
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ characterization techniques of protein corona around nanomaterials
Fangqin
Fu
ab,
Daniel
Crespy
 c,
Katharina
Landfester
*d and
Shuai
Jiang
*ab
c,
Katharina
Landfester
*d and
Shuai
Jiang
*ab
aKey Laboratory of Marine Drugs, Chinese Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, China. E-mail: jiangshuai@ouc.edu.cn
bLaboratory for Marine Drugs and Bioproducts, Qingdao Marine Science and Technology Center, Qingdao 266237, China
cDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong 21210, Thailand
dMax Planck Institute for Polymer Research, Mainz 55128, Germany. E-mail: landfester@mpip-mainz.mpg.de
First published on 18th September 2024
Abstract
Nanoparticles (NPs) inevitably interact with proteins upon exposure to biological fluids, leading to the formation of an adsorption layer known as the “protein corona”. This corona imparts NPs with a new biological identity, directly influencing their interactions with living systems and dictating their fates in vivo. Thus, gaining a comprehensive understanding of the dynamic interplay between NPs and proteins in biological fluids is crucial for predicting therapeutic effects and advancing the clinical translation of nanomedicines. Numerous methods have been established to decode the protein corona fingerprints. However, these methods primarily rely on prior isolation of NP–protein complex from the surrounding medium by centrifugation, resulting in the loss of outer-layer proteins that directly interact with the biological system and determine the in vivo fate of NPs. We discuss here separation techniques as well as in situ characterization methods tailored for comprehensively unraveling the inherent complexities of NP–protein interactions, highlighting the challenges of in situ protein corona characterization and its significance for nanomedicine development and clinical translation.
 Fangqin Fu | Fangqin Fu obtained her Master degree in Pharmaceutics from Jinan University in China in 2022. She joined the group of Prof. Shuai Jiang at the Ocean University of China for her PhD thesis. Her research interests mainly focus on the influence of protein corona on the in vivo bio-fate of nanoparticles. |
 Daniel Crespy | Daniel Crespy studied Chemistry at the University of Strasbourg where he first came into contact with heterophase polymerizations. He joined Professor Katharina Landfester in 2003 to complete a PhD at the University of Ulm where he developed novel methods to prepare nanocapsules in miniemulsions. In 2006, he held a position as project leader at Empa, working on stimuli-responsive materials for textiles. He joined the department of Professor K. Landfester in the Max Planck Institute for Polymer Research in 2009 as group leader. Daniel Crespy is now Associate Professor since 2016 in the Vidyasirimedhi Institute of Science and Technology (VISTEC) in Thailand. |
 Katharina Landfester | Katharina Landfester received her Doctoral Degree in 1995. After a postdoctoral stay at the Lehigh University, she worked at the Max Planck Institute of Colloids and Interfaces leading the miniemulsion group. From 2003 to 2008, she was Full Professor at the University of Ulm. She joined the Max Planck Society in 2008 as one of the directors of the MPIP. She was awarded the Reimund Stadler prize of the German Chemical Society and the prize of the Dr Hermann Schnell Foundation. Her research focusses on creating functional colloids for new material and biomaterial applications. |
 Shuai Jiang | Shuai Jiang studied chemistry at Yantai University. He obtained his Doctoral Degree in 2016 in the Chinese Academy of Sciences. During his PhD, he worked with Prof. K. Landfester and Dr D. Crespy at the Max Planck Institute for Polymer Research through the CAS-MPG Doctoral Promotion Program. From 2016 to 2019, he conducted postdoctoral research with Prof. K. Landfester at the Max Planck Institute for Polymer Research. From 2019, he served as group coordinator for the Max Planck-VISTEC Laboratory for Sustained Materials. Since 2021, he is a Full Professor at the Ocean University of China. His research focuses on nanomedicines and nano-bio interactions. |
Key learning points1. The concept of protein corona and its composition, biological impacts, and the challenges associated with its analysis.2. Separation techniques for isolating nanoparticle-protein complexes from protein media and the separation mechanisms. 3. In situ characterization methods for protein corona analysis and their characterization mechanisms. |
1. Introduction
Nanomedicines have drawn particular attention in cancer therapy due to their enhanced drug solubility, targeted delivery, and reduced systemic toxicity.1,2 Considerable efforts have been devoted to advancing the clinical translation of nanomedicines. Up to now, only ca. 14 nano-formulations have received FDA approval for clinical anti-tumor practice. However, no active-targeting nanomedicines have progressed through clinical trials.3 Indeed, the complex in vivo microenvironment and biological interactions present barriers to the clinical translation of nanomedicines.Upon entering blood circulation, nanoparticles (NPs) inevitably interact with biomolecules, mainly proteins, resulting in the formation of protein corona that endows a new “biological identity” to the synthetic NPs. The unique protein corona fingerprint may lead to unpredictable effects on the in vivo fate of NPs,4–6 such as the aggregation or destabilization of NPs,7,8 opsonization that accelerates NP clearance from blood,6,9–11 and potential immunological reactions due to interactions between NPs and immune cells.12–14 Conversely, specific apolipoprotein adsorption can be advantageous for NPs’ stealth behavior (by Apo A1 and clusterin)15–17 and penetration of the blood–brain barrier (by Apo E).18 Therefore, an in-depth understanding of NP–protein interactions is a prerequisite to reveal their biological effects and predict the in vivo fate of NPs.19
Protein corona formation is a thermodynamic and kinetic equilibrium process of protein adsorption and desorption.20 Depending on the binding affinity and dissociation rate of proteins to NP surface, protein corona can be divided into “hard” corona (HC) and “soft” corona (SC).21 The HC is composed of proteins with high binding affinity and low dissociation rate, tightly adsorbing on NP surface. Whereas the SC consists of proteins with low binding affinity and high dissociation rate, leading to their loose adhesion to NPs and continuous exchange with surrounding species. Notably, HC and SC are relative concepts based on protein binding affinity to NPs. Indeed, there are still no unified and specific methods to clearly distinguish SC from HC due to their unclear boundaries. The most common separation technique is centrifugation, and it is generally considered that SC can be removed from NPs after repeated washing by centrifugation. Although the general concepts of HC and SC are similar in different reports, varied compositions of HC and SC may obtain from different separation techniques.
The short residence time of SC proteins makes it challenging to distinguish them from free proteins in the medium. Conventional analysis methods of protein corona such as SDS-PAGE and LC-MS rely on isolation of the NP–protein complex from the medium by centrifugation, resulting in the loss of SC proteins that directly interact with the biological system and determines in vivo fate of NPs.22,23 Hence the obtained compositions do not accurately reflect the real protein corona fingerprints. Therefore, scientific efforts have been made for developing either efficient isolation approaches that maximumly preserve the SC proteins or for developing in situ characterization techniques capable of directly detecting NP–protein interactions in biological medium without isolation.
In this review, we summarize the recent progress on separation techniques and in situ characterization methods for precise protein corona analysis. The separation techniques are systematically discussed and classified according to their mechanisms, including centrifugation, magnetic separation and chromatographic methods. In situ characterization methods are categorized based on the analytical purpose, including the size evolution of NPs, morphology of NP–protein complex, kinetics of NP–protein interactions, conformation changes of proteins, as well as composition of the protein corona (Fig. 1).
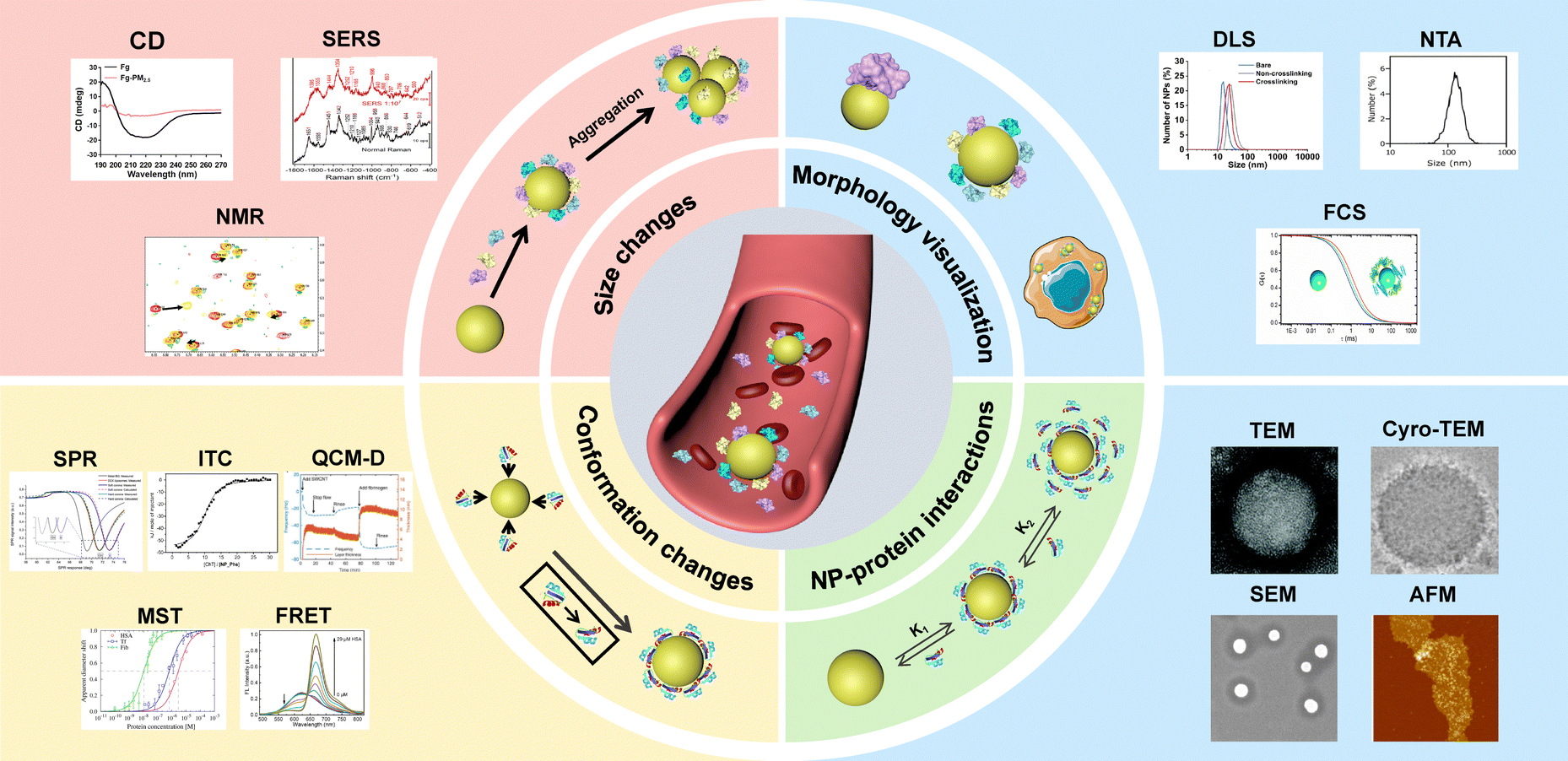 | ||
| Fig. 1 In situ characterization techniques for analyzing protein corona around NPs. DLS result was reproduced from ref. 24 with permission from American Chemical Society. NTA result was reproduced from ref. 25 with permission from Elsevier Ltd. FCS result was reproduced from ref. 26 with permission from American Chemical Society. TEM result was reproduced from ref. 27 with permission from the Royal Society of Chemistry. Cryo-TEM result was reproduced from ref. 28 with permission from Nature Publishing Group. SEM result was reproduced from ref. 29 with permission from Elsevier Ltd. AFM result was reproduced from ref. 30 with permission from American Chemical Society. SPR result was reproduced from ref. 31 with permission from Controlled Release Society. ITC result was reproduced from ref. 32 with permission from American Chemical Society. QCM-D result was reproduced from ref. 33 with permission from Nature Publishing Group. MST result was reproduced from ref. 34 with permission from American Chemical Society. FRET result was reproduced from ref. 35 with permission from Wiley-VCH. CD result was reproduced from ref. 36 with permission from Elsevier Ltd. SERS result was reproduced from ref. 37 with permission from Frontiers. NMR result was reproduced from ref. 38 with permission from American Chemical Society. | ||
2. Separation techniques for isolating NP–protein complex from protein media
Isolation of the NP–protein complex from the biological fluids is often required for ex situ analysis of protein corona. The composition of obtained protein corona could be further analyzed with protein identification techniques such as LC-MS and SDS-PAGE. However, the separation procedure critically affects the obtained composition of protein corona due to the loss of loosely bound proteins. The most commonly used separation techniques for isolating NP–protein complexes are centrifugation, magnetic separation and chromatographic methods.2.1 Centrifugation
Centrifugation is the most commonly applied separation technique for isolating NP–protein complexes,39,40 relying on the density and size differences between NPs and free proteins. It is considered an efficient means of isolating tightly adsorbed proteins on the NPs.23 The sedimentation velocity V of spherical NPs with a diameter D can be described by the Stokes equation (eqn (1)):41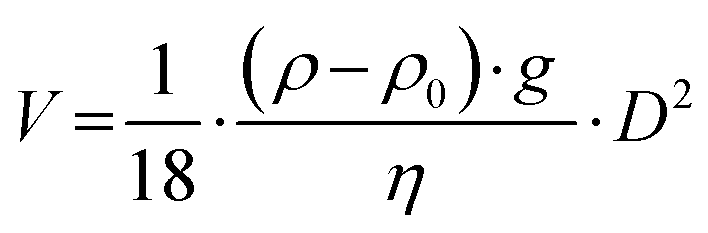 | (1) |
The composition of obtained corona proteins is significantly influenced by centrifugation parameters such as force, duration, and cycles. Increasing the number of washing cycles led to the elimination of loosely bounded proteins, favoring the formation of HC rather than SC.44 Studies have revealed that low abundance proteins such as apolipoprotein A1 and antithrombin-III became enriched in the protein corona as the number of washing cycles increased from 1 to 3. This enrichment occurred due to the easier removal of loosely bounded SC proteins, thereby concentrating the low abundance yet high affinity proteins within the obtained HC corona.45 Additionally, false positive or false negative outcomes may arise from simultaneous precipitation of free proteins or protein aggregates together with the NP–protein complexes, or detachment of loosely bound proteins.46 Recent reports indicate that a substantial amount of albumin, the most abundant protein in plasma and serum, tends to sediment during the centrifugation process for isolating the liposome–protein corona complexes.47 Moreover, the medium solutions used for protein corona preparation also affect the composition results. Brückner et al.48 compared eight different solutions and found that the obtained corona composition was dramatically affected by the washing solutions. For instance, when water was employed as a washing medium, there was an enrichment in immunoglobulins, a phenomenon not observed with other aqueous solutions. Conversely, washing with Dulbecco's Modified Eagle Medium (DMEM) favored a strong binding of serum albumin. These distinct protein corona signatures may stem from alterations in protein structure induced by variation of pH and osmotic pressure of the diverse washing media. Böhmert et al.49 systematically reviewed studies on the critical centrifugation parameters such as rotational velocity, time, and temperature, in protein corona study (Fig. 2). They concluded that these parameters should be considered in a holistic view. For specific NPs and given biological medium, the centrifugation parameters must be optimized in order to acquire accurate information.
