
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aldol condensation of mixed oxygenates on TiO2†
Brandon Elliott
Oliphant
 a,
Mathew
Rasmussen
b,
Laura
Paz Herrera
a,
Michael B.
Griffin
b and
J. Will
Medlin
*a
a,
Mathew
Rasmussen
b,
Laura
Paz Herrera
a,
Michael B.
Griffin
b and
J. Will
Medlin
*a
aDepartment of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80303, USA. E-mail: medlin@colorado.edu
bNational Renewable Energy Laboratory, Golden, CO 80401, USA
First published on 16th February 2024
Abstract
Aldol condensation of mixtures of acetaldehyde, acetone and butanone was investigated over a powder TiO2 catalyst to probe a method for coupling light oxygenates generated from biomass pyrolysis into heavier, higher-value products. Self-aldol condensation (SAC) of each component was found to produce the expected dimers (e.g., crotonaldehyde from acetaldehyde, mesityl oxide from acetone) and higher molecular weight products formed from multiple coupling reactions. Mixed-aldol condensation (MAC) reactions for acetaldehyde, acetone, and butanone yielded the same products as well as cross-condensation products such as 3-penten-2-one. In MAC reactions, acetaldehyde suppressed the reaction of the other species in the mixture, such that in low-conversion reactions with equimolar feeds, crotonaldehyde dominated product selectivity. Kinetic experiments conducted with variable partial pressures of the reactants indicated a higher dependence of reactions rates on the pressures of acetone and butanone compared to acetaldehyde, consistent with a difference in the saturation of catalytic active sites between reactant molecules. Infrared spectroscopy experiments after pyridine chemisorption showed that acetaldehyde SAC reactions decreased Lewis acid site availability more quickly than acetone SAC reactions. Results from kinetic and temperature programmed desorption studies indicated that the higher rates for acetaldehyde conversion could be attributed to both its tendency to out-compete acetone for access to adsorption sites and its effectiveness as an electrophile.
Introduction
As the transportation sector has become the largest contributor to greenhouse gas emissions in the United States,1 rapid transition towards renewable energy sources is a critical pillar of the global strategy for limiting global warming.2 Sustainable aviation fuels are of particular interest, as the aviation sector accounts for 2.5% of global greenhouse gas emissions, and current prospects for electrification are limited.3 There are several emerging processes for generating biofuels, including pyrolysis of lignocellulosic biomass and hydrotreating of liquid organic feedstocks such as vegetable oil. These processes take advantage of longer hydrocarbon chains already present in the biomass, which can be thermally or catalytically cracked into fuel grade molecules in the C8–C20 range, before being deoxygenated to form biofuel. The main byproducts from the cracking and upgrading steps are mixtures of volatile oxygenates in the C1–C4 range that are not captured in the liquid product stream.2,4Ideally, these biofuel production processes could be implemented by repurposing existing petroleum processing infrastructure, allowing use of current systems for the co-processing of biogenic feedstocks and petroleum-based feedstocks.5–9 As such, technological advancements in research areas that lend themselves toward the processing of biogenic feedstocks using petroleum refinery processes and equipment are one of the key ways to overcome current economic challenges for commercial-scale adoption of biofuel production.10 One such advancement is in the use of heterogeneous catalysis to condense light oxygenates into more valuable materials. Advancements in this space would be useful for upgrading the volatile oxygenate waste stream from biofuel production and other sources.10
There are several potential reaction pathways for condensation of light oxygenates for upgrading to value added materials. These reactions include Guerbet condensation for coupling two alcohol molecules to generate a larger alcohol molecule and Lebedev condensation for coupling of two alcohol molecules to produce larger 1,3-unsaturated alkenes.10 The key coupling step for both reaction pathways is aldol condensation, which involves the coupling of aldehydes and ketones to larger unsaturated aldehydes and ketones. Aldol condensation has been studied on a wide variety of materials, including alkaline earth metal oxides such as MgO (ref. 11–13) and SrO (ref. 11) and transition metal oxide materials such as CeO2,14–17 V2O5–P2O5 (ref. 18) and TiO2.19–28 These materials have been the subject of interest for aldol condensation based on their low cost and easy recovery. Much of the research in this space has been dedicated to determining which materials and material properties are the most important for activating this reaction. Analysis of materials with different types of active sites, such as basic alkaline earth metal oxides and transition metal oxides, which have a mixture of acid and base sites, has shown that a combination of both acidic and basic sites are necessary for maximizing catalyst performance.11–19,21–29 There are a few different proposed mechanisms for this reaction, each dependent on the types of active sites present in the material. Fig. 1 shows a widely agreed-upon mechanism for aldol condensation on transition metal oxides containing Lewis acid and Lewis/Brønsted base sites, using TiO2 as an example.
 | ||
| Fig. 1 Reaction mechanism for acetaldehyde aldol condensation on TiO2, adapted from ref. 25. | ||
Fig. 1 shows the electrophilic acidic metal cation site binding the carbonyl group of the reactant molecule.23–25 Vicinal basic oxygen sites interact with the α-methyl group, abstracting an α-hydrogen and forming a reactive enolate species. Carbon–carbon coupling occurs via nucleophilic attack by the enolate on the carbonyl carbon of a vicinal electrophile. The product molecule can desorb and undergo dehydration to form the aldol condensation product, in this case crotonaldehyde. There is strong literature support for these four steps as the main mechanistic pathway for aldol condensation on metal oxide surfaces.17,19,25–27,30,31 Previous observations of inhibition of acetone self-aldol condensation (SAC) in the presence of pyridine or acetic acid provide additional support for the concerted contributions of acidic and basic catalytic sites.25 Fourier transform infrared (FTIR) spectroscopy studies indicated adsorption of the reactant through interaction between acid sites and the carbonyl oxygen group.23,24
While aldol condensation of reaction feeds with one enolate-forming species are widely studied, less attention has been paid to reaction feeds with multiple reactive components. Mixed aldol condensation (MAC) is relevant for applications such as upgrading mixtures of volatile oxygenates which can contain a wide variety of C1–C4 alkanals and alkanones. Studies of aldol condensation in multicomponent systems largely focused on the production of unsaturated α,β-carbonyls through the combination of an enolate-forming species like acetone, and an electrophilic species like formaldehyde18,20,22 or furfural.32 The electrophilic species used in these studies are unable to form the enolate intermediate due to a lack of available α-hydrogens. This limitation results in only two possible reaction pathways, SAC of the enolate forming species, or cross aldol condensation (CAC) between the enolate forming species and the electrophilic species. Results from these studies show that the electrophile (furfural or formaldehyde) has higher reactivity than the enolate forming species,15 such that the reaction has almost complete selectivity to the CAC product over the SAC product.18,20,22,32
The work described here was aimed at elucidating competitive reaction processes for mixtures of acetaldehyde, acetone, and butanone. These three compounds are representative of the volatile oxygenate fraction produced during the catalytic fast pyrolysis of biomass to produce biofuel,33 which allows us to probe reactant activity and reaction selectivity of a potentially valuable biofuel production waste stream. The binary and ternary reaction systems discussed in this work have many possible reaction products, since all reaction components can form enolates or act as electrophiles. This added layer of complexity allows us to study differences in reactivity across molecules of different sizes with different carbonyl positioning. This paper reports on reaction and characterization studies aimed at understanding how interactions between acetaldehyde, acetone, and butanone alter the kinetics of aldol condensation reactions.
Methods
Commercial mixed-phase TiO2 catalyst (Evonik, Aerolyst 7711) was crushed and sieved to 70+ mesh size. The TiO2 catalyst we employed in this work consists of an approximately 70%/30% mixture of anatase/rutile phases. Of the three phases of TiO2, pure anatase phase TiO2 and commercial mixed-phase TiO2 (between 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 4:1 ratio of anatase to rutile) are the most studied types of TiO2 for catalyzing aldol condensation. Anatase TiO2 has been found to be the more active of the pure phases, though anatase-rich commercial mixtures of phases have been found to be more active than pure anatase,21,25 which is the reason we selected a similar mixture for detailed study. Reaction experiments used either 15 or 30 mg of powder TiO2 packed into a quartz reactor tube between two layers of quartz wool. Temperature programmed desorption (TPD) experiments used 50 mg of TiO2 packed the same way into a quartz reactor tube. Catalyst samples were pretreated in 36 sccm of H2 (99.999%, Airgas) at 450 °C for 1 hour, then purged in 36 sccm He or N2 (99.999%, Airgas) at 450 °C for 30 minutes. After pretreatment, samples were cooled to reaction temperature or, in the case of TPD experiments, to room temperature. For each reaction, catalyst samples were exposed to mixtures of acetaldehyde (99.5%, Acros Organics), acetone (ACS Reagent Grade, Macron), and 2-butanone (≥99.0%, Sigma-Aldrich) while the reaction temperature was held for 3 hours. Experiments were conducted across a broad range of temperatures. High-conversion studies were carried out between 300 °C and 400 °C, the temperature range relevant for the catalytic fast pyrolysis (CFP) downstream process. Reaction temperatures for these experiments were increased at a rate of approximately 3.3 °C min−1 between temperature set points. For mechanistic investigation, we also conducted experiments at much lower temperatures. For example, kinetic experiments for measuring activation energies involved measuring the rate of reaction at temperatures of 150 °C, 175 °C and 200 °C. For these experiments, the partial pressure of each reactant was kept constant at 0.023 bar. The pressure series experiments were conducted at 200 °C using partial pressures of reactants between 0.023 bar and 0.28 bar. The kinetic isotope experiments were also conducted at 200 °C with a constant reactant concentration of 0.025 bar.
:1 and 4:1 ratio of anatase to rutile) are the most studied types of TiO2 for catalyzing aldol condensation. Anatase TiO2 has been found to be the more active of the pure phases, though anatase-rich commercial mixtures of phases have been found to be more active than pure anatase,21,25 which is the reason we selected a similar mixture for detailed study. Reaction experiments used either 15 or 30 mg of powder TiO2 packed into a quartz reactor tube between two layers of quartz wool. Temperature programmed desorption (TPD) experiments used 50 mg of TiO2 packed the same way into a quartz reactor tube. Catalyst samples were pretreated in 36 sccm of H2 (99.999%, Airgas) at 450 °C for 1 hour, then purged in 36 sccm He or N2 (99.999%, Airgas) at 450 °C for 30 minutes. After pretreatment, samples were cooled to reaction temperature or, in the case of TPD experiments, to room temperature. For each reaction, catalyst samples were exposed to mixtures of acetaldehyde (99.5%, Acros Organics), acetone (ACS Reagent Grade, Macron), and 2-butanone (≥99.0%, Sigma-Aldrich) while the reaction temperature was held for 3 hours. Experiments were conducted across a broad range of temperatures. High-conversion studies were carried out between 300 °C and 400 °C, the temperature range relevant for the catalytic fast pyrolysis (CFP) downstream process. Reaction temperatures for these experiments were increased at a rate of approximately 3.3 °C min−1 between temperature set points. For mechanistic investigation, we also conducted experiments at much lower temperatures. For example, kinetic experiments for measuring activation energies involved measuring the rate of reaction at temperatures of 150 °C, 175 °C and 200 °C. For these experiments, the partial pressure of each reactant was kept constant at 0.023 bar. The pressure series experiments were conducted at 200 °C using partial pressures of reactants between 0.023 bar and 0.28 bar. The kinetic isotope experiments were also conducted at 200 °C with a constant reactant concentration of 0.025 bar.
Gas composition in the reaction studies was analyzed with an online Agilent 7820A gas chromatograph. Product identities were confirmed by an offline Agilent 7890a/5975c GC-MS. For all reaction experiments conducted at differential conversion, each data point was produced using fresh catalyst to minimize the effect of deactivation on the response of the reaction to changing variables. The TPD experiments were conducted using the same reactor preparation and pretreatment procedure as described above. Once the reactor was cooled to room temperature, the catalyst bed was dosed with the reactant molecules for 30 minutes. Then the reactor temperature was raised from room temperature to 700 °C at a ramp rate of 10 °C per minute. Desorption data was recorded by an online Pfeiffer Prisma quadrupole mass spectrometer. Adjacent-averaging data smoothing (using the average of 10 individual points) was used to reduce noise from TPD results.
Light oxygenate reactants were introduced into the gas phase using 30 mL graduated glass bubbler tubes (AceGlass). Each reactant was contained in a separate bubbler which was submerged in a refrigerated water bath. The water bath temperature and flow rate of inert gas, either He or N2 (99.999%, Airgas), were manipulated to deliver the desired concentration of reactant in the gas phase. Concentrations of each reactant in the gas phase were calculated by assuming that the gas flowing through each bubbler reached equilibrium with the liquid reactant, and the partial pressures of the reactant vapors exiting the bubbler were estimated using the Antoine parameters from the NIST chemistry WebBook.34 A constant inert gas flow rate of 27sccm through the reactor was maintained for all reaction experiments. Before all reaction experiments, the reaction feed was passed through a reactor bypass stream to the GC until reactant concentrations stabilized. Once the reaction feed stream reached steady state, the feed was diverted to flow through the reactor.
Experimental error for the high conversion studies was estimated by doing a triplicate experiment for one of the test conditions, then applying that relative error to the other self and cross condensation reactions. Experimental error for data points in differential reactor experiments was calculated using the standard error from triplicate experiments for each data point. The error reported for calculated activation energy and reaction order was calculated using the standard error from calculated values from triplicate experiments. Carbon balances for both the high conversion and low conversion reactions were calculated by dividing the sum of all recorded peak areas during each GC injection during the reaction by the average of the sum of the reactant peak areas recorded during the reactor bypass injections. GC peak areas were recorded manually for all experiments, so only the largest peaks were recorded for calculating carbon balances, conversion, and selectivity. Carbon balances for the differential conversion studies calculated using this method were high (between 95% and 105%). Carbon balances for the high conversion study were much lower (between 30% and 70%) likely due to product condensation between the reactor and GC, coke deposition on the catalyst surface, and the limitations of analyzing a large number of GC peaks.
The impact of mass transfer effects on reactions for this system were investigated by calculating the Weisz–Prater parameter and by changing catalyst particle size. The Weisz–Prater parameter was calculated to be 10−4, ruling out internal mass transfer limitations. As a further test of possible heat or mass transfer effects, reaction rates were measured for catalyst particles of different size and were found to be nearly invariant within error (see ESI† for details of the transport limitation tests).
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) studies of the chemisorption of pyridine (99%, Sigma Aldrich) on TiO2 were performed on a Thermo Scientific Nicolet 6700 FT-IR spectrometer equipped with an MCT-a detector and a Praying Mantis diffuse reflection accessory with an in situ high temperature reaction chamber (Harrick Scientific Products Inc.). For each DRIFTS experiment, 50 mg of powder TiO2 was loaded into the reaction chamber and purged in 200 sccm of Ar at room temperature for 15 minutes. Fresh catalysts were pretreated in 200 sccm of H2 (99.999%, Airgas) at 300 °C for 1 hour, then purged in 200 sccm of Ar at the same temperature for 30 minutes. For experiments involving adsorption of acetaldehyde and acetone, fresh TiO2 was dosed with the probe molecule for 1 hour using 200 sccm Ar flowing through a 30 mL graduated gas bubbler tube (AceGlass) kept at 1 °C. After dosing, the sample was purged in 200 sccm Ar for 30 minutes at 200 °C to simulate reaction conditions. For experiments involving spent catalysts, the sample was treated with 200 sccm of Ar at 200 °C for 30 minutes to remove moisture and any physisorbed ambient species, instead of undergoing the reductive pretreatment. After pretreatment of the catalyst sample, background scans were collected at 150 °C under 200 sccm of Ar prior to pyridine dosing. At the same temperature, pyridine was introduced into the system by flowing 200 sccm Ar through a 30 mL graduated gas bubbler tube (AceGlass) containing pyridine at room temperature for 20 minutes. After pyridine dosing, the system was purged with 200 sccm Ar while scans were collected over the course of 1 hour. All DRIFTS spectra included in the body of this work were collected after 5 minutes of purging in Ar after the pyridine dose. Duplicates of all pyridine DRIFTS spectra were obtained. The spectra were normalized by calculating the average multiplying factor required from each duplicate pair to align their intensity with that of the reference fresh TiO2 spectrum. Reported normalization values, calculated from the duplicate experiments, display the average as the center point and the range as the error bars.
