573 K with and without the electric field. The electric field was applied with 6 mA of direct current, and all flow rates were 100 mL min−1.Structural characterisation of the catalyst
Powder X-ray diffraction (XRD) measurements were performed to confirm the crystalline structures of the catalysts (SmartLab 3; Rigaku Corp.). Cu Kα radiation was applied at 40 kV and 40 mA.The specific surface areas were calculated from the N2 adsorption isotherm at 77 K using the Brunauer–Emmett–Teller (BET) method (Gemini VII; Micromeritic Instrument Corp.). Before the measurements, the samples were heated to 473 K for 1 h under a N2 atmosphere to remove adsorbates from the catalyst surface.
The exposed metal surface area and dispersion were measured by conducting CO pulse chemisorption on a catalyst analyser (MRB BELCAT II; Microtrac Inc.). The pulse measurement was carried out by introducing pulses of 10% CO in He at 323 K. The instrument was equipped with a TCD to measure the CO uptake. Calculations were based on an Rh:CO stoichiometry factor of 1:1.
The electronic states of Rh in Rh/Ce0.7Zr0.3O2 were evaluated using operando X-ray absorption fine structure (XAFS) spectroscopy at the BL14B2 beamline of SPring-8 in Japan. After the 5 wt% Rh/Ce0.7Zr0.3O2 powder mixed with BN was pressed into a 10 mmϕ disk, the sample disk was placed in the cell. Then, XAFS measurements were performed in the transmission mode. Operando XAFS measurements were carried out under 1000 ppm N2O in N2 balance with a total flow rate of 100 mL min−1 at 373, 473, 573 and 673 K with and without the electric field. The same pre-treatment to the activity test was done. The applied current was 6 mA. The response voltage was approximately 0.10–0.20 kV.
Results and discussion
Catalytic activities for N2O decomposition
Fig. 1 depicts the catalytic conversions of N2O over the 0.5 wt% Rh/Ce0.7Zr0.3O2 and 5 wt% Fe, Co, Ni, Cu/Ce0.7Zr0.3O2 catalysts with and without an electric field by application of 6 mA of direct current under 1000 ppm N2O + 10% O2. Remarkably, the application of the electric field resulted in significantly high N2O conversion to N2 for all catalysts, even at 473 K. Specifically, the 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst exhibited the highest N2O conversion of approximately 75%. In the catalytic reaction without the electric field, N2O conversion was almost zero. | ||
| Fig. 1 Catalytic decomposition of N2O over 0.5 wt% Rh/Ce0.7Zr0.3O2 and 5 wt% Fe, Co, Ni, Cu/Ce0.7Zr0.3O2 catalysts with and without the electric field at 473 K. Conditions: 1000 ppm N2O + 10%O2; catalyst weight, 200 mg; total flow rate, 100 mL min−1; SV, 50000 h−1; current, 0 or 6 mA. | ||
Fig. S3 in the ESI† shows the results of N2O conversion over 0.5 wt% Rh/Ce1−xZrxO2 (x = 0, 0.1, 0.3 and 0.5) catalysts. The 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst exhibited high N2O decomposition activity with and without the electric field. For that reason, the 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst was selected for additional analyses.
Fig. 2 shows the catalytic activities for N2O decomposition over the 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst and Ce0.7Zr0.3O2 support with and without the electric field under 1000 ppm N2O + 10% O2 + 0 or 10% H2O. Fig. 2(A) confirmed the excellent promotion of catalytic activity by application of the electric field, even at low temperatures. These plots show the steady-state activity after 90 min. The selectivity for N2 generation was almost 100% (Fig. S4 in the ESI†). Fig. 2(B) shows N2O decomposition over the Ce0.7Zr0.3O2 support (i.e. no supported metal) in the presence of 1000 ppm N2O + 10% O2. Negligible catalytic performance was observed without the electric field at temperatures lower than 700 K. Very low (approximately 10% N2O conversion) activities were achieved at 423–773 K with the electric field, which suggests that the high de-N2O catalytic performance observed at low temperatures in Fig. 2(A) is attributable mainly to Rh. Fig. 2(C) shows the N2O conversion with time on stream over the 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst. The activity without the electric field decreased gradually over time at 573 K, whereas the activity remained stable for at least 90 min at 373 K when applying the electric field. One can infer that O2 strongly adsorbs on the catalyst surface, inhibiting the reaction which occurs without the electric field. Alternatively, one can speculate that the detrimental effect of oxygen on the catalyst is mitigated by electric field application. Oxygen effects on catalytic activity are discussed later herein.
