DOI:
10.1039/D3CY01800H
(Review Article)
Catal. Sci. Technol., 2024,
14, 2064-2089
Olefins from alcohols via catalytic acceptorless dehydrogenation coupling reactions
Received
30th December 2023
, Accepted 16th February 2024
First published on 1st March 2024
Abstract
Olefins serve as essential building blocks for creating a wide range of molecular structures, including pharmaceuticals, polymers, and various natural products. In recent years, homogeneous transition-metal catalysis has emerged as a highly efficient method for synthesizing carbon–carbon double bonds in a cascade manner. Among the various approaches to introducing unsaturation, the catalytic acceptorless dehydrogenation coupling (ADC) reactions stand out due to their atom economy and eco-benign characteristics. By dehydrogenating alcohol, ADC produces aldehydes or ketones, which then undergo a condensation reaction with another molecule to yield an unsaturated product. The only byproducts of this process are H2 and H2O, which make the ADC reaction superior to the traditional way of producing chemicals. Surprisingly, despite the significance of olefins, there is currently no dedicated review article that specifically addresses catalytic routes to olefins from alcohols (A2O) via the ADC method. This review article aims to highlight the latest advancements in olefination reactions involving various substrates, including carbonyl compounds, phosphorus ylides and substrates with acidic α-hydrogens (such as sulfones, alkyl-substituted N-heteroarenes, and nitriles). Various aspects of metal–ligand cooperation (MLC) and their application are also discussed in brief detail. MLC involving bi-functional catalysts, where the metal center and the ligand jointly participate in the reaction, has shown great potential in ADC reactions. By exploring these catalytic pathways, we hope to inspire researchers to develop novel methods for olefin synthesis using abundant feedstocks.
1. Introduction
Olefins are versatile building blocks that are essential for the construction of molecular frameworks such as drugs, polymer compounds, and natural products. They are also widely used for sensitization, electroluminescence, and organic dyes.1 Many styrene derivatives exhibit critical biological activities, including resistance to cardiovascular and cancerous diseases.2 Historically, alkyl halide dehydrohalogenation and alcohol dehydration were the primary methods for producing alkenes.3 In 1953, Wittig pioneered a reliable method to form C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds that became a classic in organic chemistry to form CC bonds by treating carbonyl compounds with phosphorus ylides to give the corresponding alkenes and phosphorus oxide (Fig. 1).4 The Wittig olefination reaction has since undergone stereoselective modifications, such as the E-selective Horner–Wadsworth–Emmons (HWE) olefination and the Z-selective Still–Gennari olefination. Peterson introduced a seminal methodology for olefination, employing alpha-silyl carbanions in conjunction with ketones.5 In 1973, Julia introduced a novel approach to olefin synthesis utilizing sulfones.6 The Tebbe olefination reaction,7 a major breakthrough for the efficient synthesis of olefinic compounds, followed these advancements. Ir, Ru, Os, and Rh-based metal catalysts can also dehydrogenate abundant alkanes to produce alkenes with high efficiency.8 Furthermore, various methods for alkene functionalization have emerged, such as olefin metathesis reactions,9 which enable the synthesis of diverse and complex olefins from simple alkenes. A legacy of the era of cross-coupling is the Heck reaction,10 which inserts substituted olefins into a molecular framework in the presence of a base and a Pd-catalyst.
C bonds that became a classic in organic chemistry to form CC bonds by treating carbonyl compounds with phosphorus ylides to give the corresponding alkenes and phosphorus oxide (Fig. 1).4 The Wittig olefination reaction has since undergone stereoselective modifications, such as the E-selective Horner–Wadsworth–Emmons (HWE) olefination and the Z-selective Still–Gennari olefination. Peterson introduced a seminal methodology for olefination, employing alpha-silyl carbanions in conjunction with ketones.5 In 1973, Julia introduced a novel approach to olefin synthesis utilizing sulfones.6 The Tebbe olefination reaction,7 a major breakthrough for the efficient synthesis of olefinic compounds, followed these advancements. Ir, Ru, Os, and Rh-based metal catalysts can also dehydrogenate abundant alkanes to produce alkenes with high efficiency.8 Furthermore, various methods for alkene functionalization have emerged, such as olefin metathesis reactions,9 which enable the synthesis of diverse and complex olefins from simple alkenes. A legacy of the era of cross-coupling is the Heck reaction,10 which inserts substituted olefins into a molecular framework in the presence of a base and a Pd-catalyst.
 |
| | Fig. 1 Various approaches to access olefin. | |
Traditional synthesis of alkenes requires highly nucleophilic and reactive aldehydes, super stoichiometric amounts of strong bases, heavy transition metal catalysts, and phosphine ligands, which often face selectivity and functional group compatibility issues. In recent times, there has been an increasing demand for synthesizing effective and environmentally friendly processes for diverse chemicals (Fig. 1). Transition metal catalysis has been shown to be a potent tool for accomplishing this goal by facilitating the production of eco-friendly synthetic methods. Thus, there is considerable interest in searching for biomass outside the food chain and non-petroleum waste materials as carbon sources to establish cost-effective and environment-friendly synthetic methods.11 Lignocellulose is a readily available plant biomass, generally out of food chain competition, and can be processed into alcohol.12,13 In modern chemistry, alcohol has emerged as a crucial electrophile for the synthesis of coupling compounds via the hydrogen auto-transfer approach and the ADC (acceptorless dehydrogenation coupling) strategies.14 In 2021, Maiti and co-workers reported recent progress in the transition metal-catalyzed oxidative olefination of sp2 and sp3 C–H bonds.15 This review article focuses on the acceptorless dehydrogenation coupling strategy among various olefination methodologies. This strategy enables the formation of olefin compounds using alcohol as a reagent.
Dehydrogenation and related reactions of fundamental feedstocks16 is a valuable approach employed in organic synthesis to activate unreactive molecules (Fig. 2). In particular, ADC of alcohols involves the use of a metal catalyst to dehydrogenate alcohols, resulting in the formation of highly reactive aldehydes or ketones along with a metal hydride complex. The resulting active carbonyl intermediates can further participate in coupling reactions with other molecules, leading to the formation of complex compounds featuring unsaturated bonds. The metal hydride species releases hydrogen when it reacts with another molecule, regenerating the active catalyst and enabling continuous catalytic cycles. Recently, the ADC strategy has garnered significant attention from numerous research groups, particularly in the synthesis of olefin compounds with the generation of water as a by-product. Furthermore, the absence of external hydrogen acceptors simplifies the reaction conditions and reduces reagent costs. The ADC strategy has emerged as a versatile strategy for alkene synthesis, employing various noble transition metal catalysts such as Ru, Ir, Rh, Ru-nanoparticles, Pd heterogeneous catalysts, and base metals such as Ni, Mn, and Co. The electronic and steric properties of the coordinating ligands profoundly influence the reactivity and properties of these transition metal complexes. The concept of metal–ligand cooperation (MLC) involving bi-functional catalysts, where the metal center and the ligand jointly participate in the reaction, has shown great potential in ADC reactions.
 |
| | Fig. 2 Three main approaches to AAD reactions: (a) acceptorless dehydrogenative coupling (ADC), (b) borrowing hydrogenation strategy (BHS), and (c) interrupted borrowing hydrogen strategy. | |
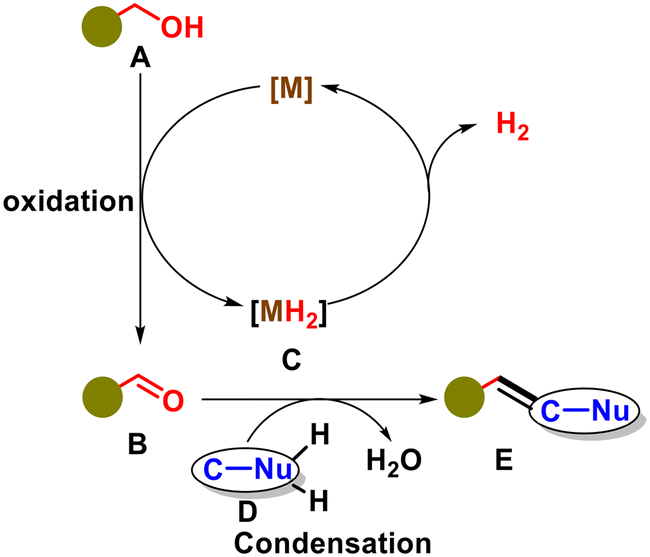
2. General reaction mechanism of olefination using alcohol via ADC pathway
The acceptorless dehydrogenative coupling of a nucleophile using alcohol as a precursor enables the olefination reaction, as illustrated in Fig. 2 and 3. This process involves the transfer of hydrogen from alcohol A to a metal catalyst, forming the corresponding carbonyl compound B and a metal hydride intermediate species C. Subsequently, the acidic hydrogen-bearing compound D loses its acidic hydrogen, transforming into a nucleophile. The nucleophile then engages in a condensation reaction with the aldehyde, leading to the formation of the olefin compound E. The metal hydride species can be protonated to release hydrogen gas or accepted by an oxidant to complete the catalytic cycle.
 |
| | Fig. 3 General procedure for olefination via acceptorless dehydrogenation coupling. | |
3. Wittig type olefination using alcohol
One of the most common and widely used CC bond-forming transformations in organic synthesis is the Wittig olefination reaction (Scheme 1a).17 Over the years, there has been considerable interest in the development of efficient and easy-to-operate alternative strategies, as Wittig transformation requires carbonyls for the ylides to react, and carbonyls show less reactivity and selectivity.18 Using alcohols instead of carbonyls, Williams and co-workers reported an indirect Wittig-type olefination reaction that yielded alkane as the final product. The iridium or ruthenium catalyst followed an elegant borrowing hydrogen pathway, but the reduction of alkene could not be controlled in the final step.19
 |
| | Scheme 1 Wittig-type olefination reactions: (1a) classical Wittig reaction. (1b and c) Wittig-type olefination reactions using alcohol by (1b) the research group of Milstein, and (1c) Srimani and co-workers. | |
Milstein and co-workers developed a Ru-catalyzed Wittig-type reaction of an alcohol with a ylide that produces an olefin and hydrogen gas without using any oxidant (Scheme 1b).20 After several optimization studies, it was observed that n-hexanol as a limiting reagent with an excess of Wittig salt n-butyl-triphenylphosphonium bromide (1.1 eq.) in the presence of 1.2 eq. of base and 1 mol% ruthenium catalyst (Cat. 1) provided the final olefin product with 85% yield. NMR studies revealed that the Z-isomer of the olefin product formed as a major diastereomer with a 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :28 ratio over the E-isomer. The reaction was compatible with various functional groups, such as aliphatic alcohols, heterocycles, amines, halides, and aromatic rings with different electronic properties. The study revealed that the formation of olefin in the benzylic position exhibited a predominant E selectivity, whereas the formation of olefin in the unstabilized position exhibited a Z selectivity. The reaction mechanism involved the dehydrogenation of alcohol by the catalyst to form aldehyde and H2, followed by a Wittig-type olefination with the non-stabilized ylide. Using a Ru-pincer complex (Cat. 2) with an acridine-based SNS ligand, Srimani and co-workers achieved a Wittig-type olefination of alcohols with phosphonium salts under oxidant-free conditions (Scheme 1c).21 They used alcohol as a limiting reagent, KOtBu as a base, and various aromatic and aliphatic phosphonium salts as coupling reagents. A diverse range of benzyl alcohols exhibiting different electronic properties, heteroaryl alcohols, aliphatic alcohols, as well as both aromatic and aliphatic phosphonium salts, have been successfully employed in the reaction resulting in the formation of the desired product with moderate to good yields. The olefin products showed a preference for the E isomer over the Z isomer in most cases, with moderate to good yields. The reaction mechanism involved the dehydrogenation of alcohol by the catalyst to form aldehyde and H2, followed by a Wittig-type olefination with the phosphonium salt.
:28 ratio over the E-isomer. The reaction was compatible with various functional groups, such as aliphatic alcohols, heterocycles, amines, halides, and aromatic rings with different electronic properties. The study revealed that the formation of olefin in the benzylic position exhibited a predominant E selectivity, whereas the formation of olefin in the unstabilized position exhibited a Z selectivity. The reaction mechanism involved the dehydrogenation of alcohol by the catalyst to form aldehyde and H2, followed by a Wittig-type olefination with the non-stabilized ylide. Using a Ru-pincer complex (Cat. 2) with an acridine-based SNS ligand, Srimani and co-workers achieved a Wittig-type olefination of alcohols with phosphonium salts under oxidant-free conditions (Scheme 1c).21 They used alcohol as a limiting reagent, KOtBu as a base, and various aromatic and aliphatic phosphonium salts as coupling reagents. A diverse range of benzyl alcohols exhibiting different electronic properties, heteroaryl alcohols, aliphatic alcohols, as well as both aromatic and aliphatic phosphonium salts, have been successfully employed in the reaction resulting in the formation of the desired product with moderate to good yields. The olefin products showed a preference for the E isomer over the Z isomer in most cases, with moderate to good yields. The reaction mechanism involved the dehydrogenation of alcohol by the catalyst to form aldehyde and H2, followed by a Wittig-type olefination with the phosphonium salt.
