
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced photocatalytic activity of Fe@UiO-66 for aerobic oxidation of N-aryl tetrahydroisoquinolines†
Kai
Zhou
ab,
Yuanyuan
Zhang
ab,
Mingjie
Liu
ab,
Zhenghua
Zhao
ab,
Xiang
Liu
*b,
Zongbi
Bao
 ab,
Qiwei
Yang
ab,
Qilong
Ren
ab and
Zhiguo
Zhang
*ab
ab,
Qiwei
Yang
ab,
Qilong
Ren
ab and
Zhiguo
Zhang
*ab
aKey Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310058, P. R. China. E-mail: zhiguo.zhang@zju.edu.cn
bInstitute of Zhejiang University–Quzhou, Quzhou 324000, P. R. China. E-mail: liu-xiang@zju.edu.cn
First published on 2nd February 2024
Abstract
Highly dispersed iron oxide has been rapidly and precisely loaded onto the Zr6 nodes of a stable metal–organic framework (MOF), UiO-66, via an atomic layer deposition (ALD) process to form novel Fe–O–Zr metal cluster sites. By varying the number of ALD cycles, three Fe-decorated UiO-66 materials (denoted as Fe@UiO-66-xc, x = 1, 2, 3) were synthesized. A series of photoelectrochemical measurements, including UV-vis diffuse reflectance spectroscopy, state photoluminescence spectroscopy, and transient photocurrent response, indicate that Fe@UiO-66-1c prepared by a single deposition cycle exhibits improved visible-light absorption ability and enhanced photo-generated charge carrier separation efficiency due to metal-to-metal charge transfer. Moreover, Fe@UiO-66-1c shows excellent photocatalytic activity for aerobic oxidation of N-aryl tetrahydroisoquinolines. This material also exhibits good stability and is capable of cycling the reaction six times and maintaining the crystal structure with a low leaching rate of iron ions. The study explores the application of the atomic layer deposition process in the preparation of advanced photocatalytic materials.
1. Introduction
Photocatalysis, a prospective technology to convert renewable solar energy into chemical energy, shows great potential in a series of chemical reactions under mild conditions.1–3 For instance, visible-light-induced photocatalytic oxidation with dioxygen has been recently recognized as a green and feasible way to produce valuable oxygenated chemicals for the manufacture of agrochemicals and pharmaceuticals.4–10 Various types of catalysts, such as organic dyes, flavins, metal complexes, heterogeneous materials, and polyoxometalates, have been developed to generate reactive oxygen species for a wide range of photo-oxidation systems. However, these photocatalytic systems still face some challenges and limitations, such as low light utilization efficiency, low charge transfer rate at the surface, and low selectivity and stability due to catalyst degradation or poisoning. Moreover, the efficiency is still far from satisfactory mainly because of the difficulty of fully utilizing the photogenerated charges and reactive oxygen species. Therefore, a rational design of photogenerated charge transfer pathways with proper functionality may be a favourable approach to realize efficient artificial photosynthesis, yet this field is still at the early stage and tremendous efforts are needed to be made.Metal–organic frameworks (MOFs) are crystalline materials consisting of metallic nodes and organic linkers, which have recently emerged as a promising class of heterogeneous photocatalysts for visible-light-induced oxidations.11–15 The organic bridging ligands of MOFs can serve as antennas to harvest light and activate metal nodes, illustrating semiconductor-like behaviour and demonstrating great potential in activating dioxygen and promoting the oxidative process.16–18 However, most functional MOFs still suffer from low efficiency of light absorption, and charge separation and transfer, restricting their further development. A variety of strategies, such as metal doping, ligand exchange, and cavity encapsulation have been explored to circumvent the challenge of charge transfer and hence to improve the photocatalytic performance.19–23 Among them, introducing active metals as photoactive species seems to be the optimal candidate to modify MOFs.24–26 In particular, the introduction of metal clusters or atom-sized metals has been shown to not only maximize the utilization of metals, but also promote the charge separation and transfer for MOFs. For example, many types of metal single atoms (e.g., Cu, Fe, etc.) have been bonded into MOFs by solvothermal or microwave-assisted post-synthetic modification methods, which act as effective electron acceptors for spatial separation and transfer of charge carriers via the ligand–metal charge transfer (LMCT), and metal-to-metal charge transfer (MMCT) occurs upon the connection of two metal centers with different valence states via an oxygen bridge, leading to enhanced photocatalytic performance.27–29 Although there are great achievements on active metal modification of MOFs, more techniques for efficient deposition of the active metals onto MOFs are still expected to be established and utilized.