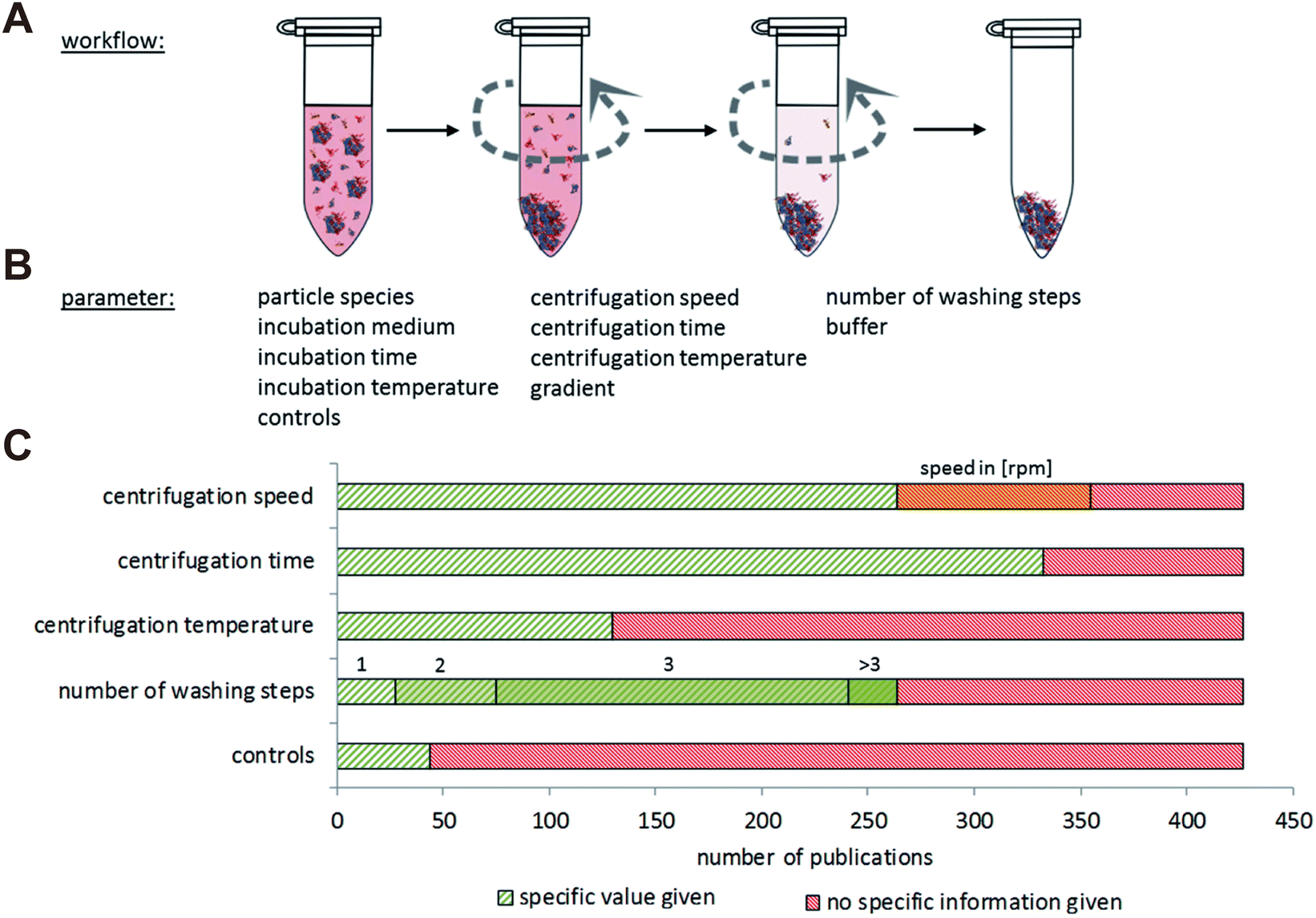 | ||
| Fig. 2 Overview of NP separation methodology based on centrifugation. (A) General procedure of centrifugation comprising repeated washing and centrifugation steps. (B) Main parameters related to centrifugation techniques for protein corona analysis. (C) Number of literatures involving the study on centrifugation speed, centrifugation time, centrifugation temperature, number of washing steps and controls. Green: the specific information is given in the publication; red: no specific information is given in the publication; orange: only the rpm values are given in the publication but not the exact centrifugation force.49 Reproduced from ref. 49 with permission from the Royal Society of Chemistry. | ||
Advanced centrifugation approaches such as using sucrose cushions and ultracentrifugation have been employed for protein corona isolation. A sucrose cushion improves centrifugation separation efficiency by stratifying the sample components based on their buoyant densities.50 This is achieved by creating a density gradient with sucrose, wherein sucrose's higher density relative to the samples allows for more precise fractionation. Unbound proteins in the incubation media can be separated from the NPs and agglomerates by the sucrose cushion method.51,52 For the NPs with low density, ultracentrifugation is a suitable approach to achieve high separation efficiency. However, NPs and proteins may aggregate upon the high centrifugation force (commonly above 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g). Therefore, it can be applied jointly with a sucrose gradient cushion for better separation efficiency.53,54 Multiple centrifugation and purification steps may result in SC loss and disrupt the adsorption equilibrium of corona proteins.
000 g). Therefore, it can be applied jointly with a sucrose gradient cushion for better separation efficiency.53,54 Multiple centrifugation and purification steps may result in SC loss and disrupt the adsorption equilibrium of corona proteins.
2.2 Magnetic separation
Magnetic separation is an important technique for preparing protein corona of magnetic NPs (e.g., iron oxide NPs).55 Compared with centrifugation, the magnetic separation process is relatively mild and capable of selectively isolating the NP–protein complex from its mixture with biological fluids components such as blood cells and biomolecules.56 Hoang et al.57 compared the composition of protein corona obtained by centrifugation and magnetic separation. It was found that more proteins were lost during centrifugation compared to magnetic separation. The magnetic force facilitated the removal of more hydrophilic proteins by overcoming van der Waals forces between proteins and NPs, whereas the intensive centrifugal force was able to overcome electrostatic attraction forces between proteins and NPs, resulting in the removal of more positively charged proteins. Similarly, Bonvin et al.58 investigated the influence of centrifugation and magnetic separation methods on the HC composition. They found that magnetic separation dramatically reduced the content of false-positive proteins resulting from protein aggregation during centrifugation process. Moreover, Blume et al.59 developed a multiple magnetic NPs-based protein separation method that relies on the differential interactions of distinct NPs with proteins (Fig. 3). Therefore, employing multiple NPs with distinct surface properties could enhance proteomic sampling and yield more comprehensive proteomics data. Nevertheless, magnetic separation cannot function as a universal method for protein corona preparation due to the limitation of NP types, i.e. they must be superparamagnetic and responsive to an external magnetic field. Additionally, such an automated method may introduce protein contamination due to protein adsorption from plasma to the well plates. Moreover, as the size of NPs increases, agglomeration occurs during magnetic separation. Therefore, NPs with diameters larger than 10 nm are not suitable for the magnetic separation.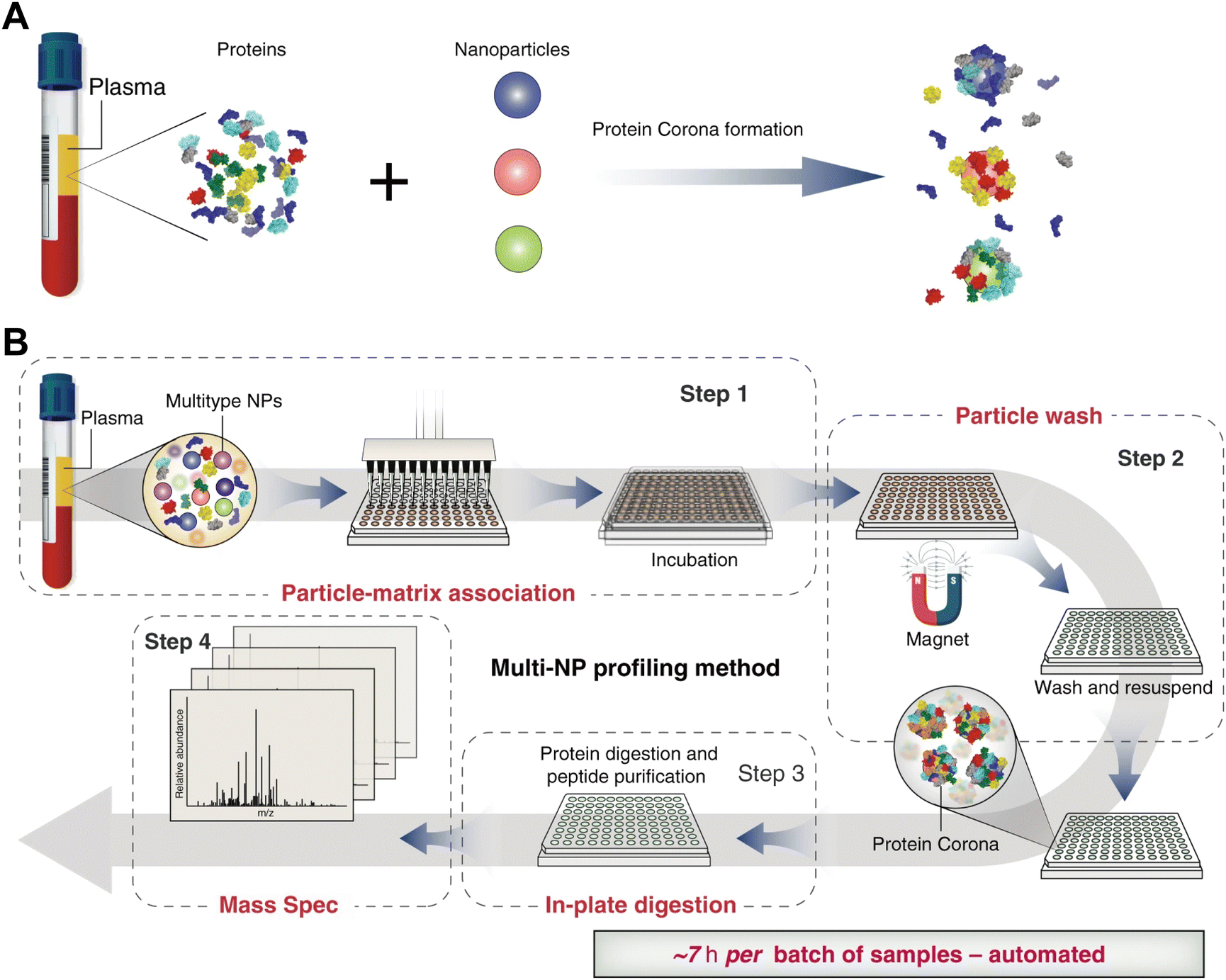 | ||
| Fig. 3 (A) Incubation of NPs with different physicochemical properties (indicated by different colors) in blood plasma results in different protein corona compositions. (B) Proteograph platform workflow based on multi-NP protein corona approach and mass spectrometry for plasma proteome analysis. The proteograph workflow includes four steps: (1) NP-plasma incubation and protein corona formation; (2) NP protein corona purification by a magnet; (3) digestion of corona proteins; and (4) LC-MS/MS analysis. In this context, each plasma-NP well is a sample, for a total of 96 samples per plate.59 Reproduced from ref. 59 with permission from Nature Publishing Group. | ||
Magnetic levitation (MagLev) has been utilized to investigate the heterogeneity of protein corona surrounding NPs.60 Unlike conventional magnetic separation, the measured NPs do not require responsiveness to magnetic fields. The principle relies on the dispersion of diamagnetic objects, such as diamagnetic NPs, in a paramagnetic medium under a non-homogenous magnetic field. The diamagnetic object, provided that the difference of magnetic susceptibilities between diamagnetic object and surrounding paramagnetic medium is negative, will levitate at a position depending on its density.50 Owing to the difference in density between bare NPs and NPs coated with protein corona, a variation of protein corona composition on a sole population of NPs was observed, corresponding to different levitation height and hence revealing heterogeneities of protein corona.61 However, it remains unclear whether the paramagnetic medium influences the composition of the protein corona.
2.3 Chromatographic methods
Chromatographic methods, including asymmetric-flow-field-flow-fractionation (AF4), size exclusion chromatography (SEC), capillary electrophoresis (CE)62–64 and hydrodynamic chromatography (HDC),65 have been utilized for isolating NP–protein corona complexes. Chromatographic methods effectively separate different species based on their hydrodynamic sizes, enabling the determination of protein affinity to NPs and the association/dissociation rates of proteins with NPs. Compared with centrifugation and magnetic separation, chromatographic separation offers a milder process that minimizes perturbation of protein corona. However, the chromatographic separation methods are usually time-consuming and costly.SEC is a conventional chromatography method that separates species based on their hydrodynamic volumes.66–70 The stationary phase of SEC is filled with porous beads that allow small species to enter the pore cavity, while larger species are eluted first by the mobile phase. NPs with tightly adsorbed proteins are eluted faster than bare NPs due to the increased size, whereas NPs with lower protein affinity result in easy dissociation of proteins from NPs and therefore are eluted at similar times as bare NPs. This separation mechanism enables to determine association and dissociation rates of proteins. However, the interactions of corona proteins with stationary phase may reduce the separation efficiency. Additionally, the shear stress occurring between the NP, protein corona and stationary phase may lead to the detachment of loosely bound proteins, consequently altering the corona composition.71,72 Besides, it is found that unbound free proteins can co-elute with liposomes during SEC isolation of liposome–protein complexes. Notably, a comparison of isolation methods including SEC, centrifugation and membrane ultrafiltration revealed that SEC is susceptible to co-eluting PEGylated liposomes with other free proteins, leading to contamination of the protein corona.25
Field flow fractionation (FFF) is a flow-assisted separation technique designed for samples with a molar weight lower than 1015 g mol−1,73 specifically for fractionating materials of nanoscale dimensions.74 Unlike traditional chromatography methods such as SEC, FFF does not use a stationary phase, which helps to avoid unwanted interactions between samples and the stationary phase.75 Instead, solutes are fractionated within layers of a continuous flow, directed by an external field perpendicular to the direction of the mobile phase. FFF offers significant advantages over other chromatography methods, particularly in reducing shearing force and accurately analyzing complex mixtures and fragile samples, such as aggregates.76 AF4 is a mild separation technique suitable for separating biological substances within the size range of 1 nm to 1 μm.77 The AF4 channel consists of a porous wall on one side, forming a perpendicular force to the sample flow that forces sample separation (Fig. 4).78–80 Species with different diffusion coefficients can be effectively separated, with smaller substances eluting faster than larger ones.81 Compared with other conventional chromatographic methods, AF4 replaces traditional gel permeation columns with a hollow channel without the stationary phase, thereby minimizing nonspecific interactions of proteins with the column materials. Moreover, the low shear forces of AF4 can minimize the perturbation to the protein corona composition.82,83 Weber et al.84 compared the protein corona patterns of polystyrene NPs obtained from centrifugation and AF4. Their results revealed that human serum albumin (HSA), typically associated with NPs as a soft corona protein, was identified as a major component in the protein corona following AF4 separation, whereas only a minimal amount of HSA was detected in the protein corona after centrifugation. These results implied that the AF4 is suitable for isolating SC proteins without disturbing the weak interactions of NPs with low affinity proteins. Although AF4 provides a mild and efficient way to isolate NP–protein complex and meanwhile preserves the SC, the establishment of AF4 methodology is usually time-consuming owning to its multiple parameters to be optimized. Moreover, the adsorption of sample to the membrane of AF4 channels has also to be considered.
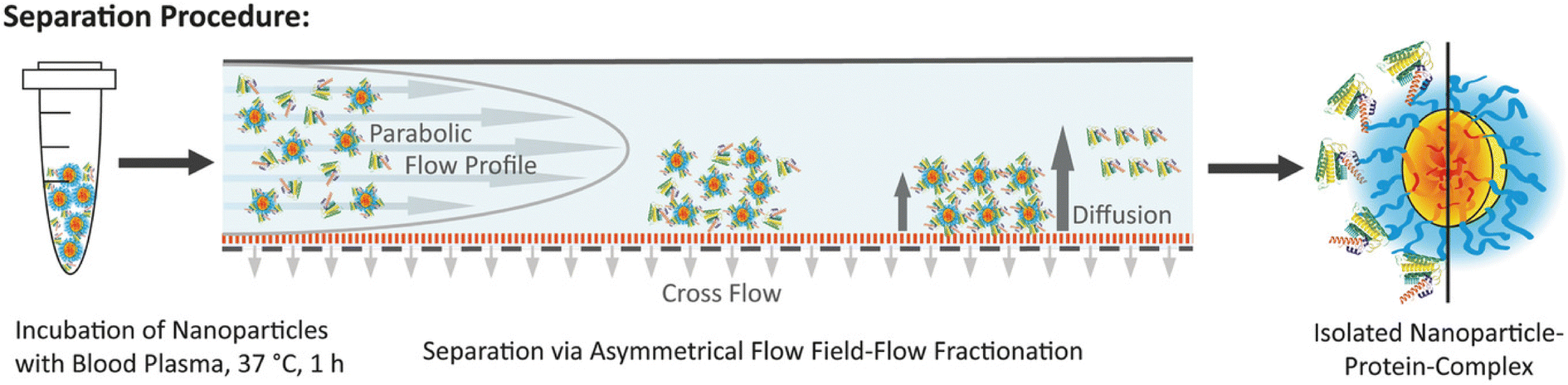 | ||
| Fig. 4 Schematic illustration of the separation process of NP–protein complex from unbound proteins via AF4 and the isolated NP–protein complex.80 Reproduced from ref. 80 with permission from Wiley-VCH. | ||
Collectively, a range of separation techniques have been developed to achieve comprehensive isolation of the protein corona while minimizing the loss of its components. Nonetheless, these techniques inevitably disrupt the formation and composition of protein corona to varying degrees, leading to potential inaccuracies such as false positives or false negatives. Table 1 provides a comprehensive overview of the diverse separation techniques discussed in this review.
| Techniques | Advantages | Disadvantages |
|---|---|---|
| Centrifugation | Commonly available | Long centrifugation times |
| Economical applicability | Disturbing NP–protein interaction | |
| High throughput85 | ||
| Magnetic separation | Economical applicability | Only for magnetic NPs with diameter less than 10 nm58 |
| High throughput | ||
| Chromatographic methods | Short measurement time78,84 | Expensive |
| Reduced perturbation of protein composition | Long time to establish standard process |
2.4 New techniques for protein corona separation
To overcome the dilemma of traditional separation methods, considerable efforts have been devoted to develop mild and specific techniques for efficient isolation of protein corona. Mohammad-Beigi et al.86 developed a novel separation technique based on strain-promoted alkyne azide cycloaddition (SPAAC) “click” chemistry (Fig. 5). This capture process enabled the identification of weakly interacting proteins along with the long-lived protein corona forming around NP in complex media. Specifically, silica and polystyrene NPs were first incubated with fetal bovine serum (FBS) to form NP-HC complexes after centrifugation. Then, the NP-HC and FBS (to form SC layer) was separated modified to carry –N3 and –DBCO groups, enabling a spontaneously click-chemistry reaction after incubation. SC proteins can be then covalently captured for further proteomic analysis. Proteomic results showed that most of captured proteins are present in both SC and HC, indicating that the same proteins can have both high and low binding affinities to NPs. This click chemistry-based method is highly versatile for the separation of protein coronas across various NPs. However, chemical modification of proteins may alter their characteristics and adsorption profiles. The modified chemical identities, i.e., –N3 and –DBCO groups, may also contribute to the interaction of NP-HC with SC proteins.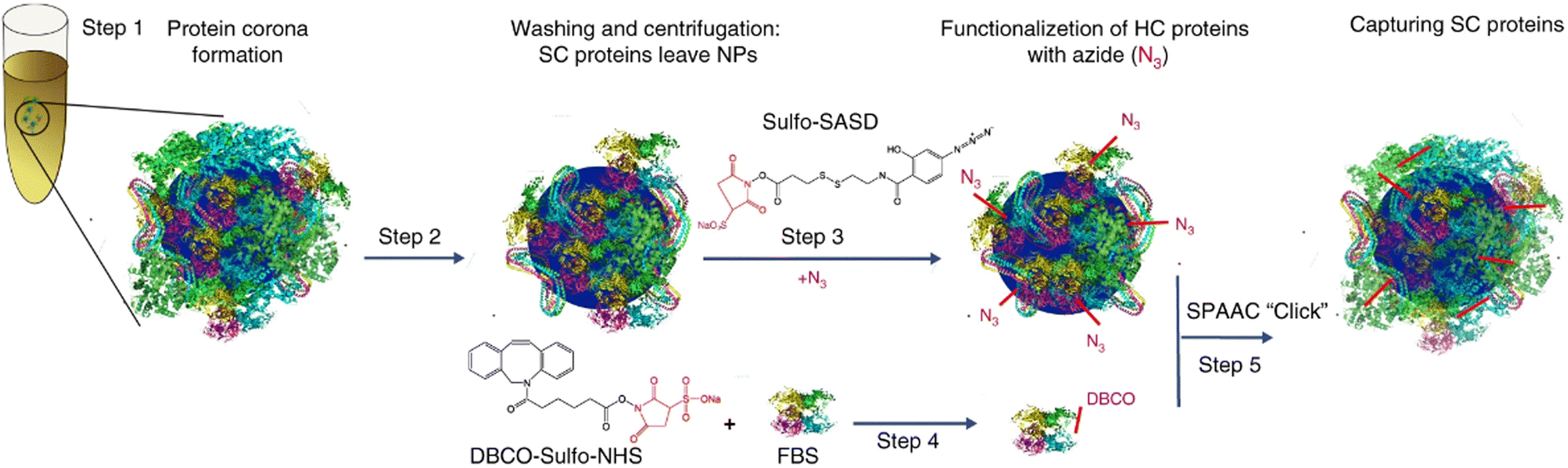 | ||
| Fig. 5 Schematic representation of capturing SC proteins. After protein corona formation (steps 1 and 2), the HC proteins were modified with N3 by reacting with sulfo-SASD (step 3) followed by a SPAAC “click” reaction (step 5) with FBS-D proteins (prepared in step 4).86 Reproduced from ref. 86 with permission from Nature Publishing Group. | ||
Due to their stereo-selective surfaces, chiral nanomaterials have been found to induce distinct biological responses including cellular uptake, energy metabolism, and cell differentiation.87 Baimanov and co-workers88 established a bio-layer interferometry (BLI)-based fishing strategy for real-time monitoring of the dynamic interactions of chiral NPs with serum proteins (Fig. 6). Specifically, IgG was initially immobilized on an amine-reactive 2nd generation biosensor (AR2G) capable of reacting with protein or peptides containing primary amine groups, followed by the binding of chiral Cu2S NPs (L-NPs and D-NPs) via their high binding affinity to the IgG. The obtained chiral biosensors were exposed to 10% mouse serum at 37 °C under continuous shaking of the microplate. Protein adsorption on NPs was monitored in real-time by measuring the change of wavelength shift (Δλ) in the biosensor, which reflects changes in bio-layer thickness due to protein adsorption. The SC and HC proteins were obtained through sequential washing steps and further subjected to LC-MS/MS analysis. The results showed that the chirality of Cu2S NPs dramatically affected the composition of SC and HC proteins, leading to distinct blood clearance profiles of NPs. In comparison with the SC of L-NPs, more opsonin proteins were detected in the SC of D-NPs that resulted in shorter blood circulation. This fishing strategy enables real-time detection of the evolution of HC and SC on NPs and reveals subtle difference in NP–protein interactions even within several minutes. However, the binding of IgG and Cu2S NPs may reduce the contact area of NPs with serum proteins, therefore reducing the number of adsorbed proteins and altering their spatial distribution. To generalize this BLI-based fishing strategy for different NPs, systematic screening of high-affinity proteins specific to each NP is required.
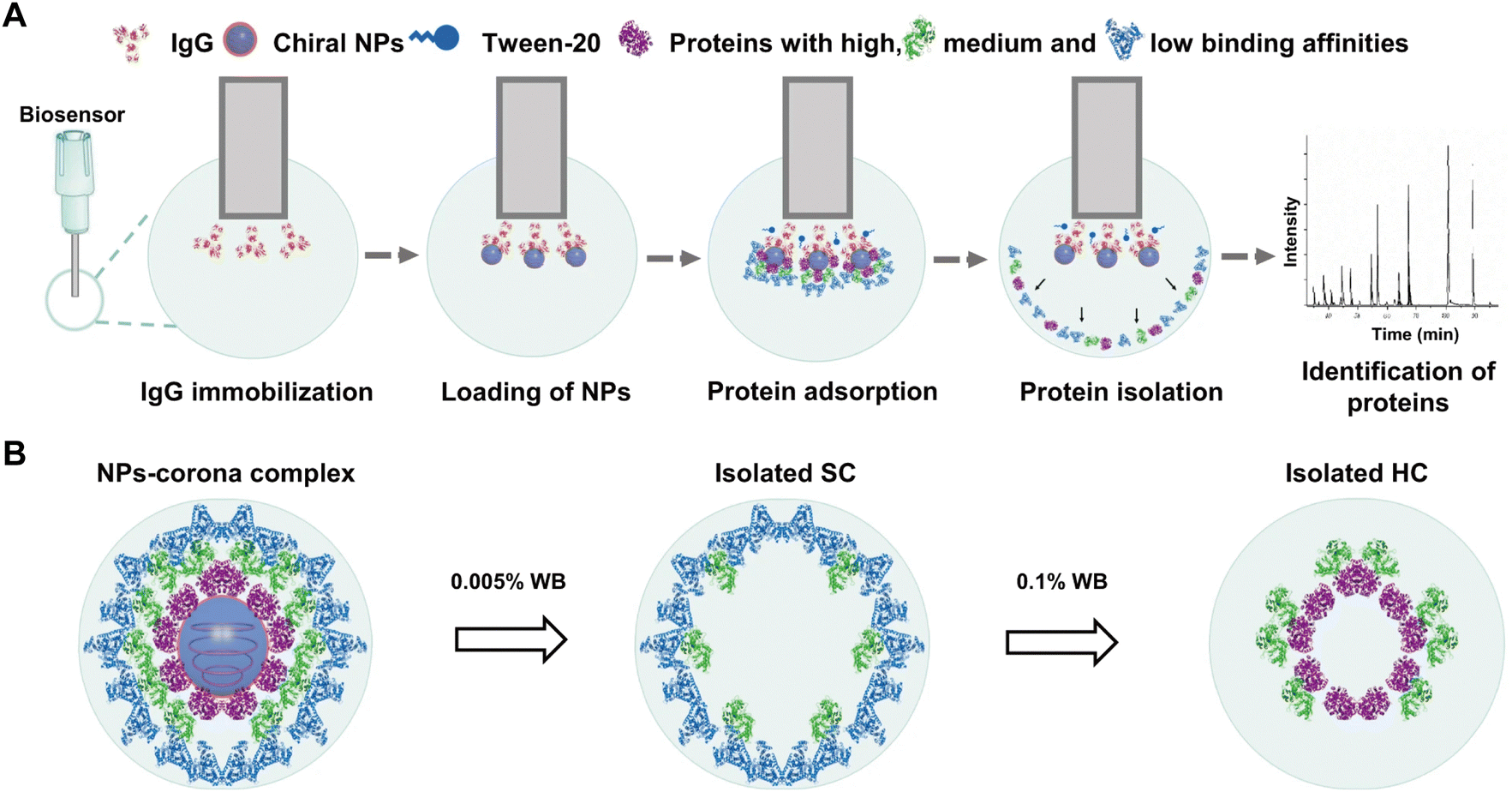 | ||
| Fig. 6 Schematic illustration of BLI-based fishing strategy including protein corona formation on the chiral NPs, isolation of corona, and identification of protein composition and abundance. (A) The whole method includes five steps: (1) coating of the biosensors with IgG; (2) loading of chiral NPs onto the biosensors; (3) incubation of NPs with biological fluids and formation of protein corona; (4) isolation of protein coronas from the NPs; (5) protein digestion and proteomic analysis for identification of corona components by LC–MS/MS. (B) Isolation process of NPs-corona complex. The spherical structures with gray blue and blue colors refer to Cu2S NPs and Tween-20 (a surfactant), respectively; while the irregular structures with purple, green, light blue, and red colors represent various types of proteins.88 Reproduced from ref. 88 with permission from Nature Publishing Group. | ||
Chu and co-workers89 developed an affinity chromatography (AfC) method using His-tagged anti-PEG single-chain variable fragment (PEG-scFv) for efficient and specific separation of protein corona formed on the PEGylated liposomes (sLip) (Fig. 7). In this method, the His-tagged PEG-scFv could readily capture sLip without affecting protein corona compositions, while also separating the sLip/protein complexes from plasma protein aggregates and endogenous vesicles through the Ni-NTA column. Briefly, the PEG-scFv was tagged with His tag for binding on the Ni-NTA column. Upon exposure in a sLip-protein solution, the His-tagged PEG-scFv successfully capture the sLip-protein complexes through specific binding of antigen (PEG on sLip) with antibody (PEG-scFv). Therefore, the protein corona on sLip can be separated from unbound free proteins, protein aggregates, and other endogenous vesicles. The AfC displayed a 43-fold increase of in vitro protein corona separation efficiency compared to centrifugation. This method avoided contamination from protein aggregates and endogenous vesicles occurring during centrifugation and effectively reserved the loosely bounded SC proteins due to the mild separation condition. Collectively, the AfC provides an unprecedented approach for deeply deciphering protein coronas and is suitable for separating in vivo protein coronas due to its high specificity and minimal sample volume requirement. However, this method is only applicable to the PEGylated samples, constraining its scope of application. Moreover, the protein corona may shield the binding of PEG chains to the PEG-scFv on the column.
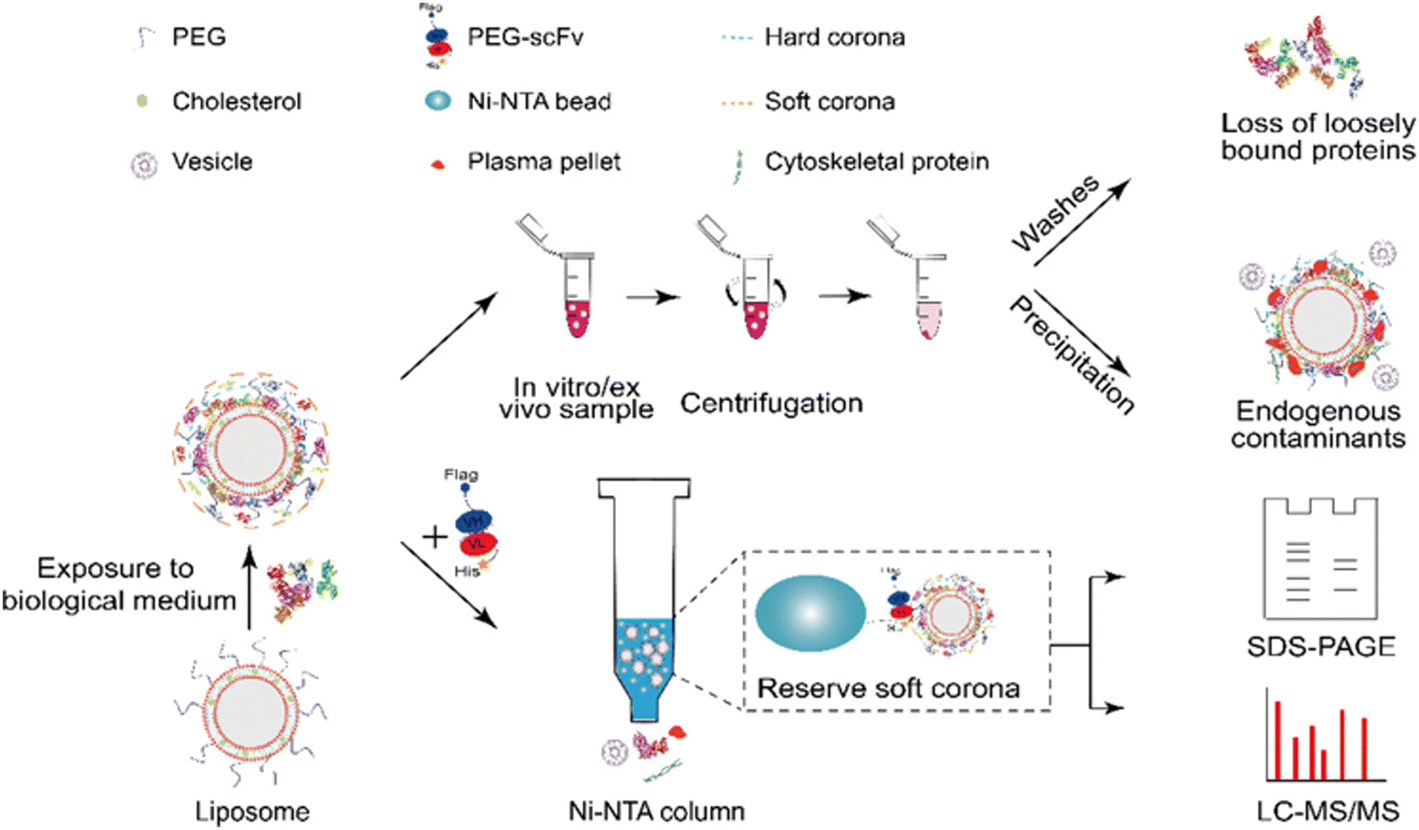 | ||
| Fig. 7 Schematic illustration of the separation procedures of protein corona by affinity chromatography (lower row) and comparison with centrifugation (upper row).89 Reproduced from ref. 89 with permission from American Chemical Society. | ||
Table 2 provides a comprehensive overview of the diverse separation techniques discussed in this review. While the delicate design of these novel methods enhances the efficiency and specificity of protein corona preparation, they also face several challenges. Often, they require complex preparatory procedures or expensive materials, impeding their universal applications. For instance, the BLI-based fishing strategy is applicable only to Cu2S NPs due to their high affinity to IgG. To broaden its application, it is essential to explore proteins with high affinity for other types of NPs, such as lipid, polymer, and silica NPs. The AfC method, based on PEG-scFv, is currently limited to separating protein coronas on PEGylated NPs. Given that PEGylation is a common modification in nanomedicine, this approach holds promise as a versatile method. Furthermore, by leveraging diverse antigen–antibody interactions, this method could be extended to other functionalized NPs, such as those modified with antibodies or glycosylation. Collectively, advancing towards direct measurement methods that circumvent the challenges of the separation step represents a promising strategy for protein corona studies.
| Techniques | Advantages | Disadvantages |
|---|---|---|
| Click chemistry-based strategy | Wide applicability | Chemical modification may affect intrinsic characteristics and adsorption profiles of proteins |
| High sensitivity | ||
| Simple operation process | ||
| BLI-based fishing strategy | High sensitivity | Costly and time-consuming |
| Real-time monitoring of the protein corona evolution | Only for Cu2S NPs with high binding affinity to IgG | |
| Protein corona evolution | ||
| Affinity chromatography | Mild separation condition | Complex preparatory procedures Expensive |
| High specificity | Only applicable to PEGylated samples | |
| Reduced perturbation of protein composition and protein contamination |
3. In situ characterization methods of protein corona
In situ characterization directly measures the NP–protein interactions in biological media without the need of an extra separation step for isolating the NP–protein complexes. Therefore the disturbance of NP–protein interactions and the loss of weakly adsorbed SC proteins during the separation procedure can be avoided.90–92 Classical in situ characterization methods include dynamic light scattering (DLS),24,93 fluorescence correlation spectroscopy (FCS),94,95 transmission electron microscopy (TEM),96 circular dichroism (CD) spectroscopy,97,98 and isothermal titration calorimetry (ITC), etc.99,100 These methods provide insights into the size and morphology of NP–protein complexes, the quantity, kinetics, and affinity of NP–protein interactions, as well as the conformational changes of proteins upon adsorption.3.1 Characterization methods for the size of NP–protein complex
Determining the size change of NPs after their incubation with proteins or in other biological media can reveal the protein adsorption and aggregation state of NPs. Common methods for this analysis include DLS, FCS, nanoparticle tracking analysis (NTA), and diffusion 19fluorine-magnetic resonance (19F-NMR) spectroscopy.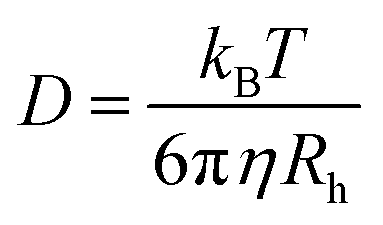 | (2) |
To eliminate background signals from unbound proteins, Balog et al.106 applied the depolarized dynamic light scattering (DDLS) to investigate size changes of differently PEGylated Au NPs (coated with PEG-NH3+, PEG-COO−, or PEG-CH3) after incubation in common biological fluids (Fig. 8A). Owing to their polycrystalline nature, spherical metallic NPs (e.g., Au and Ag NPs) exhibit an internally heterogeneous structure and deviate slightly from perfect sphericity, resulting in a subtle yet significant optical anisotropy. When excited by electromagnetic waves, the Au NPs support the coherent oscillation of the surface conduction electrons, known as localized surface plasmon resonance (LSPR). Upon scattering, the LSPR coupled with inherent optical anisotropy of Au NPs leads to a depolarized speckle pattern, whose temporal fluctuations of NPs can yield information about the size of Au NPs. Contrasted with the depolarized scattering of Au NPs, the depolarized scattering arising from biological matrices, such as unbounded proteins and lipids, was almost negligible and can be disregarded. The finding demonstrated that the sizes of PEG-COO− and PEG-CH3 coated Au NPs remained unchanged after incubation with all tested fluids (including PBS, bovine serum albumin (BSA) in PBS, DMEM, and 10% FBS in DMEM), indicating their low affinity to the protein molecules. Conversely, the size of PEG-NH3+ coated Au NPs increased by approximately 14 nm when exposed to the BSA solution, implying a strong interaction between the NPs and BSA. The DDLS efficiently mitigated irrelevant signals within complex environments, enabling real-time quantitative measurements of Au NP–protein complexes with an unprecedented signal-to-noise ratio. Nonetheless, this approach is limited to spherical metallic NPs characterized by a polycrystalline structure, such as Au NPs, Ag NPs, and Cu NPs.
