
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low temperature CO oxidation over Rh supported on N-doped carbon†
Colby A.
Whitcomb
 ,
Anukriti
Shrestha
,
Christopher
Paolucci
* and
Robert J.
Davis
*
,
Anukriti
Shrestha
,
Christopher
Paolucci
* and
Robert J.
Davis
*
Department of Chemical Engineering, University of Virginia, 351 McCormick Road, Charlottesville, VA 22904, USA. E-mail: cp9wx@virginia.edu; rjd4f@virginia.edu
First published on 18th March 2024
Abstract
Some isolated transition metals supported on nitrogen-doped carbon (M–N–C) are effective catalysts for reactions involving O2, including low temperature CO oxidation. In this work, screening of various M–N–C materials using quantum chemical calculations showed that group 9 transition metals (Co, Rh, and Ir) in nitrogen-doped carbon have similar binding affinities for CO and O2 and were able to form a stable CO–O2 intermediate, which are criteria for a low-temperature CO oxidation catalyst. A Rh–N–C catalyst was therefore synthesized and evaluated for CO oxidation. The steady-state reaction at low temperature (<403 K) over Rh–N–C had positive reaction orders in both CO and O2 with a very small apparent activation energy. Results from kinetic experiments and quantum chemical calculations are consistent with a reaction path involving weak adsorption of CO onto Rh ions with turnover coming from CO-assisted activation of weakly adsorbed O2. The reaction mechanism does not involve a redox cycle with Rh and appears to be general in nature for low temperature CO oxidation. These findings may be conceptually useful for the design of other catalysts for reactions involving dioxygen activation.
1. Introduction
Transition metal ions (M) isolated in a nitrogen-doped carbon matrix (M–N–C) have demonstrated activity for reactions involving molecular oxygen in both electrocatalytic1–6 and thermocatalytic7–11 processes, but the coordination environment around the isolated metal center and the mechanism for O2 activation remain controversial.7–9,11 Oxidation of CO through O2 activation has been studied over M–N–C catalysts since the reaction lacks complicating factors such as solvents and applied potentials.11,12 Specifically, catalytic CO oxidation was investigated on Fe–N–C, Co–N–C, and materials containing both Co and Fe (Co,Fe–N–C), with the Co-containing catalysts exhibiting activity even at dry-ice acetone temperature.12 In that work, the low-temperature activity was attributed to weak but preferential binding of CO to Co with O2 binding to the adjacent Fe.12 In a related study, results from CO pulse cryo-chemisorption showed that CO chemisorbed on Fe–N–C and Fe,Ni–N–C catalysts, whereas CO did not chemisorb on Ni–N–C.13,14 Clearly, CO adsorption and O2 activation are highly dependent on the nature of the transition metal element in M–N–C materials.We recently used kinetic measurements, quantum chemical calculations, and molecular dynamics simulations to study a Co–N–C catalyst for low temperature CO oxidation.11 Results from that work are consistent with a mechanism for O2 activation that avoids direct dissociation of O2 on the transition metal ion.11 Instead, weak adsorption of CO onto Co ions followed by CO-assisted activation of weakly adsorbed O2 with the carbon support produced CO2 in a reaction path with an apparent activation energy that is negative.11 Interestingly, the proposed mechanism did not involve a redox cycle on the isolated Co metal ion.11 The absence of a redox cycle on the metal and the weak interactions of the reactants with the isolated transition metal ion suggest that other M–N–C catalysts may also be active for CO oxidation at low temperatures.
A variety of M–N–C catalysts have been synthesized (i.e. Ag, Au, Cd, Cr, Cu, Fe, Ir, Mn, Ni, Pd, Pt, Rh, Ru, and Zn) and their activity and stability in O2 activation reactions vary accordingly.15 For example, Cr–, Co–, and Cu–N–C materials exhibited high initial catalytic activity for the oxidative dehydrogenation of benzyl alcohol, but the activity for the Cu samples was not recovered upon recycling or regeneration.7 Evidently, although M–N–C materials with various transition metals can be synthesized, not all of them may be stable under catalytic conditions. The stability of M–N–C structures was investigated by density functional theory (DFT) calculations using the formation energy of transition metals in graphite and N-doped structures.16 The study showed that many M–N–C materials in the same N-doped double vacancy motif that were previously studied are stable.16 The stability of other metals indicates that other M–N–C samples could be synthesized, explored for low temperature CO oxidation, and compared to DFT calculated mechanisms.
In this study, we computationally screened reportedly stable M–N–C catalysts to determine CO and O2 adsorption energy trends that might enable the low-temperature activation of O2 on various M–N–C catalysts. Rhodium ions in N-doped carbon (Rh–N–C) as well as Ir ions in N-doped carbon (Ir–N–C) are predicted to activate O2 through a mechanism similar to that described in our previous publication on Co–N–C.11 To confirm that the mechanism descriptors are correct in predicting a mechanism for CO oxidation on metals, we chose to synthesize Rh–N–C and tested it in low temperature CO oxidation. The low temperature CO oxidation kinetics are consistent with our computationally derived mechanism with CO assisting the activation of O2 with the participation of the carbon matrix.
2. Methods
2.1 Quantum chemical density functional theory calculations
We performed spin-polarized periodic supercell DFT calculations with a plane wave basis set and an energy cutoff of 500 eV using the Vienna ab initio simulation package (VASP)17 version 5.4.4. For all structures, convergence criteria of 10−6 eV and 0.01 eV Å−1 for self-consistent-field (SCF) energies and atomic forces, respectively, were used. Structures in various graphene supercell sizes may have different adsorbate binding energies so two different cell sizes were generated to compare to the literature. An 8.5 Å by 9.8 Å graphene supercell with 14 Å vacuum in the z-direction and two adjacent carbon vacancies added to place the metal ion (corresponding to a density of 1 M per 0.8 nm2) was generated by using a total of 30 C atoms (when the number of nitrogen atoms is zero). The carbon atoms surrounding the metal added to the defect site were then substituted with 4 pyridinic N atoms. A previous study used a supercell with less M density (1 M per 1.3 nm2),16 and thus for comparison, we generated a graphene supercell with 58 C (without N substitutions) and 15 Å vacuum in the z-direction. All the structure files for optimized geometries are included in the ESI.† Monkhorst–Pack k-points were used with a 4 × 4 × 1 mesh since they were the optimized k-points for the small cell (Fig. S1†). A variety of metals were used so the spin states for each were checked by doing an optimization starting from a low spin state on the entire structure by setting the magnetic moment to zero, and then doing a second optimization starting from a high spin state located on the metal atom. Geometry optimizations for O2 adsorption were started from both bidentate and monodentate initial configurations, and the more exothermic binding mode after geometry optimization is reported here. Only energies are reported and not free energies since the reaction intermediates have been shown previously to weakly adsorb in the case of Co ions doped in nitrogen–carbon (Co–N–C)11 and thus are not amenable to standard free energy approximations. Vibrational zero-point energy corrections were neglected to reduce computational cost as they are expected to have a negligible contribution to reaction energies for our system.Mavrikakis and co-workers16 reported binding energies of CO and O2 on a variety of metals calculated with the Perdew, Burke, and Ernzerhof (PBE)18 functional and the previously described larger supercell. Our PBE computed adsorption energies were analogous to theirs (Table S1†), except for CO binding to Fe–N–C. Our computed binding energy for CO on Fe–N–C is more exothermic but has a value that is consistent with other PBE-computed binding energies for CO on Fe–N–C.19–21 Finally, we chose to report van der Waals density functional (vdW-DF)22 computed values in the main text (PBE values are reported in Table S1† for comparison) because this functional reproduces the experimental enthalpy of the reaction for gas-phase CO oxidation,11 has been previously benchmarked for the binding energy of CO on Co–N–C,11 and accounts for nonlocal electron correlation dispersion interactions that are important for weak adsorption.
