
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Au nanoparticle decoration of nanoparticular and nanotubular TiO2 using atmospheric pressure cold plasma for photocatalytic applications†
Andjelika
Bjelajac
 *a,
Rada
Petrovic
b,
Milica
Stefanovic
c,
Adrian-Marie
Phillipe
a,
Yves
Fleming
a,
Jérôme
Guillot
a,
Jean-Baptiste
Chemin
a,
Patrick
Choquet
a,
Joris
Kadok
a and
Simon
Bulou
a
*a,
Rada
Petrovic
b,
Milica
Stefanovic
c,
Adrian-Marie
Phillipe
a,
Yves
Fleming
a,
Jérôme
Guillot
a,
Jean-Baptiste
Chemin
a,
Patrick
Choquet
a,
Joris
Kadok
a and
Simon
Bulou
a
aLuxembourg Institute of Science and Technology, 28, Avenue Des Hauts-Fourneaux, L-4365 Esch-sur-Alzette, Luxembourg. E-mail: andjelika.bjelajac@list.lu
bFaculty of Technology and Metallurgy, University of Belgrade, Karnegijeva 4, 11000 Belgrade, Serbia
cInnovation Centre of the Faculty of Technology and Metallurgy, University of Belgrade, Karnegijeva 4, 11000 Belgrade, Serbia
First published on 2nd August 2024
Abstract
Herein, we present the effectiveness of using an atmospheric pressure dielectric barrier discharge (DBD) plasma torch for gold (Au) nanoparticle (NP) decoration of TiO2 nanoparticles and nanotubes (NTs). Au NPs were synthesised using an aerosol of HAuCl4·3H2O solution that was carried with an inert gas to the near plasma post-discharge zone. Careful optimisation of the deposition parameters was done to ensure the uniform and dispersed decoration of TiO2, demonstrated by scanning and transmission electron microscopy with energy dispersive spectroscopy. The X-ray diffraction was used to confirm that the deposit was pure metallic Au. Unlike the bare TiO2 nanoparticles, the samples with Au NPs showed the plasmon resonance peak in the region of 500–600 nm. The photocatalytic property enhancement of Au NP decorated TiO2 structures was demonstrated: The TiO2 NPs@Au nanoparticular powder showed improved photocatalytic activity by enabling methyl orange dye degradation 35% faster than that of the pristine TiO2 NPs; superior photocatalytic behaviour of TiO2 NTs@Au thin films compared to bare TiO2 NTs was observed in the photodegradation of stearic acid.
Introduction
Today's society's biggest two concerns are about solving the energy crisis and reducing pollution. One of the strategies to deal with both is the use of photocatalytic active semiconductor metal-oxides1–3 that by absorbing light create photogenerated charges used further for H2 production,4 CO2 reduction,5 water pollutant degradation6 or for the self-cleaning glass coatings.7 Among many, TiO2 is advantageous as it is cheap, chemically stable and non-toxic.6,8 However, the limitation of TiO2 comes from its absorption range (anatase 3.2 eV), which is around 4% of the solar spectrum.9 To overcome this, different approaches have been studied, like making nanocomposites with nanoparticles (NPs) of a noble metal, such as gold (Au).10 This has been of interest due to the synergetic effect of the surface plasmon resonance (SPR) and the Schottky junction11 that provides minimization of electron/hole recombination and amplifies visible light absorption, thus, resulting in enhancement of photocatalytic activity of TiO2@Au compared to the bare TiO2.12–16 The common decoration technique of TiO2 with Au NPs usually implies time-consuming multisteps, and complex recovery/collection for further handling.17–19 A brief overview of the reported preparation methods for photocatalytic TiO2@Au is provided in Table 1.| TiO2@Au samples | Preparation method | Photocatalytic performances |
|---|---|---|
| 6.1–7.6 nm Au NPs on TiO2 (P25) with 0.25–1.5 wt% of Au20 | Deposition–precipitation method, using urea as a basification agent and HAuCl4·H2O as Au precursor over TiO2 (P25) | The best photocatalytic performance in methyl orange dye degradation was achieved with 0.5 wt% of Au with 6 nm Au NPs over P25 having a rate constant of 0.018 ppm min−1, while for the P25 TiO2 was 0.005 ppm min−1 |
| 80–100 nm wide and 5–10 nm thick TiO2 nanotubes (NTs) decorated with 10 nm Au NPs13 | Anodization of Ti wire to form TiO2 NTs that were used as support for electrodeposition of Au NPs using HAuCl4 solution | The best photocatalytic degradation of methyl orange (5.2 mg L−1, 68.1% in 30 min of UV-vis) was provided by Au/TiO2/Au/TiO2 that was prepared via alternating anodization and electrodeposition |
| 150 ± 10 nm wide and 1.5 μm long TiO2 NTs decorated with ∼20 nm Au NPs21 | Anodized TiO2 NTs were first soaked in a HAuCl4 solution for 16 h followed by a 30 min irradiation with a UVA lamp (500 W) | TiO2 NTs@Au with moderate Au content of 5.52 wt% showed the highest antibacterial activity and the highest anti-inflammatory efficiency under visible-light irradiation. The non-decorated NTs did not show any of these properties |
| Aggregates of ∼23 nm ball-like shaped TiO2 NPs (89% rutile + 11% anatase) with ∼12 nm Au NPs15 | Wet chemical preparation comprising sol–gel method for TiO2 synthesis followed by citrate functionalization and adding HAuCl4·3H2O | 2.5 mg of photocatalyst degraded 5 μM solution of methyl orange dye with the rate constant of 8.54 × 10−2 min−1 |
| 7–20 nm Au NPs with 2–14 wt% loading on P25 (ref. 22) | Colloidal Au NPs were presynthesized via a hydrothermal method. 3-Mercaptopropionic acid was used as a capping ligand to the TiO2 surface, which was removed further by annealing at 200 °C | Methyl orange degradation was achieved under visible light within 3 h with 10 nm Au NPs on P25 (6 wt% of Au) |
| 0.5–3 wt% of 10 nm (±4 nm) Au NPs decorating 50–75 nm TiO2 NPs23 | Aqueous sol–gel method was employed for the decoration of TiO2 with Au NPs using HAuCl4·3H2O as Au precursor, peroxo-titanate sol and NaBH4 as a reducing agent | 10 ppm of RhB and 50 mg of Au@TiO2 with 2 wt% of Au annealed at 450 °C provided ∼95% RhB degradation in 175 min |
| 4–10 nm Au NPs homogeneously dispersed on internal (5–8 nm wide) and external walls (8–12 nm wide) of TiO2 NTs24 | Hydrothermally obtained TiO2 NTs were decorated with Au using HAuCl4 as Au precursor | 50 mL methyl orange solution (40 mg L−1) degraded in 3 h using TiO2 NTs@Au catalyst |
The most convenient and well-recognized protocols to obtain Au NPs on the surface of a semiconductor, such as TiO2, require pre-synthesis of Au NPs that can further be anchored to the surface of TiO2 by simply mixing a Au colloidal solution with the semiconductor dispersion or using an appropriate amount of Au precursor for the precipitation on the TiO2 surface.22,24,25 The obvious disadvantage of this method is the aggregation of NPs during the mixing stage, thus reducing the efficiency of the desired application. Anchoring of Au NPs onto the chemically modified surface of semiconductors using linker molecules, thereby reducing the aggregation or coalescence of Au NPs, was proposed as a solution.26 However, the subsequent removal of the linker has to be made as it can inhibit the photogenerated charge transfer and thus reduce the photocatalytic activity of TiO2@Au. Therefore, herein we explore the possibility of in situ formation and direct deposition of Au NPs on the TiO2 surface using an aerosolized HAuCl4·3H2O solution and an atmospheric pressure (AP) dielectric barrier discharge (DBD) plasma torch at low temperature. The precursor solution droplets react with the plasma reactive species, leading to NP growth.27 We previously demonstrated that this method is adequate for Au NP synthesis on a solid substrate, such as Si or glass.28 Thus, by adapting the set-up, we obtained TiO2@Au nanocomposites with: i) powder of nanoparticular TiO2 and ii) thin films of vertically aligned TiO2 nanotubes (NTs) perpendicularly oriented to the substrate.
Experimental
Sample preparation
As a proof-of-concept, two forms of TiO2 were used for Au decoration: i) commercially available Degussa, P25 powder, consisting of 75% anatase and 25% rutile, that was dispersed in water underneath the plasma torch at a 5 cm distance. The time of deposition was varied to ensure a uniform and good dispersion of Au NPs on the TiO2 surface, where 1 mg of powder in 40 mL of water for 1 h of Au deposition was chosen as the optimal; ii) the TiO2 NTs films were obtained via anodization of titanium (Ti) foils. Ti foils (99.7%, Aldrich) were degreased by ultrasonication in acetone, ethanol, and deionized water (DI) for 30 min (each solvent 10 min). After that, titanium foils were rinsed with DI water and dried in a dryer at 100 °C for 1 h. The ordered TiO2 nanotubes were prepared by using 1 M H3PO4 and 0.5% HF electrolyte. The electrochemical anodization was carried out in a two-electrode cell using a DC power source (PEQLAB EV 231), where a Ti foil was used as an anode and a thin platinum foil as a counter electrode. The distance between the Ti foil and the platinum foil was 2.5 cm. After anodization, the samples were cleaned with DI water and then dried in air. The anodized TiO2 films were annealed in a furnace at 450 °C under an open atmosphere with a heating rate of 5 °C min−1 for 1 h to transform from an amorphous to a crystalline nanotubular structure. Post-calcination under these conditions was performed on the as-anodized TiO2 films to obtain the anatase structure, as it was shown to be much more photocatalytically active than rutile TiO2.12 The annealed samples were allowed to cool down to room temperature and then used as substrates for Au NP deposition. The time of deposition was varied to avoid the excessive amount of Au i.e. by clogging the NTs or complete coverage of the TiO2 surface, hindering the available surface for photocatalytic reactions.29,30 The TiO2 NT films were placed 1.5 cm below the plasma torch.The deposition experimental setup used here consisted of a vertically placed AP torch with a coaxial DBD geometry, composed of 2 concentrical hollow quartz tubes. The plasma was created between the tubes, where the inner tube was coated with Pt using the physical vapor deposition (PVD) technique and the outer tube with Al foil. To produce the plasma, 10 slm of Ar are sent in the gap between the 2 tubes, and a sinusoidal high voltage is applied to the outer electrode (AFS generator, 52 kHz, 20 W). The exact details of the setup can be found in our previous publication.28 As the Au precursor, we used a 0.25 g L−1 HAuCl4·3H2O in pure ethanol. The injection was done at 0.1 ml min−1 using a Hamilton 100 ml syringe and a syringe pump system. The microdroplets were produced thanks to an ultrasonic nebulizer (Sono-Tek®, 1 W, f = 120 kHz). Ar was used as the carrier gas (10 slm flow rate) to carry the aerosol into the plasma near post-discharge.
Structural characterization techniques
TEM analysis was conducted on a JEOL JEM-F200 cold FEG microscope operating at an acceleration voltage of 200 kV. Crystalline nanostructures were analyzed by direct spacing measurements on high-resolution TEM (HRTEM) images using Digital Micrograph Software from Gatan (version v.3.50.3584.0). The energy-dispersive spectroscopy (EDS) mapping was done in scanning mode (STEM). The scanning electron microscopy (SEM) investigations were carried out on a Hitachi SU-70 field emission SEM. X-ray diffraction (XRD) patterns were recorded at a fixed grazing incidence of 0.5° on a PAnalytical X'Pert Pro instrument equipped with a Cu Ka anode (1.54184 Å) and operated at 45 kV and 40 mA.X-Ray photoelectron spectra (XPS) were acquired using a Kratos Axis Ultra-DLD photoelectron spectrometer with a mono-chromatic Al Kα source (E = 1486.6 eV, 150 W) and a 700 × 300 mm spot size. Survey spectra were acquired using an energy resolution of 2.3 eV, whereas high-resolution spectra of the Au 4f, Ti 2p, O 1s, and C 1s regions were collected with a pass resolution of 0.6 eV. The binding energies were referenced to the position of O2− ions (at 530 eV) of the surface lattice oxygen in TiO2, as TiO2 is a certain component of the system.
Photoluminescence spectroscopy was employed to study the photoinduced charge transfer between TiO2 and Au NPs. For that purpose, a Microplate Reader Tecan INFINITE M1000 PRO spectrometer was used with 360 nm excitation light.
Photocatalytic performances analysis
To analyse the absorption properties of the samples, the diffuse reflectance spectra (DRS) were recorded using a LAMBDA 1050 UV–vis–NIR spectrophotometer from Perkin Elmer with a 100 mm integration sphere. Measurements were performed in the UV–vis spectral range (300–1500 nm).Photocatalytic performances evaluation of P25 and P25@Au was done by following the degradation of a common organic water pollutant from the textile industry, methyl orange (MO) dye (10 ml of 5 mg L−1 aqueous solution) using 7 mg of the catalyst and by measuring the intensity of the absorption peak at 468 nm. The samples were dispersed in the dye solution and first left in the dark for 30 min and then illuminated by a solar simulator (CS-100, Oriel Instruments). The sampling of the solution was done in 10 min intervals and the absorption spectra were recorded on a Microplate Reader Tecan INFINITE M1000 PRO spectrometer. To compare the samples, kinetic plots were made with t = f(A/A0), where t is time in minutes, A is the absorption at the time t, and A0 is the initial intensity of the absorption peak at 468 nm. A linear fitting of the ln(A0/A) = kt was made to approximate the kinetic rate, k.31
As a common test for TiO2-based thin film self-cleaning performances, we investigated the rate of stearic acid, CH3(CH2)16CO2H, removal. The TiO2 NTs and TiO2 NTs@Au thin films were analysed after deposition of 10 μl of stearic acid (SA) by spin coating (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 round min−1 during 30 s) after dilution in methanol (2.65 mol L−1) on the substrates.32 Fourier Transform Infrared (FTIR) spectrometry analyses were performed using a BRUKER VERTEX 70FTIR for the samples after 24 h in the dark and each 30 min under solar illumination. The photocatalytic efficiency of the films was estimated by calculating the A/A0, where A is the integrated area of the adsorption peaks at about 2919 cm−1 and 2848 cm−1 after the time t, and A0 is the initial area measured after the samples were kept in the dark. These peaks are attributed to −CH2− band asymmetric and symmetric stretching vibrations,33 and are linked to the quantity of SA. The linear regression of the area band gives a degradation rate of zero-order (cm−1 min−1)7 illustrating the photocatalytic efficiency.
000 round min−1 during 30 s) after dilution in methanol (2.65 mol L−1) on the substrates.32 Fourier Transform Infrared (FTIR) spectrometry analyses were performed using a BRUKER VERTEX 70FTIR for the samples after 24 h in the dark and each 30 min under solar illumination. The photocatalytic efficiency of the films was estimated by calculating the A/A0, where A is the integrated area of the adsorption peaks at about 2919 cm−1 and 2848 cm−1 after the time t, and A0 is the initial area measured after the samples were kept in the dark. These peaks are attributed to −CH2− band asymmetric and symmetric stretching vibrations,33 and are linked to the quantity of SA. The linear regression of the area band gives a degradation rate of zero-order (cm−1 min−1)7 illustrating the photocatalytic efficiency.
Results and discussion
In our previous study,28 we reported the optimisation of the Au NP synthesis parameters using the same plasma torch. We demonstrated how the concentration of the precursor influences the deposition, as well as the solvent composition. As the ethanol-based precursor solution provided the best dispersion and narrow size distribution of the Au NPs, herein for the decoration of TiO2, the same conditions were kept. The only parameter varied now was the deposition time as it directly influences the amount of Au NPs deposited. For the fixed amount of P25 dispersed in water (1 mg) for 0.5 h of Au decoration, not many Au NPs were observed (Fig. S1a†). For 1 h deposition, the Au NPs were well dispersed and of narrow size (Fig. S1b†), unlike for 1.5 h decoration (Fig. S1c†), where not just a higher amount of deposited Au was noticed, but also the NP size inhomogeneity appeared. We wanted to avoid the excessive amount of Au, as it is known that it can hinder photocatalysis, serving as recombination centres for photoelectrons and holes. Moreover, the excessive Au NPs can act as an irradiation shield of the TiO2 surface and prevent the photogeneration of holes necessary for the photocatalytic degradation of a pollutant. The surface of TiO2 must stay accessible for direct contact with a pollutant. Therefore, 1 h of deposition is considered enough to improve the photocatalytic performances of TiO2, but also to be detectable by means of characterisation techniques used herein (XRD, XPS, DRS). Fig. 1a presents pristine P25, whereas the representative micrographs after 1 h of Au plasma deposition are given in Fig. 1b and c. Fig. 1c shows the interplanar spacings of 0.235 nm corresponding to the Au (111) atomic plane, whereas 0.256 nm spacing corresponds to the (101) plane of the anatase phase of P25. The average Au NP size was around 5 nm. This size has been reported as beneficial for photocatalytic performances of TiO2@Au nanocomposites, compared to those with bigger Au NPs.14 | ||
| Fig. 1 TEM micrographs of: a) pristine P25, and b) and c) P25 after decoration with Au NPs. | ||
The results of EDS mapping done in STEM mode of a representative portion of the P25@Au sample are presented in Fig. 2. As it can be seen, the signal for Ti and O is homogeneously distributed, corresponding to the TiO2 from the P25 support, whereas the Au signal is very localized, confirming the formation of NPs on the TiO2 surface. It is worth noting that a certain amount of C and Cl was detected as it can originate from the ethanol-based HAuCl4·3H2O solution. Nevertheless, XRD showed tiny peaks corresponding to metallic gold, 2θ = 38.18° matching the (111) plane and 2θ = 44.39° matching the (200) planes from the JCPDS file 00-066-0091, whereas anatase and rutile identification was provided by comparing the peaks with the 04-014-8515 and 04-005-6491 files (Fig. 3). The results are in accordance with the TEM measurements.
 | ||
| Fig. 2 STEM micrograph and corresponding EDS mappings, for Ti, O and Au, respectively (bar is 100 nm), of P25 after decoration with Au NPs. | ||
 | ||
| Fig. 3 GIXRD diffractograms of: a) bare P25 and b) P25@Au powder. | ||
Another confirmation of the Au decoration of P25 was seen in the DRS spectrum of P25@Au that presented an absorption shoulder in the visible range, unlike that of the bare P25 (Fig. 4a). This is due to the SPR, characteristic for Au NPs, which can lead to an enhancement of photocatalytic activity if the amount of Au is not excessive to hinder the photon flux to the TiO2 surface.3,15,25,29,30,34
 | ||
| Fig. 4 a) Absorbance spectra of bare TiO2 (P25) and P25@Au powders, b) photodegradation of methyl orange using bare P25 and P25@Au and c) linear fitting for estimation of the kinetic constant for P25 and P25@Au photocatalytic efficiency. | ||
To evaluate the influence of the Au NP inclusion on the photocatalytic performances on the obtained TiO2@Au nanocomposites, we followed the degradation of MO. Indeed, the P25@Au sample has exhibited 85% dye degradation after 60 min, whereas pure P25 exhibited it after 80 min (Fig. 4b). The kinetic rates calculated as the slope of the linear fit of t = f(ln(A0/A)), were 0.02 min−1 for P25 and 0.027 min−1 for P25@Au. Similar findings were reported by Oros-Ruiz et al.20 who used the deposition–precipitation method to prepare P25@Au with different loadings of Au NPs, demonstrating that 0.5% of Au provided the best photodegradation of MO compared to others with lower and higher amounts of Au.
Further investigations were about the decoration of anodized TiO2 NTs with Au NPs under the same conditions. The variation of deposition time was done from 2 to 20 min (Fig. S2†). Again, to provide enough evidence of the Au presence, we chose 20 min as the optimal, as, i.e., 2, 5 and 10 min deposition did not show XRD peaks characteristic for Au because of the scarce deposition. The 20 min decoration is clearly visible by comparing the SEM micrographs before (Fig. 5a) and after deposition, as presented in Fig. 5b and c. Au NPs preserved their shape and size as in the case of the decoration of P25. The EDS analysis affirmed the presence of Au (0.34 at%) with a negligible amount of Cl (lower than the detection limit, 0.01 at%). XRD analysis confirmed the presence of Au in the metallic phase (Fig. 6) by detecting characteristic peaks at 2θ = 38.18°, 44.39°, 65.58°, 77.55° and 81.72° matching the (111), (200), (220), (311) and (222) planes (JCPDS file 04-004-8456). For anatase and Ti identification, the JCPDS files 04-014-8515 and 04-007-9313 were used, respectively.
 | ||
| Fig. 5 SEM micrographs of: a) bare TiO2 NTs, and b) and c) TiO2 NTs after Au NP decoration. | ||
 | ||
| Fig. 6 GIXRD diffractograms of: a) bare TiO2 NTs and b) TiO2 NTs@Au thin films. | ||
The bare TiO2 NTs showed the characteristics of the plasmonic structure as reported before by having the second threshold at 547 nm.35 The SPR effect of Au is visible as the absorption peak at 550 nm, whereas for pristine TiO2 NTs in that wavelength, the absorption drops. It is due to the Au deposition that the photocatalytic activity of TiO2 NTs (Fig. 7b) increases. The thin film with Au NPs (20 min deposition) demonstrated the complete photodegradation of stearic acid just after 1 h of illumination, whereas the bare TiO2 NTs reached almost 95% after a much longer time (3.5 h). The SA degradation rates are of zero-order, k = (A/A0)/t7,36 and they are calculated to be 0.11 cm−1 min−1 and 0.18 cm−1 min−1 for bare TiO2 NTs and TiO2 NTs@Au, respectively. Hence, a 60% increase in photocatalytic efficiency is achieved with Au NP decoration. Similarly, TiO2 NTs@Au structures reported by Xu et al.21 demonstrated photocatalytic enhancement compared to bare TiO2 NTs. They prepared Au@TiO2 NTs by soaking the anodized TiO2 NTs films in a 10 mg mL−1 HAuCl4 solution for 16 h. Our 20 min decoration is thus, very much competitive in terms of the preparation time.
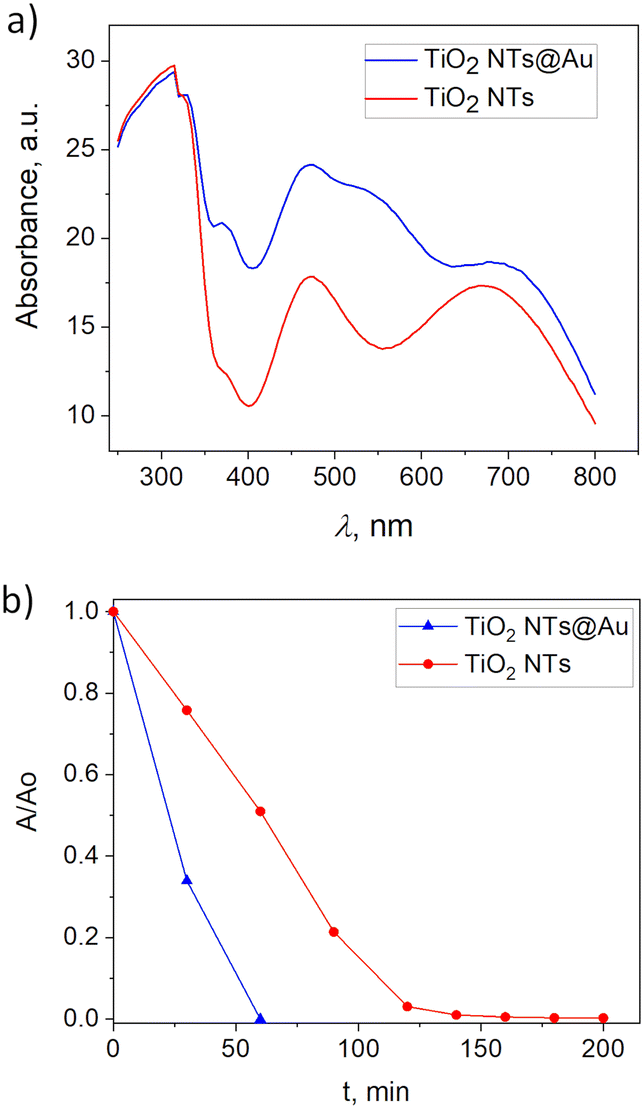 | ||
| Fig. 7 a) Absorbance spectra of bare TiO2 NTs and TiO2 NTs@Au thin films, and b) photodegradation of stearic acid using bare TiO2 NTs and TiO2 NTs@Au thin films. | ||
The XPS results revealed different mechanisms of Au deposition in the case of the decoration of P25 and TiO2 NTs. As P25 powder was dispersed in water during the Au plasma deposition, the oxidation state of Ti and O did not change, as the full width at half maximum (FHWM) remained the same for both Ti and O (spectra given in Fig. 8a and b). The Au 4f spectrum corresponds well to metallic Au, but with a 0.45 eV downshift (from 83.8 eV to 83.35 eV), which indicates significant charge transfer from TiO2 (P25) to Au and thus confirms the strong Au/TiO2 interaction and formation of the Schottky junction.22 However, in the case of a “dry” deposition of Au on TiO2 NTs films, two phenomena were observed: 1) plasma-induced reduction of Ti4+ to Ti3+ creating oxygen vacancies (Fig. 8d and 2) Au 4f position showed a slight upward shift, which can be associated to metallic Au(0) particles partially being encapsulated in carbon,37–39 which originates from the ethanol-based precursor solution. This phenomenon was previously described.28 In addition, a downward shift of the binding energy of C 1s is observed as well, confirming the Au–C interaction, as C-based capping agents could lead to the injection and trapping of electrons from Au NPs to C.37
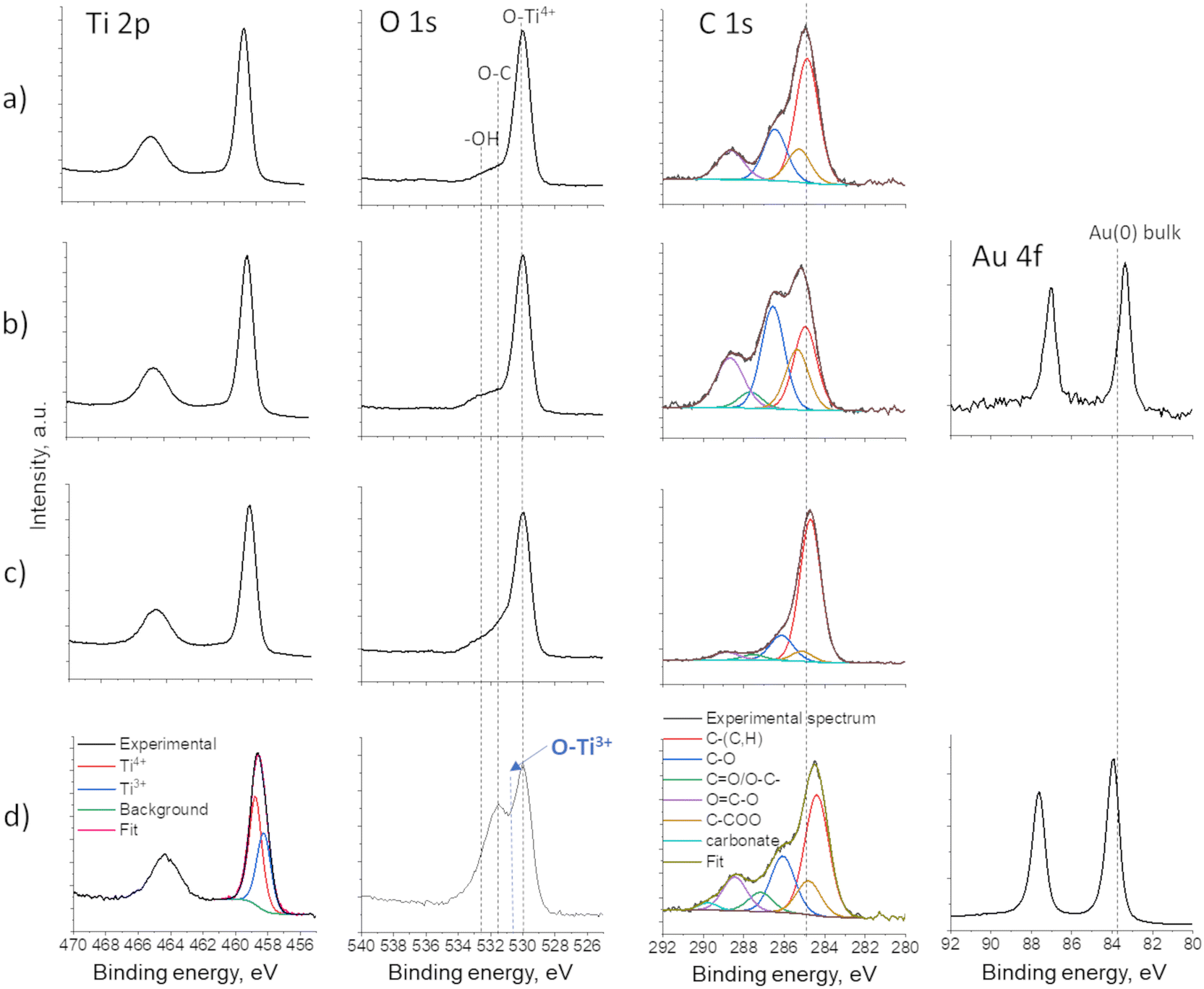 | ||
| Fig. 8 XPS spectra of Ti 2p, O 1s, C 1s and Au 4f of: a) bare P25, b) P25@Au, c) bare TiO2 NTs and d) TiO2 NTs@Au thin films. | ||
Photocatalysis mechanism
The mechanism of photocatalytic degradation of MO and SA could be explained from 3 aspects:40 1) TiO2 absorbs UV light and produced electrons are being transferred to Au, thus recombination of the charges is reduced, 2) Au generates “hot” electrons by visible-light absorption due to the SPR of Au and 3) carbon encapsulating Au NPs in the case of TiO2 NT decoration, serving as electron mediators, and providing efficient isolation of electrons in TiO2 and holes in Au (Fig. 9). | ||
| Fig. 9 Schematic illustration of the proposed mechanisms for photocatalytic degradation of MO and SA using P25@Au and TiO2 NTs@Au catalysts. | ||
1) Upon UV illumination, the electrons (e−) are being excited from the VB to the CB of TiO2, leaving behind holes (h+). As the 5 nm Au NPs created a Schottky junction with TiO2 from P25 (proven by XPS analysis and demonstrated by HRTEM) recombination of photogenerated charges within TiO2 is hindered due to the transfer of electron (e−) from the CB of TiO2 to Au.41,42 These e− can form the superoxide anions (˙O2−) from the adsorbed O2, whereas the holes (h+) in the VB of TiO2 react with H2O or OH− to produce hydroxyl radicals (˙OH). Both O2− and ˙OH are known to cause the decomposition of the azo dyes (such as MO) and SA.
2) However, having tested the photocatalytic activity of the samples just in UV light and obtaining closely the same photocatalytic response for Au@TiO2 and pristine TiO2 (results not shown here), we can conclude that the main contribution of improved photocatalytic activity of Au-decorated samples is coming from the visible light absorption by Au creating additional electrons.13 Under visible light irradiation, the Au plasmon induced the oscillations of free electrons to provide thermal electrons. These “hot” electrons have a higher potential energy than the Schottky barrier that is formed when Au NPs make direct physical contact with TiO2 at the interface. This large potential energy difference will induce a fast and efficient transfer of “hot electrons” to the CB of TiO2.14,16 The Schottky barrier at the interface also helps the transferred “hot” electrons accumulate in the TiO2 CB, preventing them from traveling back to the Au NPs. This is supported by the observation of Fu et al.13 that UV light applied on the system degradation could not produce sufficient electron–hole pairs on the photocatalyst's surface to oxidize MO molecules. Similar findings were reported by Qian et al.14 in the photocatalytic H2 production from H2O, and Verbruggen et al. in the case of photocatalytic degradation of SA,43 where they demonstrated that the photoinduced catalytic activity of TiO2@Au was driven by visible-light excitation of the Au NPs originated from their intrinsic optical properties (SPR). Hence, in our case the visible light absorption is a key factor: it is enhanced with Au deposition with regards to pure TiO2, producing more photogenerated electrons.
3) Moreover, carbon encapsulation of Au in the case of TiO2 NT decoration also contributes to the fast transfer of “hot” electrons.44 The presence of carbon forms a Au–C–TiO2 Z-scheme photocatalytic system,45 where carbon acts as an electron mediator facilitating the electron flow between the two photosystems. The “hot” electrons created due to the SPR of Au are transferred to the carbon interlayer but further recombine with holes from the VB of TiO2 (ref. 41) created upon UV light radiation. This proves efficient isolation of electrons in TiO2 and holes in Au. Thus, Au becomes hole-rich to provide the creation of ˙OH, while TiO2 accumulates electrons for the creation of ˙O2−.
In the present study, the PL spectra are compared to evaluate the interaction of photogenerated charge carriers of Au coupled with TiO2 (Fig. 10). In the case of P25@Au, the presence of a PL band in the visible range (550–700 nm) implies that, as assumed previously, the main mechanism for the photocatalytic reaction is due to the SPR of Au (2), but not due to the reduced recombination by the Schottky junction (1).29,46 However, this band is not present in the case of TiO2 NTs@Au, which could be due to carbon encapsulation of Au (mechanism 3), as noticed by Fernández-Ponce et al. when they measured the weakening of PL emission in the case of Au NPs capped with carbon.47
 | ||
| Fig. 10 PL spectra of a) P25 vs. P25@Au and b) TiO2 NTs vs. TiO2 NTs@Au catalysts. | ||
Conclusions
Herein we demonstrated the effectiveness of an AP cold plasma torch for the in situ creation of ∼5 nm Au NPs with direct decoration of TiO2 powder (nanoparticular P25) as well as nanotubular TiO2 anodized thin films. The Au deposition had a positive effect on the TiO2 photocatalytic performances that were tested by degradation of the MO dye and SA. We believe this is mainly due to the visible light absorption of Au NPs and generation of “hot” electrons thanks to the SPR effect. It is also possible that the charge recombination is reduced by transferring the photogenerated electrons from the conduction band of TiO2 to Au, as the Schottky junction. Moreover, carbon encapsulating Au NPs, in the case of TiO2 NT decoration, can serve as an electron mediator, providing efficient isolation of electrons in TiO2 and holes in Au. This study opens a new direction in TiO2@Au nanocomposite formation as the plasma deposition method provides the tuning of the deposition parameters. Moreover, other metallic NPs can be synthesised using the same method and the decoration of a support of interest can be investigated further.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Andjelika Bjelajac: conceptualization, investigation, and methodology; Rada Petrovic: formal analysis and investigation; Milica Stefanovic: methodology, formal analysis, and investigation; Adrian-Marie Phillipe: formal analysis and investigation; Yves Fleming: formal analysis and investigation; Jérôme Guillot: formal analysis and investigation; Jean-Baptiste Chemin: formal analysis; Joris Kadok: formal analysis and investigation; Patrick Choquet: funding acquisition and resources; Simon Bulou: funding acquisition, investigation, methodology, project administration, supervision, and validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors from LIST gratefully acknowledge the Luxembourg National Research Fund (FNR) for the financial support through the PlaSprayNano project grant (C20/MS/14701998/PlaSprayNano/bulou). The authors from the University of Belgrade gratefully acknowledge the Ministry of Science, Technological Development and Innovation of the Republic of Serbia for the financial support through the project contract no. 451-03-65/2024-03/200135 and 451-03-66/2024-03/200287.Notes and references
- H. Kumari, S. Suman, R. Ranga and S. Chahal, Water, Air, Soil Pollut., 2023, 234, 1–46 CrossRef PubMed.
- F. T. Geldasa, M. A. Kebede, M. W. Shura and F. G. Hone, RSC Adv., 2023, 13, 18404–18442 RSC.
- B. Xue, et al. , Chemosphere, 2023, 332, 138829 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC.
- C. Díaz, M. Segovia and M. L. Valenzuela, Photochem, 2022, 2, 609–627 CrossRef.
- S. Alofi, C. O. Rourke and A. Mills, J. Photochem. Photobiol., A, 2023, 435, 114273 CrossRef CAS.
- H. Lin, C. P. Huang, W. Li, C. Ni, S. I. Shah and Y. H. Tseng, Appl. Catal., B, 2006, 68, 1–11 CrossRef CAS.
- M. Humayun, F. Raziq, A. Khan and W. Luo, Green Chem. Lett. Rev., 2018, 11, 86–102 CrossRef CAS.
- Q. Wang, et al. , ChemNanoMat, 2022, 8, e202100396 CrossRef CAS.
- L. Jing, W. Zhou, G. Tian and H. Fu, Chem. Soc. Rev., 2013, 42, 9509–9549 RSC.
- M. Murdoch, et al. , Nat. Chem., 2011, 3, 489–492 CrossRef CAS PubMed.
- C. Fu, M. Li, H. Li, C. Li, X. Guo Wu and B. Yang, J. Alloys Compd., 2017, 692, 727–733 CrossRef CAS.
- K. Qian, et al. , J. Am. Chem. Soc., 2014, 136, 9842–9845 CrossRef CAS PubMed.
- J. Singh, K. Sahu, B. Satpati, J. Shah, R. K. Kotnala and S. Mohapatra, J. Phys. Chem. Solids, 2019, 135, 109100 CrossRef CAS.
- W. Wang, et al. , Chem. Eng. J., 2023, 455, 140909 CrossRef CAS.
- Q. Fang, et al. , ACS Appl. Energy Mater., 2019, 2, 2654–2661 CrossRef CAS.
- Z. H. N. Al-Azri, et al. , J. Catal., 2015, 329, 355–367 CrossRef CAS.
- R. J. Wong, et al. , Catal. Sci. Technol., 2016, 6, 8188–8199 RSC.
- S. Oros-Ruiz, et al. , Catal. Commun., 2012, 21, 72–76 CrossRef CAS.
- W. Xu, et al. , J. Electroanal. Chem., 2019, 842, 66–73 CrossRef CAS.
- D. Ding, K. Liu, S. He, C. Gao and Y. Yin, Nano Lett., 2014, 14, 6731–6736 CrossRef CAS PubMed.
- S. K. Khore, S. R. Kadam, S. D. Naik, B. B. Kale and R. S. Sonawane, New J. Chem., 2018, 42, 10958–10968 RSC.
- Y. Gao, et al. , Mater. Lett., 2014, 130, 1–4 CrossRef CAS.
- J. Nie, J. Schneider, F. Sieland, S. Xia and D. W. Bahnemann, J. Photochem. Photobiol., A, 2018, 2, 111–117 CrossRef.
- C. Ray and T. Pal, J. Mater. Chem. A, 2017, 5, 9465–9487 RSC.
- E. Nadal, et al. , Nanotechnology, 2021, 32, 175601 CrossRef PubMed.
- A. Bjelajac, et al. , Nanoscale Adv., 2023, 5, 2573–2582 RSC.
- C. Feng, Z. Yu, H. Liu, K. Yuan and X. Wang, Appl. Phys. A: Mater. Sci. Process., 2017, 123, 1–9 CrossRef.
- C. Liu, X. Lu, Y. Tong, J. Z. Zhang, Y. Hsu and Y. Li, Nano Lett., 2013, 13, 3817–3823 CrossRef PubMed.
- A. Bjelajac, I. Florea, M. Zamfir, S. Tusseau Nenez and C.-S. Cojocaru, Nanotechnology, 2023, 34, 495704 CrossRef PubMed.
- B. Dey, et al. , Sci. Rep., 2020, 10(21952), 1–12 Search PubMed.
- D. Li, et al. , Appl. Surf. Sci., 2019, 466, 63–69 CrossRef CAS.
- G. Yang, et al. , Appl. Catal., B, 2018, 234, 260–267 CrossRef CAS.
- A. Bjelajac, et al. , Ceram. Int., 2015, 41, 7048–7053 CrossRef CAS.
- T. W. Liao, et al. , Nanomaterials, 2018, 8, 1–9 CAS.
- A. Patnaik and C. Li, Appl. Surf. Sci., 1999, 140, 197–203 CrossRef CAS.
- F. Gao and Y. Xu, J. Appl. Polym. Sci., 1997, 65, 931–938 CrossRef CAS.
- X. Duan, et al. , Chem. Sci., 2016, 7, 3181–3187 RSC.
- D. Hao, et al. , Energy Mater. Adv., 2021, 2021, 1–12 Search PubMed.
- X. Dai, et al. , J. Phys. Chem. C, 2019, 123, 20325–20332 CrossRef CAS.
- L. G. Devi and R. Kavitha, Appl. Surf. Sci., 2016, 360, 601–622 CrossRef.
- S. W. Verbruggen, et al. , Appl. Catal., B, 2014, 156–157, 116–121 CrossRef CAS.
- S. Chen, X. Li, W. Zhou, S. Zhang and Y. Fang, Appl. Surf. Sci., 2018, 466, 254–261 CrossRef.
- B. Ng, L. K. Putri, X. Y. Kong, Y. W. Teh and P. Pasbakhsh, Adv. Sci., 2020, 7, 1903171 CrossRef CAS PubMed.
- M. Alsaidi, F. A. Azeez, L. A. Al and H. Adel, Environ. Sci. Pollut. Res., 2023, 30, 17951–17964 CrossRef CAS PubMed.
- C. Fernández-Ponce, J. P. Muñoz-Miranda, D. M. de los Santos, E. Aguado, F. García-Cozar and R. Litrán, J. Nanopart. Res., 2018, 20, 305 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00345d |
| This journal is © The Royal Society of Chemistry 2024 |