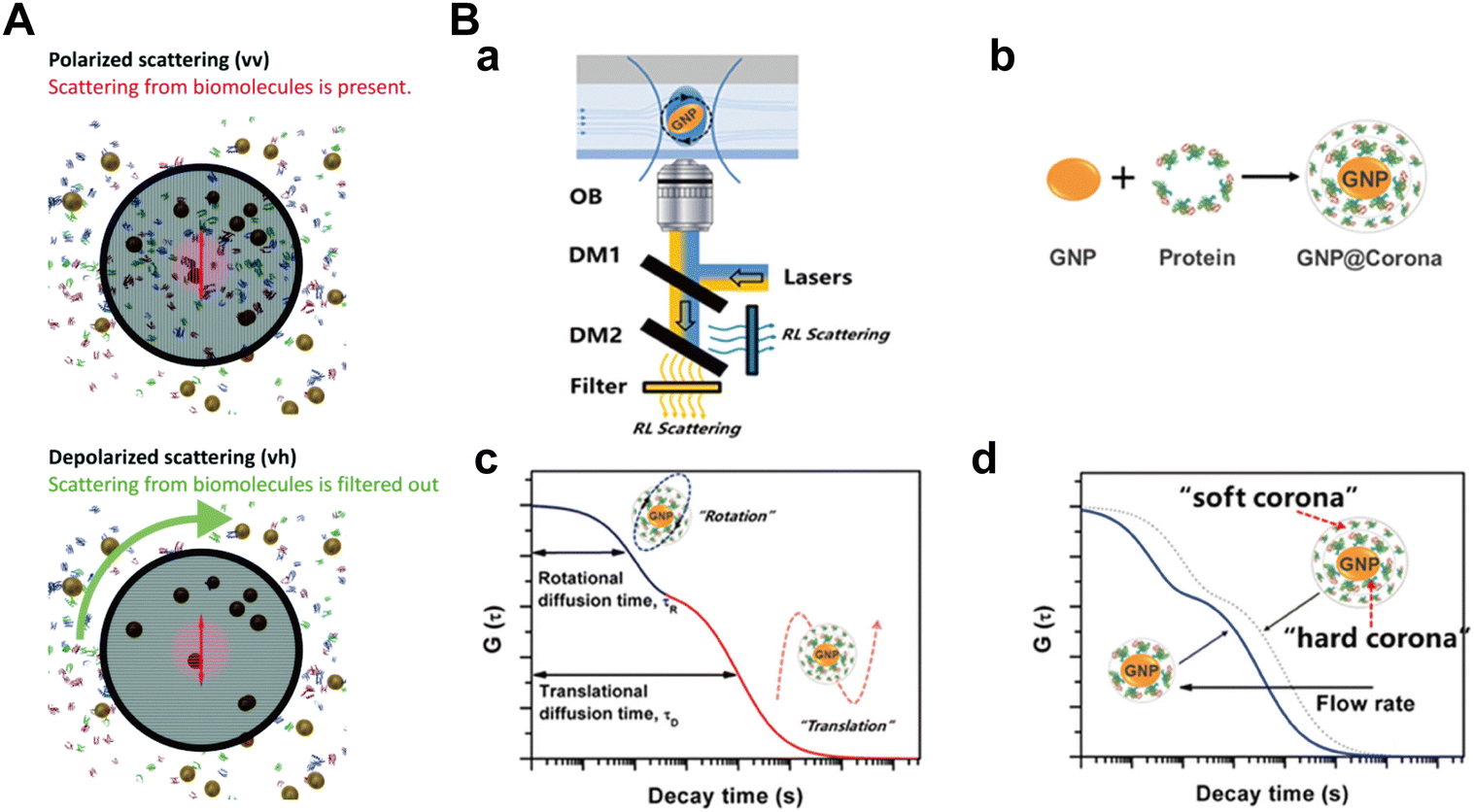 | ||
| Fig. 8 (A) Standard polarized (top) and depolarized (down) DLS experiments. In the polarized scattering experiment scattering from the biomolecules is present, while it is invisible in the depolarized scattering experiment and thus can be ignored compared to the depolarized scattering from the NPs. Therefore, scattering exclusively from the NPs on an essentially zero background is detected in the depolarized scattering experiment.106 Reproduced from ref. 106 with permission from the Royal Society of Chemistry. (B) Detection of protein corona formation in situ by D-RLSCS; (a) schematic illustration of D-RLSCS system measuring gold NPs (GNPs) in a microfluidic channel; (b) formation of protein corona on the surface of GNPs; (c) the curve of D-RLSCS consisting of rotational and translational diffusion of GNPs with protein corona; (d) D-RLSCS curves of GNPs with protein corona flowed in the microchannel varied with flow rate.107 Reproduced from ref. 107 with permission from the Royal Society of Chemistry. | ||
However, the application of FCS for in situ measurements in blood is limited by pronounced absorption and scattering of light by blood components (e.g., cells and proteins) in the visible spectrum where conventional FCS setups and common fluorescent labels operate. Negwer et al.119 addressed this limitation by either labeling NPs or loading cargos with near-infrared (NIR) dyes with excitation and emission wavelengths falling within the range of 700–1100nm. This range corresponds to the so-called NIR window in biological tissue, wherein light exhibits maximal penetration depth. Utilizing NIR labels and fitting the experimental autocorrelation functions with an analytical model that considers the presence of blood cells, they successfully monitored the size and drug loading of NPs in the blood, achieving sensitivity down to single dye molecule level. This technique enabled the investigation of protein corona formation on NPs within flowing blood. While the translational diffusion of NPs determined by FCS is directly proportional to particle size, the rotational diffusion coefficient of NPs exhibits a proportionality to the cube of particle size. This implies that the rotational diffusion of NPs should exhibit greater sensitivity to size changes than their translational diffusion. However, extracting this parameter poses challenges due to the high rotation rate and subtle optical signal fluctuations in conventional methods, resulting in diminished sensitivity to NP–protein binding. Moreover, introducing a fluorescence label on either the NPs or the proteins may alter their properties and hinder accurate measurement of rotational diffusion.
To overcome this issue, Zhang et al.107 proposed a dual-wavelength laser-irradiated differenced resonance light scattering correlation spectroscopy (D-RLSCS) technique, combined with the modified generation method of the D-RLSCS curve to in situ investigate the protein corona formation on the label-free NPs. Specifically, the dissociation constants and binding rates of proteins, including BSA and FIB, to gold nanosheets were determined by analyzing the binding-induced ratiometric diffusion changes of NPs (the ratio of characteristic rotational diffusion time to translational diffusion time) (Fig. 8B). The results showed that BSA exhibits stronger binding constant and faster binding rate to gold nanosheets compared with FIB. In contrast to conventional methods such as FCS, this novel D-RLSCS strategy provides comprehensive insights into protein corona by averaging the diffusion behavior of numerous NPs. Besides, it effectively mitigates the impact of increased viscosity resulting from the high concentration of proteins.
19F-NMR spectroscopy has also been utilized to detect size changes of the NP–protein complex. Diffusion 19F-NMR measures the diffusion coefficient of 19F-labeled NPs by identifying the chemical shift of 19F resulting from the protein binding, and then converts it to the diameter of NPs using the Stokes–Einstein equation (eqn (2)).120 Owing to its non-optical nature and the absence of interfering fluorinated molecules in physiological media, the 19F-NMR spectroscopy can be applied in turbid environments, even in the presence of cells. For example, Carril and co-workers121 employed 19F-NMR spectroscopy to determine alterations in the hydrodynamic radius of fluorinated PEG-labeled Au NPs with different head groups (–COOH and –NH2) and surface coating (poly(isobutylene-alt-maleic anhydride), PMA) in complex media. The results indicated that all three types of NPs remained colloidally stable and well dispersed in blood and plasma, without any detected agglomeration. NP-F/NH2@PMA exhibited a size increase of approximately 5.5nm after plasma incubation, while the size of NP-F/COOH and NP-F/NH2 decreased due to the shrinkage of the PEG-containing ligands in plasma. These results illustrated the suitability of 19F-NMR for in situ analysis of protein corona formation without any purification. However, 19F-NMR spectroscopy results highly depend on the concentration of fluorine atoms. The presence of fluorine head groups on NPs can repel proteins and may hence affect their adsorption. Specifically, the hydrophobic nature of fluorine head groups can affect interactions with proteins like fibrinogen. Moreover, larger-sized NPs may experience increased mobility constraints, leading to limitations in accurately detecting diffusion coefficients. Additionally, evaluating interactions between proteins and NPs with soft coatings, such as PEG, presents challenges due to the inherent shrinkage and compressibility of these coatings.
3.2 Characterization methods for the morphology of NP–protein complex
Imaging techniques, notably transmission electron microscopy (TEM), cryogenic transmission electron microscopy (Cryo-TEM), scanning electron microscopy (SEM), and atomic force microscopy (AFM), serve as intuitive and compelling means for characterizing protein corona formation on NPs.122–124 These methods are instrumental in providing a comprehensive understanding of the morphology, protein binding forms, and aggregation state of the NP-corona complexes.125–127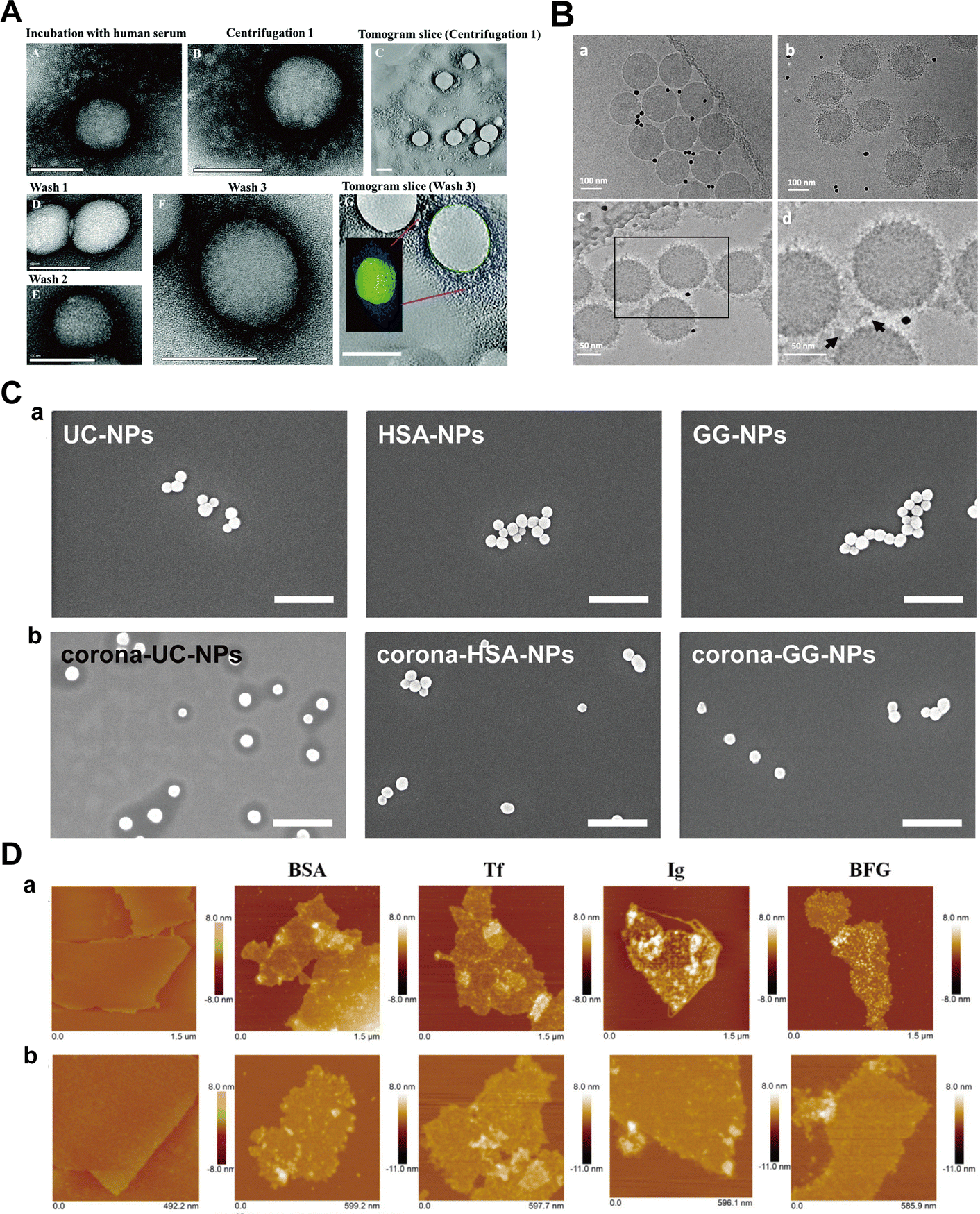 | ||
| Fig. 9 (A) TEM images of protein corona surrounding polystyrene NPs; (a) HC and SC of polystyrene NPs in human serum. (b) HC and SC of NPs after centrifugation and removal of the supernatant, but before washing steps. (c) Tomogram slice of the area in (b). (d) and (e) HC obtained after 1st wash, after 2nd wash, and after 3rd wash, respectively. (g) Electron tomogram slice of the area presented in (f). Scale bar: 100 nm.27 Reproduced from ref. 27 with permission from the Royal Society of Chemistry. (B) Cryo-TEM images of polystyrene NPs; TEM images of (a) bare NPs without corona and (b) NPs with corona; (c) and (d) Distribution of proteins and their association with NP surface at higher magnification. Black dots in the images are 10 nm gold fiducial markers. The arrows indicate proteins accumulated between NPs as large clusters.28 Reproduced from ref. 28 with permission from Nature Publishing Group. (C) SEM images of silica NPs; (a) SEM images and (b) corresponding size distribution of uncoated NPs and NPs pre-coated with HSA and γ-globulin; (c) SEM images and (d) corresponding size distribution plots of corona-coated NPs. Scale bars are 500 nm.29 Reproduced from ref. 29 with permission from Elsevier Ltd. (D) Morphology of GO-protein complexes by AFM; AFM images of bare-GO (first column) and GO-protein complexes after incubation of BSA, Tf, Ig, or BFG with GO for 5 min (a) and 60 min (b).30 Reproduced from ref. 30 with permission from American Chemical Society. | ||
While electron microscopic techniques offer valuable insights into the morphology of protein corona, they also present inherent challenges. The high vacuum conditions during sample preparation at room temperatures, required for sample drying, poses a risk of introducing artifacts and the formation of large aggregates. Moreover, high protein concentrations in biological fluids can lead to significant background levels.136 Hence, these techniques are typically used as complementary methods of in situ characterization of the protein corona.
3.3 Characterization of the kinetics of NP–protein interactions
Monitoring NP–protein interactions in the bloodstream is of crucial importance for comprehending the in vivo behavior of nanomedicines administered intravenously. Key methods for studying the kinetics of NP–protein interactions include surface plasmon resonance (SPR),137–139 isothermal titration calorimetry (ITC),140–142 quartz crystal microbalance with dissipation monitoring (QCM-D),143,144 microscale thermophoresis (MST),145 and fluorescence resonance energy transfer (FRET).146,147| Rd = A·e−kd·t + Rd0 | (3) |
| Ra = A2 − A3·e−(C·ka + kd)·t | (4) |
Kari et al.31 utilized multi-parametric SPR (MP-SPR) to monitor protein corona formation and determine the affinity (KD) of C3b with liposomes in undiluted human serum. Their observations revealed formulation-dependent HC and SC with distinct refractive indices, layer thicknesses, and surface mass densities (Fig. 10A). Upon serum injection, a SC with a thickness of 38–87 nm was formed on DOX liposomes, while the layer thickness on DOX + PEG liposomes was slightly thinner (26–60 nm). After washing with buffer, a significantly thinner HC layer remained on the surface of DOX liposomes compared to the DOX + PEG liposomes (3–7 vs. 8–13 nm). Moreover, the KD values were 3.8 × 10−8 M and 7.2 × 10−8 M for the interaction of C3b with DOX liposomes and DOX + PEG liposomes, respectively. Miclăuş et al.152 introduced an LSPR technique for quantifying both SC and HC formation on polyvinylpyrrolidone (PVP)-stabilized silver nanocubes (AgNCs). This method capitalizes on the sensitivity of the LSPR phenomenon of AgNCs to the surrounding refractive index due to coherent oscillation of surface conduction electrons, enabling a precise quantification of protein binding to the NPs. Their findings demonstrated a significantly elevated protein presence in SC compared to HC at all time points (with an 8-fold difference under 10% serum conditions), highlighting the sensitivity of SPR in probing interaction dynamics. Nevertheless, the relatively high cost and the need to tailor SPR biosensors for individual NPs limit the universal applicability of SPR.92 Additionally, the detection limit of SPR is relatively high, e.g., 1–10 nM for proteins with molecular weight smaller than 20 KDa.
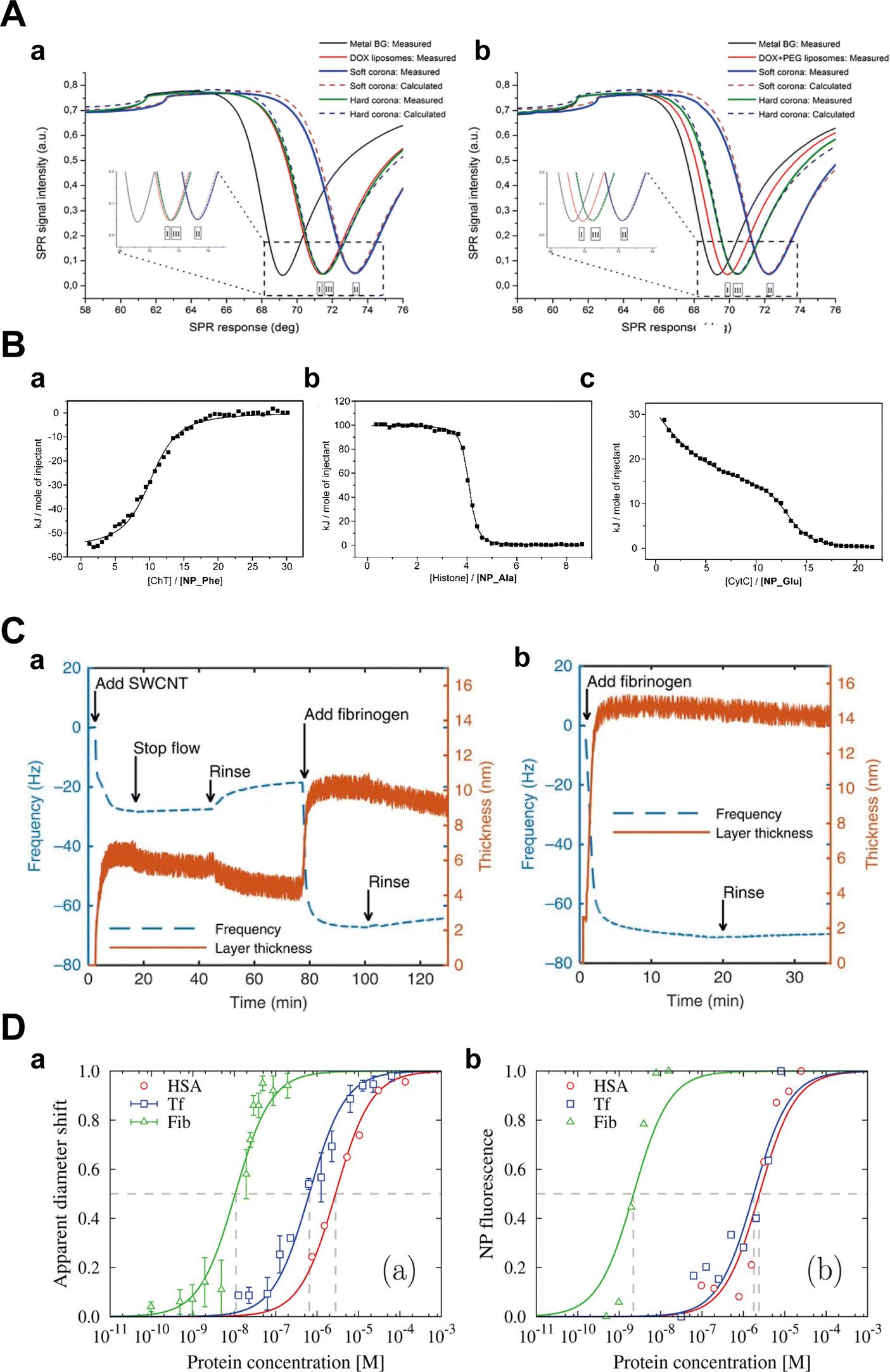 | ||
| Fig. 10 (A) Interactions between liposomes and proteins; layer modelling for (a) DOX and (b) DOX + PEG liposomes under different conditions using measured full SPR angular spectra. Solid curves and dotted curves represent measured and fitted full SPR spectra, respectively. The black line is SPR spectra of metal sensor surface. I, II, and III represent SPR spectra of pure DOX liposomes, SC, and HC, respectively.31 Reproduced from ref. 31 with permission from Controlled Release Society. (B) The binding capacity of proteins to NPs detected by ITC; ITC analysis for the binding of (a) ChT with NP_Phe, (b) histone with NP_Ala, and (c) CytC with NP_Glu in 5 mM sodium phosphate buffer (pH = 7.4). The squares represent the integrated heat changes during the formation of the NP–protein complex and the curve fit to the binding isothermal functions.32 Reproduced from ref. 32 with the permission from American Chemical Society. (C) Changes of frequency (dashed blue curve) and layer thickness (solid orange curve) for a fibrinogen layer deposited on top of a (a) DPPE-PEG5k -SWCNT layer and (b) the gold-coated quartz crystal as measured by QCM-D, and calculated by Voigt viscoelastic model, respectively.33 Reproduced from ref. 33 with permission from Nature Publishing Group. (D) Characterization of NPs in mono-component protein solutions as a function of protein molar concentration along binding isotherms; (a) normalized apparent diameter of NP–protein complexes to the value of bare NPs; (b) normalized NP fluorescence Fnorm after NPs diffusion under a thermal gradient. The symbols represent experimental data and the lines are the best fits with related equations.34 Reproduced from ref. 34 with permission from American Chemical Society. | ||
ΔG = ΔS – TΔH = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) lnK lnK | (5) |
De et al.32 investigated the interaction of Au NPs bearing various L-amino acid functionalities with α-chymotrypsin (ChT), histone and cytochrome c (CytC), and quantified their binding thermodynamics by ITC. The obtained thermodynamic parameters (ΔG, ΔH, and ΔS) revealed significant variations in the interaction mode between Au NPs and different proteins. The binding of NPs with ChT was an enthalpy-driven process, whereas binding with histones and CytC was driven by entropy (Fig. 10B). Yin et al.159 studied the mechanism of interaction between gold nanoclusters functionalized with dihydrolipoic acid (DHLA-AuNCs) with HSA and Tf. The results corroborated that the interaction of DHLA-AuNCs with HSA was an endothermic dynamic process, while it exhibited an exothermic static process with Tf. Besides, ΔH and ΔS associated with the interaction of DHLA-AuNCs with HSA were found to be approximately 64 kJ mol−1 and 327 J mol−1 K−1, respectively. In contrast, ΔH and ΔS for DHLA-AuNCs-Tf interactions were about −5 kJ mol−1 and 74 J mol−1 K−1, respectively. The positive ΔS values indicate that the interactions between DHLA-AuNCs and both proteins are entropy-driven, which is attributed to desolvation of water molecules during hydrophobic interactions. However, potential aggregation of NPs due to concentrated samples required for ITC measurements and thermal perturbations caused by protein denaturation could potentially lead to misleading outcomes. Therefore, integrating complementary methods becomes imperative to validate ITC results and retrieve additional insights into the nature of the interactions.
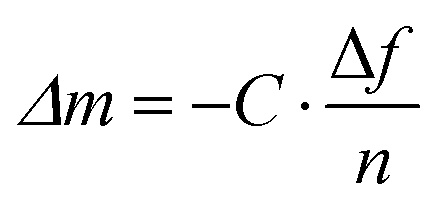 | (6) |
Bisker et al.33 monitored the adsorption of fibrinogen onto DPPE-PEG5k-labeled single-wall carbon nanotubes (SWCNTs) on a gold-coated crystal, and subsequently determined the resulting layer thickness. The adsorption process, as evidenced by a gradual decrease in the crystal's oscillation frequency (Fig. 10C), suggested an increase in layer thickness. Upon measurement, it was revealed that a fibrinogen monolayer (approximately 5 nm thick) was attached to the top of the SWCNTs, implying a parallel alignment of the protein's long axis with the nanotube surface. Kaufman et al.163 investigated the adsorption of BSA, CytC, and myoglobin (Mb) onto mercaptoundecanoic acid-stabilized Au NPs using QCM-D. The results indicated the formation of adsorption layers for all three proteins, consisting of both reversibly and irreversibly adsorbed fractions. BSA exhibited the highest affinity for AuNPs, forming a rigid, irreversibly adsorbed monolayer with a side-down orientation. The irreversibly adsorbed fraction of CytC exhibited a monolayer structure, whereas that of Mb displayed a bilayer. Nevertheless, the presence of water and ions can affect mass change during protein adsorption. Distinguishing the experimental impact of solvents and adsorbed proteins on QCM frequency is particularly challenging.165 Furthermore, the mass change detected by QCM-D is influenced by the viscoelastic properties of adsorbed proteins, complicating the interpretation of quantitative analysis.
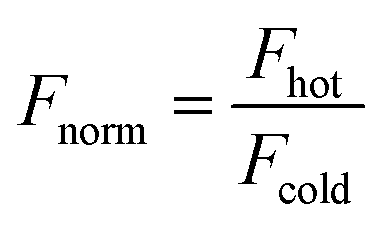 | (7) |
| Fnorm = (1 − FB)·Fnorm,unbound + Fnorm,bound | (8) |
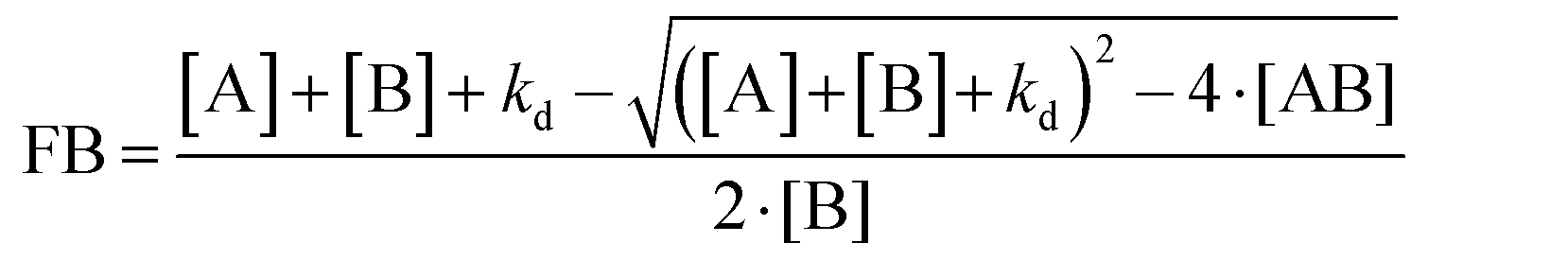 | (9) |
F hot is the fluorescence intensity after thermodiffusion and Fcold is the initial fluorescence intensity of NPs. Funbound and Fbound are the normalized fluorescence intensity of unbound and bound fluorescent proteins, respectively. FB is the bound fraction of [B] while [AB] is the concentration of the binding complexes of [A] and [B]. [A] and [B] are the concentrations of ligands and receptors, respectively.
MST avoids the immobilization procedures necessary for SPR and QCM-D, and minimizes sample consumption compared to ITC, thereby enhancing assay design flexibility.166 Vilanova et al.34 employed MST and differential centrifugal sedimentation (DCS) to assess the binding affinity of HSA, Tf, and Fib with silica NPs. By analyzing the Fnorm under thermal gradients, they calculated the kd order as: HSA > Tf ≫ Fib, corroborated by DCS results (Fig. 10D). Likewise, Wang et al.168 used MST to investigate the binding between BSA and various nanoscale metal–organic frameworks (NMOFs), and they discovered that BSA exhibited a lower binding affinity with amino-modified NMOFs compared to unmodified ones. Despite its high sensitivity to diverse binding-induced interactions, MST cannot measure binding constants in cases of substantial NP aggregation and significant adsorption on the capillaries.167 Moreover, fluorescence labelling of proteins required in MST may change the protein structure and hence influence the adsorption of protein to NPs.
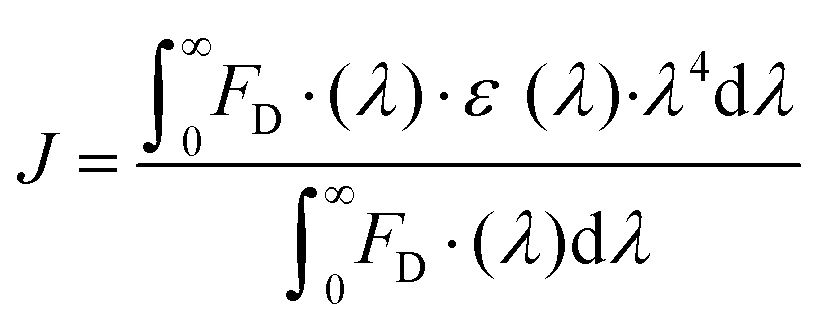 | (10) |
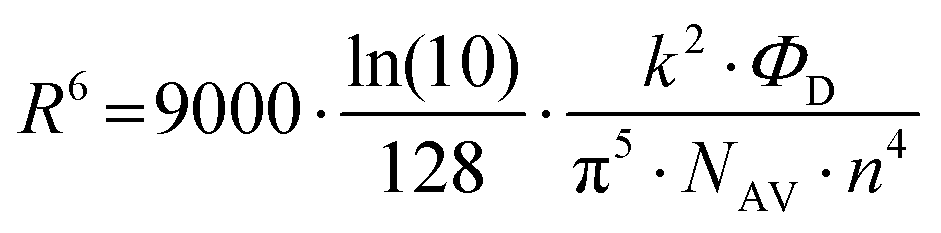 | (11) |
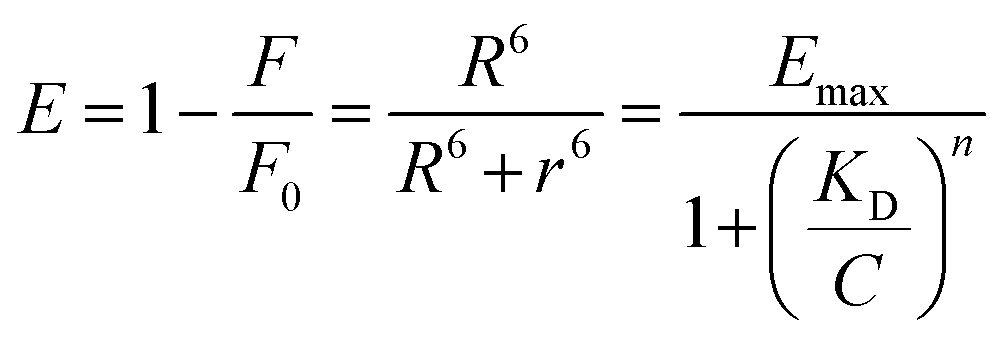 | (12) |
Using FRET, Qu et al.35 investigated HSA adsorption on InP@ZnS quantum dots (QDs) with different chirality (d- and l-penicillamine), revealing distinct binding behaviors, including the affinity and adsorption orientation related to chirality (Fig. 11A). The KD value of l-PA-QDs (0.14 × 10−6 m) was approximately 1.65 times lower than that of d-PA-QDs (0.23 × 10−6 m), indicating that the chirality of surface ligands influenced protein binding to QDs. The l-PA-QDs demonstrated a stronger binding affinity compared to d-PA-QDs. Furthermore, the Emax value for l-PA-QDs (0.70) was lower than that for d-PA-QDs (0.82), suggesting that HSA may adopt a more compact configuration on the surface of d-PA-QDs compared to l-PA-QDs. These results underscore the potential of FRET in elucidating NP–protein interactions. Nevertheless, the effectiveness of FRET highly relies on the distance between the donor and acceptor, making it challenging to detect loosely bound SC proteins on the outer layers of NPs. Moreover, while FRET analysis detects NP–protein interactions, it does not yield quantitative and qualitative insights into the protein corona, e.g., adsorption amount and corona compositions.
 | ||
| Fig. 11 (A) (a) Schematic representation of FRET between InP@ZnS QDs and fluorescently labeled HAS; (b) schematics of the FRET of chiral InP@ZnS QDs.35 Reproduced from ref. 35 with the permission from Wiley-VCH. (B) Kinetics of protein corona formation around silica particles under static particle conditions; (a) representative images obtained from 2D CLSM imaging, depicting the changes in fluorescence intensity of NPs during incubation; (b) an enlarged version of linearized data, focusing mainly on the first two phases of adsorption.172 Reproduced from ref. 172 with permission from American Chemical Society. (C) Standard workflow of DDM: bare NPs are mixed with labeled proteins followed by transferring the mixture into a glass capillary for fluorescence microscopy imaging. Time series of images are then analyzed to extract the 2D Fourier transform and the differential image correlation function (DICF).173 Reproduced from ref. 173 with permission from Wiley-VCH. | ||
Latreille et al.173 applied differential dynamic microscopy (DDM) in fluorescence imaging mode to quantitatively assess protein adsorption on NPs and monitor the resulting NP aggregation behaviors in situ. Their experimental setup involved a standard epifluorescence microscope equipped with an sCMOS camera. Upon the adsorption of fluorescently labeled proteins onto the surface of polystyrene NPs, the NP surface emitted fluorescence signals. By analyzing the fluorescence fluctuations resulting from NP Brownian diffusion, DDM quantifies the contributions of free and adsorbed proteins in NP–protein interactions (Fig. 11C). The results showed that protein adsorption promoted the NP aggregation over a wide concentration range. Moreover, the binding affinity of proteins to polystyrene NPs was dependent on NP concentration, and the composition of protein corona changed with the dilution of proteins in complex solutions. This method is primarily suitable for isotropic NPs (spherical NPs) and may require adjustments when applied to highly anisotropic NPs such as nanorods, nanocubes, and nanosheets.
3.4 Characterization of the conformation change of proteins
Protein binding to NPs can induce significant conformational or structural changes of adsorbed proteins. Given the biological significance of such structural alterations in maintaining physiological homeostasis, it is essential to investigate the impact of NP–protein interactions on protein structure.174–176 Commonly used characterization techniques include circular dichroism (CD) spectroscopy, surface-enhanced Raman scattering (SERS), and nuclear magnetic resonance spectroscopy (NMR).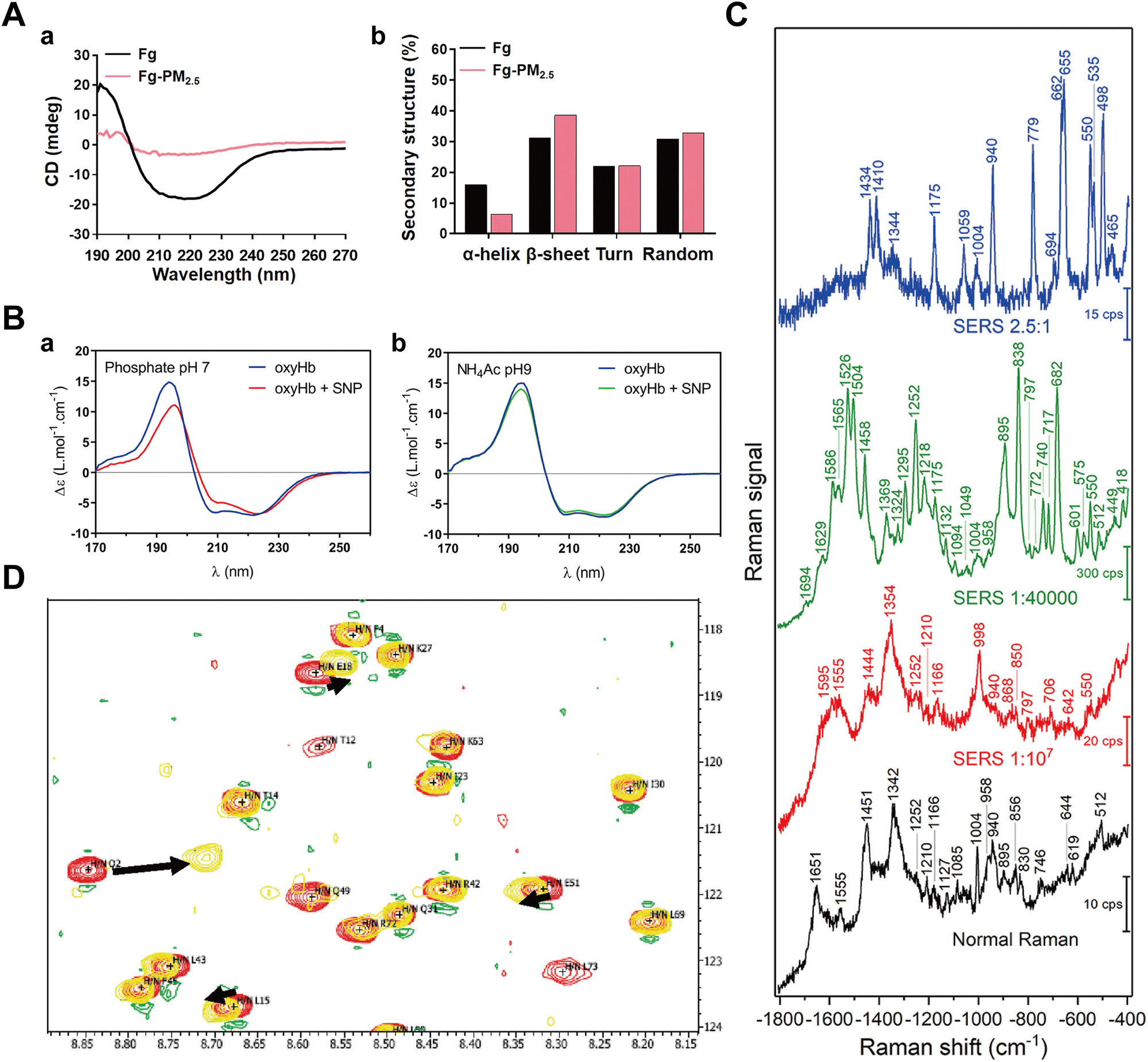 | ||
| Fig. 12 (A) Secondary structure changes detected by CD; (a) CD spectra of Fg in the absence and presence of PM2.5; (b) Content of α-helix, β-sheet, turn and random of Fg in the absence and presence of PM2.5, calculated by DichroWeb.36 Reproduced from ref. 36 with permission from Elsevier Ltd. (B) SRCD spectroscopy of oxyHb in the absence and presence of SNPs at 22 °C in (a) phosphate buffer pH 7 and (b) NH4Ac buffer pH 9.180 Reproduced from ref. 180 with permission from American Chemical Society. (C) SERS spectroscopy of BSA solution in the absence and presence of Au NPs. The blank curve is the SERS spectroscopy of pure BSA. The red, green, and blue curves represent the SERS spectroscopy of the sample with the Au NPs:BSA ratio of 1:107, 1:140000, and 2.5:1, respectively.37 Reproduced from ref. 37 with permission from Frontiers. (D) 2D [15N-1H] NMR spectroscopy of free hUbq (red) and hUbq plus Au NP (yellow) samples at pH 7. Black arrows indicate NH backbone groups with the largest chemical shift perturbations (Q2, L15, and E18) in this spectral window.181 Reproduced from ref. 38 with permission from American Chemical Society. | ||
Synchrotron-radiation circular dichroism (SRCD) is exploited as an advanced tool for characterizing protein structures.182 Leveraging the intense light of a synchrotron beam, SRCD improves the precision of CD spectroscopy and facilitates the analysis of protein structures in the low UV regions.183 Sanchez-Guzman et al.180 applied SRCD to investigate the impact of silica NPs on the secondary structure of oxyhemoglobin (oxyHb) present in SC and HC, revealing significant alterations in the secondary structure of oxyHb within the HC, while no influence on the weakly bound oxyHb in the SC (Fig. 12B). Notably, light scattering arising from aggregated or large particles can potentially interfere with CD results.
The above-mentioned methods for protein corona characterization are summarized in Table 3.
| Characteristics | Characterization methods | Abbreviations | Brief description | Ref. |
|---|---|---|---|---|
| Size changes | Dynamic light scattering | DLS | Hydrodynamic size | 102 and 104 |
| Nanoparticle tracking analysis | NTA | NP concentration and size | 108 and 110 | |
| Fluorescence correlation spectroscopy | FCS | Changes in diffusion coefficient and size of NP | 26 and 117 | |
| Diffusion 19fluorine-magnetic resonance | 19F-NMR | Changes in diffusion coefficient and size of NP | 120 | |
| Change of morphology | Transmission electron microscopy | TEM | Morphology of NP–protein complex | 128 |
| Cryogenic TEM | Cryo-TEM | 28 | ||
| Scanning electron microscopy | SEM | Thickness of protein corona | 132 | |
| Atomic force microscopy | AFM | 134 | ||
| NP–protein interactions | Surface plasmon resonance | SPR | Adsorption kinetic parameters | 138 |
| Isothermal titration calorimetry | ITC | Thermodynamic parameters of binding | 140 and 141 | |
| Quartz crystal microbalance with dissipation monitoring | QCM-D | Mass changes induced by protein adsorption | 144 | |
| Microscale thermophoresis | MST | Non-radiative transmission of excitation energy | 145 | |
| Fluorescence resonance energy transfer | FRET | Binding parameters | 147 | |
| Conformation changes of protein | Circular dichroism | CD | Secondary structural changes of proteins induced by NP–protein interaction | 177 |
| Surface-enhanced Raman scattering | SERS | 184 | ||
| Nuclear magnetic resonance | NMR | 190 |
4. Conclusions and outlook
The coverage of blood proteins reshapes the innate surface properties of NPs and thus dictates their ultimate fate in vivo, impacting critical behaviors such as pharmacokinetics, biodistribution, targetability, and therapeutic efficacy. A comprehensive understanding of NP–protein interaction along with an insight into the composition of protein corona is crucial for elucidating the suboptimal therapeutic outcomes of nanomedicines during clinical trials and advancing the clinical translation of nanomedicines. This review provides an overview of separation and in situ characterization techniques for comprehensively unravelling the intricacies inherent in NP–protein interactions.Previous techniques for characterizing the protein corona in situ are primarily focused on the size, morphology, and adsorption kinetics, yet lack robust tools for quantification of the protein corona. Although the BCA assay offers a means to quantify corona proteins, the process of detaching adsorbed proteins from NPs following the isolation of NP–protein complexes from the incubation medium may lead to the loss and alteration of protein corona fingerprints.193,194 Consequently, current research has focused on developing new methods that allow in situ quantitative analysis of protein corona. Lo Giudice et al.195 utilized flow cytometry to investigate NP–protein interactions by employing an immunoprobe (mTfQD630) designed to specifically recognize the Tf epitope AA. This approach facilitated the in situ characterization of specific motifs presenting in the biomolecular corona, thereby contributing to a better understanding and potential prediction of NP-biomolecular interactions with cells. Moreover, a key focus in current protein corona analysis lies in achieving direct and quantitative determination of NP–protein interactions in situ and at the single-NP level. For instance, Tan et al.196 developed an innovative real-time three-dimensional single-particle-tracking microscopy combined with a lock-on filtering algorithm (RTY-3D-SPT) to detect the NPs-protein interactions at the single particle level, providing insights into in situ protein corona on a per-particle basis and bridging gaps in corona characterization at the single-particle level. Although significant strides have been made in characterizing the protein corona, the direct deciphering of protein corona compositions in situ and understanding the heterogeneity among NPs remain challenging.
Nonspecific NP–protein interactions are typically highly heterogeneous, resulting in an undetectable biological behaviors for individual NPs. Therefore, in addition to detecting the NP–protein interactions in situ and at the single-NP level, it is also important to analyze the heterogeneity of protein interaction among different NPs. Niu et al.197 established an in situ approach allowing quantitative analysis of NP−blood protein adsorption at the individual NP level and also the interparticle heterogeneity of NP–protein interactions based on a dual fluorescence quantification technique. This approach not only allowed the in situ quantitative monitoring of protein adsorption kinetics and the aggregation status of NPs in the serum, it also revealed the interparticle heterogeneity of NP–protein interactions with the single-NP resolution.
In summary, achieving a precise and comprehensive analysis of the protein corona requires careful consideration of several crucial points. Firstly, gaining a holistic understanding of various aspects of the protein corona require the application of multiple complementary techniques, given the distinct focus of these methods. Secondly, standardizing both preparation and characterization procedures is a prerequisite for generating reproducible data, therefore providing reliable insights into the biological effects of nanomedicines. Thirdly, characterizing NPs at the single-particle level is essential to unveil the heterogeneity of NPs and their intricate interactions with proteins, potentially leading to varied behaviors among NPs within the same sample. Last but not least, multi-omics approaches should be applied to obtain insights into proteins as well as lipids and other relevant components present in phycological fluids associated with NPs. Looking forward, collaborative interdisciplinary efforts hold great promise of inspiring breakthroughs in the characterization of protein corona in the future.
Data avaibility
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Foundation of China (32201149) and the Shandong Provincial Natural Science Fund for Excellent Young Scientists Fund Program (Overseas) (2022HWYQ-063). Open Access funding provided by the Max Planck Society.References
- B. Pelaz, C. Alexiou, R. A. Alvarez-Puebla, F. Alves, A. M. Andrews, S. Ashraf, L. P. Balogh, L. Ballerini, A. Bestetti, C. Brendel, S. Bosi, M. Carril, W. C. W. Chan, C. Chen, X. Chen, X. Chen, Z. Cheng, D. Cui, J. Du, C. Dullin, A. Escudero, N. Feliu, M. Gao, M. George, Y. Gogotsi, A. Grünweller, Z. Gu, N. J. Halas, N. Hampp, R. K. Hartmann, M. C. Hersam, P. Hunziker, J. Jian, X. Jiang, P. Jungebluth, P. Kadhiresan, K. Kataoka, A. Khademhosseini, J. Kopeček, N. A. Kotov, H. F. Krug, D. S. Lee, C.-M. Lehr, K. W. Leong, X.-J. Liang, M. Ling Lim, L. M. Liz-Marzán, X. Ma, P. Macchiarini, H. Meng, H. Möhwald, P. Mulvaney, A. E. Nel, S. Nie, P. Nordlander, T. Okano, J. Oliveira, T. H. Park, R. M. Penner, M. Prato, V. Puntes, V. M. Rotello, A. Samarakoon, R. E. Schaak, Y. Shen, S. Sjöqvist, A. G. Skirtach, M. G. Soliman, M. M. Stevens, H.-W. Sung, B. Z. Tang, R. Tietze, B. N. Udugama, J. S. VanEpps, T. Weil, P. S. Weiss, I. Willner, Y. Wu, L. Yang, Z. Yue, Q. Zhang, Q. Zhang, X.-E. Zhang, Y. Zhao, X. Zhou and W. J. Parak, ACS Nano, 2017, 11, 2313–2381 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- M. A. Younis, H. M. Tawfeek, A. A. H. Abdellatif, J. A. Abdel-Aleem and H. Harashima, Adv. Drug Delivery Rev., 2022, 181, 114083 CrossRef CAS PubMed.
- K. E. Wheeler, A. J. Chetwynd, K. M. Fahy, B. S. Hong, J. A. Tochihuitl, L. A. Foster and I. Lynch, Nat. Nanotechnol., 2021, 16, 617–629 CrossRef CAS PubMed.
- P. L. Latreille, M. Le Goas, S. Salimi, J. Robert, G. De Crescenzo, D. C. Boffito, V. A. Martinez, P. Hildgen and X. Banquy, ACS Nano, 2022, 16, 1689–1707 CrossRef CAS PubMed.
- S. Abbina, L. E. Takeuchi, P. Anilkumar, K. Yu, J. C. Rogalski, R. A. Shenoi, I. Constantinescu and J. N. Kizhakkedathu, Nat. Commun., 2020, 11, 3048 CrossRef CAS PubMed.
- M. Safi, J. Courtois, M. Seigneuret, H. Conjeaud and J. F. Berret, Biomaterials, 2011, 32, 9353–9363 CrossRef CAS PubMed.
- F. Barbero, S. Michelini, O. H. Moriones, J. Patarroyo, J. Rosell, M. F. Gusta, M. Vitali, L. Martín, F. Canals, A. Duschl, J. Horejs-Hoeck, L. Mondragón, N. G. Bastús and V. Puntes, Bioconjugate Chem., 2022, 33, 1505–1514 CrossRef CAS.
- V. P. Vu, G. B. Gifford, F. Chen, H. Benasutti, G. Wang, E. V. Groman, R. Scheinman, L. Saba, S. M. Moghimi and D. Simberg, Nat. Nanotechnol., 2019, 14, 260–268 CrossRef CAS.
- F. Chen, G. Wang, J. I. Griffin, B. Brenneman, N. K. Banda, V. M. Holers, D. S. Backos, L. Wu, S. M. Moghimi and D. Simberg, Nat. Nanotechnol., 2017, 12, 387–393 CrossRef CAS PubMed.
- N. Bertrand, P. Grenier, M. Mahmoudi, E. M. Lima, E. A. Appel, F. Dormont, J.-M. Lim, R. Karnik, R. Langer and O. C. Farokhzad, Nat. Commun., 2017, 8, 777 CrossRef PubMed.
- F. Barbero, L. Russo, M. Vitali, J. Piella, I. Salvo, M. L. Borrajo, M. Busquets-Fité, R. Grandori, N. G. Bastús, E. Casals and V. Puntes, Semin. Immunol., 2017, 34, 52–60 CrossRef CAS PubMed.
- L. Digiacomo, D. Pozzi, S. Palchetti, A. Zingoni and G. Caracciolo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1615 CAS.
