
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidating the role of the state of Pd in the H2-SCR of NOx by operando XANES and DRIFTS†
Thomas J.
Eldridge
 *ab,
Michael
Borchers
b,
Patrick
Lott
b,
Jan-Dierk
Grunwaldt
ab and
Dmitry E.
Doronkin
*ab
*ab,
Michael
Borchers
b,
Patrick
Lott
b,
Jan-Dierk
Grunwaldt
ab and
Dmitry E.
Doronkin
*ab
aInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology, Eggenstein-Leopoldshafen 76344, Germany. E-mail: thomas.eldridge@kit.edu; dmitry.doronkin@kit.edu
bInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, Karlsruhe 76131, Germany
First published on 25th June 2024
Abstract
The selective catalytic reduction (SCR) of NOx with hydrogen is an attractive strategy for NOx removal when H2 is used as a sustainable fuel in combustion engines. However, the pathway suffers from a strong overconsumption of H2via direct oxidation to water. In order to improve the understanding of the SCR mechanism with H2 as the reductant, the state of the active metal, the reactive surface intermediates, and the conditions which are suited for efficient SCR need to be uncovered. A 1%Pd/5%V2O5/20%TiO2–Al2O3 catalyst was investigated using operando X-ray absorption spectroscopy (XAS) and diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) to track the temperature dependent structure and state of Pd, as well as its gradients and surface intermediates. XAS shows that NO reduces oxidized Pd, forming metal-support interfacial nitrates according to DRIFTS. Partially reduced Pd becomes oxidized by reducing interfacial NOx species to Pd-nitrosyls. Limitations for the H2-SCR of NOx arise from the balance of adsorbed Pd–NO and activated H2, which is dependent on Pd state. The bulk metallic Pd which forms above 250 °C causes a runaway activation of H2, further reduction of Pd, and loss of nitrosyls on Pd. The highest activity occurs when Pd is oxidized enough to promote metal-support interfacial nitrates and also reduced enough to convert these nitrates to Pd-nitrosyls. The storage of NOx and formation of NHx on vanadia–titania permits conversion of NO at high temperatures, but does not counteract the deactivation of Pd. In conclusion, activation of H2 is favored on metallic sites, but must be moderated to allow for PdO being present as well.
Introduction
In the course towards reducing carbon-based engine-out emissions and because of its potential future production from sustainable sources,1,2 hydrogen has been investigated as an attractive alternative to diesel in thermal combustion engines. Apart from being a potential sustainable energy carrier, the primary motivation for using H2 is the avoidance of carbon and particulate matter (PM) emissions in the exhaust. However, under the high temperatures of H2 combustion, N2 oxidation by O2 from air leads to NOx as a pollutant in the 1000 ppm range. While these NOx emissions can be mitigated by optimal air-to-fuel ratios and engine control methods,3–5 H2 thermal combustion engines have a higher NOx emission potential relative to traditional fuels.6In order to meet NOx emission guidelines, an emission-out catalyst for the selective catalytic reduction (SCR) of NOx is necessary. At present, SCR of NOx is largely executed using NH3 as the reductant.7–13 However, due to the direct availability from the fuel tank, H2 has been investigated as an alternative SCR agent.
Current catalysts for SCR of NOx by H2 show higher levels of NOx conversion, relative to NH3 as the reducing agent, at lower temperatures.14,15 However, the primary challenge of H2-SCR is combatting the competitive direct oxidation of H2 to H2O, which becomes the favored reaction pathway at high temperatures. Further complications include several reactions simultaneously occurring over the catalyst bed, including NH3 formation and subsequent NH3-SCR, leading to gradients in reaction product concentrations and in catalyst structure.16,17
Supported noble metal catalysts, with favor towards Pt18–23 and Pd,24–29 are most widely investigated for H2-SCR. Pt and Pd present different benefits and challenges relative to other noble metals: where Pt shows substantially higher NOx conversion but lower selectivity for N2 with respect to N2O, and where Pd has high selectivity for N2 but lower overall catalytic activity.15
There are several primary groups of catalyst supports that have been studied in H2-SCR: (1) TiO2,28–32 often combined with V2O5,26,27,33,34 (2) WO3/ZrO2,35–38 and (3) MgO–CeO2,19–21 and (4) zeolites, alone or in combination with other supports.39–43 H2-SCR catalysts with low noble metal loadings (<1 wt%) of Pt or Pd on WO3/ZrO2 or MgO-CeO2 have shown wide temperature ranges (100 to 400 °C) of NOx conversion with high (>80%) selectivity for N2. However, they have not been studied as extensively as catalysts supported on TiO2. Pd on TiO2–Al2O3 is notable for converting NOx over a wide temperatures range from 100 to 350 °C, but with a significant drop in activity at 200 °C.26,30 This phenomenon was explained by NO reduction to N2, occurring from 100 to 200 °C, NO oxidation to NO2, occurring from 200 to 400 °C, and NO2 reduction to N2 at 100 to 400 °C. The addition of V2O5 to Pd/TiO2–Al2O3 followed a study with Pd/V2O5/Al2O3 which showed high levels of NO reduction to N2 in the narrow range of 200 to 300 °C,33 which well counteracted the observed activity decrease on Pd/TiO2–Al2O3.26
In order to better understand the reactive conditions of Pd/V2O5/TiO2–Al2O3 for H2-SCR of NOx, a study was performed using a honeycomb monolith and a variety of concentrations of NO, H2, and O2.34 It was shown that the H2 reaction pathway plays a significant role, where NO reduction to N2 occurred more readily when either H2 amounts were increased or O2 concentration decreased, as O2 competes with NO for reaction with H2. Additionally, the catalyst state (e.g. Pd oxidized/reduced, Pd particles or clusters) was shown to play a significant role in SCR activity, as the pre-reduced catalyst showed significantly higher activity and N2 selectivity at temperatures below 200 °C.
In order to gain further insight into the surface reactions and the mechanism, ex situ characterization and catalytic activity studies, which use outlet gas beyond the catalyst, are not sufficient for observing what occurs within the black-box catalyst. For this purpose, operando and in situ characterization is need to unravel the species and structure of the catalyst as a function of time and space while the reaction is ongoing.44 Spectroscopic techniques, such as X-ray absorption spectroscopy (XAS) and diffuse reflectance infrared Fourier-transformed spectroscopy (DRIFTS) are able to measure the bulk and surface state of catalysts during steady-state and transient chemical reactions. These techniques also enable the measurement of spatial gradients in surface species concentration and catalyst state45–47 and are thus applied during H2-SCR in this study. Herewith, the present work aims at linking the oxidation state of Pd to the catalytic hydrogen combustion and selective catalytic reduction of NOx by H2 under dynamic reaction conditions. Both operando spatially and time-resolved XAS and operando DRIFTS measurements were conducted on a 1%Pd/V2O5/TiO2–Al2O3 catalyst.
Experimental
A 1%Pd/5%V2O5/20%TiO2–Al2O3 catalyst was prepared via the following steps: γ-Al2O3 (Puralox, Sasol) was calcined in air for 5 h at 700 °C and suspended in a solution of Ti(OBu)4 (Merck) and EtOH (≥99.8%, VWR Chemicals); Ti(OBu)4 was hydrolyzed by dropwise addition of deionized water to the suspension and rigorous stirring. The received TiO2–Al2O3 solid was dried for 2 h at 70 °C and calcined in air for 6 h at 500 °C. V2O5 was added to the 20 wt.% TiO2–Al2O3 support via incipient wetness impregnation (IWI) using a NH4VO3 (Alfa Aesar) solution with aqueous oxalic acid (Merck) and subsequent drying for 1 h at 70 °C and calcination in air for 12 h at 500 °C. Pd was added in a second IWI using an aqueous Pd(NH3)4(NO3)2-solution (abcr) and calcination of the received catalyst powder for 16 h at 500 °C. Inductively coupled plasma optical emission spectroscopy (ICP-OES) confirmed the target elemental composition of the catalyst.N2-physisorption of the catalyst powder was measured using a BELSORP-mini II instrument (BEL Japan) with pretreatment under vacuum at 300 °C for 2 h. Analysis of the adsorption–desorption curve by the Brunauer–Emmett–Teller (BET) method yielded the specific surface area and total pore volume of the catalyst.
X-ray diffraction (XRD) patterns were acquired with an X'PERT PRO diffractometer (PANalytical) using a Cu-Kα radiation source with a wavelength of 1.54 Å (2θ from 20 to 80°, step size of 0.017°, acquisition time of 0.44 s per data point).
Temperature-programmed reduction by hydrogen (H2-TPR) was performed from 40 to 600 °C at 10 K min−1 in a 50 mL min−1 flow of 10% H2 in Ar using an AutoChem II Chemisorption Analyzer (Micromeritics). A thermal conductivity detector (TCD) measured H2 consumption. Prior to H2-TPR, the sample was pretreated by heating with a temperature ramp of 10 K min−1 up to 500 °C under 20% O2 in N2.
Operando X-ray absorption spectroscopy (XAS) measurements were obtained at the PETRA III P64 beamline48 at DESY (Hamburg, Germany). Catalyst powder was pressed and sieved to 125–250 μm grains to minimize mass transport limitations. The sieved fraction was placed between two quartz wool plugs in a fixed-bed quartz capillary serving as plug-flow microreactor.49 The microreactor was heated by a hot air gas blower (Leister LE mini kit). Using capillary microreactors of 2 mm outer diameter and 0.02 mm wall thickness loaded with 12.5 mg of the granulated catalyst for a bed length of 4.1 mm, NO conversion was measured by a mass spectrometer (MS, Hiden ExQ) at the outlet, while simultaneously recording time-resolved X-ray absorption near edge structure (XANES) spectra at different positions along the catalyst bed, under a 50 mL min−1 flow (GHSV 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1) of 1000 ppm NO, 5000 ppm H2, and 10% O2 (He balance) in different gas combinations, following the sequential order as shown in Table 1; between each gas mixture, the setup pipes and the reactor itself were flushed with He. The sample was oxidized in 10% O2 at 500 °C prior to TPR. A single light-off-light-out cycle from 25 to 350 °C at 2 K min−1 was performed for each gas mixture during continuous XANES measurements.
000 h−1) of 1000 ppm NO, 5000 ppm H2, and 10% O2 (He balance) in different gas combinations, following the sequential order as shown in Table 1; between each gas mixture, the setup pipes and the reactor itself were flushed with He. The sample was oxidized in 10% O2 at 500 °C prior to TPR. A single light-off-light-out cycle from 25 to 350 °C at 2 K min−1 was performed for each gas mixture during continuous XANES measurements.
| Gas mix | NO (ppm) | H2 (ppm) | O2 (%) |
|---|---|---|---|
| TPO | 0 | 0 | 10 |
| NOA | 1000 | 0 | 10 |
| SCR | 1000 | 5000 | 10 |
| MixA | 0 | 5000 | 10 |
| NOB | 1000 | 0 | 0 |
| MixB | 1000 | 5000 | 0 |
| TPR | 0 | 5000 | 0 |
Due to time limitations at the XAS beamline, only two-point calibrations (zero gas and educts at zero conversion) of MS instrumentation were possible and allowed for only qualitative and semi-quantitative analysis. Additionally, small quantities (i.e. <100 ppm) of nitrogen species such as NH3, N2, N2O, and NO2 measured at m/z values of 15, 28, 44, and 46 were particularly difficult to detect by MS and did not produce enough counts for robust quantitative analysis.
Quick extended X-ray absorption fine structure (QEXAFS) spectra50 were obtained with a monochromator frequency of 1 Hz, resulting in 2 spectra per second. The polychromatic X-ray beam from the tapered undulator was tuned to the Pd K edge by a liquid nitrogen cooled Si(111) channel-cut crystal monochromator. With a beam size of 1.0 mm (horizontal) × 1.0 mm (vertical), 4 points along the catalyst bed were measured. For the spatially resolved XANES evaluation, the positions were continuously cycled during light-off and the constantly running QEXAFS measurements, holding at each position for 30 seconds; timestamps were recorded before and after each movement of the reactor to isolate and average spectra obtained at each position in order to improve the signal quality. Pd foil measured simultaneously with the catalyst served as the reference.
For evaluation of the XANES spectra, the QEXAFS data were read-in, split, and exported as text files using the JAQ Analyzes QEXAFS software (version 5.3).48 Using a Python (version 3.10) script empowered by the Larch (version 0.9.65) package,51 XANES spectra were calibrated for energy, sorted and averaged by position, and correlated with light-off temperature values. Normalization of spectra and subsequent linear combination analysis (LCA) were performed using the Athena software from the Demeter software package (version 0.9.26),52 across an energy range from 24340 to 24400 eV, using the as-prepared Pd/V2O5/TiO2–Al2O3 catalyst in oxygen and H2 after temperature-programmed oxidation and reduction at 350 °C, respectively, as references.
DRIFTS spectra were obtained using a Vertex 70 spectrometer (Bruker) equipped with a Praying Mantis mirror assembly (Harrick) and a liquid nitrogen cooled mercury cadmium telluride (MCT) detector with a resolution of 4 cm−1. A Praying Mantis high temperature reaction chamber (Harrick) with flat CaF2 windows was loaded with 24.5 mg granulated catalyst (125–250 μm). Steady-state DRIFTS spectra were recorded as an average of 300 scans at 40 kHz at 25 °C intervals from 50 to 325 °C, approximating a single light-off-light-out cycle, with a flow of 50 ml min−1 (GHSV 30000 h−1) using the same pretreatment, gas mixtures, and sequence as with the XAS measurements and as described in Table 1, but using argon instead of helium for balance. Background reference interferograms were collected at each temperature once during cooling down after the initial oxidation pretreatment (10% O2, 325 °C, 30 min). For DRIFTS quantitative analysis and comparison, the baseline of the pseudo-absorbance spectra was zeroed and flattened using a piecewise cubic Hermite interpolating polynomial (PCHIP) function.
Results and discussion
Characterization and catalytic activity of Pd/V2O5/TiO2–Al2O3
The characterization data, light-off conversion profiles, product selectivity, and reaction pathway selectivity for this catalyst have previously been reported in great detail on the same catalyst coated on a honeycomb.34 Hence, the results reported herein are discussed with respect to the outlet mass spectrometer measurements for the purpose of relating data from the XAS and DRIFTS measurements to the activity profile in the specific operando measurements. Fig. 1a and b show the light-off curves for NO reduction in the presence of H2 and the H2-SCR of NO, respectively.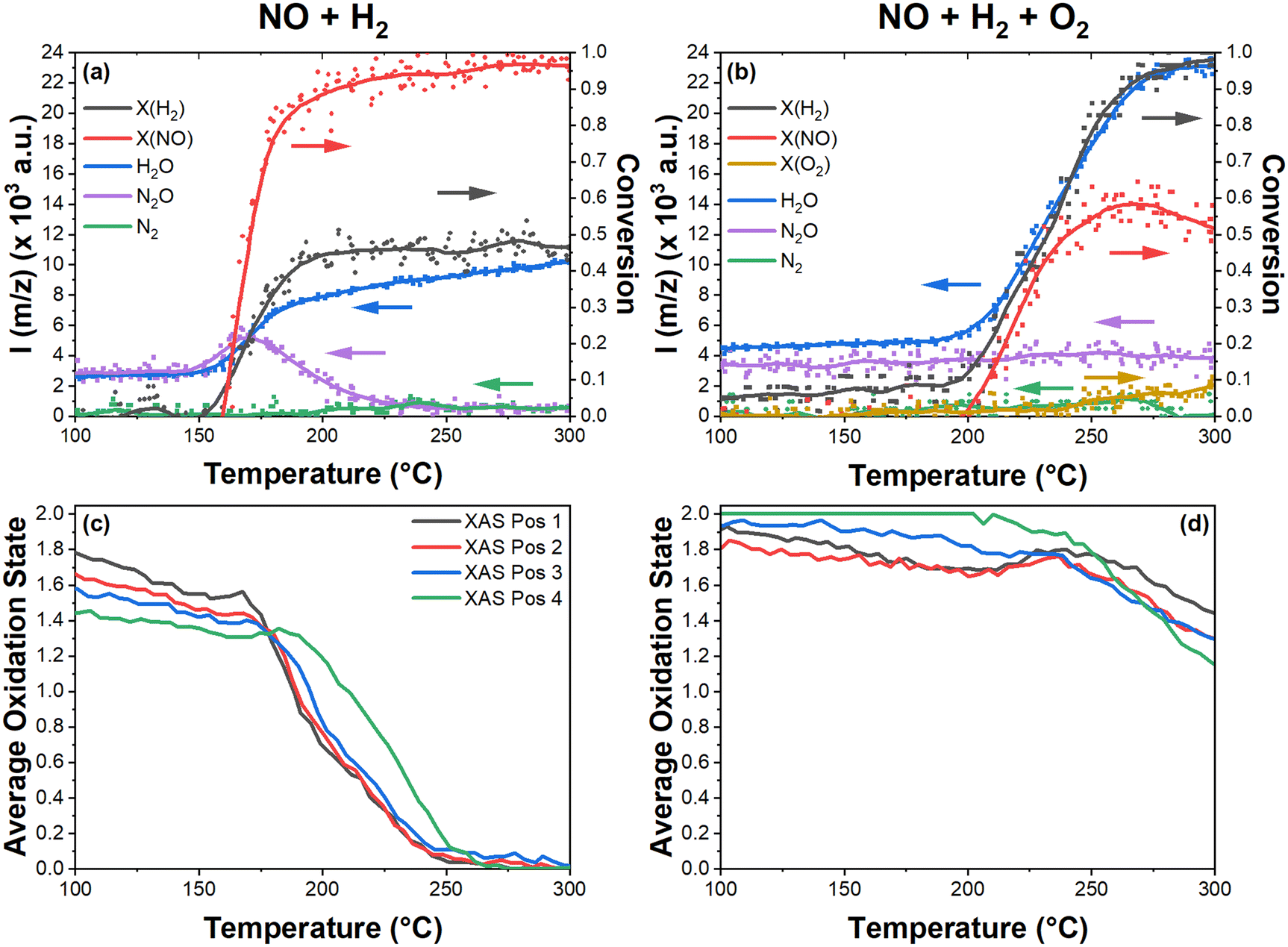 | ||
| Fig. 1 Light off curves on 1%Pd/5%V2O5/20%TiO2–Al2O3 (heating ramp of 2 K min−1, 50 mL min−1/GHSV 60000 h−1) during conversion of H2, NO, and O2 to H2O, N2, and N2O (MS intensities at m/z = 2, 30, and 32 and 18, 28, and 44, respectively) in (a) 1000 ppm NO, 5000 ppm H2 in He and (b) 1000 ppm NO, 5000 ppm H2, and 10% O2 in He (SCR); the dots are experimental data and the solid lines are locally weighted scatterplot smoothing (LOWESS) regressions; (c and d). | ||
The atypical light-off observed in Fig. 1a is due to the absence of O2, matching well with the honeycomb study which showed that the NO conversion curve broadens and shifts to lower temperatures when the ratio of O2 to H2 is lowered. Over-stoichiometric conversion of H2 and the gradual increase in H2O MS intensity in the absence of NO during NO reduction by H2, as shown in Fig. 1a, is due to the broad reduction area from 200 to 600 °C previously shown34 by H2-TPR for this catalyst. The change in the MS zero line for N2O suggests conversion of trace N2O impurities present in the NO gas cylinder available at the synchrotron.
Due to inherent differences between testing monolith and powder catalysts, under identical SCR gas conditions, the powder catalyst of this study underwent light-off for NO and H2 conversion at higher temperatures relative to the monolith catalyst (cf.Fig. 1b); however, it is possible to correlate changes in the Pd state and surface species with the light-off and relate these data to the previously reported honeycomb catalyst study. This will be discussed in more detail below in combination with DRIFTS and XAS analysis below.
With exception to N2O in Fig. 1a, nitrogen species products formed in amounts below the detection limit of the MS instrumentation; however, product selectivity under these gas conditions has been previously reported34 and is out of the scope of this study. MS data measured during other gas conditions are shown in Fig. S1.† The relatively low O2 conversion in Fig. 1b is due to the stoichiometric excess of O2 used; Fig. S2† shows the absolute concentrations of H2, NO, and O2. From these data, a slight delay between the onset of SCR activity at 200 °C and increased O2 conversion at 230 °C was observed, at which point the conversion of NO stagnated. This matches well with previous findings with the honeycomb catalyst where it was concluded that the direct oxidation of H2 predominantly opposes H2-SCR activity at elevated temperatures.
Operando XANES measurements
In order to elucidate the dynamic changes in the Pd oxidation state during H2-SCR over the 1%Pd/5%V2O5/20%TiO2-Al2O3 catalyst, QEXAFS measurements were performed with different gas mixtures. The resulting XANES spectra were quantified by LCA using corresponding reference spectra. Reference spectra for metallic/reduced Pd and oxidized Pd were obtained from 5000 ppm H2 in argon (TPR) and 10% O2 in argon (TPO) measurements, as shown in the ESI† by Fig. S3; LCA fits for these spectra were done using Pd foil and PdO references to confirm the catalyst had fully reduced and oxidized during TPR and TPO, respectively. Further analysis of the extended X-ray absorption fine structure (EXAFS) of the fully reduced Pd yielded an average Pd coordination number of 8.1 (cf. Table S1†), corresponding to nanoparticles of approximately 1 nm in diameter assuming a fcc full truncated cubooctahedral particle.53Sample XANES spectra for the H2-SCR (1000 ppm NO, 5000 ppm H2, and 10% O2 in argon) and NO reduction (1000 ppm NO and 5000 ppm H2 in argon) gas conditions along with metallic Pd and PdO reference spectra are shown in Fig. 2. The LCA fits to these spectra are shown in Fig. 1c and d for the SCR and NO reduction conditions, respectively, showing space-resolved dynamic changes in the average oxidation state of Pd.
 | ||
| Fig. 2 Temperature dependent XANES spectra at the Pd K-edge for the 1%Pd/5%V2O5/20%TiO2-Al2O3 catalyst during the (a) 1000 ppm NO, 5000 ppm H2, and 10% O2 in Ar (H2-SCR) and (b) 1000 ppm NO and 5000 ppm H2 in Ar. | ||
In the SCR reaction mixture, Pd is mostly in the oxidized state (Fig. 1d), gradually reducing with increasing temperature from 50 to 200 °C, with the beginning of the catalyst bed more reduced than the end. At the onset of SCR activity at 200 °C, the catalyst front oxidizes slightly, matching the conversion curve of NO shown in Fig. 1b. A local maxima of oxidation was reached at 250 °C, correlating with the maximum NO conversion. The subsequent Pd reduction matches the decrease in NO conversion from 250 to 300 °C. Spatially, the front of the catalyst bed reduces more from 50 to 200 °C and the end is generally more oxidized before the onset of SCR activity at 200 °C. From 200 to 300 °C, an inversion occurs such that the end of the catalyst bed is more reduced than the front. In the absence of DRIFTS data, the increase in Pd oxidation state during NO conversion suggests that Pd assists H2 in the reduction of adsorbed NO species leading to the formation of N2O or N2. When considering the competing oxidation and reduction of Pd by O2 and H2, respectively, the alternate explanation for the oxidation of Pd from 200 to 250 °C is the loss of H2 due to conversion during the H2-SCR reaction. However, this is not likely to be the case. The substantial reduction of Pd above 250 °C, which was not observed with the NO + O2 and H2 + O2 gas conditions (cf. Fig. S3†), indicates that H2 and NHx continue to reduce Pd even when participating in separate reactions. While the oxidation of Pd by O2 is sufficient to counteract PdO reduction by H2, reduced NHx species limit the reoxidation of Pd by O2. The role of NO in the oxidation of Pd will be discussed in further detail in the DRIFTS analysis below.
For the NO reduction condition (without O2), at lower temperatures (50 to 150 °C) Pd is slightly reduced yet mostly oxidized with an average oxidation state of ∼1.6 (Fig. 1c), with the end of the catalyst more reduced than the front. Pd gradually reduces in the presence of NO and H2 until 168 °C where all positions rapidly reduce to Pd0, with a temperature-dependent delay (ΔT) of 5 and 32 °C towards the end of the catalyst bed at positions 3 and 4, respectively. The onset of NO conversion occurs at 150 °C (before the rapid reduction in Pd, cf. MS data shown in Fig. 1a). Instead of correlating directly with NO reduction, the Pd oxidation state profile matches the diminishing production of N2O. This suggests that H2 partially reduces NO to N2O in the process of reducing Pd, either directly or via an intermediate. The space-resolved average oxidation states of Pd according to LCA fits for gas conditions of 1000 ppm NO in argon and 1000 ppm NO and 10% O2 in argon can be found in the ESI† (Fig. S3).
As XAS is a bulk sensitive technique, detailed mechanisms cannot be drawn solely from these data. In order to complement these measurements and provide a more complete insight into the mechanism under these dynamic conditions, the role of surface intermediates has been investigated in more detail by DRIFTS.
