
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of strong π-acceptor ligands on Cr-K-edge X-ray absorption spectral signatures and consequences for the interpretation of surface sites in the Phillips catalyst†‡
Yuya
Kakiuchi
 a,
Svetlana
Shapovalova
b,
Bogdan
Protsenko
b,
Sergey
Guda
bc,
Olga V.
Safonova
d,
Alexander
Guda
*b and
Christophe
Copéret
*a
a,
Svetlana
Shapovalova
b,
Bogdan
Protsenko
b,
Sergey
Guda
bc,
Olga V.
Safonova
d,
Alexander
Guda
*b and
Christophe
Copéret
*a
aETH Zürich, Vladimir-Prelog-Weg 2, 8093 Zurich, Switzerland. E-mail: ccoperet@ethz.ch
bThe Smart Materials Research Institute, Southern Federal University, Rostov-on-Don, 344090, Russia. E-mail: guda@sfedu.ru
cInstitute of Mathematics, Mechanics and Computer Science, Southern Federal University, Rostov-on-Don, 344090, Russia
dPaul Scherrer Institute, 5232 Villigen, Switzerland
First published on 20th May 2024
Abstract
X-ray absorption spectroscopy (XAS) has been central to the study of the Phillips polymerization catalyst (CrO3/SiO2). As Cr K-edge XAS signatures are sensitive to the oxidation state, geometry and types of ligands on surface (active) sites, the superposition of these effects makes their interpretation challenging. Notably, CO has been particularly used as a reductant to generate low valent Cr sites from CrO3/SiO2 and as a structural IR probe for analysing reduced Cr surface sites. Hence, it is essential to establish a solid understanding of the spectroscopic impact of CO on low-valent Cr sites. We thus built a series of fully characterized low-valent Cr molecular compounds bearing isoelectronic isocyanide ligands in place of CO, with the goal of understanding the effect of the coordination of π-acceptor ligands on the XANES signature of Cr sites. Cr K-edge spectra supplemented with DFT calculations elucidate the effect of the coordination of π-acceptor ligands on XAS signatures, giving a sharp resonance at the white line while modifying the fine structure due to short Cr–C distances and stability of low-spin Cr(II/III) species. The isocyanide references allow the deconvolution of the XAS spectra of the reduced CrO3/SiO2 catalyst by evaluating the types of surface species and relative amounts of bound CO at different CO pressures and temperatures.
Introduction
The Phillips ethylene polymerization catalyst, based on chromium(VI) oxide supported on silica (CrO3/SiO2), is at the heart of the polymer industry and responsible for ca. 40% of the world production of high-density polyethylene.1,2 The preparation and activation of the Phillips catalyst consists of multiple steps. First, calcination yields a pre-catalyst with isolated Cr(VI) sites.1,3 Upon introduction of ethylene (typically at 100–150 °C) or treatment under CO at high temperatures (at ca. 350 °C),2,4 low-coordinated and low-valent Cr sites are generated and initiate polymerization after an induction period (and removal of CO).2,5 While there is no doubt that active sites are low valent Cr species, there has been a longstanding debate regarding their formal oxidation state, namely Cr(II), Cr(III), or even Cr(V) sites.3Identification of the nature of these surface sites has relied on detailed spectroscopic investigations, which have often employed probe molecules; in this regard, CO has played a key role in the characterization of reduced Phillips catalysts. For instance, the IR signatures of adsorbed CO point to the presence of various types of low valent Cr sites, with blue and red shifted CO bands. It has been proposed that these sites are associated with low-coordinated and highly coordinated Cr(II) sites, known as “non-classical”6–9 and “classical” carbonyl species,4 respectively.
X-ray absorption spectroscopy (XAS) is also considered to be the method of choice to understand the nature of surface sites under various conditions, i.e. during activation, initiation and chain propagation steps.4,10–16 X-ray absorption near-edge spectroscopy (XANES) is also known to provide unique information about local symmetry (related to the coordination number) and density of unoccupied electron states (related to the oxidation state and nature of ligands).15 In addition, XAS can be readily performed in situ or operando enabling the monitoring of the spectral and structural changes of catalysts under relevant conditions.13,14,16 XAS has thus been central to discussions related to the assignment of the formal oxidation state in pristine and reduced Phillips catalysts.3,4 However, assignment of XAS features to specific oxidation states is not trivial. In fact, recent studies on Cr K-edge XAS, based on a library of molecular compounds augmented by a supervised machine learning approach, highlighted significant overlaps of spectral features between Cr(II) and Cr(III) species.17 The investigation of activated Phillips catalysts and related homogenous Cr-based oligomerization catalysts have also demonstrated the essential effect of ligand types and their geometry, by modulating the energy shift of the absorption edge spanning a few eV, thus making the attribution of a specific edge energy to a given oxidation state difficult.18 Furthermore, the coordination of π-acceptor ligands like CO (or ethylene) can potentially have a dramatic impact on the XAS signature, complicating potential interpretation.19 In fact, the reported Cr K-edge XAS spectra of Cr(0)/Cr(I) carbonyl species exhibit interesting high-energy shifted rising edges with characteristic intense peaks.20,21 These examples clearly show the importance of careful benchmarking prior to interpreting spectroscopic data, which calls for extending a spectral library based on well-defined molecular compounds to guide the analysis.5,7
When establishing experimental benchmarks for CO-bound Cr sites, one inherent challenge lies in the accessibility of molecular Cr carbonyl complexes across various oxidation states and coordination numbers in environments close to what could be expected in CO-reduced catalysts. While building a library of molecular Cr–CO adducts is difficult, their isoelectronic counterparts with isocyanide (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) NR) ligands,22–25 that are easy-handled ligands in the condensed phase, offer an unique opportunity to evaluate the effect of π-acceptor ligands on spectroscopic signatures.
NR) ligands,22–25 that are easy-handled ligands in the condensed phase, offer an unique opportunity to evaluate the effect of π-acceptor ligands on spectroscopic signatures.
In this work, we have thus developed a series of Cr(II) and Cr(III) isocyanide adducts bearing siloxide or alkoxide ligands, focused on modelling low-valent Cr–CO adducts, which are commonly proposed to exist in the reduced Phillips catalyst in contact with CO. The library of experimental spectra coupled with theoretical simulations demonstrated the striking effect of isocyanide (and CO) ligands on XANES signatures, modulating their rising edge and pre-edge features that are usually employed to discuss the nature of the Cr sites. The relationships between the spectral features and number of the isocyanide ligands, and their effect on the electronic structure of Cr complexes is addressed via density-functional theory (DFT) calculations. Finally, the experimental XAS spectra of molecular complexes 1–3 are used to decipher the surface species in CO-reduced Phillips catalysts in the presence or absence of a CO probe molecules at different temperatures.