- M. Tonigold, J. Simon, D. Estupiñán, M. Kokkinopoulou, J. Reinholz, U. Kintzel, A. Kaltbeitzel, P. Renz, M. P. Domogalla, K. Steinbrink, I. Lieberwirth, D. Crespy, K. Landfester and V. Mailänder, Nat. Nanotechnol., 2018, 13, 862–869 CrossRef CAS PubMed.
- S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailänder and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef PubMed.
- M. Li, S. Jiang, J. Simon, D. Paßlick, M.-L. Frey, M. Wagner, V. Mailänder, D. Crespy and K. Landfester, Nano Lett., 2021, 21, 1591–1598 CrossRef CAS PubMed.
- S. Jiang, D. Prozeller, J. Pereira, J. Simon, S. Han, S. Wirsching, M. Fichter, M. Mottola, I. Lieberwirth, S. Morsbach, V. Mailänder, S. Gehring, D. Crespy and K. Landfester, Nanoscale, 2020, 12, 2626–2637 RSC.
- G. Perini, F. Giulimondi, V. Palmieri, A. Augello, L. Digiacomo, E. Quagliarini, D. Pozzi, M. Papi and G. Caracciolo, Pharmaceutics, 2021, 13, 378 CrossRef CAS PubMed.
- P. C. Ke, S. Lin, W. J. Parak, T. P. Davis and F. Caruso, ACS Nano, 2017, 11, 11773–11776 CrossRef CAS PubMed.
- C. Weber, S. Morsbach and K. Landfester, Angew. Chem., Int. Ed., 2019, 58, 12787–12794 CrossRef CAS PubMed.
- S. Winzen, S. Schoettler, G. Baier, C. Rosenauer, V. Mailaender, K. Landfester and K. Mohr, Nanoscale, 2015, 7, 2992–3001 RSC.
- S. Milani, F. Baldelli Bombelli, A. S. Pitek, K. A. Dawson and J. Rädler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, M. Foy, T. Berggård, S. C. Donnelly, G. Cagney, S. Linse and K. A. Dawson, Angew. Chem., Int. Ed., 2007, 46, 5754–5756 CrossRef CAS PubMed.
- C. Wang, B. Chen, M. He and B. Hu, ACS Nano, 2021, 15, 3108–3122 CrossRef CAS PubMed.
- K. Kristensen, R. Münter, P. J. Kempen, M. E. Thomsen, A. Stensballe and T. L. Andresen, Acta Biomater., 2021, 130, 460–472 CrossRef CAS PubMed.
- L. Shang and G. U. Nienhaus, Acc. Chem. Res., 2017, 50, 387–395 CrossRef CAS PubMed.
- M. Kokkinopoulou, J. Simon, K. Landfester, V. Mailänder and I. Lieberwirth, Nanoscale, 2017, 9, 8858–8870 RSC.
- S. Sheibani, K. Basu, A. Farnudi, A. Ashkarran, M. Ichikawa, J. F. Presley, K. H. Bui, M. R. Ejtehadi, H. Vali and M. Mahmoudi, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- V. Mirshafiee, R. Kim, S. Park, M. Mahmoudi and M. L. Kraft, Biomaterials, 2016, 75, 295–304 CrossRef CAS PubMed.
- Y. Chong, C. Ge, Z. Yang, J. A. Garate, Z. Gu, J. K. Weber, J. Liu and R. Zhou, ACS Nano, 2015, 9, 5713–5724 CrossRef CAS PubMed.
- O. K. Kari, T. Rojalin, S. Salmaso, M. Barattin, H. Jarva, S. Meri, M. Yliperttula, T. Viitala and A. Urtti, Drug Delivery Transl. Res., 2017, 7, 228–240 CrossRef CAS PubMed.
- M. De, C.-C. You, S. Srivastava and V. M. Rotello, J. Am. Chem. Soc., 2007, 129, 10747–10753 CrossRef CAS PubMed.
- G. Bisker, J. Dong, H. D. Park, N. M. Iverson, J. Ahn, J. T. Nelson, M. P. Landry, S. Kruss and M. S. Strano, Nat. Commun., 2016, 7, 10241 CrossRef CAS PubMed.
- O. Vilanova, J. J. Mittag, P. M. Kelly, S. Milani, K. A. Dawson, J. O. Rädler and G. Franzese, ACS Nano, 2016, 10, 10842–10850 CrossRef CAS PubMed.
- S. Qu, F. Sun, Z. Qiao, J. Li and L. Shang, Small, 2020, 16, 1–7 Search PubMed.
- Z. Liu, Q. Zhu, E. Song and Y. Song, J. Hazard. Mater., 2021, 414, 125499 CrossRef CAS PubMed.
- G. P. Szekeres and J. Kneipp, Front. Chem., 2019, 7, 30 CrossRef CAS PubMed.
- V. P. Brahmkhatri, A. Singh, A. Chakraborty, R. S. Sharma, K. Chandra and H. S. Atreya, Appl. Surf. Sci. Adv., 2022, 11, 100272 CrossRef.
- C. Pisani, J. C. Gaillard, C. Dorandeu, C. Charnay, Y. Guari, J. Chopineau, J. M. Devoisselle, J. Armengaud and O. Prat, Nanoscale, 2017, 9, 5769–5772 RSC.
- L. Digiacomo, F. Giulimondi, A. L. Capriotti, S. Piovesana, C. M. Montone, R. Z. Chiozzi, A. Laganà, M. Mahmoudi, D. Pozzi and G. Caracciolo, Nanoscale Adv., 2021, 3, 3824–3834 RSC.
- W. W.-F. Leung, Centrifugal Separations in Biotechnology, 2007, 19–35 CrossRef.
- M. Singh, H. Chand and K. C. Gupta, Chem. Biodiversity, 2005, 2, 809–824 CrossRef CAS PubMed.
- M. Iqbal and R. E. Verrall, J. Phys. Chem., 1987, 91, 1935–1941 CrossRef CAS.
- F. Pederzoli, G. Tosi, M. A. Vandelli, D. Belletti, F. Forni and B. Ruozi, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, 1–23 Search PubMed.
- T. Lima, K. Bernfur, M. Vilanova and T. Cedervall, Sci. Rep., 2020, 10, 1–9 CrossRef PubMed.
- G. Caracciolo, L. Callipo, S. C. De Sanctis, C. Cavaliere, D. Pozzi and A. Laganà, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 536–543 CrossRef CAS PubMed.
- R. Pattipeiluhu, S. Crielaard, I. Klein-Schiphorst, B. I. Florea, A. Kros and F. Campbell, ACS Cent. Sci., 2020, 6, 535–545 CrossRef CAS PubMed.
- M. Brückner, J. Simon, S. Jiang, K. Landfester and V. Mailänder, Acta Biomater., 2020, 114, 333–342 CrossRef PubMed.
- L. Böhmert, L. Voß, V. Stock, A. Braeuning, A. Lampen and H. Sieg, Nanoscale Adv., 2020, 2, 563–582 RSC.
- T. P. Doan-Nguyen and D. Crespy, Chem. Soc. Rev., 2022, 51, 8612–8651 RSC.
- D. Docter, U. Distler, W. Storck, J. Kuharev, D. Wünsch, A. Hahlbrock, S. K. Knauer, S. Tenzer and R. H. Stauber, Nat. Protoc., 2014, 9, 2030–2044 CrossRef CAS PubMed.
- A. Perez-Potti, H. Lopez, B. Pelaz, A. Abdelmonem, M. G. Soliman, I. Schoen, P. M. Kelly, K. A. Dawson, W. J. Parak, Z. Krpetic and M. P. Monopoli, Sci. Rep., 2021, 11, 6443 CrossRef CAS PubMed.
- D. Di Silvio, N. Rigby, B. Bajka, A. Mayes, A. Mackie and F. Baldelli Bombelli, Nanoscale, 2015, 7, 11980–11990 RSC.
- M. Werwie, N. Fehr, X. Xu, T. Basché and H. Paulsen, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 1651–1656 CrossRef CAS PubMed.
- K. S. Siddiqi, A. Ur Rahman, T. Tajuddin and A. Husen, Nanoscale Res. Lett., 2016, 11, 498 CrossRef PubMed.
- U. Sakulkhu, M. Mahmoudi, L. Maurizi, G. Coullerez, M. Hofmann-Amtenbrink, M. Vries, M. Motazacker, F. Rezaee and H. Hofmann, Biomater. Sci., 2015, 3, 265–278 RSC.
- K. N. L. Hoang, K. E. Wheeler and C. J. Murphy, Anal. Chem., 2022, 94, 4737–4746 CrossRef CAS PubMed.
- D. Bonvin, D. Chiappe, M. Moniatte, H. Hofmann and M. Mionić Ebersold, Analyst, 2017, 142, 3805–3815 RSC.
- J. E. Blume, W. C. Manning, G. Troiano, D. Hornburg, M. Figa, L. Hesterberg, T. L. Platt, X. Zhao, R. A. Cuaresma, P. A. Everley, M. Ko, H. Liou, M. Mahoney, S. Ferdosi, E. M. Elgierari, C. Stolarczyk, B. Tangeysh, H. Xia, R. Benz, A. Siddiqui, S. A. Carr, P. Ma, R. Langer, V. Farias and O. C. Farokhzad, Nat. Commun., 2020, 11, 1–14 CrossRef PubMed.
- A. A. Ashkarran, N. Dararatana, D. Crespy, G. Caracciolo and M. Mahmoudi, Nanoscale, 2020, 12, 2374–2383 RSC.
- E. Quagliarini, L. Digiacomo, D. Caputo, A. Coppola, H. Amenitsch, G. Caracciolo and D. Pozzi, Nanomaterials, 2022, 12 Search PubMed.
- A. J. Chetwynd, E. J. Guggenheim, S. M. Briffa, J. A. Thorn, I. Lynch and E. Valsami-Jones, Nanomaterials, 2018, 8, 99 CrossRef PubMed.
- A. L. Capriotti, G. Caracciolo, C. Cavaliere, V. Colapicchioni, S. Piovesana, D. Pozzi and A. Laganà, Chromatographia, 2014, 77, 755–769 CrossRef CAS.
- N. Li, S. Zeng, L. He and W. Zhong, Anal. Chem., 2010, 82, 7460–7466 CrossRef CAS PubMed.
- A. K. Brewer, Chromatographia, 2021, 84, 807–811 CrossRef CAS.
- A. Oliva, M. Llabrés and J. B. Fariña, J. Pharm. Biomed. Anal., 2001, 25, 833–841 CrossRef CAS PubMed.
- P. Hong, S. Koza and E. S. P. Bouvier, J. Liq. Chromatogr. Relat. Technol., 2012, 35, 2923–2950 CrossRef CAS PubMed.
- T.-Y. Huang, L.-M. Chi and K.-Y. Chien, J. Chromatogr. A, 2018, 1571, 201–212 CrossRef CAS PubMed.
- S. Fekete, A. Beck, J.-L. Veuthey and D. Guillarme, J. Pharm. Biomed. Anal., 2014, 101, 161–173 CrossRef CAS PubMed.
- K. Kristensen, T. B. Engel, A. Stensballe, J. B. Simonsen and T. L. Andresen, J. Controlled Release, 2019, 307, 1–15 CrossRef CAS PubMed.
- M. Hadjidemetriou, Z. Al-Ahmady, M. Mazza, R. F. Collins, K. Dawson and K. Kostarelos, ACS Nano, 2015, 9, 8142–8156 CrossRef CAS PubMed.
- K. Yang, B. Mesquita, P. Horvatovich and A. Salvati, Acta Biomater., 2020, 106, 314–327 CrossRef CAS PubMed.
- P. Reschiglian, A. Zattoni, B. Roda, E. Michelini and A. Roda, Trends Biotechnol., 2005, 23, 475–483 CrossRef CAS PubMed.
- F. Quattrini, G. Berrecoso, J. Crecente-Campo and M. J. Alonso, Drug Delivery Transl. Res., 2021, 11, 373–395 CrossRef CAS PubMed.
- U. B. Kavurt, M. Marioli, W. T. Kok and D. Stamatialis, J. Chem. Technol. Biotechnol., 2015, 90, 11–18 CrossRef CAS.
- T. Otte, H. Pasch, T. Macko, R. Brüll, F. J. Stadler, J. Kaschta, F. Becker and M. Buback, J. Chromatogr. A, 2011, 1218, 4257–4267 CrossRef CAS PubMed.
- F. A. Messaud, R. D. Sanderson, J. R. Runyon, T. Otte, H. Pasch and S. K. R. Williams, Prog. Polym. Sci., 2009, 34, 351–368 CrossRef CAS.
- M. Wagner, S. Holzschuh, A. Traeger, A. Fahr and U. S. Schubert, Anal. Chem., 2014, 86, 5201–5210 CrossRef CAS PubMed.
- G. Yohannes, M. Jussila, K. Hartonen and M.-L. Riekkola, J. Chromatogr. A, 2011, 1218, 4104–4116 CrossRef CAS PubMed.
- I. Alberg, S. Kramer, M. Schinnerer, Q. Hu, C. Seidl, C. Leps, N. Drude, D. Möckel, C. Rijcken, T. Lammers, M. Diken, M. Maskos, S. Morsbach, K. Landfester, S. Tenzer, M. Barz and R. Zentel, Small, 2020, 16(18), 1907574 CrossRef CAS PubMed.
- U. Hansen and A. F. Thünemann, Langmuir, 2015, 31, 6842–6852 CrossRef CAS PubMed.
- J. Ashby, S. Pan and W. Zhong, ACS Appl. Mater. Interfaces, 2014, 6, 15412–15419 CrossRef CAS PubMed.
- J. Ashby, S. Schachermeyer, S. Pan and W. Zhong, Anal. Chem., 2013, 85, 7494–7501 CrossRef CAS PubMed.
- C. Weber, J. Simon, V. Mailänder, S. Morsbach and K. Landfester, Acta Biomater., 2018, 76, 217–224 CrossRef CAS PubMed.
- J. D. Robertson, L. Rizzello, M. Avila-Olias, J. Gaitzsch, C. Contini, M. S. Magoń, S. A. Renshaw and G. Battaglia, Sci. Rep., 2016, 6, 27494 CrossRef CAS PubMed.
- H. Mohammad-Beigi, Y. Hayashi, C. M. Zeuthen, H. Eskandari, C. Scavenius, K. Juul-Madsen, T. Vorup-Jensen, J. J. Enghild and D. S. Sutherland, Nat. Commun., 2020, 11(1), 4535 CrossRef CAS PubMed.
- K. A. Dawson and Y. Yan, Nat. Nanotechnol., 2021, 16, 229–242 CrossRef CAS PubMed.
- D. Baimanov, J. Wang, J. Zhang, K. Liu, Y. Cong, X. Shi, X. Zhang, Y. Li, X. Li, R. Qiao, Y. Zhao, Y. Zhou, L. Wang and C. Chen, Nat. Commun., 2022, 13(1), 5389 CrossRef CAS PubMed.
- Y. Chu, W. Tang, Z. Zhang, C. Li, J. Qian, X. Wei, T. Ying, W. Lu and C. Zhan, Nano Lett., 2021, 21, 2124–2131 CrossRef CAS PubMed.
- H. Wang, Y. Lin, K. Nienhaus and G. U. Nienhaus, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2018, 10, e1500 Search PubMed.
- R. García-álvarez and M. Vallet-Regí, Nanomaterials, 2021, 11(4), 888 CrossRef PubMed.
- D. Baimanov, R. Cai and C. Chen, Bioconjugate Chem., 2019, 30, 1923–1937 CrossRef CAS PubMed.
- A. Tukova, I. C. Kuschnerus, A. Garcia-Bennett, Y. Wang and A. Rodger, Nanomaterials, 2021, 11 Search PubMed.
- H. Wang, K. Nienhaus, L. Shang and G. U. Nienhaus, Chin. J. Chem., 2022, 40, 2685–2693 CrossRef CAS.
- M. Martinez-Moro, D. Di Silvio and S. E. Moya, Biophys. Chem., 2019, 253, 106218 CrossRef CAS PubMed.
- A. Maity, S. K. De and A. Chakraborty, J. Phys. Chem. B, 2021, 125, 2113–2123 CrossRef CAS PubMed.
- J. S. Yoneda and M. B. Cardoso, Nanomedicine, 2023, 18, 709–711 CrossRef CAS PubMed.
- S. Kihara, N. J. van der Heijden, C. K. Seal, J. P. Mata, A. E. Whitten, I. Köper and D. J. McGillivray, Bioconjugate Chem., 2019, 30, 1067–1076 CrossRef CAS PubMed.
- D. Prozeller, S. Morsbach and K. Landfester, Nanoscale, 2019, 11, 19265–19273 RSC.
- X. Xu and J. Dzubiella, Colloid Polym. Sci., 2020, 298, 747–759 CrossRef CAS.
- X. Zhang, H. Shi, R. Zhang, J. Zhang, F. Xu, L. Qiao and S. Yu, Part. Part. Syst. Charact., 2019, 36, 1800257 CrossRef.
- S. Dominguez-Medina, S. Chen, J. Blankenburg, P. Swanglap, C. F. Landes and S. Link, Annu. Rev. Phys. Chem., 2016, 67, 489–514 CrossRef CAS PubMed.
- Z. Li, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2009, 80, 61204 CrossRef PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS PubMed.
- K. Rausch, A. Reuter, K. Fischer and M. Schmidt, Biomacromolecules, 2010, 11, 2836–2839 CrossRef CAS PubMed.
- S. Balog, L. Rodriguez-Lorenzo, C. A. Monnier, M. Obiols-Rabasa, B. Rothen-Rutishauser, P. Schurtenberger and A. Petri-Fink, Nanoscale, 2015, 7, 5991–5997 RSC.
- T. Zhang, C. Dong and J. Ren, Anal. Chem., 2023, 95, 2029–2038 CrossRef CAS PubMed.
- K. Nienhaus and G. U. Nienhaus, Biointerphases, 2020, 15, 61201 CrossRef CAS PubMed.
- A. Malloy and B. Carr, Part. Part. Syst. Charact., 2006, 23, 197–204 CrossRef.
- H. Saveyn, B. De Baets, O. Thas, P. Hole, J. Smith and P. Van der Meeren, J. Colloid Interface Sci., 2010, 352, 593–600 CrossRef CAS PubMed.
- Z. Varga, B. Fehér, D. Kitka, A. Wacha, A. Bóta, S. Berényi, V. Pipich and J.-L. Fraikin, Colloids Surf., B, 2020, 192, 111053 CrossRef CAS PubMed.
- V. Filipe and A. Hawe, and C. evaluation of N. T. A. (NTA) by N. for the measurement of nanoparticles and protein aggregates. W. Jiskoot, Pharm. Res., 2010, 27, 796–810 CrossRef CAS PubMed.
- M. S. Bannon, A. López Ruiz, K. Corrotea Reyes, M. Marquez, Z. Wallizadeh, M. Savarmand, C. A. LaPres, J. Lahann and K. McEnnis, Part. Part. Syst. Charact., 2021, 38, 2100016 CrossRef CAS.
- A. Jedlovszky-Hajdú, F. B. Bombelli, M. P. Monopoli, E. Tombácz and K. A. Dawson, Langmuir, 2012, 28, 14983–14991 CrossRef PubMed.
- J. Y. Jeong, H. Joung, G. J. Jang and S. Y. Han, Bull. Korean Chem. Soc., 2023, 44, 551–557 CrossRef CAS.
- M. Martinez-moro, D. Di Silvio and S. E. Moya, Biophys. Chem., 2019, 253, 106218 CrossRef CAS PubMed.
- O. Krichevsky and G. Bonnet, Rep. Prog. Phys., 2002, 65, 251–297 CrossRef CAS.
- B. Pelaz, P. Del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernández, J. M. De La Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS PubMed.
- I. Negwer, A. Best, M. Schinnerer, O. Schäfer, L. Capeloa, M. Wagner, M. Schmidt, V. Mailänder, M. Helm, M. Barz, H.-J. Butt and K. Koynov, Nat. Commun., 2018, 9, 5306 CrossRef CAS PubMed.
- R. Mashiach, L. Avram and A. Bar-Shir, Nano Lett., 2022, 22, 8519–8525 CrossRef CAS PubMed.
- M. Carril, D. Padro, P. Del Pino, C. Carrillo-Carrion, M. Gallego and W. J. Parak, Nat. Commun., 2017, 8(1), 1542 CrossRef PubMed.
- D. Maiolo, P. Del Pino, P. Metrangolo, W. J. Parak and F. Baldelli Bombelli, Nanomedicine, 2015, 10, 3231–3247 CrossRef CAS PubMed.
- J. Piella, N. G. Bastús and V. Puntes, Bioconjugate Chem., 2016, 28, 88–97 CrossRef PubMed.
- H. Wang, L. Shang, P. Maffre, S. Hohmann, F. Kirschhöfer, G. Brenner-Weiß and G. U. Nienhaus, Small, 2016, 12, 5836–5844 CrossRef CAS PubMed.
- H. Yin, R. Chen, P. S. Casey, P. C. Ke, T. P. Davis and C. Chen, RSC Adv., 2015, 5, 73963–73973 RSC.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- S. Milani, F. Baldelli Bombelli, A. S. Pitek, K. A. Dawson and J. Rädler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS PubMed.
- Z. S. Al-Ahmady, M. Hadjidemetriou, J. Gubbins and K. Kostarelos, J. Controlled Release, 2018, 276, 157–167 CrossRef CAS PubMed.
- L. Li, X. Jiang and J. Gao, Adv. Mater. Interfaces, 2022, 9, 1–17 Search PubMed.
- L. S. Franqui, M. A. De Farias, R. V. Portugal, C. A. R. Costa, R. R. Domingues, A. G. Souza Filho, V. R. Coluci, A. F. P. Leme and D. S. T. Martinez, Mater. Sci. Eng., C, 2019, 100, 363–377 CrossRef CAS PubMed.
- M. Ovais, S. K. Nethi, S. Ullah, I. Ahmad, S. Mukherjee and C. Chen, Nanomedicine, 2020, 15, 1037–1061 CrossRef CAS PubMed.
- V. Serpooshan, M. Mahmoudi, M. Zhao, K. Wei, S. Sivanesan, K. Motamedchaboki, A. V. Malkovskiy, A. B. Gladstone, J. E. Cohen, P. C. Yang, J. Rajadas, D. Bernstein, Y. J. Woo and P. Ruiz-Lozano, Adv. Funct. Mater., 2015, 25, 4379–4389 CrossRef CAS PubMed.
- J. Schaefer, C. Schulze, E. E. J. Marxer, U. F. Schaefer, W. Wohlleben, U. Bakowsky and C.-M. Lehr, ACS Nano, 2012, 6, 4603–4614 CrossRef CAS PubMed.
- K. R. Kim, J. Kim, J. H. Back, J. E. Lee and D. R. Ahn, ACS Nano, 2021, 16, 7331–7343 CrossRef PubMed.
- D. Evanko, Nat. Methods, 2012, 9, 778–779 CrossRef PubMed.
- L. Treuel, K. A. Eslahian, D. Docter, T. Lang, R. Zellner, K. Nienhaus, G. U. Nienhaus, R. H. Stauber and M. Maskos, Phys. Chem. Chem. Phys., 2014, 16, 15053–15067 RSC.
- A. R. Ferhan, J. A. Jackman, T. N. Sut and N.-J. Cho, Sensors, 2018, 18, 1283 CrossRef PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- M. Reynolds, M. Marradi, A. Imberty, S. Penadés and S. Pérez, Chem. – Eur. J., 2012, 18, 4264–4273 CrossRef CAS PubMed.
- F. Fu, Z. Huang, W. Wang, W. Wang, X. Ma, L. Wang, Y. Huang, P. Hu, X. Pan and C. Wu, J. Drug Delivery Sci. Technol., 2021, 64 Search PubMed.
- K. Henzler, B. Haupt, K. Lauterbach, A. Wittemann, O. Borisov and M. Ballauff, J. Am. Chem. Soc., 2010, 132, 3159–3163 CrossRef CAS PubMed.
- S. Lindman, I. Lynch, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Nano Lett., 2007, 7, 914–920 CrossRef CAS PubMed.
- E. H. Pilkington, O. J. R. Gustafsson, Y. Xing, J. Hernandez-Fernaud, C. Zampronio, A. Kakinen, A. Faridi, F. Ding, P. Wilson, P. C. Ke and T. P. Davis, ACS Nano, 2018, 12, 6066–6078 CrossRef CAS PubMed.
- Y. Wang, Y. Sun, M. Li, L. Xiong, X. Xu, N. Ji, L. Dai and Q. Sun, Food Hydrocolloids, 2020, 102, 105615 CrossRef CAS.
- S. Lippok, S. A. I. Seidel, S. Duhr, K. Uhland, H.-P. Holthoff, D. Jenne and D. Braun, Anal. Chem., 2012, 84, 3523–3530 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 1983 Search PubMed.
- Y. Feng, L. Liu, S. Hu, P. Zou, J. Zhang, C. Huang, Y. Wang, S. Wang and X. Zhang, Luminescence, 2016, 31, 356–363 CrossRef CAS PubMed.
- B. Douzi, Protein–Protein Interactions: Surface Plasmon Resonance BT - Bacterial Protein Secretion Systems, Methods and Protocols, 2017, 257–275 Search PubMed.
- S. G. Patching, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 43–55 CrossRef CAS PubMed.
- P. G. Stockley and B. Persson, Surface Plasmon Resonance Assays of DNA-Protein Interactions BT - DNA-Protein Interactions, Principles and Protocols, 2009, 653–669 CAS.
- A. Patra, T. Ding, G. Engudar, Y. Wang, M. M. Dykas, B. Liedberg, J. C. Y. Kah, T. Venkatesan and C. L. Drum, Small, 2016, 12, 1174–1182 CrossRef CAS PubMed.
- T. Miclăuş, V. E. Bochenkov, R. Ogaki, K. A. Howard and D. S. Sutherland, Nano Lett., 2014, 14, 2086–2093 CrossRef PubMed.
- M. Zakłos-Szyda, G. Budryn, J. Grzelczyk, H. Pérez-Sánchez and D. Żyżelewicz, Molecules, 2020, 25(1), 206 CrossRef PubMed.
- M. M. Islam, S. Barik and M. Sarkar, J. Phys. Chem. B, 2019, 123, 1512–1526 CrossRef CAS PubMed.
- R. Huang and B. L. T. Lau, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 945–956 CrossRef CAS PubMed.
- J. Tellinghuisen and J. D. Chodera, Anal. Biochem., 2011, 414, 297–299 CrossRef CAS PubMed.
- A. A. Rehman, H. Ahsan and F. H. Khan, Int. J. Biol. Macromol., 2016, 83, 366–375 CrossRef CAS PubMed.
- Y. Yu, Y. Luan and W. Dai, Int. J. Biol. Macromol., 2022, 205, 731–739 CrossRef CAS PubMed.
- M.-M. Yin, W.-Q. Chen, Y.-Q. Lu, J.-Y. Han, Y. Liu and F.-L. Jiang, Nanoscale, 2020, 12, 4573–4585 RSC.
- V. Forest and J. Pourchez, Nano Today, 2016, 11, 700–703 CrossRef CAS.
- X. Sha, C. Sun, X. Xu, L. Alexander, P. J. Loll and L. S. Penn, Anal. Chem., 2012, 84, 10298–10305 CrossRef CAS PubMed.
- P. Hampitak, D. Melendrez, M. Iliut, M. Fresquet, N. Parsons, B. Spencer, T. A. Jowitt and A. Vijayaraghavan, Carbon, 2020, 165, 317–327 CrossRef CAS.
- E. D. Kaufman, J. Belyea, M. C. Johnson, Z. M. Nicholson, J. L. Ricks, P. K. Shah, M. Bayless, T. Pettersson, Z. Feldotö, E. Blomberg, P. Claesson and S. Franzen, Langmuir, 2007, 23, 6053–6062 CrossRef CAS PubMed.
- M. Gagliardi, L. Colagiorgio and M. Cecchini, Biosensors, 2023, 13(6), 607 CrossRef CAS PubMed.
- M. A. Plunkett, P. M. Claesson, M. Ernstsson and M. W. Rutland, Langmuir, 2003, 19, 4673–4681 CrossRef CAS.
- S. A. I. Seidel, P. M. Dijkman, W. A. Lea, G. van den Bogaart, M. Jerabek-Willemsen, A. Lazic, J. S. Joseph, P. Srinivasan, P. Baaske, A. Simeonov, I. Katritch, F. A. Melo, J. E. Ladbury, G. Schreiber, A. Watts, D. Braun and S. Duhr, Methods, 2013, 59, 301–315 CrossRef CAS PubMed.
- G. Wang, W. Wang, E. Shangguan, S. Gao and Y. Liu, Mater. Sci. Eng., C, 2020, 111, 110830 CrossRef CAS PubMed.
- W. Wang, H. Liu, Z. Huang, F. Fu, W. Wang, L. Wu, Y. Huang, C. Wu and X. Pan, Chin. Chem. Lett., 2022, 33(9), 4185–4190 CrossRef CAS.
- L. D. Field, S. A. Walper, K. Susumu, G. Lasarte-Aragones, E. Oh, I. L. Medintz and J. B. Delehanty, Bioconjugate Chem., 2018, 29, 2455–2467 CrossRef CAS PubMed.
- M. Raoufi, M. J. Hajipour, S. M. Kamali Shahri, I. Schoen, U. Linn and M. Mahmoudi, Nanoscale, 2018, 10, 1228–1233 RSC.
- L. X. Gao, H. Hao, Y. Q. Yu, J. L. Chen, W. Q. Chen, Z. D. Gong, Y. Liu and F. L. Jiang, Langmuir, 2023, 39, 15275–15284 CrossRef CAS PubMed.
- A. C. G. Weiss, K. Krüger, Q. A. Besford, M. Schlenk, K. Kempe, S. Förster and F. Caruso, ACS Appl. Mater. Interfaces, 2019, 11, 2459–2469 CrossRef CAS PubMed.
- P. L. Latreille, J. M. Rabanel, M. Le Goas, S. Salimi, J. Arlt, S. A. Patten, C. Ramassamy, P. Hildgen, V. A. Martinez and X. Banquy, Adv. Mater., 2022, 34(38), 2203354 CrossRef CAS PubMed.
- M. Germain, F. Caputo, S. Metcalfe, G. Tosi, K. Spring, A. K. O. Åslund, A. Pottier, R. Schiffelers, A. Ceccaldi and R. Schmid, J. Controlled Release, 2020, 326, 164–171 CrossRef CAS PubMed.
- D. Sahoo, P. Bhattacharya, H. K. Patra, P. Mandal and S. Chakravorti, J. Nanoparticle Res., 2011, 13, 6755–6760 CrossRef CAS.
- M. S. Jahan Sajib, P. Sarker, Y. Wei, X. Tao and T. Wei, Langmuir, 2020, 36, 13356–13363 CrossRef CAS PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef CAS PubMed.
- B. A. Wallace, Q. Rev. Biophys., 2009, 42, 317–370 CrossRef CAS PubMed.
- C. Ge, J. Du, L. Zhao, L. Wang, Y. Liu, D. Li, Y. Yang, R. Zhou, Y. Zhao, Z. Chai and C. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16968–16973 CrossRef CAS PubMed.
- D. Sanchez-Guzman, G. Giraudon-Colas, L. Marichal, Y. Boulard, F. Wien, J. Degrouard, A. Baeza-Squiban, S. Pin, J. P. Renault and S. Devineau, ACS Nano, 2020, 14, 9073–9088 CrossRef CAS PubMed.
- L. Calzolai, F. Franchini, D. Gilliland and F. Rossi, Nano Lett., 2010, 10, 3101–3105 CrossRef CAS PubMed.
- P. S. Kumagai, A. P. U. Araujo and J. L. S. Lopes, Biophys. Rev., 2017, 9, 517–527 CrossRef CAS PubMed.
- B. A. Wallace, Nat. Struct. Biol., 2000, 7, 708–709 CrossRef CAS PubMed.
- D. Zhang, O. Neumann, H. Wang, V. M. Yuwono, A. Barhoumi, M. Perham, J. D. Hartgerink, P. Wittung-Stafshede and N. J. Halas, Nano Lett., 2009, 9, 666–671 CrossRef CAS PubMed.
- A. Kudelski, Talanta, 2008, 76, 1–8 CrossRef CAS PubMed.
- C. Zong, M. Xu, L.-J. Xu, T. Wei, X. Ma, X.-S. Zheng, R. Hu and B. Ren, Chem. Rev., 2018, 118, 4946–4980 CrossRef CAS PubMed.
- I. Kuschnerus, K. Giri, J. Ruan, Y. Huang, N. Bedford and A. Garcia-Bennett, J. Colloid Interface Sci., 2022, 612, 467–478 CrossRef CAS PubMed.
- C. Zhang, Z. Jin, B. Zeng, W. Wang, G. Palui and H. Mattoussi, J. Phys. Chem. B, 2020, 124, 4631–4650 CrossRef CAS PubMed.
- A. L. Ginzburg, L. Truong, R. L. Tanguay and J. E. Hutchison, ACS Nano, 2018, 12, 5312–5322 CrossRef CAS PubMed.
- M. Assfalg, L. Ragona, K. Pagano, M. D’Onofrio, S. Zanzoni, S. Tomaselli and H. Molinari, Biochim. Biophys. Acta, Proteins Proteomics, 2016, 1864, 102–114 CrossRef CAS PubMed.
- O. Cala, F. Guillière and I. Krimm, Anal. Bioanal. Chem., 2014, 406, 943–956 CrossRef CAS PubMed.
- M. Lundqvist, I. Sethson and B.-H. Jonsson, Langmuir, 2004, 20, 10639–10647 CrossRef CAS PubMed.
- B. Hu, C. Wang, B. Chen and M. He, ACS Nano, 2021, 15, 3108–3122 CrossRef PubMed.
- M. Dolci, Y. Wang, S. W. Nooteboom, P. E. D. Soto Rodriguez, S. Sánchez, L. Albertazzi and P. Zijlstra, ACS Nano, 2023, 17, 20167–20178 CrossRef CAS PubMed.
- M. C. Lo Giudice, L. M. Herda, E. Polo and K. A. Dawson, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- X. Tan and K. Welsher, Angew. Chem., Int. Ed., 2021, 60, 22359–22367 CrossRef CAS PubMed.
- Y. Niu, Y. Yu, X. Shi, F. Fu, H. Yang, Q. Mu, D. Crespy, K. Landfester and S. Jiang, Nano Lett., 2024, 24, 9202–9211 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |