
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic processes for the selective hydrogenation of fats and oils: reevaluating a mature technology for feedstock diversification
Maximilian L.
Spiekermann
 and
Thomas
Seidensticker
*
and
Thomas
Seidensticker
*
Laboratory for Industrial Chemistry, Department of Biochemical and Chemical Engineering, TU Dortmund University, Emil-Figge-Straße 66, 44227 Dortmund, Germany. E-mail: thomas.seidensticker@tu-dortmund.de; Tel: +49 231 755 2310
First published on 26th June 2024
Abstract
The selective hydrogenation of polyunsaturated fatty acids (PUFAs) and their derivatives to their monounsaturated counterparts is a critical catalytic step in various disciplines, such as biofuels and food chemistry. Furthermore, the necessary transformation of the chemical industry towards sustainability and circularity demands the development of new platform chemicals from renewable resources. The selective hydrogenation of PUFAs offers the flexibility to use a variety of commodity feedstocks in existing processes in the chemical value chain; this is indispensable for staying competitive and in shaping a sustainable future-proof chemical industry. The naturally occurring differences in the composition of vegetable fats and oils, especially the degree of saturation of the carbon chain, necessitate a preparation step to provide a standardized feedstock for existing chemical processes. Since many chemical reactions towards valuable intermediates start from mono-unsaturated substrates, selectivity plays a crucial role in hydrogenation. Furthermore, standardization alleviates the need to grow speciality monocultures or to utilize edible substrates, to further reducing the negative impact on society and the environment. In this critical review, all the relevant and pioneering catalytic transformations throughout history up to this time and place are discussed and evaluated towards their applicability and usefulness for the provision of renewable-based resources for the chemical industry, with particular emphasis on the selectivity towards mono-unsaturated substrates.
1. Introduction
For more than a century, the catalytic hydrogenation of (poly-)unsaturated oils and their derivatives has been of high interest to researchers in academia, the food industry, and the energy sector. Starting with the desire to create a plant-based alternative to butter, this technology has recently been used to improve the physical properties of biodiesel. Now, it is time to reevaluate the reaction's potential in the context of the defossilization in the chemical industry.1–4As a mandatory carbon-based industry, the chemical industry has to future-proof itself urgently by diversifying the feedstock because the demand for carbon-based products is at an all-time high.5 Thereby, many valuable intermediates are based on simple short-chain olefins that allow for versatile functionalization at their double bond(s) towards alcohols, aldehydes, ketones, epoxides, amines or amides, only to name a few. Those unsaturated molecules are readily accessible from petroleum feedstocks, such as C2–C5 olefins from steam cracking and >C5 from oligomerization reactions, for instance, in the well-established shell higher olefin process (SHOP) producing linear 1-alkenes. However, the chemical industry must strive for sustainability and reduce its dependency on fossil resources. Changing the feedstock to the most abundant renewable materials, carbohydrates, brings various complications and is very demanding: different atomic composition, primarily the high content of functional groups containing oxygen in contrast to the above-described simple alkenes, obstructs their direct implementation into existing industrial processes.6 Chemicals from plant oils, so-called oleochemicals, offer great potential: they have a relatively low oxygen content and are typically unsaturated, thus containing isolated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds like fossil resource-based alkenes. Consequently, they offer similar potential for functionalization and might be used in the established processes for syntheses of value products. However, functionalization is likely to occur on each double bond, and most oleochemicals contain multiple double bonds, stated as their degree of unsaturation. Additionally, they often exhibit a longer carbon chain than the standard fossil resource-based platform chemicals.
C double bonds like fossil resource-based alkenes. Consequently, they offer similar potential for functionalization and might be used in the established processes for syntheses of value products. However, functionalization is likely to occur on each double bond, and most oleochemicals contain multiple double bonds, stated as their degree of unsaturation. Additionally, they often exhibit a longer carbon chain than the standard fossil resource-based platform chemicals.
Up to now, many reactions performed with oleochemicals utilize the terminal carboxylic function. Reduction to fatty alcohols and synthesis of fatty amines for detergents are well-known pathways that are already industrially applied, as well as the synthesis of various esters. As aforementioned, the double bonds offer further potential for functionalization as in the established processes starting from fossil resource-based alkenes. Furthermore, mono-unsaturated oleochemicals offer great opportunities for the synthesis of difunctional platform chemicals.7–11 These open pathways to renewable monomers for bioplastics, for instance, which can be accessed by metathesis or oxidative cleavage. The provision of uniform bio-based substrates opens up additional markets and can help make them more sustainable.12
Only a limited number of reactions that use the CC double bond of the fatty acid chain of monounsaturated derivatives are industrially applied since they all rely on a specific substrate on which the process is specifically tuned. Elevance had planned to carry out metathesis from its Asian site, and Matrica performs oxidative cleavage reactions with high oleic safflower oil.13,14 The processes are specialized towards these substrates, but both rely on a molecular basis on monounsaturated substrates, like methyl oleate.15
Selective partial hydrogenation of fatty acids and their derivatives solves the problem of substrate dependency by providing uniform monounsaturated substrates (Fig. 1). The composition of the corresponding products is independent of the renewable raw material, which can be converted into new intermediates and products via numerous functionalization reactions already demonstrated. The catalysts used in these functionalizations are usually prone to deactivation which is believed to be caused by the fatty acid chains with more than one double bond.10,16,17 Not only could a wider range of bifunctional intermediates be accessible, but existing processes could benefit from an extended variety of raw materials after the standardization step towards high purity monounsaturated substrates. Further benefits for cleavage applications by the aforementioned standardization could be found in the simplified downstream processing. After oxidative cleavage or ethenolysis substrates with only one double bond at the same position only yield two products, whereas polyunsaturated substrates yield at least three products and isomerized substrate mixtures yield two products per isomer.10,12,17,18
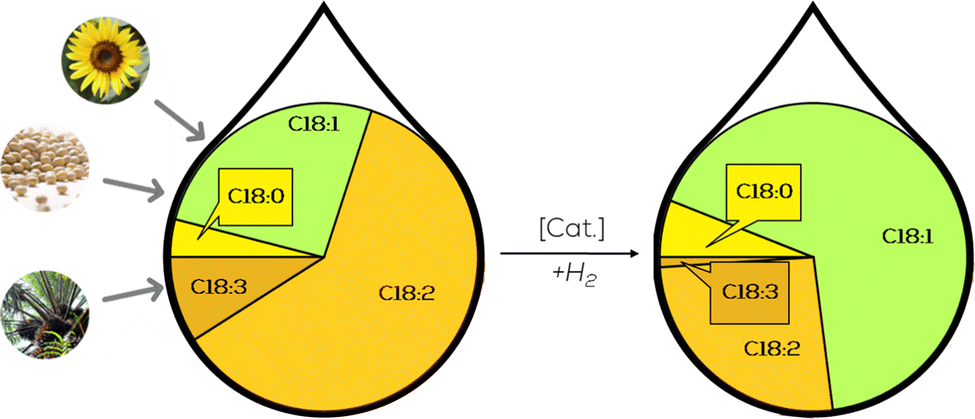 | ||
| Fig. 1 Schematical presentation of partial hydrogenation of fatty acid mixtures. | ||
Some plants have been modified to form an increased amount of oleic acid in the respective oils. Especially high oleic sunflower oil (HOSO) is a substrate with industrial potential despite being substantially more expensive as the standard sunflower oil. The high content of up to 90% of oleic acid brings much convertible substrate but even this oil could benefit from selective partial hydrogenation since the content of polyunsaturated fatty acids is still at least 3%.17,19
The selective hydrogenation technology has been known for more than 100 years and is already applied primarily in two industrial sectors:20 in the food industry, margarine became accessible by hydrogenation of polyunsaturated plant oils, and in the energy sector, the physical properties of biodiesel was improved by partial hydrogenation of seed oil-derived fatty acid methyl esters. Both technologies have been widely studied and reviewed focusing on the respective important key figures and characteristics.21,22 What is crucially missing, though, is a comprehensive comparison and critical assessment of reported catalytic processes for the (selective) hydrogenation of fatty acids and their derivatives for providing uniformed oleochemical substrates for material use in the chemical industry sector. This review addresses this important gap.
1.1. Sources of fatty acids and their potential for the chemical industry
From saturated fatty acids (SFA), for instance, capric acid (C10:0) over monounsaturated (MUFA) like oleic acid (C18:1) to polyunsaturated fatty acids (PUFA) linolenic acid (C18:3) common plant oils differ in their characteristic composition.23 They occur as triglycerides with different fatty acids in one molecule (Fig. 2). Oil plants can be grown under various conditions (climate, land). Therefore, plant oils are readily available. Global production of fats and oils has increased enormously in recent years. In the 2021/22 season, over 200 million tons of major vegetable oils have been produced.24 Within this biomass production, over 100 plant oils are known for human consumption. The amount of non-edible oils exceeds this number even further.25 | ||
| Fig. 2 An exemplary triglyceride of a plant oil and the shortcut naming.23 | ||
Median oil prices in 2020/21 in Rotterdam were $1266 per ton for soybean oil, $1350 per ton for sunflower oil, $1017 per ton for palm oil and $1306 per ton for canola oil, which makes these substrates attractive for further refining towards uniformed platform chemicals.26,27 Since these oils are still more expensive than fossil resources, the choice towards renewables is more of an environmental one than a pure economical one. The well determined composition and the more advanced functionalization of plant oils give an advantage which could come in handy depending on the final application. But feedstock can come from more resources than classic consumable plant oils. Side streams like tall oil fatty acids from paper production or non-edible plant oils like jatropha, which grows in meagre soils and arid climates, may be utilized to secure the global supply of oleochemicals.28,29 Globally, about 25% of oils were used industrially in 2019 to produce biodiesel and oleochemicals or as animal feed; 75% were used for human nutrition.10
The composition of plant oils concerning their fatty acid profile, and thus their content of CC-double bonds, differs quite considerably depending on the individual plant (Table 1). All seed oils mentioned in Table 1 can be easily transesterified into their methyl esters, the most common form of biodiesel and a well-known substrate for the chemical industry. This transesterification reaction with methanol is a key example of industrially proven reactions performed with oleochemicals.10,12,18,30,31
| Name | SFA [wt%] | MUFA [wt%] | PUFA [wt%] | Annual production 2[kt a−1] | Special characteristic |
|---|---|---|---|---|---|
| Common plant oils | |||||
| Palm | 51.4 | 38.9 | 9.7 | 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 583 583 |
|
| Soybean | 5–20.8 | 23–31 | 51–62 | 59904 |
|
| Canola | 4 | 63.5 | 29 | 24407 |
|
| Sunflower | 8.2–17.5 | 13–41.3 | 48–74.3 | 20054 |
|
| Palm kernel | 73–92.6 | 12–18 | 1–4.7 | 8226 | |
| Cottonseed | 26.8 | 15–36 | 34–55 | 4446 | |
| Peanut | 16–27 | 48–66.3 | 14–28.3 | 4228 | |
| Corn | 9.3–22.5 | 20–42.6 | 39.9–67.1 | 3541 | |
| Coconut | 95.3 | 3.9 | 0.8 | 3160 | |
| Olive | 8–25 | 55.3–86.5 | 3.5–22.5 | 3097 | |
| Sesame | 14.8 | 39.1 | 46.1 | 1034 | |
| Linseed | 5.35–11.3 | 17.8–24.3 | 51.4–75 | 738 | |
| Safflower | 8–11 | 10–20.5 | 68–83.2 | 76 | |
| Low-capacity special plant oils | |||||
| Tung | 5 | 4–10 | 81–99 | 75 | Inedible; high content in α-eleostearic acid |
| Oiticica | 7.2–12 | 6–18 | 78.3–85.8 | 20 | |
| Stillingia33 | 4–9 | 7–11 | 72–89 | ||
| Poppyseed | 8.1–15.7 | 16.8–31.6 | 62–73 | 76 | |
| Jatropha | 23.3–25.3 | 30–44 | 30.3–52.3 | 1735 | Resistant and frugal; toxic components |
| Tigernut | 17–20 | 67–73 | 6–15 | ||
| Salicornia34 | ∼10 | ∼20 | ∼70 | Halophyte | |
| Castor | 3.2 | 87.3 | 5.4 | 360 | High content in ricinoleic acid |
In the context of providing unified, monounsaturated platform chemicals with one double bond via selective partial hydrogenation, some plant oils are more suited than others. In particular, the high content of SFA in palm oil, coconut oil and palm kernel oil disqualify those plant oils. Hence, soybean oil and linseed oil, which have a high content of polyunsaturated C18:2 and C18:3 fatty acids in triglycerides, are common plant oils investigated for selective hydrogenation, which has a high content of polyunsaturated C18:2 and C18:3 fatty acids in the triglycerides. Sunflower oil, or canola oil also represent suitable raw materials since fine-tuning the degree of saturation is a promising application of selective hydrogenation as well.21
In addition to the already mentioned high-volume plant oils, non-edible alternatives with suitable acid composition could be found in jatropha, karanja, and rubber seed oil, which are predominantly composed of oleic, stearic, palmitic, and linoleic acid.35 These alternatives have already been examined as possible sources for biodiesel.36–39 Using oil from the Chinese tung oil tree, which contains primarily non-edible C18 fatty acids and a significant amount of α-eleostearic acid (see Fig. 3), might be a viable approach. This acid, featuring three conjugated double bonds, is prone to auto-oxidation, making it challenging for biodiesel use. Additionally, its high trans-configured double bonds content renders it unsuitable for human consumption. However, if the double bond configuration is not a critical factor, hydrogenated methyl esters of tung oil could serve as a promising plant-based source for producing platform chemicals through selective partial hydrogenation.35,40
 | ||
| Fig. 3 Tung oil derived methyl α-eleostearate.41 | ||
Oleochemicals can be produced globally in a wide variety under various climate conditions.12 However, their contribution as platform chemicals towards shaping a sustainable chemical industry is underestimated so far, although they offer high potential in chemical research.30 For the exclusively monounsaturated oleochemicals, quite a lot of reactions have been subject of investigation so far. Some reactions already performed with methyl oleate are hydroformylation, metathesis and oxidation. The products of these reactions and some additional ones are shown in Fig. 4.
 | ||
| Fig. 4 Selection of homogeneously catalysed reactions reported with methyl oleate.11,23,42,43 | ||
1.2. Reaction network and catalytic performance evaluation
The reaction network of exemplary C18 fatty acid methyl ester (FAME) hydrogenation could be simplified and described as three consecutive reactions, all utilizing the same co-reactant hydrogen, shown in Fig. 5. Depending on the catalyst, the double bonds in the fatty acid chain can move up and down the carbon chain and change their configuration between the naturally occurring cis- or Z-configuration and the trans- or E-configuration.44 | ||
| Fig. 5 Simplified reaction scheme of linolenate hydrogenation.30,43 | ||
When not completed to only C18:0 as a product, this reaction leads to a product mixture with numerous components. Historically, many of these components were difficult to identify (and some still are), or distinguishing between certain isomers can be quite involved. Additionally, for many applications, an entire breakdown of the product mixture is not necessary. This leads to a lack of detailed information about the composition in literature. At this point, the importance of highlighting the different components in the C18:1 fraction should be emphasized.
To evaluate this hydrogenation reaction, though, different mole fractions should be considered to obtain comprehensive information that enables a reasonable evaluation of the catalysis in any context. The formed mole fraction of xC18:1 (eqn (1)) refers to the amount of newly formed monounsaturated compounds nC18:1 − nC18:10 in the product mixture.
 | (1) |
 | (2) |
 | (3) |
The primary performance indicator discussed is the fatty acid composition, particularly maximizing C18:1 content for platform chemicals, while also considering stereo- and regioselectivity. All significant catalytic processes will be reevaluated for their feasibility in this context, using standardized figures (1)–(3), independent of their original applications.
Typically, chemical reaction performance is quantified using conversion Xi (eqn (4)), selectivity, and yield. Catalyst performance expands these metrics with turnover frequency (TOF) (eqn (6)) to indicate activity and turnover number (TON) (eqn (5)) to measure stability, including catalyst recycling. When pondering a reuse of a catalyst a summarized TON may be considered as well. For a maximum summarized TON it is crucial to recover most of the catalyst which shows also beneficial for the reduction of potentially toxic catalytic material in the product. TOF, crucial for representing reaction rate, is often calculated at a specific substrate conversion point, typically 20%, although initial values may also be measured at the start or consistently across experiments, complicating comparisons.
In industrial contexts, the focus shifts towards process-oriented metrics such as space–time yield, which measures the productivity of a process unit over time in producing a targeted product amount.45
 | (4) |
 | (5) |
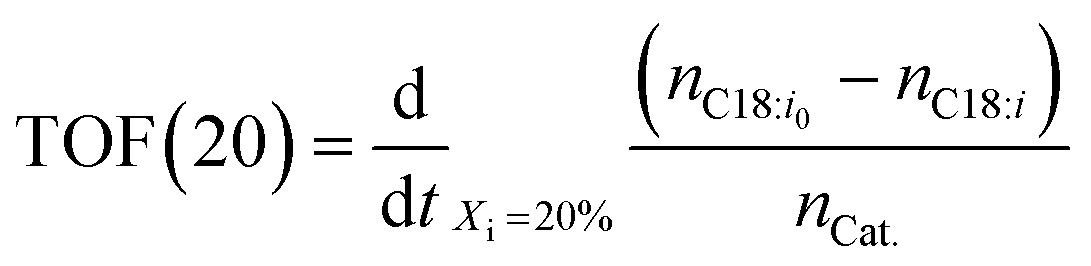 | (6) |
The numbers either fail to represent the entire composition or, by combining several components into one for more straightforward calculation, create a lack of detailed information for the single components.
Therefore, the chemistry of fats and oils has its performance indicators and usually utilizes a different way of visualization of reaction progress. The iodine value (IV) is an essential number for hydrogenation reactions at double bonds of the fatty acid chain. It gives an average degree of saturation for the entire mixture.
It can be measured by the amount of iodine added to the remaining double bonds per 100 g of fat or calculated via GC-FID measurement of the fatty acid composition. Fig. 6 shows a typical chromatogram of a GC-FID of partially hydrogenated soybean oil-derived methyl ester. The different peaks are grouped and then assigned to a specific degree of saturation. Subsequently to the measurement, the IV is calculated by multiplication with the component-specific impact factors (eqn (7)).46
| IV = f1 × xC18:1 + f2 × xC18:2 + f3 × xC18:3 | (7) |
 | ||
| Fig. 6 Typical chromatogram of a partially hydrogenated soybean oil derived methyl ester analyzed by GC-FID. Separation on a 90 m DB FastFAME column. Methylation by addition of 50% (w/w) TMSH in methanol. | ||
The impact factors of the critical C16 and C18 components are shown in Table 2.46
| Component | Impact factor fi |
|---|---|
| Methyl palmitate (C16:0) | 0 |
| Methyl stearate (C18:0) | 0 |
| Methyl oleate (c-C18:1) | 0.860 |
| Methyl elaidate (t-C18:1) | 0.860 |
| Methyl linoleate (C18:2) | 1.732 |
| Methyl linolenate (C18:3) | 2.605 |
With a known degree of saturation for the substrate of the reaction and the utilized mass of fatty substrate, the decrease in IV (eqn (7)) is proportional to the conversion of hydrogen. Although the iodine number gives a good overview of the overall reaction progress, any more detailed information about the composition of the mixture is missing. To address this circumstance, the different reaction rates or final compositions used to be related to each other, resulting in various selectivities and figures. Linolenate selectivity (SLn), linoleate selectivity (SL), oleate selectivity (SO),47 degree of oversaturation (DOO),48 selectivity isomers index (SII),49 conversion of specific components, the ratio between t-C18:1 and c-C18:1, the mass conversion ratio of C18:2 (CRC18:2),50 to name a few. For the comparison between reactions, these numbers can be utilized depending on the critical characteristics that one focuses on. Still, for a more in-depth evaluation of a specific research question, it is indispensable to regard the course of the molar fractions over the reaction progress.
For the calculation of linolenate selectivity (SLn), linoleate selectivity (SL), and oleate selectivity (SO), the simplified reaction network (Fig. 5) defines reaction rates that are utilized to calculate these selectivities. The reaction rates are defined as follows.51
 | (8) |
 | (9) |
 | (10) |
 | (11) |
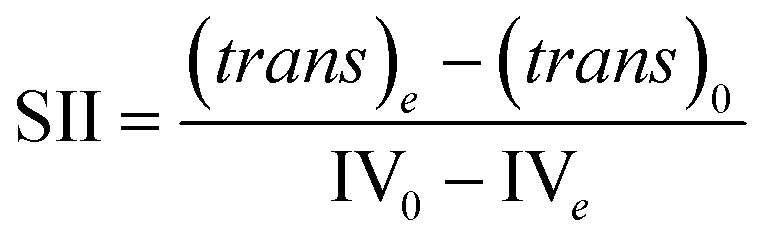 | (12) |
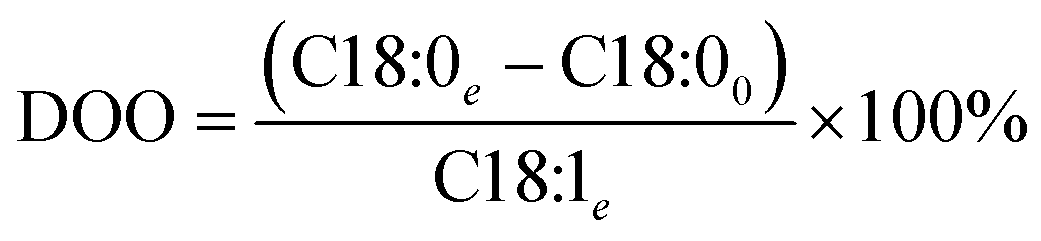 | (13) |
 | (14) |
| Variable | Meaning |
|---|---|
| n | Amount of a substance |
| Cat. | Indication for the catalyst |
| c | Concentration of a substance |
| k | Rate constant of a reaction |
| x | Mole fraction of a substance |
| x C18:1(ω−9) | Mole fraction of a specific positional isomer (oleic acid) |
| DOO | Degree of oversaturation |
| CRC18:2 | Conversion rate of C18:2 |
| S | Selectivity towards a specific substance |
| SII | Selectivity isomers index |
| IV | Iodine value |
| f | Impact factor |
| TON | Turnover number |
| TOF(X) | Turnover frequency at a specific conversion |
| E/t- | Indication for the trans-configuration |
| Z/c- | Indication for the cis-configuration |
| X i | Conversion of component i6 |
At this point, it should have become clear that these historically grown numbers and figures lack some information and do not represent all connections and actual reactions in the hydrogenation process. Therefore, the reaction network has to be described in more detail to sort all the different catalytic processes, which will be reviewed in the following chapters.
In recent years, the reaction network and the catalytic mechanism were reinvestigated based on already published data. This information is crucial to understand the possible occurring product compositions and to define the nature given boundaries of the hydrogenation process.58Fig. 7 illustrates the complexity of the reaction network. If the entire identification of the reaction mixture is necessary, the state-of-the-art technique is a specialized GC-FID analysis. All fatty acid chains in the mixture are transesterified to their respective methyl esters and separated in a specialized capillary column by their degree of saturation and the configuration and position of their double bonds. This technique is also used for the calculation, as mentioned above, of the IV, thus decreasing the overall information content of the analysis just to fit the historically given standards.
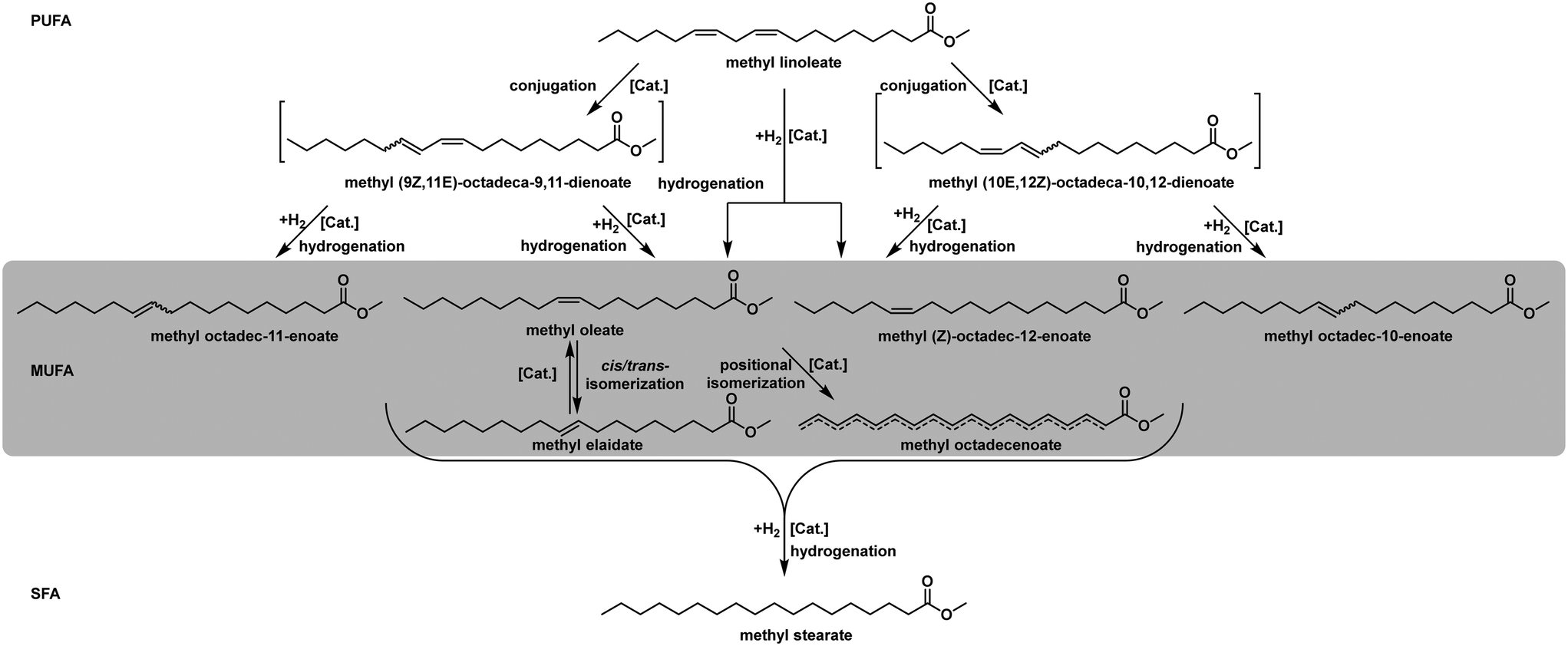 | ||
| Fig. 7 Reaction network for partial hydrogenation of methyl linoleate (isolated dienes). | ||
Since all the substances, as mentioned earlier, are usually already present in the mixture, all reactions can occur right from the beginning of the experiment. Furthermore, isomerisation is the dominant side reaction in the overall reaction network.59 This gives various intermediates/final products, depending on the purpose, some more desired than others. Some research also reports the formation of conjugated double bonds before the hydrogenation reaction. This more complex reaction scheme depends on the catalyst and is widely accepted. This mechanism is shown for the substrate methyl linoleate in Fig. 7, and all occurring value products in the monounsaturated C18:1 fraction are highlighted.60
This complexity and the application-specific demand for the product mixture insist on a context-based treatment of the individual processes and catalysts. Furthermore, a specific catalyst's performance that was insufficient for biodiesel application due to an occurring side reaction, e.g., could be considered as highly economically feasible to produce platform chemicals because the definition of value product includes more substances of the mixture, and therefore, the yield drastically increases. By creating a comprehensive overview and revisiting all catalytic processes outside their initial context, new connections might be created, and a new way of value-adding for a specific plant-based oleochemical could be found.
The choice of catalytic metal is fundamental to shaping a reaction or catalytic system towards desired or expected specifications and key figs. A brief overview of catalytic metals for hydrogenation reactions without further examination of the processes is given in the next chapter.
1.3. Catalytic metals used for hydrogenation processes
Historically, the most common catalyst metal for the hydrogenation of fatty substrates is nickel. Due to its low prize and sufficient productivity, it is an obvious choice for cheaper products like hardened fats and oils for nutrition.20 However, nickel-catalyzed processes usually suffer from low selectivity to MUFAs and form high amounts of unhealthy trans-configured acids. Therefore, food-related industrially processed oils are today typically fully saturated and mixed with unprocessed oils. Consequently, researchers focused on developing new catalysts with higher selectivity to cis-configured monounsaturated products. During the 20th century, other common catalysts metals like iron, silver, cobalt, zinc, and copper, especially in combination with chromium, were investigated.61 More rare transition metals like palladium and platinum were also used as hydrogenation catalysts. A comprehensive overview of the essential transition metals is given in Table 4.| Characteristic | Order of metals |
|---|---|
| Activity | Pd > Rh > Pt ≫ Ir > Ru ≫ Os |
| Selectivity | Pd > Rh > Pt > Ru ≫ Ir |
| cis–trans isomerisation | Pd > Rh > Ru > Ir > Pt |
| Migration of double bonds | Pd > Rh Ru > Os > Ir > Pt |
To complete the overview, gold and tin are some additional candidates that have not been studied as intensively.62 Some additives and promoters for catalysts were found in alkaline and alkaline earth metals.63 Since this review is written from an engineering perspective, it is mandatory to not only state catalysts' characteristics without giving the context of the process because reaction engineering and other process engineering can significantly influence a catalytic system's performance.64 Many of these engineering efforts evolve around the effective utilization of catalyst material to reduce the overall cost of the process. For comparison of current metal prices, please refer to Table 5.
A significant number of researchers studied the intensification of the catalytic processes by modification of catalyst placement, reactor modification or a combination of different unit operations. Some approaches increased the overall activity. Others supplied hydrogen more effectively to the catalyst. Therefore, essential process characteristics will usually be mentioned and discussed within the following chapters while exploring all influential and pioneering catalytic processes for the selective partial hydrogenation of fats and oils from the invention up to this time and place. Most importantly, this review evaluates their potential for producing platform chemicals from renewables for the chemical industry.
2. Significant research on selective partial hydrogenation
This chapter presents the existing catalytic systems for selective partial hydrogenation of plant oils and oleochemicals, respectively. We start with heterogeneous systems since those are the most common and were historically first mentioned. We will then evaluate the potential of molecular homogeneous systems and go over to colloidal nanocatalysts. We will finish with more exotic systems based on membranes and plasma.To evaluate the suitability of the given catalytic system for providing unified platform chemicals for material use in the chemical industry, we will represent the results of the hydrogenation reaction as depicted in Fig. 8. For the provision of uniformed platform chemicals, the crucial factors are a maximum amount of C18:1 depicted as a large green area in the product composition, minimum amount of C18:0 and the corresponding process conditions, pressure, temperature and catalyst loading, which should all be in a reasonable range for a hydrogenation reaction. Other important side-factors are described in the adjacent text.
 | ||
| Fig. 8 Example for the evaluation of the given catalytic hydrogenation reaction. | ||
Most researchers spent years on this topic and published many (intermediate) work. Not all steps of the respective research will be represented in such a figure, only the final results of a specific research or significant intermediate results.
3. Catalytic systems for selective partial hydrogenation
When focusing on the liquid phase hydrogenation of fats and oils, the comprehensive overview starts at the beginning of the 20th century. Margarine was invented in 1869 by Mege-Mouries by mixing beef tallow with vegetable oils to create a product with the desired texture. Normann invented the hardening of oils with heterogeneous nickel catalysts in 1901 to make cheaper resources feasible for margarine production. Consequently, no animal fats had to be added to achieve the product texture. From then on, hydrogenated vegetable oils could replace these fats and mixtures.743.1. Heterogeneous catalysts and general principles
To mimic the butter's texture and meet the customer's needs, the hardened plant oils were not hydrogenated entirely but still contained a reasonable amount of unsaturated fatty acids. Substrates for this partial hydrogenation were found in linseed, soybean and cottonseed oil. The hydrogenation reaction was initially performed at 180 °C and above, but investigations towards lower temperatures were highly anticipated. The process is displayed as a fatty acid composition before and after the reaction with the catalyst and necessary reaction conditions in between (Fig. 9).75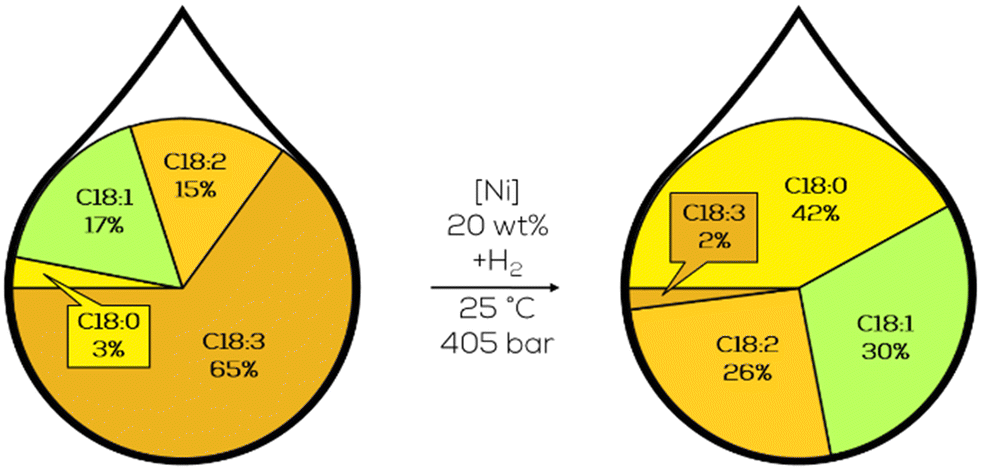 | ||
| Fig. 9 C18 fatty acid composition in the hydrogenation of linseed oil performed by Normann.75 | ||
The displayed process was performed with refined linseed oil at room temperature with a hydrogen pressure of 400 atm. The reaction mixture consisted of 50 g of substrate, 100 ml of diethyl ether and 5 g heterogeneous nickel catalysts with a nickel content of 20 wt%. No significant further hydrogenation was observed after 48 hours. Other experiments with further refined linseed oil led to completely hydrogenated products within 23 hours. Although hydrogenated oils with suitable properties for human consumption could be produced using this process, the production of selective monounsaturated fatty acids could not be achieved. These results are the historical benchmark for selective hydrogenation reactions of fats and oils. Normann mentioned the importance of incomplete hydrogenation towards C18:1 compounds in his articles but did not provide a suitable solution. Nevertheless, one can see that the lack of selectivity hinders the implementation as a continuous process. During this review, it will become clear that selectivity towards C18:1 and high catalyst loadings are still decisive challenges that continue to exist to this day.20,75,76
Horiuti and Polanyi gained mechanistic insights in 1934, which are still widely accepted. They presented a catalytic way that offered an explanation for hydrogenation and isomerisation of double bonds on metallic surfaces under hydrogen pressure, shown in Fig. 10.77
 | ||
| Fig. 10 Hydrogenation mechanism postulated by Horiuti and Polanyi.77 | ||
The unsaturated substrate adsorps to the hydrogenated catalyst surface (1), forming a σ-bond to the metal atom (2). In contrast, a hydrogen atom from the catalyst surface migrates to the β-carbon of the alkene (3). The last steps are the reductive elimination of the alkane from the catalyst (4) followed by desorption of the product from the catalysts surface (5). Isomerisation is usually caused by an inversion of the second and third step, which can result in a differently configured double bond.77
Damiani and Tonetto and co-workers performed a reaction parameter screening with commercial Ni-catalysts, concluded that a hydrogen deficiency on the catalyst's surface is the leading promoter for trans-isomerisation during the reaction.78
Konkol and co-workers studied the hydrogenation of canola oil with Ni catalysts. The research was a side-project of benzene hydrogenation, published in 2016. The focus was firstly on the effect of hydrogen pressure on hydrogenation kinetics. They found that all hydrogenation rate constants increased with increased hydrogen pressure, with a twofold increase in monoene hydrogenation. A welcome side-effect was observed with a decrease in isomerisation rate constants, leading to a decrease from 36% to 22.7% in t-C18:1 content in the product mixtures.79 In further studies, the catalyst was promoted with cerium, resulting in better E/Z-ratio. In combination with improved hydrogen pressure, the final t-C18:1 content was decreased by 48%, referring to the initial experiments. The final hydrogenation to C18:0 was not inhibited by the cerium-modified catalyst, though.80
3.1.1.1. First copper based catalysts. Koritala, Moulton, Dutton, and co-workers investigated selective hydrogenation basics dominantly on copper catalysts in the late 1960s. They structured their research in 2 different storylines: one dealt with the hydrogenation of soybean oil overall, and the other one paid particular attention to a specific catalyst based on copper and chrome.61 Parallel to them, Coenen and co-workers. Also worked on the hydrogenation of polyunsaturated fats and oils with similar catalysts.81–83
CuCr-catalysts were found to be promising in the selective hydrogenation of C18:3, which is responsible for undesired flavours in soybean oil and, therefore, the subject of further investigation.61 In the first ambitions, soybean oil was hydrogenated in batch-reactors. Despite variation of reaction conditions and different constructive approaches, almost only t-C18:1 product was formed during the decrease of C18:3 to less than 1%.84,85 A temperature-dependent effect was found in more formed conjugated dienes and subsequently more scattered positional isomers in the C18:1 with higher reaction temperatures.85,86 Investigations on the kinetics of the hydrogenation reaction revealed that with increased pressure, positional isomerisation of the remaining double bond decreased, and the hydrogenation was faster. Additionally, the C18:2 hydrogenation occurred even with still C18:3 compound in the mixture. The research also indicated the formation of conjugated dienes as an intermediate, which reacts up to 22 times faster than naturally occurring C18:2 containing isolated double bonds.87
Naturally occurring substrates like triglycerides from soybean oil and the corresponding methyl esters in technical grade purity were studied from thereon. Next, developments for the CuCr-catalysts opted towards functionalization with nickel (Fig. 11). The resulting catalyst showed increased activity in C18:3 hydrogenation whilst maintaining C18:2 concentration similar to previously reported CuCr-catalysts and was found to be interesting for further research. Despite this statement, no significant ongoing reports could be found on this topic.88
 | ||
| Fig. 11 C18 fatty acid composition in the hydrogenation of refined soybean oil with Ni-containing CuCr catalysts as performed by Griffin and co-workers.88 | ||
With the switch to natural substrates, the researchers faced catalyst poisons and activity-diminishing substances. The search for catalyst poisons for the CuCr-catalyst exposed free fatty acids, sodium soaps, phosphatides, water, monoglycerides, glycerol, β-carotene, organic phosphor and sulfur compounds. These impurities decreased the catalyst activity, which was preserved by refining and bleaching the fatty substrate.89 The activity of the catalysts was increased by changing the catalysts preparation method. The switch to a copper–silica catalyst and evaluating different precipitation and drying methods increased the activity of the catalyst to more than three times compared to the original CuCr-catalyst; however, the exact composition of fatty acids retained in the product mixture.90
Since all critical, influential factors of batch hydrogenation with various Cu-catalysts were extensively investigated, increasing the scale of the reaction to batches of 10 t of soybean oil was the next logical step. The reactions performed with 2% CuCr-catalysts were considered successful, but the larger setup changed the selectivity of the process slightly. Compared to previously performed lab reactions, the complete hydrogenation to C18:0 occurred at an IV of 112 gI/100 g. Less effective mixing than in lab-scale experiments might be the cause for this. At this iodine value, a reasonable amount of polyunsaturated compounds is still present in the mixture. For reference, a triglyceride containing only monounsaturated C18:1 fatty acids has an IV of 86.2. The total trans-configured content in these batches decreased from 27% to less than 20%, and after the removal of C18:0 of the product mixture, the leached Cu and Cr content was lower than one ppm.91
3.1.1.2. Effect of hydrogen pressure on the hydrogenation reaction. Other significant increases in the reaction performance were achieved by the NRRL by investigating the effect of the hydrogen pressure, temperature, and catalyst concentration. With an increase of each of these variables, the reaction rate increased significantly except for pressures above 1000 bar; here, no increase in the reactivity was observed. However, exceeding pressures of 68 bar had an additional effect on reaction stereoselectivity. The amount of formed t-C18:1 could be decreased by 50% and more on copper catalysts with increased hydrogen pressures (Fig. 12).92,93
 | ||
| Fig. 12 C18 fatty acid composition in the hydrogenation of soybean oil under high hydrogen pressure as performed by Koritala et al.93 | ||
Johannsson et al. reinvestigated the CuCr-catalysts from the NRRL.94 They found that the activity of the catalyst varies with time, mainly the time the catalyst is exhibited to the hydrogenation conditions. They tried to reduce the fluctuation of the catalyst's performance by a constant hydrogen flow surpassing the catalyst's surface, resulting in shorter hydrogenation times, with most other key figures remaining unchanged.95 Further insights into the parallel occurring catalyst reduction mechanism were obtained by the pressure variation during hydrogenation. The reaction rate is not the highest at the highest hydrogen pressure.96 The oxidation state of the catalytically active copper was not entirely shown, but it was found that these differences in activity occur with Cu on Silica catalysts as well.97 For heterogeneously catalyzed processes, this investigation is essential to consider to ensure constant product quality (Fig. 13).
 | ||
| Fig. 13 C18 fatty acid composition in the hydrogenation of canola oil with heterogeneous copper catalysts immobilized on SiO2 as performed by Johannson et al.97 | ||
Overall, these were some relatively new insights into the state of this heterogeneous catalyst, and good chemoselectivity to C18:1 was obtained in the experiments. But no groundbreaking performance was described here in terms of activity and complete conversion of all polyunsaturated compounds.97
3.1.1.3. Modification of copper catalysts. The last attempts in reaction optimization went in the direction of modifying the catalyst with different trialkyl silane (Fig. 14), leading to an increase in catalyst activity, but at the cost of only forming t-C18:1 compound in the monounsaturated fraction and also small amounts of the saturated methyl ester.98
 | ||
| Fig. 14 Triethylsilane as used for the activation of copper stearate-catalysts in the hydrogenation of soybean oil as used by Koritala et al.98 | ||
A particular form of copper present explains this loss in the selectivity of the active catalyst, indicated by a red colour, which was also observed before with sodium borohydride-reduced catalysts.98 The copper stearate catalyst was homogenized using trialkyl aluminium to overcome this issue. Different alkyl chains were used in the homogenization, with diisobutyl aluminium hydride forming the lowest amount of C18:0 (Fig. 15). The trans-component formation in the hydrogenation was similar for most tested catalysts. The removal of copper was achieved by washing with diluted acid.99 Changing the metal in the homogeneous catalyst to palladium led to higher activities than the copper stearate-catalyst. The reaction occurred at mild conditions but with a high t-C18:1 formation at temperatures above 60 °C. High chemoselectivity to the C18:1 fraction was achieved though.100
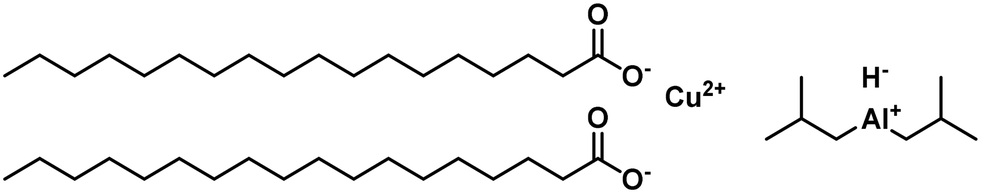 | ||
| Fig. 15 Copper stearate and diisobutyl aluminium hydride precursors for homogeneously catalyzed hydrogenation of soybean oil, as used by Koritala et al.99 | ||
3.1.1.4. Research on continualization. Lastly, the continualization of the process was the subject of investigation. Copper catalysts were considered superior compared to Ni-catalysts. However, the high hydrogen pressures necessary to reach good activity prevented an industrial application due to the high cost of high-pressure batch equipment. Flow reactors could be cheaper to produce than stirred batch reactors and utilize less reactor holdup for a comparable product amount. Benchmarking the reactor was done with nickel since it was already used in the unselective hardening of plant oils. Commercial Girdler G-15 nickel catalyst was used to hydrogenate soybean oil. As the reaction scheme suggests, reactors with a narrow residence time distribution were investigated at first to suppress consequential reactions like isomerisation or saturation of the fatty acid chain. In the lab scale, a co-current flow reactor was used. In comparison to a batch reactor, 5% less t-C18:1 compound, 19.9% absolute, was formed at similar extents of the reaction while also shortening the residence time to less than 20% of the batch reactor.101 Ongoing investigation revealed some limitations of the process. The hydrogenation generally behaved as expected, increasing with temperature and residence time. The Nysel catalyst showed increased activity compared to a sulfur-promoted Ni-catalyst and was less prone to isomerisation. Just with oil flow rates above four l h−1, no effect of the temperature was observable. The authors did not explain this instance. Low catalyst loadings, low temperature, and high hydrogen pressure are favourable conditions for sufficient hydrogenation results for nutrition purposes with less than 50% of t-C18:1 in the C18:1 fraction (Fig. 16).102
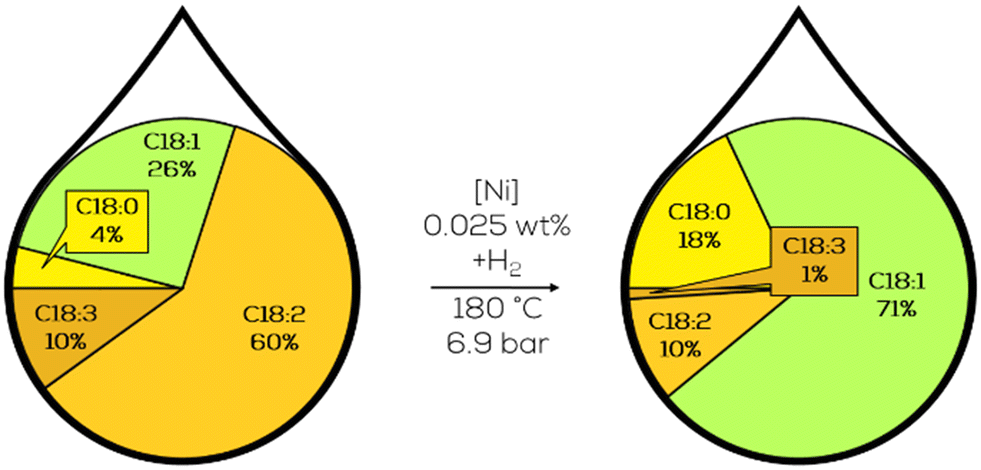 | ||
| Fig. 16 C18 fatty acid composition in the continuous slurry hydrogenation of soybean oil in a co-current tubular flow reactor as described by Koritala et al.102 | ||
Since this reactor concept was found promising with commercial Ni-catalysts, the copper, as mentioned earlier, catalysts were also used in the flow setup. Among the non-idealities which should be solved were a higher amount of conjugated dienes and a higher content of trans-monoenes than in batch experiments. Reaction pressures higher than 6.9 bar reduced conjugated dienes, depending on the reaction temperature. No increase in C18:0 was observed, even with higher pressures. However, the trans-formation relative to the extent of the reaction was still more significant than in comparable batch experiments at all pressures in the continuous reactor.103
Investigating the plug flow (PFR) slurry reactor to a greater extent by systematically varying the operating conditions revealed no reasonable progress. All parameters, pressure, temperature, or catalyst concentration, increased the system's reactivity but with no significant change in either chemoselectivity or isomerisation.104
Research on the PFR reactor was considered complete, and other continuous reactors were investigated. An ultrasonic cell was investigated with the slurry input, but no significant differences were observed apart from some increased activity. The authors could not conclusively clarify whether this increase in activity is due to the energy input in the form of ultrasound or the resulting temperature increase.105 A completely different approach was the use of a fixed-bed reactor. Here, the reaction temperature could be lowered by 40 K with a comparable product composition. Although the equipment limitations did not allow a complete conversion of the linolenic acid, the approach was classified as promising, especially for faster hydrogenation at lower temperatures with good selectivities to C18:1 due to the narrow residence time distribution. But, over 80% of the produced monoene was present in its trans-configuration.106
In general, the results achieved with copper catalysts achieved a high standard. Still, the chemoselectivity of these catalysts was not high enough, leading to an increased amount of C18:0 and, therefore, loss of valuable substrate.
3.1.1.5. Research on different catalytic metals. Additional early publications by the NRRL showed sodium borohydride-reduced catalysts containing Pt, Pd, Co, Fe, and Ag. Palladium showed the most promising results from these metals by achieving good linoleate selectivity and limited t-C18:1 formation.61 The investigation indicated that platinum might be superior regarding increased E/Z selectivity, albeit with tradeoffs due to high hydrogenation of C18:1. Cobalt formed no C18:0. Still, most formed C18:1 compound was present in trans-configuration despite trans-shares of 16.9% and less depending on the metal. This was due to very incomplete hydrogenation reactions. A compromise was found in using Pd as metal. Leading to very little increase in C18:0 content and only a limited amount of t-C18:1 formed. Since the analysis of the trans-share was performed by IR-photometry which is no state of the art measurement in present times, these results should be questioned regarding their precision.61
A step towards continualization with Pd immobilized on different polymers as fixed bed resulted in less t-C18:1 product than the Cu catalyzed continuous process at temperatures of 100 °C and only 3.4 bar of hydrogen pressure. The reported fixed bed catalysts required extensive tuning of operating conditions for each specific substrate and still produced high amounts of stearates. The authors stated that the pore-diffusion of the triglyceride is the rate-determining step in this process, which gives the catalyst enough time to completely saturate the adsorbed triglyceride.107
3.1.1.6. Research on conjugation and other isomerization. Deuterated methyl cis-9,cis-15-octadecadienoate, methyl linoleate, and the conjugated isomers were performed to explore the catalytic mechanism. All substrates contain two double bonds in the carbon chain but have different positional relations. Conjugated double bonds were believed to behave differently in the hydrogenation reaction than isolated ones. Additionally, positional isomerisation of closely positioned but isolated double bonds, as in linoleate, was found to isomerize and form conjugated double bonds and be hydrogenated after that. By evaluating the hydrogenation performance of a compound like methyl cis-9,cis-15-octadecadienoate, the effect of conjugable double bonds compared to single or non-conjugable isomers was to be quantified. This isomer of methyl linoleate has to isomerize over three positions more in the “right direction” and is, therefore, less prone to conjugation. Gained insights included the only slightly higher reactivity of the non-conjugatable C18:2 compound compared to C18:1.108
Also, deuterium was found in any positional isomers of the dienes when performing the deuteration instead of the hydrogenation (Fig. 17). The amount increased with the extent of isomerisation. This was also found for the t-C18:1 fraction. Due to the lower reactivity of non-conjugatable dienes, the amount of C18:0 was considerably higher in these reactions since the double bonds of monoenes and non-conjugable dienes are hydrogenated at a similar rate. Therefor no higher selectivity towards C18:1 could be observed since the formation of a monoene from a non-conjugatable diene is not as preferred as the formation from a conjugatable diene.109
 | ||
| Fig. 17 C18 fatty acid composition in the deuteration of methyl linoleate with Pd on carbon as performed by Koritala et al.109 | ||
Investigations including Pd, Pt, and Lindlar catalysts showed that Pt and Pd were most active when it comes to conjugated diene or triene hydrogenation to monoenes; thus, both being more active in the hydrogenation of monoenes to C18:0 compound than Ni or Lindlar catalysts.108–110
Overall, the department for heterogeneous catalysis of the NRRL provided an efficient catalyst for the heterogeneous hydrogenation of plant oils for nutrition. Different copper catalysts were studied intensively with a CuCr-cocatalyst, resulting in a very selective and active catalyst at elevated hydrogen pressures. The focus was on linolenate hydrogenation to increase the shelf-life and taste of soybean oil. At this early stage of research, essential insights were gained.
However, the catalyst utilized in an adequately tuned continuous reactor might work well to provide a uniformed platform chemical since selectivity to C18:1 is significantly enhanced compared to Ni-catalysts. On the other hand, the reaction conditions are quite harsh, and the resulting product is only suitable for subsequent processes where positional and configurational isomers are no issue. Nevertheless, these copper-based catalysts are effective when the E/Z ratio is not crucial and high activity isn't the primary concern. However, given that the platform chemicals fall into the single-digit price range, the process conditions and substantial catalyst loading, along with the high content of trans-configured products, make this approach less than ideal.
