
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism and structure–activity relationship of H2 and CO2 activation at the ZnO/Cu catalyst interface†
Xin
Xin
 ab,
Peng
Gao
*abc and
Shenggang
Li
*abcd
ab,
Peng
Gao
*abc and
Shenggang
Li
*abcd
aCAS Key Laboratory of Low-Carbon Conversion Science and Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, 100 Haike Road, Shanghai 201210, China. E-mail: gaopeng@sari.ac.cn; lisg@sari.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Low Carbon Catalysis and Carbon Dioxide Utilization, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, China
dSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China
First published on 29th July 2024
Abstract
Cu/ZnO/Al2O3 catalysts are the most well-known heterogeneous catalysts for the hydrogenation of CO and CO2 into methanol. Herein, density functional theory calculations were performed to investigate the mechanism of H2 activation and the effects of hydrogen spillover on CO2 adsorption and activation at the interfacial site of the ZnO/Cu model catalyst, which was simulated by loading ZnO ribbons of different sizes on the Cu(111), Cu(100), and Cu(211) surfaces. The ZnO/Cu interface is found to facilitate the formation of H adsorbates from the dissociation of H2 molecules, which promotes the facile formation of oxygen vacancy (VO) sites in the ZnO component due to its reducibility and the hydrogen spillover effect. The resulting interfacial structure of the ZnO/Cu model catalyst can contain perfect, hydroxylated, and oxygen-vacancy-present ZnO sites, which may act as the adsorption and activation sites for CO2. Further calculations show that molecular CO2 adsorbed at the VO site  can be efficiently activated by direct dissociation or hydrogenation to the HCOO* species. In addition, the smaller ZnO structure and less exposure of the Cu(211) facet facilitate hydrogen spillover and the formation of the interfacial VO site. This study provides important insights into the structure–activity relationship for the active sites of the ZnO/Cu model catalyst and the mechanisms of CO2 activation and hydrogenation.
can be efficiently activated by direct dissociation or hydrogenation to the HCOO* species. In addition, the smaller ZnO structure and less exposure of the Cu(211) facet facilitate hydrogen spillover and the formation of the interfacial VO site. This study provides important insights into the structure–activity relationship for the active sites of the ZnO/Cu model catalyst and the mechanisms of CO2 activation and hydrogenation.
1 Introduction
The use of fossil energy led to excessive emission of greenhouse gases especially carbon dioxide (CO2), and the resulting global warming effect is threatening the sustainable development of mankind.1–4 In recent years, CO2 capture, utilization, and storage (CCUS) technologies have received increasing attention because of their potential for mitigating the severe consequences of excessive CO2 emission and at the same time producing necessary fuels and valuable chemicals.5–7 CO2 hydrogenation is an effective approach for the synthesis of methanol,8–11 which can be directly used as a fuel or fuel additive, or indirectly used as a chemical intermediate to further produce more advanced fuels and chemicals. Cu/ZnO/Al2O3 is the most widely studied catalyst for CO2 hydrogenation to methanol due to its low cost and high activity, and the industrial Cu/ZnO/Al2O3 catalyst has been employed for over 50 years.12–15 However, CO2 conversion and methanol selectivity at low temperature and pressure still have room for improvement,16–18 and the tendency for the conventional Cu/ZnO/Al2O3 catalyst to deactivate due to sintering also makes it necessary to further improve its stability, by searching for better support materials,19–21 adding suitable promoters,22–24 and changing the synthesis method,25,26 all of which can lead to enhanced catalyst performance. In addition, the unique role of and interaction between the Cu and ZnO components27–29 were also examined to improve our understanding of the CO2 hydrogenation mechanism for the rather complex Cu/ZnO/Al2O3 catalyst system, but our understanding on its active site and reaction mechanism remains very limited.There remain debates on the nature of the active site of the Cu/ZnO/Al2O3 catalyst, although the Cu/ZnO interface or the CuZn alloy has been generally recognized as the active site for methanol synthesis.12,30–35 Despite intensive efforts, it remains difficult for experimental methods alone to provide a thorough understanding on the active site and reaction mechanism of the Cu/ZnO/Al2O3 catalyst for methanol synthesis. For instance, X-ray photoelectron spectroscopic (XPS) experiments have difficulty in clarifying the valence states of the Zn species due to its limited ability to distinguish the 2p peaks of the different Zn species.30,36,37 In addition, introducing a reactive atmosphere can lead to significant changes in the already complex structure of the Cu/ZnO/Al2O3 catalyst, which makes it greatly challenging to elucidate the structure, properties, and reaction mechanism of the catalyst active site.38–41
The metal-oxide interface has unique electronic properties due to its strong interaction,42–45 which has been proposed by experimental studies to promote the CO2 hydrogenation reaction. Kattel et al. compared the activities of the oxide-on-metal ZnO/Cu and bimetallic ZnCu catalysts for CO2 hydrogenation built from a single crystal model system, which illustrated the pivotal role of ZnO on the Cu surface and suggested the metal-oxide interface as the active site for methanol synthesis.31 Wu et al. used the higher valent metal oxide ZrO2 to construct the ZrO2/Cu model catalyst, which greatly enhanced the reactivity of the Cu catalyst and showed excellent catalytic performance in methanol synthesis.46 Recently, Liu et al. further proposed the interface of the oxygen-deficient ZnO and Cu to favour methanol synthesis from CO2 hydrogenation through the HCOO pathway,34 and ZnO1−x/Cu catalysts with abundant oxygen vacancies prepared by Zhang et al. showed excellent methanol selectivity of above 90%.47 These studies demonstrate the rational design of reverse oxide/metal configurations as efficient CO2 hydrogenation catalysts and the importance of the oxygen vacancy in the CO2 hydrogenation reaction.
Density functional theory (DFT) calculations can now provide important physical insights, and thus can potentially reduce the number of experimental trials. Previous computational studies suggest that methanol can be synthesized from CO2 hydrogenation through the HCOO pathway, or via the reverse water gas shift (RWGS) reaction followed by CO hydrogenation, known as the RWGS + CO-hydro pathway.31,34,42,48 Previous theoretical studies on the Cu/ZnO/Al2O3 catalyst for CO2 hydrogenation to methanol mainly focused on the nature of the active site, as well as the morphology and metal coordination of the catalyst.12,34,48–53 Catalyst models, including Cu-supported ZnO and ZnO-supported Cu, have been employed to simulate the ZnO/Cu interface, and the size and shape of the supported Cu cluster and ZnO cluster or nanoribbons were examined by several studies.31,34,45,47,49,51–55 Some recent theoretical investigations examined the mechanism of CO2 hydrogenation at the ZnO/Cu interface, which is generally modelled by placing ZnO clusters or nanoribbons on the Cu surface. Kattel et al.31 and Liu et al.34 constructed such ZnO/Cu catalyst models using different ZnO clusters or nanoribbons, and calculated the reaction pathway of CO2 hydrogenation to methanol through the above two reaction pathways at the perfect or oxygen-deficient ZnO/Cu interface, but only focused on the interfacial site between the ZnO and the Cu(111) surface. It is worth noting that in these reverse oxide-on-metal catalyst models, the metal is modified by the reducible oxide, and there are metal, redox, and Brønsted and Lewis acid sites,56 so the reaction mechanism can be more complex than that on the pure metal or pure oxide surface. To this end, DFT calculations can greatly improve our understanding on the properties of the active site of the Cu/ZnO/Al2O3 catalyst as well as the reaction mechanism for CO2 hydrogenation especially when combined with results from various experimental characterization techniques.
In this work, extensive DFT calculations were performed to reveal the mechanisms of H2 and CO2 activation at the interface of the ZnO/Cu model catalyst during CO2 hydrogenation. Our results show that the ZnO/Cu interface plays a vital role in H2 dissociation, which provides H adsorbates for CO2 hydrogenation. Hydrogen spillover from the Cu surface to the terminal O of the ZnO ribbons results in Brønsted acid and redox sites at the ZnO/Cu interface. The interfacial VO site formed upon hydrogen spillover from Cu to ZnO facilitates both the direct dissociation of CO2 and the formation of the HCOO intermediate by CO2 hydrogenation. The size of the ZnO ribbon and the exposed Cu crystal plane have important effects on H2 activation, which is relevant to the formation of interfacial VO sites. Our work thus provides significant physical insights on the mechanisms of H2 and CO2 activation at the ZnO/Cu interface, which are crucial towards the rational design of more active Cu-based catalysts for CO2 hydrogenation to methanol.