4. Direct olefination of alcohol with sulfones via ADC
In 1973, Marc Julia reported the multistep classical olefination via the formation of β-acyloxy alkyl sulfones from aldehydes and phenyl sulfones followed by the reductive elimination with sodium amalgam to furnish the olefin (Scheme 2a).6a In 2014, the research group of Milstein reported the single-step Julia-type olefination of alcohols using sulfones, catalyzed by the PNN-based ruthenium pincer complex (Cat. 3) via the ADC strategy (Scheme 2b).22 Following a series of optimization studies, it was determined that the reaction of dimethyl sulfones (1.06 mmol) with alcohol (1 mmol) in the presence of catalyst 3 (0.025 mmol) and KOtBu (1.1 eq.) base in refluxing dioxane for 5 hours at 125 °C resulted in the formation of desired product styrenes in good to excellent yields, accompanied by the release of hydrogen gas. A number of electron-donating and electron-withdrawing alcohols underwent this catalytic protocol successfully. The formation of E-stilbenes was observed when benzyl phenyl sulfones were utilized as alkenylating reagents in combination with various benzyl alcohols. The findings of mechanistic experiments indicate that the reactions proceed through a complex pathway that differs significantly from the conventional Julia-type olefination. The investigation of a potential reaction intermediate has suggested that the reaction may proceed via metal–ligand cooperation. However, a definitive depiction of the mechanistic pathway could not be ascertained. Dehydroxy-methylation of alcohol reagents was observed when the reaction was conducted under mild H2 pressure in the presence of dimethylsulfone.
 |
| | Scheme 2 Olefination reactions using sulfones: (2a) classical Julia olefination, (2b) Julia-type olefination reaction using alcohol by (2b) the research group of Milstein, (2c) the research group of Yan-Biao Kang, and (2d) by Shimizu and co-workers. | |
In 2015, Yan-Biao Kang and co-workers successfully implemented a novel method for base-mediated E-specific direct Julia olefination,23 utilizing aryl alcohols without the aid of any transition metal catalyst (Scheme 2c). The efficacy of several bases, including NaOtBu, KOtBu, NaH, KOH, n-BuLi, and LiHMDS, was evaluated, and it was found that NaH exhibited the most favorable conversion with excellent yield. A range of styrenes was synthesized using this base-promoted protocol in good to excellent yield by using different alcohols in conjunction with phenyl methyl sulfone. Notably, the utilization of alkyl phenyl sulfones as alkenylating reagents in combination with various aromatic alcohols resulted in the formation of diverse internal alkenes exhibiting E-stereoselectivity. Remarkably, many functional groups, including methoxy, halides, heterocycles, and amines, remained intact throughout the reaction and yielded the final product.
The proposed plausible mechanism of base-mediated olefination reaction is discussed below (Fig. 4). In the presence of a base, alcohol (A-1) generates a trace amount of aldehyde (A-2) by thermal dehydrogenation. On the other hand, the base abstracts a proton from B-1 and forms sulfone salt B-2, which subsequently reacts with aldehyde and forms A-3. Then, A-3 isomerizes to A-3′, followed by the elimination of NaOH to lead to the intermediate A-4. Later, another molecule of alcohol reacts with intermediate A-4, and a hydride transfer from alcohol to intermediate A-4 results aldehyde A-2 and alkyl phenyl sulfone intermediate A-5. Aldehyde A-2 triggers a fresh catalytic cycle initiation, while intermediate A-5 undergoes an elimination step mediated by a base to produce the ultimate product olefin.
 |
| | Fig. 4 Mechanistic cycle for base-mediated olefination using sulfone. | |
Using heterogeneous Pt nanoparticles on carbon (Pt/C), Shimizu et al. achieved the direct olefination of various primary and secondary alcohols with sulfones through heterogeneous catalysis (Scheme 2d).24 The authors examined various carbon-supported Pt-catalysts that were pre-reduced with H2 at 300 °C. Benzyl alcohol (1.5 mmol) and dimethyl sulfone (1 mmol) were chosen as model substrates. The Pt/C catalyst effectively converted dimethyl sulfone to styrene with 88% yield. Noteworthy that the reaction of benzyl alcohol and dimethyl sulfone with 0.02 mol% of Pt/C catalyst (TON = 3950) increased the efficiency by 150 times compared to the other previously reported catalyst for the olefination of benzyl alcohol with dimethyl sulfone.
The development of new methods for olefination reactions using base metal catalysts is a challenging area of research. Balaraman and co-workers reported a Ni-metal-catalyzed direct olefination of primary alcohols with sulfones, which afforded various terminal and internal olefins with the liberation of hydrogen gas.25 It was observed that a Ni(II) NNN-type complex (Cat. 4) effectively catalyzed the direct Julia-type olefination of alcohols to E-stilbenes in excellent yield in the presence of base KOtBu at 110 °C (Fig. 5, Scheme 3). A range of benzylic alcohols with diverse functional groups, such as electron-donating, electron-withdrawing, ether, aryl, alkyl, and halides (fluoro, chloro, and bromo), was successfully employed in the olefination reaction with sulfones. The reaction proceeded smoothly and delivered the desired olefins in high yields. The application of this strategy was extended to the stereo-selective synthesis of biologically relevant molecules, such as DMU-212 (used for breast cancer treatment) and resveratrol.
 |
| | Fig. 5 Single crystal X-ray structure of catalyst 4. | |
 |
| | Scheme 3 The Ni–NNN catalyzed Julia-type olefination of alcohols using sulfones. | |
A possible mechanism for the olefination of alcohols using an NNN–Ni catalyst is shown in Scheme 4. The precatalyst reacts with a base to generate complex C-1, which undergoes β-hydride elimination to produce the corresponding aldehyde and Ni–H species C-2. The aldehyde then participates in a Julia-type olefination with sulfone to afford the desired alkene product, while the Ni–H species C-2 is converted to intermediate C-4 by metal–ligand cooperation (MLC) mediated dehydrogenation of intermediate C-3, thus completing the catalytic cycle.
 |
| | Scheme 4 A plausible mechanism for Ni–NNN catalyzed olefination using sulfones. | |
Using commercially available FeCl2 and 1,10-phenanthroline as a catalytic system, KOtBu as base, and toluene as solvent at 120 °C, both primary and secondary alcohols underwent direct Julia-type olefination with dimethyl sulfone to afford various olefin products in good to excellent yields (Scheme 5a).26 This reaction was compatible with different electron-donating and withdrawing groups on the benzyl or heteroaryl alcohols. A remarkable case was the conversion of 1,2,3,4-tetrahydronaphthalene-1-ol to 1-methylnapthalene as the only product. This protocol was also applicable to other functional groups containing 1,2,3,4-tetrahydronaphthalene-1-ol, such as cyclic ether, aliphatic, and substituted benzylic groups. Control experiments and labelling studies suggested the reaction proceeded via the ADC of 1,2,3,4-tetrahydronaphthalene-1-ol followed by the formation of terminal alkene by Julia-type olefination using dimethylsulfone. In the presence of Fe-catalyst, the terminal alkene isomerized to an internal alkene, and subsequent dehydrogenations lead to the formation of an aromatic final product. The rate-determining step was found to be the C–H bond cleavage of the alcohol, with a KH/KD value of 2.53.
 |
| | Scheme 5 Ni and Fe-catalyzed olefination using sulfones. Research work conducted by (5a) Balaraman & co-workers, and (5b) Maji & co-workers. | |
Of late, Maji and co-workers also reported a similar type of olefination reactions using alcohol and sulfones with Ni catalyst (NiBr2) and bidentate nitrogen ligands (1,10-phenanthroline and neocuproine), which showed a good to excellent formation of stilbene derivatives with a predominant E-selectivity (Scheme 5b).27 Performed mechanistic studies indicated the reaction follows an ADC-type pathway.
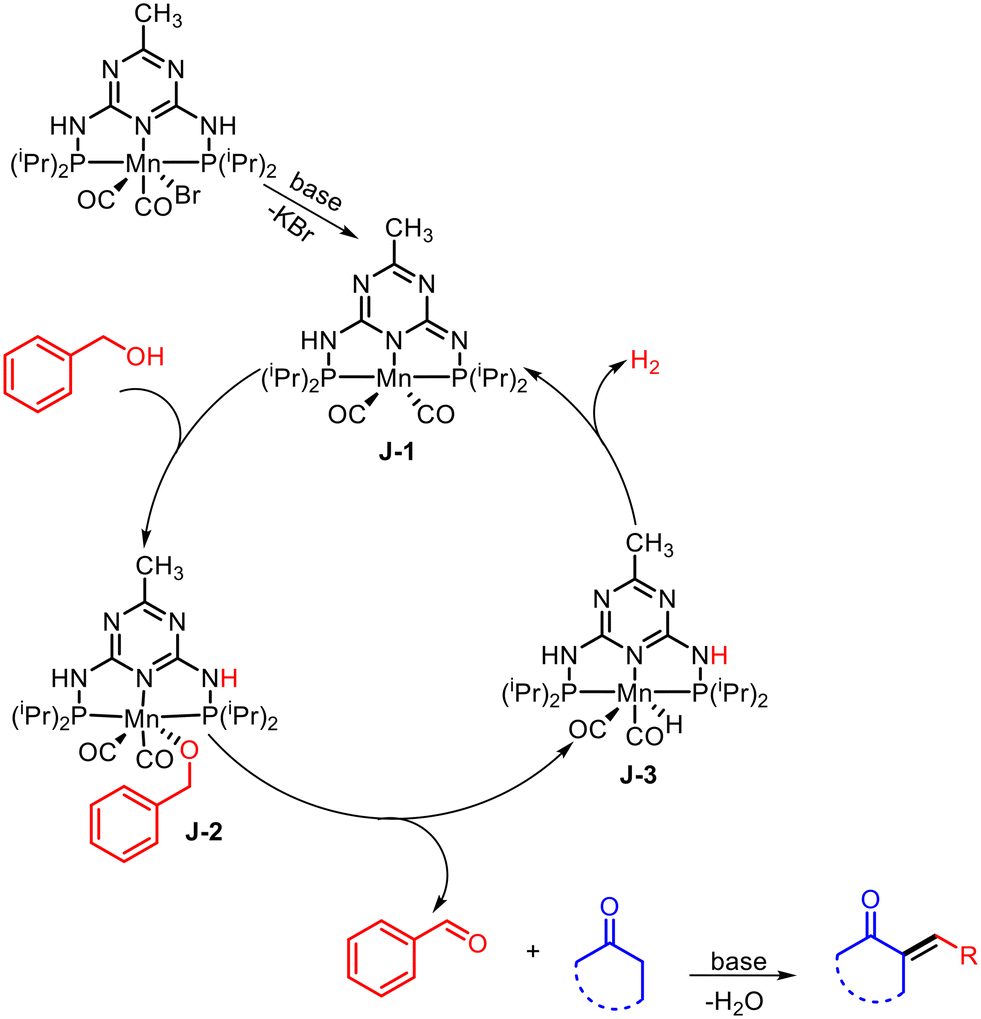
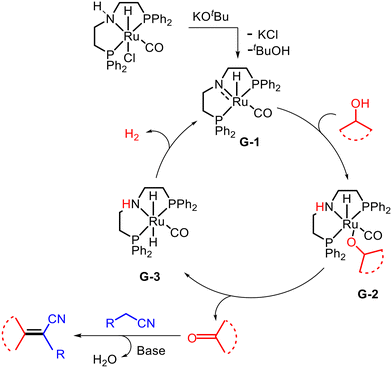
5. Olefination of alkyl substituted N-heteroarenes with alcohols
Aryl-substituted olefins bearing N-heteroarenes are important scaffolds in various biologically active drug molecules and agrochemicals.28E-selective conjugated N-heteroarenes are used in the synthesis of conducting polymers and have wide applications in material sciences as emissive layers in organic light-emitting diodes.29 An Mn-catalyzed protocol for the α-olefination of alkyl-substituted N-heteroarenes with alcohols via dehydrogenation coupling was recently reported by Kempe and co-workers (Scheme 6).30 Using an Mn complex (Cat. 5) stabilized by a triazine-based ligand (PN5P type ligand) with a para-methyl substituent as a catalyst and KOH as a base at 135 °C, the authors successfully synthesized (E)-2-styrylquinoline from 2-methyl quinoline and benzyl alcohol. In this case, the olefin/alkane selectivity was greater than 20:1, with alkane as a minor side product. The NMR analysis confirmed that the final alkene product was obtained with E-stereoselectivity. The reaction was compatible with various alkyl-substituted N-heteroarenes and alcohols bearing different functional groups, such as thiophene, methoxy, bromide, chloride, and iodide. The protocol also enabled the synthesis of an imaging agent (STB-8) for the detection of Alzheimer's disease β-amyloid plaques.