Operando DRIFTS measurements
 | ||
Fig. 3 Temperature dependent DRIFTS spectra of the 1%Pd/5%V2O5/20%TiO2–Al2O3 catalyst under a flow of 1000 ppm NO in Ar, shown at (a) high wavenumbers for OH (negative) and water bands, (b) middle wavenumbers for NO+, V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (negative), and Pd–NO bands, and (c) low wavenumbers for support nitrate and nitrite bands. O (negative), and Pd–NO bands, and (c) low wavenumbers for support nitrate and nitrite bands. | ||
The negative bands between 3500 and 3800 cm−1 are assigned to OH-species, ν(OH),54–56 with the 3733 and 3691 cm−1 bands associated with a loss of Ti4+–OH and the 3658 cm−1 band with a loss of V–OH species. The bands associated with Ti–OH are more intense than those for V–OH. These bands become less intense at higher temperature, following the gradual disappearance of the broad water peak. The negative band at 2047 cm−1 is associated with 2ν(VO) overtones and a loss of V5+ species within the catalyst.56,57
Bands at 2181 and 2111 cm−1 have generally been assigned to NO+ on TiO2 catalysts and linked to NO adsorption on Brønsted acid sites as a replacement of H+ in OH species.58,59 These bands are very weak relative to other bands (Fig. 4a) but are present in the greatest amount at temperatures where nitrates are more prevalent than Pd-adsorbed nitrosyls (Pd–NO), as described below. They are indicative of an oxidizing environment for NO. The band at 2181 cm−1 increases in intensity up to 175 °C before decreasing and disappearing by 250 °C, whereas the 2111 cm−1 band is most pronounced at 50 °C and vanishes above 175 °C. Both peaks redshift with increasing temperature, suggesting slight relaxation of the N–O bonds due to reduced constraints localized around the surface binding site.
 | ||
| Fig. 4 Absolute intensities of selected IR bands of reactive intermediates from DRIFTS pseudo-absorbance spectra for (a) NO, (b) NO and O2, (c) NO and H2, and (d) SCR gas mixtures. The V5+ and OH bands are negative in absorbance and represent the relative decrease in VO and Ti–OH species, respectively. | ||
The bands between 1725 and 1900 cm−1 are associated with linearly adsorbed NO on Pd and V, which have been previously reported at 1890 (V4+ (NO)2, νs(NO)60 and Pd2+ (NO)2, νs(NO)61), 1828 (V3+ (NO)2, νs(NO)),56 1830 (Pd2+, ν(NO)),59 1776 (V4+ (NO)2, νas(NO)62 and Pd+, ν(NO)55), and 1745 cm−1 (V4+ (NO)2, νas(NO)56 and Pd0, ν(NO)55). While there is potential overlap between Pd–NO and V–NO species, IR bands of nitrosyls are only seen on vanadium at or above ambient temperatures after substantial reduction of the catalyst.56 A previous DRIFTS study by Macleod et al. on V2O5/Al2O3 with and without Pd concluded that V3+ and V4+ centers do not form under H2-SCR conditions (4000 ppm H2, 500 ppm NO, 5% O2), while also measuring a negative V5+ band at 2047 cm−1.33 Likewise, only nitrosyl bands for the Pd loaded catalyst were observed, and hence the bands at 1819 and 1788 cm−1 were assigned to Pd+-NO. Considering these findings, it is reasonable to conclude that the bands between 1725 and 1900 cm−1 in the current study are predominantly due to NO species adsorbed on Pd.
In the case of the measurements reported herein, at low temperatures for the adsorption of NO on Pd, the peak at 1813 cm−1 (Pdn+, Pd2+(OH)NO, ν(NO))63 is the predominant band. The blueshift of this peak to 1820 cm−1, which occurred with increasing temperature, may be due to overlap among a combination of lesser bands at 1830 and 1828 cm−1 with the 1813 cm−1 band, suggesting a greater prevalence of Pd2+. This corresponds to the slight oxidizing effect of NO on Pd when Pd is sufficiently reduced as seen by XAS (cf. Fig. S2e†). However, the dominance and symmetry of the 1820 cm−1 band above 150 °C makes it difficult to assign direct contribution of additional nitrosyl states under these conditions. At lower temperatures from 100 to 150 °C, weaker bands at 1745, 1776, and 1868 cm−1 may contribute asymmetry to the 1813 cm−1 band, as they are significant features observed under other gas conditions discussed in later sections below, but are not significant features during NO adsorption on this catalyst. At approximately 250 °C, the band intensity of Pd–NO moieties begins to decrease sharply. At these high temperatures, according to previous temperature programmed desorption (TPD) measurements of NO on Pd,64 the rate of desorption of NO from Pd limits the presence of adsorbed nitrosyl species.
The negative band area at 2047 cm−1 decreases in unison with the shrinking of negative OH bands (3500–3800 cm−1) and of low frequency bands from 1200 to 1650 cm−1, which are assigned to different nitrate species (ν3(NO3)). The bands at 1647 and 1620, 1581 and 1545, and 1310 and 1270 cm−1 correspond to bridging nitrates, bidentate nitrates, and monodentate nitrates, respectively.55–58,65,66 Bands around 1680 cm−1 are not well established in the literature, but evolve in parallel with bridge nitrates, suggesting they are an additional bridging species (e.g. metal-support bridging nitrates). Additional smaller peaks near 1200 cm−1 may contribute to the broad band from 1350 to 1200 cm−1 due to v3 splitting of the bridge (Δv3 ∼ 420 cm−1) and bidentate (Δv3 ∼ 300 cm−1) species. The band at 1488 cm−1 has been previously seen in the literature, but doesn't have a clear assignment;61,66 it can be a chelating nitrate or hydrogenated NO species such as NH2NO, but unlikely to be ammonia species due to the lack of more intense bands at 1434 cm−1.
On TiO2 and V2O5 supports, it is generally understood that nitrates form via surface hydroxides and are the primary contributors towards the negative bands at 3500 to 3800 and 2047 cm−1,57,58,65 following the reaction,
| 2Ti–OH + 2NO + 2[O] → Ti–O–NO2 + Ti–O–NO + H2O | (1) |
O with increasing temperature (Fig. 4a). Among all the nitrate species, the bidentate nitrates at 1581 and 1545 cm−1 are the most stable and are still present in small amounts at 300 °C. The conditions under which nitrates are stable appear to be opposite for nitrosyl adsorption on Pd, as the bands between 1725 and 1900 cm−1 remained relatively weak until the nitrate bands diminished at 175 °C. At the point when nitrate bands become less intense than Pd–NO nitrosyls, Pd begins to reduce NOx species, supported by the disappearance of NO+ bands and the gradual increase in Pd oxidation state as measured by XAS (cf. Fig. S3e†). Here it can be inferred that at lower temperatures, NOx is more easily oxidized by the support and PdO, leading to more oxidized nitrogen species. As temperature increases, nitrates at the metal-support interface become reduced by Pd and form Pdn+–NO nitrosyl species. These findings are observed to a greater extent with other gas conditions below.
 | ||
| Fig. 5 Temperature dependent DRIFTS spectra of the 1%Pd/5%V2O5/20%TiO2–Al2O3 catalyst under a flow of 1000 ppm NO and 10% O2 in Ar, shown at (a) high wavenumbers for OH (negative) and water bands, (b) middle wavenumbers for NO+, VO (negative), and Pd–NO bands, and (c) low wavenumbers for support nitrate and nitrite bands. | ||
The most significant changes can be seen in the nitrosyl bands from 1725 to 1900 cm−1, where the main similarity between the NO and NO + O2 conditions is the desorption of NO from Pd above 250 °C. In the co-adsorption of NO and O2, the splitting among bands at 1826 and 1813 cm−1 is more pronounced, and a new band emerges at 1796 cm−1. These bands, as well as the 1776 cm−1 band, evolve in unison, increasing until 225 °C before gradually decaying with increasing temperature due to rapid desorption of NO from Pd. The maximum intensity of these bands is substantially less than with the adsorption of only NO. Also unlike in the previous gas condition, Pd gradually reduces with increasing temperature during the co-adsorption of NO and O2, as shown by XAS (cf. Fig. S3b†), which correlates both with Pd–NO gradually increasing and a substantial presence of NO+ from broad and intense bands at 2181 cm−1. The oxidation state of Pd levels out and remains constant at 225 °C, at which point Pd–NO and NO+ bands decrease.
From the combined observations of NO adsorption and NO and O2 co-adsorption, it can be inferred that nitrosyl species on Pd form more easily from nitrates which are reduced by Pd, given the stronger Pdn+–NO bands with NO alone. The slight reduction of PdO observed with the co-adsorption of NO and O2 suggests that PdO is able to some degree oxidize and store NOx. However, given the more significant oxidation of Pd and greater presence of nitrosyl species on Pd, as observed during NO adsorption, it appears Pd more efficiently reduces surface NOx.
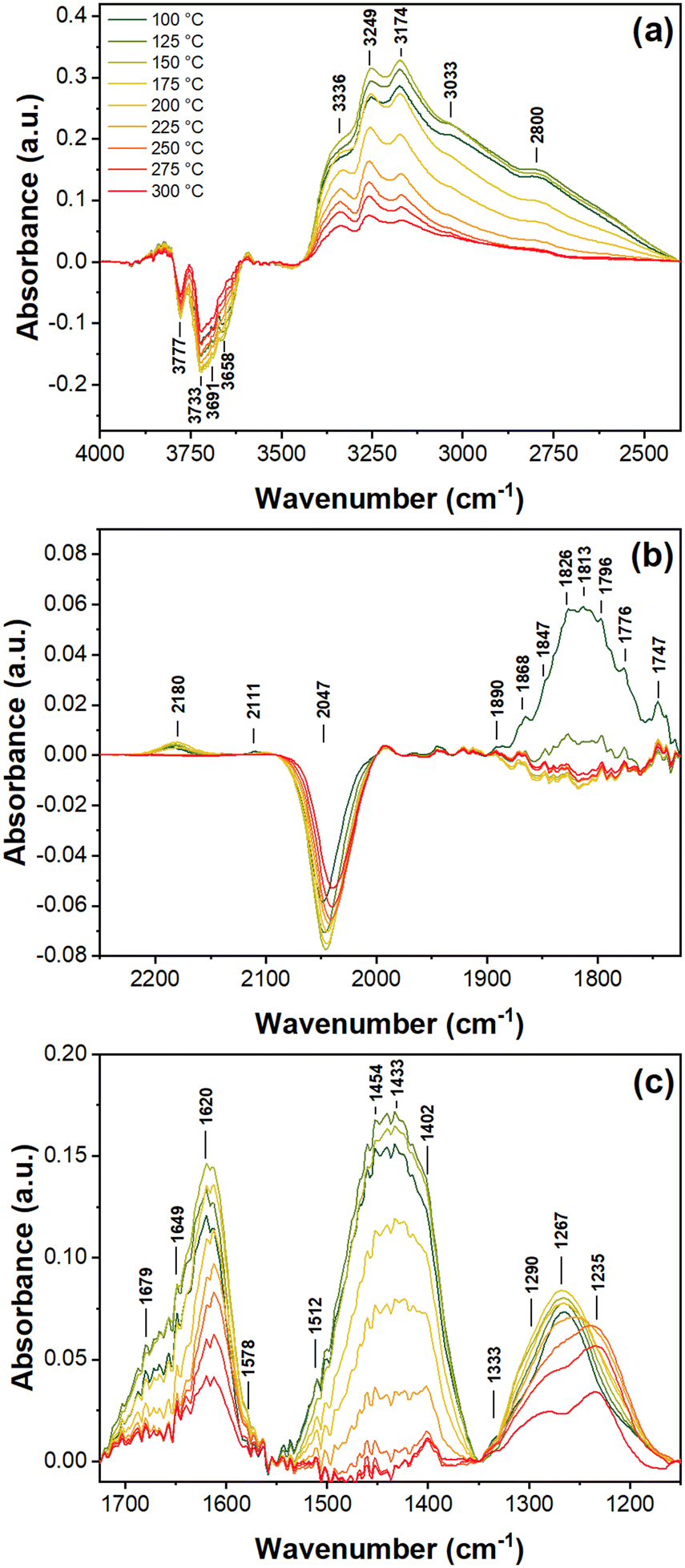 | ||
| Fig. 6 Temperature dependent DRIFTS spectra of the 1% Pd/5%V2O5/20%TiO2-Al2O3 catalyst under a flow of 1000 ppm NO and 5000 ppm H2 in Ar, shown at (a) high wavenumbers for OH (negative), ammonia, and water bands, (b) middle wavenumbers for NO+, V=O (negative), and Pd-NO bands, and (c) low wavenumbers for support nitrates, nitrites, and ammonia bands. | ||
The bridging nitrate species are present at all measured temperatures, and the bands decrease slightly with increasing temperature, matching well with the negative OH and V5+ bands. Despite the apparent stability of nitrate species under reducing conditions, the overall intensities of these bands are substantially less than during the co-adsorption of NO and O2. Additionally, the lower wavenumber bidentate nitrate species are largely absent at all temperatures, indicating their relative instability under reducing conditions. Signs of altered nitrate chemistry are given by the blueshift of the OH bands and redshift of the V5+ bands with increasing temperature. An additional sign of altered chemistry is the presence of a monodentate nitrite band ν3(NO2) at 1512 cm−1, which is relatively weak and rapidly vanishes with increasing temperature.
Unlike the previously discussed gas conditions, the Pdn+–NO band at 1813 cm−1 was observed at 100 °C and rapidly vanished by 150 °C, along with neighboring and less intense Pd2+, Pd+, and Pd0 bands at 1868, 1776, and 1747 cm−1, respectively, (Fig. 6b). The pathway through which NO adsorbs onto Pd under these reducing conditions at these temperatures is not clear, as both the average oxidation state of Pd (Fig. 1c) and nitrosyl bands (Fig. 4c) decrease simultaneously, but the overall lack of NO+ and low presence of nitrates suggests that NO is more likely reduced while adsorbing onto Pd. Additionally, the presence of H2 without O2 causes these linearly adsorbed species to be highly unstable and are likely converted into ammonia species, explained in more detail below.
New peaks from 2500 to 3500 cm−1 and 1350 to 1500 cm−1 are all associated with the formation of ammonium ions (NH4+) on Brønsted acid sites and NH3 adsorbed on Lewis acid sites. It is well reported in the literature that the 3358 and 3240 cm−1 bands belong to the νs(N–H) modes, of NH3 on Lewis sites, whereas the 3183, 3033, and 2800 cm−1 bands are for the νas(N–H), νas(N–H), and 2δas(H–N–H) modes, respectively, of NH4+ adsorbed on Brønsted acid sites.57,60,66,67 Lower frequency NH4+δas(H–N–H) modes are found at 1454, 1433, and 1402 cm−1,67 with a corresponding δs(H–N–H) mode at 1660 cm−1.33 Low frequency modes of NH3 adsorbed on Lewis acid sites, which overlap with the bands of monodentate nitrates at 1297, 1282, and 1256 cm−1, are particularly difficult to isolate but may contribute to band broadening near 1300 cm−1. A weak band at 1333 cm−1 is observed with questionable significance and may be attributed to nitro-compounds or N–N vibrations of cis-N2O2 species.58 In the case of nitro-compounds, an additional band at 1580 cm−1 would be expected, but overlaps significantly with nitrate species. N2O2 species would have counterpart bands below 1050 cm−1 which is below the sample cut-off. During the co-adsorption of NO and H2, this band is of questionable value but becomes more significant under SCR conditions, as discussed in detail below, suggesting it belongs to features of partially reduced NOx species which are not observed in the absence of H2.
NH4+ adsorbed on Brønsted acid sites remain stable until around 200 °C, where they decay following the rapid reduction of Pd, as shown in Fig. 1c and 4c. By 250 °C, Pd is fully reduced and negligible NH4+ remains on Brønsted acid sites. Beyond 250 °C and when NO approaches full conversion, only the bands at 1402, 3174, 3249, and 3336 cm−1 remain. The more intense modes of NH3 adsorbed on Lewis acid sites suggest these as the dominant surface ammonia species. The decay curve of NH4+ species matches that of N2O in Fig. 1a, suggesting that NH4+ is a critical intermediate for the formation of N2O. The dependence of ammonium ions on oxidized Pd suggest NH4+ forms at the metal-support interface via the reduction of NOx species bridging PdO and TiO2 Brønsted acid sites with activated hydrogen. These interfacial NH4+ are then able to react with nitrates bridging PdO and TiO2, forming N2O via the following mechanism:68
| NH4+ + NO3− → N2O + 2H2O | (2) |
Once bulk reduced Pd appears (cf.Fig. 1c), ammonium ions are unable to form – as indicated by the disappearance of NH4+ (1433 cm−1) bands (cf.Fig. 4c and 6c) – and activated hydrogen becomes the dominant reducing agent, fully reducing nitrosyls and nitrates to N2 and H2O and eliminating N2O as a product. Also under these conditions, trace amounts of N2O in the gas phase are easily converted.
NH3 adsorbed on Lewis acid sites becomes the dominant ammonia species at high temperatures, as indicated by lingering bands at 3249 and 3336 cm−1. This suggests that Lewis acid sites on the support are not significantly impacted by the state of Pd and that NH3 is a more stable reactive intermediate than NH4+ at high temperatures. No significant gaseous NH3 was detected in the MS data (m/z = 15), which could indicate that NH3 adsorbed at Lewis acid sites on the titania–vanadia support performs SCR of NOx in parallel with activated H2 on Pd, matching previous reports;28 however, this cannot be confidently concluded due to limitations in measuring small quantities of nitrogen species, especially in the case of NH3, which has only a minor fragmentation ion at m/z = 15.
 | ||
| Fig. 7 Temperature dependent DRIFTS spectra of the 1%Pd/5%V2O5/20%TiO2–Al2O3 catalyst under a flow of 1000 ppm NO, 5000 ppm H2, and 10% O2 in Ar, shown at (a) high wavenumbers for OH (negative), ammonia, and water bands, (b) middle wavenumbers for NO+, VO (negative), and Pd–NO bands, and (c) low wavenumbers for support nitrates, nitrites, and ammonia bands. | ||
Similar to the observations of the previously discussed gas mixtures (cf.Fig. 4), the negative OH and V5+ bands decrease in absolute area alongside the bridge nitrate bands with increasing temperature. However, the competition between NH4+ and nitrates for adsorbing on Brønsted acid sites is more pronounced; from 50 to 100 °C, where NH4+ species (1427 cm−1) are most prevalent, the bridge and bidentate nitrate bands are less intense and grow in intensity from 125 °C as adsorbed NH4+ decreases in prevalence. Additionally, the 1620 cm−1 bridge species undergoes a slight blueshift in the presence of NH4+ at 50 and 75 °C. The clash between these molecules further suggests that bridge and bidentate nitrates form at Brønsted acid sites and compete with NH4+. In the absence of O2, NH4+ species dominate at low temperatures. The presence of O2 does not appear to lower the stability of NH4+ species, considering that the band intensities at 1427 and 3031 cm−1 are equal or higher than observed with just the NO + H2 condition. O2 rather promotes the formation of nitrates and causes the low frequency bands of NH4+ adsorbed on Brønsted acid sites to redshift with increasing temperature. It is important to note that the presence of O2 also promotes NH4+ species at higher temperatures. Under the NO + H2 condition, NH4+ species nearly fully diminished by 275 °C, whereas the low frequency NH4+ band at 1427 cm−1 remains present in significant amounts at 300 °C with the addition of O2. No significant change was observed in the MS intensity of N2O which suggests O2 limits the conversion of NHx species to N2O. However, the small amounts of N2O expected from this catalyst (i.e. 50 ppm)34 during SCR of NOx was not detected by MS.
Another significant change due to the addition of O2 to the NO + H2 mixture is the more significant presence of bands between 1300 and 1400 cm−1, which undergo a blueshift with increasing temperatures. The band at 1317 cm−1, generally assigned to monodentate nitrates on TiO2, is significant at 50 °C but gradually decreases and seemingly shifts to 1334 cm−1 at 175 °C and ultimately 1365 cm−1 by 300 °C. Part of this shift can be explained by diminishing nitrate bands throughout the entire spectrum. However, the blueshift to frequencies closer to 1365 cm−1 correlates with the onset of SCR activity at 200 °C (Fig. 1b), and all bands around 1365 cm−1 begin to decrease in intensity as NO conversion decreases beyond 250 °C. Here it can be inferred that more complex partially reduced NxOy species (e.g. nitro-compounds,)55,58 are formed as SCR intermediates.
The bands for active metals from 1725 and 1900 cm−1 indicate/support three temperature regimes relevant for the performance of the H2-SCR reaction. Similar to the NO and NO + O2 conditions, the Pd–NO bands are largely absent until the bridge nitrate species at 1649 and 1620 cm−1 significantly decrease. In this low temperature regime, below 200 °C and the onset of SCR activity, the abundance of nitrates and NO+ species suggests that NO is more favorably oxidized. Given the gradual reduction of Pd, as shown by XANES in Fig. 1d and as observed during the co-adsorption of NO and O2, PdO appears able to oxidize NOx and form metal-support bridge nitrates (1680 cm−1) according to eqn (1). This then is an intermediate step towards the formation of Pd–NO nitrosyls. After the onset of SCR activity above 200 °C, nitrates and NO+ species diminish, converting to nitrosyl species on Pd and forming PdO, as observed in the adsorption of NO alone. Above 250 °C, SCR activity decreases in intensity, Pd–NO IR bands rapidly shrink, and the average oxidation state of Pd begins to significantly decline. Here, as observed with the adsorption of NO and the co-adsorption of NO and O2, the rate of desorption of NO limits the presence of NO species on the active metal. In turn, activated hydrogen on bulk reduced Pd more readily reduces Pd and combusts with impinging O2.
It is notable that the conversion of NO decreases less drastically as the desorption of NO from Pd and the reduction of Pd. This can be explained by the role of the titania–vanadia support, which has been previously shown to reduce NOx at high temperatures without the presence of active metal.26 To this end, NH3 adsorbed on Lewis acid sites, indicated by the persistent band at 3240 cm−1, is a species which exists independently of the state of Pd which is efficient at reducing NOx stored on the TiO2 support during H2-SCR.28
These findings point towards Pdn+–NO as the active surface intermediate for H2-SCR activity on the active metal and NH3 adsorbed onto Lewis acid sites as an active intermediate for SCR on the support. Notably, at 250 °C where NO conversion is maximized, the nitrosyl (1796, 1813, and 1826 cm−1) bands are also at their maximum intensity. When the temperature increases, NO conversion weakens and Pd becomes more reduced due to runaway activation of H2 and its subsequent catalytic oxidation, which correlates with the decrease in the area of all three IR bands. Despite the rapid reduction of Pd, the presence of O2 slows this reduction relative to what was observed during the co-adsorption of NO and H2. This suggests that O2 buffers the Pd state during SCR, delaying and limiting the rate of reduction.
Conclusions
The average oxidation state of Pd on a 1%Pd/5%V2O5/20%TiO2-Al2O3 catalyst create three distinct regimes and plays a pivotal role in which adsorbed species exist on the surface and which reaction pathways for NOx reduction occur during H2-SCR (cf.Scheme 1). The prevalence of PdO on the surface and in the bulk material, mediated by the presence of O2, influence how NOx is stored and reacts with adsorbed reducing agents.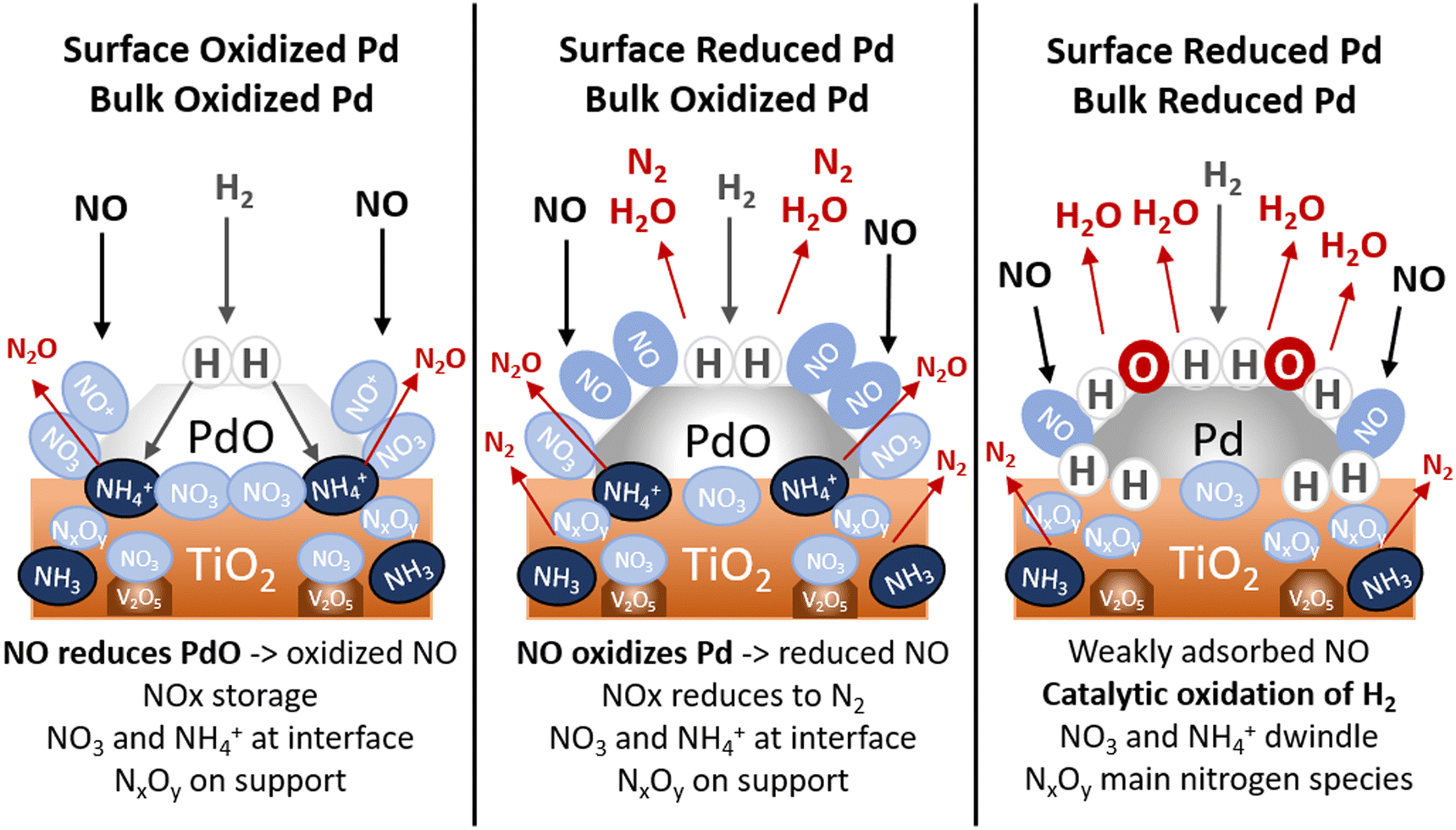 | ||
| Scheme 1 Evolution of nitrogen species on Pd/V2O5/TiO2–Al2O3 with respect to Pd oxidation state. | ||
In the case of mostly oxidized Pd, where both the surface and bulk are PdO-like, gradual reduction of Pd was observed during adsorption of NO, both with and without O2. This coincided with the formation of NO+ ions and nitrates, demonstrating that NO is able to reduce PdO. While Pdn+–NO adsorbates were observed under these conditions, oxidation of NO favors the formation of other nitrogen species (e.g. nitrates, nitrites, nitro-compounds) which store at the metal-support interface or on the metal oxide. Highly oxidized Pd is not directly conducive to SCR, either due to stable NOx adsorbates in the presence of O2 or due to the formation of undesired N2O from the combination of NH4+ and nitrates at the metal-support interfacial Brønsted acid sites.
The SCR of NO is most efficient on slightly reduced Pd, in which the surface contains a significant amount of Pd0 for NOx adsorbates to oxidize. Metal-support bridged nitrates react with Pd0 to form adsorbed Pdn+–NO. H2 then further reduces these nitrosyl species to H2O and N2. Partially reduced adsorbed NxOy species (e.g. nitro-compounds) are also present in significant amounts under SCR conditions when Pd is also partially reduced.
Once bulk PdO is reduced to metallic Pd, both O2 and H2 activate and catalytic oxidation of H2 becomes the favored reaction pathway. At temperatures above 250 °C, the fast desorption of NO from Pd outpaces the ability for nitrates to oxidize Pd, further inhibiting SCR activity and causing a runaway reduction of Pd.
Ammonium ions adsorbed on Brønsted acid sites at the Pd-support interface, are highly affected by the Pd oxidation state. In the absence of O2, NH4+ ions are not formed on reduced Pd. The presence of O2 stabilizes adsorbed ammonium and restricts the undesired conversion to N2O.
NH3 adsorbed on Lewis acid sites exists independent of the Pd state, with or without O2.
Here the vanadia–titania support plays a critical role in the SCR of NO by oxidizing impinging NO and storing it on the surface as stable nitrates and nitrites at Brønsted acid sites. VO readily oxidizes NO across all measured temperatures both in the presence and absence of O2. The Lewis acid sites on TiO2 also permit stable NH3 formation in the presence of H2, which selectively reduce absorbed NOx to N2 at high temperatures when NO desorbs from the active metal.
From the data presented herein, in order to optimize SCR of NOx by H2, metal-support bridge nitrates need to be the focus as they are a form of NOx storage as well as a critical intermediate for nitrosyls on the active metal. This would require increased Brønsted acid site prevalence near or at the active metal interface. However, it should be kept in mind that these sites are susceptible to NH4+ ions and N2O formation especially under rich exhaust conditions. With sufficient O2, the sites will ultimately increase the amount of desired reactive intermediates and therefore increase catalytic activity.
Data availability
Processed data supporting conclusions is available in the manuscript and the ESI.† Raw data can be provided by the authors upon a reasonable request.Author contributions
Thomas Eldridge: writing – original draft, visualization, validation, data curation, methodology, investigation. Michael Borchers: methodology, investigation. Patrick Lott: writing – review & editing. Jan-Dierk Grunwaldt: writing – review & editing, funding acquisition, resources, supervision. Dmitry Doronkin: writing – review & editing, validation, methodology, investigation, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work has been supported by the Helmholtz Association through the MTET (Materials and Technologies for Energy Transition) program. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III and we would like to thank Dr. Aleksandr Kalinko and Dr. Wolfgang Caliebe for assistance in using beamline P64. Beamtime was allocated for proposal I-20220246. We thank Dr. Olaf Deutschmann for provision of infrastructure, resources, and fruitful discussion.References
- G. Ertl, H. Knözinger and J. Weitkamp, Handbook of heterogeneous catalysis, VCH Weinheim, 1997 Search PubMed.
- K. F. Kalz, R. Kraehnert, M. Dvoyashkin, R. Dittmeyer, R. Gläser, U. Krewer, K. Reuter and J.-D. Grunwaldt, ChemCatChem, 2017, 9, 17–29 CrossRef CAS PubMed.
- C. M. White, R. R. Steeper and A. E. Lutz, Int. J. Hydrogen Energy, 2006, 31, 1292–1305 CrossRef CAS.
- S. Verhelst, P. Maesschalck, N. Rombaut and R. Sierens, Int. J. Hydrogen Energy, 2009, 34, 4406–4412 CrossRef CAS.
- R. Jeeragal and K. A. Subramanian, J. Therm. Sci., 2019, 28, 789–800 CrossRef CAS.
- K. W. Scholl and S. C. Sorenson, SAE Trans., 1993, 1450–1462 Search PubMed.
- I. Nova, C. Ciardelli, E. Tronconi, D. Chatterjee and B. Bandl-Konrad, Catal. Today, 2006, 114, 3–12 CrossRef CAS.
- A. Grossale, I. Nova, E. Tronconi, D. Chatterjee and M. Weibel, J. Catal., 2008, 256, 312–322 CrossRef CAS.
- M. Colombo, I. Nova and E. Tronconi, Catal. Today, 2012, 197, 243–255 CrossRef CAS.
- T. Günter, H. W. Carvalho, D. E. Doronkin, T. Sheppard, P. Glatzel, A. J. Atkins, J. Rudolph, C. R. Jacob, M. Casapu and J.-D. Grunwaldt, Chem. Commun., 2015, 51, 9227–9230 RSC.
- O. Deutschmann and J.-D. Grunwaldt, Chem. Ing. Tech., 2013, 85, 595–617 CrossRef CAS.
- A. Boubnov, H. W. P. Carvalho, D. E. Doronkin, T. Günter, E. Gallo, A. J. Atkins, C. R. Jacob and J.-D. Grunwaldt, J. Am. Chem. Soc., 2014, 136, 13006–13015 CrossRef CAS PubMed.
- Y. Ganjkhanlou, T. V. W. Janssens, P. N. R. Vennestrøm, L. Mino, M. C. Paganini, M. Signorile, S. Bordiga and G. Berlier, Appl. Catal., B, 2020, 278, 119337 CrossRef CAS.
- Z. Liu, J. Wu and C. Hardacre, Catal. Surv. Asia, 2018, 22, 146–155 CrossRef CAS.
- Z. Hu and R. T. Yang, Ind. Eng. Chem. Res., 2019, 58, 10140–10153 CrossRef CAS.
- J.-D. Grunwaldt, N. V. Vegten and A. Baiker, Chem. Commun., 2007, 4635–4637 RSC.
- E. K. Dann, E. K. Gibson, R. H. Blackmore, C. R. A. Catlow, P. Collier, A. Chutia, T. E. Erden, C. Hardacre, A. Kroner, M. Nachtegaal, A. Raj, S. M. Rogers, S. F. R. Taylor, P. Thompson, G. F. Tierney, C. D. Zeinalipour-Yazdi, A. Goguet and P. P. Wells, Nat. Catal., 2019, 2, 157–163 CrossRef CAS.
- C. N. Costa and A. M. Efstathiou, Environ. Chem. Lett., 2004, 2, 55–58 CAS.
- C. N. Costa and A. M. Efstathiou, J. Phys. Chem. B, 2004, 108, 2620–2630 CrossRef CAS.
- C. N. Costa and A. M. Efstathiou, Appl. Catal., B, 2007, 72, 240–252 CrossRef CAS.
- G. G. Olympiou and A. M. Efstathiou, Chem. Eng. J., 2011, 170, 424–432 CrossRef CAS.
- P. Wu, L. Li, Q. Yu, G. Wu and N. Guan, Catal. Today, 2010, 158, 228–234 CrossRef CAS.
- D. T. Koch, E. Eßer, S. Kureti and A. Sousa, Motortech. Z., 2020, 81, 32–39 CrossRef.
- B. Wen, Fuel, 2002, 81, 1841–1846 CrossRef CAS.
- G. Qi, R. T. Yang and L. T. Thompson, Appl. Catal., A, 2004, 259, 261–267 CrossRef CAS.
- G. Qi, R. T. Yang and F. C. Rinaldi, J. Catal., 2006, 237, 381–392 CrossRef CAS.
- L. Wang, C. Yin and R. T. Yang, Appl. Catal., A, 2016, 514, 35–42 CrossRef CAS.
- Z. Hu, X. Yong, D. Li and R. T. Yang, J. Catal., 2020, 381, 204–214 CrossRef CAS.
- V. K. Patel and S. Sharma, Catal. Today, 2021, 375, 591–600 CrossRef CAS.
- A. Ueda, T. Nakao, M. Azuma and T. Kobayashi, Catal. Today, 1998, 45, 135–138 CrossRef CAS.
- R. Burch and M. D. Coleman, Appl. Catal., B, 1999, 23, 115–121 CrossRef CAS.
- K. Duan, Z. Wang, C. Hardacre, Z. Liu, S. Chansai and C. Stere, Catal. Today, 2019, 332, 69–75 CrossRef CAS.
- N. Macleod and R. M. Lambert, Catal. Lett., 2003, 90, 111–115 CrossRef CAS.
- M. Borchers, K. Keller, P. Lott and O. Deutschmann, Ind. Eng. Chem. Res., 2021, 60, 6613–6626 CrossRef CAS.
- F. J. P. Schott, P. Balle, J. Adler and S. Kureti, Appl. Catal., B, 2009, 87, 18–29 CrossRef CAS.
- M. Leicht, F. J. P. Schott, M. Bruns and S. Kureti, Appl. Catal., B, 2012, 117–118, 275–282 CrossRef CAS.
- C. Hahn, M. Endisch, F. J. P. Schott and S. Kureti, Appl. Catal., B, 2015, 168–169, 429–440 CrossRef CAS.
- C. Hahn, M. Endisch and S. Kureti, Top. Catal., 2017, 60, 238–242 CrossRef CAS.
- T. Nanba, C. Kohno, S. Masukawa, J. Uchisawa, N. Nakayama and A. Obuchi, Appl. Catal., B, 2003, 46, 353–364 CrossRef CAS.
- H. Hamada and M. Haneda, Appl. Catal., A, 2012, 421–422, 1–13 CrossRef CAS.
- Z. Hong, X. Sun, Z. Wang, G. Zhao, X. Li and Z. Zhu, Catal. Sci. Technol., 2019, 9, 3994–4001 RSC.
- M. Borchers, P. Lott and O. Deutschmann, Top. Catal., 2023, 66, 973–984 CrossRef CAS.
- L. Zhang, Y. Shan, Z. Yan, Z. Liu, Y. Yu and H. He, J. Environ. Sci., 2024, 138, 102–111 CrossRef CAS PubMed.
- H. Topsøe, J. Catal., 2003, 216, 155–164 CrossRef.
- A. Urakawa, N. Maeda and A. Baiker, Angew. Chem., Int. Ed., 2008, 47, 9256–9259 CrossRef CAS PubMed.
- L. van Beek, D. Jain, P. Gholkar, T. J. Eldridge, H. P. Nguyen, K. Muramoto and A. Urakawa, Catal. Today, 2024, 429, 114466 CrossRef CAS.
- A. M. Gänzler, M. Casapu, D. E. Doronkin, F. Maurer, P. Lott, P. Glatzel, M. Votsmeier, O. Deutschmann and J.-D. Grunwaldt, J. Phys. Chem. Lett., 2019, 10, 7698–7705 CrossRef PubMed.
- B. Bornmann, J. Kläs, O. Müller, D. Lützenkirchen-Hecht and R. Frahm, AIP Conf. Proc., 2019, 2054, 040008 CrossRef.
- D. E. Doronkin, H. Lichtenberg and J.-D. Grunwaldt, XAFS Techniques for Catalysts, Nanomaterials, and Surfaces, 2017, pp. 75–89 Search PubMed.
- D. Lützenkirchen-Hecht, J.-D. Grunwaldt, M. Richwin, B. Griesebock, A. Baiker and R. Frahm, Phys. Scr., 2005, 2005, 831 CrossRef.
- M. Newville, Larch: an analysis package for XAFS and related spectroscopies, J. Phys.: Conf. Ser., 2013, 430, 012007 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- A. Puig-Molina, F. M. Cano and T. V. W. Janssens, J. Phys. Chem. C, 2010, 114, 15410–15416 CrossRef CAS.
- M. Kantcheva, J. Catal., 2001, 204, 479–494 CrossRef CAS.
- J. M. Watson and U. S. Ozkan, J. Catal., 2002, 210, 295–312 CrossRef CAS.
- K. Hadjiivanov, P. Concepción and H. Knözinger, Top. Catal., 2000, 11, 123–130 CrossRef.
- L. Chen, J. Li and M. Ge, J. Phys. Chem. C, 2009, 113, 21177–21184 CrossRef CAS.
- K. Hadjiivanov and H. Knözinger, Phys. Chem. Chem. Phys., 2000, 2, 2803–2806 RSC.
- R. F. Ilmasani, J. Woo, D. Creaser and L. Olsson, Ind. Eng. Chem. Res., 2020, 59, 9830–9840 CrossRef CAS.
- M. A. Centeno, I. Carrizosa and J. A. Odriozola, Appl. Catal., B, 2001, 29, 307–314 CrossRef CAS.
- Y. Zhang, S. Xu, J. Li, E. He and Z. Liu, J. Phys. Chem. C, 2023, 127, 7248–7256 CrossRef CAS.
- M. A. Matsko, I. P. Prosvirin, T. B. Mikenas, V. A. Zakharov, E. A. Paukshits, V. I. Bukhtiyarov and I. G. Danilova, J. Mol. Catal. A: Chem., 2000, 158, 443–446 CrossRef CAS.
- I. Song, K. Khivantsev, Y. Wang and J. Szanyi, J. Phys. Chem. C, 2022, 126, 1439–1449 CrossRef CAS.
- H. D. Schmick and H. W. Wassmuth, Surf. Sci., 1982, 123, 471–490 CrossRef CAS.
- K. Hadjiivanov, V. Bushev, M. Kantcheva and D. Klissurski, Langmuir, 1994, 10, 464–471 CrossRef CAS.
- N. Macleod, R. Cropley and R. M. Lambert, Catal. Lett., 2003, 86, 69–75 CrossRef CAS.
- C.-H. Lin and H. Bai, Appl. Catal., B, 2003, 42, 279–287 CrossRef CAS.
- J. Luo, Y. Tang, S. Joshi, K. Kamasamudram, N. Currier and A. Yezerets, SAE Int. J. Engines, 2017, 10, 1691–1696 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00574k |
| This journal is © The Royal Society of Chemistry 2024 |