Transition states for the CO oxidation reaction on Rh–N–C structures were determined using climbing image nudged elastic band (CI-NEB) calculations derived from the method of Henkelman et al. and are reported in Fig. S2.†23,24 To reduce computational cost, the transition state calculations were completed using the higher metal density structures (small supercell) since for Rh–N–C the binding energies were within 15 kJ mol−1 of each other (−68 and −68 kJ mol−1 for CO and O2, respectively, in the higher metal density cell and −54 and −60 kJ mol−1, respectively, for the lower metal density cell). For transition state one (TS1), we investigated multiple spin states and found that although the barrier is sensitive to spin and typically decreases with lower spin, the reactant binding energies only weakly vary with the spin state. Therefore, we report the low spin reaction coordinate (magnetic moment = 0) for the CI-NEB for TS1. Bader charge and density of states (DOS) analyses were used to investigate the oxidation state of Rh along the reaction coordinate.25 The DOS was obtained by setting LORBIT equal to 11 and using vaspkit26 to obtain the projected Rh DOS for the different configurations along the reaction coordinate.
2.2 Synthesis of Rh catalysts
The Rh–N–C catalyst was synthesized by a modified high-temperature pyrolysis method similar to previously reported methods.8,11,27 A solution of Rh nitrate (0.293 g) (Aldrich, rhodium(III) nitrate solution ∼10% (wt/wt) Rh in >5 wt% nitric acid) was added to 10 cm3 of distilled, deionized (DDI) H2O and added to a solution of 1,10 phenanthroline (0.358 g) (Sigma-Aldrich Corporation) in 15 cm3 ethanol (Sigma-Aldrich Corporation) to make a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of Rh/phenanthroline (stirred for 20 min at 353 K). This mixture was subsequently added dropwise to a 0.1 M NaOH slurry with carbon black (Carbon Black Pearls 2000, Cabot Corporation) at 353 K (stirred for 2 h). Washing of the slurry with 3000 cm3 of DDI H2O via vacuum filtration was then done before drying overnight at 343 K. A small portion of the carbon black with the Rh/phenanthroline complex (0.1 g) was then impregnated with 80 wt% dicyandiamide (Aldrich) relative to the complex. This slurry was stirred vigorously at 343 K until all the solution evaporated and the solid was dried overnight at the same temperature. This batch was then combined with another batch to reach 0.2 g before ramping in ultrahigh purity (UHP) N2 (100 cm3 min−1) (99.999%, Praxair) at 10 K min−1 and holding at 973 K for a high temperature thermal treatment. The thermally treated Rh sample was then treated in a solution of 11 M HCl and 1 wt% H2O2 for 3 h at 333 K in an attempt to remove any Rh metal nanoparticles formed during pyrolysis. Approximately 0.2 g (two combined batches of acid-washed samples) was heated at 10 K min−1 to 673 K and held for 2 h in flowing UHP H2 (100 cm3 min−1) (5% H2/Ar) to produce the as-synthesized Rh–N–C catalyst. A 0.29 wt% Rh/SiO2 catalyst was synthesized using incipient wetness impregnation. An aqueous solution of Rh nitrate (Aldrich, rhodium(III) nitrate solution ∼10% (wt/wt) Rh in >5 wt% nitric acid) (0.053 g) was added to 2.5 cm3 of DDI H2O and added dropwise to silica gel (2 g) (Davisil 636 Silica Gel).28,29 The catalyst was then dried overnight at room temperature before heating in air for 2 h at 393 K. The catalyst was then heated at 10 K min−1 to 673 K and thermally treated for 2 h in flowing air (Medical Grade, Praxair). A commercial 5 wt% Rh/C catalyst (Aldrich) was used from the bottle with no further treatment prior to going into the reactor.
:2 molar ratio of Rh/phenanthroline (stirred for 20 min at 353 K). This mixture was subsequently added dropwise to a 0.1 M NaOH slurry with carbon black (Carbon Black Pearls 2000, Cabot Corporation) at 353 K (stirred for 2 h). Washing of the slurry with 3000 cm3 of DDI H2O via vacuum filtration was then done before drying overnight at 343 K. A small portion of the carbon black with the Rh/phenanthroline complex (0.1 g) was then impregnated with 80 wt% dicyandiamide (Aldrich) relative to the complex. This slurry was stirred vigorously at 343 K until all the solution evaporated and the solid was dried overnight at the same temperature. This batch was then combined with another batch to reach 0.2 g before ramping in ultrahigh purity (UHP) N2 (100 cm3 min−1) (99.999%, Praxair) at 10 K min−1 and holding at 973 K for a high temperature thermal treatment. The thermally treated Rh sample was then treated in a solution of 11 M HCl and 1 wt% H2O2 for 3 h at 333 K in an attempt to remove any Rh metal nanoparticles formed during pyrolysis. Approximately 0.2 g (two combined batches of acid-washed samples) was heated at 10 K min−1 to 673 K and held for 2 h in flowing UHP H2 (100 cm3 min−1) (5% H2/Ar) to produce the as-synthesized Rh–N–C catalyst. A 0.29 wt% Rh/SiO2 catalyst was synthesized using incipient wetness impregnation. An aqueous solution of Rh nitrate (Aldrich, rhodium(III) nitrate solution ∼10% (wt/wt) Rh in >5 wt% nitric acid) (0.053 g) was added to 2.5 cm3 of DDI H2O and added dropwise to silica gel (2 g) (Davisil 636 Silica Gel).28,29 The catalyst was then dried overnight at room temperature before heating in air for 2 h at 393 K. The catalyst was then heated at 10 K min−1 to 673 K and thermally treated for 2 h in flowing air (Medical Grade, Praxair). A commercial 5 wt% Rh/C catalyst (Aldrich) was used from the bottle with no further treatment prior to going into the reactor.
2.3 Characterization of the catalysts
High-angle annular dark-field scanning transmission electron microscopy (STEM) imaging was performed using an aberration-corrected STEM Themis, operating at 200 kV and using a convergence semi-angle of 25 mrad.All inductively coupled plasma optical emission spectroscopy (ICP-OES) analyses were conducted at Galbraith Laboratories Inc. (2323 Sycamore Drive, Knoxville, TN 37921) using a PerkinElmer Optima 5300 V, 8300DV, or Avio 500 ICP-OES.