2 Computational methods
All periodic DFT calculations were carried out with the Vienna ab initio simulation package (VASP)57,58 using the Perdew–Burke–Ernzerhof (PBE)59 exchange–correlation functional and the projector-augmented wave (PAW) potentials.60,61 An energy cutoff of 400 eV and a Gaussian smearing width of 0.05 eV were used. The electronic energy in the self-consistent field (SCF) iteration was set to 10−4 eV, and the forces on all unconstrained atoms were converged to 0.03 eV Å−1. A vacuum space of 15 Å was used to separate adjacent slabs along the Z direction.Recently, inverse ZnO/Cu catalyst models with ZnO clusters or nanoribbons supported on the Cu slab surface were constructed to simulate the ZnO/Cu surface.31,34,49,55 Considering the ZnO overlayer observed in experiments of Cu/ZnO/Al2O3 catalysts,34,62–64 catalyst models with ZnO ribbons are used in this work. The interface between Cu and ZnO was modelled by placing a layer of (3 × 3) ZnO(0001) ribbon on top of the three-layer (4 × 8) Cu(111) slab, the three-layer (3 × 5) Cu(100) slab, or the five-layer (3 × 4) Cu(211) slab. For comparison, interface models were also built by loading the smaller one-layer (2 × 3) ZnO(0001) ribbon on the above Cu surfaces. We use Wxy(z) to represent the six models, where x, y, and z refer to the number of ZnO layers, the number of columns of ZnO ribbons, and the Miller index of the Cu surface, respectively. For instance, Fig. 1(a) shows the catalyst model denoted by W13(111), where one layer of the three columns of ZnO ribbons is supported by the Cu(111) surface. Our calculations show that catalyst models with three Cu layers can give results as accurate as with five Cu layers including adsorption energies, reaction energies, and energy barriers as shown in Fig. 4, 5 and S1.† For the ZnO/Cu catalyst models containing ZnO ribbons employed in this work, the mismatch along the extended direction of the ZnO ribbons may result in some strain between these two components, and the misfit can be calculated using the following equation:

 | ||
| Fig. 1 Top and side views of (a) perfect and (b) defective W13(111) surfaces; comparison of (c) the calculated energy barriers (Ea/eV); and (d) BEP relationship for H2 dissociation at the ZnO/Cu interface and the Cu surface. | ||
For W13(111) and W12(111), the Brillouin zone was sampled with a Monkhorst–Pack k-point grid mesh of (3 × 1 × 1), whereas for W13(100) and W12(100), it was sampled with that of (2 × 1 × 1); in these calculations, the bottom two atomic layers were fixed, whereas the top atomic layer and the adsorbed species were relaxed. For W13(211) and W12(211), the Brillouin zone was sampled with a Monkhorst–Pack k-point grid mesh of (1 × 2 × 1), and the bottom three atomic layers were fixed, whereas the top two atomic layers and the adsorbed species were relaxed. The k-points for the different ZnO/Cu catalyst models were generated using the VASPKIT program with the recommended KPT-resolved value of 0.04 2 × π/Å.65
The Hubbard U correction for the Zn atoms was included in our DFT calculations to treat the on-site Coulomb repulsion of their 3d electrons using an effective U value of 4.7 eV (Ueff = U − J) based on the previous work of Liu et al.34 The climbing-image nudged elastic band (CI-NEB) method66,67 and the improved dimer method (IDM)68 were employed to locate the transition states, which were further verified by vibrational analysis showing one and only one imaginary mode. All structures were built and visualized using Materials Visualizer from Materials Studio.69
The formation energy of a VO site (ΔEf,VO,O2) with respect to gas phase O2 is defined as the reaction energy of the thermal desorption of molecular O2:
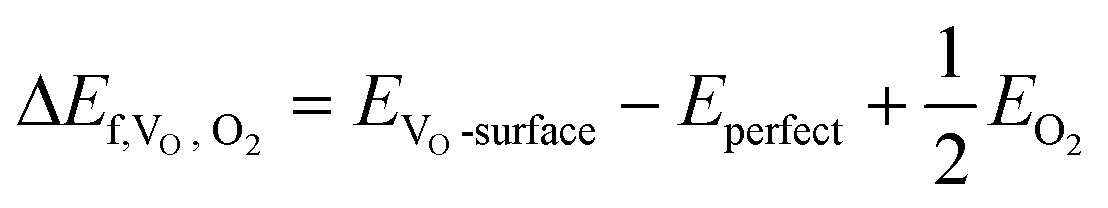
where EVO-surface, Eperfect, EO2, EH2 and EH2O denote the total energies of the defective surface, perfect surface, and gas phase O2, H2 and H2O. The adsorption energy of an adsorbate A on the ZnO/Cu slab surface S is defined as:
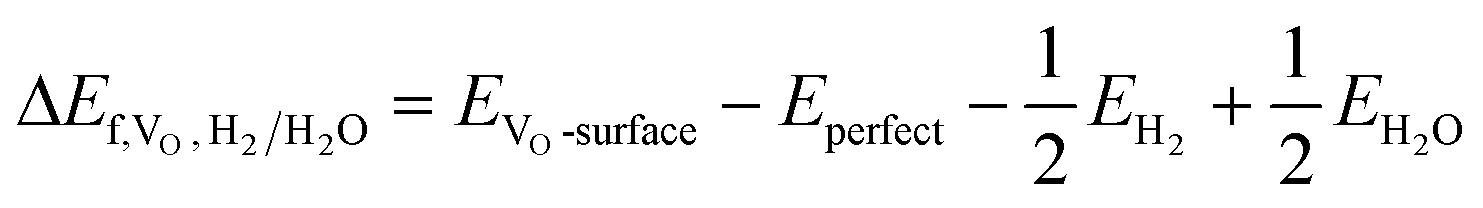
| Ead,A = Etotal − (Eslab + EA) |
3 Results and discussion
3.1 H2 dissociation at the ZnO/Cu interface and the Cu surface
We first compare the dissociative adsorption of H2 at the ZnO/Cu interface and the traditional Cu surface sites.70 As shown in Fig. 1 and 2, unlike the homolytic dissociation of H2 on the Cu surface (TS-HCu), the heterolytic dissociation of H2 at the perfect ZnO/Cu(*_P) interface (TS-Hint) leads to two H adsorbates at the O and Cu sites, where one H atom (H_O*) is adsorbed at the terminal O site of ZnO resulting in a Brønsted acid site, and the other H atom (H_Cu*) is adsorbed on the Cu surface. For the perfect W13(111) surface, the O–HA bond formed after H2 dissociation has a length of 0.98 Å, while the other H atom is adsorbed at the three-fold hollow site surrounded by three Cu atoms. H2 dissociation at this interfacial site has a lower energy barrier (Ea) of 0.26 eV than that on the Cu surface of 0.47 eV. In its transition state, the distance between HA and HB is 0.91 Å, similar to that on the Cu surface of 0.92 Å. H2 dissociation on the Cu(100) and Cu(110) surfaces was also investigated by Higham et al.,71 whose energy barriers were calculated to be 0.54 and 0.42 eV; these values are in good agreement with our results for the homolytic dissociation of H2 on the Cu surfaces. Besides, our calculated energy barrier for the heterolytic dissociation of H2 at the ZnO/Cu interface is comparable to 0.17 eV for the homolytic dissociation of H2 at the Zn sites over the perfect Zn-terminated ZnO(0001) surface.72 The energy barriers of H2 dissociation on all studied ZnO/Cu catalyst models are compared in Fig. 1(c), and those at the interface are generally much lower than those on the Cu surfaces. In addition, H2 dissociation at the interface is strongly exothermic, indicating that it is favoured both thermodynamically and kinetically. Further details in the calculated energy barriers and reaction energies (Er) are given in Table S1.† | ||
| Fig. 2 Potential energy profiles for H2 dissociation at the interface and the subsequent hydrogen spillover process with their energy barriers (Ea/eV) shown. The images display structures of the intermediates and transition states on the perfect W13(111) surface. Brown: Cu, grayish blue: Zn, red: O, white: H. | ||
Fig. 1(d) further shows the Bell–Evans–Polanyi (BEP)73–75 relationship between the energy barrier and the reaction energy for H2 dissociation at the perfect ZnO/Cu interface and on the Cu surface. The BEP relationship for H2 dissociation on the Cu surface shows a reasonably good linear fit with a coefficient of determination (COD) of R2 = 0.89, which makes it possible to estimate the energy barrier of H2 dissociation from its reaction energy on the Cu surface. However, the BEP relationship for H2 dissociation at the perfect ZnO/Cu interface is more complex than linear, as indicated by the low COD of R2 = 0.46 when a linear fit is attempted even by including only four catalyst models and excluding the perfect W12(211) interface, which is marked by the blue dashed circle and is clearly an outlier. Thus, although a roughly linear trend appears between the energy barrier and the reaction energy of H2 dissociation at the interfacial sites of ZnO and different Cu crystal planes, the linearity is not nearly as good as that on the Cu surface.
3.2 Hydrogen spillover mechanism
To study the mechanism and effect of hydrogen spillover on oxygen vacancy formation, we then calculated the formation energy of oxygen vacancies (ΔEf,VO,O2) with respect to gas phase O2 at the interfaces of the different ZnO/Cu catalyst models. For the perfect W13(111) interface model shown in Fig. 1(a), we find that the O sites from the rightmost ZnO ribbon are more likely to be hydrogenated to form oxygen vacancies due to their lower oxygen vacancy formation energies (with an average oxygen vacancy formation energy of 2.94 eV as listed in Table S2†) than those from the other two ZnO ribbons. For the other five catalyst models, the oxygen vacancy formation energies of the O sites from the rightmost ZnO ribbon are generally also lower, indicating the more likely location of oxygen vacancy formation at the ZnO/Cu interface. Thus, in our subsequent calculations, the defect site at the ZnO/Cu interface is chosen to be located at the O site from the rightmost ZnO ribbon.Fig. 2 and Tables S1 and S3† show H2 dissociation at the ZnO/Cu interface and on the Cu surface, leading to two hydrogen spillover mechanisms and oxygen vacancy formation at the interface. After H2 dissociation at the interface, the H atom adsorbed on the Cu surface is further transferred to the Brønsted acid site (OH site) (TS-Hsp2), which is itself formed from H2 dissociation. The energy barrier of this step is between 0.79 and 0.94 eV, and an adsorbed H2O molecule is formed after the hydrogen spillover. Upon H2O desorption, an oxygen vacancy site is formed leading to the defective ZnO/Cu surfaces (*_D).
In contrast, after H2 dissociation on the Cu surface, an additional hydrogen spillover step is required to first form the aforementioned Brønsted acid site followed by the further formation of the oxygen vacancy site by another hydrogen spillover step. Over the perfect ZnO/Cu surfaces, after H2 dissociation on the Cu surface, both H atoms are adsorbed on the hollow Cu sites. For the perfect W13(111) surface, the first H adsorbate diffuses from the Cu surface to the terminal O of ZnO to form a Brønsted acid site (TS-Hsp1) with an energy barrier of 0.74 eV and an exothermicity of −0.54 eV. Then, the second H atom also spills over from the Cu surface to the above Brønsted acid site to form an adsorbed H2O molecule (TS-Hsp2), desorption of which results in an oxygen vacancy and the defective surface as shown in Fig. 1(b).
As discussed in the previous section, H2 tends to dissociate at the ZnO/Cu interface. Nevertheless, the above-mentioned two hydrogen spillover processes are both likely to occur, so the ZnO/Cu interface can promote H2 dissociation to form a Brønsted acid site, as well as the formation of a Brønsted acid site and an oxygen vacancy site after hydrogen spillover. As shown in Table S2,† the formation energies of an oxygen vacancy (ΔEf,VO,H2/H2O) with respect to gas phase H2/H2O for the interfacial O atoms in ZnO are mostly negative, ranging from −0.28 to −0.04 eV, except for that in W13(211) of 0.18 eV, indicating that the formation of the interfacial VO sites in these catalyst models are usually thermodynamically favorable. Under the actual reaction conditions, H2O molecules, which are also products of CO2 hydrogenation, are also present in the reaction atmosphere, so we also consider the possible dissociation of H2O at the ZnO component to form Brønsted acid sites. As shown in Fig. S2 and Table S3,† H2O dissociation at the ZnO component involves a rather low energy barrier of <0.30 eV, leading to the formation of two hydroxyl groups (Zn–OH). This indicates that H2O produced during CO2 hydrogenation may also promote the formation of Brønsted acid sites, and as the reaction proceeds, the ZnO component will likely undergo continuous hydrogenation and redox reactions, leading to dynamic structural changes.
3.3 CO2 adsorption at the ZnO/Cu interface
CO2 can potentially adsorb at the metal site or the oxygen vacancy site on the ZnO/Cu model catalyst. For the perfect and defective W13(111) surfaces, CO2 can adsorb at the interfacial O site of ZnO on the perfect ZnO/Cu surface to form the carbonate configuration as shown in Fig. 3(a), or physisorb in a linear
as shown in Fig. 3(a), or physisorb in a linear  configuration
configuration  as shown in Fig. 3(b), or chemisorb at the interfacial oxygen vacancy site on the defective W13(111) surface to form the bent
as shown in Fig. 3(b), or chemisorb at the interfacial oxygen vacancy site on the defective W13(111) surface to form the bent 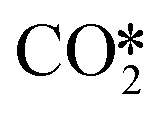 configuration
configuration 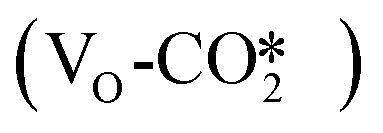 as shown in Fig. 3(c). The different CO2 adsorption structures on the different ZnO/Cu catalyst models are shown in Fig. S3 and S4.†
as shown in Fig. 3(c). The different CO2 adsorption structures on the different ZnO/Cu catalyst models are shown in Fig. S3 and S4.†
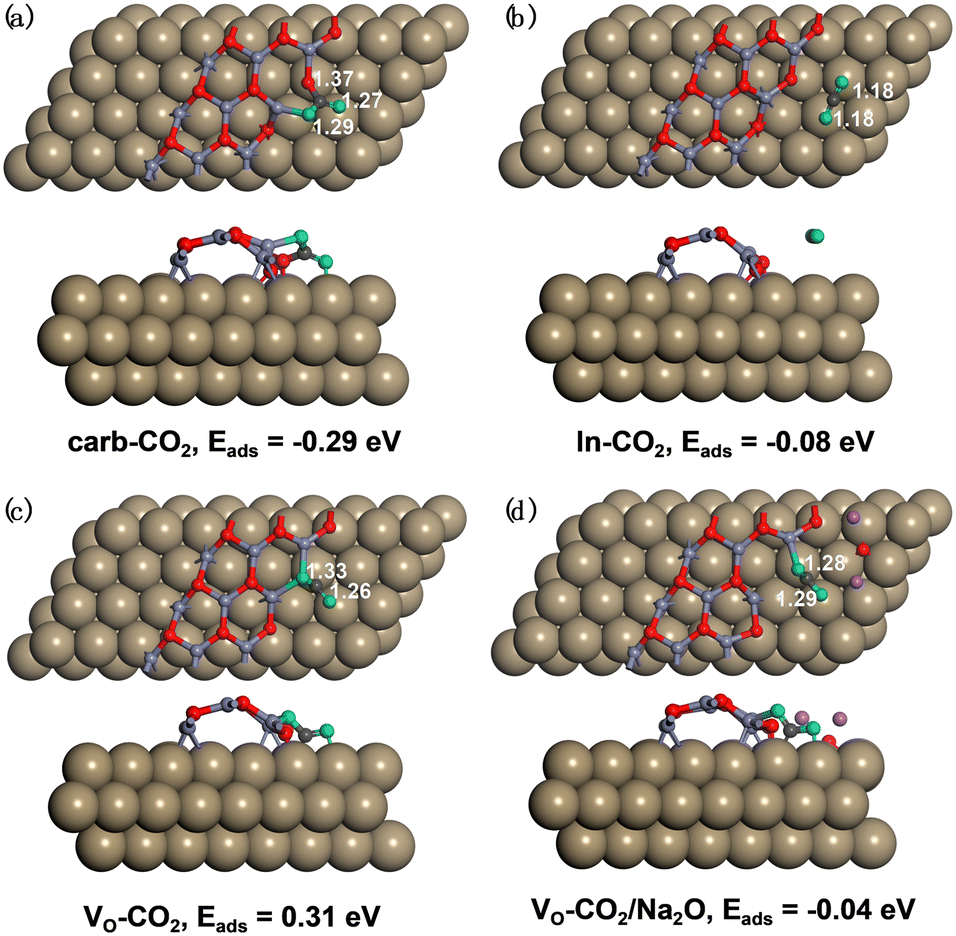 | ||
Fig. 3 Adsorption structures and adsorption energies (Eads/eV) of (a)  , (b) , (b)  on the perfect W13(111) surface, (c) on the perfect W13(111) surface, (c)  on the defective W13(111) surface, and (d) on the defective W13(111) surface, and (d) 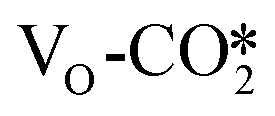 in the presence of the Na promoter. Brown: Cu, greyish blue: Zn, red: O, dark grey: C, green: O in CO2 molecules, purple: Na. in the presence of the Na promoter. Brown: Cu, greyish blue: Zn, red: O, dark grey: C, green: O in CO2 molecules, purple: Na. | ||
For the perfect W13(111) surface without an oxygen vacancy site at the interface, CO2 can combine with a terminal O atom of ZnO to form the 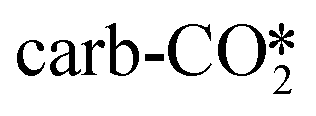 , where the C–O bond lengths in the adsorbed CO2 are elongated to 1.27 and 1.29 Å with the formation of a new C–O bond of 1.37 Å. During the adsorption, the original Zn–O bond is broken, and this Zn atom binds one O atom in the adsorbed CO2, while the other O atom in CO2 binds the surface Cu atom. CO2 can also physisorb in the
, where the C–O bond lengths in the adsorbed CO2 are elongated to 1.27 and 1.29 Å with the formation of a new C–O bond of 1.37 Å. During the adsorption, the original Zn–O bond is broken, and this Zn atom binds one O atom in the adsorbed CO2, while the other O atom in CO2 binds the surface Cu atom. CO2 can also physisorb in the 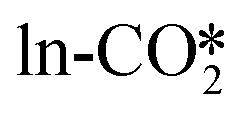 at the perfect ZnO/Cu interface, which undergoes little changes from the free CO2 molecule. For the defective W13(111) surface with an oxygen vacancy site at the interface, CO2 can chemisorb in the
at the perfect ZnO/Cu interface, which undergoes little changes from the free CO2 molecule. For the defective W13(111) surface with an oxygen vacancy site at the interface, CO2 can chemisorb in the 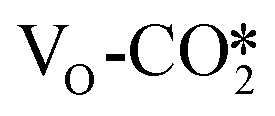 , which undergoes significant deformation compared to the free CO2 molecule, resulting in two C–O bonds of 1.26 and 1.33 Å and an O–C–O bond angle of 120.5°. In the
, which undergoes significant deformation compared to the free CO2 molecule, resulting in two C–O bonds of 1.26 and 1.33 Å and an O–C–O bond angle of 120.5°. In the 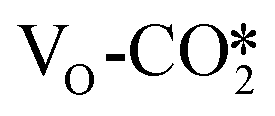 , the oxygen vacancy is essentially occupied by one O atom in CO2, while the other O and the C atoms bind different surface Cu atoms.
, the oxygen vacancy is essentially occupied by one O atom in CO2, while the other O and the C atoms bind different surface Cu atoms.
As shown in Tables 1 and S4,† after CO2 adsorption in the  on the perfect W13(111) surface, the Bader charges of the Cu and Zn atoms bound to the adsorbate increase by 0.17 and 0.11 |e|, respectively, while the Bader charges of the O atoms in the adsorbate decrease by 0.09 |e|. Furthermore, after CO2 adsorption in the
on the perfect W13(111) surface, the Bader charges of the Cu and Zn atoms bound to the adsorbate increase by 0.17 and 0.11 |e|, respectively, while the Bader charges of the O atoms in the adsorbate decrease by 0.09 |e|. Furthermore, after CO2 adsorption in the  on the defective W13(111) surface, the Bader charges of the two Cu atoms around the adsorbate increase by 0.22 and 0.13 |e|, whereas the Bader charges of the two Zn atoms around the adsorbate also increase by 0.29 and 0.23 |e|.
on the defective W13(111) surface, the Bader charges of the two Cu atoms around the adsorbate increase by 0.22 and 0.13 |e|, whereas the Bader charges of the two Zn atoms around the adsorbate also increase by 0.29 and 0.23 |e|.
| W13(111) surface | Q/|e| | Cu | Zn | O |
|---|---|---|---|---|
| *_P | Q1 (*_P) | 0.00 | 1.08 | −1.08 |
Q2 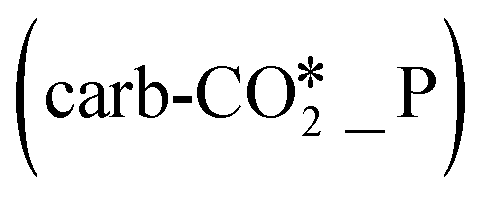 |
0.17 | 1.19 | −1.17 | |
| ΔQ1 (= Q2 − Q1) | 0.17 | 0.11 | −0.09 | |
| *_D | Q3 (*_D) | −0.03/−0.05 | 0.83/0.80 | — |
Q4  |
0.19/0.08 | 1.12/1.03 | — | |
| ΔQ2 (= Q4 − Q3) | 0.22/0.13 | 0.29/0.23 | — |
In addition, as shown in Table S5,† the negative charge on ZnO over the defective surface is significantly lower than that over the perfect surface, so the presence of the oxygen vacancy sites greatly reduces the number of electrons transferred from Cu to ZnO. We note that an opposite direction of charge transfer was predicted by Heenemann et al. for the ZnO-supported Cu nanoparticle models.76 When CO2 is adsorbed on both the perfect and defective surfaces, CO2 acquires some electrons, and that in the  obtains about one electron, which is significantly more than that in the
obtains about one electron, which is significantly more than that in the  of 0.17 electrons on average. For example, after CO2 adsorption on the defective W13(111) surface, the CO2 adsorbate obtains 0.99 |e|, and the Bader charges of the other adsorbed CO2 are shown in Table S5.† This suggests that the presence of the oxygen vacancy site can significantly promote electron transfer from the defective surface to the CO2 adsorbate. Thus, the
of 0.17 electrons on average. For example, after CO2 adsorption on the defective W13(111) surface, the CO2 adsorbate obtains 0.99 |e|, and the Bader charges of the other adsorbed CO2 are shown in Table S5.† This suggests that the presence of the oxygen vacancy site can significantly promote electron transfer from the defective surface to the CO2 adsorbate. Thus, the 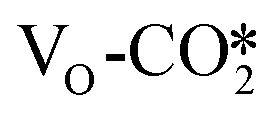 may be more likely hydrogenated to the HCOO intermediate, because it carries more negative charge and is partially reduced upon the formation of the oxygen vacancy.77,78
may be more likely hydrogenated to the HCOO intermediate, because it carries more negative charge and is partially reduced upon the formation of the oxygen vacancy.77,78
It is worth noting that although the 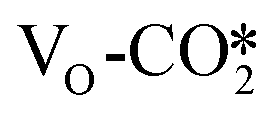 obtains more electrons than the
obtains more electrons than the  , the adsorption energy of the
, the adsorption energy of the  is significantly less negative than that of the
is significantly less negative than that of the  . As shown in Table S6,† the adsorption energy of the
. As shown in Table S6,† the adsorption energy of the  on the perfect W13(111) surface is −0.29 eV, which is much more negative than that of
on the perfect W13(111) surface is −0.29 eV, which is much more negative than that of 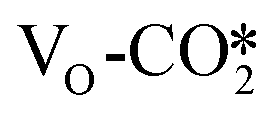 on the defective W13(111) surface of 0.31 eV. Reichenbach et al. also reported a similar observation that adsorption of the activated CO2 molecule on the ZnO cluster models with different oxidation states for Zn is relatively unstable with CO2 adsorption energies of 0.28–0.66 eV.49 We conjecture that introducing the Na2O promoter may further enhance the adsorption of the
on the defective W13(111) surface of 0.31 eV. Reichenbach et al. also reported a similar observation that adsorption of the activated CO2 molecule on the ZnO cluster models with different oxidation states for Zn is relatively unstable with CO2 adsorption energies of 0.28–0.66 eV.49 We conjecture that introducing the Na2O promoter may further enhance the adsorption of the  , and as shown in Fig. 3(d), our calculations show that adding Na2O can indeed change its adsorption energy from 0.31 to −0.04 eV, thus leading to stronger adsorption. Besides, although the carbonate adsorption structures are much stable at the ZnO/Cu interface compared to the
, and as shown in Fig. 3(d), our calculations show that adding Na2O can indeed change its adsorption energy from 0.31 to −0.04 eV, thus leading to stronger adsorption. Besides, although the carbonate adsorption structures are much stable at the ZnO/Cu interface compared to the  , recent computational works suggest that the carbonate species, which is easy to form and stable, will block the active sites of the ZnO surface;72,79 the role of the carbonate will be further discussed in the next section.
, recent computational works suggest that the carbonate species, which is easy to form and stable, will block the active sites of the ZnO surface;72,79 the role of the carbonate will be further discussed in the next section.
In summary, the CO2 adsorbates in both the  and the
and the  appear to be activated considering the decrease in the C–O–C angle and the increase of the C–O bond length, compared with that in the
appear to be activated considering the decrease in the C–O–C angle and the increase of the C–O bond length, compared with that in the  . However, the absence or presence of the oxygen vacancy at the ZnO/Cu interface shows a distinct effect on the stability of the
. However, the absence or presence of the oxygen vacancy at the ZnO/Cu interface shows a distinct effect on the stability of the  and the
and the  , in which the activated
, in which the activated  is more stable than the activated
is more stable than the activated  at the ZnO/Cu interface. Although the
at the ZnO/Cu interface. Although the  is thermodynamically more stable and the
is thermodynamically more stable and the  has an endothermic adsorption energy, the oxygen vacancy site promotes the charge transfer in the latter leading to the accumulation of more electrons on the CO2 adsorbate, which may facilitate its further conversion.
has an endothermic adsorption energy, the oxygen vacancy site promotes the charge transfer in the latter leading to the accumulation of more electrons on the CO2 adsorbate, which may facilitate its further conversion.
3.4 HCOO formation at the ZnO/Cu interface
It is generally believed that CO2 can be hydrogenated to methanol via the formate (HCOO) pathway,77,80–83 where H can either come from direct H2 dissociation or from the Brønsted acid site resulting from hydrogen spillover as discussed in the earlier section. We thus examined CO2 adsorption at the ZnO/Cu interface, and its further hydrogenation to the HCOO intermediate.As shown in Table S7,† in the absence of an oxygen vacancy site at the ZnO/Cu interface, hydrogenation of the  to the HCOO intermediate needs to overcome a very high energy barrier of >2.00 eV, and the energy barrier for the hydrogenation of the
to the HCOO intermediate needs to overcome a very high energy barrier of >2.00 eV, and the energy barrier for the hydrogenation of the  by H* from the Brønsted acid site to the HCOO intermediate remains substantial at 1.42–1.62 eV, whereas the energy barrier of the hydrogenation of the
by H* from the Brønsted acid site to the HCOO intermediate remains substantial at 1.42–1.62 eV, whereas the energy barrier of the hydrogenation of the 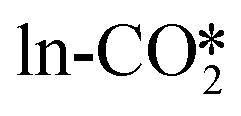 by H* adsorbed on the Zn site is the lowest at only 0.24–0.34 eV. At the perfect W13(111) interface, the detailed reaction pathway for the hydrogenation of the
by H* adsorbed on the Zn site is the lowest at only 0.24–0.34 eV. At the perfect W13(111) interface, the detailed reaction pathway for the hydrogenation of the  and
and  adsorbates to form the HCOO* is shown in Fig. 4(a). The
adsorbates to form the HCOO* is shown in Fig. 4(a). The  can react with the H* adsorbed at the hollow Cu site with a very high energy barrier of 2.02 eV and an exothermicity of −0.53 eV, where the dissociated O atom binds the Zn site, confirming the inactivity of the carbonate at the ZnO/Cu interface for methanol synthesis, similar to that on the ZnO surface.72,79 The
can react with the H* adsorbed at the hollow Cu site with a very high energy barrier of 2.02 eV and an exothermicity of −0.53 eV, where the dissociated O atom binds the Zn site, confirming the inactivity of the carbonate at the ZnO/Cu interface for methanol synthesis, similar to that on the ZnO surface.72,79 The  can react with the H* adsorbed at the O site with a substantial energy barrier of 1.62 eV and an endothermicity of 0.26 eV. The same reaction step of CO2 hydrogenation to HCOO* through H atoms in the Brønsted acid site was calculated by Liu et al. to have a lower energy barrier of 1.15 eV,34 which is likely due to the adsorption of the reactant CO2 molecule at the Cu sites near the interface, different from the physisorbed CO2 in our work. Furthermore, the
can react with the H* adsorbed at the O site with a substantial energy barrier of 1.62 eV and an endothermicity of 0.26 eV. The same reaction step of CO2 hydrogenation to HCOO* through H atoms in the Brønsted acid site was calculated by Liu et al. to have a lower energy barrier of 1.15 eV,34 which is likely due to the adsorption of the reactant CO2 molecule at the Cu sites near the interface, different from the physisorbed CO2 in our work. Furthermore, the  can also react with the H* adsorbed at the Zn site to form the HCOO* with a very low energy barrier of 0.26 eV and an exothermicity of −1.05 eV. The significant difference in the reaction barrier and reaction energy for the hydrogenation of the
can also react with the H* adsorbed at the Zn site to form the HCOO* with a very low energy barrier of 0.26 eV and an exothermicity of −1.05 eV. The significant difference in the reaction barrier and reaction energy for the hydrogenation of the  by the H_O* and H_Zn* can be attributed to the different affinity of the H* adsorbate to the O and Zn sites. This can be inferred from the rather close effective energy barriers (Ea,eff) for the hydrogenation of the
by the H_O* and H_Zn* can be attributed to the different affinity of the H* adsorbate to the O and Zn sites. This can be inferred from the rather close effective energy barriers (Ea,eff) for the hydrogenation of the 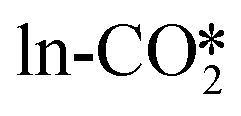 to the HCOO* in these two pathways, which are 0.94 and 0.88 eV involving the H_O* and H_Zn*, respectively.
