
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrodeoxygenation and hydrodenitrogenation of n-hexadecanamide over Pt catalysts: effect of the support†
Emma
Verkama
 *a,
Sylvia
Albersberger
b,
Aitor
Arandia‡
a,
Kristoffer
Meinander
c,
Marja
Tiitta§
b,
Reetta
Karinen
a and
Riikka L.
Puurunen
a
*a,
Sylvia
Albersberger
b,
Aitor
Arandia‡
a,
Kristoffer
Meinander
c,
Marja
Tiitta§
b,
Reetta
Karinen
a and
Riikka L.
Puurunen
a
aDepartment of Chemical and Metallurgical Engineering, School of Chemical Engineering, Aalto University, P.O. Box 16100, 00076 Aalto, Finland. E-mail: emma.verkama@aalto.fi
bNeste Corporation, P.O. Box 310, 06101 Porvoo, Finland
cDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, P.O. Box 16300, 00076 Aalto, Finland
First published on 15th December 2023
Abstract
Efficient catalysts for simultaneous hydrodeoxygenation (HDO) and hydrodenitrogenation (HDN) are needed for the production of renewable fuels. In this study, Pt catalysts supported on SiO2, γ-Al2O3, SiO2–Al2O3, ZrO2, CeO2–ZrO2, Nb2O5, and TiO2 were studied for the hydrotreatment of n-hexadecanamide (C16 amide) to n-paraffins at 300 °C and 80 bar H2. The catalysts favored HDO over HDN, and the initial differences in the nitrogen removal level were smaller than the differences in the oxygen removal level. The Lewis acid properties of the support influenced the initial C16 amide conversion route and HDO activity, which was reflected in the reaction network and condensation reaction selectivity of the catalysts. Pt/Nb2O5 and Pt/TiO2, with intermediate strength Lewis acid sites, initially favored the HDO of C16 amide to nitrogen-containing compounds. In contrast, the other catalysts converted C16 amide to oxygen- and nitrogen-containing compounds with similar selectivity. The HDO of the oxygen-containing compounds proceeded more efficiently on the Pt catalysts supported on oxides with weak Lewis acid sites (Pt/ZrO2, Pt/CeO2–ZrO2) than on the irreducible oxides with strong or no Lewis acid sites (Pt/γ-Al2O3, Pt/SiO2–Al2O3, Pt/SiO2). As the presence of oxygen-containing compounds suppressed HDN activity, the catalysts with the highest HDO activity eventually gave the highest paraffin yield, regardless of which oxygen removal pathway was favored.
1. Introduction
Renewable fuels with a high energy density are needed to mitigate the CO2 emissions from the heavy-duty transport sector and aviation industry. Major research efforts have been devoted to the hydrodeoxygenation (HDO) of renewable feedstocks to fuels due to their significant oxygen content.1–8 Hydrodenitrogenation (HDN) has received less attention, even though nitrogen-containing compounds are present in renewable feedstocks, such as animal fats and algal biocrudes.9,10 Since nitrogen-containing compounds can poison catalysts in downstream processing units and negatively impact fuel stability, the nitrogen content of feedstocks needs to be reduced along with the oxygen content.11A high HDO activity and the ability to catalyze C–N bond hydrogenolysis make supported noble metal catalysts an alternative to transition metal sulfide catalysts for the hydrotreatment of sulfur-free feedstocks.7,8,12–15 In our previous study on the co-hydrotreatment of fatty acids and alkyl amines on Pt/ZrO2, we found that HDN was inhibited by the HDO of oxygen-containing intermediates and the formation of secondary amines and amides through condensation reactions.16 An information gap remains for the application of noble metal catalysts in the simultaneous HDO and HDN of molecules that contain both oxygen and nitrogen and on the effect of the catalyst composition on the relative HDO and HDN activity. These topics are addressed in this study.
Primary amides are present in feedstocks that are used for the production of renewable fuels, which makes them relevant model compounds for studying simultaneous HDN and HDO.10,17 Nevertheless, the HDN and HDO of primary amides to different paraffins remain sparsely studied, whereas the hydrogenation of secondary and tertiary amides to amines and alcohols on supported noble metal catalysts has been reported previously.18–28 Liu et al.26 evaluated Co/SiO2, Co/γ-Al2O3 and Co/H-ZSM-22 in the co-hydrotreatment of palmitic acid and n-hexadecanamide. The reaction products comprised n-paraffins (C14–C16), iso-paraffins (C15–C16) and 1-hexadecanol, and the hydrotreatment of n-hexadecanamide was proposed to proceed through n-hexadecanal.26 The catalyst with the highest overall acidity, Co/H-ZSM-22, showed the highest activity and favored the formation of normal and branched C16 paraffins through C–O bond hydrogenolysis, while the less acidic Co/SiO2 and Co/γ-Al2O3 catalysts exhibited a lower activity level and favored decarbonylation and decarboxylation routes.26 Shimizu et al.18 likewise found that the support strongly influenced the activity of Pt-based catalysts for the selective HDO of n-acetyl piperidine to the corresponding amine.18 Pt/Nb2O5 emerged as a highly active catalyst for the reaction, which was attributed to Lewis acid–base interactions between the Lewis acid sites of the partially reduced Nb2O5 support and the carbonyl group of the amide.18
The acidity and reducibility of the catalyst support are known to affect the activity and selectivity of noble metal catalysts in the HDO of oxygen-containing compounds.14,29–31 Lewis acid sites bond to and activate carbonyl and hydroxyl groups, whereas Brønsted acid sites confer dehydration and isomerization activity.14,31–33 The enhancing effect of reducible supports on HDO activity has been attributed to the catalytic activity of oxophilic Lewis acid sites for materials such as ZrO2 and CeO2–ZrO2, and strong metal–support interactions for materials such as Nb2O5 and TiO2.29,32,34–37 Based on HDO studies on supported noble metal catalysts and the findings of Liu et al.26 and Shimizu et al.,18 the acidity and reducibility of the catalyst support can therefore be expected to influence the activity and selectivity of noble metal catalysts in the HDN and HDO of primary amides.
In this study, Pt catalysts supported on SiO2, γ-Al2O3, two SiO2–Al2O3 materials, ZrO2, CeO2–ZrO2, Nb2O5, and TiO2 were studied for the hydrotreatment of n-hexadecanamide (C16 amide, C16H33NO). C16 amide was chosen as a model compound due to the presence of primary amides in feedstock relevant to the production of renewable fuels, whereas the supports were chosen due to their diverse acid and redox properties.10,17 The purpose of this study was to describe the effect of the support on the catalytic activity and selectivity in the HDO and HDN of C16 amide and to discuss the active sites for the reactions based on the catalyst characterization and activity test data. To the best of our knowledge, the effect of the support on the activity and selectivity of Pt catalysts for the simultaneous HDO and HDN of molecules that contain oxygen and nitrogen has not been reported before.
2. Experimental
2.1 Materials
For catalyst preparation, platinum(IV) nitrate solution (Alfa Aesar, 15 wt% Pt) was used as the platinum precursor. The following materials were used as catalyst supports: TiO2 (44429, Alfa Aesar), ZrO2 (SZ 31164, Saint-Gobain Norpro), SiO2 (silica gel, Davisil Grade 646, Sigma-Aldrich), γ-Al2O3 (calcined from Pural NW Boehmite, Sasol), 5 wt% SiO2/Al2O3 (5SiO2–95Al2O3, Siralox5, Sasol), 30 wt% SiO2/Al2O3 (30SiO2–70Al2O3, Siralox30, Sasol), 25 wt% CeO2/ZrO2 (CeO2–ZrO2, XZO 1290, MEL Chemicals), and Nb2O5 (calcined from niobium oxide hydrate, Companhia Brasileira de Metalurgia e Mineração).The following chemicals were used without further purification for the reactor experiments and calibrations: n-hexadecanamide (>95%, Tokyo Chemical Industry), decalin (decahydronaphthalene, anhydrous, mixture of cis and trans, >99%, Sigma Aldrich), n-pentadecane (>99%, Aldrich), n-hexadecane (>99%, Sigma Aldrich), n-hexadecanal (>97%, Tokyo Chemical Industry), 1-hexadecanol (96%, Acros Organics), palmitic acid (>98%, Riedel de Haën), 1-hexadecylamine (>95%, Tokyo Chemical Industry), n-pentadecanonitrile (>95%, Tokyo Chemical Industry), n-heptadecanonitrile (>95%, Tokyo Chemical Industry), palmityl palmitate (>99%, Sigma Aldrich), n-dodecane (>99%, Merck), and 2-propanol (>99%, Riedel de Haën). The pyridine used for acid site characterization was obtained from Sigma Aldrich (anhydrous, 99.8%).
The gases used for the reactor experiments, catalyst characterization and product analysis (H2, He, N2 O2, Ar, and synthetic air) were all of 99.999% purity and were acquired from AGA and Woikoski. The helium and synthetic air used in the pyridine Fourier transform infrared spectroscopy (FTIR) measurements were obtained from Linde. The 5 vol% CO2/He (99.999%/99.999%) gas mixture was obtained from Woikoski, whereas the 2 vol% H2/Ar (99.999%/99.999%) gas mixture was purchased from AGA.
2.2 Catalyst preparation
The catalysts were prepared using a vacuum impregnation method with a small excess of liquid. The supports were crushed and sieved to a particle size of 0.25–0.42 mm and calcined in ambient air in a static muffle furnace for 10 h prior to impregnation (250 °C for 5SiO2–95Al2O3, 450 °C for TiO2 and CeO2–ZrO2, 500 °C for Nb2O5, and 600 °C for γ-Al2O3, SiO2, 30SiO2–70Al2O3, and ZrO2). The impregnation and calcination were carried out as described previously.162.3 Catalyst characterization
The Pt 4f spectra were fit using five doublets for metallic Pt(0), Pt(I), Pt(II), Pt(IV), and a mixed state located between Pt(I) and Pt(II). The binding energies for the Pt 4f7/2 of these components were located at approximately 71.0 eV, 72.2 eV, 73.5 eV, 74.5 eV, and 72.7 eV, respectively. For the Al2O3-containing catalysts, an additional peak at 74.9 eV was used for Al 2p due to the overlap of Pt 4f and Al 2p. The full width at half maximum (FWHM) for all Pt peaks was approximately 1.4 eV, with an energy separation between the doublets of 3.35 eV, while the FWHM for the Al component was 2.4 eV. For the Pt/SiO2, Pt/Al2O3, Pt/5SiO2–95Al2O3, and Pt/30SiO2–70Al2O3 catalysts, the O 1s spectra were deconvoluted using two Gaussian components of equal FWHM. One component was used for lattice oxygen and one for surface hydroxyls, at binding energies approximately 1.0–1.5 eV higher than the lattice oxygen. For Pt/ZrO2, Pt/CeO2–ZrO2, Pt/Nb2O5, and Pt/TiO2, an additional minor Gaussian component at approximately 533.3 eV, possibly related to oxygen bound to organic contaminants, was used. The C 1s spectra of all catalysts were fitted using four Gaussian components according to standard tabulated chemical shifts, with peak positions at 284.8 eV (C–C), 286.5 eV (C–O), 287.8 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and 288.9 eV (O–CO). The relative intensities for the carbon components corresponded to adventitious carbon.
O), and 288.9 eV (O–CO). The relative intensities for the carbon components corresponded to adventitious carbon.
The Si 2p spectra of the Pt/SiO2, Pt/5SiO2–95Al2O3, and Pt/30SiO2–70Al2O3 catalysts were fitted using a single Gaussian at approximately 103.0 eV, corresponding to silicon oxide. The Ti 2p spectra of Pt/TiO2 and the Nb 3d spectra of Pt/Nb2O5 were fitted using single component doublets, with the Ti 2p3/2 at an energy of 458.7 eV and the Nb 3d5/2 at an energy of 207.3 eV. The Zr 3d spectra of the Pt/ZrO2 and Pt/CeO2–ZrO2 catalysts were fitted using two doublets, with the 3d5/2 peaks located at 182.1 eV and 183.4 eV. For Pt/CeO2–ZrO2, a fitting scheme similar to that of Bêche et al.40 was adopted to differentiate between Ce(III) and Ce(IV) oxides, using a total of three doublets for the Ce(IV) oxide and two doublets for the Ce(III) oxide.
The catalyst samples (15–30 mg) were pressed into self-supported pellets, 1.1 cm in diameter, with a hydraulic press. The samples were heated in a vacuum to 90 °C with a 5 °C min−1 heating rate and maintained for 30 min. Next, the temperature was increased to 450 °C with a 20 °C min−1 heating rate and maintained for 60 min. The temperature was then lowered to 170 °C and held for 10 min, after which the spectra of the clean samples were recorded. Next, the samples were saturated with pyridine for 10 min using an atmospheric saturator, followed by evacuation and a 15 min hold. After that, the spectra used to quantify the acidity were recorded.
The Omnic 9.11 software was used to subtract the background and the spectra of the clean samples from the spectra of the pyridine-saturated samples to carry out a stepwise linear baseline correction. Peak integration and deconvolution were carried out using Omnic 9.11 and OriginPro, and the concentration of Lewis and Brønsted acid sites were estimated from the corresponding peak areas and sample weight using the relationships presented by Emeis.41
2.4 Catalytic activity tests
The catalytic activity tests were carried out in a 100 ml high-pressure Hastelloy batch reactor by Parr Instrument Company, which was equipped with a heated feed vessel. The catalyst (20 mg) was dried in situ at 180 °C under 10 bar of N2 for 60 min and then reduced at 350 °C under 20 bar of H2 for 60 min. The drying and reduction conditions were adopted from the work by Mäkelä and González Escobedo et al.,30 while the experimental conditions were chosen based on preliminary experiments.The feed mixture was prepared by dissolving 56.5 mg of n-hexadecanamide into 31 ml of decalin under heating (∼100 °C), targeting an initial nitrogen concentration of 100 ppm. A 1 ml zero-sample was then taken from the feed mixture, and the feed mixture was transferred to the feed vessel attached to the reactor.
The feed mixture was released from the feed vessel to the reactor, which was pre-heated to 300 °C. The reactor was pressurized to 80 bar H2, and stirring at 600 rpm was initiated, which marked the onset of the reaction time. Once the chosen reaction time of 15–300 min had elapsed, the heating and stirring were stopped, and the reactor was quenched with ice. A reaction time of 60 min was used as a reference for the activity and selectivity comparison of the catalysts and supports. The reactions were studied with respect to batch residence time τ (gcat h gamide−1), as defined in eqn (1), to take variations in the initial amounts of catalyst and reactant into account.
 | (1) |
The absence of external diffusion limitations was confirmed by conducting experiments while stirring at 200 and 1000 rpm. The repeatability of the experiments was evaluated using three 60 min repetition experiments with the Pt/ZrO2 catalyst, and a 60 min experiment was conducted with a repetition batch of the Pt/ZrO2 catalyst to ensure the repeatability of the catalyst preparation. The 180 min experiment with the Pt/ZrO2 catalyst was also repeated. Fig. S1 of the ESI† displays the product distribution of the control experiments.
2.5 Product analysis
The n-hexadecanamide model compound and some of the reaction products are sparsely soluble in decalin and other nonpolar hydrocarbon solvents at room temperature. In order to dissolve the model compound and reaction products, 10–20 vol% of a second solvent (2-propanol) was added to the samples prior to analysis. An internal standard (n-dodecane) was also introduced to the samples.The inlet of the GC was at 325 °C, as was the temperature of both the FID and the NPD. An injection volume of 2 μl and a split ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was used. The analysis program used for the HP5 column started with a 3 min hold at 40 °C, from which the temperature was increased to 100 °C at a rate of 20 °C min−1 and then held for 3 min. The temperature was increased to 150 °C at a rate of 5 °C min−1, and from there to 325 °C at a rate of 10 °C min−1, where a final 12 min hold took place. For the HP1 column, the analysis program started with a 3 min hold at 80 °C, from which the temperature was increased to 100 °C at a rate of 20 °C min−1 and then held for 3 min. The temperature was increased to 160 °C at a rate of 10 °C min−1 and from there to 325 °C at a rate of 20 °C min−1, where a final 20 min hold took place.
:1 was used. The analysis program used for the HP5 column started with a 3 min hold at 40 °C, from which the temperature was increased to 100 °C at a rate of 20 °C min−1 and then held for 3 min. The temperature was increased to 150 °C at a rate of 5 °C min−1, and from there to 325 °C at a rate of 10 °C min−1, where a final 12 min hold took place. For the HP1 column, the analysis program started with a 3 min hold at 80 °C, from which the temperature was increased to 100 °C at a rate of 20 °C min−1 and then held for 3 min. The temperature was increased to 160 °C at a rate of 10 °C min−1 and from there to 325 °C at a rate of 20 °C min−1, where a final 20 min hold took place.
Weight-based FID response factors, relative to the internal standard n-dodecane, were determined experimentally for n-pentadecane, n-hexadecane, 1-hexadecylamine, 1-hexadecanol, palmitic acid, n-hexadecanamide, n-pentadecanonitrile and n-heptadecanonitrile. The response factors for n-pentadecanonitrile and n-heptadecanonitrile were averaged to obtain an estimate for the response factor of n-hexadecanonitrile. The response factors for n-hexadecyl hexadecylamine, n-hexadecyl hexadecanamide, dipentadecyl ketone and palmityl palmitate were estimated based on their combustion enthalpy, using the procedure by de Saint Laumer et al.42
The reactant conversion XA (%) was calculated using eqn (2),
 | (2) |
The yield for each product YP (%) was calculated using eqn (3),
 | (3) |
The oxygen removal (O-removal, %) was estimated from the product distribution, using eqn (4),
 | (4) |
The molar carbon balance closure BC (%) was calculated using eqn (5),
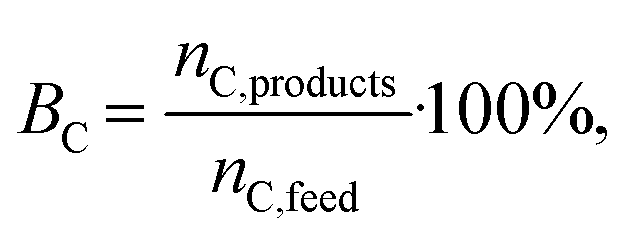 | (5) |
The molar carbon balance closure was generally above 90% for the catalytic experiments. The molar carbon balance closures for tests of the bare ZrO2, CeO2–ZrO2, and TiO2 supports were lower (∼85%), indicating that some products remained undetected or adsorbed on the surface of the supports.
 | (6) |
3. Results
3.1 Catalyst characterization
Table 1 displays the specific surface area, mean pore diameter and pore volume obtained from N2-physisorption measurements, as well as the mean Pt particle size and standard deviation of the mean Pt particle size derived from STEM images.| Catalyst | N2-physisorption | STEM | |||
|---|---|---|---|---|---|
| S BET (m2 g−1) | d pore,mean (nm) | V pore (cm3 g−1) | d Pt (nm) | Standard deviation, dPt (nm) | |
| Pt/SiO2 | 289 | 15 | 0.98 | 1.8 | 0.8 |
| Pt/γ-Al2O3 | 144 | 11 | 0.39 | 1.9 | 1.0 |
| Pt/5SiO2–95Al2O3 | 266 | 7 | 0.60 | 1.5 | 0.7 |
| Pt/30SiO2–70Al2O3 | 315 | 8 | 0.64 | 1.8 | 0.5 |
| Pt/ZrO2 | 42 | 19 | 0.21 | 2.1 | 0.7 |
| Pt/CeO2–ZrO2 | 70 | 14 | 0.24 | 1.6 | 0.5 |
| Pt/Nb2O5 | 73 | 6 | 0.13 | 2.0 | 0.5 |
| Pt/TiO2 | 100 | 13 | 0.25 | 1.7 | 0.6 |
There was considerable variability between the specific surface area, pore volume, and pore size distribution of the catalysts (Table 1). The specific surface area increased in the order of Pt/ZrO2 (42 m2 g−1) < Pt/CeO2–ZrO2 (70 m2 g−1), Pt/Nb2O5 (73 m2 g−1) < Pt/TiO2 (100 m2 g−1) < Pt/γ-Al2O3 (144 m2 g−1) ≪ Pt/5SiO2–95Al2O3 (266 m2 g−1), Pt/SiO2 (289 m2 g−1), Pt/30SiO2–70Al2O3 (315 m2 g−1). The pore volumes increased in a similar order, with the exception of Pt/Nb2O5, which had the lowest pore volume (0.13 cm3 g−1). The pore volumes of Pt/5SiO2–95Al2O3 (0.60 cm3 g−1) and Pt/30SiO2–70Al2O3 (0.64 cm3 g−1) were also lower compared with Pt/SiO2 (0.92 cm3 g−1), despite a similar surface area. Consequently, Pt/Nb2O5, Pt/5SiO2–95Al2O3 and Pt/30SiO2–70Al2O3 had the narrowest mean pore diameter (6–8 nm), while Pt/ZrO2 had the largest mean pore diameter (19 nm). Fig. S2 and S3 of the ESI† display the N2-physisorption isotherms and the BJH pore size distribution of the measured catalysts. The N2-physisorption isotherms of all catalysts corresponded to type IV(a) of the IUPAC classification.43
The semi-quantitative XRF measurements provided a Pt loading of 0.6 wt% for Pt/ZrO2, Pt/5SiO2–95Al2O3 and Pt/30SiO2–70Al2O3, while the Pt loading was 0.7 wt% for Pt/SiO2 and Pt/γ-Al2O3, and 0.8 wt% for Pt/CeO2–ZrO2, Pt/Nb2O5 and Pt/TiO2. These values are within the measurement accuracy of each other. The STEM images suggested that the Pt distribution was heterogeneous on the analyzed catalysts (Fig. S4 of the ESI†). The Pt particle size distribution derived from the STEM images is presented in Fig. S5 of the ESI.† The mean Pt particle size ranged between 1.5 and 2.1 nm for the catalysts, but the statistical significance of the differences between the mean Pt particle size of the catalysts was limited (Table 1). No reflections characteristic of Pt were identified in the X-ray diffractograms of the catalysts, which suggests that the Pt was X-ray amorphous and well dispersed, in agreement with the STEM images (Fig. S6, ESI†).44
XPS measurements were carried out to study the chemical composition of the surface of the supported Pt catalysts. The catalyst samples were reduced ex situ at 350 °C before the measurements and transferred to the equipment through air. Table 2 presents the surface concentration of Pt, the relative fraction of the different oxidation states of Pt, the binding energy of the Pt 4f7/2 component of Pt(0), and the binding energy of the lattice oxygen for all catalysts. Fig. 1 displays the Pt 4f/Al 2p region for all catalysts. The survey spectra of all catalysts are available in Fig. S7 of the ESI,† and the surface elemental composition of the catalysts is presented in Table S1 of the ESI.†
| Catalyst | Pt 4f/4da (at%) | Oxidation states of Pt | Binding energy | |||||
|---|---|---|---|---|---|---|---|---|
| Pt(0) (%) | Pt(I) (%) | Pt(mix) (%) | Pt(II) (%) | Pt(IV) (%) | Pt(0)b (eV) | Lattice Oc (eV) | ||
| a Pt 4d used for Pt/SiO2, Pt/γ-Al2O3, Pt/5SiO2–95Al2O3 and Pt/30SiO2–70Al2O3. b Pt 4f7/2. c O 1s. | ||||||||
| Pt/SiO2 | 3.4 | 75% | 12% | 3% | 6% | 4% | 71.0 | 533.4 |
| Pt/γ-Al2O3 | 5.9 | 69% | 4% | 15% | 6% | 6% | 70.8 | 531.5 |
| Pt/5SiO2–95Al2O3 | 6.2 | 71% | 15% | 4% | 5% | 5% | 71.0 | 531.5 |
| Pt/30SiO2–70Al2O3 | 3.6 | 75% | 16% | 1% | 4% | 5% | 70.9 | 531.7 |
| Pt/ZrO2 | 4.9 | 72% | 16% | 3% | 5% | 4% | 70.9 | 530.0 |
| Pt/CeO2–ZrO2 | 3.9 | 72% | 17% | 2% | 6% | 3% | 70.9 | 529.6 |
| Pt/Nb2O5 | 4.5 | 77% | 12% | 2% | 6% | 3% | 71.2 | 530.3 |
| Pt/TiO2 | 3.2 | 82% | 11% | 1% | 3% | 3% | 70.7 | 530.0 |
 | ||
| Fig. 1 High-resolution X-ray photoelectron spectra of the Pt 4f/Al 2p region of the supported Pt catalysts. The catalysts were reduced ex situ at 350 °C before the measurements. | ||
As seen from Table 2 and Fig. 1, shifts up to 0.5 eV were identified between the Pt(0) binding energies of the catalysts, potentially indicative of differences in electron transfer between Pt and the supports. Pt/TiO2 displayed the lowest Pt 4f binding energy, i.e., the highest electron density for Pt, whereas Pt/Nb2O5 had the highest Pt 4f binding energy. The oxidation state of Pt was similar for all catalysts, and most of the platinum, 69–82%, was in the form of metallic Pt(0). Pt(I) was the second most prevalent oxidation state, 11–17%, for all catalysts except for Pt/γ-Al2O3, followed by less than 6% in each of the higher oxidation states. Out of the measured catalysts, Pt/γ-Al2O3 displayed the highest amount of Pt at higher oxidation states and had the highest concentration of the mixed Pt state, 15%. The surface concentration of Pt varied between 3.2 and 6.2 at%.
As expected, the O 1s lattice oxygen binding energies differed significantly between the catalysts (Table 2). Pt/TiO2, Pt/ZrO2 and Pt/CeO2–ZrO2 exhibited the lowest lattice oxygen binding energies (529.6–530.0 eV), followed by Pt/Nb2O5 (530.3 eV), and Pt/γ-Al2O3, Pt/5SiO2–95Al2O3 and Pt/30SiO2–70Al2O3 (531.5–531.7 eV). Pt/SiO2 had the highest lattice oxygen binding energy, 533.4 eV.
The characteristics of the Al 2p and Al 2s peaks of Pt/γ-Al2O3, Pt/5SiO2–95Al2O3 and Pt/30SiO2–70Al2O3 were similar, with energies corresponding to Al2O3. The binding energy of the Si 2p spectra of Pt/SiO2, Pt/5SiO2–95Al2O3 and Pt/30SiO2–70Al2O3 (103.0 eV) suggests a sub-oxide state for the silica, and a lower O/(1.5Al + 2Si) ratio was observed with increasing amounts of Si in the catalysts (Table S2, ESI†). The sub-oxide state may be related to electronic interactions between Pt and SiO2.45 Sodium (0.4 at%) was additionally detected on the surface of Pt/SiO2.
Two components were identified from the Zr 3d spectra of the Pt/ZrO2 and Pt/CeO2–ZrO2 catalysts, with 3d5/2 peaks located at 182.1 and 183.4 eV. The lower binding energy component corresponds to ZrO2, while the higher binding energy component may be related to surface defects or Zr bound to hydroxyl species. Of the Zr atoms on the catalyst surface, 16.3% and 10.8% were found in the higher binding energy state for Pt/ZrO2 and Pt/CeO2–ZrO2, respectively. Of the Ce atoms on the surface of Pt/CeO2–ZrO2, 38.5% were found as Ce(III) and 61.5% as Ce(IV), implying that reduction of the support readily occurred, and that Ce was present in both oxidation states under the reaction conditions.
The reducibility and reducible species of the bare supports and the catalysts was qualitatively studied via H2-TPR. The H2-TPR profiles are presented in Fig. 2 for the bare supports (a) and the calcined supported Pt catalysts (b).
 | ||
| Fig. 2 H2-TPR profiles of (a) the bare supports and (b) the calcined supported Pt catalysts. | ||
No reducible species could be identified from the bare SiO2, γ-Al2O3, 5SiO2–95Al2O3, 30SiO2–70Al2O3, ZrO2 and Nb2O5 supports (Fig. 2a). The H2-TPR profile of the bare CeO2–ZrO2 support had a major reduction peak with a maximum intensity at 560 °C, which based on the XPS measurements, was related to the reduction of Ce(IV) to Ce(III). The H2 consumption was elevated in the H2-TPR profile of the bare TiO2 support between 350 °C and 550 °C, plateauing at 450 °C. This likely corresponded to the partial reduction of TiO2.46,47
The H2 consumption was slightly elevated until approximately 100 °C in the H2-TPR profiles of the Pt/SiO2, Pt/γ-Al2O3, Pt/5SiO2–95Al2O3, Pt/30SiO2–70Al2O3, Pt/Nb2O5 and Pt/TiO2 catalysts. This can likely be attributed to the reduction of bulk PtO2 species with weak interactions with the support or potentially to the chemisorption of H2 (Fig. 2b).48–50 Stabilization of the gas flow may also have been partially responsible for the elevated signal at the start of the temperature ramp. The H2-TPR profiles of Pt/SiO2, Pt/γ-Al2O3, Pt/5SiO2–95Al2O3, Pt/30SiO2–70Al2O3 and Pt/Nb2O5 contained no further peaks, suggesting that Pt was in its reduced form before the H2-TPR measurement, or that the reduction occurred before 100 °C. No metal-assisted reduction of the support appeared to occur for these catalysts in the studied temperature range. For the irreducible oxides, i.e., SiO2, γ-Al2O3, and both SiO2–Al2O3 supports, the results align with the literature, whereas the lack of peaks related to the reduction of the support was somewhat more surprising for Pt/Nb2O5.48,51–53
The H2-TPR profile of Pt/ZrO2 contained a broad reduction peak with a maximum intensity at 140 °C, likely corresponding to the reduction of Pt (Fig. 2b).30,54 The H2-TPR profile of Pt/CeO2–ZrO2 exhibited a major reduction peak between the start of the temperature ramp and 170 °C, with a maximum intensity at 83 °C and a shoulder at approximately 120 °C (Fig. 2b). A minor reduction peak was additionally observed at 335 °C. The higher reduction temperature of Pt on Pt/ZrO2 and Pt/CeO2–ZrO2 may indicate stronger electronic interactions between Pt and the support compared with the other catalysts.51,55–57 The reduction peak observed at 560 °C on the bare CeO2–ZrO2 support (Fig. 2a) was completely absent from the H2-TPR profile of the Pt/CeO2–ZrO2 catalyst. This suggests that the broad reduction peak on Pt/CeO2–ZrO2 with a maximum intensity at 83 °C involved both reduction of Pt and the support. In the literature, this has been explained by dissociative adsorption of H2 on the reduced Pt particles followed by spillover to the support, which facilitates the reduction of the support.58–62
The H2-TPR profile of the Pt/TiO2 catalyst contained a major reduction peak with a maximum intensity at 290 °C, which is 160 °C lower than the maximum intensity of the reduction peak of the bare TiO2 support (Fig. 2). The presence of Pt, therefore, lowered the reduction temperature of TiO2 on the Pt/TiO2 catalyst, similar to Pt/CeO2–ZrO2.30,55,63
The desorbed amounts of CO2 derived from the CO2-TPD measurements are presented in Table 3, whereas the CO2 desorption profiles are displayed in Fig. S8 of the ESI.† The CO2-TPD results are indicative of the basicity of the catalysts, with the amount of adsorbed CO2 describing the number of basic sites and the desorption temperature correlating with the strength of the basic sites.64
| Catalyst | Lewis aciditya (μmol g−1) | Brønsted aciditya (μmol g−1) | Total aciditya (μmol g−1) | Total basicityb (μmol g−1) | Lewis aciditya (μmol m−2) | Brønsted aciditya (μmol m−2) | Total aciditya (μmol m−2) | Total basicityb (μmol m−2) |
|---|---|---|---|---|---|---|---|---|
| a From FTIR spectroscopy using pyridine as a probe molecule. b From CO2-TPD measurements. | ||||||||
| Pt/SiO2 | ∼0 | ∼0 | ∼0 | 2 | ∼0 | ∼0 | ∼0 | <0.1 |
| Pt/γ-Al2O3 | 210 | ∼0 | 210 | 26 | 1.4 | ∼0 | 1.8 | 0.2 |
| Pt/5SiO2–95Al2O3 | 150 | ∼0 | 150 | 22 | 0.6 | ∼0 | 0.6 | 0.1 |
| Pt/30SiO2–70Al2O3 | 70 | 30 | 100 | 3 | 0.2 | 0.1 | 0.3 | <0.1 |
| Pt/ZrO2 | 30 | ∼0 | 30 | 42 | 0.7 | ∼0 | 0.7 | 0.8 |
| Pt/CeO2–ZrO2 | 240 | ∼0 | 240 | 210 | 3.4 | ∼0 | 3.4 | 3.0 |
| Pt/Nb2O5 | 210 | 90 | 300 | 2 | 2.8 | 1.3 | 4.1 | <0.1 |
| Pt/TiO2 | 140 | <10 | 140 | 15 | 1.4 | <0.1 | 1.4 | 0.2 |
Overall, the catalysts displayed relatively weak basicity. The CO2 desorption peak reached its maximum intensity at 105–110 °C for all catalysts (Fig. S8, ESI†). The CO2 adsorption capacity of Pt/γ-Al2O3 and Pt/5SiO2–95Al2O3 were similar at 26 and 22 μmol gcat−1, respectively (Table 3). The basic site concentration of SiO2–Al2O3 materials is known to decrease markedly as the SiO2 content increases, explaining the insignificant basicity of Pt/30SiO2–70Al2O3.65 SiO2 and Nb2O5 likewise had a negligible CO2 adsorption capacity. Slightly less CO2 (15 μmol gcat−1) desorbed from Pt/TiO2 than from Pt/γ-Al2O3 and Pt/5SiO2–95Al2O3. The CO2 adsorption capacity of Pt/ZrO2 and Pt/CeO2–ZrO2 was the highest out of the catalysts at 42 and 210 μmol gcat−1, respectively.
Acid site characterization was performed using FTIR spectroscopy, with pyridine as the probe molecule. Fig. 3 displays the FTIR spectra of the pyridine-saturated catalyst samples, whereas the Table 3 presents the adsorbed amounts of pyridine on Lewis acid sites and Brønsted acid sites.
 | ||
| Fig. 3 Transmission FTIR spectra for the pyridine-saturated supported Pt catalysts, with the background and spectra of the clean samples subtracted. The spectra were collected at 170 °C. The vibration bands characteristic for pyridine adsorbed on Brønsted acid sites (1540 and 1546 cm−1) and Lewis acid sites (1442–1453 cm−1) have been indicated with vertical lines. | ||
The catalysts displayed major differences in the acid site concentration and exhibited varying acid site strength, as observed from shifts in the wavenumber of the absorption bands (Table 3, Fig. 3).66–68 The catalysts were predominantly Lewis acidic (1442–1453 cm−1), and Brønsted acid sites were identified only on Pt/30SiO2–70Al2O3 (1546 cm−1) and Pt/Nb2O5 (1540 cm−1). Overall, the obtained FTIR spectra and adsorbed amounts of pyridine align with those reported for similar catalysts and supports in the literature.66–68
The catalysts were divided into four groups based on the strength of their Lewis acid sites. The first group comprises the catalysts with the strongest Lewis acid sites, Pt/30SiO2–70Al2O3, Pt/5SiO2–95Al2O3 and Pt/γ-Al2O3. The vibration bands characteristic of pyridine adsorbed on Lewis acid sites were located at 1453 cm−1 for Pt/30SiO2–70Al2O3 and at 1451 cm−1 for Pt/5SiO2–95Al2O3 and Pt/γ-Al2O3 (Fig. 3).66–68 In the following, this group is referred to as Si–Al. In the case of Pt/Nb2O5 and Pt/TiO2, the vibration band of pyridine adsorbed on Lewis acid sites was shifted toward a lower wavenumber (1446 cm−1) compared with the catalysts of the Si–Al group. This indicates the presence of Lewis acid sites of intermediate strength.66–68 Pt/Nb2O5 and Pt/TiO2, therefore, formed their own group, referred to as Ti–Nb. Pt/ZrO2 and Pt/CeO2–ZrO2, denoted Ce–Zr, contained the weakest Lewis acid sites, with the main vibration band of pyridine adsorbed on Lewis acid sites located at 1442 cm−1 for both catalysts.66–68 The FTIR spectra of Pt/CeO2–ZrO2 showed another vibration band at 1420 cm−1, possibly attributed to hydrogen-bonded pyridine or pyridine adsorbed on weaker Lewis acid sites, e.g., Ce3+ cations.68,69 Pt/SiO2 was the only catalyst where no pyridine adsorption could be detected and hence formed its own group (Si).
The Lewis acid site concentration of the catalysts of the Si–Al group decreased in the order Pt/γ-Al2O3 (210 μmol g−1, Table 3), Pt/5SiO2–95Al2O3 (150 μmol g−1), and Pt/30SiO2–70Al2O3 (70 μmol g−1). Pt/30SiO2–70Al2O3 additionally adsorbed 30 μmol g−1 pyridine on Brønsted acid sites, but its total acidity remained lower compared with the other catalysts in the Si–Al group. For SiO2–Al2O3 materials, the Lewis acidity is known to decrease and the Brønsted acidity to increase as the SiO2 content increases.65 The results are, therefore, in line with the literature.
Within the Ti–Nb group, the Lewis acid site concentration of Pt/Nb2O5 (210 μmol g−1, Table 3) exceeded the Lewis acid site concentration of Pt/TiO2 (140 μmol g−1). Pt/Nb2O5 additionally adsorbed 90 μmol g−1 pyridine on weak Brønsted acid sites and therefore had the highest total acid site concentration out of the studied catalysts.66,70 Pt/CeO2–ZrO2 had the highest Lewis acid site concentration (240 μmol g−1) out of all tested catalysts, whereas Pt/ZrO2 had the second lowest acid site concentration of the catalysts (30 μmol g−1), after the non-acidic Pt/SiO2.
3.2 Catalytic hydrotreatment of n-hexadecanamide
Hydrotreating experiments using n-hexadecanamide (C16 amide, 100 ppm N) as the model compound were carried out for the bare supports and the supported Pt catalysts at 300 °C and 80 bar H2 for 60 min. The product samples contained n-pentadecane (C15 paraffin), n-hexadecane (C16 paraffin), n-hexadecanal (C16 aldehyde), 1-hexadecanol (C16 alcohol), palmitic acid (C16 acid), n-hexadecanonitrile (C16 nitrile), 1-hexadecylamine (C16 amine), dipentadecyl ketone (C31 ketone), n-hexadecyl hexadecylamine (C32 amine), palmityl palmitate (C32 ester), and n-hexadecyl hexadecanamide (C32 amide). Fig. 4 presents the product distribution, conversion and nitrogen removal from the C16 amide hydrotreating experiments on the bare supports (a) and supported Pt catalysts (b), grouped according to their Lewis acid site strength. The oxygen removal, estimated from the product distribution, has also been indicated for the supported Pt catalysts. | ||
| Fig. 4 Product distribution, conversion and nitrogen removal of the C16 amide hydrotreating experiments on (a) the bare supports and (b) the supported Pt catalysts. The oxygen removal of the supported Pt catalysts was derived from the product distribution, while the nitrogen removal was obtained from the total nitrogen content analysis. The materials have been grouped according to their Lewis acid site strength. Reaction conditions: 300 °C, 80 bar H2, 60 min (τ = 0.37 gcat h gamide−1). | ||
Fig. 5 displays the product distribution for the hydrotreatment of C16 amide (300 °C and 80 bar H2) as a function of batch residence time for the Pt/γ-Al2O3 (a), Pt/TiO2 (b) and Pt/ZrO2 (c) catalysts, representing the Si–Al, Ti–Nb, and Ce–Zr groups, respectively. The 60 min reference experiments shown in Fig. 4 correspond to a batch residence time of 0.37 gcat h gamide−1.
 | ||
| Fig. 5 Product distribution as a function of batch residence time for (a) Pt/γ-Al2O3, (b) Pt/TiO2, and (c) Pt/ZrO2 in the hydrotreatment of C16 amide at 300 °C and 80 bar H2. The 60 min reference experiments of Fig. 4 correspond to a batch residence time of 0.37 gcat h gamide−1. The trendlines have been added to guide the eye. | ||
The product distribution of the 60 min experiments was similar for the catalysts within each group, but the groups deviated from one another, especially in conversion and oxygen removal (Fig. 4). The differences in nitrogen removal and paraffin yields were initially smaller between the groups but became more pronounced with an increasing batch residence time (Fig. 5). A reaction network, adapted and extended from a previous study conducted by Verkama et al.,16 is proposed in Scheme 1. The following paragraphs introduce the reaction network on a general level, whereas the activity and selectivity of each group are discussed in sections 3.2.1–3.2.4.
 | ||
| Scheme 1 Proposed reaction network for the hydrotreatment of C16 amide. Indicated compounds: 1 C16 amide, 2 C32 isoimide, 3 C16 nitrile, 4 C16 acid, 5 C16 hemiaminal, 6 C16 imine, 7 C16 amine, 8 C16 aldehyde, 9 C16 alcohol, 10 C32 amide, 11 C32 amine, 12 C32 ester, 13 C15 paraffin, 14 C16 paraffin, and 15 C31 ketone. The bimolecular deammoniation (BDA), direct dehydration (DHY), hydrogenation (HYD) and hydrolysis (HYDR) of the C16 amide, and the condensation (COND), HDO and HDN reactions of the intermediates have been indicated. The bimolecular ketonization of the C16 acid (KET) only occurred over the bare supports. The reaction network has been adapted and extended from a previous study conducted by Verkama et al.16 | ||
The conversion of C16 amide appeared to proceed via two main pathways on the bare supports. One main pathway comprised the bimolecular deammoniation (BDA) of C16 amide to an isoimide, which decomposed to C16 nitrile and C16 acid (Scheme 1).71 The BDA of amides has been described by Davidson and Karten.71 The direct dehydration to C16 nitrile was the other main pathway for the conversion of C16 amide, and the preference between direct dehydration and BDA depended on the support. The hydrolysis of C16 amide to C16 acid and ammonia may have occurred additionally.21,72 The BDA and dehydration of C16 amide also occurred thermally, but the thermal activity was considerably lower compared with the activity of all tested materials except for the bare SiO2 support (Fig. 4a).71 In the case of the supported Pt catalysts, the conversion of C16 amide might have concurrently proceeded through other pathways, such as HDN to C16 aldehyde or C16 alcohol, and HDO to C16 imine.18,21,25,26,73 The conversion of C16 amide to C16 alcohol and C16 imine likely proceeded via a hemiaminal intermediate, and C16 imine was rapidly hydrogenated further to C16 amine.21 The isoimide, hemiaminal and imine intermediates were not detected in the product samples. Therefore, the initial pathway selectivity of the supported Pt catalysts could not be unambiguously confirmed from the experimental data. The distribution between oxygen-containing and nitrogen-containing intermediate products was, nevertheless, similar for the supported Pt catalysts and the corresponding bare supports, which might indicate that they favored the same initial conversion routes (Fig. 4).
The bimolecular ketonization of C16 acid to C31 ketone and the reduction of C16 acid to C16 aldehyde and ultimately to C16 alcohol were observed on the bare ZrO2, CeO2–ZrO2 and TiO2 supports (Fig. 4).15,74–76 These bare supports showed the lowest carbon balance closure (∼85%) out of all tested materials, which might indicate that some reaction products remained adsorbed on the supports or that non-volatile products that could not be detected with the GC were formed, additionally. C16 aldehyde and C31 ketone were not present in the product samples of the supported Pt catalysts.
C16 nitrile, C16 acid, C16 aldehyde, C16 alcohol, and C16 amine were further converted over all of the supported Pt catalysts, eventually forming C15 paraffin and C16 paraffin (Scheme 1, Fig. 4). C16 nitrile was readily hydrogenated to C16 amine, whereas C16 acid was hydrogenated to C16 alcohol, either directly or via C16 aldehyde.32 C15 paraffin was formed via decarboxylation of C16 acid and decarbonylation of C16 aldehyde, while C16 paraffin was obtained from the HDO of C16 alcohol and the HDN of C16 amine. C32 amine, C32 amide and small amounts of C32 ester were formed via condensation reactions of the intermediates, as indicated in Scheme 1 and discussed by Verkama et al.16 For instance, C32 amide was formed through the condensation of C16 acid and C16 amine.16,32 The C32 condensation products eventually decomposed to C16 paraffin.16,77,78
The supported Pt catalysts of the Si–Al group were highly active for the formation of C16 amine through hydrogenation of C16 nitrile (Fig. 4b). In the batch residence time series experiments on Pt/γ-Al2O3, this was reflected by C16 amine dominating the nitrogen-containing intermediates, whereas the C16 nitrile yield never exceeded 2% (Fig. 5a). The HDO of C16 acid, in contrast, proceeded relatively slowly over the supported Pt catalysts of the Si–Al group, as indicated by high yields of C16 acid in the product samples of the 60 min reference experiments (Fig. 4b). Consequently, C16 acid and C16 amine were present in relatively high amounts, which favored the formation of C32 amide (Scheme 1).16 In particular, the Pt/SiO2–Al2O3 catalysts showed a high tendency towards the formation of C32 amide (12–15%). C32 amine was also formed, but to a lower extent than on the other groups. In the batch residence time series experiments on Pt/γ-Al2O3, the C32 amide and C32 amine yields first increased and then stabilized (Fig. 5a). Therefore, the decomposition of these C32 compounds to C16 paraffin did not occur readily.
The total paraffin yields from the 60 min reference experiments ranged between 8% and 12% for the supported Pt catalysts of the Si–Al group (Fig. 4b). At batch residence time point 1.86 gcat h gamide−1, the product mixture of Pt/γ-Al2O3 contained 39% C16 paraffin and 12% C15 paraffin (C16 paraffin/C15 paraffin = 3.2 mol/mol, Fig. 5a). The HDO and HDN routes, therefore, dominated over decarbonylation and decarboxylation (Scheme 1).
The high yields of C16 nitrile (60–80%) and low yields of C16 acid (<10%) on the bare supports of the Ti–Nb group indicate that C16 amide was primarily converted through direct dehydration (Fig. 4a, Scheme 1). The strong tendency towards the oxygen removal from C16 amide was reflected by C16 and C32 amines dominating the product distribution of the supported Pt catalysts of the Ti–Nb group (Fig. 4b). Notably, the yield of C32 compounds in the product samples of the reference experiments was 2–3 times higher for the supported Pt catalysts of the Ti–Nb group compared with the other catalysts. However, the steep decrease in the C32 amine yield on Pt/TiO2 at batch residence times above 0.75 gcat h gamide−1 indicated that the catalyst was active for the conversion of C32 amine to C16 paraffin, in contrast to Pt/γ-Al2O3 (Fig. 5a and b).
Considerably less oxygen-containing intermediate products (<10%) were present in the product samples of the Ti–Nb group compared with the other groups (Fig. 4a and b). All oxygen-containing compounds were converted with a batch residence time of 0.75 gcat h gamide−1 on Pt/TiO2, further reflecting the high HDO activity (Fig. 5b).
Pt/TiO2 showed a total paraffin yield of 23% in the 60 min reference experiment, which exceeded the paraffin yields of the Si–Al group and Pt/Nb2O5 (10%) significantly (Fig. 4b). The Ti–Nb group favored C16 paraffin over C15 paraffin to an even greater extent than the Si–Al group, i.e., no C15 paraffin was detected for Pt/Nb2O5, and the C15 paraffin yield was below 2% for Pt/TiO2. At the highest batch residence time point (1.86 gcat h gamide−1), the total paraffin yield of Pt/TiO2 was 80% (23 molC16 paraffin/molC15 paraffin), which is 26 percentage points higher than the total paraffin yield of Pt/γ-Al2O3 at a similar batch residence time (Fig. 5a and b).
The conversion of the oxygen-containing intermediates proceeded more efficiently on the Ce–Zr group than on the Si–Al group. In the bare support experiments, this was reflected by the Ce–Zr group exhibiting activity towards the reduction and ketonization of C16 acid to C16 aldehyde and C31 ketone, respectively (Fig. 4a). For the supported Pt catalysts, the enhanced HDO activity of the Ce–Zr group could be observed, e.g., based on the absence of C16 acid in the product samples of the 60 min reference experiments (Fig. 4b), and upon comparing the evolution of the C16 acid and C16 alcohol yields in the batch residence time series experiments (Fig. 5a and c).
Pt/CeO2–ZrO2 formed 11 percentage points more paraffins (28%) than Pt/ZrO2 in the 60 min reference experiment (Fig. 4b). Considering the overall product distribution, conversion, nitrogen removal and oxygen removal, Pt/CeO2–ZrO2 was more active than Pt/ZrO2 in both HDN and HDO, even though the corresponding bare supports exhibited a similar activity level (Fig. 4a).
In a similar way to the other supported Pt catalysts, the supported Pt catalysts of the Ce–Zr group favored C16 paraffin over C15 paraffin. The total paraffin yield of the product sample of Pt/ZrO2 at the highest batch residence time (1.86 gcat h gamide−1) was 20 percentage points higher compared with Pt/γ-Al2O3 but 9 percentage points lower compared with Pt/TiO2 (Fig. 5). The C16 to C15 paraffin ratio for the product sample of Pt/ZrO2 was 6.1 mol/mol at this batch residence time.
Both Pt/ZrO2 and Pt/CeO2–ZrO2 produced a total of 16% of C32 condensation products in the 60 min reference experiments. In contrast to the Si–Al group, C32 amine was present in higher quantities than C32 amide (Fig. 4b). The high activity towards the HDO of C16 acid likely limited the formation of C32 amide on the Ce–Zr group, whereas the formation of C32 amine via condensation of C16 alcohol and C16 amine was more preferred (Scheme 1).16 C32 amine could be further converted to C16 paraffin, but this was not favored until the oxygen-containing intermediates had been consumed (Fig. 5c).16
4. Discussion
The Si–Al (Pt/γ-Al2O3, Pt/5SiO2–95Al2O3, Pt/30SiO2–70Al2O3), Ti–Nb (Pt/TiO2, Pt/Nb2O5), Ce–Zr (Pt/ZrO2, Pt/CeO2–ZrO2), and Si (Pt/SiO2) groups exhibited clear differences in their activity and selectivity for the HDO and HDN of C16 amide. The following sections discuss the impact of the material properties on the initial C16 amide conversion route, HDO activity, HDN activity, and the role of the C32 compounds in the reaction network.4.1 Initial conversion pathway
The selectivity towards the initial C16 amide conversion route differed between the catalyst groups (Fig. 4 and 5). On the bare supports, the main pathways were the direct dehydration of C16 amide to C16 nitrile and the BDA of C16 amide to C16 acid and C16 nitrile, whereas the supported Pt catalysts additionally may have converted the C16 amide to C16 alcohol and C16 amine via HDN and HDO, respectively (Scheme 1).The Ti–Nb group was highly active for the initial oxygen removal from C16 amide; thus, the nitrogen-containing intermediates dominated the product distribution. The significant initial C16 nitrile yields suggest that the direct dehydration of C16 amide was favored both on the bare supports and the supported Pt catalysts (Fig. 4 and 5b). Based on the product distribution, the HDO of C16 amide to C16 amine could nevertheless not be excluded on the supported Pt catalysts. The direct dehydration reaction was likely initiated upon the adsorption of the amide carbonyl group on the intermediate strength Lewis acid sites of TiO2 and Nb2O5.72,79 Shimizu et al.18 found that the oxygen of the carbonyl group of acetamide interacted more strongly with the Lewis acid sites of Nb2O5 and MoO3/TiO2 compared with the Lewis acid sites of Al2O3 and ZrO2, which enabled the HDO of amides to proceed efficiently on Pt/Nb2O5 and Pt/MoO3/TiO2 catalysts. These findings are in agreement with the activity of the Ti–Nb group for the initial oxygen removal from C16 amide.
In contrast, the bare supports of the Si–Al, Ce–Zr, and Si groups preferentially converted C16 amide via BDA, as indicated by the nearly stoichiometric formation of C16 nitrile and C16 acid or C16 acid derivatives (Fig. 4).71 Based on the batch residence time series experiments, the formation of C16 amine and C16 alcohol via the HDO and HDN of C16 amide, respectively, may have occurred in parallel with BDA on the supported Pt catalysts (Fig. 5). Therefore, various oxygen-containing and nitrogen-containing intermediate products were formed on the Si–Al, Ce–Zr, and Si groups, in contrast to the Ti–Nb group, which heavily favored the nitrogen-containing intermediate products.
The conversion of C16 amide to C16 alcohol and C16 amine may have proceeded via the formation of a hemiaminal intermediate by a cooperative mechanism with activation of the amide carbonyl group by a Lewis acid site on the support and hydrogenation by Pt.18,21,28,80 The enhanced conversion on the Ce–Zr group compared with the Si–Al group might therefore be related to the weak Lewis acid sites of the Ce–Zr group facilitating the formation of a hemiaminal, whereas the low activity on the Si group can be explained by the lack of Lewis acid sites (Fig. 3 and 4, Table 3). The conversion of the hemiaminal intermediate to C16 alcohol and C16 amine was likely catalyzed by the Pt sites, as the Si, Si–Al, and Ce–Zr groups exhibited a similar selectivity, despite differences in acidity and reducibility (Fig. 2).28
4.2 Conversion of the oxygen-containing intermediate products
The oxygen-containing intermediate products were readily formed on the Ce–Zr and Si–Al groups. However, the Ce–Zr group converted them more efficiently (Fig. 4b and 5). Therefore, the enhanced HDO activity of the Ce–Zr group distinguished the Si–Al and Ce–Zr groups from each other.C16 acid and C16 alcohol were the most important oxygen-containing intermediate products in the reaction network of C16 amide. The HDO of C16 acid to C16 alcohol likely occurred through the adsorption of C16 acid onto the Lewis acid sites of the supports and hydrogenolysis catalyzed by Pt.14,29,32 The HDO of C16 alcohol to the C16 paraffin may have proceeded similarly or through dehydration via an E2 mechanism involving Lewis acid–base site pairs (Table 3) followed by Pt-catalyzed hydrogenation.81–83 The adsorption and dehydration of C16 alcohol may alternatively have occurred on Brønsted acid sites in the case of Pt/30SiO2–70Al2O3. The Brønsted acidity of Pt/30SiO2–70Al2O3 likely enhanced the dehydration activity of the catalyst, which may explain why its paraffin yield was higher compared with Pt/γ-Al2O3 and Pt/5SiO2–95Al2O3 (Fig. 4b).33,84 Nevertheless, the activity of Pt/30SiO2–70Al2O3 did not exceed the activity of the catalysts of the Ce–Zr group.
The high HDO activity of the Ce–Zr group can be explained by the catalytic properties of the weak Lewis acid sites on Pt/ZrO2 and Pt/CeO2–ZrO2 (Fig. 3–5). These weak Lewis acid sites can be oxophilic incompletely coordinated Zr or Ce cations.29,85–90 Pt/CeO2–ZrO2 was more active than Pt/ZrO2, which, considering the H2-TPR, XPS, and pyridine FTIR analysis, may be due to the enhanced reducibility of the support and higher concentration of Lewis acid sites (Fig. 2, Table 3). The smaller mean Pt particle size of Pt/CeO2–ZrO2, i.e., enhanced active surface area, may additionally explain why the catalyst was more active than Pt/ZrO2 (Table 1).
The bare supports of the Si–Al group did not convert the C16 acid further. In contrast, the bare supports of the Ce–Zr group exhibited activity for the reduction of C16 acid to C16 aldehyde and subsequent conversion to C16 alcohol (Scheme 1, Fig. 4a). The bimolecular ketonization of C16 acid to C31 ketone was also observed. The activity towards these reactions can be attributed to the Lewis acid–base and redox properties of the bare supports of the Ce–Zr group.64,74–76,91,92 The reduction of C16 acid to C16 aldehyde was likely catalyzed by the oxygen vacancies on the ZrO2 and CeO2–ZrO2 supports.75,93–96 The conversion of C16 aldehyde to C16 alcohol may have been catalyzed by the Lewis acid–base site pairs of ZrO2 and CeO2–ZrO2, which are capable of heterolytic dissociation of H2.64,91,96,97 The presence of Pt, however, markedly increased the activity towards the conversion of C16 acid to C16 alcohol (Fig. 4b). The ketonization reaction was likely Lewis acid catalyzed.74,92
The C15 paraffin yields of the Si–Al and Ce–Zr groups did not exceed 12% at full C16 amide conversion (Fig. 4b and 5). C15 paraffin could be formed via decarbonylation of C16 aldehyde or via decarboxylation of C16 acid, both catalyzed by Pt.14,98 No correlation between the support or the Pt-related properties and the C15 paraffin yield were identified for the supported Pt catalysts of the Si–Al and Ce–Zr groups (Tables 1 and 2).
In contrast to the Ce–Zr and Si–Al groups, the Ti–Nb group formed relatively low amounts of oxygen-containing intermediate products due to the high initial activity for the HDO of C16 amide (Fig. 4). The low C15 paraffin selectivity of the Ti–Nb group was a consequence, as C15 paraffin was formed only from C16 acid or C16 aldehyde (Scheme 1).
4.3 Conversion of the nitrogen-containing intermediate products
The differences in the HDN activity of the catalysts were less pronounced than the differences in the HDO activity (Fig. 4). The HDN of nitrogen-containing intermediates was inhibited by the preferential HDO of oxygen-containing intermediates and the formation of condensation products.16 Therefore, in this complex reaction network (Scheme 1), the effect of the support on the HDN activity appeared to be outweighed by the effect of the support on the initial C16 amide conversion route and HDO activity.The hydrogenation of C16 nitrile to C16 amine was catalyzed by Pt, and all supported Pt catalysts exhibited a high activity towards the reaction (Fig. 4b and 5).99,100 The HDN of C16 amine to C16 paraffin likely occurred via a mechanism involving the dissociative adsorption of C16 amine to a hydrogen-deficient surface species on the Pt sites.13,77,101,102 A cooperative mechanism with adsorption of C16 amine on a Lewis acid site and hydrogenolysis catalyzed by Pt, similar to the HDO of C16 alcohol, may also be possible. Brønsted acid site catalyzed Hofmann elimination of C16 amine to an olefin, followed by hydrogenation of the olefin on Pt, may have occurred in the case of Pt/30SiO2–70Al2O3 (Fig. 3).103
Cattenot et al.13 suggested that the support can influence the activity and selectivity for the HDN of amines by affecting the electronic properties of the Pt particles. Based on the XPS analysis, the largest difference between the Pt electron densities was between the two catalysts of the Ti–Nb group, with Pt/TiO2 displaying the highest and Pt/Nb2O5 the lowest electron density for Pt (Table 2). The selectivity of Pt/TiO2 and Pt/Nb2O5 was similar, but Pt/TiO2 showed a higher nitrogen removal and paraffin yield than Pt/Nb2O5 in the 60 min reference activity test (Fig. 4b). It is possible that the electronic properties of the Pt particles contributed to the enhanced HDN activity of Pt/TiO2, but the smaller mean Pt particle size on Pt/TiO2 than Pt/Nb2O5 (Table 1) may also have influenced the order of activity. No evident correlation emerged between the Pt properties and the HDN activity of the other catalysts, possibly due to the complex reaction network.
Nitrogen-containing compounds, such as amines and ammonia, can poison Lewis and Brønsted acid sites.11,104 Strong adsorption of the C16 and C32 amines on Lewis (or Brønsted) acid sites might have interfered with the preferred reaction pathways and inhibited the catalytic activity towards the Lewis (or Brønsted) acid site catalyzed reactions. The adsorption of the nitrogen-containing intermediate products may have been stronger on the catalysts with the strongest Lewis acid sites.11 This provides another potential explanation for the relatively low overall activity of the catalysts of group Si–Al (Fig. 3 and 4).
4.4 C32 condensation products
The formation and decomposition of the C32 condensation products played an important part in the reaction network of the supported Pt catalysts. The preference for the formation of C32 amide and C32 amine was influenced by the distribution between the oxygen-containing and the nitrogen-containing intermediate products, which in turn was determined by the initial C16 amide conversion route and the HDO activity of the catalysts. Consequently, the role of C32 amide and C32 amine in the reaction network of the Si–Al, Ti–Nb, and Ce–Zr groups differed, as discussed further in the following paragraphs.The highest amounts of C32 amine were formed on the Ti–Nb group (Fig. 4b and 5). This may have followed from the high concentrations of C16 nitrile and C16 amine, as Pt is highly active for the formation of secondary amines via the disproportionation of primary amines and the condensation of imines and primary amines (Scheme 1).12,13,100,102,105–107 In the case of the Ce–Zr group, C32 amine was of importance, too, but its formation through condensation of C16 alcohol and C16 amine was more favored than on the Ti–Nb group due to the significant intermediate C16 alcohol yields.108,109
The formation of C32 amine could be catalyzed by the Pt sites, but a mechanism involving both Pt sites and Lewis acid sites of the support may also have been possible, particularly for the pathways that involved C–O bond scission (Scheme 1).13,32,101,107,110 The HDO activity of the weak Lewis acid sites of the Ce–Zr group might thus explain why the C32 amine was formed to a higher extent over the Ce–Zr than Si–Al group (Fig. 4b and 5).
The Si–Al group favored the formation of C32 amide over C32 amine (Fig. 4b). C32 amide was formed via condensation of C16 acid and C16 amine, and the reaction could be catalyzed by the bare supports, as observed previously by Verkama et al.16 The C16 amine and C16 acid were likely adsorbed on Lewis or Brønsted acid sites, which was followed by a condensation reaction. The high C32 amide yield of the Si–Al group was related to the relatively high concentration of C16 acid compared with the other catalysts, which resulted from the poor HDO activity of this group.
The decomposition of the C32 compounds eventually accounted for a significant share of the C16 paraffin yield. The HDO of C32 amide to C32 amine involved Lewis acid and Pt sites.18,80 Similarly to the HDO of C16 acid and C16 alcohol, the HDO of C32 amide proceeded efficiently over the supported Pt catalysts of the Ce–Zr and Ti–Nb groups (Fig. 4b and 5). A poor activity for the HDO of C32 amide also inhibited HDN, as the compound had to undergo HDO before its nitrogen could be removed.78
The HDN of C32 amine to C16 paraffin could proceed on the Pt sites, or possibly the Lewis acid and Pt sites.13,77 The HDN of C32 amine did not seem to occur readily until the oxygen-containing intermediates had been converted, suggesting that the presence of oxygen-containing compounds inhibited the reaction (Fig. 5).16 Consequently, the HDN of C32 amine occurred more efficiently on the Ti–Nb and Ce–Zr groups compared with the Si–Al group, which was reflected by the slow evolution of the C16 paraffin yield and accumulation of C32 compounds on Pt/γ-Al2O3 compared with Pt/ZrO2 and Pt/TiO2 (Fig. 5). These trends further emphasize the importance of the Lewis acid properties of the supports, which accounted for the differences in HDO activity.
5. Conclusions
In this study, the catalytic hydrotreatment of C16 amide (n-hexadecanamide) was studied over Pt supported on SiO2, γ-Al2O3, 5SiO2–95Al2O3, 30SiO2–70Al2O3, ZrO2, CeO2–ZrO2, Nb2O5 and TiO2. The HDO and HDN of C16 amide proceeded through several parallel and competing reaction pathways. The bare supports exhibited activity for the conversion of C16 amide to C16 nitrile and C16 acid, but the presence of Pt was required for hydrogenation and hydrogenolysis activity. The differences in the activity and selectivity of the catalysts could primarily be attributed to the properties of the support.The Lewis acid properties of the supports influenced the selectivity towards the initial C16 amide conversion route and the activity for HDO of the oxygen-containing intermediate products. Accordingly, the catalysts were divided into four groups based on the strength of their Lewis acid sites, i.e., Si–Al (Pt/γ-Al2O3, Pt/5SiO2–95Al2O3, Pt/30SiO2–70Al2O3), Ti–Nb (Pt/TiO2, Pt/Nb2O5), Ce–Zr (Pt/ZrO2, Pt/CeO2–ZrO2), and Si (Pt/SiO2). The intermediate strength Lewis acid sites of the Ti–Nb group were decisive for the activity and selectivity towards the dehydration of C16 amide to C16 nitrile, distinguishing the reaction network of the Ti–Nb group from the other groups. The Si–Al and Ce–Zr groups initially produced oxygen-containing and nitrogen-containing intermediate products with a similar selectivity, but the oxygen-containing intermediate products were converted more efficiently on the Ce–Zr group. The HDO activity of the Ce–Zr group could be related to their oxophilic weak Lewis acid sites.29,85–90 The activity of the Si group was inferior to the other groups due to a lack of Lewis acid sites, which were required for several reactions. The preferred condensation reaction pathway and the yields of the C32 condensation products were influenced by the initial C16 amide conversion route and HDO activity and, consequently, differed between the catalyst groups.
The differences in the HDN activity of the catalyst groups were more subtle than the differences in the HDO activity. With an increasing batch residence time, it nevertheless became evident that the high HDO activity of the Ce–Zr and Ti–Nb groups was also beneficial for HDN activity, as the inhibition of HDN by the presence of oxygen-containing compounds was suppressed, regardless of the favored oxygen removal pathway. The importance of the HDO activity could be observed from the inferior paraffin yield and accumulation of C32 condensation products on Pt/γ-Al2O3 compared with Pt/ZrO2 and Pt/TiO2 in the time-series experiments. Overall, the results of this study emphasize the influence of the Lewis acid properties of the catalyst support on the activity of noble metal catalysts for the HDO and HDN of compounds that are relevant for the production of renewable fuels.
Author contributions
Emma Verkama: conceptualization, investigation, methodology, formal analysis, visualization, writing – original draft, Sylvia Albersberger: conceptualization, writing – review & editing, project administration, Aitor Arandia: conceptualization, investigation, methodology, writing – review & editing, Kristoffer Meinander: investigation, formal analysis, writing – review & editing, Marja Tiitta: conceptualization, writing – review & editing, supervision, Reetta Karinen: conceptualization, writing – review & editing, supervision, Riikka L. Puurunen: conceptualization, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
Non-disclosure agreement prevents researchers from disclosing conflict of interest.Acknowledgements
Dr. Hua Jiang is acknowledged for the STEM images, and Ellen Järvinen is acknowledged for assistance with the batch reactor experiments. Dr. Yingnan Zhao and the Research Analytics department of Neste Corporation are acknowledged for the guidance with the acid site characterization and for access to the equipment that was used for the pyridine FTIR measurements. All members of the Neste-Aalto HDN catalyst development project group are gratefully acknowledged for fruitful discussions. This study was funded by Neste Corporation (Neste-Aalto HDN catalyst development project). The Bioeconomy and Raw materials research infrastructures at Aalto University and the OtaNano Nanomicroscopy Center were used for the study.References
- A. S. Berenblyum, V. Ya. Danyushevsky, E. A. Katsman, T. A. Podoplelova and V. R. Flid, Pet. Chem., 2010, 50, 305 CrossRef.
- H. Brännström, H. Kumar and R. Alén, BioEnergy Res., 2018, 11, 592 CrossRef.
- T. Kandaramath Hari, Z. Yaakob and N. N. Binitha, Renewable Sustainable Energy Rev., 2015, 42, 1234 CrossRef.
- K. Malins, Fuel, 2021, 285, 119129 CrossRef CAS.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1 CrossRef CAS.
- P. Šimáček, D. Kubička, G. Šebor and M. Pospíšil, Fuel, 2009, 88, 456 CrossRef.
- B. Veriansyah, J. Y. Han, S. K. Kim, S.-A. Hong, Y. J. Kim, J. S. Lim, Y.-W. Shu, S.-G. Oh and J. Kim, Fuel, 2012, 94, 578 CrossRef CAS.
- X. Li, X. Luo, Y. Jin, J. Li, H. Zhang, A. Zhang and J. Xie, Renewable Sustainable Energy Rev., 2018, 82, 3762 CrossRef CAS.
- P. Biller and A. B. Ross, Bioresour. Technol., 2011, 102, 215 CrossRef CAS PubMed.
- T. Madl and M. Mittelbach, Analyst, 2005, 130, 565 RSC.
- E. Furimsky and F. E. Massoth, Catal. Rev.: Sci. Eng., 2005, 47, 297 CrossRef CAS.
- G. Meitzner, J. Catal., 1986, 98, 513 CrossRef CAS.
- M. Cattenot, E. Peeters, C. Geantet, E. Devers and J. L. Zotin, Catal. Lett., 2005, 99, 171 CrossRef CAS.
- S. Ding, C. M. A. Parlett and X. Fan, Mol. Catal., 2021, 111492 Search PubMed.
- M. Snåre, I. Kubičková, P. Mäki-Arvela, K. Eränen and D. Yu. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708 CrossRef.
- E. Verkama, P. Auvinen, S. Albersberger, M. Tiitta, R. Karinen and R. L. Puurunen, Top. Catal., 2023, 66, 1353 CrossRef CAS.
- O. Palardy, C. Behnke and L. M. L. Laurens, Energy Fuels, 2017, 31, 8275 CrossRef CAS.
- K. Shimizu, W. Onodera, A. S. Touchy, S. M. A. H. Siddiki, T. Toyao and K. Kon, ChemistrySelect, 2016, 1, 736 CrossRef CAS.
- R. Burch, C. Paun, X.-M. Cao, P. Crawford, P. Goodrich, C. Hardacre, P. Hu, L. McLaughlin, J. Sá and J. M. Thompson, J. Catal., 2011, 283, 89 CrossRef CAS.
- Y. Nakagawa, R. Tamura, M. Tamura and K. Tomishige, Sci. Technol. Adv. Mater., 2015, 16, 014901 CrossRef PubMed.
- J. R. Cabrero-Antonino, R. Adam, V. Papa and M. Beller, Nat. Commun., 2020, 11, 3893 CrossRef CAS PubMed.
- T. Toyao, S. M. a. H. Siddiki, Y. Morita, T. Kamachi, A. S. Touchy, W. Onodera, K. Kon, S. Furukawa, H. Ariga, K. Asakura, K. Yoshizawa and K. Shimizu, Chem. – Eur. J., 2017, 23, 14848 CrossRef CAS PubMed.
- J. Coetzee, H. G. Manyar, C. Hardacre and D. J. Cole-Hamilton, ChemCatChem, 2013, 5, 2843 CrossRef CAS.
- M. Stein and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 2231 CrossRef CAS PubMed.
- A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477 CrossRef CAS PubMed.
- M. Liu, Y. Shi, K. Wu, J. Liang, Y. Wu, S. Huang and M. Yang, Catal. Commun., 2019, 129, 105726 CrossRef CAS.
- J. Liang, R. Ding, Y. Wu, Y. Chen, K. Wu, Y. Meng, M. Yang and Y. Wang, J. Mol. Catal. A: Chem., 2016, 411, 95 CrossRef CAS.
- Y. Zhang, L. Li, F. Liu, H. Qi, L. Zhang, W. Guan, Y. Liu, A. Wang and T. Zhang, ACS Catal., 2022, 12, 6302 CrossRef CAS.
- O. U. Valdés-Martínez, J. N. Díaz de León, C. E. Santolalla, A. Talavera-López, H. Avila-Paredes and J. A. de los Reyes, Ind. Eng. Chem. Res., 2021, 60, 18880 CrossRef.
- E. Mäkelä, J. L. González Escobedo, J. Neuvonen, J. Lahtinen, M. Lindblad, U. Lassi, R. Karinen and R. L. Puurunen, ChemCatChem, 2020, 12, 4090 CrossRef.
- K. A. Rogers and Y. Zheng, ChemSusChem, 2016, 9, 1750 CrossRef CAS PubMed.
- K. Kon, W. Onodera, S. Takakusagi and K. Shimizu, Catal. Sci. Technol., 2014, 4, 3705 RSC.
- A. M. Robinson, J. E. Hensley and J. W. Medlin, ACS Catal., 2016, 6, 5026 CrossRef CAS.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389 CrossRef CAS.
- S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsley, Science, 1981, 211, 1121 CrossRef CAS PubMed.
- G. L. Haller and D. E. Resasco, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1989, vol. 36, p. 173 Search PubMed.
- C.-J. Pan, M.-C. Tsai, W.-N. Su, J. Rick, N. G. Akalework, A. K. Agegnehu, S.-Y. Cheng and B.-J. Hwang, J. Taiwan Inst. Chem. Eng., 2017, 74, 154 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373 CrossRef CAS.
- E. Bêche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant, Surf. Interface Anal., 2008, 40, 264 CrossRef.
- C. A. Emeis, J. Catal., 1993, 141, 347 CrossRef CAS.
- J.-Y. de Saint Laumer, E. Cicchetti, P. Merle, J. Egger and A. Chaintreau, Anal. Chem., 2010, 82, 6457 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CrossRef CAS.
- M. Behrens and R. Schlögl, in Characterization of Solid Materials and Heterogeneous Catalysts, 2012, p. 609 Search PubMed.
- L. Deng, H. Miura, T. Shishido, S. Hosokawa, K. Teramura and T. Tanaka, Chem. Commun., 2017, 53, 6937 RSC.
- N. S. de Resende, J.-G. Eon and M. Schmal, J. Catal., 1999, 183, 6 CrossRef CAS.
- J. A. Wang, A. Cuan, J. Salmones, N. Nava, S. Castillo, M. Morán-Pineda and F. Rojas, Appl. Surf. Sci., 2004, 230, 94 CrossRef CAS.
- D. A. G. Aranda, A. L. D. Ramos, F. B. Passos and M. Schmal, Catal. Today, 1996, 28, 119 CrossRef CAS.
- H. Lieske, G. Lietz, H. Spindler and J. Völter, J. Catal., 1983, 81, 8 CrossRef CAS.
- C. Bozo, N. Guilhaume, E. Garbowski and M. Primet, Catal. Today, 2000, 59, 33 CrossRef CAS.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, ACS Catal., 2017, 7, 6493 CrossRef CAS.
- C. Nico, T. Monteiro and M. P. F. Graça, Prog. Mater. Sci., 2016, 80, 1 CrossRef CAS.
- S. B. T. Tran, H. Choi, S. Oh and J. Y. Park, J. Catal., 2019, 375, 124 CrossRef CAS.
- D. L. Hoang and H. Lieske, Catal. Lett., 1994, 27, 33 CrossRef CAS.
- T. Huizinga, J. Van Grondelle and R. Prins, Appl. Catal., 1984, 10, 199 CrossRef CAS.
- G. Pacchioni, Phys. Chem. Chem. Phys., 2013, 15, 1737 RSC.
- M. Yu. Smirnov and G. W. Graham, Catal. Lett., 2001, 72, 39 CrossRef CAS.
- S. Eriksson, S. Rojas, M. Boutonnet and J. L. G. Fierro, Appl. Catal., A, 2007, 326, 8 CrossRef CAS.
- J. A. Wang, T. López, X. Bokhimi and O. Novaro, J. Mol. Catal. A: Chem., 2005, 239, 249 CrossRef CAS.
- S. Bernal, J. J. Calvino, G. A. Cifredo, A. Laachir, V. Perrichon and J. M. Herrmann, Langmuir, 1994, 10, 717 CrossRef CAS.
- L. V. Mattos and F. B. Noronha, J. Power Sources, 2005, 145, 10 CrossRef CAS.
- J. Kašpar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285 CrossRef.
- C. Zhang, H. He and K. Tanaka, Appl. Catal., B, 2006, 65, 37 CrossRef CAS.
- H. Hattori, Chem. Rev., 1995, 95, 537 CrossRef CAS.
- G. Busca, Catal. Today, 2020, 357, 621 CrossRef CAS.
- G. Busca, Phys. Chem. Chem. Phys., 1999, 1, 723 RSC.
- M. Tamura, K. Shimizu and A. Satsuma, Appl. Catal., A, 2012, 433–434, 135 CrossRef CAS.
- M. I. Zaki, M. A. Hasan, F. A. Al-Sagheer and L. Pasupulety, Colloids Surf., A, 2001, 190, 261 CrossRef CAS.
- R. C. R. Neto and M. Schmal, Appl. Catal., A, 2013, 450, 131 CrossRef.
- G. S. Foo, D. Wei, D. S. Sholl and C. Sievers, ACS Catal., 2014, 4, 3180 CrossRef CAS.
- D. Davidson and M. Karten, J. Am. Chem. Soc., 1956, 78, 1066 CrossRef CAS.
- D. T. Mowry, Chem. Rev., 1948, 42, 189 CrossRef CAS PubMed.
- M. Tamura, S. Ishikawa, M. Betchaku, Y. Nakagawa and K. Tomishige, Chem. Commun., 2018, 54, 7503 RSC.
- R. Pestman, A. van Duijne, J. A. Z. Pieterse and V. Ponec, J. Mol. Catal. A: Chem., 1995, 103, 175 CrossRef CAS.
- R. Pestman, R. M. Koster, J. A. Z. Pieterse and V. Ponec, J. Catal., 1997, 168, 255 CrossRef CAS.
- R. Pestman, R. M. Koster, A. van Duijne, J. A. Z. Pieterse and V. Ponec, J. Catal., 1997, 168, 265 CrossRef CAS.
- N. Sivasankar and R. Prins, J. Catal., 2006, 241, 342 CrossRef CAS.
- C. Zhu, O. Y. Gutiérrez, D. M. Santosa, M. Flake, R. Weindl, I. Kutnyakov, H. Shi and H. Wang, Appl. Catal., B, 2022, 307, 121197 CrossRef CAS.
- M. H. Al-Huniti and M. P. Croatt, Asian J. Org. Chem., 2019, 8, 1791 CrossRef CAS.
- I. Sorribes, S. C. S. Lemos, S. Martín, A. Mayoral, R. C. Lima and J. Andrés, Catal. Sci. Technol., 2019, 9, 6965 RSC.
- S. Roy, G. Mpourmpakis, D.-Y. Hong, D. G. Vlachos, A. Bhan and R. J. Gorte, ACS Catal., 2012, 2, 1846 CrossRef CAS.
- P. Kostestkyy, J. Yu, R. J. Gorte and G. Mpourmpakis, Catal. Sci. Technol., 2014, 4, 3861 RSC.
- E. Iglesia, D. G. Barton, J. A. Biscardi, M. J. L. Gines and S. L. Soled, Catal. Today, 1997, 38, 339 CrossRef.
- E. Peeters, M. Cattenot, C. Geantet, M. Breysse and J. L. Zotin, Catal. Today, 2008, 133–135, 299 CrossRef CAS.
- P. M. de Souza, R. C. Rabelo-Neto, L. E. P. Borges, G. Jacobs, B. H. Davis, U. M. Graham, D. E. Resasco and F. B. Noronha, ACS Catal., 2015, 5, 7385 CrossRef CAS.
- C. A. Teles, P. M. de Souza, A. H. Braga, R. C. Rabelo-Neto, A. Teran, G. Jacobs, D. E. Resasco and F. B. Noronha, Appl. Catal., B, 2019, 249, 292 CrossRef CAS.
- K. A. Resende, C. A. Teles, G. Jacobs, B. H. Davis, D. C. Cronauer, A. Jeremy Kropf, C. L. Marshall, C. E. Hori and F. B. Noronha, Appl. Catal., B, 2018, 232, 213 CrossRef CAS.
- M. J. Mendes, O. A. A. Santos, E. Jordão and A. M. Silva, Appl. Catal., A, 2001, 217, 253 CrossRef CAS.
- K. Kreuzer and R. Kramer, J. Catal., 1997, 167, 391 CrossRef CAS.
- J. Ni, W. Leng, J. Mao, J. Wang, J. Lin, D. Jiang and X. Li, Appl. Catal., B, 2019, 253, 170 CrossRef CAS.
- M. Tamura, D. Yonezawa, T. Oshino, Y. Nakagawa and K. Tomishige, ACS Catal., 2017, 7, 5103 CrossRef CAS.
- H. Idriss, K. S. Kim and M. A. Barteau, in Studies in Surface Science and Catalysis, ed. R. K. Grasselli and A. W. Sleight, Elsevier, 1991, vol. 67, p. 327 Search PubMed.
- S. Foraita, J. L. Fulton, Z. A. Chase, A. Vjunov, P. Xu, E. Baráth, D. M. Camaioni, C. Zhao and J. A. Lercher, Chem. – Eur. J., 2015, 21, 2423 CrossRef CAS PubMed.
- B. Peng, C. Zhao, S. Kasakov, S. Foraita and J. A. Lercher, Chem. – Eur. J., 2013, 19, 4732 CrossRef CAS PubMed.
- T. Yokoyama and N. Yamagata, Appl. Catal., A, 2001, 221, 227 CrossRef CAS.
- H. Tsuji and H. Hattori, ChemPhysChem, 2004, 5, 733 CrossRef CAS PubMed.
- K. Tanabe and T. Yamaguchi, Catal. Today, 1994, 20, 185 CrossRef CAS.
- A. N. Kay Lup, F. Abnisa, W. M. A. W. Daud and M. K. Aroua, Appl. Catal., A, 2017, 541, 87 CrossRef CAS.
- Y. Huang and W. M. H. Sachtler, Appl. Catal., A, 1999, 182, 365 CrossRef CAS.
- J. Volf and J. Pašek, in Studies in Surface Science and Catalysis, ed. L. Cerveny, Elsevier, 1986, vol. 27, p. 105 Search PubMed.
- G. Meitzner, W. J. Mykytka and J. H. Sinfelt, Catal. Lett., 1995, 32, 335 CrossRef CAS.
- J. H. Sinfelt, Catal. Lett., 1991, 9, 159 CrossRef CAS.
- W. E. Farneth and R. J. Gorte, Chem. Rev., 1995, 95, 615 CrossRef CAS.
- M. Marafi and E. Furimsky, Energy Fuels, 2017, 31, 5711 CrossRef CAS.
- H. Greenfield, Ind. Eng. Chem. Prod. Res. Dev., 1967, 6, 142 CAS.
- A. J. Adamczyk, Surf. Sci., 2019, 682, 84 CrossRef CAS.
- Y. Huang and W. M. H. Sachtler, Appl. Catal., A, 1999, 182, 365 CrossRef CAS.
- A. Baiker and W. Richarz, Ind. Eng. Chem. Prod. Res. Dev., 1977, 16, 261 CrossRef CAS.
- A. Baiker, W. Caprez and W. L. Holstein, Ind. Eng. Chem. Prod. Res. Dev., 1983, 22, 217 CrossRef CAS.
- S. Zhang, Z. Xia, Y. Zou, M. Zhang and Y. Qu, Nat. Commun., 2021, 12, 3382 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01480k |
| ‡ VTT Technical Research Centre of Finland Ltd. P.O. Box 1000, FI-02044 Espoo, Finland. |
| § 06650 Hamari, Finland. |
| This journal is © The Royal Society of Chemistry 2024 |