
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
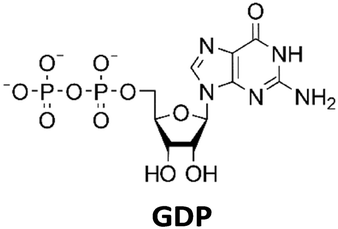
Development of a multi-enzyme cascade for 2′3′-cGAMP synthesis from nucleosides†
Martin
Becker
 b,
Isabel
Nowak
b,
Katharina
Hildebrand
b,
Stephan
Lütz
b and
Katrin
Rosenthal
*a
b,
Isabel
Nowak
b,
Katharina
Hildebrand
b,
Stephan
Lütz
b and
Katrin
Rosenthal
*a
aSchool of Science, Constructor University, Campus Ring 1, 28759 Bremen, Germany. E-mail: krosenthal@constructor.university
bDepartment of Biochemical and Chemical Engineering, Chair for Bioprocess Engineering, TU Dortmund University, Emil-Figge-Strasse 66, 44227 Dortmund, Germany
First published on 26th April 2024
Abstract
The interest in multi-enzyme cascades for the synthesis of pharmaceutically relevant active ingredients has increased in recent years. Through a smart selection of enzymes, cascades enable multi-step synthesis in a one-pot reaction without the purification of intermediates. In this study, a five-enzyme cascade for the formation of cyclic 2′3′-GMP-AMP (2′3′-cGAMP) from adenosine and guanosine in seven reaction steps was successfully developed. First, the substrate scope of kinases for the phosphorylation of nucleosides and nucleotides was investigated, which were then combined in an enzyme cascade for 2′3′-cGAMP formation from adenosine, guanosine, and polyphosphate. An overall conversion of 57% of the substrates into 2′3′-cGAMP was achieved in relation to the initial guanosine concentration.
Introduction
The synthesis of active pharmaceutical ingredients (APIs) is often complex and includes a multitude of reaction steps. Biotechnological synthesis has established itself as an indispensable tool to produce active ingredients by utilizing whole cell biocatalysts or isolated enzymes. In whole cell biocatalysts, the natural metabolic network of the microorganism is exploited to produce high value products from low-cost substrates. Suitable enzymes can also be purified and used in in vitro multi-enzyme cascades for the synthesis of the desired products with fewer side reactions and higher control compared to whole cell-based methods.1 Since enzyme cascade reactions have several benefits, their significance is steadily increasing.2 Higher product yields can be achieved by shifting the equilibrium of thermodynamically unfavorable reactions, which, combined with the avoidance of purification of intermediates, leads to a reduction in waste and costs.3,4 In addition, enzyme cascades enable the handling of unstable or toxic intermediates, as the accumulation of intermediates can be avoided by converting them directly into the end product.5In order to exploit advantages of enzyme cascades, the reaction conditions of the individual enzymes must be compatible with each other, which can lead to extensive fine-tuning to achieve optimal reactivity. For example, a suitable temperature, pH and reaction system must be found in which all enzymes used are sufficiently catalytically active.6,7 In addition, the concentrations of substrates and enzymes must be adjusted to each other to prevent accumulation of intermediates, enzyme inhibition and to avoid side reactions.8,9
The regeneration of depleted cofactors such as nicotinamide adenine dinucleotide (phosphate), coenzyme A or adenosine 5′-triphosphate (ATP) is important in multi-enzyme cascades, which themselves often require complex regeneration enzyme cascades.10 In case of ATP, one of the most used regeneration systems consists of phosphorylation from adenosine 5′-diphosphate ADP by acetate kinases (AcK), as used for example in the synthesis of glucose 6-phosphate.11,12 Electrochemically coupled ATP regeneration by a pyruvate oxidase and an AcK has in addition already been demonstrated.13 In recent years the use of polyphosphate kinases 2 (PPK2) has gained interest due to the use of low-cost polyphosphate (polyP).12 They have been successfully used for ATP regeneration in enzyme cascades for methyltransferases,14 multigram-scale aldehyde synthesis,15 formation of nucleotide sugars,16 and in the monoacylation of symmetrical diamines.17 The PPK2 enzymes can be divided into three classes: PPK2s class I catalyze the phosphorylation of nucleoside diphosphates (NDPs) into nucleoside triphosphates (NTPs), PPK2s class II catalyze the conversion of nucleoside monophosphates (NMPs) into NDPs and PPK2s class III combine both properties phosphorylating NMPs into NDPs and NDPs into NTPs. Besides PPK2 enzymes, there are nucleoside kinases (NKs), which catalyze the transfer of a γ-phosphate of a NTP to a nucleoside. Enzyme cascades consisting of NKs and PPK2s enable ATP formation from polyP and adenosine,14 and the enzymes are often used for NTP regeneration.12
Beside the role of NTPs as an energy-rich cofactor, there are also pharmaceutically relevant derivatives, which can act as APIs. For example, the antiviral derivatives molnupiravir,18 remdesivir,19 and sofosbuvir20 are prodrugs that are further metabolized into their corresponding biologically active triphosphate forms. Another exciting field of application for NTP-based APIs is the synthesis of 2′3′-cyclic GMP-AMP (2′3′-cGAMP). This second messenger is part of the mammalian innate immune system and is synthesized from ATP and guanosine 5′-triphosphate (GTP) by the enzyme cyclic GMP-AMP synthase (cGAS) in presence of double-stranded DNA.21 The cyclic dinucleotide (CDN) initiates a signalling cascade by binding to the receptor for the stimulator of interferon genes (STING) located at the endoplasmic reticulum.22 Binding to the STING receptor leads to the release of type-I interferons stimulating the innate immune system. This property makes 2′3′-cGAMP a promising candidate for drug discovery, with potential applications in cancer immunotherapy or adjuvant in vaccines.23,24 In addition to the described biocatalytic cyclization25,26 of 2′3′-cGAMP by cGAS from ATP and GTP, there are also chemical synthesis routes. The most widely used synthesis route starts with phosphoramidites and involves eight synthesis steps, achieving yields of up to 30%.27,28
To date, two enzyme cascades for 2′3′-cGAMP formation or its derivatives have been developed. In a cascade developed in our lab, adenosine was phosphorylated into ATP by one NK and two PPK2s generating 2′3′-cGAMP concentrations of 0.62 mM with stoichiometric GTP addition.29 In another cascade, the fluorinated dithio-2′3′-cGAMP derivative MK-1454 was synthesized from two non-natural nucleotide monothiophosphates30 at a preparative scale. An adenylate kinase, guanylate kinase, AcK, and cGAS were used and an isolated yield of 62% was achieved.
To utilize polyphosphate and thus avoiding the requirement of expensive acetyl phosphate as phosphate donor, we developed a five-enzyme cascade for 2′3′-cGAMP formation from nucleosides, as shown in Fig. 1. For this purpose, various NKs were tested for their ability to phosphorylate guanosine and adenosine as well as PKK2s to phosphorylate guanine and adenine nucleotides. The findings obtained were used to select reaction conditions for 2′3′-cGAMP formation by truncated human cGAS (thscGAS) from adenosine, guanosine, and polyP.
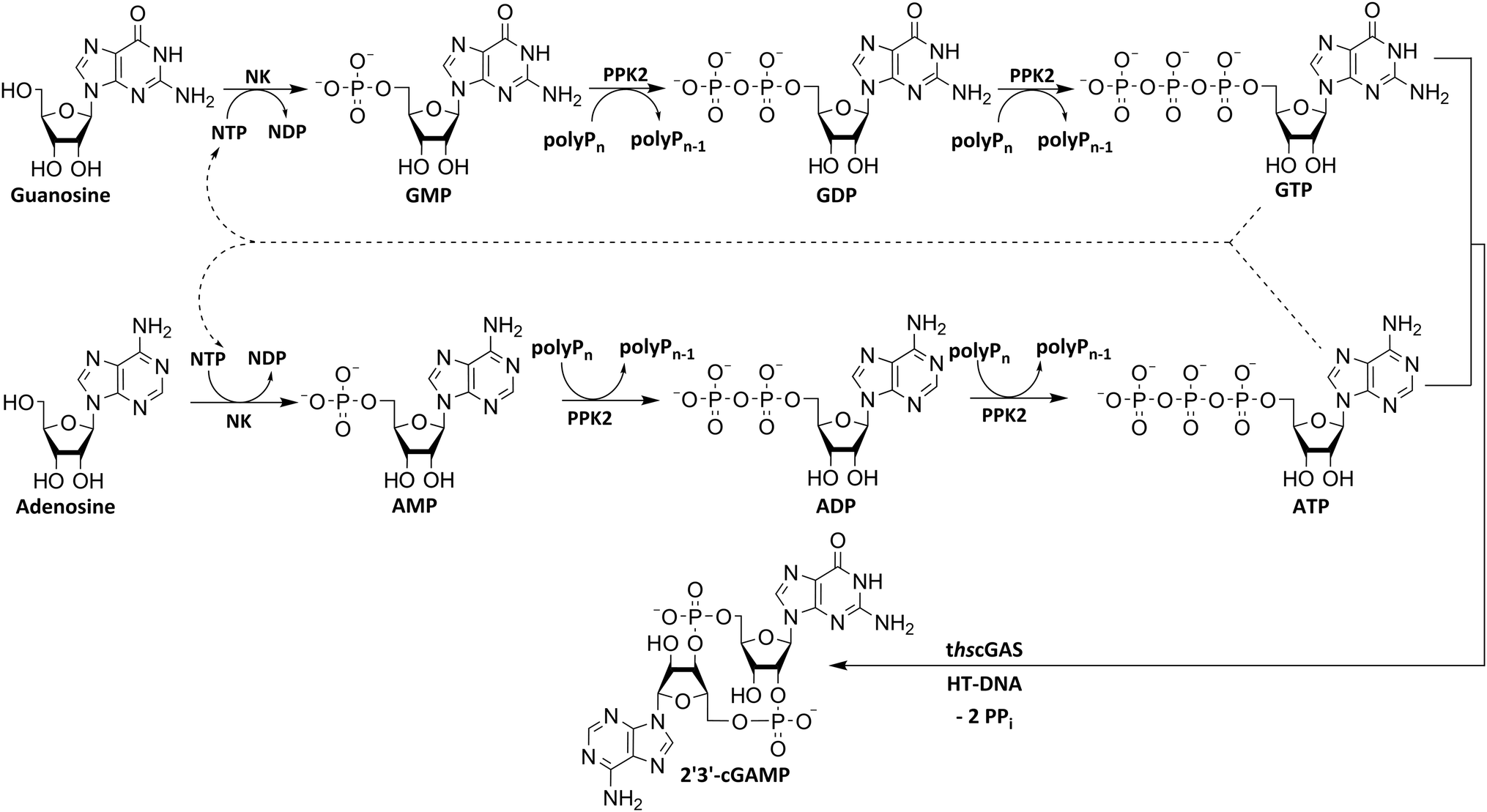 | ||
| Fig. 1 Simplified scheme for an enzyme cascade 2′3′-cGAMP synthesis from nucleosides using NKs, PPK2s and thscGAS. NK: nucleoside kinase; PPK2: polyphosphate kinase 2; polyPn: polyphosphate; polyPn−1: polyP truncated by one phosphate monomer.; GMP: guanosine monophosphate; GDP: guanosine diphosphate; GTP: guanosine triphosphate; AMP: adenosine monophosphate; ADP: adenosine diphosphate; ATP: adenosine triphosphate; NDP: nucleoside diphosphate; NTP: nucleoside triphosphate; 2′3′-cGAMP: 2′3′-cyclic GMP-AMP; HT-DNA: herring testis DNA; PPi: pyrophosphate. | ||
Results and discussion
Enzyme screening for cascade design
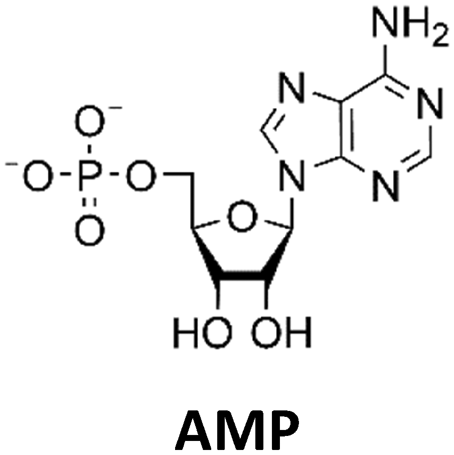
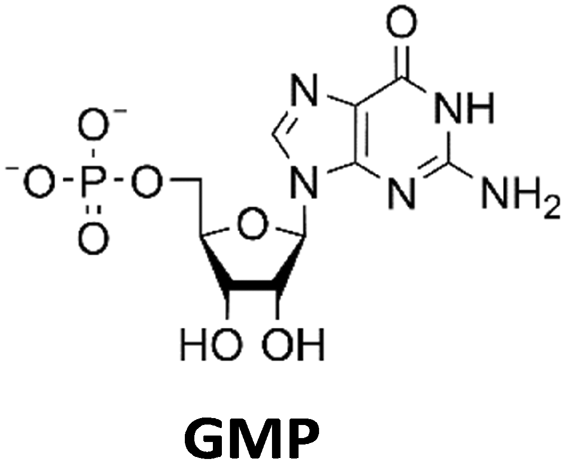
Various nucleoside kinases and polyphosphate kinases were investigated for their accepted substrate scope to identify suitable enzymes for the formation of 2′3′-cGAMP from nucleosides.The two nucleoside kinases from Saccharomyces cerevisiae (ScADK)31 and Methanocaldococcus jannaschii (MjNK)32 were tested for their ability to phosphorylate adenosine and guanosine with either ATP or GTP as phosphate donor. Adenosine was completely converted into AMP by ScADK, which is consistent with previous experiments.14,29 Since no conversion of guanosine was detected by ScADK, MjNK was investigated. However, no phosphorylation of guanosine was detected at the standard assay temperature of 37 °C, which is due to the fact that MjNK has a temperature optimum of 85 °C.33 Since an excessive hydrolysis of the co-substrates ATP can be suspected at higher temperatures, the temperature-dependent hydrolysis was investigated (ESI,† Fig. S3 and S4). After incubation for 6 h at 80 °C, only 48% of the initial ATP concentration was detected. At 70 °C, 91% of ATP was detected after 6 h incubation, which is close to the published values.34 Therefore, an initial reaction temperature of 70 °C was chosen for the phosphorylation of guanosine to decrease ATP hydrolysis. Under these conditions, a conversion of 95 ± 1% guanosine into GMP was detected after 24 h, which is of note because the pH of the Tris buffer used is temperature dependent.35 For the phosphorylation of adenosine, only 2% were converted to AMP after 24 h, which is in accordance with previous publications.33
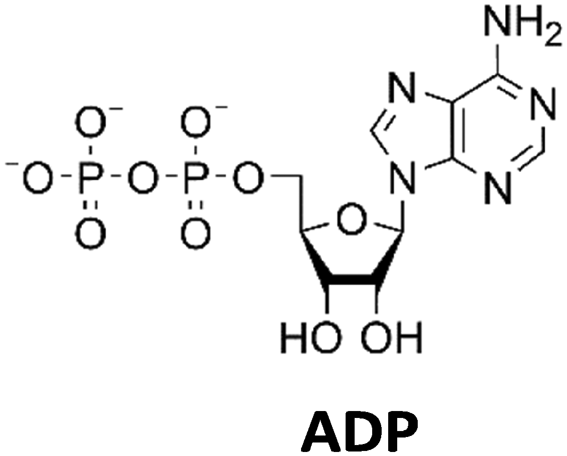
The PPK2 of Acinetobacter johnsonii (AjPPK2),36 belonging to PPK2 class II, and from Sinorhizobium meliloti (SmPPK2),37 belonging to PPK2 class I, were investigated for their ability to phosphorylate ADP/GDP and AMP/GMP, respectively. The class III PPK2s from Meiothermus ruber (MrPPK2),38Erysipelotrichaceae bacterium (EbPPK2),15 and Cytophaga hutchinsonii (ChPPK2),39 were investigated regarding their ability to convert AMP and GMP into ATP and GTP. Substrates were supplied with a concentration of 5 mM using polyP as phosphate donor. The determined conversions of substrates into the final products after 24 h are summarized in Table 1. AjPPK2 catalyzed the phosphorylation of AMP and GMP into ADP and GDP in the range already reported.40 For the SmPPK2-catalyzed phosphorylation, 29% conversion of ADP to ATP and 59% of GDP to GTP was achieved.
| Enzyme | PPK2 class | Substrate | Substrates | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Adenosine species | Guanosine species | |||||||||
|
|
|
|
|
|||||||
| Reaction | Conversion in product [%] | TON | Conversion in product [%] | TON | Conversion in product [%] | TON | Conversion in product [%] | TON | ||
| — not tested.a Performed in 1.5 mL Eppendorf tubes in triplicates, 30 mM polyP, enzyme concentrations: 0.01 mg mL−1.b Only GDP was formed.c Only the conversion of NMPs to NTPs was investigated and not the conversion to NDPs. | ||||||||||
| AjPPK2 | II | NMP → NDP | 81 ± 3 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 460 460 |
— | — | 70 ± 1 | 5124 | — | — |
| SmPPK2 | I | NDP → NTP | — | — | 29 ± 1 | 2080 | — | — | 59 ± 4 | 1869 |
| MrPPK2c | III | NMP → NTP | 29 ± 3 | 1553 | — | — | 0 ± 0 | 1446b | — | — |
| EbPPK2c | III | NMP → NTP | 31 ± 10 | 8097 | — | — | 51 ± 10 | 7884 | — | — |
| ChPPK2c | III | NMP → NTP | 82 ± 15a | 33273 |
— | — | 62 ± 1a | 20577 |
— | — |
The MrPPK2 did not catalyze the phosphorylation of GMP, but EbPPK2 and ChPPK2 were both able to convert AMP and GMP into ATP and GTP, which makes them interesting for the enzyme cascade. With 82% ATP and 62% GTP, ChPPK2 showed higher conversions for both substrates into the final products compared to the EbPPK2 after 24 h. The Km values of ChPPK2 of 620 μM for AMP and 3150 μM for GMP show a higher affinity to AMP.39
To obtain a benchmark of the catalytic efficiency of the enzymes, the turnover numbers (TONs) were calculated from the product and enzyme quantities. It should be noted that it cannot be ruled out that the reaction was limited by reaching equilibrium or that the reaction was not fully completed within the reaction time of 24 h. Therefore, the TONs determined should be considered as reference or apparent value. A TON of 2575 was determined for ScADK, 1210 for MjNK, and 1862 for thscGAS. With a TON between 33273 with AMP as substrate and 20577 with GMP as substrate, ChPPK2 stands out compared to the other PPK2s, whose TONs are in the range between 1446 and 12460 as shown in Table 1.
Enzyme cascade development for 2′3′-cGAMP formation from guanosine
After successfully screening of the enzymes regarding their substrate scope, the next goal was to develop a cascade for 2′3′-cGAMP formation from guanosine and ATP, since the phosphorylation cascade from adenosine into ATP is already established.29 2′3′-cGAMP was formed in four reaction steps from guanosine (Fig. 2). The incubation temperature for the respective reaction steps was adjusted and the enzymes were added sequentially, first only MjNK at 70 °C and after decreasing the temperature to 37 °C for the remaining enzymes.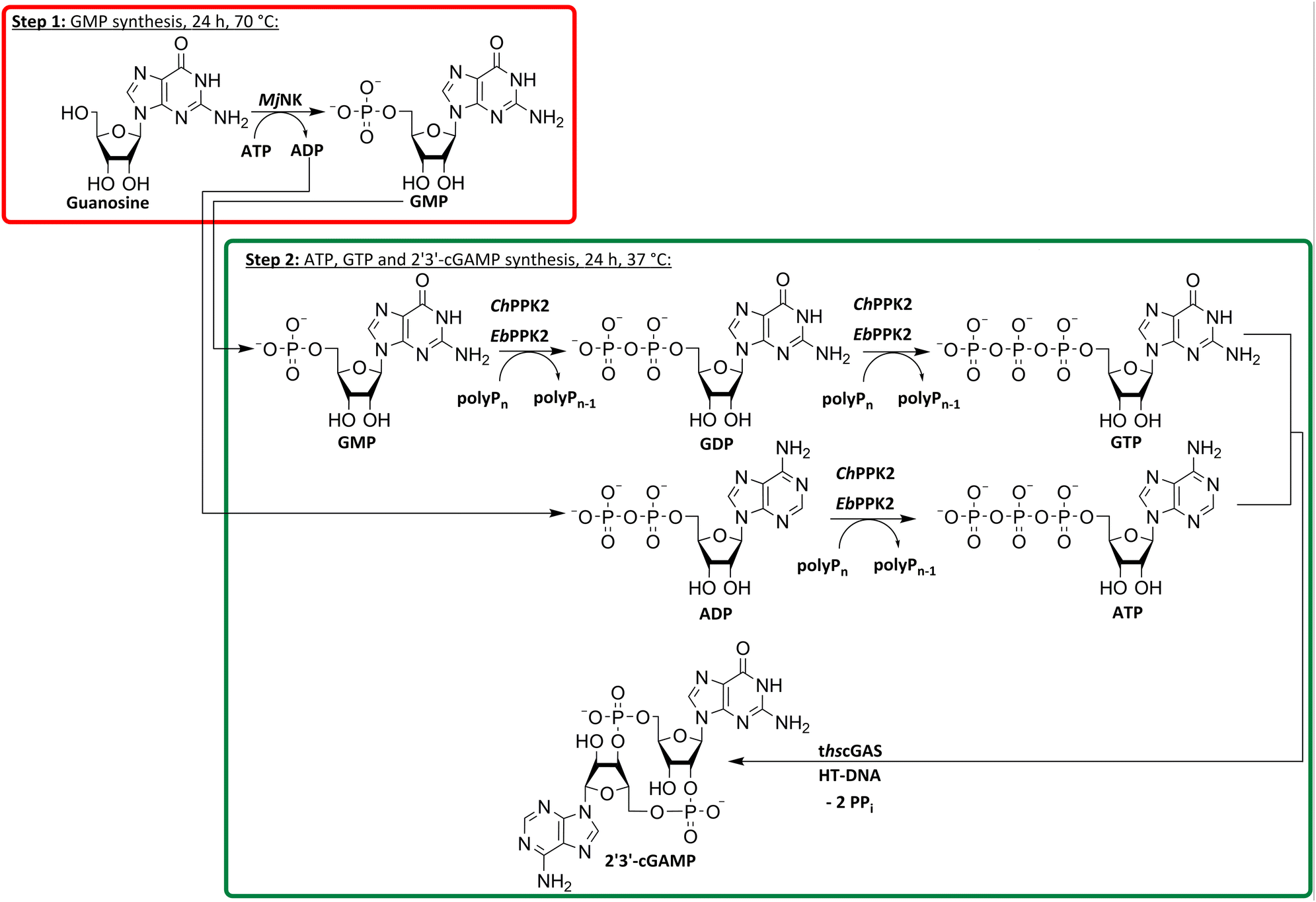 | ||
| Fig. 2 Reaction scheme of 2′3′-cGAMP formation starting from guanosine and ATP. In the first assay step, 5 mM guanosine and equimolar ATP were incubated with 0.1 mg mL−1MjNK and 10% DMSO at 70 °C for 24 h. Additional 2.5 mM ATP was added after 6 h. In the second step 50% (v/v) from the reaction solution was mixed with ChPPK2 (0.05 mg mL−1), EbPPK2 (0.05 mg mL−1), lyophilized thscGAS (0.08 mg mL−1), 30 mM polyP and 0.1 mg mL−1 HT-DNA and incubated at 37 °C for 24 h. | ||
The phosphorylation of guanosine catalyzed by MjNK was carried out with 5 mM guanosine and an equimolar ATP concentration. After 6 h, further 2.5 mM ATP were added. After 24 h, 4.70 ± 0.06 mM GMP was detected, which may have been slightly overestimated due to incomplete base line separation in the HPLC chromatograms (ESI,† Fig. S8). After decreasing the temperature, ChPPK2, EbPPK2, and thscGAS, as well as polyP and herring testis DNA (HT-DNA) (required for cGAS activation) were added and incubated at 37 °C for 24 h. The reaction progress at 37 °C is shown in Fig. 3.
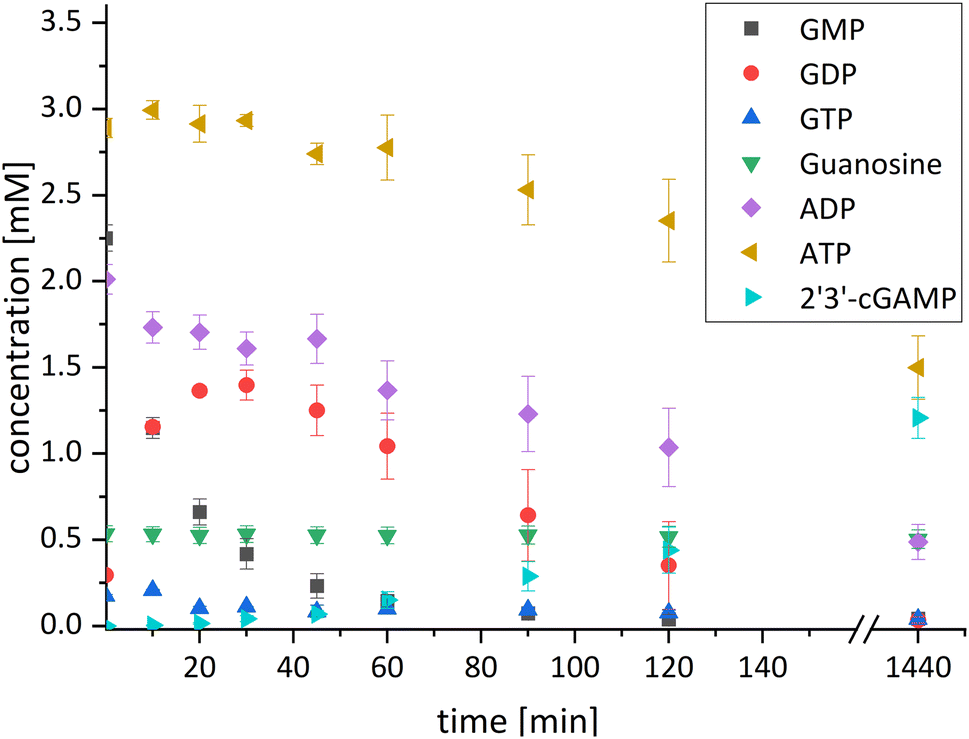 | ||
| Fig. 3 Concentration curve of the four-enzyme cascade for 2′3′-cGAMP formation starting from guanosine and ATP. Reaction conditions: the assay was performed in a reaction volume of 1 mL at 37 °C and 300 rpm for 24 h. The reaction contained 50% (v/v) of the reaction solution from the first reaction step (composition is explained in detail in the Experimental section) and an initial GMP concentration of 2.25 mM. To start the reaction, the enzymes ChPPK2 (0.05 mg mL−1), EbPPK2 (0.05 mg mL−1), lyophilized thscGAS (0.08 mg mL−1), 30 mM polyP, and 0.1 mg mL−1 HT DNA were added. The standard deviations shown are from biological triplicates. | ||
After 24 h, 1.21 ± 0.12 mM 2′3′-cGAMP was detected corresponding to a conversion of 24% of the initial guanosine concentration used. This is three times higher than the existing enzyme cascade starting from adenosine,29 demonstrating an efficient flux of guanosine substrates through the cascade. The specific thscGAS activity was 43.0 ± 13.7 mU mg−1 within the first 2 h, which is more than half of the maximum enzyme activity of 73.5 ± 7.8 mU mg−1.41 This represents the first reported 2′3′-cGAMP formation from guanosine, polyP, and ATP.
With a TON of 1445 for the MjNK (ESI,† Table S1), 120% of the catalytic potential was utilized compared to the enzyme screening. This increase of TON may result from different reactions conditions, batch-to-batch differences in the enzyme preparation, a shift in the equilibrium of the reaction or that the reaction was not fully completed within the reaction time in the initial enzyme screening. Since ChPPK2 and EbPPK2 catalyze the same reaction in the last step, a combined TON of 953 was calculated. This corresponds to 5% of the average catalytic potential of both enzymes, indicating the optimization opportunities of the reaction. The TON of these enzymes may be restricted by the rate-limiting enzyme in the cascade or by the equilibrium constraints of the system, rather than by the enzymes itself. With 847, the TON of thscGAS reached 45% of the initial screening. Since the substrates were limiting, the catalytic potential of thscGAS could not be fully exploited.
Enzyme cascade development for 2′3′-cGAMP formation from nucleosides
The next goal was to design a sequential enzyme cascade that allows the phosphorylation of adenosine and guanosine to ATP and GTP and their subsequent cyclization to 2′3′-cGAMP. Based on the different optima of the enzymes, the temperature was set to either 37 °C or 60 °C, and the enzymes were added sequentially to the assay as shown in Fig. 4(a). In the first step, ATP was to be formed within 5 h at 37 °C starting from 10 mM adenosine, 1 mM AMP and 45 mM polyP using the enzymes ScADK and ChPPK2. In the second assay step, GMP formation was to be carried out from guanosine and ATP within 5 h at 60 °C using MjNK. The third assay step for AMP and GMP phosphorylation into ATP and GTP was catalyzed by EbPPK2 and ChPPK2 within 2 h at 37 °C. The final assay step was to obtain 2′3′-cGAMP from ATP and GTP by adding and incubating at 37 °C for 24 h. Since it is known from own experiments that the stability of thscGAS is low, 2′3′-cGAMP formation was performed in a separate step to fully exploit the catalytic potential of thscGAS. The reaction courses of the four reaction steps are summarized in Fig. 4(b) and (c).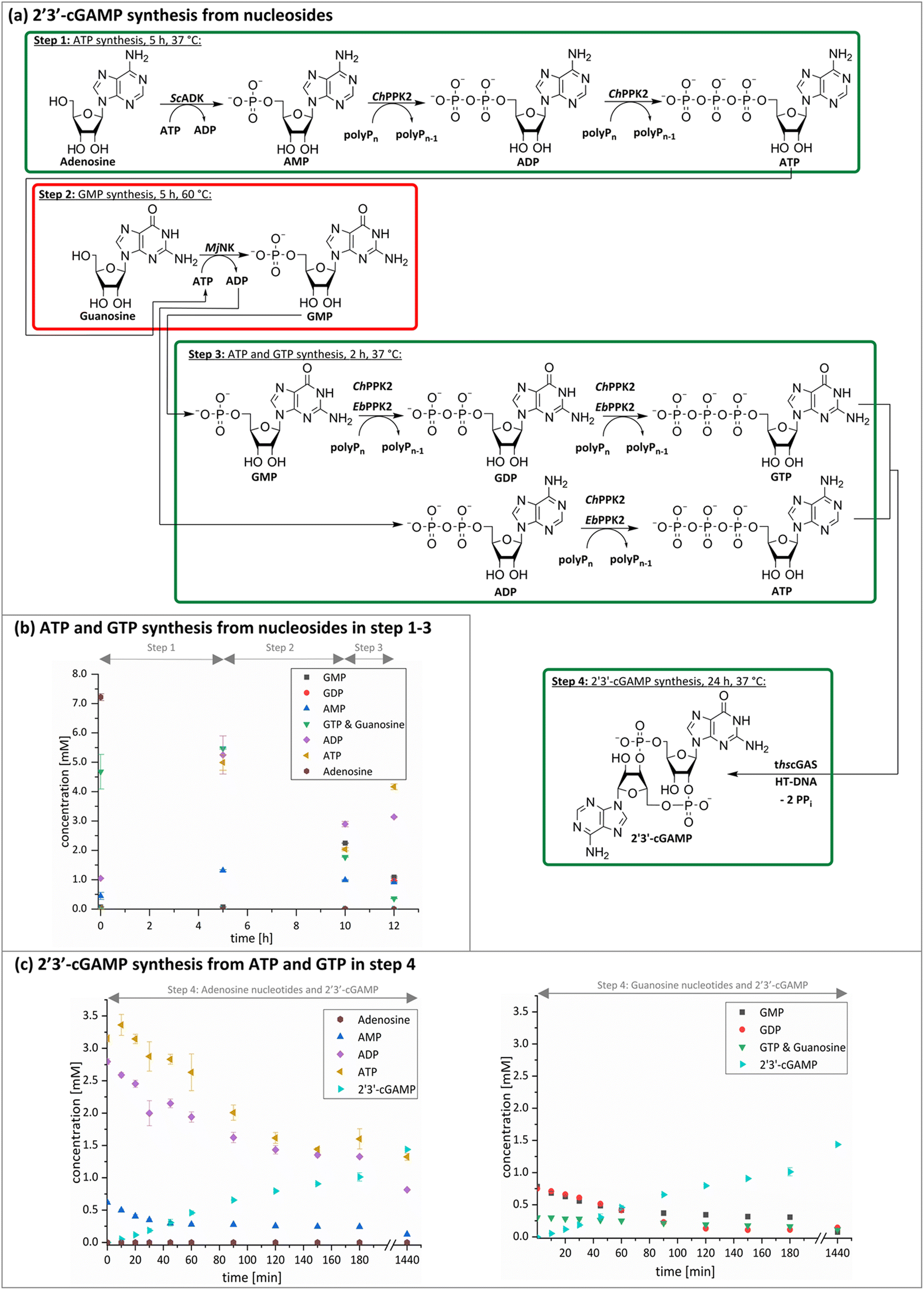 | ||
| Fig. 4 2′3′-cGAMP formation starting from guanosine and adenosine in four assay steps. (a) Reaction scheme; (b) ATP and GTP formation from nucleoside within the first three assay steps; (c) 2′3′-cGAMP formation from adenosine nucleotides (left) and guanosine nucleotides (right). In the first assay step, ATP was to be obtained within 5 h at 37 °C starting from 10 mM adenosine, 1 mM AMP and 45 mM polyP using 0.05 mg mL−1ScADK and 0.05 mg mL−1ChPPK2 in a reaction volume of 1 mL. Also, 5 mM guanosine and 10% (v/v) DMSO were supplied. In the second assay step, guanosine phosphorylation was to be carried out within 5 h at 60 °C and 0.2 mg mL−1MjNK in a reaction volume of 1.1 mL. The third assay step to obtain ATP and GTP was performed in a reaction volume of 1.3 mL for 2 h at 37 °C using 0.05 mg mL−1 of EbPPK2, 0.05 mg mL−1ChPPK2 and 45 mM polyP. The final assay step to cyclize ATP and GTP into 2′3′-cGAMP was carried out at 37 °C for 24 h with 0.12 mg mL−1 lyophilized thscGAS and 0.1 mg mL−1 HT-DNA in 1.5 mL. | ||
As can be seen in Fig. 4(b), adenosine was completely converted within the first 5 h into 1.31 ± 0.03 mM AMP, 5.25 ± 0.65 mM ADP and 4.99 ± 0.26 mM ATP. After the second step (10 h sample), 2.24 ± 0.02 mM GMP were obtained from guanosine. Due to insufficient separation of GTP and guanosine for the HPLC measurements, the concentrations of the two substances are listed together.
In the third step, 4.17 ± 0.10 mM ATP and 0.36 ± 0.01 mM GTP/guanosine were detected. After the addition of thscGAS, polyP, and HT-DNA in the last assay step, the concentrations of ATP and GTP steadily decreased. The continuous decrease of the concentrations of AMP, ADP, GMP, and GDP shows that the formation of ATP and GTP continued during the entire period monitored. After 24 h, 1.31 ± 0.05 mM ATP, 0.81 ± 0.02 mM ADP, 0.14 ± 0.00 mM GDP and 0.12 ± 0.00 mM GTP/guanosine were detected, indicating that mainly adenosine nucleotides remain due to the double initial adenosine amount. The 2′3′-cGAMP production could hence be further increased by increasing the guanosine concentration.
During the entire assay, 2′3′-cGAMP was constantly formed up to a concentration of 1.44 ± 0.01 mM after 24 h corresponding to a conversion of 57% relative to the initial guanosine. This is eight times higher than the existing enzyme cascade for 2′3′-cGAMP formation from adenosine.29 The reason for the improvement in product concentration and conversion is that both ATP and GTP are continuously formed and thus do not have an inhibitory effect on the substrate-inhibited cyclization reaction. In addition, ATP and GTP are not consumed simultaneously for the phosphorylation of the nucleosides, as these were already formed in a previous step, which is in contrast to the previously published cascade. The average specific activity of thscGAS in the cascade was 50.0 ± 2.2 mU mg−1 within the first 2 h, which is slightly lower compared to the maximum enzyme activity of 73.5 ± 7.8 mU mg−1 at equimolar substrate concentrations of 0.5 mM.41 Nevertheless, it is worth noting that the specific activity is relatively similar, as there were non-ideal conditions for the enzyme in the cascade. Substrate inhibition above 2 mM and a decrease in catalytic activity at non-equimolar substrate concentrations were reported.42 Therefore, the conditions for the enzyme were not ideal with 3.1 ± 0.1 mM ATP and 0.3 ± 0.0 mM GTP at the beginning of step 4. Furthermore, the specific activity of cGAS is higher than in the enzyme cascade starting from guanosine and ATP and even higher than in the recently published cascade for 2′3′-cGAMP formation from adenosine and GTP.29 This demonstrates an effective exploitation of the catalytic potential of the enzyme within the developed cascade.
With a TON of 7365 for ScADK (ESI,† Table S2), the TON achieved in the cascade was more than twice as high as compared to TONs obtained with the enzyme screening experiments. The TONs of the other enzymes is 36–44% of the enzyme screening indicating potential for optimizations, such as the adjustment of the enzyme concentrations to avoid rate-limiting steps or equilibriums in the system. In summary, the developed enzyme cascade for 2′3′-cGAMP formation comprises five enzymes in a total of four assay steps, which differ primarily in terms of their temperature. In further optimizations, it would be desirable to develop the cascade so that it runs at a constant temperature to parallelize several assay steps leading to a reduced reaction time. This can be achieved by increasing the assay temperature if all enzymes used are stable at higher temperatures. If this is not possible, alternative suitable enzymes could be explored. Especially an alternative for the MjNK would be interesting, which also accomplishes the guanosine phosphorylation at 37 °C such as the guanosine kinase from Trichomonas vaginalis,43 which showed guanosine phosphorylation at 30 °C.
Conclusions
In this study, 2′3′-cGAMP formation from the nucleosides adenosine and guanosine was developed using a multi-enzyme cascade. It was shown that 2′3′-cGAMP can be obtained in seven reaction steps using two NKs, two PPK2s and thscGAS under consumption of adenosine, guanosine, and polyP. The cascade design was started with an enzyme screening and the development of a cascade for 2′3′-cGAMP from guanosine, whereby necessary reaction parameters, such as temperature changes, were defined. The enzyme cascade for GTP was then combined with the formation of ATP and subsequent cyclization of these precursors to 2′3′-cGAMP. The conversion of 57% of the substrates into the final product with a formation of eight bonds by five enzymes is approaching the assay yield of 68% of an established enzyme cascade for the synthesis of a dithio-2′3′-cGAMP derivative with the formation of six bonds by four enzymes,30 demonstrating the high performance of the enzyme cascade. In comparison to the previously published cascade, the use of polyphosphate instead of acetyl phosphate as a phosphate donor would be an improvement in terms of the cost of product formation. However, when comparing the cascades, it must be mentioned that the reactions were carried out on different scales (1 mL in this work, 1 L in the literature30). It therefore remains to be tested whether the cascade developed here can be scaled, as well as an efficient product purification can be developed and thus be able to compete. Irrespective of this, the continuous supply of ATP and GTP is interesting, as the last enzyme of the cascade is substrate inhibited, which could thus be circumvented. However, the current cascade reaction is relatively complex, involving numerous enzymes, and could possibly be accomplished with fewer enzymes. Nevertheless, the current approach may be advantageous for using modified nucleosides to synthesize non-natural derivatives of 2′3′-cGAMP. The successful phosphorylation of nucleotide derivatives has already been demonstrated for other enzyme cascades with various kinases that use polyP44 or phosphoenolpyruvate45 as a phosphate donor. Further optimization of the enzyme cascade could be achieved by adjusting the enzyme ratios to allow an increased flux of nucleoside substrates and to further increase the formed 2′3′-cGAMP amount. By using a structured optimization strategy, such as a orthogonal factorial design,46 Bayesian optimization,47 or kinetic modeling,48 further optimizations are possible.49To conclude, the achieved results of the 2′3′-cGAMP formation from nucleosides confirm that the complex development of enzyme cascades is worth it. By combining multiple biocatalytic reactions, it is possible to design new, shorter pathways for drug synthesis and to support the chemical and pharmaceutical industry's goal of moving to more sustainable and environmentally friendly processes.50
Experimental section
All chemicals used for this study were purchased from VWR (VWR International GmbH, Darmstadt, Germany), ThermoFisher (ThermoFisher Scientific, Waltham, MA, USA), Merck (Merck KGaA, Darmstadt, Germany), AppliChem (AppliChem GmbH, Darmstadt, Germany) and Roth (Carl Roth, Karlsruhe, Germany) in the highest available purity.Plasmids and strains
The enzymes were produced with E. coli BL21 (DE3) pLysS and E. coli BL21 (DE3). Descriptions of the plasmids for the expression of ScADK, AjPPK2, and SmPPK2 can be found in Mordhorst et al.14 The thscGAS expression strain E. coli BL21 (DE3) pLysS pET28a-SUMOthscGAS is described by Rosenthal et al.26 A list of all bacterial strains used can be found in Table 2 as well as all DNA sequences and UniProt IDs of the enzymes in the ESI.†| Expression strains | Enzyme | Description |
|---|---|---|
| a His6-Tag. b His6SUMO-Tag. c Cloned in ref. 15. | ||
| E. coli BL21 (DE3) pET28a-MrPPK251 | MrPPK2a | Polyphosphate kinase from Meiothermus ruber |
| E. coli BL21 (DE3) pET28a-AjPPK214 | AjPPK2a | Polyphosphate kinase from Acinetobacter johnsonii |
| E. coli BL21 (DE3) pET28a-SmPPK214 | SmPPK2a | Polyphosphate kinase from Sinorhizobium meliloti |
| E. coli BL21 (DE3) pET28a-EbPPK2c | EbPPK2a | Polyphosphate kinase from Erysipelotrichaceae bacterium |
| E. coli BL21 (DE3) pET28b-ChPPK217 | ChPPK2a | Polyphosphate kinase from Cytophaga hutchinsonii |
| E. coli BL21 (DE3) pLysS pET28a-ScADK14 | ScADKa | Adenosine kinase from Saccharomyces cerevisiae |
| E. coli BL21 (DE3) pLysS pET28a-MjNK44 | MjNKa | Nucleoside kinase from Methanocaldococcus jannaschii |
| E. coli BL21 (DE3) pLysS pET28a-SUMOthscGAS26 | thscGASb | N-terminally truncated cGAS protein from Homo sapiens |
Recombinant enzyme expression
The bacterial strains were spread on LB (10 g L−1 tryptone, 5 g L−1 yeast extract, 5 g L−1 NaCl) agar plates, supplemented with 50 mg L−1 kanamycin. For expression strains containing pLysS, 25 mg L−1 chloramphenicol were added. The agar plates were incubated over night at 37 °C. The following day, a pre-culture was made by the inoculation of 10 mL LB medium in case of the expression of the kinases and 10 mL 2xYT (16 g L−1 tryptone, 10 g L−1 yeast extract, 5 g L−1 NaCl) medium in case of thscGAS with one colony. The same antibiotic concentrations were used as for agar plates. The pre-cultures were incubated for 8 h at 37 °C and 200 rpm. For the main cultures, the same media conditions were used as in the pre-cultures. They were inoculated to an OD600 of 0.05 in case of the expression of the kinases and OD600 of 0.1 for thscGAS. In case of thscGAS, 200 mL of culture were incubated in a 2 L baffled shaking flask and 400 mL in case of the expression of kinases. The main cultures were incubated at 37 °C and 200 rpm. The cultures were incubated on ice for 15 min, after an OD600 of 1 was reached. The enzyme expression was induced with 0.5 mM isopropyl-β-D-thiogalactopyranosid (IPTG). In case of thscGAS, the main cultures were incubated at 20 °C for 11 h and for 16 h in case of kinases. The cells were isolated by centrifugation (25 min, 4 °C, 4700 × g) in 50 mL aliquots and the pellets were stored at −20 °C.Enzyme purification
After resuspending two cell pellets in 20 mL of lysis buffer (kinases: 40 mM TRIS–HCl, 100 mM NaCl, 10% (v/v) glycerol, pH 8.0; thscGAS: 50 mM TRIS–HCl, 300 mM NaCl, 40 mM imidazole, 1 mM tris(2-carboxyethyl)phosphine (TCEP), pH 8.0), the cells were stored on ice. By sonication, the cells were disrupted using a Branson Digital Sonifier (BRANSON Ultrasonics Corporation, Danbury, CT, USA). Five cycles of 30 s with a pulse on time of 0.5 s and a pulse off time of 1 s with an amplitude of 10% was used. Insoluble cellular components were removed by centrifugation (20 min, 4 °C, 43000 × g). After sterile filtration (0.2 μm) of the supernatant, it was loaded onto a 1 mL HisTrap™ FF crude column (GE Healthcare, Solingen, Germany). The column was pre-equilibrated using 5 column volumes (CV) of ultrapure water and 10 CV of lysis buffer. After loading, the column was washed with 10 CV lysis buffer and incubated for 1 h at 4 °C. The enzyme has been eluted with elution buffer (20 mM TRIS–HCl, 150 mM NaCl, 300 mM imidazole, pH 7.4) in six fractions of 1 mL. Protein-containing fractions were detected using a Bradford assay and merged to a volume of 2.5 mL. By using a PD-10 SephadexTM G-25 column (GE Healthcare, Little Chalfont, UK), a size-exclusion chromatography was performed for buffer exchange with activity buffer (50 mM TRIS–HCl, 40 mM MgCl2·6H2O, pH 8.0). The column was pre-equilibrated with 25 mL ultrapure water and 25 mL activity buffer. The enzyme was eluted from the column in 3.5 mL activity buffer. The concentration of the enzyme was determined by a second Bradford assay. In the ESI,† SDS gels of the purified enzymes can be found (Fig. S5–S7†). The purified kinases were stored at −20 °C. In case of thscGAS, the enzyme was lyophilized overnight and stored at −20 °C afterwards.
In vitro enzyme assay with MjNK
The enzyme assays were performed in 50 mM TRIS–HCl containing 40 mM MgCl2·6H2O activity buffer with 0.05 mg mL−1MjNK, 5% (v/v) DMSO, 2.5 mM guanosine and 2.5 mM ATP concentrations. After 6 h, further 1.25 mM ATP was added. The assays were carried out in 1.5 mL microtubes incubated at 70 °C in an orbital shaker at 300 rpm in triplicates. The reaction was started by adding the enzyme. Samples were taken after 0 and 24 h. The reaction was stopped by heating the samples to 95 °C for 5 min.In vitro enzyme assays for enzyme screening
The enzyme assays were performed in 50 mM TRIS–HCl containing 40 mM MgCl2·6H2O activity buffer. The substrate concentrations were 5 mM unless otherwise stated. Based on a single phosphate monomer's molecular weight of 101.96 g mol−1, the concentration of 30 mM polyP utilized was calculated. The enzyme concentrations of the kinases AjPPK2, SmPPK2, MrPPK2, and EbPPK2 were 0.02 mg mL−1 and 0.01 mg mL−1 in case of ChPPK2.The assays of the AjPPK2, SmPPK2, MrPPK2, and EbPPK2 were performed with 20% (v/v) DMSO in 1 mL assay volume in deep well microplates (polypropylene square 96-deepwell microplates CR1496, vessel volume 2.2 mL, EnzyScreen BV, Heemstede, Netherlands) and incubated at 300 rpm at 37 °C in duplicates. The ChPPK2 assays were performed without DMSO in microtubes incubated at 37 °C in an orbital shaker at 300 rpm in triplicates.
The TON of thscGAS was calculated from an experiment with 0.04 mg mL−1 enzyme, 2 mM ATP and 2 mM GTP in 40 mM HEPES buffer, pH 7.2 containing 10 mM MgCl2·6H2O. The final 2′3′-cGAMP concentration was 1.33 mM after 24 h incubation at 37 °C in an orbital shaker at 300 rpm.
The assay volume was 1 mL. The reaction was started by adding the enzyme. After 0 h and 24 h samples were taken. The reaction was stopped by heating the samples to 95 °C for 5 min.
Enzyme cascade for 2′3′-cGAMP formation from guanosine
The enzyme cascade reactions were performed in 50 mM TRIS–HCl containing 40 mM MgCl2·6H2O activity buffer in a 2 mL microtube, with a reaction volume of 1 mL.The reaction was divided into two assays. In the first assay, the reaction was carried out at 70 °C and 10% (v/v) DMSO by using 0.1 mg mL−1MjNK to convert the 5 mM guanosine and 5 mM ATP to GMP in 24 h. After 6 h, 2.5 mM ATP was added to the assay. The second assay for 2′3′-cGAMP formation was performed at 37 °C for 24 h. This consisted of 50% (v/v) of the first assay step, 0.05 mg mL−1ChPPK2, 0.05 mg mL−1EbPPK2, 30 mM polyP, 0.1 mg mL−1 HT DNA, and 0.08 mg mL−1 lyophilized thscGAS. During the first assay, 85 μL of sample was collected after 0 and 24 h each. In the second assay, 85 μL sample was taken after 0, 10, 20, 30, 45, 60, 90, 120 min, 24 h, and 48 h each. The reaction was stopped by heating the samples to 95 °C for 5 min.
Enzyme cascade for 2′3′-cGAMP formation from adenosine and guanosine
The enzyme cascade reactions were performed in 50 mM TRIS–HCl containing 40 mM MgCl2·6H2O activity buffer in a 2 mL microtube, starting with a reaction volume of 1 mL. The reaction was divided into four assay steps, which can be seen in Fig. 5. In the first assay step, ATP was to be formed within 5 h at 37 °C starting from 10 mM adenosine using 0.05 mg mL−1ScADK and 0.05 mg mL−1ChPPK2, 1 mM AMP, and 45 mM polyP within 5 h. Also, 5 mM guanosine and a DMSO concentration of 10% (v/v) were supplied in the reaction volume of 1 mL. In the second assay step, GMP formation was to be carried out within 5 h at 60 °C. An MjNK concentration of 0.2 mg mL−1 in a reaction volume of 1.2 mL was set. Subsequently, the assay was placed on ice for cooling, as the third assay step for ATP and GTP formation was performed for 2 h at 37 °C using 0.05 mg mL−1 each of EbPPK2 and ChPPK2 as well as 45 mM polyP. The reaction volume was adjusted to 1.3 mL. The final assay step of 2′3′-cGAMP formation was carried out at 37 °C for 24 h with 0.12 mg mL−1 lyophilized thscGAS and 0.1 mg mL−1 HT-DNA. The reaction volume was adjusted to 1.5 mL. At the beginning of each step, 100 μL of sample was taken. During the last assay step of the 2′3′-cGAMP formation, samples were taken after 0, 10, 20, 30, 45, 60, 90, 120, 150, and 180 min and after 24 h. The reaction was stopped by heating the samples to 95 °C for 5 min.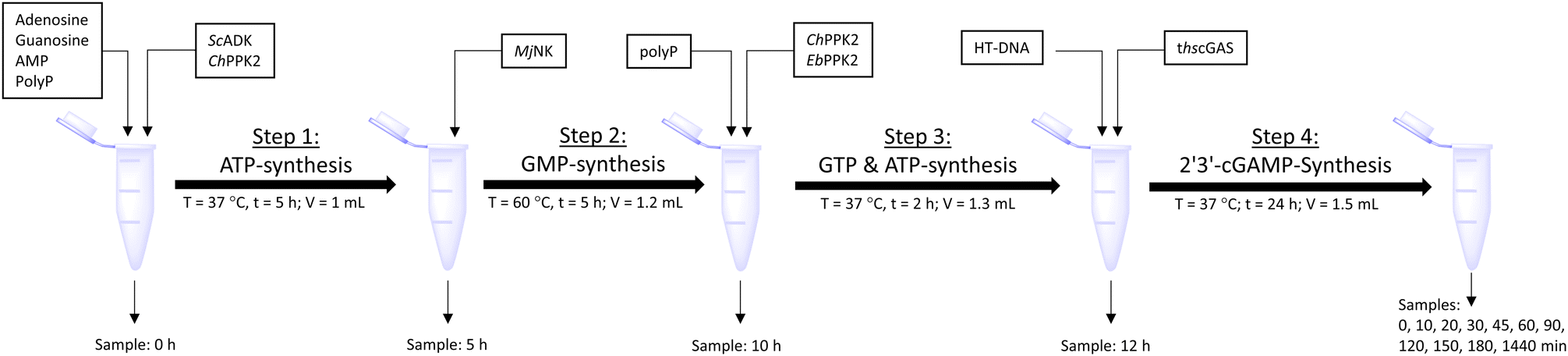 | ||
| Fig. 5 Schematic workflow of the sequential one-pot approach for 2′3′-cGAMP formation starting from adenosine and guanosine. | ||
Temperature stability of ATP and GTP
In 1.5 mL Eppendorf tubes, 1 mL of either 5 mM ATP or 5 mM GTP solution was incubated in 50 mM TRIS–HCl, 40 mM MgCl2·6H2O activity buffer in duplicates at 60 °C, 70 °C, and 80 °C. Samples were taken after 0, 2, 3.5, 4.75, 6, 7.25 and 24 h and analysed by HPLC (1260 Infinity II LC system (Agilent Technologies, Santa Clara, USA) consisting of a diode array detector (1260 DAD HS), column thermostat (1260 MCT), multisampler (1260 Multisampler), binary pump (1260 Binary Pump) and an Agilent Poroshell 120 EC-C18 (3.0 × 150 mm, 2.7 μm particle size) column). An isocratic method was used with a mobile phase consisting of 50 mM triethylammonium acetate (TEAA) with 2% acetonitrile at 0.7 mL min−1 for 10 min. The column was heated to 40 °C and the injection volume was 5 μL. Peaks were detected at 254 nm. The measured concentrations were normalized to the initial substrate concentration of 5 mM. The results can be found in the ESI† Fig. S3 and S4.Quantification of 2′3′-cGAMP, nucleosides, and nucleotides
The analysis of the samples of the enzyme assays was performed by reversed-phase high-performance liquid chromatography (RP-HPLC). An Ultimate™ 3000 HPLC system (Thermo Fisher Scientific™, Waltham, USA) consisting of a pump (UltiMate 3000 Pump), autosampler (UltiMate 3000 Autosampler), column oven (UltiMate 3000 Flow Manager), and a wavelength detector (UltiMate 3000 Variable Wavelength Detector). The separation of the analytes was performed with an ISAspher 100-5 C18 AQ column (250 × 4 mm, ISERA GmbH, Düren, Germany) column heated to 30 °C using an injection volume of 10 μL. The gradient used consisted of mobile phase A (50 mM TEAA with 2% acetonitrile) and mobile phase B (95% acetonitrile, 5% water) at a flow rate of 1 mL min−1 (0 min: 100% A; 16 min: 100% A; 24.5 min: 96% A, 4% B; 25 min: 75% A, 25% B; 27 min: 5% A, 95% B; 33 min: 5% A, 95% B; 34 min: 100% A; 49 min: 100% A). The samples and standards were analyzed at a wavelength of 254 nm. The chromatograms were evaluated using Chromeleon™ (Thermo Fisher Scientific™, Waltham, USA).LC–MS validation of 2′3′-cGAMP
A 1260 Infinity II LC system (Agilent Technologies, Santa Clara, USA) consisting of a diode array detector (1260 DAD HS), multicolumn thermostat (1260 MCT), degasser (1260 Degasser), multisampler (1260 Multisampler), binary pump (1260 Binary Pump) and an Agilent Poroshell 120 EC-C18 (4.6 × 100 mm, 2.7 μm particle size) column were used for LC–MS analysis. The gradient used consisted of mobile phase A (0.1% formic acid) and mobile phase B (100% acetonitrile) at a flow rate of 1 mL min−1 (0 min: 5% A, 95% B; 7 min: 95% A, 5% B; 9 min: 5% A, 95% B; 14 min: 5% A, 95% B). The column was heated to 40 °C and the injection volume was 5 μL. Peaks were detected at 254 nm. Using an Agilent Technologies 6120 Single Quadrupole LC–MS, the mass spectra were obtained over the mass range of m/z 100 to 1000. A temperature of 350 °C and a dry gas flow rate of 12 L min−1 were established in the electrospray chamber. The capillary voltage was 3 kV, and the nebulizer pressure was 35 psi.Author contributions
Conceptualization, Katrin Rosenthal (K. R.) and Stephan Lütz (S. L.); data curation, Martin Becker (M. B.), Isabel Nowak (I. N.), and K. R.; formal analysis, M. B., I. N. and K. R.; funding acquisition, K. R. and S. L.; investigation, I. N., Katharina Hildebrand (K. H.), and M. B.; methodology, I. N., K. H., M. B., K. R., and S. L.; project administration, K. R. and S. L.; resources, K. R. and S. L.; supervision, K. R. and S. L.; validation, M. B., I. N., K. H., and K. R.; visualization, M. B., I. N., and K. R.; writing – original draft, M. B. and K. R.; writing – review and editing, M. B., K. R., and S. L. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Jennifer N. Andexer for providing the plasmids for the enzymes ScADK, AjPPK2, SmPPK2, MrPPK2 and MjNK. The authors would also like to thank Nicholas Turner, Sabine Flitsch and Christian Schnepel for providing the plasmid of ChPPK2 and Regine Siedentop for cloning EbPPK2. Thanks go to Claudio Piselli for the determination of the TON of thscGAS. Thanks also go to Subhan Aamir, Hugo A. Martinez Alvarado, Daniela Miño, Amisha Rai, Jade V. Rivera, and Bibiazhar Suleimen for carrying out the temperature-dependent stability analyses of ATP and GTP. The project is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - 535167833.Notes and references
- N. J. Claassens, S. Burgener, B. Vögeli, T. J. Erb and A. Bar-Even, Curr. Opin. Biotechnol., 2019, 60, 221–229 CrossRef CAS PubMed.
- J. M. Sperl and V. Sieber, ACS Catal., 2018, 8, 2385–2396 CrossRef CAS.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- R. Abu and J. M. Woodley, ChemCatChem, 2015, 7, 3094–3105 CrossRef CAS.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- J. Muschiol, C. Peters, N. Oberleitner, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Commun., 2015, 51, 5798–5811 RSC.
- M. Milić, E. Byström, P. Domínguez de María and S. Kara, ChemSusChem, 2022, 15, 1–8 CrossRef PubMed.
- Y. Zhang and H. Hess, ACS Catal., 2017, 7, 6018–6027 CrossRef CAS.
- P. Dvorak, N. P. Kurumbang, J. Bendl, J. Brezovsky, Z. Prokop and J. Damborsky, ChemBioChem, 2014, 15, 1891–1895 CrossRef CAS PubMed.
- S. Mordhorst and J. N. Andexer, Nat. Prod. Rep., 2020, 37, 1316–1333 RSC.
- W. Berke, H.-J. Schüz, C. Wandrey, M. Morr, G. Denda and M.-R. Kula, Biotechnol. Bioeng., 1988, 32, 130–139 CrossRef CAS PubMed.
- J. N. Andexer and M. Richter, ChemBioChem, 2015, 16, 380–386 CrossRef CAS PubMed.
- R. Siedentop, T. Prenzel, S. R. Waldvogel, K. Rosenthal and S. Lütz, ChemElectroChem, 2023, 10, 1–9 Search PubMed.
- S. Mordhorst, J. Siegrist, M. Müller, M. Richter and J. N. Andexer, Angew. Chem., Int. Ed., 2017, 56, 4037–4041 CrossRef CAS PubMed.
- M. Tavanti, J. Hosford, R. C. Lloyd and M. J. B. Brown, Green Chem., 2021, 23, 828–837 RSC.
- H. Frohnmeyer and L. Elling, Carbohydr. Res., 2023, 523, 108727 CrossRef CAS PubMed.
- M. Lubberink, C. Schnepel, J. Citoler, S. R. Derrington, W. Finnigan, M. A. Hayes, N. J. Turner and S. L. Flitsch, ACS Catal., 2020, 10, 10005–10009 CrossRef CAS.
- G. R. Painter, M. G. Natchus, O. Cohen, W. Holman and W. P. Painter, Curr. Opin. Virol., 2021, 50, 17–22 CrossRef CAS PubMed.
- M. K. Lo, R. Jordan, A. Arvey, J. Sudhamsu, P. Shrivastava-Ranjan, A. L. Hotard, M. Flint, L. K. McMullan, D. Siegel, M. O. Clarke, R. L. Mackman, H. C. Hui, M. Perron, A. S. Ray, T. Cihlar, S. T. Nichol and C. F. Spiropoulou, Sci. Rep., 2017, 7, 1–7 CrossRef PubMed.
- E. Lawitz, F. F. Poordad, P. S. Pang, R. H. Hyland, X. Ding, H. Mo, W. T. Symonds, J. G. McHutchison and F. E. Membreno, Lancet, 2014, 383, 515–523 CrossRef CAS PubMed.
- A. Ablasser, M. Goldeck, T. Cavlar, T. Deimling, G. Witte, I. Röhl, K.-P. Hopfner, J. Ludwig and V. Hornung, Nature, 2013, 498, 380–384 CrossRef CAS PubMed.
- L. Sun, J. Wu, F. Du, X. Chen and Z. J. Chen, Science, 2013, 339, 786–791 CrossRef CAS PubMed.
- L. Corrales, L. H. Glickman, S. M. McWhirter, D. B. Kanne, K. E. Sivick, G. E. Katibah, S.-R. Woo, E. Lemmens, T. Banda, J. J. Leong, K. Metchette, T. W. Dubensky and T. F. Gajewski, Cell Rep., 2015, 11, 1018–1030 CrossRef CAS PubMed.
- T. W. Dubensky, D. B. Kanne and M. L. Leong, Ther. Adv. Vaccines, 2013, 1, 131–143 CrossRef PubMed.
- B. Novotná, L. Vaneková, M. Zavřel, M. Buděšínský, M. Dejmek, M. Smola, O. Gutten, Z. A. Tehrani, M. Pimková Polidarová, A. Brázdová, R. Liboska, I. Štěpánek, Z. Vavřina, T. Jandušík, R. Nencka, L. Rulíšek, E. Bouřa, J. Brynda, O. Páv and G. Birkuš, J. Med. Chem., 2019, 62, 10676–10690 CrossRef PubMed.
- K. Rosenthal, M. Becker, J. Rolf, R. Siedentop, M. Hillen, M. Nett and S. Lütz, ChemBioChem, 2020, 21, 3225–3228 CrossRef CAS PubMed.
- X. Zhang, H. Shi, J. Wu, X. Zhang, L. Sun, C. Chen and Z. J. Chen, Mol. Cell, 2013, 51, 226–235 CrossRef CAS PubMed.
- B. L. Gaffney, E. Veliath, J. Zhao and R. A. Jones, Org. Lett., 2010, 12, 3269–3271 CrossRef CAS PubMed.
- M. Becker, P. Nikel, J. N. Andexer, S. Lütz and K. Rosenthal, Biomolecules, 2021, 11, 1–13 CrossRef PubMed.
- J. A. McIntosh, Z. Liu, B. M. Andresen, N. S. Marzijarani, J. C. Moore, N. M. Marshall, M. Borra-Garske, J. V. Obligacion, P. S. Fier, F. Peng, J. H. Forstater, M. S. Winston, C. An, W. Chang, J. Lim, M. A. Huffman, S. P. Miller, F.-R. Tsay, M. D. Altman, C. A. Lesburg, D. Steinhuebel, B. W. Trotter, J. N. Cumming, A. Northrup, X. Bu, B. F. Mann, M. Biba, K. Hiraga, G. S. Murphy, J. N. Kolev, A. Makarewicz, W. Pan, I. Farasat, R. S. Bade, K. Stone, D. Duan, O. Alvizo, D. Adpressa, E. Guetschow, E. Hoyt, E. L. Regalado, S. Castro, N. Rivera, J. P. Smith, F. Wang, A. Crespo, D. Verma, S. Axnanda, Z. E. X. Dance, P. N. Devine, D. Tschaen, K. A. Canada, P. G. Bulger, B. D. Sherry, M. D. Truppo, R. T. Ruck, L.-C. Campeau, D. J. Bennett, G. R. Humphrey, K. R. Campos and M. L. Maddess, Nature, 2022, 603, 439–444 CrossRef CAS PubMed.
- P. Barrado, M. J. Rodríguez, A. Jiménez and M. Fernández Lobato, Yeast, 2003, 20, 1145–1150 CrossRef CAS PubMed.
- L. Arnfors, T. Hansen, P. Schönheit, R. Ladenstein and W. Meining, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 1085–1097 CrossRef PubMed.
- T. Hansen, L. Arnfors, R. Ladenstein and P. Schönheit, Extremophiles, 2007, 11, 105–114 CrossRef CAS PubMed.
- C. Moeller, C. Schmidt, F. Guyot and M. Wilke, Biophys. Chem., 2022, 290, 106878 CrossRef CAS PubMed.
- G. J. Pielak, Biochemistry, 2021, 60, 3436–3440 CrossRef CAS PubMed.
- C. F. C. Bonting, G. J. J. Kortstee and A. J. B. Zehnder, J. Bacteriol., 1991, 173, 6484–6488 CrossRef CAS PubMed.
- B. Nocek, S. Kochinyan, M. Proudfoot, G. Brown, E. Evdokimova, J. Osipiuk, A. M. Edwards, A. Savchenko, A. Joachimiaka and A. F. Yakunin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17730–17735 CrossRef CAS PubMed.
- K. Motomura, R. Hirota, M. Okada, T. Ikeda, T. Ishida and A. Kuroda, Appl. Environ. Microbiol., 2014, 80, 2602–2608 CrossRef PubMed.
- B. P. Nocek, A. N. Khusnutdinova, M. Ruszkowski, R. Flick, M. Burda, K. Batyrova, G. Brown, A. Mucha, A. Joachimiak, L. Berlicki and A. F. Yakunin, ACS Catal., 2018, 8, 10746–10760 CrossRef CAS.
- S. T. Kulmer, A. Gutmann, M. Lemmerer and B. Nidetzky, Adv. Synth. Catal., 2017, 359, 292–301 CrossRef CAS.
- J. Rolf, R. Siedentop, S. Lütz and K. Rosenthal, Int. J. Mol. Sci., 2020, 21, 105 CrossRef CAS PubMed.
- J. Hall, E. C. Ralph, S. Shanker, H. Wang, L. J. Byrnes, R. Horst, J. Wong, A. Brault, D. Dumlao, J. F. Smith, L. A. Dakin, D. C. Schmitt, J. Trujillo, F. Vincent, M. Griffor and A. E. Aulabaugh, Protein Sci., 2017, 26, 2367–2380 CrossRef CAS PubMed.
- W. H. Miller and R. L. Miller, Mol. Biochem. Parasitol., 1991, 48, 39–46 CrossRef CAS PubMed.
- P. Benčić, M. Keppler, M. Kuge, D. Qiu, L. M. Schütte, M. Häner, K. Strack, H. J. Jessen, J. N. Andexer and C. Loenarz, FEBS J., 2023, 290, 4899–4920 CrossRef PubMed.
- M. Fehlau, F. Kaspar, K. F. Hellendahl, J. Schollmeyer, P. Neubauer and A. Wagner, Front. Bioeng. Biotechnol., 2020, 8, 854 CrossRef PubMed.
- E. V. Canettieri, G. J. de M. Rocha, J. A. de Carvalho and J. B. de Almeida e Silva, Bioresour. Technol., 2007, 98, 422–428 CrossRef CAS PubMed.
- R. Siedentop, M. Siska, N. Möller, H. Lanzrath, E. von Lieres, S. Lütz and K. Rosenthal, Catalysts, 2023, 13, 468 CrossRef CAS.
- J. Engel, U. T. Bornscheuer and S. Kara, Org. Process Res. Dev., 2021, 25, 411–420 CrossRef CAS.
- R. Siedentop, C. Claaßen, D. Rother, S. Lütz and K. Rosenthal, Catalysts, 2021, 11, 1–24 CrossRef.
- K. Rosenthal, U. T. Bornscheuer and S. Lütz, Angew. Chem., Int. Ed., 2022, 61, 1–3 CrossRef PubMed.
- G. A. Strohmeier, A. Schwarz, J. N. Andexer and M. Winkler, J. Biotechnol., 2020, 307, 202–207 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00147h |
| This journal is © The Royal Society of Chemistry 2024 |