
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-temperature stable hydroxyls tuning the local environment of Pt single atoms for boosting formaldehyde oxidation†
Mingyi
Xiao‡
a,
Lina
Zhang‡
b,
Shuzhe
Zheng‡
a,
Ling
Fang
a,
Tulai
Sun
a,
Yonghe
Li
a,
Mingwu
Tan
c,
Jianghao
Zhang
d,
Yihan
Zhu
a,
Jinshu
Tian
 *a and
Haifeng
Xiong
*b
*a and
Haifeng
Xiong
*b
aCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou, 310014, China. E-mail: tianjs@zjut.edu.cn
bCollege of Chemistry and Chemical Engineering, State Key Laboratory for Physical Chemistry of Solid Surfaces, Xiamen University, Xiamen, 361005, China. E-mail: haifengxiong@xmu.edu.cn
cInstitute of Sustainability for chemicals, Energy and Environment, Agency for Science, Technology and Research (A*STAR), 1 Pesek Road, Jurong Island, 627833, Singapore
dState Key Joint Laboratory of Environment Simulation and Pollution Control, Research Center for Eco-environmental Sciences, Chinese Academy of Sciences, Beijing, 100085, China
First published on 11th March 2024
Abstract
The relationship between the structure and performance of metal single-atom catalysts remains elusive because it is a challenge to tailor the local environments of single atoms (SAs) while keeping the atomic dispersion. Here, two Pt/CeO2 catalysts with different Pt coordination environments were prepared at high temperatures, via both atom trapping (Pt1/CeO2-AT) and thermal shock (Pt1/CeO2-TS), respectively. The Pt SAs in the Pt1/CeO2-TS catalyst with an unsymmetrical structure surrounded by stable surface hydroxyls exhibit excellent HCHO oxidation activity. Pt SAs are primarily found at Ce substitution sites in the Pt1/CeO2-TS catalyst, which has a Pt1O3 configuration with hydroxyls and more active surface lattice oxygen than the Pt1/CeO2-AT catalyst. Due to their different coordination and redox properties, Pt1/CeO2-TS and Pt1/CeO2-AT show different performances in HCHO oxidation (T100 of HCHO shifts from 60 to 200 °C), confirming that the catalytic performance of Pt SACs can be tailored by optimizing the local environment via high-temperature stable hydroxyls.
1. Introduction
Environmental catalysis has been attracting much attention because of the emission of pollutants such as aromatics and hydrocarbons. Noble metal catalysts have played a vital role in reducing these environmental pollutants, but their widespread use has been curbed due to their prohibitive cost. Given that each noble metal atom can serve as an active site in catalysis, metal single-atom catalysts (SACs) provide the opportunity for using noble metals with less capital investment. Recently, some metal SACs have been reported to exhibit high reactivity in pollutant mitigation such as the oxidation of volatile organic compounds.1–3Much progress has been made in the preparation of metal SACs, such as the approaches of coprecipitation, atomic layer deposition (ALD),4 atom trapping5 and vapor-phase self-assembly.6 However, when compared to nano-cluster/particle catalysts, SACs are not always active in catalytic reactions because of the unfavorable coordination structure of metal single atoms. Fine-tuning the oxidation states or local environments of SACs has the potential to significantly improve their performance in catalysis.7–12 In CO oxidation, several strategies have been employed to generate metal single atoms with improved catalytic performance. For example, steam-treatment of a Pt1/CeO2 catalyst enabled the generation of hydroxyl groups adjacent to Pt single atoms, leading to a dramatic activity increase in CO oxidation due to the generation of surface active lattice oxygen.13 The use of a highly defective CeO2–Al2O3 support can tune the oxidation state of Pt1 and enhance catalytic activity in CO, CH4, and NO oxidation.12 Recently, Li et al. grafted isolated and defective CeOx nanoglue islands onto SiO2, hosting one Pt atom on average. This catalyst exhibits significantly increased activity for CO oxidation.14 Furthermore, using a simple calcination temperature-control strategy, Tan et al. fabricated CeO2 supported Pt1 with precisely controlled coordination environments, and its activity trend in CO oxidation and NH3 oxidation is reversed due to the different property in reactant activation and H2O desorption.8 In addition, O2 plasma leads to the formation of surface peroxo (O22−) structures that facilitate the formation of strongly bonded Pt2+ atoms, rendering them active and resistant to sintering in the CO oxidation reaction.15 These Pt1/CeO2 catalysts were prepared at low temperatures, involving the effect of various kinds of surface functionals such as hydroxyls.16 The presence of various hydroxyls therefore masked the identification of the structure–performance relationship when investigating the effect of local coordination environments of metal single atoms in catalysis. In addition, the distance between a single metal atom and a hydroxyl group is crucial in determining the formation of active intermediates, which in turn impacts the catalytic performance in catalysis. For example, Wu et al. recently discovered that the closeness between the OH group and a single Rh atom on a plane surface significantly influences the catalytic activity of the Rh1/CeO2 single-atom catalyst system in CO oxidation.17
In this work, we focus on the high-temperature preparation of Pt1/CeO2 single-atom catalysts to avoid the effect of different kinds of hydroxyl groups because high temperature treatment can remove the weakly adsorbed hydroxyls, only leaving the strongly bonded hydroxyls behind. We employed two high-temperature approaches, i.e., atom trapping (AT) and thermal shock (TS), to prepare Pt1/CeO2 catalysts. The Pt/CeO2 SAC prepared by atom trapping (Pt1/CeO2-AT) is achieved by ramping the temperature to 800 °C in air and keeping at that temperature for 10 h, and the Pt/CeO2 SAC prepared by thermal shock (Pt1/CeO2-TS) is achieved by thermally shocking the sample to ∼1000 °C for ∼0.5 s and repeating the process multiple times. The catalytic performances of the Pt1/CeO2 catalysts were then investigated in formaldehyde oxidation and the function of the local environment of the Pt single atoms was studied.
