
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient catalytic direct C–H hydroxylation of benzene by graphite-supported μ-nitrido-bridged iron phthalocyanine dimer†
Yasuyuki
Yamada
 *ab,
Yoshiki
Uno
a,
Chee-Ming
Teoh
a,
Hirotaka
Ohkita
b,
Yuka
Toyoda
b,
Akiko
Sakata
b,
Yutaka
Hitomi
c and
Kentaro
Tanaka
*ad
*ab,
Yoshiki
Uno
a,
Chee-Ming
Teoh
a,
Hirotaka
Ohkita
b,
Yuka
Toyoda
b,
Akiko
Sakata
b,
Yutaka
Hitomi
c and
Kentaro
Tanaka
*ad
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan. E-mail: yamada.yasuyuki.i6@f.mail.nagoya-u.ac.jp; kentaro@chem.nagoya-u.ac.jp
bResearch Center for Materials Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan
cDepartment of Molecular Chemistry and Biochemistry, Graduate School of Science and Engineering, Doshisha University, Kyotanabe, Kyoto, 610-0321, Japan
dResearch Institute for Quantum and Chemical Innovation, Institutes of Innovation for Future Society, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8602, Japan
First published on 31st July 2024
Abstract
Hydroxylation of benzene directly into phenol without converting it into other compounds by using a metal complex-based oxidation catalyst is challenging because of the chemical stability of benzene. We demonstrated that a graphite-supported μ-nitrido-bridged iron phthalocyanine dimer, which acts as a potent iron–oxo-based molecular methane oxidation catalyst, can efficiently catalyze the direct benzene hydroxylation at 25 °C in an aqueous acetonitrile solution containing excess H2O2. It was confirmed that the catalytic benzene hydroxidation activity of the graphite-supported μ-nitrido-bridged iron phthalocyanine dimer was significantly higher than that of the silica gel-supported μ-nitrido-bridged iron phthalocyanine dimers.
1. Introduction
High-valent iron–oxo species are an important class of reactive intermediates that are often involved in the oxidation of organic substrates by natural oxygenases such as methane monooxygenase and cytochrome P450.1–3 Inspired by the fact that these oxygenases achieve difficult C–H activation of organic molecules by utilizing high-valent iron–oxo species under mild reaction conditions, a large variety of artificial iron–oxo-based molecular catalysts have been created.3–6 Artificial iron–oxo-based molecular catalysts are expected to be applicable to the catalytic oxidation reactions of various organic compounds.μ-Nitrido-bridged iron porphyrinoid dimers such as μ-nitrido-bridged iron phthalocyanine dimers are an important class of potent artificial iron–oxo-based molecular catalysts.7–23 For instance, the iron–oxo species of a μ-nitrido-bridged iron phthalocyanine dimer 1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) O, which can be generated by treating 1 or its 1e−-oxidized product 1+ with H2O2 in an acidic aqueous solution, have such a high oxidizing ability that can even catalytically activate the inert C–H bonds of methane at temperatures lower than 100 °C (Fig. 1a).7–9,11,13,14,18 Moreover, we recently demonstrated that the catalytic activity of 1 is significantly enhanced by stacking on the π-surface of graphite.22 We considered that stacking interaction between 1 and graphite would change the electronic structure of both of 1 and 1O to affect the catalytic activity of 1. In order to confirm this assumption, we compared the catalytic methane oxidation activity of the graphite-supported μ-nitrido-bridged iron phthalocyanine dimer 1 (1/G) with that of 1 or 1+·I− on silica gel-support (1/SiO2 or 1+·I−/SiO2). Actually, 1/G showed more than 70 times higher methane oxidation activity than 1/SiO2 or 1+·I−/SiO2 in an acidic aqueous solution at 60 °C. A detailed examination of 1/G via electrochemistry and DFT calculations suggested that the high catalytic activity of 1/G is derived from the electronic interaction between the catalyst molecule and graphite, to cause the partial charge transfer from the catalyst molecule to graphite without changing the redox states of the iron ions in the μ-nitrido-bridged iron phthalocyanine dimer and also lower the singly occupied molecular orbital (SOMO) level of the catalyst molecule on graphite. As a result, electron transfer from methane to 1O/G in the proton-coupled electron transfer process could have been facilitated (Fig. 1b).