A Micromeritics ASAP 2020 adsorption system was used for H2 chemisorption. The Rh/SiO2 catalyst was evacuated for 2 h at 673 K, followed by reduction at that temperature for 2 h in flowing H2 (99.999%, Praxair UHP). After reduction, the system was cooled under vacuum to 308 K for analysis. Available metal sites were determined by extrapolating the high pressure, linear portion of the isotherm to zero pressure. The measured H/Rh ratio was 1.2 for our Rh/SiO2, consistent with 100% of the metal exposed. A prior study of the highly dispersed Rh metal particles has shown that the stoichiometric ratio of H to surface Rh atoms can be as high as 2:1.30
2.4 Oxidation of CO
The oxidation of CO was carried out in a stainless steel, down-flow, continuous packed bed reactor. Silicon carbide (2 g) (Universal Photonics, Inc., 150 mesh) was used to dilute the catalyst in each reaction. The reaction conditions involved 160 cm3 min−1 total gas flow with 1% CO (Praxair, 99.99%), 2% O2 (Praxair 99.99% O2) and balance He (Praxair UHP, 99.999%) at 3 atm total pressure. Helium was passed through an OMI-2 indicating purifier while CO and O2 were passed through silica traps immersed in a dry-ice acetone bath to remove trace water and trace carbonyls (from CO). Before initiating the reaction, the catalyst was thermally treated in situ in flowing He by ramping at 10 K min−1 to 673 K and holding for 2 h before cooling to the reaction temperature. A Balzers quadrupole mass spectrometer was used to analyze the reactor effluent with signals associated with masses (m/z) 28, 29, 44, and 45 amu recorded at a voltage of 1200 V and a dwell time of 50 ms for all masses except 44 and 45 amu which were measured at 100 ms. A cooling bath for reactions at 199 K was made by adding dry-ice to acetone.3. Results
3.1 Density functional theory screening of metal ions for low temperature CO oxidation activity
We used DFT calculations with vdW-DF (full calculation details are provided in the Methods section) to investigate the viability of O2 activation through a CO-assisted mechanism on a variety of isolated transition metal ions in N-doped carbon. Briefly, a previous mechanism for Co–N–C involved CO binding to the metal and interacting with O2 to form a CO–O2 complex, which evolves into 2 CO2 molecules with subsequent reactions.11 Limited charge transfer to Co occurred throughout the catalytic cycle, indicating that other metal ions may also be viable active centers. This low-temperature mechanism requires: 1) an exothermic enough CO binding energy to the metal to result in non-negligible CO coverage, 2) a CO binding energy that is at least competitive with the O2 binding energy (to the metal) to prevent O2 poisoning, and 3) the ability to form a stable CO–O2 complex. Consequently, for screening purposes we used a CO binding energy more exothermic than −30 kJ mol−1, CO and O2 binding energies that are within 30 kJ mol−1 of each other, and a convergence to a local minimum CO–O2 complex (additional details for how we arrived at these numbers can be found in Fig. S3 and S4†). We screened 15 candidate metal ions in four-fold coordinated pyridinic N sites with a double vacancy since this motif is reported for many synthesized catalysts11,15,16,31–34 and has been shown computationally to stabilize the metal atom.16,35,36 The transition metals for screening were chosen based on their proximity to Co in the periodic table or similar electronegativity in the case of Ga (ref. 37) and experimental evidence that they can be synthesized in the double vacancy motif (Ag and Mo were neglected since both were found to be unstable in the double vacancy motif).15,16Fig. 1 shows the vdW-DF computed CO and O2 binding energies for various metal ions coordinated to four N atoms (M–N–C catalysts) presented with periodic trends (a) and with a comparison of the binding energy of CO or O2 on each metal (b). Candidate metals in groups 10–13 bind CO weakly, except for Cd, and are unlikely to have non-negligible CO coverage. Weak binding to Ni–N–C is consistent with the observed lack of CO uptake on Ni–N–C, even at cryo-temperatures.13,14 The Cd atom seems to not follow the periodic trends for the magnitude of the binding energies of CO and O2 but can be rationalized by the weak binding energy of the Cd atom in the double vacancy site, as was previously reported.16 An inspection of Cd–N–C with adsorbates indicates that the Cd atom had increasing bond lengths with the neighboring N atoms. Conversely, transition metal ions in groups 6–9 have reasonably exothermic CO binding energy values in a wide range (−226 to −50 kJ mol−1) and therefore require application of the other criteria to differentiate. The Cr–, Mn–, and Cd–N–C cases bound O2 stronger than CO by at least 56 kJ mol−1, which indicates that O2 would poison these sites for a low temperature path initiated with CO bound to the transition metal. The preference for binding O2 over CO does not preclude low temperature CO oxidation, but only higher energy mechanisms for O2 starting on the transition metal atom have been reported.16 Previous reports from Mavrikakis and co-workers have found a higher barrier mechanism using DFT calculations where O2 dissociates on the transition metal and undergoes an Eley–Rideal type mechanism that has barriers in the range of 66 kJ mol−1, which is not consistent with low temperature (<400 K) activity.16 Thus, in the quest for metals that follow a low temperature mechanism similar to Co, application of the first two criteria narrows the search to metals in groups 8 and 9.