Results
Reaction pathway study
To provide a baseline for studies of oxygenate mixtures under conditions relevant for the CFP application, the reactions of acetaldehyde, acetone, and butanone as single components were investigated over a powder TiO2 catalyst under identical molar flowrates at 300 °C, 350 °C and 400 °C. Conversion trends show that acetaldehyde had the highest conversion at all three temperatures, while butanone had the lowest (Fig. S1†).For acetaldehyde SAC, the primary reaction product at 300 °C was the dimer crotonaldehyde (2-butenal, Fig. S2†). At higher temperatures, selectivity toward C6 and C8 condensation products increased, with the highest selectivity going to 2-methyl-2-cyclopenten-1-one. This product is likely formed through the trimerization of acetaldehyde followed by a cyclization and deoxygenation reaction. For acetone SAC, the primary reaction product at all temperatures was mesitylene, a cyclic C9 alkane molecule (Fig. S3†). Mesitylene was likely formed through the stepwise condensation of acetone to mesityl oxide (dimer) and then to 4,6-dimethylhepta-3,5-dien-2-one (trimer). The trimer then underwent internal 1,6-aldol condensation to form the final cyclic product, mesitylene.21 In contrast to acetaldehyde SAC, the product stream for acetone SAC did not appear to have any significant amount of the dimer, mesityl oxide, at the high conversions used in this study, suggesting a high reactivity for C6 species produced from acetone. 3,4-Dimethyl-3-hexen-2-one and 5-methyl-4-hepten-3-one are the two dimers formed from butanone SAC, as the reactant molecule has two distinct positions for α-hydrogen extraction and enolate formation.
A light hydrocarbon mixture was produced in moderate-to-high yields for all SAC reactions. GC-MS analysis of the condensed product mixture from the acetaldehyde and acetone SAC reactions identified products such as isobutene and butane. The production of these compounds is consistent with previous studies, which report butene formation from acetaldehyde and acetone coupling on metal oxide surfaces.21,35–38 The mechanism for the formation of these volatile aliphatic compounds is hypothesized to involve acid-catalyzed cracking of larger condensation products.21,37,38 Selectivity to cracking products tended to increase at higher temperatures for all three reactants. Additionally, selectivity towards cracking products tended to increase with the size of the reactant molecule. This may indicate that product mixtures with a higher average carbon chain length are more readily decomposed into light hydrocarbons. This correlation between carbon chain length and cracking activity is consistent with previous reports involving the cracking of linear paraffins and olefins over acidic metal oxide catalysts.39,40
Binary and ternary mixed-aldol condensation reactions
The conversion and reaction selectivity of binary mixtures of acetaldehyde, acetone and butanone MAC reactions were analyzed over the same powder TiO2 catalyst at 300 °C, 350 °C and 400 °C to understand how reactions with mixed feeds differ from reactions with pure feeds. Conversion of each molecule in the binary mixtures followed the same general trend as the single component reactions: acetaldehyde > acetone > butanone. However, some novel trends related to competitive reactivity emerged in the binary mixtures. In both binary mixtures containing acetaldehyde, the conversion of acetaldehyde was generally enhanced compared to the pure component reaction, while the conversion of the second component, either acetone or butanone, was suppressed (Fig. 2).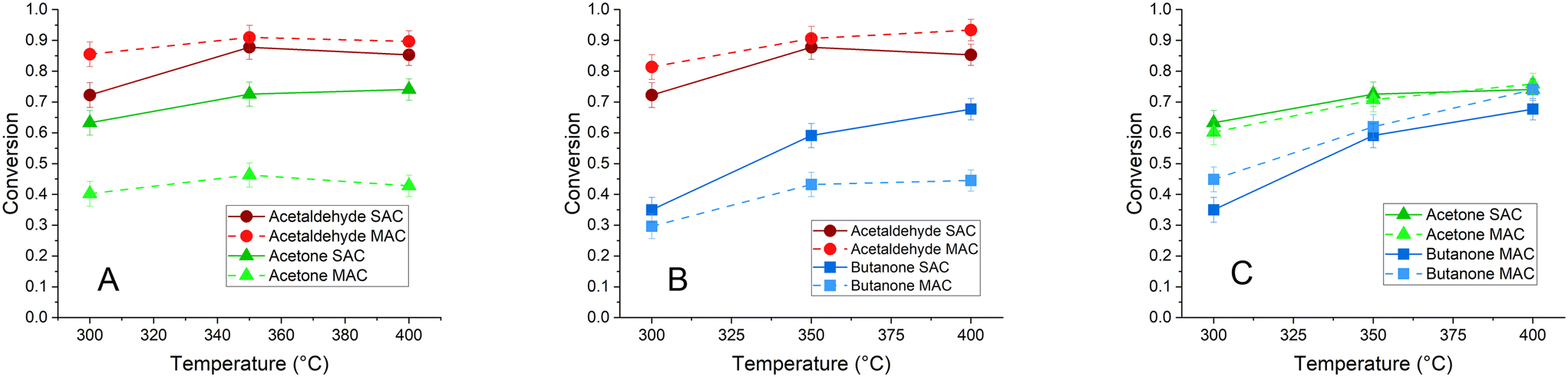 | ||
| Fig. 2 Conversion for binary, equimolar mixtures containing acetaldehyde and acetone (A), acetaldehyde and butanone (B) and acetone and butanone (C) plotted as a function of temperature. Conversions of each component in the binary mixture (dotted lines) are plotted alongside SAC reaction conversions (solid lines) for comparison. | ||
This result indicates that acetaldehyde tends to outcompete the larger, more substituted alkanones for surface active sites when the components are present at equal partial pressures in the gas phase. These high acetaldehyde conversions also suggest that the product mixture in both binary mixtures should be enriched in acetaldehyde-derived compounds. The primary identified product for acetaldehyde/acetone condensation was 3-penten-2-one. This C5 molecule is a direct cross-condensation product, which is formed when acetaldehyde reacts with enolized acetone adsorbed to the catalyst surface. Most of the other identified components in the product stream were compounds derived from acetaldehyde SAC, such as crotonaldehyde and 2-methyl-2-cyclopenten-1-one.
Ternary mixed feed reactions of acetaldehyde, acetone, and butanone over TiO2 were investigated to understand reactant mixtures relevant for downstream CFP processing. Ternary mixed feed reactions were examined using molar ratios of each component that have been reported in previous research during catalytic fast pyrolysis of biomass.33 The calculated molar ratios for this feedstock were 5.7:4.7:1 corresponding to reactant partial pressures of 7.3, 6.0 and 1.3 mbar for acetaldehyde, acetone and butanone, respectively. Conversion for this reaction is plotted as a function of reaction temperature in Fig. 3. The product mixture was highly complex, as described in further detail below, but selectivities of measurable products are shown in Fig. S15.† The product stream contained a complex mixture of condensation products, resulting in the relatively high selectivity to C5+ products (identified products are listed in the ESI†). The primary identified reaction products for this reaction were the acetaldehyde SAC derived 2-methyl-2-cyclopenten-1-one and the acetaldehyde and acetone cross condensation product 3-penten-2-one. Conversion of acetaldehyde was increased in the ternary system compared to the SAC system, while under most conditions the conversion of acetone and butanone was lower. This again suggests acetaldehyde outcompeting the ketones for surface reaction sites. More details on the ternary reaction system, including testing of an equimolar ternary composition, are described in the ESI.†
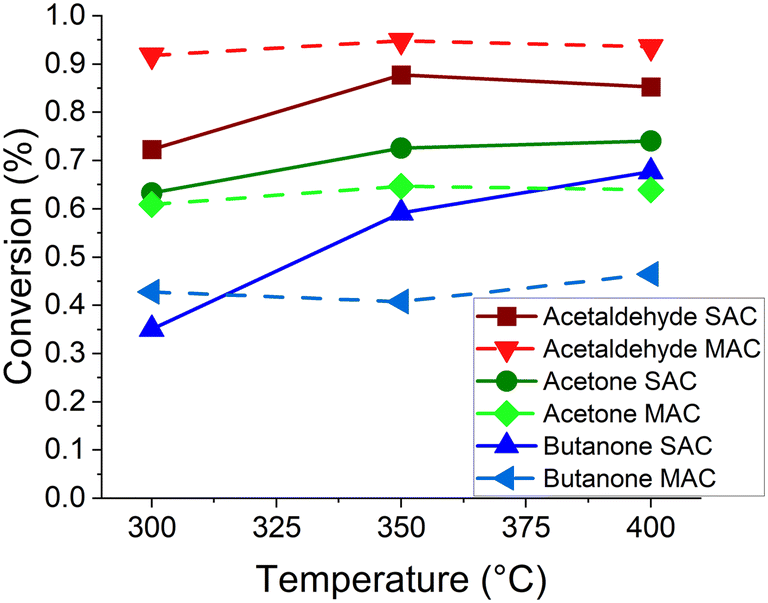 | ||
| Fig. 3 Reaction results of ternary mixtures of acetaldehyde, acetone and butanone at previously reported molar feed ratios observed during catalytic fast pyrolysis of biomass (5.7:4.7:1 for acetaldehyde:acetone:butanone). Conversions of each component in the ternary mixture (dotted lines) are plotted alongside pure-feed SAC reaction conversions (solid lines). | ||
Self-aldol condensation kinetic studies
To further investigate these trends in reactivity and selectivity, we investigated self- and cross-aldol condensation for acetaldehyde, acetone and butanone under low temperature conditions. Studying these reactions at low temperatures allowed us to focus on the initial coupling steps to better understand the drivers of mixture reactivity. Reaction experiments were initially conducted to analyze the kinetic trends for SAC reactions in which only a single oxygenated component was in the feed. Under the conditions of differential conversion (<10%) used here, the dominant SAC products were dimers: crotonaldehyde was produced from acetaldehyde condensation, mesityl oxide was produced from acetone condensation and various C8 oxygenates were produced from 2-butanone condensation. Higher molecular weight signals from multiple aldol condensation steps were detected in all cases but represented a minimal percentage (<15%) of the total product peak area at near-steady state reaction conditions. All trends reported below are based on reaction rates calculated at 3 hours on stream for the primary single condensation product; however, general trends for total condensation rates were similar throughout the 3 hour reaction time, given the high selectivity to single condensation products (Fig. S4 and S5†). Nevertheless, the catalyst deactivation caused by product accumulation complicates efforts to quantitatively assess kinetics, as discussed below. Our focus is therefore on using temperature and pressure dependencies to understand basic differences in reactivities of the various carbonyl compounds.Results from the SAC temperature series experiments (Fig. S1†) show similar activities for acetaldehyde and acetone at higher temperatures, with acetaldehyde reactivity dropping more severely as temperature decreased. Butanone was less reactive at the high end of the temperature range, however the rate of reaction was relatively insensitive to the drop in temperature. These trends in sensitivity to temperature change are characterized by acetaldehyde having the highest apparent activation energy under these conditions at 34 ± 3 kJ mol−1 (Fig. 4A), similar to the 37 kJ mol−1 value reported in previous work by Rekoske and Barteau.41 The apparent barriers for acetone and butanone were found to be very low, 10 ± 2 kJ mol−1 and 7 ± 4 kJ mol−1, respectively (Fig. 4A and S6†). Such low apparent barriers may indicate that increases in temperature lead to a decrease in active site availability, likely due to the heavier products produced from coupling reactions. A recent report utilizing single-facet dominant TiO2 (101) and (001) found activation energies close to ∼50 kJ mol−1.26 These studies were conducted with a Cu/SiO2 cocatalyst that was used to mitigate accumulation of heavy products through the saturation of product compounds.
 | ||
| Fig. 4 (A) Arrhenius plot for SAC of acetaldehyde and acetone. Reaction rates calculated based on crotonaldehyde (acetaldehyde dimer) and mesityl oxide (acetone dimer). SAC activation barriers are 34 ± 3 kJ mol−1 and 10 ± 2 kJ mol−1 for acetaldehyde and acetone condensation respectively. (B) Reaction order plots for acetaldehyde and acetone. Reaction rates calculated based on crotonaldehyde and mesityl oxide. Reaction orders are 0.5 ± 0.2 and 1.09 ± 0.01 for acetaldehyde and acetone condensation respectively. | ||
Experiments were conducted across a range of reactant pressures to determine the reaction order and to probe the kinetic relevance of the carbon–carbon coupling step. Results from the SAC reaction order experiments showed fractional reaction orders for acetaldehyde condensation and approximately first-order dependence for acetone and butanone condensation (0.5 ± 0.2 for acetaldehyde, 1.089 ± 0.008 for acetone and 1.1 ± 0.2 for butanone) (Fig. 4B and S7†). The SAC reaction orders reported in this study have relatively good agreement with those reported in literature.28 Young et al. reported near first-order reaction rates for acetaldehyde condensation on TiO2 at 553 K with acetaldehyde partial pressures under 10 kPa. As acetaldehyde pressure increased, the study reported deviation toward fractional reaction orders.28 Notably, near second order reaction rates for SAC reported previously were for reaction mixtures that included spectator species, such as the SAC of acetone co-fed with vapor phase isopropyl alcohol, where isopropyl alcohol derived species were expected to cover a substantial fraction of the surface.26
While most of the literature agrees on the general form of the aldol condensation mechanism, there is less consensus on which of the mechanistic steps is kinetically relevant. A study conducted by Zhao et al. on the SAC of valeraldehyde on TiO2 found that the carbon–carbon coupling step was the most probable rate determining step.27 Conversely, studies on the SAC of acetaldehyde over various metal oxide catalysts have determined that reactant adsorption and product desorption were most likely the kinetically relevant steps due to the absence of significant kinetic isotope effects and because of fitted values for adsorption and desorption rate constants.17,26,28,30 There is also literature evidence for the kinetic relevance of the enolate formation step, as shown through DFT analysis of acetone SAC on TiO2 by Lin et al., and through combined kinetic, isotopic and theoretical analysis of aldol condensation on TiO2 by Wang and Iglesia.19,26 Such a wide range of conclusions points toward reaction conditions having a major effect on measured reaction kinetics.
As a partial probe of kinetically relevant steps, kinetic isotope effect (KIE) experiments were conducted by comparing reaction rates for the SAC of acetone and deuterated acetone to probe the kinetic relevance of the enolate formation step. The acetone self-condensation reaction showed a weak KIE of 1.13 ± 0.02 at 200 °C, indicating that enolate formation is likely not kinetically relevant for the acetone SAC reaction. Similar KIE experiment results were reported in other studies on aldol condensation on metal oxide catalysts and other relevant materials. A study conducted by Young et al. on the aldol condensation of acetaldehyde reported a kinetic isotope effect of 0.98 on TiO2 and 1.1 on hydroxyapatite catalysts.28 Additional evidence for weak kinetic isotope effects were also reported for acetone condensation on TiO2 single crystals26 and for acetaldehyde condensation on TiO2 in the presence of ethanol.30
Acetaldehyde and acetone mixed-aldol condensation reactions
We also investigated the aldol condensation of mixed feeds containing acetaldehyde and acetone. Under the same temperatures and reactant partial pressures as were used in the self-condensation experiments, only two major products were detected: the acetaldehyde self-condensation product, crotonaldehyde, and the cross-condensation product 3-penten-2-one. 3-Penten-2-one is the product formed when an adsorbed acetone molecule forms the enolate that reacts with a vicinal acetaldehyde molecule (Fig. S8†). Interestingly, no detectable production of the acetone self-condensation product mesityl oxide or the alternate cross-condensation product 3-methyl-2-butenal was observed, indicating that acetone was ineffective in serving as an electrophile in the mixed feed.Unexpectedly, we recorded a large difference in reaction rates between crotonaldehyde (acetaldehyde SAC) and 3-penten-2-one (acetaldehyde and acetone CAC), despite partial pressures of acetaldehyde and acetone in the system being almost identical within error. At 200 °C the acetaldehyde self-condensation reaction was 11 ± 2 times more rapid than the cross-condensation of acetaldehyde and acetone. Preferential selectivity to crotonaldehyde suggests that acetaldehyde outcompetes acetone for access to active sites and is highly effective as both an electrophile and an enolate former.
Kinetic studies also show profound differences between the SAC and MAC systems. Shown in Fig. 5, the apparent activation energies for production of crotonaldehyde and 3-pentenone were 42 ± 5 kJ mol−1 and 44 ± 12 kJ mol−1, respectively. The large increase in the apparent activation energy for acetone condensation is attributed to the suppressed formation of acetone-derived heavy products, which deactivate the catalyst at higher temperature, during MAC. The reaction order for crotonaldehyde production with respect to acetone was measured to be 0.01 ± 0.03. The insensitivity of the acetaldehyde self-condensation reaction rate on acetone concentration indicates that any surface coverage effects from changing acetone concentration have no effect on acetaldehyde self-condensation. This is evidence that acetaldehyde is more abundant on the catalyst surface, such that increases in acetone concentration do not substantially lower the surface coverage of acetaldehyde. The reaction order for 3-penten-2-one formation with respect to acetone was measured to be 1.4 ± 0.2. This high reaction order with respect to acetone indicates that some interaction between acetone molecules contributed to the rate aside from the reaction between the acetone and acetaldehyde molecule. This interaction could come in the form of relatively stabilizing interactions with vicinal acetone molecules.
 | ||
| Fig. 5 (A) Arrhenius plot for the cross-aldol condensation reaction between acetaldehyde and acetone. Activation energies calculated for the two products with the highest selectivity, crotonaldehyde (acetaldehyde SAC product) and 3-penten-2-one (one of two possible CAC products). (B and C) Reaction order with respect to acetone (B) and acetaldehyde (C) for the CAC reaction between acetaldehyde and acetone. Reaction orders calculated for the two products with the highest selectivity, crotonaldehyde (acetaldehyde SAC product) and 3-penten-2-one (one of two possible CAC products). | ||
Reaction orders measured for acetaldehyde in the MAC system support the kinetic relevance of acetone for the cross-condensation reaction and point toward acetone having a small effect on acetaldehyde self-condensation chemistry. The reaction order for crotonaldehyde formation with respect to acetaldehyde in the mixed system was measured to be 0.64 ± 0.08, almost within error of the value for the pure feed system. This result again indicates that acetaldehyde condensation is minimally affected by the presence of acetone. The acetaldehyde order for MAC was measured to be −0.62 ± 0.03 (Fig. 5C), again reflecting the suppression of this reaction as acetaldehyde surface coverage is increased.
Temperature programmed desorption
TPD experiments were used to probe the differences in desorption energy of acetaldehyde, acetone, and crotonaldehyde with the catalyst surface. Acetaldehyde TPD showed two peaks in the m/z = 29 signal, with one relatively sharp peak occurring near 130 °C and one broader peak at high temperature. Tracking of other mass signals beyond m/z = 29 indicate that the high-temperature peak consists of a mixture of products, including coupling products (Fig. S18†). In contrast, acetone exhibited only a low temperature peak near 110 °C on TiO2. Adsorption energies for acetaldehyde and acetone were calculated to be 115 kJ mol−1 and 110 kJ mol−1 using the low temperature peaks.42 More importantly, the high temperature desorption feature(s) during acetaldehyde TPD, presumably originating from sites that bind acetaldehyde strongly, indicates a population of acetaldehyde adsorbates that are bound much more strongly than acetone-derived species. Crotonaldehyde TPD showed intact crotonaldehyde desorption at around 115 °C. Similar desorption temperatures between the reactants and products indicate that product desorption is likely not a rate-limiting step for this reaction. Again, however, crotonaldehyde produced a high-temperature peak that indicated a complex product mixture (Fig. S19†), indicating a population of sites that strongly adsorb crotonaldehyde (Fig. 6). | ||
| Fig. 6 TPD experiments of acetone, acetaldehyde, and crotonaldehyde on TiO2. The first desorption peaks of the black and blue traces are attributed to acetaldehyde and crotonaldehyde desorption respectively. The magnitude of the crotonaldehyde trace is multiplied by a factor of 10 for clarity. | ||
The TPD results presented in this work are consistent with differences in adsorption energy and adsorption mechanisms between acetaldehyde and acetone calculated using DFT. It has been reported that adsorption energy between the acetaldehyde carbonyl group and surface Ti5C atoms are much stronger than the adsorption energy between the acetone carbonyl group and surface Ti5C atoms (220 kJ mol−1 and 80 kJ mol−1 respectively).43 Higher energies of adsorption were also calculated for acetaldehyde across the other binding modes which involve interaction between surface O atoms and hydrogen atoms present in the reactant. Acetaldehyde and acetone TPD experiments from other studies have also recorded differences in surface coverage and reactivity between the two molecules.21 These studies have also shown acetone to desorb almost completely intact, while acetaldehyde is more likely to form large condensation products.21,44,45
Pyridine DRIFTS
Pyridine DRIFTS was used to probe the distribution of Lewis and Brønsted acid sites on the catalyst surface and the role of each of these sites during the aldol condensation reaction. The specific IR adsorption bands corresponding to Lewis acid–pyridine complexes, formed when dosed pyridine interacts with Lewis acid sites, are typically centered in the range 1440–1465 cm−1, while the bands corresponding to the pyridinium ion, formed from Brønsted acid site interaction with pyridine, are typically centered around 1540–1545 cm−1.46–48Fig. 7 shows the in situ pyridine DRIFTS spectra of TiO2 in the 1400–1700 cm−1 region. The DRIFT spectrum from fresh TiO2 after reductive pretreatment (Fig. 7.i) has bands corresponding to pyridine bound to Lewis acid sites at 1444, 1573 and 1603 cm−1 and a band at 1491 cm−1 that is attributed to both Lewis and Brønsted acid sites.49,50 The absence of a band in the 1540–1545 cm−1 region suggests that there is a low density of Brønsted acid sites present in this sample.46,51 The spectrum from TiO2 used in a 3 hour, equimolar acetaldehyde and acetone ex situ MAC reaction at 200 °C is shown in Fig. 7.iv. The intensity of all observed peaks is much lower than those observed in the fresh sample, indicating less availability of active sites after reaction. This is likely due to a buildup of carbonaceous species on Lewis acid sites. It should be noted that as MAC reactions were conducted ex situ, and the in situ pretreatment differed from that of the fresh TiO2. Consequently, the conclusions drawn here are primarily qualitative. Pyridine DRIFTS was also done on TiO2 samples that were dosed in situ with acetone (Fig. 7.ii) and acetaldehyde (Fig. 7.iii). These exposures were for one hour at 200 °C to simulate SAC reaction conditions. After dosing the catalyst with acetaldehyde (Fig. 7.iii), the intensity of all bands decreased significantly, resulting in a similar spectrum to the spent sample (Fig. 7.iv). After dosing the catalyst with acetone (Fig. 7.ii), peak intensity also decreased, however the reduction in peak intensity was much less pronounced than that of the spent, or acetaldehyde-dosed sample.
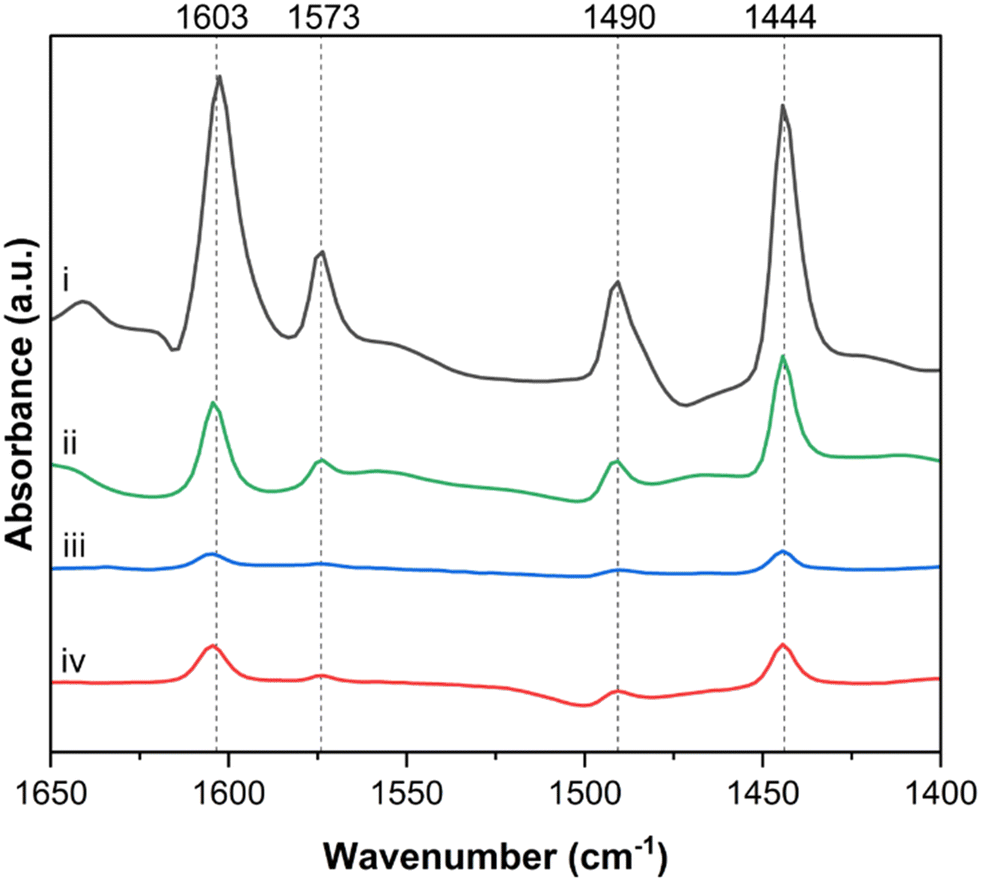 | ||
| Fig. 7 Pyridine DRIFTS spectra for i. fresh TiO2, ii. TiO2 after adsorption of acetone, iii. TiO2 after adsorption of acetaldehyde, and iv. spent TiO2. | ||
To quantify the observed decrease in band intensities, spectra ii, iii, and iv were normalized by the intensity of the 1444 cm−1 band from the fresh TiO2 sample in spectra i. As observed in Fig. S14† the 1444 cm−1 band decreased by a factor of 16.6 ± 1.7 after acetaldehyde exposure (Fig. S14.iii†), while only decreasing by a factor of 2.7 ± 0.2 after acetone exposure (Fig. S14.ii†). This difference in pyridine adsorption on TiO2 after dosing with acetaldehyde and acetone could be due to the ability of pyridine to displace acetone more readily than acetaldehyde. This trend agrees with the TPD results reported in this work which show that acetaldehyde has a higher energy of desorption than acetone on TiO2.
Discussion
Analysis of selectivity trends and reaction kinetics for SAC and MAC systems shows that the presence of acetaldehyde in a mixed-feed reaction had a major impact on the reactivity of acetone. Results from both the high- and low-conversion studies show that in mixed systems, acetaldehyde conversion increased, while acetone conversion was suppressed relative to pure-component feed reactions. Acetaldehyde appeared to impact the reaction kinetics of acetone through two effects: (i) competition for adsorption on the catalyst surface, and (ii) preferential activation of acetaldehyde as an electrophilic species. In other words, acetaldehyde appeared to outcompete acetone in formation/reaction of both enolate and electrophile species, but its suppression of acetone as an electrophile was much more severe. This results in the shifting of reaction selectivity toward crotonaldehyde and 3-penten-2-one, and away from mesityl oxide and 3-methyl-2-butenal, in the low-temperature reaction studies. Selectivity trends between crotonaldehyde and 3-penten-2-one, the two major products from the MAC of acetaldehyde and acetone under these conditions would be expected to be controlled by relative coverages of the enolates formed from acetaldehyde and acetone or differences in the energetic barrier for coupling of those enolates with an acetaldehyde electrophile. The observation that the measured apparent activation energies for crotonaldehyde and 3-penten-2-one formation were within error of one another points toward differences in surface coverage being the driving factor for higher selectivity to crotonaldehyde over 3-penten-2-one.In binary mixed aldol systems, there is literature evidence for the suppression of alkanone self-condensation in favor of the cross condensation reaction between alkanal and alkanone reactants,18–20,22,32 which is consistent with the results presented in this paper. Several of these studies attribute the suppression of alkanone self-condensation and sole activation of the alkanal as the electrophilic species to a balance of enthalpic and entropic effects related to overall molecule size and the nature of the substituents on the carbonyl carbon.14–16,19,20 The enthalpic effects include the influence of repulsive forces such as steric hindrance and attractive forces such as van der Waals interaction between molecules adsorbed on the catalyst surface.19,20 The entropic effects, specifically for molecules in the C1–C3 range, are primarily governed by the magnitude of entropy changes upon the formation of the transition state during the nucleophilic attack of an enolate on an electrophile.20
Applying these findings to the current study, the acetaldehyde carbonyl group likely has a lower energetic barrier for aligning with an adsorbed enolate species than the acetone carbonyl group due to acetaldehyde having a terminal carbonyl group and a smaller tail group. Additionally, the carbonyl carbon of acetaldehyde is more electrophilic than that of acetone due to the lack of stabilizing electron density from α-methyl groups attached to the carbonyl group.52 This combination of enthalpic and entropic effects results in acetaldehyde acting as the sole electrophile in this system in the low-temperature reaction study, and likely explains why acetaldehyde is consumed to a greater extent at higher temperatures and conversions.
The effect of acetaldehyde on the activity of alkanones in mixed feed aldol condensation reactions is attributed in large part to the relatively high affinity of acetaldehyde for the catalyst surface, and to the preferential activation of acetaldehyde as an electrophile. At low temperatures, acetaldehyde likely occupies a relatively large percentage of the active sites on the catalyst surface, leading to the high selectivity of crotonaldehyde. At high temperatures, the difference in surface coverage may be less pronounced. Under these conditions, the sole action of acetaldehyde as the electrophile results in reaction selectivity being dominated by cross condensation to products where acetaldehyde undergoes nucleophilic attack by adsorbed enolates. As acetaldehyde acts as at least one half of each condensation reaction, its conversion is enhanced while the conversion of the other reactants in the feed is suppressed. These effects are observed both at low temperatures and conversions and at the much higher temperatures and conversions that are most relevant for upgrading of oxygenates present in CFP vapors. Design of reactor units for application will thus need to consider that acetaldehyde consumption will occur at earlier residence times, with more substantial incorporation of acetaldehyde into coupled products than acetone and butanone.
Conclusion
This study was aimed at understanding interaction effects among mixtures of light oxygenates relevant for the upgrading of catalytic fast pyrolysis vapors over TiO2 catalysts. Kinetic analysis of self-aldol condensation reactions with acetaldehyde and acetone showed a lower reaction order for acetaldehyde, indicating that acetaldehyde has higher surface coverage under these conditions. This conclusion was supported by TPD results showing that acetaldehyde was bound more strongly to the catalyst surface and more readily participated in coupling reactions. Similar desorption temperatures between products and reactants as well as the absence of a significant kinetic isotope effect from the use of deuterated acetone in the SAC regime suggest that reactant adsorption, product desorption and enolate formation were not kinetically relevant, leaving C–C bond formation as the most likely rate determining step for this reaction under these conditions. Experiments conducted at high temperatures and high conversions showed that in mixed feeds, acetaldehyde suppressed the activity of acetone and butanone. Kinetic analysis of the acetaldehyde/acetone cross-condensation reaction showed a negative reaction order for acetaldehyde; given the observation of high selectivity for 3-penten-2-one as the mixed aldol product, this suggests that acetone reaction was limited by competition with acetaldehyde for vacant enolate adsorption sites.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding for this research, provided by the U.S. Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy (EERE), Bioenergy Technologies Office (BETO) under award number EE0008921. This work was performed in collaboration with the Chemical Catalysis for Bioenergy Consortium (ChemCatBio), a member of the Energy Materials Network, at Oak Ridge National Laboratory (ORNL) under Contract No. DE-AC05-00OR22725, and at the National Renewable Energy Laboratory (NREL) under Contract No. DE-AC36-08-GO28308.References
- Environmental Protection Agency, Inventory of U.S Greenhouse Gas Emissions and Sinks: 1990-2011, 2019 Search PubMed.
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, The Role of Renewable Energy in the Global Energy Transformation, Energy Strategy Reviews, 2019, 24, 38–50, DOI:10.1016/j.esr.2019.01.006.
- N. A. Huq, G. R. Hafenstine, X. Huo, H. Nguyen, S. M. Tifft, D. R. Conklin, D. Stück, J. Stunkel, Z. Yang, J. S. Heyne, M. R. Wiatrowski, Y. Zhang, L. Tao, J. Zhu, C. S. McEnally, E. D. Christensen, C. Hays, K. M. Van Allsburg, K. A. Unocic, H. M. Meyer, Z. Abdullah and D. R. Vardon, Toward Net-Zero Sustainable Aviation Fuel with Wet Waste–Derived Volatile Fatty Acids, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(13), e2023008118, DOI:10.1073/pnas.2023008118.
- IATA, Sustainable Avaition Fuel Roadmap, 2015.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Catalytic Conversion of Biomass to Biofuels, Green Chem., 2010, 12(9), 1493, 10.1039/c004654j.
- D. A. Ruddy, J. A. Schaidle, J. R. Ferrell Iii, J. Wang, L. Moens and J. E. Hensley, Recent Advances in Heterogeneous Catalysts for Bio-Oil Upgrading via “Ex Situ Catalytic Fast Pyrolysis”: Catalyst Development through the Study of Model Compounds, Green Chem., 2014, 16(2), 454–490, 10.1039/C3GC41354C.
- E. Furimsky, Catalytic Hydrodeoxygenation, Appl. Catal., A, 2000, 199(2), 147–190, DOI:10.1016/S0926-860X(99)00555-4.
- M. H. Langholtz, B. J. Stokes and M. L. Eaton, 2016 Billion-Ton Report: Advancing Domestic Resources for a Thriving Bioeconomy, Volume 1: Economic Availability of Feedstocks, Oak Ridge National Lab, U.S. Department of Energy, Oak Ridge, TN, 2016 Search PubMed.
- J. C. Serrano-Ruiz and J. A. Dumesic, Catalytic Routes for the Conversion of Biomass into Liquid Hydrocarbon Transportation Fuels, Energy Environ. Sci., 2011, 4(1), 83–99, 10.1039/C0EE00436G.
- R. G. Grim, A. T. To, C. A. Farberow, J. E. Hensley, D. A. Ruddy and J. A. Schaidle, Growing the Bioeconomy through Catalysis: A Review of Recent Advancements in the Production of Fuels and Chemicals from Syngas-Derived Oxygenates, ACS Catal., 2019, 9(5), 4145–4172, DOI:10.1021/acscatal.8b03945.
- W. Shen, G. A. Tompsett, R. Xing, W. Curtis Conner and G. W. Huber, Vapor Phase Butanal Self-Condensation over Unsupported and Supported Alkaline Earth Metal Oxides, J. Catal., 2012, 286, 248–259, DOI:10.1016/j.jcat.2011.11.009.
- W. Ji, Y. Chen and H. H. Kung, Vapor Phase Aldol Condensation of Acetaldehyde on Metal Oxide Catalysts, Appl. Catal., A, 1997, 161(1–2), 93–104, DOI:10.1016/S0926-860X(96)00390-0.
- V. V. Ordomsky, V. L. Sushkevich and I. I. Ivanova, Study of Acetaldehyde Condensation Chemistry over Magnesia and Zirconia Supported on Silica, J. Mol. Catal. A: Chem., 2010, 333(1–2), 85–93, DOI:10.1016/j.molcata.2010.10.001.
- G. Li, B. Wang and D. E. Resasco, Oxide-Catalyzed Self- and Cross-Condensation of Cycloketones. Kinetically Relevant Steps That Determine Product Distribution, J. Catal., 2020, 391, 163–174, DOI:10.1016/j.jcat.2020.07.019.
- P. Mäki-Arvela, N. Shcherban, C. Lozachmeur, K. Eränen, A. Aho, A. Smeds, N. Kumar, J. Peltonen, M. Peurla, V. Russo, K. P. Volcho and D. Yu. Murzin, Aldol Condensation of Cyclopentanone with Valeraldehyde Over Metal Oxides, Catal. Lett., 2019, 149(5), 1383–1395, DOI:10.1007/s10562-019-02701-1.
- W. An, The Role of Synergic Interaction in Transition State Formation for the Aldol Reaction on a Metal Oxide Catalyst: A DFT Investigation, Phys. Chem. Chem. Phys., 2015, 17(35), 22529–22532, 10.1039/C5CP03478G.
- S. Bhasker-Ranganath, Md. S. Rahman, C. Zhao, F. Calaza, Z. Wu and Y. Xu, Elucidating the Mechanism of Ambient-Temperature Aldol Condensation of Acetaldehyde on Ceria, ACS Catal., 2021, 11(14), 8621–8634, DOI:10.1021/acscatal.1c01216.
- M. Ai, The Production of Methyl Vinyl Ketone by the Vapor-Phase Aldol Condensation between Formaldehyde and Acetone, J. Catal., 1987, 106(1), 273–279, DOI:10.1016/0021-9517(87)90231-4.
- S. Wang and E. Iglesia, Substituent Effects and Molecular Descriptors of Reactivity in Condensation and Esterification Reactions of Oxygenates on Acid–Base Pairs at TiO 2 and ZrO 2 Surfaces, J. Phys. Chem. C, 2016, 120(38), 21589–21616, DOI:10.1021/acs.jpcc.6b07304.
- S. Wang and E. Iglesia, Entropy-Driven High Reactivity of Formaldehyde in Nucleophilic Attack by Enolates on Oxide Surfaces, J. Am. Chem. Soc., 2018, 140(2), 775–782, DOI:10.1021/jacs.7b11749.
- S. Luo and J. L. Falconer, Acetone and Acetaldehyde Oligomerization on TiO2 Surfaces, J. Catal., 1999, 185(2), 393–407, DOI:10.1006/jcat.1999.2511.
- Y. Zhao, X. Zhu, H. Wang, J. Han, D. Mei and Q. Ge, Aqueous Phase Aldol Condensation of Formaldehyde and Acetone on Anatase TiO2 (101) Surface: A Theoretical Investigation, ChemCatChem, 2020, 12(4), 1220–1229, DOI:10.1002/cctc.201901736.
- B. I. Stefanov, Z. Topalian, C. G. Granqvist and L. Österlund, Acetaldehyde Adsorption and Condensation on Anatase TiO2: Influence of Acetaldehyde Dimerization, J. Mol. Catal. A: Chem., 2014, 381, 77–88, DOI:10.1016/j.molcata.2013.10.005.
- M. Singh, N. Zhou, D. K. Paul and K. J. Klabunde, IR Spectral Evidence of Aldol Condensation: Acetaldehyde Adsorption over TiO2 Surface, J. Catal., 2008, 260(2), 371–379, DOI:10.1016/j.jcat.2008.07.020.
- S. Wang, K. Goulas and E. Iglesia, Condensation and Esterification Reactions of Alkanals, Alkanones, and Alkanols on TiO2: Elementary Steps, Site Requirements, and Synergistic Effects of Bifunctional Strategies, J. Catal., 2016, 340, 302–320, DOI:10.1016/j.jcat.2016.05.026.
- F. Lin, H. Wang, Y. Zhao, J. Fu, D. Mei, N. R. Jaegers, F. Gao and Y. Wang, Elucidation of Active Sites in Aldol Condensation of Acetone over Single-Facet Dominant Anatase TiO 2 (101) and (001) Catalysts, JACS Au, 2021, 1(1), 41–52, DOI:10.1021/jacsau.0c00028.
- L. Zhao, H. An, X. Zhao and Y. Wang, TiO2-Catalyzed n-Valeraldehyde Self-Condensation Reaction Mechanism and Kinetics, ACS Catal., 2017, 7(7), 4451–4461, DOI:10.1021/acscatal.7b00432.
- Z. D. Young, S. Hanspal and R. J. Davis, Aldol Condensation of Acetaldehyde over Titania, Hydroxyapatite, and Magnesia, ACS Catal., 2016, 6(5), 3193–3202, DOI:10.1021/acscatal.6b00264.
- C. Zhao, C. Watt, P. R. Kent, S. H. Overbury, D. R. Mullins, F. C. Calaza, A. Savara and Y. Xu, Coupling of Acetaldehyde to Crotonaldehyde on CeO2–x (111): Bifunctional Mechanism and Role of Oxygen Vacancies, J. Phys. Chem. C, 2019, 123(13), 8273–8286, DOI:10.1021/acs.jpcc.8b08535.
- H. Zhang, M. Y. S. Ibrahim and D. W. Flaherty, Aldol Condensation among Acetaldehyde and Ethanol Reactants on TiO2: Experimental Evidence for the Kinetically Relevant Nucleophilic Attack of Enolates, J. Catal., 2018, 361, 290–302, DOI:10.1016/j.jcat.2018.02.030.
- S. Herrmann and E. Iglesia, Elementary Steps in Acetone Condensation Reactions Catalyzed by Aluminosilicates with Diverse Void Structures, J. Catal., 2017, 346, 134–153, DOI:10.1016/j.jcat.2016.12.011.
- L. Hora, V. Kelbichová, O. Kikhtyanin, O. Bortnovskiy and D. Kubička, Aldol Condensation of Furfural and Acetone over MgAl Layered Double Hydroxides and Mixed Oxides, Catal. Today, 2014, 223, 138–147, DOI:10.1016/j.cattod.2013.09.022.
- A. Dutta, K. Lisa, M. Talmadge, C. Mukarakate, M. Griffin, E. Tan and N. Wilson, Ex Situ Catalytic Fast Pyrolysis of Lignocellulosic Biomass to Hydrocarbon Fuels: 2019 State of Technology and Future Research, Technical Report TP-5100-76269, National Renewable Energy Laboratory, 2020, https://www.nrel.gov/docs/fy20osti/76269.pdf Search PubMed.
- NIST WebBook, https://webbook.nist.gov/.
- S. Luo and J. L. Falconer, Aldol Condensation of Acetaldehyde to Form High Molecular Weight Compounds on TiO2, Catal. Lett., 1999, 57(3), 89–93, DOI:10.1023/A:1019003817314.
- H. Idriss and M. A. Barteau, Selectivity and Mechanism Shifts in the Reactions of Acetaldehyde on Oxidized and Reduced TiO2(001) Surfaces, Catal. Lett., 1996, 40(3–4), 147–153, DOI:10.1007/BF00815275.
- L. Kubelkovå, J. Čjka, J. Nováková, V. Boszácek, I. Jirka and P. Jíaru, Acetone Conversion and Deactivation of Zeolites, in Studies in Surface Science and Catalysis, Elsevier, 1989, vol. 49, pp. 1203–1212, DOI:10.1016/S0167-2991(08)62006-6.
- S. Lippert, W. Baumann and K. Thomke, Secondary Reactions of the Base-Catalyzed Aldol Condensation of Acetone, J. Mol. Catal., 1991, 69(2), 199–214, DOI:10.1016/0304-5102(91)80145-S.
- J. Abbot and P. R. Dunstan, Catalytic Cracking of Linear Paraffins: Effects of Chain Length, Ind. Eng. Chem. Res., 1997, 36(1), 76–82, DOI:10.1021/ie960255e.
- J. S. Buchanan, J. G. Santiesteban and W. O. Haag, Mechanistic Considerations in Acid-Catalyzed Cracking of Olefins, J. Catal., 1996, 158(1), 279–287, DOI:10.1006/jcat.1996.0027.
- J. E. Rekoske and M. A. Barteau, Kinetics, Selectivity, and Deactivation in the Aldol Condensation of Acetaldehyde on Anatase Titanium Dioxide, Ind. Eng. Chem. Res., 2011, 50(1), 41–51, DOI:10.1021/ie100394v.
- P. A. Redhead, Thermal Desorption of Gases, Vacuum, 1962, 12(4), 203–211, DOI:10.1016/0042-207X(62)90978-8.
- M. Yao, Y. Ji, H. Wang, Z. Ao, G. Li and T. An, Adsorption Mechanisms of Typical Carbonyl-Containing Volatile Organic Compounds on Anatase TiO 2 (001) Surface: A DFT Investigation, J. Phys. Chem. C, 2017, 121(25), 13717–13722, DOI:10.1021/acs.jpcc.7b02964.
- S. A. Larson, J. A. Widegren and J. L. Falconer, Transient Studies of 2-Propanol Photocatalytic Oxidation on Titania, J. Catal., 1995, 157(2), 611–625, DOI:10.1006/jcat.1995.1326.
- D. Brinkley and T. Engel, Photocatalytic Dehydrogenation of 2-Propanol on TiO 2 (110), J. Phys. Chem. B, 1998, 102(39), 7596–7605, DOI:10.1021/jp981901l.
- B. H. Davis, R. A. Keogh, S. Alerasool, D. J. Zalewski, D. E. Day and P. K. Doolin, Infrared Study of Pyridine Adsorbed on Unpromoted and Promoted Sulfated Zirconia, J. Catal., 1999, 183(1), 45–52, DOI:10.1006/jcat.1998.2387.
- A. Platon and W. J. Thomson, Quantitative Lewis/Brönsted Ratios Using DRIFTS, Ind. Eng. Chem. Res., 2003, 42(24), 5988–5992, DOI:10.1021/ie030343g.
- Y. Wu, F. Gao, H. Wang, L. Kovarik, B. Sudduth and Y. Wang, Probing Acid–Base Properties of Anatase TiO 2 Nanoparticles with Dominant {001} and {101} Facets Using Methanol Chemisorption and Surface Reactions, J. Phys. Chem. C, 2021, 125(7), 3988–4000, DOI:10.1021/acs.jpcc.0c11107.
- A. P. Kulkarni and D. S. Muggli, The Effect of Water on the Acidity of TiO2 and Sulfated Titania, Appl. Catal., A, 2006, 302(2), 274–282, DOI:10.1016/j.apcata.2006.01.033.
- M. I. Zaki, M. A. Hasan, F. A. Al-Sagheer and L. Pasupulety, In Situ FTIR Spectra of Pyridine Adsorbed on SiO2–Al2O3, TiO2, ZrO2 and CeO2: General Considerations for the Identification of Acid Sites on Surfaces of Finely Divided Metal Oxides, Colloids Surf., A, 2001, 190(3), 261–274, DOI:10.1016/S0927-7757(01)00690-2.
- J. Lu, K. M. Kosuda, R. P. Van Duyne and P. C. Stair, Surface Acidity and Properties of TiO 2/SiO2 Catalysts Prepared by Atomic Layer Deposition: UV−visible Diffuse Reflectance, DRIFTS, and Visible Raman Spectroscopy Studies, J. Phys. Chem. C, 2009, 113(28), 12412–12418, DOI:10.1021/jp902200c.
- R. M. LoPachin and T. Gavin, Molecular Mechanisms of Aldehyde Toxicity: A Chemical Perspective, Chem. Res. Toxicol., 2014, 27(7), 1081–1091, DOI:10.1021/tx5001046.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01798b |
| This journal is © The Royal Society of Chemistry 2024 |