 |
| | Scheme 6 An Mn–PNP catalyzed α-olefination of alkyl-substituted N-heteroarenes. | |
According to previous literature reports and mechanistic studies, the base-mediated dehalogenation of the Mn-precatalyst (Scheme 7) leads to the dearomatization of the triazine ring and the formation of the active catalyst D-1. The active catalyst D-1 then reacts with alcohol via MLC-mediated O–H activation to yield the Mn–alkoxy complex D-2, which undergoes β-hydride elimination to produce aldehyde and the manganese–hydride complex D-3. The aldehyde then participates in a base-mediated aldol condensation-type reaction to afford the desired olefin product, while the complex D-3 releases hydrogen gas and regenerates the active catalyst D-1.
 |
| | Scheme 7 Plausible catalytic cycle for Mn–PNP catalyzed α-olefination of alkyl-substituted N-heteroarenes. | |
Maji and co-workers reported the catalytic olefination of methyl-substituted N-heteroarenes with primary alcohols.31 2-Methylpyrazine and benzyl alcohol were used as model substrates to optimize the reaction condition. The reaction of these substrates with Mn(CO)5Br (5 mol%) and NNN type ligand L-3 (5 mol%) in the presence of KOtBu at 110 °C for 16 h afforded the olefinated product (E)-2-styrylpyrazine with 96% yield (Scheme 8a). The reaction was compatible with various N-heteroarenes and primary alcohols bearing different functional groups and steric and electronic effects. Using this catalytic protocol, the author also synthesized the in vivo imaging reagent STB-8 and the core structures of molnekulast and heparin blue. The mechanistic finding revealed that the reaction follows an ADC-type pathway; H2 and water are the only two by-products of this synthetic protocol.
 |
| | Scheme 8 Mn and Ni-catalyzed α-olefination of alkyl-substituted N-heteroarenes. Research work conducted by (8a) Maji & co-workers, (8b) Banerjee & co-workers, (8c) Baidva & co-workers, and (8d) Ramesh & co-workers. | |
Recently, the research groups of Banerjee,32 Baidya,33 and Ramesh34 independently reported alternative procedures for the α-olefination of alkyl-substituted N-heteroarenes with the aid of Ni catalysts. Banerjee and coworkers have used 2-methylquinoline, methylpyrazine, and other N-heteroarenes as substrates. They observed that 5 mol% of NiBr2 and 6 mol% of 1,10-phenanthroline (L1) ligand effectively catalyzed the reaction of alcohols with 2-methyl N-heteroarenes to give the corresponding olefin products in good yields (Scheme 8b).32 Several alcohol-containing natural products, such as oleyl alcohol, and citronellol, successfully delivered the corresponding olefin product upon reaction with alkyl-substituted N-heteroarenes in good yield. The synthetic utility of this ADC reaction was showcased in the synthesis of STB-8 and galipinine (shows antimalarial activities). The mechanistic study indicated that the reaction was proceeding through an enamine intermediate by dearomatization of 2-alkyl heteroarenes with the generation of water and dihydrogen as by-products.
Baidya and his co-workers have used Ni-catalysts in combination with different N-based amide ligands for the olefination reaction. In a typical example, 2-methylquinoline reacted with benzyl alcohol in the presence of base KOtBu and in situ generated catalyst from NiBr2 and 8-aminoquinoline picolinic amide ligand L4 at 140 °C gave the final product (E)-2-styrylquinoline with 88% yield (Scheme 8c).33 This protocol provided the olefinated products with excellent E-selectivity and good to very high yields. Mechanistic studies suggested that the catalytic protocol follows the ADC-type pathway. Recently, Ramesh and co-workers also achieved olefination of 2-methylheteroarenes with benzyl alcohols using Ni(II)–N, S chelating complex (Cat. 6) as a catalyst which provided the product yield up to 93% (Scheme 8d).34
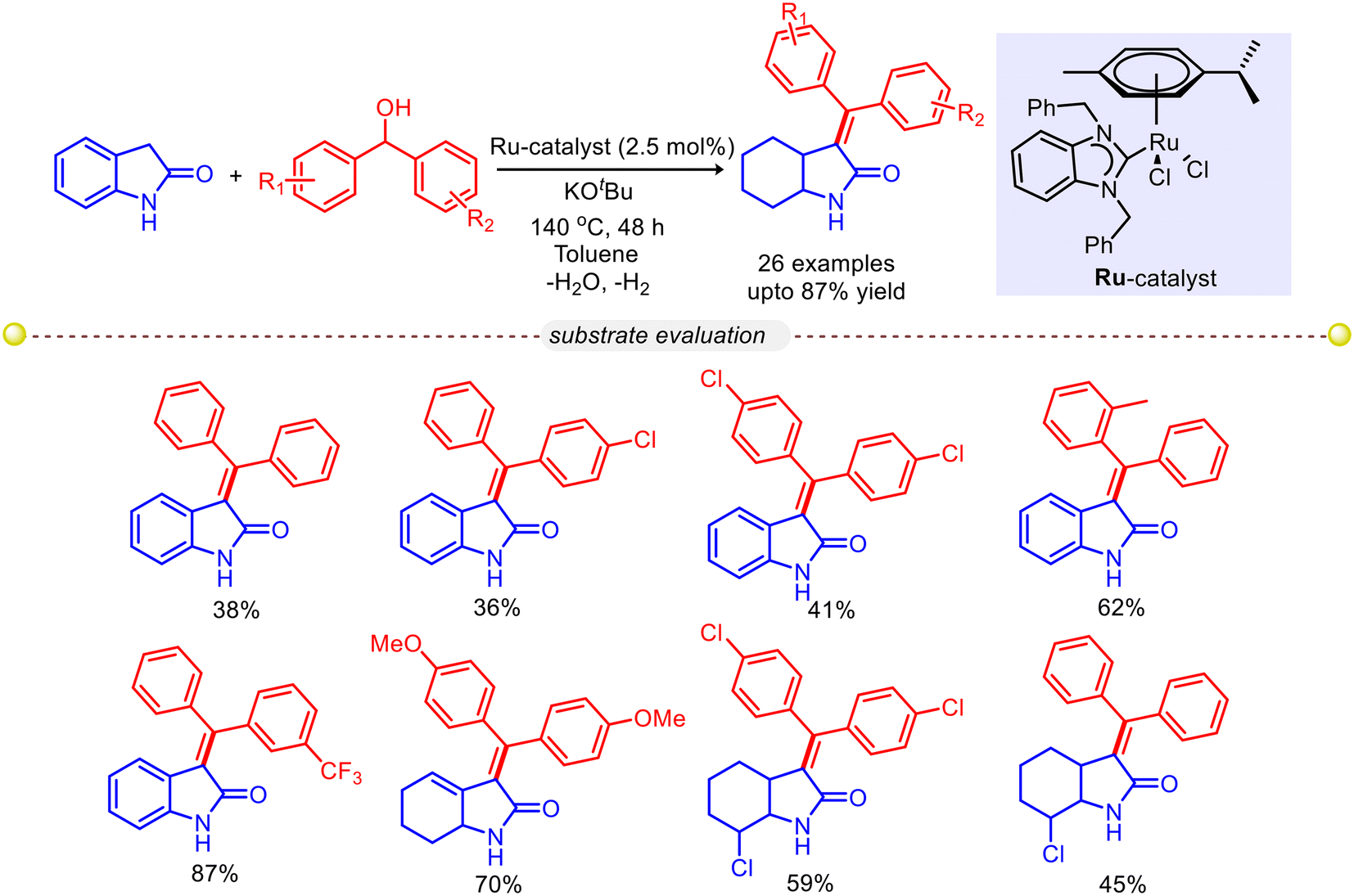
Gnanaprakasam and co-workers reported an acceptorless dehydrogenative α-olefination of 2-oxindole with diaryl methanols using Ru–NHC as a catalyst to synthesize a wide variety of arylidene-2-oxindole derivatives (Scheme 9).35 This methodology was successfully applied for the synthesis of a bioactive drug i.e. TAS-301. The biological activities of the synthesized 3-(diphenylmethylene)indolin-2-one derivatives were investigated against the plasmodium falciparum parasite and showed significant activity with IC50 = 2.24 μM.
 |
| | Scheme 9 Ru–NHC-catalyzed α-olefination of 2-oxindole with diaryl methanols.35 | |
6. Alkenation of alcohols via ADC with hydrazine/hydrazone
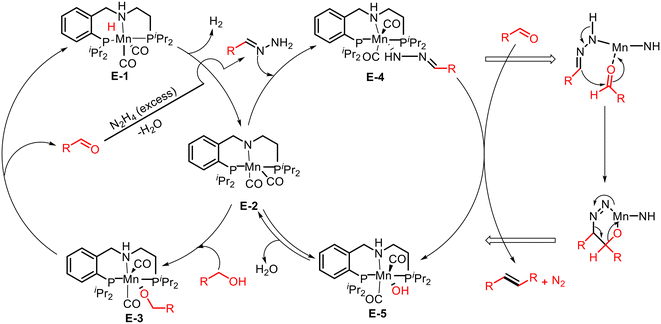
Li and co-workers successfully introduced a new strategy for deoxygenation reaction using alcohols and hydrazine by Ru or Ir catalyst.36 The research group of Milstein reported a similar reaction using a manganese pincer complex, which produced alkenes from alcohols and hydrazine/hydrazone under base-free conditions with the formation of H2, N2, and water as the only byproducts.37 In a closed Schlenk tube, benzyl alcohol and hydrazine in THF were treated with catalyst 7 (3 mol%) at 120 °C, resulting in the conversion of alcohol to stilbene (91% yield) after 24 h (Scheme 10a). A variety of benzyl alcohols bearing electron-donating and -withdrawing groups participated in the Mn-catalyzed olefination reaction and gave good to excellent yields, whereas heteroaryl alcohols and nitro-substituted alcohol failed to produce the desired product. Notably, the protocol was further extended to synthesize mixed alkenes from the coupling of alcohol and hydrazone (Scheme 10b). A broad range of aromatic and aliphatic alcohols reacted with the THF solution of benzylidenehydrazine in the presence of catalyst 7 at 120 °C, providing E-selective mixed alkenes in moderate to good yields.
 |
| | Scheme 10 Olefination of (10a) hydrazine and (10b) hydrazone via ADC strategy.37 | |
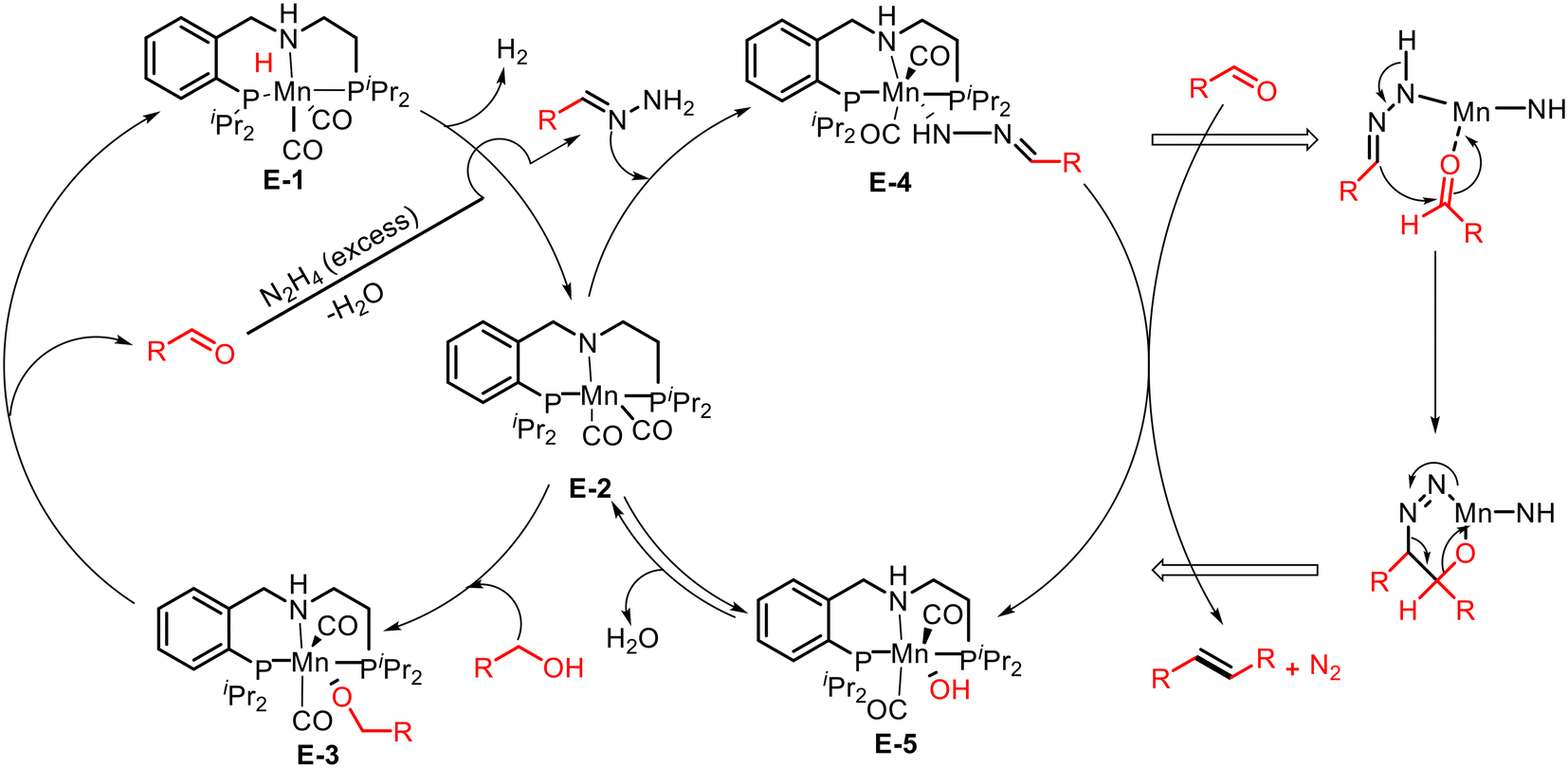
The plausible pathway for olefination by coupling of alcohols with hydrazones in the presence of an Mn catalyst is discussed in Scheme 11. Under thermal conditions, the precatalyst E-1 liberates hydrogen and forms complex E-2, which activates the O–H bond of alcohol via MLC and produces an alkoxy intermediate E-3. This intermediate undergoes β-hydride elimination and generates the corresponding aldehyde. The aldehyde, synthesized de novo, reacts with excess hydrazine to form a hydrazone. The hydrazone then reacts with intermediate E-2via N–H activation and forms complex E-4. This complex undergoes an Mn-catalyzed concerted reaction with an aldehyde and yields the final product of olefin, N2, and Mn–hydroxo complex E-5. Finally, the complex E-5 releases H2O regenerates the intermediate complex E-2, and continues the catalytic cycle.
 |
| | Scheme 11 Plausible catalytic cycle for olefination of hydrazine and hydrazone via ADC strategy. | |
7. Transition metal-catalyzed α-olefination of nitriles
α,β-Unsaturated acrylonitriles are used as building blocks to synthesize a variety of drug molecules, herbicides, and natural products.38 Notably, vinyl nitriles have applications in optoelectronic materials and the synthesis of light-emitting diodes.39 The conventional approach to make vinyl nitriles is a base-mediated Knoevenagel reaction using carbonyl compounds and nitriles as starting materials. Base-mediated condensation reactions have several drawbacks like side reactions including self-condensation, Cannizzaro reaction, and aldol condensation, etc.40,41
In 2017, Milstein and co-workers reported an α-olefination of nitriles with alcohols via a dehydrogenation coupling pathway. The reaction is catalyzed by a molecularly defined Mn–PNP pincer complex (Cat. 7).42 A reaction mixture of benzyl alcohol (0.25 mmol), benzyl cyanide (0.25 mmol), and catalyst 7 in 1 mL of toluene at 135 °C produced 2,3-diphenylacrylonitrile in 87% isolated yield with complete Z-isomer selectivity. The reaction is general and various benzyl alcohols bearing electron-donating and withdrawing groups at the para position and substituted benzyl cyanides were employed under reaction conditions. The protocol was successfully extended to aliphatic alcohols. The by-products of these reactions are H2 and H2O (Scheme 12).
 |
| | Scheme 12 An Mn–PNP catalyzed α-olefination of nitriles with alcohols.42 | |
The proposed mechanism for α-olefination of alcohols is illustrated in Scheme 13. The proposed mechanism for α-olefination of alcohols involving Mn–pincer complex F-1 is initiated with hydrogen liberation and formation of amido complex F-2. After C–H activation complex F-2 transformed to thermodynamically more stable complex F′-2. The O–H activation of alcohol through proton transfer to either the nitrogen of complex F-2 or benzylic carbon of F′-2 giving alkoxy complex following β-hydride elimination releases the aldehyde. In a simultaneous pathway α-hydrogen bearing nitrile generates a nitrile carbanion via deprotonation. Control experiments and NMR studies suggested that complex F-2 abstracts a proton from nitriles bearing α-hydrogen and generates a nitrile carbanion. The nucleophilic attack of carbanion F-4 at the aldehyde leads to the formation of α,β-unsaturated nitrile, and amido complex with the liberation of water.
 |
| | Scheme 13 Plausible catalytic cycle for Mn–PNP complex catalyzed α-olefination of nitriles with alcohols. | |
Wang and co-workers observed that vinyl nitriles can be synthesized via dehydrogenative coupling of alkylnitriles and alcohols using bi-nuclear Rh-complex as a catalyst.43 Notably, an atmosphere-controlled chemoselectivity was observed. Reaction under inert Argon atmosphere resulted in alkylation via borrowing hydrogen pathway whereas, reaction under oxygen atmosphere assisted in dehydrogenation process and noticed the formation of olefins as final product. The isolated yield of olefinated product was found to be optimum using binuclear rhodium catalyst (Cat. 8) and base NaOH in toluene at 110 °C (Scheme 14). The reaction was carried out in a Radleys Carousel tube equipped with an O2 balloon.
 |
| | Scheme 14 Rh-binuclear complex catalyzed α-olefination of nitriles using alcohols.43 | |
After the initial report of olefination of nitriles using primary alcohol by Milstein and Wang groups,42,43 Gunanathan and co-workers reported Ru(II)-pincer complex (Cat. 9) catalyzed synthesis of β-disubstituted vinyl nitriles using secondary alcohols (Scheme 15).44 One equiv. of phenyl acetonitrile with a two equiv. of cyclohexanol in the presence of Ru-MACHO (1 mol%), KOtBu as a base in toluene at 135 °C resulted in the formation of a final olefin product with 84% yield. Electron-donating, electron-withdrawing, halo-substituted phenyl acetonitriles, nitriles bearing heterocycles, and aliphatic side chains provided the corresponding alkene products in moderate to good yield under the optimized reaction conditions. The protocol was further extended to various cyclic and acyclic secondary alcohols. Notably, cycloheptanol, cyclooctanol, and sterically bulky 2-adamantanol are amenable and enable product formation under the optimized reaction condition. From the kinetic studies, it was confirmed that the reaction follows first-order kinetics. Mechanistic findings suggested a plausible catalytic cycle (Scheme 16) where in the presence of base precatalyst forms coordinatively unsaturated complex G-1. Complex G-1 reacts with secondary alcohol and generates alkoxy-ligated Ru-complex G-2 upon MLC-mediated O–H bond activation of a secondary alcohol. Subsequently, the ketone is generated through a β-hydride elimination reaction, concomitant with the release of a ruthenium dihydride complex G-3. Hydrogen liberation from complex G-3 regenerates active catalyst G-1.
 |
| | Scheme 15 Ru–PNP complex catalyzed synthesis of alkenyl nitriles using alcohols.44 | |
 |
| | Scheme 16 Plausible catalytic cycle for Ru–PNP complex catalyzed synthesis of substituted alkenyl nitriles using alcohols. | |
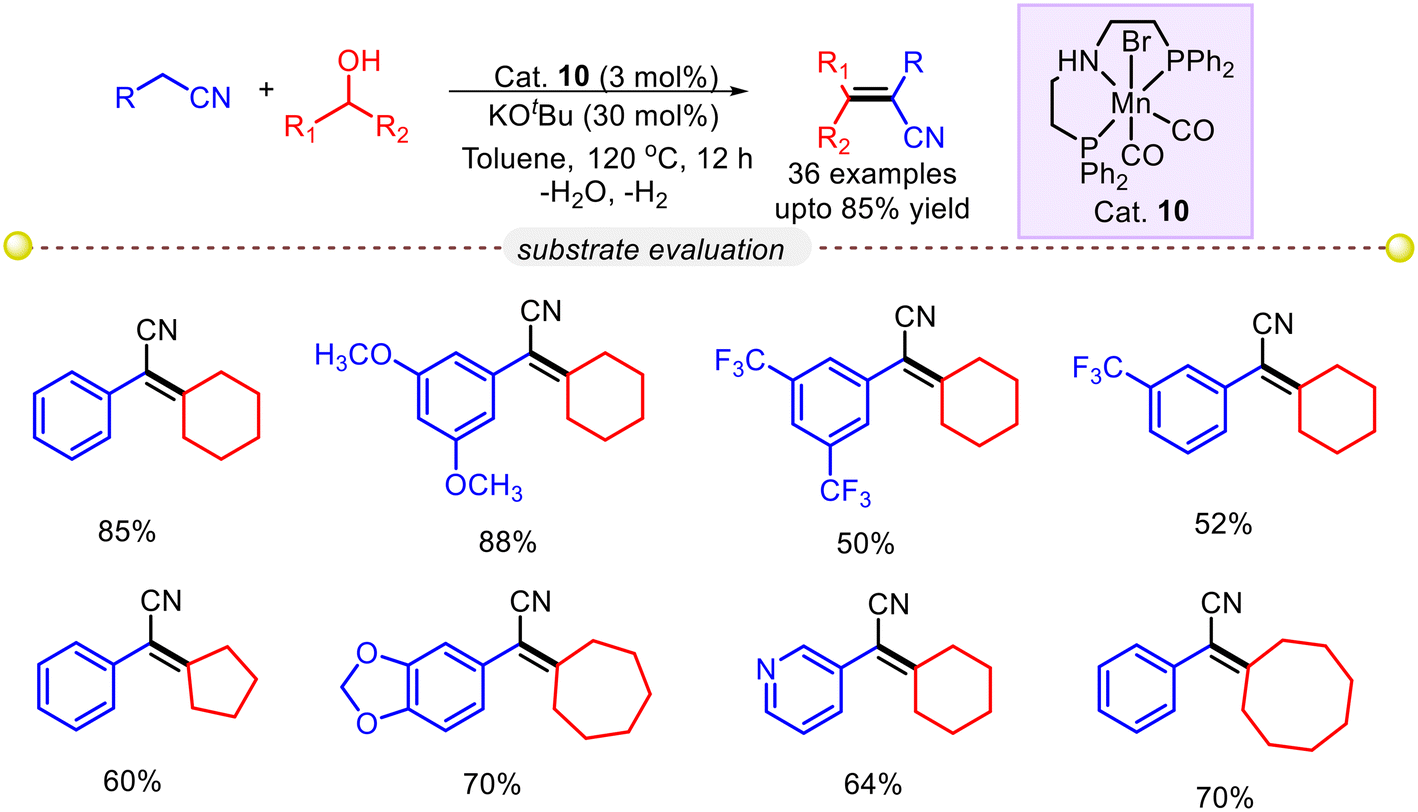
Since the replacement of precious metals with non-precious metals is one of the prime goals in modern chemistry, Balaraman and co-workers came up with an elegant method for synthesizing vinyl nitriles from nitriles and secondary alcohols using a well-defined manganese catalyst. KOtBu was used as a base, and Mn(I)–PNP pincer precatalyst (Cat. 10) was used in toluene solvent at 120 °C to selectively olefinate a number of acetonitriles with different secondary alcohols (Scheme 17).45 The olefination reaction of acetonitrile, incorporating both electron-donating and electron-withdrawing groups, as well as halides, exhibited excellent reactivity, yielding the desired products in good to excellent isolated yields. The kinetic studies indicated that the reaction follows first-order kinetics concerning both phenyl acetonitrile and cyclohexanol, while the catalyst showed a fractional order. Additionally, a kinetic deuterium-labeling experiment revealed a kH/kD ratio of 2.53, indicating that the rate-determining step involves the activation of the C–H bond in secondary alcohol.