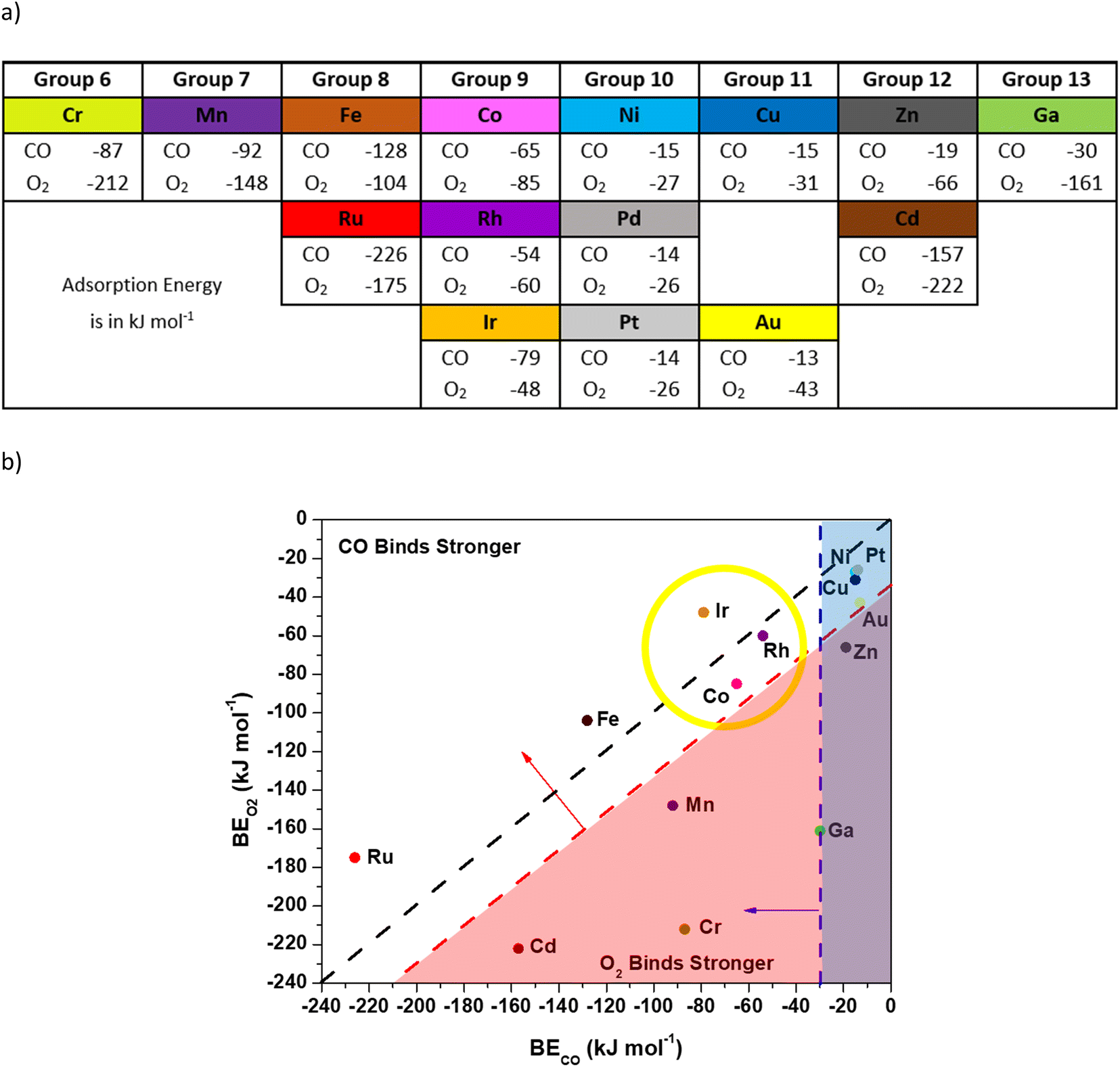 | ||
| Fig. 1 Adsorption energies of CO and O2 computed with vdW-DF (PBE results are in Table S1†). All metal ions were bound to four pyridinic nitrogen atoms. For O2 binding, the more stable binding motif (bidentate or monodentate O2) was reported. All structure files can be viewed in the ESI† and the metal magnetic moments are reported in Fig. S6.† a) Binding energies for CO or O2 on various metals in the order they appear in the periodic table. b) Binding energies for CO or O2 on various metals presented with criteria for the mechanism study. The black line bisecting the figure represents equal binding energies for CO and O2 on the metal. The vertical blue line indicates the −30 kJ mol−1 binding energy needed for CO to adsorb on the metal atom (criteria 1). The diagonal red line indicates the −30 kJ mol−1 excess O2 binding energy relative to CO (criteria 2). The yellow circle indicates the metals that fit all criteria 1–3. | ||
Optimization of structures, starting from our previously reported CO–O2 complex that forms on Co, was used to differentiate groups 8 and 9 further. The transition metals in group 8 (Fe, Ru) do not form a CO–O2 complex. Optimization of the structures starting with CO bound to the metal atom in a bent configuration and O2 coordinated to the CO (e.g.Fig. 2 structure 3) resulted in the CO relaxing to a linear orientation with a large distance (3.5 Å) between the carbon atom in CO and either of the oxygen atoms in the O2 molecule (see the ESI†). The inability of Fe–N–C to form this complex, and our assumption that this precludes participation in low-temperature CO-oxidation, is consistent with prior experimental results where an Fe–N–C catalyst was reported to be catalytically inactive for low temperature (200 K) CO oxidation.12 Although there are no experimental data for CO oxidation on Ru–N–C, it appears to lack a local minimum where the metal is in the plane after adsorption of CO or O2, precluding our low temperature mechanism.
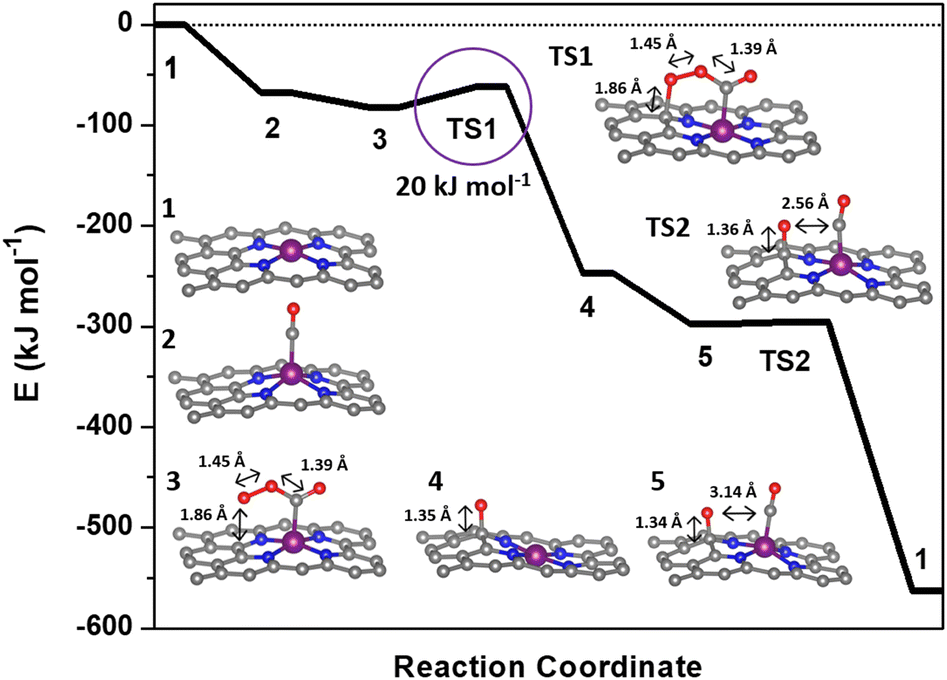 | ||
| Fig. 2 Reaction coordinate for the low-temperature CO oxidation mechanism over Rh–N–C computed with vdW-DF. The molecular structures corresponding to the reaction coordinates are included in the ESI.† | ||
Rebarchik and co-workers previously proposed a mechanism where adsorbates were present on both sides of a metal site, which modified the binding energy during the oxygen reduction reaction.38 To investigate if a similar phenomenon could occur in our system, we examined the binding energy of CO and O2 with an extra adsorbate below the Rh–N–C structure. We found that the addition of an O2 adsorbate resulted in changes in the relevant CO or O2 binding energy of less than 10 kJ mol−1. The addition of an extra CO adsorbate decreased the exothermicity of the topside CO by 46 kJ mol−1 (i.e. −68 to −21 kJ mol−1). When attempting to form the CO–O2 complex, the addition of an extra CO adsorbate caused the binding energy of O2 to the bound CO to be −24 kJ mol−1 more exothermic. The presence of an extra O2 adsorbate did not significantly affect this binding energy. We also examined if addition of CO or O2 to the other side of the metal would enable Fe–N–C and (or) Ru–N–C to satisfy the third criterion and form a stable CO–O2 complex; however, we found that this was not the case. These results are summarized in and below Fig. S5† and suggest that the addition of an extra adsorbate to the bottom of the metal does not significantly affect which metals could fit our criteria.
Therefore, the application of all three criteria leaves only the transition metal atoms in group 9, which can form stable CO–O2 complexes with reasonable bond distances between the carbon atom in CO and an oxygen atom in O2 (1.5 Å). Group 9 includes not only Co, which we used to establish the selection criteria, but also Ir–N–C and Rh–N–C.11 Rhodium–N–C catalysts have previously been experimentally synthesized27,39 so the Rh–N–C catalyst was chosen instead of Ir–N–C for subsequent calculations of the reaction coordinate and comparison with experimental data.
The Rh–N–C catalyst provides an opportunity for both experimental validation and an exploration of potential intermediates in a catalytic cycle using structures from the previously described Co–N–C mechanism.11 The first step in the catalytic cycle involves adsorption and as discussed earlier, calculations for CO and O2 revealed that they had similar binding energies on the Rh so either structure could be the starting structure for the catalytic cycle. Although Rh is known to form Rh gem-dicarbonyl structures,40 optimization of Rh gem-dicarbonyl structures resulted in the desorption of one molecule of CO, leaving only one CO bound to the metal. The next step involves interaction of both CO and O2 and the mechanism could start with either CO or O2 initially adsorbed but calculations with O2 bound to the metal ion and CO in the gas phase did not result in formation of a CO–O2 complex. The inability to form the CO–O2 complex with O2 initially on the metal suggests that CO binding to the Rh metal is the first step in the catalytic cycle and is denoted in the potential energy diagram in Fig. 2 as step 2. The CO molecule then interacts with a gas phase O2 molecule to form a CO–O2 complex on the Rh (step 3). The complex then goes through a transition state where the bound O2 molecule interacts with the carbon support (step TS1) before dissociating and forming O* on the carbon support and producing a gas phase CO2 molecule (step 4). We cannot completely rule out the migration of the O* atom to the metal site but the estimated barrier is high (∼39 kJ mol−1) relative to the reaction with CO (Fig. S7†). The reaction sequence is then closed by the binding of a new CO molecule to Rh (step 5) before interacting with the O* on the carbon surface through a negligible barrier of ∼0 kJ mol−1 (TS2) to form a gas CO2 molecule. The bond distance between the second adsorbed CO and the O on the surface is 2.56 Å during TS2 which is longer than the typical 1.16 Å for C–O bonds in CO2 and reflects the early transition state for this reaction (Fig. S2b†). Apparently, the distance still allows for the low barrier of formation for the second CO2 which suggests that the binding of CO to the metal may not be necessary since it is possible that the O on the surface is reactive enough to form CO2 directly. Since the metal site likely adsorbs CO throughout the reaction process, it is difficult to deconvolute whether CO would bind first. Thus, the only significant barrier during the overall reaction sequence is the 20 kJ mol−1 activation energy (TS1). This barrier was similar regardless of the use of vdW-DF or the PBE functional (18 kJ mol−1 for PBE, Fig. S7†). The barriers associated with this mechanism are similar to those of our previously calculated barriers for CO oxidation on a Co–N–C catalyst, with its most significant barrier equal to 16 kJ mol−1.11 The mechanism for Co–N–C did not involve a redox cycle on the Co and thus redox on the Rh atom was further explored.11
The oxidation state of Rh was investigated by Bader charge and density of states (DOS) analyses. Bader charge analysis (Table S2†) indicated small differences in the charge density of Rh (0.0–0.3 e) throughout the mechanism. Calibration of the charge densities to Rh structures with known oxidation states41,42 yielded inconclusive results for Rh oxidation states because the assignments varied widely depending on the precise choice of reference structures, which can be attributed to the larger variations in Bader-derived computed charges than other charge-partitioning schemes.43 To assess charge transfer on Rh throughout the mechanism in more detail, we performed projected density of states analysis (PDOS) for Rh. Although the PDOS analysis (Fig. S8a†) shows variations in the energy levels of the Rh d-states for different reaction intermediates, the integrated Rh PDOS at the Fermi level (Fig. S8b†) shows an approximately equal number of electrons on Rh for all structures along the reaction coordinate. The total number of occupied states for Rh does not change during the CO oxidation reaction, which suggests that there is no significant change in the Rh oxidation state throughout the cycle. The invariance of the Rh oxidation state during CO oxidation is consistent with the analogous reaction coordinate for Co–N–C.11
3.2 Catalyst synthesis and characterization
The DFT calculations suggest that Rh–N–C sites may utilize a mechanism that involves weak binding of reactants and a low transition state barrier for low temperature CO oxidation. To investigate isolated Rh in N-doped carbon for low temperature CO oxidation, a 1.26 wt% Rh–N–C catalyst was synthesized via a modified high-temperature pyrolysis method similar to previous Co– (ref. 8 and 11) and Rh–N–C (ref. 27) catalysts. To confirm the presence of isolated Rh ions on Rh–N–C, we examined the Rh–N–C by atomic-resolution HAADF-STEM, as shown in Fig. 3. Although atomically dispersed Rh ions are apparent and are likely stable since they are present after treatments in acid-peroxide solution and high-temperature H2, some nanoparticles are still observed. The coexistence of isolated Rh with Rh nanoparticles prevents detailed characterization by a bulk averaging technique such as X-ray absorption spectroscopy. Nevertheless, the similar content of Rh before and after treatment (1.23 versus 1.26 wt%) indicates that a significant amount of Rh single atoms is not removed and thus can be investigated for low temperature CO oxidation. | ||
| Fig. 3 Atomic-resolution HAADF-STEM images of Rh–N–C show isolated Rh atoms and small Rh nanoparticles. The left panel shows a small area where the isolated atoms of Rh are visible and the right panel shows an expanded view with few nanoparticles of Rh present. Some isolated Rh atoms in the image are circled in yellow. Rhodium nanoparticles are circled in red. | ||
3.3 Oxidation of CO over Rh catalysts
The steady-state activity of 1.26 wt% Rh–N–C for CO oxidation was compared to those measured over a nitrogen-free commercially available 4.63 wt% Rh/C catalyst and a highly dispersed 0.29 wt% Rh/SiO2 catalyst. All of these catalysts were evaluated over a range of temperatures (199 to 460 K) at a total pressure of 3 atm. Temperatures were alternated between high and low to ensure that any potential hysteresis could be explored. Fig. 4 shows the catalytic activity of Rh–N–C at temperatures <418 K, with activity being observed at as low as 199 K. The high activity for the Rh–N–C contrasts with the other Rh catalysts which contain metal particles that are known to be covered in CO at the lower temperatures used here, blocking the O2 adsorption needed for turnover. Indeed, the other Rh containing catalysts exhibited no detectable activity at low temperatures (273 K and lower). As the N–C support exhibited negligible conversion at the temperatures studied, it is likely that the activity comes from the isolated Rh.11 Interestingly, a Rh/TiO2 catalyst was previously reported to have low temperature CO oxidation activity and its performance was attributed to the formation of a CO–O2 complex similar to the one proposed in this study.44 In that work, CO bound on Rh was used to activate O2 with the reducible TiO2 support. Although our Rh–N–C catalyst does not have a reducible support, its low temperature CO oxidation activity is consistent with the CO-assisted activation of O2 as described earlier. In the higher temperature regime (>418 K), the activity begins to increase with temperature. The presence of nanoparticles, as confirmed by STEM, could account for the observed high temperature activity of Rh–N–C. Fortunately, reaction kinetics can be used to discriminate between the participation of metal nanoparticles versus isolated Rh cations in the CO oxidation reaction.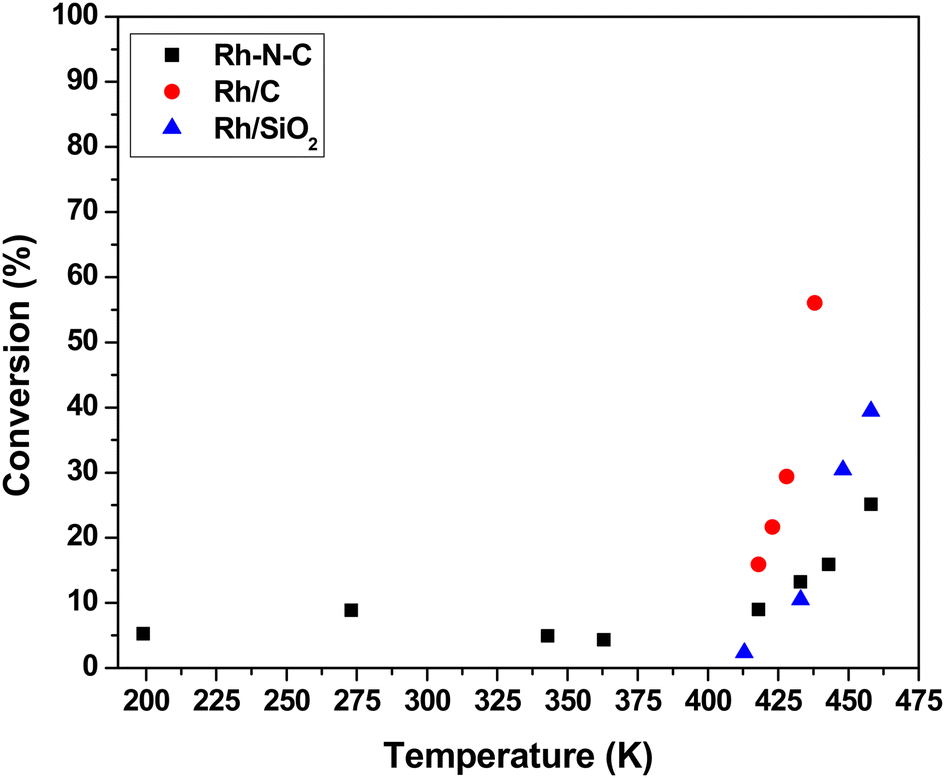 | ||
| Fig. 4 Influence of temperature on the steady-state CO conversion over Rh catalysts. The reactions were run with 0.1 g of catalyst diluted in SiC with 160 cm3 min−1 total flow, 1% CO, 2% O2 and balance He at 3 atm. | ||
The observed steady-state orders of reaction during CO oxidation catalyzed by Rh–N–C as shown in Table 1 were strikingly different at low temperature (273 K) compared to high temperature (498 K). The positive order dependence of the rate on CO for the low temperature CO oxidation over Rh–N–C contrasts with the inhibition by CO observed over Rh/C and Rh/SiO2 in this study (Table 1) and other reported platinum-group metal catalysts.45,46 The positive CO reaction order is consistent with the weak adsorption of CO on the active site, as suggested by DFT. The calculated binding energy of CO on Rh–N–C is significantly lower than that on Rh metal (−120 kJ mol−1), which has a negative CO order under similar concentrations of CO and O2.47 As CO oxidation does not occur to any measurable extent on Rh nanoparticles at low temperature, the reactivity results are attributed solely to the isolated Rh ions in Rh–N–C and the neighboring Rh nanoparticles are merely spectators on the Rh–N–C. While the reaction order for CO in the high temperature regime (>418 K) is different than that at low temperature, the nearly zero order dependence in CO is consistent with high temperature catalytic activity being a convolution of the reaction occurring on the nanoparticles (observed using STEM) and on the isolated Rh ions. Evidently, inhibition by CO on isolated Rh nanoparticles and the positive order in CO on isolated Rh combine to a nearly zero-order dependence at the temperature used here. A previous study on catalysts with various ratios of both nanoparticles and isolated atoms (Ir–MgAl2O4) reported changes in the reaction order with reaction conditions being used to probe isolated atoms versus nanoparticles.48 By increasing the partial pressure of CO, which inhibited the reaction on nanoparticles, the kinetics of the isolated atoms could be studied.48 In our case, the higher partial pressures of CO did not result in a low enough contribution of the inhibited nanoparticles. The reaction order in O2 is positive on both nanoparticles and isolated Rh at both low and high temperatures, so it cannot be used to discriminate between the different types of sites.
| Catalyst | E apparent (kJ mol−1) | Orders of reactionb | |
|---|---|---|---|
| CO | O2 | ||
| a Apparent activation energies were determined from Arrhenius-type plots (Fig. S9†). b Kinetic orders were evaluated under steady-state differential conversion (<15%) and were determined by varying the partial pressure of one while holding the other constant (Fig. S10a and b†). It should be noted that at higher partial pressures of CO and O2 for the high temperature regime for the Rh–N–C catalyst, the conversion goes as high as 28% but does not appear to affect the linearity. c Kinetic orders for O2 on Rh/C and Rh/SiO2 were calculated under constant 3% CO instead of 1% CO reaction conditions to remain in the differential conversion regime. | |||
| Rh–N–C | |||
| Low-T regime <403 K | ∼0 | 0.6 | 0.6 |
| High-T regime >403 K | 40 | 0 | 1.1 |
| Rh/C | 98 | −1.0 | 1.5c |
| Rh/SiO2 | 110 | −1.0 | 1.5c |
Table 1 shows the apparent activation energies for CO oxidation over the Rh containing catalysts. The rate varies little with temperature in the low temperature regime for Rh–N–C (<413 K), which is consistent with a very small apparent activation energy (∼0 kJ mol−1). The low temperature regime contrasted with the high temperature regime (413–460 K) wherein the rate increased significantly with temperature. Similar to the reported orders of reaction in this high temperature regime, the apparent activation energy is likely a convolution of the rate occurring on both the isolated Rh atoms and the Rh nanoparticles. The apparent activation energies measured in the high temperature regime (413–460 K) for the other Rh containing catalysts, Rh/C and Rh/SiO2, are consistent with CO oxidation on Rh nanoparticles.45 Thus, the reaction orders and apparent activation energy for Rh–N–C at high temperatures are not consistent with the CO-assisted O2 activation mechanism depicted in Fig. 2, whereas the low temperature CO oxidation kinetics over Rh–N–C are completely consistent with that mechanism.