2. Experimental section
2.1 Material preparation
Cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O, ≥99.99%) was purchased from Alfa Aesar. Chloroplatinic acid hexahydrate (H2PtCl6·6H2O, Pt ≥ 37.5%) was purchased from Sinopharm Chemical Reagent Limited Corporation. Briefly, 1 g of chloroplatinic acid hexahydrate powder was dissolved in 10 mL of deionized water to prepare a chloroplatinic acid solution. Ce(NO3)3·6H2O was calcined in air at 350 °C for 2 h to obtain CeO2, which was used as the support to prepare Pt1/CeO2 catalysts. All the reagents were used as received.The Pt1/CeO2-AT single-atom catalyst was prepared via atom trapping (AT).13 A 1.0 wt% Pt/CeO2 catalyst was synthesized by the incipient wetness impregnation method. An appropriate amount of chloroplatinic acid solution was added dropwise to CeO2 while being kept grinding in a mortar and pestle. Then, the obtained mixture was dried at 80 °C for 12 h in static air and calcined in a muffle furnace at 800 °C for 6 h at a temperature increase rate of 10 °C min−1. The resulting catalyst powder was denoted as Pt1/CeO2-AT.
The Pt1/CeO2-TS single-atom catalyst was prepared via thermal-shock (TS).11 The TS synthesis features a high temperature, but very short heating duration operated in an inert atmosphere. Specifically, a thin layer of the dried Pt/CeO2 precursor powder was uniformly spread out into a graphite plate and rapidly heated to a high temperature and then quenched through a carbon heater using Joule heating equipment (Hefei in situ Technology Co., Ltd. China). The temperature was monitored to be around 1000 °C for ∼0.5 s. The pulse power was about 300 Å × 40 V. This thermal shock process was repeated 6 times, and before each heating, the Pt/CeO2 precursor powder was mixed to ensure uniform heating and single atom formation. The resulting catalyst was denoted as Pt1/CeO2-TS.
2.2 Catalytic activity measurements
The HCHO oxidation reaction was carried out in a fixed-bed quartz flow reactor (i.d. = 7.0 mm) under atmospheric pressure. Briefly, 80 mg catalyst (40–60 mesh) was mixed with 120 mg quartz sand. After placing in the quartz tube reactor, the feed gas composition of 400 ppm HCHO/20% O2/N2 was introduced and the total flow rate was kept at 50 mL min−1. Water vapor was generated by introducing liquid water into a vaporizer using a Cole Parmer 74900 syringe pump and carried into the reactor by N2. To investigate the moisture effect, relative humidity (20%) was achieved by adjusting the injection speed of the pump. The products in the effluent gas were analyzed using an online gas chromatograph (GC2060, Shanghai Ruimin GC Instruments Inc.), which was described in previous work.182.3 Characterization
Details of the characterization of the catalysts are described in the ESI.†3. Results and discussion
3.1 Microstructure characterization of materials
Atom trapping (AT) and thermal shock (TS) methods were used to prepare Pt1/CeO2-AT and Pt1/CeO2-TS catalysts with a Pt loading of 1.0 wt%. The powder X-ray diffraction (XRD) patterns of the two catalysts (Fig. S1†) only show the diffraction peaks of ceria (JCPDS No.34-0394), without the diffraction peaks of metallic Pt or PtO2. X-ray photoelectron spectroscopy (XPS) in the Pt 4f region revealed that Pt remained as the ionic form of Pt2+ (72.4 eV and 75.8 eV) over both catalysts (Fig. S2†) without the existence of Pt0 and Pt4+ species.19,20 High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images revealed the presence of Pt single atoms dispersed on the CeO2 support in the Pt1/CeO2-AT and Pt1/CeO2-TS (Fig. 1A and B and Fig. S3†) catalysts, without the presence of Pt clusters or nanoparticles, confirming the high dispersion of Pt single atoms (SAs). This suggests that the atomic dispersion of Pt2+ was present on both Pt1/CeO2-AT and Pt1/CeO2-TS catalysts. | ||
| Fig. 1 Representative AC-STEM images of the obtained Pt1/CeO2-AT (A) and Pt1/CeO2-TS (B) catalysts, and the CO-DRIFT spectra of the Pt1/CeO2-AT (C) and Pt1/CeO2-TS (D) catalysts at 120 °C (CO-DRIFT experiments were performed by the following steps. Step 1: CO/O2/He was first introduced into the cell for 10 min; step 2: the CO flow was stopped while O2/He was kept flowing for 10 min). | ||
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) using CO as a probe molecule (CO-DRIFTS) was used to figure out the local environment of the two Pt1/CeO2 SACs. Fig. 1C and D reveal that the two catalysts present isolated Pt species, as confirmed by the CO bands at 2086 and 2089 cm−1, which is consistent with the XPS and HAADF-STEM results. The spectra do not show the lower-wavenumber features (<2000 cm−1) assigned to the bridge or three-fold adsorbed CO on Pt clusters or nanoparticles,21,22 further indicating the atomic dispersion of the Pt species on the two Pt1/CeO2 catalysts. The different peaks between 2000 and 2090 cm−1 of the two catalysts indicate the different local environments (e.g., location or coordination structure) of Pt single atoms. Pt1/CeO2-TS shows a more intense CO–Pt2+ (∼2089 cm−1) peak and a broader CO–Ptδ+ (∼2052 cm−1) peak, suggesting the wide distribution of local environments of Pt2+ with an unsaturated coordination configuration,8,22 which is also evidenced by density functional theory (DFT) simulation below. The simulations in Fig. S4† showed that as the Pt–O coordination number decreased on the Pt1/CeO2 (111) model, the vibrational frequency decreased from 2123.7 to 2028.2 cm−1. This corroborates that the Pt atoms on Pt1/CeO2-TS are likely coordinated with less adjacent heteroatoms. Along with the peak at ∼2086 cm−1 assigned to CO on Pt2+ species with a square-planar Pt1O4 configuration, there is no obvious peak at 2052 cm−1 for Pt1/CeO2-AT, implying that Pt1/CeO2-AT contains very few less-coordinated Pt single atoms, which agrees well with the previous result.11,22
