
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Primary amines from alkenes and carbonyl compounds: highly selective hydrogenation of oximes using a homogeneous Ru-catalyst†
Kevin
Hares‡
,
Hannes W.
Wegener‡
 ,
Thomas F. H.
Roth
,
René
Reichert
,
Dieter
Vogt
and
Thomas
Seidensticker
*
,
Thomas F. H.
Roth
,
René
Reichert
,
Dieter
Vogt
and
Thomas
Seidensticker
*
Laboratory of Industrial Chemistry, Department of Biochemical and Chemical Engineering, TU Dortmund University, Emil-Figge-Str. 66, 44227 Dortmund, Germany. E-mail: thomas.seidensticker@tu-dortmund.de; Web: https://tc.bci.tu-dortmund.de/ Tel: +49 231 7552310
First published on 17th April 2024
Abstract
The efficient production of aliphatic primary amines is still a major challenge despite their production on a large scale. Particularly when considering the overall production route starting from alkenes, current strategies suffer in at least one reaction step from poor regio- or chemoselectivity. This work presents an efficient and selective synthesis protocol for primary aliphatic amines via their corresponding aldoximes. These are readily produced from the condensation of hydroxylamine and the respective aldehydes. This straightforward condensation can be carried out either with isolated aldehydes or with crude reaction solutions from the hydroformylation of alkenes. It allows a straightforward separation of the aldoximes via their precipitation, which then serve as intermediates for the final reduction. In a newly developed protocol for aldoxime reduction, yields of up to 90% of the desired primary amines from several different aldoximes are achieved. The ruthenium/triphos catalyst system showed activities exceeding 7500 h−1, which is significantly faster than other amination protocols, and complete conversion is achieved within minutes. Our approach allows the synthesis of the polyamide-12 precursor (methyl 12-aminododecanoate) from the unsaturated, renewable oleochemical methyl 10-undecenoate via the hydroformylation, condensation, reduction sequence on up to 6 g scale with up to 68% overall selectivity.
Introduction
Today, primary amines play a vital role in producing pharmaceuticals, agrochemicals, polymers, and other chemical intermediates, which contribute to many everyday products.1–3 Especially the synthesis of primary aliphatic amines remains challenging, as most routes suffer from low selectivities or poor atom economy.4 This is a significant drawback for industrial-scale production due to the immense waste and byproducts generated. Hence, there is a large environmental and economic interest in new, more efficient synthesis protocols to produce primary amines. The most industrially abundant chemicals are alkenes, making them desirable starting materials. However, the direct transformation into amines is complex and remains one of the critical challenges in catalysis (Scheme 1).4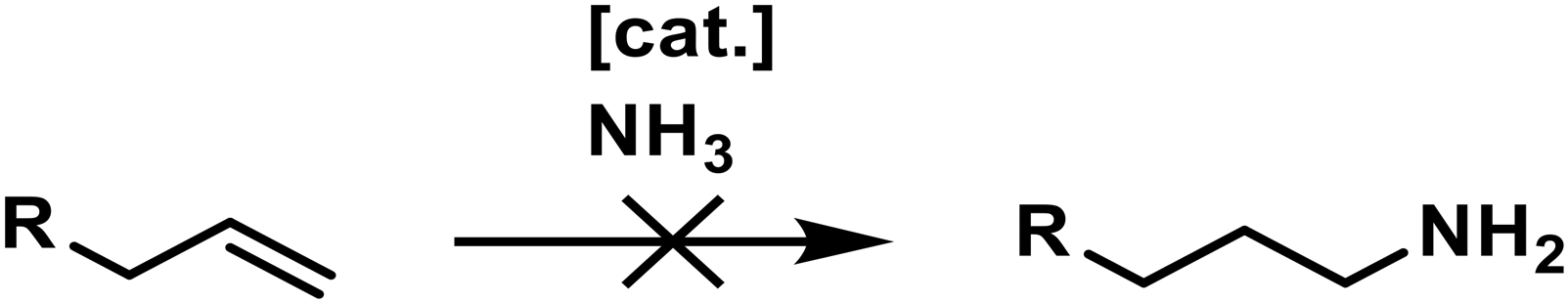 | ||
| Scheme 1 Catalytic, regioselective hydroamination of non-activated alkenes with ammonia towards primary amines remains a dream reaction. | ||
The direct hydroamination of non-activated alkenes with ammonia would yield amines in a 100% atom economic reaction and was titled the “holy grail of catalysis”.4 The main challenges are the high dissociation energy required for activating the N–H bond in ammonia,5 the fact that the reaction is thermoneutral, and the controlled transformation into the anti-Markovnikov product.4 Therefore, a wide variety of synthesis routes have been explored to address these problems (Scheme 2).
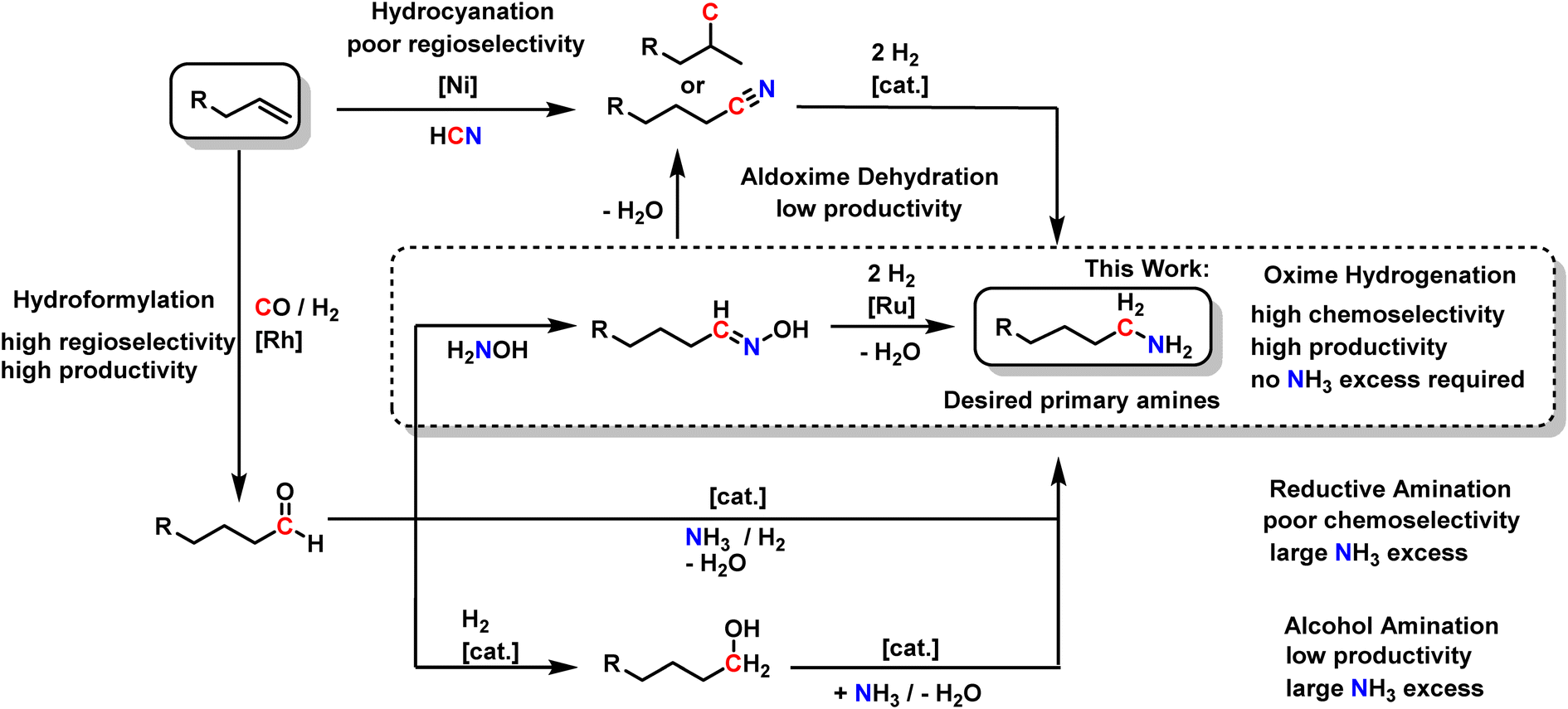 | ||
| Scheme 2 Reaction pathways toward primary amines starting from alkenes. | ||
Since the direct hydroamination of non-activated aliphatic 1-alkenes with ammonia to produce primary amines is still one of the major challenges in organic chemistry,4 the chemical industry commonly uses related hydrofunctionalizations. For instance, Ni-catalyzed hydrocyanation transforms alkenes into nitriles, a highly atom-economic process using hydrogen cyanide.6–8 However, the control of regioselectivity is challenging since the desired linear amine resembles the anti-Markovnikov product and is, therefore, not the thermodynamically favored product.7 Thus, hydrocyanation is only established for the synthesis of adiponitrile from 1,3-butadiene.9 For linear, aliphatic 1-alkenes, poor regioselectivity is obtained. In a consecutive hydrogenation, the corresponding nitriles are transformed into the desired aliphatic primary amines with high yields and selectivities, a process that is well understood and already highly optimized.10,11
Alternatively, the Rh-catalyzed hydroformylation of aliphatic 1-alkenes affords aldehydes with the highest regioselectivity at high productivity.12 These aldehydes are valuable platform intermediates for a range of different products, among them aliphatic primary amines. Three different routes generally exist: the reductive amination of aliphatic aldehydes directly with ammonia is still suffering from poor chemoselectivity.13,14 Secondary and tertiary amines are formed by consecutive reactions of the primary amines since the produced amines are more reactive than ammonia.14–17 Typically, a large excess of ammonia is thus used to shift the product distribution toward the primary amine, hampering productivity.