Atomic layer deposition (ALD) is a chemical vapor deposition technique that uses sequential pulses of precursor gases to react with the surface in a self-limiting manner. ALD is a unique method that offers precise control of atomic-scale thickness, excellent three-dimensional conformality, and large-area uniformity, making it increasingly popular for the direct synthesis and post-synthetic modification of advanced catalysts.30–36 For instance, controlling the precursors and adjusting the number of Fe ALD cycles enables the selective deposition of either Fe single atoms or an ultrathin Fe2O3 film onto the surface of TiO2.37,38 Omar Farha's group has loaded a variety of metals on Zr6-based MOF NU-1000 using ALD and found that the ALD reaction occurs at inorganic nodes.39–41 Herein, we first applied the ALD process to incorporate iron oxide onto the Zr6 nodes of UiO-66. Through controlling the vapor deposition cycle program, we successfully prepared a series of Fe@UiO-66 photocatalysts with different iron loadings. Fe@UiO-66 samples inherit the high stability of UiO-66 and maintain the morphology and porosity. Meanwhile, the Fe–O–Zr sites constructed on the metal nodes extend the visible light absorption due to MMCT effects. Among this series of photocatalysts, the Fe@UiO-66-1c material produced by a single deposition cycle showed the best photocatalytic activity and could achieve aerobic oxidation of N-aryl tetrahydroisoquinolines with excellent yield. Photoelectrochemical measurements indicated that Fe@UiO-66-1c possesses better separation efficiency of photogenerated electron–hole pairs and could activate oxygen efficiently to generate superoxide radicals. In addition, benefiting from the highly dispersed isolated iron sites and the stable Fe–O–Zr bonds, Fe@UiO-66-1c could be reused six times with a high yield of over 80%, and exhibited a low ion leaching rate in the reaction cycles.
2. Experimental
2.1. Synthetic methods
2.2. Characterization
UiO-66 and Fe@UiO-66 samples were degassed at 423 K for 12 h before measuring the N2 sorption isotherms on a Micromeritics 3Flex 3.01 instrument at 77 K. Powder X-ray diffraction (XRD) patterns were obtained using a PANalytical X'Pert PRO diffractometer with Cu Kα radiation operated at 40 kV, 40 mA. 1H NMR and 13C NMR spectra were measured using a Bruker Avance III device with a frequency of 400 MHz. Thermogravimetric analysis (TGA) was conducted on a TA Q500 instrument under a 100 mL min−1 flow of N2, ramping from 50 °C to 700 °C at a rate of 10 °C min−1. Scanning electron microscopy (SEM) images were recorded on a Hitachi SU-8010 instrument. Transmission electron microscopy (TEM) images were recorded on a Hitachi HT-7700 instrument. X-ray photoelectron spectroscopy (XPS) spectra were obtained on a Thermo Scientific K-Alpha analyzer with an Al Kα (1486.6 eV) X-ray source, and the binding energy was calibrated by the C 1s peak (284.8 eV). Electron paramagnetic resonance (EPR) spectra were recorded using a Bruker (A300) EPR spectroscopy. UV-vis diffuse reflectance spectra were recorded on a SHIMADZU UV-2600i spectrometer, using BaSO4 as the reference. Inductively coupled plasma optical emission spectrometry (ICP-OES) was performed using a Varian-730ES atomic absorption spectrometer. The samples for ICP-OES measurement were digested in HNO3 aqueous solution at 160 °C for 4 h. The state photoluminescence (PL) spectrum was recorded at room temperature using an Edinburgh FLS1000.Photoelectrochemical measurements were obtained on a CHI 660E electrochemical workstation (Chenhua Instrument, Shanghai, China). The working electrodes were prepared by dropping the sample suspension (10 μL), which was obtained from the mixture of the as-synthesized samples (5 mg), 30 μL Nafion, and 1 mL ethanol under sonication for 30 min, onto the surface of a glassy carbon electrode. After drying at room temperature, electrochemical measurements were performed in a standard three-electrode system with the photocatalyst-coated glassy carbon electrode as the working electrode, Pt plate as the counter electrode, and Ag/AgCl as a reference electrode. A 300 W xenon lamp (HDL-II, Bobei Light Co. Ltd) was used as the light source. A 0.2 M Na2SO4 solution was used as the electrolyte. The photoresponsive signals of the samples were measured at 0.3 V. And the electrochemical impedance spectroscopy (EIS) was performed in the frequency range from 10−1 to 105 Hz with a bias potential of 0.2 V.
2.3. Aerobic oxidation of N-aryl tetrahydroisoquinolines
N-Aryl tetrahydroisoquinolines (0.1 mmol), and the MOF catalyst (5 mg) were added into a Schlenk tube equipped with a magnetic stir bar. MeCN (2.0 mL) and DBN (0.15 mmol, 18 μL) were then injected with a syringe. The reaction tube was flushed three times with oxygen gas and kept under an oxygen atmosphere by using a balloon. After the reaction was completed, the catalyst was removed by centrifugation and the product was purified by column chromatography.3. Results and discussion
3.1. The fabrication and characterizations of Fe@UiO-66
Iron oxide was effectively loaded in UiO-66 through a two-step ALD process with varying cycles (Fe@UiO-66-xc, where x = 1, 2, 3). In brief, the initial step involved the reaction of gaseous Fe(Cp)2 with μ3-OH on Zr6 nodes, where it competed for coordination with the terminal carboxyl of the H2BDC ligand. Subsequently, the remaining cyclopentadienyl groups reacted with O3 to create a stable Fe–O bond, anchoring iron oxide at Zr6 nodes (Fig. 1a). | ||
| Fig. 1 (a) A schematic depiction of the process used to anchor Fe to the Zr6 node of UiO-66 via ALD. Color code: Zr (green), O (red), C (gray), H (white), Fe (brown). (b) SEM image of Fe@UiO-66-1c. (c) TEM and EDS mapping images of Fe@UiO-66-1c. (d) Powder XRD patterns of Fe@UiO-66 samples. (e) N2 isotherms of UiO-66 and Fe@UiO-66 samples. | ||
Scanning electron microscopy (SEM) images (Fig. 1b and S1†) showed that Fe@UiO-66 samples maintained the morphology of UiO-66. The element mapping images indicate that the Fe element is uniformly distributed on the surface of Fe@UiO-66 samples (Fig. 1c and S2†). Further, the cross-sectional EDS mapping images of Fe@UiO-66-1c indicated a uniform distribution of Fe inside the particles (Fig. S3†). And there were no nanoparticles or thin films observed in the HRTEM images (Fig. S4†). Powder X-ray diffraction (XRD) patterns indicate that the crystal structure of UiO-66 is well maintained after three cycles of the ALD process and no obvious XRD peaks of Fe sites are observed, owing to their high dispersion (Fig. 1d). Thermogravimetric analysis (TGA) showed that the thermal stability of these materials can still be maintained at around 450 °C (Fig. S5†). The surface area was analyzed by the Brunauer–Emmett–Teller (BET) method, which was measured as 1128.5 m2 g−1 for UiO-66, 1082.7 m2 g−1 for Fe@UiO-66-1c, 1072.3 m2 g−1 for Fe@UiO-66-2c, and 849.9 m2 g−1 for Fe@UiO-66-3c (Fig. 1e). This result is consistent with observations in previous studies.43 Furthermore, a significant reduction of surface area after the third deposition cycle was observed, implying the multi-layer deposition of Fe on the Zr6 nodes. In addition, the pore size distributions of the samples, analyzed by nonlinear density functional theory (NL-DFT), remain unchanged. A slight decrease in pore volume is attributed to the occupation of iron oxide (Fig. S6†).
X-ray photoelectron spectroscopy (XPS, Fig. 2a) confirms the coexistence of building elements. The Fe 2p3/2 binding energy of 710.8 eV demonstrates the Fe(III) state (Fig. 2c), and the satellite features of Fe 2p (2p3/2, 715.6 eV; 2p1/2, 729.6 eV) verify the Fe(II) state. Upon introducing Fe species into UiO-66, the peak of Zr 3d5/2 shifts to lower binding energy from 182.95 to 182.81 eV, which could be due to the electron-donating effect of the iron oxide sites (Fig. S7†). Electron paramagnetic resonance (EPR) was used to analyze the dispersion of iron sites.44,45 The EPR peak at g = 4.3 is assigned to high-spin Fe(III), which mainly exists in highly isolated Fe(III) in tetrahedral and distorted tetrahedral coordinations. The signal at g = 2.0 is ascribed to isolated Fe(III) in a high-symmetry octahedral coordination or FexOy oligomers (Fig. 2d). The iron content of the samples was measured by inductively coupled plasma optical emission spectrometry (ICP-OES). The results showed that the iron loading was 1.60%, 3.24%, and 4.94% wt for the samples with 1, 2, and 3 cycles of atomic layer deposition (corresponding to ∼0.58, 1.17, and 1.79 iron atom per Zr6 node), respectively, suggesting that the amount of iron oxide could be well controlled by varying the number of ALD cycles.
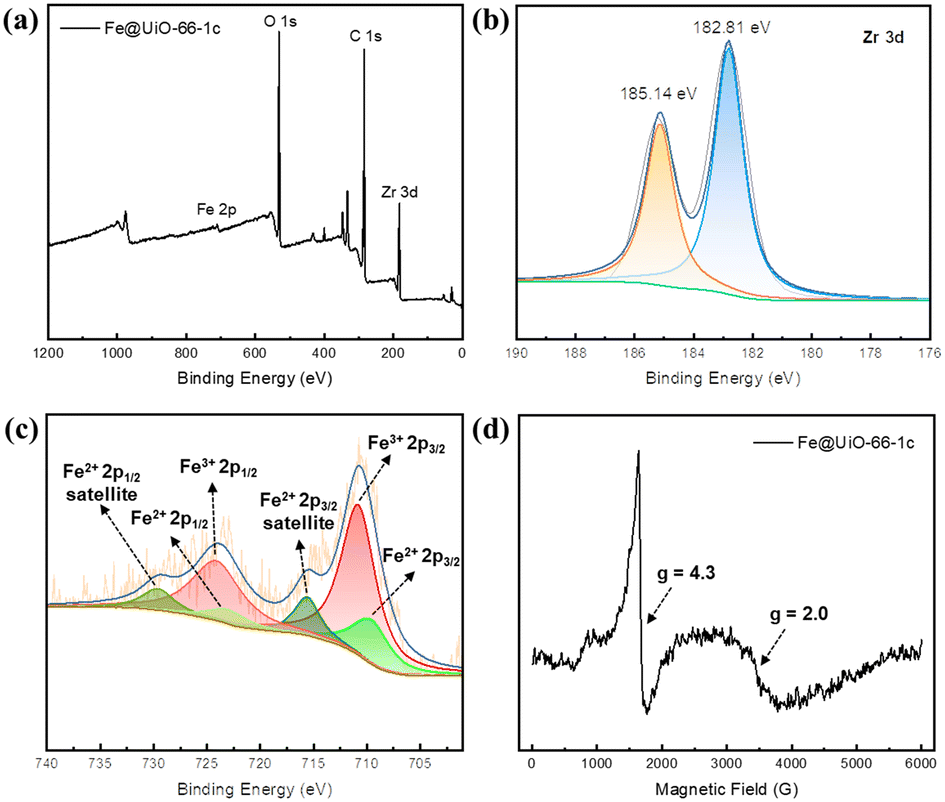 | ||
| Fig. 2 XPS spectra of (a) a survey scan of Fe@UiO-66-1c, (b) Zr 3d and (c) Fe 2p. (d) EPR spectra of Fe@UiO-66-1c at room temperature. | ||
3.2. Photocatalytic oxidation of N-aryl tetrahydroisoquinolines
The photocatalytic activity of Fe@UiO-66 samples was evaluated by the aerobic oxidation of N-aryl tetrahydroisoquinolines. 2-(4-Methoxyphenyl)-tetrahydroisoquinoline (1a) was employed as a model substrate to compare the catalytic efficiency of these materials under an oxygen atmosphere. As shown in Table 1, the reaction rarely proceeded without any catalyst (entry 1), and UiO-66 gave only a low yield of the corresponding amide (2a) (35.3%, entry 2). To our delight, a great improvement in the yield was observed with Fe@UiO-66-1c, indicating that the introduction of iron sites noticeably enhanced the photocatalytic activity. However, with an increase in the number of ALD cycles, the yield of 2a gradually decreased from 87.5% to 48.9%. This result could be attributed to the coverage of Fe–O–Zr sites, formed by the first Fe deposition cycle, and by subsequent Fe deposition in the second and third cycles. This would reduce the catalytic sites of Fe@UiO-66-2c and Fe@UiO-66-3c.43,46 In contrast, the catalytic activity of commercial nano Fe2O3 was much inferior, yielding only 7.8% of 2a.|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb [%] |
| a Reaction conditions: 1a (0.1 mmol), catalyst (5 mg), DBN (0.15 mmol), O2 (1 bar), CH3CN (2 mL), white LEDs (10 W), 30 °C, 8 h. b Yields were calculated by 1H NMR with 1,3,5-trimethoxybenzene as the internal standard. | ||
| 1 | Trace | |
| 2 | UiO-66 | 35.3 |
| 3 | Fe@UiO-66-1c | 87.5 |
| 4 | Fe@UiO-66-2c | 60.3 |
| 5 | Fe@UiO-66-3c | 48.9 |
| 6 | Fe2O3 | 7.8 |

Next, we focused on the optimization of reaction parameters, including solvents, base additives, and reaction atmosphere (Table 2). Alcoholic solvents showed extremely poor selectivity towards product 2a. However, changing to alternative polar non-protonic solvents, such as DMF or 1,4-dioxane, could provide competitive yields (entries 1–6, Table 2). Additionally, we examined the influence of other bases, including DBU, DIPEA, and Cs2CO3, but none of them led to an improvement in the yield of 2a (entries 7–10, Table 2). It is worth noting that the reaction proceeded smoothly under an air atmosphere, giving a 75% yield of the desired 2a product (entry 11).
|
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yieldb [%] |
| a Reaction conditions: 1a (0.1 mmol), Fe@UiO-66-1c (5 mg), base (1.5 equiv.), solvent (2 mL), O2 (1 bar), white LEDs (10 W). b Yields were calculated by HPLC with naphthalene as the internal standard. c 1 bar of air. d Under N2 atmosphere. | |||
| 1 | DBN | MeOH | Trace |
| 2 | DBN | EtOH | Trace |
| 3 | DBN | CF3CH2OH | Trace |
| 4 | DBN | CHCl3 | 6.1 |
| 5 | DBN | DMF | 80 |
| 6 | DBN | 1,4-Dioxane | 83 |
| 7 | DBN | CH3CN | 87 |
| 8 | DBU | CH3CN | 86 |
| 9 | DIPEA | CH3CN | Trace |
| 10 | Cs2CO3 | CH3CN | 37 |
| 11 | DBN | CH3CN | 75c |
| 12 | DBN | CH3CN | Traced |

With the optimized reaction conditions in hand, the substrate scope for the aerobic oxidation of amines was next examined (Table 3). Both electron-donating and electron-withdrawing groups on the N-phenyl ring were well-tolerated, affording corresponding products 2a–2f in good to excellent yields (77–94%). In general, electron-donating substituents exhibited superior activity than electron-withdrawing ones. Furthermore, 6,7-dimethoxyl N-aryl tetrahydroisoquinolines are also synthesized efficiently (2g–2h). In contrast, the activation of the α-position C–H of N-alkyl tetrahydroquinoline proved to be more challenging, resulting in a lower yield of 43% (2i). These catalytic results indicated that Fe@UiO-66-1c had comparable performance to the reported organic small molecule photocatalysts, such as rose bengal and eosin Y, in the photocatalytic oxidation of N-aryl tetrahydroisoquinolines (Table S1†).
|
|
||
|---|---|---|
| a Reaction conditions: 1 (0.1 mmol), Fe@UiO-66-1c (5 mg), DBN (0.15 mmol), O2 (1 bar), CH3CN (2 mL), white LEDs (10 W), 30 °C. Yield of isolated product. | ||
 2a, 8 h, 87%
2a, 8 h, 87% |
 2b, 24 h, 97%
2b, 24 h, 97% |
 2c, 12 h, 77%
2c, 12 h, 77% |
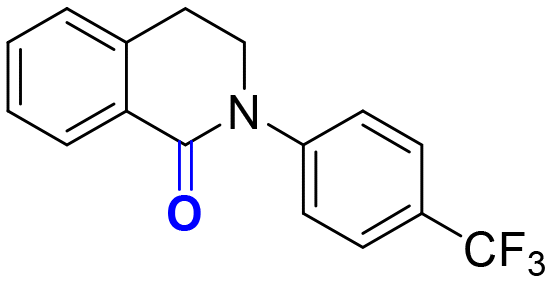 2d, 90 h, 80%
2d, 90 h, 80% |
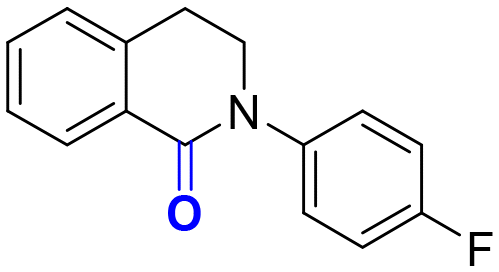 2e, 12 h, 85%
2e, 12 h, 85% |
 2f, 24 h, 82%
2f, 24 h, 82% |
 2g, 12 h, 87%
2g, 12 h, 87% |
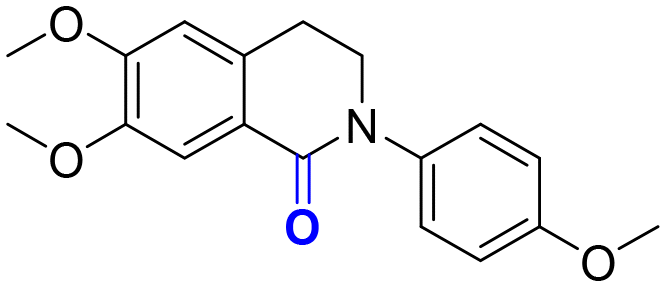 2h, 12 h, 71%
2h, 12 h, 71% |
 2i, 24 h, 43%
2i, 24 h, 43% |

Subsequently, we delved into the mechanism of this photocatalytic aerobic oxidation process. UV-vis diffuse-reflectance spectroscopy (UV-DRS) was utilized to verify the optical features of the Fe@UiO-66 samples. A promising photocatalyst is expected to have a broad absorption in the visible light region. As revealed in the obtained spectra (Fig. 3a), UiO-66 showed initial UV absorption at around 330 nm, attributed to the ligand-based absorption affected by the nearby metal clusters. In contrast, Fe@UiO-66 samples exhibited a wide range of visible light absorption, extending to 700 nm. Notably, Fe@UiO-66-1c exhibited a new absorption peak at 646 nm, liking arising from the MMCT process between Fe and Zr sites. The optical band gap of the as-prepared Fe@UiO-66 samples was calculated by Tauc plots based on UV-DRS curves (Fig. 3b). The estimated band gaps were found to be 3.97 and 3.72 eV for UiO-66 and Fe@UiO-66-1c, respectively. The band structure was determined over Mott–Schottky analysis to evaluate its oxidative potential. The positive slopes of the Mott–Schottky plots revealed n-type behavior for UiO-66 and Fe@UiO-66-1c (Fig. S8† and 3c). Typically, in n-type semiconductors, the conduction band (CB) is more negative by about 0.10 V than the flat band potential (Vfb). Therefore, the CB values of UiO-66 and Fe@UiO-66-1c were determined from the intersection with values of −1.02 and −0.86 V vs. NHE, respectively, which are more negative than the reduction potential of O2 to O2˙− (−0.33 V vs. NHE).47 Based on the estimated bandgap values, the valence band (VB) positions were then estimated as 2.95 V (UiO-66) and 2.86 V (Fe@UiO-66-1c) vs. NHE. Moreover, a photocurrent test indicated that Fe@UiO-66-1c had a stronger photocurrent response than UiO-66, suggesting its superior charge transfer efficiency (Fig. 3d). This result was further verified by the electrochemical impedance spectroscopy (EIS) study, which measured the interfacial charge-transfer resistance of the materials. Fe@UiO-66-1c exhibited a smaller radius, indicating lower charge transfer resistance than UiO-66 (Fig. S9a†). Furthermore, the reduced intensity of Fe@UiO-66-1c in photoluminescence (PL) emission spectroscopy corresponded to the higher separation rate of photoinduced charges (Fig. S9b†).
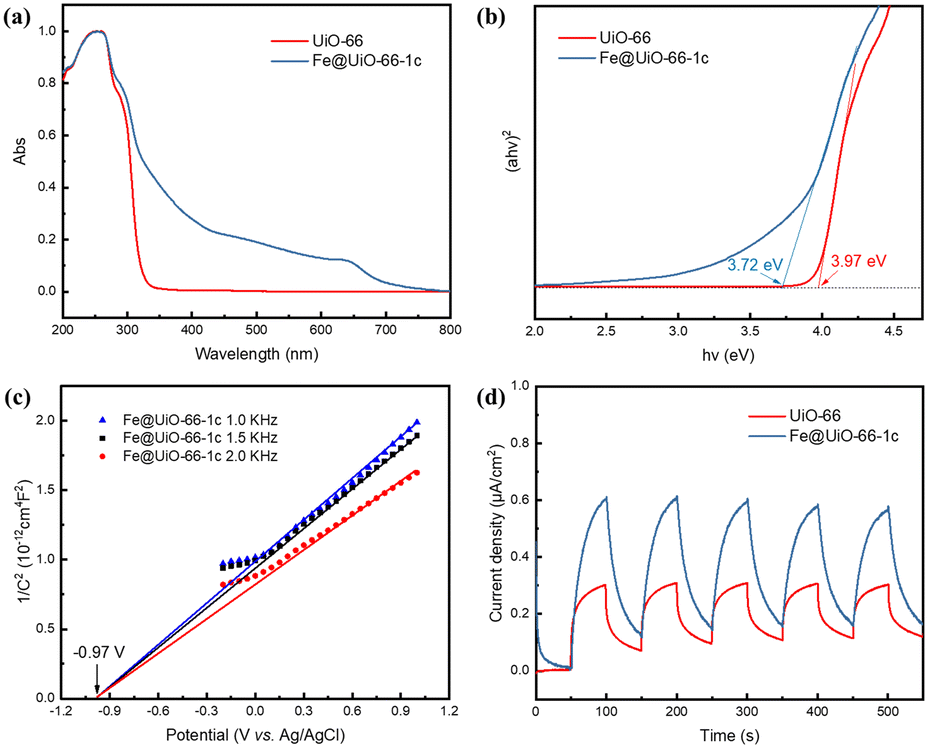 | ||
| Fig. 3 (a) UV-vis DRS spectra of UiO-66 and Fe@UiO-66-1c. (b) Tauc plots of UiO-66 and Fe@UiO-66-1c. (c) Mott–Schottky plots of Fe@UiO-66-1c. (d) Transient photocurrent responses of UiO-66 and Fe@UiO-66-1c. | ||
To gain deeper insights into the main active species in the reaction, several radical quenching experiments were carried out under the standard reaction conditions.48,49 The presence of Na2C2O4 (a scavenger for h+) and isopropanol (a scavenger for OH·) did not affect the occurrence of this reaction. However, when 1,4-benzoquinone (BQ, 3 equiv.) was added, the oxidation reaction was markedly inhibited, providing clear evidence for the presence of the superoxide radical (O2˙−).50 Moreover, we employed 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the radical trapping agent. Under the visible-light irradiation, in the presence of Fe@UiO-66-1c in MeCN and an air atmosphere, in situ EPR spectra revealed a characteristic signal of superoxide radical, confirming that O2˙− was indeed the main active species (Fig. 4b). These findings aligned with the results obtained from the electrochemical analysis of the material potentials. Based on all of the above experimental results, we propose a plausible mechanism in Fig. 4c, where O2˙− serves as reactive oxygen species.
 | ||
| Fig. 4 Catalytic mechanism. (a) Radical quenching experiments. (b) EPR spectra of Fe@UiO-66-1c in the presence of DMPO in MeCN, under an air atmosphere under dark and under visible-light irradiation. (c) Plausible mechanism for Fe@UiO-66-1c catalyzing the visible-light-driven aerobic oxidation. | ||
As a heterogeneous photocatalyst, recyclability and reusability are prominent features in industrial and practical processes. After the photocatalytic reactions, Fe@UiO-66-1c was recovered by centrifugation and reused for six consecutive runs under identical conditions. To our delight, the yield of 2a remained almost unchanged throughout these cycles (Fig. S10a†). In addition, the crystal structure of Fe@UiO-66-1c remained intact, as evidenced by the PXRD patterns (Fig. S10b†) and the SEM image (Fig. S11†). Additionally, there was only a slight decrease in the iron loading, from 1.60 to 1.42 wt%, after six runs, indicating the stable anchoring of iron oxide to the Zr6 nodes.
4. Conclusions
In summary, we have successfully employed the ALD technique to incorporate iron oxide onto the Zr6 nodes of UiO-66. This method was proven to be a facile, reliable, and controllable approach to prepare Fe@UiO-66 photocatalysts. Notably, our investigations revealed that the Fe@UiO-66-1c catalyst, with just a single deposition cycle, exhibited the highest photocatalytic activity for the aerobic oxidation of N-aryl tetrahydroisoquinolines under visible-light irradiation. The superior photocatalytic efficiency could be attributed to the MMCT effect of Fe–O–Zr open sites, which significantly improve the separation efficiency of the photo-produced carriers and broaden the visible light absorption region. Moreover, thanks to the well-dispersed isolated iron sites and the stable Fe–O–Zr bonds, Fe@UiO-66-1c demonstrated minimal ion leaching rate during reaction cycles, allowing for reuse at least 6 times with a consistently high yield of over 80%. We envision that this vapor deposition strategy may open new avenues for the application of MOF-based photocatalysts in more complex and synthetically valuable organic transformations.Author contributions
Kai Zhou: methodology, data curation, formal analysis, investigation, and writing – original draft preparation. Yuanyuan Zhang: formal analysis. Mingjie Liu and Zhenghua Zhao: validation. Xiang Liu: writing – reviewing and editing. Zongbi Bao, Qiwei Yang, and Qilong Ren: supervision and validation. Zhiguo Zhang: conceptualization, supervision, funding acquisition, project administration, and writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (Grant No. 2021YFC2103704), the National Natural Science Foundation of China (Grant No. 22078288, 22225802, 22288102, 22271248, and 22208294), and the Key R&D Program Projects in Zhejiang Province (Grant No. 2021C03005).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
.
- C. Han, X. Zhu, J. S. Martin, Y. Lin, S. Spears and Y. Yan, ChemSusChem, 2020, 13, 4005–4025 CrossRef CAS PubMed
- W. Zhang, W. Huang, B. Wu, J. Yang, J. Jin and S. Zhang, Coord. Chem. Rev., 2023, 491, 215235 CrossRef CAS
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC
- Y. Zhang, D. Riemer, W. Schilling, J. Kollmann and S. Das, ACS Catal., 2018, 8, 6659–6664 CrossRef CAS
- K. C. C. Aganda, B. Hong and A. Lee, Adv. Synth. Catal., 2019, 361, 1124–1129 CrossRef CAS
- G. Claassen, E. Brin, C. Crogan-Grundy, M. T. Vaillancourt, H. Z. Zhang, S. X. Cai, J. Drewe, B. Tseng and S. Kasibhatla, Cancer Lett., 2009, 274, 243–249 CrossRef CAS PubMed
- J. Jin, K. Zhang, F. Dou, C. Hao, Y. Zhang, X. Cao, L. Gao, J. Xiong, X. Liu, B.-F. Liu, G. Zhang and Y. Chen, Eur. J. Med. Chem., 2020, 207, 112709 CrossRef CAS PubMed
- T. Shiro, T. Fukaya and M. Tobe, Eur. J. Med. Chem., 2015, 97, 397–408 CrossRef CAS PubMed
- S. Gan, Y. Zeng, J. Liu, J. Nie, C. Lu, C. Ma, F. Wang and G. Yang, Catal. Sci. Technol., 2022, 12, 1202–1210 RSC
- L. Qi, J. Chen, B. Zhang, R. Nie, Z. Qi, T. Kobayashi, Z. Bao, Q. Yang, Q. Ren, Q. Sun, Z. Zhang and W. Huang, ACS Catal., 2020, 10, 5707–5714 CrossRef CAS
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed
- K. G. Liu, Z. Sharifzadeh, F. Rouhani, M. Ghorbanloo and A. Morsali, Coord. Chem. Rev., 2021, 436, 213827 CrossRef CAS
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
- Y. Qian, F. Zhang and H. Pang, Adv. Funct. Mater., 2021, 31, 2104231 CrossRef CAS
- N. Kolobov, M. G. Goesten and J. Gascon, Angew. Chem., Int. Ed., 2021, 60, 26038–26052 CrossRef CAS PubMed
- C. G. Silva, I. Luz, F. Xamena, A. Corma and H. Garcia, Chem. – Eur. J., 2010, 16, 11133–11138 CrossRef CAS PubMed
- P. Rassu, X. Ma and B. Wang, Coord. Chem. Rev., 2022, 465, 214561 CrossRef CAS
- Y. Qin, M. Hao, D. Wang and Z. Li, Dalton Trans., 2021, 50, 13201–13215 RSC
- H. Lin, Y. Xu, B. Wang, D.-S. Li, T. Zhou and J. Zhang, Small Struct., 2022, 3, 2100176 CrossRef CAS
- X. Feng, Y. Song and W. Lin, Trends Chem., 2020, 2, 965–979 CrossRef CAS
- I. A. Lázaro, H. Szalad, P. Valiente, J. Albero, H. García and C. Martí-Gastaldo, ACS Appl. Mater. Interfaces, 2022, 14, 21007–21017 CrossRef PubMed
- J.-D. Xiao, Q. Shang, Y. Xiong, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 9389–9393 CrossRef CAS PubMed
- L. Xiao, Q. Zhang, P. Chen, L. Chen, F. Ding, J. Tang, Y.-J. Li, C.-T. Au and S.-F. Yin, Appl. Catal., B, 2019, 248, 380–387 CrossRef CAS
- T. Wang, X. Tao, Y. Xiao, G. Qiu, Y. Yang and B. Li, Catal. Sci. Technol., 2020, 10, 138–146 RSC
- Y. Liu, J. Wang, K. Ji, S. Meng, Y. Luo, H. Li, P. Ma, J. Niu and J. Wang, J. Catal., 2022, 416, 149–156 CrossRef CAS
- C. Xu, Y. Pan, G. Wan, H. Liu, L. Wang, H. Zhou, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2019, 141, 19110–19117 CrossRef CAS PubMed
- S. Wang, F. Meng, X. Sun, M. Bao, J. Ren, S. Yu, Z. Zhang, J. Ke and L. Zeng, Appl. Surf. Sci., 2020, 528, 147053 CrossRef CAS
- S. De, G. C. Quan, B. Gikonyo, C. Martineau-Corcos, C. Bousige, L. Veyre, T. Devic, C. Marichy and A. Fateeva, Inorg. Chem., 2020, 59, 10129–10137 CrossRef CAS PubMed
- I. S. Kim, S. Ahn, N. A. Vermeulen, T. E. Webber, L. C. Gallington, K. W. Chapman, R. L. Penn, J. T. Hupp, O. K. Farha, J. M. Notestein and A. B. F. Martinson, J. Am. Chem. Soc., 2020, 142, 242–250 CrossRef CAS PubMed
- I. Utke, P. Swiderek, K. Höflich, K. Madajska, J. Jurczyk, P. Martinović and I. B. Szymańska, Coord. Chem. Rev., 2022, 458, 213851 CrossRef CAS
- J. Fonseca and J. Lu, ACS Catal., 2021, 11, 7018–7059 CrossRef CAS
- N. Pastukhova, A. Mavrič and Y. Li, Adv. Mater. Interfaces, 2021, 8, 2002100 CrossRef CAS
- R. Li, N. Li, J. Hou, Y. Yu, L. Liang, B. Yan and G. Chen, J. Hazard. Mater., 2021, 402, 123513 CrossRef CAS PubMed
- J. Plutnar and M. Pumera, Small, 2021, 17, 2102088 CrossRef CAS PubMed
- X. Wang, B. Jin, Y. Jin, T. Wu, L. Ma and X. Liang, ACS Appl. Nano Mater., 2020, 3, 2867–2874 CrossRef CAS
- Y.-Q. Cao, T.-Q. Zi, X.-R. Zhao, C. Liu, Q. Ren, J.-B. Fang, W.-M. Li and A.-D. Li, Sci. Rep., 2020, 10, 13437 CrossRef CAS PubMed
- I. S. Kim, J. Borycz, A. E. Platero-Prats, S. Tussupbayev, T. C. Wang, O. K. Farha, J. T. Hupp, L. Gagliardi, K. W. Chapman, C. J. Cramer and A. B. F. Martinson, Chem. Mater., 2015, 27, 4772–4778 CrossRef CAS
- Z. Li, N. M. Schweitzer, A. B. League, V. Bernales, A. W. Peters, A. B. Getsoian, T. C. Wang, J. T. Miller, A. Vjunov, J. L. Fulton, J. A. Lercher, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 1977–1982 CrossRef CAS PubMed
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC
- Z. Li, A. W. Peters, J. Liu, X. Zhang, N. M. Schweitzer, J. T. Hupp and O. K. Farha, Inorg. Chem. Front., 2017, 4, 820–824 RSC
- H. Liu, J. Wang, T. Yu, S. Fan and M. Shen, Catal. Sci. Technol., 2014, 4, 1350–1356 RSC
- V. Cnockaert, I. Bellemans, T. Crivits, H. Vrielinck, B. Blanpain and K. Verbeken, J. Sustain. Metall., 2021, 7, 519–536 CrossRef
- P. C. Lemaire, D. T. Lee, J. Zhao and G. N. Parsons, ACS Appl. Mater. Interfaces, 2017, 9, 22042–22054 CrossRef CAS PubMed
- L. Xiong and J. Tang, Adv. Energy Mater., 2021, 11, 2003216 CrossRef CAS
- J. Wang and S. Wang, Chem. Eng. J., 2020, 401, 126158 CrossRef CAS
- Y. Guo, J. Long, J. Huang, G. Yu and Y. Wang, Water Res., 2022, 215, 118275 CrossRef CAS PubMed
- E. D. Kerver, I. M. C. Vogels, K. S. Bosch, H. VreelingSindelarova, R. J. M. VandenMunckhof and W. M. Frederiks, Histochem. J., 1997, 29, 229–237 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01768k |
| This journal is © The Royal Society of Chemistry 2024 |