 | ||
| Fig. 2 Catalytic conversion of N2O with and without the electric field. (A) 1000 ppm N2O + 10% O2 for 0.5 wt% Rh/Ce0.7Zr0.3O2. (B) 1000 ppm N2O + 10% O2 for Ce0.7Zr0.3O2. (C) Conversion of N2O with time on stream over the 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst. (D) 1000 ppm N2O + 10% O2 + 10% H2O for 0.5 wt% Rh/Ce0.7Zr0.3O2. (E) Effect of H2O on N2O conversion over 0.5 wt% Rh/Ce0.7Zr0.3O2 with the electric field at 433 K. Weight of the catalyst, 100 or 200 mg; total flow rate, 100 mL min−1; SV, 50000 or 100000 h−1; current, 0 or 6 mA. | ||
To investigate the H2O effects on the steady-state activity, N2O decomposition with the introduction of H2O was conducted as shown in Fig. 2(D). Although the presence of 10% H2O in the feed adversely affected the catalyst activity, approximately 30% N2O conversion was achieved at 435 K when using the electric field. In addition, H2O-switching experiments for N2O decomposition were performed as a function of time-on-stream, as shown in Fig. 2(E) with the electric field and in Fig. S5 in the ESI† without the electric field. As shown clearly in those figures, the N2O conversion is lower with the introduction of 10% H2O, both with and without the application of the electric field, but it recovers immediately as soon as H2O is removed from the feed gas stream, which indicates that H2O has a completely reversible detrimental effect on de-N2O catalytic performance.
During these tests using the electric field, the catalyst bed temperature was measured directly with a thermocouple to verify the effects of Joule heating by the applied direct current on catalytic activity. All graphs showing the temperature dependence of activity show N2O conversion against the actual catalyst bed temperature. Remarkable enhancement of catalytic activity was observed, suggesting that the high activity and selectivity are not solely attributable to Joule heating. Moreover, structural analyses of the 0.5 wt% Rh/Ce0.7Zr0.3O2 catalysts and Ce0.7Zr0.3O2 support before and after the reaction are presented in Fig. S6 and Table S3 in the ESI.† It is noteworthy that the crystal structure, BET surface area, and Rh particle size were equal before and after the activity tests. None changed.
Reaction site in the N2O decomposition mechanism
To elucidate the reaction site for N2O decomposition, we evaluated the turnover frequency (TOF) with various amounts of Rh-supported catalysts. The TOF values determined by the Rh surface area (TOF-s) and by the perimeter of the Rh–support interface (TOF-p) are calculated based on the following equations: | (7) |
 | (8) |
Data from Table S4† were used to calculate TOF. Fig. S7 in the ESI† shows that the TOF-s is independent of Rh particle size, which suggests that N2O decomposition proceeds mainly on the Rh surface with and without the electric field. In addition, to confirm the electronic state of Rh loaded on Ce0.7Zr0.3O2 both with and without the application of the electric field during N2O decomposition, operando XAFS measurements were performed. As shown in Fig. 3, the oxidation state of the supported Rh species is Rh2O3, indicating that the active sites for the catalytic N2O decomposition are the Rh2O3 surface.
 | ||
| Fig. 3 Operando Rh K-edge XANES spectra of 5 wt% Rh/Ce0.7Zr0.3O2 under N2O decomposition (A) with and (B) without the electric field. Conditions: 1000 ppm N2O in N2 balance; total flow rate, 100 mL min−1; current, 0 or 6 mA. | ||
Elucidation of N2O decomposition mechanisms
 | ||
| Fig. 4 Dependence of the conversion rate of N2O decomposition over the 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst on the pressures of (A) N2O, (B) O2, and (C) O2. Conditions: 500–2000 ppm N2O + 10% O2 + 10% H2O, or 1000 ppm N2O + 0.05–15% O2 + 10% H2O in Ar balance; weight of the catalyst, 100 mg; total flow rate, 100 mL min−1; SV, 100000 h−1; current, 0 or 6 mA; catalyst bed temperature, 423 K (with EF) and 638 K (without EF). | ||
| With EF | Without EF | |
|---|---|---|
| Reaction order (500–2000 ppm N2O) | 0.70 | 1.10 |
| Reaction order (500–2000 ppm O2) | −0.02 | −0.21 |
| Reaction order (5–15% O2) | −0.06 | −0.51 |
:1 ratio. Therefore, adsorbed oxygen is involved actively in N2O decomposition, such that direct N2O decomposition proceeds at low temperatures even in an excess oxygen atmosphere.
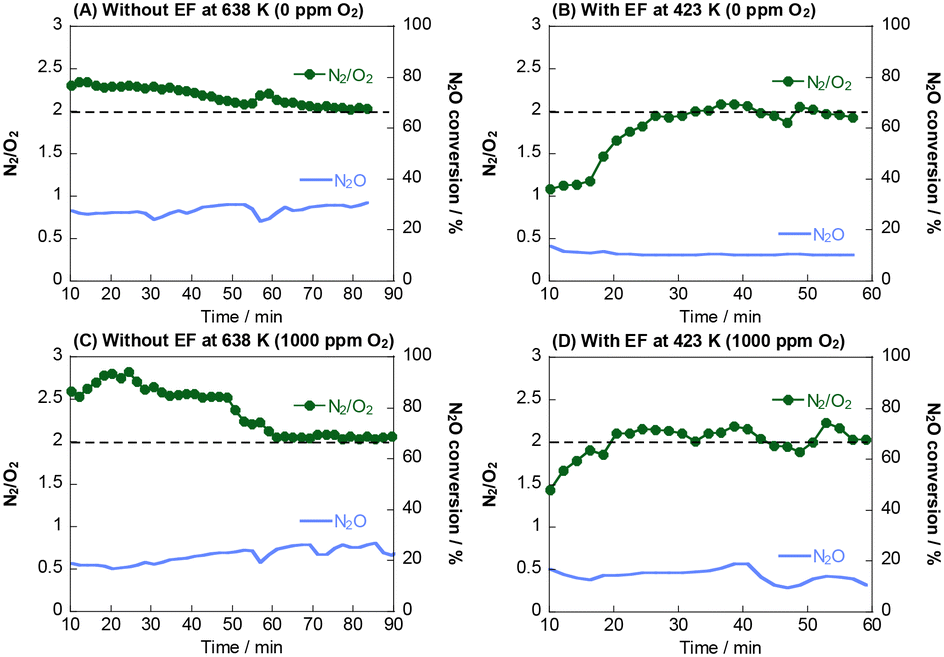 | ||
| Fig. 5 N2/O2 molar ratio under a flow of (A) and (B) 1000 ppm N2O + 10% H2O and (C) and (D) 1000 ppm N2O + 1000 ppm O2 + 10% H2O over the 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst without the electric field at 638 K and with the electric field at 423 K; weight of the catalyst, 100 mg; total flow rate, 100 mL min−1; current, 0 or 6 mA. | ||
 | ||
| Fig. 6 Isotopic responses of m/z = 28, 34, 36 and 44, obtained after switching from 18O2 to N216O + 16O2 over the 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst; weight of the catalyst, 200 mg; total flow rate, 100 mL min−1; SV, 50000 h−1; current, 0 or 6 mA. | ||
Based on the results described above, we propose the catalytic N2O decomposition scheme on the Rh/Ce0.7Zr0.3O2 catalyst with and without the electric field, as depicted in Fig. 7.
 | ||
| Fig. 7 Presumed reaction scheme for N2O decomposition with and without the electric field over the Rh/Ce0.7Zr0.3O2 catalyst. | ||
For N2O decomposition without the electric field (conventional system), we were able to propose that N2O decomposition occurs through the following steps: (i) N2O molecules are adsorbed onto the active sites of the Rh oxide species (marked as Rh*). They subsequently decompose into N2(g) and adsorbed oxygen (eqn (9)). (ii) O2 desorbs to regenerate the active sites (eqn (10)), thereby completing the catalytic N2O decomposition cycle.
| N2O + Rh* → N2(g) + Rh*–O | (9) |
| 2Rh*–O ↔ O2(g) + 2Rh* | (10) |
At low temperatures, the associative desorption of oxygen species is regarded as the rate-determining step.18,38,51 Our findings confirm the inhibitory effect of oxygen on N2O decomposition. Furthermore, Fig. S17† shows the O2-TPD profile for the 0.5 wt% Rh/Ce0.7Zr0.3O2 catalyst without the electric field. The figure shows the desorption of oxygen occurring around 573 K, corresponding to the temperature at which N2O decomposition becomes active without the electric field.