4. Discussion
To understand if the mechanism and elementary steps reasonably approximate the experimental kinetics, a rate expression for the reaction path was derived. The reaction path involves the weak adsorption of reactants on the surface, which is consistent with the observed reaction kinetics (observed reaction orders and the approximately zero apparent activation energy). The deviations from first-order dependence suggest non-zero coverage of CO or competition between CO and O2, which is apparent with the similar computationally calculated binding energies for the structure (−68 kJ mol−1 for both CO and O2).The overall rate of reaction should be proportional to the reaction step involving dioxygen activation (TS1 in Fig. 2, CI-NEB shown in Fig. S2a†) as the sequential oxidation of CO with the second oxygen atom from Fig. 2 is nearly barrierless (CI-NEB for TS2 in Fig. S2b†). Thus, we write the expression for the observed rate as:
| rate = k[CO–O2]ads | (1) |
| rate = kθCOγO2 | (2) |
 | (3) |
 . To compare with the proposed rate expression with experiment, the Langmuir isotherm can be approximated as a power law expression to give an observed rate expression as:
. To compare with the proposed rate expression with experiment, the Langmuir isotherm can be approximated as a power law expression to give an observed rate expression as: | (4) |
 | (5) |
| Eapparent = Ea + 0.6ΔHCO + 0.6ΔHCO–O2 − 0.4ΔHO2 = −1 kJ mol−1 | (6) |
The proposed low-temperature mechanism of CO oxidation on Rh–N–C does not preclude other potential mechanisms for low or high temperature CO oxidation occurring on the isolated Rh sites. As mentioned in previous sections, other screened metals had various binding energies of CO and O2 to the metal ion. Metal ions that bound CO more weakly than the binding energy of CO to Co or Rh were unable to form the O–O–C–O transition state, while transition metals such as Fe have been previously investigated with O2 binding first to give a higher barrier transition state.16 This indicates that other mechanisms are possible, but the one studied here seems amenable to this particular column of the periodic table and thus catalysts containing metals in group 9 appear to be active at low temperatures for CO oxidation.
A simplified kinetic analysis was applied to the low-temperature regime but the model accounts for the change in binding energy and potential competition of CO and O2 on the surface of the Rh ion. The model does still account for the positive order behavior of both CO and O2 along with the nearly zero apparent activation energy. This similarity implies that there is a small barrier for O2 activation, and the rate is instead dominated by the number of adsorbed intermediates leading to the product and the competition of CO and O2 for the same surface sites. This contrasts with the literature on platinum group metals and supported Rh nanoparticles where CO inhibits adsorption of O2 to adjacent surface vacancies and thus prevents dissociative O2 chemisorption, leading to a negative order for CO.46,49
Interestingly, similar transition states have previously been attributed to low temperature CO oxidation on Rh and Ir. For example, Rh/TiO2 was found to have a low barrier with the reducible support enabling the formation of O–O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and activation through vacancies in the TiO2.44 The similarity to other Rh systems indicates that other isolated Rh ions may be able to activate O2 in a similar mechanism. Indeed, another transition metal from the same group as Co, Ir, was previously investigated as an isolated Ir-on-MgAl2O4 catalyst.50 The proposed mechanism for CO oxidation involved a spectator CO molecule that enables an Eley–Rideal-type mechanism with surface oxygen.50 Isolated atoms may enable interesting interactions with surface groups surrounding the transition metal site.
O and activation through vacancies in the TiO2.44 The similarity to other Rh systems indicates that other isolated Rh ions may be able to activate O2 in a similar mechanism. Indeed, another transition metal from the same group as Co, Ir, was previously investigated as an isolated Ir-on-MgAl2O4 catalyst.50 The proposed mechanism for CO oxidation involved a spectator CO molecule that enables an Eley–Rideal-type mechanism with surface oxygen.50 Isolated atoms may enable interesting interactions with surface groups surrounding the transition metal site.
After screening metals for reaction paths similar to the previously derived CO-assisted mechanism, it appears that the group 9 metals containing Co, Rh, and Ir have characteristics that enable low temperature CO oxidation through a CO-assisted mechanism. While other metals may be active for low temperature O2 activation, the group 9 transition metals appear to activate O2 through a CO-assisted mechanism involving the graphene structure as a second site.
5. Conclusions
Our results suggest that the group 9 transition metal ions in N–C have similar characteristics of preferential binding of CO, exothermic enough binding energy of CO on the transition metal ion, and the ability to form a CO–O2 complex similar to our previous report on Co–N–C. Detailed DFT calculations on the Rh–N–C catalyst suggest that the Rh–N–C catalyst could proceed through a low temperature mechanism involving CO-assisted O2 activation with the carbon support. Experimental studies with a synthesized Rh–N–C catalyst showed low temperature activity and reaction orders that are consistent with the DFT calculations. Application of the kinetic parameters with the DFT derived adsorption and barrier values resulted in a predictive rate expression. The results suggest that O2 could be activated through a CO assisted method on the transition metal atom in group 9 and this could be general in nature.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Financial support was provided by the U.S. National Science Foundation under grant #CBET-1802482. C. P. acknowledges the donors of the American Chemical Society Petroleum Research Fund #65153-DNI6 for the partial support of this research. The scanning transmission emission electron microscopy research was assisted by Dr. Helge Heinrich at UVA's Nanoscale Materials Research Facility.References
- H. W. Liang, W. Wei, Z. S. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed.
- Q. Liu and J. Zhang, Langmuir, 2013, 29, 3821–3828 CrossRef CAS.
- D. Malko, A. Kucernak and T. Lopes, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil, S. Mukerjee, E. Fonda and F. Jaouen, Nat. Commun., 2017, 8, 957 CrossRef PubMed.
- Q. Zhang, K. Mamtani, D. Jain, U. Ozkan and A. Asthagiri, J. Phys. Chem. C, 2016, 120, 15173–15184 CrossRef CAS.
- S. Vijay, W. Ju, S. Brückner, S. C. Tsang, P. Strasser and K. Chan, Nat. Catal., 2021, 4, 1024–1031 CrossRef CAS.
- J. Xie, K. Yin, A. Serov, K. Artyushkova, H. N. Pham, X. Sang, R. R. Unocic, P. Atanassov, A. K. Datye and R. J. Davis, ChemSusChem, 2017, 10, 359–362 CrossRef CAS PubMed.
- J. Xie, J. D. Kammert, N. Kaylor, J. W. Zheng, E. Choi, H. N. Pham, X. Sang, E. Stavitski, K. Attenkofer, R. R. Unocic, A. K. Datye and R. J. Davis, ACS Catal., 2018, 8, 3875–3884 CrossRef CAS.
- K. Huang, H. Fu, W. Shi, H. Wang, Y. Cao, G. Yang, F. Peng, Q. Wang, Z. Liu, B. Zhang and H. Yu, J. Catal., 2019, 377, 283–292 CrossRef CAS.
- J. Li, J. Liu, J. Zhang, T. Wan, L. Huang, X. Wang, R. Pan, Z. An and D. G. Vlachos, Appl. Catal., A, 2020, 591 Search PubMed.
- C. A. Whitcomb, A. Sviripa, M. I. Schapowal, K. Mamedov, R. R. Unocic, C. Paolucci and R. J. Davis, ACS Catal., 2022, 12, 15529–15540 CrossRef CAS.
- J. Wang, R. You, C. Zhao, W. Zhang, W. Liu, X. P. Fu, Y. Li, F. Zhou, X. Zheng, Q. Xu, T. Yao, C. J. Jia, Y. G. Wang, W. Huang and Y. Wu, ACS Catal., 2020, 10, 2754–2761 CrossRef CAS.
- F. Luo, C. H. Choi, M. J. M. Primbs, W. Ju, S. Li, N. D. Leonard, A. Thomas, F. Jaouen and P. Strasser, ACS Catal., 2019, 9, 4841–4852 CrossRef CAS.
- F. Luo, S. Wagner, I. Onishi, S. Selve, S. Li, W. Ju, H. Wang, J. Steinberg, A. Thomas, U. I. Kramm and P. Strasser, Chem. Sci., 2021, 12, 384–396 RSC.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- T. Kropp and M. Mavrikakis, ACS Catal., 2019, 9, 6864–6868 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. Zhang, X. F. Chen, J. S. Lian and Q. Jiang, J. Phys. Chem. C, 2012, 116, 17572–17579 CrossRef CAS.
- Q. Wang, B. Jin, M. Hu, C. Jia, X. Li, E. Sharman and J. Jiang, J. Phys. Chem. C, 2021, 125, 5616–5622 CrossRef CAS.
- B. L. He, J. S. Shen and Z. X. Tian, Phys. Chem. Chem. Phys., 2016, 18, 24261–24269 RSC.
- M. Dion, H. Rydberg, E. Schroder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Josson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Josson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- Z. Luo, C. A. Whitcomb, N. Kaylor, Y. Zhang, S. Zhang, R. J. Davis and T. B. Gunnoe, ChemCatChem, 2021, 13, 260–270 CrossRef CAS.
- N. Kaylor, J. Xie, Y. S. Kim, H. N. Pham, A. K. Datye, Y. K. Lee and R. J. Davis, J. Catal., 2016, 344, 202–212 CrossRef CAS.
- Y. L. Lam and M. Boudart, J. Catal., 1977, 50, 530–540 CrossRef CAS.
- F. B. M. Duivenvoorden and D. C. Koningsberger, J. Catal., 1987, 38, 26–38 Search PubMed.
- N. Zhang, T. Zhou, M. Chen, H. Feng, R. Yuan, C. A. Zhong, W. Yan, Y. Tian, X. Wu, W. Chu, C. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 111–118 RSC.
- E. Haque, A. Zavabeti, N. Uddin, Y. Wang, M. A. Rahim, N. Syed, K. Xu, A. Jannat, F. Haque, B. Y. Zhang, M. A. Shoaib, S. Shamsuddin, M. Nurunnabi, A. I. Minett, J. Z. Ou and A. T. Harris, Chem. Mater., 2020, 32, 1384–1392 CrossRef CAS.
- R. Ma, G. Lin, Q. Ju, W. Tang, G. Chen, Z. Chen, Q. Liu, M. Yang, Y. Lu and J. Wang, Appl. Catal., B, 2020, 265, 118593 CrossRef CAS.
- J. Liu, X. Kong, L. Zheng, X. Guo, X. Liu and J. Shui, ACS Nano, 2020, 14, 1093–1101 CrossRef CAS PubMed.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- D. Jain, Q. Zhang, V. Gustin, J. Hightower, S. Gunduz, A. C. Co, J. T. Miller, A. Asthagiri and U. S. Ozkan, J. Phys. Chem. C, 2020, 124, 10324–10335 CrossRef CAS.
- J. C. A. Boeyens, Z. Naturforsch., B: J. Chem. Sci., 2008, 63, 199–199 CrossRef CAS.
- M. Rebarchik, S. Bhandari, T. Kropp and M. Mavrikakis, ACS Catal., 2020, 10, 9129–9135 CrossRef CAS.
- Y. Xiong, J. Dong, Z. Q. Huang, P. Xin, W. Chen, Y. Wang, Z. Li, Z. Jin, W. Xing, Z. Zhuang, J. Ye, X. Wei, R. Cao, L. Gu, S. Sun, L. Zhuang, X. Chen, H. Yang, C. Chen, Q. Peng, C. R. Chang, D. Wang and Y. Li, Nat. Nanotechnol., 2020, 15, 390–397 CrossRef CAS PubMed.
- J. T. Yates and K. Kolasinski, J. Chem. Phys., 1983, 79, 1026–1030 CrossRef CAS.
- Y. Shimizu, H. Mitsuhashi and E. Caspi, Tetrahedron Lett., 1966, 7, 4113–4116 CrossRef.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- Z. Wang, J. Jin and M. Liu, J. Phys. Chem. C, 2023, 127, 17345–17354 CrossRef CAS.
- H. Guan, J. Lin, B. Qiao, X. Yang, L. Li, S. Miao, J. Liu, A. Wang, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 2820–2824 CrossRef CAS.
- S. M. McClure and D. W. Goodman, Chem. Phys. Lett., 2009, 469, 1–13 CrossRef CAS.
- G. Krenn, I. Bako and R. Schennach, J. Chem. Phys., 2006, 124, 144703 CrossRef CAS PubMed.
- A. Guerrero-Ruiz, A. Maroto-Valiente, B. Bachiller-Baeza and I. Rodriguez-Ramos, Top. Catal., 2002, 19, 303–311 CrossRef CAS.
- Y. Lu, C. T. Kuo, L. Kovarik, A. S. Hoffman, A. Boubnov, D. M. Driscoll, J. R. Morris, S. R. Bare and A. M. Karim, J. Catal., 2019, 378, 121–130 CrossRef CAS.
- A. D. Allian, K. Takanabe, K. L. Fujdala, X. Hao, T. J. Truex, J. Cai, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 4498–4517 CrossRef CAS PubMed.
- Y. Lu, J. Wang, L. Yu, L. Kovarik, X. Zhang, A. S. Hoffman, A. Gallo, S. R. Bare, D. Sokaras, T. Kroll, V. Dagle, H. Xin and A. M. Karim, Nat. Catal., 2019, 2, 149–156 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01518a |
| This journal is © The Royal Society of Chemistry 2024 |