Secondly, aldehydes can be reduced to the corresponding alcohols, which can be transformed into amines by alcohol amination.18–23 Excellent chemoselectivities towards primary amines, even with linear aliphatic alcohols, can be achieved using ammonia.19 However, the high chemoselectivity comes with the price of high ammonia excess and extended reaction times. The reaction time often exceeds 12 hours, resulting in low productivity.
A third but underrepresented way of synthesizing primary amines is via oximes as intermediates. Aldehydes stoichiometrically and highly selectively react with hydroxylamine to form aldoximes, which is why this reaction is used to identify aldehydes in complex mixtures.24 In this way, nitrogen is introduced into molecules without risking consecutive reactions to secondary or tertiary amines, thus avoiding a high excess of the nitrogen source. Recently, the potential of combining Rh-catalyzed hydroformylation with aldoxime formation has been demonstrated in the context of nitrile formation via biocatalytic aldoxime dehydration.25–28 We recently showed that an aqueous hydroxylamine solution does not inhibit biphasic Rh-catalyzed hydroformylation so that aldoximes can be obtained in a one-pot reaction starting from alkenes.29 This offers an elegant way to produce aldoximes since the isolation of the reactive aldehydes is circumvented. Interestingly, it is possible to increase the already high regioselectivity from the hydroformylation step by aldoxime formation: in some cases, the linear oximes selectively crystallize from the reaction mixture.28 Therefore, rendering the otherwise difficult separation of linear and branched products very efficient, making aldoximes very interesting intermediates on the way to primary amines.
However, as interesting as the biocatalytic transformation of aldoximes into nitriles for circumventing the use of hydrogen cyanide is, this reaction sequence has two major drawbacks for the synthesis of primary aliphatic amines: firstly, aldoxime dehydration has relatively low reaction rate, and secondly, starting from aliphatic 1-alkenes, four reaction steps are required in total.
Therefore, we envisage that direct aldoxime hydrogenation is more straightforward and could thus be an essential step to the highly desired primary aliphatic amines. It perfectly complements the established reaction sequence of Rh-catalyzed hydroformylation and aldoxime formation. In contrast to hydrocyanation, high regioselectivity and high productivity from the initial hydroformylation can be at least preserved. In contrast to reductive amination and alcohol amination, minimal to no excess of the N-source hydroxylamine is required, and consecutive reactions may be efficiently suppressed, resulting in high chemoselectivity.
On laboratory scale hydrides are often used as reducing agent. However, they produce quantitative amounts of waste.30 The cleanest reducing agent is molecular hydrogen in combination with a catalyst, and dates to 1956; Reeve and Christian showed excellent activity for RANEY® cobalt.31 The challenge here is to selectively stop the reaction at the primary amine and avoid consecutive reactions. The reaction mechanism for the heterogeneously catalyzed reaction was investigated by Müller et al.32 They used oxide-supported Ni and Co catalysts, which are less active but show higher selectivity for primary amines, as well as Pd and Rh on activated carbon, which are more active but yield the secondary amine as the main product. This study revealed that the selectivity of the oxime reduction is highly dependent on the nature of the substrate.32 More branched, thus, more sterically demanding substrates yield higher selectivities than entirely linear oximes, and ketoximes produce the highest primary amines selectivity.32
Very recently, the reduction of aldoximes was used to synthesize heterocyclic primary amines, which can be used to produce polymers and liquid crystal coordinating agents. With a heterogeneous Ni–Mo/ZrO2 catalyst, Bai-cheng et al. achieved yields above 90% after two to four hours of reaction time on a 60 g scale. However, excess ammonia was necessary for high yields, and the products were beta-cyclic compounds.33
In 2019, the group of Wang reported excellent hydrogenation results using a palladium-based catalyst consisting of nanoparticles with nitrogen-containing ligands.34 They obtained a 99% yield of benzylamine and its derivatives. The yield of aliphatic amines was reduced to 83% in the case of octylamine and 71% in the case of butylamine. Remarkably, the conversion takes place at an atmospheric pressure of H2 and room temperature, and even after 10 recycling runs, the performance did not decrease significantly. However, the reaction is limited to a highly diluted substrate on an extremely small scale, namely 0.05 mmol in 10 ml H2O.34
To our best knowledge, there are no examples of molecular catalysts for the reduction of aldoximes with molecular hydrogen as the reductant, particularly for aliphatic linear oximes. We are convinced this is a reaction with high potential for the chemical industry and the synthesis of pharmaceuticals since it makes a wide variety of primary amines accessible selectively. Additionally, combined with our previous work, this reaction offers an efficient synthesis of primary aliphatic amines starting from alkenes, which remains a significant challenge. A highly relevant example would be the synthesis of green PA 12 starting from the renewable methyl 10-undecenoate, which can be sourced from the oil of the Ricinus plant.35,36
Results and discussion
The introduction of nitrogen into a molecule via hydroxylamine at a carbonyl group is a well-known and straightforward reaction, as they usually readily react and form the desired oxime intermediate. This reaction is highly selective to carbonyls and can target them directly from a mixture of compounds. However, the subsequent hydrogenation still requires a well-optimized catalyst for selective conversion.In their pure form, aldoximes (1) are usually more stable compared to the corresponding aldehydes (8). However, several side reactions may occur in contact with the catalyst or the reaction solution (Scheme 3), e.g., acidic conditions result in a Beckmann rearrangement, forming amides (4), which are relatively stable and not readily hydrogenated.
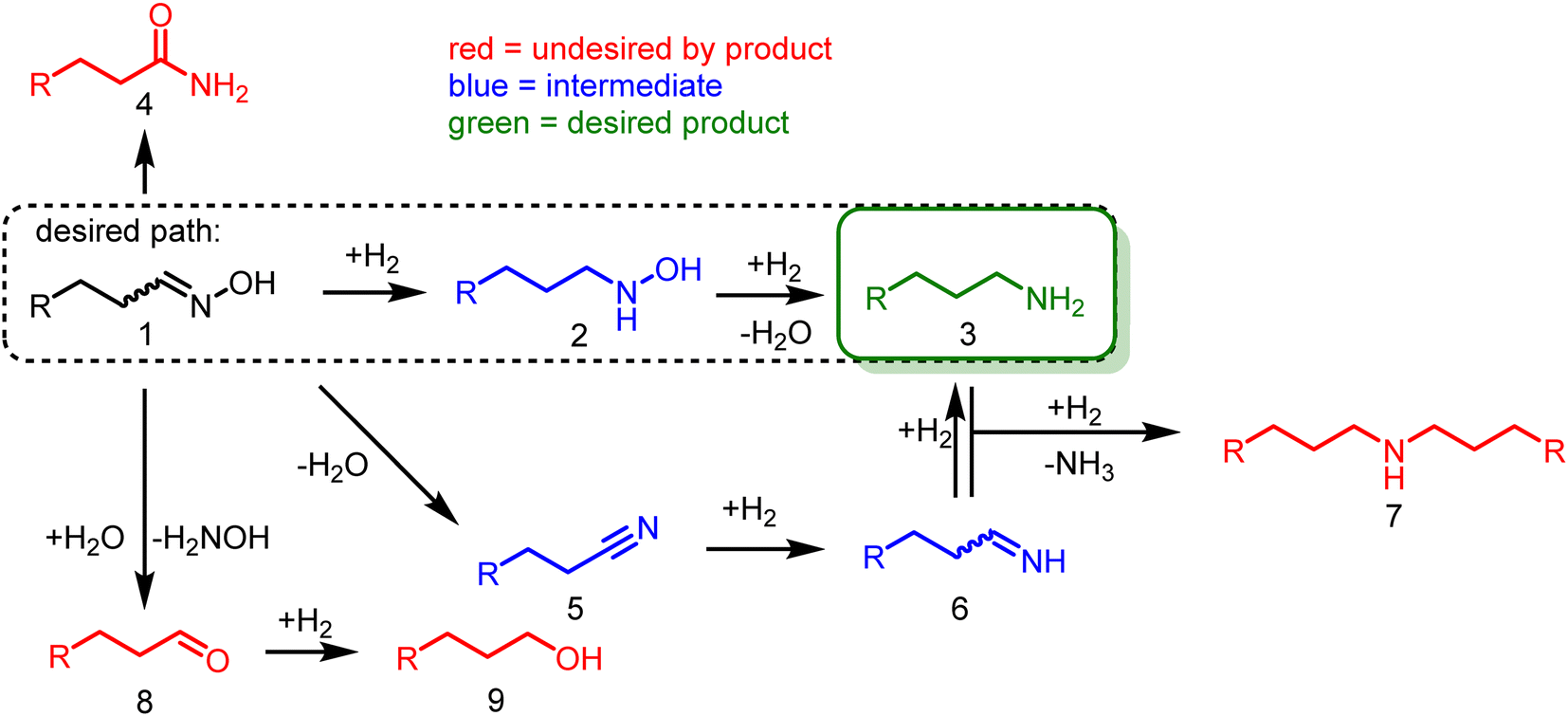 | ||
| Scheme 3 Possible side reactions in the hydrogenation of aldoximes. | ||
The oxime (1) can also be hydrolyzed at elevated temperatures to the aldehyde (8) and hydroxylamine, which are rapidly converted into the corresponding alcohol (9) and ammonia under reducing conditions. Theoretically, the alcohol (9) can react with ammonia to form the desired primary amines (3) via alcohol amination. However, secondary amine (7) formation is more likely since the more reactive primary amine (3) is already present in the reaction mixture and will preferably react with the alcohol (9).