O, which can be generated by treating 1 or its 1e−-oxidized product 1+ with H2O2 in an acidic aqueous solution, have such a high oxidizing ability that can even catalytically activate the inert C–H bonds of methane at temperatures lower than 100 °C (Fig. 1a).7–9,11,13,14,18 Moreover, we recently demonstrated that the catalytic activity of 1 is significantly enhanced by stacking on the π-surface of graphite.22 We considered that stacking interaction between 1 and graphite would change the electronic structure of both of 1 and 1O to affect the catalytic activity of 1. In order to confirm this assumption, we compared the catalytic methane oxidation activity of the graphite-supported μ-nitrido-bridged iron phthalocyanine dimer 1 (1/G) with that of 1 or 1+·I− on silica gel-support (1/SiO2 or 1+·I−/SiO2). Actually, 1/G showed more than 70 times higher methane oxidation activity than 1/SiO2 or 1+·I−/SiO2 in an acidic aqueous solution at 60 °C. A detailed examination of 1/G via electrochemistry and DFT calculations suggested that the high catalytic activity of 1/G is derived from the electronic interaction between the catalyst molecule and graphite, to cause the partial charge transfer from the catalyst molecule to graphite without changing the redox states of the iron ions in the μ-nitrido-bridged iron phthalocyanine dimer and also lower the singly occupied molecular orbital (SOMO) level of the catalyst molecule on graphite. As a result, electron transfer from methane to 1O/G in the proton-coupled electron transfer process could have been facilitated (Fig. 1b).
 | ||
| Fig. 1 (a) Formation of a high-valent iron–oxo species 1O from 1 or 1+ for C–H activation of methane. (b) Efficient C–H activation of methane by a graphite-supported high-valent iron–oxo species 1O/G generated from a μ-nitrido-bridged iron phthalocyanine dimer stacked on graphite 1/G. (c) Direct benzene hydroxylation reaction. (d) μ-Nitrido-bridged iron phthalocyanine dimers 2 and 3+·I− that catalyze benzene hydroxylation. | ||
In this manuscript, we report the direct C–H bond activation of benzene by 1/G. As benzene is a chemically stable compound having a high C–H bond dissociation energy of 113 kcal mol−1, direct C–H bond activation of benzene at lower temperature is considered as a difficult C–H activation.24–30 A. B. Sorokin et al. confirmed that 2 can catalytically oxidize benzene to afford a mixture of phenol and p-quinone in a homogeneous aqueous CH3CN solution containing H2O2 at 60 °C.10 Our group also demonstrated that a monocationic μ-nitrido-bridged iron–phthalocyanine dimer having 16 electron-donating peripheral methyl groups on SiO2 (3+·I−/SiO2) showed 10 times higher catalytic benzene oxidation activity than at 40 °C in an aqueous CH3CN solution containing H2O2.20 Considering that 1/G showed considerably higher catalytic methane oxidation activity than those of 1 or 1+, we became interested in comparing the catalytic benzene oxidation activity of 1/G with that of 3+·I−/SiO2. Herein, we compared the catalytic activity of 1/G with that of 3+·I−/SiO2 under the same reaction conditions and demonstrated that 1/G is effective for the efficient catalytic oxidation of benzene at 25 °C.
2. Results and discussion
The graphite-supported catalyst 1/G was prepared according to our previous report; that is, a monocationic form of μ-nitrido-bridged iron phthalocyanine dimer (1+·I−) was heated with graphite in pyridine at 80 °C to adsorb the catalyst molecule on graphite, followed by washing with aqueous trifluoroacetic acid to remove pyridine.22,31 In our previous report, we confirmed that the adsorbed μ-nitrido-bridged iron phthalocyanine dimer was reduced by one electron by graphite to become its neutral form (1) using X-ray photoelectron spectroscopy (XPS) spectroscopy and electrochemical analysis.22Benzene oxidation reactions were performed in a CH3CN solution containing excess amounts of benzene (450 mM), TFA (141 mM), and H2O2 (1.04 M) at 25 °C in the presence of 1+·I−/SiO2, 3+·I−/SiO2 or 1/G (49 μM as 1+, 3+, or 1). A sufficient amount of TFA was added because acidic conditions are effective in producing high-valent iron–oxo species efficiently.7–23 The reaction was monitored and the oxidized compounds were detected by 1H-NMR spectroscopy.
