
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt-catalysed hydroformylation of epoxides in the presence of phosphine oxides†
Fábio G.
Delolo
 ab,
Christoph
Kubis
b,
Baoxin
Zhang
b,
Helfried
Neumann
b,
Eduardo N.
dos Santos
a,
Elena V.
Gusevskaya
*a and
Matthias
Beller
*b
ab,
Christoph
Kubis
b,
Baoxin
Zhang
b,
Helfried
Neumann
b,
Eduardo N.
dos Santos
a,
Elena V.
Gusevskaya
*a and
Matthias
Beller
*b
aDepartamento de Química, Universidade Federal de Minas Gerais, Belo Horizonte, MG, Brazil. E-mail: elena@ufmg.br
bLeibniz-Institut für Katalyse, Albert-Einstein-Straße 29a, Rostock, 18059, Germany. E-mail: Matthias.Beller@catalysis.de
First published on 2nd February 2024
Abstract
A novel cobalt-catalysed hydroformylation of epoxides to β-hydroxyaldehydes was developed. Compared to previous works this methodology proceeds under significantly milder conditions (70 °C; 40 bar of CO/H2 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Crucial for the activity of the cobalt catalyst is the use of phosphine oxides, especially tricyclohexylphosphine oxide, as cheap and easily available promoters. The hydroformylation reaction can be easily combined with a consecutive one-pot hydrogenation of the in situ generated β-hydroxyaldehydes to produce directly 1,3-diols (important intermediates for polyesters and fibres) starting from epoxides. The rapid formation of a mononuclear acyl cobalt carbonyl complex as an intermediate was observed by in situ FTIR spectroscopy.
:1). Crucial for the activity of the cobalt catalyst is the use of phosphine oxides, especially tricyclohexylphosphine oxide, as cheap and easily available promoters. The hydroformylation reaction can be easily combined with a consecutive one-pot hydrogenation of the in situ generated β-hydroxyaldehydes to produce directly 1,3-diols (important intermediates for polyesters and fibres) starting from epoxides. The rapid formation of a mononuclear acyl cobalt carbonyl complex as an intermediate was observed by in situ FTIR spectroscopy.
Introduction
Hydroformylation of olefins, also called oxo-process, is the largest scale transition metal complex catalysed industrial process and is responsible for the global production of more than 10 million metric tons of aldehydes per year.1 This transformation was discovered by Otto Roelen in 1938 and consists of the addition of “syngas” (carbon monoxide and hydrogen mixture) to olefins resulting in aldehydes that can be used directly or further transformed into a variety of bulk or fine chemicals.2While the hydroformylation of olefins is well-known, the use of non-olefinic substrates in this reaction has been much less explored. For example, the hydroformylation of epoxides, which can be produced by olefin epoxidation,3 offers an elegant and atom-economic pathway to access β-hydroxyaldehydes. These bifunctional compounds are important intermediates for the synthesis of polyesters and fibres4 as well as many bioactive compounds.5
The first catalytic hydroformylation of epoxides was described by Watanabe and co-workers in 1964,6,7 but only in the 90s the investigations were intensified. The increased interest on this transformation relies on the manufacture of 1,3-propanediol (PDO) from ethylene oxide. Here, initially 3-hydroxypropanal (HPA) is formed in the hydroformylation step, which subsequently can be hydrogenated to PDO, an important intermediate in the production of polyester fibres and films (Fig. 1A).
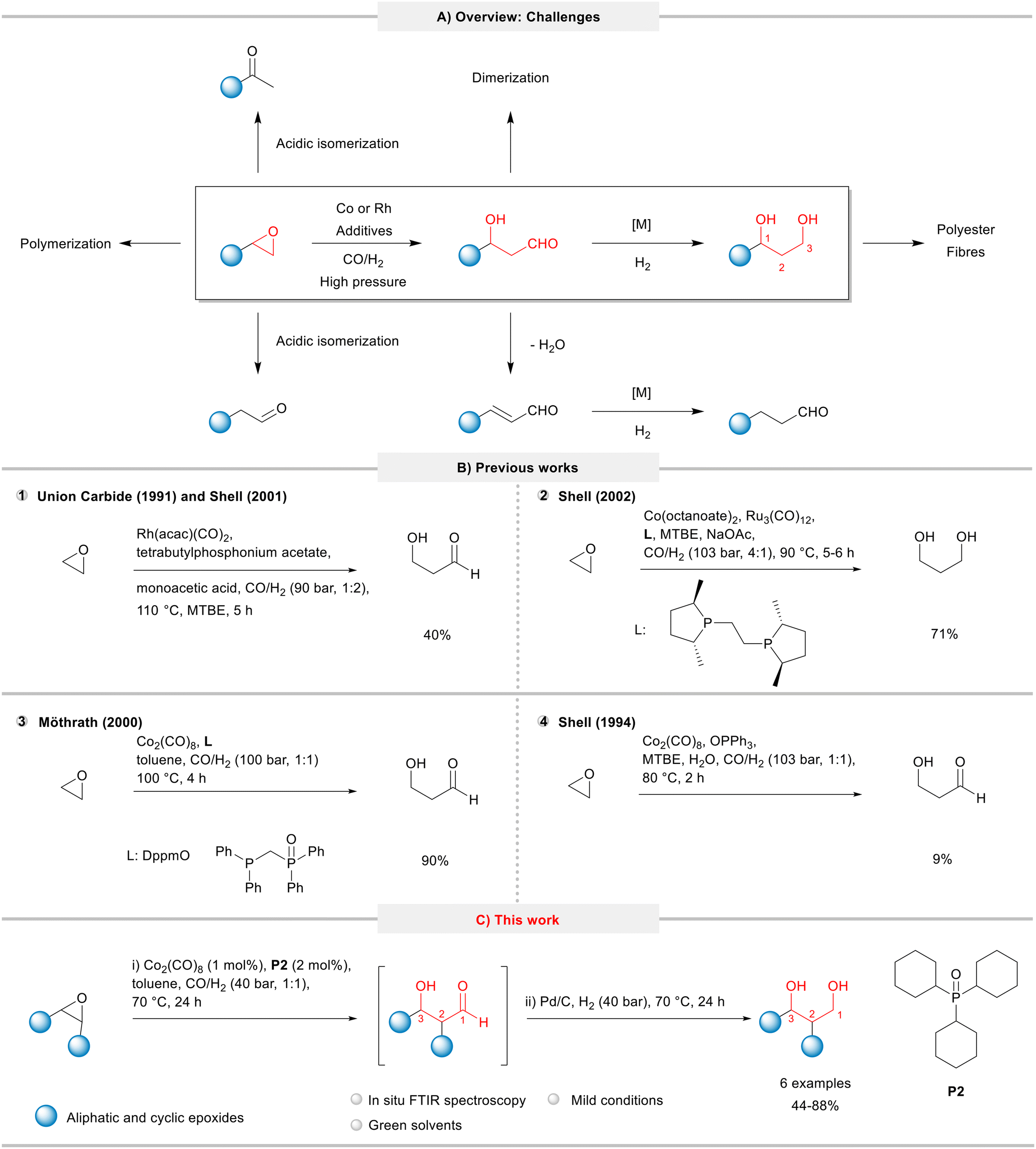 | ||
| Fig. 1 Hydroformylation of epoxides: A) overview: applications and undesired side and consecutive reactions. B) Reaction conditions and product yields in previous works. C) This work. | ||
In terms of reaction conditions, the hydroformylation of epoxides requires high pressures of syngas and a close look at temperature to manage the balance between a high catalyst activity and the extent of undesired side reactions such as isomerization, polymerization, oligomerization, elimination, and hydrogenation (Fig. 1A). Therefore, these processes are generally characterized by poor selectivity and poor yields of the desired products.
Due to the industrial interest, the literature related to the hydroformylation of epoxides is currently dominated by patents. Shell filed several patents for the hydroformylation of ethylene oxide based on cobalt as a catalyst precursor and a huge library of ligands (diphosphines, phosphines, arsines, etc.) on an industrial scale.8 In order to promote as well as to improve catalyst recycling, the HPA intermediate or PDO product can be recovered from a non-water-soluble solvent system by water extraction. This two-step process allows to recycle a majority of the cobalt carbonyl catalyst with the organic solvent phase, as desired.9 Suitable promoters include sources of mono- and multivalent metal cations of weak bases such as salts of carboxylic acids,10 tertiary amines,11 quaternary ammonium salts,12 quaternary phosphonium salts,13 quaternary arsonium salts,14 dihydroxyarenes (hydroquinones),15 and porphyrins.16 In addition, Shell explored bimetallic catalysts in the epoxide hydroformylation, combining cobalt with other metals, such as Fe,17 Rh18 and Ru.19 Other companies, such as Hoechst Celanese,20 Union Carbide,21 and Eastman Kodak,22 also filed patents on this theme. Unlike the Shell process, their methods are mainly based on rhodium precursors and have not been commercialized yet to the best of our knowledge.23
Union Carbide (now Dow) and Shell filed patents (1991 and 2001, respectively) for the hydroformylation of ethylene oxide using a rhodium catalyst promoted with tetrabutylphosphonium acetate under 90 bar (CO/H2: 1/2) and 110 °C to yield HPA in 40% (Fig. 1, item 1).13,24 In 2002, Shell combined in a tandem process the hydroformylation of ethylene oxide followed by the hydrogenation using cobalt and diphosphine-modified ruthenium catalysts to give the desired PDA product in a 71% yield (Fig. 1B, item 2).25 Later, Möthrath and collaborators reported the use of cobalt complexes with hemilabile P–O chelating ligands in the synthesis of β-hydroxyaldehydes; however, the method required harsh reaction conditions (CO/H2 = 100 bar, 100 °C) and provided only moderate product yields (Fig. 1B, item 3).26
In attempts to improve the production of HPA, Shell claimed the use of phosphine oxides (in particular, triphenylphosphine oxide) to “accelerate the hydroformylation reaction and to permit the recycle of essentially all the cobalt catalyst in the organic phase following water extraction of product HPA”.27 Although the patent protected a wide range of phosphine oxides, no examples with the use of tricyclohexylphosphine oxide (P2) were provided. Using Co2(CO)8 and OPPh3 under relatively hash conditions (CO/H2 = 103 bar, 1:1, 80 °C), the desired HPA product was obtained in only 9% (Fig. 1B, item 4).27,28
In the classical hydroformylation of olefins, the systems based on rhodium and phosphine oxides have been also described. For example, Abu-Gnim and Amer showed that the systems with mixed amino phosphine oxide ligands displayed exceptionally high activity and regioselectivity in the hydroformylation of styrene to give branched aldehyde as compared to the phosphine analogues.29 It was also shown that the systems with pyridylphosphine oxides (P(O)–N) were remarkably more active than those containing pyridylphosphine analogues.30
Satisfactory results were obtained at the hydroformylation/hydrogenation of C8 olefins to produce isononyl aldehydes and alcohols using a rhodium–triphenylphosphine oxide system.31 (Hydroxy(phenyl)methyl)diphenylphosphine oxide, a phosphine oxide compound, was successfully employed by Alper et al. as a ligand in the rhodium-catalysed hydroformylation of olefins, allowing for good conversions and high regioselectivities.32
We have recently showed that phosphine oxides are able to efficiently promote some cobalt catalysed processes under mild conditions, such as the hydroformylation of olefins,33 reductive etherification of benzaldehyde derivatives,34 and ring expansion/ring opening of oxetanes.35 Based on these works, herein, we report the results of the systematic evaluation of simple, relatively cheap, and commercially available phosphine oxides in the cobalt-catalysed hydroformylation of epoxides aiming to synthesize β-hydroxyaldehydes under mild conditions. The developed methodology for a model system was combined with the consecutive one-pot hydrogenation, which was then applied to produce 1,3-diols starting from epoxides (Fig. 1C).
Results and discussion
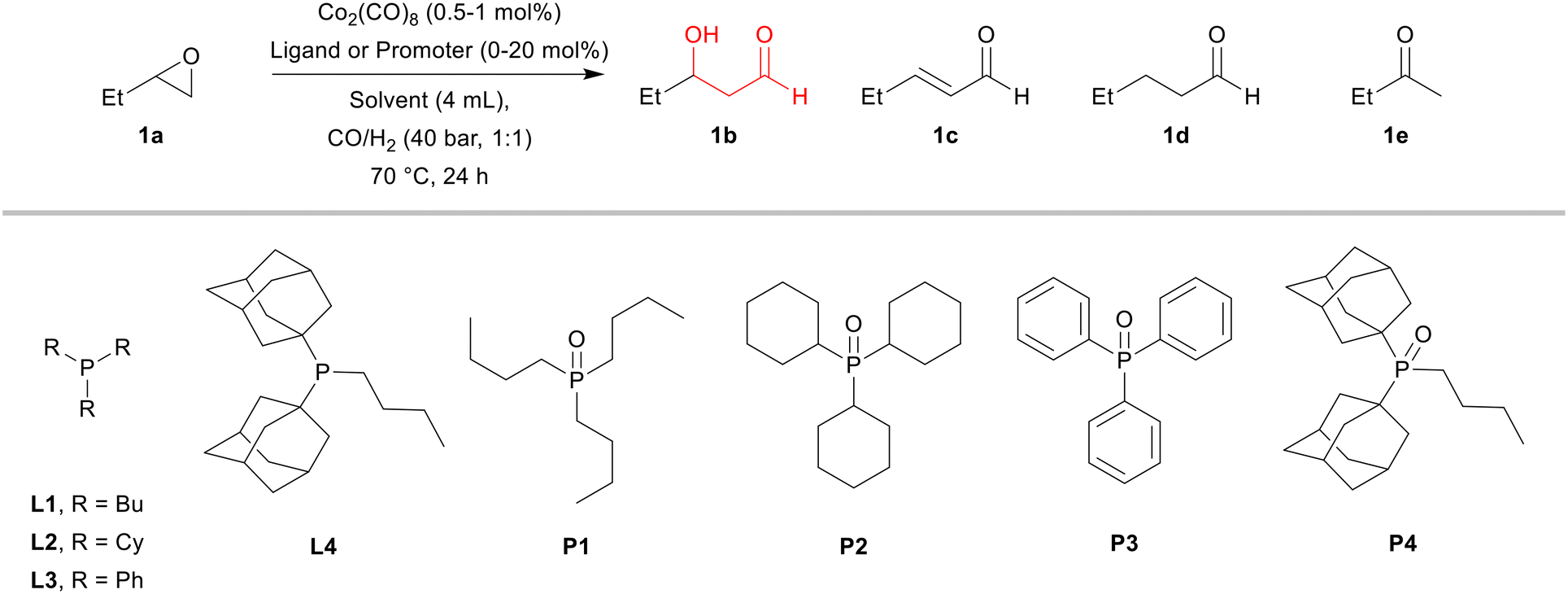
At the start of this work, 1,2-epoxybutane (1a) was chosen as a model substrate due to its physical properties (easy handling) and similarity to the commercial process. To overcome the harsh conditions of previously reported hydroformylation of epoxides, we focused on the development of a catalytic system that operates at low temperature and pressure (70 °C and 40 bar CO/H2 – 1:1). Typically, the reactions were performed for 24 h using Co2(CO)8 as the catalyst precursor.
For comparison, we started the study using an unmodified system, i.e. the system without any phosphorus ligand present (Table 1, entry 1). Under these conditions only 8% of the substrate was converted to give exclusively β-hydroxyaldehyde 1b. In the hydroformylation of olefins, the modification of the catalytic system with phosphorus ligands plays an important role in improving not only the reaction rate, but also its selectivity. For this reason, we evaluated several common, relatively cheap, and commercially available phosphines as auxiliaries in the hydroformylation of 1a. However, the results were disappointing. In the presence of phosphines L1–L3 the corresponding catalyst systems were completely inactive (Table 1, entries 2–4). Only in the presence of phosphine L4, little conversion of the epoxide was detected (8%, Table 1, entry 5). These results are in principle aligned with the reported observations that phosphines, as well as phosphites and arsines, reduce the catalytic activity of cobalt carbonyl complexes in hydroformylation.36 Nevertheless, these ligands are often applied to improve the regioselectivity for linear aldehydes and in tandem processes to promote the hydrogenation of aldehydes into corresponding alcohols.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Ligand or promoter | Solvent | Conversionb (%) | Selectivityb (%) | ||||
| 1b | 1c | 1d | 1e | Others | ||||
|
a Reaction conditions: 1a (2 mmol), Co2(CO)8 (0.5 mol%), ligand (L1–L4) or promoter (P1–P4) (2.0 mol%), toluene (4 mL), 70 °C, gas phase – CO/H2 (1:1) 40 bar, 24 h. Dimethyl carbonate (DMC); diethyl carbonate (DEC).
b Determined by GC analysis using isooctane as internal standard.
c Co2(CO)8 (1 mol%).
d
P2 (4 mol%).
e
P2 (20 mol%).
|
||||||||
| 1 | — | Toluene | 8 | >99 | 0 | 0 | 0 | 0 |
| 2 | L1 | Toluene | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | L2 | Toluene | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | L3 | Toluene | 1 | >99 | 0 | 0 | 0 | 0 |
| 5 | L4 | Toluene | 8 | 72 | 0 | 0 | 9 | 19 |
| 6 | P1 | Toluene | 52 | 54 | 0 | 0 | 7 | 39 |
| 7 | P2 | Toluene | 65 | 72 | 0 | 0 | 7 | 21 |
| 8 | P3 | Toluene | 24 | 64 | 0 | 0 | 13 | 23 |
| 9 | P4 | Toluene | 60 | 76 | 0 | 0 | 3 | 21 |
| 10c | P2 | Toluene | 81 | 77 | 0 | 2 | 4 | 17 |
| 11c,d | P2 | Toluene | 74 | 60 | 5 | 10 | 3 | 22 |
| 12c,e | P2 | Toluene | 40 | 22 | 17 | 46 | 4 | 11 |
| 13c | P2 | Anisole | 81 | 56 | 0 | 5 | 9 | 30 |
| 14c | P2 | DMC | 75 | 35 | 6 | 4 | 24 | 31 |
| 15c | P2 | DEC | 67 | 29 | 6 | 8 | 24 | 33 |
| 16c | P2 | 1,4-Dioxane | 80 | 65 | 0 | 0 | 11 | 24 |
| 17c | P2 | THF | 62 | 29 | 16 | 26 | 4 | 25 |
| 18c | P2 | MeCN | 15 | 43 | 0 | 0 | 5 | 52 |
| 19c | P2 | DMF | 1 | 0 | 0 | 0 | 0 | >99 |
| 20c | P2 | DMSO | 3 | 0 | 0 | 0 | 29 | 71 |
A remarkable increase in the activity of the cobalt catalyst was achieved using phosphine oxides P1–P4 as promoters instead of the respective phosphines L1–L4 (Table 1, entries 6–9 vs. entries 2–5). In the presence of phosphine oxides, the hydroformylation was highly regioselective resulting exclusively in the linear product, β-hydroxyaldehyde 1b. As minor products were detected ketone 1e (formed due to the acid catalysed isomerization of the substrate) and saturated aldehyde 1d (formed due to the dehydration of 1b to give unsaturated aldehyde 1c and its subsequent hydrogenation). The isomerization of epoxides has been recently reviewed by Jat and Kumar.37 In particular, these reactions can be promoted by carbonyl cobalt complexes, as reported by the Coates group.38 The best results were obtained with tricyclohexylphosphine oxide P2 (Table 1, entry 7), which was the best promoter for the cobalt-catalysed ring expansion/ring opening of oxetanes, too.35 Although the reaction in the presence P4, a structurally more complex compound, showed similar results (Table 1, entry 9), we decided to choose P2 for further studies. It is worthwhile to highlight the lower performance of triphenylphosphine oxide P3 (Table 1, entry 8), claimed in the Shell patent as an efficient promoter for the cobalt catalysed hydroformylation of ethylene oxide under more severe conditions.27
For the further process optimization, we varied the concentrations of the cobalt precursor and the promoter. With the increase in the cobalt amounts from 0.5 to 1 mol%, both the substrate conversion, and selectivity for 1b were improved (Table 1, entry 10 vs. entry 7). Conversely, increasing the P2 concentration not only significantly decelerated the reaction but also dramatically decreased its selectivity. The drop in the selectivity for 1b was due to its further transformation into the dehydration and dehydration/hydrogenation products 1c and 1d, respectively (Table 1, entries 10–12). The kinetic profile for the substrate conversion and product formation in the reaction given in entry 10 in Table 1 are presented in Fig. S1.†
Next, different cobalt sources were tested as catalyst precursors in the combination with tricyclohexylphosphine oxide P2, such as Co(acac)2, Co(acac)3, Co(OAc)2, and Co(NO3)2·6H2O (Table S1 in ESI†). Unfortunately, none of these salts promoted a detectable conversion of the epoxide under the conditions in which the system with Co2(CO)8 gave 81% conversion (entries 1–5 in Table S1†vs. entry 10 in Table 1). Similar results were obtained with the catalysts based on other metals, i.e., iridium, palladium, iron, and manganese (Table S1,† entries 7–11). Even rhodium, known as the most active metal in the hydroformylation of olefins, was completely inefficient for the hydroformylation of epoxide 1a under these reaction conditions (Table S1,† entry 6). These results suggest that the acidic nature of cobalt species derived from Co2(CO)8 under the reaction conditions plays a key role in the activation of the epoxide towards the interaction with carbon monoxide and hydrogen.39 No precipitation of the metallic cobalt was observed under the applied conditions.
It is well known that the nature of solvent can drastically affect the outcome of the catalytic reaction. The important role of solvents in catalysis have been recently reviewed by Dyson and Jessop.40 In the monophasic hydroformylation of olefins, the replacement of conventional solvents by greener alternatives proved to be a viable approach allowing to match catalytic efficiency and process sustainability.41 In the Shell patent,42 methyl tertiary-butyl ether (MTBE) was used as the main solvent but problems related to low solubility, instability under acidic conditions and a low flash point were considered disadvantages for using this solvent.43
Based on the modern solvent sustainability guides,44 we selected the following compounds: anisole, dimethyl carbonate (DMC), and diethyl carbonate (DEC). However, it was found that the use of these solvents instead of toluene had no positive effect on the reaction rate; instead, it decreased the reaction selectivity to hydroxyaldehyde 1b (Table 1, entries 13–15 vs. entry 10). The loss in selectivity was due to the formation of products 1c, 1d, and 1e along with several unidentified compounds. Interestingly, the reactions in DMC and DEC showed a relatively strong tendency to the substrate isomerization pathway to give a significant amount of ketone 1e. As the results obtained using the sustainability criteria were not satisfactory, we decided to investigate the solvent influence in terms of polarity and dielectric constant. See Fig. S2† for more details.
The reactions in dioxane and THF (relatively low polarity and dielectric constant, ε = 2.25 and 7.58, respectively) gave similar results in terms of the substrate conversion as compared to other non-polar solvents: toluene (ε = 2.38), anisole (ε = 4.33), DMC (ε = 3.09) and DEC (ε = 3.10) (Table 1, entries 16 and 17). However, the selectivity for the desired hydroxyaldehyde was lower than in the reaction performed in toluene (Table 1, entry 10). The loss in selectivity was particularly high in THF. Hydroxyaldehyde 1b was found to be much less stable in THF than in other solvents, being transformed into the dehydration and dehydration/hydrogenation products 1c and 1d (ca. 40% of the mass balance, Table 1, entry 17). Finally, the runs were performed in highly polar solvents: MeCN (ε = 37.5), DMF (ε = 36.7), and DMSO (ε = 46.7) (Table 1, entries 18–20). In all these reactions very low or even no conversion of the epoxide was observed for 24 h.
Mechanistic aspects and in situ FTIR-spectroscopic investigations
To obtain a better understanding about the promoting effect of phosphine oxide on the cobalt catalyst system in situ FTIR-experiments have been performed. The reaction conditions (n(1a) = 8 mmol, n(Co2(CO)8) = 0.04 mmol, n(P2) = 0.16 mmol, 80 °C, p(CO/H2) = 40 bar, vol. (toluene) = 16 mL) have been slightly modified to meet the requirements of the HP FTIR experiment. In a first step the hydroformylation of 1,2-epoxybutane 1a with the unmodified catalyst using Co2(CO)8 as a precursor in the absence of the phosphine oxide was conducted. From the series of infrared spectra covering 20 h of the reaction only a slow increase of bands assigned to the β-hydroxyaldehyde product 1b (ν(CO) = 1726 cm−1) was observed which is in agreement with the low activity found in the catalytic experiments (see Fig. 2). The pre-catalyst Co2(CO)8 (ν(CO) = 1850, 1858, 2022, 2039, and 2068 cm−1) was detected as the only dominant species during the entire experiment. No further intermediate complex was identified.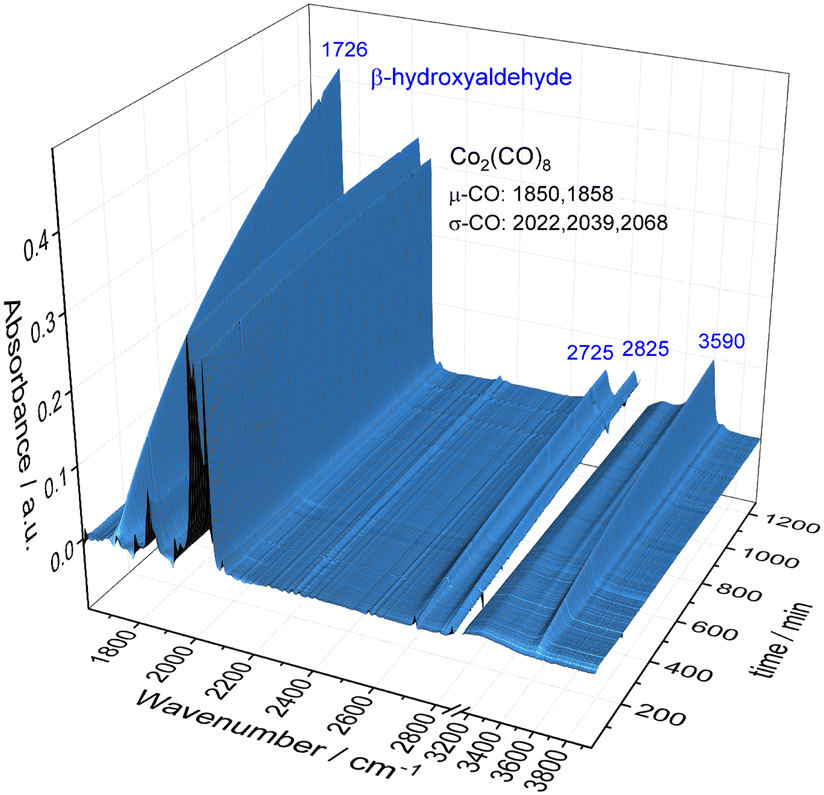 | ||
| Fig. 2 HP FTIR-spectra series collected during the hydroformylation of 1,2-epoxbutane 1a with Co2(CO)8 used as a pre-catalyst at 80 °C and p(CO/H2) = 40 bar in toluene as a solvent. Further reaction conditions: n(1a) = 8 mmol, n(Co2(CO)8) = 0.04 mmol, vol. (toluene) = 16 mL. | ||
In the next step, a HP IR experiment was performed in the presence of tricyclohexylphosphine oxide P2, with [P2]/[Co] = 2, at otherwise identical conditions. Notably, during the preparation of the reaction solution a precipitation of a solid material took place, which is explained by disproportionation of the starting cobalt carbonyl complex to a salt of the type [Co(B)6][Co(CO)4]2 in the presence of a base (B) (see discussion below). From the first measured spectra after the transfer of the catalyst mixture into the reactor system and pressurization with synthesis gas at the desired temperature a rapid formation of a certain amount of aldehyde was detected, see Fig. 3a. The conversion of 1a seem to be completed within ca. 23 h after which almost a plateau of the bands for the product was reached. Interestingly, the initial infrared spectra in the region for transition metal carbonyls show a pattern with band positions at ν(CO) = 2004, 2024, 2044, and 2105 cm−1 which can be assigned to an acyl complex of the type RC(O)Co(CO)4, see Fig. 3b.45 At higher conversions these bands were decreasing and those assigned to Co2(CO)8 increased in intensity. Finally, Co2(CO)8 is the only detectable cobalt carbonyl species, see Fig. 3c. Control experiments have been performed without the epoxide substrate. For the phosphine oxide free system with Co2(CO)8 as the pre-catalyst, a very slow partial formation of the hydride complex HCo(CO)4 (ν(CO) = 2024, 2048, 2114 cm−1) was observed (Fig. S3 and S4†). For the phosphine oxide (P2) containing system, the hydride formation took place to a similar extent but was significantly quicker (Fig. S5 and S6†). Based on the catalytic results obtained with 1,2-epoxybutane and the FTIR experiments, a simplified mechanistic scheme for the transformation of epoxides under the hydroformylation conditions is proposed in Fig. 4. Initially, phosphine oxide acts as a Lewis base,46 thereby promoting disproportionation of the Co2(CO)8 dimer providing the active monometallic HCo(CO)4 species.47 Further roles of phosphine oxides, such as generating positive solvation effects48 cannot be ruled out. The unsuccessful attempts of using other metal precursors in the hydroformylation of epoxides (see Table S1†) shows the importance of the acidic properties of HCo(CO)4 for the substrate activation.34,39 The nucleophilic attack of [Co(CO)4]− (step A) occurs preferentially on the side of lower steric hindrance in monosubstituted epoxide a to give exclusively linear products.49 Next, the linear cobalt–hydroxyalkyl intermediate I can undergo β-hydride elimination followed by tautomerization to give ketone e, the isomer of the original epoxide a. Alternatively, intermediate I can be involved in the well-established and desired hydroformylation sequence (steps B and C). This route consists of the CO insertion step (step B) to give cobalt–acyl intermediate II followed by the oxidative addition of H2, reductive elimination of β-hydroxyaldehyde b, and regeneration of the active HCo(CO)4 species (step C). Under the reaction conditions applied, β-hydroxyaldehyde can undergo acid catalysed dehydration resulting in α,β-unsaturated aldehyde c, which can be further hydrogenated to give aliphatic aldehyde d.
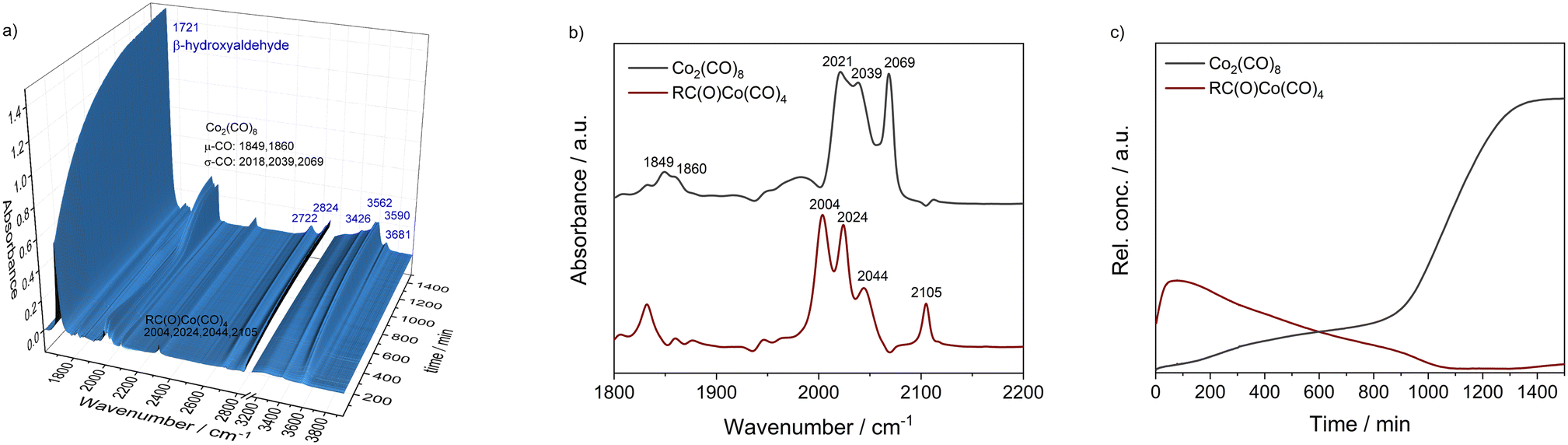 | ||
| Fig. 3 a) HP FTIR-spectra series collected during the hydroformylation of 1,2 epoxbutane 1a with Co2(CO)8 in the presence of P2 with [Co]/[P2] = 2/1 at 80 °C and p(CO/H2) = 40 bar in toluene as a solvent. Further reaction conditions: n(1a) = 8 mmol, n(Co2(CO)8) = 0.04 mmol, n(P2) = 0.04 mmol, vol.(toluene) = 16 mL. b and c) Pure component spectra and relative concentration profiles obtained via the chemometric treatment of spectroscopic data with peak group analysis (PGA). | ||
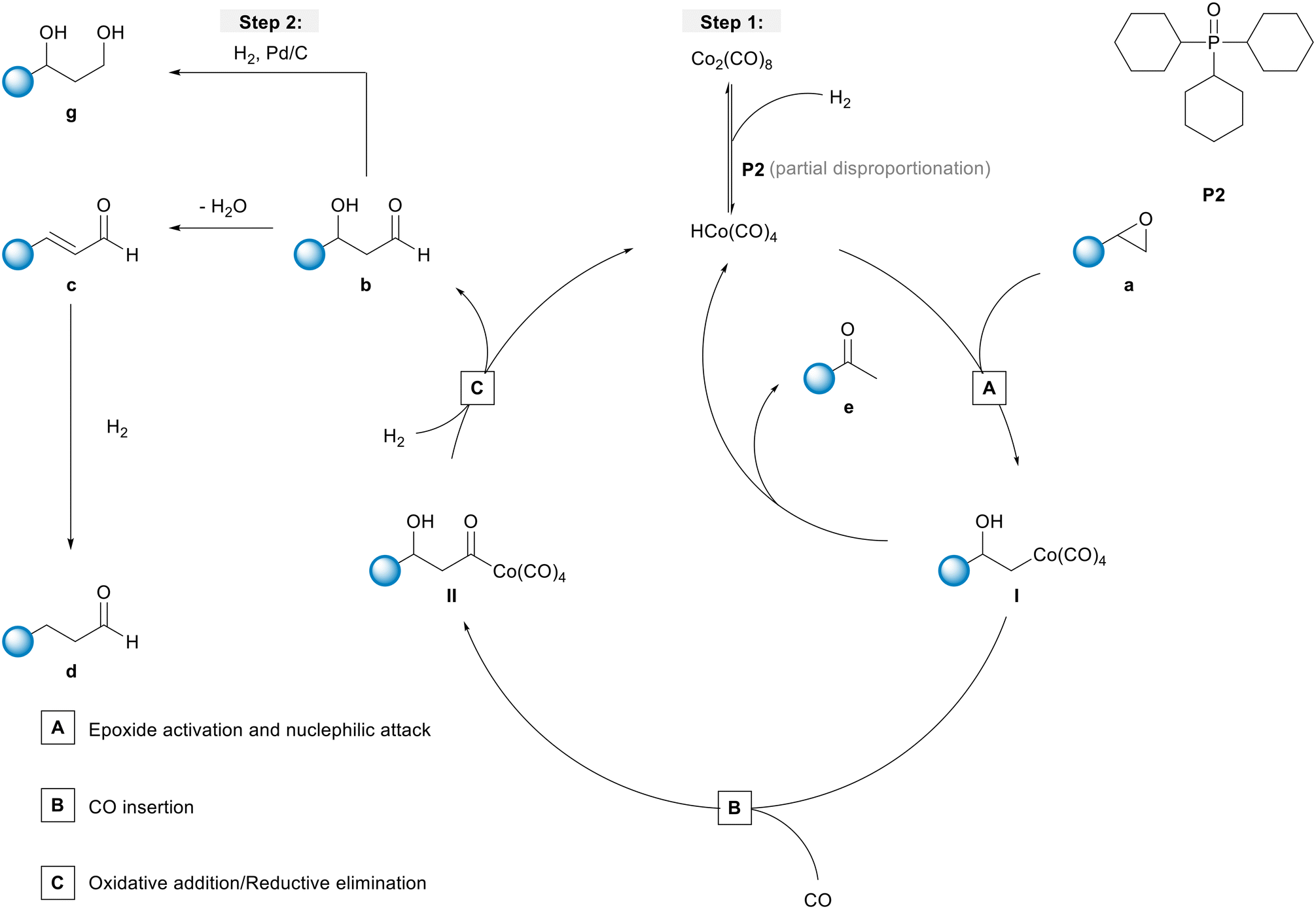 | ||
| Fig. 4 Simplified mechanistic proposal for the cobalt-catalysed hydroformylation of epoxide a using phosphine oxide (P2) (step 1) and the hydrogenation of β-hydroxyaldehyde b to afford the corresponding 1,3-diol g (step 2). | ||
Substrate scope
In general, β-hydroxyaldehydes are unstable and isolation is often tedious. To overcome this problem, further reaction of one of the functional groups is possible. As an example, silylformylation, which allows in situ protection of the alcohol group to give β-silyloxyaldehydes, have been performed.50–52 The strategies to protect the formyl group involve, for example, its acetalization. Nozaki and co-workers described the synthesis of β-hydroxy dimethyl acetals from epoxides by Co2(CO)8-catalysed tandem hydroformylation/acetalization performed in the presence of an excess of trimethyl orthoformate.53As mentioned in the introduction, an alternative to obtain stable and useful products from epoxides under the hydroformylation conditions is the consecutive hydrogenation of β-hydroxyaldehydes into 1,3-diols. However, such cobalt-catalysed hydrogenations proceed only under harsh conditions of temperature and pressure.54 Indeed, in all the runs presented in Table 1, no formation of even trace amounts of the corresponding 1,3-diol was observed. To hydrogenate the aldehyde in the presence of the cobalt catalyst under mild conditions, we tried to perform the process in two consecutive steps. In the first step, the hydroformylation of 1a was run under the conditions of entry 10 in Table 1 (70 °C, CO:H2 = 1:1, 40 bar, 24 h). After that, the gas phase was replaced by hydrogen (40 bar) and the reaction was allowed to proceed for another 24 h. Nevertheless, no formation of the desired diol in the reaction mixture was detected.
Then, we decided to use an additional catalyst capable to perform the hydrogenation step under mild conditions (Fig. 5). After the hydroformylation step, Pd/C was added to the reaction vial and the syngas was replaced by hydrogen (40 bar). As a result, β-hydroxyaldehyde 1b was completely converted into the corresponding 1,3-diol 1g, which then was isolated from the reaction mixture in 60% yield (Fig. 5). Alternatives for the hydrogenation process of aldehydes can be found in the literature.55
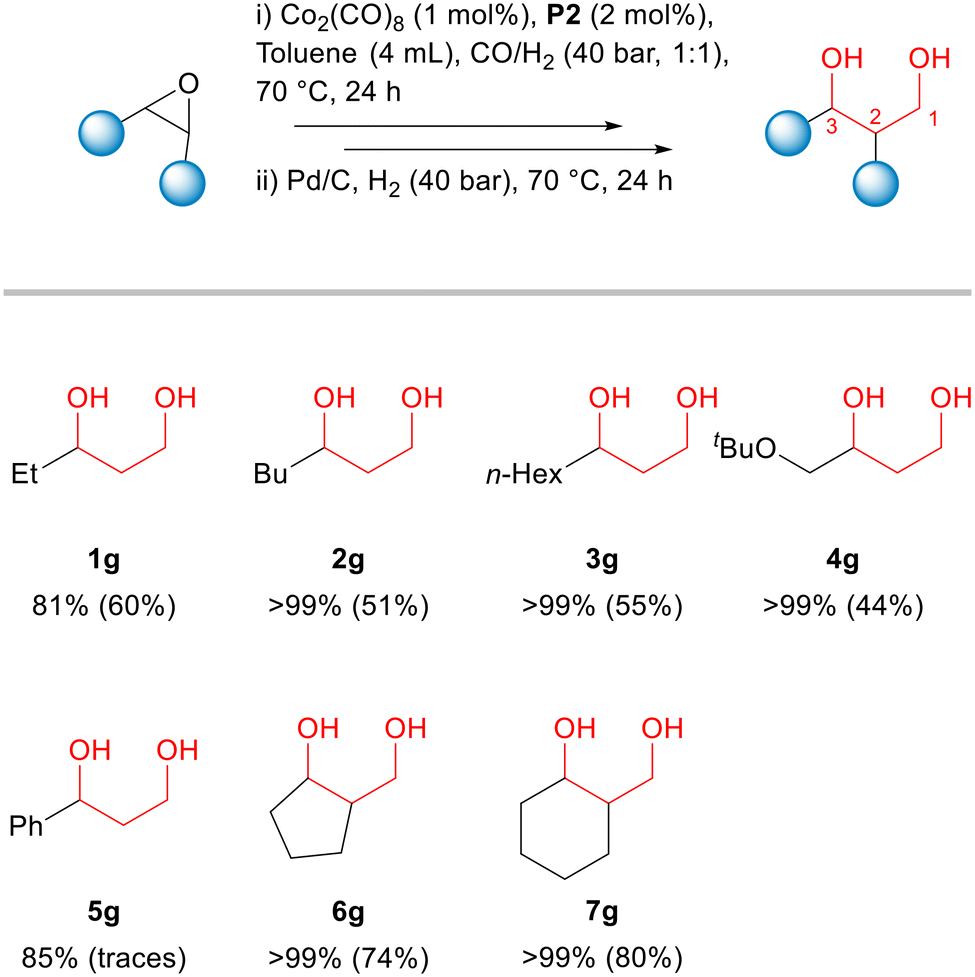 | ||
| Fig. 5 Cobalt-catalysed hydroformylation of epoxides: substrate scope. Reaction conditions: i) substrate (2 mmol), Co2(CO)8 (1 mol%), P2 (2 mol%), toluene (4 mL), 70 °C, gas phase – CO/H2 (1:1) 40 bar, 24 h. ii) Pd/C (10.6 mg), gas phase – H2 40 bar, 24 h. Conversion (related to the first step) determined by GC analysis using isooctane as internal standard. Numbers in parentheses correspond to the isolated yield. | ||
This one-pot two-step process could be applied for several other aliphatic epoxides using the previously optimized conditions (hydroformylation: 70 °C, CO:H2 = 1:1, 40 bar, 24 h; hydrogenation: 70 °C, H2, 40 bar, 24 h). Conversions of epoxides in the hydroformylation step and isolated yields of 1,3-diols are presented in Fig. 5. The detailed data on product selectivities as well the characterization data for the products of these reactions along with their structures are given in the ESI.† (Tables S2–S6).
The monosubstituted terminal epoxides 2a, 3a and 4a led to the corresponding 1,3-diols 2g, 3g and 4g in 51, 55 and 44% isolated yields, respectively (Fig. 5). The main side products in these reactions were the ketones (2e, 3e and 4e) and aldehydes (2f, 3f, and 4f) formed due to the acidic isomerization of the original epoxides. Utilizing styrene oxide 5a, its acidic isomerization to give acetophenone 5e and phenylethanal 5f became the main reaction pathway, with only trace amounts of the hydroformylation products being detected (Fig. 5). On the other hand, cyclic epoxides 6a and 7a reacted smoothly and provided 74 and 80% yield of the corresponding 1,3-diols 6g and 7g. The hydroformylation of cyclohexene oxide 7a was studied also in more details (see ESI,† Tables S7 and S8). The trends were similar to those obtained with terminal epoxide 1a (Table 1). The reactions employing the unmodified system or in the presence of phosphines L1–L4 showed no or poor conversions (Table S7,† entries 1–5). Conversely, the addition of phosphine oxides P1–P4 significantly accelerated the hydroformylation reaction to give β-hydroxyaldehyde 7b with ca. 80% selectivity (Table S7,† entries 6–9). Tricyclohexylphosphine oxide P2 also showed the best performance, as in the case of 1,2-epoxybutane 1a. It is important to note that the reactions with cyclohexene oxide were faster than those with 1,2-epoxybutane under the same conditions (cf. corresponding entries in Tables 1 and S7†).
In the reaction with P2, for example, cyclic epoxide 7a was fully converted, whereas the conversion of terminal epoxide 1a was only 65% (entry 7 in Table 1vs. entry 7 in Table S8†). The reactivity of olefins in carbonylation reactions usually follows the order determined by the steric hindrance of their C–C double bond: terminal > internal > cyclic.36 However, we have found that the reactivity of epoxides in hydroformylation does not follow this tendency. The enhanced reactivity of cyclic epoxides vs. terminal epoxides can be explained by the higher ring strain of the formers that is released by hydroformylation, which overcomes the steric hindrance effect.49
It is important to note that the reaction temperature during the hydroformylation of epoxides must be maintained low (up to 70 °C) to ensure high selectivity for hydroxyaldehydes. Otherwise, the dehydration of β-hydroxyaldehydes occurs followed by the C–C double bond hydrogenation (Fig. 4). The data on the hydroformylation of cyclohexene oxide 7a at different temperatures are presented in Table S8.† At 60 °C, the reaction showed 85% selectivity for β-hydroxyaldehyde 7b and 6% combined selectivity for the dehydration and dehydration/hydrogenation products 7c and 7d (Table S8,† entry 4). The temperature increase had a dramatic impact on the product distribution due to the consecutive transformations of β-hydroxyaldehyde related to the strong acidic properties of the cobalt species. The reaction at 100 °C gave compounds 7c and 7d as the main products (72% of the mass balance: 27 and 45%, respectively) along with only 24% of the aldehyde 7b (Table S8,† entry 1). The substrates which showed low conversions or/and selectivity towards hydroformylation of epoxides under standard conditions are presented in Fig. S3 (ESI†).
Experimental
General procedure for the hydroformylation of epoxides
In a typical experiment, the reactions were performed in 10 mL glass vial containing a stirring bar that was sequentially charged with Co2(CO)8 (0.5–1 mol%), ligand (L1–L4) or promoter (P1–P4) (2–20 mol%) and solvent (4 mL) in the glove box. The vials were closed using rubber septum/phenolic cap, and then removed from the glove box. Substrates (2 mmol) were added under argon atmosphere, then the vials were pierced with a syringe needle and set on a metal plate inside a Parr 4560 series reactor (300 mL). The reactor was closed and flushed three times with syngas. After the last release, the autoclave was pressurized with syngas (40 bar) and then heated to 70 °C for 24 h in an aluminium block. At the end of the reaction, the autoclave was placed into an ice bath to cool down and stop the reaction. Finally, the pressure was released, and the reactor flushed with N2 and opened. The reaction mixture was analysed by GC using isooctane as internal standard.General procedure for the hydrogenation of β-hydroxyaldehydes
After the hydroformylation reaction, the vials were opened and charged with 10.6 mg of Pd/C (10% Pd in charcoal). Afterward, the reaction vials were pierced with a syringe needle and set in a metal plate inside a Parr 4560 series reactor (300 mL). The reactor was closed, and the gas line was purged with N2 (about 20 bar). The procedure was repeated two times and three times with H2 (about 20 bar). After the last release, the autoclave was pressurized with 40 bar of H2 and then heated to 70 °C for 24 h inside an aluminium block. At the end of the reaction, the autoclave was placed into an ice bath to cool down and stop the reaction. Pure products were obtained by silica gel column chromatography (using 50% pentane/ethyl acetate as eluent) and isolated yields were calculated. Spectroscopic data for the products are presented in the ESI.†General procedure for the HP in situ FTIR-experiments
The hydroformylation reactions for in situ FTIR-investigations have been performed using a cylindric 25 mL stainless steel reactor system connected to a pressurizable transmission flow-through infrared spectroscopic cell placed in the optical pathway of a modified Bruker Matrix spectrometer equipped with an MCT detector. The liquid reaction solution have been circulated between the reactor and IR-cell with the help of a micro-gear-pump. A solution of all components has been prepared using standard Schlenk-techniques. The mixture has been transferred into the IR-reactor system and it was set to the desired temperature under circulation in the presence of argon. In the next step, the reaction was started by the addition of synthesis gas (40 bar) and the FTIR-monitoring was started simultaneously. Further details on the technical components of the IR-reactor system and individual experiments are given in the ESI.†Conclusions
In this work, we have developed a novel protocol for the cobalt-catalysed hydroformylation of epoxides under comparably mild conditions using simple and commercially inexpensive phosphine oxides as promoters. This hydroformylation was combined with the in situ hydrogenation of β-hydroxyaldehydes in a one-pot two-step process for the synthesis of 1,3-diols. The method was applied successfully to both terminal and cyclic aliphatic epoxides allowing to obtain the corresponding 1,3-diols in relatively good yields.Crucial for this novel transformation is the use of phosphine oxides, in particular, tricyclohexylphosphine oxide (P2), to promote the in situ generation of the active catalyst species HCo(CO)4 from Co2(CO)8 under syngas atmosphere. In the presence of the epoxide substrate the rapid formation of an acyl intermediate of the type RC(O)Co(CO)4 was observed by in situ FTIR spectroscopy only for the phosphine oxide modified catalyst system. We also evaluated the use of various solvents in this reaction, including green solvents. The results suggest that solvents with low polarity are better for the performance of the catalytic system.
We believe that this research can inspire further investigations in the hydroformylation of non-olefinic substrates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) – Finance Code 001 (88887.364603/2019-00), CNPq, INCT-Catálise, FAPEMIG, (Brazil), as well as the State of Mecklenburg-Western Pomerania (Germany). The authors thank the analytical staff of the Leibniz Institute for Catalysis, Rostock, especially Sandra Leiminger for their excellent service.Notes and references
- (a) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed; (b) E. V. Gusevskaya, J. Jiménez-Pinto and A. Börner, ChemCatChem, 2014, 6, 365 CrossRef CAS.
- (a) M. Beller, B. Cornils, C. D. Frohning and C. W. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17–85 CrossRef CAS; (b) P. W. Van Leeuwen and C. Claver, Rhodium catalyzed hydroformylation, Springer Science & Business Media, 2002 CrossRef.
- M. Du, Y. Sun, J. Zhao, H. Hu, L. Sun and Y. Li, Chin. Chem. Lett., 2023, 34, 108269 CrossRef CAS.
- G. A. Kraus, Clean: Soil, Air, Water, 2008, 36, 648–651 CAS.
- A. Börner and R. Franke, in Hydroformylation, ed. A. Börner and R. Franke, 2016, pp. 525–626, DOI:10.1002/9783527677931.ch6.
- C. Yokokawa, Y. Watanabe and Y. Takegami, Bull. Chem. Soc. Jpn., 1964, 37, 677–679 CrossRef CAS.
- Y. Takegami, C. Yokokawa, Y. Watanabe and H. Masada, Bull. Chem. Soc. Jpn., 1964, 37, 672–676 CrossRef CAS.
- C. W. Smith, G. N. Schrauzer, R. J. Windgassen and K. F. Koetitz, US3463819A, 1965; F. R. Lawrence and R. H. Sullivan, US3456017A, 1968; P. R. Weider, J. B. Powell and K. T. Lam, US5684214A, 1995, See also patent family.
- J. B. Powell, L. H. Slaugh, T. C. Forschner, J.-J. Lin, T. B. Thomason, P. R. Weider, T. C. Semple, J. P. Arhancet, H. L.-H. Fong, S. B. Mullin, K. D. Allen, D. C. Eubanks and D. W. Johnson, US5777182A, 1996.
- J. B. Powell, L. H. Slaugh, T. C. Forschner, T. C. Semple and P. R. Weider, US5545766A, 1994; P. R. Weider, J. B. Powell, L. H. Slaugh, T. C. Forschner and T. C. Semple, US5545767A, 1994; L. H. Slaugh and J. P. Arhancet, US5304686A, 1993; J. F. Knifton, T. G. James, K. D. Allen, P. R. Weider, J. B. Powell and L. H. Slaugh, US6576802B2, 2002, See also patent family.
- (a) J. B. Powell, L. H. Slaugh, T. C. Semple and P. R. Weider, US5585528A, 1994; (b) J. B. Powell, S. B. Mullin, P. R. Weider, D. C. Eubanks and J. P. Arhancet, US5770776A, 1996, See also patent family.
- J. B. Powell, L. H. Slaugh, T. C. Forschner, T. B. Thomason, T. C. Semple, P. R. Weider and J. P. Arhancet, US5463144A, 1994, See also patent family.
- J. B. Powell, L. H. Slaugh, T. C. Forschner, T. C. Semple and P. R. Weider, US5463145A, 1994, See also patent family.
- L. H. Slaugh, J. B. Powell, T. C. Forschner, T. C. Semple and P. R. Weider, US5545765A, 1994; L. H. Slaugh, J. B. Powell, T. C. Forschner, T. C. Semple and P. R. Weider, US5463146A, 1994, See also patent family.
- T. C. Semple, J. B. Powell, L. H. Slaugh, T. C. Forschner and P. R. Weider, US5576471A, 1994, See also the patent family.
- L. H. Slaugh, P. R. Weider, J. B. Powell and J. P. Arhancet, US5731478A, 1996; L. H. Slaugh, P. R. Weider, J. B. Powell and J. P. Arhancet, US5723389A, 1996, See also patent family.
- K. D. Allen, T. G. James, J. F. Knifion, J. B. Powell, L. H. Slaughm and P. R. Weider, EP1409132A1, 2002.
- J. B. Powell, W. R. Pledger, A. N. Matzakos, P. R. Weider and J. P. Arhancet, US5786524A, 1996; P. R. Weider, L. H. Slaugh and J. B. Powell, US5689016A, 1996.
- L. H. Slaugh, P. R. Weider and J. B. Powell, US5841003A, 1996; J. P. Arhancet and L. H. Slaugh, US5304691A, 1993; K. D. Allen, J. B. Powell, P. R. Weider and J. F. Knifton, US6545190B2, 2001.
- Selected patents: (a) M. A. Murphy, B. L. Smith, A. Aguilo and K. D. Tau, US4873378A, 1987; (b) M. A. Murphy, A. Aguilo and B. L. Smith, EP0257967, 1987; (c) M. A. Murphy, EP0343944, 1989; (d) M. A. Murphy, US4873379, 1989; (e) M. A. Murphy, B. L. Smith, A. Aguilo and K. D. Tau, US4935554, 1990; (f) K. D. Tau, US5053562, 1991, See also patent families.
- Selected patents: (a) J. R. Briggs, J. M. Maher and A. M. Harrison, US5030766, 1991; (b) J. R. Briggs, J. M. Maher and A. M. Harrison, US5225387, 1993; (c) J. R. Briggs, J. M. Maher and A. M. Harrison, US5210318, 1993, See also patent families.
- Selected patents: (a) W. A. Beavers, US4973741, 1990; (b) W. A. Beavers, US5043480, 1991, See also patent families.
- T. Haas, B. Jaeger, R. Weber, S. Mitchell and C. King, Appl. Catal., A, 2005, 280, 83–88 CrossRef CAS.
- J. R. Briggs, J. M. Maher and A. M. Harrison, US5449653, 1995.
- K. D. Allen, J. F. Knifton, J. B. Powell and L. H. Slaugh, WO02/098887, 2002.
- R. Weber, U. Englert, B. Ganter, W. Keim and M. Möthrath, Chem. Commun., 2000, 1419–1420 RSC.
- (a) L. H. Slaugh and J. P. Arhancet, US5344993A, 1993; (b) P. R. Weider, J. B. Powell, L. H. Slaugh, T. C. Forschner and T. C. Semple, US5563302A, 1994.
- L. H. Slaugh and P. R. Weider, US5256827A, 1993.
- (a) C. Abu-Gnim and I. Amer, J. Chem. Soc., Chem. Commun., 1994, 115–117 RSC; (b) C. Abu-Gnim and I. Amer, J. Organomet. Chem., 1996, 516, 235–243 CrossRef CAS.
- C. Basoli, C. Botteghi, M. A. Cabras, G. Chelucci and M. Marchetti, J. Organomet. Chem., 1995, 488, C20–C22 CrossRef CAS.
- (a) Rhodium-Catalyzed Isononyl alcohol Mfg Process (2), Mitsubishi Chemical, Nippon Chemtech Consulting Report I-89–2, 1989; (b) T. Onoda, CHEMTECH, 1993, 23(9), 34 Search PubMed; (c) K. Tano, K. Sato and T. Okoshi, US4528403, 1985; (d) K. Tano, K. Sato and T. Okoshi, Ger. Pat., 3338340, Mitsubishi Chemical, 1984; Chem. Abstr., 1984, 101, 170705 Search PubMed; (e) D. Hue, D. Pang, T.-E. Wang, Y. Chen, Y. Liu, J. Liu and Q. Zhu, J. Mol. Catal. A: Chem., 2001, 174, 21–28 CrossRef; (f) D. Hue, J. Liu, Y. Liu, T. Wang, D. Pang, Y. Chen, Y. Liang and Q. Zhu, Catal. Lett., 2001, 73, 2–4 Search PubMed.
- H. J. Clark, R. Wang and H. Alper, J. Org. Chem., 2002, 67, 6224–6225 CrossRef CAS PubMed.
- F. G. Delolo, J. Yang, H. Neumann, E. N. dos Santos, E. V. Gusevskaya and M. Beller, ACS Sustainable Chem. Eng., 2021, 9(14), 5148–5154 CrossRef CAS.
- F. G. Delolo, J. Fessler, H. Neumann, K. Junge, E. N. dos Santos, E. V. Gusevskaya and M. Beller, Chem. – Eur. J., 2022, 28, e202103903 CrossRef CAS PubMed.
- F. G. Delolo, J. Fessler, H. Neumann, K. Junge, E. N. dos Santos, E. V. Gusevskaya and M. Beller, Mol. Catal., 2022, 530, 112621 CrossRef CAS.
- A. Börner and R. Franke, Hydroformylation: fundamentals, processes, and applications in organic synthesis, John Wiley & Sons, 2016 Search PubMed.
- J. L. Jat and G. Kumar, Adv. Synth. Catal., 2019, 361, 4426 CrossRef CAS.
- (a) J. R. Lamb, Y. Jung and G. W. Coates, Org. Chem. Front., 2015, 2, 346–349 RSC; (b) J. R. Lamb, M. Mulzer, A. M. LaPointe and G. W. Coates, J. Am. Chem. Soc., 2015, 137(47), 15049–15054 CrossRef CAS PubMed.
- E. J. Moore, J. M. Sullivan and J. R. Norton, J. Am. Chem. Soc., 1986, 108, 2257–2263 CrossRef CAS PubMed.
- P. J. Dyson and P. G. Jessop, Catal. Sci. Technol., 2016, 6, 3302–3316 RSC.
- F. G. Delolo, E. N. dos Santos and E. V. Gusevskaya, Green Chem., 2019, 21, 1091–1098 RSC.
- J. B. Powell, L. H. Slaugh, T. C. Forschner, J.-J. Lin, T. B. Thomason, P. R. Weider, T. C. Semple, J. P. Arhancet, H. L.-H. Fong, S. B. Mullin, K. D. Allen, D. C. Eubanks and D. W. Johnson, US5777182, 1996.
- K. Watanabe, N. Yamagiwa and Y. Torisawa, Org. Process Res. Dev., 2007, 11(2), 251–258 CrossRef CAS.
- (a) D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288 RSC; (b) C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879 RSC.
- (a) R. Tuba, L. T. Mika, A. Bodor, Z. Pusztai, I. Tóth and I. T. Horváth, Organometallics, 2003, 22, 1582–1584 CrossRef CAS; (b) L. T. Mika, R. Tuba, I. Tóth, S. Pitter and I. T. Horváth, Organometallics, 2011, 30, 4751–4764 CrossRef CAS.
- (a) F. Hebrard and P. Kalck, Chem. Rev., 2009, 109, 4272–4282 CrossRef CAS PubMed; (b) H. W. Sternberg, I. Wender, R. A. Friedel and M. Orchin, J. Am. Chem. Soc., 1953, 75, 2717–2720 CrossRef CAS; (c) I. Wender, H. W. Sternberg and M. Orchin, J. Am. Chem. Soc., 1952, 74, 1216–1219 CrossRef CAS; (d) I. Ryoji, Bull. Chem. Soc. Jpn., 1962, 35, 865–869 CrossRef.
- (a) D. Darensbourg, M. Y. Darensbourg and N. Walker, Inorg. Chem., 1981, 20, 1918–1921 CrossRef CAS; (b) D. Darensbourg, N. Walker and M. Y. Darensbourg, J. Am. Chem. Soc., 1980, 102, 1213–1214 CrossRef CAS.
- Z. Huang, Y. Cheng, X. Chen, H.-F. Wang, C.-X. Du and Y. Li, Chem. Commun., 2018, 54, 3967–3970 RSC.
- K. Nakano and K. Nozaki, in Catalytic Carbonylation Reactions, ed. M. Beller, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 223–238, DOI:10.1007/3418_023.
- Y. Fukumoto, N. Chatani and S. Murai, J. Org. Chem., 1993, 58, 4187–4188 CrossRef CAS.
- Y. Seki, S. Murai, I. Yamamoto and N. Sonoda, Angew. Chem., Int. Ed. Engl., 1977, 16, 789 CrossRef.
- S. Park, Chem. – Asian J., 2019, 14, 2048–2066 CrossRef CAS PubMed.
- K. Nakano, M. Katayama, S. Ishihara, T. Hiyama and K. Nozaki, Synlett, 2004, 1367–1370 CAS.
- G. M. Torres, R. Frauenlob, R. Franke and A. Börner, Catal. Sci. Technol., 2015, 5, 34–54 RSC.
- (a) X. Lan and T. Wang, ACS Catal., 2020, 10(4), 2764–2790 CrossRef CAS; (b) S. Cattaneo, S. Capelli, M. Stucchi, F. Bossola, V. Dal Santo, E. Araujo-Lopez, D. I. Sharapa, F. Studt, A. Villa, A. Chieregato, B. D. Vandegehuchte and L. Prati, J. Catal., 2021, 399, 162–169 CrossRef CAS; (c) P. Mäki-Arvela, J. Hájek, T. Salmi and D. Yu. Murzin, Appl. Catal., A, 2005, 292, 1–49 CrossRef; (d) R. A. Farrar-Tobar, A. Dell'Acqua, S. Tin and J. G. de Vries, Green Chem., 2020, 22, 3323–3357 RSC; (e) Y. Song, X. Pan, D. He, Y. Liu, L. Yunhai, J. Hu and Wanhua Chemical Group Co., Ltd., China, CN115433065, 2022; (f) L. Yunhai, H. Jianglin, J. Yuxin, S. Yanfang, Y. Yang, C. Yong, H. Cunhe, Z. Hongliang, W. Lei and L. Yuan, CN111908999A, 2020; (g) J.-P. Lange, WO2001070658A1, 2001.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00109e |
| This journal is © The Royal Society of Chemistry 2024 |