
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium nanoparticle deposition on spherical carbon supports for heterogeneous catalysis in continuous flow†
Sarah L.
Boyall
 a,
Phoebe
Berman
a,
Anthony
Griffiths
a,
Alexander
Massey
a,
Thomas
Dixon
a,
Thomas
Shaw
a,
Joanna
Miller
a,
Jonathan P.
White
a,
Robert
Menzel
a,
Kevin
Leslie
b,
Graeme
Clemens
b,
Frans L.
Muller
a,
Richard A.
Bourne
a and
Thomas W.
Chamberlain
*a
a,
Phoebe
Berman
a,
Anthony
Griffiths
a,
Alexander
Massey
a,
Thomas
Dixon
a,
Thomas
Shaw
a,
Joanna
Miller
a,
Jonathan P.
White
a,
Robert
Menzel
a,
Kevin
Leslie
b,
Graeme
Clemens
b,
Frans L.
Muller
a,
Richard A.
Bourne
a and
Thomas W.
Chamberlain
*a
aInstitute of Process Research and Development, School of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: T.W.Chamberlain@leeds.ac.uk
bChemical Development, Pharmaceutical Technology & Development, Operations, AstraZeneca, Macclesfield, SK10 2NA, UK
First published on 26th March 2024
Abstract
Heterogeneous catalysis is widely exploited by the chemical industry, both in batch reactors and in continuous flow, the latter via the use of packed bed reactors. Unfortunately, the transfer of commercially available heterogeneous catalysts to high pressure flow systems is often difficult, with challenges such as catalyst deactivation through metal leaching, and the crushing of pelleted supports. Thus, the limited availability of suitable catalysts for heterogeneous flow processes, which can satisfy all the requirements for its application, is a major bottle neck in the commercial implementation of these systems. Polymer-based spherical activated carbon beads (diameter = 474 ± 96 μm) offer a promising solution: these small, spherical and monodisperse beads have high mechanical strengths and large surface areas (1583 ± 8 m2 g−1), offering desirable properties for this task, such as reproducible packing and low pressure drops across packed catalyst beds. Two series of Pd/C spherical bead catalysts were synthesised and compared to a commercial catalyst from Johnson Matthey (1 wt% Pd/C pellets), in small scale screenings (20 mg) via a recirculating batch platform, for their activity in a model nitro reduction reaction. It was observed that small, robust, highly active palladium nanoparticles (PdNPs) supported on spherical carbon beads with a narrow size distribution (e.g.1e – Pd – dNP = 5.0 ± 1.4 nm) can be synthesised via solution phase deposition. In contrast, the NP catalysts made via gas phase deposition were much larger (e.g.2e – Pd – dNP = 22.8 ± 13.1 nm), less active and unstable due to metal leaching. The applicability of these NP catalysts for use in continuous flow was subsequently demonstrated on a larger scale (0.5–1 g), with a high activity and stability achieved over a two day operating period. This work demonstrates the production of an active, stable heterogeneous catalyst suitable to be employed in a pilot scale continuous flow packed bed reactor, for the production of APIs.
Introduction
Catalysis has become fundamental and essential to the production of a vast number of chemical processes and is involved in at least one step for the production of over 90% of all chemical products.1 Catalysis is exploited in a wide variety of industrial applications from fine chemicals (i.e. pharmaceuticals, agrochemicals etc.) to the production of fuels and polymers,2 offering an alternative chemical pathway which requires a lower activation energy toward product formation.3 Creating novel, active, but also stable catalysts is a pivotal and growing area of research, which has led to the enhancement of new and existing chemical processes that maximise yields and reduce waste in order to make industrial processes more efficient and use fewer raw materials.4 The majority of applied catalysis performed, homo- and heterogeneous, uses precious metals due to their activity and selectivity for key reaction steps.5,6 The immobilisation of these metal catalysts onto supports promotes catalyst stability and recyclability, with the ease of separation and regeneration of the catalysts, improving the purity of products in many cases. This ease of separation is particularly beneficial when the catalysts are used in continuous flow, with packed bed reactors being well documented for their use in the pharmaceutical industry for the production of active pharmaceutical ingredients (APIs).7–9 The advantages of flow chemistry are well defined in the literature,10 these include improved heat and mass transfer, easier scale up and better process control, leading to less waste and improved safety of the reactor systems. However, the transfer of heterogeneous catalysis to high pressure flow systems is known to cause significant problems including metal leaching and crushing of the catalyst support, ultimately leading to blockages, and reducing the efficiency of the overall process.Ideally, catalysts need to be supported on an appropriate material to benefit fully from the advantages of continuous flow operation. Supports are required to have a large active surface area and maintain chemical and physical stability, whilst also limiting the pressure drop across the reactor bed.11 Carbon-based supports, especially when used with precious metals such as Pd, Pt or Rh, have been reported to provide a highly active and chemically stable catalyst support surface.12–14 Many types of carbon supports have been documented in the literature, including powders, foams, and pellets.15–17 Larger supports lead to lower pressure drops across the reactor but typically cause a decrease in the active surface area, as well as inhomogeneity in the flow path through the catalyst bed, both decreasing the efficiency of the catalyst.18 Pressure drops have been modelled for different flow regimes and guidelines, outlined by Hickman et al. in 2016, suggest that a maximum pressure drop of 20% of the reactor inlet pressure is acceptable for successful operation.19 The ratio between particle size and reactor column internal diameter has been identified as an important factor to consider when designing reactor systems.20
Polymer-based spherical activated carbon beads (PBSAC) are a relatively new and promising support; with highly tuneable and controlled pore sizes, a high mechanical strength, and large surface areas.21,22 These polystyrene–divinylbenzene based materials are available commercially and a number of studies have reported their use for the adsorption of dyes and toxins in the treatment of water23 and as catalyst supports for reactions in continuous flow.24,25 They pack regularly and show comparatively smaller pressure drops across packed bed reactors, as well as a high activity when combined with metal NP catalysts.26 In order to improve upon existing catalysts, the NPs must be stable and available for reactions on the support material.27 For this to be achieved, the NPs must be effectively bound to the support surface. Munoz et al. reported the development of 1 wt% Pd/PBSAC catalysts for application in hydrodechlorination to treat contaminants in wastewater. The PBSAC support was first oxidised with H2SO4 to remove their hydrophobic properties, allowing for their use in aqueous conditions and providing oxygen rich sites for Pd anchorage before functionalisation of the beads with Pd via ion-exchange. The group reported reaction kinetics similar to commercial powdered catalysts and noted their long-term stability in continuous conditions.28
This work aims to investigate how the method of PdNP deposition affects the catalyst activity for a known, well understood, pharmaceutically relevant model reaction. Additionally, the stability of the catalyst will be monitored and characterised for extended use, to study their industrial applicability. A heterogeneous hydrogenation will be used to investigate activity of the catalysts, as this reaction is prevalent in industry and used in the production of APIs, with GSK reporting that 77% of their fundamental gas–liquid transformations are heterogeneous hydrogenations.26 Hydrogen gas has been utilised in this three-phase hydrogenation which is atom efficient, and safer when used in continuous flow, with the benefit of smaller amounts of hydrogen being used at one time.29
Materials and methods
Materials
The unfunctionalized PBSAC beads used in this work were supplied by IBUtec, Germany. A commercial egg-shell catalyst with PdNPs supported on extrudate carbon pellets (product code: 113784) (1 wt%) was supplied by Johnson Matthey (JM). Tris(dibenzylideneacetone)dipalladium, palladium(II)hexafluoroacetylacetonate, methanol (>99%) and biphenyl (99.5%) were purchased from Sigma Aldrich. Nitrobenzene and aniline (99.5%) were purchased from Acros Organics. Hydrogen gas (99.9%) was purchased from BOC. All reagents were used without further purification.Pd/C bead catalyst preparation
Two methods of deposition on to the carbon beads were evaluated: solution phase deposition (impregnation); and gas phase deposition (sublimation–deposition). These procedures were adapted from those reported by Aygün et al. for the deposition of palladium on graphitised carbon nanofibers (GNFs).30Material characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) was added to dissolve the Pd and the solution was diluted with ultrapure (99%) water (45 mL). Calibration solutions (2, 4, 6, 8, 10 mg L−1) were prepared from a Pd standard purchased from Sigma Aldrich.
:1 ratio) was added to dissolve the Pd and the solution was diluted with ultrapure (99%) water (45 mL). Calibration solutions (2, 4, 6, 8, 10 mg L−1) were prepared from a Pd standard purchased from Sigma Aldrich.
The data has been processed using CasaXPS software. The spectra were calibrated against the adventitious C 1s peak, which was set to a binding energy of 284.8 eV. The angular distribution correction was set to 54.7°. Survey spectrum quantification was performed using the quantify tool in CasaXPS. Shirley-type backgrounds were applied to the elemental peaks, subsequent integration of the peaks enabled quantification of the atomic concentrations at the surface of the sample. Default RSF values for each element contained within the software were used to scale the peak areas. A Shirley background was also defined for the high-resolution spectra. When deconvolving the Pd 3d high-res spectra, the Pd(0) contributions were assigned an asymmetric LA (1.9, 7, 2) line shape. All other peaks were then fitted with GL(30) line shapes. The area of the Pd 3d3/2 peaks were constrained to two-thirds the area of the Pd 3d5/2 peaks. The position of the Pd 3d3/2 peaks was constrained to +5.3 eV of the corresponding Pd 3d5/2 peaks.
Catalyst testing
Results and discussion
The PdNPs were formed on the carbon beads using solution phase deposition and gas phase deposition with a range of loadings (1–5 wt%) as described above and shown in Fig. 1. This was based on previous work by Aygün et al., who used similar deposition techniques to deposit Pd and Pt on GNFs.30 The catalysts for ease of identification are labelled 1 for solution, and 2 for gas deposition, and a–e for the various loadings for each deposition technique, with a being the lowest Pd wt% and e being the highest. The Pd content of the beads produced was quantified by ICP-MS with the expected and actual loadings for each batch illustrated in Fig. 2. Gas phase deposition produced catalyst particles with a greater Pd content than solution phase deposition at all Pd loadings, however, both contained less Pd than theorised. When forming catalysts 2a–e, the ampule containing the beads and Pd precursor is rapidly cooled, and the Pd is deposited where it is located within the ampule at that time, including on the inside of the glass. This results in some of the Pd precursor being deposited on other surfaces, decreasing the Pd loading on the carbon beads. The Pd loading on catalysts 1a–e was much lower than expected, possibly due to a low affinity of the Pd precursor to the carbon beads, however, this is not well documented in the literature. No optimisation of either technique was performed in this work, but variables such as temperature and the amount, and type of solvent used could be varied to increase the Pd loadings onto the beads. | ||
| Fig. 1 Deposition techniques used to form PdNPs on to the surface of carbon beads. a) Solution phase, dissociative adsorption of Pd2(dba)3 from CHCl3, and b) gas phase deposition of Pd(hexafluoroacetylacetonate)2, followed by thermal decomposition. | ||
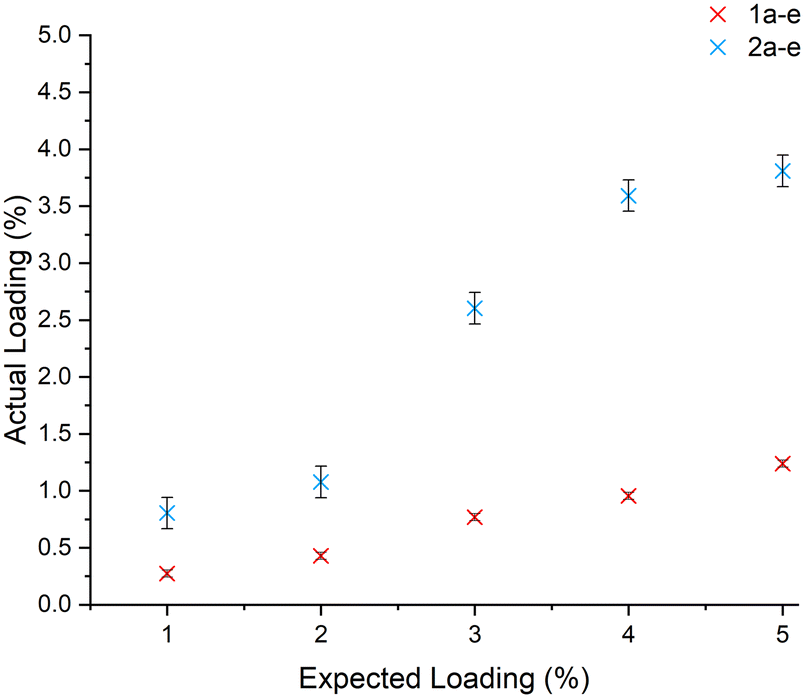 | ||
| Fig. 2 ICP-MS data showing the actual vs. the expected PdNP loadings for the different deposition techniques, black = 1a–e, and red = 2a–e. | ||
Catalyst characterisation
N2 adsorption/desorption was used to characterise the pore structure and surface area of the carbon beads and supported catalysts. BET analysis showed a high surface area for the unfunctionalized carbon beads of 1583 m2 g−1, with other details including pore size and crush strength summarised in Table 1. The surface area of both the supports and Pd loaded catalysts were comparable to other PBSAC beads in the literature (1160–1946 m2 g−1).23,33 However, their surface area was much larger than alternative carbon supports (such as the activated carbon supports – 890 m2 g−1 and carbon nanotube supports – 531 m2 g−1 reported by Pikna et al.).15 This shows there is a potentially larger active surface area with the possibility for more catalytically active sites upon NP deposition. The BET surface area of the catalysts decreased with the deposition of PdNPs, likely due to the blocking of some of the smaller micropores within the carbon bead, causing these to become inaccessible. This is in keeping with observed decrease in pore volume and an increase in the average pore size measured for all functionalised catalysts, which both occur to a larger extents with increasing Pd loading (see ESI† Section S6 for summary).| Catalyst | Average particle sizea (μm)32 | Pore volumeb (cm3 g−1) | SSAb,c (m2 g−1) | Average pore diameterb (nm) | Crush strength (N) |
|---|---|---|---|---|---|
| a See Section S5 in ESI† for size distribution. b See Section S6 in ESI† for all catalyst BET data. c SSA = specific surface area. | |||||
| 1 wt% JM Pd/C pellets | Length – 307, diameter – 165 | 0.16 | 1102 | 3.9 | 1.12 ± 0.55 |
| PBSAC beads | 465 | 0.66 | 1582 | 12.8 | 0.69 ± 0.27 |
| 1e | 465 | 0.57 | 1136 | 17.3 | 0.79 ± 0.33 |
| 2e | 465 | 0.62 | 1275 | 20.7 | 1.09 ± 0.34 |
The crush strength of the PBSAC beads, functionalized PBSAC catalysts, and commercial Pd/C pellets was tested to assess their applicability for use in high pressure packed bed reactors (Table 1). The data shows that the mechanical strength of all four materials were similar and within the error calculated by taking the standard deviation of 50 samples. However, the advantage of using catalyst supports such as PBSAC beads for continuous flow applications is that, due to their spherical shape, they pack into a column more uniformly than extruded pellets, and therefore the catalyst bed performance should, in principle, be more reproducible.
SEM was utilised to show the topography and characteristics of the surface and inner pore network of the carbon beads – shown in Fig. 3a (see ESI† Section S7 for more SEM images of beads). Cracks on the surface can be seen that are created during the production of the carbon beads. An energy dispersive X-ray spectroscopy (EDS) map shows that the PdNPs are homogeneously distributed over the surface of the carbon beads but are more concentrated at the defects (i.e., cracks) on the surface (see Section S8 in ESI† for EDS map). The cracks offer beneficial sites for the nucleation and growth of the NPs with strong binding to the Pd achieved at these defects.34 Whilst this is anticipated to enhance the stability of the PdNPs located here, it may also have a negative effect on the mechanical strength of the catalyst, thus making it an important parameter to control during manufacture.35 Cracks in PBSAC beads are well documented in the literature, with An et al. investigating PBSAC preparation for supercapacitor electrodes, noting that an increase in activator dosage and activation temperature caused more cracks in the surface.36 Singh et al. developed a method for producing activated carbon spheres using resole-type cross-linked phenolic beads and showed the correlation with an increase in surface cracks with an increase in burn-off, leading to a detrimental effect on their mechanical strength (120 N mm−2 to 3 N mm−2).37
 | ||
| Fig. 3 Characterisation data from SEM and TEM for 1a–e and 2a–e. a) SEM images of the external surface and internal pore structure of an unfunctionalized PBSAC bead. b) TEM images showing the size of the nanoparticles made via the two deposition techniques, showing the smaller nanoparticles of catalyst 1e, than 2e. c) TEM data showing the size of Pd nanoparticles for different Pd wt% loadings for 1a–e (left) and 2a–e (right) showing the increase in the average size and size distribution of the PdNPs made via the two deposition techniques. d) SEM image, EDS maps and line scans for carbon and palladium elemental content for 1e. * = surface of the bead. | ||
TEM and STEM were used to investigate the size distribution of the NPs deposited on the carbon beads. The results of the TEM and STEM analysis are shown in Fig. 3 (more TEM images of each catalyst are shown in Section S9 in the ESI†). The data shows catalysts 1a–e possess NPs with much smaller diameters and narrower size distributions than 2a–e. The average size of the NPs and size distribution increased slightly with an increase in loading, but all loadings gave NPs of less than 10 nm in diameter for catalysts 1a–e. TEM data for 2a–e also showed an increase in the average size and size distribution of the NPs as the Pd loading increased. The increase in Pd content of the gas phase deposition catalysts seems to suggest that any increase in Pd content, preferentially promotes the growth of existing NPs rather than the nucleation of new NPs. Aygün et al. also found this trend with the deposition of 0.5 wt% Pd on GNFs using these two techniques. The Pd-GNFs made via gas phase deposition were larger in size and had a greater size distribution (10.79 ± 3.86 nm) than those made with solution phase deposition (2.26 ± 0.56 nm), suggesting that the increase in size of the NPs is not due to the nature of the support, but due to the deposition technique used.30
Plasma focussed ion beam SEM-EDS (PFIB-SEM-EDS) was used to investigate the location and depth of the PdNPs on both the PBSAC-based catalysts (PFIB data for catalyst 1e shown in Fig. 3d and see Section S10 in ESI† for 2e). Catalysts 1e and 2e were analysed using this technique and both showed only a slight decrease in Pd concentration below the surface of the carbon bead; however, it showed that the Pd concentration did not decrease to zero, showing consistent amounts of Pd present throughout the entire carbon support. FIB-SEM-EDX has previously been reported for the commercial pellets by White et al. showing that the location of the Pd is limited to the top 6 μm of the pellets.32,38 BET data shows the pore diameter is much smaller for the commercial pellets than the beads, likely making deposition of Pd more difficult in the pores of the pellets.
Activity of the catalysts
Nitrobenzene hydrogenation experiments were performed in a recirculating batch platform (Fig. 4a and b) designed to enable screening of relatively small amounts of catalysts (20 mg) before they are scaled up and used in a larger packed bed flow reactor system (0.5–1 g). The principle of our new testing unit is to benefit from advantages of a continuous flow of gaseous reagents (constant replenishing of the H2 in the system over the course of the reaction), whilst maintaining control the reaction conditions (pressure, temperature, and both gas and liquid flow rates) and the versatility of operation in batch. This platform allows the nitrobenzene solution to pass over the catalyst bed multiple times, and benefits from the ease of sampling at ambient temperature and pressure. The column also contains a hydrogen pre-saturation section of only glass beads before the reactants meet the catalysts, to ensure that the reaction is not mass transfer limited. The system and instrumentation are detailed in Section S1 of the ESI.† | ||
| Fig. 4 a) Nitrobenzene reduction with Pd/C catalyst and hydrogen gas to aniline. b) Schematic diagram of the recirculating batch platform for the continuous hydrogenation of nitrobenzene for catalyst screening. c) A comparison of the activities of the catalysts 1a–e (red) 2a–e (blue) and the commercial catalyst pellets (grey). d) Relationship between rate constant k and the palladium loading % on each catalyst, with solution phase catalysts 1a–e in red, gas phase catalysts 2a–e in blue, and the commercial pellets in grey. Reaction conditions – 7 bar pressure, 35 °C, 1 mL min−1 liquid flow rate (0.08 M), 20 mL min−1 hydrogen flow rate, Pd/C catalyst (20 mg). | ||
Pressure drop data were collected across the catalyst bed to quantify the pressure drop for the smaller carbon bead support compared to the commercial pelleted carbon support (data shown in Section S11 of ESI†). The reactor pressure is a highly influential factor on hydrogen solubility and reaction rate, thus effecting reaction yields and selectivity.39 It was found that there was a comparable pressure drop for both the PBSAC and commercial pelleted catalysts, showing a 0.1 bar pressure difference (5%) and demonstrating the applicability of carbon beads as a catalyst support for industrial use and scale-up. This value also satisfies the maximum pressure drop stated in the recommended guidelines by Hickman et al. (<20%).19
Fig. 4c illustrates the difference in activities for the catalysts at different wt% loadings. The data points were fitted using a two-component model, the initial period (0–60 min) incorporates a hydrogen limited regime, as previously been reported by White et al.32 A pseudo first order reaction model is then fitted for the final five hours, with the combined model enabling calculation of observed rate constants for each synthesised catalyst, as well as the commercial catalysts (Fig. 4d). The reactions with catalysts 1a–e all reached completion within the first five hours showing a high activity for those synthesised via solution phase deposition. The size of the metal catalyst nanoparticles has been shown to be directly related to the catalyst activity.40 The smaller the nanoparticle size, the larger the active surface area to volume ratio, providing more sites for catalysis. The smaller NPs present in catalysts 1a–e were shown to be much more active in the reduction of nitrobenzene (cf. Kobs = 0.46–1.41 mol L−1 h−1), presumably due to their larger catalytically active surface area. The larger NPs observed in catalysts 2a–e have a smaller active surface area, which resulted in the decrease in activity for the reduction of nitrobenzene, with their calculated rate constants (Kobs = 0.13–0.24 mol L−1 h−1), much smaller than those made via solution phase deposition. The difference in catalytic activities appears to be primarily a result of the Pd NP size in the two catalyst types, with smaller NPs (<5 nm) shown to be more active for the reduction of nitrobenzene, as there was no difference in the location of the PdNPs made via the two deposition techniques.
The commercial pelleted JM catalysts showed a similar activity to catalysts 2a–e over the 6-hour period. This is likely due to a difference in the catalyst support and location of the NPs, rather than NP size, as TEM analysis previously reported by White et al. for the commercial JM catalysts showed similar NP sizes and distribution s (dNP = 6.0 ± 2.0) to the solution phase deposition catalysts (cf. dNP = 3.2–5 nm, see ESI† Section S12 for TEM data).32,41 N2 adsorption/desorption showed that the carbon bead support exhibited a larger BET surface area and a greater pore volume and diameter than the commercial pellets, which could mean more sites are available for catalysis. The PdNPs are also not limited to the surface of the catalyst for 1a–e and 2a–e (as they are with the JM commercial pellets), which may indicate that the pores are accessible for catalysis and offer beneficial sites for catalysis during the reaction.
Turnover numbers (TON) and turnover frequencies (TOF) were calculated using the equations outlined in Section S13 of the ESI,† allowing for a direct comparison between the catalysts for each deposition technique (summary of all TON and TOF for all catalysts in Section S14 of ESI†). The TONs showed a decrease with an increase in Pd loading, with 1a having the largest TON at 2246 and a TOF after six hours of reaction of 374.3 h−1. This can be compared to other catalysts in the literature (summarised in Section S15 in ESI†). Wang et al. reported a TOF of 108.8 h−1 for a Pd/C that used activated carbon as a catalyst support.42 This is three times less than the catalyst 1a reported here, but greater than 2a–e (which displayed a TOF of 78.5–21.1 h−1 respectively). The data shows that the TON for the commercial pelleted catalysts was much lower than 1a–c, reflecting their lower activity. The greater activity of catalysts 1a–c is beneficial as this allows similar, or more importantly, a decrease in the amount of Pd required. This leads to a reduction in the use of the limited precious metal resources, while maintaining the same catalytic activity and more reproducible packing in packed bed reactors due to the spherical shape of the support.
Stability tests
It is also important for industrial application that the catalysts prepared in this work are stable for extended periods of time and research is still required in this area to minimise the waste of precious metals used in catalysis due to catalyst deactivation.43 The stability of both PBSAC catalysts 1e and 2e was tested in the recirculating batch platform; the reactor configuration was altered so the reaction solution and hydrogen would only pass over the catalyst bed once (see details in Section S16 of the ESI†). The stability of the two catalysts was recorded over an 11-hour period and is shown in Fig. 5. | ||
| Fig. 5 Comparison of the stability of the two different deposition catalysts, left – aniline concentration over 11 hours for 1e and 2e sampled every 30 minutes, right – concentration of nitrobenzene and aniline over 24 hours using 1e sampled every hour. Reaction conditions for all experiments – 7 bar pressure, 35 °C, 1 mL min−1 liquid flow rate (0.08 M), 20 mL min−1 hydrogen flow rate, Pd/C catalysts (20 mg). | ||
The data shows no significant decrease in the activity of 1e with the catalyst staying stable for the entire 11-hour experiment. However, a decrease in activity was seen for 2e over the 11-hour reaction, indicative of slow deactivation of the catalyst.
XPS was used to determine the oxidation state of the PdNPs on the surface of the carbon beads before and after the reaction to probe the deactivation mechanism of the catalysts (see Section S17 in ESI† for survey and high res XPS spectra). Samples 1e and 2e (the highest loading for each of the different catalysts) were chosen for analysis to ensure the signal to noise ratio was high enough for good fitting of the data to be achieved. The results showed that in all samples, the most abundant form of Pd is Pd(0), indicated by the large peaks at 335.4 eV and 340.7 eV in the high-resolution Pd 3d spectra, which correspond to characteristic binding energies for the Pd(0) 3d5/2 and 3d3/2 peaks, respectively.44 The high-resolution Pd 3d spectrum for catalyst 1e shows a relatively large amount of Pd(II), as shown by the deconvoluted peaks at higher binding energies. This is most likely in the form of PdO; due to the small size of the nanoparticles, their surface is prone to oxidation.45,46 Catalyst 2e exhibited a much lower concentration of Pd(II). The Pd 3d3/2 peak has a long tail compared to 1e, which was attributed to PdF2 from the Pd(HFAc)2 precursor and the presence of a plasmon loss peak.44 Following use in the catalytic reaction, the overall concentration of Pd(II) decreases similar to 1e. This showed there was no significant change in oxidation state for catalyst 2e showing mostly Pd(0), which is characteristic of PdNPs, both before and after the reaction.47 Upon using the catalyst in the reaction, the concentration of Pd(II) decreases as it is reduced via the catalytic mechanism with H2 gas. This is shown in the increase in the peak area of the Pd(0) species, see ESI† Section S17. As the oxidation state of the PdNPs on catalyst 2e did not change significantly during the reaction, ICP-MS was subsequently used to investigate the deactivation mechanism of the catalyst. This showed a large decrease in the Pd% loading for the 2e catalyst from 3.8 to 1.5 wt% Pd, showing the deactivation mechanism was likely to be due to metal leaching form the carbon support. ICP-MS was also performed for 1e and no change in Pd metal content was seen, showing no loss of Pd via metal leaching from this catalyst. Therefore, the stability of 1e was assessed for 24 hours at the same concentration and reaction conditions as used previously. Catalyst 1e showed no deactivation, and the data shows that under these mild reaction conditions, the catalyst could be used with no user intervention for 24 hours with no decrease in activity, generating 1.33 g of aniline using only 20 mg of catalyst in relatively dilute concentrations. TEM analysis was also performed to look at the PdNP size before and after this reaction and showed no change in either the size or size distribution (see ESI† Section S9). The data showed that the most suitable catalyst candidate for scale-up was 1a due to the high activity and stability of this catalyst.
Transfer and scale-up into packed bed reactor system
The activity of the catalysts for the reduction of nitrobenzene was also evaluated on a large scale (0.5–1 g of catalyst) in a packed bed reactor platform (Fig. 6a), information on the system and instrumentation is explained further in Section S4 the ESI.† This demonstrates the use of the catalysts on a scale that is similar to that used in industry and is capable of producing products on the kg per day scale.9 The most appealing catalyst candidate for scale-up was chosen to be 1a as it showed the a good activity and had the lowest Pd loading, therefore limiting the volume of precious metal used when the catalysts were made on a larger scale. The catalysts could then be compared to the commercial JM pellets used at the same reaction conditions (Fig. 6b). There is an initial increase in the yield of aniline observed for both catalysts before steady state is achieved. This is possibly due to an initial reduction of the surface upon pre-treatment with H2, however, we believe it to be result of the initial pre-treatment with hydrogen cleaning the catalyst surface before the surface becomes covered in adsorbed reactant, product and intermediates, and an equilibrium is established. The data shows that catalyst 1a was more active than the commercial catalyst as more aniline was produced under the same reaction conditions. This result agrees with those seen on a smaller scale in the recirculating batch platform, with a TOF of 261 h−1 achieved over the 10-hour experiment in the packed bed reactor. This is lower than that seen in the recirculating batch platform (374 h−1) and may be due to other scale-up factors, such as a change in the reactor pressure, hydrogen flow rate or temperature of the system, or a change in the reactor dimensions, affecting mass transfer rates. However, it is much greater than the TOF observed with the commercial pellets when they were scaled up into the packed bed platform – 59 h−1.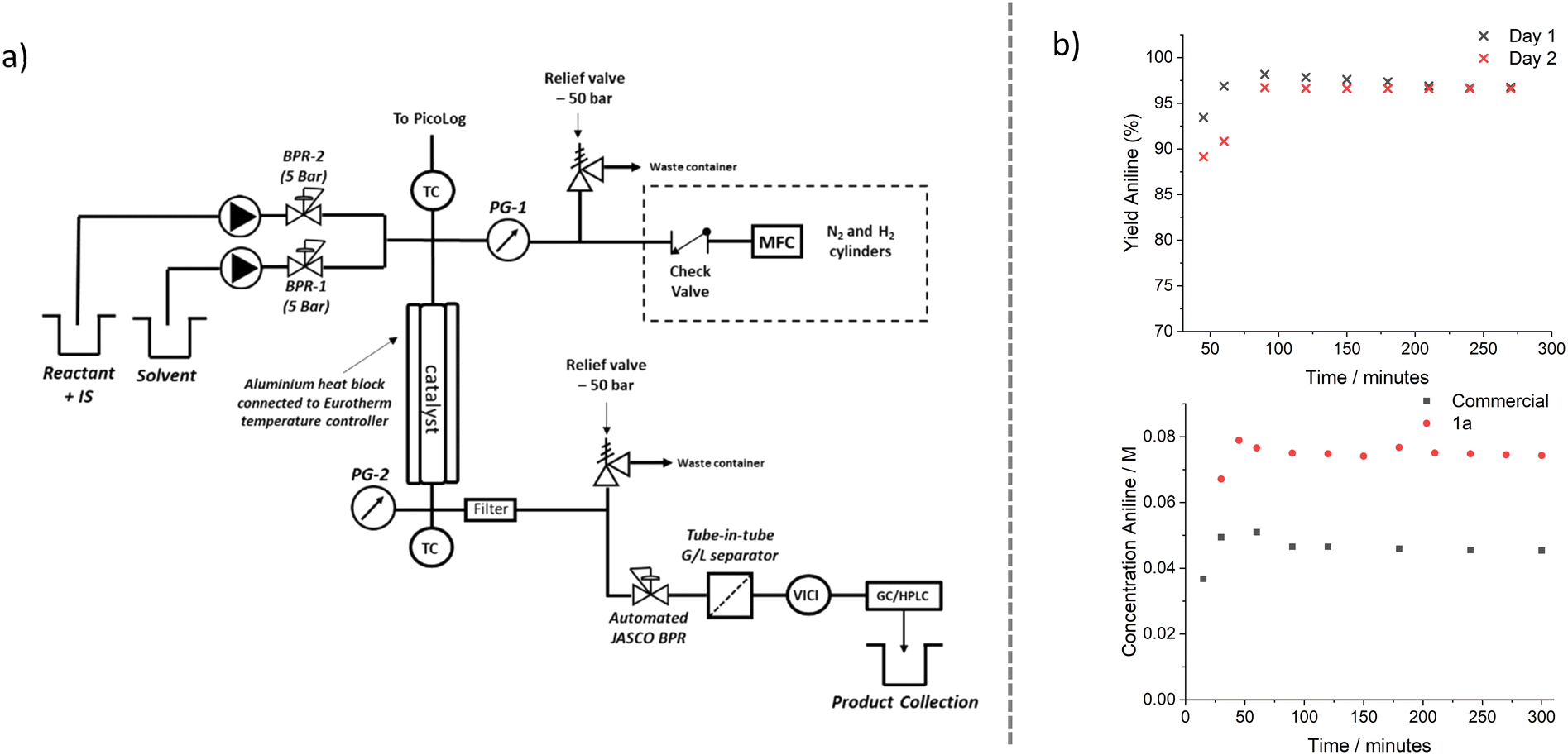 | ||
| Fig. 6 a) Schematic diagram of the automated packed bed reactor platform for continuous flow hydrogenations with hydrogen gas. b) Top – Yield of aniline produced in packed bed reactor platform with 1a showing the stability of the catalyst over two days, sampled every 30 minutes. Bottom – Comparison of yield of aniline produced using commercial 1 wt% Pd/C and 1a. Reaction conditions – 40 bar pressure, 50 °C, 1 mL min−1 liquid flow rate (0.08 M), 50 mL min−1 hydrogen flow rate, Pd/C catalysts (500 mg). | ||
The stability of 1a was also evaluated (Fig. 6b), to investigate if the harsher reaction conditions caused any catalyst deactivation. The catalysts were used in a way to mimic how they would be used on plant in industry, with a solvent flush between days and the bed was kept under a nitrogen atmosphere at pressure overnight. The 1a catalyst showed no decrease in activity over both days, with TEM confirming no change in NP size (see ESI† Fig. S13), showing their applicability to be used for extended periods of time, maintaining their productivity. Over the 10 hours, 4.6 g of aniline was made, with the possibility of producing 11 g per day under these reaction conditions. Increasing the flow rate and concentration of the starting material would increase the reaction throughput. However, no optimisation of reaction conditions was performed for this study, with reaction conditions kept constant to enable differences between reaction performances relating to the catalyst used to be determined.
Conclusions
Pd-PBSAC catalysts were made via two different deposition techniques of varying Pd loadings and compared to commercial JM 1 wt% Pd/C pellets. Catalysts 1a–e, prepared by solution-phase deposition, had a greater activity for the reduction of nitrobenzene than 2a–e, prepared by gas-phase deposition. Characterisation of the catalysts showed that the PdNPs made via solution phase deposition (e.g., 1e – dNP = 5.0 ± 1.4 nm) were much smaller than those made via gas phase deposition (e.g., 2e – dNP = 22.8 ± 13.1 nm). This smaller NP size led to a higher TON and rate constant for this test reaction and outperformed commercial pelletized Pd/C catalysts.The catalysts synthesised by solution phase deposition were shown to be stable under both mild conditions (7 bar pressure, 35 °C) for 24 hours (1e), and when transferred into a larger scale packed bed reactor, where they were used for two days under harsher conditions (40 bar, 50 °C), a productivity of 0.46 g h−1 was observed (1a). Catalyst 2e showed a decrease in activity when assessed under mild conditions for 11 hours, with the deactivation mechanism found to be metal leaching of the PdNPs from the carbon bead support.
This work demonstrates the high potential for PBSAC supports to be used industrially to perform continuous heterogeneous hydrogenations for the production of APIs and enable telescoping of reactions, leading to process intensification. This is all in trend with the paradigm shift towards the large-scale implementation of continuous manufacturing within the pharmaceutical industry.
Author contributions
Sarah Boyall and Phoebe Berman performed the experiments and analysis. Anthony Griffiths help with BET, TEM and crush strength testing. Alexander Massey for XPS analysis. Thomas Dixon for help running overnight experiments. Thomas Shaw and Robert Menzel for help writing and editing paper. Joanna Miller and Jonathan White for initial catalyst formulation and pressure drop testing – and TEM on C pellets. Kevin Leslie, Graeme Clemens, Frans Muller, Richard Bourne and Thomas Chamberlain for supervision and guidance during the project and drafting of this paper. Thomas Chamberlain conceived the original idea and secured funding to support the project.Conflicts of interest
Kevin Leslie and Graeme Clemens are employees of, and shareholders in, AstraZeneca, who funded the research.Acknowledgements
We would like to that AstraZeneca and the EPSRC for the funding of this work (EP/R032807/1). The authors would like to thank John Harrington, Zabeada Aslam, Stuart Micklethwaite of Leeds Electron Microscopy and Spectroscopy Centre (LEMAS) for their support and assistance in this work in performing FIB-SEM-EDX, Plasma FIB-EDX and TEM, and Alexander Kulak for SEM and EDX analysis. The authors would also like to thank Simon Lloyd, Karina Alves Thorne and Adrian Cunliffe for ICP-MS analysis, and Andrew Britton for XPS analysis.Notes and references
- J. R. Ludwig and C. S. Schindler, Chem, 2017, 2, 313–316 CAS.
- J. A. Dumesic, G. W. Huber and M. Boudart, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. kGaA, Weinheim, Germany, 2008 Search PubMed.
- S. S. Joshi and V. V. Ranade, in Industrial Catalytic Processes for Fine and Specialty Chemicals, Elsevier, Amsterdam, 2016 Search PubMed.
- M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS PubMed.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- M. Irfan, E. Petricci, T. N. Glasnov, M. Taddei and C. O. Kappe, Eur. J. Org. Chem., 2009, 2009, 1327–1334 CrossRef.
- M. O'Brien, N. Taylor, A. Polyzos, I. R. Baxendale and S. V. Ley, Chem. Sci., 2011, 2, 1250–1257 RSC.
- M. T. Rahman, S. Wharry, M. Smyth, H. Manyar and T. S. Moody, Synlett, 2020, 31, 581–586 CrossRef CAS.
- T. Ouchi, C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Process Res. Dev., 2014, 18, 1560–1566 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- H. Ishitani, Y. Saito, B. Laroche, X. Rao and S. Kobayashi, in Flow Chemistry: Integrated Approaches for Practical Applications, ed. S. V. Luis and E. Garcia-Verdugo, The Royal Society of Chemistry, Cambridge, 2020, ch. 1, pp. 1–49 Search PubMed.
- H. Jüntgen, Fuel, 1986, 65, 1436–1446 CrossRef.
- E. Lam and J. H. T. Luong, ACS Catal., 2014, 4, 3393–3410 CrossRef CAS.
- A. Pascu, E. M. Stanciu, C. Croitoru, I. C. Roată and M. H. Tierean, Adv. Mater. Sci. Eng., 2018, 2018, 4730192 Search PubMed.
- Ľ. Pikna, M. Heželová, S. Demčáková, M. Smrčová, B. Plešingerová, M. Štefanko, M. Turáková, M. Králik, P. Puliš, P. Lehocký and A. Duslo, Chem. Pap., 2014, 68, 591–598 Search PubMed.
- J. Aumo, S. Oksanen, J. P. Mikkola, T. Salmi and D. Yu. Murzin, Ind. Eng. Chem. Res., 2005, 44, 5285–5290 CrossRef CAS.
- Á. Prekob, Á. Szamosvölgyi, G. Muránszky, J. Lakatos, Z. Kónya, B. Fiser, B. Viskolcz and L. Vanyorek, Int. J. Mol. Sci., 2022, 23, 6423 CrossRef PubMed.
- A. Bansal, R. K. Wanchoo and S. K. Sharma, Chem. Eng. Commun., 2008, 195, 1085–1106 CrossRef CAS.
- D. A. Hickman, J. C. Degenstein and F. H. Ribeiro, Curr. Opin. Chem. Eng., 2016, 13, 1–9 CrossRef.
- E. Masson, E. M. Maciejewski, K. M. P. Wheelhouse and L. J. Edwards, Org. Process Res. Dev., 2022, 26, 2190–2223 CrossRef CAS.
- J. B. Yang, L. C. Ling, L. Liu, F. Y. Kang, Z. H. Huang and H. Wu, Carbon, 2002, 40, 911–916 CrossRef CAS.
- H. Klefer, M. Munoz, A. Modrow, B. Böhringer, P. Wasserscheid and B. J. M. Etzold, Chem. Eng. Technol., 2016, 39, 276–284 CrossRef CAS.
- B. Böhringer, O. Guerra Gonzalez, I. Eckle, M. Müller, J. M. Giebelhausen, C. Schrage and S. Fichtner, Chem. Ing. Tech., 2011, 83, 53–60 CrossRef.
- M. J. Schneider, M. Lijewski, R. Woelfel, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2013, 52, 6996–6999 CrossRef CAS PubMed.
- A. Feiz, A. Bazgir, A. M. Balu and R. Luque, Sci. Rep., 2016, 6, 32719 CrossRef PubMed.
- E. Fernandez-Puertas, A. J. Robinson, H. Robinson, S. Sathiyalingam, H. Stubbs and L. J. Edwards, Org. Process Res. Dev., 2020, 24, 2147–2156 CrossRef CAS.
- I. C. Gerber and P. Serp, Chem. Rev., 2020, 120, 1250–1349 CrossRef CAS PubMed.
- M. Munoz, V. Kolb, A. Lamolda, Z. M. de Pedro, A. Modrow, B. J. M. Etzold, J. J. Rodriguez and J. A. Casas, Appl. Catal., B, 2017, 218, 498–505 CrossRef CAS.
- C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2016, 20, 327–360 CrossRef CAS.
- M. Aygün, T. W. Chamberlain, M. del C. Gimenez-Lopez and A. N. Khlobystov, Adv. Funct. Mater., 2018, 28, 1802869 CrossRef.
- D4179 Standard Test Method for Single Pellet Crush Strength of Formed Catalysts and Catalyst Carriers, https://www.astm.org/d4179-22.html, (accessed 28 June 2023).
- J. P. White, T. W. Chamberlain, R. A. Bourne, D. Taylor, C. Brennan and F. L. Muller, Chem. Eng. J., 2020, 394, 124290 CrossRef CAS.
- S. Yenisoy-Karakaş, A. Aygün, M. Güneş and E. Tahtasakal, Carbon, 2004, 42, 477–484 CrossRef.
- W. J. Lee, S. Bera, C. M. Kim, E. K. Koh, W. P. Hong, S. J. Oh, E. A. Cho and S. H. Kwon, NPG Asia Mater., 2020, 12, 1–13 CrossRef.
- N. K. Tripathi, AIMS Mater. Sci., 2018, 5, 1016–1052 Search PubMed.
- X. An, G. Xing, J. Wang, Y. Tian, Y. Liu and Q. Wan, Carbon Lett., 2021, 31, 667–676 CrossRef.
- A. Singh and D. Lal, J. Appl. Polym. Sci., 2008, 110, 3283–3291 CrossRef CAS.
- A. Lekhal, B. J. Glasser and J. G. Khinast, Chem. Eng. Sci., 2001, 56, 4473–4487 CrossRef CAS.
- M. R. Mohammadi, F. Hadavimoghaddam, M. Pourmahdi, S. Atashrouz, M. T. Munir, A. Hemmati-Sarapardeh, A. H. Mosavi and A. Mohaddespour, Sci. Rep., 2021, 11, 17911 CrossRef CAS PubMed.
- X. Zhou, W. Xu, G. Liu, D. Panda and P. Chen, J. Am. Chem. Soc., 2010, 132, 138–146 CrossRef CAS PubMed.
- J. P. White, PhD thesis, University of Leeds, 2022 Search PubMed.
- J. Wang, C. Du, Q. Wei and W. Shen, Energy Fuels, 2021, 35, 4358–4366 CrossRef CAS.
- J. Gao, C. Ren, X. Huo, R. Ji, X. Wen, J. Guo and J. Liu, ACS ES&T Eng., 2021, 1, 562–570 Search PubMed.
- NIST X-ray Photoelectron Spectroscopy (XPS) Database, Version 3.5, https://srdata.nist.gov/xps/, (accessed 7 June 2023).
- E. Sutter and P. Sutter, J. Phys. Chem. C, 2012, 116, 20574–20578 CrossRef CAS.
- V. P. Zhdanov and B. Kasemo, Chem. Phys. Lett., 2008, 452, 285–288 CrossRef CAS.
- L. J. Wang, J. Zhang, X. Zhao, L. L. Xu, Z. Y. Lyu, M. Lai and W. Chen, RSC Adv., 2015, 5, 73451–73456 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01718d |
| This journal is © The Royal Society of Chemistry 2024 |