After a 2 h oxidation by 1/G at 25 °C, the production of phenol was observed, as shown in Fig. 2a. A low concentration of p-quinone, which can be generated by the oxidation of 1,4-dihydroxybenznene, was also observed, whereas other types of overoxidized products, such as o-quinone and 1,2-, 1,3-, or 1,4-dihydroxybenzenes, which can be produced by hydroxylation of phenol, were not found. It is considered that the peak observed at 8.14 ppm, which is assignable to HCOOH, was derived from CH3CN because it was observed in the absence of benzene, as shown in Fig. S1,† suggesting that 1/G has such a high oxidizing ability that it can oxidize chemically inert CH3CN even at room temperature. We also performed benzene oxidation using H2O as the solvent instead of CH3CN at 25 °C for 4 h (a two-phase reaction; for details, see ESI† page S4) to confirm that a significant amount of phenol was produced, but p-quinone was not observed under these reaction conditions (entry 14 in Table 1 and Fig. S2†). In an aqueous solution, the graphite surface can be surrounded by benzene molecules because of the hydrophobicity of both the graphite surface and benzene. We assumed that this might have caused the lower production rate of p-quinone by maintaining a high effective concentration of benzene around the graphite surface.
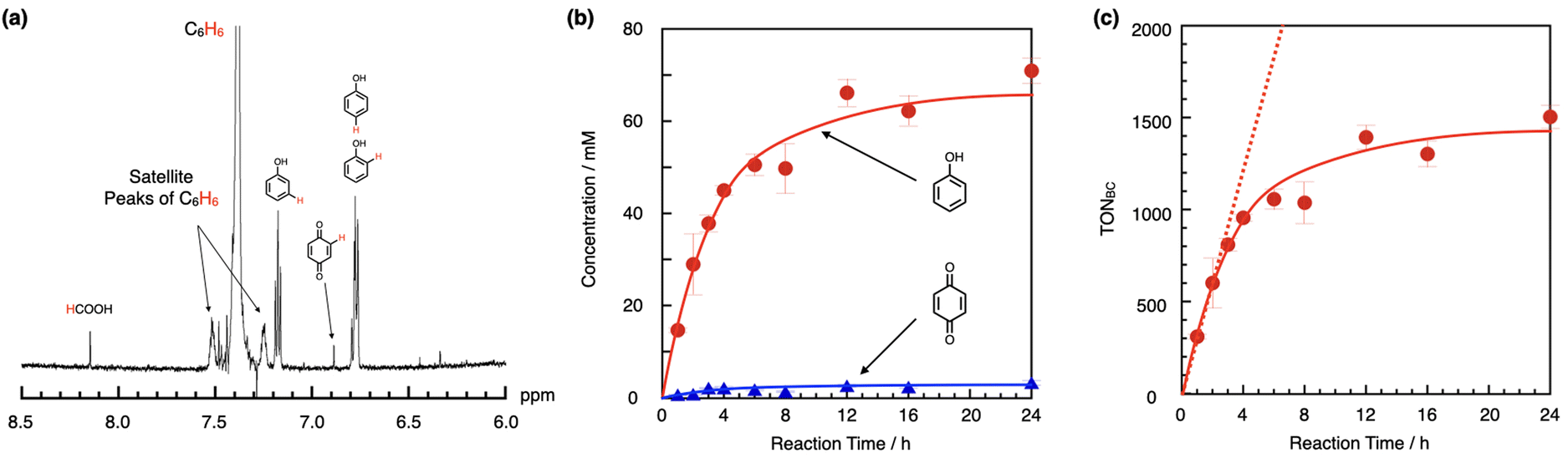 | ||
| Fig. 2 (a) 1H-NMR spectrum of a reaction mixture of benzene oxidation by 1/G at 25 °C for 2 h. (b) Time dependence of the concentrations of phenol (filled red circle) and p-quinone (filled blue triangle) observed in the benzene oxidation reaction performed at 25 °C. (c) Time dependence of TONBC (definition is shown in the main text) for the benzene oxidation reaction at 25 °C. Initial reaction condition: [benzene] = 450 mM, [TFA] = 141 mM, [H2O2] = 1.04 M in CH3CN (1.0 mL) containing 10 mg of 1/G ([1] = 49 μM). Error bars indicate standard deviations of the three independent reactions. | ||
| Entry | Catalyst | Reaction time/h | Solvent | Additive | [Phenol]/mM | [p-Quinone]/mM | TONBC |
|---|---|---|---|---|---|---|---|
| 1 | 1/G | 1 | CH3CN | ⋯ | 14.7 (0.53) | 0.6 (0.1) | 310 (13) |
| 2 | 1/G | 2 | CH3CN | ⋯ | 28.9 (6.6) | 0.8 (0.2) | 601 (137) |
| 3 | 1/G | 3 | CH3CN | ⋯ | 37.8 (1.8) | 2.1 (0.1) | 851 (34) |
| 4 | 1/G | 4 | CH3CN | ⋯ | 45.0 (1.0) | 2.1 (0.4) | 955 (18) |
| 5 | 1/G | 6 | CH3CN | ⋯ | 50.6 (2.4) | 1.8 (0.2) | 1057 (54) |
| 6 | 1/G | 8 | CH3CN | ⋯ | 49.8 (5.4) | 1.4 (0.1) | 1037 (114) |
| 7 | 1/G | 12 | CH3CN | ⋯ | 66.1 (2.9) | 2.6 (0.3) | 1393 (66) |
| 8 | 1/G | 16 | CH3CN | ⋯ | 62.2 (3.3) | 2.3 (0.2) | 1303 (70) |
| 9 | 1/G | 24 | CH3CN | ⋯ | 71.0 (2.8) | 3.3 (0.5) | 1504 (63) |
| 10 | 1/G | 2 | CH3CN | 102 mM DMPO | 18.7 | n.d. | 378 |
| 11 | 3 +·I−/SiO2 | 2 | CH3CN | ⋯ | 3.43 (0.88) | n.d. | 69 (18) |
| 12 | 1 +·I−/SiO2 | 2 | CH3CN | ⋯ | n.d. | n.d. | 0 (0) |
| 13 | 1/SiO2 | 2 | CH3CN | ⋯ | n.d. | n.d. | 0 (0) |
| 14 | 1/G | 4 | H2O/benzene | ⋯ | 11.3 (2.4) | n.d. | 239 (51) |
The time dependence of the concentrations of phenol and p-quinone produced in the reaction at 25 °C in CH3CN was summarized in Fig. 2b and Table 1. Because benzene oxidation can proceed in a stepwise manner from phenol to p-quinone, we calculated the turnover number for benzene consumption (TONBC), according to the following eqn (i), where Cphenol and Cp-quinone, and CCat represent the concentrations of phenol, p-quinone, and the catalyst molecule, respectively.
| TONBC = (Cphenol + Cp-quinone)/CCat | (i) |
As shown in Fig. 2c, the amount of TONBC increased almost linearly during the initial stage of the reaction (up to 2 h) and then gradually saturated. The initial linear increase in TONBC suggests that 1/G worked stably under these reaction conditions, as in the case of CH4 oxidation in an acidic aqueous solution containing excess H2O2.22 We also confirmed that 1/G was reusable after the reaction at 25 °C for 1 h with its catalytic activity almost unchanged from that before use (95%) as summarized in page S6 in the ESI.† The gradual saturation of TONBC was presumably due to the over-oxidation of p-quinone and a decrease in the concentration of H2O2. In our previous study, we confirmed that the concentration of H2O2 gradually decreases through the catalase reaction by 1/G in an acidic aqueous solution containing excess H2O2.22 Oxidation of CH3CN could also contribute to the decrease in the H2O2 concentration. At an elevated temperature (80 °C), the concentrations of over-oxidized products, including p-quinone, maleic acid, oxaloacetic acid, and formic acid, increased, as shown in Fig. S3.† These results indicate that 1/G has such a high oxidizing ability that it can even decompose the benzene ring in the presence of H2O2, as observed with 3+·I−/SiO2.
The catalytic benzene oxidation rate of 1/G was compared with those of 1+·I−/SiO2 and 3+·I−/SiO2 at 25 °C for 2 h of oxidation (entries 2, 11, and 12 in Table 1). 1+·I−/SiO2 did not show apparent benzene oxidation activity under these reaction conditions (entry 12 in Table 1). While it was observed that 3+·I−/SiO2 catalyzed benzene oxidation even at room temperature (entry 11 in Table 1), 1/G showed a considerably higher benzene oxidation activity (entry 2 in Table 1). We previously reported that the catalytic CH4 oxidation activity of 1/SiO2 was nearly identical to that of 1+·I−/SiO2 in an aqueous solution containing excess H2O2 and TFA at 60 °C.22 Moreover, in this research, we confirmed that both of 1+·I−/SiO2 and 1/SiO2 did not show benzene oxidation activity at 25 °C in aqueous CH3CN solutions containing excess H2O2 and TFA (see entries 12 and 13 in Table 1). Considering that small amounts of HCOOH, which were generated from CH3CN, were observed for both of the reactions of entry 12 and 13, the reaction without using CH3CN and/or a longer reaction time should be necessary for 1+·I−/SiO2 and 1/SiO2 to show apparent benzene oxidation activities at room temperature. Although it was difficult to check the benzene oxidation activity of 3/SiO2, where a neutral complex 3 is used as the catalyst molecule, because 3 is easily oxidized in the air, it seems that the high catalytic activity of 1/G is not derived from the difference in the oxidation state of the μ-nitrido-bridged iron phthalocyanine dimer but from the significant effect of the graphite substrate on the electronic structure of the μ-nitrido-bridged iron phthalocyanine dimer.
To further understand the reaction mechanism of benzene oxidation by 1/G, the initial oxidation rates of several benzene derivatives were examined and compared with those of benzene, as shown in Fig. 3 and Table 2. A linear correlation with a negative slope was observed in the Hammett plot.32 This result apparently indicates the electrophilic nature of the reactive intermediate in the oxidation reaction of the benzene derivatives. In our previous paper, we determined the Hammett ρ value of 3+·I−/SiO2 in a similar acidic aqueous CH3CN solution at 40 °C to be −1.14, which is less negative than that for 1/G at 25 °C (−3.79).20 These results could suggest that the high-valent iron oxo species of 1/G (1O/G) become more electrophilic in the benzene oxidation than that of 3+·I−/SiO2 because of the interaction between 1 and graphite(G).33
 | ||
| Fig. 3 Hammett plot of the turnover numbers (TONs) for the oxidation of benzene derivatives by 1/G. In the Hammett plot, the following substrates were used: PhH (σp = 0.00), PhCl (σp = 0.23), PhBr (σp = 0.23), PhCF3 (σm = 0.43), PhNO2 (σm = 0.71). (Ref. 32) for benzene (PhH) oxidation, TONBC at 2 h oxidation (entry 2 in Table 1) was used as the TON. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Benzene derivative | P1/mM | P2/mM | P3/mM | Q1/mM | TON (total) |
| 1 | PhCl (X = Cl) | 2.67 (0.29) | 0.88 (0.25) | 2.33 (0.11) | n.d. | 122 (13) |
| 2 | PhBr (X = Br) | 2.11 (0.45) | 1.30 (0.25) | 1.93 (0.10) | n.d. | 111 (15) |
| 3 | PhCF3 (X = CF3) | 0.20 (0.28) | 1.61 (0.07) | 0.72 (0.14) | n.d. | 52 (3) |
| 4 | PhNO2 (X = NO2) | 0.04 (0.06) | 0.01 (0.01) | 0.00 (0.00) | n.d. | 1 (1) |
We also examined the kinetic isotope effects (KIE) by comparing the initial oxidation rates (1 h oxidation) of C6H6 and C6D6. The obtained KIE value (kH/kD) was 1.0 (for details, see ESI† page S12), implying that the C–H bond cleavage is not involved in the turnover-limiting step. It is known that Fenton-type reaction, where a hydroxyl radical (·OH) is the reactive intermediate, shows KIE values of 1.7–1.8,34 which are apparently higher than that of the benzene oxidation by 1/G (1.0). Additionally, it was confirmed that the benzene oxidation by 1/G was not significantly quenched by the addition of an excess amount of 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), a well-known radical scavenger (entry 10 in Table 1).27,29 These results also imply that the high-valent iron–oxo species 1O/G generated from 1/G should be the reactive intermediate, as observed in the case of methane oxidation.22
In the case of CH4 oxidation, oxidation by high-valent iron–oxo species generally proceeds through a proton-coupled electron transfer (PCET) mechanism, wherein proton and electron abstractions from the substrate occur in a concerted manner.35,36 The results of benzene oxidation suggest that the abstraction of the π-electron of the benzene ring by 1O/G could be involved in the turnover-limiting step of benzene oxidation, whereas the opposite should be observed for the proton abstraction process. Possible reaction mechanisms are summarized in Fig. 4. First, the high-valent terminal iron–oxo species 1O/G is produced through the reaction of 1/G with H2O2. 1O/G act as the reactive intermediate to cause the successive C–O bond formation coupled with electron transfer between 1O/G and benzene. Our previous DFT calculation on 1/G suggested that interaction of 1 with graphite support cause SOMO stabilization and partial charge transfer from 1 to graphite. Since it is reported that 1O also has a SOMO distributed mainly over O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Fe–NFe center,7,37 stabilization of SOMO of 1O with partial charge transfer could also occur on graphite support as described in Fig. 4.22 This electronic state can enhance the electron abstraction ability of the high-valent iron–oxo species compared to those of 1+·I−/SiO2 and 3+·I−/SiO2 to facilitate the successive C–O bond formation coupled with electron transfer between 1O/G and benzene.38 This assumption does not contradict the fact that the Hammett ρ value of 1/G (−3.79) was much more negative than that observed for 3+·I−/SiO2 (−1.14).
Fe–NFe center,7,37 stabilization of SOMO of 1O with partial charge transfer could also occur on graphite support as described in Fig. 4.22 This electronic state can enhance the electron abstraction ability of the high-valent iron–oxo species compared to those of 1+·I−/SiO2 and 3+·I−/SiO2 to facilitate the successive C–O bond formation coupled with electron transfer between 1O/G and benzene.38 This assumption does not contradict the fact that the Hammett ρ value of 1/G (−3.79) was much more negative than that observed for 3+·I−/SiO2 (−1.14).
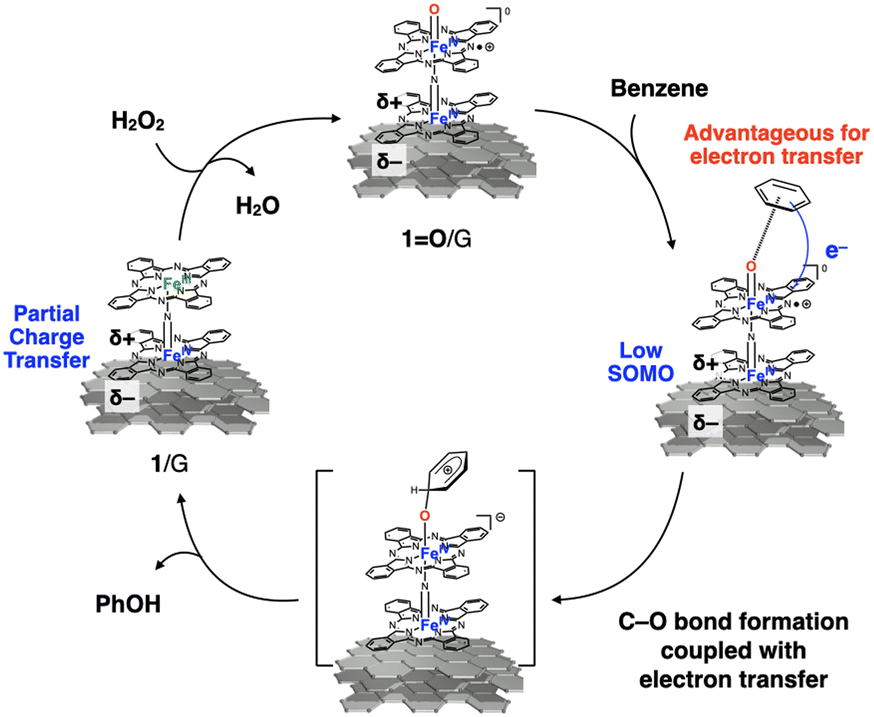 | ||
| Fig. 4 Possible reaction mechanism of the benzene oxidation by 1/G. | ||
Conclusions
In conclusion, we investigated the oxidation reaction of benzene in an aqueous CH3CN solution containing excess H2O2 and TFA by using a graphite-supported μ-nitrido-bridged iron phthalocyanine dimer (1/G) as a catalyst. It was demonstrated that 1/G efficiently oxidized benzene into phenol and p-benzoquinone even at 25 °C and its catalytic activity was much higher than that of silica gel-supported catalysts 1+·I−/SiO2 and 3+·I−/SiO2 in the same reaction conditions. These results apparently indicate the significant effect of the graphite support on the catalytic oxidation activity of μ-nitrido-bridged iron phthalocyanine dimer, which can facilitate the electron transfer from benzene to 1/G.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by a JSPS KAKENHI Grant-in-Aid for Scientific Research (B) (22H02094) awarded to KT, a JSPS KAKENHI Grant-in-Aid for Challenging Exploratory Research (Number 22 K19045), a Grant-in-Aid for Scientific Research (B) (Number 22H02156), and a Grant-in-Aid for Transformative Research Areas (A) Green Catalysis Science for Renovating Transformation of Carbon-Based Resources “Green Catalysis Science” (Number 24H01844) awarded to YY. YY thanks to the financial support by Okumura Corporation. This work was supported by “Quantum-Based Frontier Research Hub for Industry Development”, Nagoya University, Japan.References
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed.
- S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS PubMed.
- V. C.-C. Wang, S. Maji, P. P.-Y. Chen, H. K. Lee, S. S.-F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- A. R. McDonald and L. Que Jr., Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS.
- V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404–419 CrossRef CAS PubMed.
- R. A. Baglia, J. P. T. Zaragoza and D. P. Goldberg, Chem. Rev., 2017, 117, 13320–13352 CrossRef CAS PubMed.
- P. Afanasiev and A. B. Sorokin, Acc. Chem. Res., 2016, 49, 583–593 CrossRef CAS PubMed.
- A. B. Sorokin, Catal. Today, 2021, 373, 38–58 CrossRef CAS.
- A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562–2564 RSC.
- E. V. Kudrik and A. B. Sorokin, Chem. – Eur. J., 2008, 14, 7123–7126 CrossRef CAS PubMed.
- E. V. Kudrik, P. Afanasiev, L. X. Alvarez, P. Dubourdeaux, M. Clémancey, J.-M. Latour, G. Blondin, D. Bouchu, F. Albrieux, S. E. Nefedov and A. B. Sorokin, Nat. Chem., 2012, 4, 1024–1029 CrossRef CAS PubMed.
- L. X. Alvarez and A. B. Sorokin, J. Organomet. Chem., 2015, 793, 139–144 CrossRef CAS.
- Ü. Ísci, A. S. Faponle, P. Afanasiev, F. Albrieux, V. Briois, V. Ahsen, F. Dumoulin, A. B. Sorokin and S. P. de Visser, Chem. Sci., 2015, 6, 5063–5075 RSC.
- M. G. Quesne, D. Senthilnathan, D. Singh, D. Kumar, P. Maldivi, A. B. Sorokin and S. P. de Visser, ACS Catal., 2016, 6, 2230–2243 CrossRef CAS.
- N. Mihara, Y. Yamada, H. Takaya, Y. Kitagawa, K. Igawa, K. Tomooka, H. Fujii and K. Tanaka, Chem. – Eur. J., 2019, 25, 3369–3375 CrossRef CAS PubMed.
- Y. Yamada, K. Morita, N. Mihara, K. Igawa, K. Tomooka and K. Tanaka, New J. Chem., 2019, 43, 11477–11482 RSC.
- Y. Yamada, J. Kura, Y. Toyoda and K. Tanaka, New J. Chem., 2020, 44, 19179–19183 RSC.
- Y. Yamada, J. Kura, Y. Toyoda and K. Tanaka, Dalton Trans., 2021, 50, 6718–6724 RSC.
- Y. Yamada, Y. Miwa, Y. Toyoda, T. Yamaguchi, S. Akine and K. Tanaka, Dalton Trans., 2021, 50, 16775–16781 RSC.
- Y. Yamada, C.-M. Teoh, Y. Toyoda and K. Tanaka, New J. Chem., 2022, 46, 955–958 RSC.
- Y. Yamada, Y. Miwa, Y. Toyoda, Q. M. Phung, K. Oyama and K. Tanaka, Catal. Sci. Technol., 2023, 13, 1725–1734 RSC.
- Y. Yamada, K. Morita, T. Sugiura, Y. Toyoda, N. Mihara, M. Nagasaka, H. Takaya, K. Tanaka, T. Koitaya, N. Nakatani, H. Ariga-Miwa, S. Takakusagi, Y. Hitomi, T. Kudo, Y. Tsuji, K. Yoshizawa and K. Tanaka, JACS Au, 2023, 3, 823–833 CrossRef CAS PubMed.
- Y. Yamada, Y. Miwa, Y. Toyoda, Y. Uno, Q. M. Phung and K. Tanaka, Dalton Trans., 2024, 53, 6556–6567 RSC.
- R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, Appl. Organomet. Chem., 2020, 34, e5900 CrossRef CAS.
- S. Fukuzumi and K. Ohkubo, Asian J. Org. Chem., 2015, 4, 836–845 CrossRef CAS.
- K. Yoshizawa, Y. Shiota and T. Yamabe, J. Am. Chem. Soc., 1999, 121, 147–153 CrossRef CAS.
- M. Yamada, K. D. Karlin and S. Fukuzumi, Chem. Sci., 2016, 7, 2856–2863 RSC.
- Y. Morimoto, S. Bunno, N. Fujieda, H. Sugimoto and S. Itoh, J. Am. Chem. Soc., 2015, 137, 5867–5870 CrossRef CAS PubMed.
- T. Tsuji, A. A. Zaoputra, Y. Hitomi, K. Mieda, T. Ogura, Y. Shiota, K. Yoshizawa, H. Sato and M. Kodera, Angew. Chem., Int. Ed., 2017, 56, 7779–7782 CrossRef CAS PubMed.
- Y. Shimoyama, T. Ishizuka, H. Kotani and T. Kojima, ACS Catal., 2019, 9, 671–678 CrossRef CAS.
- Y. Yamada, T. Sugiura, K. Morita, H. Ariga-Miwa and K. Tanaka, Inorg. Chim. Acta, 2019, 489, 160–163 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- S. P. de Visser, K. Oh, A.-R. Han and W. Nam, Inorg. Chem., 2007, 46, 4632–4641 CrossRef CAS PubMed.
- R. Augusti, A. O. Dias, L. L. Rocha and R. M. Lago, J. Phys. Chem. A, 1998, 102, 10723–10727 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4019 CrossRef CAS PubMed.
- J. E. M. N. Klein and G. Knizia, Angew. Chem., Int. Ed., 2018, 57, 11913–11917 CrossRef CAS PubMed.
- C. Comonban, E. V. Kudrik, V. Briois, J. C. Shwarbrick, A. B. Sorokin and P. Afanasiev, Inorg. Chem., 2014, 53, 11517–11530 CrossRef PubMed.
- M. Asaka and H. Fujii, J. Am. Chem. Soc., 2016, 138, 8048–8051 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental data of oxidation experiments. See DOI: https://doi.org/10.1039/d4cy00661e |
| This journal is © The Royal Society of Chemistry 2024 |