
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot synthesis of iron-doped ceria catalysts for tandem carbon dioxide hydrogenation†
Albert
Gili
 *ab,
Maged F.
Bekheet
c,
Franziska
Thimm
d,
Benjamin
Bischoff
c,
Michael
Geske
d,
Martin
Konrad
d,
Sebastian
Praetz
e,
Christopher
Schlesiger
e,
Sören
Selve
f,
Aleksander
Gurlo
c,
Frank
Rosowski
dg and
Reinhard
Schomäcker
a
*ab,
Maged F.
Bekheet
c,
Franziska
Thimm
d,
Benjamin
Bischoff
c,
Michael
Geske
d,
Martin
Konrad
d,
Sebastian
Praetz
e,
Christopher
Schlesiger
e,
Sören
Selve
f,
Aleksander
Gurlo
c,
Frank
Rosowski
dg and
Reinhard
Schomäcker
a
aTechnische Universität Berlin, Faculty II Mathematik und Naturwissenschaften, Institut für Chemie, Straße des 17. Juni 135, 10623 Berlin, Germany
bHelmholtz-Zentrum Berlin für Materialien und Energie, 14109, Berlin, Germany. E-mail: albert.gili@helmholtz-berlin.de
cTechnische Universität Berlin, Faculty III Process Sciences, Institute of Materials Science and Technology, Chair of Advanced Ceramic Materials, Straße des 17. Juni 135, 10623 Berlin, Germany
dBasCat-UniCat BASF JointLab, D-10623Berlin, Germany
eTechnische Universität Berlin, Faculty II Mathematics and Natural Science, Institute for Optic and Atomic Physics, Straße des 17. Juni 135, 10623 Berlin, Germany
fCenter for Electron Microscopy (ZELMI), Technische Universität Berlin, Straße des 17. Juni 135, 10623 Berlin, Germany
gBASF SE, Catalysis Research, Ludwigshafen, Germany
First published on 25th June 2024
Abstract
We report on the one-pot synthesis of inexpensive and abundant CeO2 and 1.5, 4.5, and 9 mol% Fe-doped ceria (Ce1−xFexO2−δ) systems and their catalytic activity for tandem CO2 hydrogenation. XAFS and XRD demonstrate that oxygen vacancies are generated via two mechanisms: firstly, by the substitution of Ce4+ by Fe3+ in the lattice and the subsequent loss of oxygen anions. Secondly, by the partial reduction of Ce4+ to Ce3+, which is enhanced by the presence of Fe. All the samples tested show high activity for CO2 hydrogenation and the production of CO, CH4, and light (C2–C4) alkanes and alkenes, with the 9 mol% Fe-doped CeO2 showing the best performance in terms of CO2 reaction rate and product selectivity. During reaction, Fe exsolves/seggregates from the ceria, resulting in particles decorating the surface of the catalyst and increasing the reaction rates of CO2. This system is composed of two functionalities, the oxygen vacancy and the Fe, whose close vicinity results in a high selectivity toward CO and CH4 detrimental to the more valuable hydrocarbons. A rather complex interplay between the two functionalities, their interface, and the particle size of the catalysts exists for this tandem reaction network on this catalytic system and deserves further studies.
Introduction
Ceria (CeO2) is an abundant, inexpensive, easy to process, and non-toxic material that finds application in solid-oxide fuel cell technology,1 membrane technology,2 and in three-way catalysts.3 Ceria has native oxygen vacancies originated by the partial reduction of two moles of Ce4+ into one mole of Ce3+, and the loss of one mole of O2− anions to maintain charge neutrality. The concentration of vacancies strongly depends on the temperature and the gas atmosphere:4 moreover, it is enhanced by the presence of metallic nanoparticles on the surface of ceria.5 The resulting vacancies can activate CO2 (ref. 2 and 6) or disproportionate H2O2.7 The fluorite-type crystal structure of ceria can form substitutional solid solutions with a wide range of compounds and to high concentrations. Additional oxygen vacancies are generated upon substitution of Ce4+ by trivalent cations such as Sm3+, Gd3+, Fe3+, In3+, etc. Since these cations have different ionic radii compared to Ce4+, their doping in the CeO2 lattice results in a change in its unit cell volume. Ceria is the paradigm of non-innocent support: the metal–ceria interface is considered to have a prominent role in several chemical reactions like the water–gas shift (WGS), the partial oxidation of methane (POM), and fuel cells.3 Indeed, CO oxidation happens at the interface between supported metals and CeO2, not on the metals themselves,8 with some of the Ce3+ being simple expectator.5 Ceria displays strong metal support interactions (SMSI) with several metals relevant for CO2 activation, like Pt or Ni, enhancing their stability.Doping of Fe into CeO2 (FDC, Ce1−xFexO2−δ) has been done with different goals: as a photocatalyst, 1.5 mol% FDC performed the best for methyl-orange visible light degradation.9 Solid state studies of the FDC system were reported, and the structural, optical, magnetic, and electronic properties upon doping were studied.10 Fe- and Sm-doped ceria (FSDC) has also been applied as electrocatalyst for CO2 reduction in SOFC.11 Fe is used as an additive12 in ceria-based systems to scavenge impurities and enhance the oxygen ion conductivity,2 finding application in SOFC13 and dual-phase membranes technologies.2 As heterogeneous catalyst, Fe/CeO2 has been applied for catalytic diesel soot combustion,14 Fenton heterogeneous oxidation of rhodamine B,15 and carbon dioxide hydrogenation.16
Tandem CO2 hydrogenation to hydrocarbons (HCs) has recently attracted interest within the heterogeneous catalysis scientific community as a tool to mitigate anthropogenic greenhouse gas emissions.17 The initial hydrogenation reaction is coupled with a second functionality in a single reaction vessel to further react the intermediates of the first reaction to the final desired products. Three main routes have been suggested: (i) a modified Fischer–Tropsch synthesis (m-FTS), (ii) the methanol-mediated route, and (iii) the CO-mediated route,18 in which the current work focuses. The advantages of a tandem system compared to single reactions are several, including the increase of the reaction rates due to Le Chatelier's principle, the achievement of unprecedented selectivity, avoiding the handling of dangerous intermediates of reaction (like CO), the decrease of equipment and operation cost, etc. For CO2 tandem hydrogenation, the most desired products are C2–C4 paraffins and olefins,18–20 oxygenated compounds like dimethyl ether (DME),17 and gasoline (C5–C11).21 Current trends in tandem catalysis focus on the determination of the structure–activity correlations, elucidating reaction mechanisms and the detection of transient intermediates of reaction, system performance optimization, the mitigation of deactivation phenomena by catalyst interaction,22 and developing new and low-cost methods of catalyst synthesis.18
The CO-mediated route relies upon the initial reduction of CO2 to CO by means of the reverse water–gas shift (RWGS) reaction (reaction (1)). This endothermic reaction is favored at high temperatures, which are necessary to achieve significant CO2 reaction rates. The intermediate CO further hydrogenates via the FTS (reaction (2)) to produce hydrocarbons. This highly exothermic reaction is favored at lower temperatures, imposing a discrepancy regarding which operating conditions window is suitable for the integration. Furthermore, other reactions like CO2 methanation (reaction (4)) and methanol synthesis (reaction (5)) occur to some extent under these conditions.
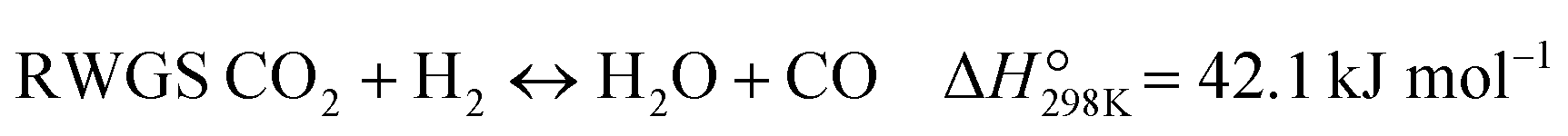 | (R1) |
 | (R2) |
 | (R3) |
 | (R4) |
 | (R5) |
In this work, a one-pot synthesis of CeO2 and FDC catalysts with different Fe content has been done and applied as catalysts for CO2 tandem hydrogenation via the CO-route for the first time. Solid solutions of Ce1−xFexO2−δ have been prepared using the citrate method and characterized with XRD, XAFS, and STEM-EDX, which reveal differences in the oxygen vacancy concentration and ex-solution of Fe during reaction, allowing to stablish the structure–activity correlations.
Experimental methods
Catalyst preparation
Ceria (CeO2) and Fe-doped ceria (FDC, Ce1−xFexO2−δ) were prepared using the liquid citrate method.2,31 Depending on the desired composition, the appropriate amounts (0.02 mol basis) of Ce(NO3)3·6H2O (Alfa Aesar, 99.5%, Haverhill, MA, USA) and Fe(NO3)3·9H2O (Riedel de Haën, 99.95%, Seelze, Germany) were dissolved in 300 mL of deionized water together with 7.68 g of citric acid (C6H8O7, Roth, 99.5% anhydrous, Karlsruhe, Germany). The solution was stirred and heated to the boiling point for 4 h to induce polymerization and prevent evaporation. After, the solution was uncovered and allowed to evaporate for 3–4 h until a gel was formed. This solution was dried at 110 °C for 24 h. The resulting gel was self-ignited in a furnace at 400 °C under flowing air to ensure total oxidation. The resulting powder was calcined at 600 °C for 12 h with heating and cooling rates of 2 °C min−1. Subsequently, the powder was sieved, and the desired fraction of 100–200 μm was used for catalytic tests. For simplicity, the different catalysts described will be referred to as CeO2 or XFDC, being X = 1.5, 4.5, or 9 mol weight%.Catalytic testing
The catalysts were tested simultaneously in a 4-fold parallel setup manufactured by ILS-Integrated Lab Solutions (GmbH, Germany), with each reactor tube being a packed-bed plug-flow unit. Around 50 mg of sample, sieved to the 100–200 μm fraction, was weighted and diluted with 0.95 mL (100 mg) of Davisil gel (Steatite, CeramTec Steatite Inert Material 400–700 μm). Davisil was also packed before and after the catalyst + davisil bed. A gas dosing unit with mass flow controllers (MFCs, Bronkhorst) controls the gas flow rate and composition. To decompose possible present volatile metal carbonyls like Fe carbonyls, a carbonyl trap is built in front of the CO MFC. This carbonyl trap consists of a U-shaped tubing filled with Al2O3 which is heated to a temperature of 180 °C. Each reactor consists of a reactor tube made of 316 L with an inner diameter of 9.4 mm and a length of 645 mm, a reactor head with inlet connections for the feed, and a hull for the thermocouple with an outer diameter of 3.0 mm. Hence, the effective diameter is 6.4 mm. The temperature is measured at three different points along the reactor length. The reactors have an isothermal zone with a length of 12.5 cm, whereupon the catalyst bed is positioned so that it ends at the end of the isothermal zone. The bypass consists of a 1/8′′ tube. The pressure inside each reactor tube is controlled individually using an Equilibar (USA) and a Bronkhorst EL-Press back pressure controller. After the reactor and to prevent condensation, the effluent gases are diluted with low-pressure N2. Through a multi-position valve and heated tubing (180 °C), the diluted effluent gas is led to the gas chromatograph (Agilent 7890B). For the separation and quantification of H2, Ar, and N2 an HP-Plot 5A column (30 m × 0.53 mm × 50 μm) with a thermal conductivity detector (TCD) is used. In front of the HP-Polt 5A column positioned in the GC oven is a Hayesep Q pre-column. To separate and quantify the carbon-containing gasses, an HP-Plot Q column (30 m × 0.53 m × 40 μm) from Agilent with a Polyarc® reactor and a flame ionization detector (FID) is used. For the separation of waxes and oils in front of the HP-Plot Q column, a few centimeters of a Hayesep Q backflush are positioned in the valve box which is heated to 200 °C. He is used as the GC carrier gas, while Ar is used as the internal standard. A hot sampling unit with a coalescent filter sits behind the connection to the GC followed by a cold sampling unit where the effluent gas is directed through water to remove condensable components. GC allows for determination of carbon dioxide, carbon monoxide, hydrogen, nitrogen, argon, methane, ethane, propane, n-butane, n-pentane, n-hexane, n-heptane, ethylene, propylene, n-butane, methanol, ethanol, n-propanol, n-butanol, dimethyl ether, acetaldehyde, propanal, butanal, formic acid, acetic acid, methyl formate, methyl acetate, and ethyl acetate. An unidentified peak in the GC analysis is shown in Fig. S5.† Due to the N2 dilution, the real mole fractions (xi,corr) must be corrected by multiplying the measured fractions in the GC (xi,GC) by the Ar fraction (xAr,bypass) divided by the Ar fraction in the reactor (xAr,reactor) according to eqn (6). The product-based conversion of CO2 (XCO2) was determined according to eqn (7), with (CNi) being the carbon number and (XCO2,0) the mole fraction of CO2 in the inlet gas. Product selectivity is calculated according to eqn (8). Yields were calculated according to eqn (9). The reaction conditions are described in the Table S1.† | (6) |
 | (7) |
 | (8) |
 | (9) |
Thermodynamic calculations
The change in the Gibbs free energy for the chemical reactions was calculated using eqn (10) (Gibbs–Helmholtz equation), eqn (11), and eqn (12), and using different sources for the enthalpies and entropies of formation of the different compounds.32,33 | (10) |
 | (11) |
 | (12) |
Catalyst characterization
Element composition was measured by inductively coupled plasma optical emission spectroscopy (ICP-OES) with a Horiba Scientific ICP Ultima2 (Horiba, Kyoto, Japan). The samples were digested in an aqueous solution of concentrated nitric acid at 200 °C for 5 h in a Teflon-lined autoclave before the analysis. The Series 744 Elemental Analyzer (LECO, USA) is used to test the mass fractions of sulfur based on the non-dispersive infrared (NDIR) principle of scanning the SO2 released during the heating of the sample to obtain the mass fraction. 10 mg are tested three times and the result is the average of the weight percentages. The specific surface area (SSA) of the samples was measured by nitrogen sorption analysis in a QuadraSorb Station 4 apparatus (Quantachrome, USA). Isotherms were recorded at 77 K after degassing for 12 hours at 200 °C under vacuum. The surface area was calculated using Brunauer–Emmett–Teller (BET) calculations. The Quantachrome/QuadraWin software version 5.05 was used to analyze all nitrogen sorption data.The crystal structure of the catalysts was determined using powder X-ray diffraction (pXRD) in the P02.1 beamline of the DESY (Deutsche Synchrotron) in Hamburg, Germany. Small amounts of the fresh and spent catalysts were introduced in the tip of quartz capillaries (Hilgenberg GmbH, Germany) and sealed. The sample was mounted in the beamline and rotated to improve statistics. The sample-to-detector distance (SDD) was determined using the CeO2 sample, and a LaB6 660b standard from NIST was used to quantify the instrument's peak broadening. The incident beam energy was set to 60 keV (λ = 0.2073 Å), and the acquisition was performed with varying times (60–180 s) on a Varex 4343CT area detector. The 2D resulting images were azimuthally integrated using Dioptas.34 Rietveld refinement (RR) was performed to obtain values of lattice parameters and crystallite size using the FULLPROF program35 with profile function 7 (Thompson-Cox-Hastings pseudo-Voigt convoluted with an axial divergence asymmetry function).36 The resolution function of the instrument was obtained from the structure refinement of a LaB6 standard. The theoretical lattice parameter of Ce1−xFexO2−δ is calculated using eqn (13), assuming that all the Fe3+ is incorporated into the ceria lattice. This equation is derived from,37 with ionic radii of oxygen (rO = 1.38 Å), oxygen vacancy (rVo = 1.164 Å), Fe cation (rFe3+ = 1.78 Å) and Ce cations (rCe3+ = 1.143 Å and rCe4+ = 0.97 Å) obtained from references.37–39
 | (13) |
Results and discussion
Fig. 1 includes the full catalytic experiment showing the XCO2 (panel B) and the ratio of CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO (panel A) of all samples as a function of the TOS. Detailed experimental conditions for each data point are shown in Table S1 and Fig. S1.† During the initial activation step of approximately 50 h under reaction conditions, the XCO2 of all samples increases. This is most probably caused by (i) the generation of oxygen vacancies in the (both doped and un-doped) CeO2 under reductive atmosphere3,4,43 and/or by (ii) the exsolution/segregation of the atomic Fe from the bulk of the FDC samples (which most probably reduces on the CeO2 surface to produce metallic Fe or a FexC carbide) which can catalyze both CO2 methanation (reaction (4)) and the FTS (reaction (2)). The increase in the product CH4:CO ratio, as shown in the top panel of Fig. 1, suggests the occurrence of the second hypothesis. An increase in the Fe-particle size can also contribute to higher SCH4 and lower SCO.30 The increase in the CH4:CO ratio is more notorious in the Fe-containing samples compared to the pure CeO2. The oxygen vacancies in CeO2 are speculated to be the active site for CO2 reduction to CO over this catalyst viareaction (1).44 The addition of Fe to the CeO2 results in an increase in the XCO2, being the 9FDC the most active over the full experiment, followed closely by the 1.5 and 4.5FDC. Interestingly, the 1.5FDC sample shows higher XCO2 after 130 h compared to the 4.5FDC (Fig. 1B). Moreover, the 9FDC sample displays a higher ratio of CH4:CO over the full experiment, followed by the 1.5FDC. If segregated, more Fe in the surface of the catalyst is expected to (i) produce CH4viareaction (4) and (ii) consume CO viareaction (2) and (3), resulting in the increase of the CH4:CO ratio. The size of the Fe-particles does play a role in both reactions,30,45,46 and sintering of the segregated particles at this temperature is expected.
:CO (panel A) of all samples as a function of the TOS. Detailed experimental conditions for each data point are shown in Table S1 and Fig. S1.† During the initial activation step of approximately 50 h under reaction conditions, the XCO2 of all samples increases. This is most probably caused by (i) the generation of oxygen vacancies in the (both doped and un-doped) CeO2 under reductive atmosphere3,4,43 and/or by (ii) the exsolution/segregation of the atomic Fe from the bulk of the FDC samples (which most probably reduces on the CeO2 surface to produce metallic Fe or a FexC carbide) which can catalyze both CO2 methanation (reaction (4)) and the FTS (reaction (2)). The increase in the product CH4:CO ratio, as shown in the top panel of Fig. 1, suggests the occurrence of the second hypothesis. An increase in the Fe-particle size can also contribute to higher SCH4 and lower SCO.30 The increase in the CH4:CO ratio is more notorious in the Fe-containing samples compared to the pure CeO2. The oxygen vacancies in CeO2 are speculated to be the active site for CO2 reduction to CO over this catalyst viareaction (1).44 The addition of Fe to the CeO2 results in an increase in the XCO2, being the 9FDC the most active over the full experiment, followed closely by the 1.5 and 4.5FDC. Interestingly, the 1.5FDC sample shows higher XCO2 after 130 h compared to the 4.5FDC (Fig. 1B). Moreover, the 9FDC sample displays a higher ratio of CH4:CO over the full experiment, followed by the 1.5FDC. If segregated, more Fe in the surface of the catalyst is expected to (i) produce CH4viareaction (4) and (ii) consume CO viareaction (2) and (3), resulting in the increase of the CH4:CO ratio. The size of the Fe-particles does play a role in both reactions,30,45,46 and sintering of the segregated particles at this temperature is expected.
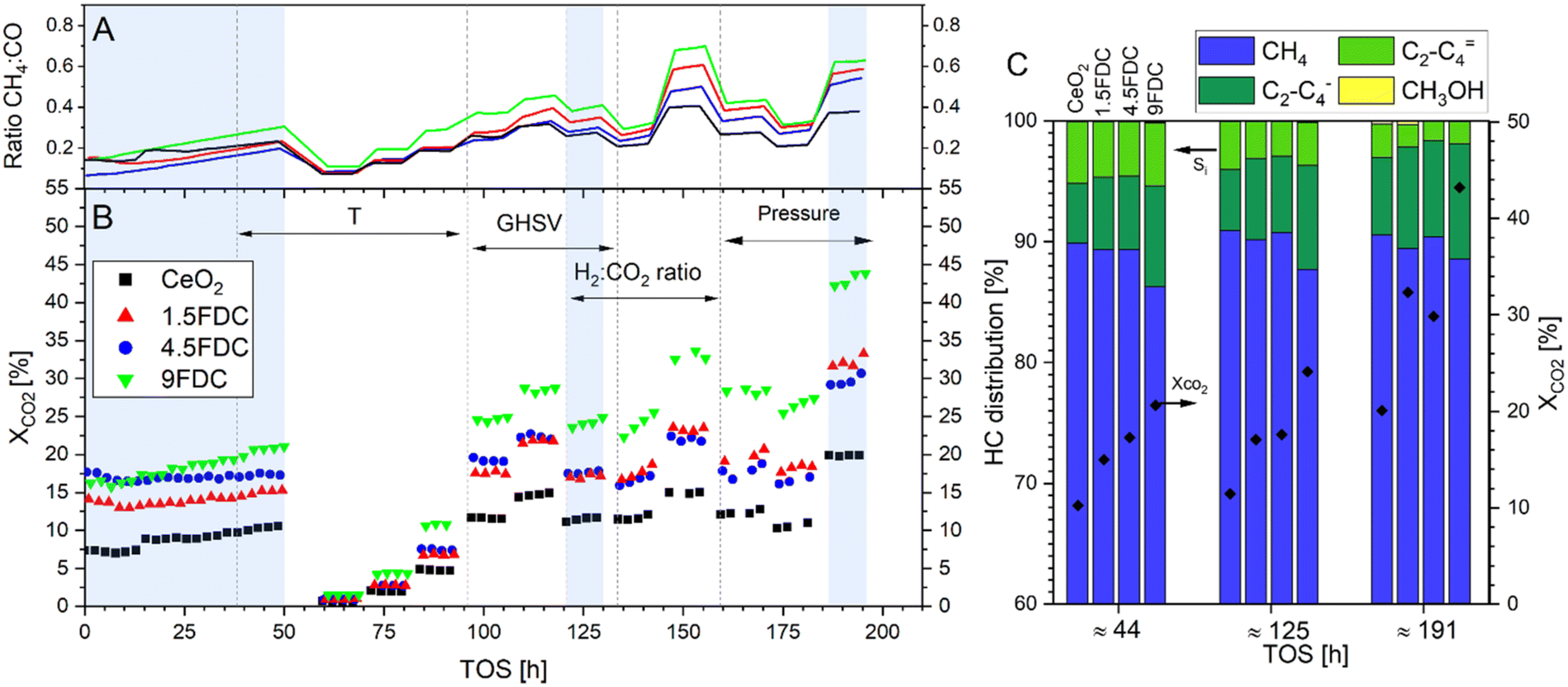 | ||
| Fig. 1 Full experiment displaying the XCO2 (bottom-panel B) and CH4:CO ratio (top panel-A) with TOS for all the samples. Arrows within the bottom panel display the variation periods for specific parameters, while the regions with blue backgrounds are obtained at the same operating conditions (500 °C, H2:CO2:Ar = 3:1:1, 20 bar and 50000 mL h−1 gcat−1). The detailed experimental conditions of each data point can be found in the ESI.† Panel C displays the selectivity toward HCs for all the samples in the blue regions in panel B (same operating conditions as panel B). | ||
