
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBulky olefin epoxidation under mild conditions over Mo-based oxide catalysts†
Diana M.
Gomes
a,
Xingyu
Yao
b,
Patrícia
Neves
*a,
Nicola
Pinna
 b,
Patrícia A.
Russo
b and
Anabela A.
Valente
*a
b,
Patrícia A.
Russo
b and
Anabela A.
Valente
*a
aDepartment of Chemistry, CICECO-Aveiro Institute of Materials, University of Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal. E-mail: pneves@ua.pt; atav@ua.pt
bDepartment of Chemistry, IRIS Adlershof & The Center for the Science of Materials Berlin, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
First published on 2nd January 2024
Abstract
Molybdenum-based epoxidation catalysts are among the most investigated since the homogeneous Mo-based catalytic process claimed by Halcon International Inc. for liquid phase olefin epoxidation (EPO). While homogeneous Mo-based catalytic technologies reached industrial implementation for olefin EPO, the same does not apply for heterogeneous Mo-based ones, which have not reached industrial implementation (e.g., to meet catalyst productivity and stability requirements). In this work, EPO nanocatalysts consisting of oxides possessing molybdenum and M = Ta, Nb or W were prepared via a simple, versatile methodology. The influence of the material synthesis conditions on the material properties was investigated to meet superior catalytic performances. Promising Mo-based solid catalysts were obtained which promoted the EPO of relatively bulky olefins such as fatty acid methyl esters (methyl oleate, methyl linoleate), using tert-butyl hydroperoxide as oxidant under mild conditions; e.g., the materials Mo(75D)M-0.3 with M = Nb, W (75 at% Mo relative to M, and MoO2Cl2 as precursor), obtained in a fast synthesis of 0.3 h, led to 92–96% epoxide selectivity at 84–95% methyl oleate conversion, at 70 °C. To the best of our knowledge, these are the first Mo,M oxides reported for these reactions.
Introduction
The chemical valorisation of olefins via epoxidation (EPO) routes is of great industrial relevance because epoxides are important intermediates to many end use industries.1,2 In the search for more efficient epoxides production routes, EPO technologies have evolved from hazardous stoichiometric processes to greener catalytic processes, and the latter are shifting from homogeneous to heterogeneous catalysis.3,4 For example, the industrial heterogeneous catalytic EPO process known as hydrogen peroxide–propylene oxide technology (HPPO) uses a titanium silicalite-1 (TS-1) type catalyst, which is a crystalline microporous material possessing an MFI topology consisting of (medium pore) 10-memberred ring channels with ca. 5.5 Å width.1,5–7 While this catalyst is suitable for small reactant molecules, such as light olefins and H2O2 as oxidant, it may be impractical for reactions involving relatively bulky olefins and/or oxidant molecules (e.g., organic hydroperoxides) due to its reduced pore sizes.8 This drawback gains considerable relevance in times where the chemical industry faces challenges to use renewable sources of organic carbon, such as relatively bulky vegetable biomass components9,10 which includes fatty acid esters with varying degrees of unsaturation.11Relatively bulky fatty acid methyl esters (FAMEs) are important industrial chemicals with a growing market (Scheme 1).9,12,13 The EPO of FAMEs gives epoxy fatty acid methyl esters (EFAMEs) with environmentally friendly characteristics such as biodegradability and nontoxicity.14 EFAMEs have a growing global market with a broad applications profile, e.g., solvents, plasticizers, lubricants, biofuels.5,12,15–24 However, heterogeneous catalytic EPO of FAMEs presents challenges, such as the development of highly active, selective and stable catalysts, using catalyst synthesis methodologies which are timesaving and versatile (e.g., possibility of introducing different metals to meet superior performances).14 In the choice of the types of catalytic materials, fully inorganic metal oxides may be preferable to materials possessing organic components, in what concerns thermal and chemical stabilities. Moreover, some monometallic or mixed metal oxides may be prepared via synthesis strategies which only require the metal precursors and an appropriate solvent (advantageous in relation to ordered mesoporous metal/metalloid oxides synthesized using organic templates which need to be subsequently destroyed), with the possibility of offering some control over the crystallinity, size and morphology of the materials.25,26
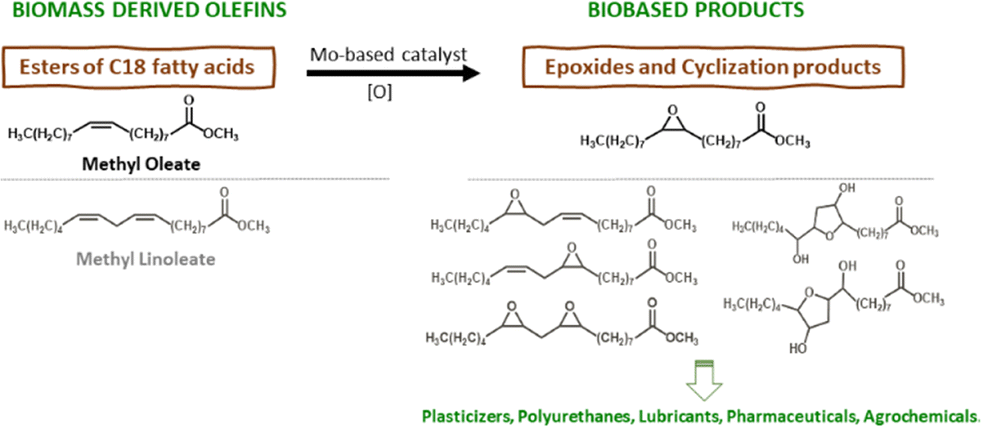 | ||
| Scheme 1 Chemical valorisation of fatty acid methyl esters (FAMEs) obtainable from food/agricultural/industrial waste or surpluses, via catalytic epoxidation (EPO) routes to useful epoxides. | ||
Molybdenum-based EPO catalysts are promising and among the most investigated since the homogeneous Mo-based catalytic EPO process claimed by Halcon International Inc. (USA) in the 1960s.27–30 While homogeneous Mo-based catalytic technologies reached industrial implementation for olefin EPO, the same does not apply for heterogeneous Mo-based ones, which continue of great interest, albeit challenging, e.g. to meet the catalyst productivity and stability requirements.31–33
Besides molybdenum, different metal oxides were reported for olefin EPO, such as niobium, tantalum and tungsten oxides.34 To the best of our knowledge, mixtures of Mo oxide with oxides of Nb, Ta or W were not investigated for liquid phase EPO. According to the literature, precursors of group 5 metals (e.g., niobic acid, Ta2O5) may react with several other precursors, such as molybdenum of group 6 (e.g., (NH4)6Mo7O24·4H2O, MoO3), to form multimetallic oxides,35,36 which may result in improved catalytic performance for different oxidation systems.37 For example, it was reported that the presence of niobium in mixed oxide materials containing molybdenum (which is redox flexible) may enhance the material's stability towards oxidation/reduction. Moreover, niobium and tantalum may confer high mechanical and corrosion resistance to materials.38
In this work, EPO catalysts consisting of Mo and M = Ta, Nb or W oxides were prepared via a simple and versatile non-aqueous sol–gel synthesis, using acetophenone as solvent. According to ECHA, acetophenone presents biodegradability and low aquatic toxicity properties towards the environment.39 These catalytic materials promoted the EPO of relatively bulky olefins. The influence of the type and concentration of the molybdenum precursor and synthesis time on the material properties and catalytic performances were firstly investigated, based on the model reaction of cis-cyclooctene (Cy) using tert-butylhydroperoxide as oxidant, at 70 °C. The catalytic stability was studied by performing consecutive catalytic runs and characterising the recovered solids. The best-performing catalysts were explored for the EPO of the biobased FAMEs methyl oleate and methyl linoleate. The Mo,M oxides were more effective than the monometallic oxides MxOy, and relatively stable Mo,Nb- and Mo,W oxides promoted the conversion of the FAMEs; e.g., Mo(75D)M-0.3 with M = Nb, W (material synthesis time of 0.3 h, using 75 at% Mo relative to M, and MoO2Cl2 as precursor), led to 92–93% epoxide selectivity at 90–95% methyl oleate conversion.
Experimental
Materials
The following reagents and chemicals were purchased from Sigma-Aldrich, unless otherwise stated, and used as received. Synthesis: acetophenone (99.9%), molybdenum(VI) dichloride dioxide, tungsten(VI) chloride (≥99.9%), tantalum(V) chloride (99.8%, Alfa Aesar), and niobium(V) chloride (99.95%, ABCR). Catalysis: cis-cyclooctene (95%, Alfa Aesar), methyl oleate (99%), methyl linoleate (95%, Alfa Aesar), tert-butylhydroperoxide (5.5 M in decane, containing ca. 4% water), anhydrous α,α,α-trifluorotoluene (≥99%), acetone (99.5%, Honeywell, Riedel de Häen), 1,2-dichloroethane (≥99%), methyl decanoate (99%), undecane (>99%), and the crystalline oxides MoO2 (99%), MoO3 (99.5%, VWR), WO3 (99%, Fluka), Nb2O5 (99.99%, Fluka) and Ta2O5 (99.99%, PI-KEM).Synthesis of the catalysts
In a typical synthesis, 20 mL of acetophenone and a total of 0.5 mmol of the metal precursors (TaCl5, NbCl5, WCl6, and/or MoCl5 or MoO2Cl2) in the desired molar proportions, were added to a microwave glass vial in a glovebox. The vial was heated in an Anton Paar Monowave 300 microwave reactor, at 220 °C for 20 min. The solid product was separated by centrifugation, washed four times with 20 mL of acetone and ethanol, and dried at 65 °C for 12 h. The 24 h reactions (for selected materials) were carried out in a similar fashion, albeit using a Teflon-cup-lined stainless steel autoclave.25,40 The Mo,M oxide materials are denoted Mo(xD)M-t or Mo(xP)M-t where x is the normalized at% of Mo relative to M of the synthesis mixture (i.e., x = 25, 50 and 75 for a ratio Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M of 25:75, 50:50 and 75:25, respectively), t is the material synthesis time, and D corresponds to the use of MoO2Cl2 as precursor and P corresponds to the use of MoCl5 as precursor.
:M of 25:75, 50:50 and 75:25, respectively), t is the material synthesis time, and D corresponds to the use of MoO2Cl2 as precursor and P corresponds to the use of MoCl5 as precursor.
Characterization of the catalysts
Powder X-ray diffraction (PXRD) data were collected on an Empyrean PANalytical diffractometer (Cu-Kα X-radiation, λ = 1.54060 Å) equipped with a spinning flat sample holder and a PIXcel 1D detector set at 240 mm from the sample, in a Bragg–Brentano para-focusing optics configuration (45 kV, 40 mA) at ambient temperature. Samples were prepared in a spinning flat plate sample holder and step-scanned from 3 to 70° (2θ) in 0.026° 2θ steps with a counting time of 90 s per step.Scanning electron microscopy (SEM) images and EDS analyses were obtained on a Hitachi SU-70 SEM microscope equipped with a Bruker Quantax 400 detector operating at 15 kV. Samples were prepared by deposition on aluminum sample holders followed by carbon coating using an Emitech K 950 carbon evaporator. Transmission electron microscopy (TEM), high resolution TEM (HRTEM), high angle annular dark field scanning transmission electron microscopy (HAADF-STEM), and elemental mapping analyses were carried out on a FEI Talos F200S scanning/transmission electron microscope (S/TEM) operated at 200 kV, and a Hitachi HD2700 microscope equipped with a Bruker Quantax SVE 6 EDS detector operating at 80–200 kV.
The textural properties were determined from the N2 sorption isotherms at −196 °C, which were measured using a Quantachrome instrument (automated gas sorption data using Autosorb IQ2, Quantachrome Instruments). The sample was pre-treated at 170 °C for 3 h, under vacuum (<4 × 10−3 bar). The specific surface area was calculated using the Brunauer, Emmett, Teller equation (SBET).
ICP-OES analyses (for Mo, W, Nb and Ta) were performed at the Central Analysis Laboratory (University of Aveiro); the measurements were carried out on a Horiba Jobin Yvon Activa M spectrometer (detection limit of ca. 20 μg dm−3; experimental range of error of ca. 5%). Prior to analysis, 10 mg of solid sample was digested using 0.5 mL HF and 0.5 mL HNO3, and microwave heating at 180 °C.
Attenuated total reflectance (ATR) FT-IR spectra were measured on a Bruker Tensor 27 spectrophotometer equipped with a Specac® Golden Gate Mk II ATR accessory having a diamond top plate and KRS-5 focusing lenses (resolution 4 cm−1, 256 scans). Diffuse reflectance (DR) UV-vis spectra were recorded using a JASCO V-780 spectrophotometer equipped with a JASCO ISV-469 integrating sphere coated with barium sulfate, with light detection by a built-in photomultiplier tube attached to the base of the sphere. The spectra were collected in reflectance mode with a wavelength scan speed of 200 nm min−1, step size of 0.5 nm, and a slit width of 2.0 nm.
Catalytic tests
The catalytic reactions were carried out in borosilicate reactors equipped with a Teflon valve for sampling and a magnetic stirrer. Initially, the catalyst (5.6 gcat per mole of olefin), substrate and solvent (α,α,α-trifluorotoluene (TFT)) were added to the reactor, which was then immersed in a temperature-controlled oil bath at 70 °C, under stirring (1000 rpm, optimized to avoid mass transfer limitations). After 10 min, the preheated oxidant, namely tert-butylhydroperoxide (TBHP), was added to the reactor, and this moment was taken as the initial instant of the catalytic reaction. For the model reaction of cis-cyclooctene (Cy), the initial reaction conditions were ca. 1 M Cy and mole ratio TBHP:Cy = 1.5. For the biobased FAMEs, namely methyl oleate (MeOle) and methyl linoleate (MeLin), the initial reaction conditions were ca. 0.5 M FAME, and TBHP:FAME = 1.5 for MeOle or 2.5 for MeLin.
The evolution of the reactions was monitored by analyzing freshly prepared samples by gas chromatography (GC), using a Varian 450 GC instrument equipped with a BR-5 capillary column (30 m × 0.25 mm × 0.25 μm) and an FID detector with H2 as carrier gas. The quantifications of the reactants and products were based on calibrations (the internal standard was undecane for Cy, and methyl decanoate for bioolefins). The initial activity (mmol gcat−1 h−1) was calculated based on olefin conversion at 1 h reaction. The reaction products were identified by GC-MS (GC MS QP2010 Ultra Shimadzu), using He as the carrier gas; the product identifications were based on commercial mass spectrometry databases (Wiley229, NIST14, NIST Chemistry WebBook, MAINLIB) and mass spectra similarities. The products' mass spectra were reported previously.41,42
The catalyst stability was evaluated by reusing the recovered solids in consecutive batch runs, keeping constant the initial mass ratio of catalyst:Cy:TBHP between runs. After each run, the solids were separated from the reaction mixture by centrifugation (3500 rpm), thoroughly washed with acetone, dried overnight under air atmosphere, and finally vacuum-dried (ca. 0.1 bar) at 60 °C for 1 h.
Results and discussion
Characterization of the materials
The mole ratios of the materials (Mo/M), as well as those of the respective synthesis (Syn) mixtures ((Mo/M)Syn = 0.3, 1 or 3 for x = 25, 50 and 75, respectively) are indicated in Fig. S1.† In general, Mo(xD)M-0.3 and Mo(xP)M-0.3 possessed lower Mo/M than the respective (Mo/M)Syn, indicating that not all metal content of the synthesis mixture was incorporated in these materials. Increasing the synthesis time from 0.3 h to 24 h led to higher Mo/M; e.g., the Mo/M ratios of Mo(50D)M-0.3 versus Mo(50D)M-24 (both synthesized using (Mo/M)Syn = 1) were in the ranges 0.10–0.23 and 0.54–0.86, respectively.
On the other hand, the influence of the type of Mo precursor (D or P) was studied for the Mo,M oxides with M = W, Nb and x = 25, 50, or with M = Ta and x = 50. The D precursor led to higher Mo/M than the P one, e.g., Mo(50D)W-0.3 possessed approximately double the Mo/M ratio of Mo(50P)W-0.3 (0.23 and 0.12, respectively). Hence, MoO2Cl2 seemed more favorable for introducing molybdenum in the materials.
ICP-OES gave roughly comparable results to EDS (please see Table S1 and Fig. S2,† for selected materials).
 | ||
| Fig. 1 PXRD patterns of the Mo,M oxides with M = Ta (A), Nb (B), or W (C): (b) Mo(50D)M-24, (c) Mo(75D)M-0.3, (d) Mo(50D)M-0.3, (e) Mo(50P)M-0.3, (f) Mo(25D)M-0.3, (g) Mo(25P)M-0.3 (excluding for M = Ta). For comparison, bulk (a) MoO2 (A–C) and (h) MxOy (M = Ta (A), Nb (B), W (C)). | ||
The PXRD pattern of monometallic MoO2 exhibits very broad reflections, which are indicative of the presence of very small crystallites. A 10-fold amplification of the diffractogram of MoO2 showed very weak peaks centered at ca. 37, 54 and 66° 2θ (Fig. S3†), assignable to the (100), (102) and (110) crystal planes, respectively, of the hexagonal phase of molybdenum(IV) dioxide (ICDD PDF card no. 00-050-0739).43 Koziej et al.25 reported the solvothermal synthesis of MoO2 nanoparticles possessing hexagonal crystal structure, using MoO2Cl2 as precursor and a solvent mixture of acetophenone and benzyl alcohol at 200 °C for 10 min (in the present study, solely acetophenone was used as solvent at 220 °C). Commercial molybdenum(VI) trioxide (MoO3-com) possessed an orthorhombic crystal structure (ICDD PDF card no. 01-074-7909).
The PXRD patterns of Ta2O5, Nb2O5, Mo,Nb and Mo,Ta oxides resultant from solvothermal syntheses with a duration of t = 0.3 h, also show broad reflections indicative of small crystallites (Fig. 1A and B).
For the Mo,M oxides with M = Ta, Nb (x = 50), increasing the synthesis time from 0.3 h to 24 h led to the appearance of narrow reflections from the hexagonal MoO2 phase (ca. 36.7, 38, 41.5, 53.8, 66° 2θ; ICDD PDF card no. 00-050-0739). This suggests that increasing the synthesis time resulted in the growth of the MoO2 particles, but not of those of Ta2O5 or Nb2O5. It is worth noting that the presence of MoO2 in the Mo,M oxides indicates that they may possess reduced Mo(IV) sites.
The PXRD pattern of the W oxide nanoparticles showed a main peak at ca. 24° 2θ, and additional weak broad peaks centered at ca. 27.4, 36, 47.6 and (very weak) 56° 2θ, assignable to WO2.72 with monoclinic crystal structure (ICDD PDF card no. 04-005-4539) (Fig. 1C).
The Mo,W oxides Mo(xP)W-0.3 (x = 25, 50) and Mo(25D)W-0.3 exhibited comparable PXRD patterns to WO2.72. For the remaining materials (with greater x and/or t values, using the D precursor), namely Mo(50D)W-0.3, Mo(75D)W-0.3 and Mo(50D)W-24, the peaks become more intense and narrower, suggesting particle growth. Besides WO2.72, these materials seem to possess the orthorhombic phase of (W1−xMox)O3 (x ≤ 1; based on ICDD PDF card numbers 00-054-1012 and 00-046-1048); peaks at ca. 14, 18.2, 23, 27, 28.2 (main peak), 36.7, 47.1, 49.3, 50, 53.7, 55.7° 2θ. A longer synthesis time of 24 h (Mo(50D)W-24) led to enhanced crystallinity.
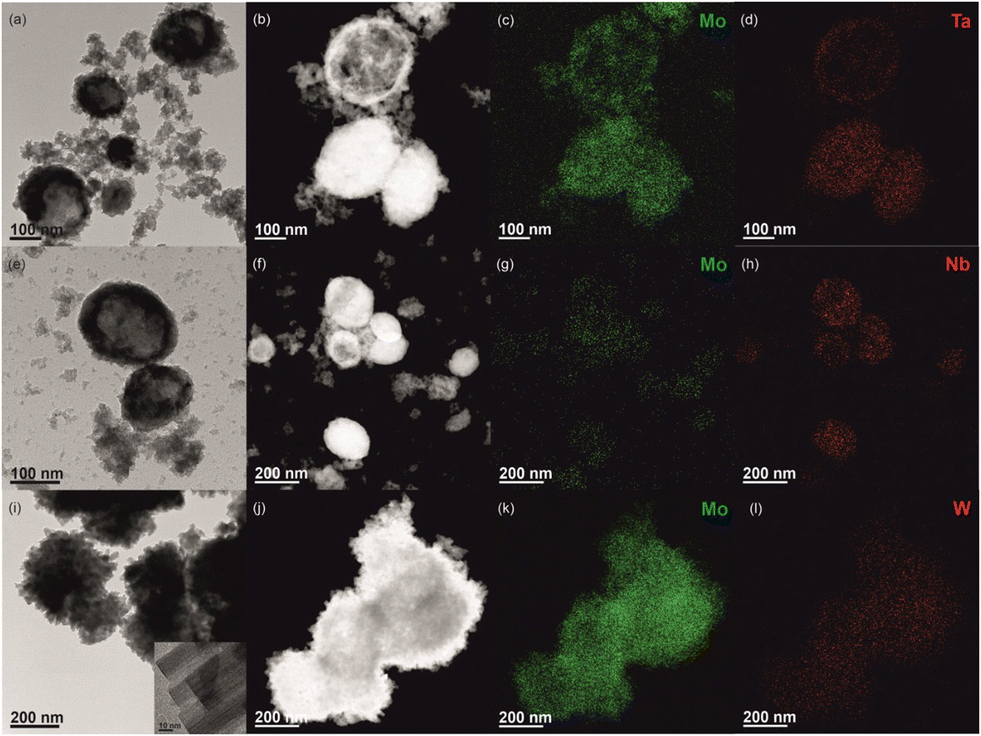
The morphology was studied by electron microscopy for selected materials, namely Mo(75D)M-0.3 and Mo(50D)M-24 with M = Ta, Nb or W, which were synthesised using the highest (Mo/M)Syn of 3 or longer synthesis time of 24 h, respectively (Fig. 2 and 3). The TEM, HAADF-STEM, and elemental maps showed that the materials consisted of agglomerates of intimately mixed nanoparticles of different morphology. For Mo,M oxides with t = 0.3 h, the Ta and Nb oxide components consist of irregular platelet-like nanoparticles with the size of ca. 19 nm and 3–5 nm, respectively; the W oxide component consists of agglomerated nanobelts of ca. 7–12 nm width; and the Mo oxide component is made of relatively large layered-like particles of ca. 57 nm. For t = 24 h, the Ta and Nb oxide components tend to form spherical agglomerates (Fig. 3(a, b, e and f)) of particles with sizes about 25 nm and 5–7 nm, respectively; the width of the W oxide nanobelts increases to ca. 25 nm, as well as their crystallinity (Fig. 3(i)); and the Mo oxide component becomes made of irregularly shaped interconnected particles of about 23 nm size. In general, elemental mapping suggested that the different particles are well mixed and in close proximity at the nanoscale (Fig. 2 and 3 and S4†). The pure MoO2 sample consists of large sphere-like particles in the size range 350 to 500 nm.
 | ||
| Fig. 2 TEM (a, e and i), HAADF-STEM and elemental maps (b–d, f–h and j–l) of Mo(75D)M-0.3 with M = Ta (a–d), Nb (e–h) or W (i–l). | ||
 | ||
| Fig. 3 TEM (a, e and i), HAADF-STEM and elemental maps (b–d, f–h and j–l) of Mo(50D)M-24 with M = Ta (a–d), Nb (e–h) or W (i–l). The inset in (i) shows a HRTEM image of the WO2.72 nanobelts. | ||
Regarding the type of Mo precursor, for the materials Mo(xD)M-0.3 with the same M and synthesized using the D precursor, SBET decreased with increasing Mo/M ratio of the materials (Fig. S5(a)†), whereas the opposite was verified for the related materials synthesized using the P precursor (Fig. S5(b)†). Hence, SBET does not solely depend on the type of Mo precursor (or Mo/M) and may be due to interplay of different factors. The type of M metal may also influence SBET; e.g., Mo(50D)M-24 possessing (different) M = Ta and Nb, and synthesized using the same D precursor, possessed somewhat comparable PXRD features (Fig. 1) and chemical compositions (Table S1†), albeit considerably different SBET (54 and 402 m2 g−1, M = Ta and Nb, respectively). Possible differences in the size/density/structure of the nanoparticle ensembles or agglomerates,44,45 may at least partly influence the interparticle void space and total specific surface area. For example, for the Mo(50D)M-0.3 family, SBET (Table S2†) increased with decreasing particle size: M = Ta (19 nm width; 98 m2 g−1); M = W (7–12 nm; 118 m2 g−1); M = Nb (3–5 nm; 254 m2 g−1). On the other hand, the morphology also seems to affect SBET; e.g., particle sizes were slightly larger for Mo(50D)Nb-24 than (morphologically different) Mo(50D)Nb-0.3, but the former possessed higher SBET (402 m2 g−1versus 254 m2 g−1).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, and a broad band centred at ca. 660 cm−1 which may be associated with polynuclear Mo–O–Mo groups.46–48
O groups, and a broad band centred at ca. 660 cm−1 which may be associated with polynuclear Mo–O–Mo groups.46–48
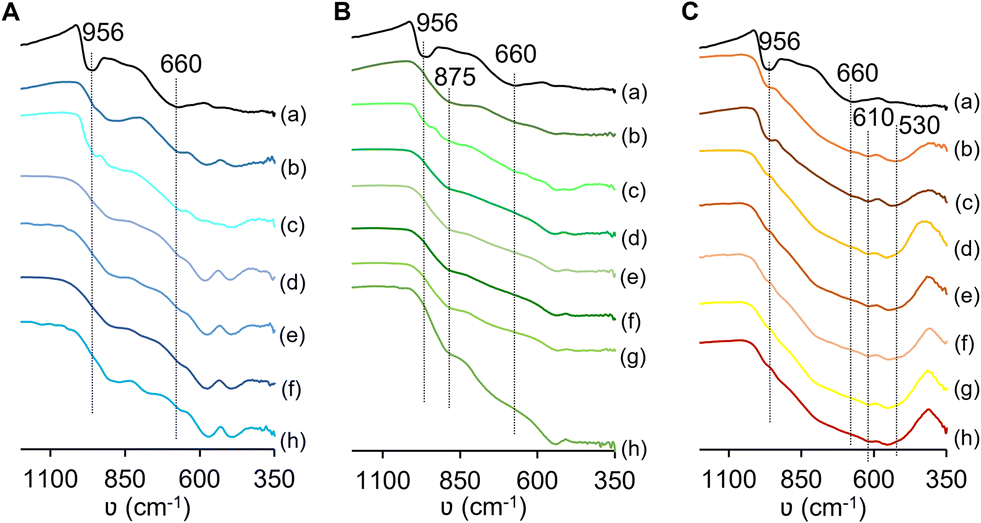 | ||
| Fig. 4 ATR FT-IR spectra of the materials possessing M = Ta (A), Nb (B), or W (C) and, for comparison, (a) MoO2 is included in the three plots; (b) Mo(50D)M-24, (c) Mo(75D)M-0.3, (d) Mo(50D)M-0.3, (e) Mo(50P)M-0.3, (f) Mo(25D)M-0.3, (g) Mo(25P)M-0.3 (excluding for M = Ta) and (h) MxOy (M = Ta (A), Nb (B), W (C)). | ||
Bulk Nb2O5 exhibited a poorly defined spectrum; a broad band centred at ca. 875 cm−1 may be due to stretching vibrations of NbO groups (Fig. 4B).49 Ta2O5 exhibited several broad bands below 1000 cm−1 (Fig. 4A), which may be due to vibrational modes of Ta–O–Ta and Ta–O type groups with different bond lengths/angles/chemical environments (band assignments are not consensual in the literature).50–56 A comparative study for the Mo,M oxides with M = Ta, Nb, indicated that an absorption band at ca. 956 cm−1 (assignable to ν(MoO)) was distinguishable for Mo(75D)M-0.3 (M = Ta, Nb) and Mo(50D)Ta-24 and hardly distinguishable for the remaining materials (Fig. 4(A and B)). A broad band centred at ca. 660 cm−1 (assignable to polynuclear Mo–O–Mo) was somewhat distinguishable for Mo,Ta oxides, and not clearly distinguishable for the Mo,Nb-mixed oxides (possibly due to overlapping of broad bands).
Regarding the tungsten-based materials, WO2.72 exhibited poorly defined bands at ca. 956, 610 and 530 cm−1 (Fig. 4(C)). The first band may be due to terminal WO groups57 and the others to polynuclear W–O–W groups possessing different bond lengths/angles/chemical environments.57,58 Similarly, the Mo,W oxides exhibited a band at ca. 956 cm−1 assignable to ν(MO) (M = Mo or W), and bands at ca. 610 and 530 cm−1. The band at ca. 660 cm−1, associated with polynuclear Mo–O–Mo groups (verified for monometallic MoO2), was not clearly distinguishable in the spectra of the Mo,W oxides.
The M metals belong to group 5 (Nb, Ta) and 6 (W), and were in their highest oxidation states in the corresponding synthesis precursors (Nb(V), Ta(V), W(VI)). The metals in these oxidation states may be present in the Mo,M oxides (e.g., group 5 metals may be less prone to changes in oxidation state). On the other hand, molybdenum (group 6) may have different oxidation states. DR UV-vis spectroscopy may give insights into the coordination numbers and oxidation states of the metal sites. Fig. S6† shows the DR UV-vis spectra of the materials MxOy, MoO2 and selected Mo,M oxides, namely Mo(75D)M-0.3 and Mo(50D)M-24 (M = Ta, Nb, W). In general, the mixed metal oxides exhibited bands centred at ca. 210–215, 240–250 and 310–320 nm. Rigorous assignments of these bands are difficult, partly due to possible superimposable contributions from M and/or Mo containing groups. Some literature studies suggested that bands below ca. 250 nm may be due to isolated molybdenum sites,59–62 albeit (polymeric) MoO2 also exhibited bands at ca. 215 and 240 nm, which were also verified for Ta2O5, Nb2O5 and WO2.72. Different literature studies suggested that bands below ca. 250 nm may be partly due to distorted four-coordinated {TO4} sites (T = Mo or M);59–61,63,64,67–71 In the cases of MoO2 and Mo,M oxides (for which PXRD identified the presence of MoO2), the bands below 250 nm may be associated with reduced Mo(IV) groups. The relatively low oxidation state of molybdenum in MoO2 (Mo(IV)) results in an increasing absorption above 350 nm, which was also verified for crystalline (commercial) MoO2-com (together with very weak absorptions at ca. 210 and 240 nm, Fig. S6D†) and in agreement with literature data.60,70–73 The presence of Mo(IV) sites in the Mo,M oxide nanomaterials was further supported by the fact that the very dark color of MoO2 and MoO2-com was also verified for the Mo,M oxides, especially Mo(75D)M-0.3 and Mo(50D)M-24.
The nanomaterials Nb2O5 and WO2.72 exhibited a band centered at ca. 320 nm which may be partly due to six-coordinated metal sites.63,65,67,74 The Mo,M oxides (M = Ta, Nb, W) also exhibited a band at ca. 310–330 nm, which at least for the Mo,Ta oxides may be attributed to molybdenum containing groups (e.g., six-coordinated Mo(VI) sites or Mo–O–Mo groups),59–62,70,75,76 because Ta2O5 did not exhibit bands in this spectral range. In relation to the defective nanomaterials, crystalline (commercial) MoO3-com (Mo(VI)), Nb2O5-com (Nb(V)) and WO3-com (W(VI)) exhibited a higher wavelength band (ca. 355 nm) which may be associated with their infinitely (long range ordered) stacked octahedra of Mo(VI), Nb(V) and W(VI), respectively (Fig. S6-D†).71,75,77–79 A lower wavelength band (ca. 260 nm) was predominant for crystalline Ta2O5-com which may be associated with its distinct coordination features (shared distorted polyhedra of Ta(V), i.e., {TaO6} octahedra and {TaO7} pentagonal bipyramides).66,69 In summary, the above results suggested that decreasing the particle sizes down to the nanoscale influences the surface chemistry, and the nanomaterials may possess molybdenum sites with different oxidation states (e.g., Mo(IV), Mo(VI)).
Catalytic studies
TFT was chosen as solvent since it is readily available, relatively inexpensive, and has good capacity to dissolve a wide range of organic compounds.80 Its relatively high boiling point (ca. 102 °C) makes it more appealing (e.g., avoiding atmospheric emissions) than other more volatile halogenated solvents. Additionally, its poor coordinating properties avoids competitive reactions with reactant molecules in the coordination to active metal species, which, together with the remaining aspects, has contributed to its successful use as solvent in several catalytic epoxidation systems.81,82
Cyclooctene oxide (CyO) was the sole product (100% selectivity), formed in up to 100% yield within 24 h, at 70 °C. Blank tests carried out without catalyst or without oxidant, gave negligible olefin conversion.
The bulk MxOy materials led to sluggish results; conversion at 24 h was 25%, 7% and 4% for M = Nb, Ta and W, respectively. On the other hand, MoO2 led to 100% conversion at 1 h, suggesting that molybdenum plays an important catalytic role. According to the literature, the performance of molybdenum oxides may strongly depend on the type of crystalline structure.83 For example, commercial orthorhombic MoO3-com (Fig. S7†) was far less active (conversion at 1 h/4 h/24 h was 33%/79%/100%) than MoO2. However, MoO2 presents stability issues; the hexagonal crystal structure may suffer phase transition;25,84 and the epoxidation reaction in the presence of MoO2 led to yellow-coloured liquid phase, characteristic of soluble oxidized molybdenum species. This was further confirmed by a catalyst filtration test, which indicated a major homogeneous catalytic contribution (please see the ESI† for details).
Based on the mechanistic studies reported in the literature for Mo-catalysed EPO of olefins with hydroperoxide oxidants (ROOH), the active oxidizing species may be formed via a heterolytic mechanism involving the coordination of the oxidant (ROOH) to a molybdenum centre.85–88 This leads to the formation of a moiety of the type {Mo-OOR} responsible for the oxygen atom transfer step to the olefin, giving the epoxide product (plus the coproduct of TBHP, namely tert-butanol). These mechanistic considerations may also apply for W-catalysed olefin epoxidation,89–92 and other metals (e.g., active oxidizing species possessing the moiety {M-OOR} were reported for Ta-containing catalysts in olefin epoxidation93). Hence, one cannot exclude the possible roles of Mo and M sites of the Mo,M oxides, as discussed below.
Concerning the type of Mo precursor (keeping constant the type of M), the Mo,M oxides synthesized using MoO2Cl2 (D) were, in general, more active than those synthesized using MoCl5 (P), under similar conditions (Fig. 5(a–c)); exceptionally, Mo(50D)Ta-0.3 and Mo(50P)Ta-0.3 possessed somewhat comparable activities.
 | ||
| Fig. 5 Influence of the material synthesis conditions on the catalytic performances: type of Mo precursor ((a) Mo,Ta oxides, (b) Mo,Nb oxides, (c) Mo,W oxides); relative amount of Mo (x) in the material synthesis mixture ((d) Mo,Ta oxides; (e) Mo,Nb oxides; (f) Mo,W oxides); and type of M metal (Mo(xD)M-0.3 with x = 25 (g), 50 (h) or 75 (i)). Reaction conditions: mole ratio TBHP:Cy = 1.5, 5.6 gcat molCy−1, 70 °C. Each set of three bars indicates Cy conversion at 1 h (green), 4 h (red) or 24 h (blue) reaction. Epoxide (CyO) selectivity was always 100%. | ||
The influence of x (or (Mo/M)Syn) was investigated for the two families of materials Mo(xP)M-0.3 and Mo(xD)M-0.3, keeping constant the type of Mo precursor, type of M metal and synthesis time (t = 0.3 h) (Fig. 5(d–f) and S8†). In general, olefin conversion (Fig. 5(d–f)) and initial activity (Fig. S8†) increased with increasing x. For example, for the Mo(xD)M-0.3 family (Fig. S8(a)†), initial activity for M = Ta increased from 18 mmolCy gcat−1 h−1 (x = 25) to 157 mmolCy gcat−1 h−1 (x = 75); for M = Nb, from 25 mmolCy gcat−1 h−1 (x = 25) to 157 mmolCy gcat−1 h−1 (x = 75); and for M = W, from 27 mmolCy gcat−1 h−1 (x = 25) to 157 mmolCy gcat−1 h−1 (x = 75). The highest conversion at 1 h was 87%, reached for the materials with x = 75 (Fig. 5(d–f)).
On the other hand, increasing the synthesis time from 0.3 h to 24 h (i.e., Mo(50D)M-0.3 versus Mo(50D)M-0.24 with the same M) favoured the epoxidation reaction kinetics (Fig. 6). The differences in reaction kinetics were very pronounced, especially for M = Ta (for the material synthesis time of t = 0.3 h and 24 h, the conversion was 14 and 100%, respectively, at 4 h). Initial activity increased from 20 mmolCy gcat−1 h−1 (t = 0.3 h) to 175 mmolCy gcat−1 h−1 (t = 24 h) for M = Ta; from 27 mmolCy gcat−1 h−1 (t = 0.3 h) to 110 mmolCy gcat−1 h−1 (t = 24 h) for M = Nb; and from 52 mmolCy gcat−1 h−1 (t = 0.3 h) to 103 mmolCy gcat−1 h−1 (t = 24 h) for M = W (Fig. S9†).
 | ||
| Fig. 6 Influence of the material synthesis time (0.3 h or 24 h) on the performances of the Mo(50D)M catalysts. Reaction conditions: mole ratio TBHP:Cy = 1.5, 5.6 gcat molCy−1, 70 °C. Each set of three bars indicates Cy conversion at 1 h (green), 4 h (red) or 24 h (blue) reaction. Epoxide (CyO) selectivity was always 100%. | ||
In general, based on the characterisation studies for the materials with the same M metal, increasing x and t led to higher Mo/M of the materials formed, which may partly explain the faster catalytic reaction kinetics, as shown in Fig. 7, i.e., conversion somewhat increased with Mo/M, especially for Mo/M > 0.1.
 | ||
| Fig. 7 Dependency of Cy conversion at 1 h (Δ), 4 h (◊) or 24 h (○) reaction, on the Mo/M ratio (EDS) of the Mo,M oxides with M = Ta (a), Nb (b) or W (c). Reaction conditions: mole ratio TBHP:Cy = 1.5, 5.6 gcat molCy−1, 70 °C. Epoxide (CyO) selectivity was always 100%. | ||
Concerning the influence of the type of M metal, a comparative study for the materials Mo(xD)M-0.3 with the same x, indicated differences in catalytic activities. Specifically, for x = 25 or 50, olefin conversion (Fig. 5(g–i)) and initial activity (Fig. S10†) increased in the order Ta < Nb < W. For x = 75, the differences in catalytic results were not pronounced because of the very high activity of these materials (ca. 87% conversion at 1 h, and initial activity = 157 mmolCy gcat−1 h−1 for the three materials Mo(75D)M-0.3). These results did not correlate directly with SBET which increased in the order Ta (78 m2 g−1) < W (104 m2 g−1) < Nb (222 m2 g−1) (Table S2†).
In order to gain further insight into the influence of the type of M, a comparative study was carried out for the Mo(50D)M-0.3 materials, keeping constant the initial mole ratio Mo:olefin of the catalytic reaction mixture (Fig. S11†). If all the molybdenum sites of the different catalysts were equivalent (i.e., possessing equal intrinsic activity), one could expect the reaction rate to be similar because the initial mole ratio Mo:olefin was kept constant in these catalytic tests. However, this was not the case, i.e., the initial activity followed the order Nb < Ta < W and the conversion at 24 h followed the order Ta < Nb < W, indicating differences in reaction kinetics, and suggesting that not all Mo sites were equivalent. The characterisation studies of the materials with different M, indicated structural differences, e.g., Mo(50D)W-0.3 is crystalline, whereas the corresponding materials with M = Ta and Nb did not exhibit distinguishable crystalline domains (Fig. 1). Additionally, the characterisation studies suggested that the nanomaterials may possess different types of Mo sites (non-equivalent sites), which may have different intrinsic activities. Moreover, the coordination environment and/or redox properties of M species may be different; e.g., according to the literature, Ta and Nb with similar coordination spheres may possess significantly different redox properties.94 From the characterization studies of the MoM oxides, one cannot exclude the possible existence of proximal Mo and M species. Accordingly, the M species could influence the electronic/structural features of vicinal Mo species, and consequently the intrinsic activities. Overall, the catalytic performances seem to be due to a complex interplay of different material properties, such as structure, surface chemistry and composition.
:TBHP:catalyst, and characterized after use. In general, the olefin conversion remained roughly comparable in consecutive runs, excluding the materials with M = Ta which suffered partial drop of activity (e.g., for Mo(50D)Ta-24, conversion at 4 h decreased from 100% in run 1 to 75% in run 3; Fig. S12(a)†).
The characterisation studies of the used Mo,M catalysts indicated that, in general, their structural (PXRD, Fig. S13†) and morphological features (electron microscopy, Fig. S14–S17†), metal distributions (elemental mappings, Fig. S14 and S15†), chemical compositions (Mo:M ratio, Fig. S18†) and surface chemistry (ATR FT-IR (Fig. S19†) and DR UV-vis (Fig. S20†) spectroscopy) were essentially preserved. Exceptionally, the MoO2 hexagonal phase remained present in Mo(50D)Nb-24-used, but not in Mo(50D)Ta-24-used (Fig. S13†), which may be due to an interplay of several factors such as stability issues. MoO2 nanoparticles may be susceptible to partial oxidation of interfacial Mo(IV) sites,95–97 and small variations in the molybdenum valence may affect the physical properties.83 Accordingly, differences in redox properties and coordination features of Ta and Nb sites (discussed in the literature for different coordination compounds of these metals94,98) may have implications on the stability of immobilized MoO2.
C double bond)), parallels that reported in the literature for Nb-silica catalysts (although for the latter, olefin conversion tended to a plateau after ca. 1 h reaction at 90 °C due to partial catalytic deactivation99). Since MeLin is a polyunsaturated olefin, its conversion may be faster (two CC double bonds are available) than of the monoene MeOle. Moreover, it was proposed in the literature that polyunsaturated FAMEs may be more reactive than monounsaturated ones, due to possible electron-donating effects of adjacent allyl groups (H2CCH–CH2).100
 | ||
| Fig. 8 Epoxidation of methyl oleate (MeOle) with TBHP, at 70 °C, in the presence of Mo(75D)M-0.3 with M = Ta (○), M = Nb (Δ) and M = W (□): (a) kinetic profiles and (b) dependency of product selectivity on conversion for the monoepoxide. Reaction conditions: mole ratio TBHP:FAME = 1.5, 5.6 gcat molFAME−1, 70 °C. | ||
 | ||
| Fig. 9 (a) Kinetic profiles of methyl linoleate (MeLin) epoxidation, in the presence of Mo(75D)M-0.3 with M = Ta (○), M = Nb (Δ) and M = W (□), and (b–d) dependency of selectivity on conversion (monoepoxides (○), diepoxides (□) and furan type products (Δ)). Reaction conditions: mole ratio TBHP:FAME = 2.5, 5.6 gcat molFAME−1, 70 °C. | ||
| M | Reaction conditionsa | Conv.b (%) | Productc | Select.d (%) | Yieldd (%) | |
|---|---|---|---|---|---|---|
| FAME | t/h | |||||
|
a Reaction conditions: mole ratio TBHP:FAME = 1.5 for MeOle or 2.5 for MeLin, 5.6 gcat molFAME−1, 70 °C.
b Olefin conversion at the specified reaction time (t).
c MeOleEp = methyl 9,10-epoxyoctadecanoate, MeLinEp = methyl 9,10-epoxy-12-octadecenoate and methyl 12,13-epoxy-9-octadecenoate, MeLinDiEp = methyl 9,10-12,13-diepoxyoctadecanoate, CFur = furan type cyclic products.
d Selectivity or yield of products, at 6 h/24 h.
e Reaction performed at 90 °C.
|
||||||
| Ta | MeOle | 6 | 67 | MeOleEp | 98 | 66 |
| 24 | 84 | 96 | 81 | |||
| Nb | 6 | 83 | MeOleEp | 94 | 78 | |
| 24 | 95 | 92 | 87 | |||
| W | 6 | 78 | MeOleEp | 100 | 78 | |
| 24 | 90 | 93 | 84 | |||
| Ta | MeLin | 6 | 86 | MeLinEp | 70 | 60 |
| MeLinDiEp | 30 | 26 | ||||
| CFur | 0 | 0 | ||||
| 24 | 97 | MeLinEp | 40 | 39 | ||
| MeLinDiEp | 45 | 44 | ||||
| CFur | 15 | 15 | ||||
| Nb | 6 | 93 | MeLinEp | 45 | 42 | |
| MeLinDiEp | 35 | 33 | ||||
| CFur | 20 | 19 | ||||
| 24 | 100 | MeLinEp | 18 | 18 | ||
| MeLinDiEp | 30 | 30 | ||||
| CFur | 52 | 52 | ||||
| W | 6 | 91 | MeLinEp | 64 | 58 | |
| MeLinDiEp | 35 | 32 | ||||
| CFur | 1 | 1 | ||||
| 24 | 97 | MeLinEp | 40 | 39 | ||
| MeLinDiEp | 48 | 47 | ||||
| CFur | 12 | 12 | ||||
| Nbe | 6 | 93 | MeLinEp | 20e | 20e | |
| MeLinDiEp | 61e | 61e | ||||
| CFur | 19e | 19e | ||||
| 24 | 100 | MeLinEp | 8e | 8e | ||
| MeLinDiEp | 64e | 64e | ||||
| CFur | 28e | 28e | ||||
For each FAME, the initial catalytic activity (mmol gcat−1 h−1) followed the order Nb (94 mmolMeOle gcat−1 h−1; 132 mmolMeLin gcat−1 h−1) > W (88 mmolMeOle gcat−1 h−1; 119 mmolMeLin gcat−1 h−1) > Ta (69 mmolMeOle gcat−1 h−1; 96 mmolMeLin gcat−1 h−1). These results somewhat correlated with the Mo/M ratio which decreased in the same order (Fig. 8 and 9, Table S1†), and with decreasing specific surface area (222 m2 g−1 (M = Nb) > 104 m2 g−1 (M = W) > 78 m2 g−1 (M = Ta), Table S2†). However, the initial activities expressed per unit of surface area (mmol m−2 h−1) followed a different trend: Nb (0.42 mmolMeOle m−2 h−1; 0.59 mmolMeLin m−2 h−1) < W (0.85 mmolMeOle m−2 h−1; 1.14 mmolMeLin m−2 h−1) ≅ Ta (0.88 mmolMeOle m−2; 1.23 mmolMeLin m−2 h−1). Possibly, the catalytic activity may be partly influenced by the density of active sites.
The MeOle reaction in the presence of Mo(75D)M-0.3 gave mainly the epoxide product, methyl 9,10-epoxyoctadecanoate (MeOleEp) (Scheme 1), formed in 96% selectivity at 84% conversion for M = Ta, and 92–93% selectivity at 90–95% conversion for M = Nb and W (Fig. 8, Table 1).
The reaction of MeLin gave mono- and diepoxides (Fig. 9, Scheme 2). The monoepoxides were methyl 12,13-epoxy-9Z-octadecanoate and methyl 9,10-epoxy-12Z-octadecanoate (MeLinEp), and the diepoxides were diastereoisomers of methyl 9,10,12,13-diepoxy-octadecanoate (MeLinDiEp). The selectivity to the total epoxides (MeLinEp plus MeLinDiEp) at 6 h/24 h reaction was 99%/88% at 91%/97% conversion for M = W; 100%/85% at 86%/97% conversion for M = Ta; and 80%/48% at 93%/100% conversion for M = Nb.
 | ||
| Scheme 2 Conversion of methyl linoleate (MeLin) in the presence of Mo(75D)M-0.3 (M = W, Nb and Ta), leading to epoxides and furan type products. | ||
For the three catalysts with MeLin, the monoepoxides were formed in approximately equimolar amounts (negligible regioselectivity), suggesting that the two CC double bonds are similarly reactive. The kinetic profiles suggest that the monoepoxides are intermediates of the conversion of MeLin to diepoxides (Scheme 2). The latter are converted to (cyclic) furan type products (CFur), namely methyl 10,13-dihydroxy-9,12-epoxy-octadecanoate and methyl 9,12-dihydroxy-10,13-epoxy-octadecanoate. According to the literature, these furan type products may be formed via acid-catalyzed ring opening of one epoxide group (giving diol intermediates) and subsequent cyclisation involving the other epoxide group.42,101,102 However, diol intermediates were not detected in measurable amounts under these conditions, suggesting that their cyclisation may be relatively fast.
For Mo(75D)Ta-0.3 and Mo(75D)W-0.3, the product distributions at 24 h were very similar: at 97% MeLin conversion, the selectivities were 40% MeLinEp, 45–48% MeLinDiEp and 12–15% CFur (Fig. 9, Table 1). The Mo(75D)Nb-0.3 catalyst led to higher CFur selectivity; 52% selectivity, compared to 18% and 30% selectivity to mono and diepoxides, respectively, at 100% conversion, 24 h. Although the latter material possessed higher SBET, which may be favorable for the adsorption of intermediates and corresponding consecutive reactions, this does not seem to be the sole factor influencing the products distribution because no direct correlation between SBET and the product distributions (for the different materials) could be established. The differences in product distributions may be partly associated with differences in structure/electronic properties of the materials' active sites (suggested by the characterization studies). On the other hand, water (TBHP may contain up to 4 wt% water) may interact with metal sites and induce the formation of Brønsted acidity,103 which may contribute to epoxide ring opening reactions. Moreover, according to the literature, in situ water adsorption may affect the coordination number and oxidation state of molybdenum.71
Increasing the MeLin reaction temperature from 70 to 90 °C in the presence of Mo(75D)Nb-0.3, led to higher initial activity (increased from 132 to 170 mmolMeLin gcat−1 h−1) and the reaction was complete within 4 h (Fig. S21†). Moreover, the ratio of diepoxides/monoepoxides was enhanced (i.e., MeLinDiEp/MeLinEp = 64%/8%, at 24 h), as well as the ratio of (total epoxides)/CFur, (72%/28%). According to the above mechanistic considerations (conversion of epoxide to CFur via the intermediate formation of diol), the presence of water (which was added together with the oxidant) may cause epoxide ring opening. Possibly, at the higher reaction temperature, competitive adsorption effects may be unfavourable for water adsorption, avoiding consecutive reactions.
To gain further insights into the influence of water on the products distributions, the following catalytic tests were carried out for MeLin conversion, in the presence of Mo(75D)Nb-0.3, at 70 °C (Fig. S21†): (i) adding (dehydrated) molecular sieves to the reaction mixture (to reduce the water content in the liquid bulk); and (ii) adding water to the catalytic reaction mixture (ca. 35 wt% relative to the initial mass of MeLin). For test (i), CFur yield was lower (20% at 24 h) than for the normal catalytic test (52%), and MeLinDiEp yield was higher (57% MeLinDiEp yield at 24 h, compared to 30% for the normal catalytic test). Hence the removal of water enhanced diepoxides yields and was unfavorable for CFur formation. This was further confirmed by test (ii) which gave mainly CFur (87%/98% yield at 24 h/48 h), and MeLinDiEp was not formed in measurable amounts. In parallel to that verified for test (ii), the addition of water to the reaction of MeLin at 90 °C led to faster formation of CFur (88%/98% yield at 6 h/24 h, compared to 29%/55% at 24 h/48 h for the normal catalytic test at 90 °C) (Fig. S21†). Water may react with epoxides leading to ring-opening and formation of diol intermediates, and, as discussed above, the latter may undergo fast cyclisation to CFur.
To the best of our knowledge, these are the first Mo,M oxides (M = W, Nb or Ta) reported as catalysts for the target reactions. A literature survey for Mo-containing silica/silicates tested in the epoxidation of MeOle or MeLin, indicated two previous studies.104,105 Gao et al.105 reported the epoxidation of MeOle with H2O2, in the presence of molybdenum supported on the external surface of TS-1, which led to ca. 65% MeOleEp selectivity at ca. 52% conversion, 80 °C, 12 h; e.g., Mo(75D)M-0.3 led to 100% MeOleEp selectivity at 78% conversion, 70 °C, 6 h (Table 1). In a different study, molybdenum containing mesoporous silica of the type TUD-1 led to 100% MeOleEp selectivity at 89% MeOle conversion, using TBHP as oxidant at 70 °C, 24 h.104 The two literature studies reported deactivation of the silica-based catalysts.
A literature survey covering fully inorganic heterogeneous catalysts possessing different transition metals or oxide supports, tested for the target reactions is presented in Table S3.† There are few literature studies, and they are mostly focused on titanium-containing silicas/silicates. With MeOle as substrate, the results for Mo(75D)M-0.3 (entries 1–3) compared favorably to those reported for other catalysts. The highest epoxide yield was reported for MoO3–Al2O3, albeit at a higher reaction temperature of 115 °C (99% MeOleEp yield) and catalytic stability was not reported (entry 4).106 Ti–SiO2 and Ti-MCM-41 led to 86% at 24 h and >95% at 12 h, respectively, at 90 °C (entries 7, 11), but catalytic stability was also not reported.107 With MeLin as substrate, the yields of total epoxide products for the Mo(75D)M-0.3 catalysts were intermediate (entry 15) of those reported for Ti-silicas, albeit the catalytic stability was not reported.107,108
Conclusions
The challenge of developing selective, stable epoxidation solid catalysts for epoxidation (EPO) of relatively bulky olefins with relatively bulky tert-butylhydroperoxide under mild conditions, was studied by developing nanocatalysts consisting of Mo,M oxides (namely, Mo(xD)M-t and Mo(xP)M-t with M = Ta, Nb or W). The catalysts were synthesized via versatile solvothermal methodology, simply using the desired metals precursors and (biodegradable) acetophenone. In order to meet superior catalytic performances, the material synthesis conditions were optimised, namely, the type (D or P) and amount (x) of molybdenum precursor, type of M metal, and synthesis time (t = 0.3 or 24 h). The obtained Mo,M oxides possessed Mo/M ratios in the range 0.01–0.97, specific surface areas in the range 54–402 m2 g−1 and different structural features. Catalytic screening tests (cis-cyclooctene model reaction) indicated that EPO activity and epoxide yields can be enhanced by: (i) using MoO2Cl2 (D) as Mo precursor instead of MoCl5 (P), (ii) increasing the mole ratio (Mo/M)Syn of the synthesis mixture, and/or (iii) increasing the synthesis time. Importantly, the catalysts Mo(75D)M-0.3 and Mo(50D)M-24 with M = Nb, W were the most stable.The Mo(75D)M-0.3 nanocatalysts effectively promoted the EPO of relatively bulky biobased fatty acid methyl esters (methyl oleate, methyl linoleate) with TBHP; e.g., Mo(75D)M-0.3 led to 92–96% epoxide selectivity at 84–95% methyl oleate conversion. To the best of our knowledge, these are the first Mo,M-mixed oxides reported for the target FAMEs reactions.
The simple and versatile material synthesis methodology can be further explored to prepare multifunctional catalysts, e.g., for integrated acid-oxidation reaction systems. Moreover, these nanomaterials may be interesting for preparing formulated or composite materials for diverse applications.
Author contributions
DMG (formal analysis, investigation, validation, visualization, writing – original draft), PN (supervision, validation, visualization, writing – original draft, resources), XY (formal analysis, investigation), NP (resources, validation, visualization), PAR (conceptualization, methodology, investigation, supervision, validation, resources), AAV (conceptualization, project administration, resources, supervision, validation, visualization, writing – review & editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020, UIDP/50011/2020 & LA/P/0006/2020, financed by national funds through the FCT/MEC (PIDDAC), and by Project POCI-01-0145-FEDER-030075 (COMPETE 2020 Operational Thematic Program for Competitiveness and Internationalization), co-financed by national funds through the FCT/MCTES and the European Union through the European Regional Development Fund under the Portugal 2020 Partnership Agreement. D. M. G. (grant ref. 2021.04756.BD) acknowledges the FCT for PhD grant (State Budget, European Social Fund (ESF) within the framework of Portugal 2020, namely through the Centro 2020 Regional Operational Program). The authors are very grateful to Doctor M. Rosário Soares (CICECO-Aveiro Institute of Materials) for the discussions and kind assistance with the PXRD studies. C. Erdmann is acknowledged for the TEM measurements. X. Y. thanks the China Scholarship Council (CSC).References
- Y. Meng, F. Taddeo, A. F. Aguilera, X. Cai, V. Russo, P. Tolvanen and S. Leveneur, Catalysts, 2021, 11, 765–787 CrossRef CAS.
- Global Epoxides Market – Industry Trends and Forecast to 2029, https://www.databridgemarketresearch.com/reports/global-epoxides-market, (accessed 6 June 2023).
- P. Bassler, H. G. Göbbel and M. Weidenbach, Chem. Eng. Trans., 2010, 21, 571–576 Search PubMed.
- Development of New Propylene Oxide Process, https://www.sumitomo-chem.co.jp/english/rd/report/files/docs/20060100_ely.pdf, (accessed 17 July 2023).
- V. Russo, R. Tesser, E. Santacesaria and M. Di Serio, Ind. Eng. Chem. Res., 2013, 52, 1168–1178 CrossRef CAS.
- F. Schmidt, M. Bernhard, H. Morell and M. Pascaly, Chim. Oggi, 2014, 32, 31–35 CAS.
- Propylene Oxide Market - Global Forecast to 2026, https://www.marketsandmarkets.com/Market-Reports/propylene-oxide-market-55659975.html?gclid=Cj0KCQiA1NebBhDDARIsAANiDD3AJLp8tZRYiAyJKcKsAT-YsJKubp9n_IqsaveJNZvRyVTZFPWqsQ4aAhzxEALw_wcB, (accessed 6 June 2023).
- V. Smeets, E. M. Gaigneaux and D. P. Debecker, ChemCatChem, 2022, 14, e202101132 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- P. Bajpai, in Biermann's Handbook of Pulp and Paper, Elsevier, 3rd edn, 2018, pp. 19–74 Search PubMed.
- A. H. N. Armylisasa, M. F. S. Hazirahb, S. K. Yeonga and A. H. Hazimaha, Grasas Aceites, 2017, 68, e174 CrossRef.
- Fatty Acid Methyl Ester (FAME) Market Share, Size, Trends, Industry Analysis Report, By Source (Vegetable Oils, Animal Fats, and Used Cooking Oils); By Application; By Region; Segment Forecast, 2022–2030, https://www.polarismarketresearch.com/industry-analysis/fatty-acid-methyl-ester-fame-market, (accessed 5 June 2023).
- W. Yan, Z. Wang, C. Luo, X. Xia, Z. Liu, Y. Zhao, F. Du and X. Jin, ACS Sustainable Chem. Eng., 2022, 10, 7426–7446 CrossRef CAS.
- H. Hosney, B. Nadiem, I. Ashour, I. Mustafa and A. El-Shibiny, J. Appl. Polym. Sci., 2018, 135, 46270–46281 CrossRef.
- A. Ghosh-Dastidar, S. Kaujalgikar and B. I. Chaudhary, US Pat., 9499681B2, 2016 Search PubMed.
- Y.-Q. Xu and J.-P. Qu, J. Appl. Polym. Sci., 2009, 112, 3185–3191 CrossRef CAS.
- R. Belhassen, F. Vilaseca, P. Mutjé and S. Boufi, Ind. Crops Prod., 2014, 53, 261–267 CrossRef CAS.
- N. Lv, W. He, Z. Fang, Q. Sun, C. Qiu and K. Guo, Eur. J. Lipid Sci. Technol., 2018, 120, 1700257–1700264 CrossRef.
- V. Pantone, C. Annese, C. Fusco, P. Fini, A. Nacci, A. Russo and L. D. Accolti, Molecules, 2017, 22, 333–345 CrossRef PubMed.
- W. F. Hölderich, L. A. Rios, P. P. Weckes, H. Schuster, W. F. Holderich, L. A. Rios and P. P. Weckes, J. Synth. Lubr., 2004, 20, 289–301 CrossRef.
- W. He, G. Zhu, Y. Gao, H. Wu, Z. Fang and K. Guo, Chem. Eng. J., 2020, 380, 122532–122537 CrossRef.
- C. Kongyai and M. Hunsom, J. Renewable Sustainable Energy, 2012, 4, 053108–053119 CrossRef.
- K. Wadumesthrige, S. O. Salley and K. Y. S. Ng, Fuel Process. Technol., 2009, 90, 1292–1299 CrossRef CAS.
- D. Koziej, M. D. Rossell, B. Ludi, A. Hintennach, P. Novák, J. D. Grunwaldt and M. Niederberger, Small, 2011, 7, 377–387 CrossRef CAS PubMed.
- R. Deshmukh and M. Niederberger, Chem. – Eur. J., 2017, 23, 8542–8570 CrossRef CAS PubMed.
- J. M. Brégeault, Dalton Trans., 2003, 3289–3302 RSC.
- T. A. Nijhuis, M. Makkee, J. A. Moulijn and B. M. Weckhuysen, Ind. Eng. Chem. Res., 2006, 45, 3447–3459 CrossRef CAS.
- G. R. Haas and J. W. Kolis, Organometallics, 1998, 17, 4454–4460 CrossRef CAS.
- J. Kollar, assignor to Halcon International, Inc., US Pat., 3351635, 1967 Search PubMed.
- K. R. Jain, W. A. Herrmann and F. E. Kühn, Coord. Chem. Rev., 2008, 252, 556–568 CrossRef CAS.
- Y. Shen, P. Jiang, P. T. Wai, Q. Gu and W. Zhang, Catalysts, 2019, 9, 31–57 CrossRef.
- S. Shylesh, M. Jia and W. R. Thiel, Eur. J. Inorg. Chem., 2010, 28, 4395–4410 CrossRef.
- I. W. C. E. Arends and R. A. Sheldon, Appl. Catal., A, 2001, 212, 175–187 CrossRef CAS.
- N. Deligne, D. Bayot, M. Degand and M. Devillers, J. Solid State Chem., 2007, 180, 2026–2033 CrossRef CAS.
- C. Tagusagawa, A. Takagaki, K. Takanabe, K. Ebitani, S. Hayashi and K. Domen, J. Catal., 2010, 270, 206–212 CrossRef CAS.
- T. Ushikubo, Catal. Today, 2000, 57, 331–338 CrossRef CAS.
- Niobium and tantalum, https://www.usgs.gov/publications/niobium-and-tantalum, (accessed 27 April 2023).
- Acetophenone, https://echa.europa.eu/pt/registration-dossier/-/registered-dossier/14683/6/2/1, (accessed 14 July 2023).
- K. Skrodczky, M. M. Antunes, X. Han, S. Santangelo, G. Scholz, A. A. Valente, N. Pinna and P. A. Russo, Commun. Chem., 2019, 2, 1–11 CrossRef.
- P. Neves, A. C. Gomes, F. A. A. Paz, A. A. Valente, I. S. Gonçalves and M. Pillinger, Mol. Catal., 2017, 432, 104–114 CrossRef CAS.
- G. J. Piazza, A. Nuñez and T. A. Foglia, J. Am. Oil Chem. Soc., 2003, 80, 901–904 CrossRef CAS.
- L. C. Yang, Q. S. Gao, Y. H. Zhang, Y. Tang and Y. P. Wu, Electrochem. Commun., 2008, 10, 118–122 CrossRef CAS.
- L. Wang, K. J. Dong, C. C. Wang, R. P. Zou, Z. Y. Zhou and A. B. Yu, Powder Technol., 2022, 401, 117217–117226 Search PubMed.
- D. Vollath, Beilstein J. Nanotechnol., 2020, 11, 854–857 CrossRef CAS PubMed.
- A. Chithambararaj, N. S. Sanjini, A. C. Bose and S. Velmathi, Catal. Sci. Technol., 2013, 3, 1405–1414 RSC.
- N. Dukstiene, D. Sinkeviciute and A. Guobiene, Cent. Eur. J. Chem., 2012, 10, 1106–1118 CAS.
- Materials Explorer - MoO2, https://materialsproject.org/materials/mp-1094123/, (accessed 19 April 2023).
- J. A. da Cruz, E. A. Volnistem, R. F. Ferreira, D. B. Freitas, A. J. M. Sales, L. C. Costa and M. P. F. Graça, Therm. Sci. Eng. Prog., 2021, 25, 101015–101020 CrossRef CAS.
- H. Ono and K. I. Koyanagi, Appl. Phys. Lett., 2000, 77, 1431–1433 CrossRef CAS.
- J. Y. Zhang, Q. Fang and I. W. Boyd, Appl. Surf. Sci., 1999, 138–139, 320–324 CrossRef CAS.
- M. R. Mohammadi, D. J. Fray, S. K. Sadrnezhaad and A. Mohammadi, Mater. Sci. Eng., B, 2007, 142, 16–27 CrossRef CAS.
- F. Z. Tepehan, F. E. Ghodsi, N. Ozer and G. G. Tepehan, Sol. Energy Mater. Sol. Cells, 1999, 59, 265–275 CrossRef CAS.
- J. Y. Zhang and I. W. Boyd, Appl. Phys. Lett., 2000, 77, 3574–3576 CrossRef CAS.
- H. Sun, S. Liu, S. Liu and S. Wang, Appl. Catal., B, 2014, 146, 162–168 CrossRef CAS.
- R. R. Krishnan, K. G. Gopchandran, V. P. MahadevanPillai, V. Ganesan and V. Sathe, Appl. Surf. Sci., 2009, 255, 7126–7135 CrossRef CAS.
- N. Sharma, M. Deepa, P. Varshney and S. A. Agnihotry, J. Non-Cryst. Solids, 2002, 306, 129–137 CrossRef CAS.
- O. Rezaee, H. Mahmoudi Chenari and F. E. Ghodsi, J. Sol-Gel Sci. Technol., 2016, 80, 109–118 CrossRef CAS.
- A. C. Faro, P. Grange and A. C. B. dos Santos, Phys. Chem. Chem. Phys., 2002, 4, 3997–4007 RSC.
- B. Zhang, S. Xiang, A. I. Frenkel and I. E. Wachs, ACS Catal., 2022, 12, 3226–3237 CrossRef CAS.
- K. S. Chung and F. E. Massoth, J. Catal., 1980, 64, 320–331 CrossRef CAS.
- L. Wang and W. K. Hall, J. Catal., 1982, 77, 232–241 CrossRef CAS.
- E. I. Ross-Medgaarden and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 15089–15099 CrossRef CAS.
- C. Tiozzo, C. Bisio, F. Carniato, A. Gallo, S. L. Scott, R. Psaro and M. Guidotti, Phys. Chem. Chem. Phys., 2013, 15, 13354–13362 RSC.
- S. Dworakowska, C. Tiozzo, M. Niemczyk-Wrzeszcz, P. Michorczyk, N. Ravasio, R. Psaro, D. Bogdał and M. Guidotti, J. Cleaner Prod., 2017, 166, 901–909 CrossRef CAS.
- E. L. Lee and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 14410–14425 CrossRef CAS.
- M. Anilkumar and W. F. Hölderich, J. Catal., 2008, 260, 17–29 CrossRef CAS.
- F. Hashemzadeh, R. Rahimi and A. Ghaffarinejad, Ceram. Int., 2014, 40, 9817–9829 CrossRef CAS.
- M. Baltes, A. Kytökivi, B. M. Weckhuysen, R. A. Schoonheydt, P. Van Der Voort and E. F. Vansant, J. Phys. Chem. B, 2001, 105, 6211–6220 CrossRef CAS.
- J. H. Praliaud, J. Less-Common Met., 1977, 54, 387–399 CrossRef.
- H. Aritani, T. Tanaka, T. Funabiki, S. Yoshida, K. Eda, N. Sotani, M. Kudo and S. Hasegawa, J. Phys. Chem., 1996, 100, 19495–19501 CrossRef CAS.
- W. Dong, S. A. Zhou, Y. Ma, D. J. Chi, R. Chen, H. M. Long, T. J. Chun, S. J. Liu, F. P. Qian and K. Zhang, Rare Met., 2023, 42, 1195–1204 CrossRef CAS.
- Y. Chang, Y. Zhang, T. Hu, W. Chen, T. Tang, E. Luo and J. Jia, Molecules, 2023, 28, 4739 CrossRef CAS PubMed.
- N. Scotti, N. Ravasio, C. Evangelisti, R. Psaro, M. Penso, P. S. Niphadkar, V. V. Bokade and M. Guidotti, Catalysts, 2019, 9, 334–347 CrossRef.
- A. Uchagawkar, A. Ramanathan, Y. Hu and Bala Subramaniam, Catal. Today, 2020, 343, 215–225 CrossRef CAS.
- S. Patnaik, G. Swain and K. M. Parida, Nanoscale, 2018, 10, 5950–5964 RSC.
- J. N. Kondoa, Y. Hiyoshi, R. Osuga, A. Ishikawa, Y.-H. Wang and T. Yokoi, Materials, 2018, 262, 191–198 Search PubMed.
- M. Gancheva, I. Szafraniak-Wiza, R. Iordanova and I. Piroeva, J. Chem. Technol. Metall., 2019, 54, 1233–1239 Search PubMed.
- X. Yang, N. Wu, Y. Miao and H. Li, Nanomaterials, 2018, 8, 781–797 CrossRef PubMed.
- A. Ogawa and D. P. Curran, J. Org. Chem., 1997, 62, 450–451 CrossRef CAS PubMed.
- T. R. Amarante, P. Neves, A. A. Valente, F. A. A. Paz, A. N. Fitch, M. Pillinger and I. S. Gonçalves, Inorg. Chem., 2013, 52, 4618–4628 CrossRef CAS PubMed.
- Y. Kobayashi, S. Inukai, N. Asai, M. Oyamada, S. Ikegawa, Y. Sugiyama, H. Hamamoto, T. Shioiri and M. Matsugi, Tetrahedron: Asymmetry, 2014, 25, 1209–1214 CrossRef CAS.
- N. Wazir, C. Ding, X. Wang, X. Ye, X. Lingling, T. Lu, L. Wei, B. Zou and R. Liu, Nanoscale Res. Lett., 2020, 15, 156–164 CrossRef CAS PubMed.
- A. Bento, A. Sanches, E. Medina, C. D. Nunes and P. D. Vaz, Appl. Catal., A, 2015, 504, 399–407 CrossRef CAS.
- L. F. Veiros, Â. Prazeres, P. J. Costa, C. C. Romão, F. E. Kühn and M. José Calhorda, Dalton Trans., 2006, 1383–1389 RSC.
- P. J. Costa, M. J. Calhorda and F. E. Kühn, Organometallics, 2010, 29, 303–311 CrossRef CAS.
- M. J. Calhorda and P. J. Costa, Curr. Org. Chem., 2012, 16, 65–72 CrossRef CAS.
- P. Sözen-Aktaş, E. Manoury, F. Demirhan and R. Poli, Eur. J. Inorg. Chem., 2013, 15, 2728–2735 CrossRef.
- F. E. Kühn, W. M. Xue, A. Al-Ajlouni, A. M. Santos, S. Zang, C. C. Romão, G. Eickerling and E. Herdtweck, Inorg. Chem., 2002, 41, 4468–4477 CrossRef PubMed.
- J. Pisk and D. Agustin, Molecules, 2022, 27, 6011–6036 CrossRef CAS PubMed.
- P. Jin, D. Wei, Y. Wen, M. Luo, X. Wang and M. Tang, J. Mol. Struct., 2011, 992, 19–26 CrossRef CAS.
- A. Comas-Vives, A. Lledós and R. Poli, Chem. – Eur. J., 2010, 16, 2147–2158 CrossRef CAS PubMed.
- M. Fadhli, I. Khedher and J. M. Fraile, C. R. Chim., 2017, 20, 827–832 CrossRef CAS.
- M. H. Furigay, S. Chaudhuri, S. M. Deresh, A. B. Weberg, P. Pandey, P. J. Carroll, G. C. Schatz and E. J. Schelter, Inorg. Chem., 2022, 61, 23–27 CrossRef CAS PubMed.
- Q. Zhang, X. Li, Q. Ma, Q. Zhang, H. Bai, W. Yi, J. Liu, J. Han and G. Xi, Nat. Commun., 2017, 8, 14903–14911 CrossRef PubMed.
- W. Zhang, L. Xing, J. Chen, H. Zhou, S. Liang, W. Huang and W. Li, J. Alloys Compd., 2020, 822, 153530–153536 CrossRef CAS.
- Y. Sun, X. Hu, W. Luo and Y. Huang, J. Mater. Chem., 2012, 22, 425–431 RSC.
- P. A. Abramov and M. N. Sokolov, Molecules, 2023, 28, 4912–4923 CrossRef CAS PubMed.
- C. Tiozzo, C. Bisio, F. Carniato, L. Marchese, A. Gallo, N. Ravasio, R. Psaro and M. Guidotti, Eur. J. Lipid Sci. Technol., 2013, 115, 86–93 CrossRef CAS.
- Y. B. Huang, M. Y. Yao, P. P. Xin, M. C. Zhou, T. Yang and H. Pan, RSC Adv., 2015, 5, 74783–74789 RSC.
- G. B. Bantchev, K. M. Doll, G. Biresaw and K. E. Vermillion, J. Am. Oil Chem. Soc., 2014, 91, 2117–2123 CrossRef CAS.
- K. Herman and J. Foodtechn, Fat Sci. Technol., 1995, 97, 269–273 Search PubMed.
- T. Kitano, S. Okazaki, T. Shishido, K. Teramura and T. Tanaka, J. Mol. Catal. A: Chem., 2013, 371, 21–28 CrossRef CAS.
- D. M. Gomes, P. Neves, M. M. Antunes, A. J. S. Fernandes, M. Pillinger and A. A. Valente, Catalysts, 2022, 12, 1513 CrossRef CAS.
- X. Gao, Y. Zhang, Y. Hong, B. Luo, X. Yan and G. Wu, Microporous Mesoporous Mater., 2022, 333, 111731–111740 CrossRef CAS.
- E. Ucciani, A. Debal and G. Rafaralahitsimba, Fat Sci. Technol., 1993, 95, 236–239 Search PubMed.
- M. Guidotti, N. Ravasio, R. Psaro, E. Gianotti, L. Marchese and S. Coluccia, Green Chem., 2003, 5, 421–424 RSC.
- M. Guidotti, I. Batonneau-Gener, E. Gianotti, L. Marchese, S. Mignard, R. Psaro, M. Sgobba and N. Ravasio, Microporous Mesoporous Mater., 2008, 111, 39–47 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation of Mo,M oxides (composition, textural properties, DR UV-vis), MxOy (PXRD, TEM, SEM, ATR FT-IR) and used catalysts (PXRD, TEM, SEM, STEM, composition, DR UV-vis, ATR FT-IR). Catalytic results (influence of material properties on the initial activities, kinetic profiles for MeLin). See DOI: https://doi.org/10.1039/d3cy01299a |
| This journal is © The Royal Society of Chemistry 2024 |