 |
| | Scheme 17 An Mn–PNP complex catalyzed the synthesis of substituted alkenyl nitriles using secondary alcohols.45 | |
The proposed catalytic cycle for the Mn–PNP complex catalyzed synthesis of substituted alkenyl nitriles using secondary alcohols is shown in Scheme 18. Initially, the Mn-precatalyst is subjected to a base and transforms to a coordinatively unsaturated intermediate, denoted as H-1. Subsequently, the alcohol undergoes O–H activation via a proton transfer to the amido nitrogen of intermediate H-1, leading to the formation of an intermediate of the alkoxy-type, referred to as H-2. Next, the dehydrogenation reaction proceeds through the favorable pathway of base-promoted β-hydrogen abstraction. This intramolecular metal–ligand cooperation facilitates the formation of ketones and the manganese hydride complex H-3, which subsequently liberates hydrogen gas to regenerate the catalytically active intermediate H-1. Eventually, through Knoevenagel condensation between the in situ generated ketones and nitriles, vinyl nitriles are formed, with the elimination of a water molecule.
 |
| | Scheme 18 Plausible catalytic cycle for Mn–PNP complex catalyzed synthesis of substituted alkenyl nitriles using secondary alcohols. | |
In 2021, Balaraman and co-workers disclosed an unprecedented olefin (E) synthesis using nickel-catalyzed acceptorless dehydrogenative coupling of alcohols with nitriles via a decyanation pathway,46 where previous reports had shown α-alkylated/alkenylated products with the same starting materials. After screening several reaction conditions, the optimization experiments revealed that 4-methoxybenzyl alcohol and 2-phenylacetonitrile underwent a Ni-catalyzed (Cat. 4) dehydrogenative coupling to form (E)-1-methoxy-4-styrylbenzene with 87% yield when KOtBu, n-octane solvent and Cat. 4 was used under refluxing temperature at 120 °C (Scheme 19). The reaction conditions were well tolerated and provided good to excellent yield for ortho, para, and meta-substituted benzyl alcohol and phenyl acetonitriles. The reaction protocol was equally viable for heteroaryl systems, sterically demanding ortho-substituted alcohols, and sterically hindered secondary alcohols. The reaction methodology was applied to synthesize DMU-212 (a drug for breast cancer treatment) and resveratrol (pan-assay interference compound) with an excellent yield of 83% and 74%, respectively under the present Ni-catalyzed conditions. To get a mechanistic elucidation, deuterium labeling experiments, and several control experiments were performed. The experimental results suggested that the reaction proceeds via a tandem acceptorless dehydrogenative coupling decyanation pathway.
 |
| | Scheme 19 Ni–NNN complex catalyzed decyanative olefination of nitriles using alcohol.46 | |
Based on the mechanistic insights, the catalytic cycle (Scheme 20) started with a displacement reaction where the alcohol displaced the chloride ligand from the Ni-metal center of the pre-catalyst in the presence of KOtBu followed by a β-hydride elimination which led to an aldehyde and Ni–H species. The Ni–H species regenerated the active catalyst I-1via H2 liberation followed by reacting with the alcohol. The in situ generated aldehyde underwent the Knoevenagel condensation with a nitrile and led to vinyl-nitrile by eliminating a water molecule, and subsequent hydrogenation in the presence of Ni-catalyst followed by condensation resulted in α-alkylated arylacetamide. In the final step, the elimination of formamide from α-alkylated arylacetamide resulted in (E)-stilbene product.
 |
| | Scheme 20 Plausible catalytic cycle for Ni–NNN complex catalyzed decyanative olefination of nitriles using alcohol. | |
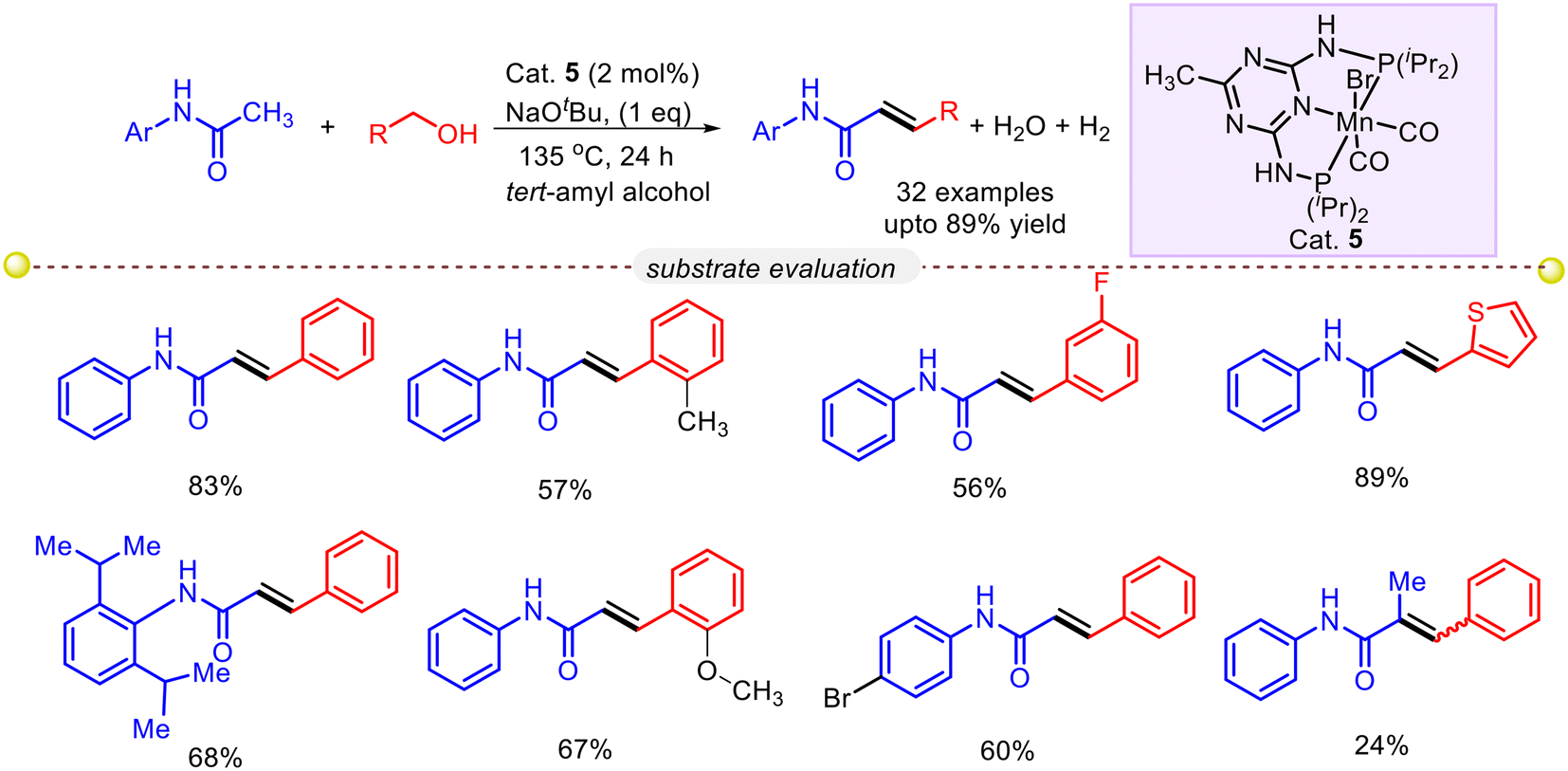
8. Synthesis of α,β-unsaturated ketone using alcohol
α,β-Unsaturated ketones have broad applications in the synthesis of pharmaceuticals, biologically active compounds, pesticides, food additives, and other functional materials.47 Conventionally, α,β-unsaturated ketones are synthesized via aldol condensation using ketones or aldehydes with a stoichiometric amount of strong bases under cryogenic conditions.48 As an alternative, the acceptorless dehydrogenative coupling method of alcohols with ketones is one of the most suitable methods to form α,β-unsaturated ketones. In recent years, compared to precious noble metal catalysts, the use of earth-abundant, economically feasible, and less toxic base metals has been highly demanded.
Recently Gunanathan and co-workers reported an atom-economical dehydrogenative coupling reaction of alcohols and ketones to α,β-unsaturated ketone using a manganese pincer complex (Cat. 5).49 In a representative example, 1-tetralone was reacted with a small excess of benzyl alcohol at 135 °C for 12 hours using catalyst 5 in the presence of a catalytic amount of base Cs2CO3. Spectroscopic analyses revealed that 1-tetralone effectively converted to 80% alkenyl product (Scheme 21) with a selectivity of 97:3 over alkane product. Under these catalytic conditions, heteroaryl, aliphatic, and benzylic primary alcohols featuring various functional groups (such as chloride, bromide, and methoxy) and various activated ketones furnished the desired product in good to moderate yield. Deuterium labelling studies and other experimental analyses predicted the reaction protocol follows the ADC pathway (Scheme 22). In the presence of a base, the precatalyst generates coordinatively unsaturated dearomatized intermediate J-1. The formation of alkoxy-ligated complexes J-2 is observed upon the reaction of intermediate J-1 with primary alcohols. Subsequently, a β-hydride elimination from J-2 resulted in an aldehyde with an Mn–hydride complex J-3. The de novo synthesized aldehyde underwent a base-promoted olefination with 1-tetralone to yield the final α,β-unsaturated ketone product, whereas Mn–hydride complex J-3 liberates hydrogen and regenerates dearomatized intermediate J-1.
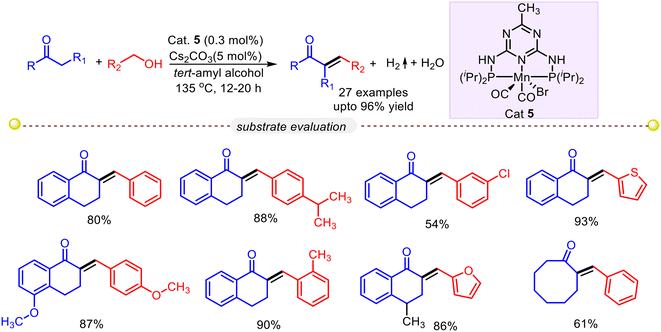 |
| | Scheme 21 An Mn–PNP complex catalyzed synthesis of α,β-unsaturated ketones via the ADC strategy.49 | |
 |
| | Scheme 22 Plausible catalytic cycle for Mn–PNP complex catalyzed synthesis of α,β-unsaturated ketones using alcohol. | |
The research group of Banerjee reported iron-catalyzed dehydrogenative coupling of alcohols and ketones to obtain α,β-unsaturated ketones.50 Iron(II) acetate in combination with 1,10-phenanthroline and base tBuONa in toluene at 110 °C provided the corresponding E-selective (>25:1) olefins from α-tetralone and 4-methoxybenzyl alcohol with 75% yield. A number of cyclic (C7, C8, C15 membered cyclic ketones) and acyclic ketones, as well as benzyl alcohols with delicate functional groups such as nitro, nitrile, trifluoromethyl, fluoro, chloro, 1,3-dioxolone, alkyl, and alkoxy, including heterocycles (furan functionality), were tolerated by the established catalytic process. The protocol was also applied for the synthesis of a sunscreen component (Scheme 23). Mechanistic experiments such as deuterium labeling studies, determination of the rate and order of the reaction, and quantitative determination of H2 gas suggested that the reaction followed the ADC-type pathway.
 |
| | Scheme 23 Iron-catalyzed synthesis of α,β-unsaturated ketones using alcohol.50 | |
Allylic alcohols are versatile C-3 precursors that can be used to synthesize various organic compounds with different applications. In 2021, Gunanathan and co-workers devised a method to produce α-alkenyl and α-alkyl ketones by directly cross-coupling primary alcohols and secondary allylic alcohols with a PNP–Mn(I) pincer complex catalyst. The best yield of the olefinated product was obtained with precatalyst 1 and a mild base Cs2CO3 for 24 h, giving a 97:3 ratio of alkenyl and alkylation products (Scheme 24).51
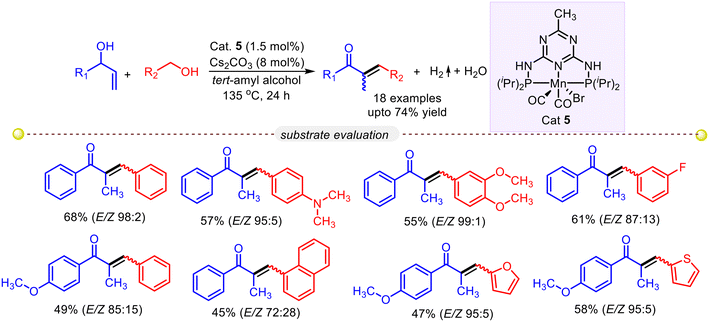 |
| | Scheme 24 Mn-complex catalyzed cross-coupling of secondary allylic alcohols and primary alcohols.51 | |
The cross-coupled olefin product was formed in moderate to good yield as a mixture of E and Z isomers from benzyl alcohols with electron-donating, electron-withdrawing, or halo substituents, as well as activated alcohols with heterocycles and secondary allyl alcohols with electron-donating groups. Based on several experimental studies, a plausible catalytic pathway for the PNP–Mn(I) pincer complex catalyzed olefination reaction is shown in Scheme 25. In the presence of a base, the Mn-precatalyst generates coordinatively unsaturated dearomatized intermediate K-1. The formation of alkoxy-ligated complexes K-2 and K-3 is observed upon the reaction of intermediate K-1 with secondary allylic alcohols and primary alcohols, respectively. This reaction proceeds through the facile O–H activation of alcohol functionalities. Subsequently, a β-hydride elimination from K-2 and K-3 resulted in α,β-unsaturated ketone and aldehyde, respectively with an Mn–hydride complex K-4. The Mn–hydride complex K-4 has been found to facilitate the selective hydrogenation of α,β-unsaturated ketone and produces propiophenone which subsequently reacts (in an aldol condensation pathway) with aldehyde and yields a final olefin product in the presence of base.
 |
| | Scheme 25 Plausible catalytic cycle for Mn–PNP complex catalyzed cross-coupling of secondary allylic alcohols and primary alcohols. | |
9. α-Olefination of alkenyl phosphine oxides using alcohol
Alkenylphosphine oxides are widely used in synthetic chemistry as the unsaturated C–C bond can be tamed to various modifications further. The unsaturation-containing phosphine oxides have widespread utility in agricultural, medicinal, and industrial chemistry.52 Pandia et al. devised an excellent method for the synthesis of alkenylphosphine oxides via dehydrogenative coupling of methyldiphenylphosphine oxide and arylmethyl alcohols using PNP–Mn(I) pincer complex (Scheme 26).53 A broad range of neutral, electron-donating, electron-withdrawing aromatic systems underwent the reaction using KOtBu as a base in tert-amyl alcohol solvent at 135 °C. In the presence of a base, the Mn-precatalyst generates coordinatively unsaturated dearomatized intermediate L-1 (Scheme 27). The formation of alkoxy-ligated complexes L-2 is observed upon the reaction of intermediate L-1 with primary alcohols. Subsequently, a β-hydride elimination from L-2 resulted in an aldehyde with a Mn–hydride complex L-3. The de novo synthesized aldehyde underwent a base-promoted olefination to yield the final alkenylphosphine oxides, whereas the Mn–hydride complex liberates hydrogen and generates dearomatized intermediate L-1.
 |
| | Scheme 26 Mn-catalyzed α-olefination of alkenylphosphine oxides using alcohol.53 | |
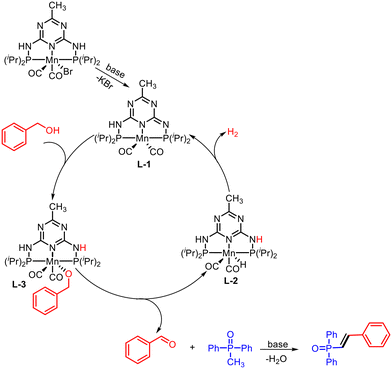 |
| | Scheme 27 Plausible catalytic cycle for Mn–PNP complex catalyzed α-olefination of alkenylphosphine oxides using alcohol. | |
10. α-Olefination of alkylamides using alcohol
Unsaturated amides play a crucial role in a wide range of applications, including natural products, bioactive molecules, polymeric materials, and pharmaceuticals.54 The traditional method for synthesizing α,β-unsaturated amides involves nucleophilic substitution of α,β-unsaturated carboxylic acids with amines, which often requires excessive reagents and suffers from limited compatibility with different functional groups. To address these challenges and pave the way for a sustainable future, chemists have been exploring various catalytic methods to introduce unsaturation alongside the amide functional group. However, there is a pressing need for a simple, selective, cost-effective, and efficient synthesis strategy. Gunanathan and co-workers developed a synthetic protocol for α-alkenylation of amides using primary alcohol as an alkenylation reagent and earth-abundant base metal manganese complex as a catalyst. Precatalyst 1 and base NaOtBu were determined to be the best combination for the isolated yield of olefinated product when used for 24 hours at 135 °C in a tert-amyl alcohol solvent (Scheme 28).55 A wide range of C(α)-olefinated amides were prepared using this Mn-catalyzed protocol by employing various primary alcohols and amides. Electron-donating benzyl alcohols and heteroaryl alcohols produced E-selective α,β-unsaturated amides with moderate to good yield. In the case of benzyl alcohols bearing electron-withdrawing group the yield of the expected olefinated product was only moderate, whereas secondary alcohols and aliphatic alcohols failed to provide desired alkene product. The catalytic reaction was found to be applicable to tertiary amides, although it resulted in a combination of alkenylation and alkylation products. A series of control experiments and deuterium labeling experiments were performed to get a mechanistic insight (Scheme 29).
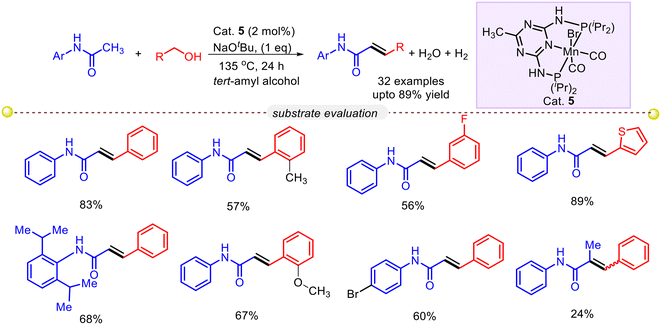 |
| | Scheme 28 An Mn–PNP complex catalyzed α-olefination of alkenylamides using alcohol.55 | |
 |
| | Scheme 29 Plausible catalytic cycle for Mn–PNP complex catalyzed α-olefination of alkenylamides using alcohol. | |
Mechanistic studies indicate, in the presence of a base, precatalyst generates coordinatively unsaturated dearomatized intermediate M-1. The formation of alkoxy-ligated complexes M-2 is observed upon the oxidative addition of primary alcohols with intermediate M-1. Subsequently, a β-hydride elimination from M-2 resulted in an aldehyde with an Mn–hydride complex M-3. The in situ generated aldehyde underwent a base-promoted olefination with corresponding amide to yield the final α,β-unsaturated amide product, whereas the Mn–hydride complex liberates hydrogen and generates dearomatized intermediate M-1.
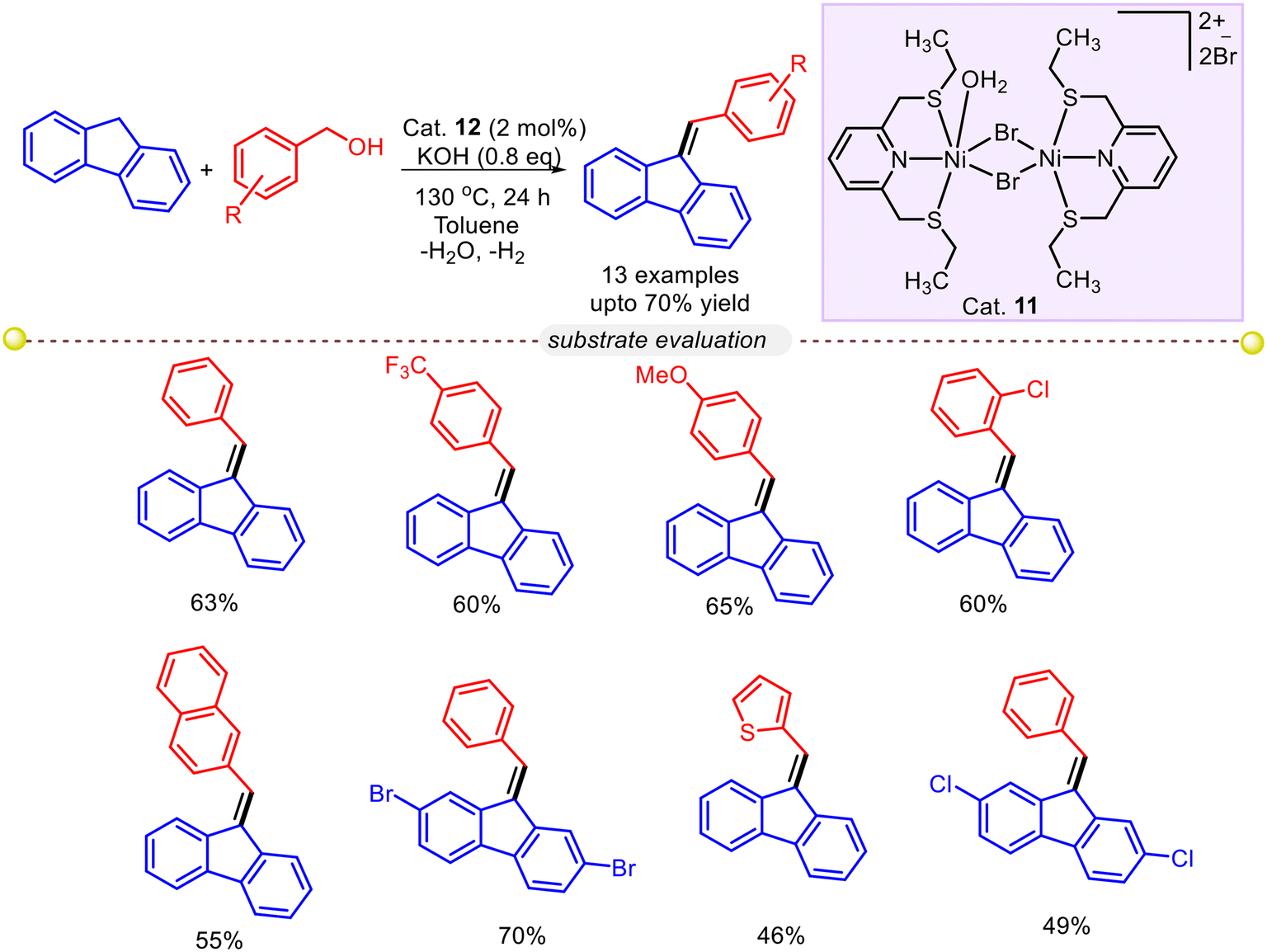
11. C(sp3)–H olefination of 9H-fluorene
Organic compounds generated from fluorene molecules have potential use in optoelectronics, solar cell, and semiconductor properties and have medicinal applications.56 Realising the importance of alkylated and olefinated fluorene derivatives, Gnanaprakasam and co-workers reported the sp3 C−H alkylation of 9H-fluorene using primary alcohol and a Ru catalyst via the borrowing hydrogen strategy.57 However, this reaction with secondary alcohols in the absence of any external oxidants furnished the tetrasubstituted alkene as the major product. Hence, a reaction of 9H-fluorene 1 with substituted benzhydrols such as methyl-, methoxy-, chloro and trifluoromethyl in the presence of 5 mol% [Ru(p-cymene)Cl2]2 and 2 equiv. of KOtBu at 140 °C afforded the respective alkene products in good yield. A higher selectivity for the formation of alkene in the case of secondary alcohols, can be due to the fact that it is difficult for the sterically hindered alkene to coordinate with the Ru catalyst for a further hydrogenation reaction (Scheme 30).
 |
| | Scheme 30 Reaction scheme and substrate scopes for Ru-catalyzed α-olefination of alkenylamides using benzhydrols.57 | |
Srimani and co-workers devised the BH (borrowing hydrogen) and ADC method where benzyl alcohols were used as alkylating and alkenylating sources (Scheme 31).58 After careful investigation of several reaction parameters, it was found that treating fluorene and 3-methoxybenzylalcohol in the presence of Cat. 11 and toluene with base KOH at 130 °C provided corresponding 9-alkylidenefluorene product with 65% yield. The formation of an alkylated side product (after the reduction of the olefin bond) was also observed. Electron-donating, electron-withdrawing, halo-substituted benzyl alcohols, activated alcohols bearing heterocycles, and substituted fluorene derivatives were well-acquainted with the reaction protocol and delivered the desired product with moderate to excellent yield. Control experiments and kinetic studies suggested that the reaction protocol follows an ADC-type pathway, whereas the reduced alkylated product was formed via the BH approach.
 |
| | Scheme 31 Reaction scheme for Ni-catalyzed C(sp3)–H olefination of 9H-fluorene. | |
Outlook and conclusion
Olefination stands out as one of the paramount reactions for the formation of CC bonds. Acceptorless dehydrogenation (ADH) is a novel strategy for synthesizing alkenes from various substrates, such as nitriles, sulfones, phosphorus ylides, and carbonyl compounds. This review discusses the environmental benefits of ADH-based olefinations that use alcohols as the hydrogen source, producing only H2O and H2 as the waste. This review holds significant importance, as olefins serve as crucial feedstocks or intermediates in both industrial and laboratory. We also showcase the diverse catalytic systems that have been developed for this purpose and their industrial relevance. However, there are still many challenges and opportunities for further improvement. This review aims to inspire researchers in this area to design more effective catalysts that can lower the reaction temperature, base amount, and reaction time, and increase the selectivity. By overcoming the current drawbacks and promoting innovative solutions, we can make the field of acceptorless dehydrogenation coupling for direct olefination more efficient and practical.
Conflicts of interest
The authors declare that they have no known competing financial interests.
Acknowledgements
This work is supported by CSIR (Project No: 01/(3030)/21/EMR-II) and SERB (Award No: CRG/2022/006504). E. B. is a Swarnajayanti Fellow (Award No.: SERB/F/5892/20202021) and gratefully acknowledges support from the Alexander-von-Humboldt (AvH) Foundation. R. B. acknowledges PMRF and G. S. thanks IISER-Tirupati for their fellowships.
References
-
(a) A. Kraft, A. C. Grimsdale and A. B. Holmes, Electroluminescent Conjugated Polymers-Seeing Polymers in a New Light, Angew. Chem., Int. Ed., 1998, 37, 402–428 CrossRef;
(b) U. Mitschke and P. Bäuerle, The Electroluminescence of Organic Materials, J. Mater. Chem., 2000, 10, 1471–1507 RSC;
(c) C. P. Nicholas, Applications of Light Olefin Oligomerization to the Production of Fuels and Chemicals, Appl. Catal., A, 2017, 543, 82–97 CrossRef CAS;
(d) S. Yadav, K. S. Tiwari, C. Gupta, M. K. Tiwari, A. Khan and S. P. Sonkar, A Brief Review on Natural Dyes, Pigments: Recent Advances and Future Perspectives, Results Chem., 2023, 5, 100733 CrossRef CAS.
-
(a) K. Hagiwara, N. Kosaka, Y. Yoshioka, R. Takahashi, F. Takeshita and T. Ochiya, Stilbene Derivatives Promote Ago2-Dependent Tumour-Suppressive MicroRNA Activity, Sci. Rep., 2012, 2, 314 CrossRef;
(b) J.-C. Jung, E. Lim, Y. Lee, J.-M. Kang, H. Kim, S. Jang, S. Oh and M. Jung, Synthesis of Novel Trans-Stilbene Derivatives and Evaluation of Their Potent Antioxidant and Neuroprotective Effects, Eur. J. Med. Chem., 2009, 44, 3166–3174 CrossRef CAS PubMed;
(c) D. Simoni, M. Roberti, F. P. Invidiata, E. Aiello, S. Aiello, P. Marchetti, R. Baruchello, M. Eleopra, A. Di Cristina, S. Grimaudo, N. Gebbia, L. Crosta, F. Dieli and M. Tolomeo, Stilbene-Based Anticancer Agents: Resveratrol Analogues Active toward HL60 Leukemic Cells with a Non-Specific Phase Mechanism, Bioorg. Med. Chem. Lett., 2006, 16, 3245–3248 CrossRef CAS.
-
(a) J. Martí-Rujas and F. Guo, Dehydrohalogenation reactions in second-sphere coordination complexes, Dalton Trans., 2021, 50, 11665–11680 RSC;
(b) A. Le Coq and A. Gorgues, Alkynes via Phase Transfer–Catalyzed Dehydrohalogenation: Propiolaldehyde Diethyl Acetal, Org. Synth., 1979, 59, 10 CrossRef;
(c)
J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, New York, 3rd edn, 1985 Search PubMed.
- G. Wittig and G. Geissler, Zur Reaktionsweise Des pentaphenyl-Phosphors Und Einiger Derivate, Justus Liebigs Ann. Chem., 1953, 580, 44–57 CrossRef CAS.
- D. J. Peterson, Carbonyl Olefination Reaction Using Silyl-Substituted Organometallic Compounds, J. Org. Chem., 1968, 33, 780–784 CrossRef CAS.
-
(a) M. Julia and J.-M. Paris, Syntheses a l'aide de Sulfones v(+)-Methode de Synthese Generale de Doubles Liaisons, Tetrahedron Lett., 1973, 14, 4833–4836 CrossRef;
(b) Y. Zhao, B. Gao and J. Hu, From Olefination to Alkylation: In-Situ Halogenation of Julia–Kocienski Intermediates Leading to Formal Nucleophilic Iodo- and Bromodifluoromethylation of Carbonyl Compounds, J. Am. Chem. Soc., 2012, 134, 5790–5793 CrossRef CAS PubMed.
- F. N. Tebbe, G. W. Parshall and G. S. Reddy, Olefin Homologation with Titanium Methylene Compounds, J. Am. Chem. Soc., 1978, 100, 3611–3613 CrossRef CAS.
-
(a)
P. G. Nandi, V. Arora, E. Yasmin and A. Kumar, in Pincer-Metal Complexes, ed. A. Kumar, Elsevier, 2022, ch. 2, pp. 69–122 Search PubMed;
(b) A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117(19), 12357–12384 CrossRef CAS PubMed;
(c)
K. Das and A. Kumar, in Advances in Organometallic Chemistry, ed. P. J. Pérez, Academic Press, 2019, ch. 1, vol. 72, pp. 1–57 Search PubMed.
-
R. H. Grubbs, Handbook of Metathesis, Wiley-VCH, Weinheim, 2003, vol. 1 Search PubMed.
- R. F. Heck and J. P. J. Nolley, Palladium-Catalyzed Vinylic Hydrogen Substitution Reactions with Aryl, Benzyl, and Styryl Halides, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Valorization of Biomass: Deriving More Value from Waste, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Renewable Chemical Commodity Feedstocks from Integrated Catalytic Processing of Pyrolysis Oils, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Complete Lignocellulose Conversion with Integrated Catalyst Recycling Yielding Valuable Aromatics and Fuels, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
-
(a) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Borrowing Hydrogen in the Activation of Alcohols, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS;
(b) G. Guillena, D. J. Ramón and M. Yus, Alcohols as Electrophiles in C-C Bond-Forming Reactions: The Hydrogen Autotransfer Process, Angew. Chem., Int. Ed., 2007, 46, 2358–2364 CrossRef CAS;
(c) R. Yamaguchi, K. I. Fujita and M. Zhu, Recent Progress of New Catalytic Synthetic Methods for Nitrogen Heterocycles Based on Hydrogen Transfer Reactions, Heterocycles, 2010, 81, 1093–1140 CrossRef CAS;
(d) Y. Obora and Y. Ishii, Iridium-Catalyzed Reactions Involving Transfer Hydrogenation, Addition, N-Heterocyclization, and Alkylation Using Alcohols and Diols as Key Substrates, Synlett, 2011, 1, 30–51 CrossRef;
(e) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, The Catalytic Amination of Alcohols, ChemCatChem, 2011, 3, 1853–1864 CrossRef;
(f) C. Gunanathan and D. Milstein, Applications of Acceptorless Dehydrogenation and Related Transformations in Chemical Synthesis, Science, 2013, 341, 1229712 CrossRef PubMed;
(g) L. Guo, Y. Liu, W. Yao, X. Leng and Z. Huang, Iridium-Catalyzed Selective α-Alkylation of Unactivated Amides with Primary Alcohols, Org. Lett., 2013, 15, 1144–1147 CrossRef CAS PubMed;
(h) G. Guillena, D. J. Ramón and M. Yus, Hydrogen Autotransfer in the N-Alkylation of Amines and Related Compounds Using Alcohols and Amines as Electrophiles, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed;
(i) G. E. Dobereiner and R. H. Crabtree, Dehydrogenation as a Substrate-Activating Strategy in Homogeneous Transition-Metal Catalysis, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed;
(j) A. J. A. Watson and J. M. J. Williams, The Give and Take of Alcohol Activation, Science, 2010, 329, 635–636 CrossRef CAS PubMed;
(k) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Transition Metal Catalysed Reactions of Alcohols Using Borrowing Hydrogen Methodology, Dalton Trans., 2009, 753–762 RSC;
(l) T. Suzuki, Organic Synthesis Involving Iridium-Catalyzed Oxidation, Chem. Rev., 2011, 111, 1825–1845 CrossRef CAS;
(m) R. K. Takeuchi Satoko, Iridium-Catalyzed Formation of Carbon-Carbon and Carbon-Heteroatom Bonds, Synthesis, 2006, 20, 3349–3366 CrossRef;
(n) R. Crabtree, Iridium Compounds in Catalysis, Acc. Chem. Res., 1979, 12, 331–337 CrossRef CAS;
(o) S. Pan and T. Shibata, Recent Advances in Iridium-Catalyzed Alkylation of C–H and N–H Bonds, ACS Catal., 2013, 3, 704–712 CrossRef CAS;
(p)
O. Saidi and J. M. J. Williams, Iridium-Catalyzed Hydrogen Transfer Reactions, in Iridium Catalysis, ed. P. G. Andersson, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 77–106 Search PubMed;
(q) B. Blank, M. Madalska and R. Kempe, An Efficient Method for the Selective Iridium-Catalyzed Monoalkylation of (Hetero)Aromatic Amines with Primary Alcohols, Adv. Synth. Catal., 2008, 350, 749–758 CrossRef CAS;
(r) B. Blank, S. Michlik and R. Kempe, Selective Iridium-Catalyzed Alkylation of (Hetero)Aromatic Amines and Diamines with Alcohols under Mild Reaction Conditions, Chem. – Eur. J., 2009, 15, 3790–3799 CrossRef CAS PubMed;
(s) B. Blank, S. Michlik and R. Kempe, Synthesis of Selectively Mono-N-Arylated Aliphatic Diamines via Iridium-Catalyzed Amine Alkylation, Adv. Synth. Catal., 2009, 351, 2903–2911 CrossRef CAS;
(t) J. Schranck, A. Tlili and M. Beller, More Sustainable Formation of C-N and C-C Bonds for the Synthesis of N-Heterocycles, Angew. Chem., Int. Ed., 2013, 52, 7642–7644 CrossRef CAS PubMed;
(u) R. H. Crabtree, An Organometallic Future in Green and Energy Chemistry?, Organometallics, 2011, 30, 17–19 CrossRef CAS;
(v) G. W. Lamb and J. M. J. Williams, Borrowing hydrogen-C-N bond formation from alcohols, Chim. Oggi, 2008, 26, 17–19 CAS.
- W. Ali, G. Prakash and D. Maiti, Recent Development in Transition Metal-Catalysed C-H Olefination, Chem. Sci., 2021, 12, 2735 RSC.
-
(a) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Dehydrogenation and Related Reactions Catalyzed by Iridium Pincer Complexes, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed;
(b) R. H. Crabtree, Homogeneous Transition Metal Catalysis of Acceptorless Dehydrogenative Alcohol Oxidation: Applications in Hydrogen Storage and to Heterocycle Synthesis, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed.
-
A. D. Abell and M. K. Edmonds, in Organophosphorus Reagents, Oxford University Press, Oxford, UK, 2004, pp. 99–127 Search PubMed.
-
A. D. Abell and M. K. Edmonds, in Modern Carbonyl Olefination, Wiley-VCH, Weinheim, Germany, 2004, ch. 1 Search PubMed.
- M. G. Edwards and J. M. J. Williams, Catalytic Electronic Activation: Indirect “Wittig” Reaction of Alcohols, Angew. Chem., Int. Ed., 2002, 41, 4740–4743 CrossRef CAS PubMed.
- E. Khaskin and D. Milstein, Catalytic, Oxidant-Free, Direct Olefination of Alcohols Using Wittig Reagents, Chem. Commun., 2015, 51, 9002–9005 RSC.
- N. Biswas, K. Das, B. Sardar and D. Srimani, Acceptorless Dehydrogenative Construction of C-N and C-C Bonds through Catalytic Aza-Wittig and Wittig Reactions in the Presence of an Air-Stable Ruthenium Pincer Complex, Dalton Trans., 2019, 48, 6501–6512 RSC.
- D. Srimani, G. Leitus, Y. Ben-David and D. Milstein, Direct Catalytic Olefination of Alcohols with Sulfones, Angew. Chem., Int. Ed., 2014, 53, 11092–11095 CrossRef CAS PubMed.
- C.-Z. Yao, Q.-Q. Li, M.-M. Wang, X.-S. Ning and Y.-B. Kang, (E)-Specific Direct Julia-Olefination of Aryl Alcohols without Extra Reducing Agents Promoted by Bases, Chem. Commun., 2015, 51, 7729–7732 RSC.
- S. M. A. H. Siddiki, A. S. Touchy, K. Kon and K. Shimizu, Direct Olefination of Alcohols with Sulfones by Using Heterogeneous Platinum Catalysts, Chem. – Eur. J., 2016, 22, 6111–6119 CrossRef CAS PubMed.
- V. G. Landge, V. Yadav, M. Subaramanian, P. Dangarh and E. Balaraman, Nickel(II)-Catalyzed Direct Olefination of Benzyl Alcohols with Sulfones with the Liberation of H2, Chem. Commun., 2019, 55, 6130–6133 RSC.
- V. G. Landge, R. Babu, V. Yadav, M. Subaramanian, V. Gupta and E. Balaraman, Iron-Catalyzed Direct Julia-Type Olefination of Alcohols, J. Org. Chem., 2020, 85, 9876–9886 CrossRef CAS PubMed.
- S. Waiba, A. Das, M. K. Barman and B. Maji, Base Metal-Catalyzed Direct Olefinations of Alcohols with Sulfones, ACS Omega, 2019, 4, 7082–7087 CrossRef CAS PubMed.
-
(a) T. Endo, M. Tsuda, T. Okada, S. Mitsuhashi, H. Shima, K. Kikuchi, Y. Mikami, J. Fromont and J. Kobayashi, Nagelamides A−H, New Dimeric Bromopyrrole Alkaloids from Marine Sponge Agelas Species, J. Nat. Prod., 2004, 67, 1262–1267 CrossRef CAS PubMed;
(b) J. Dai, Z.-Q. Liu, X.-Q. Wang, J. Lin, P.-F. Yao, S.-L. Huang, T.-M. Ou, J.-H. Tan, D. Li, L.-Q. Gu and Z.-S. Huang, Discovery of Small Molecules for Up-Regulating the Translation of Antiamyloidogenic Secretase, a Disintegrin and Metalloproteinase 10 (ADAM10), by Binding to the G-Quadruplex-Forming Sequence in the 5′ Untranslated Region (UTR) of Its MRNA, J. Med. Chem., 2015, 58, 3875–3891 CrossRef CAS PubMed;
(c) S. Blanchard, A. D. William, A. C.-H. Lee, A. Poulsen, E. L. Teo, W. Deng, N. Tu, E. Tan, K. L. Goh, W. C. Ong, C. P. Ng, K. C. Goh, Z. Bonday and E. T. Sun, Synthesis and Evaluation of Alkenyl Indazoles as Selective Aurora Kinase Inhibitors, Bioorg. Med. Chem. Lett., 2010, 20, 2443–2447 CrossRef CAS.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Light-Emitting Diodes Based on Conjugated Polymers, Nature, 1990, 347, 539–541 CrossRef CAS.
- G. Zhang, T. Irrgang, T. Dietel, F. Kallmeier and R. Kempe, Manganese-Catalyzed Dehydrogenative Alkylation or α-Olefination of Alkyl-Substituted N-Heteroarenes with Alcohols, Angew. Chem., Int. Ed., 2018, 57, 9131–9135 CrossRef CAS.
- M. K. Barman, S. Waiba and B. Maji, Manganese-Catalyzed Direct Olefination of Methyl-Substituted Heteroarenes with Primary Alcohols, Angew. Chem., Int. Ed., 2018, 57, 9126–9130 CrossRef CAS PubMed.
- J. Das, M. Vellakkaran and D. Banerjee, Nickel-Catalysed Direct α-Olefination of Alkyl Substituted N-Heteroarenes with Alcohols, Chem. Commun., 2019, 55, 7530–7533 RSC.
- B. M. Ramalingam, I. Ramakrishna and M. Baidya, Nickel-Catalyzed Direct Alkenylation of Methyl Heteroarenes with Primary Alcohols, J. Org. Chem., 2019, 84, 9819–9825 CrossRef CAS PubMed.
- G. Balamurugan and R. Ramesh, Nickel(II)-Catalyzed Selective (E)-Olefination of Methyl Heteroarenes Using Benzyl Alcohols via Acceptorless Dehydrogenative Coupling Reaction, ChemCatChem, 2022, 14, e202101455 CrossRef CAS.
- G. S. Bisht, A. M. Pandey, M. B. Chaudhari, S. G. Agalave, A. Kanyal, K. Karmodiya and B. Gnanaprakasam, Ru-Catalysed Dehydrogenative Synthesis of Antimalarial Arylidene Oxindoles, Org. Biomol. Chem., 2018, 16, 7223–7229 RSC.
- X.-J. Dai and C.-J. Li, En Route to a Practical Primary Alcohol Deoxygenation, J. Am. Chem. Soc., 2016, 138, 5433–5440 CrossRef CAS PubMed.
- U. K. Das, S. Chakraborty, Y. Diskin-Posner and D. Milstein, Direct Conversion of Alcohols into Alkenes by Dehydrogenative Coupling with Hydrazine/Hydrazone Catalyzed by Manganese, Angew. Chem., Int. Ed., 2018, 57, 13444–13448 CrossRef CAS.
-
(a) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, Nitrile-Containing Pharmaceuticals: Efficacious Roles of the Nitrile Pharmacophore, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed;
(b) F. F. Fleming, Nitrile-Containing Natural Products, Nat. Prod. Rep., 1999, 16, 597–606 RSC.
-
(a) R. Gómez, J. L. Segura and N. Martín, New Optically Active Polyarylene Vinylenes: Control of Chromophore Separation by Binaphthyl Units, Chem. Commun., 1999, 619–620 RSC;
(b) J. L. Segura, N. Martín and M. Hanack, Oligo-2,6-Naphthylenevinylenes – New Building Blocks for the Preparation of Photoluminescent Polymeric Materials, Eur. J. Org. Chem., 1999, 3, 643–651 CrossRef.
- F. Ladhar and R. El Gharbi, Direct Synthesis of α,β-Unsaturated Nitriles in Solid/Liquid Heterogeneous Medium, Synth. Commun., 1991, 21, 413–417 CrossRef CAS.
- S. Arseniyadis, K. Skyler and D. S. Watt, Addition and Substitution Reactions of Nitrile-Stabilized Carbanions, Org. React., 1984, 31, 40–42 Search PubMed.
- S. Chakraborty, U. K. Das, Y. Ben-David and D. Milstein, Manganese Catalyzed α-Olefination of Nitriles by Primary Alcohols, J. Am. Chem. Soc., 2017, 139, 11710–11713 CrossRef CAS PubMed.
- J. Li, Y. Liu, W. Tang, D. Xue, C. Li, J. Xiao and C. Wang, Atmosphere-Controlled Chemoselectivity: Rhodium-Catalyzed Alkylation and Olefination of Alkylnitriles with Alcohols, Chem. – Eur. J., 2017, 23, 14445–14449 CrossRef CAS.
- S. Thiyagarajan and C. Gunanathan, Ruthenium-Catalyzed α-Olefination of Nitriles Using Secondary Alcohols, ACS Catal., 2018, 8, 2473–2478 CrossRef CAS.
- V. Yadav, V. G. Landge, M. Subaramanian and E. Balaraman, Manganese-Catalyzed α-Olefination of Nitriles with Secondary Alcohols, ACS Catal., 2020, 10, 947–954 CrossRef CAS.
- S. P. Midya, M. Subaramanian, R. Babu, V. Yadav and E. Balaraman, Tandem Acceptorless Dehydrogenative Coupling–Decyanation under Nickel Catalysis, J. Org. Chem., 2021, 86, 7552–7562 CrossRef CAS PubMed.
-
(a) M. J. Climent, A. Corma, S. Iborra and A. Velty, Activated Hydrotalcites as Catalysts for the Synthesis of Chalcones of Pharmaceutical Interest, J. Catal., 2004, 221, 474–482 CrossRef CAS;
(b) R. Li, G. L. Kenyon, F. E. Cohen, X. Chen, B. Gong, J. N. Dominguez, E. Davidson, G. Kurzban, R. E. Miller, E. O. Nuzum, P. J. Rosenthal and J. H. McKerrow, In Vitro Antimalarial Activity of Chalcones and Their Derivatives, J. Med. Chem., 1995, 38, 5031–5037 CrossRef CAS PubMed.
-
(a)
D. N. Dhar and D. H. R. Barton, The chemistry of chalcones and related compounds, Wiley, New York, 1981 Search PubMed;
(b)
J. Chopin, M. L. Bouillant and E. Besson, in Flavonoids, Advances in Research, Chapman and Hall, London, 1982 Search PubMed.
- S. S. Gawali, B. K. Pandia and C. Gunanathan, Manganese(I)-Catalyzed α-Alkenylation of Ketones Using Primary Alcohols, Org. Lett., 2019, 21, 3842–3847 CrossRef CAS PubMed.
- M. Sk, A. Kumar, J. Das and D. Banerjee, A Simple Iron-Catalyst for Alkenylation of Ketones Using Primary Alcohols, Molecules, 2020, 25, 1590 CrossRef CAS PubMed.
- B. K. Pandia, S. Pattanaik and C. Gunanathan, Manganese(I) Catalyzed Cross-Coupling of Secondary Allylic Alcohols and Primary Alcohols, Tetrahedron, 2021, 101, 132472 CrossRef CAS.
-
(a) K. Moonen, I. Laureyn and C. V. Stevens, Synthetic Methods for Azaheterocyclic Phosphonates and Their Biological Activity, Chem. Rev., 2004, 104, 6177–6216 CrossRef CAS PubMed;
(b)
D. E. C. Corbridge, Phosphorus: Chemistry, Biochemistry and Technology, CRC Press, London, 6th edn, 2013 Search PubMed;
(c) G. P. Horsman and D. L. Zechel, Phosphonate Biochemistry, Chem. Rev., 2017, 117, 5704–5783 CrossRef CAS PubMed;
(d) H. Ni, W.-L. Chan and Y. Lu, Phosphine-Catalyzed Asymmetric Organic Reactions, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS.
- B. K. Pandia, S. Pattanaik and C. Gunanathan, Manganese(I) Catalyzed Alkenylation of Phosphine Oxides Using Alcohols with Liberation of Hydrogen and Water, J. Org. Chem., 2021, 86, 17848–17855 CrossRef CAS PubMed.
-
(a) V. J. Pattabiraman and J. W. Bode, Rethinking amide bond synthesis, Nature, 2011, 480, 471–479 CrossRef CAS;
(b) E. Valeur and M. Bradley, Amide bond formation: beyond the myth of coupling reagents, Chem. Soc. Rev., 2009, 38, 606–631 RSC;
(c) C. A. G. N. Montalbetti and V. Falque, Amide bond formation and peptide coupling, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS;
(d)
Handbook of Reagents for Organic Synthesis: Activating Agents and Protecting Groups, ed. A. J. Pearson and W. R. Roush, Wiley-VCH, New York, 1999 Search PubMed.
- B. K. Pandia and C. Gunanathan, Manganese(I) Catalyzed α-Alkenylation of Amides Using Alcohols with Liberation of Hydrogen and Water, J. Org. Chem., 2021, 86, 9994–10005 CrossRef CAS PubMed.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chemical Sensors Based on Amplifying Fluorescent Conjugated Polymers, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed.
- M. A. Shaikh, S. G. Agalave, A. S. Ubale and B. Gnanaprakasam, J. Org. Chem., 2020, 85, 2277–2290 CrossRef CAS.
- R. Sharma, A. Mondal, A. Samanta and D. Srimani, A Strategic Approach for Csp3–H Functionalization of 9H-Fluorene: an Acceptorless Dehydrogenation and Borrowing Hydrogen Approach, Catal. Sci. Technol., 2023, 13, 611–617 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b and
Ekambaram
Balaraman
*b and
Ekambaram
Balaraman