Fig. 1C and S2† display the change in XCO2 test over the 200 h period, resembling a stability test. The XCO2 of all the samples was still increasing at the end of the experiment, reaching as much as 45% for the 9FDC (rCO2 = 157.3 mmol h−1 gcat−1). The desirable increase in XCO2 with TOS occurs while HC selectivity remains fairly constant, with the overall selectivity toward CO decreasing with TOS for all the samples tested. Fig. S3† shows the paraffin and olefin selectivity over the full experiment: while the SC2–C4 = slightly decreases over TOS for all samples, the SC2–C4 – notoriously increases. This observation might be a result of the change in the particle size of the Fe-containing segregated particles.30
The XCO2 and Si trends must be evaluated with care as the catalysts continue to activate during the full experiment. Nevertheless, the variation of the operating conditions blocks, i.e. temperature variation, pressure variation, etc., occurred subsequently, minimizing the masking effect that the structural changes of the catalysts might have on the performance. Each data point of HC distribution and XCO2 in Fig. 1C and 2 is an average of 4–5 data points obtained over 2 h at the same operating conditions. Fig. 2A shows the effect of temperature on the XCO2 and the HC distribution for all the catalysts. As expected, an increase in the temperature results in an increase in the XCO2. At lower temperature, the yield of methanol is enhanced compared to light HCs following the thermodynamics of the reaction (Fig. S4†). Interestingly, the 9FDC displays a lower selectivity of methanol compared to the 4.5FDC: there is a significant difference in the structure of the catalysts which influences the production of methanol. As the temperature increases so does the yield toward C2–C4, especially toward the paraffin fraction (Fig. S3†), being the selectivity of the 9FDC the highest of all samples. Fig. 2B shows the effect of the pressure in the HC distribution: an increase results in higher XCO2, which can be explained by Le Chatelier's principle and the observation of the number of moles change in reactions (1–5).47 The CH4:CO ratio increases with increasing pressure and can be explained by a combination of an increase in the rate of the FTS (reactions (2) and (3), increase of the consumption rate of CO) and a more pronounced shift with the pressure of the equilibrium of the methanation reaction compared to the RWGS. The overall selectivity towards C2–C4 increases with increasing pressure concurrent with an increase in the ratio of paraffins to olefins. The H2:CO2 ratio effect is shown in Fig. 2C: an increase in the ratio results in an increase in the XCO2, achieving a highest rCO2 = 187.8 mmol h−1 gcat−1 for the 9FDC sample at H2:CO2:Ar = 4:1:1. An increase in the ratio results in worse selectivity toward C2–C4: an increase toward CH4 concurrent with less olefins production can be explained by higher extend of hydrogenation caused by the enhanced availability of molecular H2. Fig. 2D shows how a change in the GHSV has a minor effect on both the XCO2 and HC distribution, although the SC2–C4 – is notoriously increased at the lower GHSV. An unidentified peak in the gas chromatograph is shown in Fig. S5.†
 | ||
| Fig. 2 Hydrocarbon (HC) distribution as a function of temperature (A-top left), pressure (B-top right), H2:CO2 ratio (C-bottom left, Ar ratio is always 1), and the GHSV (D-bottom right). Standard conditions are T = 500 °C, P = 20 bar, H2:CO2:Ar = 3:1:1 and GHSV = 50 × 103 mL h−1 gcat−1. | ||
Table S2† shows the Fe mol% obtained from ICP-OES, which are slightly below the expected values, resulting in CeO2 (CeO2), Ce0.986Fe0.014O2−δ, Ce0.958Fe0.042O2−δ and Ce0.912Fe0.088O2−δ samples. The Fe-impurities in the CeO2 sample are probably introduced via the Ce-precursor or by contamination during synthesis/calcination. The same Table S2† displays values of SSA obtained from N2-adsoprtion BET. The addition of Fe results in an increase of the SSA compared to the CeO2 after the calcination step at 600 °C up to the 4.5FDC sample, with a later decrease in the 9FDC sample. Similar trends of SSA were already reported.9
Fig. 3A shows the Ce L3 X-ray absorption data of the fresh samples: panels A1 and A2 allow for better visualization and comparison of the absorption pre-edge and whiteline (XANES region). Fig. 3A1 shows a shift of the absorption pre-edge and edge toward lower energies with increasing content of Fe. This shift of the Ce-L3 pre-edge and edge has been previously observed and is associated with a partial reduction of Ce4+ → Ce3+,3,48,49 which results in the generation of oxygen vacancies.43 Absorption edges are quantified as CeO2: 5728.75 ± 0.14 eV, 1.5FDC: 5728.64 ± 0.13 eV, 4.5FDC: 5728.35 ± 0.15 eV (4.5FDC), and 9FDC: 5728.27 ± 0.16 (edge fittings are shown in Fig. S6,† the specified uncertainty of the edge positions is the std. of the gaussian fits). Fig. 3A2 shows the maxima of the absorption edge, with a shift toward lower intensities with the increase of the Fe content. This reduction of the intensity is associated with a partial reduction of the Ce4+ to Ce3+.3,7 Other studies reported an increase in intensity with an increase in the oxidation state of Ce.49
 | ||
| Fig. 3 Panel A: XAFS data of the fresh catalysts showing the normalized Ce L3 absorption edge, with the pre-edge XANES region extended (insight A1) and the edge (insight A2). Panel B: XAFS data of the normalized Fe K-edge, the fresh Fe-containing samples and a reference Fe2O3. | ||
Following the location of Fe before and after catalysis has been done applying a combination of XAFS, XRD and TEM. After synthesis, Fe can substitute Ce in the lattice or be present as FeOx. Fig. 3B shows XANES of the Fe K-edge for the Fe-containing samples and a reference Fe2O3, demonstrating that the incorporated Fe is in a +3 oxidation state for all the samples, agreeing with previous reports.9 The low content of Fe in the 1.5FDC sample combined with the high absorption of the heavy Ce yields data with high noise level. Other reference materials, including oxidation states of Fe2+, Fe1+, and Fe0 are disregarded (see Fig. S7†). For the 9FDC no additional oxidation state of Fe can be determined, meaning that any Fe present outside of the CeO2-solid solution is also in a +3 state or below the detection limit. No XAFS data of the spent samples could be obtained due to the dilution of the samples, the heavy absorption of Ce, and the relatively low number of photons of the lab instrument.
Fig. 4A shows the XRD patterns of the 4 fresh samples. All samples' intensity has been normalized to the (111) reflection at 2θ ≈ 3.8 (@60 keV). For all the fresh catalysts, the unique present crystal structure is a fluorite-type Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (CeO2 PDF 00-034-0394), agreeing with previous literature10 and suggesting the formation of a unique crystalline phase of CexFe1−xO2−δ: Fe2O3 could still be present as amorphous side phases and/or with domain size below the detection limit. As shown in Fig. 4A-1, the magnification of the normalized (220) reflection displays both a shift of the reflection position and a change of the FWHM with the amount of Fe in the samples. After catalysis, all the “spent” samples maintain the Fmm structure with the appearance of some small additional reflections (Fig. S8†), being such presence more notorious in the spent 9FDC sample. However, these additional reflections cannot be indexed to any of the Ce- or Fe-based crystalline phases. Thus, these might be caused by contamination of the catalysts during the testing or the recovery from the reactor. Besides, the graphitic carbon (PDF 00-041-1487) with its main XRD reflection (002) at 2Θ = 3.52 could not also be identified in any sample.
m (CeO2 PDF 00-034-0394), agreeing with previous literature10 and suggesting the formation of a unique crystalline phase of CexFe1−xO2−δ: Fe2O3 could still be present as amorphous side phases and/or with domain size below the detection limit. As shown in Fig. 4A-1, the magnification of the normalized (220) reflection displays both a shift of the reflection position and a change of the FWHM with the amount of Fe in the samples. After catalysis, all the “spent” samples maintain the Fmm structure with the appearance of some small additional reflections (Fig. S8†), being such presence more notorious in the spent 9FDC sample. However, these additional reflections cannot be indexed to any of the Ce- or Fe-based crystalline phases. Thus, these might be caused by contamination of the catalysts during the testing or the recovery from the reactor. Besides, the graphitic carbon (PDF 00-041-1487) with its main XRD reflection (002) at 2Θ = 3.52 could not also be identified in any sample.
 | ||
| Fig. 4 Panel A: XRD patterns of the fresh catalysts (data normalized to the CeO2(111) intensity, patterns are stacked). The insight (panel A1) shows a magnification of the CeO2(220) reflection (also normalized, not stacked). Panel B: Rietveld results of (B2) lattice parameter and (B1) crystallite size of Ce1−xFexO2−δ as a function of the Fe-content. | ||
Fig. 4B shows the lattice parameter obtained from refinement (see Table S3†) of the fresh and spent samples and the theoretical lattice parameter of FDC if all the Fe3+ (detected from ICP) was included in the lattice. In doped CeO2, changes in the lattice parameter can occur due to: (i) the substitution of larger Ce4+ cations by smaller Fe2+ or Fe3+ cations and/or, (ii) the partial reduction of smaller Ce4+ cations into larger Ce3+ cations, generating oxygen vacancies in the CeO2−δ lattice.4,10 For the experimental values, a significant decrease in the lattice parameter occurs by the substitution of Ce (Ce4+(VIII) = 0.97 Å)50 by Fe (Fe3+(VIII) = 0.78 Å, or Fe2+(VIII) = 0.92 Å)50 (XAFS data has disregarded the presence of Fe2+). As shown in Fig. 4B2, the experimental values of the lattice parameter of the FDC samples are higher than the theoretical values calculated according to eqn (13), assuming the full substitution of Fe dopant in the CeO2−δ lattice. These results suggest that most of the Fe seem to not be present as a solid solution in the Ce1−xFexO2−δ lattice: as appears invisible to the XRD technique, this Fe must be as amorphous Fe2O3 phases or with crystallite sizes below the techniques' detection limit. Fe is a scavenger of grain boundary impurities and could be accumulating in the grain boundary,12 being the mechanism of such process still not fully understood. The presence of Fe induces the Ce4+ → Ce3+ transition, as shown in Fig. 3: the decrease of the lattice parameter caused by Fe-substitution occurs simultaneously with an increase of the lattice parameter resulting from Ce-reduction, masking each contribution. After catalysis, a noticeable increase of the lattice parameter (see Fig. 4B2) occurs for all samples. This is caused by the lattice exsolution of Fe and the further partial reduction of Ce4+ → Ce3+ during the reaction. For all samples, the lattice parameter increases above the value of the pure CeO2, being higher for the 4.5FDC compared to the 9FDC sample. The resulting vacancies are not reoccupied after exposure to air and reach the bulk of the particles, as proved by the XRD analysis. Different levels of reducibility of CeO2 with the size and presence of metal particles on the surface have been demonstrated.3Fig. 4B1 shows the crystallite size before and after catalysis: for the fresh samples, smaller crystallites are present with increasing Fe-content due to a smaller interfacial contact area between grains occurring during self-ignition.31 There is a mild increase in the crystallite size after catalysis for all the samples.
Fig. 5 shows the STEM-EDX characterization of all samples before and after catalysis (the EDX-spectra are shown in Fig. S10†). Similar images at lower magnification can be found in Fig. S9.† All samples display very similar morphology, with a minor presence of nanorod structures in the 4.5FDC (Fig. S11†). For the fresh samples, no particles composed of Fe can be observed, and EDX-analysis shows that Fe is homogeneously distributed over the CeO2 (Fig. 5). After catalysis, exsolved nanoparticles containing Fe clearly appear in the 1.5FDC, 4.5FDC (Fig. S12†), and 9FDC (Fig. 6) samples, with a higher concentration of these with increasing content of Fe in the fresh catalysts. Few particles containing Fe could also be observed in the spent CeO2 sample, explaining the impurities detected with ICP-OES (Table S2†).
 | ||
| Fig. 5 STEM-EDX characterization of the (A.1 and A.2) CeO2, (B.1 and B.2) 1.5FDC, (C.1 and C.2) 4.5FDC, and (D.1 and D.2) and 9FDC before catalysis (left column) and after catalysis (right column). All the STEM images show the SEI-compo signal. The EDX mappings represent atomic%. EDX full spectra are shown in Fig. S10.† | ||
 | ||
| Fig. 6 STEM of the spent 9FDC. Panel A: HAADF image of a particle, with EDX-mappings on the right side. Panel B.1: magnification of the region highlighted in the red frame in panel A, showing a core–shell particle. Panel B.2: extracted line-scan quantification, with color coding as in panel B.1 (C is not displayed) and blue being Ce. All EDX-mappings are shown in counts. | ||
Fig. 6A-1 shows a typical STEM-HAADF image of the spent 9FDC sample. It displays regions with CexFe1−xO2−δ representing non-exsoluted FDC particles. The SiO2-diluent can easily be distinguished. Clearly, exsoluted particles (sometimes with a high amount of carbon) also appear on the catalyst surface with both spherical and elongated shapes. A core–shell exsoluted Fe-containing particle can be seen (Fig. 6B-1), confirming the prior assumption of exsolution of Fe from the FDC material during catalysis. The EDX mapping shows a Fe- and C-rich core, a very thin S-containing intermediate shell (contamination of 0.5 wt%, similar values for Fig. S11 and S12†), and a most external O- and Fe-rich shell. Both the EDX mapping and the line-scan (Fig. 6B-1 and 2) demonstrate such a structure. Most probably, the metallic Fe nanoparticles segregate during catalysis, eventually growing on the surface of the catalyst. The presence during catalysis of a FeOx phase under such a reductive atmosphere is not likely, and most of the FTS published data reports on the Fe or FexC composition of the catalyst.51 Indeed, several phases of FexC can coexist in Fe-particles, with different performances for the FTS,28 rendering their identification very complex.51 Therefore, the oxidic outer shell is most probably formed upon exposure of the recovered catalysts to air after the reaction. The presence of S has also been determined in the fresh samples using the combustion method (Table S2†), and is therefore introduced by the catalyst precursors or during the synthesis/self-ignition/calcination steps. The core of these core–shell particles is a Fe-rich single crystal (see Fig. S13†), which d-spacings obtained using FFT of 0.225 nm and 0.198 nm. These spacings best fit the Fe3C (Pnma, PDF 00-035-0772) (002) and (112) planes, although the first could belong to a Fe5C2 (020) (C2/c, 00-089-7272), and the second could as well fit the Fe7C3 (300) (P63mc, 00-017-0333). The measured angles between the reflections differ from the theoretic values for each possible phase, so the precise phase could not be determined and is part of future experiments. The O-containing (external) shell is amorphous. Fig. S14† displays TEM-SAED of a broader area containing particles: the SAED of Panel A clearly depicts interplanar distances belonging to the Fe3C (Pnma, PDF 00-035-0772) phase. Identification of the crystal structure of Fe-containing catalysts is not trivial and is better done using in situ methods.51 A very similar exsoluted structure is observed in the 4.5FDC spent sample (see Fig. S11†).
Our experiments show that the presence of Fe results in improved performance in terms of a higher reaction rate of CO2 and STY of C2–C4 HCs. Amongst all the tested catalysts, the 9FDC operating at 500 °C, 20 bar, 50 × 103 mL h−1 gcat−1 and H2:CO2:Ar = 2:1:1 displayed the best performance in terms of rates and selectivity. The addition of Fe to the CeO2 does improve performance, although the un-doped CeO2 already catalyzes the desired tandem CO2 hydrogenation to HCs. In terms of XCO2, the FDC catalytic systems are extremely active and do not deactivate over the tested period: conversion was still increasing after the 200 h tested period. The reason is the combination of the segregation of Fe-containing particles on the CeO2 surface and the generation of oxygen vacancies during the reaction. Despite the high activity, the selectivity toward CO and CH4 is still a drawback, a general limitation of this tandem route: high CO partial pressure is necessary to produce HCs beyond CH4, and both Fe and Co-based catalysts for the FTS are also Sabatier-active, resulting in high selectivity toward CH4.18,19 Our catalysts show rather low rates for the FTS, the second part of the desired tandem reaction, in part caused by the relatively low content of Fe (∼3 wt%) compared to commercial catalysts and/or Co-containing tandem systems exploiting the same route (5.8 wt%19 and 12 wt%25). Increasing the Fe-content in this work results in an increase of FTS-products. For some tandem systems, the closest does not mean the best, and an optimal distance between two active sites exist,17,22 concept precisely controlled in core–shell systems containing Pt and Co-catalysts.19,25 The material system reported in this work uses available and non-toxic compounds prepared using a one-pot procedure, far away from core–shell complex structures that make use of novel and scarce materials. A proper techno-economic assessment is necessary to determine which catalysts are more suitable for large-scale industrial applications.18
Determining the location of Fe before and after catalysis is not trivial: after synthesis, Fe in +3 oxidation state can be substituting Ce atoms in the lattice and as Fe2O3, which appears invisible to XRD, i.e. must be amorphous or with domain size below the technique's detection limit. After catalysis, Fe exsolves (from the lattice) or segregates to the surface of the CeO2. The combined XRD and XAFS data demonstrate that the addition of Fe to CeO2 induces oxygen vacancies via two different mechanisms: (1) by substituting Ce atoms and inducing the material to lose O-atoms to maintain neutrality (as proven by XRD) and (2) by inducing the partial reduction of Ce, generating native oxygen vacancies (as proven by XAFS).
There is a non-linear correlation of the performance (conversion and selectivity) with the Fe-content: the reasons are not fully disentangled, and further research is necessary. These are most probably related to (i) the size of the exsolved Fe particles, (ii) the reducibility of the CeO2 to form oxygen vacancies (also affected by the size of the Fe-particles), and (iii) the distance of CeO2 catalytic centers to the Fe-centers. Distance between functionalities plays a crucial role in selective tandem systems. The size of the metallic particles decorating the surface of CeO2 has an effect on the reducibility of CeO2 (generation of oxygen vacancies).3,5,8 Moreover, the size of the particles determines the amount of the metal–CeO2 interface, of high importance for different chemical reactions.8 Finally, the FTS reaction rate and product selectivity are also size-dependent: previous studies have shown an increase in the yield of CH4 for very small (∼2 nm) iron carbide particles;29 for 2.5–12.9 nm Fe-particles, the selectivity to C2+ HCs increases continuously while the selectivity toward CO decreases with increasing size.30 The complex interplay between all these factors deserves further studies, preferably using in situ methods, to disentangle the structure–activity relationships for this catalytic system. Closing the pressure gap in microscopic and spectroscopic in situ techniques is still not possible and the produced information shall be rationalized carefully. From a fundamental point of view, it would be very interesting to exploit the capacity of CeO2 to form solid solutions with heteroatoms that marginally exsolve like Sm3+. This should allow us to disentangle the contribution of the oxygen vacancy and Fe displayed in the current work. Furthermore, metals other than Fe, like Co, Cu, or Ni, should be explored to catalyze specific reactions more selectively.
Conclusions
We report the one-pot synthesis of CeO2 and solid solutions of Ce1−xFexO2−δ for CO2 hydrogenation via the CO-mediated route as an abundant, inexpensive, and non-hazardous catalyst. This method exploits the capacity of CeO2 to form solid solutions with trivalent cations, the generation of oxygen vacancies, and the exsolution/segregation of metallic particles under reaction conditions to result in catalysts with high activity and stability. Catalytic data shows the dominance of CO production via the RWGS reaction, concurrent with CH4 generation and the production of C2–C4 olefins and paraffins. Minor amounts of CH3OH were also produced under some conditions.Initially, Fe partially occupies the lattice of the fluorite-type CeO2 crystal structure, as proved with XRD by the reduction of the lattice parameter due to the substitution of the bigger Ce4+ by the smaller Fe3+ concomitant with the partial reduction of Ce4+ → Ce3+ as demonstrated with XAFS. Fe2O3, although elusive to the techniques applied, must also be present as amorphous and/or with very small domain size. The concentration of oxygen vacancies increases with the content of Fe in the fresh samples. STEM-EDX characterization reveals a similar morphology amongst the fresh samples and the presence of exsolved core–shell particles composed of FexC (and an oxidic outer shell most probably generated upon exposure to air) after catalysis.
The best performance in terms of conversion and selectivity is achieved by the 9FDC sample followed by the 1.5FDC sample, although an undesired high selectivity toward CH4 and CO occurs for all systems. The complex interplay between size and reducibility of the two functionalities in this system deserves further studies to shed light on the structure–activity correlations, which will allow to design catalysts more rationally. Future steps should focus on the use of non-exsolving metals to disentangle the contributions of the oxygen vacancies (e.g. Sm) and the metal particles. The doping of metals other than Fe should be studied to aim at different reaction products. As a tandem system, distance control between functionalities can dramatically enhance product selectivity. The techno-economic assessment shall be performed to determine the suitability of this low-cost, abundant, and non-toxic catalytic system for CO2 hydrogenation in the process scale.
Data availability
The data set containing: the catalytic tests raw data in a .xlsx file; the XAFS integrated raw data; the XRD integrated raw data; and the S-determination LECO output file can be accessed in the DOI: https://doi.org/10.5281/zenodo.11580844.Author contributions
A. Gili: conceptualization, investigation, project administration, writing – original draft preparation, visualization, formal analysis; M. F. Bekheet: investigation, formal analysis, writing – review, and editing; F. Thimm: writing – review and editing; B. Bischoff: investigation; M. Geske: supervision; M. Konrad: investigation, methodology, formal analysis; S. Praetz: formal analysis, investigation, writing – review and editing. C. Schlesiger: supervision; S. Selve: investigation, methodology, writing – review, and editing; A. Gurlo: writing – review and editing, project administration; F. Rosowski: supervision; R. Schomäcker: conceptualization, resources, supervision, funding acquisition, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of this work was funded by the Deutsche Forschungsgemeinschaft (DFG German Research Foundation) under Germany's Excellence Strategy - EXC2008-390540038 – UniSysCat. STEM analyses were performed on a probe-corrected JEM-ARM300F2, funded by DFG – project number 403371556 – GZ: INST 131/789-1 FUGG. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities under LTP II-20210010. A. Gili acknowledges support from the German Federal Ministry of Education and Research in the framework of the project Catlab (03EW0015A) and Jan Simke from ZELMI for support in TEM and SAED analysis.Notes and references
- S. McIntosh and R. J. Gorte, Direct hydrocarbon solid oxide fuel cells, Chem. Rev., 2004, 104(10), 4845–4865, DOI:10.1021/cr020725g.
- A. Gili, B. Bischoff, U. Simon, F. Schmidt, D. Kober, O. Görke, M. F. Bekheet and A. Gurlo, Ceria-Based Dual-Phase Membranes for High-Temperature Carbon Dioxide Separation: Effect of Iron Doping and Pore Generation with MgO Template, Membranes, 2019, 9(9), 108, DOI:10.3390/membranes9090108.
- A. M. Gänzler, M. Casapu, F. Maurer, H. Störmer, D. Gerthsen, G. Ferré, P. Vernoux, B. Bornmann, R. Frahm, V. Murzin, M. Nachtegaal, M. Votsmeier and J.-D. Grunwaldt, Tuning the Pt/CeO2 Interface by in Situ Variation of the Pt Particle Size, ACS Catal., 2018, 8(6), 4800–4811, DOI:10.1021/acscatal.8b00330.
- M. F. Bekheet, M. Grünbacher, L. Schlicker, A. Gili, A. Doran, J. D. Epping, A. Gurlo, B. Klötzer and S. Penner, On the structural stability of crystalline ceria phases in undoped and acceptor-doped ceria materials under in situ reduction conditions, CrystEngComm, 2019, 38, 439, 10.1039/C8CE01726C.
- R. Kopelent, J. A. van Bokhoven, J. Szlachetko, J. Edebeli, C. Paun, M. Nachtegaal and O. V. Safonova, Catalytically Active and Spectator Ce3+ in Ceria-Supported Metal Catalysts, Angew. Chem., Int. Ed., 2015, 54(30), 8728–8731, DOI:10.1002/anie.201503022.
- Y. Liu, Z. Li, H. Xu and Y. Han, Reverse water–gas shift reaction over ceria nanocube synthesized by hydrothermal method, Catal. Commun., 2016, 76, 1–6, DOI:10.1016/j.catcom.2015.12.011.
- T.-S. Wu, Y. Zhou, R. F. Sabirianov, W.-N. Mei, Y.-L. Soo and C. L. Cheung, X-ray absorption study of ceria nanorods promoting the disproportionation of hydrogen peroxide, Chem. Commun., 2016, 52(28), 5003–5006, 10.1039/c5cc10643e.
- M. Cargnello, V. V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts, Science, 2013, 341(6147), 771–773, DOI:10.1126/science.1240148.
- D. Channei, B. Inceesungvorn, N. Wetchakun, S. Ukritnukun, A. Nattestad, J. Chen and S. Phanichphant, Photocatalytic degradation of methyl orange by CeO2 and Fe-doped CeO2 films under visible light irradiation, Sci. Rep., 2014, 4, 5757, DOI:10.1038/srep05757.
- S. Soni, V. S. Vats, S. Kumar, B. Dalela, M. Mishra, R. S. Meena, G. Gupta, P. A. Alvi and S. Dalela, Structural, optical and magnetic properties of Fe-doped CeO2 samples probed using X-ray photoelectron spectroscopy, J. Mater. Sci.: Mater. Electron., 2018, 29(12), 10141–10153, DOI:10.1007/s10854-018-9060-x.
- L. Ye, K. Zhu, Y. Jiang, S. Zhang, R. Peng and C. Xia, Carbon dioxide reduction processes on a samarium doped ceria electrocatalyst with exsolved Fe particles, J. Mater. Chem. A, 2023, 11(20), 10646–10658, 10.1039/D3TA01519J.
- T. Zhang, J. Ma, L. B. Kong, S. H. Chan, P. Hing and J. A. Kilner, Iron oxide as an effective sintering aid and a grain boundary scavenger for ceria-based electrolytes, Solid State Ionics, 2004, 167(1–2), 203–207, DOI:10.1016/j.ssi.2004.01.006.
- Y. Zheng, M. Zhou, L. Ge, S. Li, H. Chen and L. Guo, Effect of Fe2O3 on Sm-doped ceria system solid electrolyte for IT-SOFCs, J. Alloys Compd., 2011, 509(2), 546–550, DOI:10.1016/j.jallcom.2010.09.103.
- P. Sudarsanam, B. Hillary, M. H. Amin, N. Rockstroh, U. Bentrup, A. Brückner and S. K. Bhargava, Heterostructured Copper-Ceria and Iron-Ceria Nanorods: Role of Morphology, Redox, and Acid Properties in Catalytic Diesel Soot Combustion, Langmuir, 2018, 34(8), 2663–2673, DOI:10.1021/acs.langmuir.7b03998.
- N. Zhang, E. P. Tsang, J. Chen, Z. Fang and D. Zhao, Critical role of oxygen vacancies in heterogeneous Fenton oxidation over ceria-based catalysts, J. Colloid Interface Sci., 2020, 558, 163–172, DOI:10.1016/j.jcis.2019.09.079.
- L. Torrente-Murciano, R. S. L. Chapman, A. Narvaez-Dinamarca, D. Mattia and M. D. Jones, Effect of nanostructured ceria as support for the iron catalysed hydrogenation of CO2 into hydrocarbons, Phys. Chem. Chem. Phys., 2016, 18(23), 15496–15500, 10.1039/c5cp07788e.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst, Nat. Chem., 2017, 9(10), 1019–1024, DOI:10.1038/nchem.2794.
- Z. Ma and M. D. Porosoff, Development of Tandem Catalysts for CO2 Hydrogenation to Olefins, ACS Catal., 2019, 9(3), 2639–2656, DOI:10.1021/acscatal.8b05060.
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Tandem Catalysis for CO2 Hydrogenation to C2-C4 Hydrocarbons, Nano Lett., 2017, 17(6), 3798–3802, DOI:10.1021/acs.nanolett.7b01139.
- W. Wang, X. Jiang, X. Wang and C. Song, Fe–Cu Bimetallic Catalysts for Selective CO2 Hydrogenation to Olefin-Rich C2+ Hydrocarbons, Ind. Eng. Chem. Res., 2018, 57(13), 4535–4542, DOI:10.1021/acs.iecr.8b00016.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Directly converting CO2 into a gasoline fuel, Nat. Commun., 2017, 8, 15174, DOI:10.1038/ncomms15174.
- G. Brösigke, J.-U. Repke, R. Schomäcker and S. Matera, The closer the better? Theoretical assessment of the impact of catalytic site separation for bifunctional core–shell catalyst particles, Chem. Eng. J., 2022, 446, 136891, DOI:10.1016/j.cej.2022.136891.
- H. Yan, K. He, I. A. Samek, D. Jing, M. G. Nanda, P. C. Stair and J. M. Notestein, Tandem In2O3-Pt/Al2O3 catalyst for coupling of propane dehydrogenation to selective H2 combustion, Science, 2021, 371(6535), 1257–1260, DOI:10.1126/science.abd4441.
- Z. Wang, J. Qi, N. Yang, R. Yu and D. Wang, Core–shell nano/microstructures for heterogeneous tandem catalysis, Mater. Chem. Front., 2021, 5(3), 1126–1139, 10.1039/D0QM00538J.
- E. Gioria, L. Duarte-Correa, N. Bashiri, W. Hetaba, R. Schomaecker and A. Thomas, Rational design of tandem catalysts using a core-shell structure approach, Nanoscale Adv., 2021, 3(12), 3454–3459, 10.1039/d1na00310k.
- M. E. Dry, Practical and theoretical aspects of the catalytic Fischer-Tropsch process, Appl. Catal., A, 1996, 138(2), 319–344, DOI:10.1016/0926-860X(95)00306-1.
- David A. Hardy, Edward T. Nguyen, Sara E. Parrish, Elyse A. Schriber, Lukas Schlicker, Albert Gili, J. Franz Kamutzki, Nathan Hohman and Geoffrey F. Strouse, Prussian blue iron/cobalt mesocrystals as a template for the growth of Fe/Co carbide (cementite) and Fe/Co nanocrystals, Chem. Mater., 2019, 31(19), 8163–8173, DOI:10.1021/acs.chemmater.9b02957.
- E. de Smit and B. M. Weckhuysen, The renaissance of iron-based Fischer-Tropsch synthesis: on the multifaceted catalyst deactivation behaviour, Chem. Soc. Rev., 2008, 37(12), 2758–2781, 10.1039/b805427d.
- H. M. Torres Galvis, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. Jong, Iron particle size effects for direct production of lower olefins from synthesis gas, J. Am. Chem. Soc., 2012, 134(39), 16207–16215, DOI:10.1021/ja304958u.
- J. Zhu, G. Zhang, W. Li, X. Zhang, F. Ding, C. Song and X. Guo, Deconvolution of the Particle Size Effect on CO2 Hydrogenation over Iron-Based Catalysts, ACS Catal., 2020, 10(13), 7424–7433, DOI:10.1021/acscatal.0c01526.
- P. Arunkumar, S. Preethi and K. Suresh Babu, Role of iron addition on grain boundary conductivity of pure and samarium doped cerium oxide, RSC Adv., 2014, 4(84), 44367–44376, 10.1039/C4RA07781D.
- D. R. Lide, CRC Handbook of Chemistry and Physics, Internet Version 2005, CRC Press, 2005 Search PubMed.
- https://www.chemeo.com .
- C. Prescher and V. B. Prakapenka, DIOPTAS : a program for reduction of two-dimensional X-ray diffraction data and data exploration, High Pressure Res., 2015, 35(3), 223–230, DOI:10.1080/08957959.2015.1059835.
- J. Rodríguez-Carvajal, Recent advances in magnetic structure determination by neutron powder diffraction, Phys. B, 1993, 192(1–2), 55–69, DOI:10.1016/0921-4526(93)90108-i.
- L. W. Finger, D. E. Cox and A. P. Jephcoat, A correction for powder diffraction peak asymmetry due to axial divergence, J. Appl. Crystallogr., 1994, 27(6), 892–900 CrossRef CAS.
- S. J. Hong and A. V. Virkar, Lattice Parameters and Densities of Rare-Earth Oxide Doped Ceria Electrolytes, J. Am. Ceram. Soc., 1995, 78(2), 433–439, DOI:10.1111/j.1151-2916.1995.tb08820.x.
- M. Grünbacher, L. Schlicker, M. F. Bekheet, A. Gurlo, B. Klötzer and S. Penner, H2 reduction of Gd- and Sm-doped ceria compared to pure CeO2 at high temperatures: effect on structure, oxygen nonstoichiometry, hydrogen solubility and hydroxyl chemistry, Phys. Chem. Chem. Phys., 2018, 20(34), 22099–22113, 10.1039/c8cp04350g.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32(5), 751–767, DOI:10.1107/S0567739476001551.
- C. Schlesiger, S. Praetz, R. Gnewkow, W. Malzer and B. Kanngießer, Recent progress in the performance of HAPG based laboratory EXAFS and XANES spectrometers, J. Anal. At. Spectrom., 2020, 35(10), 2298–2304, 10.1039/D0JA00208A.
- C. Schlesiger, L. Anklamm, H. Stiel, W. Malzer and B. Kanngießer, XAFS spectroscopy by an X-ray tube based spectrometer using a novel type of HOPG mosaic crystal and optimized image processing, J. Anal. At. Spectrom., 2015, 30(5), 1080–1085, 10.1039/C4JA00303A.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(Pt 4), 537–541, DOI:10.1107/S0909049505012719.
- L. R. Shah, B. Ali, H. Zhu, W. G. Wang, Y. Q. Song, H. W. Zhang, S. I. Shah and J. Q. Xiao, Detailed study on the role of oxygen vacancies in structural, magnetic and transport behavior of magnetic insulator: Co-CeO2, J. Phys.: Condens. Matter, 2009, 21(48), 486004, DOI:10.1088/0953-8984/21/48/486004.
- Y. Liu, Z. Li, H. Xu and Y. Han, Reverse water–gas shift reaction over ceria nanocube synthesized by hydrothermal method, Catal. Commun., 2016, 76, 1–6, DOI:10.1016/j.catcom.2015.12.011.
- A. E. Rashed, A. Nasser, M. F. Elkady, Y. Matsushita and A. A. El-Moneim, Fe Nanoparticle Size Control of the Fe-MOF-Derived Catalyst Using a Solvothermal Method: Effect on FTS Activity and Olefin Production, ACS Omega, 2022, 7(10), 8403–8419, DOI:10.1021/acsomega.1c05927.
- Y. Liu, J.-F. Chen and Y. Zhang, The effect of pore size or iron particle size on the formation of light olefins in Fischer–Tropsch synthesis, RSC Adv., 2015, 5(37), 29002–29007, 10.1039/C5RA02319J.
- B. Todic, L. Nowicki, N. Nikacevic and D. B. Bukur, Fischer–Tropsch synthesis product selectivity over an industrial iron-based catalyst: Effect of process conditions, Catal. Today, 2016, 261, 28–39, DOI:10.1016/j.cattod.2015.09.005.
- L. Zheng, A. Zimina, M. Casapu and J.-D. Grunwaldt, Hydrocarbon and Soot Oxidation over Cerium and Iron Doped Vanadium SCR Catalysts, ChemCatChem, 2020, 12(24), 6272–6284, DOI:10.1002/cctc.202001314.
- D. Eggart, X. Huang, A. Zimina, J. Yang, Y. Pan, X. Pan and J.-D. Grunwaldt, Operando XAS Study of Pt-Doped CeO2 for the Nonoxidative Conversion of Methane, ACS Catal., 2022, 12(7), 3897–3908, DOI:10.1021/acscatal.2c00092.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- Q.-Y. Liu, C. Shang and Z.-P. Liu, In Situ Active Site for Fe-Catalyzed Fischer-Tropsch Synthesis: Recent Progress and Future Challenges, J. Phys. Chem. Lett., 2022, 13(15), 3342–3352, DOI:10.1021/acs.jpclett.2c00549.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00439f |
| This journal is © The Royal Society of Chemistry 2024 |