
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A transaminase-mediated aldol reaction and applications in cascades to styryl pyridines†
Yu
Wang
 a,
Yiwen
Li
b,
Yeke
Ni
a,
Dejan-Krešimir
Bučar
a,
Paul A.
Dalby
b,
John M.
Ward
b,
Jack W. E.
Jeffries
*b and
Helen C.
Hailes
*a
a,
Yiwen
Li
b,
Yeke
Ni
a,
Dejan-Krešimir
Bučar
a,
Paul A.
Dalby
b,
John M.
Ward
b,
Jack W. E.
Jeffries
*b and
Helen C.
Hailes
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: h.c.hailes@ucl.ac.uk
bDepartment of Biochemical Engineering, University College London, Gower Street, London, WC1E 6BT, UK. E-mail: jack.jeffries.12@ucl.ac.uk
First published on 1st March 2024
Abstract
Transaminase enzymes are well established biocatalysts that are used in chemical synthesis due to their beneficial sustainability profile, regio- and stereoselectivity and substrate specificity. Here, the use of a wild-type Chromobacterium violaceum transaminase (CvTAm) in enzyme cascades revealed the formation of a novel hydroxystyryl pyridine product. Subsequent studies established it was a transaminase mediated reaction where it was exhibiting apparent aldolase reactivity. This promiscuous enzyme reaction mechanism was then explored using other wild-type transaminases and via the formation of CvTAm mutants. Application of one pot multi-step enzyme cascades was subsequently developed to produce a range of hydroxystyryl pyridines.
Introduction
Biocatalysis has become an important aspect of modern organic synthesis, both in academia and across the chemical and pharmaceutical industries. Its success has been largely due to a rapid expansion of the range of chemical reactions accessible and substrates accepted. Applications of naturally occurring enzymes, however, can be limited by narrow substrate specificities. Some classes of enzymes do show good native enzyme substrate promiscuity where the same transformation is performed and is due to active site plasticity or alternative substrate binding modes. Recently, interest has also focused on discovering functional enzyme promiscuity where enzymes can additionally catalyse a non-physiological reaction.1 Examples include hydrolytic enzymes such as porcine pancreatic lipase and a peptidase from Sulfolobus tokodaii that have been reported to catalyse aldol reactions.1–4 Enzyme mutagenesis has been reported to modify enzyme functionality and an early example was a single mutation in alanine racemase which converted it into an aldolase.5,6 More recently, a range of new to nature enzyme functionalities have been reported such as photoinduced enzyme catalysis with ene reductases and variants via radical mechanisms for asymmetric reduction, hydroalkylation and conjugate addition.7–9 Enzyme engineering to produce other new enzyme functionalities such as efficient Morita–Baylis–Hillman and cyclopropanes have also been described, highlighting the potential of new biocatalysts for use in synthesis.10–12Transaminases (TAms) are well established biocatalysts that reversibly transform a ketone or aldehyde group into an amine moiety using an amine donor and the co-factor pyridoxal 5′-phosphate (PLP) 1.13,14 When using prochiral ketones, the products can be single enantiomers and TAms have been used in numerous applications towards single isomer pharmaceutical ingredients, biologically active compounds, and small molecule amines not readily accessible via traditional synthetic routes.15–20
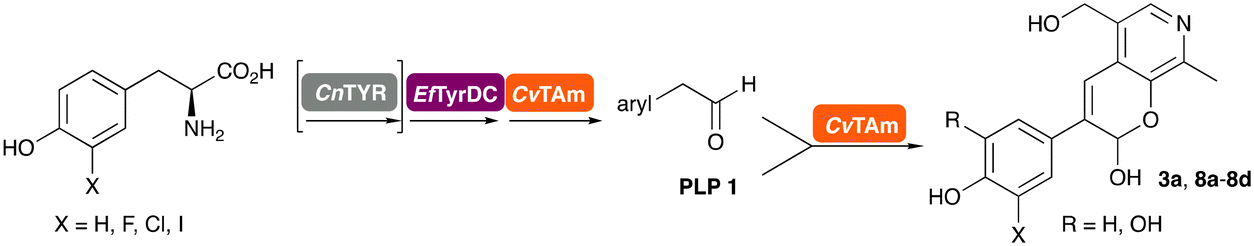
In this study, while incorporating the transaminase from Chromobacterium violaceum (CvTAm)21 into biocatalytic cascades to give benzylisoquinoline alkaloids (BIAs), a novel product was identified, arising from one of the aldehydes formed in situ from a CvTAm reaction and subsequent aldol condensation. Investigations revealed that CvTAm appeared to catalyse the aldol reaction, and this was explored further via in silico enzyme modelling and the production of CvTAm variants. Applications of this new CvTAm functionality were then applied in multistep one-pot enzyme cascades incorporating a tyrosinase (TYR), tyrosine decarboxylase (TyrDC) and CvTAm to generate complex styryl pyridines (Scheme 1), which are a promising class of molecules for several biological targets.
 | ||
| Scheme 1 Previous work using cascades to alkaloids and novel aldolase activity mediated by CvTAm in this work giving rise to styryl pyridines. | ||
Results and discussion
Initial hydroxystyryl pyridine formation
In previous work one-pot enzyme cascades were developed to generate new BIAs, starting from L-tyrosine 2a and analogues. Several recombinant enzymes in E. coli were used, a tyrosinase from Candidatus Nitrosopumilus salaria BD31 (CnTYR), tyrosine decarboxylase from Enterococcus faecalis DC32 (EfTyrDC), CvTAm and Thalictrum flavum norcoclaurine synthase (TfNCS) to produce, in vitro, alkaloids such as norlaudanosoline in high yields and stereoselectivities.22,23 In subsequent work, parallel cascades were constructed using CnTYR and variants together with a EfTyrDC to produce the arylethylamine component, and EfTyrDC and CvTAm to produce arylacetaldehydes, enabling the use of two different amino acid starting materials. Addition of TfNCS was then able to generate novel doubly halogenated BIAs.24 When generating arylacetaldehydes in situ from 2a and analogues, it was noted that an unknown side-product 3a was formed. While these could readily be separated from the BIA products, we were intrigued to understand how they were formed to streamline future cascade design.NMR spectroscopic analysis confirmed that the new product 3a, formed when using 2a, was not a BIA with the characteristic tetrahydroisoquinoline signals, ruling out the involvement of TfNCS. The presence of a deshielded proton at ∼8.2 ppm suggested that it could be a pyridinium species arising from 1. Ortho-aryl couplings indicated that hydroxylation using CnTYR had not occurred, and that intermediates present including tyramine 4a, formed by an in situ decarboxylation with EfTyrDC, or 4-hydroxylphenylacetaldehyde 5a generated by a subsequent reaction with CvTAm, may be involved. Accurate mass spectrometry (MS) data (m/z 286.1072) corresponded to the addition of 1 and 5a and loss of H2O (and hydrolysis of the phosphate group), suggesting an aldol addition. Analysis of the NMR data indicted key NOEs between a CH2 group and alkene CH, the later also giving rise to an NOE with the aryl group (Scheme 2). Together, this suggested that 3a was 5-(hydroxymethyl)-3-(4-hydroxyphenyl)-8-methyl-2H-pyrano[2,3-c]pyridin-2-ol, and this was consistent with results of single crystal X-ray diffraction studies of a trifluoroacetate salt of 3a-OEt – an ethoxy hemiacetal of 3a obtained through recrystallization from ethanol.
 | ||
| Scheme 2 Formation of 3a and key characterisation data. A. COSY NMR spectra of 3a with a key long-range coupling indicated with double headed arrows and assignments using coloured circles. Also, the molecular structure of the 3a-OEt trifluoroacetate cation, as derived from single crystal X-ray diffraction analyses; B NOESY NMR spectra of 3a showing key NOEs with double headed arrows, and proton assignments. | ||
Recent studies have reported that some (E)-4-(substituted styryl)pyridines, structurally related to resveratrol, inhibited the formation of the vascular endothelial growth factor (VEGF) from HT-29 cells and expression of the telomerase-related hTERT and c-Myc genes.25 Other hydroxystyryl pyridines have been determined to act as aldose reductase inhibitors for type II diabetes applications.26 The hydroxystyryl pyridine 3a was therefore a potentially interesting pharmacophore, and also due to the unusual mode of formation the reaction was explored in more detail to give a range of styryl pyridines.
Exploring the reaction conditions required for styryl pyridine formation
To establish whether the aldol addition to produce 3a was enzymatically catalysed or due to the presence of particular buffers, phenylacetaldehyde 5b and 1 were initially reacted together in different buffers or water (Table 1a), including 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) buffer (50 mM, pH 5.5 and pH 7.5), potassium phosphate (KPi) buffer (300 mM, pH 5.5 and pH 7.5) and 2-amino-2-hydroxymethyl-propane-1,3-diol (Tris) buffer (100 mM, pH 5.5 and pH 7.5). While all reactions gave rise to three new peaks (P1–P3) (P4 is 5b) after 16 h by HPLC analysis, they were mixtures of uncharacterized side products (potentially formed via the enol of 5b) confirming that the formation of the styryl pyridine 3b was enzyme mediated.|
|
|---|
| Reaction conditions: (a) 5b (10 mM, 1 equiv.) and 1 (15 mM, 1.5 equiv.) was added to reaction buffers, and 10% (v/v) DMSO as a co-solvent; (b and c) 10% (v/v) enzyme lysates (4 mg mL−1 total protein) were added. Reactions were performed at 37 °C for 16 h and monitored by analytical HPLC at 280 nm. Note: P4 is 5b. |

|

Enzymes used in the original enzyme cascade were then investigated including an empty cell vector lysate, lysates of CnTYR, EfTyrDC, CvTAm and TfNCS. In agreement with the preliminary observations, CnTYR and TfNCS were not involved in the styryl pyridine formation, giving rise to peaks P1–P4 only, and this was also the case for EfTyrDC. However, the reaction with CvTAm gave a new product (P5, Table 1b) and separation by preparative HPLC followed by characterisation by NMR spectroscopy (COSY, NOESY, Fig. 1) indicated the structure as the aldol product 3b. Single crystal X-ray diffraction analysis also confirmed the structure of 3b (Fig. 1A).
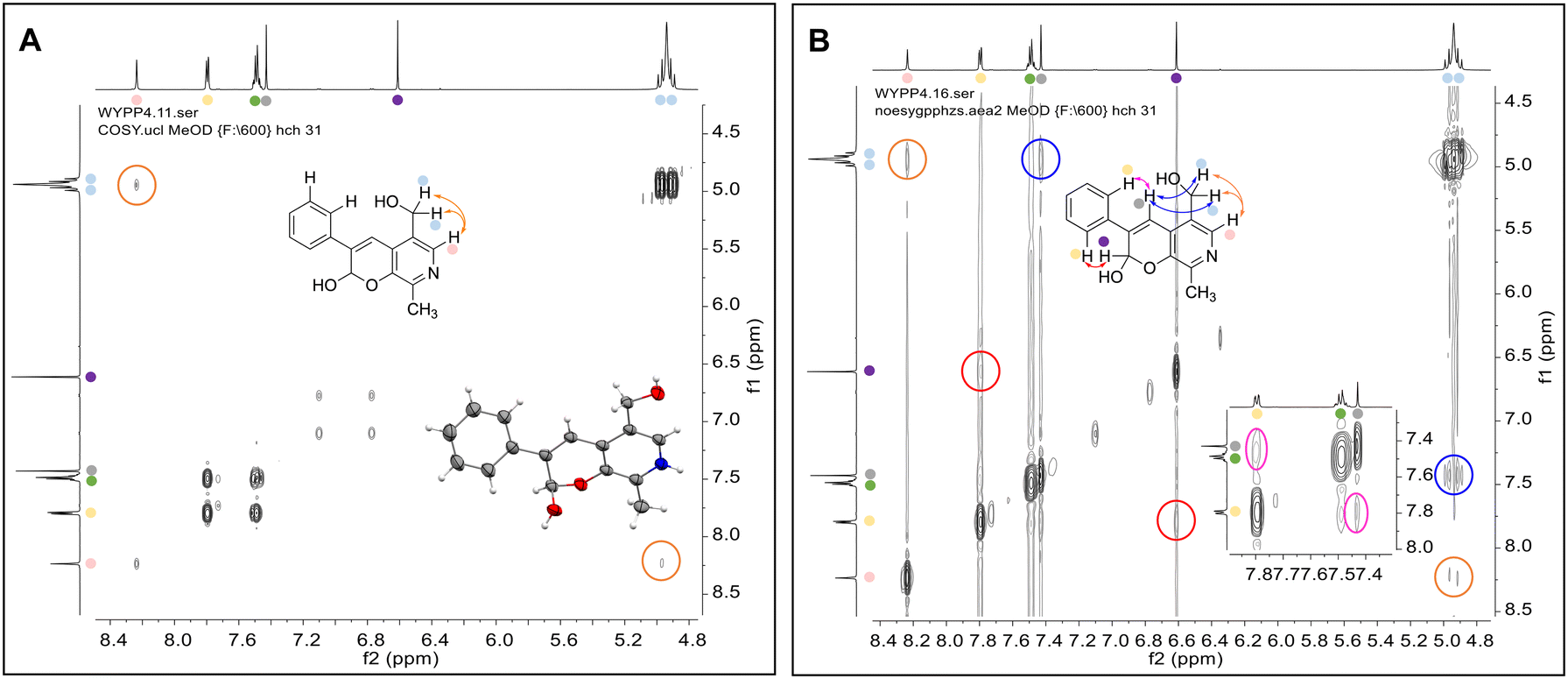 | ||
| Fig. 1 Key characterisation data for 3b. A. COSY NMR spectra of 3b with a key long-range coupling indicated with the double headed arrows and assignments using coloured circles. Also, the molecular structure of the 3b trifluoroacetate cation, as derived from single crystal X-ray diffraction analyses;† B NOESY NMR spectra of 3b showing key NOEs with double headed arrows, and proton assignments. | ||
The apparent promiscuous ability of CvTAm to catalyze an aldol addition of arylacetaldehydes 5a and 5b to 1 was intriguing and so initially studies were carried out to determine whether other transaminases could also catalyze the reaction. Using 5b and 1 again as starting materials, the (R)-selective transaminase from Mycobacterium vanbaalenii (MvTAm)27 and Arthrobacter sp. (ArRmut11),15 and (S)-selective transaminases from Vibrio fluvialis (VfTAm),28Klebsiella pneumoniae (KpTAm; from UCL plasmid pQR1005)29,30 and Rhodobacter sphaeroides (RsTAm; from UCL plasmid pQR 1019)30,31 were selected due to their previous biocatalytic applications. Several other transaminases from a drain metagenome, expressed from plasmids pQR2189, pQR2191, and pQR2208 which have shown good activity towards aromatic amines, were also selected to test in the reaction.32,33CvTAm gave a reasonable yield (42%) of 3b. Notably, none of the other transaminases gave 3b (Table 1c). This data indicated that CvTAm was distinctive in being able to effectively catalyse the aldol addition to give styryl pyridines.
To try and establish the key residues in CvTAm that gave rise to the aldolase activity, molecular docking studies with apo CvTAm (PDB: 4BA4)34 and 3b were performed using AutoDock Vina.35,36 Analysis of the liganded structures revealed that 3b (for five conformations out of nine with the lowest energies) was located in a cleft between the dimeric subunits of CvTAm (Fig. 2 and Table S2†). Thus, it is likely that the catalytic site for the aldol condensation is distinct from the catalytic site for transaminase activity (Fig. 2F). Type I aldolases such as 2-deoxyribose-5-phosphate aldolase (DERA), contain two key lysine residues in the catalytic site, one which forms a Schiff base intermediate, with a second residue nearby to perturb the pKa of the reactive lysine.37–39 Inspired by this concept it was considered that key residues in CvTAm for the aldol addition could be two lysines, positioned at the cleft between the subunits with one lysine residue located on each subunit and one lysine forming an imine with one of the substrates. Modelling positioned 3b in close proximity to Lys288.A and Lys90.B (the distance between Lys288.A and Lys90.B is 14.2 Å, and we recognise that this is a larger distance than in native and evolved aldolases,37–39 Fig. S7A†) suggesting that these may potentially be involved in the promiscuous aldolase activity. Other transaminases including VfTAm (PDB:5ZTX,40 Lys285.A and Lys126.B, Fig. S7B†) ArRmut11 (PDB: 3WWJ,41 Lys188.A and Lys142.B, Fig. S7C†) have the equivalent residues. However, no aldol additions were observed, possibly due to the greater separation of the two lysine residues (24.4 Å for ArRmut11), although for VfTAm the distance was similar to CvTAm (14.6 Å), suggesting other factors may be important. No equivalent residues were found for KpTAm (PDB: 3I5T,42 Fig. S7D†).
 | ||
| Fig. 2 Molecular docking studies using CvTAm (dimer)with styryl pyridine 3b. A. For aldolase activities, ligand 3b (orange) was located between Lys288.A and Lys90.B (subunit A in blue and subunit B in pink, active sites in red square). For transaminase activities, 1 interacts with Lys288. B and C. Transaminase active sites are located at the central of each subunit. D, E and G. Potential aldolase active sites located in a cleft between the dimeric subunits. F. Potential catalytic sites for the aldol condensation and (known) transamination are distinct. Figures were generated using UCSF ChimeraX.43,44 | ||
To establish whether these are the key catalytic residues, the CvTAm variants K288T, K90T and double mutant K90T/K288T were generated, as threonine has a smaller size compared to lysine, but retains the polar residue characteristics and is uncharged. The single mutants were generated by site directed mutagenesis and the K90T mutant was then used as a template for the creation of the double mutant. Reactions using phenylacetaldehyde 5b and 1 were performed with purified CvTAm WT, K288T, K90T, K90T/K288T (0.1 mg mL−1) and an empty vector control. WT CvTAm only gave the product 3b (Fig. 3A). When reacting tyramine 4a (to produce 5ain situ) and 1, the aldol product 3a again was only formed with WT CvTAm (Fig. 3B). The experiments with K288T resulted in the loss of transamination activity with no formation of 5a as expected, as Lys288 is a key mechanistic lysine. In addition, while the transamination reactivity was retained in K90T giving 5a, the aldol addition activity was lost. The double mutant K90T/K288T also lost all transaminase and aldol activities (Fig. 3). These experiments supported the hypothesis that Lys288 and Lys90 have key mechanistic roles in the aldol reaction.
 | ||
| Fig. 3 HPLC traces of CvTAm reactions showing key HPLC product peaks. A. HPLC traces of CvTAm reaction with 5b and 1; B. HPLC traces of the CvTAm reaction with 4a and 1. Reaction conditions: 5b or 4a (10 mM, 1 equiv.) and 1 (15 mM, 1.5 equiv.) were added to reaction buffers, and 10% (v/v) DMSO as a co-solvent. To initiate the reactions, 0.1 mg mL−1 purified enzyme was added. Reactions were performed at 37 °C for 16 h and monitored by analytical HPLC at 280 nm. | ||
A possible mechanism is shown in Scheme 3. Firstly, aldehyde 5b could be protonated by Lys288.A and nucleophilic attack by Lys90.B, would give the carbinolamine. Protonation and subsequent loss of water would give the imine and subsequent deprotonation of the α-proton by Lys288.A, would give the key enamine. This can then attack the carbonyl carbon in 1 with protonation from Lys288.A to give the aldol product and subsequent imine hydrolysis would give the corresponding aldehyde. Intramolecular hemiacetal formation followed by phosphate hydrolysis would generate 3b (Scheme 3). It was considered that phosphate hydrolysis likely occurred during the purification step and this is discussed further in the following section. Other potential aldol mechanisms are also possible such as switching which aldehyde forms the Lys imine: first PLP imine formation and then the aldol addition of 5b, with subsequent elimination of the Lys.
 | ||
| Scheme 3 Proposed mechanism for the aldol addition by CvTAm to give (after purification) styryl pyridine 3b. Note that the roles of Lys288 and Lys90 could be reversed. | ||
To investigate the potential competition between transamination activity and aldol activity, kinetic studies were performed. Firstly, the concentration of pyruvate was optimised. The results showed that pyruvate did not participate in the aldol addition but instead acted as an amine accepter for transamination activity. Therefore, pyruvate was used at 1 eq. to 4a (1 eq., 10 mM, Fig. S9†). Different ratios of 1 (0–2.5 eq.) to 4a (1 eq., 10 mM) were also tested with purified WT CvTAm (0.1 mg mL−1). With lower equivalents of 1 (<0.1 eq.), only the transamination product 5a was formed. The aldol product 3a was observed at higher equivalents of PLP 1 (>0.2 eq.) and reached its maximum at 1.5 eq. of 1 (48% yield by HPLC analysis, Fig. S10†). Therefore, for the kinetic study of CvTAm for aldolase activity, 1 was used at 1.5 eq. to the substrate 4a and 1 eq. pyruvate was used. The CvTAm kinetic studies were performed with 0.1 eq. of 1 and 1 eq. of pyruvate, revealing that the transaminase activity (kcat.app/Km.app = 3.57 s−1 mM−1, Km.app = 1.69 mM and kcat.app = 6.04 s−1) is about 20 times higher than the aldolase activity (kcat.app/Km.app = 0.18 s−1 mM−1, Km.app = 9.84 mM and kcat.app = 1.75 s−1). This also suggests that the aldolase activity has little effect on the transaminase reaction.
Exploring the substrate scopes for the aldol addition to give styryl pyridines by CvTAm
The substrate scope for the production of styryl pyridines using CvTAm was then investigated. Tyramine 4a as before, dopamine 4c, meta-tyramine 4d and ortho-tyramine 4e were selected, with a view to them forming the corresponding aldehydes in situ and reacted with 1. Both 4a and 4e gave the products 3a and 3e, respectively (Table 2). While 4c and 4d can readily form the corresponding aldehydes,23,24 the presence of the meta-OH appeared to inhibit the aldol reaction. The scope of the aldehydes accepted was then investigated using 2-pyridinecarboxaldehyde 6a, 4-pyridinecarboxaldehyde 6b, pyrrole-2-carboxaldehyde 6c and pyrrole-2-carboxaldehyde 6d which were reacted with 4a and 5b. As shown in Table 2, the pyrrole carboxaldehydes were not accepted, while reactions using pyridine carboxaldehydes 6a and 6b gave the (E)-styryl pyridines 7a–7d in 35–46% (by analytical HPLC against product standards). It was noted that as previously reported,33,45 pyridine and pyrrole carboxaldehydes/ketones can be accepted by transaminases. Here, the pyridine-and pyrrole-substituted methylamine peaks had HPLC retention times that overlapped with 1 and sodium pyruvate, so competing aminations were not monitored. Benzaldehyde and analogues substituted with electron withdrawing groups, 2-, 3-, and 4-nitrobenzaldehyde, were also used with 5b, but no aldol products were observed. These results indicated that a pyridine structure appeared to be required in the electrophile for the aldol addition, perhaps to assist in the orientation of the substrate into a productive conformation.|
|
|||
|---|---|---|---|
| X | Y | Product | Isolated yield (yield by HPLC) |
| a Reaction conditions: starting materials X (10 mM, 1 equiv.) and Y (15 mM, 1.5 equiv.) were added to HEPES buffer (50 mM, pH 7.5), and 10% (v/v) DMSO was used as a co-solvent – 0.1 mmol reactions were performed. For reactions with 4a, 4c–4e, 1 mM 1 was also added for maintaining the transamination activity of CvTAm and 10 mM pyruvate. To initial reactions, 10% (v/v) CvTAm lysates (4 mg mL−1) were added. Reactions were then carried out at 37 °C for 16 h and monitored by analytical HPLC at 280 nm against product standards. | |||
 4a
4a
|
1 |
 3a
3a
|
36% (44%) |
 4c
4c
|
1 | No aldol product | — |
 4d
4d
|
1 | No aldol product | — |
 4e
4e
|
1 |
 3e
3e
|
29% (37%) |
 4a
4a
|
 6a
6a
|
 7a
7a
|
24% (35%) |
 4a
4a
|
 6b
6b
|
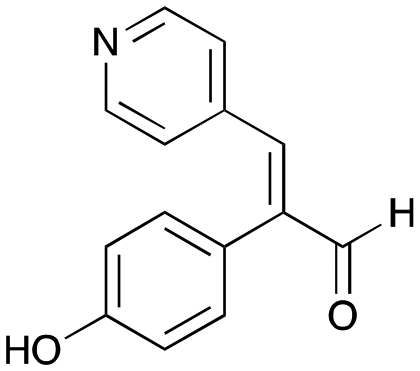 7b
7b
|
25% (36%) |
 4a
4a
|
 6c
6c
|
No aldol product | — |
 4a
4a
|
 6d
6d
|
No aldol product | — |
 5b
5b
|
 6a
6a
|
 7c
7c
|
26% (39%) |
 5b
5b
|
 6b
6b
|
 7d
7d
|
35% (46%) |

The phosphate hydrolysis step was then explored as the loss of phosphate was potentially due to the acidic work-up and purification method used. For the reaction between 1 and 4e (using the same conditions as previously), when this was quenched and purified in the absence of acid, it gave 3e-phosphate as the isolated product in 28% yield (ESI† section S3). This indicated that the phosphate hydrolysis was due to the work-up and purification conditions used, rather than a transaminase-mediated step.
Enzyme cascades to halogenated hydroxystyryl pyridines
Enzyme cascades to halogenated hydroxystyryl pyridines were then developed, combining EfTyrDC and CvTAm and using 2a and the halogenated tyrosines 2b–2d as the starting materials as halogenated arylacetaldehydes are not readily available. First, the cascade to 3a was optimised from 2a using EfTyrDC and CvTAm to give the arylacetaldehyde, which directly condensed with 1 using CvTAm giving 3a in 39% isolated yield. Similarly, 8a was formed from 2b in 37% yield (51% HPLC yield, Table 2). As 2b can also be hydroxylated by CnTYR from previous work,23,24 this was also included in the cascade to the arylacetaldehyde, which with 1 gave the catechol styryl pyridine 8b in 35% yield. Finally, application of the cascade without wild-type CnTYR, which does not readily accept 2c and 2d, gave products 8c and 8d in 36% and 29% isolated yields, respectively (Table 3). The ability to start from readily available amino acids, and through one pot two-enzyme three-step, or three-enzyme four-step cascades, to produce complex hydroxylated styryl pyridines is a valuable route to such compounds in 29–39% yields (41–51% yields by HPLC).|
|
|||
|---|---|---|---|
| Amino acid | Cascade route | Product | Isolated yielda (yield by HPLC)b |
| a Reaction conditions: 1 (10 mM, 1 equiv.) and substrate 2a–d (15 mM, 1.5 equiv.) were added to HEPES buffer (50 mM, pH 7.5) – 0.1 mmol reactions were performed. To initial reactions, 10% (v/v) enzyme lysates (4 mg mL−1 total enzyme) of CnTYR (where used), EfTyrDC and CvTAm were added into one-pot. Reactions were performed at 37 °C for 16 h. b Reactions were monitored by analytical HPLC at 280 nm against product standards. | |||
 2a
2a
|

|
 3a
3a
|
39% (48%) |
 2b
2b
|

|
 8a
8a
|
37% (51%) |
 2b
2b
|

|
 8b
8b
|
35% (42%) |
 2c
2c
|

|
 8c
8c
|
36% (48%) |
 2d
2d
|

|
 8d
8d
|
29% (41%) |
As hydroxystyryl pyridines have an interesting pharmacophore, initial molecular dynamics modelling with 3a was investigated with human pancreatic amylase (HPA) (ESI† section S6), which suggested that they could be a potential inhibitor and useful scaffold for use in future studies.
Conclusions
Here, a transaminase from Chromobacterium violaceum was identified as being able to catalyse the aldol addition between aryl acetaldehydes and pyridine carboxaldehydes, giving styryl pyridines and hydroxystyryl pyridines. This C–C bond formation is believed to be catalysed by two lysine residues Lys288.A and Lys90.B in the dimeric enzyme. Given the importance of hydroxystyryl pyridines as promising inhibitors for type II diabetes, enzyme cascades to various hydroxystyryl pyridines were established starting from commercially available amino acids. Above all, our data illustrates the potential of native enzymes to achieve promiscuous activities and produce novel products.Author contributions
Y. W. and Y. N. performed chemical syntheses, chemical characterizations, and enzymatic assays. D-K. B. carried out the X-ray crystallography experiments and J. W. E. J. generated the enzyme variants. P. A. D., Y. L. and Y. W. performed the molecular dynamic simulations. The manuscript was written through contributions of all authors. The project was conceived by Y. W., J. W. E. J. and H. C. H., and was supervised by J. M. W., J. W. E. J. and H. C. H. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by a UCL Dean's Prize to Y. W. Funding from the Biotechnology and Biological Sciences Research Council (BBSRC) to J. W. E. J. (BB/N01877X/1) is gratefully acknowledged. We also acknowledge 700 MHz NMR equipment support by EPSRC (EP/P020410/1). We thank K. Karu (UCL Mass Spectrometry Facility) and A. E. Aliev (UCL NMR Facility) in the Department of Chemistry at UCL. Molecular graphics and analyses were performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.Notes and references
- R. D. Gupta, Sustainable Chem. Processes, 2016, 4, 2 CrossRef.
- A. Patti and C. Sanfilippo, Int. J. Mol. Sci., 2022, 23, 2675 CrossRef CAS PubMed.
- C. Li, X. W. Feng, N. Wang, Y. J. Zhou and X. Q. Yu, Green Chem., 2008, 10, 616–618 RSC.
- R. Li, B. Perez, H. Jian, M. M. Jensen, R. Gao, M. Dong, M. Glasius and Z. Guo, Appl. Microbiol. Biotechnol., 2015, 99, 9625–9634 CrossRef CAS PubMed.
- F. P. Seebeck and D. Hilvert, J. Am. Chem. Soc., 2003, 125, 10158–10159 CrossRef CAS PubMed.
- M. D. Toscano, M. M. Müller and D. Hilvert, Angew. Chem., Int. Ed., 2007, 46, 4468–4470 CrossRef CAS PubMed.
- Y. Nakano, M. J. Black, A. J. Meichan, B. A. Sandoval, M. M. Chung, K. F. Biegasiewicz, T. Zhu and T. K. Hyster, Angew. Chem., Int. Ed., 2020, 59, 10484–10488 CrossRef CAS PubMed.
- X. Huang, B. Wang, Y. Wang, G. Jiang, J. Feng and H. Zhao, Nature, 2020, 584, 69–74 CrossRef CAS PubMed.
- P. T. Cesana, C. G. Page, D. Harris, M. A. Emmanuel, T. K. Hyster and G. S. Schlau-Cohen, J. Am. Chem. Soc., 2022, 144, 17516–17521 CrossRef CAS PubMed.
- P. C. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- R. Crawshaw, A. E. Crossley, L. Johannissen, A. J. Burke, S. Hay, C. Levy, D. Baker, S. L. Lovelock and A. P. Green, Nat. Chem., 2022, 14, 313–320 CrossRef CAS PubMed.
- D. C. Miller, S. V. Athavale and F. H. Arnold, Nat. Synth., 2022, 1, 18–23 CrossRef PubMed.
- S. Mathew and H. Yun, ACS Catal., 2012, 2, 993–1001 CrossRef CAS.
- F. Guo and P. Berglund, Green Chem., 2017, 19, 333–360 RSC.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- T. Sehl, H. C. Hailes, J. M. Ward, U. Menyes, M. Pohl and D. Rother, Green Chem., 2014, 16, 3341–3348 RSC.
- C. S. Fuchs, J. E. Farnberger, G. Steinkellner, J. H. Sattler, M. Pickl, R. C. Simon, F. Zepeck, K. Gruber and W. Kroutil, Adv. Synth. Catal., 2018, 360, 768–778 CrossRef CAS.
- D. Baud, J. W. E. Jeffries, T. S. Moody, J. M. Ward and H. C. Hailes, Green Chem., 2017, 19, 1134–1143 RSC.
- S. J. Novick, N. Dellas, R. Garcia, C. Ching, A. Bautista, D. Homan, O. Alvizo, D. Entwistle, F. Kleinbeck, T. Schlama and T. Ruch, ACS Catal., 2021, 11, 3762–3770 CrossRef CAS.
- A. Dunbabin, F. Subrizi, J. M. Ward, T. D. Sheppard and H. C. Hailes, Green Chem., 2017, 19, 397–404 RSC.
- U. Kaulmann, K. Smithies, M. E. B. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628–637 CrossRef CAS.
- B. R. Lichman, M. C. Gershater, E. D. Lamming, T. Pesnot, A. Sula, N. Keep, H. C. Hailes and J. M. Ward, FEBS J., 2015, 282, 1137–1151 CrossRef CAS PubMed.
- Y. Wang, N. Tappertzhofen, D. Méndez-Sánchez, M. Bawn, B. Lyu, J. M. Ward and H. C. Hailes, Angew. Chem., Int. Ed., 2019, 58, 10120–10125 CrossRef CAS PubMed.
- Y. Wang, F. Subrizi, E. M. Carter, T. D. Sheppard, J. M. Ward and H. C. Hailes, Nat. Commun., 2022, 13, 5436 CrossRef CAS PubMed.
- R. Marti-Centelles, J. Murga, E. Falomir, M. Carda and J. A. Marco, Med. Chem. Commun., 2015, 6, 1809–1815 RSC.
- H. Chen, X. Zhang, X. Zhang, W. Liu, Y. Lei, C. Zhu and B. Ma, Molecules, 2020, 25, 5135 CrossRef CAS PubMed.
- M. Höhne, S. Schätzle, H. Jochens, K. Robins and U. T. Bornscheuer, Nat. Chem. Biol., 2010, 6, 807–813 CrossRef PubMed.
- J. S. Shin, H. Yun, J. W. Jang, I. Park and B. G. Kim, Appl. Microbiol. Biotechnol., 2003, 61, 463–471 CrossRef CAS PubMed.
- B. R. Lichman, E. D. Lamming, T. Pesnot, J. M. Smith, H. C. Hailes and J. M. Ward, Green Chem., 2015, 17, 852–855 RSC.
- D. Baud, N. Tappertzhofen, T. S. Moody, J. M. Ward and H. C. Hailes, Adv. Synth. Catal., 2022, 364, 1564–1572 CrossRef CAS.
- M. F. Villegas-Torres, R. J. Martinez-Torres, A. Cázares-Körner, H. Hailes, F. Baganz and J. M. Ward, Enzyme Microb. Technol., 2015, 81, 23–30 CrossRef CAS PubMed.
- F. Subrizi, L. Benhamou, J. M. Ward, T. D. Sheppard and H. C. Hailes, Angew. Chem., Int. Ed., 2019, 58, 3854–3858 CrossRef CAS PubMed.
- L. Leipold, D. Dobrijevic, J. W. E. Jeffries, M. Bawn, T. S. Moody, J. M. Ward and H. C. Hailes, Green Chem., 2018, 21, 75–86 RSC.
- C. Sayer, M. N. Isupov, A. Westlake and J. A. Littlechild, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 564–576 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- T. D. Machajewski and C. H. Wong, Angew. Chem., Int. Ed., 2000, 39, 1352–1374 CrossRef CAS PubMed.
- H. Andreas, J. G. Luz, C. H. Wong and I. A. Wilson, J. Mol. Biol., 2004, 343, 1019–1034 CrossRef PubMed.
- L. Giger, S. Caner, R. Obexer, P. Kast, D. Baker, N. Ban and D. Hilvert, Nat. Chem. Biol., 2013, 9, 494 CrossRef CAS PubMed.
- Y. C. Shin, H. Yun and H. H. Park, Sci. Rep., 2018, 8, 11454 CrossRef PubMed.
- L. J. Guan, J. Ohtsuka, M. Okai, T. Miyakawa, T. Mase, Y. Zhi, F. Hou, N. Ito, A. Iwasaki, Y. Yasohara and M. A. Tanokura, Sci. Rep., 2015, 5, 10753 CrossRef CAS PubMed.
- Y. Patskovsky, R. Toro, J. Freeman, J. Do, J. M. Sauder, S. K. Burley and S. C. Almo, RCSB Protein Data Bank, https://www.rcsb.org/structure/3I5T, (accessed Feb 04, 2023).
- E. F. Pettersen, T. D. Goddard, C. C. Huang, E. C. Meng, G. S. Couch, T. I. Croll, J. H. Morris and T. E. Ferrin, Protein Sci., 2021, 30, 70–82 CrossRef CAS PubMed.
- T. D. Goddard, C. C. Huang, E. C. Meng, E. F. Pettersen, G. S. Couch, J. H. Morris and T. E. Ferrin, Protein Sci., 2018, 27, 14–25 CrossRef CAS PubMed.
- C. E. Paul, M. Rodríguez-Mata, E. Busto, I. Lavandera, V. Gotor-Fernández, V. Gotor, S. García-Cerrada, J. Mendiola, Ó. de Frutos and I. Collado, Org. Process Res. Dev., 2014, 18, 788–792 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, supporting figures and tables and chemical characterization, dynamic simulations (Movies S1–S3, mpgs). CCDC 2271746 contains the supplementary crystallographic data for 3b. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy01370g |
| This journal is © The Royal Society of Chemistry 2024 |