The local coordination environment and the oxidation state of Pt single atoms were further investigated by X-ray absorption spectroscopy (XAS) at the Pt L3-edge.23,24Fig. 2A shows the XANES spectra of the reference Pt foil and Pt1/CeO2 catalysts. The spectral analysis of Pt1/CeO2-TS closely resembles that of Pt1/CeO2-AT, which has been previously confirmed to consist of isolated platinum species carrying a +2 state.11,15,22 Because of the absence of Pt–Pt scattering in the EXAFS spectrum (Fig. 2C), the Pt species on Pt1/CeO2-TS is therefore the isolated Pt2+ cation, which is also confirmed by XPS (Fig. S2†). It is noted that Pt1/CeO2-TS shows a slightly lower white line intensity as compared to Pt1/CeO2-AT (Fig. 2B), indicating that the ionic Pt2+ on the former is a less saturated coordination.13 Furthermore, in the XANES absorption edge of Pt1/CeO2-TS, a rising-edge feature was observed, demonstrated by a bump in the first-order derivative curve (Fig. 2B). Such a rising edge is due to the decreased local coordination symmetry of the Pt.25 This confirms that the TS technique produces the Pt1/CeO2-TS catalyst having isolated Pt2+ species with an unsaturated coordination configuration, which were analyzed by fitting the EXAFS spectra (Fig. S5 and Table S2†). The Pt1/CeO2-AT catalyst presents a square-planar Pt1O4 configuration with four equivalent Pt–O bonds, as revealed in previous work.26,27 However, Pt1/CeO2-TS has a relatively low Pt–O coordination number (CN) of approximately 3.6 (Table S2†), indicating a defective Pt1Ox (x < 4) configuration. It is known that the square-planar PtO4 has high thermal stability, whereas the low-coordinated Pt1Ox in the air-exposed state is less stable. The improved stability of Pt1Ox is therefore proposed to be related to the hydroxyl existing on the surface at high temperature.13,16 To verify the existence of the high-temperature stable hydroxyl, we performed the deconvolution of the O 1s XPS spectra for both samples, which included three peaks (Fig. S6†). The peaks at ∼528.9, 530.8 and 531.6 eV are assigned to the lattice oxygen (OL), defect-related oxygen (OD) and surface hydroxyl oxygen (OOH), respectively. Therefore, we deduced that compared to the Pt1/CeO2-AT sample with a square-planar Pt1O4 coordination, a non-equilibrium Pt1Ox configuration stabilized by the surface hydroxyl on the CeO2 (111) facet was formed on the Pt1/CeO2-TS catalyst during the TS treatment, which is confirmed by the FTIR result (Fig. S7†). The data shown in Fig. S7† clearly indicate that the Pt1/CeO2-TS catalyst exhibits unique vibrational peaks around 3500 cm−1, which we can associate with surface hydroxyl groups. This finding further supports and confirms our initial theory.
 | ||
| Fig. 2 The XAS analysis of the Pt samples demonstrating the different coordination environments of Pt single atoms over the Pt1/CeO2 SACs. (A) Normalized XANES spectra, (B) the first derivative of the normalized XANES spectra in (A) and (C) EXAFS magnitude of the Fourier transformed k2-weighted χ(k) data for Pt1/CeO2-AT (black) and Pt1/CeO2-TS (red) at the Pt-L3 edge. The reference Pt foil (blue) was used for XANES analysis. | ||
The oxygen-containing surface species on the catalysts were further characterized by CO temperature-programmed reduction (CO-TPR) (Fig. 3A). As for Pt1/CeO2-AT, the surface lattice oxygen in the vicinity of Pt (Pt–O–Ce bond) can react with CO at ∼90 °C, while on the Pt1/CeO2-TS catalyst, it can not only react with CO at a lower temperature (∼50 °C) but also produce more CO2, suggesting that surface oxygen species are more active and abundant on the latter. Meanwhile, more surface hydroxyl groups were generated on the Pt1/CeO2-TS catalyst, which can react with CO at ∼175 °C. The effect of surface hydroxyls was rarely considered in previous studies on Pt1/CeO2-AT,23,28,29 primarily because those formed by AT are far away from the active sites and have little influence on the local environment of the active single atoms.13,16 Surface hydroxyls were found to be more abundant in Pt1/CeO2-TS, correlating with the XPS results. Simultaneously, these surface hydroxyls had a lower reaction temperature than those formed from AT and a similar reaction temperature to hydroxyls formed by high-temperature steam treatment,13,16,30 implying that the hydroxyl groups on Pt1/CeO2-TS could affect the local environment of Pt single atoms. This lends credence to the hypothesis that surface hydroxyls produced from TS can stabilize single atoms. We also found the presence of more oxygen defects at Pt1/CeO2-TS (Fig. S8†), which agrees well with the results of EXAFS and CO-DRIFTS. Raman spectroscopy (Fig. S9†) revealed the formation of the Pt–O–Ce bond, as evidenced by the feature of the peak at 550 cm−1 (the one at 657 cm−1 corresponding to Pt–O). The significant decrease in the peak strength of the Pt1/CeO2-TS catalyst is due to the looser bonds of Ce–O–Ce and Pt–O–Ce as compared to the Pt1/CeO2-AT sample, which also confirms that the surface oxygen is more active via TS treatment.31,32 Models of Pt1/CeO2-TS and Pt1/CeO2-AT were built based on the characterization results and are shown in Fig. 3B. The model for the Pt1/CeO2-AT catalyst is a symmetric square-planar Pt1O4 coordination, whereas the structure model of Pt1O3 on CeO2 (111) stabilized with surface hydroxyls is built for the Pt1/CeO2-TS catalyst.11 For the PtO3 model, the formation energy of the oxygen vacancy is lower than that of the square-planar Pt1O4 ensemble (1.09 vs. 1.61 eV). Since the formation energy of the O-vacancy of the low-coordination PtO2 is greater than that of the square-planar Pt1O4 coordination (2.21 vs. 1.61 eV), which contradicts the CO-TPR and EXAFS results, the local environment of the Pt single atoms in the Pt1/CeO2-TS catalyst is therefore determined as the non-equilibrium PtO3 configuration surrounded with the surface hydroxyls existing at high temperature.
 | ||
| Fig. 3 (A) CO-TPR profiles of the Pt1/CeO2-AT and Pt1/CeO2-TS catalysts, CO2 (m/z = 44) and H2 (m/z = 2) signals. (B) A top-down view of the PtOx active site on the surface of CeO2 (111), with atoms close to the Pt atom and the corresponding formation energy of the O-vacancy. | ||
3.2 Evaluation performance of HCHO oxidation
We tested the two Pt1/CeO2 catalysts prepared at high temperatures in HCHO oxidation to evaluate their distinct catalytic performance in environmental catalysis. The Pt1/CeO2-AT catalyst has a high onset temperature of ∼120 °C and achieves only 50% HCHO conversion at 170 °C (Fig. 4A). However, the Pt1/CeO2-TS catalyst exhibits low-temperature activity, achieving 100% HCHO conversion at 60 °C. The turnover frequencies (TOFs) of the Pt1/CeO2-TS catalyst in formaldehyde catalytic oxidation can reach up to approximately 8.5 per hour at room temperature. This level of activity is relatively impressive compared to other catalytic systems based on Pt for similar reaction conditions, as indicated in Table S3.† Moreover, the Pt1/CeO2-TS catalyst demonstrated excellent stability in a time-on-stream run of 50 h in the presence of 400 ppm HCHO at 30 °C (Fig. S10†). The spent Pt1/CeO2-TS catalyst after 50 h of HCHO oxidation (Pt1/CeO2-TS-spent) was characterized, and the HAADF-STEM (Fig. S11A, B and S13†), XRD (Fig. S11C†), Raman (Fig. S11D†) and XPS results (Fig. S11E and F†) confirmed that there are no significant changes in the CeO2 morphology and Pt local environments. Furthermore, the surface Ce species (Fig. S11E†), oxygen defects and hydroxyl groups (Fig. S12†) have no obvious changes after the reaction, which is consistent with the in situ HCHO-DRIFTS results below (Fig. 5), demonstrating that the surface hydroxyls were maintained.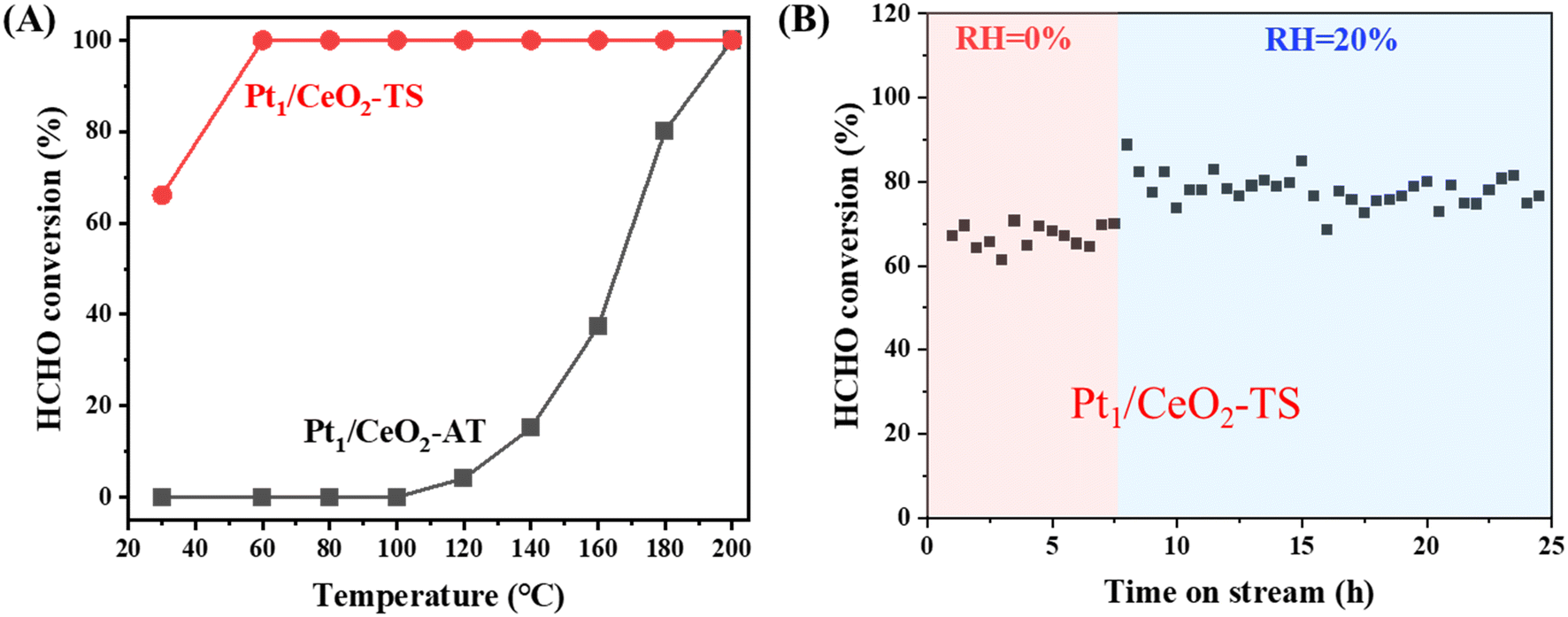 | ||
| Fig. 4 (A) HCHO conversion as a function of temperature over the Pt1/CeO2-AT and Pt1/CeO2-TS single-atom catalysts. (B) Relative humidity effect (20%) on the activity of the Pt/CeO2-TS catalyst at 30 °C. The reaction conditions were described in the Experimental section. | ||
 | ||
| Fig. 5 In situ HCHO-DRIFTS of the Pt1/CeO2-TS catalyst carried out in a flow of HCHO/N2 (solid line) or HCHO/O2/N2 (dotted line) at 30 °C. | ||
In addition, the effect of relative humidity (RH) on HCHO catalytic oxidation was investigated. As seen in Fig. 4B, under 20% relative humidity, the reactivity of Pt1/CeO2-TS is higher than that under dry conditions (∼80% vs. ∼65%). The previous studies have reported that the presence of H2O significantly improves the transformation of dioxymethylene (DOM, H2COO) into formate species (HCOO) or promotes the adsorption of formaldehyde and the formation of HCOO species. Therefore, the presence of an appropriate amount of water vapor can enhance the oxidation of HCHO, indicating the good tolerance of the Pt1/CeO2-TS catalyst against moisture. Therefore, the Pt1/CeO2-TS catalyst exhibits superior activity and excellent durability in the mitigation of HCHO.
3.3 Reaction mechanism and theoretical calculation
To investigate the reaction mechanism and the efficient activation of HCHO and O2 molecules over the Pt1/CeO2-TS catalyst, we performed in situ HCHO-DRIFTS experiments and DFT calculations. The in situ HCHO-DRIFTS experiments were carried out on the Pt1/CeO2-TS catalyst in a flow of HCHO/N2 or HCHO/O2/N2 (Fig. 5) at room temperature to gain insight into the formation of intermediates during the reaction. As shown in Fig. 5, with the introduction of HCHO/N2 or HCHO/O2/N2, there was no peak associated with the adsorbed HCHO (1722 cm−1) on the Pt1/CeO2-TS catalyst.33 The assignments of other peaks appeared are listed in Table S4.† It should be noted that with the introduction of HCHO/N2 from 20 to 40 min (Fig. 5), the contents of HCOO and DOM species gradually increase, while the adsorption peak at 3695 cm−1 becomes more negative (Fig. 5), which is assigned to the consumption of the hydroxyl species residing on the catalyst at high temperature.34,35 This indicates that the formation of surface DOM and HCOO consumes the surface hydroxyls. At the same time, we found that the intensity of the DOM peak formed during the entire HCHO/N2 process was significantly higher compared to that of the HCOO species. This result contradicts the previous findings on a steam-treated Pt1/CeO2 catalyst having surface hydroxyls,16 indicating a different reaction mechanism. When the reactant gas was switched to HCHO/O2/N2, the DOM and HCOO species were still present, whereas their intensity was much lower than that in HCHO/N2. The decrease of DOM and HCOO peaks indicates that oxygen is needed for further dehydrogenation. The peak position and intensity of the surface hydroxyl group did not change significantly throughout the catalytic cycle, which suggests that the consumption of the surface hydroxyl group was a reversible process that primarily facilitated the adsorption of intermediate species rather than breaking the C–H bond. We propose that the main reaction path in the Pt1/CeO2-TS catalyst is HCHO* → DOM* → HCOO* → COO*. Previous studies have already proposed that the reaction mechanism involves the decomposition of HCOO* into *CO, followed by its subsequent oxidation into *CO2. To reinforce the credibility and validity of our proposed mechanism, we have obtained additional supporting evidence from our in situ infrared analysis, specifically the absence of CO peaks. The activation of the HCHO* molecule is a two-step process, with the first step being the breaking of the C–H bond in DMO to form HCOO, mainly by surface active oxygen species, and the second step being the further breaking of the C–H bond to form COO* with the assistance of O2 molecules. Based on the in situ experiments, we propose that the high-temperature stable hydroxyl on the Pt1/CeO2-TS catalyst promotes the adsorption activation of DMO and HCOO, and the subsequent dehydrogenation of DOM to HCOO is the rate-limiting step of the whole reaction. Furthermore, the O2 molecule is easily activated by the Pt1/CeO2-TS catalyst to rapidly break C–H bonds.DFT simulation is used to further validate the reaction mechanism and the activation of reactants over the Pt1/CeO2-TS catalyst in HCHO oxidation. Based on the characterization, the Pt1/CeO2-TS model has a Pt single atom bonding with three oxygen atoms on CeO2 (111), as well as two neighboring active hydroxyls located adjacently to the Pt atom (PtO3–Ce(OH)2), as shown in Fig. S14.† The energy barrier of each step on the optimized Pt1/CeO2-TS model in the reaction was calculated and is shown in Fig. 6. HCHO oxidation starts with the adsorption of HCHO at the Pt1 site over the PtO3–Ce(OH)2 ensemble. The adsorbed HCHO reacts with the adjacent active oxygen species from the Pt–O bond to form  (DMO*), and meanwhile the bridging OH group interacts with O from HCHO, which stabilizes the
(DMO*), and meanwhile the bridging OH group interacts with O from HCHO, which stabilizes the 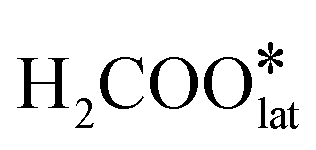 species. This is consistent with the results on the formation of DMO and the consumption of hydroxyls in the in situ DRIFTS experiment. The DFT calculation shows that this step is exothermic (−0.38 eV) with no activation barrier. Then, the formed
species. This is consistent with the results on the formation of DMO and the consumption of hydroxyls in the in situ DRIFTS experiment. The DFT calculation shows that this step is exothermic (−0.38 eV) with no activation barrier. Then, the formed  starts the first C–H bond breaking reaction, which involves the interaction of C–H with the activated Pt–Olat bond to form
starts the first C–H bond breaking reaction, which involves the interaction of C–H with the activated Pt–Olat bond to form  and OH species. Despite being exothermic (−0.36 eV), this step has a high energy barrier of 0.88 eV. This suggests that
and OH species. Despite being exothermic (−0.36 eV), this step has a high energy barrier of 0.88 eV. This suggests that  species are more difficult to form as compared to
species are more difficult to form as compared to  species, which explains the reason that the peak intensity of DMO is significantly higher than that of HCOO in the in situ DRIFTS experiment (Fig. 5). The activation of an oxygen molecule (
species, which explains the reason that the peak intensity of DMO is significantly higher than that of HCOO in the in situ DRIFTS experiment (Fig. 5). The activation of an oxygen molecule ( ) at the surface vacancy is highly exothermic (−1.93 eV), and the
) at the surface vacancy is highly exothermic (−1.93 eV), and the  species reacts with
species reacts with  to break the second C–H bond. This is a highly exothermic step (−2.41 eV) with a low energy barrier, indicating that oxygen activation is not the rate-limiting step in the reaction. This finding is consistent with the results of the in situ DRIFTS experiment. Finally, the interaction of COO*–H and hydroxyl species results in the formation of COO*, H2O*, and Pt–O bonds on the CeO2 surface, completing the reaction cycle. As a comparison, we also examined the PtO3 structure with no hydroxyls (Fig. 6, red line and S15†). The study discovered that the first step of the C–H bond activity requires a very high activation energy (∼2.5 eV), which is similar to the catalyst obtained through the AT method.16 As confirmed by the in situ experiment and DFT calculation, the high-temperature stable surface hydroxyl existing from the TS synthesis primarily facilitated the adsorption activation of DMO and HCOO* and did not participate in the C–H bond breaking. The activation of C–H bonds during the HCHO oxidation reaction is influenced by the activity of surface oxygen species. The initial C–H bond activation is facilitated by the surface lattice oxygen present in the Pt–O–Ce bond, whereas the subsequent C–H bond activation is facilitated by the adsorbed oxygen species produced by molecular oxygen dissociation.
to break the second C–H bond. This is a highly exothermic step (−2.41 eV) with a low energy barrier, indicating that oxygen activation is not the rate-limiting step in the reaction. This finding is consistent with the results of the in situ DRIFTS experiment. Finally, the interaction of COO*–H and hydroxyl species results in the formation of COO*, H2O*, and Pt–O bonds on the CeO2 surface, completing the reaction cycle. As a comparison, we also examined the PtO3 structure with no hydroxyls (Fig. 6, red line and S15†). The study discovered that the first step of the C–H bond activity requires a very high activation energy (∼2.5 eV), which is similar to the catalyst obtained through the AT method.16 As confirmed by the in situ experiment and DFT calculation, the high-temperature stable surface hydroxyl existing from the TS synthesis primarily facilitated the adsorption activation of DMO and HCOO* and did not participate in the C–H bond breaking. The activation of C–H bonds during the HCHO oxidation reaction is influenced by the activity of surface oxygen species. The initial C–H bond activation is facilitated by the surface lattice oxygen present in the Pt–O–Ce bond, whereas the subsequent C–H bond activation is facilitated by the adsorbed oxygen species produced by molecular oxygen dissociation.
 | ||
| Fig. 6 The calculated energy profile and the corresponding structures of the intermediates and transition states of the key elemental steps on Pt1O3–Ce(OH)2 on the CeO2 (111) surface in HCHO oxidation. | ||
4. Conclusions
In this work, we tune the local environment of the Pt single atoms on two Pt1/CeO2 single-atom catalysts (SACs) prepared at elevated temperatures (>800 °C), i.e., via atom trapping (Pt1/CeO2-AT) and thermal shock (Pt1/CeO2-TS). The use of the high-temperature preparation avoids the effect of various surface hydroxyls existing at low temperatures, exclusively maintaining the high-temperature stable hydroxyls. The Pt single atoms in the Pt1/CeO2-TS catalyst present the non-equilibrium local environments surrounded by high-temperature stable hydroxyls, exhibiting excellent activity in HCHO oxidation and lowering the T100 to 60 °C, as compared to the T100 of 200 °C for the Pt1/CeO2-AT catalyst.For the Pt1/CeO2-TS catalyst, Pt single atoms substituting the Ce site of CeO2 (111) have a Pt1O3 configuration coordinated with hydroxyls (Pt–O CN of 3.6), showing more active surface lattice oxygen than the Pt1/CeO2-AT catalyst (symmetric square-planar Pt1O4 coordination). The high-temperature stable hydroxyl existing on Pt1/CeO2-TS promotes the adsorption of formaldehyde, DMO and HCOO, but does not participate in the C–H bond activation. This confirms that the catalytic performance of Pt SACs can be tailored by tuning the local environment of Pt single atoms with the help of high-temperature stable hydroxyls. This work provides insight into preparing efficient single-atom catalysts for environmental catalysis.
Data availability statement
The data that support this study are available from the corresponding author upon reasonable request.Author contributions
Jinshu Tian: conceptualization, investigation, methodology, writing – original draft. Mingyi Xiao and Lina Zhang: investigation, formal analysis, methodology, writing – review & editing. Shuzhe Zheng and Ling Fang: investigation, methodology, validation, visualization. Tulai Sun: visualization, investigation, writing – review & editing. Yonghe Li: writing – review & editing. Yihan Zhu: writing – review & editing. Jianghao Zhang: writing – review & editing. Haifeng Xiong: conceptualization, investigation, methodology, writing – review and editing, supervision.Conflicts of interest
All the authors declare no conflicts of interest.Acknowledgements
The authors thank the Shanghai Synchrotron Radiation Facility (SSRF) for providing the beamtime at the BL11B beamline. This work was financially supported by the National Natural Science Foundation of China (Grant No. 22279115), the National High-Level Talent Fund (22072118) and the Fundamental Research Funds for the Provincial Universities of Zhejiang (RF-A2023005).References
- S. Zhao, Y. Wen, X. Liu, X. Pen, F. Lü, F. Gao and X. Tang, Formation of active oxygen species on single-atom Pt catalyst and promoted catalytic oxidation of toluene, Nano Res., 2020, 13, 1544–1551 CrossRef CAS.
- K. Yang, Y. Liu, J. Deng, X. Zhao, J. Yang, Z. Han, Z. Hou and H. Dai, Three-dimensionally ordered mesoporous iron oxide-supported single-atom platinum: Highly active catalysts for benzene combustion, Appl. Catal., B, 2019, 244, 650–659 CrossRef CAS.
- M. Jiang, D. Yan, X. Lv, Y. Gao and H. Jia, Recognition of water-dissociation effect toward lattice oxygen activation on single-atom Co catalyst in toluene oxidation, Appl. Catal., B, 2022, 319, 121962 CrossRef CAS.
- Q. Q. Guan, C. W. Zhu, Y. Lin, E. I. Vovk, X. H. Zhou, Y. Yang, H. C. Yu, L. N. Cao, H. W. Wang, X. H. Zhang, X. Y. Liu, M. K. Zhang, S. Q. Wei, W. X. Li and J. L. Lu, Bimetallic monolayer catalyst breaks the activity-selectivity trade-off on metal particle size for efficient chemoselective hydrogenations, Nat. Catal., 2021, 4, 840–849 CrossRef CAS.
- R. Alcala, A. Delariva, E. J. Peterson, A. Benavidez, C. E. Garcia-Vargas, D. Jiang, X. I. Pereira-Hernandez, H. H. Brongersma, R. Ter Veen and J. Stanek, Atomically Dispersed Dopants for Stabilizing Ceria Surface Area, Appl. Catal., B, 2021, 284, 119722 CrossRef CAS.
- H. C. Li, Q. Wan, C. Du, J. Zhao, F. Li, Y. Zhang, Y. Zheng, M. Chen, K. H. L. Zhang, J. Huang, G. Fu, S. Lin, X. Huang and H. Xiong, Layered Pd oxide on PdSn nanowires for boosting direct H(2)O(2) synthesis, Nat. Commun., 2022, 13, 6072 CrossRef CAS PubMed.
- J. Yang, Y. Huang, H. Qi, C. Zeng, Q. Jiang, Y. Cui, Y. Su, X. Du, X. Pan, X. Liu, W. Li, B. Qiao, A. Wang and T. Zhang, Modulating the strong metal-support interaction of single-atom catalysts via vicinal structure decoration, Nat. Commun., 2022, 13, 4244 CrossRef CAS PubMed.
- W. Tan, S. Xie, D. Le, W. Diao, M. Wang, K. B. Low, D. Austin, S. Hong, F. Gao, L. Dong, L. Ma, S. N. Ehrlich, T. S. Rahman and F. Liu, Fine-tuned local coordination environment of Pt single atoms on ceria controls catalytic reactivity, Nat. Commun., 2022, 13, 7070 CrossRef CAS PubMed.
- F. Dvorak, M. Farnesi Camellone, A. Tovt, N. D. Tran, F. R. Negreiros, M. Vorokhta, T. Skala, I. Matolinova, J. Myslivecek, V. Matolin and S. Fabris, Creating single-atom Pt-ceria catalysts by surface step decoration, Nat. Commun., 2016, 7, 10801 CrossRef CAS PubMed.
- J. Zhang, X. Qin, X. Chu, M. Chen, X. Chen, J. Chen, H. He and C. Zhang, Tuning Metal-Support Interaction of Pt-CeO2 Catalysts for Enhanced Oxidation Reactivity, Environ. Sci. Technol., 2021, 55, 16687–16698 CrossRef CAS PubMed.
- D. Jiang, Y. Yao, T. Li, G. Wan, X. I. Pereira-Hernandez, Y. Lu, J. Tian, K. Khivantsev, M. H. Engelhard, C. Sun, C. E. Garcia-Vargas, A. S. Hoffman, S. R. Bare, A. K. Datye, L. Hu and Y. Wang, Tailoring the Local Environment of Platinum in Single-Atom Pt1/CeO2 Catalysts for Robust Low-Temperature CO Oxidation, Angew. Chem., Int. Ed., 2021, 60, 26054–26062 CrossRef CAS PubMed.
- H. Jeong, D. Shin, B. S. Kim, J. Bae, S. Shin, C. Choe, J. W. Han and H. Lee, Controlling the Oxidation State of Pt Single Atoms for Maximizing Catalytic Activity, Angew. Chem., Int. Ed., 2020, 59, 20691–20696 CrossRef CAS PubMed.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- X. Li, X. I. Pereira-Hernandez, Y. Chen, J. Xu, J. Zhao, C. W. Pao, C. Y. Fang, J. Zeng, Y. Wang, B. C. Gates and J. Liu, Functional CeOx nanoglues for robust atomically dispersed catalysts, Nature, 2022, 611, 284–288 CrossRef CAS PubMed.
- W. Wan, J. Geiger, N. Berdunov, M. Lopez Luna, S. W. Chee, N. Daelman, N. Lopez, S. Shaikhutdinov and B. Roldan Cuenya, Highly Stable and Reactive Platinum Single Atoms on Oxygen Plasma-Functionalized CeO2 Surfaces: Nanostructuring and Peroxo Effects, Angew. Chem., Int. Ed., 2022, 61, e202112640 CrossRef CAS PubMed.
- L. Zhang, Q. Bao, B. Zhang, Y. Zhang, S. Wan, S. Wang, J. Lin, H. Xiong, D. Mei and Y. Wang, Distinct Role of Surface Hydroxyls in Single-Atom Pt1/CeO2 Catalyst for Room-Temperature Formaldehyde Oxidation: Acid-Base Versus Redox, JACS Au, 2022, 2, 1651–1660 CrossRef CAS PubMed.
- D. Wu, S. Zhou, C. Du, J. Li, J. Huang, H.-X. Shen, A. K. Datye, S. Jiang, J. T. Miller, S. Lin and H. Xiong, The proximity between hydroxyl and single atom determines the catalytic reactivity of Rh1/CeO2 single-atom catalysts, Nano Res., 2024, 17, 397–406 CrossRef CAS.
- L. Zhang, B. Zhang, P. Xue, J. Li, Z. Zhang, Y. Yang, S. Wang, J. Lin, H. Liao, Y. Wang, Y. Yao, S. Wan and H. Xiong, The Effect of Pretreatment on the Reactivity of Pd/Al2O3 in Room Temperature Formaldehyde Oxidation, ChemCatChem, 2021, 13, 4133–4141 CrossRef CAS.
- P. Bera, K. R. Priolkar, A. Gayen, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro, V. Jayaram and G. N. Subbanna, Ionic Dispersion of Pt over CeO2 by the Combustion Method: Structural Investigation by XRD, TEM, XPS, and EXAFS, Chem. Mater., 2003, 15, 2049–2060 CrossRef CAS.
- H.-H. Liu, Y. Wang, A.-P. Jia, S.-Y. Wang, M.-F. Luo and J.-Q. Lu, Oxygen vacancy promoted CO oxidation over Pt/CeO2 catalysts: A reaction at Pt–CeO2 interface, Appl. Surf. Sci., 2014, 314, 725–734 CrossRef CAS.
- H. A. Aleksandrov, K. M. Neyman, K. I. Hadjiivanov and G. N. Vayssilov, Can the state of platinum species be unambiguously determined by the stretching frequency of an adsorbed CO probe molecule?, Phys. Chem. Chem. Phys., 2016, 18, 22108–22121 RSC.
- X. I. Pereira-Hernandez, A. DeLaRiva, V. Muravev, D. Kunwar, H. Xiong, B. Sudduth, M. Engelhard, L. Kovarik, E. J. M. Hensen, Y. Wang and A. K. Datye, Tuning Pt-CeO2 interactions by high-temperature vapor-phase synthesis for improved reducibility of lattice oxygen, Nat. Commun., 2019, 10, 1358 CrossRef PubMed.
- D. Kunwar, S. Zhou, A. DeLaRiva, E. J. Peterson, H. Xiong, X. I. Pereira-Hernández, S. C. Purdy, R. ter Veen, H. H. Brongersma, J. T. Miller, H. Hashiguchi, L. Kovarik, S. Lin, H. Guo, Y. Wang and A. K. Datye, Stabilizing High Metal Loadings of Thermally Stable Platinum Single Atoms on an Industrial Catalyst Support, ACS Catal., 2019, 9, 3978–3990 CrossRef CAS.
- P. Xie, T. Pu, A. Nie, S. Hwang, S. C. Purdy, W. Yu, D. Su, J. T. Miller and C. Wang, Nanoceria-supported single-atom platinum catalysts for direct methane conversion, ACS Catal., 2018, 8, 4044–4048 CrossRef CAS.
- A. Ankudinov, J. Rehr, J. J. Low and S. R. Bare, Sensitivity of Pt X-ray absorption near edge structure to the morphology of small Pt clusters, J. Chem. Phys., 2002, 116, 1911–1919 CrossRef CAS.
- J. Resasco, L. DeRita, S. Dai, J. P. Chada, M. Xu, X. Yan, J. Finzel, S. Hanukovich, A. S. Hoffman and G. W. Graham, Uniformity is key in defining structure–function relationships for atomically dispersed metal catalysts: the case of Pt/CeO2, J. Am. Chem. Soc., 2019, 142, 169–184 CrossRef PubMed.
- H. A. Aleksandrov, K. M. Neyman and G. N. Vayssilov, The structure and stability of reduced and oxidized mononuclear platinum species on nanostructured ceria from density functional modeling, Phys. Chem. Chem. Phys., 2015, 17, 14551–14560 RSC.
- Y. Lu, S. Zhou, C.-T. Kuo, D. Kunwar, C. Thompson, A. S. Hoffman, A. Boubnov, S. Lin, A. K. Datye, H. Guo and A. M. Karim, Unraveling the Intermediate Reaction Complexes and Critical Role of Support-Derived Oxygen Atoms in CO Oxidation on Single-Atom Pt/CeO2, ACS Catal., 2021, 11, 8701–8715 CrossRef CAS.
- H. N. Pham, A. DeLaRiva, E. J. Peterson, R. Alcala, K. Khivantsev, J. Szanyi, X. S. Li, D. Jiang, W. Huang, Y. Sun, P. Tran, Q. Do, C. L. DiMaggio, Y. Wang and A. K. Datye, Designing Ceria/Alumina for Efficient Trapping of Platinum Single Atoms, ACS Sustainable Chem. Eng., 2022, 10, 7603–7612 CrossRef CAS.
- D. Donadio, L. M. Ghiringhelli and L. Delle Site, Autocatalytic and cooperatively stabilized dissociation of water on a stepped platinum surface, J. Am. Chem. Soc., 2012, 134, 19217–19222 CrossRef CAS PubMed.
- Y. Lee, G. He, A. J. Akey, R. Si, M. Flytzani-Stephanopoulos and I. P. Herman, Raman analysis of mode softening in nanoparticle CeO2−δ and Au-CeO2−δ during CO oxidation, J. Am. Chem. Soc., 2011, 133, 12952–12955 CrossRef CAS PubMed.
- D.-Y. Wei, M.-F. Yue, S.-N. Qin, S. Zhang, Y.-F. Wu, G.-Y. Xu, H. Zhang, Z.-Q. Tian and J.-F. Li, In situ Raman observation of oxygen activation and reaction at platinum–ceria interfaces during CO oxidation, J. Am. Chem. Soc., 2021, 143, 15635–15643 CrossRef CAS PubMed.
- X. Sun, J. Lin, H. Guan, L. Li, L. Sun, Y. Wang, S. Miao, Y. Su and X. Wang, Complete oxidation of formaldehyde over TiO2 supported subnanometer Rh catalyst at ambient temperature, Appl. Catal., B, 2018, 226, 575–584 CrossRef CAS.
- X. Chen, G. He, Y. Li, M. Chen, X. Qin, C. Zhang and H. He, Identification of a Facile Pathway for Dioxymethylene Conversion to Formate Catalyzed by Surface Hydroxyl on TiO2-Based Catalyst, ACS Catal., 2020, 10, 9706–9715 CrossRef CAS.
- Y. Huo, X. Wang, Z. Rui, X. Yang and H. Ji, Identification of the Nearby Hydroxyls' Role in Promoting HCHO Oxidation over a Pt Catalyst, Ind. Eng. Chem. Res., 2018, 57, 8183–8189 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00104d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |