
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spatiotemporal insights into forced dynamic reactor operation for fast light-off of Pd-based methane oxidation catalysts†
Kevin
Keller‡
a,
Daniel
Hodonj‡
a,
Lukas
Zeh
a,
Lachlan
Caulfield
b,
Eric
Sauter
b,
Christof
Wöll
 b,
Olaf
Deutschmann
a and
Patrick
Lott
*a
b,
Olaf
Deutschmann
a and
Patrick
Lott
*a
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstr. 20, 76131 Karlsruhe, Germany. E-mail: patrick.lott@kit.edu
bInstitute of Functional Interfaces (IFG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 18th June 2024
Abstract
Forced dynamic reactor operation by means of short reducing pulses (SRPs) during otherwise lean operation was investigated as a strategy for enhancing the catalytic activity of a monolithic Pd/CeO2 methane oxidation catalyst. Compared to static reactor operation under lean conditions, the application of SRPs during light-off experiments enables light-off at significantly lower temperatures both in dry and humid gas streams. In the presence of 10 vol% H2O, full CH4 conversion was achieved at 420 °C in dynamic operation, whereas in static lean conditions only 40% CH4 conversion were achieved at 600 °C. In addition, the results suggest that SRP operation of Pd/CeO2 is a feasible strategy to overcome long-term deactivation during hydrothermal aging. Combining in situ spatial profiling (SpaciPro) experiments with ex situ X-ray photoelectron spectroscopy (XPS) reveals that dynamic operation results in the formation of two reaction zones in the catalyst sample, with a highly active rear zone that exhibits a PdO/Pd mixed phase with highly active PdO sites that do not suffer from water inhibition due to the continuous removal of hydroxyl groups during the short rich phases. Kinetic activity tests in realistic gas mixtures demonstrate that forced dynamic reactor operation enhances the catalytic activity over the entire temperature window relevant for exhausts from lean-operated natural gas engines.
Introduction
Due to their high efficiency and their compared to gasoline or diesel applications advantageous emission characteristics, lean-burn natural gas engines (NGEs) are nowadays widely used in combined heat and power plants (CHPs) and enjoy growing popularity in the maritime and heavy-duty sector. In order to minimize emissions, a modern exhaust gas after treatment system is required, which particularly converts methane (CH4) slippage into carbon dioxide (CO2) and water (H2O).1,2Despite decades of research, palladium-based (Pd) catalysts, which commonly exhibit the highest activity for catalytic total oxidation of CH4, are still in the center of scientific attention.3–6 In particular, the inhibition of CH4 oxidation due to the inevitable combustion product H2O7–10 and catalyst poisoning due to the presence of sulfur species11–13 that are either added as odorants or that are present in fossil natural gas or biogas by default remain major challenges that need to be tackled.
Our present study aims at overcoming the inhibition by water vapor, which is most pronounced in the low-temperature regime14 and hereby particularly hampers not only fast light-off, e.g. during cold-start, but also reduces the catalytic activity of Pd-based converters in the typically rather cold exhaust gas during steady-state operation of stationary lean-burn applications, where temperatures of 500 °C and less are common.15 Previous studies uncovered hydroxyl accumulation on both the noble metal and the support material,16–18 as well as the formation of Pd(OH)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19–21 as predominant origin for the strong inhibition of CH4 oxidation by H2O.22 In the past few years, optimization of the support material,23–27 tuning of the electronic state or the morphology of the noble metal particles,28–31 and advanced catalyst operation procedures32–34 have been considered as most promising strategies for mitigating H2O inhibition.
19–21 as predominant origin for the strong inhibition of CH4 oxidation by H2O.22 In the past few years, optimization of the support material,23–27 tuning of the electronic state or the morphology of the noble metal particles,28–31 and advanced catalyst operation procedures32–34 have been considered as most promising strategies for mitigating H2O inhibition.
Especially in the context of technical catalyst systems, i.e. honeycomb-like structured catalysts as commonly applied for pollutant conversion in clean air processes, experiments with spatiotemporal resolution are of high value for understanding and controlling catalytically active sites, as these are typically heavily influenced by a multitude of physico-chemical phenomena that take place on different length and time scales.35 Capillary-based spatial profiling, better known as SpaciMS or SpaciPro technique,33,36 allows for direct in situ measurement of temperature and gas phase species concentrations in a single channel of a monolithic catalyst. Such axially resolved insights enabled to monitor and understand a wide variety of phenomena relevant for methane oxidation catalysts, i.e. water inhibition,9,37 aging-induced deactivation,38 or the formation of potentially beneficial temperature and particle size gradients along the catalyst.33,39
Our present study joins the latter corpus of work and demonstrates how short reducing pulses (SRP) can ensure a fast light-off by substantial enhancement of the low-temperature performance of Pd-based methane oxidation catalysts. By combining kinetic tests, catalyst characterization, and spatial profiling (SpaciPro) experiments with monolithic Pd/CeO2 catalyst samples, our results uncovered the SRP-induced formation of two zones in the catalyst: a front zone where metallic Pd that is less prone to H2O inhibition governs CH4 conversion to some extent and a downstream zone where highly active palladium oxide (PdO) ensures high catalytic activity. Ultimately, kinetic activity tests with simulated exhaust gas compositions that mimic real-world conditions demonstrated that forced dynamic reactor operation enhances the catalytic activity over the entire temperature window relevant for exhausts from lean-operated natural gas engines even in complex gas mixtures and under high mass flow rates.
Experimental section
Catalyst materials
Pd/CeO2 catalyst powder was prepared by incipient wetness impregnation. For this, an aqueous solution of (NH3)4Pd(NO3)2 (ChemPUR Chemicals) was used as precursor and was added dropwise to the support material CeO2 (MEL Chemicals; calcined for 5 h at 700 °C in static air prior to preparation). Five impregnation steps with intermittent drying periods (2 h/75 °C in static air) were necessary to obtain the target Pd-loading of 2 wt% (confirmed by elemental analysis via inductively coupled plasma optical emission spectroscopy). Finally, the dried catalyst powder was calcined in static air (5 h/550 °C) and coated onto cordierite honeycomb substrates (Corning; 3.0 cm length, 1.6 cm diameter, 300 cpsi, 76 cells) analogous to the procedure described by Karinshak et al.:33 an aqueous slurry containing the catalyst powder and AlO(OH) (Disperal P2, SASOL) in a ratio of 9:1 was prepared and coated onto the honeycomb substrate until the target Pd-loading of 110 g ft−3 was reached. The received monolithic sample was calcined in static air (5 h/550 °C).
Catalyst testing and test bench procedure
Coated honeycombs were tested under static and dynamic conditions in an in-house built catalyst testing bench described elsewhere.40–43 The monolithic catalyst sample was wrapped in quartz glass wool and mounted between two thermocouples (TC direct; 1 mm outer diameter, type N) that were held in the middle of the quartz glass tubular reactor (1.6 cm inner diameter) by inert honeycombs (1.0 cm in length) within the reactor for temperature control. In addition to lambda (λ) sensors (LSU 4.9, Bosch) installed at the inlet and outlet of the reactor, the concentrations of the gas stream exiting the reactor were analyzed in a Fourier-transform infrared (FTIR) spectrometer (MG2030, MKS Instruments). In order to obtain spatially resolved concentration and temperature profiles, a quartz glass capillary (180 μm outer diameter, 100 μm inner diameter) and a thin thermocouple (TC direct; 250 μm outer diameter) were placed in different channels of the honeycomb (center channels) and precisely moved by a motorized stage (LSM100A, Zaber). A mass spectrometer (HPR-20, Hiden Analytical) connected to the capillary was used for fast gas species concentration data acquisition.After an initial degreening (3200 ppm CH4, 10 vol% O2, bal. N2 at 600 °C for 1 h), light-off tests in 3200 ppm CH4, 10 vol% O2 and either 0 vol% H2O (dry) or 10 vol% H2O (wet) with N2 as inert carrier gas were conducted with a temperature ramp of 3 °C min−1 to evaluate the activity of the catalysts in a lean gas mixture. In addition, the same light-off tests were conducted under dynamic conditions by changing the gas mixture every 5 min for 10 s to rich conditions, hereinafter referred to as short reducing pulses (SRP). The gas hourly space velocity (GHSV) of 20000 h−1 was kept constant at all times by balancing with N2. Furthermore, the catalyst samples were aged for 24 h at 600 °C in the above-mentioned humid reaction gas mixture and subsequently the light-off tests as described above were repeated. Additionally, tests at two constant temperatures of 620 °C (static mode) and 400 °C (SRP mode) were conducted either under static or dynamic conditions for 4 h. Subsequently, spatially resolved profiles of temperature and gas phase species concentration were obtained by moving the quartz glass capillary stepwise through a single channel of the honeycomb (step size: 2 mm).
Catalyst characterization
X-ray photoelectron spectroscopy (XPS) was performed as described in the literature44 in a UHV setup with a high-resolution RG Scienta 4000 analyzer, with a catalyst sample from the inlet and outlet of the monolith. Al Kα (1486.6 eV) X-rays were used for excitation. X-ray diffraction (XRD) was conducted with a D8 Advance diffractometer (Bruker) using Cu Kα radiation, a Ni filter for a 2θ range of 8–120° and a step size of 0.0165°. The catalyst was additionally characterized by scanning transmission electron microscopy (STEM) using a probe-corrected Themis 300 TEM device (Thermofisher Scientific), which was operated at 300 kV. CO-chemisorption measurements were conducted with the powder catalyst.45 The samples were first placed in a tubular quartz glass reactor (QSIL AG). The catalysts underwent a pretreatment process with air for 30 min at a temperature of 550 °C. This was followed by a reduction process using 5 vol% H2 in N2 at 400 °C for a duration of 60 min and a cool-down of the reactor under continuous N2 flow to −78 °C with a mixture of dry ice and isopropanol surrounding the reactor, which prevents CO-spillover from the noble metal onto the support. Subsequently, the catalyst was saturated with a CO-containing gas mixture (100 ppm CO in N2) for 60 min while monitoring the CO uptake by means of measuring the concentrations of CO and CO2 using an infrared detector (X-Stream, Emerson). For calculating the dispersion of the noble metal from the CO uptake, an adsorption stoichiometry of 1:1 was assumed.46 Evaluation of N2-physisorption measurements according to the Brunauer–Emmett–Teller (BET) method47 that were conducted in a BELSORP Mini II analyzer (MicrotracBEL) provided information on the specific surface area and pore volume. In order to clean the surface, i.e. removal from adsorbates, the catalyst sample underwent a degassing process for a duration of 2 h at a temperature of 300 °C.
In order to obtain information on the oxygen storage capacity of the catalyst washcoat, oxygen storage capacity complete (OSCC) measurements of the Pd/CeO2 powder catalyst (300 mg) were performed in the catalyst testing setup described above using a quartz glass tubular reactor (10 mm outer diameter, 8 mm inner diameter, 720 mm length) with a total flow of 1 lSTP min−1. Based on the aftertreatment protocols for catalyst characterization suggested by Rappé et al.,48 the powder was oxidized at 600 °C with 2 vol% O2 in N2 for 20 min prior to cooling the sample in N2 to the temperature of 600 °C, 400 °C, and 200 °C, respectively, and subsequent reduction with 1 vol% CO in N2. The OSCC can be calculated by the amount of CO2 formed during the reduction of the catalyst with CO and is specified per mass of catalyst. Additionally, dynamic oxygen storage capacity (DOSC) measurements were performed under model gas conditions (3200 ppm CH4, 10 vol% O2, 10 vol% H2O in N2) by operating the catalyst during static mode or SRP mode at the designated temperature before switching oxygen on and off with a frequency of 0.01 Hz. The DOSC was calculated by subtracting the excess CO2 from the steady-state CO2 concentration after switching off oxygen.
Results and discussion
Catalyst characterization
Prior to activity testing, the catalyst was characterized by means of CO chemisorption, N2 physisorption, and oxygen storage capacity experiments according to the procedures described in the previous section. The respective results are summarized in Table 1.| Method | Value | Measured at 200 °C/400 °C/600 °C |
|---|---|---|
| a Theoretical value for complete reduction from Pd+II/Ce+IV to Pd0/Ce+III. b Max. reduction of 3200 ppm CH4 for 10 s assuming complete oxidation during a single pulse. c Assuming a hemispherical noble metal particle shape.53 | ||
| OSCC (μmol g−1) | 2849 (max. capacity)a | 191/309/482 |
| 317 (SRP)b | ||
| DOSC w/ SRP (μmol g−1) | — | NA/36/36 |
| BET surface area (m2 g−1) | 102 | — |
| Pore volume (mL g−1) | 0.27 | — |
| Noble metal dispersion (%) | 36 | — |
| Calculated mean particle diameter53 (nm)c | 3.1 | — |
Overall, the temperature dependency of OSCC observed herein – it increases with increasing temperature – is in good agreement with the literature.49,50 However, the theoretical maximum storage capacity of 2849 μmol g−1 is not reached during the measurement, indicating that in line with earlier results in the context of three-way catalysts51 not all oxygen stored in the support material is available under reaction conditions. Additionally, the OSCC measured by CO oxidation over a completely oxidized catalyst sample is of comparable magnitude to the capacity of a rich reducing pulse using CH4. Nevertheless, changing the reducing agent might further influence the measurement.51,52 In comparison, the DOSC is smaller for all temperatures studied herein, with DOSC values for applied SRPs being also slightly smaller as in static mode. However, during the following catalytic light-off measurements oxygen remains accessible, as the length of an SRP (10 s) is negligible compared to the 50 s rich phase during DOSC. X-ray diffraction (XRD) measurements showed no significant Pd-reflections (c.f. ESI† Fig. S1), suggesting small, well-dispersed nanoparticles on the ceria support. Further analysis with scanning transmission electron microscopy (STEM) in combination with energy dispersive X-ray spectroscopy (EDXS) confirmed small and well dispersed noble metal particles (c.f. ESI† Fig. S2). A palladium dispersion of 36% was obtained by means of CO-chemisorption measurements, which under the assumption of hemispherical particles53 corresponds to a mean particle diameter of 3.1 nm.
Catalytic activity in static and dynamic reactor operation
The methane conversion of the Pd/CeO2 catalyst was evaluated in three consecutive light-off measurements in a gas mixture consisting of 3200 ppm CH4 and 10 vol% O2 in N2 in the absence (Fig. 1a) and presence of 10 vol% H2O (Fig. 1b) under lean static conditions (plotted as solid lines) and under dynamic SRP conditions (plotted as dashed lines). As depicted in Fig. 1a, the Pd/CeO2 catalyst is able to convert 92%, 74%, and 66% of the methane at 600 °C during the first, second, and third consecutive light-off, respectively, under dry lean static conditions. Catalyst operation under humid conditions, i.e. dosing 10 vol% of water, results in a significant shift of the light-off curves to higher temperatures and less than 40% CH4 conversion already during the first light-off (Fig. 1b). This pronounced water inhibition is a well-known phenomenon and is commonly attributed to a blockage of active noble metal surface sites by hydroxyl groups that form during CH4 oxidation or that originate from gas phase water vapor.16,19,21,22 In addition, hydroxylation of the support has been observed, particularly for alumina-supported catalysts.8,16,23 It is consensus that lean CH4 oxidation over PdO follows a Mars–van-Krevelen mechanism,41 which strongly relies on dynamic Pd0 ⇔ PdO transformations. According to recent findings, these redox dynamics are hampered by hydroxylation18 and several studies in the field of lean methane oxidation underscored the importance of an oxygen exchange between the noble metal particles and the metal oxide support material.23,54,55 Therefore, using CeO2 as a support material with its high oxygen mobility has been suggested for improving water tolerance.16,54,56 Despite these beneficial properties, ceria-based Pd catalysts still suffer from the presence of steam if operated under static lean conditions.27,29 Nevertheless, catalysts in dynamic operation, with dynamics typically aiming at optimizing the noble metal oxidation state as well as the removal of adsorbates that block active surface sites, may benefit from the unique properties of ceria.57 Both in dry and humid conditions, the introduction of short reducing phases by means of SRPs (plotted as dashed lines in Fig. 1) results in a significant shift of the light-off curves to lower temperatures. While the first light-off in the dry environment with SRPs (dashed black line in Fig. 1a) is congruent to the third one in static operation in the beginning (blue line in Fig. 1a), CH4 conversion increases almost instantaneously at 440 °C and reaches full conversion at around 470 °C. The activity during the second and third light-off in SRP operation is even higher: The temperature of 50% CH4 conversion, T50, is found at 295 °C, which is an improvement by 255 °C compared to the light-off in static operation, and full conversion is achieved already at 340 °C. Notably, this substantial activity boost is maintained also during the third light-off. | ||
| Fig. 1 Conversion of CH4 during light-off experiments conducted in static or dynamic (SRP; every 5 min for 10 s) operation at a GHSV of 20000 h−1 in 3200 ppm CH4, 10 vol% O2, and balance N2 a) in the absence of water, b) in the presence of 10 vol% H2O, and c) in the presence of 10 vol% H2O in the fresh state and after 24 h of aging at 600 °C in humid reaction gas mixture, d) during 24 h of operation at 600 °C in in 3200 ppm CH4, 10 vol% O2, 10 vol% H2O, and balance N2 (GHSV = 20000 h−1). | ||
An analogous performance is observed in the presence of water. After an initial light-off in SRP operation that seems to activate the catalyst once reaching a certain threshold temperature, in this case approx. 470 °C, an overall better and stable catalytic performance is found during the second and third light-off (Fig. 1b): with a temperature of 50% conversion of 400 °C, the T50 is approx. 220 °C lower compared to the activity data in static operation (data under static conditions were extrapolated, because the light-off was terminated at 600 °C to prevent the sample from damage due to hydro-thermal sintering).
In general, SRPs influence the active state of the noble metal particles, i.e. the degree of Pd oxidation,32,33 and it is commonly accepted that a temporary (partial) reduction of PdO particles improves the catalytic activity under otherwise static operation.58–60 The partial reduction of PdO, however, requires a certain threshold temperature,34 which is 460 °C in dry conditions and at around 470 °C in the presence of water for the monolithic Pd/CeO2 catalyst studied herein. In this regard, it is worth mentioning that this temperature is highly dependent on the chosen support material, the size of the active noble metal particles, and the reaction gas atmosphere.27
In summary, the forced dynamic reactor operation by means of SRPs during otherwise lean conditions allows to activate the Pd/CeO2 methane oxidation catalyst and significantly reduces the light-off temperature compared to static operation. Although CH4 conversion temporarily drops during the rich phase due to a lack of oxygen, the overall performance is substantially better. Approximately 39 mmol of CH4 are dosed into the reactor during a single light-off measurement as shown in Fig. 1a and b. During the first light-off in static operation, a cumulative amount of 32 mmol CH4 is emitted in the dry atmosphere and 36 mmol in the humid atmosphere. In contrast, only 9 mmol and 19 mmol CH4 remain in the effluent gas stream for dry and humid conditions, respectively, if SRPs are applied during the light-off. Therefore, the catalyst efficiency regarding CH4 conversion can be more than two times higher due to forced dynamic reactor operation, hereby essentially halving the emission of greenhouse-active gas species. Furthermore, no relevant CO formation (typically less than 10 ppm) was observed during the short reducing phases during the light-off tests (c.f. ESI† Fig. S3), which points to an efficient exploitation of the high OSC of the ceria support even at low temperatures (c.f.Table 1). That the second and third light-off curves are congruent if SRPs are applied suggests that the durability of the catalyst is enhanced as well. Therefore, the role of catalyst aging in the context of static and SRP operation is discussed next.
Although water inhibition is an instantaneous effect, it is well-known that the accumulation of hydroxyl groups on the catalyst surface as well as noble metal particle sintering induced by hydrothermal aging can result in long-term deactivation.8,16,54,61 In order to evaluate the effect of dynamic catalyst operation during time on stream, SRPs were applied to the monolithic sample at elevated temperatures of 600 °C in the presence of 10 vol% H2O in a reactive gas atmosphere for 24 h and were compared to the data obtained in static mode (Fig. 1c and d). Despite the presence of steam, a high methane oxidation activity is observed over the whole period of the 24 h experiment under pulsing conditions, whereas the catalytic activity declines with time on stream in static operation (Fig. 1d). After only 5 h of static operation, the CH4 conversion at 600 °C drops from initially approx. 50% to only 14%. As shown in Fig. 1c that depicts the corresponding light-off curves in the fresh (= degreened) and aged catalyst state, this pronounced deactivation persists also during the light-off test. In contrast, dynamic operation in SRP mode enables light-off curves with a T50 of approx. 400 °C that match the initial activity even after 24 h of catalyst operation fairly well. Considering that previous studies on similar PdO-based systems reported that depending on the temperature an almost complete loss in catalytic activity can occur during long-term exposure to humid reaction gas mixtures,8,18,27,61 the findings in this study underscore the tremendous value of dynamic catalyst operation for activity enhancement.