As for N2O decomposition with the electric field, the assumed N2O decomposition in the presence of excess O2 with the electric field can be outlined as explained hereinafter: (i) N2O molecules react with adsorbed oxygen species on the Rh oxide surface. Subsequently, N2(g) and O2(g) are released (eqn (11): ER mechanism). (ii) The catalytic N2O decomposition cycle is completed by the adsorption of oxygen on the Rh oxide surface according to eqn (9).
| N2O + Rh*–O → N2(g) + O2(g) + Rh* | (11) |
It is worth noting that the Ce0.7Zr0.3O2 support is also involved in the N2O decomposition when applying the electric field, as shown in Fig. 2(B). Earlier reports have suggested that the oxidised support can transfer oxygen to the noble metal (eqn (12)) because of strong metal support interactions.30,37,52–54
| Rh* + Ce*–O → Rh*–O + Ce* | (12) |
This oxygen migration step engenders the creation of new N2O decomposition active sites on the Rh oxide surface (denoted as Rh*–O) for the ER mechanism, further driving the reaction presented in eqn (11).
Our observations suggest that the application of the electric field enhances the redox properties of the Ce0.7Zr0.3O2 support and facilitates the reaction in eqn (12), leading to high N2O decomposition activity achieved at low temperatures, even in the presence of excess O2.
Conclusions
The catalytic decomposition of N2O in an electric field at low temperatures has been investigated. The reaction involves adsorption onto Rh oxide sites, followed by desorption of oxygen to regenerate the active sites. At low temperatures, oxygen desorption is the rate-limiting step, indicating an inhibitory effect of oxygen. When an electric field is applied, the N2O decomposition is accelerated by oxygen migration from the Ce0.7Zr0.3O2 support, where N2O reacts with the oxygen adsorbed onto the Rh oxide surface. Increasing the Zr content in the support improves the reduction properties, which correlates with catalytic performance. Transient response tests have demonstrated that the electric field enhanced the oxygen mobility, leading to the migration of surface-lattice oxygen to the catalyst surface and the formation of the active sites (Rh*–O), ultimately increasing the N2O decomposition activity achieved at low temperatures.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualisation: AS, KM and YS; funding acquisition: KM and YS; investigation: AS, YI, and CU; project administration: TH, KM and YS; supervision: YS; validation: AS, YI, CU, TH, and YS; visualization: AS; writing – original draft: AS; writing – review & editing: YS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Seiji Yamazoe (Tokyo Metropolitan University) for great assistance in the XAFS analysis. A part of this work was supported by JSPS KAKENHI (grant no. 22K20484, 23H05404 and 23K20034) from the Japan Society for the Promotion of Science.References
- Climate Change 2021: The Physical Science Basis, Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change.
- Y. Zhang, Z. Tian, L. Huang, H. Fan, Q. Hou, P. Cui and W. Wang, Catalysts, 2023, 13, 943 CrossRef CAS.
- R. Li, Y. Li and Z. Liu, Fuel, 2024, 355, 129405 CrossRef CAS.
- M. J. Prather, J. Hsu, N. M. DeLuca, C. H. Jackman, L. D. Oman, A. R. Douglass, E. L. Fleming, S. E. Strahan, S. D. Steenrod, O. A. Søvde, I. S. A. Isaksen, L. Froidevaux and B. Funke, J. Geophys. Res., 2015, 120, 5693–5705 CrossRef CAS PubMed.
- https://ourworldindata.org/emissions-by-sector, accessed 29th May 2024.
- E. Meloni, M. Martino, S. Renda, O. Muccioli, P. Pullumbi, F. Brandani and V. Palma, Catalysts, 2022, 12, 1405 CrossRef CAS.
- M. Konsolakis, ACS Catal., 2015, 5, 6397–6421 CrossRef CAS.
- M. Jabłońska and R. Palkovits, Catal. Sci. Technol., 2016, 6, 7671–7687 RSC.
- X. Tan, H. Chen, L. Shi, Q. Lu, S. Qi, C. Yi and B. Yang, Catal. Lett., 2023, 153, 3724–3733 CrossRef CAS.
- O. Muccioli, E. Meloni, S. Renda, M. Martino, F. Brandani, P. Pullumbi and V. Palma, Processes, 2023, 11, 1511 CrossRef CAS.
- A. R. Hanna and E. R. Fisher, ACS Catal., 2020, 10, 6546–6560 CrossRef CAS.
- Z. Liu, F. He and S. Peng, Catal. Surv. Asia, 2016, 20, 121–132 CrossRef CAS.
- B. Bromley, C. Pischetola, L. Nikoshvili, F. Cárdenas-Lizana and L. Kiwi-Minsker, Molecules, 2020, 25, 3867 CrossRef CAS PubMed.
- M. Xu, H. X. Wang, H. Ouyang, L. Zhao and Q. Lu, J. Hazard. Mater., 2021, 401, 123334 CrossRef CAS PubMed.
- S. Hinokuma, T. Iwasa, Y. Kon, T. Taketsugu and K. Sato, Sci. Rep., 2020, 10, 21605 CrossRef CAS PubMed.
- N. Richards, J. H. Carter, E. Nowicka, L. A. Parker, S. Pattisson, Q. He, N. F. Dummer, S. Golunski and G. J. Hutchings, Appl. Catal., B, 2020, 264, 118501 CrossRef.
- E. Sadovskaya, L. Pinaeva, V. Skazka and I. Prosvirin, Materials, 2023, 16, 929 CrossRef CAS PubMed.
- Y. Jing, C. He, N. Zhang, Y. Murano, R. Toyoshima, H. Kondoh, Y. Kageyama, H. Inomata, T. Toyao and K. Shimizu, ACS Catal., 2023, 13, 12983–12993 CrossRef CAS.
- T. Zhang, Y. Qiu, G. Liu, J. Chen, Y. Peng, B. Liu and J. Li, J. Catal., 2020, 392, 322–335 CrossRef CAS.
- F. Lin, T. Andana, Y. Wu, J. Szanyi, Y. Wang and F. Gao, J. Catal., 2021, 401, 70–80 CrossRef CAS.
- B. Kang, R. Zhang, M. Guo, X. Guo, Z. Di, Y. Wei and J. Jia, Energy Fuels, 2023, 37, 18019–18029 CrossRef CAS.
- N. Russo, D. Mescia, D. Fino, G. Saracco and V. Specchia, Ind. Eng. Chem. Res., 2007, 46, 4226–4231 CrossRef CAS.
- N. Richards, J. H. Carter, L. A. Parker, S. Pattisson, D. G. Hewes, D. J. Morgan, T. E. Davies, N. F. Dummer, S. Golunski and G. J. Hutchings, ACS Catal., 2020, 10, 5430–5442 CrossRef CAS.
- N. Richards, L. A. Parker, J. H. Carter, S. Pattisson, D. J. Morgan, N. F. Dummer, S. E. Golunski and G. J. Hutchings, Catal. Lett., 2022, 152, 213–226 CrossRef CAS.
- L. Obalová, K. Jirátová, F. Kovanda, K. Pacultová, Z. Lacný and Z. Mikulová, Appl. Catal., B, 2005, 60, 289–297 CrossRef.
- M. Jabłońska, M. A. Arán, A. M. Beale, G. Delahay, C. Petitto, M. Nocuń and R. Palkovits, Appl. Catal., B, 2019, 243, 66–75 CrossRef.
- S. Li, J. Zhao, Z. Song, H. Wang, T. Zhang, J. Liu and Q. Jiang, Fuel, 2024, 362, 130745 CrossRef CAS.
- B. Li, X. Duan, T. Zhao, B. Niu, G. Li, Z. Zhao, Z. Yang, D. Liu, F. Zhang, J. Cheng and Z. Hao, Environ. Sci. Technol., 2024, 58, 2153–2161 CrossRef CAS PubMed.
- M. J. Kim, H. J. Kim, S. J. Lee, I. S. Ryu, H. C. Yoon, K. B. Lee and S. G. Jeon, Catal. Commun., 2019, 130, 105764 CrossRef CAS.
- Y. Li, A. Sundermann, O. Gerlach, K. B. Low, C. C. Zhang, X. Zheng, H. Zhu and S. Axnanda, Catal. Today, 2020, 355, 608–619 CrossRef CAS.
- Y. Jing, K. Taketoshi, N. Zhang, C. He, T. Toyao, Z. Maeno, T. Ohori, N. Ishikawa and K. Shimizu, ACS Catal., 2022, 12, 6325–6333 CrossRef CAS.
- E. R. S. Winter, J. Catal., 1974, 34, 431–439 CrossRef CAS.
- F. Kapteijn, J. Rodriguez-Mirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS.
- T. Yamashita and A. Vannice, J. Catal., 1996, 161, 254–262 CrossRef CAS.
- H. Dandl and G. Emig, Appl. Catal., A, 1998, 168, 261–268 CrossRef CAS.
- E. V. Kondratenko and J. Pérez-Ramírez, Catal. Lett., 2003, 91, 211–216 CrossRef CAS.
- S. Parres-Esclapez, I. Such-Basañez, M. J. Illán-Gómez, C. Salinas-Martínez de Lecea and A. Bueno-López, J. Catal., 2010, 276, 390–401 CrossRef CAS.
- S. Parres-Esclapez, M. J. Illán-Gómez, C. Salinas-Martínez de Lecea and A. Bueno-López, Int. J. Greenhouse Gas Control, 2012, 11, 251–261 CrossRef CAS.
- Y. Omori, A. Shigemoto, K. Sugihara, T. Higo, T. Uenishi and Y. Sekine, Catal. Sci. Technol., 2021, 11, 4008–4011 RSC.
- A. Shigemoto, T. Higo, Y. Narita, S. Yamazoe, T. Uenishi and Y. Sekine, Catal. Sci. Technol., 2022, 12, 4450–4455 RSC.
- A. Shigemoto, Y. Inoda, C. Ukai, T. Higo, K. Oka and Y. Sekine, Chem. Commun., 2024, 60, 1563–1566 RSC.
- K. Sugiura, S. Ogo, K. Iwasaki, T. Yabe and Y. Sekine, Sci. Rep., 2016, 6, 25154 CrossRef CAS PubMed.
- S. Ogo, H. Nakatsubo, K. Iwasaki, A. Sato, K. Murakami, T. Yabe, A. Ishikawa, H. Nakai and Y. Sekine, J. Phys. Chem. C, 2018, 122, 2089–2096 CrossRef CAS.
- K. Takise, A. Sato, K. Muraguchi, S. Ogo and Y. Sekine, Appl. Catal., A, 2019, 573, 56–63 CrossRef CAS.
- C. Sui, F. Yuan, Z. Zhang, C. Zhang, X. Niu and Y. Zhu, Catalysts, 2016, 6, 173 CrossRef.
- S. S. Kim, S. J. Lee and S. C. Hong, Chem. Eng. J., 2011, 169, 173–179 CrossRef CAS.
- A. Wącław, K. Nowińska, W. Schwieger and A. Zielińska, Catal. Today, 2004, 90, 21–25 CrossRef.
- P. J. Smeets, M. H. Groothaert, R. M. van Teeffelen, H. Leeman, E. J. M. Hensen and R. A. Schoonheydt, J. Catal., 2007, 245, 358–368 CrossRef CAS.
- G. D. Pirngruber and J. A. Z. Pieterse, J. Catal., 2006, 237, 237–247 CrossRef CAS.
- M. Zabilskiy, B. Erjavec, P. Djinović and A. Pintar, Chem. Eng. J., 2014, 254, 153–162 CrossRef CAS.
- S. Tanaka, K. Yuzaki, S. Ito, S. Kameoka and K. Kunimori, J. Catal., 2001, 200, 203–208 CrossRef CAS.
- C. Moreau, Á. Caravaca, P. Vernoux and S. Gil, ChemCatChem, 2020, 12, 3042–3049 CrossRef CAS.
- S. S. Kim, S. J. Lee and S. C. Hong, J. Ind. Eng. Chem., 2012, 18, 1263–1266 CrossRef CAS.
- G. S. Zafiris and R. J. Gorte, J. Catal., 1993, 139, 561–567 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00698d |
| This journal is © The Royal Society of Chemistry 2024 |