
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A cobalt molecular catalyst for hydrogen evolution reaction with remarkable activity in phosphate buffered water solution†
Caterina
Trotta
a,
Pardeep
Dahiya
b,
Lorenzo
Baldinelli
 a,
Gabriel
Menendez Rodriguez
*a,
Priyanka
Chakraborty
b,
Giovanni
Bistoni
*a,
Filippo
De Angelis
a,
Basker
Sundararaju
*b and
Alceo
Macchioni
*a
a,
Gabriel
Menendez Rodriguez
*a,
Priyanka
Chakraborty
b,
Giovanni
Bistoni
*a,
Filippo
De Angelis
a,
Basker
Sundararaju
*b and
Alceo
Macchioni
*a
aDepartment of Chemistry, Biology and Biotechnology and CIRCC, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123-Perugia, Italy. E-mail: gabriel.menendezrodriguez@unipg.it; giovanni.bistoni@unipg.it; alceo.macchioni@unipg.it
bDepartment of Chemistry, Indian Institute of Technology Kanpur, 208 016 Kanpur, Uttar Pradesh, India. E-mail: basker@iitk.ac.in
First published on 29th May 2024
Abstract
Herein, we show that [Cp*Co(2-ampy)I]I (2-ampy = 2-aminomethyl-pyridine) is an extremely active catalyst for HER, exhibiting a TOF of 109![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 in phosphate buffered water solution (pH 7). The key to this remarkable activity stems from the establishment of a network of weak interactions in the second coordination sphere. As a matter of fact, both experimental and theoretical studies strongly suggest that the –NH2 functionality of the 2-ampy ligand acts as an anchoring and orienting group for H2PO4− through the establishment of an intermolecular hydrogen bonding with it that, in turn, intermolecularly donates a proton to Co–H liberating H2.
000 s−1 in phosphate buffered water solution (pH 7). The key to this remarkable activity stems from the establishment of a network of weak interactions in the second coordination sphere. As a matter of fact, both experimental and theoretical studies strongly suggest that the –NH2 functionality of the 2-ampy ligand acts as an anchoring and orienting group for H2PO4− through the establishment of an intermolecular hydrogen bonding with it that, in turn, intermolecularly donates a proton to Co–H liberating H2.
The production of green hydrogen by renewable energy-powered water electrolysis is considered a viable solution to the problems of fossil solar-fuels depletion and the dramatic environmental consequences of their massive use.21–24 Catalysis plays an essential role in the realization of an efficient electrolysis apparatus making it possible to minimize the energy expenditure required, close to that established by thermodynamics, for both reductive and oxidative processes.25–29 With regard to the hydrogen evolution reaction (HER) significant advances have been achieved in the identification of molecular catalysts based on earth-abundant metals, such as iron,30–32 cobalt,33–38 nickel,39–42 copper43,44 and molybdenum.45,46 Several key factors contributing to improve HER catalysis have been identified.47–55 It has been understood that having a hanging functionality acting as proton source in the proximity of the metal active site is essential to facilitate the protonation of the M–H intermediate, thus leading to H2 evolution (D–H⋯H–M, Scheme 1a). This is exactly what occurs in [Fe,Fe] or [Ni,Fe] hydrogenases,56–59 which efficiently catalyze the reduction of protons to H2 [turnover frequency (TOF) values up to ca. 104 s−1], where an –NH moiety of the thiolate bridging ligand is responsible for intramolecularly relaying the proton from the external source to the M–H fragment. This teaching of Nature inspired the development of many mono- and dinuclear complexes, having a basic group in the second coordination sphere, as structural and functional models of hydrogenase.39,60–68 For instance, Rauchfuss and co-workers demonstrated that the presence of a pendant amine, in a structural model of [Fe,Fe] hydrogenase, enhances the HER rate.69 DuBois and co-workers reported a nickel complex [Ni(PPh2NPh)2](BF4)2, (PPh2NPh = 1,3,6-triphenyl-1-aza-3,6-diphosphacycloheptane) with remarkable activity (TOF = 106
000 s−1, in acetonitrile with 1.2 M of water) in HER attributed to the key role played by a pendant amine acting as intramolecular proton relay.39 Analogously, Nocera and co-workers showed that the presence of a carboxylic acid hanging group of a cobalt “hangman” porphyrin facilitates HER by mediating, intramolecularly, a proton-coupled electron transfer (PCET) process.66–68
 | ||
| Scheme 1 Second coordination sphere proton relay processes (a–c) facilitating HER catalysis (M = transition metal, D–H = proton donor, A–H = anchoring group). | ||
Interestingly, there are now some pieces of evidence clearly indicating that HER reaction can be also facilitated by intermolecular protonation (D–H⋯H–M, Scheme 1b). Particularly, Bren and co-workers recently reported insightful studies on the effects of pKa and buffer nature on the activity of cobalt-based catalysts.70–72 They found an acceleration of HER rates of 2 to 4 orders of magnitude moving from water to buffered water solution, at the same pH, demonstrating an active role of the buffer as a proton donor. Sakai and co-workers attributed the remarkable activity of a cobalt catalyst with a pentadentate macrocyclic ligand in HER to the key role played by H2PO4− derived from the phosphate buffer as a proton source.73
Herein, we show that [Cp*Co(2-ampy)I]I (2-ampy = 2-aminomethyl-pyridine; 1, Scheme 2) is an extremely active catalyst for HER exhibiting a remarkable TOF of 109000 s−1 in phosphate buffered water solution (PBS, pH 7). Mechanistic studies suggest that the key to success of 1 lies in the occurrence of a crucial hydrogen bond network in the second coordination sphere: the –NH2 functionality of the 2-ampy ligand acts as an anchoring and orienting group for H2PO4− through the establishment of an intermolecular hydrogen bonding with it, which, in turn, donates a proton to the M–H group (D–H⋯A–H⋯H–M, Scheme 1c).74 This hypothesis has been supported by i) contrasting the electrocatalytic performances of 1 to those of selected members of the novel family of potentially active [Cp*CoL3] catalysts (Scheme 2), deliberately turning of or altering the crucial intermolecular hydrogen bonds by a proper choice of the bidentate ligand; ii) in depth quantum DFT calculations aimed at understanding the ligand effect on the catalytic performance implicitly taking into account the interactions with the buffer.
 | ||
| Scheme 2 Structure of cobalt complexes. | ||
Novel complexes 1 and 2 were synthesized by the reaction of Cp*Co(CO)I2 (ref. 75) with commercially available inexpensive 2-ampy and characterized both in solution and in the solid state (ESI†) (Scheme 3). Complexes 3 (ref. 76) and 4 (ref. 77) were reported by one of us and prepared as per the procedure described.
 | ||
| Scheme 3 Synthesis of cobalt complexes 1 and 2 (ORTEP diagram of complexes with thermal ellipsoids are shown at the 50% probability level, H atoms are omitted for clarity). | ||
Complexes 1–4 were characterized electrochemically by cyclic voltammetry (CV) and square wave voltammetry (SWV) (Fig. 1). Each compound displays a redox event in the −0.2 and −0.5 V potential range attributed to the CoIII/II reduction. Peak current versus square root of scan rate is always linear, indicating a diffusional nature of the process (Fig. S2–S5†).
 | ||
| Fig. 1 a) CVs of 1–4 (500 μM) at 100 mV s−1 scan rate and b) SWVs of 1–4 (500 μM) recorded in 0.1 M PBS at pH 7 under N2. | ||
As shown in Table 1, the half-wave potential (E1/2) and redox behavior of the CoIII/CoII couple of 1–4 are sensitive to the nature of the ligand coordinated at the Cp*Co moiety. 1–2 show a quasi-reversible reduction as indicated by the large peak-to-peak separation (ΔEp), with a peak current ratio (iox/ired) nearly one. Notably, a positive shift of 58 mV is observed when comparing the E1/2 of 1 (−0.350 V) to that of 2 (−0.292 V). Considering the greater electron donation ability of methyl relative to hydrogen, a shift in the opposite direction should be observed suggesting that a different ligand is coordinated at Cp*Co(N,N) moiety in 1 and 2.
| Complex | E 1/2 (V) | ΔEp (mV) | i ox/ired |
|---|---|---|---|
| 1 | −0.350 | 95.2 | 0.93 |
| 2 | −0.292 | 88.2 | 0.89 |
| 3 | −0.345 | 65.9 | 1.49 |
| 4 | −0.375 | 64.1 | 1.01 |
To validate this possibility, CVs and SWVs of 1 and 2 were recorded in a 0.1 M NaClO4 (pH 7) electrolyte solution instead that in PBS. This led to an anodic shift of 30 mV of the CoIII/II redox potential of 1, while had no (or a negligible) effect on that of 2 (Fig. S6 and S7†), confirming the hypothesis that 1 and 2 in PBS differ in the nature of the third ligand coordinated at Co, which is most likely phosphate in 1 and water in 2. Consistently, a cathodic shift of E1/2 was observed for 1 upon increasing pH (i.e. increasing the HPO42−/H2PO4− ratio, Fig. S8†).78,79 The CoIII/II redox couple of 3 and 4 are observed at E1/2 = −0.345 (ΔEp = 65.9 mV) and −0.375 V (ΔEp = 64.1 mV), respectively, indicating the higher electron donor properties of the carboxylate ligand.
At more cathodic potentials, an additional irreversible wave, characterized by a significant current increase, was observed (Fig. 2). It was ascribed to the electrocatalytic hydrogen evolution reaction: 2H+ + 2e− → H2 (HER). H2 production was confirmed by rotating ring disk electrochemistry (ESI†). 1 and 2 show similar onset potentials (−1.13 V and −1.11 V, respectively). For 2 the catalytic wave is preceded by an irreversible wave that might be ascribed to the transformation of the precatalyst into the active species or the formation of a stable catalytic intermediate.78,793 and 4 exhibit a considerably more anodic onset potential of −0.88 V and −0.95 V, respectively. As shown in Table 2, the largest current increase (icat/id = 136, where icat is the maximum catalytic current and id is the cathodic peak current of the CoIII/II redox couple) was observed for 1, suggesting an extremely fast catalytic reaction, followed by 2 (icat/id = 77), 4 (icat/id = 15) and 3 (icat/id = 10). Remarkably, moving from 1 to 2, i.e. substituting a hydrogen atom with a methyl group, strongly affects the catalytic activity, causing a decrease of approximately one-half of the icat/id ratio.
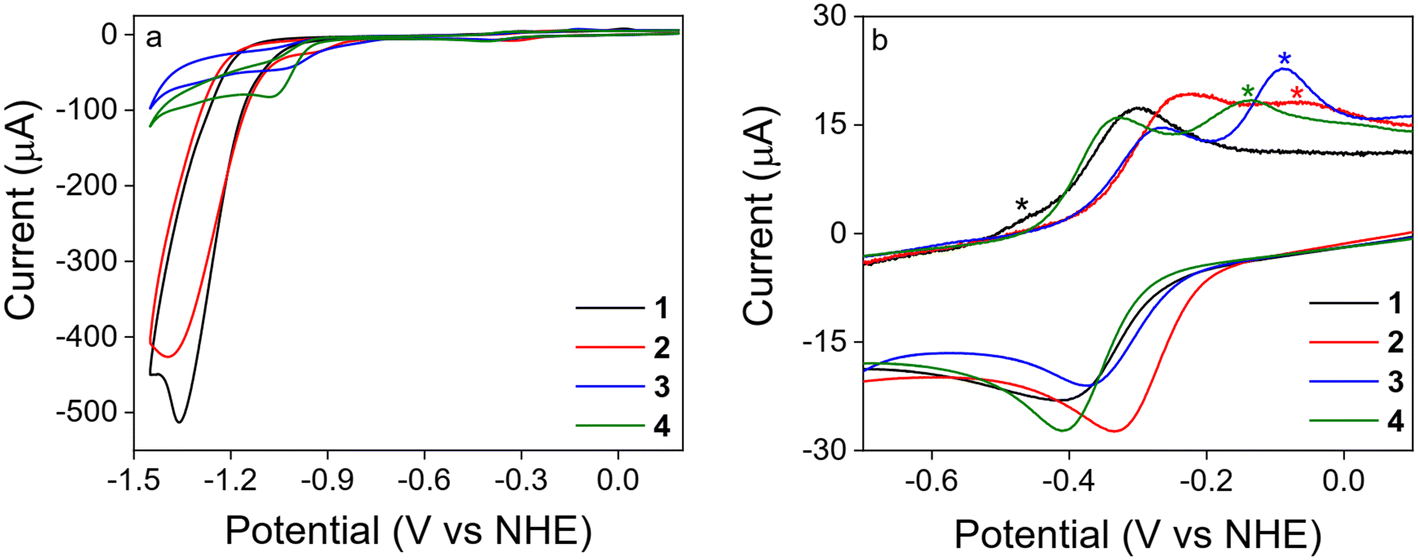 | ||
| Fig. 2 a) CVs of 1–4 (500 μM) at 100 mV s−1 scan rate recorded in 0.1 M PBS at pH 7 under N2. b) Enlarged view of CoIII/II redox region of the first CV of 1–4 (500 μM) recorded at 1 V s−1 from 0.19 to −1.45 V in 0.1 M PBS at pH 7 under N2 (asterisks denote the new oxidation features observed on the return sweep ascribed to catalyst degradation products). | ||
| Complex | i cat/id | η(Ecat/2) (V) | TOF (s−1) |
|---|---|---|---|
| 1 | 136 | 1.034 | 39000 ± 3000 |
| 2 | 77 | 0.964 | 16000 ± 2500 |
| 3 | 10 | 0.635 | 900 ± 50 |
| 4 | 15 | 0.689 | 2000 ± 250 |
Before conducting further mechanistic studies, we investigated whether the electrocatalytic activity of 1–4 was indeed ascribed to the homogeneous catalyst in solution, rather than to any electrocatalytically active species bound to the surface. The possible formation of any deposited species under catalytic conditions was evaluated by continuously recording 50 CV scans from 0.19 to −1.45 V at 1 V s−1 scan rate on a 500 μM solution of catalyst. In all cases, the catalytic current at −1.45 V gradually loses intensity as new oxidation waves appear in the −0.7 to 0.2 V potential range (Fig. S13–S16†). This behaviour is consistent with the formation of new deposited species derived from partial catalyst decomposition. The intensity of the CoIII/II redox couple of 1–4 also decreased over the same period, further indicating that the surface composition of the glassy carbon electrode was changing. However, the rinse test, performed after 50 scans, clearly shows a negligible activity of the catalyst decomposition products. Controlled-potential electrolysis (CPE) measurements carried out at −1.35 V confirmed the rather short live of the catalytic systems, as well as the substantial inactivity of deposited species, since the recorded current abruptly decreases, reaching the one obtained in the absence of catalyst in less than 3 minutes (Fig. S17†). Exception made for complex 4, whose degradation somehow leads to a slightly active species (Fig. S17†). It is worth noting that a significant difference was found between 1 and 2–4 in terms of catalyst stability. In particular, the degradation of 2–4 is more pronounced than that of 1. This can be deduced by the noticeable appearance of new oxidative waves in the first CV of the solution of 2–4. Unlike 1, for which additional redox events due to catalyst decomposition are almost negligible (Fig. 2b). This result demonstrates that 1, not only is the most active catalyst, but also the one that is less prone to degradation. More importantly, it was found that such degradation processes are only relevant in those experiments for which the catalyst is exposed to reductive potentials for a substantial period of time. As a matter of fact, in CV experiments of 1 recorded at high scan rates (>6 V s−1), i.e. where the time scale of the experiment is considerably reduced, no sign of degradation is observed (Fig. S18†). Under such conditions, pure catalytic regime is reached, allowing the TOF to be determined.
Considering the above results, to prevent that any deposited species negatively influences the current response of the studied complexes, only the first scan, recorded on a freshly polished electrode, was considered. Moreover, this work focuses only on the evaluation of the activity of the molecular complexes in solution and the related HER mechanism, not on long-term activity since in that case the formation of heterogeneous deposits on the electrode surface cannot be neglected.
In order to benchmark our catalysts, the HER overpotential, η(Ecat/2),80,81 and TOF82,83 of 1–4 were determined (ESI†). As summarized in Table 2, 1–2 operates at higher overpotentials (1.034 and 0.964 V, respectively) than 3–4 (0.635 V and 0.689 V, respectively). Catalysts with somewhat lower overpotential have been reported in the literature.63,84–86 For 1, a remarkable TOF of 39000 s−1 was observed (Fig. S20†). Whereas TOF values of 16000 s−1, 900 s−1 and 2000 s−1 were obtained for 2, 3 and 4, respectively (Fig. S21–S23†). It can be hypothesized that the superior performances of 1 is due to the presence of the –NH2 functionality that binds H2PO4− anion, properly orienting it in a way to facilitate the proton transfer to Co–H (vide infra).87,88 The significant lower activity of 2 compared to 1 is also consistent with such interpretation since having a –NHMe in place of –NH2 clearly disfavors hydrogen bonding with H2PO4−, at least statistically. The higher activity of 4 with respect to 3 might be attributed to the greater ability of the former to undergo hydrogen bonding with H2PO4−, using the lone pairs of the coordinated carboxylic oxygen atom.
Interestingly, the Log(TOF) versus overpotential plot for 1–4 exhibits a nice linear trend (Fig. 3) suggesting that the same reaction mechanism is reasonably active and the structural differences of complexes systematically tune the free energies of intermediates and reaction steps of the catalytic cycle.89–94 Our DFT computational results are in line with this hypothesis (vide infra).
 | ||
| Fig. 3 Plot of Log(TOF) as a function of catalyst overpotential. | ||
Additional electrochemical mechanistic studies were carried out for 1 exhibiting the highest catalytic performances. TOF remains constant (TOFmean = 42000 s−1) changing catalyst concentration in the 100–500 μM range (Fig. 4a), consistently with a first order on 1. By varying the pH of PBS from 5 to 6 does not significantly affect the activity of 1 (TOF = 49000 ± 2000 s−1, pH 5; TOF = 55000 ± 4000 s−1, pH 6) (Fig. 4b). However, a further increase of pH leads to a significant decrease of TOF (42000 ± 2000 s−1, pH 7; 2400 ± 250 s−1, pH 8). This peculiar behavior suggests that H2 evolution rate might be also influenced by the concentration of H2PO4−, whose concentration decreases as pH increases.38,95–97 To corroborate this hypothesis, electrocatalytic experiments were performed at constant pH varying buffer strength. It was observed a marked increase of TOF with increasing buffer strength (Fig. 4c), up to a value of 109000 ± 7000 s−1, obtaining another strong hint that H2PO4− plays an active role in hydrogen evolution, likely acting as proton source. Consistently, a significantly lower current was observed using NaClO4 or K2SO4 as supporting electrolytes instead of PBS (Fig. 4d).
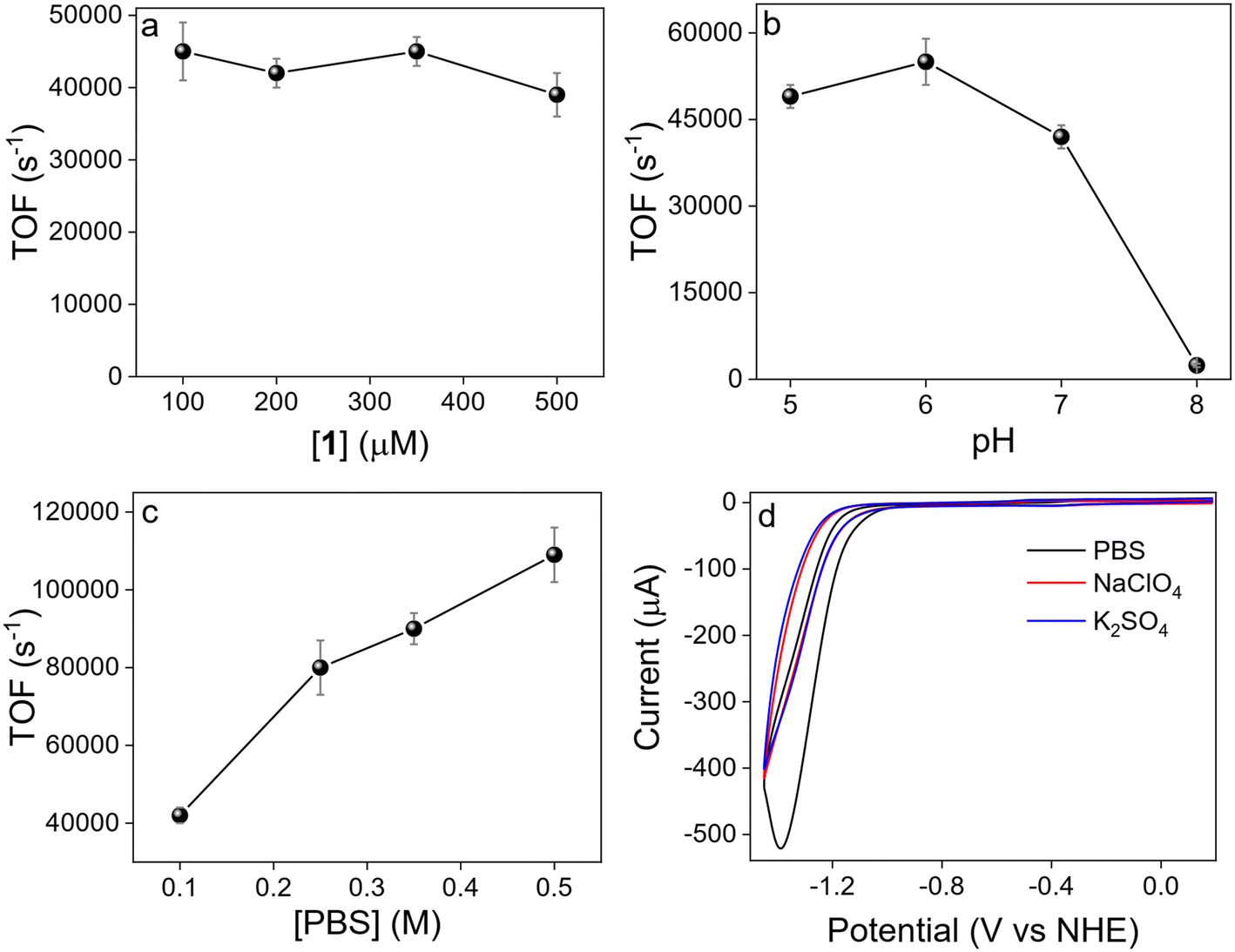 | ||
| Fig. 4 Dependence of TOF on a) [1] (0.1 M PBS at pH 7 under N2), b) pH ([1] = 200 μM, 0.1 M PBS under N2), and c) [PBS] ([1] = 200 μM, pH 7 under N2). d) CVs of 1 (200 μM) at 100 mV s−1 scan rate using PBS (black trace), NaClO4 (red trace) and K2SO4 (blue trace) as supporting electrolyte at pH 7 (0.1 M). | ||
The experimental results reported above were complemented by extensive DFT computational studies for 1 (Fig. 5) and 3 (as reference, ESI†) aimed at providing insights into the mechanistic aspects of HER catalysis, focusing on understanding the intimate reason why 1 exhibit such a remarkable activity. Our results indicate that HER catalysis by 1 and 3 follows the same reaction mechanism (Fig. 5a and S33†), which is consistent with the observed scaling relationships for these catalysts. In the first step, [Co(III)–H]+n [where Co = Cp*Co(N,N)] forms through a PCET pathway by far at lower energy (+0.95 eV for 1 and +0.78 eV for 3) compared to the two-step reduction-protonation counterpart (+1.66 eV for 1 and +1.58 eV for 3). Importantly, the calculated energy associated to the formation of the hydride species of 3 (0.78 eV), which is responsible for the measured overpotential, is significantly lower than that of 1 (0.95 eV). This trend nicely correlates with the experimentally measured overpotentials of 1 and 3, which amounts to 1.03 V and 0.63 V, respectively. Once [Co(III)–H]+n is formed, the presence of the phosphate becomes crucial for the HER catalytic mechanism. It was found that the most favorable pathway for H2 generation involves the coordination of H2PO4− in the second sphere of [Co(III)–H]+n resulting in the formation of the key intermediate [Co(III)–H]+n(H2PO4−). It is in this context that the difference between 1 and 3 appear the most. The optimized [Co(III)–H]+nH2PO4− structures shown in Fig. 5b, reveal that while a strong Co–H⋯HOPO3H dihydrogen bond98,99 is present both in 1 (distance = 1.59 Å) and 3 (distance = 1.56 Å), a strong –NH2⋯OPO3H− establishes in 1 (distance = 1.78 Å). The cooperativity of those two types of H-bonding in 1 contributes to a more facile association of H2PO4− in the second coordination sphere in 1 (+0.08 eV) than in 3 (+0.29 eV), thus favoring hydrogen donation. Once H2PO4− is in the second coordination sphere, the kinetic barrier to transfer a proton to Co–H is almost zero for both 1 and 3, and the increase of energy passing from [Co(III)–H]+n(H2PO4−) to [Co(III)–OH2]+n and H2, through a PCET pathway, is similar (+0.15 eV for 1, +0.12 eV for 3; ESI†).
 | ||
| Fig. 5 a) Proposed catalytic HER cycle for 1 and energetics at 0.0 V (blue) and 0.95 V (red). At potentials higher than 0.95 V, all steps involving an electron transfer from the electrode become exergonic. b) DFT-optimized structures for [1(III)–H]+(H2PO4−) and [3(III)–H](H2PO4−). Hydrogen-bond distances are reported in Å. | ||
In conclusion, we have reported some molecular cobalt catalysts highly active in hydrogen evolution reaction. Particularly, complex [Cp*Co(2-ampy)I]I 1, whose second coordination sphere was rationally designed for having a –NH2 moiety that attracts and orients H2PO4− of the buffer, which, in turn, acts as proton donor to the metal hydride functionality, exhibits extremely high TOF (109000 s−1), importantly, working in water exclusively. To the best of our knowledge, only an organometallic [2Fe–2S] mimic of the active site of an [FeFe]-hydrogenase enzyme shows considerably higher TOF in water solution (250000 s−1).65 Considering that 1 belongs to a broad class of compounds, we believe that a rational design of the ancillary ligands, not only in terms of desired electronic and steric features but also of proper functionalities in the second coordination sphere, will likely lead to the discovery of other catalysts with even better performances.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 VITALITY. BS acknowledge SERB (CRG/2020/001282) for to support this re-search. PD and PC thank CSIR and IITK for their fellowship. BS thanks Dr. Manoj K. Gangwar for initial study in isolation of these complexes. We acknowledge Università degli Studi di Perugia and MUR for support within the project Vitality, Progetti Fondo Ricerca di Ateneo 2021 and PON Ricerca e Innovazione DM 1062 (J91B21003250006). We also thank the Fondazione Perugia (project 21101, 2022.0405) for supporting our research.References
- E. Higuchi, H. Uchida and M. Watanabe, J. Electroanal. Chem., 2005, 583, 69–76 CrossRef CAS.
- Y. Bao, K. Nagasawa, Y. Kuroda and S. Mitsushima, Electrocatalysis, 2020, 11, 301–308 CrossRef CAS.
- J. G. Vos and M. T. M. Koper, J. Electroanal. Chem., 2019, 850, 113363 CrossRef CAS.
- J. Zheng, Y. Yan and B. Xu, J. Electrochem. Soc., 2015, 162, F1470–F1481 CrossRef CAS.
- S. K. M. Padavala and K. A. Stoerzinger, ACS Catal., 2023, 13, 4544–4551 CrossRef CAS.
- Q. Wang, J. Guo and P. Chen, Joule, 2020, 4, 705–709 CrossRef.
- F. Neese and J. Wiley, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132 DOI:10.1063/1.3382344.
- S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211–228 CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. Trasatti, Pure Appl. Chem., 1986, 58, 955–966 CrossRef CAS.
- I. A. Topol, G. J. Tawa, S. K. Burt and A. A. Rashin, J. Chem. Phys., 1999, 111, 10998–11014 CrossRef CAS.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, J. V. Coe and T. R. Tuttle, J. Phys. Chem. A, 1998, 102, 7787–7794 CrossRef CAS.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- R. Eisenberg and D. G. Nocera, Inorg. Chem., 2005, 44, 6799–6801 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, Angew. Chemie Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- G. Menendez Rodriguez and A. Macchioni, Eur. J. Inorg. Chem., 2023, 26, e202200625 CrossRef CAS.
- W. T. Eckenhoff, Coord. Chem. Rev., 2018, 373, 295–316 CrossRef CAS.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- F. Zaccaria, G. Menendez Rodriguez, L. Rocchigiani and A. Macchioni, Front. Catal., 2022, 2, 892183 CrossRef.
- R. Mejia-Rodriguez, D. Chong, J. H. Reibenspies, M. P. Soriaga and M. Y. Darensbourg, J. Am. Chem. Soc., 2004, 126, 12004–12014 CrossRef CAS PubMed.
- F. Quentel, G. Passard and F. Gloaguen, Energy Environ. Sci., 2012, 5, 7757–7761 RSC.
- Y. Na, M. Wang, K. Jin, R. Zhang and L. Sun, J. Organomet. Chem., 2006, 691, 5045–5051 CrossRef CAS.
- Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS PubMed.
- K. M. Waldie, S. K. Kim, A. J. Ingram and R. M. Waymouth, Eur. J. Inorg. Chem., 2017, 2017, 2755–2761 CrossRef CAS.
- W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. N. Ma, H. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., 2012, 124, 6043–6046 CrossRef.
- A. Call, F. Franco, N. Kandoth, S. Fernández, M. González-Béjar, J. Pérez-Prieto, J. M. Luis and J. Lloret-Fillol, Chem. Sci., 2018, 9, 2609–2619 RSC.
- C. R. Carr, A. Taheri and L. A. Berben, J. Am. Chem. Soc., 2020, 142, 12299–12305 CrossRef CAS PubMed.
- J. W. Wang, K. Yamauchi, H. H. Huang, J. K. Sun, Z. M. Luo, D. C. Zhong, T. B. Lu and K. Sakai, Angew. Chemie Int. Ed., 2019, 58, 10923–10927 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- L. Gan, T. L. Groy, P. Tarakeshwar, S. K. S. Mazinani, J. Shearer, V. Mujica and A. K. Jones, J. Am. Chem. Soc., 2015, 137, 1109–1115 CrossRef CAS PubMed.
- O. R. Luca, S. J. Konezny, J. D. Blakemore, D. M. Colosi, S. Saha, G. W. Brudvig, V. S. Batista and R. H. Crabtree, New J. Chem., 2012, 36, 1149–1152 RSC.
- E. S. Wiedner, A. M. Appel, S. Raugei, W. J. Shaw and R. Morris Bullock, Chem. Rev., 2022, 122, 12427–12474 CrossRef CAS PubMed.
- D. Khusnutdinova, B. L. Wadsworth, M. Flores, A. M. Beiler, E. A. Reyes Cruz, Y. Zenkov and G. F. Moore, ACS Catal., 2018, 8, 9888–9898 CrossRef CAS.
- P. Zhang, M. Wang, Y. Yang, T. Yao and L. Sun, Angew. Chemie Int. Ed., 2014, 53, 13803–13807 CrossRef CAS PubMed.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed.
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698–702 CrossRef CAS PubMed.
- K. Koshiba, K. Yamauchi and K. Sakai, Angew. Chemie Int. Ed., 2017, 56, 4247–4251 CrossRef CAS PubMed.
- B. D. Stubbert, J. C. Peters and H. B. Gray, J. Am. Chem. Soc., 2011, 133(45), 18070–18073 CrossRef CAS PubMed.
- H. Lei, A. Han, F. Li, M. Zhang, Y. Han, P. Du, W. Lai and R. Cao, Phys. Chem. Chem. Phys., 2014, 16, 1883–1893 RSC.
- A. Mahammed, B. Mondal, A. Rana, A. Dey and Z. Gross, Chem. Commun., 2014, 50, 2725–2727 RSC.
- B. Mondal, K. Sengupta, A. Rana, A. Mahammed, M. Botoshansky, S. G. Dey, Z. Gross and A. Dey, Inorg. Chem., 2013, 52, 3381–3387 CrossRef CAS PubMed.
- H. Lei, H. Fang, Y. Han, W. Lai, X. Fu and R. Cao, ACS Catal., 2015, 5, 5145–5153 CrossRef CAS.
- D. L. Dubois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS PubMed.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- P. Wang, G. Liang, C. E. Webster and X. Zhao, Eur. J. Inorg. Chem., 2020, 2020, 3534–3547 CrossRef CAS.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- A. Adamska-Venkatesh, D. Krawietz, J. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2014, 136, 11339–11346 CrossRef CAS PubMed.
- Y. Nicolet, A. L. De Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS PubMed.
- R. Tatematsu, T. Inomata, T. Ozawa and H. Masuda, Angew. Chemie Int. Ed., 2016, 55, 5247–5250 CrossRef CAS PubMed.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. R. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed.
- M. Fang, E. S. Wiedner, W. G. Dougherty, W. S. Kassel, T. Liu, D. L. Dubois and R. M. Bullock, Organometallics, 2014, 33, 5820–5833 CrossRef CAS.
- A. Dutta, S. Lense, J. Hou, M. H. Engelhard, J. A. S. Roberts and W. J. Shaw, J. Am. Chem. Soc., 2013, 135, 18490–18496 CrossRef CAS PubMed.
- K. E. Clary, M. Karayilan, K. C. McCleary-Petersen, H. A. Petersen, R. S. Glass, J. Pyun and D. L. Lichtenberger, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32947–32953 CrossRef CAS PubMed.
- W. P. Brezinski, M. Karayilan, K. E. Clary, N. G. Pavlopoulos, S. Li, L. Fu, K. Matyjaszewski, D. H. Evans, R. S. Glass, D. L. Lichtenberger, J. Pyun, P. Brezinski, M. Karayilan, K. E. Clary, N. Avlopoulos, R. S. Glass, D. L. Lichtenberger, J. Pyun, S. Li, L. Fu, K. Matyjaszewski and D. H. Evans, Angew. Chemie Int. Ed., 2018, 57, 11898–11902 CrossRef CAS PubMed.
- C. H. Lee, D. K. Dogutan and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 8775–8777 CrossRef CAS PubMed.
- M. M. Roubelakis, D. K. Bediako, D. K. Dogutan and D. G. Nocera, Energy Environ. Sci., 2012, 5, 7737–7740 RSC.
- D. K. Bediako, B. H. Solis, D. K. Dogutan, M. M. Roubelakis, A. G. Maher, C. H. Lee, M. B. Chambers, S. Hammes-Schiffer and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15001–15006 CrossRef CAS PubMed.
- J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2012, 4, 26–30 CrossRef CAS PubMed.
- J. M. Le, G. Alachouzos, M. Chino, A. J. Frontier, A. Lombardi and K. L. Bren, Biochemistry, 2020, 59, 1289–1297 CrossRef CAS PubMed.
- J. L. Alvarez-Hernandez, A. E. Sopchak and K. L. Bren, Inorg. Chem., 2020, 59, 8061–8069 CrossRef CAS PubMed.
- J. L. Alvarez-Hernandez, J. W. Han, A. E. Sopchak, Y. Guo and K. L. Bren, ACS Energy Lett., 2021, 6, 2256–2261 CrossRef CAS.
- J. W. Wang, K. Yamauchi, H. H. Huang, J. K. Sun, Z. M. Luo, D. C. Zhong, T. B. Lu and K. Sakai, Angew. Chemie Int. Ed., 2019, 58, 10923–10927 CrossRef CAS PubMed.
- J. N. H. Reek, B. de Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Chem. Rev., 2022, 122, 12308–12369 CrossRef CAS PubMed.
- B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Adv. Synth. Catal., 2014, 356, 1491–1495 CrossRef CAS.
- P. Dahiya, A. Sarkar and B. Sundararaju, Adv. Synth. Catal., 2022, 364, 2642–2647 CrossRef CAS.
- P. Dahiya, M. K. Gangwar and B. Sundararaju, ChemCatChem, 2021, 13, 934–939 CrossRef CAS.
- C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC.
- K. J. Lee, B. D. McCarthy and J. L. Dempsey, Chem. Soc. Rev., 2019, 48, 2927–2945 RSC.
- D. H. Pool, M. P. Stewart, M. O'Hagan, W. J. Shaw, J. A. S. Roberts, R. M. Bullock and D. L. DuBois, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15634–15639 CrossRef CAS PubMed.
- A. Dutta, S. Lense, J. Hou, M. H. Engelhard, J. A. S. Roberts and W. J. Shaw, J. Am. Chem. Soc., 2013, 135, 18490–18496 CrossRef CAS PubMed.
- C. P. Andrieux, J. M. Dumas-Bouchiat and J. M. Savéant, J. Electroanal. Chem. Interfacial Electrochem., 1980, 113, 1–18 CrossRef CAS.
- J. M. Saveant and E. Vianello, Electrochim. Acta, 1965, 10, 905–920 CrossRef CAS.
- C. Tsay and J. Y. Yang, J. Am. Chem. Soc., 2016, 138, 14174–14177 CrossRef CAS PubMed.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS PubMed.
- M. Van Der Meer, E. Glais, I. Siewert, B. Sarkar, M. Van Der Meer, E. Glais, B. Sarkar and I. Siewert, Angew. Chemie Int. Ed., 2015, 54, 13792–13795 CrossRef CAS PubMed.
- L. Tensi and A. Macchioni, ACS Catal., 2020, 10, 7945–7949 CrossRef CAS.
- L. Tensi, L. Rocchigiani, G. Menendez Rodriguez, E. Mosconi, C. Zuccaccia, F. De Angelis and A. Macchioni, Catal. Sci. Technol., 2023, 13, 6743–6750 RSC.
- M. L. Pegis, B. A. McKeown, N. Kumar, K. Lang, D. J. Wasylenko, X. P. Zhang, S. Raugei and J. M. Mayer, ACS Cent. Sci., 2016, 2, 850–856 CrossRef CAS PubMed.
- D. J. Martin, C. F. Wise, M. L. Pegis and J. M. Mayer, Acc. Chem. Res., 2020, 53, 1056–1065 CrossRef CAS PubMed.
- Y. H. Wang, B. Mondal and S. S. Stahl, ACS Catal., 2020, 10, 12031–12039 CrossRef CAS.
- B. D. Groff and J. M. Mayer, ACS Catal., 2022, 12, 11692–11696 CrossRef CAS.
- K. Teindl, B. O. Patrick and E. M. Nichols, J. Am. Chem. Soc., 2023, 145, 17176–17186 CrossRef CAS PubMed.
- M. Langerman, P. H. van Langevelde, J. J. van de Vijver, M. A. Siegler and D. G. H. Hetterscheid, Inorg. Chem., 2023, 62(48), 19593–19602 CrossRef CAS PubMed.
- J. L. Alvarez-Hernandez, A. E. Sopchak and K. L. Bren, Inorg. Chem., 2020, 59, 8061–8069 CrossRef CAS PubMed.
- K. E. Clary, M. Karayilan, K. C. McCleary-Petersen, H. A. Petersen, R. S. Glass, J. Pyun and D. L. Lichtenberger, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(52), 32947–32953 CrossRef CAS PubMed.
- J. L. Alvarez-Hernandez, J. Won Han, A. E. Sopchak, Y. Guo and K. L. Bren, ACS Energy Lett., 2022, 6(6), 2256–2261 CrossRef.
- R. Custelcean and J. E. Jackson, Chem. Rev., 2001, 101, 1963–1980 CrossRef CAS PubMed.
- R. H. Crabtree, Science, 1998, 282, 2000–2001 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The authors have cited additional references within the supporting information.1–20 CCDC 1939352 and 2297771. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cy00209a |
| This journal is © The Royal Society of Chemistry 2024 |