Additionally, there are two main pathways to form the desired primary amine (3). On the one hand, the aldoxime (1) can be dehydrated to form a nitrile (5), which in turn is hydrogenated into the corresponding imine (6). Subsequent hydrogenation yields the desired primary amine (3). The imine (6) must be hydrogenated rapidly to obtain high selectivities since it is prone to react with the primary amine (3) to form secondary amines (7).
On the other hand, the pathway promising the highest selectivities is via the direct oxime hydrogenation to the organic hydroxylamine intermediate (2), which is subsequently hydrogenated to the desired primary amine (3), liberating water.
Due to all these side reactions, the reaction system must be finely tuned, ideally favoring the organic hydroxylamine path (Scheme 3, dashed box), as it has a lower potential for side reactions.
Therefore, the reaction conditions were optimized in a single-point optimization manner.
The precursor, ligand, catalyst amount, solvent, temperature, and pressure were chosen as the optimization parameters, with a starting point selected from common hydrogenation conditions (see ESI†). The model reaction was the reduction of hexanal oxime (1a) to hexylamine (3a) (Scheme 4).
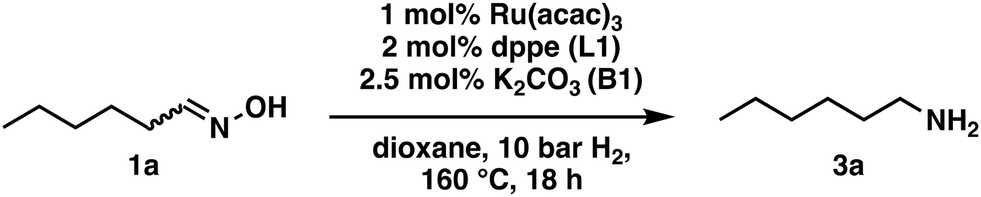 | ||
| Scheme 4 Starting conditions for optimizing the reaction conditions in the hydrogenation of hexanal oxime (1a). | ||
At first, the reaction was carried out on a 1 mmol scale in a 20 ml autoclave with the above-stated reaction conditions (Scheme 4). However, this setup was not ideal for this reaction since the selectivity was low, and reaction monitoring was impossible due to the small reaction volume and the lack of suitable periphery. The main side product was hexanol (9a), indicating a reverse reaction of the oxime 1a with water, yielding hexanal (8a), which is readily reduced under the present conditions. Additionally, a 12% yield of hexanamide (4a) was observed, resulting from a Beckmann rearrangement (see ESI†).
In previous works of alcohol amination and the amination of esters, ruthenium reached high primary amine selectivity. Therefore, we did focus on ruthenium. For hydrogenation reactions, there is a wide variety of ligands available, which were screened first.
In all cases, the oxime conversion was complete, but the distribution of the products varied. The results suggest that tridentate ligands favor the hydrogenation into the primary amine (Table 1), which is, in all cases, the main product. However, L2 and L7 (Fig. 1) stand out with more than 65% selectivity towards the primary amine. With the mono and bidentate ligands, the nitrile was the main product. This suggests that the tridentate ligands have higher activity in the hydrogenation of oximes and nitrile since less nitrile remains in the product mixture. Alternatively, the reaction route via the hexyl hydroxylamine (2a) is the preferred route, resulting in less nitrile formation in the first place.
| Ligand | X [%] | S (hexylamine 3a) [%] | S (dihexylamine 7a) [%] | S (hexanitrile 5a) [%] | S (hexanamide 4a) [%] | S (hexanol 9a) [%] |
|---|---|---|---|---|---|---|
| Conditions: 8.33 mmol hexanal oxime (1a), 0.5 mol% Ru(acac)3, 2.5 mol% KOH, 160 °C, 5 h, 700 rpm, 1,4-dioxane (30 mL), 10 bar H2, monodentate: 1.65 mol%, bidentate: 0.825 mol%, tridentate: 0.55 mol%. Determined via GC with dodecane as an internal standard. | ||||||
| L1 | 98 | 6 | 36 | 42 | 4 | 7 |
| L2 | >99 | 69 | 8 | 2 | 2 | 3 |
| L3 | >99 | 12 | 23 | 45 | 5 | 5 |
| L4 | 98 | 1 | 26 | 57 | 9 | 7 |
| L5 | >99 | 30 | 26 | 11 | 6 | 16 |
| L6 | >99 | 34 | 15 | 22 | 4 | 10 |
| L7 | >99 | 68 | 7 | 2 | 9 | 1 |
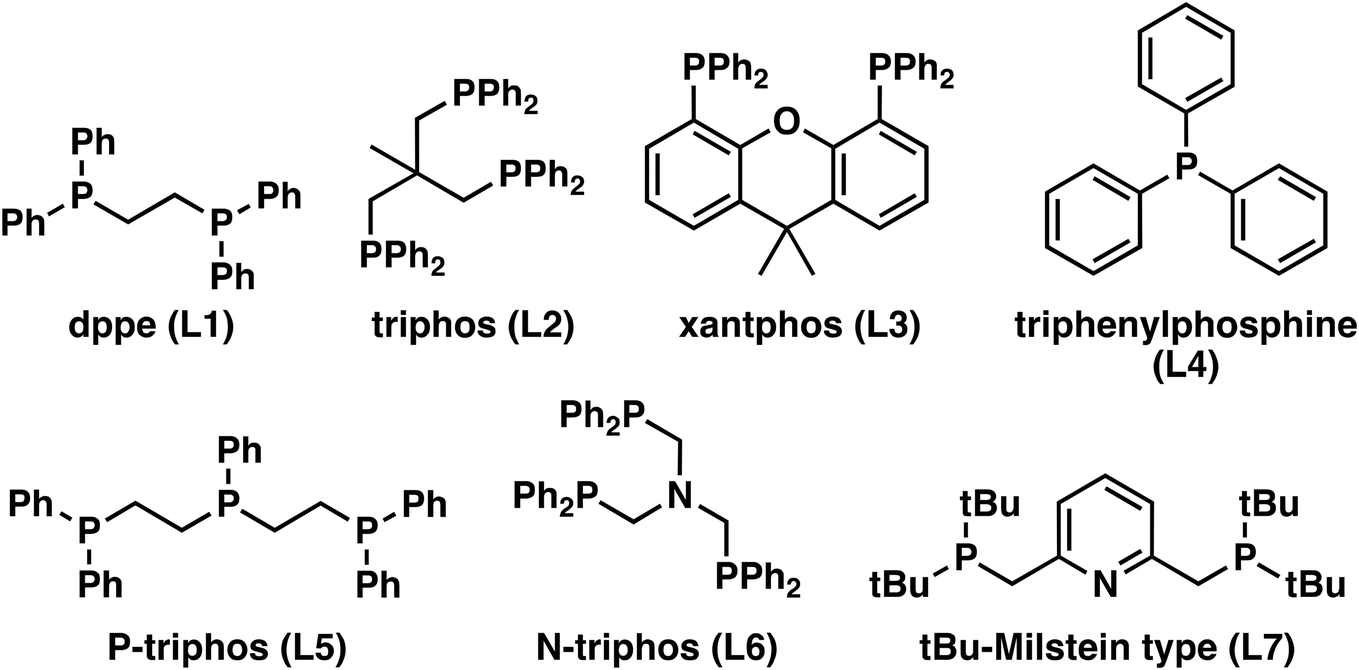 | ||
| Fig. 1 Ligands used in the reduction of hexanal oxime (1a). | ||
The triphos ligand was selected for further optimization after the ligand screening. In situ formation of the active catalyst species during preforming was based on the works of Beller et al. who reported excellent abilities to form the desired active complex.37
A range of ruthenium precursors have been tested, yet no further improvement to the existing Ru(acac)3–triphos system could be achieved (see ESI†). The Ru–triphos system is often used in combination with KOtBu (B1) as a basic activation agent. In the reduction of aldoximes, it reached a selectivity of 75% and a TOF20 of 359 h−1 (Fig. 2). KOH (B3) scored higher in selectivity with the price of a slower reaction. This could be caused by the low solubility of KOH (B3) in dioxane. A few drops of water were necessary to dissolve the required amount of base, which could interfere with the catalyst or trigger the backward reaction of hexanal oxime (1a) into hexanal (8a). Additionally, the amine bases 1,4-diazabicyclo[2.2.2]octane (DABCO) (B2) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU (B4)) were tested. Both have shown a higher reaction rate, but the best results were achieved with DBU (B4), i.e., a selectivity of 89% and a TOF20 of 575 h−1. The higher selectivity is caused by less nitrile (see Fig. S4†) formed during the reaction, thus probably favoring the hydroxylamine path and the lack of water in the system inhibits the backward reaction of hexanal oxime (1a).
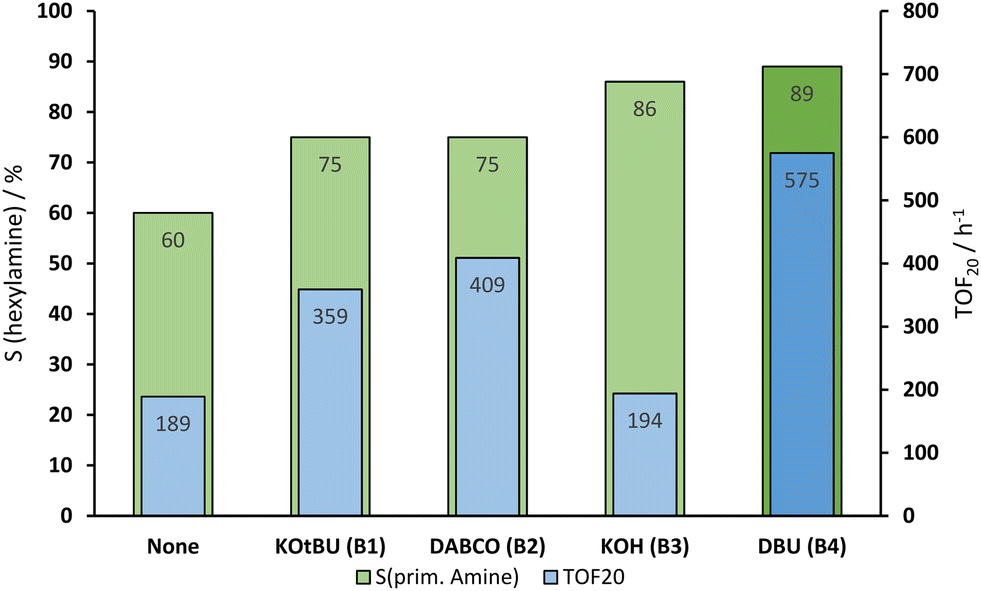 | ||
| Fig. 2 Variation of the base additives. Conditions: 8.33 mmol hexanal oxime (1a), 0.5 mol% Ru(acac)3, 0.55 mol% triphos (L1), 2.5 mol% base, 160 °C, 50 bar H2, t = 5 h, 700 rpm, 1,4-dioxane (30 mL). Determined via GC with dodecane as an internal standard. | ||
For the optimization, the setup was changed to a 100 ml reactor using a reaction volume of 30 ml. While periodically drawing samples, it became visible that the reaction system seems to undergo a relatively slow catalyst-forming process. The reaction mixture's color changed over 2 h from red, the color of the dissolved Ru(acac)3, to yellow, which is typically the color of Ru–phosphine complexes in solution (see in ESI†). After opening the reactor upon completion of the reaction under the non-optimized conditions, the typical odor of ammonia was present. A handheld ammonia detector could confirm relatively large amounts of ammonia in the reactor, indicating the reverse reaction of the oxime (1a), resulting in hydroxylamine, which is readily reduced to ammonia under the present conditions. This might be caused by slow hydrogenation of the hexanal oxime (1a) due to the not yet formed active catalyst, allowing the back reaction to hexanal (8a) and hydroxylamine.
Therefore, a preforming stage was added to the reaction procedure. This inhibited the amide (4a) formation and suppressed the backward reaction to hexanal (8a), and thus the formation of the alcohol (9a) and ammonia. The optimization showed that nitrogen based bases such as diazabicycloundecene (DBU (B4)) promote a different reaction pathway compared to oxygen bases such as KOtBu (B1), thus increasing the selectivity of the reaction. Our next goal was an improvement of the reaction speed to decrease the reaction time further.
When using basic additives that contain nitrogen, a significantly smaller amount of nitrile was detected during the reaction compared to experiments with oxygen-based bases. Thus, there is a change in the reaction pathway from the formation of the nitrile through the dehydration of water to the hydrogenation of hydroxylamine, followed by hydrogenation to the amine.
Temperature – a key factor
Generally, the reaction rate is highly dependent on the reaction temperature. Preferably, the desired reaction path is favored at elevated temperatures, further increasing the selectivity. The results of the temperature screening are presented in Fig. 3.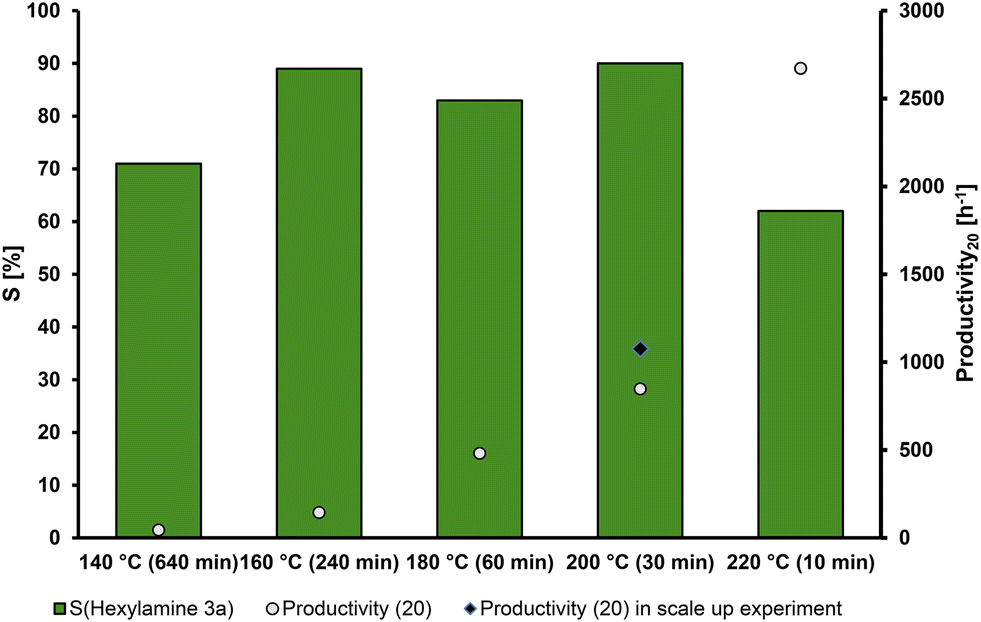 | ||
| Fig. 3 Temperature dependency of the hydrogenation of hexanal oxime (1a). Conditions: hexanal oxime (1a, 8.33 mmol), Ru(acac)3 (0.5 mol%), triphos (L2) (0.55 mol%), DBU (B4) (2.5 mol%), 1,4-dioxane (30 mL), H2 (50 bar at rt), 700 rpm, preforming 2 h under reaction conditions without substrate. The selectivity (S) of hexylamine (3a) was calculated by GC-FID using n-dodecane as an external standard. All data points for the selectivity are from time-resolved measurements and were chosen for the highest selectivity (compare in ESI†). Productivity (20) calculated at 20% yield of hexylamine (3a). | ||
We chose the productivity for comparison of the different reaction parameters because the hexanal oxime (1a) is converted extremely fast in most cases and does not indicate a better performance of the reaction, so comparing the TOF20 could be misleading, e.g., a high TOF20 could be reached via the nitrile path, but the overall reaction would be slow. In contrast, the productivity is calculated via the yield and, therefore, includes the selectivity of the conversion.
As expected, the reaction speed increases with increasing temperatures. The selectivity also increases with the temperature reaching a maximum of 90% at 200 °C. At 220 °C, the consecutive reaction takes over and reduces the selectivity towards the primary amine. The formation of 7a (Scheme 3) after complete conversion of 1a can occur either via dimerization of the primary amine (3a) or in a reaction of the imine (6) and (3), both liberating ammonia. At this temperature, the highest selectivity of 62% was reached after 10 min, and the secondary amine formation (7a) occurred faster (compared with the X/t-plot in the ESI†). At 200 °C, the highest selectivity was accomplished after 30 min. At 160 °C, a similar selectivity was reached, but it took significantly longer, as the maximum was reached after only 240 min. The maximum selectivity at 200 °C is probably the result of two factors. First, a higher reaction rate reduces the time of intermediates being present in the reaction mixture, thus suppressing side reactions and increasing selectivity. Second, at higher temperatures than 200 °C, the activation energy of the dehydrogenation of primary amine (3a) to the corresponding imine (6a) is overcome. Hence, so-called deaminative coupling proceeds, in which two primary amines react to form the corresponding secondary amine (7a) under the liberation of ammonia, thus decreasing the selectivity at higher temperatures.38
Comparing the productivity at 20% yield at the presented temperatures reveals an almost exponential relation: from 44 h−1 at 140 °C to an increased productivity of 2672 h−1 at 220 °C. A reaction temperature of 200 °C was chosen as a very high selectivity (90%) is achieved, while the productivity (20) remains high (847 h−1).
Scale up of the reaction
Next, we performed the reaction in a 300 ml autoclave with 25 mmol hexanal oxime (1a), representing a scale-up factor of 3 (Fig. 4B). Additionally, the reactor was equipped with a gas inlet stirrer to improve the hydrogen availability for the reaction. In this setup, the productivity (20) could be increased to 1074 h−1 (from 847 h−1) at 200 °C (Fig. 3). The selectivity, however, was lower at 83%. Longer preparation time with the larger reactor could have led to more side products. Noteworthy is the decay of the amine (3a) yield after it reached a maximum at 40 min of reaction time. Simultaneously, an increase in the dihexylamine (7a) yield can be observed, which suggests a deaminative coupling occurs, despite the pressurized hydrogen atmosphere. Interestingly, more nitrile (5a) was observed with this reaction setup. Therefore, the secondary amine (7a) could result from the imine (6a) intermediate.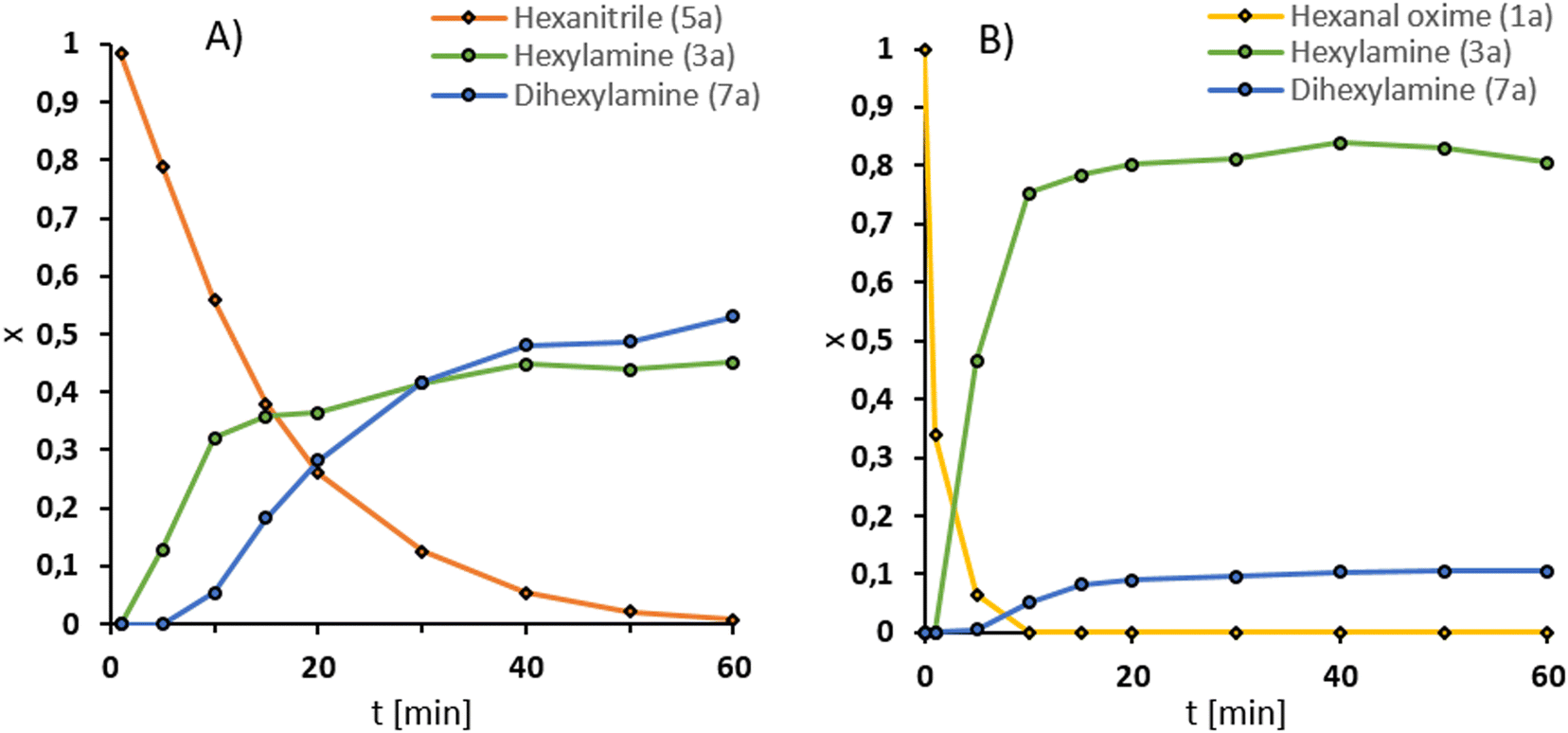 | ||
| Fig. 4 Comparison of the hydrogenation of hexanenitrile (5a) A) hexanal oxime (1a) B) under optimized conditions. Conditions: substrate (25 mmol), Ru(acac)3 (0.5 mol%), triphos (L2) (0.55 mol%), DBU (B4) (2.5 mol%), 1,4-dioxane (100 mL), H2 (50 bar at RT), 750 rpm at 200 °C. Amounts as a fraction of the substrate at the reaction start were calculated by GC-FID using dodecane as an external standard. | ||
In contrast, the alcohol (9a) and amide (4a) could only be detected in trace or not at all, respectively. Theoretically, the amide could also be converted under the present conditions.
Enlightening the reaction path
For a better understanding of the results, the path of the reaction can reveal more details. Possible reaction paths can proceed either via the dehydration of the aldoxime forming the nitrile (5a), which is subsequently hydrogenated to the imine (6a), or via a direct hydrogenation resulting in hydroxyl hexylamine (2a). As previously mentioned, the type of base added influences the reaction pathway, with oxygen bases favoring nitrile formation. To investigate whether the intermediate affects selectivity, additional experiments were carried out comparing nitrile hydrogenation and oxime hydrogenation with nitrogen bases.Both intermediates are then hydrogenated to the desired primary amine. Due to the lack of an imine species (6a), the direct hydrogenation path should be more selective.
To obtain more details, we compared the hydrogenation of hexanenitrile (5a) with the hydrogenation of the hexanal oxime (1a). Both reactions are displayed in Fig. 4.
In comparison, the nitrile reduction (Fig. 4A) is slower than the oxime hydrogenation (Fig. 4B). While the nitrile reaches full conversion only after 60 min, the oxime (1a) is already converted after 10 min under the same conditions. This trend is also reflected by the TOF20, 7940 h−1 is reached in the oxime hydrogenation and 501 h−1 in the nitrile hydrogenation, respectively. Therefore, the main reaction path is likely through the hexyl hydroxylamine (2a). Another hint is the selectivity of the reaction. As mentioned, the imine intermediate 6a is prone to side reactions, leading to an increased dihexylamine (7a) formation. The test reaction confirms this hypothesis since the selectivity of the nitrile reduction after full conversion of 45% is significantly lower than the 83% of the oxime hydrogenation.
These findings underline the importance of a fast hydrogenation reaction for the selectivity toward the desired primary amine (3a).
Scope of the reaction
Next, we have investigated the scope of the reaction (Table 2).|
|
||||
|---|---|---|---|---|
| Entry | Substrate | t [min] | Product | Yield [%] |
| Conditions: oximes (8.33 mmol), Ru(acac)3 (0.5 mol%), triphos (L2) (0.55 mol%), DBU (B4) (2.5 mol%), 1,4-dioxane 30 mL, H2 (50 bar at RT), 700 rpm at 200 °C. The yields of the amines were calculated by GC-FID by using dodecane as an internal standard.a 25 mmol scale in 100 mL 1,4-dioxane.b Determined via1H NMR.c Time until highest yield. | ||||
| 1a |
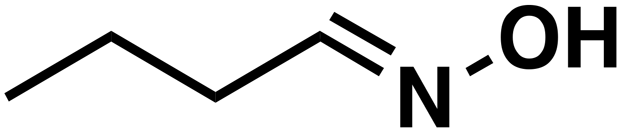 (1b) (1b) |
5 |
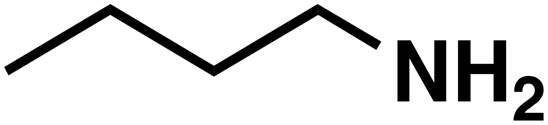 (3b) (3b) |
51 |
| 2 |
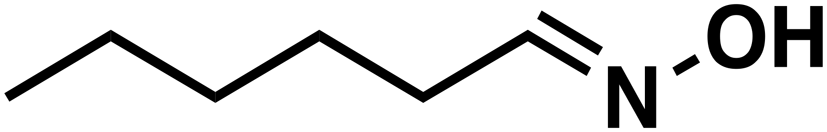 (1a) (1a) |
30 |
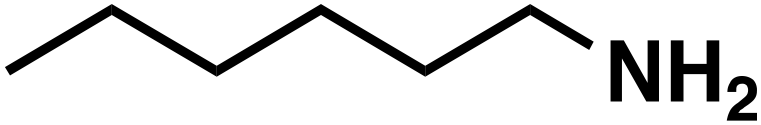 (3a) (3a) |
90 |
| 3a |
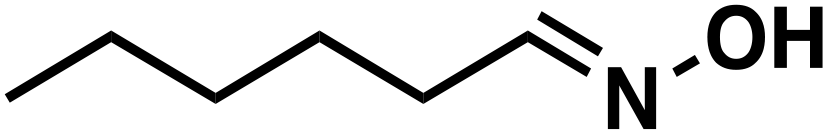 (1a) (1a) |
40 |
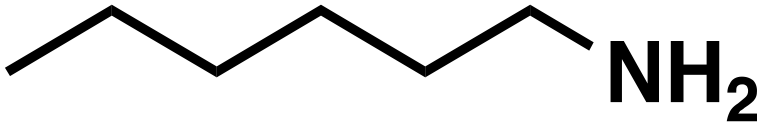 (3a) (3a) |
83 |
| 4 |
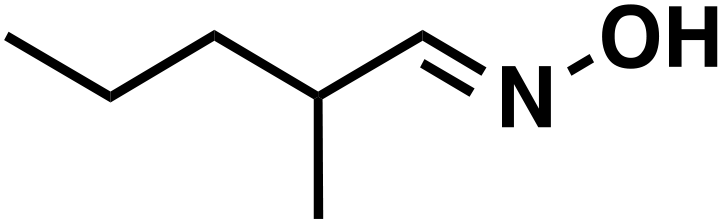 (1c) (1c) |
120 |
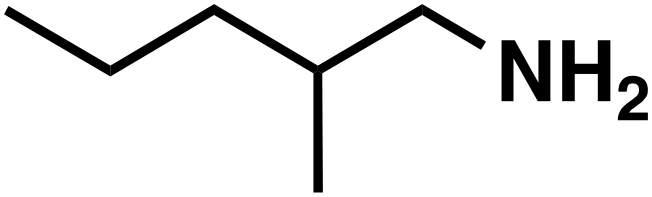 (3c) (3c) |
80 |
| 5 |
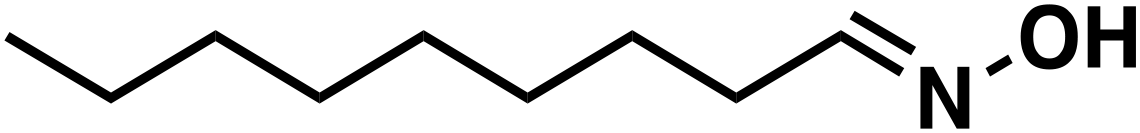 (1d) (1d) |
120 |
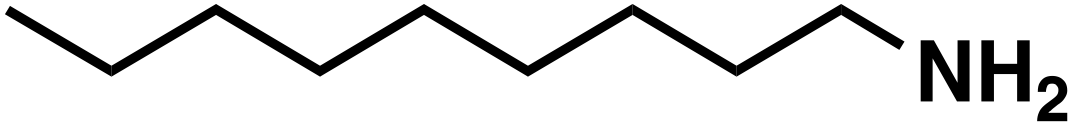 (3d) (3d) |
77 |
| 6 |
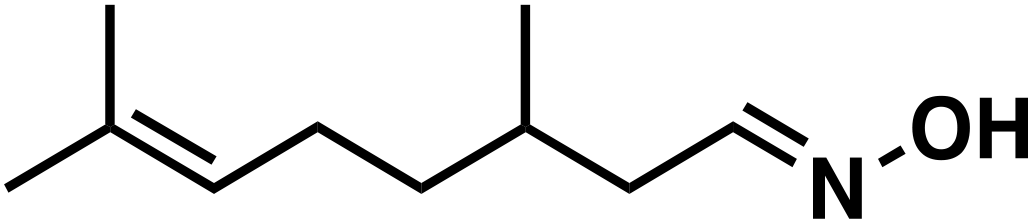 (1e) (1e) |
120 |
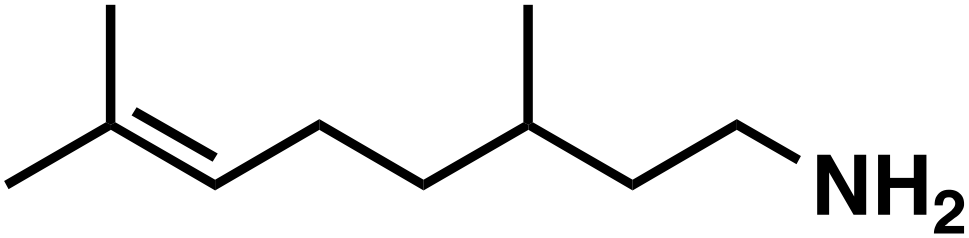 (3e) (3e) |
47 (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)b :1)b |
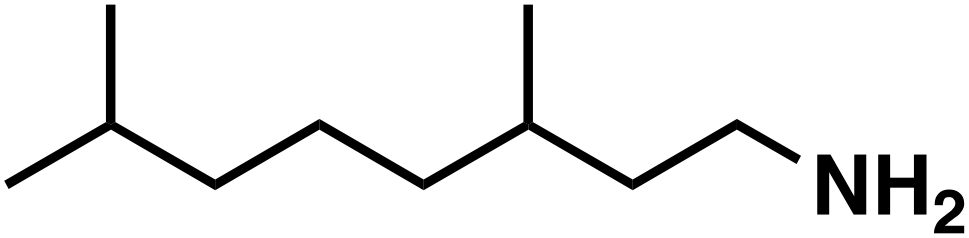 (3e′) (3e′) |
||||
| 7a |
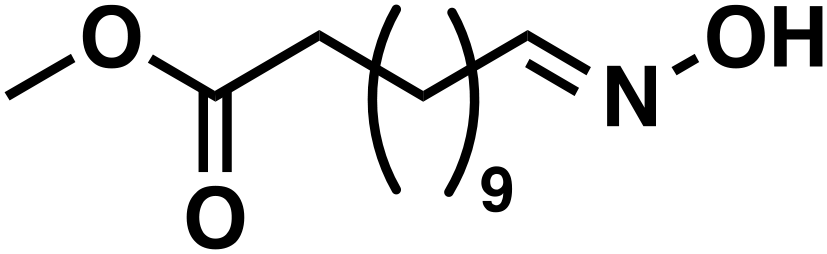 (1f) (1f) |
15 |
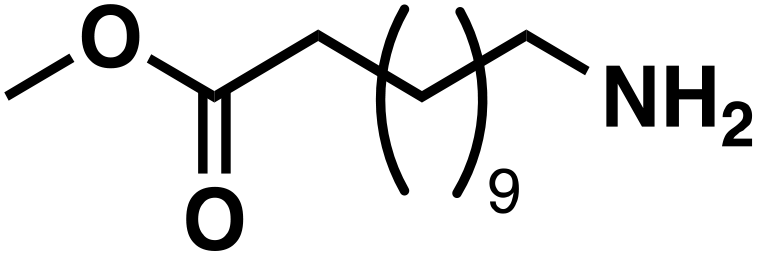 (3f) (3f) |
80 |
| 8 |
 (1g) (1g) |
120 |
 (3g) (3g) |
67 |
| 9 |
 (1h) (1h) |
120 |
 (3h) (3h) |
68 |
| 10a |
 (1i) (1i) |
15 |
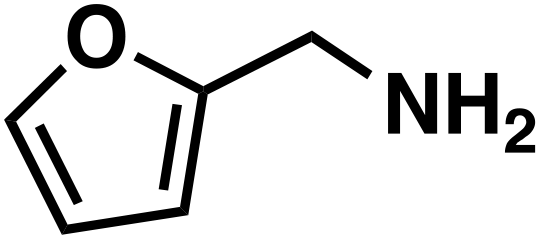 (3i) (3i) |
53 |
| 11 |
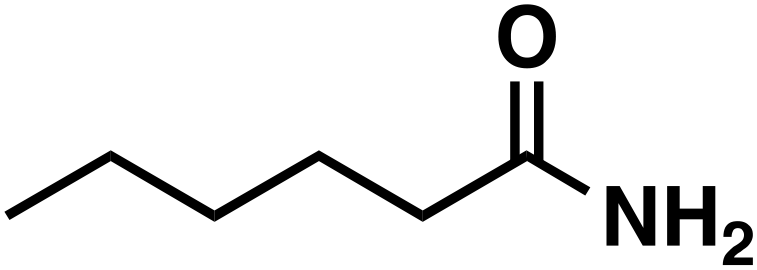 (4a) (4a) |
60 |
 (3a) (3a) |
0 |

All tested oximes could be fully converted with up to 90% selectivity. Since hexylamine (3a) was used for optimization, it reached the highest yield of 90%, with the secondary amine (7a) being the main side-product. The branched (1c) and other chain length oximes (1b, 1d) could be converted successfully with slightly less yield (Table 2, entry 1–5). The main side products were the secondary amine and the alcohol, indicating a lower selectivity toward the organic alkyl-hydroxylamine (2) path.
To our surprise, the NMR spectrum revealed that the double bond in citronellal oxime (1e) stayed 90% intact (Table 2, entry 6). Other functional groups are also tolerated, such as bromide, benzyl and furfuryl moieties (Table 2, entry 8–10), showcasing the high chemoselectivity of the Ru/triphos system towards carbonyl compounds. Again, the secondary amine is the main side product.
Regarding entry 11 no conversion was observed after 1 h of reaction time, confirming that no amide (4) was reduced under the given reaction conditions. Therefore, the amide (4) represents a dead end in this reaction setup.
Butanaloxime (1b) was more difficult to dry, since it did not crystallize and removal of water are complicated with the developed workup. Even small amounts of water lead to hydrolysis of the oxime to the aldehyde, which is then hydrogenated to the alcohol. Compared to the heterogeneous system of Reeve and Christian, the selectivity of butylamine (3b) and benzylamine (3g) are lower, as in contrast the results for the 2-furaldoxime (1i) clearly improved (43% and 53%). Since the focus of our system was on linear substrates, with or without further functional groups, a comparison is only valid to certain point. Also the pressure differences should be mentioned here, where the heterogeneous system applied 200–220 atm of H2.
In comparison to other amination reactions, our newly developed protocol combines great chemoselectivity with high reaction speed. Therefore, adding a valuable tool for the synthesis of primary amines. Noteworthy, the best results were achieved for aliphatic aldoximes, which are synthesized from aldehydes usually produced via hydroformylation.35
In order to demonstrate the feasibility and direct applicability of the catalyst system for industrial processes, further investigations were carried out with readily available bulk chemicals.
Combination with hydroformylation
To realize the desired alkene-to-primary amine path, we added hydroformylation to the reaction cascade, now consisting of three reaction steps. At first, we chose a cyclodextrin-assisted biphasic hydroformylation with a Rh/sulfoxantphos catalyst system, which yields a relatively pure product phase (Table 3, entries 1–3).39,40 This product phase was then combined with an aqueous hydroxylamine solution to form the oximes. In the last step, the oximes were hydrogenated with our newly developed protocol.|
|
||||
|---|---|---|---|---|
| Entry | Corresponding alkene (10) | Yield 8 [%] | Yieldb1 [%] | Yieldb3 [%] |
| Conditions: hydroformylation: preforming: CO/H2 (30 bar), nCO/nH2 = 1, 12 h, at 120 °C; reaction: 32 mmol 10 CO/H2 (30 bar), nCO/nH2 = 1, 6 h, at 120 °C mwater/msubstrate = 0.2, nsubstrate/nRh = 500, nCD/nRh = 100, nP/nRh = 7. Oximation: product phase of hydroformylation, aq. H2NOH (1.1 eq.), HCl (0.1 mol%) 2 h at 30 °C. Hydrogenation: 8.33 mmol 1, Ru(acac)3 (0.5 mol%), triphos (L2) (0.55 mol%), DBU (B4) (2.5 mol%), 1,4-dioxane 30 mL, H2 (50 bar at RT), 700 rpm at 200 °C. The yields were calculated by GC-FID by using dodecane as an internal standard.a Rh(acac)(CO)2, Xantphos in toluene.b Cumulated yield with previous reactions. | ||||
| 1 |
 (10a) (10a) |
76 | 71 | 60 |
| 2 |
 (10d) (10d) |
80 | 76 | 57 |
| 3 |
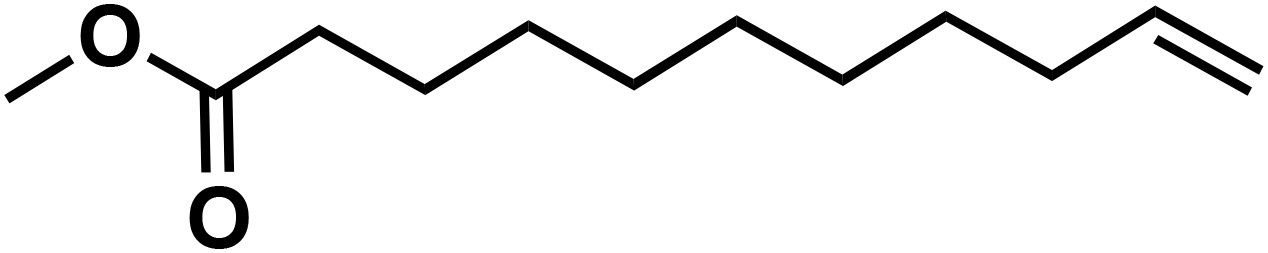 (10f) (10f) |
83 | 79 | 63 |
| 4a |
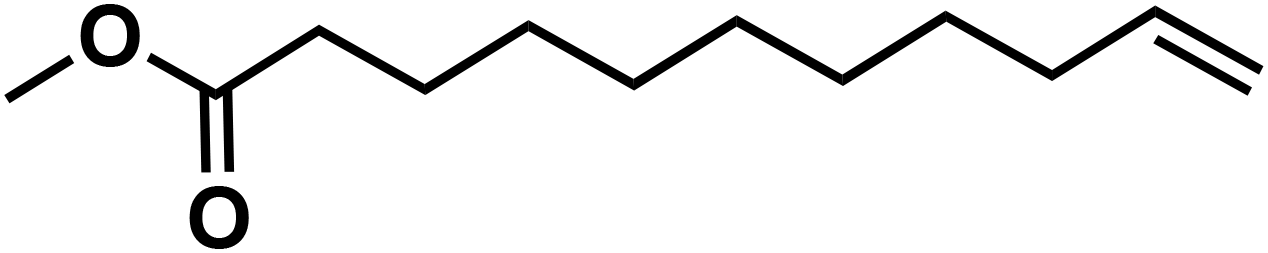 (10f) (10f) |
91 | 85 | 68 |
The hydroformylation is already established, and the combined oximation reaction was carried out with excellent yields in all cases (Table 3, yields 1). The cumulated yields starting from the alkene the yields were between 71% and 79%.
The oximation yields crystalline products, which were filtrated off readily. With hexanal oxime (1a) as an exception, which had to be crystallized from diethyl ether.
All side products and remaining substrate from the hydroformylation step were separated by washing the oxime crystals. Only for hexanal oxime (1a) a small portion of the branched oxime would co-crystallize. Afterward, the oximes were investigated in the hydrogenation reaction under optimized conditions, which resulted in good to very good yields of the primary amine and moderate overall yields (Table 3, yields 3).
The yield obtained from the hydrogenation of hexanal oxime (1a) after the whole reaction cascade starting from the alkene was slightly lower than those synthesized from the purchased aldehyde (Table 3, entry 1, yield 3). This could be caused by the presence of branched oxime or other small impurities resulting from the hydroformylation. In terms of l/b-ratio, the performance was increased by two points to 29.2. For nonylamine (3d) and methyl 12-aminododecanoate (3f), the linearity improves significantly due to an easier separation, as the crystals of the linear product already form during the oximation reaction, and the branched products remain liquid.28 Therefore, the intermediate oximation reaction also allows easy separation of linear and branched products, which otherwise requires costly separation methods for the aldehyde or amine products.
To improve the overall yield of the amine, we carried out the same reaction sequence with a monophasic hydroformylation reaction. The upside is its better performance, and the usual downside is the more difficult separation of the products since it is a monophasic mixture. With our reaction sequence, the separation of the oxime intermediate remains easy and achieves an increased cumulated yield of 85% for methyl 12-oxododecanoate oxime (1f). With the monophasic hydroformylation an increase of the total amine (3f) yield to 68% could be achieved (Table 3, entry 4).
As a proof-of-concept, we used a sample of a hydroformylation reaction solution of a continuously operated mini plant.41 The result is a diluted aldehyde 8f solution, which was then converted into the amine 3fvia its oxime 1f (Scheme 5).
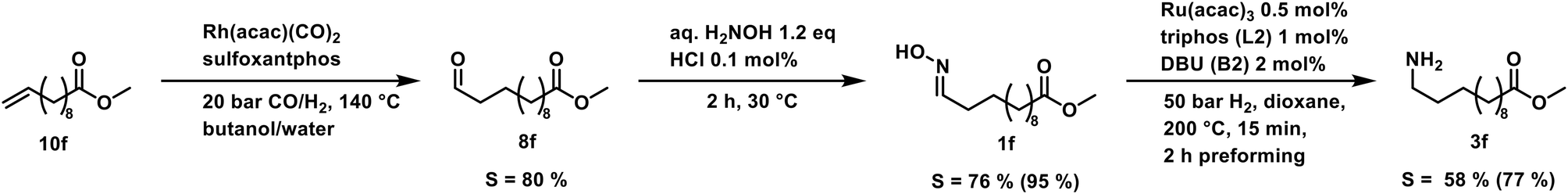 | ||
| Scheme 5 Reaction sequence starting with a reaction solution sample of a continuously operated miniplant. Selectivity combined for the alkene and each reaction in parentheses. | ||
The oximation was carried out with 145 g of the product phase, which contained 58 wt% aldehydes. Hydroxylamine was directly added to this mixture, and in a two-step crystallization, the oxime 1f was obtained in 95% yield. Noteworthy, only 1.2 eq. of the amines source were necessary for the conversion which is significantly lower compared to the large ammonia excess in reductive amination and alcohol amination. This reaction step allows a decoupling of both reactions and can convert even diluted aldehyde solutions. The hydrogenation of the oxime can then be carried out independently.
The final hydrogenation was carried out in a 300 ml autoclave with 25 mmol oxime 1f. Full conversion was achieved after only 5 min, resulting in a high TOF20 of 8934 h−1. After 15 min of reaction time, the highest selectivity of 77% was reached. Continuing the reaction after that point favors the deaminative coupling, reducing the amount of primary amine while the secondary amine is built up (see in ESI†). The productivity (at 20% yield) was high with 1004 h−1. With our protocol, we can accomplish the fast and selective conversion of methyl 10-undecanoate 10f into 12-aminododecanoate 3fvia oxime 1f in a multigram scale, thus offering a new and efficient alternative to the desired but not feasible hydroamination reaction of aliphatic alkenes.
Conclusion and outlook
We present the first homogeneously catalyzed hydrogenation of aliphatic aldoximes into primary aliphatic amines with high selectivity of up to 90% with a productivity (20) of up to 1074 h−1. The reaction is significantly faster than existing protocols and does not require a large excess of the amine source. In addition, the hydrogenation procedure is selective for the conversion of oximes and tolerates reduceable moieties such as double bonds, aromatic groups and esters. Especially the latter provides an appealing route for the preparation of bifunctional polymer precursors.Using this protocol, we show a new reaction sequence that starts from common alkenes via hydroformylation, utilizes the resulting aldehydes for oxime synthesis, and ultimately yields aliphatic primary amines. Our reaction sequence offers a viable alternative to the highly desired but not feasible hydroamination of standard alkenes and is a new approach for synthesizing primary aliphatic amines from alkenes.
The cumulated yield from alkene to the primary amine in the three-step reaction setup reaches up to 68%, with water being the only byproduct. The reaction sequence can be directly applied to current hydroformylation processes, no matter whether mono or biphasic. Since the oxime intermediate can be easily separated, even a purification of linear and branched oxime intermediates is readily possible due to their crystallization behavior, allowing greatly improved l/b ratios of the final products. We converted methyl 10-undecenoate into the PA-12 precursor methyl 12-aminododecanoate with water as the only byproduct. The high l/b ratio of the product is especially remarkable. Up to 77% yield of pure linear ω-amino ester was achieved on a 25 mmol scale with a productivity of 1004 h−1 in the hydrogenation step starting from the oxime.
Our next goal is the conversion of dioximes to obtain diamines such as hexamethylene diamine. Due to the poor solubility in dioxane, further reaction engineering and a change of the reactor setup are required. Additionally, we want to improve the reaction's selectivity in continuous reaction setups.
Associated content
The following files are available free of charge General consideration, experimental procedures, analytical data (1H, 13C NMR, HRMS, GC) (PDF).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the lab staff for their continuous support. Special appreciation to Fabian Niefer for his contribution during his time with the work group. We would also like to thank the group of Prof. Dr. Hiller for the NMR measurements, the group of Dr. Zühlke for the HRMS measurements, and the group of Prof. Dr. Hansmann for the synthesis of 6-bromopicolinaldehyde oxime. Finally, we are very thankful to the German Federal Ministry of Food and Agriculture (Bundesministerium für Ernährung und Landwirtschaft), represented by the FNR (Fachagentur Nachwachsende Rohstoffe) for financial support of the young research group “Renewlysis” (project number 2219NR355). Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 421173804.References
- A. Tullo, C&EN Global Enterprise, 2018, vol. 96, pp. 22–23 Search PubMed.
- S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8126–8129 CrossRef CAS PubMed.
- H. Tobita and A. E. Hamielec, in Ullmann's Encyclopedia of Industrial Chemistry, 2015, pp. 1–50 Search PubMed.
- S. Streiff and F. Jérôme, Chem. Soc. Rev., 2021, 50, 1512–1521 RSC.
- J. Hoover, Science, 2016, 354, 707–708 CrossRef CAS PubMed.
- C. A. Tolman, R. J. McKinney, W. C. Seidel, J. D. Druliner and W. R. Stevens, Adv. Catal., 1985, 33, 1–46 CAS.
- L. Bini, C. Müller and D. Vogt, ChemCatChem, 2010, 2, 590–608 CrossRef CAS.
- L. Bini, C. Müller and D. Vogt, Chem. Commun., 2010, 46, 8325–8334 RSC.
- P. Pollak, G. Romeder, F. Hagedorn and H. Gelbke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2000 Search PubMed.
- Q. Lu, J. Liu and L. Ma, J. Catal., 2021, 404, 475–492 CrossRef CAS.
- D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2015, 357, 883–900 CrossRef CAS.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- T. Gross, A. M. Seayad, M. Ahmad and M. Beller, Org. Lett., 2002, 4, 2055–2058 CrossRef CAS PubMed.
- G. Hahn, P. Kunnas, N. de Jonge and R. Kempe, Nat. Catal., 2018, 2, 71–77 CrossRef.
- K. Murugesan, Z. Wei, V. G. Chandrashekhar, H. Neumann, A. Spannenberg, H. Jiao, M. Beller and R. V. Jagadeesh, Nat. Commun., 2019, 10, 5443 CrossRef PubMed.
- K. Murugesan, Z. Wei, V. G. Chandrashekhar, H. Jiao, M. Beller and R. V. Jagadeesh, Chem. Sci., 2020, 11, 4332–4339 RSC.
- T. Senthamarai, K. Murugesan, J. Schneidewind, N. V. Kalevaru, W. Baumann, H. Neumann, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Nat. Commun., 2018, 9, 4123 CrossRef PubMed.
- X. Ye, P. N. Plessow, M. K. Brinks, M. Schelwies, T. Schaub, F. Rominger, R. Paciello, M. Limbach and P. Hofmann, J. Am. Chem. Soc., 2014, 136, 5923–5929 CrossRef CAS PubMed.
- E. J. Derrah, M. Hanauer, P. N. Plessow, M. Schelwies, M. K. da Silva and T. Schaub, Organometallics, 2015, 34, 1872–1881 CrossRef CAS.
- S. Imm, S. Bähn, M. Zhang, L. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599–7603 CrossRef CAS PubMed.
- D. Pingen, C. Müller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130–8133 CrossRef CAS PubMed.
- D. Pingen, J. B. Schwaderer, J. Walter, J. Wen, G. Murray, D. Vogt and S. Mecking, ChemCatChem, 2018, 10, 3027–3033 CrossRef CAS.
- C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661–8664 CrossRef CAS PubMed.
- I. Damljanović, M. Vukićević and R. D. Vukićević, Monatsh. Chem., 2006, 137, 301–305 CrossRef.
- A. Hinzmann, T. Betke, Y. Asano and H. Gröger, Chem. – Eur. J., 2021, 27, 5313–5321 CrossRef CAS PubMed.
- T. Betke, M. Maier, H. Gruber-Wölfler and H. Gröger, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- C. Plass, A. Hinzmann, M. Terhorst, W. Brauer, K. Oike, H. Yavuzer, Y. Asano, A. J. Vorholt, T. Betke and H. Gröger, ACS Catal., 2019, 9, 5198–5203 CrossRef CAS.
- A. Hinzmann, S. S. Druhmann and H. Gröger, Sustainable Chem., 2020, 1, 275–289 CrossRef.
- M. Terhorst, C. Plass, A. Hinzmann, A. Guntermann, T. Jolmes, J. Rösler, D. Panke, H. Gröger, D. Vogt, A. J. Vorholt and T. Seidensticker, Green Chem., 2020, 22, 7974–7982 RSC.
- P. Kumari, R. Gautam, H. Yadav, V. Kushwaha, A. Mishra, S. Gupta and V. Arora, Catal. Lett., 2016, 146, 2149–2156 CrossRef CAS.
- W. Reeve and J. Christian, J. Am. Chem. Soc., 1956, 78, 860–861 CrossRef CAS.
- E. Gebauer-Henke, W. Leitner, A. Prokofieva, H. Vogt, T. E. Muïler and T. E. Müller, Catal. Sci. Technol., 2012, 2, 2539–2548 RSC.
- F. Bai-cheng, X. Bao-hu, Z. Zhen-chao, H. Yu-hui, J. Yan and Y. Qing-lin, Chem. Eng. Technol., 2022, 45, 1027–1035 CrossRef.
- Y. Liu, Z. Quan, S. He, Z. Zhao, J. Wang and B. Wang, React. Chem. Eng., 2019, 4, 1145–1152 RSC.
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- A. Thomas, B. Matthäus and H.-J. Fiebig, in Ullmann's Encyclopedia of Industrial Chemistry, 2015, pp. 1–84 Search PubMed.
- Y. Li, I. Sorribes, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed.
- P. T. K. Arachchige, H. Lee and C. S. Yi, J. Org. Chem., 2018, 83, 4932–4947 CrossRef CAS PubMed.
- F. Hapiot, L. Leclercq, N. Azaroual, S. Fourmentin, S. Tilloy and E. Monflier, Curr. Org. Synth., 2008, 5, 162–172 CrossRef CAS.
- K. U. Künnemann, L. Schurm, D. Lange, T. Seidensticker, S. Tilloy, E. Monflier, D. Vogt and J. M. Dreimann, Green Chem., 2020, 22, 3809–3819 RSC.
- T. Gaide, J. M. Dreimann, A. Behr and A. J. Vorholt, Angew. Chem., Int. Ed., 2016, 55, 2924–2928 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00368c |
| ‡ These authors contributed equally to this work. All authors have given approval to the final version of the manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |