Nickel-catalyzed tandem conversion of paraformaldehyde![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :methanol to hydrogen and formate/chemo- and stereoselective hydrogenation of alkynes under neutral conditions†
:methanol to hydrogen and formate/chemo- and stereoselective hydrogenation of alkynes under neutral conditions†
Received
8th December 2023
, Accepted 27th March 2024
First published on 28th March 2024
Abstract
The development of new catalytic protocols for clean and COx-free hydrogen generation from fundamental feedstocks is always interesting and challenging. Herein, we disclose nickel-catalyzed dihydrogen generation from a mixture of paraformaldehyde–methanol under base-free and activator-free conditions. The dihydrogen generation from this redox combination under neutral, oxidative coupling conditions has been integrated with the hydrogen transfer reactions such as chemo- and stereoselective hydrogenation of alkynes in a tandem manner. This unprecedented strategy provides diverse highly stereoselective olefins with excellent tolerance of reducible functional groups such as ether, silyl ether, aldehyde, keto, ester, nitrile, halides including bromo and iodo groups, and heteroarenes. Additionally, we demonstrated catalytic stereo-interconversion of alkenes under benign conditions. The affordable gram-scale synthesis of some important pharmaceutical bioactive molecules has further enhanced their synthetic value.
1. Introduction
Methanol and paraformaldehyde are platform chemicals that are abundantly available and manufactured on a massive scale from synthesis gas. The global methanol market reached US$34.5 billion in 2022 and is projected to grow to US$47.0 billion by 2028, exhibiting a compound annual growth rate (CAGR) of 5.1% during 2023–2028.1 Additionally, the formaldehyde market was valued at US$7.92 billion in 2022 and is anticipated to grow at a CAGR of 5.7% from 2023 to 2030.2 Formaldehyde plays a significant role in living cell metabolism,3 serving as a C1 feedstock for the synthesis of carbohydrates as well as amino acids.4,5 The use of paraformaldehyde (polymer form of formaldehyde) has become more convenient and versatile in synthetic chemistry due to its ease of handling, storage, and transportation.6,7 The use of paraformaldehyde as both carbonyl electrophile and terminal reductant as well as in formaldehyde-mediated hydrogen auto-transfer in catalytic C–C couplings with alkynes is well established.8,9 Although synthetic applications of paraformaldehyde are manifold, the role of formaldehyde in the energy sector is limited.10 Of late, significant progress has been made in the generation of hydrogen from renewables.11–24 Notably, formaldehyde is an interesting hydrogen-rich molecule containing 6.7 wt% H2 in the gaseous phase and 8.4 wt% in the aqueous phase. The dihydrogen-releasing reaction of aqueous formaldehyde (H2CO + H2O ↔ CO2 + 2H2; ΔH = −35.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kJ mol−1) is exergonic. This contrasts with dihydrogen release from aqueous methanol (12.5 wt%; ΔH = +53.3 kJ mol−1).25 Thus, formaldehyde can act as a potential hydrogen carrier molecule under suitable catalytic conditions.25–27 The research groups of Prechtl,25,26 Grützmacher,27 Xu,28 Himeda,29 Singh,30–32 and Fukuzumi33 have explored the water-mediated complete decomposition of paraformaldehyde into molecular hydrogen and carbon dioxide using transition-metal complexes. Indeed, new protocols for hydrogen generation from organic feedstocks are always interesting and challenging.34,35 It is postulated that the redox mixture of paraformaldehyde and methanol will produce dihydrogen and methyl formate under a suitable catalytic condition. We envisioned that the dihydrogen generation from a mixture of paraformaldehyde and methanol could be effectively integrated with the hydrogen transfer reactions for fundamentally important synthetic organic transformation. This process involves clean and COx-free hydrogen production under oxidative coupling conditions followed by chemo- and stereoselective hydrogenation of unsaturated organic compounds under base-metal catalysis is unprecedented. Donohoe and co-workers reported a reductive C3 functionalization of pyridinium and quinolinium salts under iridium catalysis using a mixture of methanol and formaldehyde.36 Zhang and co-workers applied a similar strategy for diastereoselective annulation of azo-arenes under Ru catalysis.37
kJ mol−1) is exergonic. This contrasts with dihydrogen release from aqueous methanol (12.5 wt%; ΔH = +53.3 kJ mol−1).25 Thus, formaldehyde can act as a potential hydrogen carrier molecule under suitable catalytic conditions.25–27 The research groups of Prechtl,25,26 Grützmacher,27 Xu,28 Himeda,29 Singh,30–32 and Fukuzumi33 have explored the water-mediated complete decomposition of paraformaldehyde into molecular hydrogen and carbon dioxide using transition-metal complexes. Indeed, new protocols for hydrogen generation from organic feedstocks are always interesting and challenging.34,35 It is postulated that the redox mixture of paraformaldehyde and methanol will produce dihydrogen and methyl formate under a suitable catalytic condition. We envisioned that the dihydrogen generation from a mixture of paraformaldehyde and methanol could be effectively integrated with the hydrogen transfer reactions for fundamentally important synthetic organic transformation. This process involves clean and COx-free hydrogen production under oxidative coupling conditions followed by chemo- and stereoselective hydrogenation of unsaturated organic compounds under base-metal catalysis is unprecedented. Donohoe and co-workers reported a reductive C3 functionalization of pyridinium and quinolinium salts under iridium catalysis using a mixture of methanol and formaldehyde.36 Zhang and co-workers applied a similar strategy for diastereoselective annulation of azo-arenes under Ru catalysis.37
Chemical production through redox transformations, such as catalytic (de)hydrogenation and related reactions, provides innovative solutions to classical approaches (Fig. 1).27,38–46 Recently, increasing interest has been in utilizing earth-abundant metal-based catalysts and sustainable feedstock for (de)hydrogenation reactions.47,48 Among the 3d transition metal-based catalysts, nickel has received significant attention due to its demanding structures as well as remarkable reactivity with maximum alterability, resulting in numerous fascinating applications in both academic and applied research.49–51 The seminal discovery of hydrogenation of unsaturated compounds, mostly using heterogeneous nickel catalysts, was made by Sabatier, Senderens, Ipatieff, and Raney.52,53 By contrast, (transfer) partial hydrogenation of alkynes to alkenes catalyzed by the first-row 3d transition metal catalysts under homogeneous conditions is rare and less investigated.54–57 There are limited reports on catalytic semihydrogenation of alkynes to stereoselective olefins under nickel catalysis;58–63 nevertheless, most of the methods suffer due to the use of strong reductants and basic or acidic conditions. Hence, the development of a general and operationally simple strategy by using readily available platform chemicals is highly desirable in synthetic chemistry. Herein, we developed a nickel-catalyzed dihydrogen generation from a mixture of paraformaldehyde–methanol under base-free and activator-free conditions. The dihydrogen generation from this redox combination under neutral, oxidative coupling conditions has been integrated with hydrogen transfer reactions such as chemo- and stereoselective hydrogenation of alkynes in a tandem manner. This unprecedented strategy provides diverse highly stereoselective olefins with excellent tolerance of reducible functional groups such as ether, silyl ether, aldehyde, keto, ester, nitrile, halides including bromo and iodo groups, and heteroarenes. Additionally, we demonstrated catalytic stereo-interconversion of alkenes (Z to E) under benign conditions. We successfully applied this nickel catalysis for affordable gram-scale synthesis of life science molecules.
 |
| | Fig. 1 Various approaches of hydrogen involving reactions under transition metal catalysis. (a) Dehydrogenation of formaldehyde into H2 and CO2.25–33 (b) Direct hydrogenation reactions using H2 gas.38–40,47,48 (c) Catalytic transfer hydrogenation using various hydrogen sources.41–43 (d) Borrowing hydrogenation strategy for various C–C and C–N bond-forming reactions.43–46 (e) Dehydrogenative hydrogenation through oxidative coupling (this work). | |
2. Results and discussion
2.1. Selective conversion of paraformaldehyde![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :methanol to hydrogen and methyl formate
:methanol to hydrogen and methyl formate
In transition-metal catalysis, bidentate phosphine ligands are excellent spectators and can be tuned to deliver effective electronic and steric properties required for a given synthetic reaction occurring at the metal center.64 In this regard, we began our initial studies on the generation of hydrogen from the combination of paraformaldehyde and methanol with the formation of methyl formate using a catalytic amount of commercially available [1,3-bis(diphenylphosphino)propane]dichloronickel(II) as a catalyst. To get an insight into the formation of dihydrogen, deuterium labeling experiments were conducted. Initial labeling studies confirmed that methanol acts as a protic source and paraformaldehyde acts as a source of a hydride ion. The formation of methyl formate under the present oxidative coupling conditions was qualitatively analyzed using GC-MS and NMR spectra (ESI† Fig. S3–S5, S8 and S9). Similarly, different combinations of deuterated methanol/lower alcohols and deuterated paraformaldehyde were performed under standard reaction conditions, and the corresponding alkyl formates were identified in the chromatographic analysis (Fig. 2). These results demonstrate that the mixture of paraformaldehyde and methanol generates hydrogen gas in the presence of a nickel catalyst. The excess methanol served as a reaction medium. A noteworthy point is that the production of dihydrogen does not require any base and results in methyl formate as a co-product and dimethyl acetals as irrelevant side products. The liberation of gaseous molecules such as H2, HD, and D2 was qualitatively identified using an evolved gas analyzer technique (ESI† Fig. S1 and S2). We envisaged that dihydrogen generation from the redox combination of paraformaldehyde with methanol under nickel-catalyzed oxidative coupling conditions could be effectively integrated with hydrogen transfer reactions for fundamentally important synthetic organic transformations. In our continuous efforts, under optimized catalytic conditions, alkynes were efficiently hydrogenated in a tandem manner and offered the corresponding E-alkenes with high chemo- and stereoselectivity by the same single-site nickel catalyst (Table 1).
 |
| | Fig. 2 Hydrogen gas generation under nickel-catalyzed oxidative coupling conditions using different combinations of paraformaldehyde, methanol, and ethanol. Reaction conditions: paraformaldehyde (1 mmol), [1,3-bis(diphenylphosphino)propane]dichloronickel(II) (NiCl2·dppp; 10 mol%) and methanol or ethanol (1 mL) heated at 110 °C for 16 h. n.r. = no reaction. | |
Table 1 Optimization conditions for the E-selective transfer semihydrogenation under oxidative coupling conditions
| Reaction conditions: diphenylacetylene 1a (0.5 mmol), (HCHO)n (1.5 mmol), [1,3-bis(diphenylphosphino)propane]dichloronickel(II) (NiCl2·dppp, 10 mol%) and methanol (1 mL) heated at 110 °C for 16 h. Yields and selectivity of E/Z were determined using GC analysis. 12 mol% of ligand (L4). n.r – no reaction. |

|
2.2. Tandem oxidative coupling and transfer semihydrogenation of alkynes: optimization of reaction conditions
Based on initial studies on the selective conversion of paraformaldehyde:methanol to hydrogen and formate, we envisioned that the in situ generated H2 can be utilized for chemo- and stereoselective hydrogenation of alkynes under neutral conditions in a tandem manner. Thus, heating a reaction mixture of NiCl2·dppp (10 mol%), paraformaldehyde, and diphenyl acetylene 1a in methanolic solution at 110 °C under closed argon conditions yielded a stereoselective E-alkene 1 in 78% yield with a 99:1 E/Z ratio (Table 1, entry 1). To achieve better catalytic activity, several key reaction parameters such as solvent (other alcohols), the equivalents of paraformaldehyde, different nickel sources, ligands, and temperature were systematically studied (Tables 1 and S1–S4†). It is very interesting and important to note that among the commonly used bisphosphine ligands in nickel catalysis, only dppp (ligand L4) yielded the desired product in excellent yields with selectivity (Table 1). Such ligand-enabled reactivity and selectivity in the production formation is fascinating in chemical synthesis and rarely reported under nickel catalysis.64–66
Performing the present catalytic reaction in ethanol also showed a similar reactivity and selectivity of the product (Table 1, entry 2). However, other alcohols failed to yield the semitransfer hydrogenated product under the optimized reaction conditions (Table 1, entry 3). Notably, less reactivity was observed in the case of aqueous formaldehyde in methanol or using a paraformaldehyde:H2O mixture (Table 1, entry 4). This result underlines that the nickel catalyst is barely suitable to be used in the presence of water and shows much lower activity than the Ru-catalyzed transfer hydrogenation of alkynes using a paraformaldehyde:H2O mixture as the hydrogen source applying an air- and water-stable catalyst.67 A few examples of base-mediated aqueous formaldehyde as a hydrogen surrogate under transition metal catalysis for the hydrogenation of aldehydes68,69 and α,β-enone derivatives70 were also documented. In the absence of paraformaldehyde, there is no formation of hydrogen gas (Table 1, entry 5). This result indicates that methanol alone is not the source of dihydrogen. Recently, methanol was used as a hydrogen donor under basic conditions in the case of Mn-catalyzed semi-transfer hydrogenation of alkynes to (Z)-olefins.71 Furthermore, the in situ generated nickel(II) complex obtained by reacting [1,3-bis(diphenylphosphino)propane] ligand with NiCl2 also gave similar results (Table 1, entry 6). Decreasing the catalyst loading to 5 mol% yielded only 52% of 1 (Table 1, entry 7). Thus, the optimum amount of catalyst required for this tandem process is 10 mol%. Other nickel(II) sources such as nickel(II) chloride ethylene glycol dimethyl ether complex, Ni(acac)2, and Ni(Cp)2 with 1,3-bis(diphenylphosphino)propane ligand were unsuccessful and did not yield the desired product under standard reaction conditions (Table 1, entries 8–10).
Under optimized reaction conditions, nickel(II) triflate with 1,3-bis(diphenylphosphino)propane gave 43% of 1 with excellent E/Z selectivity (Table 1, entry 11). Importantly, the absence of nickel catalyst as well as the absence of ligand afforded no conversion (Table 1, entry 12). Temperature plays an essential role in the present semihydrogenation reaction under the present oxidative coupled conditions. Significantly, increasing the reaction temperature did not influence the product formation. Lowering the temperature to 80 °C yielded only 48% of 1 (Table 1, entry 13), and no product formation was observed at room temperature. In all the cases, no over-reduced alkane was observed. Furthermore, the results from the Hg-dropping experiment and hot filtration tests confirm the predominantly homogeneous nature of the catalytic system (see the ESI†). Indeed, the reaction mixture is primarily homogeneous with no noticeable precipitate.
2.3. Transfer semihydrogenation of alkynes: substrate scope
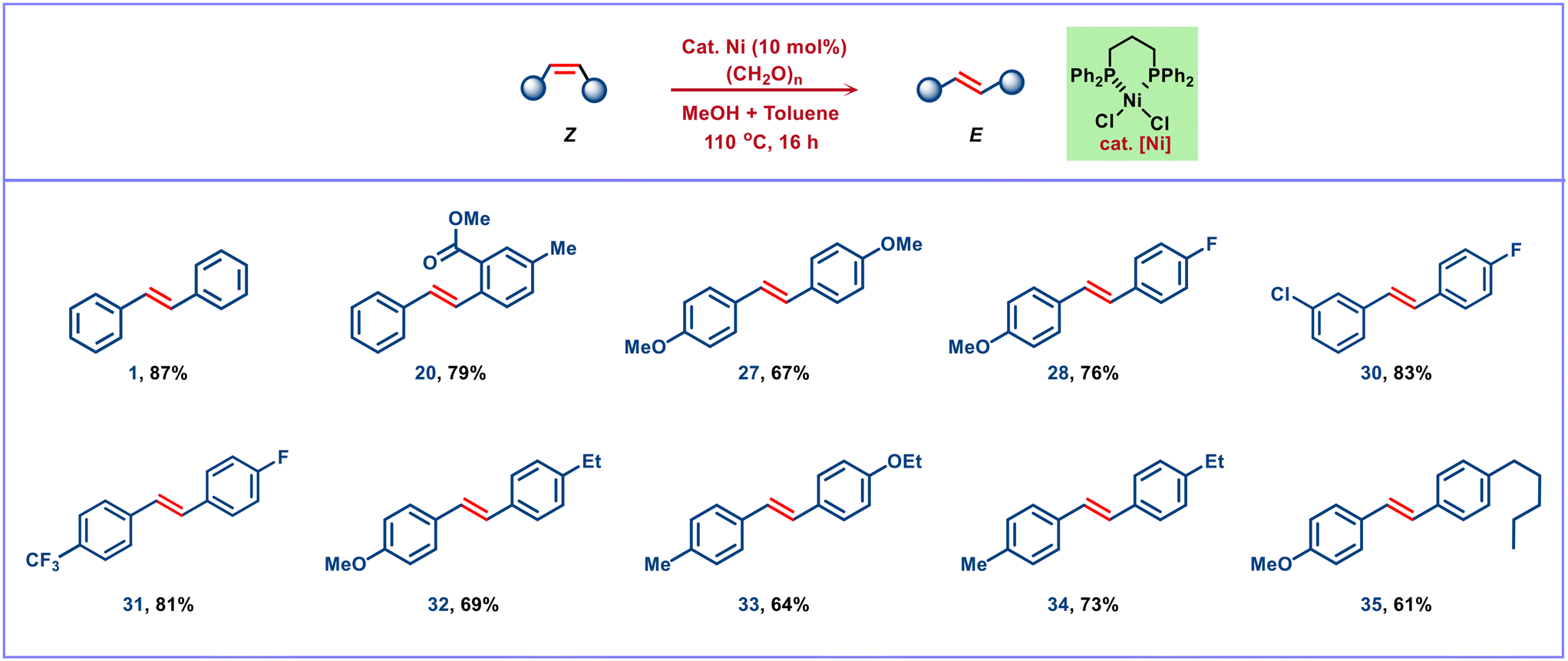
After establishing the optimized reaction conditions (Table 1), the developed synthetic methodology was applied to the broad range of internal alkynes under neutral conditions. The scope and potential utility of this method have been established and illustrated in Fig. 3. An extensive range of substrates with diverse functionalities has been studied and smoothly underwent semitransfer hydrogenation to give E-alkenes as the major product under the present ligand-enabled nickel-catalyzed conditions. The substrate bearing different steric and electronic environments provided the corresponding E-alkenes in moderate to good yield with excellent E/Z selectivity (up to 99%). The transfer semihydrogenation proceeds smoothly for the internal alkynes with substituted aryl groups such as phenyl, biphenyl (products 1–2 in 61–78% yield), and naphthalenes (product 3 in 75% yield). However, the biphenyl system 2b gave a moderate yield of 56% with an 85:15 E/Z ratio. The lowering of E/Z selectivity is probably due to the rotational flexibility of the biphenyl functional group. In particular, these methods were executed with chemo- and stereoselectivity, resulting in the reduction of substituted diaryl acetylenes featuring an array of electron-donating and electron-withdrawing substituents. The electronic environment does not affect the E- selective semihydrogenation, and therefore the electron-donating groups such as methyl, methoxy, ether (1,3 dioxolane, benzyloxy, and silyl ether), and strong electron-withdrawing groups (halides, cyano, trifluoromethyl) containing diarylalkynes gave good yields with excellent selectivity under standard conditions (products 4–14 in 66–82% yield). It was noted that easily reducible carbonyl-containing (aldehyde, ketone, and ester) diphenyl acetylene affords the corresponding semihydrogenated E-isomers 15–20 in 66–79% yields with up to 99:1 E/Z ratio. In addition, heterocyclic internal alkynes such as thiophenyl, pyridyl, bipyridyl, and quinoline derivatives provided the targeted products 21–25 in 60–82% yields with excellent stereoselectivity, respectively. Nevertheless, symmetrically as well as unsymmetrically disubstituted diphenyl acetylene derivatives with various electron-donating and electron-withdrawing substituents resulted in the corresponding E-alkenes 26–35 in moderate to good yields (up to 82%) with excellent E-selectivity under neutral conditions. Control experiments reveal that facile alkyne insertion with a Ni–H intermediate and subsequent alkene isomerization lead to the observed selectivity. It was expected that nickel hydride species are formed through the oxidation of the hemiacetal intermediate (generated from the reaction of paraformaldehyde and methanol) to give methyl formate. As a special highlight, these hydrogenation conditions did not affect the other easily reducible functional groups such as bromo (11), alkoxy ether (5–8, 14, 27–28, 32, 33, 35), cyano (12), and carbonyl (15–20) moiety and provided only the alkyne reduced semihydrogenated products with excellent selectivity of E-isomer. Unlike the other known transfer hydrogenation of alkynes using various reducing agents, the current study allows higher chemoselectivity with exceptional functional group tolerance under mild and neutral conditions.
 |
| | Fig. 3 Chemo- and stereoselective semihydrogenation of alkynes. Reaction conditions: alkyne (0.5 mmol), (HCHO)n (1.5 mmol), NiCl2·dppp (10 mol%) and methanol (1 mL) heated at 110 °C under an argon atmosphere for 16 h. †Methanol (2 mL) heated at 150 °C for 24 h. ‡Ethanol (2 mL) heated at 150 °C for 24 h. Yields in parentheses are isolated yields. | |
2.4. Applications in the synthesis of bioactive molecules
The synthetic utility of transfer semihydrogenation reaction using paraformaldehyde and methanol is further demonstrated for the E-selective synthesis of biologically relevant molecules. To our delight, a prominent biologically active molecule, resveratrol, was prepared using a two-step protocol. The hydrogenation of 36a under standard reaction conditions yielded the 36 intermediate (52% isolated yield) and was followed by demethylation using BBr3 in dichloromethane, offering resveratrol (36′′) in 65% yield. The methoxylated analogs of resveratrol 36 possess increased lipophilicity and pharmacological activity than resveratrol.72,73 This strategy was extended to the synthesis of natural products such as piceatannol (37) and pinosylvin (38). Piceatannol is a hydroxylated analog of resveratrol and is produced from resveratrol by microsomal cytochrome P450.74 Furthermore, we have also synthesized a drug for breast cancer treatment DMU-212 (39) in a single-step process from the corresponding alkyne 39avia the present transfer semihydrogenation strategy. Besides, DMU-212 has strong anti-cancer activity with higher chemoprotective activity.75,76 Finally, this protocol was successfully utilized for the synthesis of estrogen receptor intermediate 40 under our optimal reaction conditions (Scheme 1).
 |
| | Scheme 1 Application of nickel-catalyzed transfer semihydrogenation. Synthesis of biologically important molecules. | |
2.5. Mechanistic studies
A series of control experiments were performed under optimized reaction conditions to obtain insights into the reaction mechanism. The treatment of 1a under standard reaction conditions generates hydrogen gas which was qualitatively analyzed using gas chromatographic analysis (Scheme 2a, ESI† Fig. S1 and S2). To verify the source of hydrogen, a deuterium labeling experiment was conducted using deuterated paraformaldehyde as well as deuterated methanol (Scheme 2b). It was observed that among the two hydrogens in E-stilbene, in the first case, one of the hydrogen atoms was completely replaced by deuterium (1-D1) using methanol-D1 and in the second case, both hydrogen atoms were completely replaced by deuterium (1-D2) in the presence of deuterated paraformaldehyde and methanol-D1 (Scheme 2b). It was expected that nickel hydride species is formed through the oxidation of the hemiacetal intermediate (generated from the reaction of paraformaldehyde and methanol) to give methyl formate. Gratifyingly, no over-reduced products were observed in this strategy; thus, the catalytic system selectively hydrogenates only the internal triple bonds.
 |
| | Scheme 2 Mechanistic insights. | |
2.6. Stereo-interconversion of alkenes: Z → E isomerization sequence
It is important to note that the reaction of Z-stilbene under optimized reaction conditions selectively yielded its stereo-interconverted product. Hence it was confirmed that initially the formation of Z-isomer occurs. Then, the formed Z-isomer is subsequently isomerized to give an E-isomer under nickel catalysis. The deuterium labelling isomerization experiment indicates the incorporation of deuterium ion in the stilbene molecule follows addition followed by an elimination-type mechanism with the help of Ni–H species (Scheme 3).
 |
| | Scheme 3 Isomerization of Z-stilbene. | |
The mechanistic studies of the present catalytic system revealed that the formation of Z-alkenes followed by subsequent isomerization reaction catalyzed by a Ni–H species offered the E-alkene as the major product. A similar Z → E-isomerization sequence in the semihydrogenation of alkynes was observed under nickel catalysis.58–60 Based on these results, this redox combination of paraformaldehyde and methanol was used as a green hydrogen source under Ni catalysis for the alkene isomerization reaction. Thus, various symmetrical and unsymmetrical Z-alkenes were subjected to our standard reaction conditions and selectively provided the corresponding E-isomers with good to excellent yields (Fig. 4).
 |
| | Fig. 4 Nickel catalyzed stereo-intercoversion of alkenes under neutral conditions. Reaction conditions: Z-alkene (0.5 mmol), (HCHO)n (1.5 mmol), NiCl2·dppp (10 mol%), toluene (0.5 mL), and methanol (2 mL) heated at 150 °C under an argon atmosphere for 24 h. All are isolated yields. | |
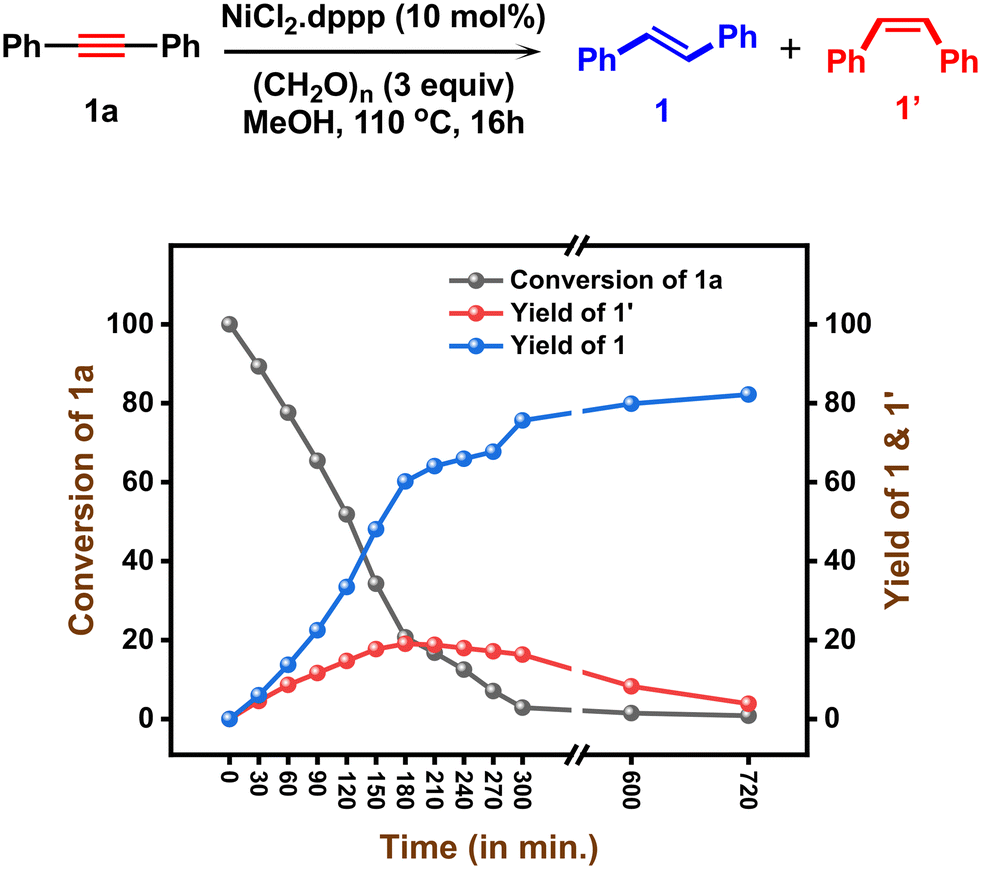
As a result of chemoselective semihydrogenation of internal alkynes and stereo-interconversion of alkenes under nickel catalysis, we applied this protocol for the selective removal of alkyne as well as Z-alkene impurities from E-alkenes. This is an important and highly demanding process in industrial applications. In general, it plays a crucial role in the valorization of bulk feedstocks, e.g., the steam cracking process.77 Under standard reaction conditions, a mixture containing alkyne 1a, Z-alkene 1′, and an excess of E-alkene 1 (in a ratio of 1:1:100) was subjected to experimental procedures. Notably, in all cases, the alkyne 1a and Z-alkene 1′ conversion was observed quantitatively, and the alkyne and Z-alkene impurities were selectively removed under standard conditions (Fig. 5).
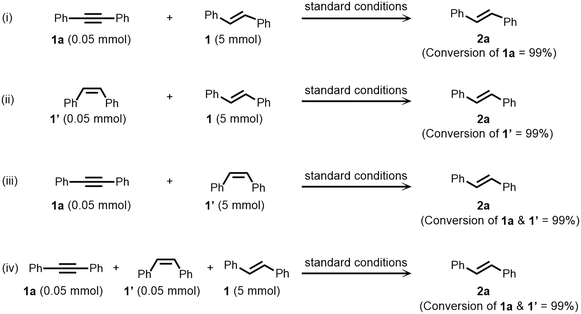 |
| | Fig. 5 Selective removal of alkyne impurities. Standard conditions: starting material (x mmol), (HCHO)n (y mmol), NiCl2·dppp (10 mol%), toluene (0.5 mL) and methanol (2 mL) heated at 150 °C for 24 h under an argon atmosphere. All are isolated yields. | |
2.7. Kinetic studies
Time-dependent experiments were performed in an oven-dried screw-capped tube under an argon atmosphere at 110 °C. The conversion of diphenylacetylene (1a) and yield of stilbene (1 and 1′) were determined by gas chromatography at regular intervals. All reported values represent the average of three independent experiments. As shown in the kinetic profile (Fig. 6), the starting material, alkyne 1a, was completely consumed after 16 h. Initially, the E-olefin formation rate was relatively higher than that of its corresponding Z-isomer. However, only a negligible amount of Z-isomer was formed and further isomerized into the corresponding E-isomer. Notably, no over-reduced products were observed during the entire reaction process.
 |
| | Fig. 6 Kinetic profile for the E-selective semihydrogenation of alkynes. | |
Next, kinetic studies were carried out using the initial rate approximation to determine the order of each component in the nickel-catalyzed partial hydrogenation of alkynes using a paraformaldehyde:methanol mixture as a hydrogen source. (Fig. 7). The reaction rate increases with the initial concentration of 1a at lower concentrations. However, this rate of increase diminishes and may even reverse at higher concentrations of 1a. A plot of log(rate) versus log(concentration of 1a) reveals a fractional order dependence on the concentration of alkyne 1a throughout the semihydrogenation process, observable at both low and high concentration ranges. Similarly, upon increasing the concentration of (CH2O)n, the rate of the reaction is independent, and a negative slope of 0.05 was obtained from the plot of log (rate) vs. log (conc. of (CH2O)n), establishing that the reaction follows zeroth-order behavior in the concentration of (CH2O)n. Next, the dependence of reaction rate on different loadings of nickel catalyst was studied. A slope of 0.081 was obtained from the plot of log (rate) vs. log (conc. of Ni), suggesting a zeroth-order reaction in the loading of a nickel catalyst. Thus, the reaction rate is independent with respect to the concentration of alkyne 1a, paraformaldehyde, and nickel catalyst.
 |
| | Fig. 7 Kinetic study. The graph of (A1) Product concentration vs. time with different low concentrations of 1a. (B1) log (rate) vs. log (conc. of 1a at low conc.). (A2) Product concentration vs. time with different high concentrations of 1a. (B2) log (rate) vs. log (conc. of 1a at high conc.). (C) Product concentration vs. time with increasing concentration of (CH2O)n. (D) log (rate) vs. log (conc. of (CH2O)n). (E) Graph of product concentration vs. time with increasing concentration of Ni catalyst. (F) log (rate) vs. log (conc. of cat. Ni). All the reactions were carried out individually with the respective time intervals. | |
To gain insights into the nature of nickel species involved in the present catalytic reaction, X-ray photoelectron spectroscopy (XPS) analysis was performed to determine the oxidation state of nickel species. Fig. 8a shows the Ni 2p core level spectrum of the catalyst before the reaction. The 2p core level shows a single symmetric peak located at 854 eV, which matches well with Ni(II) where nickel is coordinated with two chloride ligands and phosphine. The spectra collected after completing the reaction were analyzed using XPS without exposure to the atmosphere using a vacuum transfer module. The spectrum obtained after the reaction drastically differs from that of the precursor (Fig. 8b). The deconvoluted spectra showed two well-resolved species in the Ni 2p region. The one observed at a lower binding energy at ∼853 eV is slightly higher than that of Ni(0), which is generally reported at 852.6 eV.78 In our case, this species between Ni(0) and Ni(II) is assigned to a nickel coordinated to a methoxy or hydride ligand. The one observed at higher binding energy is assigned to nickel which is coordinated to an alkene or alkyne complex. Hence, it shifts the binding energy on nickel from 854 eV in the precursor to 855.4 eV in the catalyst.
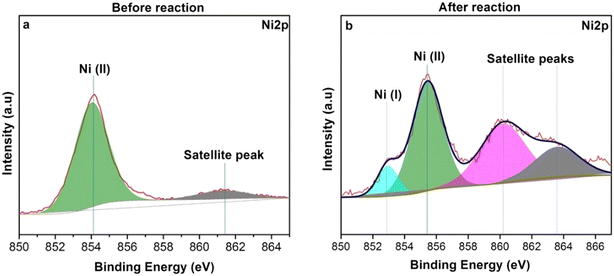 |
| | Fig. 8 XPS data. (a) Before reaction of the pre-catalyst. (b) After reaction of the catalyst. | |
Full quantum chemical calculations were performed using density functional theory (DFT) at the dispersion and solvent-corrected PBE/TZVP level of theory to understand the mechanism of the alkyne hydrogenation reaction in the presence of the Ni(II)·dppe catalyst I. The values (in kcal mol−1) were calculated at the PBE/TZVP level of theory with DFT, with methanol (ε = 32.5) modeled as the solvent (see the ESI†). The reaction is initiated at the square planar complex A (precatalyst Ni(II)). In the presence of a methanol solvent, one of the chloride ligands is replaced by the methoxy group of MeOH (complex II). In the next step, complex II reacts with paraformaldehyde. The methoxy ligand of complex II is substituted by hydride via intermediate III to form a new Ni(II) catalyst I (Scheme 4, cycle A), which acts as the catalyst in the entire diphenylacetylene to (E)-stilbene transfer hydrogenation cycle.
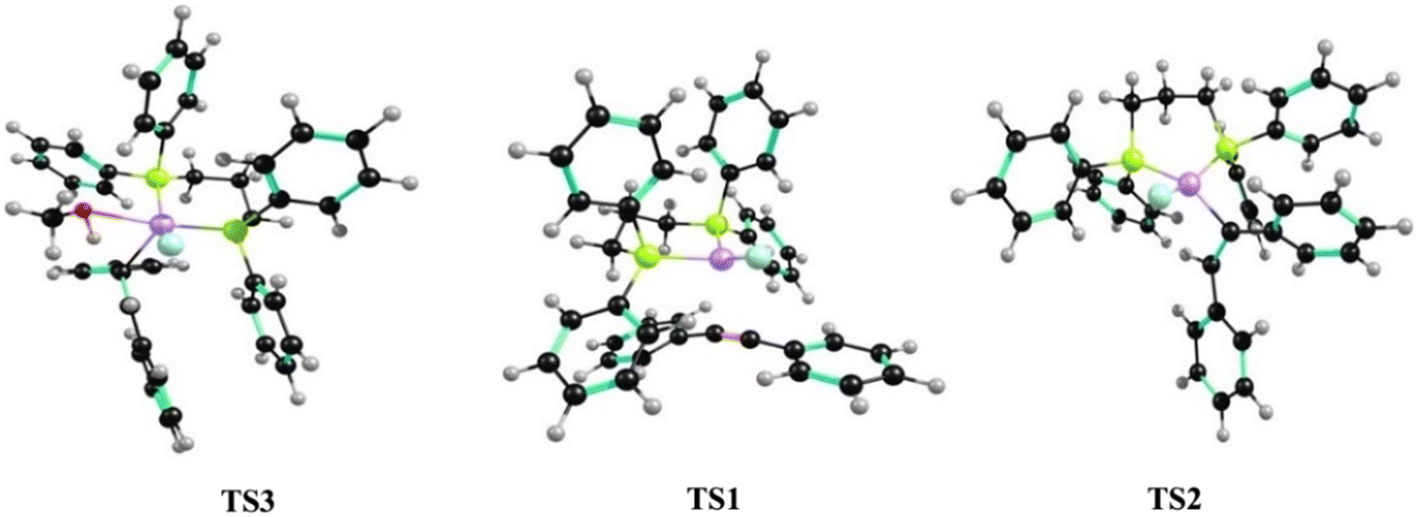
In the first step of the catalytic cycle, diphenylacetylene 1a approaches the square planar catalyst I and makes a square pyramidal π-complex, Int-1, through the transition state TS1 (Scheme 4, cycle B). In the next step, the reaction proceeds through TS2 with an intramolecular rearrangement occurring, with the hydride being transferred from the Ni(II) center to one of the alkyne carbons. Consequently, the π-complex is broken, and the covalent Ni(II)–C bond (Int-2) is formed. The next step, corresponding to the interaction of methanol with the Ni(II) center, leads to the formation of Int-3. This step proceeds via a four-membered transition state (TS3), where the proton is transferred to the alkyne carbon along with the simultaneous breaking of the Ni(II)–C bond and the formation of the Ni(II)–O bond. Thus, (Z)-stilbene is formed, and catalyst II is regenerated. The transition-state geometries for the nickel catalyst (TS1 to TS3) are illustrated in Fig. 9. The formation of (E)-stilbene may also be explained via the alkene isomerization cycle shown in Scheme 4 (cycle C).
 |
| | Scheme 4 The proposed reaction mechanism for nickel(II)-catalyzed reductive hydrogenation of diphenylacetylene into (Z)-stilbene (cycle B) and isomerization of (Z)-stilbene into (E)-stilbene (cycle C). | |
 |
| | Fig. 9 Transition-state geometries for the nickel catalyst. | |
In the first step of cycle C, the catalyst species I interacts with (Z)-stilbene, leading to Int-4. In the next step, the hydride is transferred from Ni(II) of Int-4 to the closest carbon of (Z)-stilbene, leading to Int-5. Rotation along the C–C bond of this complex leads to the conversion of Int-5 to Int-6 (Scheme 4, cycle C). Now, in the next step, there are two possibilities: either β-H transfer to the Ni center from Int-6 and leading to (E)-stilbene (Pdt) and the regeneration of the catalyst I, or the complete hydrogenation via interaction of methanol with Int-6, where methanol transfers its proton to the carbon center and the Ni(II)–C bond is broken at the same time, leading to the formation of the alkane (Pdt′) and the regeneration of the catalyst II. Nevertheless, the reaction exhibited a preference for β-H elimination, yielding exclusively E-alkenes as a final product.
In conclusion, a mixture of paraformaldehyde and methanol has been successfully used for COx-free hydrogen generation under nickel-catalyzed conditions. The hydrogen generated from this redox combination is effectively used for chemo- and stereoselective partial transfer hydrogenation of alkynes in a tandem manner. Unlike classical hydrogen donors, the paraformaldehyde–methanol mixture generates dihydrogen and methyl formate under base-free and activator-free conditions. Mechanistic studies reveal that paraformaldehyde acts as a source of hydride ions and methanol as a proton source. The affordable gram-scale synthesis of some important bioactive molecules has further enhanced their synthetic value. Further hydrogenation and related reactions are in progress in our laboratory using this strategy. We believe that the present strategy opens a new avenue in COx-free hydrogen generation in the context of the ‘hydrogen economy’ to the advancement in chemical syntheses.
Author contributions
EB conceived and directed the project. MS, SSP, and CG contributed equally to this work. MS, SSP, and CG performed the synthetic experiments and analyzed the data. TD and KV performed the computational studies. MS and EB designed and directed the project and prepared the manuscript with feedback from all authors.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
This work is supported by SERB, India (file no.: SERB/F/8407/2022-2023) and DST-Nanomission (DST/NM/TUE/EE-03/2019–1G-IISERTp). EB is a Swarnajayanti Fellow (Award No.: SERB/F/5892/20202021) and gratefully acknowledges support from the Alexander-von-Humboldt (AvH) Foundation. GS and RK thank IISER-Tirupati for their fellowships. We thank IISER-Tirupati for providing excellent analytical facilities. We thank Dr. C. P. Vinod, CSIR-NCL, Pune for the XPS analysis.
References
- IMARC Group, “Methanol Market: Global Industry Trends, Share, Size, Growth, Opportunity and Forecast 2023–2028” (Report SR112024A650, 2023); https://www.imarcgroup.com/methanol-technical-material-market-report.
- Grand View Research, “Formaldehyde Market Size, Share & Trends Analysis Report By Derivatives (Urea Formaldehyde, Melamine Formaldehyde), By End-use (Building & Construction, Furniture, Automotive), By Region, And Segment Forecasts, 2023–2030 (Report 978-1-68038-204-4, 2023); https://www.grandviewresearch.com/industry-analysis/formaldehyde-market.
- J. L. Cereghino and J. M. Cregg, Heterologous protein expression in the methylotrophic yeast Pichia pastoris, FEMS Microbiol. Rev., 2000, 24, 45–66 CrossRef CAS PubMed.
- J. D. Sutherland, The origin of life-out of the blue, Angew. Chem., Int. Ed., 2016, 55, 104–121 CrossRef CAS PubMed.
- S. W. Fox and C. R. Windsor, Synthesis of amino acids by the heating of formaldehyde and ammonia, Science, 1970, 170, 984–986 CrossRef CAS PubMed.
- B. Sam, B. Breit and M. J. Krische, Paraformaldehyde and methanol as C1-feedstocks in metal-catalyzed C-C couplings of π-unsaturated reactants: Beyond hydroformylation, Angew. Chem., Int. Ed., 2015, 54, 3267–3274 CrossRef CAS PubMed.
-
X.-F. Wu, B. Han, K. Ding and Z. Liu, Chemical transformations of C1 compounds, Wiley-VCH, 1st edn, 2022 Search PubMed.
- C. C. Bausch, R. L. Patman, B. Breit and M. J. Krische, Divergent regioselectivity in the synthesis of trisubstituted allylic alcohols by nickel- and ruthenium-catalyzed alkyne hydrohydroxymethylation with formaldehyde, Angew. Chem., Int. Ed., 2011, 50, 5687–5690 CrossRef CAS PubMed.
- C. C. Meyer and M. J. Krische, Iridium-, ruthenium-, and nickel-catalyzed C–C couplings of methanol, formaldehyde, and ethanol with π-unsaturated pronucleophiles via hydrogen transfer, J. Org. Chem., 2023, 88, 4965–4974 CrossRef CAS PubMed.
- L. E. Heim, H. Konnerth and M. H. G. Prechtl, Future perspectives for formaldehyde: Pathways for reductive synthesis and energy storage, Green Chem., 2017, 19, 2347–2355 RSC.
- J. A. Turner, Sustainable hydrogen production, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- D. R. Palo, R. A. Dagle and J. D. Holladay, Methanol steam reforming for hydrogen production, Chem. Rev., 2007, 107, 3992–4021 CrossRef CAS PubMed.
- M. Nielsen,
et al., Low-temperature aqueous-phase methanol dehydrogenation to hydrogen and carbon dioxide, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- L. Lin,
et al., Low-temperature hydrogen production from water and methanol using Pt/alpha-MoC catalysts, Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- J. Kothandaraman, S. Kar, A. Goeppert, R. Sen and G. K. S. Prakash, Advances in homogeneous catalysis for low temperature methanol reforming in the context of the methanol economy, Top. Catal., 2018, 61, 542–559 CrossRef CAS.
- R. E. Rodríguez-Lugo,
et al., A homogeneous transition metal complex for clean hydrogen production from methanol–water mixtures, Nat. Chem., 2013, 5, 342–347 CrossRef PubMed.
- S. Zhang, Y. Liu, M. Zhang, Y. Ma, J. Hu and Y. Qu, Sustainable production of hydrogen with high purity from methanol and water at low temperatures, Nat. Commun., 2022, 13, 5527 CrossRef CAS PubMed.
- A. Boddien,
et al., Efficient dehydrogenation of formic acid using an iron catalyst, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Formic acid as a hydrogen storage material: Development of homogeneous catalysts for selective hydrogen release, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- J. Guo, Y. Zhang, A. Zavabeti, K. Chen, Y. Guo, G. Hu, X. Fan and G. K. Li, Hydrogen production from the air, Nat. Commun., 2022, 13, 5046 CrossRef CAS PubMed.
- Y.-Q. Zou, N. von Wolff, A. Anaby, Y. Xie and D. Milstein, Ethylene glycol as an efficient and reversible liquid-organic hydrogen carrier, Nat. Catal., 2019, 2, 415–422 CrossRef CAS PubMed.
- T. He, P. Pachfule, H. Wu, Q. Xu and P. Chen, Hydrogen carriers, Nat. Rev. Mater., 2016, 1, 16059 CrossRef CAS.
- P. Megía, A. Vizcaíno, J. A. Calles and A. Carrero, Hydrogen production technologies: From fossil fuels toward renewable sources. A mini review, Energy Fuels, 2021, 35, 16403–16415 CrossRef.
- A. Kumar, P. Daw and D. Milstein, Homogeneous catalysis for sustainable energy: Hydrogen and methanol economies, fuels from biomass, and related topics, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- L. E. Heim, N. E. Schlörer, J.-H. Choi and M. H. G. Prechtl, Selective and mild hydrogen production using water and formaldehyde, Nat. Commun., 2014, 5, 3621 CrossRef PubMed.
- L. E. Heim, S. Vallazza, D. van der Waals and M. H. G. Prechtl, Water decontamination with hydrogen production using microwave-formed minute-made ruthenium catalysts, Green Chem., 2016, 18, 1469–1474 RSC.
- M. Trincado, V. Sinha, R. E. Rodriguez-Lugo, B. Pribanic, B. de Bruin and H. Grützmacher, Homogeneously catalysed conversion of aqueous formaldehyde to H2 and carbonate, Nat. Commun., 2017, 8, 14990 CrossRef CAS PubMed.
- Y. Zhan, S. Zhou and Y. Xu, Catalytic hydrogen production from formaldehyde over immobilized ruthenium complexes, Catal. Lett., 2024, 154, 808–815 CrossRef CAS.
- L. Wang, M. Z. Ertem, R. Kanega, K. Murata, D. J. Szalda, J. T. Muckerman, E. Fujita and Y. Himeda, Additive-free ruthenium-catalyzed hydrogen production from aqueous formaldehyde with high efficiency and selectivity, ACS Catal., 2018, 8, 8600–8605 CrossRef CAS.
- M. K. Awasthi and S. K. Singh, Ruthenium catalyzed hydrogen production from formaldehyde−water solution, Sustainable Energy Fuels, 2021, 5, 549–555 RSC.
- S. Patra and S. K. Singh, Hydrogen production from formic acid and formaldehyde over ruthenium catalysts in water, Inorg. Chem., 2020, 59, 4234–4243 CrossRef CAS PubMed.
- S. Patra, A. Kumar and S. K. Singh, Hydrogen production from formaldehyde and paraformaldehyde in water under additive-free conditions: Catalytic reactions and mechanistic insights, Inorg. Chem., 2022, 61, 4618–4626 CrossRef CAS PubMed.
- T. Suenobu, Y. Isaka, S. Shibata and S. Fukuzumi, Catalytic hydrogen production from paraformaldehyde and water using an organoiridium complex, Chem. Commun., 2015, 51, 1670–1672 RSC.
- D. Wie, X. Shi, H. Junge, C. Du and M. Beller, Carbon neutral hydrogen storage and release cycles based on dual-functional roles of formamides, Nat. Commun., 2023, 14, 3726 CrossRef PubMed.
- R. Sang, Z. Wie, Y. Hu, E. Alberico, D. Wie, X. Tian, P. Ryabchuk, A. Spannenberg, R. Razzaq, R. Jackstell, J. Massa, P. Sponholz, H. Jiao, H. Junge and M. Beller, Methyl formate as a hydrogen energy carrier, Nat. Catal., 2023, 6, 543–550 CrossRef CAS.
- A. Grozavu, H. B. Hepburn, P. J. Smith, H. K. Potukuchi, P. J. Lindsay-Scott and T. J. Donohoe, The reductive C3 functionalization of pyridinium and quinolinium salts through iridium-catalysed interrupted transfer hydrogenation, Nat. Chem., 2019, 11, 242–247 CrossRef CAS PubMed.
- H. Zhao, Y. Wu, C. Ci, Z. Tan, J. Yang, H. Jiang, P. H. Dixneuf and M. Zhang, Intermolecular diastereoselective annulation of azaarenes into fused N-heterocycles by Ru(II) reductive catalysis, Nat. Commun., 2022, 13, 2393 CrossRef CAS PubMed.
-
Catalytic Hydrogenation, ed. L. Cerveny, Elsevier, Amsterdam, 1986 Search PubMed.
-
The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed.
-
Modern Reduction Methods, ed. P. G. Andersson and J. J. Munslow, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008 Search PubMed.
- G. Brieger and T. J. Nestrick, Catalytic transfer hydrogenation, Chem. Rev., 1974, 74, 51974 CrossRef.
- D. Wang and D. Astruc, The golden age of transfer hydrogenation, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- B. Taleb, R. Jahjah, D. Cornu, M. Bechelany, M. Al Ajami, G. Kataya, A. Hijazi and M. H. El-Dakdouki, Exploring hydrogen sources in catalytic transfer hydrogenation: A review of unsaturated compound reduction, Molecules, 2023, 28, 7541 CrossRef CAS PubMed.
- P. A. Slatford, M. K. Whittlesey and J. M. J. Williams, C-C Bond formation from alcohols using a Xantphos ruthenium complex, Tetrahedron Lett., 2006, 47, 6787–6789 CrossRef CAS.
- O. Saidi, A. J. Blacker, G. W. Lamb, S. P. Marsden, J. E. Taylor and J. M. J. Williams, Borrowing hydrogen in water and ionic liquids: Iridium-catalyzed alkylation of amines with alcohols, Org. Process Res. Dev., 2010, 14, 1046–1049 CrossRef CAS.
- A. Corma, J. Navas and M. J. Sabater, Advances in one-pot synthesis through borrowing hydrogen catalysis, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Catalytic (de)hydrogenation promoted by non-precious metals – Co, Fe and Mn: Recent advances in an emerging field, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC.
- F. Kallmeier and R. Kempe, Manganese complexes for (de)hydrogenation catalysis: A comparison to cobalt and iron catalysts, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Recent advances in homogeneous nickel catalysis, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- V. P. Ananikov, Nickel: The “spirited horse” of transition metal catalysis, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS.
- V. M. Chernyshev and V. P. Ananikov, Nickel and palladium catalysis: Stronger demand than ever, ACS Catal., 2022, 12, 1180–1200 CrossRef CAS.
-
S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, John Wiley & Sons Inc., 2001 Search PubMed.
-
P. N. Rylander, Catalytic hydrogenation in organic syntheses, Academic Press, 1979 Search PubMed.
- K. C. Kumara Swamy, A. S. Reddy, K. Sandeep and A. Kalyani, Advances in chemoselective and/or stereoselective semihydrogenation of alkynes, Tetrahedron Lett., 2018, 59, 419–429 CrossRef.
- D. Baidilov, D. Hayrapetyan and A. Y. Khalimon, Recent advances in homogeneous base-metal-catalyzed transfer hydrogenation reactions, Tetrahedron, 2021, 98, 132435 CrossRef CAS.
- Y. Wang, X. Cao, L. Zhao, C. Pi, J. Ji, X. Cui and Y. Wu, Generalized chemoselective transfer hydrogenation/hydrodeuteration, Adv. Synth. Catal., 2020, 362, 4119–4129 CrossRef CAS.
- D. Decker, H.-J. Drexler, D. Heller and T. Beweries, Homogeneous catalytic transfer semihydrogenation of alkynes – an overview of hydrogen sources, catalysts and reaction mechanisms, Catal. Sci. Technol., 2020, 362, 6449–6463 RSC.
- D. J. Hale, M. J. Ferguson and L. Turculet, (PSiP)Ni-Catalyzed (E)-Selective semihydrogenation of alkynes with molecular hydrogen, ACS Catal., 2022, 12, 146–155 CrossRef CAS.
- N. O. Thiel, B. Kaewmee, T. Ngoc and J. F. Teichert, A simple nickel catalyst enabling an E-selective alkyne semihydrogenation, Chem. – Eur. J., 2020, 26, 1597–1603 CrossRef CAS PubMed.
- K. Murugesan, C. B. Bheeter, P. R. Linnebank, A. Spannenberg, J. N. H. Reek, R. V. Jagadeesh and M. Beller, Nickel-catalyzed stereodivergent synthesis of E- and Z-alkenes by hydrogenation of alkynes, ChemSusChem, 2019, 12, 3363–3369 CrossRef CAS PubMed.
- K. Li, C. Yang, J. Chen, C. Pan, R. Fan, Y. Zhou, Y. Luo, D. Yang and B. Fan, Anion controlled stereodivergent semi-hydrogenation of alkynes using water as hydrogen source, Asian J. Org. Chem., 2021, 10, 2143–2146 CrossRef CAS.
- X. Wen, X. Shi, X. Qiao, Z. Wu and G. Bai, Ligand-free nickel-catalyzed semihydrogenation of alkynes with sodium borohydride: A highly efficient and selective process for cis-alkenes under ambient conditions, Chem. Commun., 2017, 53, 5372–5375 RSC.
- A. Reyes-Sánchez, F. Cañavera-Buelvas, R. Barrios-Francisco, O. L. Cifuentes-Vaca, M. Flores-Alamo and J. J. García, Nickel-catalyzed transfer semihydrogenation and hydroamination of aromatic alkynes using amines as hydrogen donors, Organometallics, 2011, 30, 3340–3345 CrossRef.
- A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Trends in the usage of bidentate phosphines as ligands in nickel catalysis, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS PubMed.
- D.-M. Wang, L.-Q. She, Y. Wu, C. Zhu and P. Wang, Ligand-enabled Ni-catalyzed hydroarylation and hydroalkenylation of internal alkenes with organoborons, Nat. Commun., 2022, 13, 6878 CrossRef CAS PubMed.
- W.-T. Zhao, J.-X. Zhang, B.-H. Chen and W. Shu, Ligand-enabled Ni-catalysed enantioconvergent intermolecular alkyl-alkyl cross-coupling between distinct alkyl halides, Nat. Commun., 2023, 14, 2938 CrossRef CAS PubMed.
- M. N. A. Fetzer, G. Tavakoli, A. Klein and M. H. G. Prechtl, Ruthenium-catalyzed E -selective partial hydrogenation of alkynes under transfer-hydrogenation conditions using paraformaldehyde as hydrogen source, ChemCatChem, 2021, 13, 1317–1325 CrossRef CAS.
- J. Cook and P. M. Maitlis, Formaldehyde as a hydrogen-donor to aldehydes and ketones in metal-catalysed reactions in water, J. Chem. Soc., Chem. Commun., 1981, 924–925 RSC.
- K. Natte, W. Li, S. Zhou, H. Neumann and X.-F. Wu, Iron-catalyzed reduction of aromatic aldehydes with paraformaldehyde and H2O as the hydrogen source, Tetrahedron Lett., 2015, 56, 1118–1121 CrossRef CAS.
- W. Li and X.-F. Wu, Ruthenium-catalyzed conjugate hydrogenation of α,β-enones by in situ generated dihydrogen from paraformaldehyde and water, Eur. J. Org. Chem., 2015, 2015, 331–335 CrossRef CAS.
- J. Sklyaruk, V. Zubar, J. C. Borghs and M. Rueping, Methanol as the hydrogen source in the selective transfer hydrogenation of alkynes enabled by a manganese pincer complex, Org. Lett., 2020, 22, 6067–6071 CrossRef CAS PubMed.
- J. J. Heynekamp, W. M. Weber, L. A. Hunsaker, A. M. Gonzales, R. A. Orlando, L. M. Deck and D. L. Vander Jagt, Substituted trans-Stilbenes, including analogues of the natural product resveratrol, inhibit the human tumor necrosis factor alpha-induced activation of transcription factor nuclear factor kappa B, J. Med. Chem., 2016, 49, 7182–7189 CrossRef PubMed.
- W. Zhang and M. L. Go, Quinone reductase induction activity of methoxylated analogues of resveratrol, Eur. J. Med. Chem., 2007, 42, 841–850 CrossRef CAS PubMed.
- Y.-J. Surh and H.-K. Na, Therapeutic potential and molecular targets of piceatannol in chronic diseases, Adv. Exp. Med. Biol., 2016, 928, 185–211 CrossRef CAS PubMed.
- Z. Ma, O. Molavi, A. Haddadi, R. Lai and R. A. Gossage, Resveratrol analog trans 3,4,5,4′-tetramethoxystilbene (DMU-212) mediates anti-tumor effects via mechanism different from that of resveratrol, Cancer Chemother. Pharmacol., 2008, 63, 27–35 CrossRef CAS PubMed.
- V. P. Androutsopoulos, I. Fragiadaki and A. Tosca, Activation of ERK1/2 is required for the antimitotic activity of the resveratrol analogue 3,4,5,4′-tetramethoxystilbene (DMU-212) in human melanoma cells, Exp. Dermatol., 2015, 24, 630–641 CrossRef PubMed.
-
G. C. Bond, Metal-catalysed reactions of hydrocarbons, Springer Science, USA, 2005 Search PubMed.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, The role of the Auger parameter in XPS studies of nickel metal, halides and
oxides, Phys. Chem. Chem. Phys., 2012, 14, 2434–2442 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Ekambaram
Balaraman
b and
Ekambaram
Balaraman