
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigations into the influence of nickel loading on MoO3-modified catalysts for the gas-phase hydrodeoxygenation of anisole†
Simon
Haida
 ,
Sebastian
Löbner
,
Henrik
Lund
,
Stephan
Bartling
,
Carsten
Kreyenschulte
,
Hanan
Atia
,
Ali M.
Abdel-Mageed
,
Christoph
Kubis
* and
Angelika
Brückner
*
,
Sebastian
Löbner
,
Henrik
Lund
,
Stephan
Bartling
,
Carsten
Kreyenschulte
,
Hanan
Atia
,
Ali M.
Abdel-Mageed
,
Christoph
Kubis
* and
Angelika
Brückner
*
Leibniz-Institute for Catalysis e.V, Albert-Einstein Str. 29a, 18059 Rostock, Germany. E-mail: Christoph.Kubis@catalysis.de; Angelika.Brueckner@catalysis.de
First published on 5th March 2024
Abstract
Molybdenum oxide-based catalysts are promising catalysts for the gas-phase hydrodeoxygenation (HDO) of lignocellulosic pyrolysis oils with high selectivities to arenes. The gas-phase HDO is conducted at temperatures between 300 °C and 400 °C at ambient pressure, resulting in a low hydrogen consumption and high energy efficiency. The loading of nickel forming a binary structure consisting of MoO3 and nickel molybdate (NiMoO4) during calcination results in a significant increase of the catalytic activity connected with a drastic shortening of the induction period observed for the unpromoted catalyst system. The promotional effect of nickel seems to be related to its enhanced reduction behaviour and hydrogen dissociation leading eventually to an improved formation of a molybdenum oxycarbohydride phase (MoOxCyHz). The MoOxCyHz phase plays an important role in stabilizing active Mo5+ sites and prevents over-reduction to inactive MoO2. The activity and benzene selectivity are maximal when pure NiMoO4 is used as a catalyst. However, the selectivity towards undesired methane increases significantly indicating the decomposition of arenes. This effect is considerably reduced for catalysts with lower nickel contents (3–5%) which still exhibit a highly improved activity compared to MoO3. In situ XRD studies revealed that the population of the MoOxCyHz phase is strongly affected by the nickel content, the structure of the hydrocarbon substrate and the hydrogen content during pre-reduction and catalysis.
Introduction
Global warming and its consequences for our environment are one of the most challenging problems to date. For the development of a sustainable and circular economy a substitution of crude oil by renewable feedstocks is an important aspect.1 One alternative feedstock to oil is lignin, as it is produced naturally in large amounts and can provide a wide range of different possible products.2,3 Lignin is a complex polymer out of phenolic monomers of which the majority consists of sinapyl alcohol, coniferyl alcohol and p-coumaryl alcohol. For valorization, a pyrolysis step can be applied to convert biomass into its monomers, yielding a bio-oil. This can be performed by fast pyrolysis at T = 400–600 °C to obtain bio-oils in high yields.4 However, due to the acidic properties of these pyrolysis-oils, there is a tendency for repolymerization. Further, their oxygen content is too high for direct usage as a feedstock for the production of fuels or chemicals.5,6 Therefore, further process steps are required to reduce the oxygen content and thus gain a higher energy density. This can be achieved by the hydrodeoxygenation (HDO) reaction by using hydrogen. Apart from the utilization of respective products as renewable fuels, especially the production of BTX-aromatics (benzene, toluene and xylenes) seems to be an attractive route, as lignin is the most abundant polymer with aromatic rings in nature.7,8 In Scheme 1 a simplified reaction path for the HDO of anisole, which is often used as a model substrate, to BTX-aromatics is presented. | ||
| Scheme 1 Hydrodeoxygenation of anisole to BTX-aromatics and selected possible by-products. | ||
The HDO reaction can be conducted in the liquid phase at moderate temperatures (200–400 °C) and higher partial hydrogen pressures (40–200 bar). The early generation of HDO catalysts consisted of typical hydroprocessing catalysts such as sulfided NiMo/Al2O3 and CoMo/Al2O3 or transition metal based catalysts.9,10 Meanwhile, a large variety of catalyst types, e.g. supported mono- and bimetallic catalysts (noble and base metal based), supported and unsupported metal oxide catalysts (e.g. MoO3) and nitrides and phosphides have been developed and tested.11,12 Transition metal catalysts often show a high activity, but lower arene selectivity. In contrast, Mo based catalysts (NiMo, CoMo, MoO3) tend to possess lower activities, but higher arene selectivities. Alternatively, the process can be operated at moderate temperatures (300–400 °C) but low hydrogen pressures (p(H2) ≤ 1 atm) in the gas-phase. This not only has the advantage of higher arene selectivity, but also the gas-phase HDO as a downstream process is an opportunity to use the energy of the previous pyrolysis step. Thus, a process design with an optimized energy balance is possible, as no cooldown and compression are necessary. Respective economic and ecological benefits should increase the overall process efficiency. Prominent catalysts which are investigated for gas-phase HDO are supported transition metal catalysts as well as pristine and promoted MoO3. Due to the tendency of monometallic transition metal catalysts for ring hydrogenation, the arene selectivity is often relatively low. MoO3 is an interesting alternative, as it shows good activity in the HDO reaction with comparatively high selectivity towards arenes.12,13
It is widely accepted that the catalytically active sites for MoO3-based catalysts are Mo5+ ions, which are generated via partial reduction accompanied by the formation of oxygen vacancies at the surface of the catalyst on which the oxygenate substrate adsorbs via its oxygen atoms (Scheme 2a). The catalytic cycle of the HDO reaction can be described as a reverse Mars–van Krevelen type mechanism.16–21 Several reports suggests also the in situ formation of a molybdenum oxycarbohydride phase (MoOxCyHz) which seems to stabilize the oxidation state (V) of the molybdenum ions (Scheme 2b).14,15,22,23 The mechanism of the formation of such a MoOxCyHz phase from MoO3 in a hydrogen/hydrocarbon feed consists of several steps: 1) formation of MoO3−xvia partial reduction and generation of oxygen vacancies, 2) formation of molybdenum oxyhydride (MoOxHy) by insertion of dissociated hydrogen into the solid, and 3) incorporation of carbon into the MoOxHy intermediate to form MoOxCyHz.24,25 The dissociation of molecular hydrogen is catalyzed by MoO2 which is partly formed during reduction. The presence of a transition metal might promote the formation of a MoOxCyHz phase because of its enhanced activity in hydrogen dissociation.24 A significant over-reduction into MoO2 (Mo4+) leads to a significant decrease of catalytic activity. A MoOxCyHz phase cannot be formed from MoO2 because its precursor molybdenum oxyhydride (MoOxHy) species are not formed from MoO2. Coke formation is another important cause for the deactivation of HDO catalysts. A regeneration step consisting of in situ calcination followed by pre-reduction recovers the catalytic activity.14
 | ||
| Scheme 2 a) Catalytic cycle of the hydrodeoxygenation of anisole based on the reverse Mars–van Krevelen type mechanism; b) sequence of phase transformation steps for the generation of a molybdenum oxycarbohydride phase (adapted and slightly modified from Richard et al. (a)16 and Ledoux et al. (b)24). | ||
Due to the importance of oxygen vacancies for substrate adsorption and activation, it is expected that changes in the electronic properties of the catalyst have a significant impact on the catalytic performance. To achieve this, oxide materials can be modified by the introduction of secondary metal atoms. For hydrodenitrogenation (HDN) and hydrodesulfurization (HDS), it was demonstrated that NiMoO4 and CoMoO4 have altered catalytic properties compared to bulk MoO3. Also, it is known that the substitution of lattice metal atoms by low-valence dopants (LVD) does influence the redox properties of metal oxides and turn the oxide surface into a Lewis acid which in turn lowers the energy required for the formation of oxygen vacancies.26,27 For HDO, there are several reports in which molybdenum oxide-based materials doped or modified by transition metals such as Ni, Co or Pt showed improved activities compared to pure MoO3.28–33 When nickel and cobalt are used as modifiers most often the respective metal molybdate phases (NiMoO4 and CoMoO4) are obtained after catalyst preparation and calcination.29,32,34,35 Interestingly, no comparative studies have been performed to elucidate the impact of metal molybdate phases on catalyst performance and phase composition depending on their content.
Based on the discussed aspects vide supra, in this study we focus on the behavior of pure metal molybdates NiMoO4 and CoMoO4 as well as mixtures with minor amounts of these molybdate phases (1–5 wt% metal content) in a MoO3 matrix in the gas-phase hydrodeoxygenation of anisole. The influence of the content of molybdate phases in MoO3 on the catalytic performance and phase composition are investigated. A specific focus is laid on the formation and decomposition of a molybdenum oxycarbohydride phase (MoOxCyHz) under the variation of reaction conditions for different catalysts. Structure–performance relationships are derived based on the results from in situ XRD, XPS and DRIFTS investigations leading to a comprehensive understanding of the promoting effect of metal molybdates on the catalytic HDO reaction.
Experimental section
Catalyst preparation
Commercial MoO3 (Carl Roth, ≥98%) was used as a catalyst, as well as for the synthesis. For the synthesis of NiMoO4 and CoMoO4, 13.303 g (45.7 mmol) Ni(NO3)2·6H2O (Sigma Aldrich, 99.1%) or 13.297 g (45.7 mmol) Co(NO3)2·6H2O (Carl Roth, ≥98%), respectively, and 8.076 g (6.5 mmol) (NH4)6Mo7O24·4H2O (Carl Roth, ≥99%) were dissolved in water. After complete solvation, the respective solution was stirred for 0.5 h and the water was evaporated at T = 80 °C afterwards. The sample was dried at 100 °C overnight and calcined at 450 °C for 5 h in syn. air. Samples with 1, 3 and 5 wt% content of Co or Ni were prepared by the same procedure using respective amounts of the components to achieve the desired metal content. An overview about the catalyst materials used in this study is given in Table 1.| Description | ICP (M/wt%) | Denotation |
|---|---|---|
| Pure MoO3 | — | MoO3 |
| Pure NiMoO4 | 27.34 | NiMoO4 |
| Pure CoMoO4 | 26.73 | CoMoO4 |
| 5 wt% Ni in MoO3 | 4.91 | Ni(5)MoO3 |
| 3 wt% Ni in MoO3 | 2.79 | Ni(3)MoO3 |
| 1 wt% Ni in MoO3 | 1.02 | Ni(1)MoO3 |
| 5 wt% Co in MoO3 | 4.73 | Co(5)MoO3 |
According to the protocol of Glemser et al.,36 MoOxHy was prepared by forming a suspension of 5.00 g MoO3 and adding 1.8 g (27.5 mmol) zinc granules (Thermo Scientific, d = 1–5 mm, 99.999%). Hydrochloric acid was added 3.5 mL (4 N) and the mixture was stirred overnight. Afterwards, the suspension was filtered, the remaining zinc has been removed and the slurry was washed with deionized water until no chloride was detected with AgNO3. The product was dried at 60 °C.
Catalyst characterization
In situ XRD studies were performed on a Stoe Stadi P equipped with a Stoe ht2-in situ oven and a Mythen 1K detector in Debye–Scherrer geometry using monochromatized Mo Kα1 radiation (50 kV, 40 mA, 0.70930 Å). The sample investigated was ground, pressed to pellets at 10 tons, crushed, and sieved to fractions of 100–150 μm. A specimen was filled into a quartz glass capillary (approx. 2 mm outer diameter, 1 mm inner diameter, opened on both sides) until a height of approx. 6 mm was achieved and fixed using quartz glass wool. After mounting the capillary into the oven, the capillary was flushed with He (10 mL min−1) and the sample was heated to the desired temperature (350 °C or 325 °C). After equilibration, the gas feed was changed to the reaction gas mixture (details in the corresponding description) with a total flow of 10 mL min−1 and the reaction was monitored using static data collection over a 17° angular region (Mo-radiation). The gas dosage was determined using a set of Bronkhorst mass flow controller units. The applied temperature correction function was obtained by observation of well-known phase transitions (AgNO3, KClO4, Ag2SO4, SiO2, K2SO4, K2CrO4, WO3, BaCO3). Peak positions and profiles were fitted with the pseudo-Voigt function using the HighScore Plus software package (PANalytical). Phase identification was performed by using the PDF-2 database of the International Center of Diffraction Data (ICDD). Because different X-ray sources were used (Mo, Cu, Ag), the diffraction data were converted to the Q-vector for comparison and representation purposes by using eqn (1).37
 | (1) |
The specimen was dry deposited onto a Si wafer. The accelerating voltage was set to 10 kV for imaging and spectroscopy, with spot size selected for optimum resolution or current, respectively. Elemental maps were calculated from the spectral imaging data set using the net counts fitting method provided in the Pathfinder software (Thermo Fisher).
Catalytic testing
The catalytic test experiments have been conducted in a gas-phase packed-bed flow reactor. The tube reactor had an inner diameter of 11.9 mm which was fixed in a furnace. A thermocouple was used for temperature regulation and placed inside the catalyst bed. The catalyst material was pressed and sieved to achieve particle sizes between 600 and 800 μm. Before the catalyst was placed in the reactor held with quartz wool, 200 mg of the material was mixed with approximately 2.3 g of SiO2 for diluting to a fixed volume. The SiO2 had a particle size of 500–800 μm. The anisole was transferred into the reactor using a saturator where the H2–N2-mixture was flushed through. The saturator temperature was kept at 20 °C which results in a total anisole flow of 0.236 mL min−1 in the gas-phase and a WHSV = 0.31 ganisole gcat−1 h−1. During the reaction, 40 mL min−1 N2 and 20 mL min−1 H2 at ambient pressure were used. Behind the reactor, 6.5 mL min−1 ethane was added as an internal standard. All gas flows have been controlled by Bronkhorst mass flow controller units. Prior to the addition of the substrate, the catalysts were heated up in N2 to 325 °C and pre-reduced in 100% H2 for 2 h at ambient pressure afterwards. After the pre-reduction step the catalysts were flushed with N2 and bypass measurements were performed for feed analysis. The organic reactants and products were monitored continuously during the reaction on stream with an online GC (see technical details below).Product analysis by online GC and calculation of performance data
The transfer line to the online GC system (Agilent 7890A) was kept at a temperature of 200 °C and the valve box of the GC was tempered at 210 °C. The sample loop had a volume of 250 μL. The GC column was a Quadrex 624 30 m × 250 μm × 2 μm. Helium was used as a carrier gas. The temperature program started at 40 °C for 2 min followed by heating up to 160 °C with 10 °C min−1 and a hold time of 5 min. Afterwards the oven was heated to 240 °C with 20 °C min−1 keeping the target temperature for additional 20 min. A FID was used as a detector.The calculation of the conversion and selectivity is based on the corrected integral values of respective GC peaks using the “Effective Carbon Number Concept”.38 Thus, the integrals of the compounds were normalized by its corresponding response factor ECN (2).
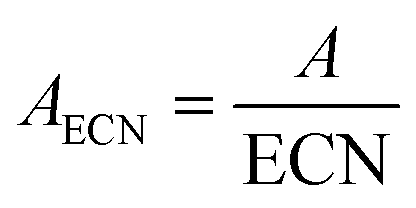 | (2) |
Conversion
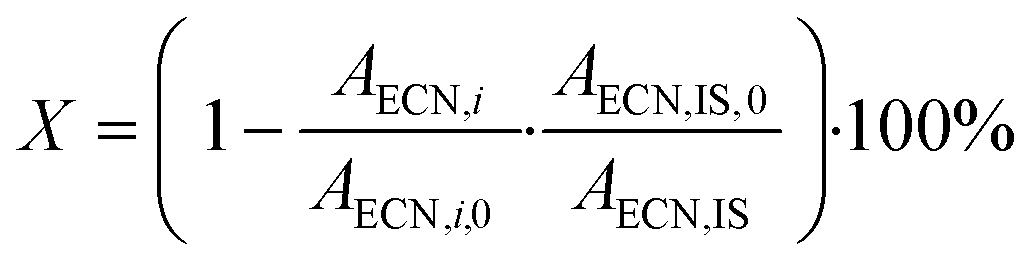 | (3) |
Selectivity
 | (4) |
Results and discussion
Selected material properties of fresh catalysts
Powder X-ray diffraction and Raman spectroscopy measurements were conducted to analyze the phase composition of fresh calcined catalyst materials (Fig. 1). The intense reflections of pure phase metal molybdates at 11 nm−1 and 21 nm−1 indicate the presence of NiMoO4, while reflections at 17 nm−1 and 20.5 nm−1 prove the existence of the CoMoO4 phase (Fig. 1a). For the nickel loaded catalysts (Ni(5)MoO3, Ni(3)MoO3 and Ni(1)MoO3), the respective reflections assigned to NiMoO4 can be observed at the same positions besides the major reflections from MoO3. This observation indicates that a binary structure consisting of NiMoO4 and MoO3 is present. The same result was observed for the cobalt containing sample (Co(5)MoO3) for which the CoMoO4 phase has been identified. | ||
| Fig. 1 Characterization of the fresh calcined samples. a) Normalized PXRD patterns; b) Raman spectra measured with a Renishaw inVia Raman microscope with λ = 633 nm and a laser power of 0.085 mW. | ||
These results were confirmed by Raman spectroscopy (Fig. 1b). The intense band for NiMoO4 observed at 961 cm−1 is characteristic of this phase and is attributed to the symmetric stretching mode of the terminal M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group.39 At the same time, bands at 818 cm−1 and 993 cm−1 belonging to the MoO3 phase are visible in the nickel loaded catalysts. The characteristic bands for the Mo–O–Co stretching vibration at ca. 937 cm−1 of CoMoO4 are visible together with respective band contributions for MoO3 in the cobalt containing catalyst.40
O group.39 At the same time, bands at 818 cm−1 and 993 cm−1 belonging to the MoO3 phase are visible in the nickel loaded catalysts. The characteristic bands for the Mo–O–Co stretching vibration at ca. 937 cm−1 of CoMoO4 are visible together with respective band contributions for MoO3 in the cobalt containing catalyst.40
Catalytic results
Prior to the catalytic testing a pre-reduction step was conducted (325 °C, 1 bar H2) for 2 h. The catalytic performance over 17 h time-on-stream (TOS) has been tested under atmospheric pressure at 325 °C for each catalyst revealing noticeable differences in their activity and product distribution (Table 2 and Fig. 2).| X(anisole) | S(methane) | S(benzene) | S(toluene) | S(xylenes) | S(phenol) | S(cresols) | S(arenes)/S(phenols) | |
|---|---|---|---|---|---|---|---|---|
| MoO3 | 35.1 | 3.4 | 27.7 | 6.1 | 4.9 | 18.9 | 8.9 | 1.36 |
| NiMoO4 | 97.9 | 20.2 | 41.8 | 10.1 | 3.0 | 4.7 | 1.5 | 8.85 |
| Ni(5)MoO3 | 80.6 | 4.2 | 32.1 | 8.7 | 1.0 | 17.7 | 13.1 | 1.36 |
| Ni(3)MoO3 | 80.3 | 3.7 | 32.7 | 9.6 | 3.5 | 15.4 | 12.0 | 1.67 |
| Ni(1)MoO3 | 55.9 | 3.7 | 31.6 | 7.8 | 2.7 | 18.5 | 12.0 | 1.38 |
| CoMoO4 | 44.6 | 3.2 | 25.3 | 5.7 | 1.6 | 27.9 | 14.5 | 0.77 |
| Co(5)MoO3 | 49.3 | 3.6 | 27.8 | 5.6 | 2.0 | 28.9 | 16.1 | 0.79 |
 | ||
| Fig. 2 Conversion of anisole HDO over 17 h TOS at 325 °C and P = 1 bar. Pre-reduced at 325 °C in 100% H2 for 2 h. | ||
Catalyst activity and induction periods
For the unpromoted MoO3 catalyst an induction period for ca. 5 h TOS was observed until the catalyst system reaches its maximum activity (equivalent to 37.7% conversion) followed by a moderate deactivation with time on stream. This type of catalytic behaviour is in accordance with previously reported results.14 In contrast to this observation, the NiMoO4 catalyst shows already in the beginning a very high activity with conversions of about 99% which only decreased slightly to 98% during the first 4 h. Surprisingly, no further deactivation is observed after reaching a steady-state within 6 h TOS until 17 h TOS. Compared to this behaviour, CoMoO4 only reaches 41.6% conversion after an induction period of ca. 5 h TOS.Ni(5)MoO3 and Ni(3)MoO3 are comparatively more active with conversions of 80.6% and 80.3% after 10 h TOS, respectively, without showing a distinct induction period and with only a low degree of deactivation. Conversely, for Ni(1)MoO3 the maximum conversion drops to 59.0% (6 h TOS) after passing through a noticeable induction period. Thereafter, a subsequent deactivation has been observed (55.9%, 10 h TOS).
The induction period for Co(5)MoO3 is significantly longer (10 h) after which a conversion value of 50.0% is achieved. Interestingly, with ongoing time-on-stream the conversion increases slightly.
The pronounced induction periods for MoO3, Ni(1)MoO3, CoMoO4 and Co(5)MoO3 might indicate complex phase transformations which follow different individual time dependencies. For the unpromoted MoO3 catalyst it is known from the literature that such an induction period is related to the formation of a MoOxCyHz phase.14 It is assumed that the lattice carbon in the oxycarbohydride phase stabilizes the active Mo5+ oxidation state and hinders the over-reduction into less active Mo4+ sites.
Further aspects regarding the dynamics of the induction period are the pre-reduction step prior to substrate addition and the hydrogen to substrate ratio during the HDO reaction which effects the reducibility in the presence of the oxygenate substrate. The generation of MoOxCyHz is significantly improved when MoO3 is pre-reduced in pure hydrogen at elevated temperatures (300–400 °C).14,24,25
Based on these considerations, one hypothesis for the absent induction period for NiMoO4, Ni(5)MoO3 and Ni(3)MoO3 is the improved reduction behaviour, when nickel is present in significant amounts, which can promote the MoOxCyHz formation proposed as an active intermediate (see discussion vide infra).24 In this context, it seems likely that MoO3 portions are reduced to a certain extent during the pre-reduction and the second step of partial carburisation is accomplished at such a short time scale, and that the effect of formation is not visible in the catalytic data.
The induction period of the cobalt containing catalysts might be caused by a much more complex reduction process of CoMoO4 which can consist of several intermediate steps (e.g. Co2Mo3O8 and CoMoO3), which are not observed for NiMoO4.41 Possibly, the reduction temperature might not be high enough to reduce CoMoO4 to Co0, which could act as a hydrogen activator. In addition, the hydrogenation capability of Co is lower compared to Ni, which will influence the activation of hydrocarbons. Which one of these properties dominates with respect to the catalytic performance is difficult to determine.
Product selectivity
For the unmodified molybdenum trioxide catalyst (MoO3), the selectivity after 10 h TOS (Table 2) towards aromatic hydrocarbons is moderate with 27.7% (benzene), 6.1% (toluene) and 4.9% (xylenes). Phenolic intermediates were formed with selectivities of 18.9% (phenol) and 8.9% (cresol), which results in an S(arenes)/S(phenols) ratio (R(A/P)) of 1.36. The selectivity to methane of 3.4% is at a low level. Hydrogenation products such as cyclohexane or cyclohexenes were not detected. It is noted that product selectivities strongly depend on the exact reaction conditions such as temperature, hydrogen![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate:catalyst ratios and residence times. A systematic study of the impact of respective parameters goes beyond the scope of this study. The product selectivity profiles over time are given in the ESI† (SI-B).
:substrate:catalyst ratios and residence times. A systematic study of the impact of respective parameters goes beyond the scope of this study. The product selectivity profiles over time are given in the ESI† (SI-B).
The cobalt containing catalysts CoMoO4 and Co(5)MoO3 showed similar selectivities in comparison to MoO3, except the phenol and cresol selectivities which increased, resulting in a R(A/P) of ≈0.78 for both.
For Ni(5)MoO3 and Ni(3)MoO3, the content of aromatic products increased to ca. 32–33% (benzene), ca. 9–10% (toluene) and ca. 1–3% (xylene). However, as the selectivities to cresols increased, the R(A/P) for Ni(5)MoO3 remained at the same level compared to MoO3. Ni(3)MoO3 showed a slightly increased R(A/P) of 1.67. For Ni(1)MoO3, the selectivity towards aromatic compounds dropped slightly while the selectivity to phenolic products increased compared to Ni(3)MoO3 (R(A/P) = 1.38). The selectivity to methane for these three nickel containing catalysts was ca. 4%.
Utilization of nickel molybdate NiMoO4 leads to even higher selectivities towards benzene (41.8%) and toluene (10.1%) accompanied by lower values for the phenolic compounds. The low selectivity towards oxygenates leads to a high R(A/P) ratio of 8.85. This could be explained by the high activity of NiMoO4. Surprisingly, a very high value for the selectivity towards methane of 20.2% was obtained using this nickel molybdate catalyst.
Such a high methane selectivity for NiMoO4 cannot be explained solely by the HDO of anisole, as it would provide approximately 14.2% methane for the selective reaction towards benzene. In addition, transmethylated products, e.g. xylenes, were obtained, which are formed by the C-transfer from the methoxy group followed by oxygen removal. From this it can be concluded that the aromatic ring must be decomposed to a significant extent. It can be assumed that besides the MoOxCyHz phase, other effects have an influence on the selectivity.
To test whether nickel has a dominant impact on the product selectivities, a catalytic control experiment was conducted with 3 wt% Ni on α-Al2O3. This catalyst showed, after 10 h TOS, a conversion of 16.7% with a methane selectivity of 44.2% and benzene selectivity of 25.8% (SI-B, Table SI-8†). In comparison to the performance data obtained for Ni(3)MoO3 (Table 2), it is shown that the selectivity and activity is not dominated by Ni but by a molybdenum species. Further, for the Ni/α-Al2O3 catalyst a strong deactivation was observed starting with a conversion of 55.0% at 1 h TOS decreasing to 11.0% after 17 h TOS. Such a deactivation behaviour was not observed for the Ni(X)MoO3 catalysts.
Characterization of spent catalysts
The spent catalysts were analysed by XRD, TG-MS, SEM and XPS to gain a better understanding about the respective phase compositions. BET surface areas (SA) have been determined as well for selected spent catalyst materials and no significant increase was observed compared to the fresh ones (SI-C†). For MoO3 the SA increased from 3 m2 g−1 to 7 m2 g−1. For Ni(5)MoO3 the SA was increased during the reaction only from 15 m3 g−1 to 18 m2 g−1, while for NiMoO4 a decrease from 40 m2 g−1 to 32 m2 g−1 was found. This indicates that SA effects are perhaps not crucial for the altered catalytic performance between unpromoted and promoted catalysts.Nevertheless, for Ni(5)MoO3, scanning electron microscopy (SEM) gave evidence for the increased SA for nickel containing systems (SI-D, Fig. SI 8 and 9†). For the fresh sample, it was observed that a binary structure of small scale NiMoO4 combined with larger MoO3 crystallites is present. While the MoO3 crystallites form planar surfaces with sharp edges, the NiMoO4 phase is distinguished by a significantly rough surface. The analysis of spent Ni(5)MoO3 showed that after the reaction the microstructure was not changed and has a comparable structure as the fresh catalyst.
XRD measurements were conducted after the reaction only for the unpromoted and the nickel systems because it is hardly possible to distinguish between all possible phases for the cobalt systems on the basis of their complex diffractograms.41 The MoOxCyHz phase could be identified in each catalyst, except the NiMoO4 sample (Fig. 3).
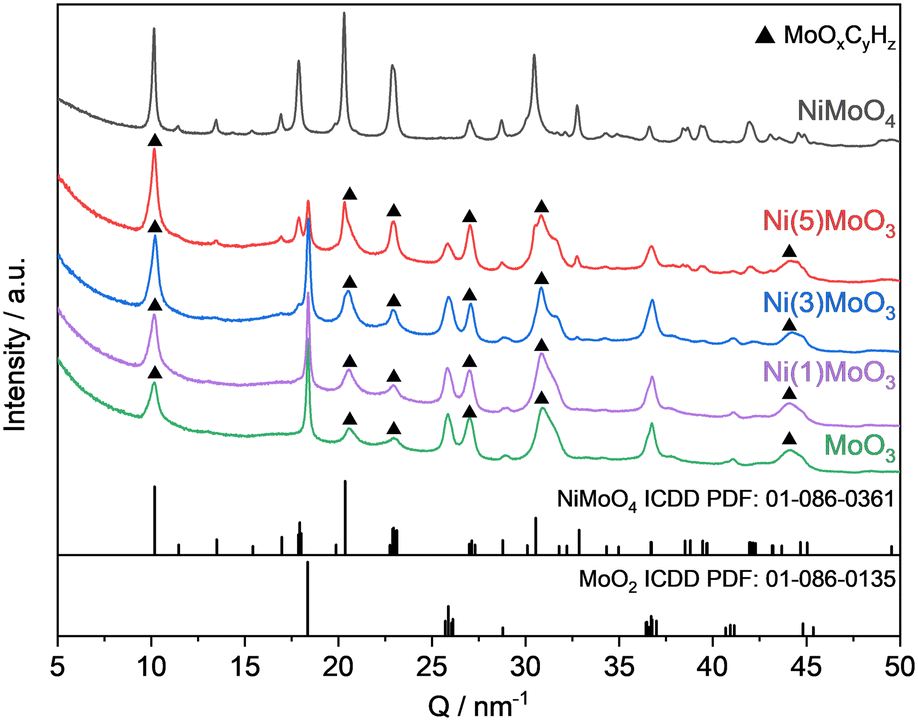 | ||
| Fig. 3 Normalized XRD patterns of the spent catalysts after the HDO reaction of anisole for 17 h at 325 °C. | ||
The respective reflections for this MoOxCyHz phase are clearly observable at 10.1 nm−1, 22.9 nm−1, 27.1 nm−1, 31.5 nm−1 and 44.2 nm−1.24,25,48,49 This was expected for the unmodified MoO3 catalyst, as such an oxycarbohydride phase was already identified for the HDO of oxygenates.14,22 The reflections assignable to MoO2 were identified as well. Interestingly, while no MoO3 was found in all spent catalysts, the NiMoO4 phase for the nickel containing systems is present. Also, for the pure nickel molybdate catalyst only the respective reflections for NiMoO4 were observed in the spent specimen. The reflections for NiMoO4 and MoOxCyHz phases tend to overlap and are not easily distinguishable. For NiMoO4, no indication for the presence of a MoOxCyHz phase has been found by TG-MS as shown later. This together confirms that the formation of a MoOxCyHz phase occurred only for Ni(5)MoO3, Ni(3)MoO3, Ni(1)MoO3 and MoO3 in measurable amounts.
There are several possible reasons for the detection of the NiMoO4 phase in the spent catalysts after 17 h TOS, although a considerable amount of reduced nickel species was expected. One option is the potential incomplete reduction of the bulk core. Another possibility is the lower reduction degree of the catalyst in the presence of anisole at 30 vol% hydrogen during the reaction compared to the pre-reduction step performed in pure hydrogen. Also, the cooling phase after the reaction with the remaining anisole inside the reactor could lead to reoxidation. The exposition of the catalyst to air after demounting the reactor at elevated temperatures probably has an impact due to the pyrophoric properties of nickel.
To support our assignments with respect to the oxycarbohydride phase, TG-MS measurements have been conducted to check for deposited coke and the decomposition of the MoOxCyHz phase. For the spent NiMoO4 catalyst the evolution of CO2 starts at 350 °C with a small peak and a second one starting at 400 °C with its maximum at ≈440 °C but at the same time no correlated water formation can be observed (Fig. 4). It can be concluded that only graphitic coke is decomposed during the calcination and no or marginal amounts of MoOxCyHz are present in the sample. In comparison to this, the decomposition of a MoOxCyHz phase starting with fast CO2 and water evolution at 380 °C and 400 °C is shown for MoO3 and Ni(5)MoO3, respectively. The decomposition of soft coke also takes place for Ni(1)MoO3 and Ni(3)MoO3 (SI-E, Fig. SI 10†). The amount of formed water should be correlated with the amount of the oxycarbohydride phase in the respective temperature regime. However, due to several experimental uncertainties regarding the possible lability when exposed to air, the quantification of the MoOxCyHz phase is hardly possible. Overall, these obtained results agree qualitatively with those obtained from XRD and indicate that Ni might have an influence on the formation and decomposition of the MoOxCyHz phase.
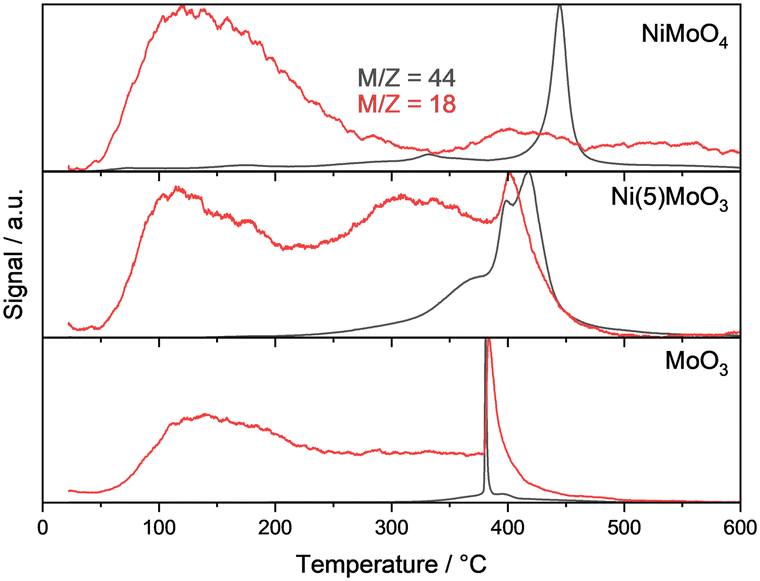 | ||
| Fig. 4 Normalized TG-MS profiles of the spent catalysts treated in syn. air, 10 °C min−1. | ||
XP spectra of selected catalysts (NiMoO4, Ni(5)MoO3 and MoO3) were recorded to elucidate surface properties and Mo oxidation states. The Mo 3d region was deconvoluted using three components Mo6+, Mo5+ and Mo4+ using a similar fitting model to Murugappan et al.15 For fresh calcined catalysts after preparation, almost exclusively, Mo6+ could be detected. This was concluded from the Mo 3d5/2 and Mo 3d1/2 doublet signals at E ≈ 232.6 and 235.9 eV, respectively (Fig. 5).42 Only for MoO3 a minor fraction of approximately 2% Mo5+ (E ≈ 231.3 and 234.4 eV) was found.
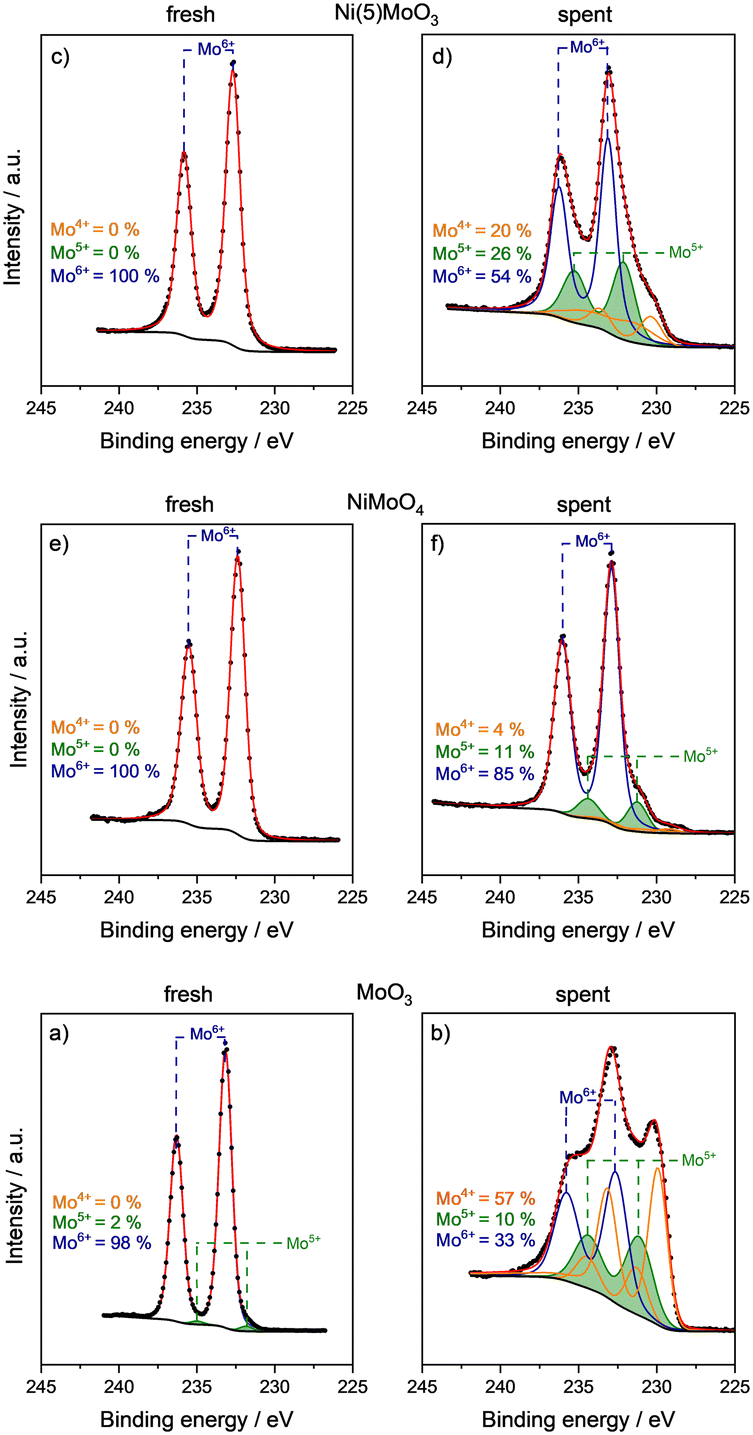 | ||
| Fig. 5 Mo 3d XP spectra of the fresh and used catalysts after TOS = 17 h. a) Fresh MoO3; b) spent MoO3; c) fresh Ni(5)MoO3; d) spent Ni(5)MoO3; e) fresh NiMoO4; f) spent NiMoO4. | ||
The spent catalysts showed significant differences with respect to their molybdenum oxidation states. The unmodified MoO3 catalyst surface contains ca. 33% Mo6+ after 17 h TOS, while Mo5+ and Mo4+ were present with 10% and 57%, respectively. The fraction of Mo5+ is of special relevance since it is ascribed to be involved in the substrate activation. As already discussed, the formation of an oxycarbohydride phase can lead to the stabilization of Mo5+ sites. Ni(5)MoO3 contained the largest fraction of Mo5+ (26%), and 54% Mo6+ and 20% Mo4+are also observed. NiMoO4 contained ca. 11% Mo5+, 85% Mo6+ and 4% Mo4+.
The increased amount of Mo5+ in Ni(5)MoO3 is consistent with the results from the literature, in which for Ni and Co modified molybdenum oxide an enhanced accumulation of Mo5+ was reported.28,29,32 It is surprising that the spent NiMoO4 catalyst is reduced only to a small degree and the majority of the used material consists of Mo6+, while it is expected that the reducibility is higher compared to pure MoO3.43 Nevertheless, the presented XPS data are in agreement with the XRD results, which showed the presence of NiMoO4 as the major phase.
To study the reduction behaviour of the fresh catalyst materials in the absence of the anisole substrate, pseudo in situ XPS experiments were performed. The samples were treated at 325 °C and 1 bar of hydrogen for 2 h. The reduction degree of Ni and Mo on the surface, of all three materials, was significantly higher compared with the spent catalysts in the expected order NiMoO4 > Ni(5)MoO3 > MoO3 (SI-G, Fig. SI 13–17†).
The observed behaviour based on the molybdenum XP spectra is also reflected in the Ni 2p spectra of the spent and in situ treated nickel containing catalysts (Ni(5)MoO3, NiMoO4). The spectroscopic data are given in the ESI† (SI-F and G). For the spent catalysts, Ni2+ was identified as the major nickel species for each catalyst. Only for NiMoO4 a small amount of Ni0 was detected. For both catalysts, during the pseudo in situ reduction the formation of mainly Ni0 was observed (SI-G, Fig. SI 16 and 17†). Additionally, H2-TPR-experiments were carried out, where the results agree with the observed reduction behaviour based on in situ XPS in the absence of oxygenates where the reduction degree increases with the nickel content. Further details are given in the ESI† (SI-H, Fig. SI 18).
The most plausible explanation of the discrepancy between the XPS results of the spent and reduced catalysts is the reoxidation process which might take place due to the presence of an oxygenate substrate under a lower partial pressure of hydrogen during the reaction and/or the exposition to air in the course of disassembling the reactor. Further, water as a coupling product can cause a reoxidation. This effect might be especially relevant for nickel, which is known for its pyrophoric properties if being present in its metallic state with small particle size.
Adsorption behaviour of anisole and benzene
The adsorption behaviour of anisole (substrate) and benzene (product) was investigated by DRIFTS experiments. After heating up in a He flow to the reaction temperature (325 °C), the catalyst sample was reduced for 2 h in 100% H2. Afterwards, the liquid component was injected into an evaporator (230 °C) using a syringe pump (2.5 μL min−1) and the vapor was then introduced via gas-phase transport with He (25 mL min−1) as a carrier into the DRIFTS cell. The flow was stopped after no intensity changes of the vibrational bands in the DRIFT spectra were observed. In the next step, the system was first flushed with He (10 min), and then the atmosphere was switched to H2. After 5 min TOS, no major differences regarding respective band positions between the different catalysts were observed (Fig. 6).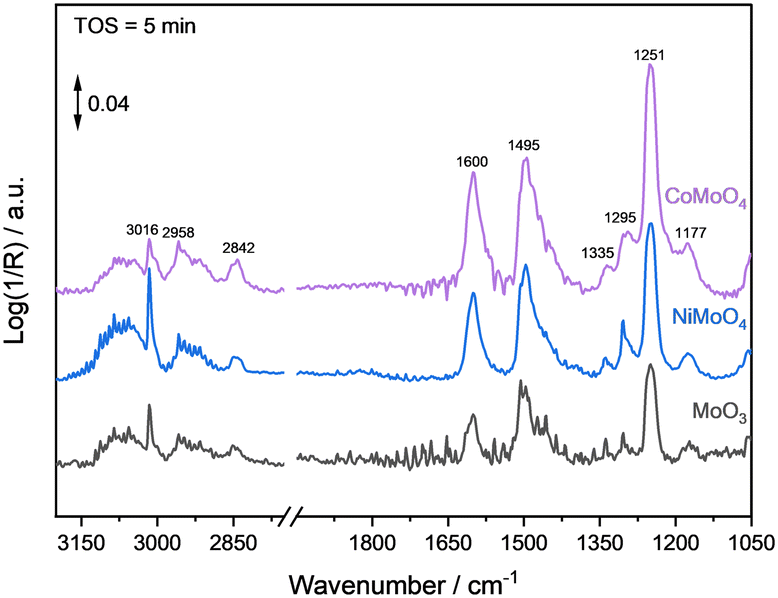 | ||
| Fig. 6 DRIFT spectra of the catalysts with adsorbed anisole at 325 °C and H2 (25 mL min−1). | ||
In each system, the strong ν(Ph–O–CH3) vibrational band at 1251 cm−1, the bands for ν(C=C) at 1495 and 1600 cm−1, and for ν(CH3) at 2842 cm−1 as well as bands for ν(C–H) at around 3000–2850 cm−1 are visible.44 In addition, each catalyst shows the formation of methane with a typical roto-vibrational band centered at 3016 cm−1. The formation of methane is expected, because from a thermodynamic point of view, the hydrogenation of the O–CH3 bond is favoured due to a lower binding energy compared to the Ph–OCH3 bond. The bond dissociation energy (BDE) of the O–CH3 bond is at about 424 kJ mol−1.12,45 However, the adsorption of anisole on each catalyst seems to be quite strong, as the decrease of the corresponding signals were slow (>40 min), see SI-I (Fig. SI 21†).
The relative integral values for the ν(Ph–O–CH3) vibrational band decreased to ca. 50% after ca. 20 min. Because the molybdenum oxide and metal molybdates form oxygen vacancies under reducing conditions, this strong adsorption behaviour can be expected for oxygen containing substrates (e.g. ethers, alcohols etc.).16,46
Besides the chemisorption of the educts, fast product desorption is crucial for active catalysts. Thus, the adsorption behaviour of benzene was tested following the same experimental procedure. Once the addition of benzene vapour was stopped after reaching stationarity in the DRIFT spectra, the respective vibrational bands of benzene started to immediately decrease for the MoO3 catalyst (Fig. 7a). After four minutes of flushing with He, the vibrational bands nearly completely vanished, thus the majority of benzene seems to be desorbed. After the gas was switched to H2, no changes in the DRIFT spectra could be observed (Fig. 7b).
 | ||
| Fig. 7 a) Adsorption experiment of benzene in He on MoO3 at 325 °C; b) switch to H2 after benzene adsorption on MoO3 at 325 °C; c) adsorption experiment of benzene in He on NiMoO4 at 325 °C; d) switch to H2 after benzene adsorption on NiMoO4 at 325 °C. | ||
For the NiMoO4 catalyst the comparatively fast desorption of benzene was also observed (Fig. 7c). After four minutes of purging with He only very weak intensity bands at 3083 cm−1 and 3057 cm−1 (ν(CH) vibrational bands) were visible. However, after switching to H2 the roto-vibrational band of CH4 centered at 3016 cm−1 was observed after 2 min (Fig. 7d). The band intensity increased further within an additional 2 min and after passing through a maximum it decreased slowly. A comparable behaviour was obtained for the CoMoO4 catalyst. However, the evolution of methane started a few minutes later and a lower intensity for the band of methane was observed compared to the NiMoO4 catalyst (SI-I, Fig. SI 19†).
Similar trends were also found in the online MS signals (m/z = 15) registered simultaneously during the DRIFTS measurements (Fig. 8). In the case of the NiMoO4 catalysts, the formation of methane started immediately after H2 was introduced into the system. The signal increased very fast and reached a short temporary plateau after about 3 min. The signal intensity increased further until a maximum appeared within 12 min. After going through the maximum, the methane evolution decreased over a longer period of more than 60 min. The CoMoO4 catalyst behaved similarly with some minor differences. The first plateau was reached also after 3 min, but the intensity of methane evolution of this plateau is significantly lower compared to the NiMoO4 catalyst. However, after ca. 20 min the maximum of methane evolution has reached a comparable intensity as for the NiMoO4 catalyst, but its decrease appeared to be slower. Contrary to the DRIFTS results, also for the MoO3 catalyst, methane formation could be observed, but to a significantly less extent compared to the metal molybdate systems. The maximum of the IC signal was reached after approximately 20 min and its intensity decreased slowly afterwards. Besides the molybdenum based catalysts, a control experiment with silica was performed to exclude other unknown methane sources. As expected, no methane formation was detected, and the signal remained very low.
 | ||
| Fig. 8 MS signal of methane evolution (m/z = 15) after benzene adsorption and switch to a H2 atmosphere at 325 °C. | ||
The formation of methane after the adsorption of benzene is quite interesting as it seems that benzene is not just simply adsorbed on the catalyst surface but underwent some chemical conversion. To further prove the decomposition of aromatic rings with NiMoO4 as a catalyst, a catalytic experiment in the fixed-bed reactor was performed with benzene as a substrate following the same protocol used for the HDO of anisole. The only products were methane and toluene and minor amounts of xylenes (SI-B, Table SI-9†). Note that the conversion is lower because of the increased WHSV compared to the experiments with anisole. However, the selectivity to methane of 21.0% after 10 h TOS is the same as found for the anisole HDO. With Ni(5)MoO3 as a catalyst methane with a selectivity of 2.8% is also formed in the reaction with benzene under the applied reaction conditions (SI-B, Table SI-10†). One hypothesis for this behaviour could be the formation of a molybdenum oxycarbohydride phase (MoOxCyHz) on a pre-reduced catalyst in the presence of benzene followed by the hydrogenolytic decomposition of this phase after the addition of hydrogen. As already mentioned, the appearance of a MoOxCyHz phase was described by Román-Leshkov et al. in the context of molybdenum oxide based HDO for the first time.14 However, its formation was already published before by Ledoux and co-workers for alkane isomerization using MoO3 as a catalyst.47–49 This phase is formed at temperatures between 300 and 380 °C in a H2/HC stream. To the best of our knowledge, there are no reports about such a phase in pure metal molybdates and only one for a cobalt promoted MoO3 system.32
Several aspects with respect to the observed methane formation need to be considered. It is known that during the reduction of NiMoO4 and CoMoO4 the formation of a mixed-phase material occurs.41,50,51 When NiMoO4 is used, one fraction of metallic Ni and a second fraction of MoO3/MoO2 might be formed during the pre-reduction step. The molybdenum oxide phase could be transformed into the oxycarbohydride phase by the benzene/hydrogen mixture. Subsequently, hydrogen could be activated at the Ni center, which then diffuses to MoOxCyHz by hydrogen spillover.25,29,52–54 This activated hydrogen could lead to the hydrogenation of carbon atoms and formation of methane (Scheme 3). However, the catalytic experiment under HDO conditions with Ni/α-Al2O3 as a catalyst and anisole as a substrate showed that nickel alone could decompose aromatic rings accompanied by the formation of methane. This was also proven by using benzene as a substrate in another catalytic control experiment (SI-B, Table SI-11†). It can be concluded that both discussed events can occur at the same time and contribute to the high CH4 selectivity with NiMoO4.
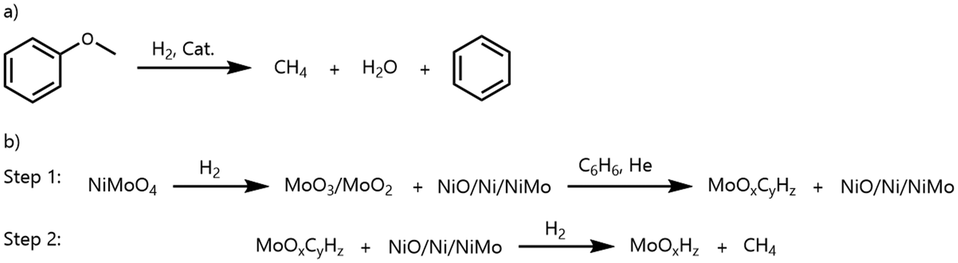 | ||
| Scheme 3 Simplified scheme for a possible pathway of the methane formation on a nickel molybdate catalyst. a) CH4 formation by the HDO reaction and b) CH4 formation by MoOxCyHz decomposition. | ||
Acidity properties and their relevance to the HDO reaction
The acidity properties of MoO3 and NiMoO4 were determined by NH3-adsorption FTIR measurements. The samples have been pre-reduced in H2 at 325 °C for 2 h to prevent differences between the analysis and HDO reaction. At room temperature, 5% NH3 in He was pulsed on the samples until saturation. Afterwards, they were heated up to 300 °C and with 50 °C steps a spectrum was recorded.Noticeable differences in the nature of the acidic sides between MoO3 and NiMoO4 have been observed (the spectroscopic data are presented in SI-J, Fig. SI 22–25†). MoO3 shows a strong vibrational band at 1425 cm−1 which is assigned to Brønsted-acid (BA) sites.55,56 Vibrational bands assignable to Lewis acid (LA) sites could not be detected.
For NiMoO4 the vibrational band at 1425 cm−1 associated with BA sites can be observed as well, however it is significantly less intense compared to MoO3. Additionally, vibrational bands related to LA sites at 3350 cm−1 and 3270 cm−1 can be observed up to 300 °C. These LA sites could play a role in the improved catalytic properties, as they can act as adsorption sites for the oxygen containing substrates. BA sites are involved in the hydrogenolysis of the substrate. NiMoO4 combines both properties, while MoO3 only has BA sites. In addition, the influence of LA sites on the formation of MoOxCyHz has been discussed in the literature before and could be beneficial for its formation.32
In situ XRD studies on the formation of MoOxCyHz phases
In situ XRD experiments were carried out to gain a deeper understanding of the MoOxCyHz phase formation and the effect of nickel on this formation.Influence of the feed composition on the MoOxCyHz phase formation starting from MoO3
To acquire profound knowledge on the reaction behaviour, the experiment series was started with the generation of MoOxCyHz in MoO3 with a H2/CH4 gas mixture. Methane was chosen because it represents a well-defined gaseous hydrocarbon, suitable for long-term in situ XRD experiments. The temperature for this experiment was set to 350 °C, because it was initially not clear, whereas the lower temperature used in the catalysis (325 °C) is sufficient for the formation of the MoOxCyHz phase from a less reactive substrate such as methane. However, it was expected that the generation of a molybdenum oxycarbohydride phase should be feasible, as it is reported in previous works that MoOxCyHz formation is possible with a variety of hydrocarbon substrates including n-alkanes and several oxygenates.14,24,25,49,57 To understand the importance of the pre-reduction step for this phase formation and the influence of the H2/CH4 ratio, the ratio was systematically varied in the following order H2/CH4 = 9:1, 1:1, 1:9.
The series was started with a ratio of H2:CH4 = 9:1 and after the introduction of the reactive gas, the transformation of MoO3 to MoO2 was initiated. With some time-shift the appearance of reflections which can be duly assigned to a MoOxCyHz phase could be clearly observed at Q ≈ 27 nm−1 and 31.5 nm−1 after ca. 200 min (Fig. 9). Those signals increased further over time within the entire investigated time span of 1200 min (SI-K, Fig. SI 26†).
 | ||
| Fig. 9 Normalized in situ XRD patterns for the MoO3 treatment at 350 °C in a H2/CH4 mixture (flow: 10 mL min−1) at TOS = 300 min for H2:CH4 = 9:1 and 1:9, TOS = 305 min for H2:CH4 = 1:1. Note that the broad reflection at Q ≈ 15 nm−1 belongs to the SiO2 capillary reactor. Ex situ diffractogram for MoOxHy treated in H2:CH4 = 9:1 at 325 °C for 1200 min forming a MoOxCyHz phase (▲) for comparison with the in situ data. | ||
To prove the assignment of the evolved new reflections to a MoOxCyHz phase, molybdenum blue was treated with H2:CH4 = 9:1 at 325 °C for 1200 min. Molybdenum blue is a pre-reduced MoO3 which might consist of several MoOxHy intermediate phases and tend to selectively form a MoOxCyHz phase when treated with hydrocarbons. The material was synthesized as described by Glemser et al.36 The obtained reflections assigned to the molybdenum oxycarbohydride phase for the in situ treated MoO3 material are in agreement those obtained for the treated molybdenum blue (Fig. 9).
When the feed ratio was altered to H2:CH4 = 1:1, the observed trends were the same and the MoOxCyHz phase was formed after approximately 200 min. However, for the experiment with a feed ratio of H2:CH4 = 1:9 a completely different behaviour was found. In this case, the starting material MoO3 was practically not reduced with no changes in the diffractograms between the starting and end point (SI-K, Fig. SI 28 and 29†). It can be concluded that a certain lower limiting amount of hydrogen in the feed is required for the reduction of Mo6+ to Mo5+ and the succeeding formation of the oxycarbohydride phase.
Formation of the MoOxCyHz phase with NiMoO4 and Ni(5)MoO3 as catalysts
In the next step, an in situ XRD experiment with NiMoO4 was performed. The temperature was slightly lowered to 325 °C because a higher activity with respect to catalyst reduction was expected. Considering the methane evolution, observed in the DRIFTS experiments, a question was raised, whether the MoOxCyHz phase can be populated, is directly decomposed, or reached equilibrium when the treatment is not carried out stepwise. At the beginning, a reduction step with pure hydrogen was performed to get more insight into the reduction behaviour of NiMoO4. For this sample after the reduction step for 120 min the dominant reflections are assigned to Ni and NiO (Fig. 10). The results from pseudo in situ XPS measurement of the reduction with hydrogen suggested that Ni0 is the dominant nickel species on the surface (SI-G, Fig. SI 16†). Reflections with a lower intensity of MoO2, which overlap in part with those of Ni, are also visible at Q ≈ 18.4 nm−1, 26.0 nm−1 and 36.7 nm−1 (Fig. 10).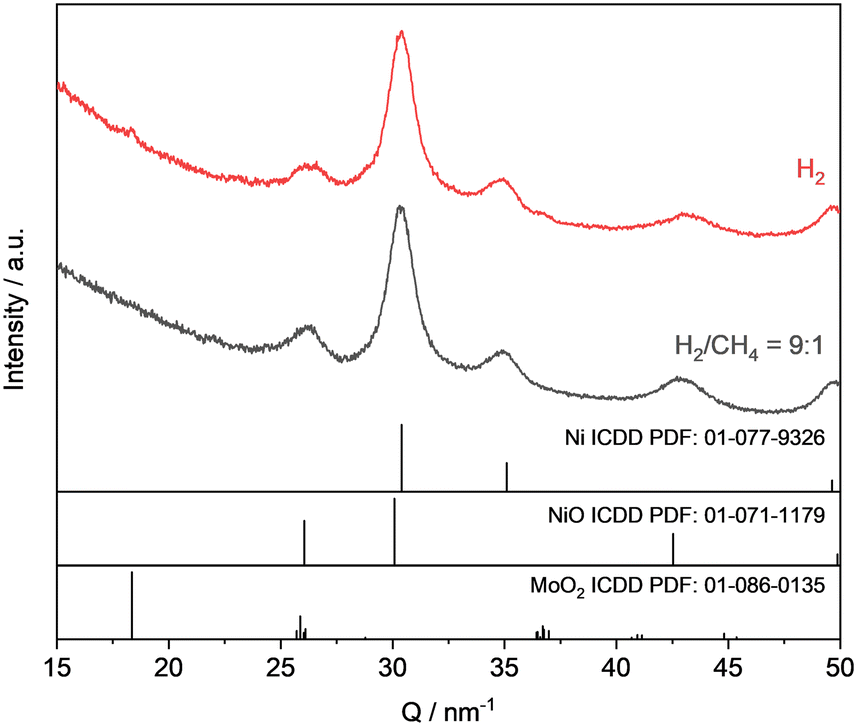 | ||
| Fig. 10 Normalized long scan time ex situ XRD patterns for NiMoO4 after in situ reduction in pure H2 for 120 min and treatment in H2/CH4 (1:1) at 325 °C for 1200 min after no further changes in the diffractograms over time were observed. | ||
When NiMoO4 was treated with the H2/CH4 (1:1) gas mixture, the result is nearly identical. The only difference is the absence of the reflections which belong to the MoO2 phase. Possible reasons for this absence could be a decrease in particle size and amorphousness compared to the nickel phases. It remains unclear whether a certain amount of a MoOxCyHz phase has been formed, for which also only weak and broad reflections (Fig. 9) are obtained.
In the next step, an in situ XRD measurement was carried out with the Ni(5)MoO3 catalyst. Due to the low Ni loading of 5 wt% nickel phases (Ni, NiO) should not dominate the diffractogram. This system is of specific interest, because the material was much more efficient with respect to the catalytic performance regarding the HDO of anisole compared to MoO3. The experiment was conducted in a modified stepwise manner. After a pre-reduction step for 2 h in hydrogen, the system was treated with methane for several hours. The reflections for the MoOxCyHz phase were visible after 150 min TOS directly after the switch to the CH4 atmosphere (SI-K, Fig. SI 30†). It is noted that the respective reflection signals registered during the in situ measurements in the capillary were quite low in intensity. Therefore, a long-time scan measurement was performed afterwards (Fig. 11). The reflections at Q ≈ 27 nm−1 and 31.5 nm−1 and 44.2 nm−1 also fully match with those acquired for the reference sample obtained from the molybdenum blue material (Fig. 9).
 | ||
| Fig. 11 Normalized XRD results from the stepwise treatment of Ni(5)MoO3 with H2 and CH4 at 325 °C compared with MoO3. | ||
Even in the case when reflections from nickel species contribute to the overall diffractogram and overlap with those of the MoOxCyHz phase, the major contribution of the three most intense reflections should stem from the MoOxCyHz phase, since the Ni content is only 5 wt%. Besides these reflections, those for MoO2 can also be observed at Q ≈ 26 nm−1 and 37 nm−1. Compared with the synthesis of the MoOxCyHz phase starting from MoO3, the Ni(5)MoO3 catalyst seems to form a significantly larger amount of the oxycarbohydride phase in agreement with an improved ratio between the integral values for MoOxCyHz/MoO2.
Effect of using more reactive propene instead of methane
The oxycarbohydride formation starting from MoO3 was also investigated with propene to elucidate the effect of using a more reactive hydrocarbon compound compared to methane. It was found that not only the formation of the MoOxCyHz phase starts earlier, also a bigger portion of the MoOxCyHz phase was built up, indicated by a larger ratio between integral values for MoOxCyHz/MoO2. For comparison, Fig. 12 presents the in situ collected diffraction data of both experiments where MoO3 is exposed to H2:C3H6 = 9:1 at 325 °C and H2:CH4 = 9:1 at 350 °C. Note the unequal time scales, as the important changes occurred after different time intervals and previous diffractograms are not shown for better clarity.
 | ||
| Fig. 12 Results from in situ XRD investigations on MoOxCyHz formation on MoO3 with a) H2:C3H6 = 9:1 at 325 °C and b) H2:CH4 = 9:1 at 350 °C. | ||
During the in situ XRD experiment, in which C3H6 was used at 325 °C, after 120 min the reflections at Q ≈ 31 nm−1 and Q ≈ 27 nm−1 which belong to the oxycarbohydride phase can be identified (Fig. 12a). After 150 min a steady state seems to have been reached and comparatively strong intensity reflections of the MoOxCyHz phase, besides the reflections for MoO2, were observable.
During the experiment with CH4 at 350 °C (Fig. 12b) the formation began noticeably later after 190 min. Quasi-stationarity was reached after 230 min. Apparently, significantly more MoO2 was formed compared to the experiment with propene.
For the interpretation of these results two aspects need to be considered. First, the slightly increased temperature during the experiment with methane could enhance the rate for the reduction of MoO3 to MoO2 to such an extent that the rate of carburisation is much lower in relation to this reduction step. Once the catalytically inactive MoO2 is formed, it cannot be transferred into MoOxCyHz.24 This is because MoO2 does not form molybdenum oxyhydride species (MoOxHy) which represents the precursor intermediate for the generation of MoOxCyHz. Additionally, the lower reactivity of methane could be one reason for the significant over-reduction to MoO2 because the carburisation of the partially reduced MoOxHy intermediate phase(s) might be slower with less reactive substrates. As already discussed for the nickel containing system Ni(5)MoO3, improved reduction properties could be responsible for the enhanced formation of the MoOxCyHz phase, which is also feasible when using propene.58 Thus, an activation treatment with a reactive hydrocarbon/hydrogen mixture could be promising to form significant amounts of such an oxycarbohydride phase to generate a catalyst with enhanced activity and stability.
Catalyst regeneration by oxidative treatment
Another important aspect is catalyst regeneration by in situ oxidative treatment. It is known from the literature that the unpromoted MoO3 catalyst possesses good regeneration properties when a calcination step is conducted.14,46,48 Thus, we were interested to study the reoxidation behaviour of a nickel molybdate modified catalyst. During the reduction, the formation of a mixed-phase composition of the NiMoO4 fraction occurs and the Ni0 phase forms particles at the surface, a reformation of the nickel molybdate phase is possible in the course of oxidative treatment according to some reports.39A sequential in situ XRD experiment was conducted with the Ni(1)MoO3 catalyst material in which an intermittent reoxidation step was included. The sample was treated with a H2/C3H6 = 9:1 mixture at 325 °C and after the formation of the MoOxCyHz phase, a gas mixture consisting of O2/He = 1:9 was added for 4 h and then switched back to the initial hydrogen/propene mixture. The diffractogram of the starting material Ni(1)MoO3 shows the reflections of MoO3 and low intensity signals, related to the NiMoO4 phase (Fig. 13). After the treatment with H2/C3H6 = 9:1, pronounced broad reflections for the MoOxCyHz phase can be observed besides low intensity reflections for MoO2. The reflection signals for NiMoO4 and MoO3 are not visible anymore.
 | ||
| Fig. 13 Normalized in situ XRD-results from the monitoring of a stepwise active phase formation from Ni(1)MoO3 with an intermittent reoxidation step at 325 °C for 4 h with O2/He (1:9) and an H2/C3H6 (9:1) mixture. | ||
After 4 h of oxidation, the reflections of MoO3 and NiMoO4 were detected again. However, tiny reflections of MoO2 are still observable. Presumably, the temperature of 325 °C is not high enough for complete oxidation. Also, the reflections are less intense and sharp, which could be caused by an alteration in the microstructure during the reduction and oxidation, respectively. Nonetheless, by the second activation step the MoOxCyHz phase with similar large reflection intensities was formed. This is a good indication that regeneration of these catalysts is possible at least for a second cycle. Further experiments are required to study the impact of the temperature on the number of reoxidation cycles and the possible accumulation of inactive MoO2. Also, the influence of the nickel content on catalyst recycling by reoxidation needs to be studied in more detail within the framework of succeeding research studies.49,58
Promotion of the MoOxCyHz phase formation by nickel
To acquire more insights about the beneficial role of lower nickel contents in the formation of the oxycarbohydride phase, a series of in situ XRD experiments were performed using Ni(5)MoO3 and MoO3 as reference materials in comparison to a physical mixture of NiO/MoO3 and layers of NiO and MoO3.To carry out this experimental approach, we would like to discuss the following points. The obtained results with Ni(5)MoO3, which showed the enhanced formation of the MoOxCyHz phase, could be related to the improved reducibility caused by nickel. It is important to mention that in the case of pure NiMoO4 the population of the oxycarbohydride phase declined, presumably because of the significantly higher nickel content which could lead to the fast hydrogenolysis of the MoOxCyHz phase. Thus, moderately enhanced reduction properties caused by a lower nickel content seem to be beneficial, as this improves the formation of a molybdenum bronze (MoOxHy) in which the carbon is intercalated from the feed gas. For the potential case, activated hydrogen provided by a spillover effect would be responsible for the improved MoOxCyHz phase formation; also a local separation of MoO3 from the H2 activator (nickel) could result in a better formation of the oxycarbohydride phase.24,52
To address this hypothesis, the first in situ XRD experiment was conducted with the following procedure: a two-layer catalyst bed consisting of NiO (first layer) and MoO3 (second layer) was prepared in the quartz capillary, in which the activated hydrogen created in the first layer could possibly lead to reduction and MoOxCyHz phase formation in the second layer by H2-spillover. As a second control experiment, a physical mixture (MoO3 + 5 wt% NiO) was also tested. Additionally, the reference catalysts Ni(5)MoO3 and MoO3 were investigated. The samples were treated for 5 h in a H2/C3H6 mixture (9:1) at 325 °C.
For the sake of better comparison, the ratios of integral values for reflections of the target MoOxCyHz phase and the MoO2 phase, which results from over-reduction, were calculated. In all samples, the formation of the oxycarbohydride phase began with the start of the reduction, resulting eventually in a mixture of MoOxCyHz with reflections at Q ≈ 26.7 nm−1 and 30.7 nm−1, and for MoO2 at Q ≈ 25.7 nm−1 and 36.5 nm−1, respectively (Fig. 14). As already discussed for the MoO3 system, when C3H6 is used instead of CH4, a significantly larger amount of the oxycarbohydride phase seems to be formed and the ratio of integral values for reflections at Q ≈ 26.7 nm−1 denoted as A(MoOxCyHz) and at Q ≈ 25.7 nm−1 denoted as A(MoO2), R = A(MoOxCyHz)/A(MoO2) increases to R = 1.5 (for CH4 this ratio was R = 0.1, see Table 3). When Ni(5)MoO3 was used, considerably more MoOxCyHz phase is formed and a ratio of R = 4.4 was determined (Fig. 14a). Also, with methane the formation of MoOxCyHz with Ni(5)MoO3 was promoted, resulting in a ratio of R = 2.8.
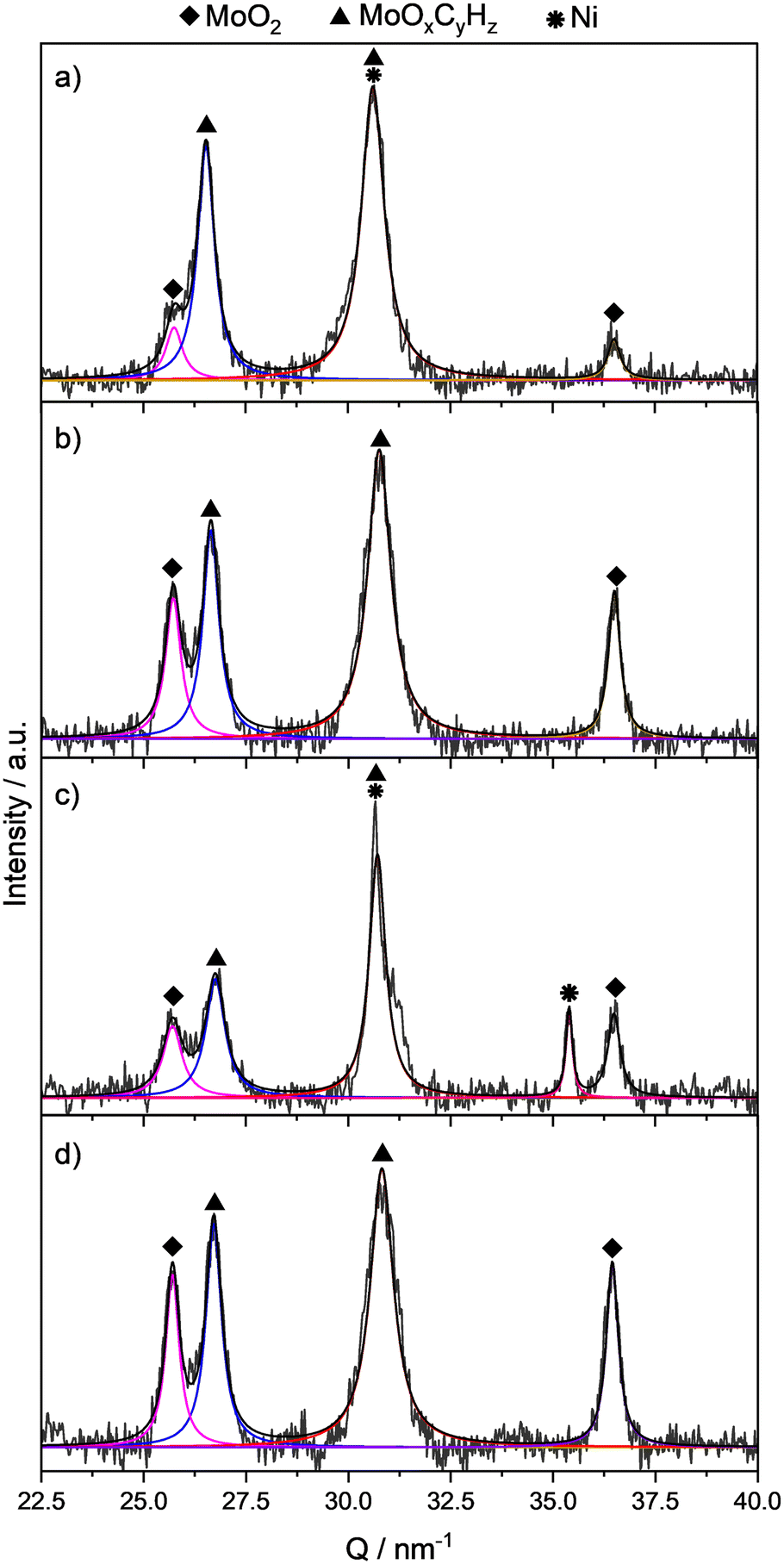 | ||
| Fig. 14
In situ XRD experiments of different samples at 325 °C in a gas mixture of H2/C3H6 = 9:1 at TOS = 310 min. a) Ni(5)MoO3; b) two layers of NiO/MoO3; c) phys. mix of NiO/MoO3; d) MoO3. | ||
| Catalyst | Gas mixture | Ratio |
|---|---|---|
| A(MoOxCyHz)/A(MoO2) | ||
| MoO3 | H2/CH4 (9:1) |
0.1 |
| MoO3 | H2/C3H6 (9:1) |
1.5 |
| MoO3 + NiO | H2/C3H6 (9:1) |
1.5 |
| MoO3 + NiO (phys. mix) | H2/C3H6 (9:1) |
1.7 |
| Ni(5)MoO3 | H2/C3H6 (9:1) |
4.4 |
| Ni(5)MoO3 | H2/CH4 (9:1) |
2.8 |
Interestingly, when the nickel oxide was added as a layer above the MoO3, the amount of the oxycarbohydride phase remained constant (R = 1.5), compared to the pure MoO3 in H2/C3H6 (Fig. 14bvs.14d). Because the H* migration could be hindered and not last until the end of the catalyst bed, the physical mixture of NiO and MoO3 was tested. For this purpose, MoO3 and 5 wt% of NiO have been mixed and pressed together to a particle size of 100–150 μm. From this experiment an integral ratio of R = 1.7 was obtained (Fig. 14c). In addition to the signals of MoOxCyHz and MoO2, a reflection of metallic Ni was observed at Q = 35.4 nm−1. It can be concluded that for both the layered system and the physical mixture, an improved oxycarbohydride phase formation could not be observed in these XRD experiments.
It is noteworthy that a reflection signal of metallic nickel was only observed for the physical mixture of NiO and MoO3. Interestingly, such a nickel reflection was absent in the Ni(5)MoO3 system, even though a reduction of the NiMoO4 phase might lead to the formation of metallic nickel.41 One reason for the absence of observable Ni reflections could be the formation of very small nickel particles which cannot be detected by XRD. A high dispersion of very small particles of Ni could be an explanation for the improved MoOxCyHz formation for nickel molybdate modified samples. One further effect could be the formation of an intermediate mesoporous defective MoO3−x structure after the reduction, which could induce the formation of mixed-phases, increasing the tendency for an improved MoOxCyHz formation.25,50
These results reveal that improved H2 activation by nickel is probably not the only reason for the enhancement of the oxycarbohydride phase formation. They rather indicate that the effects of different catalyst compositions (modification by NiO or NiMoO4) and corresponding structural properties together with substrate properties, even though being different in nature, are most likely cumulative.
Conclusion
In molybdenum oxide based catalysts loaded with nickel or cobalt the transition metals are present as a binary structure of NiMoO4 and CoMoO4 combined with MoO3, respectively. Catalysts with lower amounts of nickel up to a metal content of 5 wt% show higher activities towards the hydrodeoxygenation (HDO) of anisole with a slightly improved selectivity towards the aromatic products compared to pure phase MoO3. The pronounced induction period observed for the unpromoted MoO3 catalyst was increasingly shortened with increasing nickel content. Pure phase NiMoO4 is the most active catalyst system and showed a further improvement of the selectivity for aromatic products. However, for NiMoO4 a significantly high methane selectivity was also observed which indicates the decomposition of aromatic rings.In situ XRD experiments showed that the formation of a molybdenum oxycarbohydride phase (MoOxCyHz), which is found to stabilize the active Mo5+ sites preventing the over-reduction to inactive Mo4+, is improved for the Ni modified catalysts with lower nickel contents (up to 5 wt%). For the pure nickel molybdate, the population of a MoOxCyHz phase could not be observed for samples treated in the presence of a hydrocarbon/oxygenate substrate and molecular hydrogen. Stepwise in situ DRIFTS-MS adsorption/hydrogenation experiments on NiMoO4 indicated the decomposition of anisole and benzene over the pre-reduced nickel molybdate which could lead to a respective carbon containing phase such as MoOxCyHz, which is hydrogenated to methane when exposed to pure hydrogen.
Catalytic control experiments in a fixed-bed reactor with NiMoO4, Ni(5)MoO3 and Ni(3)/α-Al2O3 as catalysts and anisole and benzene as substrates revealed that the decomposition of arenes and the formation of methane could take place on a molybdenum and a nickel species simultaneously.
The formation of the molybdenum oxycarbohydride phase is effected by the substrate:hydrogen:molybdenum molar ratios as well as the reactivity of the substrate. The moderate reduction properties caused by a lower nickel content seem to balance the interplay between the enhanced reduction of molybdenum MoO3 to MoOxHy phases – from which the MoOxCyHz phase is readily generated and stabilizes the active Mo5+ sites – and hydrogenolysis activity without leading to excessive over-reduction.
In Scheme 4, an extended catalytic cycle of the HDO of anisole with MoO3 (a) and the formation steps of the oxycarbohydride phase MoOxCyHz starting from a nickel molybdate modified catalyst (b) are proposed. The established catalytic cycle can be complemented by the hydrogenation of incorporated carbon of MoOxCyHz in the formation of methane to MoOxHy which can be carburized again. In addition, the HDO via the reverse Mars–van Krevelen mechanism takes place at the Mo5+ sites which are distributed over MoOxHy/MoOxCyHz and are stabilized by the molybdenum oxycarbohydride phase.
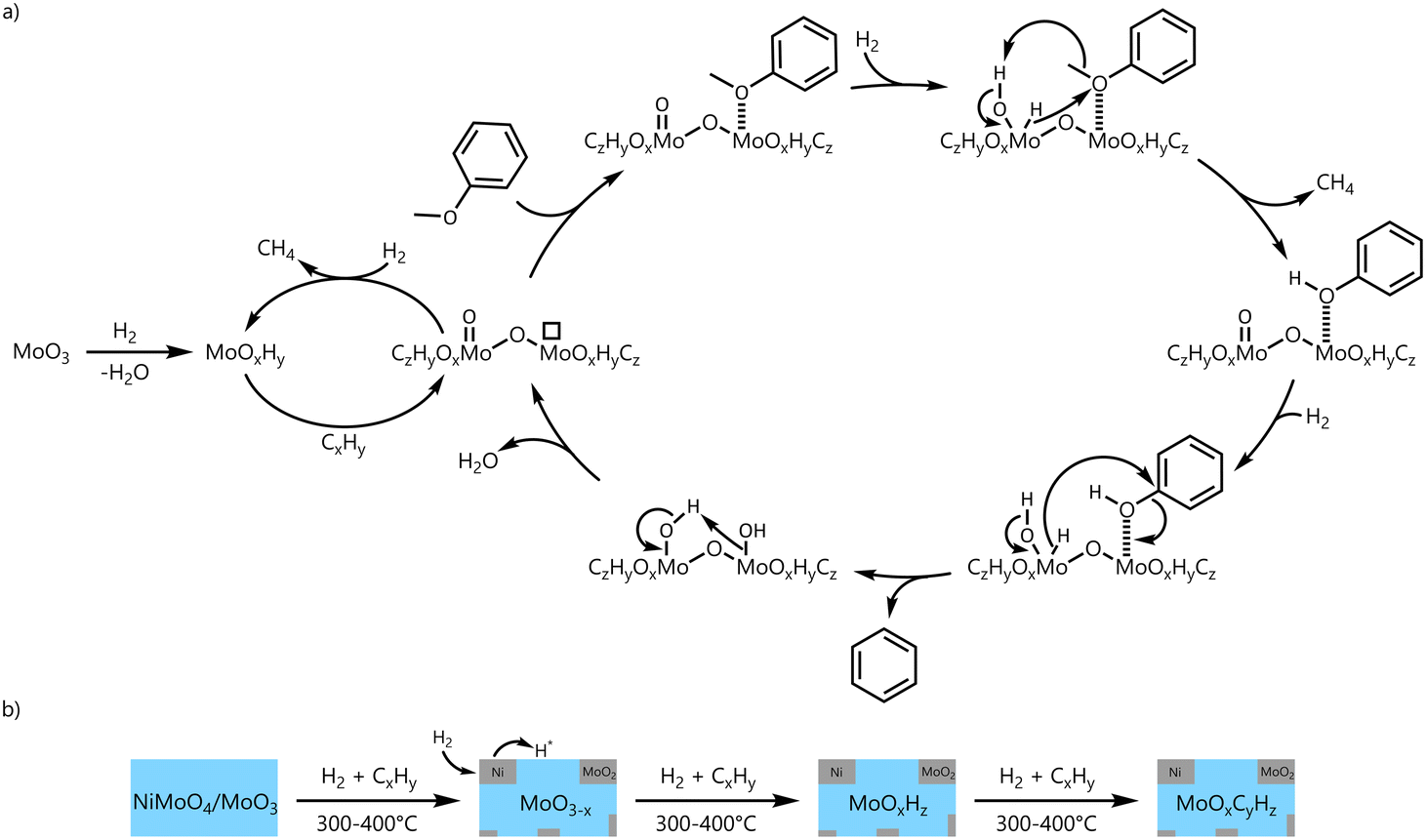 | ||
| Scheme 4 Conclusive overall scheme for the HDO of anisole with MoO3 (a) and a nickel molybdate modified catalyst (b). | ||
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research study was funded by the Deutsche Forschungsgemeinschaft (DFG, project number: 442613239). We thank Christine Rautenberg for the transmission-FTIR measurements, Anja Simula for the ICP measurements, Reinhard Eckelt for the BET measurements and Carl Julius Mussweiler for his support in the catalyst preparation and Raman measurements.References
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- E. A. Agustiany, M. Rasyidur Ridho, M. Rahmi D. N., E. W. Madyaratri, F. Falah, M. A. R. Lubis, N. N. Solihat, F. A. Syamani, P. Karungamye, A. Sohail, D. S. Nawawi, A. H. Prianto, A. H. Iswanto, M. Ghozali, W. K. Restu, I. Juliana, P. Antov, L. Kristak, W. Fatriasari and A. Fudholi, Polym. Compos., 2022, 43, 4848–4865 CrossRef CAS.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- R. G. Ramirez Brenes, E. M. Alhadeff, N. Bojorge, L. E. M. Trales and G. A. D. Pazos, Biofuels, Bioprod. Biorefin., 2023, 17, 664–681 CrossRef CAS.
- J. L. Brito, A. L. Barbosa, A. Albornoz, F. Severino and J. Laine, Catal. Lett., 1994, 26, 329–337 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- W. Jin, L. Pastor-Perez, D. K. Shen, A. Sepulveda-Escribano, S. Gu and T. R. Reina, ChemCatChem, 2019, 11, 924–960 CrossRef CAS.
- J. H. Zhang, J. M. Sun and Y. Wang, Green Chem., 2020, 22, 1072–1098 RSC.
- M. W. Nolte and B. H. Shanks, Energy Technol., 2017, 5, 7–18 CrossRef CAS.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Román-Leshkov, Energy Environ. Sci., 2014, 7, 2660–2669 RSC.
- K. Murugappan, E. M. Anderson, D. Teschner, T. E. Jones, K. Skorupska and Y. Román-Leshkov, Nat. Catal., 2018, 1, 960–967 CrossRef CAS.
- V. O. O. Gonçalves, C. Ciotonea, S. Arrii-Clacens, N. Guignard, C. Roudaut, J. Rousseau, J.-M. Clacens, S. Royer and F. Richard, Appl. Catal., B, 2017, 214, 57–66 CrossRef.
- D. R. Moberg, T. J. Thibodeau, F. G. Amar and B. G. Frederick, J. Phys. Chem. C, 2010, 114, 13782–13795 CrossRef CAS.
- M. Shetty, B. Buesser, Y. Román-Leshkov and W. H. Green, J. Phys. Chem. C, 2017, 121, 17848–17855 CrossRef CAS.
- M. Rellán-Piñeiro and N. López, ACS Sustainable Chem. Eng., 2018, 6, 16169–16178 CrossRef.
- D. Mei, A. M. Karim and Y. Wang, J. Phys. Chem. C, 2011, 115, 8155–8164 CrossRef CAS.
- M. Shetty, E. M. Anderson, W. H. Green and Y. Román-Leshkov, J. Catal., 2019, 376, 248–257 CrossRef CAS.
- G. B. Báfero, G. B. Strapasson, D. S. Leite and D. Zanchet, ChemCatChem, 2023, 15, e202300663 CrossRef.
- A. J. Kohler, C. H. Walter and B. H. Shanks, ACS Catal., 2023, 13, 14813–14827 CrossRef CAS.
- C. Bouchy, C. Pham-huu and M. J. Ledoux, J. Mol. Catal. A: Chem., 2000, 162, 317–334 CrossRef CAS.
- P. Delporte, F. d. r. Meunier, C. Pham-Huu, P. Vennegues, M. J. Ledoux and J. Guille, Catal. Today, 1995, 23, 251–267 CrossRef CAS.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- H. Metiu, S. Chrétien, Z. Hu, B. Li and X. Sun, J. Phys. Chem. C, 2012, 116, 10439–10450 CrossRef CAS.
- F. Yang, N. J. Libretto, M. R. Komarneni, W. Zhou, J. T. Miller, X. Zhu and D. E. Resasco, ACS Catal., 2019, 9, 7791–7800 CrossRef CAS.
- J. Zhang, C. Li, W. Guan, X. Chen, X. Chen, C.-W. Tsang and C. Liang, J. Catal., 2020, 383, 311–321 CrossRef CAS.
- A. A. Smirnov, S. A. Khromova, D. Y. Ermakov, O. A. Bulavchenko, A. A. Saraev, P. V. Aleksandrov, V. V. Kaichev and V. A. Yakovlev, Appl. Catal., A, 2016, 514, 224–234 CrossRef CAS.
- D. Raikwar, M. Munagala, S. Majumdar and D. Shee, Catal. Today, 2019, 325, 117–130 CrossRef CAS.
- J. Zhang, C. Li, W. Guan, X. Chen, X. Chen, C.-W. Tsang and C. Liang, Ind. Eng. Chem. Res., 2020, 59, 4313–4321 CrossRef CAS.
- V. Itthibenchapong, P. Chakthranont, C. Sattayanon, T. Butburee, K. Faungnawakij and S. Namuangruk, Appl. Surf. Sci., 2021, 547, 149170 CrossRef CAS.
- C. Ranga, V. I. Alexiadis, J. Lauwaert, R. Lødeng and J. W. Thybaut, Appl. Catal., A, 2019, 571, 61–70 CrossRef CAS.
- L. M. Madeira, M. F. Portela and C. Mazzocchia, Catal. Rev.: Sci. Eng., 2004, 46, 53–110 CrossRef.
- O. Glemser and G. Lutz, Z. Anorg. Allg. Chem., 1951, 253, 17–33 CrossRef.
- V. K. Pecharsky and P. Y. Zavalij, Fundamentals of Powder Diffraction and Structural Characterization of Materials, Springer Science & Business Media, New York, 2009 Search PubMed.
- J. T. Scanlon and D. E. Willis, J. Chromatogr. Sci., 1985, 23, 333–340 CAS.
- H. M. Abdel-Dayem, Ind. Eng. Chem. Res., 2007, 8(46), 2466–2472 CrossRef.
- J. E. Herrera and D. E. Resasco, J. Phys. Chem. B, 2003, 107(16), 3738–3746 CrossRef CAS.
- J. A. Rodriguez, J. Y. Kim, J. C. Hanson and J. L. Brito, Catal. Lett., 2002, 82, 103–109 CrossRef CAS.
- Z. Song, T. Cai, Z. Chang, G. Liu, J. A. Rodriguez and J. Hrbek, J. Am. Chem. Soc., 2003, 125, 8059–8066 CrossRef CAS PubMed.
- J. L. Brito, J. Laine and K. C. Pratt, J. Mater. Sci., 1989, 24, 425–431 CrossRef CAS.
- W. J. Balfour, Spectrochim. Acta, Part A, 1983, 39, 795–800 CrossRef.
- H. Taghvaei, M. Kheirollahivash, M. Ghasemi, P. Rostami and M. R. Rahimpour, Energy Fuels, 2014, 28, 2535–2543 CrossRef CAS.
- T. Prasomsri, T. Nimmanwudipong and Y. Román-Leshkov, Energy Environ. Sci., 2013, 6, 1732–1738 RSC.
- P. Del Gallo, F. Meunier, C. Pham-Huu, C. Crouzet and M. J. Ledoux, Ind. Eng. Chem. Res., 1997, 36, 4166–4175 CrossRef.
- C. Bouchy, C. Pham-Huu, B. Heinrich, C. Chaumont and M. J. Ledoux, J. Catal., 2000, 190, 92–103 CrossRef CAS.
- C. Bouchy, C. Pham-Huu, B. Heinrich, E. G. Derouane, S. B. Derouane-Abd Hamid and M. J. Ledoux, Appl. Catal., A, 2001, 215, 175–184 CrossRef CAS.
- R. B. Patil, S. D. House, A. Mantri, J. C. Yang and J. R. McKone, ACS Catal., 2020, 10, 10390–10398 CrossRef CAS.
- C. Wang, L. Guo, K. Wu, X. Li, Y. Huang, Z. Shen, H. Yang, Y. Yang, W. Wang and C. Li, J. Energy Chem., 2023, 84, 122–130 CrossRef CAS.
- F. Roessner and U. Roland, J. Mol. Catal. A: Chem., 1996, 112, 401–412 CrossRef CAS.
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed.
- R. Erre and J. J. Fripiat, in Studies in Surface Science and Catalysis, ed. G. M. Pajonk, S. J. Teichner and J. E. Germain, Elsevier, 1983, vol. 17, pp. 285–293 Search PubMed.
- G. Busca, Phys. Chem. Chem. Phys., 1999, 1, 723–736 RSC.
- D. Nicosia, I. Czekaj and O. Kröcher, Appl. Catal., B, 2008, 77, 228–236 CrossRef CAS.
- K. Cui, L. Yang, Z. Ma, F. Yan, K. Wu, Y. Sang, H. Chen and Y. Li, Appl. Catal., B, 2017, 219, 592–602 CrossRef CAS.
- T. Ressler, J. Wienold, R. E. Jentoft and T. Neisius, J. Catal., 2002, 210, 67–83 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01488f |
| This journal is © The Royal Society of Chemistry 2024 |