3.1.1.7. Theoretical reevaluation of NRRL results. The results of the NRRL research regarding copper as a hydrogenation catalyst were newly interpreted by Albert Dijkstra in the early 2000s. He focused mainly on mechanistic proposals for the positional isomerisation of the C18:3 compound that Koritala and Johannsson intensively reported. He proposed a different reaction mechanism of a copper catalyst, which includes the addition and abstraction of hydrogen, including mesomeric stabilized intermediates with conjugated dienes, which lead to more intense isomerisation and hydrogen addition at not only the two carbon atoms at the specific double bond but also other atoms in the mesomeric system (Fig. 18).58
 | ||
| Fig. 18 Products of hydrogen abstraction and addition mechanism for methyl linolenate as proposed by Dijkstra.58 | ||
Later, he discussed the formation of trans-compounds by different catalysts and various chemical and engineering modifications to trick the cis–trans-equilibrium. Homogeneous catalysts, supercritical solvents, use of zeolites, variation of the catalytic active metal, and other changes in the catalyst were reviewed, all of them coming to the same conclusion that either the goal of low trans-formation could not be achieved, or the invention did not cope with the costs of the process. This usually led to the situation that these developments were either not industrially used or only in a very specialized application, but they did not replace the classic Ni-catalyzed hardening of oils.62,111,112
3.1.1.8. Adaptation of NRRL catalysts to lubricant and biodiesel research. The research of Koritala et al. was taken up by 2002 by Zaccheria and co-workers and reinvestigated in the context of producing biodegradable lubricants. Different supported copper catalysts were initially tested towards their hydrogenation capabilities of canola oil (Fig. 19). The goal was to produce an oil with enhanced oxidative stability that is still liquid at low temperatures. The reduction of polyunsaturated compounds while not producing C18:0 and limiting the isomerisation towards t-C18:0 were in focus. They found that, in general, Cu on SiO2 catalysts showed good performance, with the best one reaching the entire reduction of C18:3 and C18:2 reduced to 5% of mass fraction while not producing any methyl stearate and keeping methyl elaidate mass fraction below 18%. This result was achieved by a catalyst supported on non-porous SiO2 and prepared by chemisorption hydrolysis. The experiment was performed with a catalyst loading of 2.5% with a Cu fraction of 7.4% on the overall catalyst mass, but it still took three hours to complete.113
 | ||
| Fig. 19 Composition of the C18 fatty acid fraction in the selective hydrogenation of canola oil as performed by Zaccheria.113 | ||
Other catalysts prepared by incipient-wetness-impregnation showed no activity in soybean oil-derived methyl ester hydrogenation at similar conditions.114 Focusing on biofuels and, in particular, the production from a wider variety of crops, the hydrogenation of non-edible seed oil-derived methyl esters was investigated. Tobacco seed oil and other substrates were hydrogenated to IV between 108 gI/100 g and 118 gI/100 g without producing saturated compounds. The synthesis of similar biodiesel from different sources works well, and a proper concept was introduced. Still, the competitiveness towards fossil resource-based diesel or platform chemicals is questionable due to reaction temperatures of at least 160 °C to activate the copper catalyst for hydrogenation. Additionally, no proof for the recyclability of the catalyst has been reported so far, which is a crucial step in catalytic processes to be economically competitive.115–117
 | ||
| Fig. 20 Composition of the C18 fatty acid fraction in the selective hydrogenation of sunflower oil as performed by Romanenko.121 | ||
Ongoing research included different carbon-based support structures of the subunit family which were applied to the substrates as a powder in a slurry-type reactor. The newly prepared catalysts were intensively studied and classified in terms of pore size and Pd dispersion and compared to other catalysts with carbon-based support structures based on nickel. The different catalyst designs led to halved reaction times and kept a similar activity level during the first eight recycling runs. Additionally, the formation of trans-isomers was about 10% lower than the benchmark nickel catalysts, showing a comparable level of complete saturation.122
Another approach towards heterogeneously catalyzed hydrogenation was taken by Korkut et al. from 2015 on, deriving activated carbon as a catalyst from mistalea (Viscum album L.).123 They increased the oxidation stability of corn oil-derived biodiesel from 33 minutes to 81 minutes, which sounds like a minor success. Upon closer examination of the analytics, it was clear that the biodiesel's composition before and after the reaction differed mainly in the t C18:2 content, which increased from 0.045% to 0.45%. However, this change might still fall within the GC FID method's margin of error, as there was no noticeable decrease in the c C18:3 or c C18:2 content that could account for the additional t C18:2.124 Therefore, this catalyst is unsuitable for producing a uniform platform chemical, as there was no significant difference in the product observed. These results should be seriously questioned.
3.1.3.1. Initial research by the APEC. Intense work on integrating plant-based diesel alternatives into the Asia-Pacific Economic Cooperation (APEC) mobility sectors was done by Luengnaruemitchai and her group. Their work was mainly motivated by the political push towards harmonizing biodiesel blends within the APEC. Substrates and catalysts were investigated and rated concerning their biodiesel refinement applicability. These goals were the partial hydrogenation towards monounsaturated FAME while maintaining a low amount of trans-configured compounds and a high overall catalytic activity to secure the economic feasibility.125
The general approach was based on heterogeneous Pd catalysts supported on different acidic structures based on SiO2 or Al2O3. A substrate of choice was canola oil-derived biodiesel, hydrogenated in batch mode at 80 °C and 3 bar of hydrogen pressure. With Pd on SiO2 showing the highest activity and the general observation that the addition of sulfur compounds to the substrate and a higher concentration of weak acidic sites increase the selectivity towards c-C18:1 compounds in the product mixture at the cost of decreasing activity (Fig. 21).126
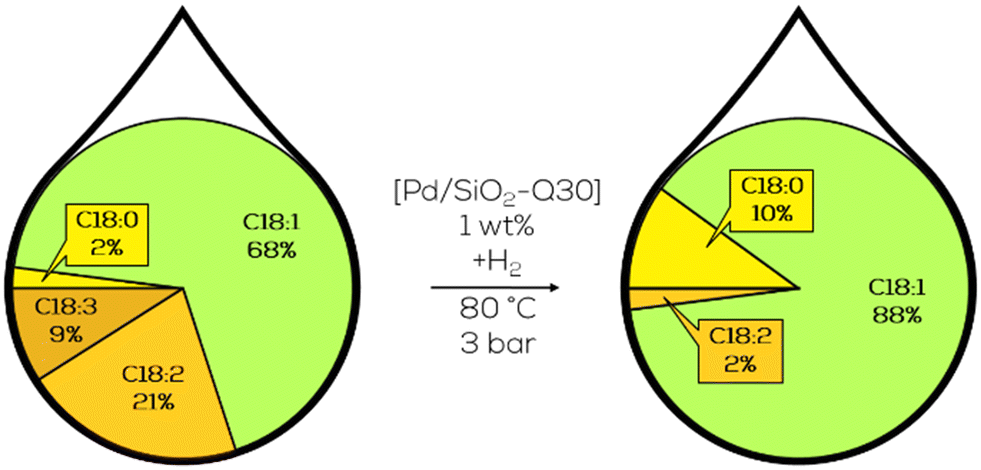 | ||
| Fig. 21 C18 fatty acid composition in the hydrogenation of canola oil-derived biodiesel over macroporous Pd on SiO2 as performed by Luengnaruemitchai.127 | ||
Pursuing the most active catalyst in the following work, variations of pore diameters of 3, 16, 45, and 68 nm were investigated. The results were quantified as TOF (moles of converted FAMEs/moles of Pd on the surface at a reaction time of 30 minutes), selectivity towards monounsaturated FAMEs and selectivity towards cis-compounds. The results showed the highest activity and C18:1 selectivity at a pore size of 46 nm. The cis–trans-isomerisation was less pronounced using the catalyst with a pore diameter of 3 nm because the reaction was assumed to take part only at the outer particle surface. The “microporous” Pd/Q50 catalyst showed similar trans-formation. Therefore, a suitable catalyst should be microporous or nonporous to decrease the undesired t-C18:1.127 Up to this point, no TOFs above 10000 h−1 were achieved. Hydrogenation of soybean methyl esters by Pd on nonporous SiO2 supports showed the expected higher TOF per active catalytic site of almost 30000 h−1. Interestingly, the tolerance towards deactivating sulfur compounds increased with a decrease in particle size.128
Changing the reactor concept to a fixed bed flow reactor with Pd on carbon increased the catalytic activity by more than 400% while retaining similar product compositions up to 80% conversion. From 80% conversion to complete conversion, the proportion of t-C18:1 and C18:0 increases disproportionately strongly.129 Smaller particle sizes and the location outside of the pores showed the highest hydrogenation activity similar to the SiO2-supported Pd particles.130
Subsequent variation of the Pd particle size focused on the increase of c-C18:1 and less formation of t-C18:1 during hydrogenation which was achieved with nanoparticles with 4 nm in diameter. This was achieved from 38% to 55% of the mass fraction but to the cost of overall activity. These particles showed a higher affinity towards polyunsaturated substrates and an even higher affinity to conjugated double bonds.131 The higher activity of larger nanoparticles was additionally confirmed by subsequent work, where different precursors were used to synthesise Pd particles of more variation in size (Fig. 22).132
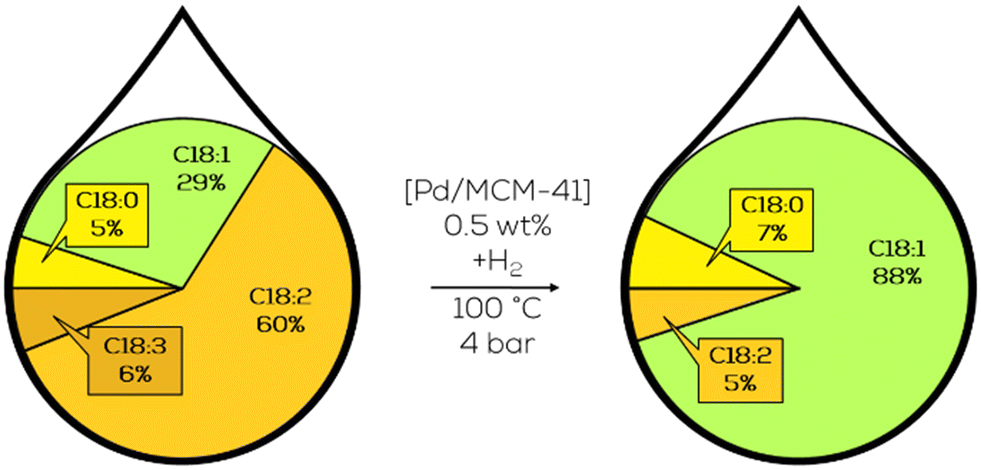 | ||
| Fig. 22 C18 fatty acid composition in the hydrogenation of soybean oil-derived methyl ester focusing on increased c-C18:1 formation by 2 nm sized Pd-nanoparticles on MCM-41 as performed by Luengnaruemitchai.133 | ||
Additionally, palm oil-derived biodiesel was used in hydrogenation reactions. Since the composition of the raw material only contains 10% of PUFAMEs, the yield towards C18:1 was lower at the complete conversion of C18:2 than at the beginning of the reaction. Interestingly, the activity of the Pd catalyst on SBA-15 had initial activities up to 8 times higher than the before mentioned Pd/γ-Al2O3.134 Later, these initial activities could be quantified to an initial TOF of almost 80000 h−1. The particle size of Pd was in the range of 5 to 10 nm in these experiments, with particles only present at the outer surface of the support.135
In ongoing work, MCM-41 frameworks with different pore sizes were investigated by Luengnaruemitchai and her group as catalyst support for Pd. In the hydrogenation of canola oil-derived biodiesel, the TOF at 30 minutes of reaction time exceeded 5700 h−1 while utilizing the largest average pore sizes of 7.55 nm and average Pd particle sizes of 2.5 nm.133 Further modification of the MCM-41 supported catalyst led to a TOF of 35000 h−1 with better selectivity to monounsaturated FAMEs when using lower Pd loadings and, therefore, smaller particles. The reaction's chemoselectivity was increased slightly by changing the silica source for the MCM-41 support structures. Introducing parts of the Si by the addition of silatrane decreased the trans-formation slightly and the C18:0 formation by half while still maintaining a comparable TOF of 25000 h−1 (Fig. 24).136
 | ||
| Fig. 23 Regeneration of heterogeneous Pd catalysts by oxygen from gaseous feed stream as proposed by research on sulphur Yoshimura.137 | ||
 | ||
| Fig. 24 C18 fatty acid composition in the hydrogenation of soybean oil-derived methyl ester over heterogeneous Pd catalysts with silatrane doped SiO2 support structure as performed by Luengnaruemitchai.136 | ||
Accumulated oxygen on the surface of the catalyst is reduced to water by the present hydrogen and leaves a free catalyst's surface. This mechanism decreases the deactivation rate by more than 75% compared to the reaction without added oxygen.137
 | ||
| Fig. 25 Composition of the C18 fatty acid fraction in the selective hydrogenation of sunflower oil as performed by Belkacemi.142 | ||
Since 2005, the partial hydrogenation of sunflower oil has been the focus of research by the group of Damiani and Tonetto. They used heterogeneous catalysts based on palladium, which were tuned with different co-metals and utilized innovative supports. The influence of the co-metals, Mo, V and Pb, and the Pd particle size was investigated in the early stages. Mo-doped catalysts showed the highest hydrogenation performance of all the screened co-metals (Fig. 26). Pd-Mo sol–gel prepared catalyst showed the highest selectivity to the c-C18:1 component. In contrast, the wet-impregnated Pd–Mo catalyst showed the highest activity with increased t-C18:1 content in the final mixture.144 Particle size studies revealed that the smallest Pd-particle size of 2 nm showed the highest selectivity to c-C18:1 but also the undesired final C18:0 component.145
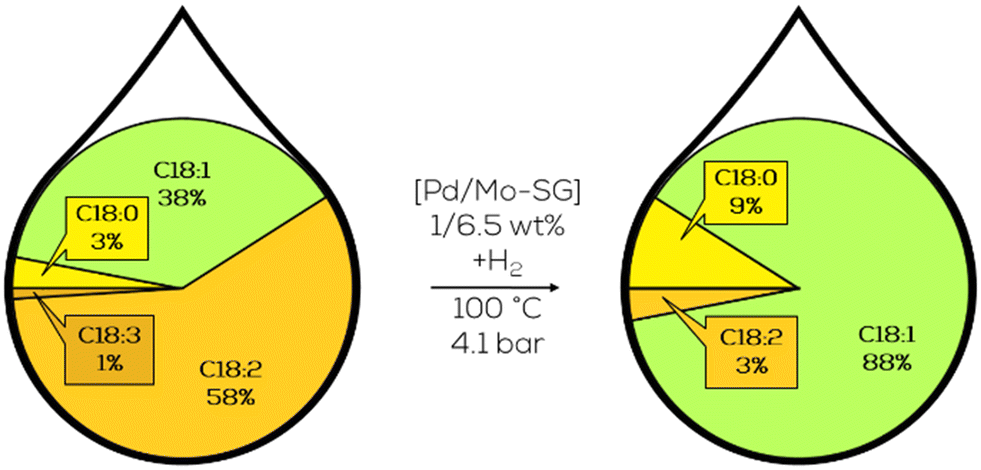 | ||
| Fig. 26 C18 fatty acid composition in the hydrogenation of sunflower oil with Mo-doped Pd catalyst immobilized in sol–gel as performed by Tonetto.144 | ||
Six different Pd-catalysts were compared to a commercial Ni-catalyst. In general, Pd showed higher activity and similar selectivity to the t-C18:1 compound with reduced selectivity of undesired C18:0. For alumina-supported Pd-catalysts, an investigation of the kinetics revealed that the cis–trans-isomerisation has higher activation energy than the hydrogenation of C18:2 to C18:1 components.146 One specific Pd-catalyst on alumina support with a metal loading of 0.78% and 60% dispersion showed significantly higher activity than the others while still producing lower t-C18:1 amount and showed higher selectivity to the monounsaturated compound. Nevertheless, the t-C18:1 content is still too high for food applications, but the initial TOF reached values of a maximum of 120.000 h−1.147 Further efforts to decrease the cis–trans-isomerisation involved using edible modifiers. Adding ethyl-benzoate showed a higher production of t-C18:1, whereas magnesium glycinate diminished the cis–trans-isomerisation. Yet, the overall activity of the catalysts decreased with the addition of both modifiers, which were subsequently not further investigated.148 Miscellaneous investigations on different solvents and catalytic metals showed slight differences in the product compositions. Namely lower cis–trans-isomerisation but increased total hydrogenation to C18:0 when using supercritical propane and platinum precursors. Modifying the catalyst with potassium increased the c-C18:1 selectivity even further. But these results were not taken up further.149
Alshaibani and co-workers investigated the hydrogenation of palm oil from 2012 onward. Their research included heterogeneous palladium catalysts, which were boron-modified. They compared the catalyst to commercial and self-prepared Pd catalysts without boron modification. They exhibited higher activity and reduced selectivity to trans-configures compounds (31% boron-doped; 58% pure supported palladium). These results were found for SiO2, Al2O3 and carbon as support structures for the catalyst.150–152
They also observed similar effects of changing reaction conditions on their reactions. Increased agitation, hydrogen pressure and temperature led to faster reactions. In contrast, the content of trans-configured components increased with higher temperature and lower pressure and agitation, presumably lowering the hydrogen supply on the catalyst's surface.153 Overall, these experiments reproduce the core statements already published by the NRRL and provide no substantial added value for the state of the art.
Luengnaruemitchai and co-workers investigated different supported metals beside Pd and subsequently combinations. The observed overall activity was Pd > Pt ≫ Ni, which is in line with the already reported activities by Dijkstra.62 The lower yield in t-C18:1 with Pt catalysts is said to be a side-effect of the higher formation of saturated FAMEs since the monounsaturated intermediate is directly converted to C18:0.154 Consistent improvement was achieved by the addition of Mg modifiers to the Pd catalyst. The formation of t-C18:1 was always lower than in experiments without Mg added to the catalyst.155 This promising result motivated further research in modifying Pd catalysts with alkaline and alkaline earth metal promoters. It was found that the lower selectivity to t-C18:1 can be maintained by Na, Ca, and Ba with Ba-modified catalysts reaching a TOF(11) at 11% conversion of more than 90000 h−1.63
Luengnaruemitchai and co-workers investigated also bimetallic Pt/Pd catalysts in equal shares, resulting in a biodiesel that meets international standards. The reported TOF(20) did not exceed 200 h−1.48 This was increased to more than 3900 h−1 with more than 50% trans-share in the monounsaturated fraction at 99% conversion of PUFAMEs while maintaining the catalyst's sulfur resistance.156
Overall, some critical insights were gained in the work of Luengnaruemitchai, Alshaibani, and Damiano and Tonetto and co-workers. The promotion with different elements, especially Ba, to boost selectivity towards C18:1 and cis-configured components and the information about the dependence of overall activity and selectivity by particle size and the affinity of smaller particles towards conjugated double bonds and polyunsaturated substrates in general.
The industrial feasibility of the described catalytic reactors was evaluated from thereon. This included a model of reaction parameters and investigations towards better catalyst recycling. Depending on the prize development of noble metals, Pd in particular, an industrial application could become feasible since the net present value (NPV) could become higher than the ones of the conventional processes. Improvements to the catalytic constructions and changing to Pt as a noble metal leading to increased recyclability, up to 40 times, works in favour of the applicability as well.160–164
A neckbreaker for applying this technology might be the labour-intensive production of catalyst-precursors and the placement within the reactors since the recyclability is still limited and the platinum-catalyzed process operates at 180 °C.
 | ||
| Fig. 27 Ni-catalyzed, microwave-heated transfer hydrogenation of methyl linoleate to methyl oleate with isopropanol as hydrogen donor as performed by Wei et al.165 | ||
The produced C18:1 was dominantly methyl elaidate, resulting in a high E/Z-ratio in the product. However, high catalyst loadings of 6–14 wt% referring to the biodiesel mass were used (Fig. 28).165
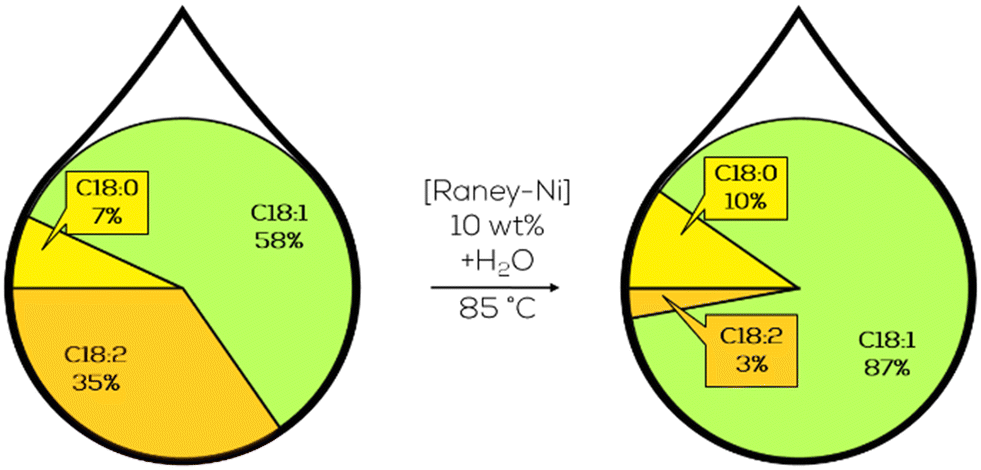 | ||
| Fig. 28 Fatty acid composition of the C18 fraction in the partial hydrogenation of jatropha oil-derived methyl ester as performed by Wei.165 | ||
An increase in the microwave intensity from 50 W up to 150 W significantly increased the reaction rate and selectivity to the monounsaturated compounds. Along with these benefits is an increase in the formation of t-C18:1 methyl esters by a factor of three as a product of the isomerisation of methyl oleate, leading to an even higher E/Z-ratio in the product. This was observed with increased microwave power and a higher amount of catalyst or isopropanol relative to the substrate.166
Further insights into this reaction were gained by intensively studying the mechanism of the transfer hydrogenation process under the influence of microwaves. It was shown that the intensifying effect on the reaction was not mainly achieved due to the heating of the catalyst but by effects on water and isopropanol providing more hydrogen to the reaction. The authors did not provide more in-depth information on this aspect, though. Also, increased deactivation by FAMEs on the catalyst surface was observed due to enhanced microwave power.167
In ongoing work, the group used a heterogeneous Ni–La–B catalyst and exchanged isopropanol for sodium borohydride as the hydrogen donor. This approach utilized ten wt% of catalyst in relation to the fatty substrate but did not achieve complete conversion of C18:2 after 150 minutes of reaction time. Additionally, they exceeded an E/Z-ratio of more than 1.5 in the monounsaturated fraction.46
Lastly, Ni/bentonite was used under direct hydrogenation conditions with 0.3 MPa of hydrogen pressure. The amount of catalyst was also decreased to 3.33 wt%, and with optimal reaction conditions, 75% of C18:2 was hydrogenated with a final E/Z ratio of less than 0.66 (Fig. 29). Catalyst recycling was also successful, with five performed cycles showing no loss in catalytic activity.50
 | ||
| Fig. 29 Fatty acid composition of the C18 fraction in the hydrogenation of jatropha oil-derived methyl ester as performed by Wei.50 | ||
Despite the achieved performance gains in microwave-assisted hydrogenation reactions, the research still leaves a few boxes unchecked: the system's power consumption, in addition to the high catalyst concentration and amount of solvent and relatively high formation of saturates and trans-components without achieving full conversion of C18:2 makes it not suitable for economically providing platform chemicals.
3.2. Homogeneous catalysts
Homogeneous catalysts for hydrogenation reactions are usually dissolved metal salts. They can be dissolved in the same phase as the substrate or in a different immiscible liquid phase by functionalisation or using different ligands. Other approaches “immobilize” the catalyst in polymeric structures either by covalent bonds, making the catalyst heterogeneous, or by intensively coordinating to polymeric structures, which can be seen as a different type of ligand. These approaches must usually deal with mass transport limitations across the interphase but can provide good catalytic results while ensuring catalyst separation after the reaction.Parallel to the research mentioned above in heterogeneous catalysis, some investigations were performed by the NRRL towards homogeneous catalysts for the selective hydrogenation of fats and oils. Frankel and co-workers used homogeneous catalysts to hydrogenate polyunsaturated methyl esters and triglycerides from linseed and soybean oil. Compared to the heterogeneous catalysts investigated by Koritala, the homogeneous acetylacetonate complexes operated at lower temperatures but higher hydrogen pressures and showed reasonable activity at low catalyst loadings. Nickel, copper, cobalt and iron, usually dissolved in methanol, were initially used for hydrogenation application.169 Besides the reaction performance, the catalyst recycling and product purification must be considered for homogeneous catalysts Since they are present in the same phase as the reactants and can, therefore, contaminate the product when not handled properly. Recovery of the catalytic metal should also be considered for economic reasons since commonly used transition metals and ligands are costly, and recycling might be critical to making a process economically feasible.42,168
The catalytic activity increased from Fe over Cu and Co to Ni; however, no hydrogenation activity was observed in the absence of solvents. Analysis of the C18:1 fraction of the product mixture revealed a scattering of the double bond in the t-C18:1 share and only naturally occurring positions in the cis-configured share.169 Since the tested acetylacetonate complexes also formed some undesired C18:0, using PtSn-complexes was the subject of further research. The complexes were formed with triphenylphosphine (TPP) or triphenylarsine (TPA) ligands and showed activity from 30 °C upwards without forming any C18:0. But in the monoenic fraction, 57% to 78% were present as undesired methylelaidate.170 To tackle the high t-C18:1 formation, carbon monoxide was used as a ligand from thereon. Different carbonyl complexes were used as catalysts in the hydrogenation of soybean methyl esters. The most active Cr(CO)3 catalyst completely hydrogenated C18:3 at higher temperatures, forming up to 67% monoene with only 3–7% of t-C18:1 content (Fig. 30).171,172
 | ||
| Fig. 30 Chromium carbonyl complex coordinating to conjugated diene used for hydrogenation of unsaturated fatty esters by Frankel et al.171 | ||
Very little C18:0 was formed during hydrogenation as well. Digging deeper in this direction included screening various substrates, ranging from safflower oil to soybean oil, keeping the trans-content usually below 10% while producing no additional saturates and reducing the IV to values below 60. This was caused by the reducing mechanism, which is a 1,4 addition of hydrogen to a conjugated diene, which the addition of deuterium could trace. However, the used chromium carbonyl complexes showed little activity below 165 °C and utilized initial hydrogen pressures of 35 bar (Fig. 31). Further optimization should address to reduce the operating conditions and include catalyst recycling.172
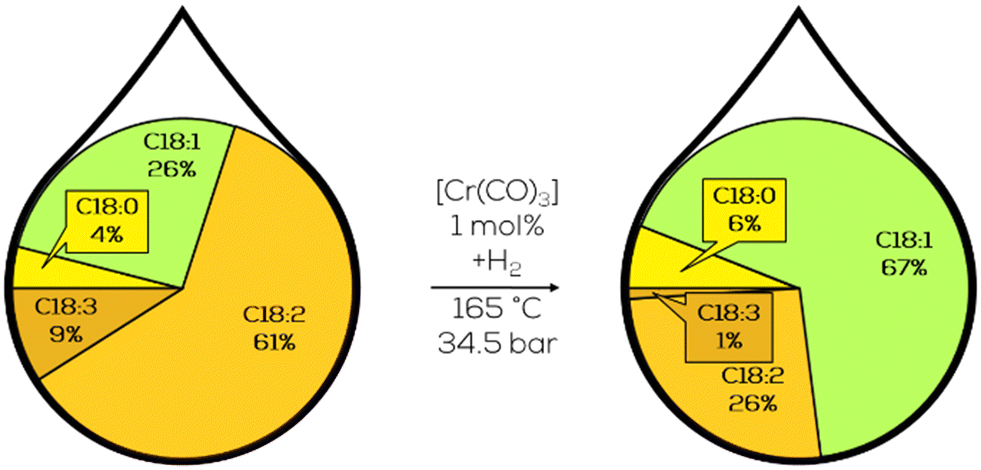 | ||
| Fig. 31 C18 fatty acid composition in the hydrogenation of soybean oil by chromium carbonyl complexes as performed by Frankel et al.172 | ||
Recyclability was approached by polymer binding the chromium carbonyls. This resulted in a catalyst that initially showed similar activity but was not recyclable more than two times due to the unstable polymer structure at the reaction temperature.173 Screening different metals on these polymer structures revealed Pd to be active below 50 °C. PdCl2 was a precursor, resulting in activities slightly below comparable heterogeneous Pd on alumina catalysts.174 These results closed the research in vegetable oil hydrogenation in the NRRL. Among the investigated homogeneous catalysts, Cr(CO)3 showed superior hydrogenation activity towards cis-configured monoenes at the expense of harsh hydrogenation conditions and a lack of feasible recycling concepts.
Bailar and Itatani researched homogeneous catalysts, and different transition metal complexes were more intensively studied. They focused on platinum and palladium catalysts.
Adding SnCl2 to the Pt-complexes boosted the activity while maintaining high chemoselectivity to monounsaturated compounds (Fig. 32). However, almost all formed monoenes were present in the trans-configuration besides some conjugated trans,trans-dienes.
 | ||
| Fig. 32 Coordination of PtSn-TPP-complex to an unsaturated fatty acid chain as proposed by Bailar and Itatani.175 | ||
The mechanism proposed included the isomerisation of the double bonds into conjugation. Product composition and activity changed slightly depending on the Pt- or Pd-precursor and the additives, which include Si, Ge, Pb, or triphenylphosphines (Fig. 33).175–178 Additionally, the authors found that in the absence of hydrogen, isomerisation of C18:2 takes place, but isomerisation of oleic acid does not occur.179
 | ||
| Fig. 33 C18 fatty acid composition in the selective hydrogenation of soybean methyl ester as performed by Bailar and Itatani.176 | ||
Graydon and Diosady, and co-workers investigated the hydrogenation of canola oil with homogeneous RuCl2(CO)2(PPh3)2, which proved to be at least very active but lacks chemoselectivity towards C18:1 components. Important to mention is that the ruthenium catalysts form very low t-C18:1 during hydrogenation processes; only a 10% maximum is reported during the hydrogenation from an IV of 122 to 45 due to the formation of saturates.180 Since the work is motivated by nutrition purposes, the low formation of trans-configured components is highly beneficial. The activity was increased by the combination with a heterogeneous nickel catalyst, which maintained the low level of trans-isomer formation.181 For an application in the provision of platform chemicals, the high reactivity to completely saturated compounds is not desired since these products are inert compounds for the production of, e.g., difunctionalized compounds.
Tucker and Riley from Procter & Gamble investigated a homogeneous (naphthalene)(tricarbonyl)chromium catalyst in the highly selective hydrogenation of different model substrates towards cis-configured isomers of methyl oleate (Fig. 34). They found only cis-configured isomers in the product mixture independent of the substrate, either methylene-interrupted or conjugated cis-configured dienes. Notably, even trans,trans-configured conjugated dienes only formed cis-isomers of methyl oleate. The reaction temperature of 130 °C was crucial for achieving these results, and no positional isomerisation occurred. Still, due to the substrates and the mechanism, four positional isomers of oleic acid were detected in the product mixture. Important to mention is that stainless steel reactors were found to be not inert since product mixtures substantially differed between glass-lined and pure steel reactors.182 Overall, these results are impressive due to the superior stereoselectivity to cis-configured monoenes. However, the use in synthesising platform chemicals is questionable due to the extremely high catalyst loading of 10% and the use of solvents, leading to comparably high processing costs. The choice of solvent is somehow essential in this catalytic system, not only because of specific operating conditions but also because an even slightly coordinating solvent competes for the active sites of the metal center. Also, the interaction of the catalyst and the stainless-steel reactor is counterproductive for industrial application.
 | ||
| Fig. 34 Coordination of a homogeneous Cr(CO)3 – complex to cis–trans-conjugated dienes as proposed by Tucker and Riley.182 | ||
000 h−1 were achieved at reasonable reaction conditions of 120 °C, 50 bar of hydrogen pressure and a molar catalyst ratio of 15000 per double bond in sunflower-derived methyl ester. The literature lacks information about the exact calculation of these values. However, complete conversion of polyunsaturates was never achieved, but at the end of the reaction, almost to t-C18:1. The catalyst was recyclable, but only one consecutive run was described (Fig. 35).184–187
 | ||
| Fig. 35 Concept for reactions with a homogeneous catalyst immobilized in the second aqueous phase. | ||
Ongoing research included different organometallic catalysts. Common single-atom platinum-based complexes and ligand-modified palladium nanoparticles showed similar reactivity (palladium, TOF = 110000 h−1) and higher selectivity to cis-configured monoenes (platinum, 5% of t-C18:1 per c-C18:1 formed, TOF = 6820 h−1 or less) (Fig. 36).188,189
 | ||
| Fig. 36 Fatty acid composition of the C18 fraction in the partial hydrogenation of linseed oil-derived methyl ester as performed by Papadogianakis.189 | ||
Overall, this system led to good results and little t-C18:1, and the authors presented an approach to catalyst recycling. However, this recycling needs to be performed more often to be considered viable, especially considering the high catalyst loadings and lower TOFs with platinum catalysts.
 | ||
| Fig. 37 Fatty acid composition of the C18 fraction in the partial hydrogenation of linseed oil-derived methyl ester as performed by Schumann and Bayer.190 | ||
In general, the different homogeneous catalytic systems up to this point showed either impressive selectivities, which are not proven to be necessary for the provision of platform chemicals, or did not gain any significant advantage over their heterogeneous counterparts. The Ru complexes investigated by Graydon and Diosady for example did not show any enhanced selectivity to monounsaturates. Furthermore, the highly selective catalysts reported by Tucker and Riley showed low overall activity, making an industrial application for cheap intermediates less likely. Ru-catalysts may present an attractive option for more specialized applications, though. Even though some research showed recyclability in principle, a long-time evaluation of catalytic activity and stability was not shown, and therefore, no recommendation for an industrial application for the provision of platform chemicals can be given for any homogeneously catalyzed process so far.