to the HCOO* in these two pathways, which are 0.94 and 0.88 eV involving the H_O* and H_Zn*, respectively.
 | ||
| Fig. 4 Potential energy profiles for CO2 hydrogenation to the HCOO* intermediate at the interface of the (a) perfect and (b) defective W13(111) surfaces. Structures of the transition states on the perfect and defective W13(111) surfaces are shown. | ||
In the presence of an oxygen vacancy site at the interface, hydrogenation of the 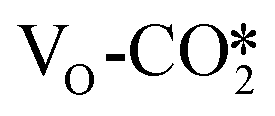 to form the HCOO* only needs to overcome a very low energy barrier of 0.18–0.30 eV, indicating that CO2 can be easily converted into the HCOO* intermediate at the oxygen vacancy site. The detailed energy barriers and reaction energies are listed in Table S7,† and for the defective W13(111) surface, the detailed reaction pathway for the hydrogenation of the
to form the HCOO* only needs to overcome a very low energy barrier of 0.18–0.30 eV, indicating that CO2 can be easily converted into the HCOO* intermediate at the oxygen vacancy site. The detailed energy barriers and reaction energies are listed in Table S7,† and for the defective W13(111) surface, the detailed reaction pathway for the hydrogenation of the  adsorbed at the interface to form the HCOO* is shown in Fig. 4(b). The
adsorbed at the interface to form the HCOO* is shown in Fig. 4(b). The 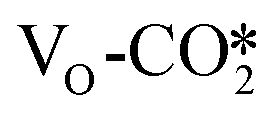 can react with the H* adsorbed at the hollow Cu site to form the HCOO* with a very low energy barrier of only 0.18 eV and an exothermicity of −0.84 eV, where the C–H distance shrinks from 2.54 Å in the initial state to 1.66 Å in the transition state, and the resulting C–O bond lengths in the HCOO* are 1.25 and 1.29 Å. Over other catalyst models such as Cu supported ZnO clusters, similar energy barriers of no more than 0.30 eV were predicted for hydrogenation to HCOO* of the bent CO2 adsorbates with one O atom adsorbed at the Zn site,31,51 comparable with that on the ZnO surface of 0.11 eV,72,79 while hydrogenation to HCOO* of the linear CO2 adsorbates with one O atom adsorbed at the Zn sites needs to overcome a slightly higher energy barrier49 of about 0.60 eV. Thus, these catalytically active sites all exhibit higher activity for HCOO* formation than the Cu surface.71,81
can react with the H* adsorbed at the hollow Cu site to form the HCOO* with a very low energy barrier of only 0.18 eV and an exothermicity of −0.84 eV, where the C–H distance shrinks from 2.54 Å in the initial state to 1.66 Å in the transition state, and the resulting C–O bond lengths in the HCOO* are 1.25 and 1.29 Å. Over other catalyst models such as Cu supported ZnO clusters, similar energy barriers of no more than 0.30 eV were predicted for hydrogenation to HCOO* of the bent CO2 adsorbates with one O atom adsorbed at the Zn site,31,51 comparable with that on the ZnO surface of 0.11 eV,72,79 while hydrogenation to HCOO* of the linear CO2 adsorbates with one O atom adsorbed at the Zn sites needs to overcome a slightly higher energy barrier49 of about 0.60 eV. Thus, these catalytically active sites all exhibit higher activity for HCOO* formation than the Cu surface.71,81
3.5 CO2 dissociation at the ZnO/Cu interface
Methanol may also form from hydrogenation of the CO intermediate, which is produced from the reverse water gas shift (RWGS) reaction. CO can form either by the direct dissociation of the adsorbed to CO* and O*, or by the indirect dissociation pathway via the carboxylic acid (COOH*) intermediate, which can further dissociate into CO* and OH*. We examined the direct dissociation and the COOH* pathways from the different CO2 adsorption structures, and the detailed energy barriers and reaction energies are listed in Tables S8 and S9.† In the absence of an oxygen vacancy on the surface, the
to CO* and O*, or by the indirect dissociation pathway via the carboxylic acid (COOH*) intermediate, which can further dissociate into CO* and OH*. We examined the direct dissociation and the COOH* pathways from the different CO2 adsorption structures, and the detailed energy barriers and reaction energies are listed in Tables S8 and S9.† In the absence of an oxygen vacancy on the surface, the 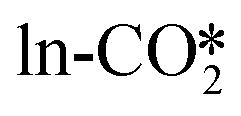 can be directly hydrogenated to the COOH*, while the
can be directly hydrogenated to the COOH*, while the  must be first hydrogenated into the bicarbonate (CO3H*) intermediate prior to its further conversion to the COOH*. On the other hand, in the presence of an oxygen vacancy on the surface, the
must be first hydrogenated into the bicarbonate (CO3H*) intermediate prior to its further conversion to the COOH*. On the other hand, in the presence of an oxygen vacancy on the surface, the  can be converted into CO by both the direct and indirect pathways.
can be converted into CO by both the direct and indirect pathways.
For the perfect W13(111) surface, the detailed reaction pathways for the hydrogenation of the  and
and  to form the COOH* are shown in Fig. 5(a). The O in the
to form the COOH* are shown in Fig. 5(a). The O in the  can be first hydrogenated with the H* adsorbed on the Cu site to form the CO3H* species with a relatively low energy barrier of 0.54 eV and an exothermicity of −0.27 eV. However, breaking the C–O bond in the CO3H* to form the COOH* must overcome a very high energy barrier of 1.82 eV, indicating that converting the
can be first hydrogenated with the H* adsorbed on the Cu site to form the CO3H* species with a relatively low energy barrier of 0.54 eV and an exothermicity of −0.27 eV. However, breaking the C–O bond in the CO3H* to form the COOH* must overcome a very high energy barrier of 1.82 eV, indicating that converting the 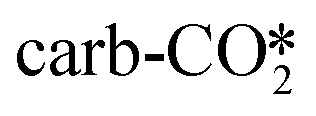 to CO* by hydrogenation is difficult. Similarly, our calculations show that it also seems difficult for hydrogenation of the
to CO* by hydrogenation is difficult. Similarly, our calculations show that it also seems difficult for hydrogenation of the  by an H atom adsorbed at the Zn site to form the COOH* to occur due to the rather high energy barrier of 1.42 eV. Therefore, in the absence of an oxygen vacancy on the surface, the adsorbed CO2 molecule can only be converted into CO by the indirect pathway, which is expected to be slow due to the high energy barrier, and similar conclusions can be drawn for the indirect dissociation of CO2 at the ZnO/Cu interface for the other studied catalyst models. This suggests that the perfect ZnO/Cu interface is rather inactive for the conversion of CO2 into either the HCOO or COOH intermediate, so the absence of oxygen vacancy sites may lead to low CO2 activity.
by an H atom adsorbed at the Zn site to form the COOH* to occur due to the rather high energy barrier of 1.42 eV. Therefore, in the absence of an oxygen vacancy on the surface, the adsorbed CO2 molecule can only be converted into CO by the indirect pathway, which is expected to be slow due to the high energy barrier, and similar conclusions can be drawn for the indirect dissociation of CO2 at the ZnO/Cu interface for the other studied catalyst models. This suggests that the perfect ZnO/Cu interface is rather inactive for the conversion of CO2 into either the HCOO or COOH intermediate, so the absence of oxygen vacancy sites may lead to low CO2 activity.
 | ||
| Fig. 5 Potential energy profiles for CO2 direct and indirect dissociations at the interface of the (a) perfect and (b) defective W13(111) surfaces. Structures of the transition states on the perfect and defective W13(111) surfaces are shown. | ||
In the presence of an oxygen vacancy at the interface, our calculations using the different catalyst models show that CO2 dissociation to form CO easily occurs. For the defective W13(111) surface, Fig. 5(b) shows the detailed potential energy diagrams for the direct dissociation of the adsorbed  at the interface and for its hydrogenation to form the COOH* intermediate. As indicated by the red line in this figure, the
at the interface and for its hydrogenation to form the COOH* intermediate. As indicated by the red line in this figure, the  can directly dissociate at the oxygen vacancy site to yield CO* and O* occupying the oxygen vacancy site with a very low energy barrier of 0.25 eV and an exothermicity of −0.23 eV. In this reaction, one O atom in the
can directly dissociate at the oxygen vacancy site to yield CO* and O* occupying the oxygen vacancy site with a very low energy barrier of 0.25 eV and an exothermicity of −0.23 eV. In this reaction, one O atom in the  binds two Zn atoms in the ZnO ribbon, and the C–O distance in the transition state is 1.74 Å. The resulting CO molecule is adsorbed at the Cu site with C–Cu and C–O bond lengths of 1.85 and 1.16 Å, respectively. There is a lack of theoretical investigation on direct dissociation of CO2, especially on the inverse ZnO/Cu catalyst models. Our results show that the VO sites at the interface favor this process with an energy barrier of 0.25 eV, which is much lower than those of about 0.60–0.79 eV on the CuZn alloy.31,48 In addition, the
binds two Zn atoms in the ZnO ribbon, and the C–O distance in the transition state is 1.74 Å. The resulting CO molecule is adsorbed at the Cu site with C–Cu and C–O bond lengths of 1.85 and 1.16 Å, respectively. There is a lack of theoretical investigation on direct dissociation of CO2, especially on the inverse ZnO/Cu catalyst models. Our results show that the VO sites at the interface favor this process with an energy barrier of 0.25 eV, which is much lower than those of about 0.60–0.79 eV on the CuZn alloy.31,48 In addition, the 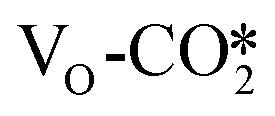 can also be hydrogenated by the H* adsorbed at the Zn site to form the COOH* with an exothermicity of −0.39 eV, and its energy barrier of 0.63 eV is significantly higher than that to the HCOO* of 0.18 eV likely due to the oxophilicity of the Zn site. Thus, based on the above analysis, oxygen vacancy formation is not only conducive to CO2 hydrogenation to form the HCOO* intermediate, but can also promote the direct dissociation of CO2 to form CO, which is crucial for the subsequent CO hydrogenation reaction pathway. Furthermore, our calculations show that the presence of oxygen vacancies in ZnO is mostly to increase CO2 reactivity, whereas its effect on the product selectivity may be rather limited.
can also be hydrogenated by the H* adsorbed at the Zn site to form the COOH* with an exothermicity of −0.39 eV, and its energy barrier of 0.63 eV is significantly higher than that to the HCOO* of 0.18 eV likely due to the oxophilicity of the Zn site. Thus, based on the above analysis, oxygen vacancy formation is not only conducive to CO2 hydrogenation to form the HCOO* intermediate, but can also promote the direct dissociation of CO2 to form CO, which is crucial for the subsequent CO hydrogenation reaction pathway. Furthermore, our calculations show that the presence of oxygen vacancies in ZnO is mostly to increase CO2 reactivity, whereas its effect on the product selectivity may be rather limited.
3.6 Effects of the ZnO size and Cu crystal plane on H2 activation
In our simulations, we considered catalyst models with different sizes of ZnO ribbons loaded on the Cu surface, including ZnO ribbons of three and two columns with all Zn atoms exposed. For ZnO ribbons of different sizes loaded on the Cu(111) surface, namely, the W13(111) and W12(111) surfaces, we further compared the energy barriers of H2 dissociation, hydrogen spillover, CO2 hydrogenation, and dissociation. As shown in Fig. 6(a), ZnO size is shown to mainly influence H2 dissociation and hydrogen spillover during CO2 hydrogenation. When decreasing the ZnO size, the energy barrier of H2 dissociation at the ZnO/Cu interface slightly decreases from 0.26 to 0.18 eV, but that on the Cu surface slightly increases, indicating that a better exposure of the Zn atoms may benefit H2 dissociation at the ZnO/Cu interface. In addition, for oxygen vacancy formation, the energy barrier of the first hydrogen spillover step decreases from 0.74 to 0.50 eV, while that of the second hydrogen spillover step to form an H2O molecule also slightly decreases. Thus, reducing the ZnO size is also conducive to the occurrence of hydrogen spillover. | ||
| Fig. 6 (a) Comparison of the energy barriers of H2 dissociation at the interface, Cu surface, and two hydrogen spillover processes on the W13(111) and W12(111) surfaces. (b) Comparison of energy barriers of the two hydrogen spillover processes at the interface of ZnO and different Cu crystal planes. | ||
Using model catalysts with well-defined structures, such as specific metal surfaces,12,51,84,85 may simplify the mechanistic investigation of complex catalysts. We thus also inspected the influence of the Cu crystal plane at the ZnO/Cu interface on the CO2 hydrogenation reaction. As shown in Fig. 6(b), we find that at the interface between the Cu(211) facet and ZnO, the energy barrier of the first hydrogen spillover step is significantly higher, while that of the second hydrogen spillover step is slightly lower than those for the other studied Cu crystal facets. This indicates that to some extent, interfaces formed on the Cu(211) facet is less favourable for spillover of the H adsorbates formed by H2 dissociation on the Cu surface, leading to less efficient formation of the Brønsted acid site (OH site).
3.7 Discussion
At present, our theoretical understanding on the ZnO/Cu interface for CO2 hydrogenation to methanol remains lacking, and based on previous experimental and theoretical studies, we constructed a series of catalyst models with ZnO ribbons of different sizes loaded on different Cu crystal planes and examined the influence of the ZnO/Cu interfacial structure on the CO2 hydrogenation reaction. Previously, Kattel et al. compared the activities of the ZnCu and ZnO/Cu model catalysts for methanol synthesis from CO2 hydrogenation, and their DFT calculations on the ZnCu(211) and ZnO/Cu(111) surfaces identified the formate pathway and the RWGS + CO-hydro pathway for methanol synthesis from CO2.31 In this work, besides the Cu(111) facet, we also examined interfaces formed between ZnO ribbons and the slightly less stable Cu(211) and Cu(100) facets.86 In addition, their studies suggest that O* formed by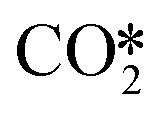 dissociation accumulates at the Zn sites on the ZnCu(211) surface, and hydrogenation of the resulting ZnO is hindered due to the relatively high energy barrier of 1.21 eV. Our calculations show that hydrogenation of the terminal O at the ZnO/Cu(211) interface to form the Zn-OH species also incurs a relatively high energy barrier of 1.08 eV, which is significantly higher than our other studied catalyst models. This suggests that the Cu(211) facet is less favourable for spillover of the H adsorbate at the ZnO/Cu interface. Furthermore, our results show more favourable CO2 hydrogenation to the HCOO* than that to the COOH*, consistent with the theoretical studies of Kattel et al.31 and Xiong et al.51 using ZnO cluster models of the ZnO/Cu(111), ZnO/Cu(110), and ZnO/Cu(100) surfaces.
dissociation accumulates at the Zn sites on the ZnCu(211) surface, and hydrogenation of the resulting ZnO is hindered due to the relatively high energy barrier of 1.21 eV. Our calculations show that hydrogenation of the terminal O at the ZnO/Cu(211) interface to form the Zn-OH species also incurs a relatively high energy barrier of 1.08 eV, which is significantly higher than our other studied catalyst models. This suggests that the Cu(211) facet is less favourable for spillover of the H adsorbate at the ZnO/Cu interface. Furthermore, our results show more favourable CO2 hydrogenation to the HCOO* than that to the COOH*, consistent with the theoretical studies of Kattel et al.31 and Xiong et al.51 using ZnO cluster models of the ZnO/Cu(111), ZnO/Cu(110), and ZnO/Cu(100) surfaces.
Recent experimental studies showed that the reverse ZnO/Cu model catalyst prepared by atomic layer deposition exhibits excellent performance for CO2 hydrogenation to methanol, which is much higher than that using the Cu catalyst itself.34In situ X-ray absorption spectroscopy (XAS) further showed that the Zn species in the highly active catalyst are present as oxygen-deficient ZnO aggregates. To elucidate the promotional effect of the Zn species on the Cu-based catalysts, theoretical calculations were performed to show that the oxygen-deficient ZnO1−x/Cu(111) interface has high activity for methanol formation from CO2 hydrogenation through the formate pathway, while indirect CO2 dissociation incurs a relatively high energy barrier. In addition to the above-mentioned indirect CO2 dissociation pathway, we also considered direct CO2 dissociation at the defective ZnO/Cu interface. Our calculations show that the direct dissociation of the  has the same low energy barrier as its hydrogenation to form the HCOO* intermediate. Thus, the defective ZnO/Cu interface can be expected to have rather similar reactivity to the ZnCu(211) surface for the competition between the direct dissociation and hydrogenation of CO2.31,48 HCOO*, the key reaction intermediate for methanol synthesis from CO2 hydrogenation, can also be easily formed at the ZnO/Cu interface. With the accumulation of CO in the gas phase, it may undergo further hydrogenation to give methanol. However, previous isotope-labelling studies show that methanol is formed from the hydrogenation of CO2 rather than CO,82,87–89 whereas CO is regarded as a mere reducing agent. As Shi et al. reported,90 under different H2, CO, CO2 pressure conditions, structures of the ZnO/Cu catalyst undergo dynamic changes, and CO can even reduce the Zn species. Furthermore, the HCO* species, which is involved in the CO hydrogenation route, are demonstrated as an unstable adsorbate, as the hydrogenation of HCO* usually incurs a higher energy barrier than the reverse reaction of CO* hydrogenation to HCO*. Many of the aforementioned studies reached the conclusion that the HCO* species should quickly dissociate to CO* and H* using quite different ZnO/Cu catalyst models, including the ZnO/Cu interface, ZnCu alloy, and pure Cu surfaces.12,48,49,80,81,90–92 Besides, Liu et al. reported that CO2 hydrogenation to methanol at the stoichiometric ZnO/Cu(111) interface through the H adatom strongly adsorbed at the edge oxygen site encounters rather high energy barriers. Although we find hydrogenation of the
has the same low energy barrier as its hydrogenation to form the HCOO* intermediate. Thus, the defective ZnO/Cu interface can be expected to have rather similar reactivity to the ZnCu(211) surface for the competition between the direct dissociation and hydrogenation of CO2.31,48 HCOO*, the key reaction intermediate for methanol synthesis from CO2 hydrogenation, can also be easily formed at the ZnO/Cu interface. With the accumulation of CO in the gas phase, it may undergo further hydrogenation to give methanol. However, previous isotope-labelling studies show that methanol is formed from the hydrogenation of CO2 rather than CO,82,87–89 whereas CO is regarded as a mere reducing agent. As Shi et al. reported,90 under different H2, CO, CO2 pressure conditions, structures of the ZnO/Cu catalyst undergo dynamic changes, and CO can even reduce the Zn species. Furthermore, the HCO* species, which is involved in the CO hydrogenation route, are demonstrated as an unstable adsorbate, as the hydrogenation of HCO* usually incurs a higher energy barrier than the reverse reaction of CO* hydrogenation to HCO*. Many of the aforementioned studies reached the conclusion that the HCO* species should quickly dissociate to CO* and H* using quite different ZnO/Cu catalyst models, including the ZnO/Cu interface, ZnCu alloy, and pure Cu surfaces.12,48,49,80,81,90–92 Besides, Liu et al. reported that CO2 hydrogenation to methanol at the stoichiometric ZnO/Cu(111) interface through the H adatom strongly adsorbed at the edge oxygen site encounters rather high energy barriers. Although we find hydrogenation of the  to the HCOO* by the weakly adsorbed H_Zn* to incur a very low energy barrier, the effective energy barrier for CO2 hydrogenation by the H_Zn* is still quite high. Furthermore, our study shows that CO2 can also adsorb on the perfect ZnO/Cu surface to form a carbonate species, although hydrogenation of this carbonate configuration is rather difficult, suggesting that the carbonate species is likely just a spectator.
to the HCOO* by the weakly adsorbed H_Zn* to incur a very low energy barrier, the effective energy barrier for CO2 hydrogenation by the H_Zn* is still quite high. Furthermore, our study shows that CO2 can also adsorb on the perfect ZnO/Cu surface to form a carbonate species, although hydrogenation of this carbonate configuration is rather difficult, suggesting that the carbonate species is likely just a spectator.
Conclusions
In this study, we performed a systematic study on the effect of the interfacial structure of the ZnO/Cu catalysts for methanol synthesis from CO2 hydrogenation using the reverse oxide-metal catalyst models with different sizes of ZnO ribbons loaded on different Cu crystal planes. The interfacial oxygen vacancy (VO) sites on the ZnO/Cu model catalysts, formed by H2 dissociation and subsequent hydrogen spillover at the ZnO/Cu interface, were found to be the effective active site for CO2 activation and hydrogenation. Our main observations are the following:(1) The formation of interfacial VO sites from H2 dissociation and hydrogen spillover is investigated. Our results show that H2 molecules tend to dissociate at the ZnO/Cu interface with lower energy barriers instead of the Cu surface, and hydrogen spillover from Cu surfaces to the terminal O atoms of the ZnO occurs with moderate energy barriers to form VO sites. These processes are kinetically favorable on most of the catalyst models with different sizes of ZnO ribbons and Cu crystal planes, consistent with the experimental observation of the promotion of methanol production by oxygen-deficient ZnO at the interface.
(2) Studies on the further conversion of the CO2 adsorbates at the ZnO/Cu interface, including the hydrogenation, indirect, and direct dissociation, indicate that the interfacial VO site accelerates the hydrogenation of the 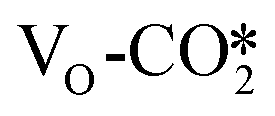 through the HCOO* pathway as well as the direct dissociation of CO2, whereas the carbonate adsorption structure may act as a spectator. Thus, the ZnO/Cu interface containing VO sites promotes the formation of the HCOO* intermediates and CO, similar to the CuZn alloy in previous studies.
through the HCOO* pathway as well as the direct dissociation of CO2, whereas the carbonate adsorption structure may act as a spectator. Thus, the ZnO/Cu interface containing VO sites promotes the formation of the HCOO* intermediates and CO, similar to the CuZn alloy in previous studies.
(3) Catalyst models with smaller ZnO ribbons are more conducive to the formation of the interfacial VO active site due to the lower reaction barrier of interfacial H2 dissociation and hydrogen spillover from the Cu surface to the ZnO ribbon. However, the presence of the Cu(211) facet disfavours the first hydrogen spillover step during the formation of the Brønsted acid site, which may result in a lower coverage of the interfacial VO site.
Our work reveals the mechanism of oxygen vacancy formation at the ZnO/Cu interface, and demonstrates the promotion of CO2 activation and reaction at the ZnO/Cu interface, thus providing significant insights into the crucial role of ZnO/Cu interface sites in the CO2 hydrogenation reaction.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22293023, 22172188, 22172189, 22293025), CAS Youth Interdisciplinary Team, Program of Shanghai Academic Research Leader (22XD1424100), and Science and Technology Commission of Shanghai Municipality (23YF1453400, 23ZR1481700).References
- C. F. Shih, T. Zhang, J. Li and C. Bai, Joule, 2018, 2, 1925–1949 CrossRef CAS.
- D. A. Lashof and D. R. Ahuja, Nature, 1990, 344, 529–531 CrossRef CAS.
- P. Friedlingstein, R. M. Andrew, J. Rogelj, G. P. Peters, J. G. Canadell, R. Knutti, G. Luderer, M. R. Raupach, M. Schaeffer, D. P. van Vuuren and C. Le Quere, Nat. Geosci., 2014, 7, 709–715 CrossRef CAS.
- A. G. Olabi and M. A. Abdelkareem, Renewable Sustainable Energy Rev., 2022, 158, 112111 CrossRef CAS.
- J. F. D. Tapia, J. Y. Lee, R. E. H. Ooi, D. C. Y. Foo and R. R. Tan, Sustain. Prod. Consum., 2018, 13, 1–15 CrossRef.
- K. Jiang, P. Ashworth, S. Y. Zhang, X. Liang, Y. Sun and D. Angus, Renewable Sustainable Energy Rev., 2020, 119, 109601 CrossRef.
- S. Y. Chen, J. F. Liu, Q. Zhang, F. Teng and B. C. McLellan, Renewable Sustainable Energy Rev., 2022, 167, 112537 CrossRef CAS.
- R. P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y. G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- S.-T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Chem. Soc. Rev., 2021, 50, 4259–4298 RSC.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. L. Kniep, M. Tovar, R. W. Fischer, J. K. Norskov and R. Schlogl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- A. Alvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef CAS PubMed.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- A. Beck, M. A. Newton, L. G. A. van de Water and J. A. van Bokhoven, Chem. Rev., 2024, 124, 4543–4678 CrossRef CAS PubMed.
- Z. Chen, J. Wen, Y. Zeng, M. Li, Y. Tian, F. Yang, M. M.-J. Li, P. Chen, H. Huang, D. Ye and L. Chen, Appl. Catal., B, 2024, 340, 123192 CrossRef CAS.
- J. Wang, Y. Song, J. Li, F. Liu, J. Wang, J. Lv, S. Wang, M. Li, X. Bao and X. Ma, Appl. Catal., A, 2024, 674, 119618 CrossRef CAS.
- Y. Xu, Z. Gao, Y. Xu, X. Qin, X. Tang, Z. Xie, J. Zhang, C. Song, S. Yao, W. Zhou, D. Ma and L. Lin, Appl. Catal., B, 2024, 344, 123656 CrossRef CAS.
- J. Zhu, D. Ciolca, L. Liu, A. Parastaev, N. Kosinov and E. J. M. Hensen, ACS Catal., 2021, 11, 4880–4892 CrossRef CAS PubMed.
- Y. Ou, T. Zhang, L. Lv, W. Tang and S. Tang, Colloids Surf., A, 2023, 676, 132167 CrossRef CAS.
- C. Han, L. Qin, P. Wang, H. Zhang, Y. Gao, M. Zhu, S. Wang and J. Li, Fuel, 2024, 357, 129945 CrossRef CAS.
- T. Qi, W. Li, H. Li, K. Ji, S. Chen and Y. Zhang, Mol. Catal., 2021, 509, 111641 CrossRef CAS.
- R. Dalebout, L. Barberis, N. L. Visser, J. E. S. van der Hoeven, A. M. J. van der Eerden, J. A. Stewart, F. Meirer, K. P. de Jong and P. E. de Jongh, ChemCatChem, 2022, 14, e202200451 CrossRef CAS PubMed.
- F. Chen, W. Gao, K. Wang, C. Wang, X. Wu, N. Liu, X. Guo, Y. He, P. Zhang, G. Yang and N. Tsubaki, Fuel, 2022, 315, 123272 CrossRef CAS.
- B. Hu, Y. Yin, Z. Zhong, D. Wu, G. Liu and X. Hong, Catal. Sci. Technol., 2019, 9, 2673–2681 RSC.
- Y. Xu, Z. Gao, L. Peng, K. Liu, Y. Yang, R. Qiu, S. Yang, C. Wu, J. Jiang, Y. Wang, W. Tan, H. Wang and J. Li, J. Catal., 2022, 414, 236–244 CrossRef CAS.
- R. M. Palomino, P. J. Ramírez, Z. Liu, R. Hamlyn, I. Waluyo, M. Mahapatra, I. Orozco, A. Hunt, J. P. Simonovis, S. D. Senanayake and J. A. Rodriguez, J. Phys. Chem. B, 2017, 122, 794–800 CrossRef PubMed.
- A. Arandia, J. Yim, H. Warraich, E. Leppäkangas, R. Bes, A. Lempelto, L. Gell, H. Jiang, K. Meinander, T. Viinikainen, S. Huotari, K. Honkala and R. L. Puurunen, Appl. Catal., B, 2023, 321, 122046 CrossRef CAS.
- C. Li, K. Chen, X. Wang, N. Xue and H. Yang, Acta Phys.-Chim. Sin., 2020, 37, 2009101 Search PubMed.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjaer, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- S. Kattel, P. J. Ramirez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- J. Nakamura, T. Fujitani, S. Kuld, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2017, 357, eaan8074 CrossRef PubMed.
- R. Dalebout, L. Barberis, G. Totarella, S. J. Turner, C. La Fontaine, F. M. F. de Groot, X. Carrier, A. M. J. van der Eerden, F. Meirer and P. E. de Jongh, ACS Catal., 2022, 12, 6628–6639 CrossRef CAS PubMed.
- X. Liu, J. Luo, H. Wang, L. Huang, S. Wang, S. Li, Z. Sun, F. Sun, Z. Jiang, S. Wei, W. X. Li and J. Lu, Angew. Chem., Int. Ed., 2022, 61, e202202330 CrossRef CAS PubMed.
- X. Liu, H. Wang and J. Lu, J. Catal., 2024, 436, 115561 CrossRef CAS.
- P. Amann, B. Klotzer, D. Degerman, N. Kopfle, T. Gotsch, P. Lomker, C. Rameshan, K. Ploner, D. Bikaljevic, H. Y. Wang, M. Soldemo, M. Shipilin, C. M. Goodwin, J. Gladh, J. H. Stenlid, M. Borner, C. Schlueter and A. Nilsson, Science, 2022, 376, 603–608 CrossRef CAS PubMed.
- D. Laudenschleger, H. Ruland and M. Muhler, Nat. Commun., 2020, 11, 3898 CrossRef PubMed.
- W. Tu, P. Ren, Y. Li, Y. Yang, Y. Tian, Z. Zhang, M. Zhu, Y. C. Chin, J. Gong and Y. F. Han, J. Am. Chem. Soc., 2023, 145, 8751–8756 CrossRef CAS PubMed.
- L. Xu, K. G. Papanikolaou, B. A. J. Lechner, L. Je, G. A. Somorjai, M. Salmeron and M. Mavrikakis, Science, 2023, 380, 70–76 CrossRef CAS PubMed.
- R. Wang, H. Wang, X. Weng, J. Dai, Z. Gong, C. Zhao, J. Lu, Y. Cui and X. Bao, J. Energy Chem., 2021, 60, 150–155 CrossRef CAS.
- S. Jensen, M. H. R. Mammen, M. Hedevang, Z. Li, L. Lammich and J. V. Lauritsen, Nat. Commun., 2024, 15, 3865 CrossRef CAS PubMed.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, ACS Catal., 2015, 5, 6696–6706 CrossRef CAS.
- J. Y. Han, J. Y. Yang, Z. X. Zhang, X. Z. Jiang, W. Liu, B. T. Qiao, J. J. Mu and F. Wang, J. Am. Chem. Soc., 2023, 145, 22671–22684 CrossRef CAS PubMed.
- C. R. A. Catlow, S. A. French, A. A. Sokol, M. Alfredsson and S. T. Bromley, Faraday Discuss., 2003, 124, 185–203 RSC.
- C. Wu, L. Lin, J. Liu, J. Zhang, F. Zhang, T. Zhou, N. Rui, S. Yao, Y. Deng, F. Yang, W. Xu, J. Luo, Y. Zhao, B. Yan, X.-D. Wen, J. A. Rodriguez and D. Ma, Nat. Commun., 2020, 11, 5767 CrossRef CAS PubMed.
- F. Zhang, B. Li, X. Quan, K. Wang, J. Xu, T. Wu, Z. Li, M. Yan, S. Liu, Y. He, Y. Shi, Y. Su and P. Xie, ACS Catal., 2024, 14, 7136–7148 CrossRef CAS.
- X.-K. Wu, G.-J. Xia, Z. Huang, D. K. Rai, H. Zhao, J. Zhang, J. Yun and Y.-G. Wang, Appl. Surf. Sci., 2020, 525, 146481 CrossRef CAS.
- T. Reichenbach, K. Mondal, M. Jäger, T. Vent-Schmidt, D. Himmel, V. Dybbert, A. Bruix, I. Krossing, M. Walter and M. Moseler, J. Catal., 2018, 360, 168–174 CrossRef CAS.
- K. Mondal, Megha, A. Banerjee, A. Fortunelli, M. Walter and M. Moseler, J. Phys. Chem. C, 2021, 126, 764–771 CrossRef.
- W. Xiong, Z. F. Wu, X. Y. Chen, J. Q. Ding, A. A. I. Ye, W. H. Zhang and W. X. Huang, Sci. China: Chem., 2023, 67, 715–723 CrossRef.
- X. Z. Wang, H. Zhang, H. Qin, K. M. Wu, K. Wang, J. F. Ma and W. D. Fan, Fuel, 2023, 346, 128381 CrossRef CAS.
- Z.-J. Zuo, P.-D. Han, Z. Li, J.-S. Hu and W. Huang, Appl. Surf. Sci., 2012, 261, 640–646 CrossRef CAS.
- S. T. Bromley, S. A. French, A. A. Sokol, C. R. A. Catlow and P. Sherwood, J. Phys. Chem. B, 2003, 107, 7045–7057 CrossRef CAS.
- K. Yao, S.-S. Wang, X.-K. Gu, H.-Y. Su and W.-X. Li, Chin. J. Catal., 2013, 34, 1705–1711 CrossRef CAS.
- J. Fu, S. Liu, W. Zheng, R. Huang, C. Wang, A. Lawal, K. Alexopoulos, S. Liu, Y. Wang, K. Yu, J. A. Boscoboinik, Y. Liu, X. Liu, A. I. Frenkel, O. A. Abdelrahman, R. J. Gorte, S. Caratzoulas and D. G. Vlachos, Nat. Catal., 2022, 5, 144–153 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlögl and M. G. Willinger, Angew. Chem., Int. Ed., 2015, 54, 4544–4548 CrossRef CAS PubMed.
- T. Song, R. Li, J. Wang, C. Dong, X. Feng, Y. Ning, R. Mu and Q. Fu, Angew. Chem., Int. Ed., 2023, 63, e202316888 CrossRef PubMed.
- M. Mahapatra, J. Kang, P. J. Ramírez, R. Hamlyn, N. Rui, Z. Liu, I. Orozco, S. D. Senanayake and J. A. Rodriguez, J. Phys. Chem. C, 2018, 122, 26554–26562 CrossRef CAS.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- A. Heyden, A. T. Bell and F. J. Keil, J. Chem. Phys., 2005, 123, 224101 CrossRef PubMed.
- Materials Studio, Biovia Software Inc., 5005 Wateridge Vista Drive, San Diego, CA 92121 USA, 2018 Search PubMed.
- Q.-L. Tang, W.-T. Zou, R.-K. Huang, Q. Wang and X.-X. Duan, Phys. Chem. Chem. Phys., 2015, 17, 7317–7333 RSC.
- M. D. Higham, M. G. Quesne and C. R. A. Catlow, Dalton Trans., 2020, 49, 8478–8497 RSC.
- Y.-F. Zhao, R. Rousseau, J. Li and D. Mei, J. Phys. Chem. C, 2012, 116, 15952–15961 CrossRef CAS.
- V. Pallassana and M. Neurock, J. Catal., 2000, 191, 301–317 CrossRef CAS.
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206–217 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- M. Heenemann, M.-M. Millet, F. Girgsdies, M. Eichelbaum, T. Risse, R. Schlögl, T. Jones and E. Frei, ACS Catal., 2020, 10, 5672–5680 CrossRef CAS.
- K. Larmier, W. C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Coperet, Angew. Chem., Int. Ed., 2017, 56, 2318–2323 CrossRef CAS PubMed.
- Y. H. Wang, S. Kattel, W. G. Gao, K. Z. Li, P. Liu, J. G. G. Chen and H. Y. Wang, Nat. Commun., 2019, 10, 1166 CrossRef PubMed.
- A. J. Medford, J. Sehested, J. Rossmeisl, I. Chorkendorff, F. Studt, J. K. Nørskov and P. G. Moses, J. Catal., 2014, 309, 397–407 CrossRef CAS.
- Y. Yang, J. Evans, J. A. Rodriguez, M. G. White and P. Liu, Phys. Chem. Chem. Phys., 2010, 12, 9909–9917 RSC.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- Y. Yang, C. A. Mims, D. H. Mei, C. H. F. Peden and C. T. Campbell, J. Catal., 2013, 298, 10–17 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- L. Jiang, K. Liu, S.-F. Hung, L. Zhou, R. Qin, Q. Zhang, P. Liu, L. Gu, H. M. Chen, G. Fu and N. Zheng, Nat. Nanotechnol., 2020, 15, 848–853 CrossRef CAS PubMed.
- T. Yang, T. Gu, Y. Han, W. Wang, Y. Yu, Y. Zang, H. Zhang, B. Mao, Y. Li, B. Yang and Z. Liu, J. Phys. Chem. C, 2020, 124, 27511–27518 CrossRef CAS.
- L. Song, X. Tian, Y. Yang, J. Qin, H. Li and X. Lin, Front. Chem., 2020, 8, 607 CrossRef CAS PubMed.
- K. C. Waugh, Catal. Lett., 2012, 142, 1153–1166 CrossRef CAS.
- G. C. Chinchen, P. J. Denny, D. G. Parkera, M. S. Spencer and D. A. Nhan, Appl. Catal., 1987, 30, 333–338 CrossRef CAS.
- G. C. Chinchen, P. J. Denny, J. R. Jennings, M. S. Spencer and K. C. Waugh, Appl. Catal., 1988, 36, 1–65 CrossRef CAS.
- Y.-F. Shi, P.-L. Kang, C. Shang and Z.-P. Liu, J. Am. Chem. Soc., 2022, 144, 13401–13414 CrossRef CAS PubMed.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- Y.-F. Zhao, Y. Yang, C. Mims, C. H. F. Peden, J. Li and D. Mei, J. Catal., 2011, 281, 199–211 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00604f |
| This journal is © The Royal Society of Chemistry 2024 |