Results and discussion
Synthesis and characterization of molecular Cr-isocyanide adducts
We first developed a series of molecular Cr(II) and Cr(III) compounds, with siloxide or aryloxide ligands as well as the isocyanide ligands, to mimic potential surface sites in the Phillips catalyst in contact with CO (Scheme 1). A bis(isocyanide) adduct of Cr(II) was obtained by treatment of Cr(OAr)2(THF)2 (Ar = 2,6-bis(tert-butyl)phenyl) with 2 equiv. of XyNC (Xy = 2,6-dimethylphenyl), resulting in an exchange of THF ligands to yield Cr(OAr)2(XyNC)2 (1) in 85% yield. Treatment of the less sterically encumbered dimeric [Cr(TBOS)2]2 (TBOS = tris(tert-butoxy)siloxy) with 8 equiv. of XyNC afforded an octahedral complex Cr(TBOS)2(XyNC)4 (2) bearing 4 equatorial isocyanide ligands in 77% yield. Using a smaller amount of XyNC only results in formation of 2 in lower yield along with the starting material. Similarly, addition of 2 equiv. of XyNC to Cr(TBOS)3(THF)2 yielded Cr(TBOS)3(XyNC)2 (3) as a pale blue powder in 78% yield, along with a change from trigonalbipyramidal to octahedral geometry with one κ2-coordinating siloxide ligand. Addition of excess XyNC did not lead to further coordination reactions.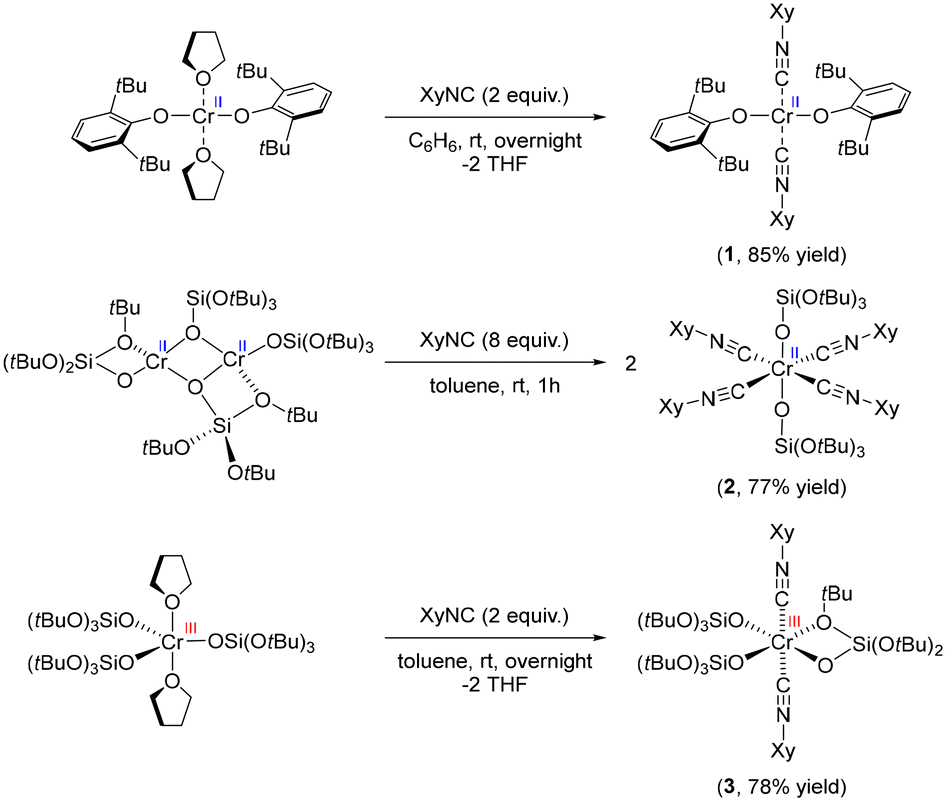 | ||
| Scheme 1 Synthesis of the Cr-isocyanide adduct series. | ||
All these complexes were first studied using single-crystal XRD analysis to unambiguously determine the structure (Fig. S4†). The bonding between the Cr centre and isocyanides can be described by the Dewar–Chatt–Duncanson (DCD) model, originally designed for carbonyl complexes.26,27 The interplay between σ-donation in the form metal ← isocyanide and π-backdonation in the form metal → isocyanide determines the bond length between the metal and isocyanide ligand, as well as the C–N bond length within the ligand. The change in C–N bond length can be observed in the IR spectrum as a red- or blue-shift of the C–N stretching band. C–N bonds in complex 1 were found to be 1.161(4) Å, which is almost identical to free isocyanide (1.160 Å)28 indicating negligible π-back donation. Conversely, tetrakis adduct 2 exhibited elongated C–N bond lengths of 1.164(16) Å and 1.166(16) Å, implying developed π-back donation from the Cr(II) center. Two distinct C–N bond lengths in 2 are likely due to packing effects in the crystal. The C–N bond length of 3 was, on the contrary, shortened to 1.141(10) Å, clearly indicating the poor π-back donation from the Cr(III) siloxide core resulting in the formation of a stronger C–N bond.29,30 Evidence of these findings was further observed in the C–N absorption band in the IR spectra (Fig. S5–S8†). While the parent 2,6-dimethylphenyl isocyanide has its C–N band at 2116 cm−1, siloxide complex 2 exhibits red-shifted C–N bands at 2077 cm−1, which is consistent with an elongated C–N bond length. Conversely, complexes 1 and 3 have blue-shifted C–N bands at 2183 and 2196 cm−1, respectively, indicating the lack of π-back donation in these species. It is noteworthy that, considering that complexes 1 and 2 have formally the same oxidation state, namely Cr(II), they exhibit appreciable differences in terms of C–N bond lengths and C–N stretching bands in their IR spectra.
Notably, compound 2, which has a six-coordinated Cr(II) centre with four isocyanide ligands, displays a red-shifted ṽCN band, while compounds 1 and 3, which have only two isocyanide ligands, show blue-shifted ṽCN bands. These observations are analogous to what has been observed for the IR spectrum of the CO-reduced Phillips catalyst under a CO atmosphere at low temperature. The Phillips catalyst spectrum shows blue- and red-shifted CO bands, attributed to the corresponding low and high coordinated Cr carbonyl species, respectively.4
The magnetic susceptibility of complexes 1–3 was studied using the Evans method,31–34 giving μeff = 5.46μB (1, S = 2), 3.04μB (2, S = 1) and 3.92μB (3, S = 3/2), respectively. The result indicates that 1 and 3 are high-spin (HS) Cr(II)/Cr(III) species, while 2 was found to possess a low-spin (LS) Cr(II) configuration. Although a high-spin state is usually dominant for low-valent Cr species, the presence of strong π-acceptor ligands including isocyanides stabilizes the low-spin state enabling their observation and/or isolation.35–37 In fact, a similar Cr(II) alkoxide complex, Cr(OCtBu2Ph)2(XyNC)4, bearing 4 isocyanide ligands was also reported to have a LS configuration, while the Cr(II) bis(isocyanide) analogue, Cr(OSi(tBu)3)2(tBuNC)2, was found to occupy a HS state, consistent with our results for complexes 1 and 2.38,39
The herein synthesized siloxide/alkoxide species draw some parallels with the potential surface Cr sites interacting with π-acceptor ligands, implying a relevance to the reduced Phillips catalyst probed in the presence of CO (vide infra).
Cr K-edge X-ray absorption spectroscopy
With this series of Cr-isocyanide complexes 1–3 in hand, their experimental XAS spectra were collected and compared to the corresponding oxygen-based analogues which were reported earlier.17 We also recorded the spectrum of Cr(CO)6 as an example of a simple carbonyl complex.20 Cr(CO)6 can be characterized by an intense white line (peak C), originating from the interplay between the Cr center and CO ligand (vide infra). An additional broad peak E appears at ca. 6020 eV due to the scattering of photoelectrons by linearly bonded CO fragments.20 These spectral features were also identified in isocyanide adducts 1–3 suggesting that carbonyl and the isocyanide ligands cause similar effects in the Cr K-edge XAS signatures (see also Fig. S11†). Notably, the position of the white line of Cr(0) (CO)6 coincides with the region expected for the Cr(II)/Cr(III) species, clearly highlighting the impact of CO, a π-acceptor ligand on the XANES signature. Cr(II) bis-isocyanide adduct 1 exhibited negligible pre-edge features, which is typical for a centrosymmetric metal centre (Fig. 1, bottom-blue). Feature D is common for complexes with alkoxide/siloxide ligands among the series, whether or not they have coordinated isocyanide ligands. Compared to the square-planar Cr(II) complex, Cr(OAr)2(THF)2, new peaks C and E develop in the rising edge and high-energy region, paralleling what was found for Cr(CO)6. This suggests that the coordination of π-acceptor ligands is the origin of these spectral features. Another difference is found in shoulder B on the rising edge, which is characteristic of planar complexes and has previously been assigned to the presence of a vacant Cr 4pz orbital aligned perpendicularly to the molecular plane.17 In fact, this shoulder disappears in octahedral compounds 2 and 3. This shoulder B was split in compound 1 compared to Cr(OAr)2(THF)2, which can be attributed to a less symmetric coordination environment.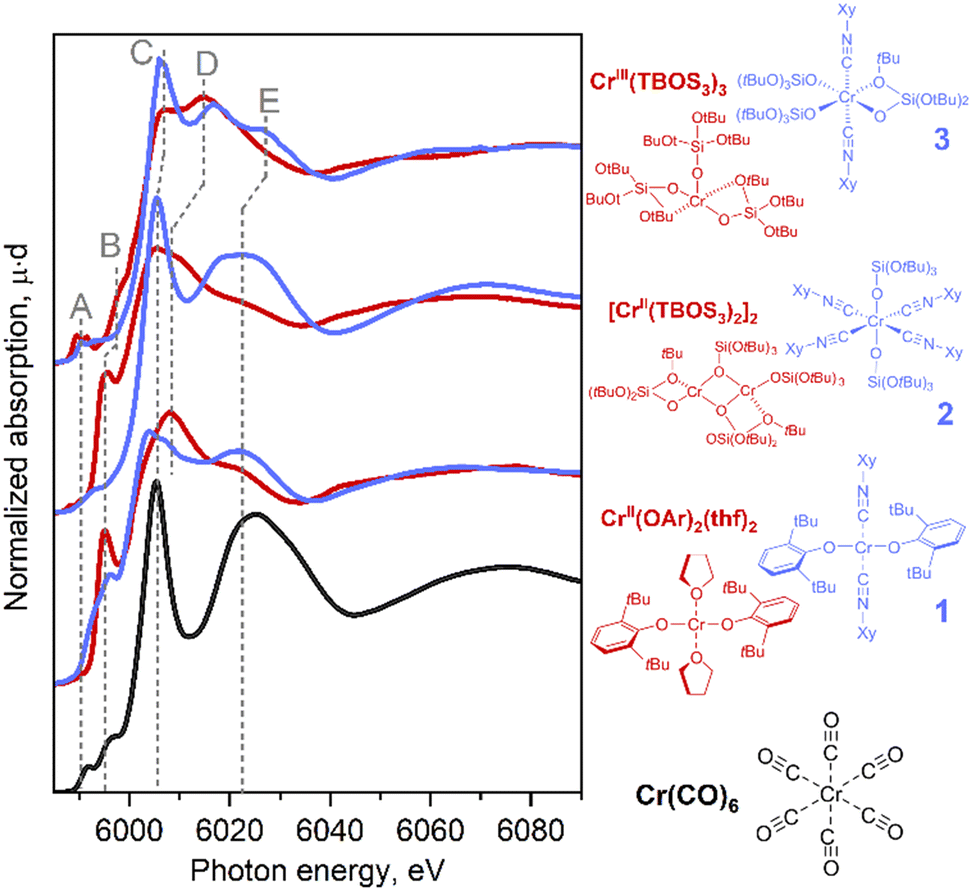 | ||
| Fig. 1 Experimental Cr K-edge XANES spectra for Cr(CO)6 (black) and the series of isocyanide complexes (blue) compared with similar reference complexes without isocyanide ligands (red). | ||
Tetrakis(isocyanide) adduct 2 exhibited a significant shift of its rising edge toward the high-energy region with a concomitant emergence of pre-edge feature A. Using the positions of the pre-edge and absorption edge alone, complex 2 could be assigned to Cr in a higher oxidation state than Cr(II). Indeed, similar energy shifts are also found in Cr(III) complexes coordinated by purely oxygen-based ligands, e.g., Cr(TBOS)3 or the polyhedral-oligosilsesquioxane complex Cr(POSS)(THF)3 (= [(cC5H9)7Si7O9(O)3Cr(THF)3]) (Fig. S13†). This high-energy shift can be interpreted as a consequence of sharp feature C that dominates the rising edge, as well as the loss of shoulder peak B upon structural change from the square planar to octahedral geometry. Further development of feature E supports the growing dominance of the isocyanide ligand coordination on the spectrum.
In summary, the introduction of the isocyanide ligands primarily causes an increase of the intensity of sharp peak C at the rising edge and feature E. In addition, for the Cr(III) species, a shift of the pre-edge towards the higher energy region was observed. Furthermore, geometrical changes cause a modulation of the spectra, as found for shoulder feature B. These effects can result in an apparent high-energy shift of the rising edge. The results imply that similar situations found for the Cr surface sites interacting with CO can potentially obscure the assignment of the oxidation state in Phillips-based polymerization catalysts in contact with a CO probe.
When analyzing surface metal sites dispersed on the support, EXAFS is another powerful method that delivers information on neighboring atoms; this is especially valuable for the assessment of metal–metal interactions that would be challenging otherwise. However, the quantitative interpretation can become ambiguous due to an overlap of the spectral features with scattering from strongly bonded organic ligands and the presence of the support.40 As CO is often used as a reductant and probe molecule, the understanding of its potential impact on the EXAFS signature is worth investigating. Note, however, that the region of distances above 2 Å in FT-EXAFS is often not informative in the reduced Phillips catalyst due to the heterogeneity of surface sites, but can contain contributions from metal–metal scattering and multiple scattering from ligands, if such species are present in the sample.
In fact, the presence of the isocyanide ligands introduces significant changes in spectral shape, not only in the XANES region but also in the higher energy region of the spectrum. Fig. 2 shows a comparison between amplitudes of Fourier transformed oscillations above the Cr K-edge in bulk Cr2O3, molecular complex 2 and Cr(CO)6. The first maximum of the modulus of the Fourier transform at 1.5 Å corresponds to the scattering from the first coordination shell consisting of oxygen and carbon atoms. Both materials show a prominent signal for distances higher than 2 Å (without phase correction), whereas this feature arises from Cr–Cr scattering in bulk oxide; contributions from single/multiple scatterings from Cr–N(O), Cr–C–N(O), Cr–C–N(O)–C in isocyanide adduct 2 or Cr(CO)6 appear in the same region. These results demonstrate the overlap of the Fourier-transformed EXAFS signature of the Cr–ligand bond with Cr–Cr and Cr–support signals, which will be an additional factor that complicates characterization. Such an effect should be taken into account when interpreting experimental XAS data from a real catalyst that contains Cr–CO interactions, in addition to metal–support and metal–metal interactions.
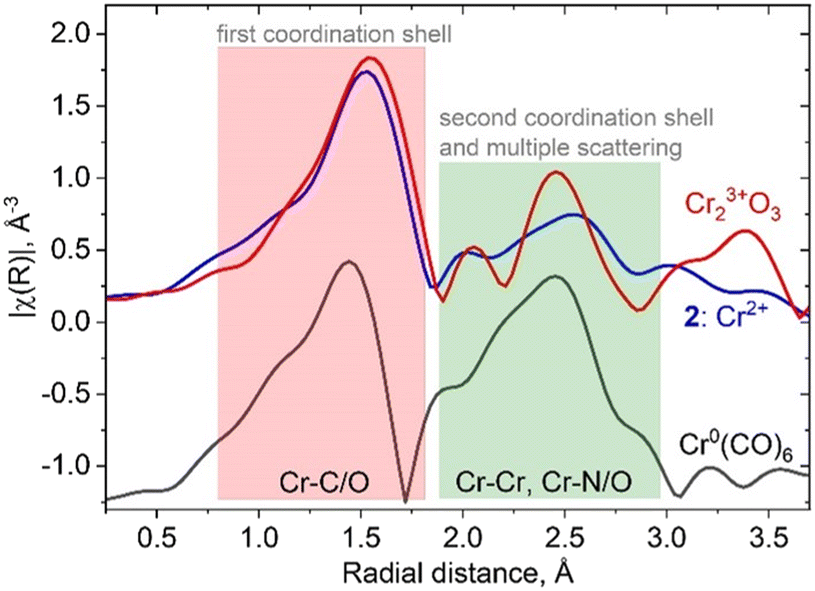 | ||
| Fig. 2 Comparison of the moduli of Fourier transformed EXAFS oscillations for bulk and molecular references containing isocyanide and carbonyl ligands. The green region demonstrates overlap between Cr–Cr and Cr-isocyanide/carbonyl scattering. | ||
Theoretical study of the origin of spectroscopic features
To reveal the origin of observed spectral features, we next carried out computations of the electronic structure and transition probabilities via molecular orbital and finite difference methods (see ESI† for details). The calculation method was first benchmarked using the XAS signature of complexes 1–3, Cr(CO)6 and a complementary siloxide series (see Fig. 1). Theoretical XAS signatures, obtained by MO-based as well as finite-difference methods nicely reproduced experimental line shapes thus validating the computational approach (Fig. S9 and S10†). Computational substitution of the isocyanide ligands in 2 with CO yielded a similar spectral signature, supporting the use of isocyanide as a model of CO (Fig. S11†). Among the isocyanide-adduct series, including Cr(CO)6, peak C on the white line was commonly found as a characteristic feature responsible for the apparent edge shift (Fig. 1). Cr(CO)6 was selected as the simplest model for the computational investigation of this feature. The origin of C was identified as transition to unoccupied molecular orbitals, composed of Cr 4p orbitals matching the anti-bonding, π*(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and σ*(CO) orbitals upon hybridization, which are shown in Fig. 3 (left and right, respectively, showing only one out of three counterparts directed towards x, y, and z axes). Such sharp molecular resonance overlapping with rising edge complicates the analysis of absorption edge shifts. In fact, the edge position of 2, a formal Cr(II) species, coincides with that of zero-valent Cr(CO)6 and some Cr(III) species, such as Cr(III)(POSS)(THF)3 or Cr2O3 (Fig. S13†). This implies difficulties in attributing XAS edge-energies to specific Cr oxidation states when π-acceptor ligands such as CO or isocyanide are coordinated to Cr.
O) and σ*(CO) orbitals upon hybridization, which are shown in Fig. 3 (left and right, respectively, showing only one out of three counterparts directed towards x, y, and z axes). Such sharp molecular resonance overlapping with rising edge complicates the analysis of absorption edge shifts. In fact, the edge position of 2, a formal Cr(II) species, coincides with that of zero-valent Cr(CO)6 and some Cr(III) species, such as Cr(III)(POSS)(THF)3 or Cr2O3 (Fig. S13†). This implies difficulties in attributing XAS edge-energies to specific Cr oxidation states when π-acceptor ligands such as CO or isocyanide are coordinated to Cr.
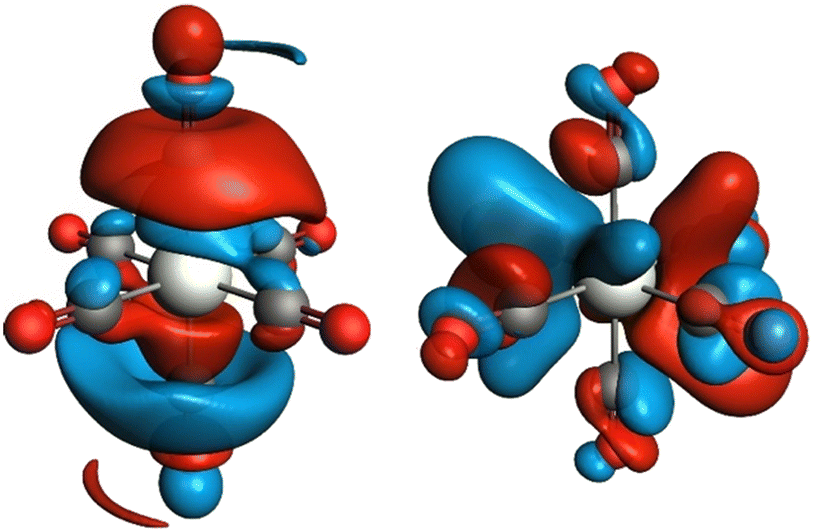 | ||
| Fig. 3 Unoccupied molecular orbitals in Cr(CO)6 contributing to peak C of the X-ray absorption spectrum in Fig. 1. Only one out of three degenerate orbitals is shown for each subset. Electron-density isosurfaces were plotted with an isovalue of ±0.03 e au−3. | ||
To examine the electronic modulation upon formation of a chemical bond between the isocyanide ligand and Cr center, we calculated the deformation electron density maps for complexes 2 and 3, using truncated MeCN ligands instead of XyNC for simplicity. Fig. 4 shows the difference between the molecular electron density and sum of fragment electron densities. The HOMO orbital of the isocyanide provides a lone pair localized on carbon, while the LUMO represents the antibonding π*(C–N) orbital (panel 4A). Upon complexation with Cr, the emergence of a σ-bond can be identified in both projections (Fig. 4B and C), while the contribution of π-back-bonding between Cr and isocyanide ligand orbitals can be identified in the equatorial projection (Fig. 4B). Hybridization of the isocyanide HOMO with the Cr dx2−y2 orbital results in an increase of electron density between Cr and C atoms and the formation of σ-bonds, as well as a decrease of electron density around the N atom. The effect of π-backdonation is observed as an increase in electron density on the isocyanide LUMO orbital (compare blue contour lines in the CN fragment with the shape of LUMO in Fig. 4A). Such a scenario is consistent with the experimentally observed elongated C–N bond lengths in 2 as well as the red-shifted CN stretching band in the IR spectrum, both pointing to the presence of an efficient π-back donation effect. On the contrary, we did not observe a population increase in the antibonding LUMO for the isocyanide ligands in Cr(III) complex 3; while the deformation electron density in Fig. 4D contains signatures of a σ-bond between the Cr center and isocyanide ligands, π-back donation is not evident (compare with 4B). The lack of electron transfer to the antibonding orbital explains the experimental observation of shortened CN bond lengths and their blue-shifted CN stretching band in the IR spectrum of 3.
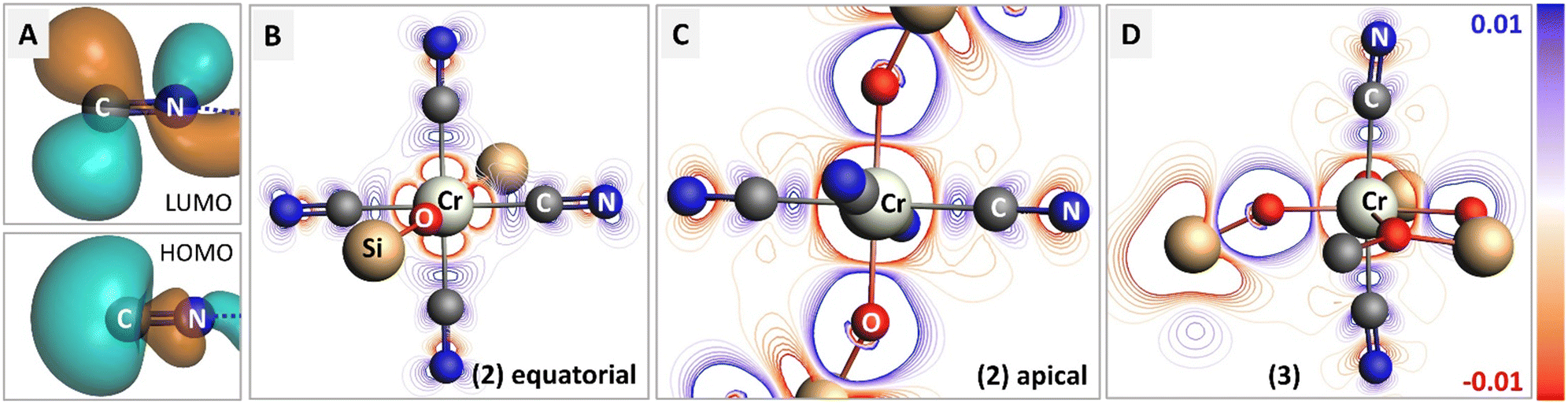 | ||
| Fig. 4 Charge transfer in complexes 2 and 3 upon bond formation with isocyanide adducts. (A) Isosurfaces for HOMO and LUMO molecular orbitals in bare isocyanide. (B and C) Deformation density maps for 2 in two orthogonal planes, containing isocyanides or isocyanide and siloxide ligands. The regions with increased electron density after bond formation are shown in blue. (D) Deformation density map for 3. Remarks on representation: isocyanide (XyNC) and siloxide (TBOS) ligands were simplified into methylisocyanide (CN–Me) and –OSiF3 for calculation. For clarity, only first and second coordination spheres are shown. The whole CN–Me molecule was used as a fragment for the deformation density calculation, while remaining parts were treated as single atom fragments. Electron-density isosurfaces were plotted with an isovalue of ±0.03 e au−3. | ||
Additionally, orbital analysis performed with natural orbitals for chemical valence (NOCV), focused on Cr-isocyanide bonding from valence deformation densities, further confirmed the conclusion above pointing to the efficient π-back donation in 2, but not in 3 (see ESI,† section 4.10).
The presence of strongly π-accepting ligand (e.g., isocyanide or CO) has a significant impact on the electronic state of the metal center and modulates the spin-state. Calculated bond energies of tetrakis(isocyanide) adduct 2 revealed a preferred LS configuration compared to HS with a −1.4 eV lower bond energy. On the contrary, Cr(II) and Cr(III) bis(isocyanide) adducts 1 and 3 preferred a HS configuration with ca. 0.6 eV and 1.2 eV higher bond energies, respectively. These stable spin configurations are further confirmed at the DFT B3LYP-D3(BJ)/QZ4P and M06-D3(BJ)/QZ4P levels of theory, consistent with experimental observations (vide supra).41 Benchmarking the calculated bond lengths of 2 in the LS state compared with distances obtained experimentally via single-crystal XRD and EXAFS analysis further confirmed the dominance of the LS state, corroborating the unique capability of the isocyanide ligands to stabilize the LS configuration (Table 1). To gain a quantitative insight into the effect of the isocyanide ligands on the spin-state, the bond energy was systematically calculated for a series of complexes with the general formula Cr(II)(TBOS)2(XyNC)n (n = 0–4) with a focus on evaluating a preference over spin states (Table 2). Starting from the DFT-optimized coordinates of 2, designated as 4 L, we removed isocyanides without changing the rest of the geometry of the complex. Each complex was denoted as nL, where n = the number of isocyanide ligands in the complex. While the experimentally investigated complex Cr(TBOS)2(XyNC)4 (2, or 4L) strongly preferred the LS state, the removal of the isocyanide ligands resulted in a relative energy rise of the LS state over the HS state; the HS state was preferred when n = 0, 1 and 2. Similar calculations for the Cr(III) complex 3 favor only the HS state with a decrease in the stabilization energy when n = 0, 1 and 2 isocyanide ligands are attached. These results indicate that the introduction of the isocyanide ligands stabilizes the low-valent metal center in the LS configuration, which directly implies that a similar scenario exists on the low-valent Cr sites of reduced Phillips catalysts in the presence of π-acceptor ligands (CO).
| XRD | EXAFS | DFT (LS) | |
|---|---|---|---|
| Cr–C1 | 2.023(11) | 2.03 ± 0.02 | 2.019 |
| Cr–C2 | 2.009(12) | 2.006 | |
| Cr–C3 | 2.023(11) | 2.019 | |
| Cr–C4 | 2.009(12) | 2.006 | |
| Cr–O1 | 1.934(8) | 1.94 ± 0.01 | 1.935 |
| Cr–O2 | 1.933(8) | 1.935 |
| Entry | Siloxides | Isocyanides | E LS − EHS, eV |
|---|---|---|---|
| 4L | 2 | 4 | −1.4 |
| 3L | 2 | 3 | −0.2 |
| 2L-cis | 2 | 2 (cis) | 0.3 |
| 2L-trans | 2 | 2 (trans) | 1.0 |
| 1L | 2 | 1 | 1.5 |
| 0L | 2 | 0 | 1.2 |
The LS Cr(II)(TBOS)2(XyNC)n (n = 0–4) series was further applied to a simulation of Cr K-edge XAS spectra, as seen in Fig. 5a (for the Cr(III) series, see Fig. S16†), revealing the systematic development of isocyanide-based signatures in the spectra upon complexation. As suggested by the calculated energy, the LS configuration of 2 was found to represent experimentally observed XAS data with nicely reproduced spectral features (Fig. 5a, 4L). The calculations show an increase in the intensities of C and E features as the amount of XyNC molecules surrounding the Cr center increases too. Theoretical spectra reproduce the higher intensity of shoulder B in the planar structures and increased intensity of pre-edge feature A for non-centrosymmetric configurations.
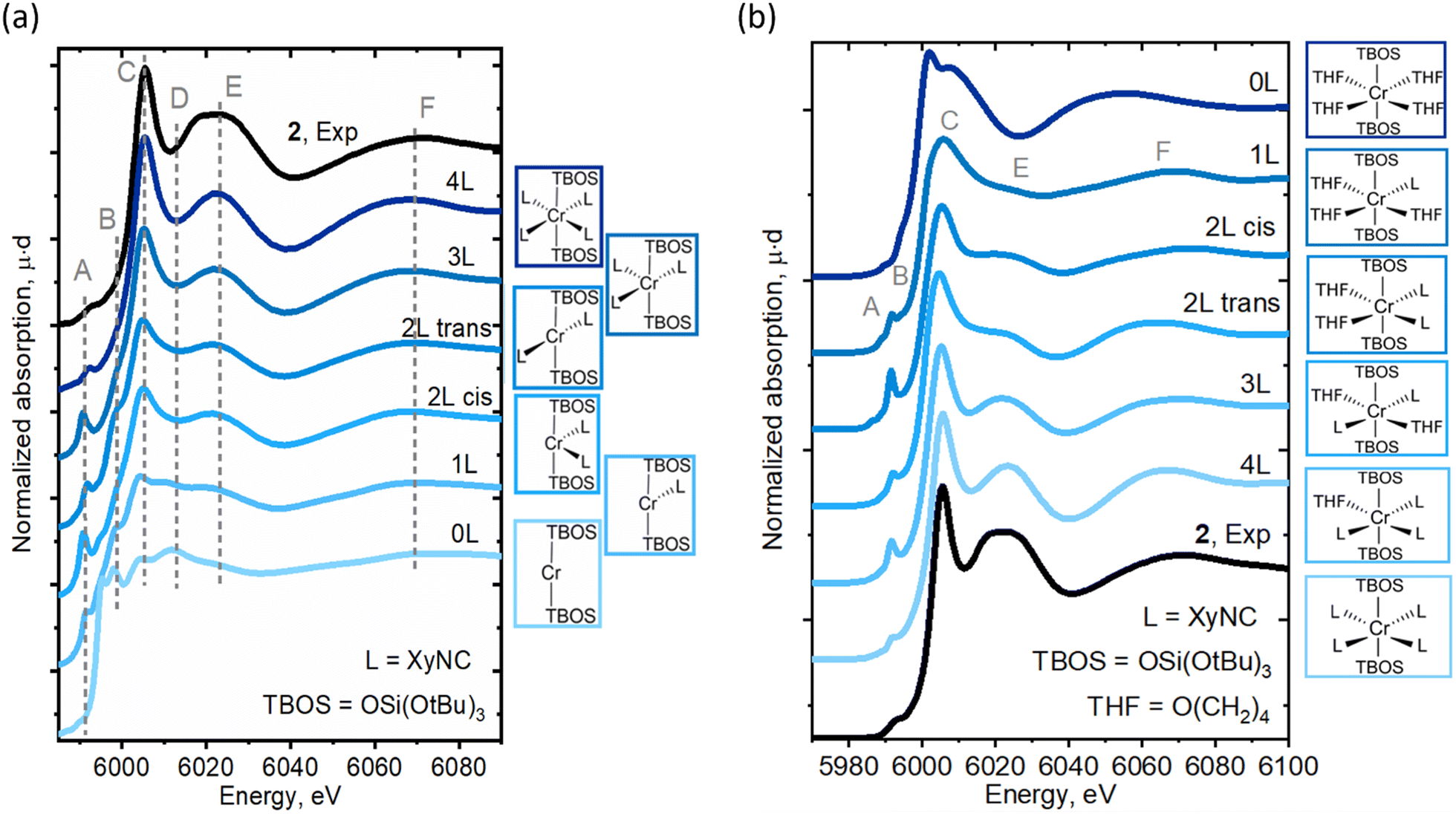 | ||
| Fig. 5 Experimental Cr K-edge XAS spectra for 2 compared to (a) the calculated spectra for Cr(TBOS)2(XyNC)n (n = 0–4). The structures were obtained by removing XyNC ligands from the DFT-optimized Cr(TBOS)2(XyNC)4 structure in a LS state without subsequent relaxation. (b) Calculated Cr K-edge XAS spectra for Cr(TBOS)2(XyNC)4−n(THF)n (n = 0–4). The structures with different ligand environments were obtained by replacing THF with XyNC ligands in the initial structure with subsequent DFT geometry optimization in the LS state. | ||
Furthermore, to isolate the influence of the isocyanide ligands from geometric effects, similar calculations were conducted for another series of complexes, namely Cr(TBOS)2(THF)4−n(XyNC)n (n = 0–4), where the THF ligands were gradually replaced by XyNC ligands while keeping the overall pseudo-octahedral geometry (Fig. 5b). While the calculated spectrum for the oxygen-based environment lacks C/E features (top), the addition of isocyanides systematically develops these features driving the apparent edge position to a higher energy whilst shifting the broad F peak. This shows that not only the geometry but the isocyanide ligand itself can also induce significant line-shape modulation.
Overall, Cr K-edge XANES signatures are significantly affected by the presence of additional (π-acceptor) ligands, e.g., CO/isocyanide. Theoretical simulations illustrated the sensitivity of the spectral signature (e.g. peak intensities, rising edge shift) to small structural changes identifying relationships with the number of such ligands. These results indicate that attributing these signatures to specific Cr sites is not trivial and can even be misleading without the use of appropriate references.
Deconvolution of experimental spectra for the Phillips catalyst
With insights taken from molecular isocyanide adducts in hand, we first compared the Cr K-edge XAS spectra of CO-reduced Phillips catalysts, depending on the specific treatment and measurement conditions. Fig. 6a shows striking differences in the Cr K-edge spectral signatures of the CO-reduced Phillips catalysts recorded at 300 K under vacuum (red) versus under a CO atmosphere at 300 K (green) and 100 K (blue).7 Notably, the XAS spectrum recorded under vacuum corresponding to a material having no residual CO, according to IR spectroscopy, displays a significant shoulder peak at the rising edge, typical of Cr(II) silicates with a square planar geometry.7 The spectra recorded under a CO atmosphere loses shoulder feature B associated with low-coordinated sites, consistent with CO binding. Additional and distinct peaks C and E appear at ca. 6005 eV and 6020 eV, respectively, whose intensities are largest at 100 K. These features correlate with the increased number of carbonyl species, as evidenced by IR.11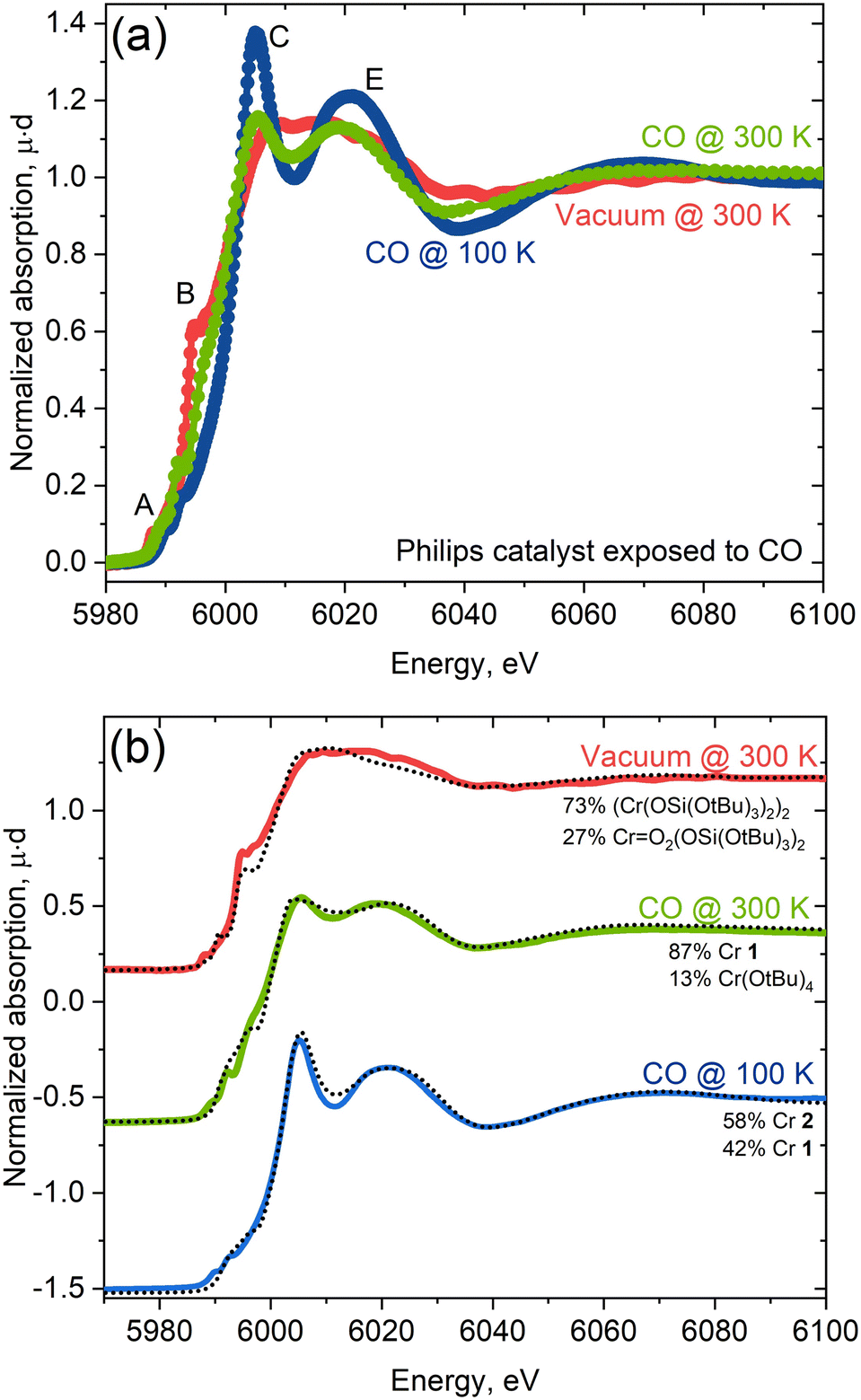 | ||
| Fig. 6 (a) Experimental spectra of the Phillips catalyst measured under vacuum (red) and CO atmosphere at 300 K (green) and at 100 K (blue). (b) Linear combination fit of experimental spectra with the library of experimental references. Best fit is obtained for linear combination of 58% of 2 (Cr(TBOS)2(XyNC)4) and 42% of 1 (Cr(OAr)2(XyNC)2). | ||
The changes in the spectral features resemble what is observed for molecular Cr(II) isocyanide adducts 1 and 2 with two or four isocyanide ligands (Fig. 1). We further analyzed quantitatively the spectra of the CO-reduced Phillips catalysts by applying linear combination fittings (LCF) using a molecular Cr K-edge XAS spectral library constituted of previously reported Cr-compounds with O-based ligands17 and the isocyanide-adduct series reported here. LCF results regarding the spectrum of the CO-reduced Phillips catalysts recorded under vacuum predicts the coexistence of square planar Cr(II) (73%) along with a small amount of Cr(VI) or Cr(IV) species (27%; Cr(VI) and Cr(IV) gave similar fit quality) (Fig. 6b), suggesting that CO makes a negligible contribution to the spectrum. The XAS spectrum recorded under a CO atmosphere at 300 K, on the other hand, can be reproduced with a linear combination of Cr(OAr)2(XyNC)2 (1, 87%) and Cr(O)2(TBOS)2 or Cr(OtBu)4 (13%) spectra, suggesting the presence of mostly low-coordinated Cr(II) carbonyl adducts at this temperature, consistent with IR data (vide infra). Finally, the XAS spectrum recorded at 100 K under CO can be fitted with two species, namely Cr(TBOS)2(XyNC)4 (2, 58%) and Cr(OAr)2(XyNC)2 (1, 42%), present in a similar amount and clearly indicating the presence of low and highly coordinated low-valent Cr(II) sites (Fig. 6b). The quantitative contribution from different oxidation states to LCF and other fits with similar L2 norms can be found in the Jupyter Notebook42 attached as ESI† to this manuscript.
The good quality of XAS LCF fits further indicates that synthesized complexes are good references for describing Cr sites upon interaction with CO. Predictions regarding the coordination of Cr(II) with CO are consistent with what has been reported from the IR spectra. At 300 K, the blue-shifted CO band is dominant in the IR spectrum and is consistent with the presence of a low coordinated “non-classical” Cr(II) carbonyl, paralleling what is observed for 1, a low-coordinated bis(isocyanide) adduct with a blue-shifted CN band and sharp distinct features in the white line in XAS. The emergence of pronounced red-shifted CO bands at 100 K, assigned to a highly coordinated “classical” carbonyl11 matches quite well with 2, a highly-coordinated tetrakis(isocyanide) adduct with red-shifted CN bands with a more pronounced white line feature in XAS.
Overall, this study shows that the white line and spectral features C and E, which were commonly found in the spectra from molecular references and the reduced Phillips catalyst, are very sensitive to the number of π-acceptor CO or isocyanide ligands. They correspond to transitions associated with the unoccupied molecular orbitals (σ*/π*) derived from the interaction of the Cr 4p orbitals with (CX) ligands (X = O or NR) and the photoelectron scattering along linear CX bonds.
Conclusions
In summary, we have developed molecular Cr(II)/(III)-isocyanide adducts as isoelectronic models of low-valent Cr sites in CO-reduced Phillips catalysts interacting with π-acceptor ligands (e.g., CO). The goal was to better understand the impact of bound π-acceptor ligands on the electronic structure and associated XANES and IR spectral signatures. The experimentally obtained Cr K-edge XAS spectra revealed the profound effect that the isocyanide ligands have on XANES and EXAFS energy regions. Compared to the characteristic signatures of square planar Cr(II) complexes, we observed the loss of the shoulder at the rising edge and appearance of two characteristic sharp features C and E at higher energy near the white line. The sharp peak C is identified as a transition to a hybridized Cr 4p orbital and to a σ* orbital and π*(CX) orbital (X = O or NR). As a result, the apparent edge energy is shifted towards high-energy and overlaps with the higher Cr oxidation state region. Furthermore, single and multiple scattering contributions from rigid linear Cr–C–O/N bonds in the Fourier-transformed EXAFS spectrum fall into the region of Cr–O–Cr distances, making the assignment of peaks in the second coordination shell in Cr-based heterogeneous catalysts when π-acceptor ligands are present even more challenging. Finally, the experimental XAS spectra for the molecular series were applied to the CO-reduced Phillips catalysts, measured in CO-rich or CO-free atmospheres at 100 K and 300 K. Linear combination fit analysis based on the molecular library confirms the dominance of the Cr(II) species in the purely oxygen-ligand environment when recorded under vacuum. In the presence of CO at 300 K, LCF points to the presence of low coordinated Cr(II) carbonyl species associated with blue-shifted IR CO bands. Highly coordinated species, associated with red-shifted IR CO bands, are observed at 100 K. While consistent with the known adsorption/desorption dynamics of CO on Phillips catalyst surfaces,3,4,43–45 this study shows that the number of π-acceptor (CO or isocyanide) ligands bound to Cr(II) has a dramatic effect, not only on the IR (blue vs. red shifted) spectral signatures but also on XAS spectral signatures, where the appearance of novel features induces a shift of the absorption edge to the higher energy values and an overlap of Cr(II) and Cr(III) signatures. This study further highlights how π-acceptor ligands like CO (and potentially ethylene) could impact XAS signatures; hence, there is a need to develop spectral libraries based on well-defined molecular analogues to identify the structure and exact coordination chemistry of surface sites. Our work also provides open access to the library of the Cr K-edge spectra along with user-friendly programming tools for analysis.
Author contributions
Y. K., A. G. and C. C. conceptualized the project. Y. K. performed synthesis of molecular Cr complexes, spectroscopic characterization, and analysed relevant diffraction data. Y. K. and O. S. collected experimental Cr K-edge XAS data for molecular complexes. S. G. and B. P. pre-processed the spectra and have developed the Jupyter Notebook providing transferability of the results and allowing researchers apply the whole library of Cr K-edge XAS to the quantitative analysis of their spectra. A. G. and S. S. have performed DFT calculations and simulated Cr K-edge spectra.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. C. and Y. K. acknowledge the Swiss National Science Foundation (grants 200021_169134 and 200020B_192050). B. P., A. G. and S. G. acknowledge the financial support from the Strategic Academic Leadership Program of the Southern Federal University (“Priority 2030”). The authors acknowledge Dr. Michael D. Wörle (ETHZ) for his assistance in processing the scXRD data. We acknowledge Prof. Elena Groppo for kindly providing the original experimental Cr K-edge spectra on CO-reduced Phillips catalysts published in ref. 11.Notes and references
- M. P. McDaniel, A Review of the Phillips Supported Chromium Catalyst and Its Commercial Use for Ethylene Polymerization, Elsevier Inc., 1st edn, 2010, vol. 53 Search PubMed.
- M. P. McDaniel, in Handbook of Transition Metal Polymerization Catalysts, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2018, pp. 401–571 Search PubMed.
- E. Groppo, G. A. Martino, A. Piovano and C. Barzan, ACS Catal., 2018, 8, 10846–10863 CrossRef CAS.
- E. Groppo, C. Lamberti, S. Bordiga, G. Spoto and A. Zecchina, Chem. Rev., 2005, 105, 115–183 CrossRef CAS PubMed.
- E. Groppo, A. Damin, F. Bonino, A. Zecchina, S. Bordiga and C. Lamberti, Chem. Mater., 2005, 17, 2019–2027 CrossRef CAS.
- A. J. Lupinetti, G. Frenking and S. H. Strauss, Angew. Chem., Int. Ed., 1998, 37, 2113–2116 CrossRef CAS.
- S. H. Strauss, J. Chem. Soc., Dalton Trans., 2000, 1–6 RSC.
- J. A. van Bokhoven and C. Lamberti, X-Ray Absorption and X-Ray Emission Spectroscopy: Theory and Applications, John Wiley & Sons, 2016 Search PubMed.
- M. Botavina, C. Barzan, A. Piovano, L. Braglia, G. Agostini, G. Martra and E. Groppo, Catal. Sci. Technol., 2017, 7, 1690–1700 RSC.
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736–1850 CrossRef CAS PubMed.
- D. Gianolio, E. Groppo, J. G. Vitillo, A. Damin, S. Bordiga, A. Zecchina and C. Lamberti, Chem. Commun., 2010, 46, 976–978 RSC.
- B. M. Weckhuysen, R. A. Schoonheydt, J.-M. Jehng, I. E. Wachs, S. J. Cho, R. Ryoo, S. Kijlstra and E. Poels, J. Chem. Soc., Faraday Trans., 1995, 91, 3245–3253 RSC.
- C. Brown, J. Krzystek, R. Achey, A. Lita, R. Fu, R. W. Meulenberg, M. Polinski, N. Peek, Y. Wang, L. J. van de Burgt, S. Profeta Jr, A. E. Stiegman and S. L. Scott, ACS Catal., 2015, 5, 5574–5583 CrossRef CAS.
- L. Zhong, M.-Y. Lee, Z. Liu, Y.-J. Wanglee, B. Liu and S. L. Scott, J. Catal., 2012, 293, 1–12 CrossRef CAS.
- E. Groppo, C. Prestipino, F. Cesano, F. Bonino, S. Bordiga, C. Lamberti, P. C. Thüne, J. W. Niemantsverdriet and A. Zecchina, J. Catal., 2005, 230, 98–108 CrossRef CAS.
- A. Chakrabarti, M. Gierada, J. Handzlik and I. E. Wachs, Top. Catal., 2016, 59, 725–739 CrossRef CAS.
- D. Trummer, K. Searles, A. Algasov, S. A. Guda, A. V. Soldatov, H. Ramanantoanina, O. V. Safonova, A. A. Guda and C. Copéret, J. Am. Chem. Soc., 2021, 143, 7326–7341 CrossRef CAS PubMed.
- S. A. Bartlett, J. Moulin, M. Tromp, G. Reid, A. J. Dent, G. Cibin, D. S. McGuinness and J. Evans, Catal. Sci. Technol., 2016, 6, 6237–6246 RSC.
- P. Glatzel, G. Smolentsev and G. Bunker, J. Phys.: Conf. Ser., 2009, 190, 012046 CrossRef.
- C. Engemann, J. Hormes, A. Longen and K. H. Dötz, Chem. Phys., 1998, 237, 471–481 CrossRef CAS.
- S. Chavan, J. G. Vitillo, M. J. Uddin, F. Bonino, C. Lamberti, E. Groppo, K.-P. Lillerud and S. Bordiga, Chem. Mater., 2010, 22, 4602–4611 CrossRef CAS.
- F. A. Cotton and F. Zingales, J. Am. Chem. Soc., 1961, 83, 351–355 CrossRef CAS.
- A. C. Sarapu and R. F. Fenske, Inorg. Chem., 1972, 11, 3021–3025 CrossRef CAS.
- A. E. Carpenter, C. C. Mokhtarzadeh, D. S. Ripatti, I. Havrylyuk, R. Kamezawa, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Inorg. Chem., 2015, 54, 2936–2944 CrossRef CAS PubMed.
- A. C. Sarapu and R. F. Fenske, Inorg. Chem., 1975, 14, 247–253 CrossRef CAS.
- G. Frenking, I. Fernández, N. Holzmann, S. Pan, I. Krossing and M. Zhou, JACS Au, 2021, 1, 623–645 CrossRef CAS PubMed.
- J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939–2947 RSC.
- T. Mathieson, A. Schier and H. Schmidbaur, J. Chem. Soc., 2001, 1196–1200 CAS.
- A. S. Goldman and K. Krogh-Jespersen, J. Am. Chem. Soc., 1996, 118, 12159–12166 CrossRef CAS.
- A. J. Lupinetti, S. Fau, G. Frenking and S. H. Strauss, J. Phys. Chem. A, 1997, 101, 9551–9559 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- E. M. Schubert, J. Chem. Educ., 1992, 69, 62 CrossRef CAS.
- D. H. Grant, J. Chem. Educ., 1995, 72, 39 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- E. M. Zolnhofer, A. A. Opalade, T. A. Jackson, F. W. Heinemann, K. Meyer, J. Krzystek, A. Ozarowski and J. Telser, Inorg. Chem., 2021, 60, 17865–17877 CrossRef CAS PubMed.
- W. S. Mialki, D. E. Wigley, T. E. Wood and R. A. Walton, Inorg. Chem., 1982, 21, 480–485 CrossRef CAS.
- C. C. Scarborough, K. M. Lancaster, S. DeBeer, T. Weyhermüller, S. Sproules and K. Wieghardt, Inorg. Chem., 2012, 51, 3718–3732 CrossRef CAS PubMed.
- M. Yousif, D. J. Tjapkes, R. L. Lord and S. Groysman, Organometallics, 2015, 34, 5119–5128 CrossRef CAS.
- O. L. Sydora, D. S. Kuiper, P. T. Wolczanski, E. B. Lobkovsky, A. Dinescu and T. R. Cundari, Inorg. Chem., 2006, 45, 2008–2021 CrossRef CAS PubMed.
- V. L. Sushkevich, O. V. Safonova, D. Palagin, M. A. Newton and J. A. van Bokhoven, Chem. Sci., 2020, 11, 5299–5312 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Kakiuchi, S. Cherkasova, B. Protsenko, S. Guda, O. Safonova, A. Guda and C. Copéret, XAS spectra of Cr K-edge and code for analysis, Mendeley Data, V3, 2023, DOI:10.17632/yjdsjvctcb.2.
- S. Bordiga, S. Bertarione, A. Damin, C. Prestipino, G. Spoto, C. Lamberti and A. Zecchina, J. Mol. Catal. A: Chem., 2003, 204–205, 527–534 CrossRef CAS.
- B. Rebenstorf, J. Catal., 1989, 117, 71–77 CrossRef CAS.
- B. Rebenstorf, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 1949–1953 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on sample synthesis, crystallographic information, Cr K-edge XAS measurements, DFT calculations and linear combination fit analysis. CCDC 2286290–2286292. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy01692g |
| ‡ Source code for reproducing LCF figures and library of Cr K-edge spectra are available also from at Mendeley Data repository doi: 10.17632/yjdsjvctcb.1. |
| This journal is © The Royal Society of Chemistry 2024 |