3.3. Colloidal catalysts
A different approach to the common heterogeneous and homogeneous catalyst is the utilization of unsupported colloidal nanoparticles. The common conception is that nanocatalysts combine the advantages of both conventional catalytic methods. This includes high activity, enormous surface area, modifiability by ligands, and good accessibility for substrates. Depending on the approach of nanoparticle synthesis, the catalytic properties can be tuned to shape selectivities in the specific reaction.191 There are drawbacks reported to these systems, though. Usually, the preparation is complex, and one could run into stability and separation problems due to their lack of support structure.192 In general, nanoparticles need to be stabilized because thermodynamics force the particles towards aggregation, resulting in different morphology leading to inferior catalytic performance. Different stabilization methods exist, including adding polymers, surfactants, or ligands as stabilizing agents, which could alter the catalytic properties due to their intense interaction with the particle. Finally, polar solvents can stabilize nanoparticles in a colloidal state that can remain stable for several months. These polar solvents also act as some kind of ligand with the surface of the particles, but the interaction is less intense. It does not alter the catalytic properties similarly to classic ligands like triphenylphosphines.168,193,194 In the following part, all relevant research regarding the partial hydrogenation of fatty substrates is portrayed and critically reviewed towards the applicability in the provision of platform chemicals for the chemical industry.Behr and co-workers conducted the first catalytic investigations featuring unsupported colloidal nanoparticles in the early 1990s. The authors synthesized Pd nanoparticles by the reduction of different precursors, PdCl2 and Pd(OAc)2, among others, in propylene carbonate (Fig. 38) and used them in the hydrogenation of different oils and their respective methyl esters. The product mixtures contained less than 1% polyunsaturated compounds at reaction times below 30 minutes while maintaining the content of C18:0 at the level of the initial composition (Fig. 39). The colloidal particles were utilized in a biphasic approach for easier catalyst recycling, performed up to 8 times. The formation of trans-configured components was an issue that increased up to a trans–cis-ratio of 2 with reaction temperature, though.195
 | ||
| Fig. 38 Solvent stabilized colloidal Pd nanoparticles in propylene carbonate as proposed by Behr.194 | ||
 | ||
| Fig. 39 Fatty acid composition of the C18 fraction in the partial hydrogenation of sunflower oil-derived methyl ester as performed by Behr et al.195 | ||
Notably, this selective synthesis runs under moderate reaction conditions while maintaining extremely high chemoselectivity at almost full conversion of polyunsaturated components.195 The catalyst also showed significant hydrogenation activity with other dienes than oleochemicals.196 These processes were also patented by Henkel KGaA, but no specific application was given for this technology.197–199 Interestingly, this work has received little to no attention from the community despite being over 30 years old and showing some of the highest activities and selectivities for monounsaturated fatty acid generation.
 | ||
| Fig. 40 Fatty acid composition of the C18 fraction in the partial hydrogenation of sunflower oil-derived methyl ester as performed by Wei Liu.201 | ||
This research does not check any boxes for an effective provision of monounsaturated intermediates since recyclability is limited and rhodium is too expensive as a catalyst if the recycling is still to be solved.
Ultimately, colloidal catalysts from nanoparticles might represent a suitable alternative to hydrogenate fatty substrates selectively to the monounsaturated compound. Since the number of active surface atoms is far higher than in classic heterogeneous porous catalysts, overall activity is higher while maintaining the same metal loading. Tuning the selectivity of a system is possible by changing the particle size and shape represents an exciting option, too. Nevertheless, the additional components necessary to realize nanoparticle catalysis diminish the advantages. The amount of work that must go into preparation is still obstructing. Similar to heterogeneous catalysts in general, as well as the separation and limited recyclability similar to homogeneous catalysts.
4. New reaction concepts for the selective partial hydrogenation
In addition to the aforementioned homogeneous, heterogeneous and colloidal catalytic systems most recently research was performed on different concepts. Not only the introduction of structured membrane reactors for well-defined substrate interaction on the catalytic material, but also the use of catalyst free plasma-powered systems will be presented in the following chapters.203,2044.1. Membrane supported reactions
This chapter focuses on partial hydrogenations performed in a membrane reactor. These membranes have a modified top layer that contains the catalyst. Two different approaches were reported. The flow configurations make the differentiation of the specific reactors. These systems are a specification of a classic heterogeneous fixed bed catalytic reactor, which should lead to a more defined flow profile and mass transport and be ideal for consecutive reactions compared to traditional fixed bed reactors.204 Or, as in other contributions described, should ensure a proper hydrogen saturation on the catalyst surface.205–207The following part goes into deeper detail and reviews the published research utilizing a membrane for catalytic partial hydrogenation of fatty substrates in the context of the production of chemical intermediates. The two main contributors in the 2000s were Mary Rezac and Reinhard Schomäcker, who worked on different approaches.
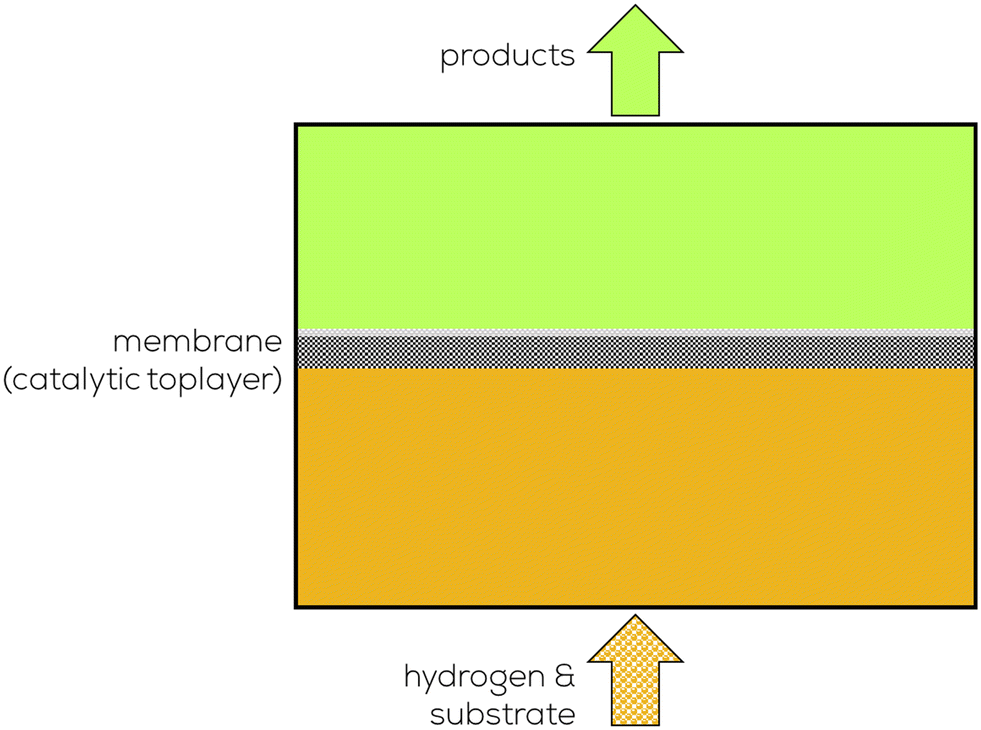 | ||
| Fig. 41 Parallel-flow configuration for membrane reactors as used by Schomäcker.208 | ||
High selectivity towards the partially hydrogenated product was expected by sharp residence time distribution in the membrane reactor. But this could not be proved by the experiments. In this work, the membrane reactors showed shorter reaction times than the fixed bed reactors.204 These results motivated to work on the hydrogenation of sunflower oil. Pd and Pt were used as catalytic material on the active sites on the membrane, with Pd showing exceptionally higher hydrogenation rates of C18:2. The membrane setup also worked as expected towards the partially hydrogenated C18:1 compound, leading to less than half the C18:0 content than the slurry reactor but was not able to match the selectivity of many other reported catalytic systems (Fig. 42). The authors reported increased isomerisation to t-C18:1 compounds, which they believed was caused by too low provision of hydrogen over the entire layer of catalyst. They argued that probably all solved hydrogen was already consumed before the liquid substrate completely passed the catalytic active parts of the membrane.209
 | ||
| Fig. 42 Fatty acid composition of the C18 fraction in the hydrogenation of sunflower oil in a membrane reactor with a Pd-modified membrane in parallel flow configuration as performed by Schomäcker.209 | ||
At least this system could be successfully applied to the partial hydrogenation of cyclooctadiene with a selectivity of more than 92% at complete conversion.208 Without the ability to increase the hydrogen concentration excessively, the lack of control for trans-compound formation forbids the application for nutrition purposes. The applicability in the production of platform chemicals is questionable as well since the general activity of the hydrogenation is not on the same level as the slurry reactor.
Rezac and her group worked on membrane reactors in the contacting crossflow configuration (Fig. 43).207 The research was focused on nutrition applications; therefore, the goal was to keep the t-C18:1 content as low as possible during the hydrogenation of soybean oil. The used catalyst was Pt supported on a polyetherimide-membrane and produced only 40% of the amount of elaidic acid that was generated in the compared slurry reactor under similar conditions. The hydrogenation was performed at 70 °C of reaction temperature and 4.5 bar hydrogen pressure. The increased selectivity towards cis-configured products came at the cost of lower overall activity compared to the slurry reactor (Fig. 44).205 This might be caused by the crossflow configuration of the reactor, which decreases the probability of oil molecules interacting with the catalyst surface for the benefit of a high hydrogen saturation to keep isomerisation low. In ongoing research, different membranes and Pt particle sizes were evaluated. All tested membranes resulted in similar low t-C18:1 content between 2% and 5% of mass fraction. It was also shown that an increase in catalyst loading on the membrane led to higher selectivity towards monounsaturated products, and an increase in particle size reduced the t-C18:1 content to the extent of an increase in stearic acid concentration.211 Switching from Pt as a catalytic metal to Pd resulted in halved reaction times, similar chemoselectivity towards C18:1, and higher isomerisation.
 | ||
| Fig. 43 Crossflow configuration for membrane reactors as used by Rezac.205 | ||
 | ||
| Fig. 44 Fatty acid composition of the C18 fraction in the partial hydrogenation of soybean oil in a cross-flow membrane reactor as performed by Rezac.205 | ||
Increasing the pressure did not change the product mixture composition since all catalytic sites were probably already saturated with H2. On the other hand, the temperature increased overall activity without changes in isomerisation. This is another indication that the mechanistic suggestion of hydrogen saturation on the catalyst surface might be particularly important in comparison to the influence of the temperature on the E/Z-equilibrium.64 Further kinetic investigations quantified the superior state of the membrane reactor operated at 2 bar of hydrogen pressure compared to a slurry reactor at 50 bar based on the overall hydrogenation activation energy Ea. It was also achieved to operate a membrane reactor with a palladium-modified catalytic membrane prepared from commercially available membranes, which makes a possible application more viable.212
4.2. Plasma-powered systems
An entirely new way of upgrading FAME or triglycerides with hydrogen is applying non-thermal parallel-plate dielectric barrier discharge (DBD) plasma (Fig. 45). The main difference is the ionization and, therefore, hydrogen activation, making it more readily available to the hydrogenation reaction. | ||
| Fig. 45 Concept of the FAME hydrogenation under the influence of DBD plasma as performed by Wongsawaeng.213 | ||
Under atmospheric conditions, the fatty substrate is hydrogenated catalyst-free, which is eco-friendly and needs no catalyst preparation step or any additional separation step like in catalyzed processes. Wongsawaeng and co-workers first described plasma-powered fat hydrogenations in the context of biodiesel production. The reaction was unselective towards the monounsaturated compound and formed t-C18:1 as well.213 Further studies optimized the reaction conditions towards higher overall reactivity and decrease of the t-C18:1 content, which shifted the purpose more towards the production of trans-fat-free margarine since the reaction remained very unselective for partial hydrogenation. In addition, cracking of the carbon chain was observed as a side reaction, leading to an increased amount of C16:0 (Fig. 46).203
 | ||
| Fig. 46 Composition of the fatty acids in the hydrogenation of palm oil as performed by Wongsawaeng.203 | ||
Finally, the reaction system was applied to refined palm oil, resulting in a production method for trans-free margarine with a similar texture to the commercial alternative.214 Despite this process being highly impressive from a technological perspective, the lack of selectivity towards C18:1 prevents an application for producing a platform chemical which has maximized C18:1 content.
DBD was used in a catalyst-containing system as well, as described by Wang in 2021.215 Soybean oil-derived methyl ester was hydrogenated over RANEY®-Nickel catalysts in a flow reactor with ionized hydrogen passed through a microwave-induced DBD plasma. Plasma led to drastically reduced reaction conditions of 25 °C and atmospheric hydrogen pressure compared to other heterogeneous Ni-based processes. The high formation of saturates and carbon chain addition side-reactions was the dominating issue in the reported research.215
5. Closing remarks
Various catalytic systems have been explored for the selective hydrogenation of FAMEs (fatty acid methyl esters) and triglycerides to produce monounsaturated analogues. Over time, the motivation for performing research on this reaction has changed substantially. From the production of margarine in the first half of the 20th century to the provision of biodiesel from different plant oils, research groups have exploited this chemistry to obtain product properties tailored to their specific application needs. This review critically surveyed the reported technologies and evaluated their potential towards the provision of platform chemicals. Particular focus was on the activity, chemo- and stereoselectivity to c-C18:1 and general applicability of the respective catalytic systems for producing a platform chemical from plant oils with maximized C18:1 content. The relatively recent restrictions in Europe and US on trans-fats for food use has necessitated a re-evaluation of historical research done on fat hydrogenation. It is important to note that for some technical (chemical industry) applications the formation of such trans-isomers may not be limiting, and this historical research therefore remains valid and potentially valuable.As revealed by this review, some aspects generally apply to most catalytic systems for selective partial hydrogenation of polyunsaturated fatty acid derivatives: increasing hydrogen pressure boosts hydrogenation activity and reduces the formation of t-C18:1 components. However, a drawback is the increased formation of saturated chains, which leads to a loss of usable substrate for subsequent reactions. Lower catalyst amounts and lower reaction temperatures improve stereoselectivity to c-C18:1, whereas raising the reaction temperature typically reduces reaction times. A sufficient hydrogen supply at the catalytic site is essential for hydrogenation to dominate over any isomerisation. This can be ensured by increasing hydrogen pressure or by limiting fatty reactant availability using optimal reactor configurations. Palladium is generally seen as having superior hydrogenation activity (though it sometimes lacks regioselectivity) when compared to platinum catalysts. Copper and cobalt catalysts require temperatures above 150 °C and are not as active and, therefore, require high catalyst loadings.
When looking at heterogeneously catalyzed systems, the choice of support, pore size, and particle size significantly influences hydrogenation activity. Some modifications are done to catalysts, such as addition of stabilizers or co-metals, to favour cis-configured monoenes. Overall, Luengnaruemitchai's group is well advanced with good results for palladium in terms of activity and chemoselectivity to C18:1. However, the complexity of catalysts is considerable and further efforts should be made on how exactly to control the partial hydrogenation better, as some results point in different research directions. Zaccheria and her group have progressed well in exploring heterogeneous copper catalysts, which show promising selectivity, but the high reaction temperatures and limited activity limit industrial application.
Nanoparticles have shown potential, with palladium nanoparticles working under ambient conditions, leading to very high chemoselectivities with acceptable activity. However, reducing metal salts to nanoparticles remains effort-intensive, and the examples shown include too-expensive solvents, given the slim profit margins. While some processes using homogeneous catalysts produce desirable product compositions with exceptionally high chemo- and regioselectivity to c-C18:1, such as the results by Schumann and Bayer achieved with immobilized Pd-complexes on PVP, others are merely comparable to heterogeneously catalyzed methods. Comparing the results achieved with Pt-TPPTS complexes by Papadogianakis et al. with the results achieved by Koritala et al. with Pt on celite or with Pt on PEI investigated by Rezac et al. it is obvious that all Pt-based systems have a tendency to oversaturate the substrates, despite the use of ligands. Overall, the used amounts of catalyst loadings exceed acceptable quantities for a product in the single dollar range per kg. Additionally, no existing current method has demonstrated efficient catalyst recycling without measurable loss of activity after more than five runs or eight runs for slurry-operated nanoparticles.
Membrane-based reactor concepts have encountered issues like mass transport limitations, affecting selectivities. However, using membranes for retaining catalysts could be a promising avenue for future exploration. Catalyst-free plasma-driven methods might emerge as alternatives, but there's a need for more research to adjust selectivities and optimize product compositions. Additionally, the energy consumption for the constant provision of plasma is so far a barrier to achieving economic feasibility.
Some systems may have more potential than was reported by the respective individuals. In our opinion, this is due in particular to the fact that not all equally important sub-aspects are given appropriate consideration in the development of the various processes. These include, for example, the selection of the catalyst metal, the setting of suitable reaction conditions, and the consideration of tailored reactor and process concepts.
Despite extensive research, none of the studied catalytic methods is suitable for producing standardized platform chemicals. The low cost of these chemicals and competition with established crude oil-based processes mean that selective partial hydrogenation needs to be very cost-efficient. Current challenges include the requirement of high temperatures and pressures or the excessive use of costly noble metals.
Even though the use of palladium seems promising, most heterogeneous methods do not meet the desired selectivity to oleic acid isomer derivatives. This could be due to suboptimal support structures. Homogeneously catalyzed methods have not provided a viable alternative, especially given the lack of an effective recycling concept and the high cost of precious metal catalysts.
Despite extensive knowledge of the various catalysts available for these substrates, none have proven adequate so far. There's a clear need for a deeper molecular understanding, which could lead to a merging of the positive attributes of the different catalytic concepts reported.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are very thankful to the German Federal Ministry of Food and Agriculture (Bundesministerium für Ernährung und Landwirtschaft), represented by the FNR (Fachagentur Nachwachsende Rohstoffe) for financial support of the young research group “Renewlysis” (project number 2219NR355).References
- A. E. Bailey, J. Am. Oil Chem. Soc., 1949, 26, 644–648 CrossRef CAS.
- A. Philippaerts, P. A. Jacobs and B. F. Sels, Angew. Chem., 2013, 125, 5328–5334 CrossRef.
- A. E. W. Beers, Lipid Technol., 2007, 19, 56–58 CrossRef CAS.
- J. W. Veldsink, M. J. Bouma, N. H. Schöön and A. A. C. M. Beenackers, Catal. Rev.: Sci. Eng., 1997, 39, 253–318 CrossRef CAS.
- Statista, Produktionswert der chemisch-pharmazeutischen Industrie in Deutschland in den Jahren 2007 bis 2021, available at: https://de.statista.com/statistik/daten/studie/242736/umfrage/produktionswert-der-chemisch-pharmazeutischen-industrie-in-deutschland/, accessed 23 December 2022.
- G. H. Vogel, Chem. Eng. Technol., 2008, 31, 730–735 CrossRef CAS.
- N. Herrmann, J. Bianga, M. Palten, T. Riemer, D. Vogt, J. M. Dreimann and T. Seidensticker, Eur. J. Lipid Sci. Technol., 2020, 122, 1900166 CrossRef CAS.
- J. Vondran, T. Benninghoff, A. Emminghaus and T. Seidensticker, Eur. J. Lipid Sci. Technol., 2022, 124, 2200041 CrossRef CAS.
- J. Vondran, J. Pela, D. Palczewski, M. Skiborowski and T. Seidensticker, ACS Sustainable Chem. Eng., 2021, 9, 11469–11478 CrossRef CAS.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021, 60, 20144–20165 CrossRef CAS PubMed.
- U. Biermann, U. T. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., 2011, 123, 3938–3956 CrossRef.
- U. Biermann, U. T. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- J. H. Phillips, in Organometallic Chemistry in Industry: A Practical Approach, ed. T. J. Colacot, J. Seechurn, C. C. Carin and R. H. Grubbs, Wiley-VCH, Weinheim, 2020, pp. 259–282 Search PubMed.
- A. Soutelo-Maria, J.-L. Dubois, J.-L. Couturier and G. Cravotto, Catalysts, 2018, 8, 464 CrossRef.
- Statista, Produktionsvolumen ausgewählter petrochemischer Primärprodukte in Deutschland im Jahr 2021, available at: https://de.statista.com/statistik/daten/studie/868074/umfrage/produktion-von-petrochemikalien-in-deutschland/, accessed 23 December 2022.
- J. O. Metzger and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2010, 112, 1–2 CrossRef CAS.
- T. F. H. Roth, A. Kühl, M. L. Spiekermann, H. W. Wegener and T. Seidensticker, ChemSusChem, 2024, e202400036 CrossRef PubMed.
- U. Biermann, W. Friedt, S. Lang, W. Lühs, G. Machmüller, J. O. Metzger, M. Rüsch Gen. Klaas, H. J. Schäfer and M. P. Schneider, Angew. Chem., Int. Ed., 2000, 39, 2206–2224 CrossRef CAS PubMed.
- P. Angeloni, M. M. Echarte, G. Pereyra Irujo, N. Izquierdo and L. Aguirrezábal, Field Crops Res., 2017, 202, 146–157 CrossRef.
- W. Normann, Chem. Umsch. Geb. Fette, Oele, Wachse Harze, 1922, 29, 277–278 CrossRef.
- D. Adu-Mensah, D. Mei, L. Zuo, Q. Zhang and J. Wang, Fuel, 2019, 251, 660–668 CrossRef CAS.
- H. P. Kaufmann, Fette, Seifen, Anstrichm., 1962, 64, 1168–1178 CrossRef CAS.
- D. Garbe, S. Reiße and T. Brück, Chem. Unserer Zeit, 2014, 48, 284–295 CrossRef CAS.
- Y. Lee, T. Tang, E. Phuah and O. Lai, Recent Advances in Edible Fats and Oils Technology, Springer Singapore, Singapore, 2022 Search PubMed.
- S. Krist, Lexikon der Pflanzlichen Fette Und Öle, Springer Wien, Vienna, 2nd edn, 2013 Search PubMed.
- Statista, Monatlicher Preis für Sonnenblumenöl im globalen Handel von November 2019 bis November 2022, available at: https://de.statista.com/statistik/daten/studie/1296737/umfrage/monatlicher-preis-sonnenblumenoel/, accessed 23 December 2022.
- Statista, Durchschnittlicher Preis der wichtigsten pflanzlichen Öle im Welthandel von November 2020 bis November 2022, available at: https://de.statista.com/statistik/daten/studie/1312207/umfrage/preis-von-pflanzlichen-oelen-im-welthandel-art-monate/, accessed 23 December 2022.
- P. Bajpai, Raw material and pulp making, Elsevier Science, Amsterdam, Oxford, Cambridge, MA, 2018, vol. 1 Search PubMed.
- P. C. Abhilash, P. Srivastava, S. Jamil and N. Singh, Environ. Sci. Pollut. Res., 2011, 18, 127–131 CrossRef CAS PubMed.
- A. Behr and T. Seidensticker, Chemistry of Renewables, Springer Berlin Heidelberg, Berlin, Heidelberg, 2020 Search PubMed.
- Multiphase homogeneous catalysis, ed. B. Cornils, W. A. Herrmann, I. T. Horváth, W. Leitner, S. Mecking, H. Olivier-Bourbigou and D. Vogt, Wiley-VCH, Weinheim, 2005 Search PubMed.
- H. Göppel and S. Kirschner, Handbuch der Pflanzenöle. Für Praxis, Wellness und Hausapotheke, Param Verlag, Ahlerstedt, 1st edn, 2014 Search PubMed.
- Y. Liu, H. Xin and Y. Yan, Ind. Crops Prod., 2009, 30, 431–436 CrossRef CAS.
- R. El-Araby, A. I. Rezk, S. A. A. El-Enin, O. A. Nofal and A. B. El-Nasharty, Bull. Natl. Res. Cent., 2020, 44, 133 CrossRef.
- S. Krist, Lexikon der pflanzlichen Fette und Öle, Springer Vienna, Vienna, 2008 Search PubMed.
- PlantFAdb, Jatropha curcas L. - Barbados nut, available at: https://plantfadb.org/plants/19098, accessed 2 August 2022.
- Handbook of plant-based biofuels, ed. A. Pandey, CRC Press, Boca Raton, FL, 2009 Search PubMed.
- A. V. S. L. S. Bharadwaj, M. Singh, N. Subramaniapillai, M. S. B. Khadhar Mohamed and A. Narayanan, Green Process. Synth., 2019, 8, 430–442 CAS.
- A. V. S. L. S. Bharadwaj, N. Subramaniapillai, M. S. B. Khadhar Mohamed and A. Narayanan, Fuel, 2021, 296, 120708 CrossRef.
- M. Abdul Hakim Shaah, M. S. Hossain, F. A. Salem Allafi, A. Alsaedi, N. Ismail, M. O. Ab Kadir and M. I. Ahmad, RSC Adv., 2021, 11, 25018–25037 RSC.
- W. Karrer, Konstitution und Vorkommen der Organischen Pflanzenstoffe (exclusive Alkaloide), Springer Basel AG, Basel, 1958, vol. 12 Search PubMed.
- A. Behr and A. J. Vorholt, in Catalysis by Metal Complexes, ed. D. J. Cole-Hamilton and P. W. N. M.van Leeuwen, 2017 Search PubMed.
- A. Behr and T. Seidensticker, Einführung in die Chemie nachwachsender Rohstoffe, Springer, Berlin, Heidelberg, 2018 Search PubMed.
- A. E. W. Beers, R. Ariaansz and D. Okonek, in Trans fatty acids, ed. A. J. Dijkstra, R. J. Hamilton, W. Hamm and R. J. Hamilton, Blackwell Pub, Oxford, Ames, Iowa, 2008, pp. 147–180 Search PubMed.
- M. Baerns, A. Behr, A. Brehm, J. Gmehling, K.-O. Hinrichsen, H. Hofmann, R. Palkovits, U. Onken and A. Renken, Technische Chemie, Wiley-VCH; Ciando, Weinheim, München, 2013 Search PubMed.
- Z. Xin, G. Wei, L. Zhang, L. Gao, Z. Li and W. Zhao, Fuel, 2021, 299, 120877 CrossRef CAS.
- A. J. Dijkstra, J. Am. Oil Chem. Soc., 2009, 87, 115–117 CrossRef.
- A. Na Rungsi, A. Luengnaruemitchai, N. Chollacoop, S.-Y. Chen, T. Mochizuki, H. Takagi and Y. Yoshimura, Appl. Catal., A, 2020, 590, 117351 CrossRef CAS.
- U. P. Laverdura, L. Rossi, F. Ferella, C. Courson, A. Zarli, R. Alhajyoussef and K. Gallucci, ACS Omega, 2020, 5, 22901–22913 CrossRef CAS PubMed.
- T. Zhu, L. Zhang, Z. Li, G. Wei, Z. Xin, D. Xiong and Z. Ou, Waste Biomass Valorization, 2021, 12, 465–474 CrossRef CAS.
- L. F. Albright, J. Am. Oil Chem. Soc., 1965, 42, 250–253 CrossRef CAS.
- D. Y. Leung, X. Wu and M. Leung, Appl. Energy, 2010, 87, 1083–1095 CrossRef CAS.
- G. Knothe, Green Chem., 2011, 13, 3048 RSC.
- T. Issariyakul and A. K. Dalai, Renewable Sustainable Energy Rev., 2014, 31, 446–471 CrossRef CAS.
- M. Suresh, C. P. Jawahar and A. Richard, Renewable Sustainable Energy Rev., 2018, 92, 38–49 CrossRef CAS.
- K. Zahan and M. Kano, Energies, 2018, 11, 2132 CrossRef.
- D. N. Thoai, C. Tongurai, K. Prasertsit and A. Kumar, Biocatal. Agric. Biotechnol., 2019, 18, 101023 CrossRef.
- A. J. Dijkstra, Eur. J. Lipid Sci. Technol., 2002, 104, 29–35 CrossRef CAS.
- D. Jovanović, Ž. Čupić, M. Stanković, L. Rožić and B. Marković, J. Mol. Catal. A: Chem., 2000, 159, 353–357 CrossRef.
- A. Bernas, J. Myllyoja, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2009, 353, 166–180 CrossRef CAS.
- S. Koritala and H. J. Dutton, J. Am. Oil Chem. Soc., 1966, 43, 86–89 CrossRef CAS.
- A. J. Dijkstra, Eur. J. Lipid Sci. Technol., 2006, 108, 249–264 CrossRef CAS.
- C. Thunyaratchatanon, A. Luengnaruemitchai, J. Jitjamnong, N. Chollacoop, S.-Y. Chen and Y. Yoshimura, Energy Fuels, 2018, 32, 9744–9755 CrossRef CAS.
- D. Singh, P. H. Pfromm and M. E. Rezac, AIChE J., 2011, 57, 2450–2457 CrossRef CAS.
- GOLD.DE, Aktueller Kupferpreis in Euro mit Rechner, available at: https://www.gold.de/kurse/kupferpreis/, accessed 25 June 2023.
- GOLD.DE, Nickelpreis für heute in Euro pro Kg, available at: https://www.gold.de/kurse/nickelpreis/, accessed 25 June 2023.
- ▷ Kobaltpreis Heute pro Kilogramm in Euro & Dollar, available at: https://www.kobaltpreis.eu/, accessed 25 June 2023.
- GOLD.DE, Rutheniumpreis Heute in Euro und Dollar, available at: https://www.gold.de/kurse/rutheniumpreis/, accessed 25 June 2023.
- GOLD.DE, Aktueller Platinpreis in Euro und Dollar, Charts, Zahlen und Fakten, available at: https://www.gold.de/kurse/platinpreis/, accessed 25 June 2023.
- GOLD.DE, Palladiumpreis aktuell in Euro und Dollar, available at: https://www.gold.de/kurse/palladiumpreis/, accessed 25 June 2023.
- GOLD.DE, Iridiumpreis Heute in Euro und Dollar, available at: https://www.gold.de/kurse/iridiumpreis/, accessed 25 June 2023.
- GOLD.DE, Rhodiumpreis heute in Euro und Dollar, available at: https://www.gold.de/kurse/rhodiumpreis/, accessed 25 June 2023.
- OSMIUM Deutschland, Aktueller OSMIUM-Preis, das seltenste und wertvollste Edelmetall der Welt, available at: https://osmium-deutschland.de/preis/, accessed 25 June 2023.
- A. Heiduschka, in Öle und Fette in der Ernährung, ed. A. Heiduschka, Springer, Berlin, Heidelberg, 1923, pp. 23–26 Search PubMed.
- W. Normann, Chem. Umsch. Geb. Fette, Oele, Wachse Harze, 1931, 38, 289–290 CrossRef CAS.
- W. Normann, Fette Seifen, 1937, 44, 330–336 CrossRef CAS.
- I. Horiuti and M. Polanyi, Trans. Faraday Soc., 1934, 30, 1164 RSC.
- M. B. Fernández, G. M. Tonetto, G. H. Crapiste and D. E. Damiani, J. Food Eng., 2007, 82, 199–208 CrossRef.
- M. Konkol, W. Wróbel, R. Bicki and A. Gołębiowski, Catalysts, 2016, 6, 55 CrossRef.
- M. Konkol, R. Bicki, M. Kondracka, K. Antoniak-Jurak, P. Wiercioch and W. Próchniak, React. Kinet., Mech. Catal., 2016, 119, 595–613 CrossRef CAS.
- C. Okkerse, A. de Jonge, J. W. E. Coenen and A. Rozendaal, J. Am. Oil Chem. Soc., 1967, 44, 152–156 CrossRef CAS.
- A. de Jonge, J. W. E. Coenen and C. Okkerse, Nature, 1965, 206, 573–574 CrossRef CAS PubMed.
- J. W. E. Coenen, J. Am. Oil Chem. Soc., 1976, 53, 382–389 CrossRef CAS.
- S. Koritala, J. Am. Oil Chem. Soc., 1968, 45, 197–200 CrossRef CAS PubMed.
- S. Koritala and H. J. Dutton, J. Am. Oil Chem. Soc., 1966, 43, 556–558 CrossRef CAS.
- S. Koritala and H. J. Dutton, J. Am. Oil Chem. Soc., 1969, 46, 245–248 CrossRef CAS.
- P. Y. Vigneron, S. Koritala, R. O. Butterfield and H. J. Dutton, J. Am. Oil Chem. Soc., 1972, 49, 371–375 CrossRef CAS.
- K. J. Moulton, R. E. Beal and E. L. Griffin, J. Am. Oil Chem. Soc., 1973, 50, 450–454 CrossRef CAS.
- S. Koritala, J. Am. Oil Chem. Soc., 1975, 52, 240–243 CrossRef CAS.
- S. Koritala, J. Am. Oil Chem. Soc., 1977, 54, 267–268 CrossRef CAS.
- G. R. List, C. D. Evans, R. E. Beal, L. T. Black, K. J. Moulton and J. C. Cowan, J. Am. Oil Chem. Soc., 1974, 51, 239–243 CrossRef CAS.
- T. L. Mounts, S. Koritala, J. P. Friedrich and H. J. Dutton, J. Am. Oil Chem. Soc., 1978, 55, 402–406 CrossRef CAS.
- S. Koritala, J. P. Friedrich and T. L. Mounts, J. Am. Oil Chem. Soc., 1980, 57, 1–5 CrossRef CAS.
- L. E. Johansson and S. T. Lundin, J. Am. Oil Chem. Soc., 1979, 56, 974–980 CrossRef CAS.
- L. E. Johansson and S. T. Lundin, J. Am. Oil Chem. Soc., 1979, 56, 981–986 CrossRef CAS.
- L. E. Johansson, J. Am. Oil Chem. Soc., 1979, 56, 987–991 CrossRef CAS.
- L. E. Johansson, J. Am. Oil Chem. Soc., 1980, 57, 16–22 CrossRef CAS.
- S. Koritala, J. Am. Oil Chem. Soc., 1981, 58, 701–702 CrossRef CAS.
- S. Koritala, J. Am. Oil Chem. Soc., 1982, 59, 309–312 CrossRef CAS.
- S. Koritala, J. Am. Oil Chem. Soc., 1985, 62, 517–520 CrossRef CAS.
- J. M. Snyder, H. J. Dutton and C. R. Scholfield, J. Am. Oil Chem. Soc., 1978, 55, 383–386 CrossRef CAS.
- S. Koritala, K. J. Moulton and E. N. Frankel, J. Am. Oil Chem. Soc., 1984, 61, 1470–1471 CrossRef CAS.
- J. M. Snyder, T. L. Mounts, C. R. Scholfield and H. J. Dutton, J. Am. Oil Chem. Soc., 1982, 59, 19–22 CrossRef CAS.
- S. Koritala, K. J. Moulton, J. P. Friedrich, E. N. Frankel and W. F. Kwolek, J. Am. Oil Chem. Soc., 1984, 61, 909–913 CrossRef CAS.
- K. J. Moulton, S. Koritala, K. Warner and E. N. Frankel, J. Am. Oil Chem. Soc., 1987, 64, 542–547 CrossRef CAS.
- K. J. Moulton and W. F. Kwolek, J. Am. Oil Chem. Soc., 1982, 59, 333–337 CrossRef CAS.
- J. A. Heldal, K. J. Moulton and E. N. Fronkel, J. Am. Oil Chem. Soc., 1989, 66, 979–982 CrossRef CAS.
- S. Koritala, J. Am. Oil Chem. Soc., 1973, 50, 11 CrossRef CAS.
- S. Koritala, J. Am. Oil Chem. Soc., 1973, 50, 317 CrossRef CAS.
- S. Koritala, J. Am. Oil Chem. Soc., 1973, 50, 307 CrossRef CAS.
- A. J. Dijkstra, Edible Oil Processing from a Patent Perspective, Springer, Boston, MA, 2013 Search PubMed.
- A. J. Dijkstra, Eur. J. Lipid Sci. Technol., 2009, 111, 857–864 CrossRef CAS.
- N. Ravasio, F. Zaccheria, M. Gargano, S. Recchia, A. Fusi, N. Poli and R. Psaro, Appl. Catal., A, 2002, 233, 1–6 CrossRef CAS.
- A. F. Trasarti, D. J. Segobia, C. R. Apesteguía, F. Santoro, F. Zaccheria and N. Ravasio, J. Am. Oil Chem. Soc., 2012, 89, 2245–2252 CrossRef CAS.
- F. Zaccheria, R. Psaro and N. Ravasio, Green Chem., 2009, 11, 462–465 RSC.
- F. Zaccheria, R. Psaro, N. Ravasio and P. Bondioli, Eur. J. Lipid Sci. Technol., 2012, 114, 24–30 CrossRef CAS.
- P. Pecchia, I. Galasso, S. Mapelli, P. Bondioli, F. Zaccheria and N. Ravasio, Ind. Crops Prod., 2013, 51, 306–309 CrossRef CAS.
- N. Hsu, L. L. Diosady, W. F. Graydon and L. J. Rubin, J. Am. Oil Chem. Soc., 1986, 63, 1036–1042 CrossRef CAS.
- A. Wright, A. Wong and L. L. Diosady, Food Res. Int., 2003, 36, 1069–1072 CrossRef CAS.
- I. L. Simakova, O. A. Simakova, A. V. Romanenko and D. Y. Murzin, Ind. Eng. Chem. Res., 2008, 47, 7219–7225 CrossRef CAS.
- R. M. Abdullina, I. N. Voropaev, A. V. Romanenko, V. A. Chumachenko, A. S. Noskov and A. S. Mashnin, Russ. J. Appl. Chem., 2012, 85, 1204–1211 CrossRef CAS.
- A. V. Romanenko, I. N. Voropaev, R. M. Abdullina and V. A. Chumachenko, Solid Fuel Chem., 2014, 48, 356–363 CrossRef CAS.
- Ö. Korkut and S. Erentürk, Biofuels, Bioprod. Biorefin., 2015, 9, 326–334 CrossRef.
- S. Erentürk and Ö. Korkut, Renewable Energy, 2018, 117, 374–379 CrossRef.
- N. Chollacoop, Thai Biodiesel Industry in ASEAN: Opportunities or Threats, 2013 Search PubMed.
- N. Numwong, A. Luengnaruemitchai, N. Chollacoop and Y. Yoshimura, J. Am. Oil Chem. Soc., 2012, 89, 2117–2120 CrossRef CAS.
- N. Numwong, A. Luengnaruemitchai, N. Chollacoop and Y. Yoshimura, Appl. Catal., A, 2012, 441–442, 72–78 CrossRef CAS.
- C. Thunyaratchatanon, A. Luengnaruemitchai, N. Chollacoop and Y. Yoshimura, Fuel, 2015, 163, 8–16 CrossRef.
- N. Numwong, A. Luengnaruemitchai, N. Chollacoop and Y. Yoshimura, Chem. Eng. J., 2012, 210, 173–181 CrossRef CAS.
- N. Numwong, N. Thachuangtumle and A. Luengnaruemitchai, International Journal on Advanced Science, Engineering and Information Technology, 2016, 4, 195 Search PubMed.
- N. Numwong, P. Prabnasak, P. Prayoonpunratn, P. Triphatthanaphong, C. Thunyaratchatanon, T. Mochizuki, S.-Y. Chen, A. Luengnaruemitchai and T. Sooknoi, Fuel Process. Technol., 2020, 203, 106393 CrossRef CAS.
- P. Udomsap, A. Eiad-Ua, S.-Y. Chen, T. Mochizuki, N. Chollacoop, Y. Yoshimura, M. Nishi, H. Tateno and H. Takagi, Int. J. Mol. Sci., 2021, 22, 1256 CrossRef CAS PubMed.
- P. Hongmanorom, A. Luengnaruemitchai, N. Chollacoop and Y. Yoshimura, Energy Fuels, 2017, 31, 8202–8209 CrossRef CAS.
- S.-Y. Chen, L. Attanatho, T. Mochizuki, Y. Abe, M. Toba, Y. Yoshimura, C. Kumpidet, P. Somwonhsa and S. Lao-ubol, C. R. Chim., 2016, 19, 1166–1173 CrossRef CAS.
- S.-Y. Chen, A. Chang, A. N. Rungsi, L. Attanatho, C.-L. Chang, J.-H. Pan, A. Suemanotham, T. Mochizuki, H. Takagi, C.-M. Yang, A. Luengnaruemitchai and H.-H. Chou, Appl. Catal., A, 2020, 602, 117707 CrossRef CAS.
- A. Na Rungsi, A. Luengnaruemitchai, S. Wongkasemjit, N. Chollacoop, S.-Y. Chen and Y. Yoshimura, Appl. Catal., A, 2018, 563, 80–90 CrossRef CAS.
- T. Mochizuki, Y. Abe, S.-Y. Chen, M. Toba and Y. Yoshimura, ChemCatChem, 2017, 9, 2633–2637 CrossRef CAS.
- K. Belkacemi, A. Boulmerka, S. Hamoudi and J. Arul, Int. J. Chem. React. Eng., 2006 DOI:10.2202/1542-6580.1245.
- K. Belkacemi and S. Hamoudi, Ind. Eng. Chem. Res., 2008, 48, 1081–1089 CrossRef.
- K. Belkacemi, A. Boulmerka, J. Arul and S. Hamoudi, Top. Catal., 2006, 37, 113–120 CrossRef CAS.
- M. Plourde, K. Belkacemi and J. Arul, Ind. Eng. Chem. Res., 2004, 43, 2382–2390 CrossRef CAS.
- K. Belkacemi, N. Kemache, S. Hamoudi and J. Arul, Int. J. Chem. React. Eng., 2007, 5 Search PubMed.
- N. Kemache, S. Hamoudi, J. Arul and K. Belkacemi, Ind. Eng. Chem. Res., 2010, 49, 971–979 CrossRef CAS.
- M. B. Fernández, C. M. Piqueras, G. M. Tonetto, G. H. Crapiste and D. E. Damiani, J. Mol. Catal. A: Chem., 2005, 233, 133–139 CrossRef.
- C. M. Piqueras, M. B. Fernández, G. M. Tonetto, S. Bottini and D. E. Damiani, Catal. Commun., 2006, 7, 344–347 CrossRef CAS.
- G. M. Tonetto, G. Crapiste and D. E. Damiani, Int. J. Chem. React. Eng., 2007 DOI:10.2202/1542-6580.1380.
- M. B. Fernández, J. F. M. Sánchez, G. M. Tonetto and D. E. Damiani, Chem. Eng. J., 2009, 155, 941–949 CrossRef.
- G. M. Tonetto, J. F. M. Sánchez, M. L. Ferreira and D. E. Damiani, J. Mol. Catal. A: Chem., 2009, 299, 88–92 CrossRef CAS.
- C. M. Piqueras, G. Tonetto, S. Bottini and D. E. Damiani, Catal. Today, 2008, 133–135, 836–841 CrossRef CAS.
- A. M. Alshaibani, Z. Yaakob, A. M. Alsobaai and M. M. Sahri, Rasayan J. Chem., 2012, 463–467 CAS.
- A. M. Alshaibani and Z. Yaakob, Int. J. Adv. Sci. Eng. Inf. Technol., 2013, 3, 164 CrossRef.
- A. M. Alshaibani and Z. Yaakob, Asian J. Chem., 2015, 27, 630–634 CrossRef CAS.
- A. M. Alshaibani, Z. Yaakob and A. Ghaleb, Orient. J. Chem., 2014, 30 Search PubMed.
- N. Numwong, A. Luengnaruemitchai, N. Chollacoop and Y. Yoshimura, J. Am. Oil Chem. Soc., 2013, 90, 1431–1438 CrossRef CAS.
- C. Thunyaratchatanon, J. Jitjamnong, A. Luengnaruemitchai, N. Numwong, N. Chollacoop and Y. Yoshimura, Appl. Catal., A, 2016, 520, 170–177 CrossRef CAS.
- A. Na Rungsi, T. H. Truong, C. Thunyaratchatanon, A. Luengnaruemitchai, N. Chollacoop, S.-Y. Chen, T. Mochizuki, H. Takagi and Y. Yoshimura, Fuel, 2021, 298, 120658 CrossRef CAS.
- J. F. M. Sánchez, O. J. González Bello, M. Montes, G. M. Tonetto and D. E. Damiani, Catal. Commun., 2009, 10, 1446–1449 CrossRef.
- J. F. M. Sánchez, D. E. Boldrini, G. M. Tonetto and D. E. Damiani, Chem. Eng. J., 2011, 167, 355–361 CrossRef.
- D. E. Boldrini, J. F. M. Sánchez, G. M. Tonetto and D. E. Damiani, Ind. Eng. Chem. Res., 2012, 51, 12222–12232 CAS.
- D. E. Boldrini, D. E. Damiani and G. M. Tonetto, AIChE J., 2014, 60, 3524–3533 CrossRef CAS.
- D. E. Boldrini, G. M. Tonetto and D. E. Damiani, Chem. Eng. J., 2015, 270, 378–384 CrossRef CAS.
- D. E. Boldrini, Chem. Eng. Process., 2018, 132, 229–240 CrossRef CAS.
- D. E. Boldrini and G. M. Tonetto, Chem. Eng. Process., 2020, 149, 107895 CrossRef.
- F. D. Troncoso and G. M. Tonetto, Chem. Eng. Process., 2022, 170, 108669 CrossRef CAS.
- G. Wei, Z. Liu, L. Zhang and Z. Li, Energy Convers. Manage., 2018, 163, 208–218 CrossRef CAS.
- L. Zhang, Z. Xin, Z. Liu, Y. Ou, Z. Ye, Z. Li and G. Wei, Fuel, 2020, 270, 117510 CrossRef CAS.
- L. Zhang, Z. Xin, Z. Liu, G. Wei, Z. Li and Y. Ou, Renewable Energy, 2020, 147, 695–704 CrossRef CAS.
- A. Behr, Angewandte homogene Katalyse, Wiley-VCH, Weinheim, 2008 Search PubMed.
- E. A. Emken, E. N. Frankel and R. O. Butterfield, J. Am. Oil Chem. Soc., 1966, 43, 14–18 CrossRef CAS.
- E. N. Frankel, E. Emken, H. Itatani and J. C. Bailar, J. Org. Chem., 1967, 32, 1447–1452 CrossRef.
- E. N. Frankel and F. L. Little, J. Am. Oil Chem. Soc., 1969, 46, 256–261 CrossRef CAS.
- E. N. Frankel, J. Am. Oil Chem. Soc., 1970, 47, 11–14 CrossRef CAS.
- R. A. Awl, E. N. Frankel, J. P. Friedrich and E. H. Pryde, J. Am. Oil Chem. Soc., 1978, 55, 577–582 CrossRef CAS.
- E. N. Frankel, J. P. Friedrich, T. R. Bessler, W. F. Kwolek and N. L. Holy, J. Am. Oil Chem. Soc., 1980, 57, 349–354 CrossRef CAS.
- J. C. Bailar and H. Itatani, J. Am. Oil Chem. Soc., 1967, 89, 1592–1599 CAS.
- H. Itatani and J. C. Bailar, J. Am. Oil Chem. Soc., 1967, 44, 147–151 CrossRef CAS.
- J. C. Bailar and H. Itatani, J. Am. Oil Chem. Soc., 1966, 43, 337–341 CrossRef CAS.
- E. N. Frankel, H. Itatani and J. C. Bailar, J. Am. Oil Chem. Soc., 1972, 49, 132–133 CrossRef CAS.
- H. Itatani and J. C. Bailar, J. Am. Oil Chem. Soc., 1967, 89, 1600–1602 CAS.
- C. Bello, L. L. Diosady, W. F. Graydon and L. J. Rubin, J. Am. Oil Chem. Soc., 1985, 62, 1587–1592 CrossRef CAS.
- A. J. Wright, A. L. Mihele and L. L. Diosady, Food Res. Int., 2003, 36, 797–804 CrossRef CAS.
- J. R. Tucker and D. P. Riley, J. Organomet. Chem., 1985, 279, 49–62 CrossRef CAS.
- L. J. Rubin, S. S. Köseoglu, L. L. Diosady and W. F. Graydon, J. Am. Oil Chem. Soc., 1986, 63, 1551–1557 CrossRef CAS.
- A. Bouriazos, K. Mouratidis, N. Psaroudakis and G. Papadogianakis, Catal. Lett., 2007, 121, 158–164 CrossRef.
- G. Papadogianakis, A. Bouriazos, K. Mouratidis and N. Psaroudakis, EP1918358B1, 2006.
- N. Nikolaou, C. E. Papadopoulos, A. Lazaridou, A. Koutsoumba, A. Bouriazos and G. Papadogianakis, Catal. Commun., 2009, 10, 451–455 CrossRef CAS.
- A. Bouriazos, S. Sotiriou, C. Vangelis and G. Papadogianakis, J. Organomet. Chem., 2010, 695, 327–337 CrossRef CAS.
- A. Bouriazos, S. Sotiriou, P. Stathis and G. Papadogianakis, Appl. Catal., B, 2014, 150–151, 345–353 CrossRef CAS.
- P. Stathis, D. Stavroulaki, N. Kaika, K. Krommyda and G. Papadogianakis, Appl. Catal., B, 2017, 209, 579–590 CrossRef CAS.
- E. Bayer and W. Schumann, J. Chem. Soc., Chem. Commun., 1986, 949–952 RSC.
- V. Pandarus, G. Gingras, F. Béland, R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2012, 16, 1307–1311 CrossRef CAS.
- H.-U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, ChemInform, 2003, 34 Search PubMed.
- A. Lux, PhD Thesis, TU Dortmund University, 2012 Search PubMed.
- A. Behr and A. Lux, Chem. Ing. Tech., 2014, 86, 1933–1940 CrossRef CAS.
- A. Behr, N. Döring, S. Durowicz-Heil, B. Ellenberg, C. Kozik, C. Lohr and H. Schmidke, Fat Sci. Technol., 1993, 95, 2–12 CAS.
- A. Behr and H. Schmidke, Chem. Ing. Tech., 1993, 65, 568–569 CrossRef CAS.
- A. Behr, N. Döring, C. Kozik, H. Schmidtke and S. Durowicz, Deutschland Pat., DE4012873A1, 1990 Search PubMed.
- A. Behr, C. Lohr and B. Ellenberg, Deutschland Pat., DE4109246A1, 1991 Search PubMed.
- A. Behr and H. Schmidtke, Deutschland Pat., DE4211126A1, 1992 Search PubMed.
- W. Liu, L. Xu, G. Lu and H. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 1368–1375 CrossRef CAS.
- W. Liu and G. Lu, J. Oleo Sci., 2017, 66, 1161–1168 CrossRef CAS PubMed.
- B. Léger, A. Ponchel, F. Hapiot and E. Monflier, Chem. Eng. Trans., 2014, 37, 337–342 Search PubMed.
- W. Wongjaikham, G. Kongprawes, D. Wongsawaeng, K. Ngaosuwan, W. Kiatkittipong, P. Hosemann and S. Assabumrungrat, Fuel, 2021, 305, 121524 CrossRef CAS.
- A. Schmidt, R. Haidar and R. Schomäcker, Catal. Today, 2005, 104, 305–312 CrossRef CAS.
- D. Singh, M. E. Rezac and P. H. Pfromm, J. Am. Oil Chem. Soc., 2008, 86, 93–101 CrossRef.
- D. Fritsch and G. Bengtson, Catal. Today, 2006, 118, 121–127 CrossRef CAS.
- D. Singh, B. Dringenberg, M. E. Rezac and P. H. Pfromm, in Advances in Food Engineering, ed. R. Paul Singh, CRC Press, 2005 Search PubMed.
- A. Schmidt, A. Wolf, R. Warsitz, R. Dittmeyer, D. Urbanczyk, I. Voigt, G. Fischer and R. Schomäcker, AIChE J., 2008, 54, 258–268 CrossRef CAS.
- A. Schmidt and R. Schomäcker, J. Mol. Catal. A: Chem., 2007, 271, 192–199 CrossRef CAS.
- J. W. Veldsink, J. Am. Oil Chem. Soc., 2001, 78, 443–446 CrossRef CAS.
- D. Singh, M. E. Rezac and P. H. Pfromm, J. Membr. Sci., 2010, 348, 99–108 CrossRef CAS.
- M. D. Wales, L. B. Joos, W. A. Traylor, P. Pfromm and M. Rezac, Catal. Today, 2016, 268, 12–18 CrossRef CAS.
- G. Kongprawes, D. Wongsawaeng, P. Hosemann, K. Ngaosuwan, W. Kiatkittipong and S. Assabumrungrat, Int. J. Energy Res., 2021, 45, 4519–4533 CrossRef CAS.
- K. Puprasit, D. Wongsawaeng, K. Ngaosuwan, W. Kiatkittipong and S. Assabumrungrat, J. Food Eng., 2022, 334, 111167 CrossRef CAS.
- W. Zhao, C. Hua, X. Zhang, X. Qi, K. Tanongkiat and J. Wang, Plasma Sci. Technol., 2021, 23, 95506 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |