
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogenolysis of isosorbide to diols and triols over a heterogeneous SiO2-supported Rh catalyst†
Pengru
Chen
 ,
Wataru
Onodera
,
Masato
Akatsuka
,
Yusuke
Kita
and
Masazumi
Tamura
*
,
Wataru
Onodera
,
Masato
Akatsuka
,
Yusuke
Kita
and
Masazumi
Tamura
*
Graduate School of Engineering, Osaka Metropolitan University, 3-3-138, Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: mtamura@omu.ac.jp
First published on 13th March 2024
Abstract
Silica-supported Rh (Rh/SiO2) was an effective and reusable heterogeneous catalyst for the hydrogenolysis of isosorbide, providing diols and triols in 58% total yield. The yield was higher than those obtained by hydrogenolysis of glucose and sorbitol, confirming the high potential of isosorbide as a biomass-derived intermediate for the synthesis of polyols.
Introduction
Lignocellulose biomass is the most abundant carbon-based resource on our planet, which has a potential to replace traditional fossil fuels because biomass has renewable and carbon-neutral properties.1 Development of transformation methods of biomass and biomass-derived intermediates to versatile platform chemicals is critically important to construct a sustainable economy and carbon-neutral society. Among various platform chemicals, diols and triols, carbohydrates with several hydroxyl groups, are fascinating target compounds because they are widely applicable to the synthesis of polymer resins, antifreeze, additives in cosmetics, and transport fuels.2–5 However, biomass-derived intermediates such as monosaccharides and sugar alcohols, easily available from biomass degradation,6–10 have high O/C ratios, and therefore, the selective removal of the OH groups is required for the synthesis of the target polyols.Glucose and xylose, typical monosaccharides, have been applied to the production of C2- or C3-diols such as 1,2-propanediol and ethylene glycol.11–13 However, these processes are complex and challenging, comprising isomerization, retro-aldol condensation and hydrogenation.14,15 Extensive focus is on using versatile sugar alcohols such as glycerol, erythritol, xylitol and sorbitol to synthesize polyols via selective C–O hydrogenolysis with H2 as a reducing agent (Scheme 1). For example, the hydrogenolysis of glycerol can produce 1,2-propanediol via the cleavage of terminal OH groups by using noble metal catalysts (Pt, Pd, Rh-based)16,17 or non-noble metal catalysts (Cu, Ni, Co-based).18–20 Metal-oxide-modified noble-metal catalysts, such as Pt–WOx-based and Ir–ReOx-based catalysts, prefer the formation of 1,3-propanediol by the activation of secondary OH groups.21–23 Erythritol, a C4 molecule produced by the fermentation of glucose,24 could be converted to butanediols (such as 1,4-, 1,3-, 1,2- and 2,3-butanediol) via selective hydrogenolysis over diverse metal-supported catalysts.25–27 When starting from C5 and C6 sugar alcohols, such as xylitol and sorbitol, which are produced by the hydrogenation of xylose and glucose,28 C–O dissociation along with C–C hydrogenolysis occurs, producing C2–C6 polyols.29–32 Moreover, C4–C6 sugar alcohols could be transformed to diols and triols by deoxydehydration in conjunction with hydrogenation over a ReOx–Pd/CeO2 catalyst, where the vicinal OH groups were simultaneously removed.33,34
 | ||
| Scheme 1 The synthesis of valuable diols and triols from typical sugar alcohols and isosorbide (this work) by hydrogenolysis. | ||
Cyclic sugar derivatives such as alkyl glucosides35 and anhydrated sugars are also potential starting materials for the synthesis of diols and triols. Deoxydehydration combined with hydrogenation of methyl glycosides provides methyl dideoxyglycosides via selectively removing cis-vicinal OH groups, which were then converted to hexanetriols via methoxy bond hydrolysis and hydrogenation.36–38 Anhydrosugars are useful feedstocks because one OH group in sugar-derived polyols can be selectively reduced by dehydration. 1,4-Anhydroerythritol, a dehydrated C4 product of erythritol, could undergo selective hydrogenolysis to synthesize 1,3- and 1,4-butanediols in various catalyst systems.39–41 Similarly, C6 anhydrosugars such as isosorbide and sorbitans are potential candidates, however, the transformation has not yet been studied (Scheme 1).
Isosorbide, a stable and commercially available compound derived from glucose, has a peculiar rigid V-shaped structure that incorporates four oxygens in β-position to each other.42 This unique structure endows isosorbide high thermal stability,43 decreasing the possibility of the formation of humins. In the transformation of biomass-derived polyols, humin formation from side reactions, which is liable to occur during C6 sugar alcohols hydrogenolysis, is a serious problem because high-temperature reactions are required for the hydrogenolysis of C–O bonds due to the strong bond even with noble metals.44,45 Moreover, the selective transformation of polyols such as sorbitol and glucose to diols and triols is difficult because polyols have many OH groups with similar reactivities in the flexible alkyl chain. In contrast, isosorbide has two different functional groups, two secondary OH groups and four ether bonds in the rigid structure, and the reactivity difference of these functional groups can be used for the selective transformation. In addition, considering the unique structure of isosorbide, the reactivity of the two OH groups is a little different due to the intramolecular hydrogen bonding, and that of the four ether bonds is also different due to steric hindrance. However, isosorbide is mainly used as the precursor for the formation of solvents and polymers without changing its pristine framework,46 whereas to our best knowledge, the direct transformation of isosorbide as a starting material to diols and triols has not been reported despite the above potential advantages. Wang et al. reported that isosorbide was an important intermediate during sorbitol hydrogenolysis at 523 K over a Pt/NbOPO4 catalyst, where isosorbide could be transformed to hexane via the first ring opening to hexanol and further C–O hydrogenolysis.47 One of the most favorable target products is α,ω-diols such as 1,6-hexanediol and 1,5-pentanediol, which are the most used diols in present chemical industry and are important monomers for the synthesis of polyesters and polycarbonates.48 Other C6 linear diols such as 1,2-, 1,4- and 1,5-hexanediols and C5 linear diols such as 1,2-, 1,3- and 1,4-pentanediols are also used for polymer syntheses.49 Therefore, linear diols, particularly, α,ω-diols such as 1,6-hexanediol and 1,5-pentanediol, are desirable target diols from isosorbide. Considering the above high potentials of isosorbide, we anticipated the selective transformation of isosorbide would be possible by the development of effective metal-supported catalysts differentiating the functional groups.
Here, we report the synthesis of diols and triols from isosorbide by hydrogenolysis. Silica-supported Rh was found to be an effective and reusable heterogeneous catalyst with high catalytic activity and selectivity to diols and triols.
Results and discussion
To begin with, the stability of isosorbide was tested at 453 K, since high temperature generally causes the formation of humic products from monosaccharides and sugar alcohols. Negligible conversion (<0.1%) was detected after 4 h in a H2SO4 aqueous solution (pH = 2), suggesting a high thermal stability of isosorbide under reaction conditions.Next, various SiO2-supported noble metal catalysts were applied to isosorbide hydrogenolysis in H2O at 453 K for 4 h (Fig. 1). The loading amount of the noble metal was 4 wt%. The formation of isosorbide isomers, tetraols, triols, diols and mono-ols as well as that of alkanes by excessive hydrogenolysis was observed. Triols and diols were mainly composed of cyclic and linear C6 and C5 compounds. In the absence of a catalyst or using SiO2 as a catalyst, low conversion was observed (<3%). Among the screened metal catalysts, Ru/SiO2 and Rh/SiO2 exhibited higher conversion than other catalysts. 68% selectivity to total diols and triols was obtained at 85% conversion over Rh/SiO2, while Ru/SiO2 gave priority to the formation of alkanes (51%) and isomers of isosorbide (34%). The higher activity of Rh/SiO2 could be ascribed to the superior hydrogenolysis activity of C–OH and C–O–C bonds than those in the case of other noble metals.50–55 Moreover, it was reported that the ring-opening hydrogenolysis of tetrahydrofurfuryl alcohol (THFA) to 1,2-pentanediol proceeds with comparatively high selectivity (>60%) over the Rh/SiO2 catalyst,54 facilitating the ring opening of isosorbide to form cyclic triols. The product formation rate over Rh/SiO2 (9.69 mmol g−1 h−1 at 453 K, 8 MPa H2 for 4 h) is higher than that reported by Wang et al. (3.55 mmol g−1 h−1, Pt/NbOPO4 at 523 K, 4 MPa H2 for 4 h)47 despite of the lower reaction temperature.
 | ||
| Fig. 1 Catalytic performances of 4 wt% M/SiO2 (M = Ru, Rh, Ir, Pt, Pd) catalysts on isosorbide hydrogenolysis. ○: conversion, bars: selectivities (stripe: alkanes, white: mono-ols, gray: diols, dark gray: triols, gray gride: tetraols, across stripe: isomers of isosorbide, black: others). Reaction conditions: 4 g isosorbide, 2 g H2O, 0.6 g catalyst, 8 MPa H2 (at 453 K), 453 K, 4 h. | ||
The support used for the metal-loaded catalysts plays an important role in the formation of the active sites via interaction between metal species and supports, which can affect the catalytic performance. Various Rh/support catalysts were synthesized by using typical metal oxides as supports (SiO2, ZrO2, Al2O3, CeO2, MgO), and the performance was compared (Fig. 2). Rh/SiO2 showed the highest conversion with diols and triols as main products, followed by Rh/ZrO2. Low conversion of isosorbide was observed when using Rh/γ-Al2O3, Rh/CeO2 and Rh/MgO with more generation of isosorbide isomers. Generally, OH groups can be adsorbed on basic sites strongly56,57 probably to suppress the access to the active Rh metal sites or destabilize the transition state, leading to the low conversion. Moreover, the reducibility of the Rh metal is also important, and inert supports such as SiO2 and C are suitable for the complete reduction of Rh species.53 Therefore, inert SiO2 which has almost no basicity and acidity is a suitable support for the reaction.
 | ||
| Fig. 2 Catalytic performances of Rh/support (Rh = 4 wt%, support = SiO2, ZrO2, Al2O3, CeO2, MgO) catalysts on isosorbide hydrogenolysis. ○: conversion, bars: selectivities (stripe: alkanes, white: mono-ols, gray: diols, dark gray: triols, gray gride: tetraols, across stripe: isomers of isosorbide, black: others). Reaction conditions: 4 g isosorbide, 2 g H2O, 0.6 g catalyst, 8 MPa H2 (at 453 K), 453 K, 4 h. | ||
The Rh/SiO2 catalysts were characterized by H2-TPR, XRD and TEM analyses. Fig. 3(A) shows the TPR profile of Rh/SiO2. The signal due to the reduction of Rh species was observed below the reaction temperature of 453![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K with a Rh reduction degree >99%, indicating that Rh species in Rh/SiO2 was in the metallic state.
K with a Rh reduction degree >99%, indicating that Rh species in Rh/SiO2 was in the metallic state.
 | ||
| Fig. 3 Characterization of Rh/SiO2. (A) TPR profile of Rh/SiO2, (B) XRD patterns of Rh/SiO2, (a) SiO2, (b) RhOx/SiO2 after calcination, (c) Rh/SiO2 at 0 h reaction, (d) Rh/SiO2 at 1 h reaction, (e) Rh/SiO2 at 4 h reaction, (f) Rh/SiO2 at 6 h reaction, and (g) Rh/SiO2 at 8 h reaction. (C) TEM image of used Rh/SiO2 catalyst after 4 h reaction. (D) Rh particle distribution. | ||
Fig. 3(B) shows the XRD patterns of Rh/SiO2 catalysts. The catalyst after calcination (b) showed a broad signal around 34°, which was assigned to RhOx species. After the reaction ((c)–(h)), signals at 41°, 47° and 70° due to the Rh metal and no signals due to RhOx were observed, indicating Rh species were reduced to the metallic state. These results are in agreement with the abovementioned H2-TPR results. The particle size of the Rh metal was calculated to be 3.9 nm from XRD, which was further confirmed by TEM analysis (Fig. 3(C) and (D)), where the aggregation of Rh particles did not occur.
Next, the effect of reaction parameters such as H2 pressure, reaction temperature and isosorbide concentration was investigated over Rh/SiO2. The H2 pressure was in the range of 2–8 MPa (Fig. S1 and Table S1†). At a low H2 pressure of 2MPa, only 37% of isosorbide was converted even after 12h reaction. The conversion linearly increased with increasing the H2 pressure, and at 8 MPa H2 it reached 96% after 8 h. The selectivity to diols and triols increased with increasing H2 by comparing them at the same conversion level (Table S1,† selectivity: 48% (2 MPa) to 69% (8 MPa)), indicating that H2 pressure exhibited a positive effect on the activity and selectivity towards diols and triols over Rh/SiO2. This phenomenon is consistent with previous reports where the activation of H2 is an important step in the hydrogenolysis process.21,23
The reaction temperature also exhibited a positive effect on the conversion of isosorbide in the range of 433 K to 473 K (Table S2†). The maintained high carbon balance (94% after 4 h) even at 473 K implied the good stability of isosorbide during the reaction. Besides, the selectivity to diols and triols was rarely influenced by temperatures. Considering the conversion and yield, the reaction at 453 K was selected for the following study.
The isosorbide concentration dependence on the hydrogenolysis over Rh/SiO2 was investigated (Fig. S2 and Table S3†). The conversion dramatically increased with the increase in the concentration from 3 to 7 M along with the increased selectivity to diols and triols. At higher concentrations ranging from 7 to 27 M, the conversion and selectivity were almost the same, suggesting that the adsorption of isosorbide was saturated over Rh/SiO2. Considering that the concentration in the standard reaction conditions was set at 14 M, isosorbide is adequately supplied to the catalyst surface under the standard reaction conditions.
We further compared the hydrogenolysis activity of other C6 sugars such as isomannide, sorbitol and glucose with that of isosorbide at similar conversion levels (Fig. S3†). The hydrogenolysis of isomannide also selectively formed diols and triols but with a much lower conversion rate of 0.8 mmol h−1 compared to that of isosorbide (19 mmol h−1). The low reactivity was also observed when using sorbitol (0.8 mmol h−1) and glucose (0.9 mmol h−1) as starting materials, where tetraols by random removal of OH groups in the structure, as well as small alkanes by the C–C cleavage, were obtained as main products, while the selectivity to diols and triols was low. Moreover, carbon balance obviously decreased in the cases of sorbitol (91%) and glucose (72%), which would be due to the poor thermal stability under reaction conditions and the formation of humins. These results confirmed that isosorbide is a promising substrate for the synthesis of diols and triols with higher reaction activity.
The reusability of Rh/SiO2 was evaluated in the hydrogenolysis of isosorbide (Table S4†). There were no significant differences in the conversion and product yields between fresh and spent Rh/SiO2 catalysts. The particle size of Rh species was maintained around 3.7–3.9 nm (Fig. S4†), suggesting good stability of Rh/SiO2.
The time course of isosorbide hydrogenolysis over Rh/SiO2 at 453 K and 8 MPa H2 is shown in Fig. 4 (Table S5†). The reaction smoothly proceeded to reach 96% conversion at 8 h. At the initial stage of 26% conversion at 0 h, the isomers of isosorbide were mainly produced in 10% yield (selectivity: 41%), which gradually decreased at longer reaction times. The yield of triols first increased from 0 to 1 h and then leveled off (around 27–29%) when prolonging the reaction time. A monotonously increased tendency was observed in diol yield with respect to reaction time. The maximum total yield of diols and triols was obtained at 8 h reaction with 58% (29% for each), and their distributions are shown in Fig. 4(b) and (c). A mixture of cyclic and linear C6 compounds was observed in triols, which can be formed by the partial and complete tetrahydrofuran ring open, respectively. Determination of the structure of C6 triols is difficult due to the similar mass patterns. 1,6-Hexanediol (5%) and 3,4-hexanediol (7%) were identified as the main C6 linear diols, although cyclic and linear C6 diols were also formed.
 | ||
| Fig. 4 (a) The time course of hydrogenolysis of isosorbide over Rh/SiO2 under optimized conditions. ○: conversion, colored circles: selectivities (blue: alkanes, gray: mono-ols, yellow: diols, red: triols, green: tetraols, purple: isomers of isosorbide). (b) Triol distribution at 8 h reaction, 1,2,6-HxT = 1,2,6-hexanetriol. (c) Diol distribution at 8 h reaction, 1,6-HxD = 1,6-hexanediol, 3,4-HxD = 3,4-hexanediol, 1,2-PeD = 1,2-pentanediol, 1,2-PrD = 1,2-propandiol, EG = ethylene glycol. Reaction conditions: 4 g isosorbide, 2 g H2O, 0.6 g Rh/SiO2 (Rh = 4 wt%), 8 MPa H2 (at 453 K), 453 K. | ||
From the time trend of products (Fig. 4), isosorbide is transformed into triols by hydrogenolysis and then further hydrogenolysis of the triols occurs to provide diols (probably some compounds are produced via the formation of tetraols). Based on previous reports on the hydrogenolysis of polyols and ethers over Rh/SiO2,50–53,58,59 the possible formation route of the identified main products of 1,6-hexanediol and 3,4-hexanediol is proposed (Scheme 2): the first step will be mainly the hydrogenolysis of either one of the C–O bonds in the ether bond (C–O–C) of isosorbide to provide triols with one tetrahydrofuran ring (cyclic C6 triols),50,51,58 and then further hydrogenolysis of the ether bond (C–O–C) of the tetrahydrofuran ring in the cyclic C6 triols provides the corresponding tetraols (1,2,5,6-hexanetetraol and 2,3,4,5-hexentetraol). 1,2,5,6-Hexanetetraol will be mainly converted to 1,6-hexenediol because the selectivity of the C–O bond dissociation of the secondary OH group is quite high (∼87%) in the hydrogenolysis of propylene glycol over Rh/SiO2.52 2,3,4,5-Hexentetraol can be converted to 3,4-hexanediol. Considering the higher reactivity of the linear vicinal OH groups than that of the ether bond in the cyclic ethers,53 the small formation amount of tetraols can be explained by the reactivity of linear vicinal OH groups. Moreover, the formation of C2–C5 diols was also observed, suggesting that degradation of C6 products such as C–C bond hydrogenolysis occurred in some contents, which is in good accordance with the previous report on hydrogenolysis of polyols over Rh catalysts.53,59 A more detailed reaction route is under investigation.
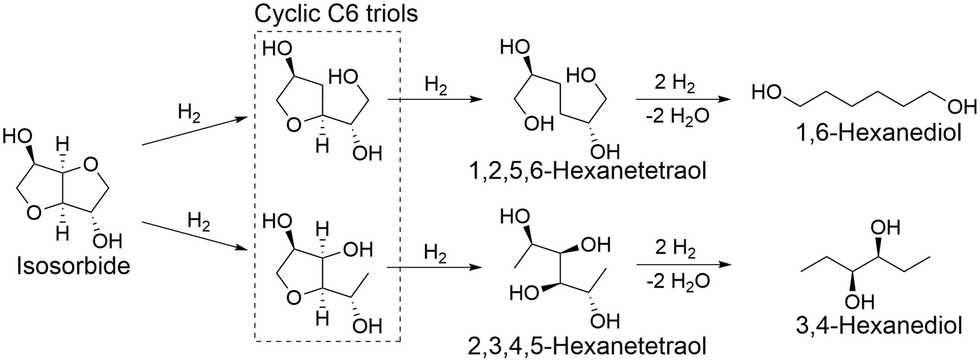 | ||
| Scheme 2 Plausible formation routes of main C6 diols (1,6-hexanediol and 3,4-hexanediol) from isosorbide by hydrogenolysis over Rh/SiO2. | ||
Conclusion
We found that isosorbide is a promising compound for the synthesis of valuable diols and triols due to its high stability. Rh/SiO2 acted as an effective and reusable heterogeneous catalyst for the hydrogenolysis of isosorbide, providing 58% total yield of target products. The characterization of the catalysts by XRD, TPR and TEM analyses suggested that metallic Rh species with 3.9 nm metal particles on the catalyst work as active sites. Moreover, Rh/SiO2 showed good reusability and stability after four cycles without significant change in the particle size of Rh species.Author contributions
Pengru Chen: investigation, formal analysis, and writing – original draft. Wataru Onodera: investigation and formal analysis. Masato Akatsuka: formal analysis. Yusuke Kita: formal analysis. Masazumi Tamura: conceptualization, funding acquisition, formal analysis, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Environment Research and Technology Development Fund [JPMEERF21S11910].Notes and references
- P. Sudarsanam, R. Zhong, S. V. Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed.
- Y. Wang, F. Wang, Q. Song, Q. Xin, S. Xu and J. Xu, J. Am. Chem. Soc., 2013, 135, 1506 CrossRef CAS PubMed.
- D. B. Klinedinst, I. Yilgör, E. Yilgör, M. Zhang and G. L. Wilkes, Polymer, 2012, 53, 5358 CrossRef CAS.
- Y. Gu, M. Tamura, Y. Nakagawa, K. Nakao, K. Suzuki and K. Tomishige, Green Chem., 2021, 23, 5786 RSC.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326 RSC.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51, 761 CrossRef CAS.
- P. Chung, A. Charmot, O. A. Olatunji-Ojo, K. A. Durkin and A. Katz, ACS Catal., 2014, 4, 302 CrossRef CAS.
- P. Chen, A. Shrotri and A. Fukuoka, ChemSusChem, 2019, 12, 2576 CrossRef CAS PubMed.
- P. H. Galebach, D. J. McClelland, N. M. Eagan, A. M. Wittrig, J. S. Buchanan, J. A. Dumesic and G. W. Huber, ACS Sustainable Chem. Eng., 2018, 6, 4330 CrossRef CAS.
- Y. Liu, Y. Liu and Y. Zhang, Appl. Catal., B, 2019, 242, 100 CrossRef CAS.
- M. Gu, Z. Shen, W. Zhang, M. Xia, J. Jiang, W. Dong, X. Zhou and Y. Zhang, ChemCatChem, 2020, 12, 3447 CrossRef CAS.
- A. Aho, S. Engblom, K. Eränen, V. Russo, P. Mäki-Arvela, N. Kumar, J. Wärnå, T. Salmi and D. Y. Murzin, Chem. Eng. J., 2021, 405, 126945 CrossRef CAS.
- M. Zheng, J. Pang, R. Sun, A. Wang and T. Zhang, ACS Catal., 2017, 7, 1939 CrossRef CAS.
- M. Lv, Y. Zhang, Q. Xin, D. Yin, S. Yu, S. Liu, L. Li, C. Xie, Q. Wu, H. Yu and Y. Liu, Chem. Eng. J., 2020, 396, 125274 CrossRef CAS.
- Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2011, 1, 179 RSC.
- Q. Sun, S. Wang and H. Liu, ACS Catal., 2017, 7, 4265 CrossRef CAS.
- S. Wang, Y. Zhang and H. Liu, Chem. – Asian J., 2010, 5, 1100 CrossRef CAS PubMed.
- X. Guo, Y. Li, R. Shi, Q. Liu, E. Zhan and W. Shen, Appl. Catal., A, 2009, 371, 108 CrossRef CAS.
- E. Ryneveld, A. S. Mahomed, P. S. Heerden, M. J. Green and H. B. Friedrich, Green Chem., 2011, 13, 1819 RSC.
- Y. Amada, Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2011, 105, 117 CrossRef CAS.
- J. Wang, X. Zhao, N. Lei, L. Li, L. Zhang, S. Xu, S. Miao, X. Pan, A. Wang and T. Zhang, ChemSusChem, 2016, 9, 784 CrossRef CAS.
- L. Liu, S. Kawakami, Y. Nakagawa, M. Tamura and K. Tomishige, Appl. Catal., B, 2019, 256, 117775 CrossRef.
- Y. Nakagawa, T. Kasumi, J. Ogihara, M. Tamura, T. Arai and K. Tomishige, ACS Omega, 2020, 5, 2520 CrossRef CAS.
- M. Gu, L. Liu, Y. Nakagawa, C. Li, M. Tamura, Z. Shen, X. Zhou, Y. Zhang and K. Tomishige, ChemSusChem, 2021, 14, 642 CrossRef CAS.
- L. Liu, J. Cao, Y. Nakagawa, M. Betchaku, M. Tamura, M. Yabushita and K. Tomishige, Green Chem., 2021, 23, 5665 RSC.
- W. Wang, K. Nakagawa, T. Yoshikawa, T. Masuda, E. Fumoto, Y. Koyama, T. Tago and H. Fujitsuka, Appl. Catal., A, 2021, 619, 118152 CrossRef CAS.
- H. Kobayashi, Y. Hosaka, K. Hara, B. Feng, Y. Hirosaki and A. Fukuoka, Green Chem., 2014, 16, 637 RSC.
- Q. Xia, G. Zhang, J. Wang, W. Zhang, M. Liu, Y. Li, B. Yin, C. Yang, J. Shen and X. Jin, Ind. Eng. Chem. Res., 2020, 59, 13879 CrossRef CAS.
- H. Liu, Z. Huang, H. Kang, X. Li, C. Xia, J. Chen and H. Liu, Appl. Catal., B, 2018, 220, 251 CrossRef CAS.
- X. Jin, J. Shen, W. Yan, M. Zhao, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, ACS Catal., 2015, 5, 6545 CrossRef CAS.
- Y. Yu, Q. Zhang, X. Chen, Y. Liu, Y. Qin, S. Qiu, H. Li and T. Wang, Fuel Process. Technol., 2020, 197, 106195 CrossRef CAS.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 1897 CrossRef CAS PubMed.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 3213 CrossRef CAS.
- A. Karam, K. D. O. Vigier, S. Marinkovic, B. Estrine, C. Oldani and F. Jérôme, ACS Catal., 2017, 7, 2990 CrossRef CAS.
- S. H. Krishna, J. Cao, M. Tamura, Y. Nakagawa, M. D. Bruyn, G. S. Jacobson, B. M. Weckhuysen, J. A. Dumesic, K. Tomishige and G. W. Huber, ACS Sustainable Chem. Eng., 2020, 8, 800 CrossRef CAS.
- M. Tamura, N. Yuasa, J. Cao, Y. Nakagawa and K. Tomishige, Angew. Chem., Int. Ed., 2018, 57, 8058 CrossRef CAS PubMed.
- J. Cao, M. Tamura, R. Hosaka, A. Nakayama, J. Hasegawa, Y. Nakagawa and K. Tomishige, ACS Catal., 2020, 10, 12040 CrossRef CAS.
- T. Wang, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2019, 12, 3615 CrossRef CAS PubMed.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2020, 22, 2375 RSC.
- K. Tomishige, M. Yabushita, J. Cao and Y. Nakagawa, Green Chem., 2022, 24, 5652 RSC.
- F. Aricò, A. S. Aldoshin and P. Tundo, ChemSusChem, 2017, 10, 53 CrossRef PubMed.
- J. Hong, D. Radojčić, M. Ionescu, Z. S. Petrović and E. Eastwood, Polym. Chem., 2014, 5, 5360 RSC.
- S. Luo, J. Li, J. Ran, R. Yangcheng, Y. Cui, Y. Zhang and J. Wang, Catal. Commun., 2023, 175, 106614 CrossRef CAS.
- A. Aho, S. Engblom, K. Eränen, V. Russo, P. Mäki-Arvela, N. Kumar, J. Wärnåa, T. Salmi and D. Murzin, J. Chem. Eng., 2021, 405, 126945 CrossRef CAS.
- W. Qian, X. Tan, Q. Su, W. Cheng, F. Xu, L. Dong and S. Zhang, ChemSusChem, 2019, 12, 1169 CrossRef CAS PubMed.
- J. Xi, Q. Xia, Y. Shao, D. Ding, P. Yang, X. Liu, G. Lu and Y. Wang, Appl. Catal., B, 2016, 181, 699 CrossRef CAS.
- Y. Gu, M. Tamura, Y. Nakagawa, K. Nakao, K. Suzukic and K. Tomishige, Green Chem., 2021, 23, 5786 RSC.
- B. M. Stadler, A. Brandt, A. Kux, H. Beck and J. G. Vries, ChemSusChem, 2020, 13, 556 CrossRef CAS PubMed.
- S. Koso, N. Ueda, Y. Shinmi, K. Okumura, T. Kizuka and K. Tomishige, J. Catal., 2009, 267, 89 CrossRef CAS.
- S. Koso, Y. Nakagawa and K. Tomishige, J. Catal., 2011, 280, 221 CrossRef CAS.
- A. Shimao, S. Koso, N. Ueda, Y. Shinmi, I. Furikado and K. Tomishige, Chem. Lett., 2009, 38, 540 CrossRef CAS.
- I. Furikado, T. Miyazawa, S. Koso, A. Shimao, K. Kunimori and K. Tomishige, Green Chem., 2007, 9, 582 RSC.
- Y. Nakagawa and K. Tomishige, Catal. Today, 2012, 195, 136 CrossRef CAS.
- K. Chen, K. Mori, H. Watanabe, Y. Nakagawa and K. Tomishige, J. Catal., 2012, 294, 171 CrossRef CAS.
- M. Tamura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 864 CrossRef CAS PubMed.
- D. R. Mullins, S. D. Senanayake and T. L. Chen, J. Phys. Chem. C, 2010, 114, 17112 CrossRef CAS.
- T. Arai, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2016, 9, 1680 CrossRef CAS PubMed.
- Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2010, 94, 318 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00266k |
| This journal is © The Royal Society of Chemistry 2024 |