Experiments with spatiotemporal resolution
Since high activity and long-term stability of catalytic converters are decisive factors for real-world applications, more detailed insights were gained by means of spatial profiling (SpaciPro) experiments. For this, the catalyst was operated in a representative humid gas atmosphere (3200 ppm CH4, 10 vol% O2, 10 vol% H2O in N2; GHSV = 20000 h−1) and at a constant temperature. For the experiments in dynamic operation, the T50 of 400 °C was chosen; previous studies suggest that the reducing periods do not cause relevant sintering of the palladium particles at such moderate temperature.28,33 Due to the strong water inhibition, spatial profiling under static lean conditions was conducted at a higher inlet temperature of 620 °C, which corresponds to an end-of-pipe methane conversion of 34%. Despite an entirely lean operation, which is considered less critical with respect to sintering than rich conditions,62 the choice of an even higher temperature – in analogy to the experiments in static operation preferentially T50 – would have accelerated hydrothermal sintering of the noble metal particles and would have biased the data. Fig. 2 summarizes the outcome of the experiments, namely spatially resolved profiles of the gas temperature and of the gas species in static operation (Fig. 2a) and in dynamic operation (Fig. 2b), respectively, as well as spatiotemporal gas species data obtained at different positions along a single channel of the monolithic catalyst sample during dynamic operation (Fig. 2c). The position of the monolith with a total length of 30 mm is highlighted by the blue area (Fig. 2a and b). According to the data in Fig. 2a, the declining CH4 concentration (yellow triangles) along the channel follows an almost linear trend. As a direct consequence of the low conversion rates, the temperature increase during the exothermic methane oxidation reaction is negligible, which results in an almost horizontal temperature profile inside the channel (red circles). These profiles agree with findings reported in earlier studies using the same measurement technique22,50–52 and clearly underscore the pronounced water inhibition already mentioned above.
 | ||
| Fig. 2 Spatially resolved CH4 concentration and temperature profiles for a) static catalyst operation at 620 °C and b) dynamic catalyst operation in SRP mode at 400 °C in 3200 ppm CH4, 10 vol% O2, 10 vol% H2O in N2; GHSV = 20000 h−1; c) temporal evolution of the CH4 concentration after applying a short reducing pulse at 0 s for 10 s at different axial positions, including the end-of-pipe concentration. | ||
As demonstrated by the data depicted in Fig. 2b, the profiles change substantially upon forced dynamic reactor operation by means of SRP mode. The axially resolved CH4 concentration data (yellow/blue triangles) suggest the formation of two different reaction zones (zone I colored in light blue, zone II colored in dark blue in Fig. 2b). The nearly horizontal concentration profile in the first 18 mm of the channel (zone I, light blue) uncovers a comparably low catalytic activity in the front zone of the catalyst: after 18 mm, still 2811 ppm CH4 remain. This low-activity zone is followed by a sudden and pronounced drop of the CH4 concentration (zone II, dark blue), that, in the best case, results in a CH4 concentration of less than 1000 ppm and a final conversion of over 70% downstream of the catalyst sample. The temperature profile (Fig. 2b, red circles) for SRP-mode directly corresponds with the activity and matches the two zones as well: once significant CH4 conversion takes place, the temperature rises up to 435 °C, which is an increase by 35 °C compared to the inlet temperature. Notably, Fig. 2b shows two CH4 concentration profiles: one represents the activity right before the SRP and thus after the catalyst sample was activated by SRP operation but was already operated under lean conditions for 5 min (yellow triangles), the other one represents the catalyst performance right after the reductive phase of 10 s (blue triangles). Both profiles are congruent in zone I, whereas the profile is steeper and the overall activity is higher right after the reducing period. At the catalyst outlet, a CH4 concentration of 601 ppm was monitored right after the reducing pulse, whereas 945 ppm CH4 remain after 5 min of lean operation.
In order to elaborate on this observation in more detail, Fig. 2c shows the temporal evolution of the CH4 concentration during a typical rich-lean cycle at different axial positions within the catalytic channel, including the end-of-pipe FTIR data. While only minor changes occur in the front zone, the CH4 concentration at positions 20, 24, and 26 mm rises slightly during the 5 min interval of lean operation that follows the short reducing pulse. The deactivation is most pronounced in the first 150 s after the reductive phase, then the CH4 concentration curves start to flatten over time. The end-of-pipe signal measured by the FTIR spectrometer follows the general trend observed for the axial positions in the rear part of the catalyst.
Overall, the spatially resolved data allow to draw three essential conclusions. First, the slopes of the CH4 concentration profiles in static (Fig. 2a) and dynamic SRP (Fig. 2b) operation strongly resemble in the front zone of the catalyst, which underscores that the reductive period activates already the first 18 mm of the catalyst because the reactor temperature is 220 °C lower in dynamic operation but the activity is similar. However, the activity boost is far more pronounced in the rear zone, which becomes even clearer when the resolved turnover frequencies (TOFs, Fig. 3) as discussed in the following are taken into consideration. Second, the SRP-induced zones persist in lean operation although the activity in the highly active rear zone has its maximum right after the reducing phase and then declines slightly over time (Fig. 2c). Third, the activation caused by the forced dynamic reactor operation is a somewhat self-amplifying effect since the significant heat evolution over the activated catalyst additionally promotes high CH4 conversion, particularly towards the end of the monolith. This enables conversion rates of up to 82% although the reactor temperature is set to T50 = 400 °C as obtained from the light-off tests (c.f.Fig. 1).
 | ||
| Fig. 3 Spatially resolved TOF alongside the catalytic channel for static mode (operation at 620 °C) and SRP mode (operation at 400 °C) as depicted in Fig. 2a and b. | ||
In order to decently quantify how these combined effects translate into catalyst performance, Fig. 3 shows the spatially resolved TOFs that were calculated from the initial CH4 mole fraction xCH4,0 and in consideration of the noble metal dispersion DPd of 36% according to eqn (1).27 Inhere, ![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) it the volumetric flow rate with the methane conversion ΔXCH4 in the respective axial section. Furthermore, p is the pressure, R the gas constant, and T the temperature. Lastly, mPdO is the used weight of PdO and MPdO is the molar weight of PdO.
it the volumetric flow rate with the methane conversion ΔXCH4 in the respective axial section. Furthermore, p is the pressure, R the gas constant, and T the temperature. Lastly, mPdO is the used weight of PdO and MPdO is the molar weight of PdO.
 | (1) |
Correlation between the catalytic activity and the oxidation state
The pronounced gradients observed in the spatial profiling experiments can be explained in more detail by taking the extensive fundamental atomic-level knowledge on the microkinetics of methane oxidation into account that became available during decades of intensive research in the field. Commonly, CH4 oxidation in lean gas atmospheres is believed to proceed via a Mars–van-Krevelen mechanism that relies on PdO as active species,64–66 although some studies suggested that CH4 conversion can be maximized by creating partially reduced or mixed PdO/Pd-phases.31,67,68 Similarly, the crucial influence of the Pd/PdO ratio for catalytic activity was also described by Khudorozhkov et al.,69 who found a linear correlation between the ratio of Pd2+ and Pd0 species on Pd/Al2O3 catalysts and the turnover frequency measured during propane oxidation. In addition to the reaction environment (lean versus rich), the noble metal particles redox potential is also dependent on the size of the particles.The SRPs as applied herein are known to influence the oxidation state of Pd-based catalysts.32–34,70 For instance, Franken et al.34 investigated a Pd/Al2O3 powder catalyst and demonstrated that in general an increasing number of reducing pulses increasingly reduces the catalyst and lowers the PdO/Pd ratio. However, as discussed in the context of Fig. 2, the monolithic Pd/CeO2 catalyst sample used in our present study undergoes an initial activation and then reaches a stable performance level during the 24 h long-term experiment that points to a relatively stable Pd–PdO equilibrium. Notably, the reduction of PdO caused by a (temporary) rich atmosphere is highly temperature-dependent71,72 and it has been suggested that an oxygen exchange between the support and the noble metal particles can take place that can ultimately facilitate the subsequent reoxidation.54,66,73
In this respect, CeO2 has been identified as particularly interesting since it exhibits a high oxygen storage capacity as well as a high oxygen mobility.24,56,74 For the monolithic Pd/CeO2 catalyst sample used herein, these properties were confirmed by the OSCC and dynamic OSC measurements (c.f.Table 1). According to our calculations, the total molar amount of the oxygen stored in the lattice of CeO2 is theoretically ten times larger than the capacity of a single rich pulse occurring for 10 s. Consequently, the support material could donate oxygen to the noble metal during the rich phase and may hereby change and stabilize the oxidation state of palladium, which would be a similar mechanism as reported for Pd-based three-way catalyst formulations containing ceria as oxygen storage component.75,76 Since the spatially resolved data strongly suggest the formation of two relatively stable zones along the monolith sample, XPS measurements were conducted for washcoat obtained from the less active front zone as well as from the highly active rear zone of the catalyst sample. Not only the respective X-ray photoelectron spectra shown in Fig. 4a and b point to differences in the oxidation state, but also the appearance of the monolithic catalyst sample after 24 h of dynamic operation in SRP mode itself. As depicted in Fig. 4c, a gradient evolved in dynamic operation that can already be seen by eye: while the inlet region exhibits a darker brown color, the rear part towards the catalyst outlet exhibits a lighter brown color. The color transition nicely correlates with the onset of the TOF increase after approx. half of the monolith sample as shown in Fig. 3.
 | ||
| Fig. 4 Pd3d X-ray photoelectron spectra of the Pd/CeO2 washcoat obtained after 24 h of forced dynamic reactor operation at 600 °C from a) the front zone (zone I in Fig. 2b) and b) the rear zone (zone II in Fig. 2b) of the monolith sample depicted in c). | ||
The analysis of the peak volume found by means of XPS for the Pd2+ and Pd0 species reveals a different degree of oxidation in the two monolith zones. Since according to the XPS data the degree of oxidation is lower in the front zone (Fig. 4a; approx. 70% Pd2+), which was found to be less active during the spatially resolved measurements, than in the rear zone (Fig. 4b; approx. 86% Pd2+), we assume that chemisorbed oxygen is more relevant and largely present in the first half of the slightly more reduced sample, whereas actual surface oxidation becomes more important in the second half of the sample. These distinct differences in the oxidation state underscore the importance of a careful fine-tuning of the PdO/Pd-ratio for boosting the catalytic activity during methane oxidation. Considering that in a lean reaction environment CH4 oxidation over PdO proceeds via a Mars–van-Krevelen mechanism,22,41,77 the correlation of the XPS data with the axially resolved activity data points to an overreduction in the front zone of the monolith. Similarly, the spatiotemporal CH4 concentration data depicted in Fig. 2c uncovers that in the rear part of the catalyst sample, i.e. axial positions 24 mm and 26 mm, the CH4 concentration starts to decline approximately 10–15 s after the reducing pulse; hence, it takes some time until the zone reaches its maximum activity. Although the response of the mass spectrometer used for gas sampling may contribute to this delayed CH4 concentration drop due to the residence time of the gas in the capillary, the dynamics of reoxidation as well as mechanistic considerations can explain the catalyst behavior observed in our experiments.
The lower surface tension of PdO compared to metallic Pd78 is one of the reasons that reoxidation of supported Pd particles is believed to start from the surface,71,72 which can result in the formation of biphasic particles with a metallic Pd core and a PdO shell.70 Typically, the first step of reoxidation, which has been reported to be a rather slow process,32 is the chemisorption of oxygen atoms onto the metallic Pd surface, followed by actual surface and bulk oxidation.71 Since according to the XPS data the degree of oxidation is lower in the front zone (approx. 70% Pd2+) than in the rear zone (approx. 86% Pd2+), the gradient directly impacts the C–H bond activation and thus the underlying CH4 oxidation reaction mechanism: By means of density functional theory (DFT) calculations a radical-like (O*⋯CH3˙⋯*OH)‡ transition state was found for Pd surfaces covered with chemisorbed oxygen, whereas a more stable four-center transition state (H3Cδ−⋯Pdox⋯Hδ+⋯Oox)‡ was found over PdO, which implies higher turnover rates over PdO.65 While in line with these fundamental results it is true that in forced dynamic reactor operation the front zone of the catalyst is less active than its rear zone, it still exhibits a similar TOF at 400 °C as the catalyst operated in static lean conditions at 620 °C (Fig. 3). We attribute this performance to the mitigation of the water inhibition along the entire length of the monolithic sample. During the rich phase hydroxyl groups both from the noble metal as well as from the support are desorbed. During the subsequent lean phase, the support material can act as a reservoir for hydroxyl groups and hereby protects the noble metal from hydroxylation until the support surface is saturated and hydroxyl groups adsorb on the noble metal particles as well.
In conclusion, two effects account for high methane conversion: continuous removal of surface adsorbates that would otherwise block active surface sites on the one hand, and a sufficient PdO content on the surface of the noble metal particles enabling CH4 oxidation under lean conditions on the other hand. The lean-rich cycling as chosen herein, namely 5 min of lean operation and 10 s of reducing conditions, seems to result in a quasi-equilibrium of the catalyst states in the front zone and the rear zone that result in the pronounced gradient uncovered by means of spatial profiling. Notably, the coexistence of Pd and PdO after reducing phases has been observed even during longer periods of lean catalyst operation, also at elevated temperatures,79 and the beneficial effect of (short) reducing phases on the catalytic activity has been observed to persist for quite some time in lean reaction mixtures.29,33 In addition, both the redox properties of Pd/PdO27,34 as well as the water inhibition effect16,19 are known to be temperature-dependent. Consequently, the frequency as well as the duration of the reducing pulses likely depend on the temperature, thus more systematic studies are desirable. Ultimately, also the gas mixture can change the requirements for optimal conditions, i.e. low versus high humidity and/or the presence of other species like nitrogen oxides (NOx) that may potentially inhibit methane oxidation.21,22
Catalytic performance in a simulated real-world exhaust gas mixture
Transferring the fundamental knowledge gained throughout the experiments presented above into real-world applications is key for designing highly efficient reactor operation strategies. Therefore, the forced dynamic reactor operation investigated herein is applied in a complex exhaust gas mixture that mimics real conditions representative for natural gas engines80,81 as summarized in Table 2. With the species concentrations listed in Table 2, an air-to-fuel equivalence ratio λ of 1.2 for the lean phase and of 0.98 for the rich phase was calculated according to eqn (2).82 Analogous to the light-off tests presented above, the catalyst was tested in three consecutive light-off experiments either in static feed conditions or in dynamic SRP operation (Fig. 5).000 h−1
| Gas unit | [O2] % | [CO] % | [CO2] % | [H2O] % | [H2] ppm | [NO] ppm | [CH4] ppm | λ |
|---|---|---|---|---|---|---|---|---|
| Lean | 3 | 0.1 | 8 | 13 | 330 | 1000 | 800 | 1.2 |
| Rich | 0.5 | 1 | 8 | 13 | 3300 | 1000 | 800 | 0.98 |
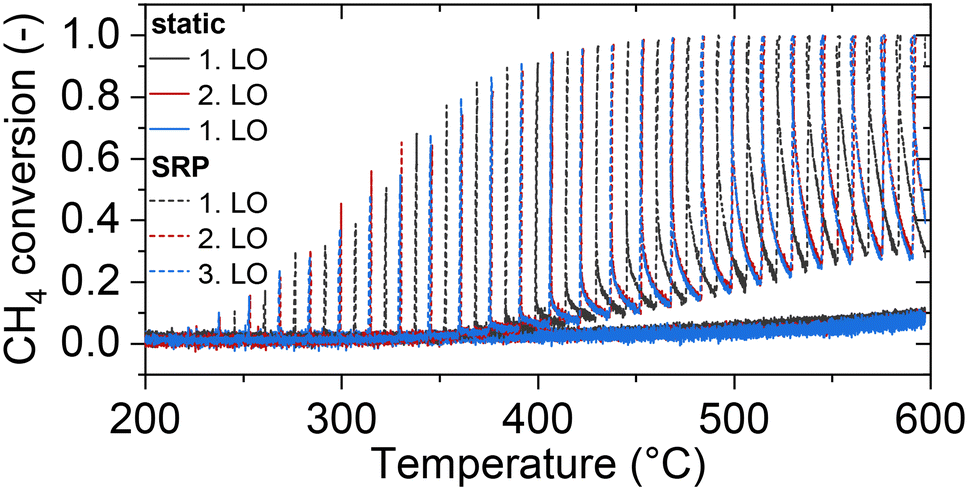 | ||
| Fig. 5 Conversion of CH4 during light-off experiments conducted in static lean or dynamic (SRP; every 5 min 10 s of rich conditions) operation at a GHSV of 60000 h−1 with the gas composition(s) listed in Table 2. | ||
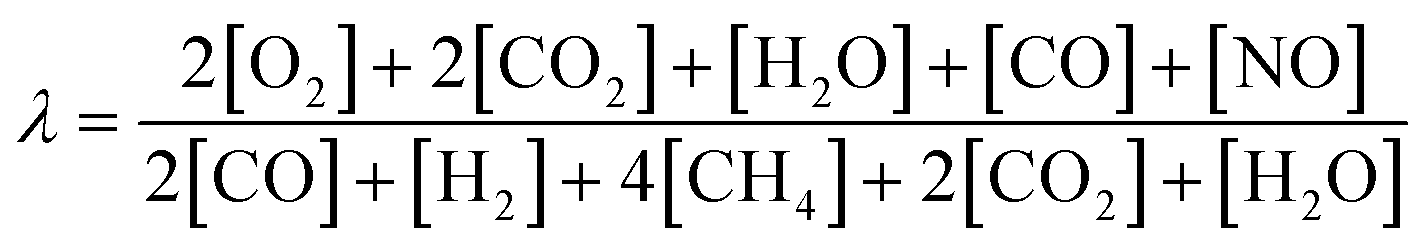
| (2) |
With regard to secondary emission formation that is typically provoked by rich conditions and which may even occur as a consequence of hydrocarbon–NOx interactions over oxidation catalysts in lean conditions,40,83 NH3 evolution is most relevant, whereas only very minor amounts of N2O form in the absence of oxygen (c.f. ESI† Fig. S5). However, NH3 formation occurs as sharp spikes only during the rich phase, whereas the beneficial effect on CH4 conversion persists also during the 5 min of lean operation (c.f. ESI† Fig. S6). Notably, the overall catalytic activity and species formation trends remain constant during three consecutive light-off measurements, which in analogy to the model gas mixture experiments suggests a fairly stable performance despite demanding conditions.
Considering the overall CH4 content entering and exiting the reactor during one light-off test, only 2% of methane are oxidized in static mode, whereas more than 18% of methane are oxidized in SRP-mode, which saves 0.24 g of CO2 equivalents during the duration of a single light-off. Although admittedly the absolute performance of the catalyst used herein is insufficient for real-world application, it should be noted that we used a well-defined and non-optimized Pd/CeO2 model catalyst formulation for the present study that allows us to deconvolute the fundamental phenomena playing a key role during forced dynamic reactor operation. For full-scale applications, more advanced formulations and a higher noble metal loading should be considered. Ultimately, the combination of robust catalyst materials and corresponding highly efficient operation strategies are not only relevant for fast light-off in the low-temperature regime, but also at elevated temperatures that typically accelerate aging and deactivation. Therefore, the experimental data shown in Fig. 5 should be considered as a proof-of-principle, which has its full effect once not only the catalyst operation strategy, but also the catalyst formulation is optimized.
Conclusions
In this study we investigated forced dynamic reactor operation for enhancing the catalytic activity of monolithic Pd/CeO2 methane oxidation catalyst. The introduction of short reducing pulses (SRPs) during otherwise lean reactor operation, namely 10 s of rich operation after 5 min of lean operation, results in a substantial increase of the catalytic activity. For instance, only 34% CH4 conversion were observed at 600 °C in a static lean, humid model reaction mixture, whereas dynamic SRP operation enables complete methane conversion already at 420 °C. This improved catalytic performance was also maintained during long-term experiments conducted at 600 °C under humid conditions, which underscores that the short reductive phases do not cause catalyst deactivation, i.e. due to sintering of the noble metal particles.Spatially resolved gas phase species concentration and temperature profiles uncovered the formation of two reaction zones along the catalytic converter: dynamic SRP operation results in a less active front zone and a highly active zone in the rear part of the monolith. We attribute the particularly high activity in the rear zone to the larger content of highly active PdO that was detected by means of XPS, whereas less active metallic Pd, likely covered with chemisorbed oxygen species, governs the activity in the front zone. Notably, water inhibition is mitigated along the entire length of the catalyst, resulting in a promotion of the turnover frequency even in the front zone. Finally, kinetic activity tests with more complex exhaust gas compositions that mimic real-world conditions demonstrated that forced dynamic reactor operation can serve as an efficient measure for boosting the catalytic activity also in real applications where complex gas mixtures and demanding high mass flow rates occur.
In conclusion, the results of our work help to understand the influence of short reducing pulses on palladium-based catalyst systems during forced dynamic reactor operation. The insights gained by combining kinetic testing, catalyst characterization, and in situ spatial profiling enable to establish operation procedures that ensure excellent low-temperature performance of methane oxidation catalysts. On the long run, exploiting oxidation state gradients and surface coverage effects as subject to our present study can enable the efficient conversion of various pollutants in a single converter. Consequently, the approach chosen herein can serve as a blueprint for a variety of studies in the future.
Data availability statement
The data supporting this article have been included as part of the ESI.†Author contributions
K. Keller: data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft; D. Hodonj: data curation, formal analysis, investigation, methodology, validation, visualization, writing – review & editing; L. Zeh: formal analysis, investigation, writing – review & editing; L. Caulfield: formal analysis, investigation, writing – review & editing; E. Sauter: formal analysis, investigation, writing – review & editing; C. Wöll: funding acquisition, resources, writing – review & editing; O. Deutschmann: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing; P. Lott: conceptualization, data curation, methodology, project administration, supervision, validation, visualization, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge S. Lichtenberg and S. Bastian (both ITCP, KIT) for technical support, T. Bergfeldt (IAM-AWP, KIT) for elemental analysis, T. Delrieux and J. Czechowsky for preparation of the catalyst, and J.-D. Grunwaldt for fruitful scientific discussion. We thank C. B. Maliakkal (INT, KIT) for TEM imaging, which was conducted with the financial support of the Karlsruhe Nano Micro Facility (KNMFi), a Helmholtz Research Infrastructure at KIT. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SFB 1441 – Project-ID 426888090.References
- P. Lott, M. Casapu, J.-D. Grunwaldt and O. Deutschmann, Appl. Catal., B, 2024, 340, 123241 CrossRef CAS.
- P. Lott and O. Deutschmann, Emiss. Control Sci. Technol., 2021, 7, 1–6 CrossRef CAS.
- M. Monai, T. Montini, R. J. Gorte and P. Fornasiero, Eur. J. Inorg. Chem., 2018, 2018, 2884–2893 CrossRef CAS.
- D. Jiang, K. Khivantsev and Y. Wang, ACS Catal., 2020, 10, 14304–14314 CrossRef CAS.
- F. Nkinahamira, R. Yang, R. Zhu, J. Zhang, Z. Ren, S. Sun, H. Xiong and Z. Zeng, Adv. Sci., 2023, 10, 2204566 CrossRef CAS PubMed.
- Z. Tang, T. Zhang, D. Luo, Y. Wang, Z. Hu and R. T. Yang, ACS Catal., 2022, 12, 13457–13474 CrossRef CAS.
- N. Sadokhina, F. Ghasempour, X. Auvray, G. Smedler, U. Nylén, M. Olofsson and L. Olsson, Catal. Lett., 2017, 147, 2360–2371 CrossRef CAS.
- P. Velin, M. Ek, M. Skoglundh, A. Schaefer, A. Raj, D. Thompsett, G. Smedler and P.-A. Carlsson, J. Phys. Chem. C, 2019, 123, 25724–25737 CrossRef CAS.
- C. Coney, C. Stere, P. Millington, A. Raj, S. Wilkinson, M. Caracotsios, G. McCullough, C. Hardacre, K. Morgan, D. Thompsett and A. Goguet, Catal. Sci. Technol., 2020, 10, 1858–1874 RSC.
- A. Boucly, L. Artiglia, M. Roger, M. Zabilskiy, A. Beck, D. Ferri and J. A. van Bokhoven, Appl. Surf. Sci., 2022, 606, 154927 CrossRef CAS.
- M. S. Wilburn and W. S. Epling, Appl. Catal., B, 2017, 206, 589–598 CrossRef CAS.
- P. Auvinen, N. M. Kinnunen, J. T. Hirvi, T. Maunula, K. Kallinen, M. Keenan, R. Baert, E. van den Tillaart and M. Suvanto, Appl. Catal., B, 2019, 258, 117976 CrossRef CAS.
- P. Lott, M. Eck, D. E. Doronkin, A. Zimina, S. Tischer, R. Popescu, S. Belin, V. Briois, M. Casapu, J.-D. Grunwaldt and O. Deutschmann, Appl. Catal., B, 2020, 278, 119244 CrossRef CAS.
- R. Kikuchi, S. Maeda, K. Sasaki, S. Wennerström and K. Eguchi, Appl. Catal., A, 2002, 232, 23–28 CrossRef CAS.
- P. Gélin and M. Primet, Appl. Catal., B, 2002, 39, 1–37 CrossRef.
- D. Ciuparu, E. Perkins and L. Pfefferle, Appl. Catal., A, 2004, 263, 145–153 CrossRef CAS.
- W. R. Schwartz, D. Ciuparu and L. D. Pfefferle, J. Phys. Chem. C, 2012, 116, 8587–8593 CrossRef CAS.
- P. Velin, F. Hemmingsson, A. Schaefer, M. Skoglundh, K. A. Lomachenko, A. Raj, D. Thompsett, G. Smedler and P. A. Carlsson, ChemCatChem, 2021, 13, 3765–3771 CrossRef CAS.
- D. Roth, P. Gélin, M. Primet and E. Tena, Appl. Catal., A, 2000, 203, 37–45 CrossRef CAS.
- W. Barrett, J. Shen, Y. Hu, R. E. Hayes, R. W. J. Scott and N. Semagina, ChemCatChem, 2020, 12, 944–952 CrossRef CAS.
- A. Boubnov, A. Gremminger, M. Casapu, O. Deutschmann and J.-D. Grunwaldt, ChemCatChem, 2022, 14, e202200573 CrossRef CAS.
- K. Keller, P. Lott, H. Stotz, L. Maier and O. Deutschmann, Catalysts, 2020, 10, 922 CrossRef CAS.
- W. R. Schwartz and L. D. Pfefferle, J. Phys. Chem. C, 2012, 116, 8571–8578 CrossRef CAS.
- K. Murata, D. Kosuge, J. Ohyama, Y. Mahara, Y. Yamamoto, S. Arai and A. Satsuma, ACS Catal., 2020, 10, 1381–1387 CrossRef CAS.
- S. Chen, S. Li, R. You, Z. Guo, F. Wang, G. Li, W. Yuan, B. Zhu, Y. Gao, Z. Zhang, H. Yang and Y. Wang, ACS Catal., 2021, 11, 5666–5677 CrossRef CAS.
- A. W. Petrov, D. Ferri, O. Kröcher and J. A. van Bokhoven, ACS Catal., 2019, 9, 2303–2312 CrossRef CAS.
- K. Keller, P. Lott, S. Tischer, M. Casapu, J.-D. Grunwaldt and O. Deutschmann, ChemCatChem, 2023, 15, e202300366 CrossRef CAS.
- Y. Mahara, K. Murata, K. Ueda, J. Ohyama, K. Kato and A. Satsuma, ChemCatChem, 2018, 10, 3384–3387 CrossRef CAS.
- P. Lott, P. Dolcet, M. Casapu, J.-D. Grunwaldt and O. Deutschmann, Ind. Eng. Chem. Res., 2019, 58, 12561–12570 CrossRef CAS.
- H. Xiong, D. Kunwar, D. Jiang, C. E. García-Vargas, H. Li, C. Du, G. Canning, X. I. Pereira-Hernandez, Q. Wan, S. Lin, S. C. Purdy, J. T. Miller, K. Leung, S. S. Chou, H. H. Brongersma, R. t. Veen, J. Huang, H. Guo, Y. Wang and A. K. Datye, Nat. Catal., 2021, 4, 830–839 CrossRef CAS.
- H. Xiong, M. H. Wiebenga, C. Carrillo, J. R. Gaudet, H. N. Pham, D. Kunwar, S. H. Oh, G. Qi, C. H. Kim and A. K. Datye, Appl. Catal., B, 2018, 236, 436–444 CrossRef CAS.
- A. W. Petrov, D. Ferri, F. Krumeich, M. Nachtegaal, J. A. van Bokhoven and O. Kröcher, Nat. Commun., 2018, 9, 2545 CrossRef PubMed.
- K. A. Karinshak, P. Lott, M. P. Harold and O. Deutschmann, ChemCatChem, 2020, 12, 3712–3720 CrossRef CAS.
- T. Franken, M. Roger, A. W. Petrov, A. H. Clark, M. Agote-Arán, F. Krumeich, O. Kröcher and D. Ferri, ACS Catal., 2021, 11, 4870–4879 CrossRef CAS.
- S. Sharma, F. Maurer, P. Lott and T. L. Sheppard, ChemCatChem, 2024, e202301655 CrossRef.
- J. Sá, D. L. A. Fernandes, F. Aiouache, A. Goguet, C. Hardacre, D. Lundie, W. Naeem, W. P. Partridge and C. Stere, Analyst, 2010, 135, 2260–2272 RSC.
- J. Ratcliff, K. Karinshak and M. P. Harold, Chem. Eng. Sci., 2023, 282, 119269 CrossRef CAS.
- Y. Wang, C. Stere, G. McCullough, M. Li and A. Goguet, Catal. Sci. Technol., 2023, 13, 1802–1817 RSC.
- F. Maurer, A. Gänzler, P. Lott, B. Betz, M. Votsmeier, S. Loridant, P. Vernoux, V. Murzin, B. Bornmann, R. Frahm, O. Deutschmann, M. Casapu and J.-D. Grunwaldt, Ind. Eng. Chem. Res., 2021, 60, 6662–6675 CrossRef CAS.
- D. Hodonj, M. Borchers, L. Zeh, G. T. Hoang, S. Tischer, P. Lott and O. Deutschmann, Appl. Catal., B, 2024, 345, 123657 CrossRef CAS.
- H. Stotz, L. Maier, A. Boubnov, A. T. Gremminger, J.-D. Grunwaldt and O. Deutschmann, J. Catal., 2019, 370, 152–175 CrossRef CAS.
- H. Stotz, L. Maier and O. Deutschmann, Top. Catal., 2017, 60, 83–109 CrossRef CAS.
- M. Borchers, K. Keller, P. Lott and O. Deutschmann, Ind. Eng. Chem. Res., 2021, 60, 6613–6626 CrossRef CAS.
- J. Wang, E. Sauter, A. Nefedov, S. Heißler, F. Maurer, M. Casapu, J.-D. Grunwaldt, Y. Wang and C. Wöll, J. Phys. Chem. C, 2022, 126, 9051–9058 CrossRef CAS.
- J. Schütz, H. Störmer, P. Lott and O. Deutschmann, Catalysts, 2021, 11, 300 CrossRef.
- H. Yoshida, T. Nakajima, Y. Yazawa and T. Hattori, Appl. Catal., B, 2007, 71, 70–79 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- K. G. Rappé, C. DiMaggio, J. A. Pihl, J. R. Theis, S. H. Oh, G. B. Fisher, J. Parks, V. G. Easterling, M. Yang, M. L. Stewart and K. C. Howden, Emiss. Control Sci. Technol., 2019, 5, 183–214 CrossRef.
- P. Li, X. Chen, Y. Li and J. W. Schwank, Catal. Today, 2019, 327, 90–115 CrossRef CAS.
- R. Di Monte and J. Kašpar, Top. Catal., 2004, 28, 47–57 CrossRef CAS.
- J. Rink, N. Meister, F. Herbst and M. Votsmeier, Appl. Catal., B, 2017, 206, 104–114 CrossRef CAS.
- D. Duprez, C. Descorme, T. Birchem and E. Rohart, Top. Catal., 2001, 16/17, 49–56 CrossRef CAS.
- L. Spenadel and M. Boudart, J. Phys. Chem., 1960, 64, 204–207 CrossRef CAS.
- D. Ciuparu, F. Bozon-Verduraz and L. Pfefferle, J. Phys. Chem. B, 2002, 106, 3434–3442 CrossRef CAS.
- J. Lee, T. H. Lim, E. Lee and D. H. Kim, ChemCatChem, 2021, 13, 3706–3712 CrossRef CAS.
- M. Cargnello, J. J. Delgado Jaén, J. C. Hernández Garrido, K. Bakhmutsky, T. Montini, J. J. Calvino Gámez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed.
- S. Fouladvand, S. Schernich, J. Libuda, H. Grönbeck, T. Pingel, E. Olsson, M. Skoglundh and P.-A. Carlsson, Top. Catal., 2013, 56, 410–415 CrossRef CAS.
- D. Bounechada, G. Groppi, P. Forzatti, K. Kallinen and T. Kinnunen, Appl. Catal., B, 2012, 119–120, 91–99 CrossRef CAS.
- X. Li, X. Wang, K. Roy, J. A. van Bokhoven and L. Artiglia, ACS Catal., 2020, 10, 5783–5792 CrossRef CAS.
- Y.-H. Chin, M. García-Diéguez and E. Iglesia, J. Phys. Chem. C, 2016, 120, 1446–1460 CrossRef CAS.
- R. L. Mortensen, H.-D. Noack, K. Pedersen, M. A. Dunstan, F. Wilhelm, A. Rogalev, K. S. Pedersen, J. Mielby and S. Mossin, Appl. Catal., B, 2024, 344, 123646 CrossRef CAS.
- A. K. Neyestanaki, F. Klingstedt, T. Salmi and D. Y. Murzin, Fuel, 2004, 83, 395–408 CrossRef CAS.
- D. H. Coller, B. C. Vicente and S. L. Scott, Chem. Eng. J., 2016, 303, 182–193 CrossRef CAS.
- J. Au-Yeung, K. Chen, A. T. Bell and E. Iglesia, J. Catal., 1999, 188, 132–139 CrossRef CAS.
- Y.-H. Chin, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2013, 135, 15425–15442 CrossRef CAS PubMed.
- D. P. Ciuparu and L. Pfefferle, Catal. Today, 2002, 77, 167–179 CrossRef CAS.
- M. Lyubovsky, L. Pfefferle, A. Datye, J. Bravo and T. Nelson, J. Catal., 1999, 187, 275–284 CrossRef CAS.
- J. Xu, L. Ouyang, W. Mao, X.-J. Yang, X.-C. Xu, J.-J. Su, T.-Z. Zhuang, H. Li and Y.-F. Han, ACS Catal., 2012, 2, 261–269 CrossRef CAS.
- A. K. Khudorozhkov, I. A. Chetyrin, A. V. Bukhtiyarov, I. P. Prosvirin and V. I. Bukhtiyarov, Top. Catal., 2017, 60, 190–197 CrossRef CAS.
- J. Nilsson, P.-A. Carlsson, N. M. Martin, E. C. Adams, G. Agostini, H. Grönbeck and M. Skoglundh, J. Catal., 2017, 356, 237–245 CrossRef CAS.
- P. Légaré, L. Hilaire, G. Maire, G. Krill and A. Amamou, Surf. Sci., 1981, 107, 533–546 CrossRef.
- Y.-S. Ho, C.-B. Wang and C.-T. Yeh, J. Mol. Catal. A: Chem., 1996, 112, 287–294 CrossRef CAS.
- S. Colussi, A. Trovarelli, G. Groppi and J. Llorca, Catal. Commun., 2007, 8, 1263–1266 CrossRef CAS.
- D. J. Fullerton, A. V. K. Westwood, R. Brydson, M. V. Twigg and J. M. Jones, Catal. Today, 2003, 81, 659–671 CrossRef CAS.
- A. Fujiwara, H. Yoshida, J. Ohyama and M. Machida, J. Phys. Chem. C, 2020, 124, 12531–12538 CrossRef CAS.
- Y. Liu, J. Yang, J. Yang, L. Wang, Y. Wang, W. Zhan, Y. Guo, Y. Zhao and Y. Guo, Appl. Surf. Sci., 2021, 556, 149766 CrossRef CAS.
- P. Hurtado, S. Ordóñez, H. Sastre and F. V. Díez, Appl. Catal., B, 2004, 51, 229–238 CrossRef CAS.
- E. Ruckenstein and J. J. Chen, J. Catal., 1981, 70, 233–236 CrossRef CAS.
- D. Ciuparu and L. Pfefferle, Appl. Catal., A, 2001, 218, 197–209 CrossRef CAS.
- M. D. Feist, M. Landau and E. Harte, SAE Int. J. Fuels Lubr., 2010, 3, 100–117 CrossRef CAS.
- L. De Simio, S. Iannaccone, C. Guido, P. Napolitano and A. Maiello, Int. J. Hydrogen Energy, 2024, 50, 743–757 CrossRef CAS.
- L. Padeste and A. Baiker, Ind. Eng. Chem. Res., 1994, 33, 1113–1119 CrossRef CAS.
- P. Lott, S. Bastian, H. Többen, L. Zimmermann and O. Deutschmann, Appl. Catal., A, 2023, 651, 119028 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00625a |
| ‡ K. K. and D. H. contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |