DOI:
10.1039/D4CS00497C
(Review Article)
Chem. Soc. Rev., 2024,
53, 9718-9737
Heavier group 14-transition metal π-complex congeners
Received
22nd May 2024
First published on 27th August 2024
Abstract
Since the dawn of organometallic chemistry, transition metal complexes of unsaturated organic molecules, namely π-complexes, have remained a central focus: our thorough understanding of the electronic nature of such species, and their importance in countless reactive processes continues to drive research in their synthesis and utilisation. Since the late 1900s, research regarding the related chemistry for the heavier group 14 elements has become increasingly more fervent. Today, heavier congeners of a vast array of classical π-complexes have been realised, from alkene to arene systems, involving Si, Ge, Sn, and Pb. This has given deeper insights into the bonding observed for these heavier elements, which typically involves a lessened degree of π-bonding and an increased polarisation. This review aims to summarise this field, identifying these disparities, and highlighting areas which we believe may be exciting for future exploration.

Terrance J. Hadlington
| Terrance J. Hadlington obtained his MChem(Hons) degree from the University of Bath (2011). He then moved to Melbourne, Australia, where he attained his PhD from Monash University (2016) under the guidance of Prof. Cameron Jones. After a postdoctoral stay at TU Berlin with Prof. Matthias Driess, he established his independent research group at TU Munich. Since that time, he has been awarded the ADUC Pries and Wöhler-BASF Nachwuchspreis, both of the GDCh, as well as an ERC Starting Grant. His research broadly involves the discovery of unique catalytic processes, including the development of cooperative low-valent p-block ligands, viz. Single-Centre Ambiphiles, and the utilisation of bespoke s-block hydride systems. |
1. Introduction
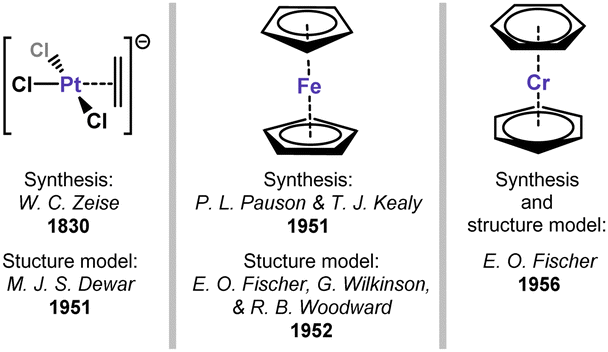
Transition metal (TM) complexes binding organic π-systems represent a foundational aspect of organometallic chemistry, being among the earliest organometallic species to be recognised and understood. The first true example of an organometallic complex was the so-called Zeise's salt,1 an ethylene adduct of the [PtCl3]− anion, discovered in 1830 but only crystallographically characterised in 1969.2 Between these two reports, Dewar correctly described the structure of this and related complexes,3 introducing the concept of π-bonding in organyl transition metal complexes. The description of the sandwich-structure of ferrocene, independently by E. O. Fischer and G. Wilkinson,4–6 in which the full π-system of two Cp− ligands coordinate FeII, cemented the importance of π-interactions between unsaturated organics and TMs: this bonding mode leads to a remarkably stable 18-electron iron species. Ultimately, these and related discoveries confirmed the ubiquitous Dewar–Chatt–Duncanson model for π-bonding,7 which is now understood to be central in broader transition metal-mediated syntheses.8,9 Very soon after the aforementioned advances, the same logic was applied in arene complexation: 18-electron bis(benzene)chromium was isolated and its structural data reported in 1956, demonstrating that TM0 centres can be stabilised exclusively by a neutral aromatic π-system.10,11 This collection of landmark systems (Fig. 1), formal alkene, cyclopentadienide, and benzene complexes of TMs, were truly transformative on our view and understanding of organometallic chemistry.
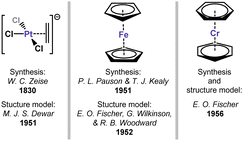 |
| | Fig. 1 Key initial discoveries and developments in π-complexation. | |
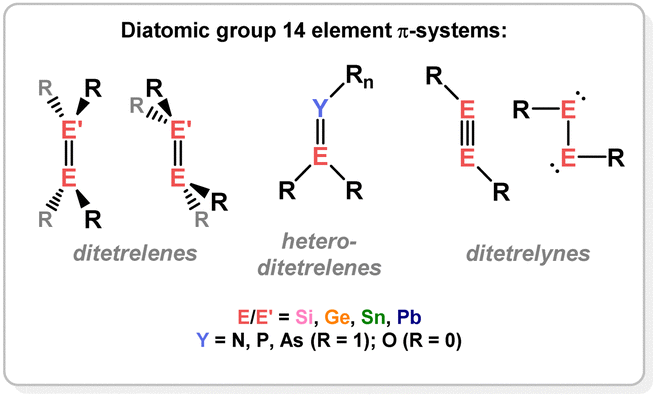
Naturally, the aforementioned discoveries gave rise to questions regarding the bonding distinctions between those carbon-centred π-systems, and the heavier group 14 congeners. These questions would invoke methods for the stabilisation of/access to such π-systems for Si, Ge, Sn, and Pb, which is not trivial given the reduced propensity of these heavier elements to partake in multiple bonding, due to reduced hybridisation of their frontier orbitals.12,13 For the same reasons, π-complexation may be expected to be perturbed in those systems, for example leading to metallacyclic complexes. This review aims to summarise and discuss these aspects of the field of low-oxidation state group 14 – transition metal chemistry. Specifically, this will focus on the formation of TM-complexes of ditetreleness (i.e. heavier alkenes), tetrenes (i.e. heavier carbonyls and imines), ditetrelynes (i.e. heavier alkynes), and heavier carbocycle π-complexes. Given that the formation of these ligand classes is in itself remarkable, a brief discussion of their syntheses will be included where appropriate, for completeness.
2. π-Complexes of low oxidation-state group 14 dimers
Dimeric low oxidation-state species of the heavier group 14 elements (viz.Fig. 2) are now relatively plentiful, being realised as stable systems in the 1970s (viz. Lappert's distannene).14,15 In the absence of electronic stabilisation by a TM, they typically rely on bulky ligands for kinetic stabilisation. A number of tetrene and ditetrelenes complexes have been accessed through generation of the low-oxidation state group 14 fragment in the coordination sphere of the TM, averting the necessity of bulky ligands. The bonding in these complexes can be compared with the classical Dewar–Chatt–Duncanson model (Fig. 3(a)); as mentioned above, as the degree of π-bonding is reduced in the heavier group 14 elements, structural deviations are expected here. In the free ditetrelenes, this leads to trans-bent structures, with bending becoming more pronounced with increasing atomic number. This is explained by the Carter–Goddard–Malrieu–Trinquier (CGMT) model:16,17 the singlet ground-state of the tetrylenes becomes more stable with increasing atomic number, which disallows orbital planar orbital overlap. The observed trans-bending in heavier ditetrelenes, then, allows for donor–acceptor double bonds, which are known to dissociate in solution. This bonding orbital also leads to weaker E–E bonding for the heavier group 14 elements, given that HOMO–LUMO separation becomes larger, i.e. the E-centred electron pair becomes lower in energy, and less likely to partake in bonding. The case is similar for ditetrelynes, but rather considering the more stable doublet and less stable quartet ground states.18,19 The result of these bonding modes, which leads to significant trans-bending, can be described as a second-order Jahn Teller distortion, due to electrostatic repulsion of residual non-bonding electron density at each E-centre.20,21 As this increases with atomic number, an increased deviation from linearity is seen moving to the heavier elements. This pertains to weaker E–E bonding interactions for these heavier elements. As such, considering alkene analogues, deviations from a classical π-complex (viz. (b), Fig. 3) is expected on descending group 14, to a metallacyclic form (viz. (c), Fig. 3), and ultimately towards bis(tetrylene) complexes (viz. (d), Fig. 3) through complete E–E bond scission. In line with weakening of E–E bonding on descending group 14, formation of these latter species would be expected to be more favoured for Pb, and for formal π-complexes to be energetically more favourable for Si, particularly given that a number of disilenes are in fact planar,22 and hence bear a high degree of classical π-bonding in the Si2 moiety. Generally, deviations from a π-complex are borne out by significant elongation of the E–E bond, and pyramidalisation/bending at the E-centres; true π-complexes of heavier group 14 multiple bonds are extremely rare.
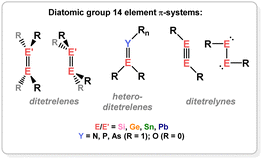 |
| | Fig. 2 The diatomic systems discussed in this review. | |
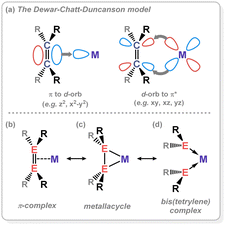 |
| | Fig. 3 Resonance extremes for the alkene complexes of transition metals. | |
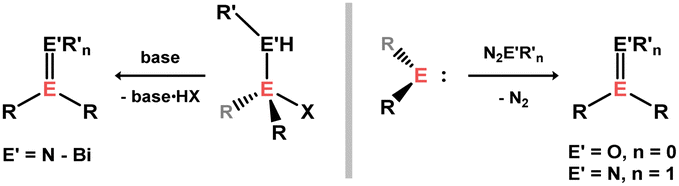
Beyond E–E and E–E′ double and triple bonds (i.e. heavier alkene and alkyne complexes, respectively; E′ = C–Pb; E ≠ E′), a number of transition metal complexes of E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bonds are known (X = O, S, N, P, As), which can be seen as heavier carbonyl and imine analogues, which will also be discussed here.
X bonds are known (X = O, S, N, P, As), which can be seen as heavier carbonyl and imine analogues, which will also be discussed here.
2.1. Homoatomic ditetrelenes complexes
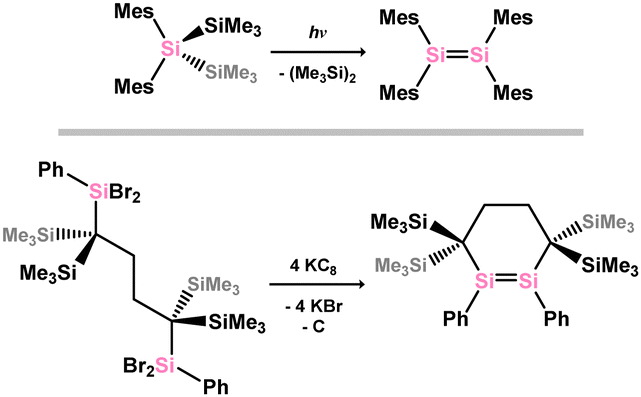
The synthesis of stable ditetrelenes represents a key milestone in modern main-group chemistry. Though Lappert reported the synthesis of the distannene [{(SiMe3)2CH}2Sn]2 in 1976,14,15 the initial example of a structurally authenticated heavier alkene analogue was the disilene {(Mes)2Si}2, reported by West and co-workers in 1981.23,24 This system was synthesised through irradiation of (Mes)2(Me3Si)2Si, on elimination of Me6Si2 (Scheme 1). A number of free disilenes have also been accessed via chemical reduction, exemplified by the disila-cyclohexene derivative shown in Scheme 1. Although there are now in excess of 300 structurally characterised ditetreleness, the majority of reported ‘heavier alkene’ complexes are generated in situ, through e.g. silane dehydrogenation, or salt-metathesis with ditetryl-1,2-diides.
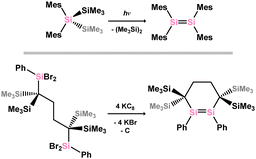 |
| | Scheme 1 Example of the photochemical (above) and reductive (below) formation of disilenes. | |
2.1.1. Disilene complexes.
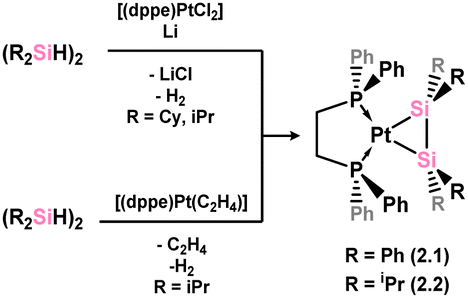
The initial examples of disilene-TM complexes were reported in 1989, utilising platinum as the TM (Scheme 2), aligning with the historical precedent of Zeise's salt, which also featured platinum. These early disilene complexes were accessed via the in situ reduction of [(dppe)PtCl2] in the presence of [{R2(H)Si}2] (R = Ph, iPr; dppe = [(Ph2P)2C2H4]), leading to dehydrogenation and formation of 2.1 and 2.2.25,26 Though in this initial publication the solid-state structure of these species was not reported, a combination of multi-nuclear NMR and IR spectroscopy, and mass spectrometry was used to determine their molecular formulas. Here, the bis-silylene form was not strictly ruled out.
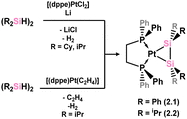 |
| | Scheme 2 Synthesis of the first heavier disilene complexes of a transition metal. | |
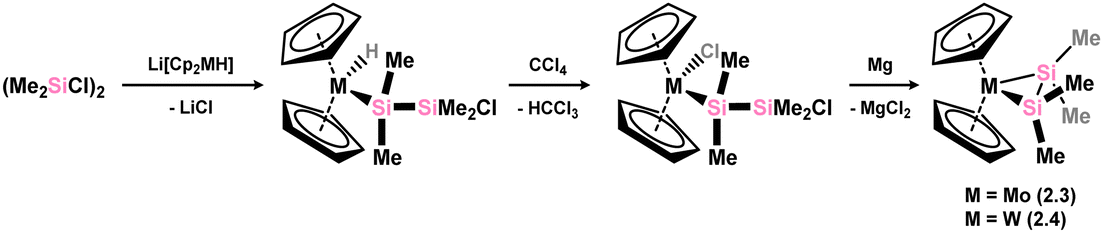
Soon after the above described publication, Berry and co-workers reported on the first structurally characterised examples of disilene-TM complexes, in Mo and W centred 2.3 and 2.4 (Scheme 3). These species were accessed through an initial salt-metathesis between metallates Li[Cp2MH] (M = Mo, W) and 1,2-dichlorodisilane [{Me2(Cl)Si}2], followed by chlorination of M with CCl4, and finally a ring-closing reduction.27 Metrical parameters would suggest that these complexes sit between formal π-complexes and metallacycles, with Si–Si distances between known single and double bonds, and a degree of pyramidalisation at each Si centre. These structural observations are in keeping with similar observations for classical alkene complexes of electron deficient early transition metals.
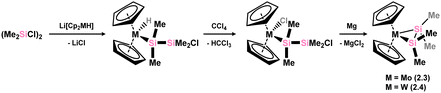 |
| | Scheme 3 Accessing molybdenum and tungsten complexes featuring the tetramethyl-disilene ligand. | |
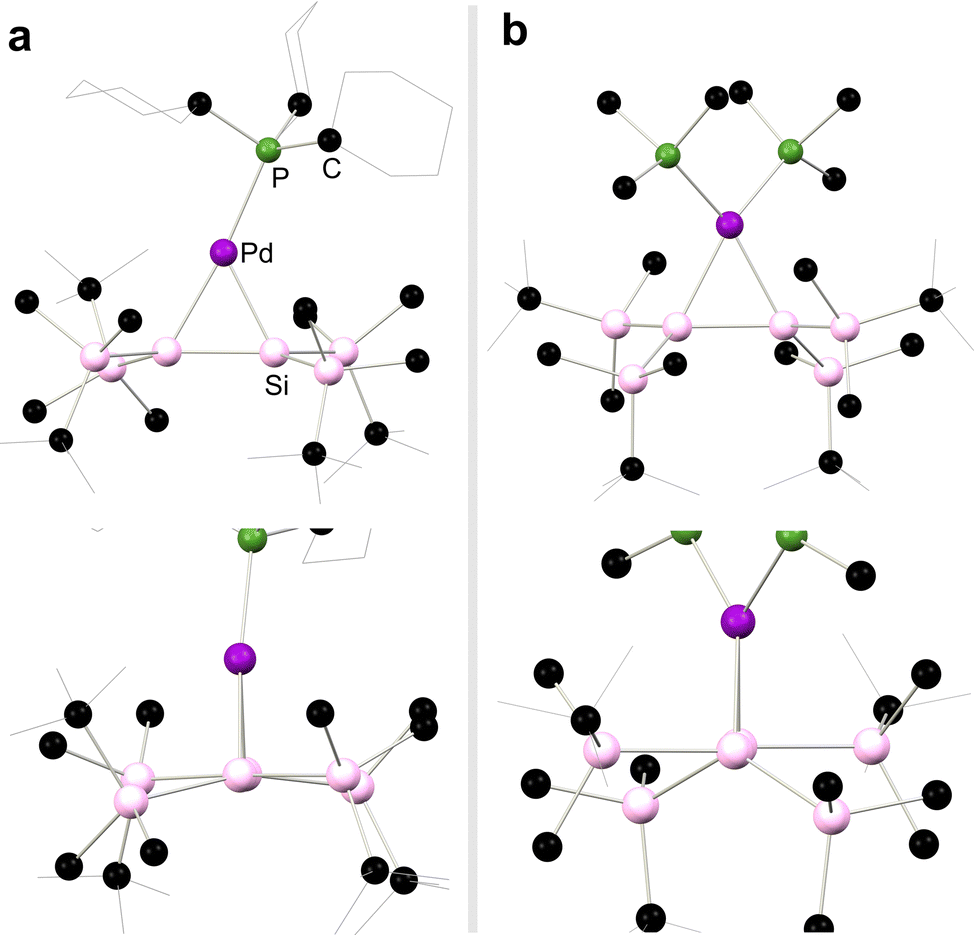
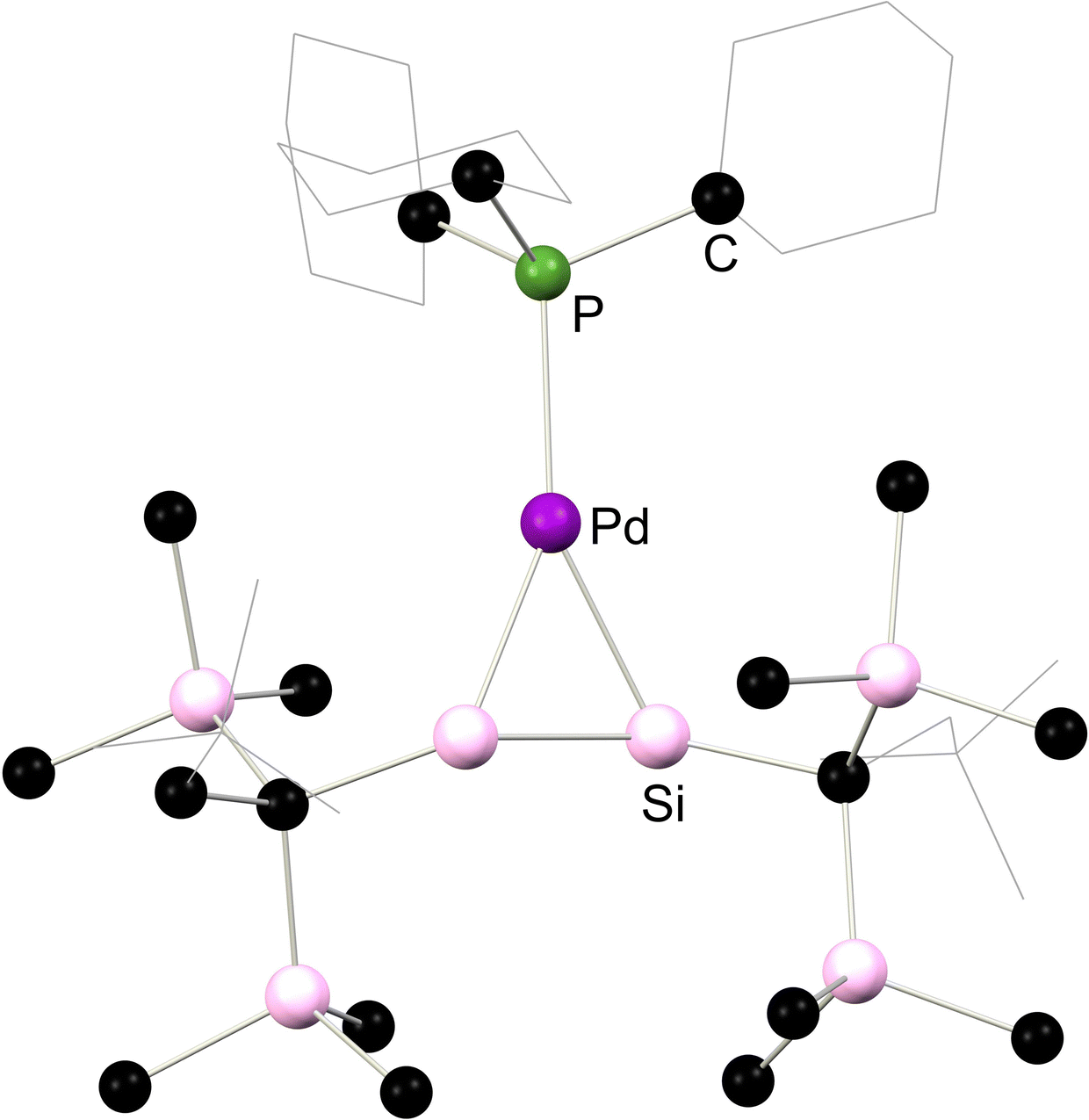
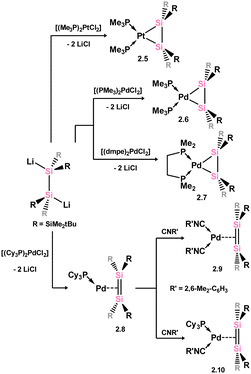
Moving again to group 10, Kira and co-workers demonstrated that complexes akin to those reported by West and co-workers can be accessed through the addition of 1,2-dilithio-disilanediide, Li2[{(tBuMe2Si)2Si}2] to [(Me3P)2PtCl2], directly forming 2.5 (Scheme 4).28 This methodology was later extended to the closely related Pd complexes 2.6 and 2.7.29 In all cases, these complexes are best described as metallacycles. However, employing the bulky phosphine ligand Cy3P gave contrasting results: presumably due to ligand bulk, the 14-electron palladium complex, 2.8, is isolated in this reaction. This reduces Pd → [Si2] back-donation, and leads to a structure akin to classical π-complexes. That is, minimal Si–Si bond elongation is observed when bound to Pd relative to the calculated Si–Si distance in the free disilene (4.3%), and negligible pyramidalisation at the Si centres is observed, with cis-bending angles at each Si centre of 4.41° and 9.65° (Scheme 4).30 The addition of isocyanide donor ligands to 2.8 generates 16-electron complexes 2.9 and 2.10, which align more closely with the metalladisilacyclopropane resonance form, offering valuable experimental insights into the electronic nature of these unique complexes.25 This effect is seen for all complexes here bearing a 4-coordinate Pd centre, as shown with the solid-state structures of 2.8 and 2.6 in Fig. 4, whereby an increase in out-of-plane bending in seen in the latter, in which the Pd centre is 4-coordinate.
 |
| | Scheme 4 Accessing disilene complexes which are best described as π-complexes. | |
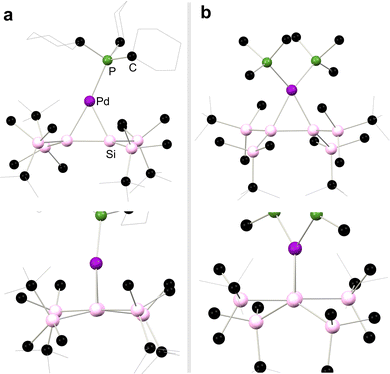 |
| | Fig. 4 Solid-state structures of (a) 2.8 and (b) 2.6, with front view shown above, and side view shown above, depicting an increase in out-of-plane bending in 2.6. | |
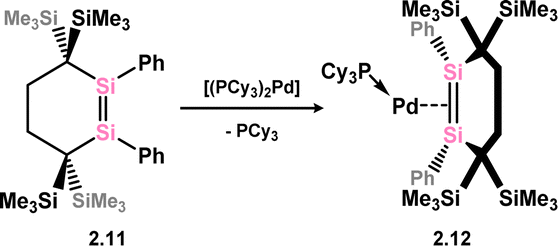
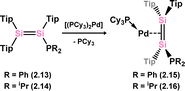
Further examples of disilene-Pd complexes which are best described as π-complexes were later reported, again by Kira and co-workers, through the direct addition of disila-cyclohexene analogue 2.11 to [(Cy3P)2Pd], forming complex 2.12 (Scheme 5).31 Scheschkewitz and co-workers also demonstrated that P-substituted disilenes 2.13 and 2.14 form related 14-electron Pd complexes 2.15 and 2.16 when combined with [(Cy3P)2Pd], leading to a greater degree of back-donation from Pd relative to 2.8 and 2.12. This is best borne out by the elongation of the Si–Si distance in these species relative to the free disilene: a value of 4.8% is found for 2.15, comparing to 3.2% and 1.9% for 2.8 and 2.12, respectively (Scheme 6).32
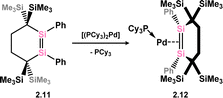 |
| | Scheme 5 Complexation of a cyclic disilene ligand to palladium(0). | |
 |
| | Scheme 6 Complexation of a phosphine-appended disilene to palladium(0). | |
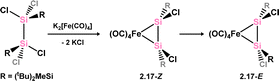
Kira and co-workers have also demonstrated the synthesis of Fe0 complexes bearing 1,2-dichlorodisilene ligands (Scheme 7).25 This was achieved by reacting dipotassium tetracarbonylferrate with tetrachlorodisilane [(tBu2MeSi)Cl2Si]2, resulting initially in the Z-isomer, 2.17-Z. Over time, this compound undergoes isomerisation to the thermodynamically favoured E-isomer, 2.17-E, with a first-order rate constant of 4.66 × 10−7 s−1 at 323 K (Scheme 7). The initial formation of 2.17-Z is justified mechanistically through the generation of a bis(silyl)silylene intermediate, followed by β-chloride migration, a pathway preferred for generating the Z-isomer. Both isomers are best described as metallacyclic complexes.
 |
| | Scheme 7 Formation of a Z-1,2-dichlorodisilene iron complex, described as a metallacycle, and its isomerisation to the E-isomer. | |
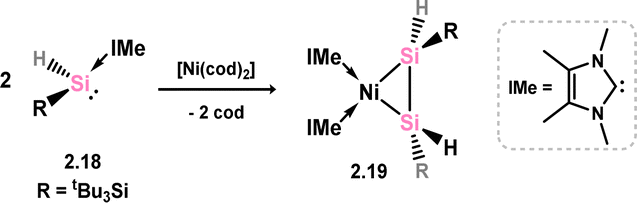
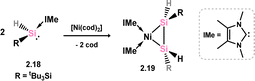
A 1,2-dihydrodisilene Ni0 complex closely related to that described above has been reported, synthesised through a ligand migration reaction upon combining the N-heterocyclic carbene (NHC)-stabilised hydridosilylene [(tBu3Si)(H)Si]·IMe (viz.2.18) with [Ni(cod)2] (Scheme 8; cod = 1,5-cyclooctadiene; IMe = [{(Me)CN(Me)}2C:]).33 In this instance, the E-isomer is exclusively formed (viz.2.19), and the Si–Si bond length and out-of-plane bending align with metallacycle character.
 |
| | Scheme 8 A nickel-mediated silylene transfer reaction, in formation of an E-1,2-dihydrodisilene complex of nickel. | |
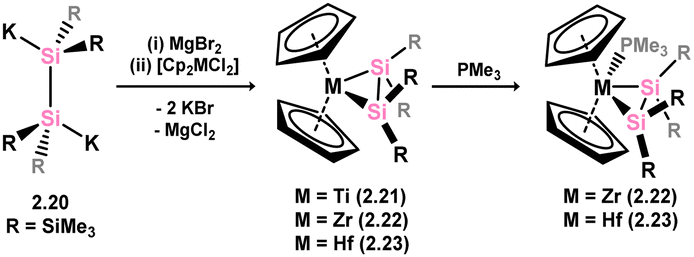
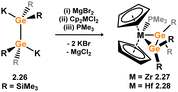
Moving towards early d-block species, a general pathway to heavier alkene complexes of electron-poor group 4 metallocene fragments has been developed. This method involves the combination of the 1,2-dipotassiodisilane 2.20 with magnesium dibromide, followed by reaction with dichlorometallocenes [Cp2MCl2] (M = Ti, Zr, Hf), resulting in the desired disilene complexes 2.21, 2.22, and 2.23, which for Zr and Hf could be crystallised only as their PMe3 adducts 2.24 and 2.25 (Scheme 9).34,35 In the initial publication from Marschner and co-workers,34 describing the synthesis of Hf complex 2.23, computational bonding analysis revealed a long Si–Si distance (dSiSi = 2.56 Å) in this species which could not be crystallographically characterised. Alongside a strongly deshielded 29Si NMR resonance (132.8 ppm), this led to the conclusion that this species sits between the metallacyclic and bis-silylene resonance forms, driven by the electron-poor nature of the Hf centre. Addition of PMe3 leads to reversion to a species better described as a metallacycle (viz.2.23), which could be structurally characterised, with a now slightly contracted Si–Si bond distance (dSiSi = 2.343(4) Å), and dramatically more shielded 29Si NMR shifts (135.7 and −159.7 ppm), pertaining to significant Zr → Si back-bonding. This species is still regarded as a metallacycle. Indeed, the remainder of structurally characterised species in this family of group 4 metal complexes best represent the metallacycle resonance form, primarily due to significant [Si2] → M donation. It is noted that in PMe3-coordinated complexes this donation is reduced, leading to Si–Si bond contraction, although this is demonstrated exclusively with DFT methods.
 |
| | Scheme 9 Accessing group 4 metal complexes of disilenes, best described as metallacycles. | |
2.1.2. Digermene complexes.
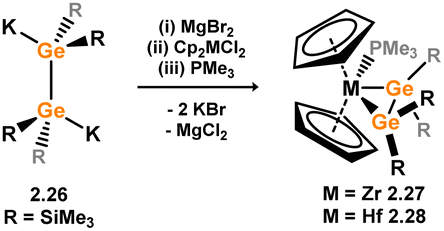
Only two digermene complexes of transition-metals are known, accessed as above, through addition of dipotassio-digermanediide K2[{(Me3Si)2Ge}2] (viz.2.26) to [Cp2MCl2] (M = Zr, Hf), in forming complexes 2.27 and 2.28 (Scheme 10).35 As per the Si systems, only the PMe3 adducts could be isolated as crystalline species. In both cases, the complexes demonstrate significant metallacycle character, with elongated Ge–Ge distances (dGeGe: 2.27 = 2.4337(14) Å; 2.28 = 2.4538(9) Å), similar to that found in hexakis(trimethylsilyl)digermane (dGeGe = 2.48 Å).36 The solid state structure of 2.27, as shown in Fig. 5, shows a significant degree of out-of-plane bending, also aligning with a metallcycle resonance form.
 |
| | Scheme 10 Access to group 4 complexes featuring digermene ligands. | |
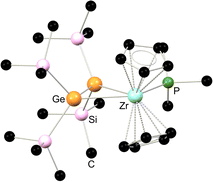 |
| | Fig. 5 Solid-state structure 2.27. | |
It is interesting to note the absence of a greater number of digermene-TM complexes, given the vast number of germylene, and specifically bis- and poly-germylene complexes which are known.37,38 It is likely that ligand sterics, in combination with electronic saturation at Ge through strongly π-donating ligands (e.g. N-heterocyclic systems), prevents Ge–Ge bonding in those complexes. This relatively unexplored field would thus benefit greatly from further synthetic developments, to uncover the electronic nature and synthetic utility of digermene complexes.
2.1.3. Distannene complexes.
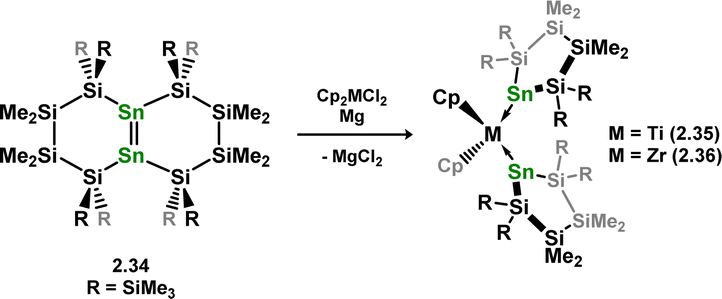
As per the above described digermene-TM complex class, distannene-TM complexes are rather rare, in contrast to the vast number of stannylene-TM complexes known.37,38 Despite success in accessing this compound class for Si and Ge in combination with the group 4 metals, no such species are known for tin. In fact, reaction of K2[{(Me3Si)2Sn}2] (viz.2.29) with [Cp2MCl2] (M = Zr, Hf) instead leads to chelating bis(stannyl) complexes 2.30 and 2.31, through a postulated disproportionation reaction which liberates the free stannylene, [(Me3Si)2Sn], which is hypothesised to be trapped in 2.32/2.33 (Scheme 11).35 Circumstantial evidence for this was later given through the addition of distannene 2.34 to [Cp2MCl2] (M = Ti, Zr) under reducing conditions, which led to complete Sn–Sn bond scission, and formation of bis(stannylene) complexes 2.35 and 2.36 (Scheme 12).39 This is perhaps not surprising, given that stannenes are known to dissociate in solution,14,15 due the typically weak Sn–Sn bonding brought about by factors discussed in the introduction of this review (e.g. the CGMT model).
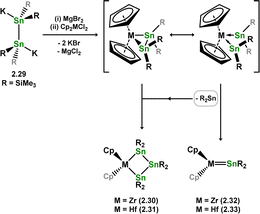 |
| | Scheme 11 Attempted synthesis of group 4 complexes bearing distannene ligands. | |
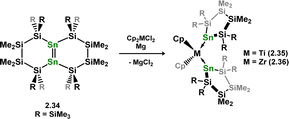 |
| | Scheme 12 Formation bis(stannylene) complexes on addition of distannene ligands to group 4 complexes. | |
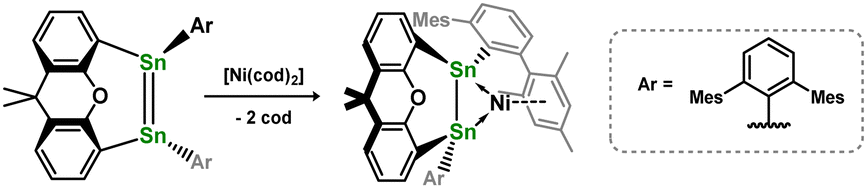
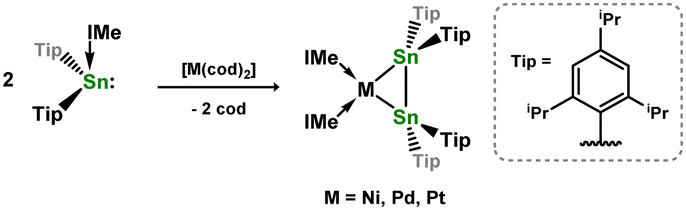
In contrast to those reactions described above, Wesemann and co-workers demonstrated that addition of the xanthene-bridged distannene 2.37 to [Ni(cod)2] leads to the formation of a unique distannene-Ni0 complex, 2.38, marking the first example of a distannene-TM complex (Scheme 13). Notably, the distannene ligand in this species remains trans-bent, due to an interesting 1,2-donor–acceptor binding mode of this ligand, presumably enforced by the geometric constraint provided by the xanthene bridge.40 The same group later showed that closely related distannene complexes for all three group 10 metals could be could be accessed (viz.2.39, 2.40, and 2.41), through coupling of two stannylene ligands in the coordination sphere of the low-valent d-block metal (Scheme 14).41 This carbene transfer reaction is reminiscent of that used by Inoue and co-workers in the synthesis of the related 1,2-dihydrodisilene complex 2.19.33 All distannene complexes are cis-bent, and are best classified as metallacycles.
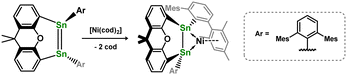 |
| | Scheme 13 ‘Push–pull’ bonding in a nickel(0) complex of tethered distannene ligand. | |
 |
| | Scheme 14 Stannylene-transfer chemistry in the formation of distannene complexes of the group 10 metals. | |
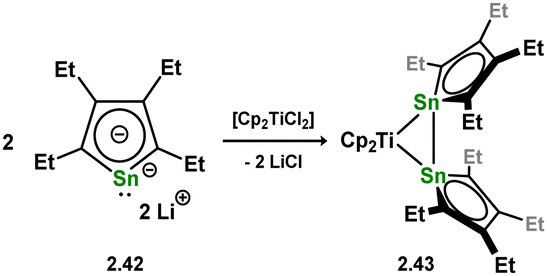
One further example of a formal distannene complex is known, and is a rather intriguing case, accessed through the addition of dilithiostannole 2.42 to [Cp2TiCl2], in forming 2.43 (Scheme 15).42 If negating the Sn–Sn bond, this species holds two trigonal planar tin centres, both in the plane of the titanium centre, which would ordinarily indicate a formal bis-stannylene complex. Yet, a short Sn–Sn distance of 3.0576(11) Å is observed indicative of a clear Sn–Sn bond, corroborated by DFT calculations. This also suggested that the bonding in 2.43 features a delocalised electron system over the central [TiSn2] ring.
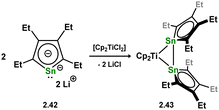 |
| | Scheme 15 A unique distannene complex titanium. | |
No diplumbene-TM complexes are known.
3. Hetero-ditetrelenes complexes
Isolable hetero-ditetreleness (i.e. R2E = E′R2; E/E′ = C–Pb; E ≠ E′), since the discovery of the first silene in 1981 by Brook and co-workers,43 are now a well-established compound class. These have been accessed with a variety of methods: photo-isomerisation of acyl silanes (Scheme 16(a)),43 the so-called sila-Peterson reaction (Scheme 16(b)),44 and salt-elimination from (alkyl)(halo)tetrylanes (Scheme 16(c))45 have all been effective in generating these species.46 Again, however, very few π-complexes of EE′ bonds have been accessed through direct reaction of isolable derivatives with TM fragments.
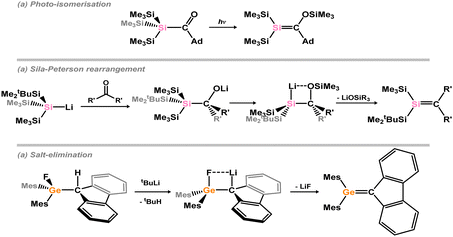 |
| | Scheme 16 Exemplary syntheses of tetrenes. | |
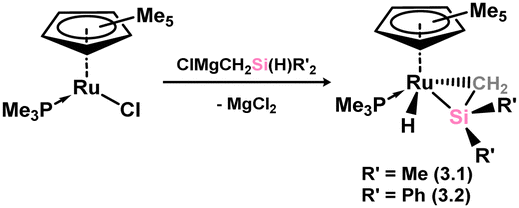
The first examples of hetero-ditetrelenes TM complexes were reported some years after the initial report of a stable silene by Brook and co-workers, in complexes 3.1 and 3.2 reported by Tilley and co-workers (Scheme 17).47,48 These species were readily accessed through the salt-metathesis of [ClMgCH2SiMe2H] with [Cp*Ru(PiPr3)Cl], leading to hydride migration and formation of the target silene complexes. The dimethyl derivative was found to be thermally unstable, decomposing rapidly at room temperature to reportedly form Me3SiH alongside a ruthenium hydride complex. In contrast, the diphenyl derivative could be crystallographically characterised, revealing a short Si–C bond of 1.78(2) Å indicative of some double-bond character. Still, a strong degree of pyramidalisation at Si (cis-bending angle = 35.2°) suggests a resonance form between π-complex and metallacycle. Soon after this initial report, two further examples, silene complexes of Ir (3.3) and W (3.4), were reported by the groups of Tilley and Berry, respectively, Tilley employing a similar synthetic route as for 3.2, and Berry employing reductive methods (Scheme 18, routes (a) and (b), respectively).49,50 All complexes were shown to undergo ring-opening reactions on the addition of polar H–X species. Tilley and co-workers also reported a closely related cationic Ir complex, 3.5, which demonstrates spontaneous silene-silyl isomerisation through group migration in solution (Scheme 19), with resonance ‘extremes’ being trapped through the addition of different donor ligands: π-donor/acceptor ligands CO and ethylene bind at Ir in complexes 3.6 and 3.7, whilst classical Lewis bases Et2O and Py bind at Si in 3.8 and 3.9.51
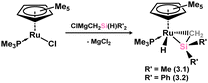 |
| | Scheme 17 The initial example of a transition metal complex of a silene. | |
 |
| | Scheme 18 Access to examples of iridium and titanium complexes bearing silene ligands. | |
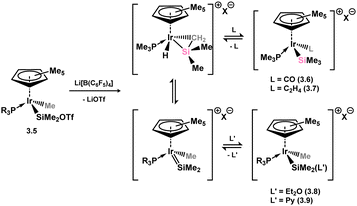 |
| | Scheme 19 Fluctional coordination chemistry in a disilene complex of iridium. Py = pyridine; OTf = triflate. | |
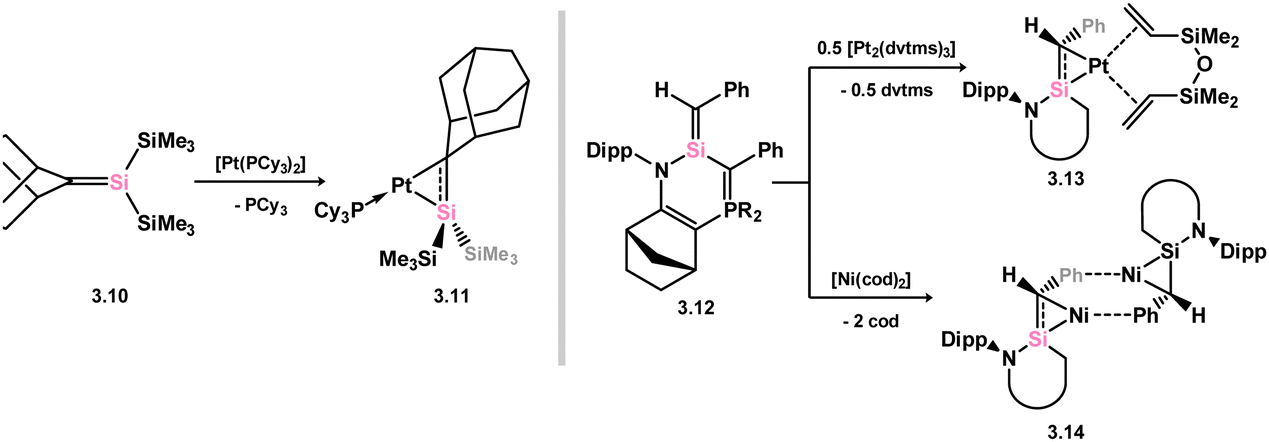
In contrast to those species described above, where the silene is generated in the coordination sphere of a TM, it has been demonstrated that free silene 3.10 readily forms a complex with Pt0 upon reaction with [(PCy3)2Pt] (viz.3.11, Scheme 20, Fig. 6).52 This product, closely related to disilene complex 2.8, exhibits a significant degree of π-character, as observed through its geometrical features: relative to the free silene, a modest elongation of the SiC bond by 5.6%, and cis-bending angles of just 25.8° (C) and 19.6° (Si) are only slightly greater than those observed in, for example, the first well-defined complex of a disilene.30 It is nevertheless characterised as a hybrid between a π-complex and a metallacycle, reflecting differences in geometry at the Si and C centers. Similarly, π-stabilised silene 3.12, formed through thermal or photolytic rearrangement of a silacyclopropane, readily undergoes complexation with Ni0 and Pt0, resulting in complexes 3.13 and 3.14, both displaying geometrical parameters similar to 3.11, and thus also described as hybrids of the π-complex and metallacycle resonance forms.53
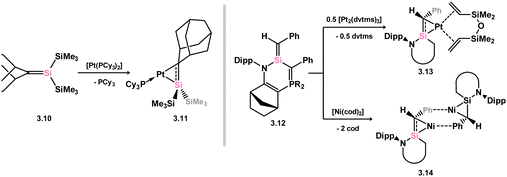 |
| | Scheme 20 Synthetic access to a group 10 complexes of silenes through combination of free silenes with M0 fragments (M = Ni, Pt). | |
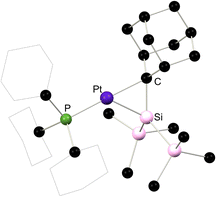 |
| | Fig. 6 Solid-state structure 3.11. | |
In addition to the above described silene complexes, a small number of Si–E ditetrelenes complexes are known (E = Ge, Sn). Marschner and co-workers reported that the addition of 1,2-dipotassio-silagermane 3.15 to [Cp2HfCl2] led to the formation of the germasilene complex 3.16, which could be isolated as phosphine adduct 3.17 (Scheme 21).35 As per closely related disilene and digermene complexes of the group 4 metals, phosphine adduct 3.17 is best described as a metallacycle, whilst it is believed that intermediate 3.16, with a lower coordinate Hf centre, may bare a greater degree of π-character.
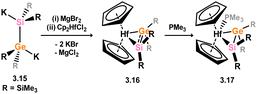 |
| | Scheme 21 Formation of a hafnium complex bearing a germasilene ligand. | |
The Marschner group later reported stannasilene complexes of Pd and Pt. The novel synthesis of these species, which presumably targeted stannylene complexes, involved the addition of the phosphine-coordinated cyclic bis(silyl)stannylene 3.18 to either in situ reduced [(dppe)PtCl2], or [Pd(PR3)3] (R = Ph, Et), resulting in the ring-opening of the stannylene and silyl migration to form 3.19 and 3.20 (Scheme 22).54 Once again, the geometrical parameters in these complexes suggest metallacycle character. Similar reactivity was observed when [{(Me3Si)3Si}2Sn·PEt3] (viz.3.21) reacted with in situ reduced [(dppe)PtCl2], yielding the acyclic stannasilene complex 3.22.
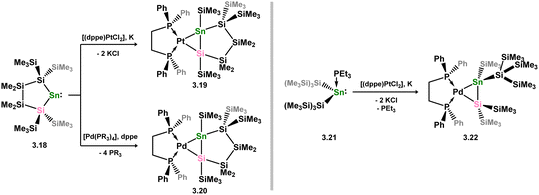 |
| | Scheme 22 Synthesis of a stannasilene complexes of group 10 metals. | |
Finally, the group of Scheskewitz have reported a number of interesting Ni complexes which can be broadly defined as germasilene complexes. In an initial publication, they demonstrated that the addition of the heavier vinylidene analogue 3.23 to [Ni(cod)2], in the presence of the NHC [{(Me)CN(iPr)}2C:] led to a migratory ring-closing reaction, in forming formal heavier cyclopropene complex 3.24 (Scheme 23).55 Further salt-methathesis reactions allow for the facile exchange of the Ge-Cl substituent, giving access to Ph- (3.25), PPh2- (3.26), and OtBu-functionalised (3.27) derivatives of this compound class. These represent the only well-defined examples of heavier metalla-cyclopropene TM complexes reported to date. Long Ge–Si bonds in these complexes are indicative of significant metallacycle character, whilst the butterfly geometry of the central [Si2GeNi] motif suggests that these species are best described as bicyclo[1.1.0]metallabutanes. It was later shown that the addition of vinylidene derivative 3.23 to [Ni(cod)2] in the absence of additional NHC leads to the heavier-allylic complex 3.28, through formal oxidative addition to Ni, with subsequent addition of NHC to this complex directly forming bicyclo[1.1.0]metallabutane 3.24. Finally, amido-functionalised heavier vinylidene analogue 3.29 was shown to react with NHC/[Ni(cod)2] to form the germasilene-Ni complex 3.30, involving C–H activation of one NHC ligand. The Ge–Si bond length in this species is intermediate between known single and double bonds, and as such this complex is again described best as having some degree of π-complex character, corroborated by DFT calculations which reveal a degree of π-bonding in the coordinated Ge–Si unit.
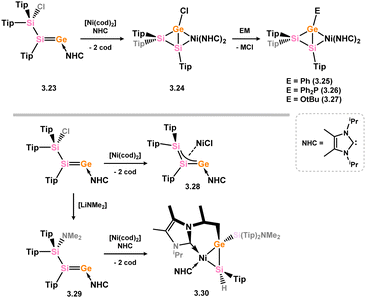 |
| | Scheme 23 Formation of a nickel(0) complex of a disilagerma-cyclopropene (above), and a closely related disilagerma-allyl complex of nickel(II). | |
4. Heavier imine and carbonyl complexes
A vast number of stable heavy imine analogues are known,56 typically accessed through oxidative EN formation upon addition of an azide to a tetrylene,57 or via salt elimination (amino)(halo)tetranes (Scheme 24).58 More recently, through bespoke ligand development, stable heavier ketone analogues have also been realised, generally accessed through oxidation of tetrylenes, e.g. with Py–O or N2O.59–61 The complexation chemistry of these isolated systems is however almost entirely unexplored. That is, heavier imine and carbonyl TM complexes are largely generated in situ. In all cases discussed here, congeners of π-complexes are ascertained to be so based on close E-TM and E′-TM contacts in structurally characterised species.
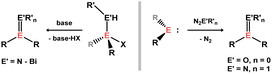 |
| | Scheme 24 General routes to doubly bonded group 14-group 15/16 species. | |
4.1. Silapnictene complexes
Transition metal complexes involving doubly-bonded silicon–pnictogen species are relatively uncommon, with early instances of silaimine complexes of zirconocene, initially reported by Berry in 1991, being among the limited examples.62 This initial example was accessed through a series of salt-metathesis and halogenation steps. Specifically, an initial salt-metathesis of [Cp2Zr(H)Cl] with Li[N(tBu)SiMe2H] led to a (silyl)amido zirconocene hydride complex. The Zr–H fragment undergoes halide-exchange with MeI, in loss of methane, and finally the addition of Li[CH2SiMe3] leads ring-closing salt-elimination in generating the formal silaimine complex 4.1 (Scheme 25). A similar approach was later utilised by Okazaki and co-workers in accessing the Ti congener 4.2.63 More recently, it was demonstrated that a (hydrido)(silylenyl)tungsten complex reacts with nitriles, leading to concurrent insertion into the W–H bond and Si–N coupling. This reaction yields complexes 4.3 and 4.4, which may be described as tungsten silaimine species (Scheme 26).64
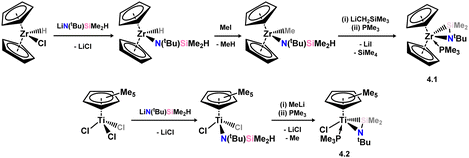 |
| | Scheme 25 Formation of the initial example of a silaimine transition metal complex (above), and it later published titanium congener (below). | |
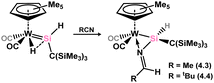 |
| | Scheme 26 Addition of nitriles to a hydrido-silylene complex of tungsten, leading to what may be described as silaimine complexes. | |
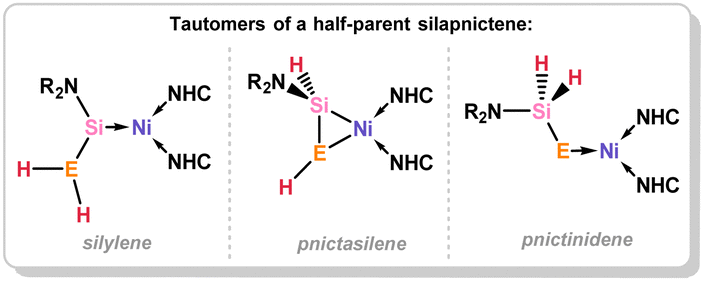
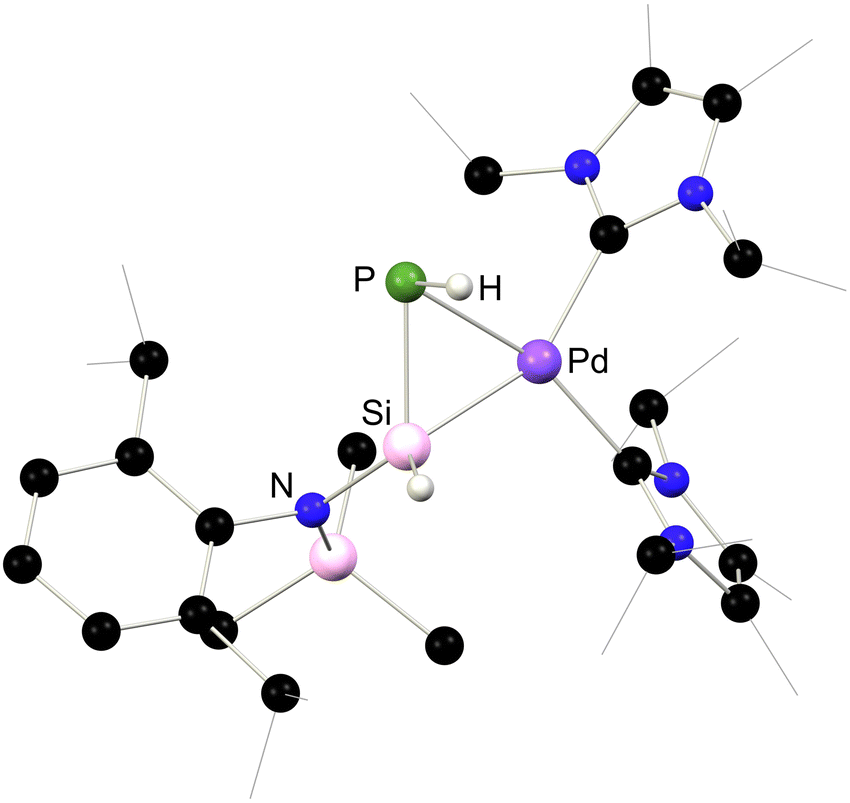
A series of 1,2-dihydrosilapnictene complexes of nickel were recently synthesised and found to readily tautomerise through hydrogen migration between silylene, silapnictene, and silyl-pnictindene complexes, depending on the identity of the pnictogen atom (Fig. 7 and Scheme 27).65 The half-parent bis(amido)silylene complex 4.5 was successfully isolated as a stable compound, synthesised through simple salt-metathesis of the chloro-silylene complex 4.6 with [LiNH2]. However, attempts to produce the phosphorus derivative resulted in the formation of the 1,2-dihydrosilaphosphene complex 4.7, though H-migration in an initially formed half-parent phosphasilene complex. Though the P–H ligand in this system was shown to fluctuate between the E- and Z-isomer, the solid state structure revealed only the E-isomer (Fig. 8), which is thus likely the most stable form. Remarkably, there is no known counterpart of such a complex for classical carbon derivatives, such as 1,2-dihydroimine complexes. Complex 4.7 was shown to coordinate BPh3 at its phosphorus centre in the formation of E-1,2-dihydrosilaphosphene complex 4.8. In the absence of borane-coordination, 4.7 undergoes further P to Si proton migration in the formation of intermediary phosphinidene complex 4.9, which dimerises in the generation of 4.10. The corresponding arsenic derivative of 4.11 was found to be unstable at ambient temperatures and disproportionates, giving rise to an intriguing nickela-silaarsene complex, 4.12, while concomitantly forming a diarsene-Ni0 complex 4.13. Nevertheless, the formation of 4.11 could be demonstrated by its deprotonation at low temperatures, leading to the room-temperature stable metallated silaarsene complex 4.14, in which H-migration is prevented. Based on their geometrical parameters, these species were determined to exhibit significant metallacycle character. However, the As–Si bond in 4.14 falls within the range of known double bonds for these elements, likely due to the high charge localisation on arsenic. As a whole, this study gives key insights into the relative stability of π-complexes of the heavier parent imine analogues, through dynamic proton migration processes.
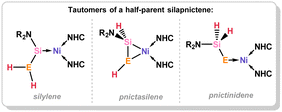 |
| | Fig. 7 Resonance forms of 1,2-dihydrosilapnictene transition metal complexes, through H-migration between E and Si (E = N–Sb). | |
 |
| | Scheme 27 Formation of silylene, silapnictene, and pseudo-pnictinidene tautomers in nickel complexation, stemming from a chloro-silylene complex of nickel(0). TMEDA = N,N-tetramethylethylenediamine. | |
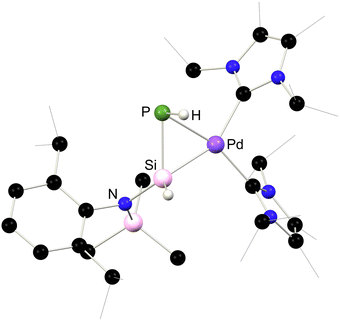 |
| | Fig. 8 Solid-state structure 4.7. | |
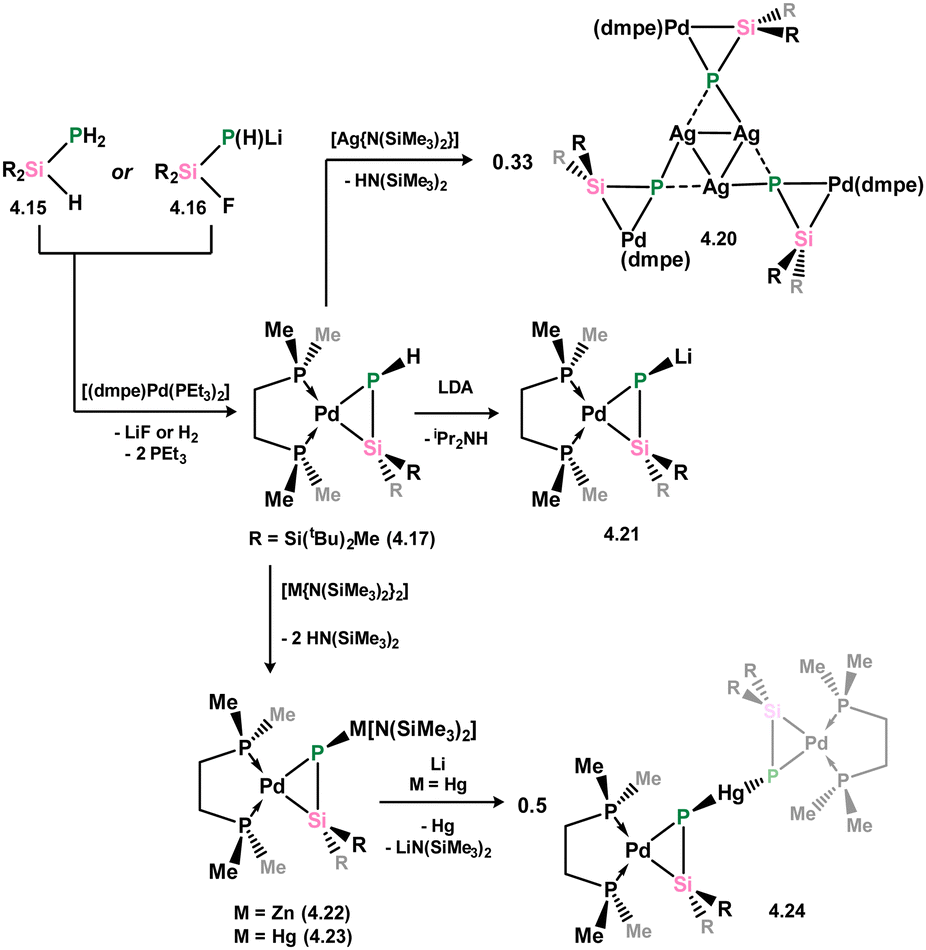
More recently, Apeloig and co-workers described the synthesis of platinum phosphasilenes akin to 4.7, which can be accessed via two pathways: phosphino-silane 4.15 or silylated lithium-phosphide 4.16 react with [(L)2Pt(PEt3)2] leading to loss of H2 or LiF, respectively (Scheme 28).66 This was primarily achieved with dmpe complex 4.17, and extended to the dppe and (PR3)2 complexes (R = Et (4.18), Ph (4.19)). All species are classified as metallacycles, due to a significant decrease in the calculated Wiberg bond indices for the P–Si bonds upon complexation, from ∼1.9 to ∼1.2. Further chemistry demonstrated that complex can be readily metallated at its P-centre, through deprotonation with Li, Zn, Ag, or Hg amides, leading to the first examples of P-metallated phosphasilene–TM complexes, 4.20 (Ag), 4.21 (Li), 4.22 (Zn), and 4.23 (Hg), which are closely related to the As-metallated species 4.14 described above. Notably, the silver system 4.20 forms a unique trimer in the solid state (Fig. 9), whilst the reduction of mercury derivative 4.23 with element lithium leads to presumed reductive disproportionation, forming bis(silaphosphenyl)mercury species 4.24.
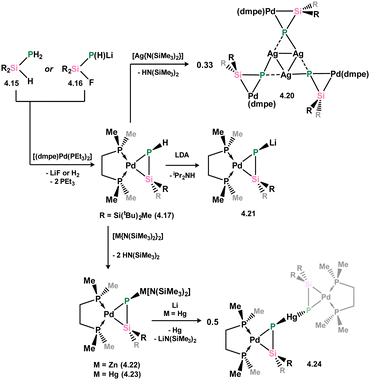 |
| | Scheme 28 Synthesis and metallation of a phosphasilene complex of palladium(0). | |
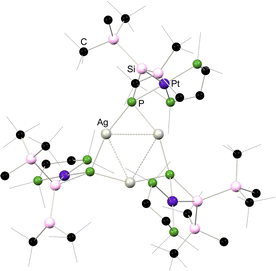 |
| | Fig. 9 Solid-state structure 4.20. | |
Heavier imine complexes are not known for the remainder of the group 14 elements.
4.2. Silanone complexes
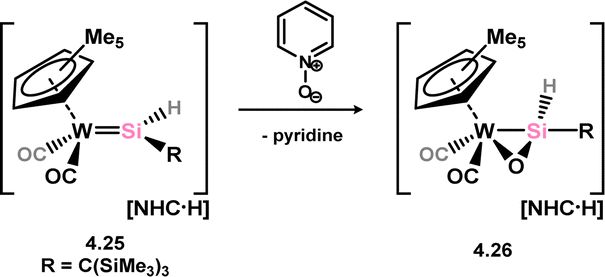
Only one structurally characterised example of a complex which can formally be described as a heavy aldehyde π-complex has been reported, by Tobita and co-workers. They demonstrated that anionic hydrido-silylene complex of tungsten 4.25 can be oxidised by Py–O, with the loss of pyridine and the formation of a sila-aldehyde complex 4.26 (Scheme 29).67 A long Si–O bond length (1.637(7) Å) and a short Si–W bond length (2.475(3) Å) are indicative of formal metallacycle character. Driess and co-workers later reported a related complex via the oxidation of (amino)(chloro)silylene complex 4.6, forming the corresponding silanone complex 4.27 at temperatures below −60 °C (Scheme 30).68 This species could be trapped by reaction with a borane, which led to addition across the Si–O bond in 4.28. On warming, silanone complex 4.28 undergoes a cascade rearrangement in the formation of an Si-metallated nickela-iminosilane, 4.29.
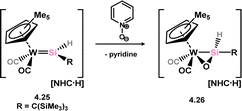 |
| | Scheme 29 Synthesis of a tungsten complex of a silanone. | |
 |
| | Scheme 30 Formation of a low-temperature stable silanone complex, its reactivity towards a borane, and rearrangement to a metalla-silaimine. | |
A compound closely related to 4.27 has also been reported by Kato and co-workers. They reported that the oxidation of manganese metallosilylene 4.30 leads to the formation of a strained [MnOSi] ring in 4.31, which is best described as a Si-metallated silanone (Scheme 31).69 The corresponding manganese-silanone complex is formed on insertion of ethylene into the Si–Mn bond, though no coordination of silicon to the metal centre is observed, in complex 4.32.
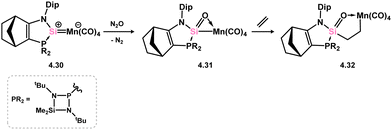 |
| | Scheme 31 Formation of a low-temperature stable silanone complex, its reactivity towards a borane, and rearrangement to a metalla-silaimine. | |
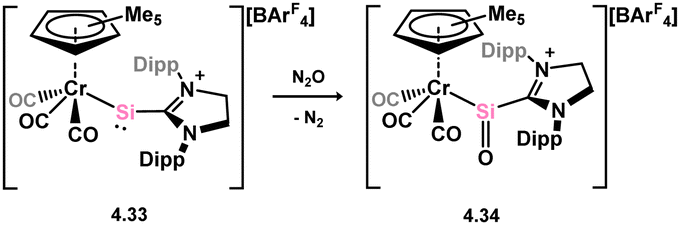
Additionally, in deviation from the above described analogues of carbonyl-TM complexes, Filippou and co-workers reported the oxidation of what is best described a metallosilylene (viz.4.33) with N2O, leading to chromium-silanone complex 4.34 (Scheme 32).70 The oxygen centre in this system shows no contact with the Cr centre, hence demonstrating the effect of metal and ligand substitution on driving or disfavouring for formation of π-complex congeners.
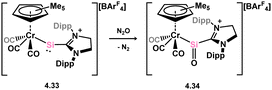 |
| | Scheme 32 Oxidative formation of metallosilanone complex. | |
4.3. Germanone complexes and derivatives
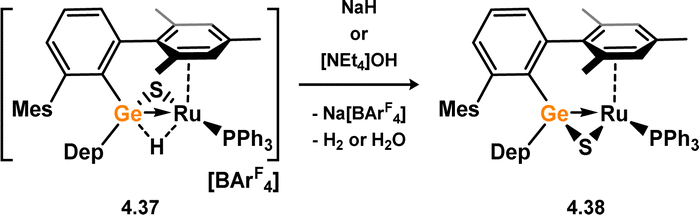
No stable TM-complexes of germanone species are known. However, germathione complexes have been reported, the first examples from Holl and co-workers.71 These were accessed in a straight forward manner: the oxidation of germylene complexes 4.35 and 4.36 with carbonyl sulphide leads to S-atom transfer, with concomitant loss of CO. This forms germathione-derivatives 4.37 and 4.38 (Scheme 33). The observed Ge–S bond distances in 4.35 (dGeS = 2.1830(1) Å) and 4.36 (dGeS = 2.1925(10) Å) are in keeping with known Ge–S single bonds, classifying these species as formal metallacycles. Closely related examples for ruthenium were later reported by Tatsumi and co-workers, through the dehydrogenation of cationic ruthenium complex 4.39 with NaH or NaOH, forming 4.40 (Scheme 34).72,73 These were studied in regards to their similarities to hydrogenase enzymes, given the formation of dihydrogen from nucleophilic hydride sources.
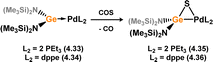 |
| | Scheme 33 Oxidative access to palladium-germathione complexes. | |
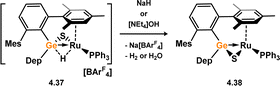 |
| | Scheme 34 Reductive formation of a ruthenium-germathione complex. | |
5. Ditetrelyne complexes
Despite the extensive knowledge of alkyne-TM complexes, there are a limited number of isolated heavier derivatives. This is perhaps not surprising given that the first reported example of a ditetrelyne was reported only in 2000 by Power and co-workers, in the diplumbyne [Ar′PbPbAr′] (Ar′ = 2,6-Tip2C6H3; Tip = 2,4,6-iPr3-C6H3).74 Further, this species was found to bear a long Pb–Pb interaction and strong trans-bending, indicative of a single Pb–Pb bond and hence negligible π-character. The situation is significantly different for the related silicon(I) species: in 2004 Sekiguchi and co-workers reported the first example of a crystallographically characterised disilyne, which features a short Si–Si bond (2.0622(9) Å) and significantly lessened trans-bending.75 We note here that earlier the same year, Wiberg and co-workers reported a closely related 1,2-disilyldisilyne which was stable in the solid state at ambient temperature, but rearranged to a trisila-cyclopropene in solution.76 The former disilyne was thus not crystallographically characterised. Nevertheless, DFT studies for both systems demonstrate that two π-bonding interactions are present in the Si–Si bond, making this much closer to bonding observed in alkynes.75,77 Such species have been utilised directly for the synthesis of π-complexes (vide infra). Metallic reductive methods are used in the synthesis of the heavier alkyne analogues, typically from tetrahalo-disilanes for the silicon derivatives,75 or from the halo-tetrylenes for germanium, tin, and lead (Scheme 35).78,79
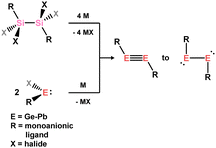 |
| | Scheme 35 General reductive routes to the heavier alkyne analogues. | |
5.1. Disilyne complexes
As it stands, only three disilyne TM-complexes have been reported. The first were 5.1 and 5.2, illustrated in Scheme 36.80 These complexes were formed through the combination of a stable disilyne with [(Cy3P)2M] (M = Pd, Pt). Both of these compounds exhibit metallacycle character attributed to significant M → (Si2) back-donation. The Si–Si distances in both cases align with known double bonds (for 5.1: 2.170(1) Å; for 5.2: 2.167(3) Å). A degree of out-of-plane bending is also observed, as seen in the molecular structure of 5.1 (Fig. 10, Scheme 36). A DFT analysis of the bonding in 5.1 suggests that this mirrors that known for the corresponding alkyne complexes, with two π → d donor interactions, and one d → π* back-bonding interaction, giving key insights into the electronic nature of these heavier main-group complexes. The closely related NHC-adduct of a disilyne 5.3 was shown to form an adduct with [ZnCl2], whereby no π-complex formation was observed (viz.5.4, Scheme 37).81
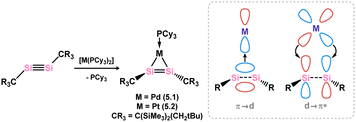 |
| | Scheme 36 Synthesis of and bonding in group 10 metal(0) complexes of a disilyne. | |
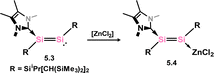 |
| | Scheme 37 Reactivity of an NHC-coordinated disilyne towards to zinc(II) chloride. | |
 |
| | Fig. 10 Solid-state structure 5.1, demonstrating a degree of out-of-plane bending. | |
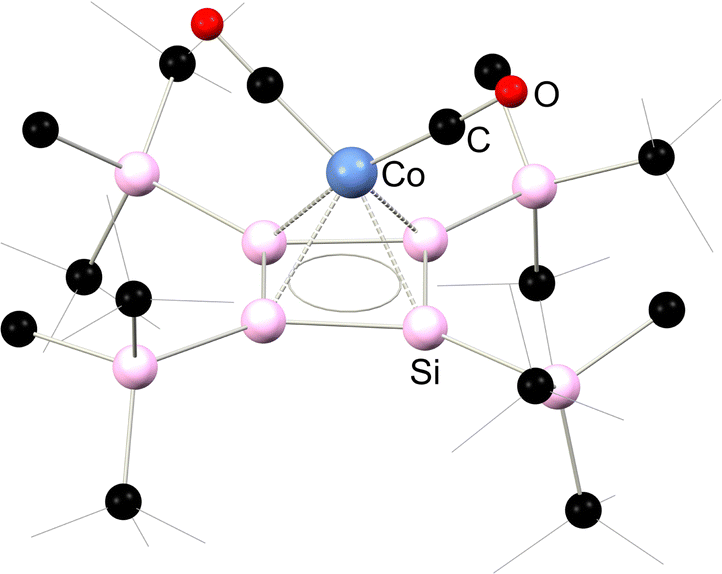
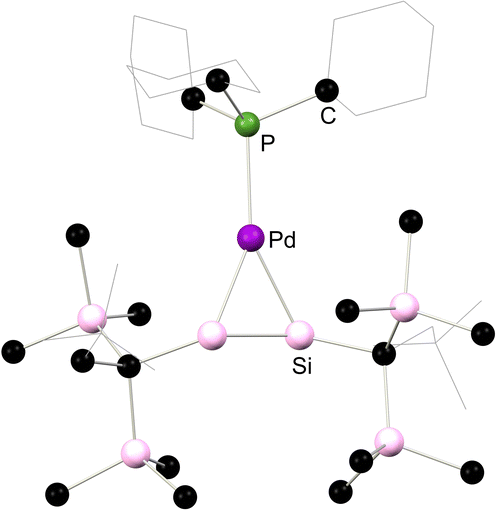
More recently, complex 5.5 was reported, which is best described as an iron-[bis(silylene)]. This species was accessed via the reduction of bis(chloro-silylene) complex 5.6 with an excess of KC8 (Scheme 38).82 As per the bis-silylene character of the free ligand,83 the [Si2] fragment does not bind Fe0 through a Si–Si π-bond, but rather through two lone pairs, one located at each Si centre. Significant Fe → [Si2] π-backdonation is also present, from the dxz orbital on Fe to a vacant π-orbital of the Si–Si bond, formed from a pz-orbital from each Si centre. As such, a long Si–Si single bond is observed in this complex. This leads to complete Si–Si bond scission on reaction with unsaturated organic molecules: (Me3Si)N3 leads to nitrene insertion, whilst benzophenone undergoes and an interesting [2+4] cycloaddition with the Si–Si bond.82
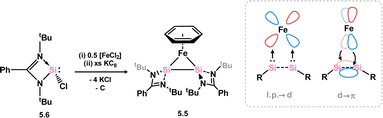 |
| | Scheme 38 Synthesis of and bonding in a bis(silylene) complex of iron. | |
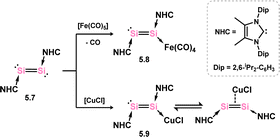
Though not a formal π-complex, Robinson and co-workers reported the reactivity of their bis-NHC complex 5.7 towards [Fe(CO)6], leading to coordination at one Si centre, as opposed to coordinating the Si–Si π-system (viz.5.8, Scheme 39).84 In contrast, the related [CuCl] complex 5.9 is proposed to form the π-complex in solution, borne out by to the observation of a single signal in the 29Si NMR for this species, despite the solid-state structure demonstrating binding at one Si centre.85
 |
| | Scheme 39 Reactivity of a formally silicon(0) dimer towards transition metal fragments. | |
5.2. Digermyne complexes
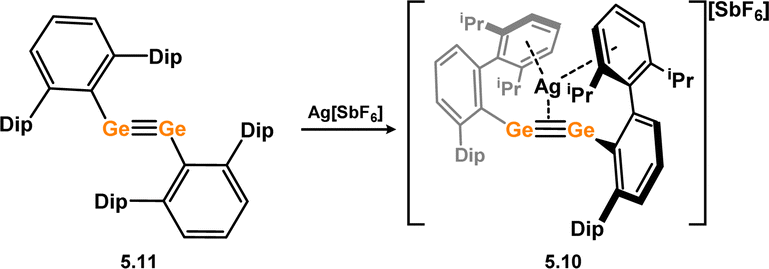
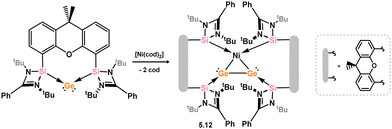
One formal digermyne-TM complex is known, that is the cationic Ag species 5.10, formed through the addition of Ag[SbF6] to digermyne 5.11 at temperatures below 0 °C (Scheme 40).86 The Ag ion in 5.10 is coordinated by the Ge–Ge bond and two flanking aryl rings, with no further ligands at Ag. A computational analysis of this complex highlights significant π-donation from the Ge–Ge triple bond to both 5s and 5p orbitals on Ag, with concomitant back-bonding from filled 4d orbitals on Ag to LUMO+1 of the digermyne, which represents an out-of-plane π* orbital, as per the above described disilyne complexes. One additional complex which may be considered a π-complex of a digermyne has been reported by Driess and co-workers, namely 5.12. Here, reaction of an isolable germylone towards [Ni(cod)2] results in Ge–Ge dimerisation, whereby the newly formed Ge–Ge interaction coordinates Ni in a side-on fashion (Scheme 41).87
 |
| | Scheme 40 Complexation of a digermyne with silver(I). | |
 |
| | Scheme 41 Formation of a Ge2–Ni π-complex from a germylone. | |
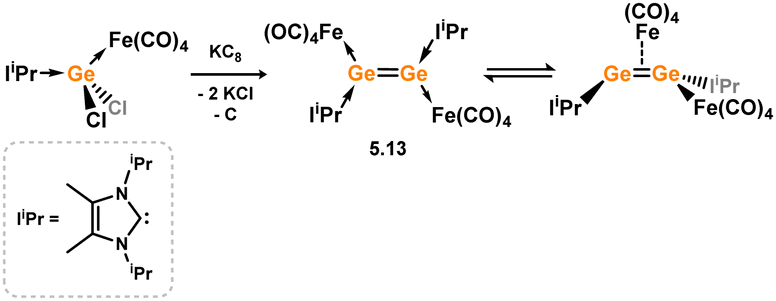
Scheschkewitz and co-workers have additionally reported a Ge species which is closely related to the above described complexation of NHC-stabilised [Si2] allotrope 5.7 with [Fe(CO)4]. Reduction of [IiPr·GeCl2·Fe(CO)4] with potassium graphite led to the formation of dimeric 5.13 (Scheme 42).88 In solution this species is hypothesised to form the symmetrical isomer, borne out by a single NHC environment as determined by 1H NMR spectroscopy. In the solid state, however, the unsymmetrical isomer was identified, which is coordinated terminally at one Ge centre by a [Fe(CO)4], and additionally features a [Fe(CO)4] moiety bridging the two Ge centres. As such, this can be seen as a π-complex of the [Ge2] dimer. As per Si complex 5.9, these two isomers are proposed to be in equilibrium in solution, with 1H NMR spectra collected at 233 K indicating now differing environments for the NHC ligands.
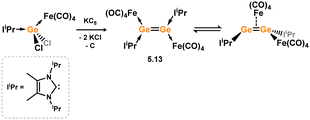 |
| | Scheme 42 Reactivity of a formally silicon(0) dimer towards transition metal fragments. | |
To the best of our knowledge, no further TM-complexes of heavier alkyne analogues are known.
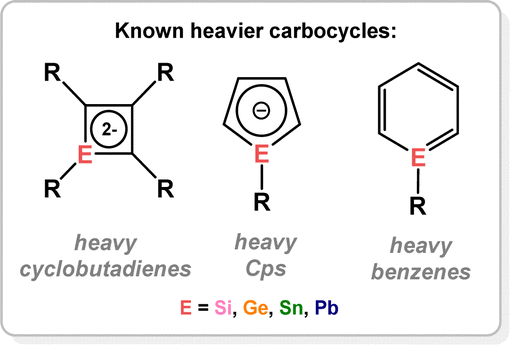
6. Heavier carbocycle derivatives
Closely related to the aforementioned ditetrelenes and tetrene complexes are TM-complexed heavier-tetrel element-substituted cyclobutene, cyclopentadiene, and benzene species (Fig. 11). ‘Free’ (i.e. non-TM complexed) derivatives of these species are known in most instances.89–95 As such, their TM complexes are typically accessed through direct combination with a suitable TM source. Though examples here are sparse compared to congeners of heavier group 14 π-complexes described earlier, those that are known allow for comparisons with the classical carbon derivatives.
 |
| | Fig. 11 The 4-, 5-, and 6-membered heavier carbocyle derivatives considerd in this review. | |
Heavier cyclobutadiene dianions have been accessed by reduction of di- or tetra-halo heavier cyclobutene derivatives (Scheme 43),89 which were utilised in salt-metathesis reactions for the formation or π-complexes. Though a number of neutral systems are known which can be described as heavier cyclobutadienes,90–92 albeit with distorted geometries, as yet the complexation chemistry of such species has not been explored. Heavier cyclopentadienide ligands have been synthesised directly from their protonated starting materials, as per classical Cp chemistry,93 which can be utilised directly in TM-complexation. For the heavier benzene derivatives, salt-elimination through reaction of heavier cyclohexadiene analogues with a suitable base has been demonstrated as an efficient method for the generation of these heavier aromatic systems.94,95 Though rare, these unique molecules can be directly utilised in complexation chemistry.
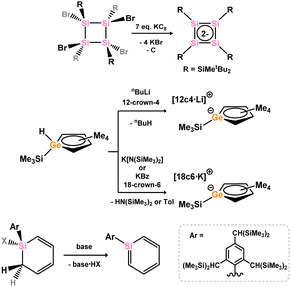 |
| | Scheme 43 Synthetic routes to potentially aromatic 4-, 5-, and 6-membered heavier carbocycle derivatives. | |
6.1. Silacycle complexes
The initial instance of an η5-silacyclopentadienyl, commonly referred to as silolyl, complex was first reported in 1998 by Tilley (Scheme 44).96 These species were accessed through the reaction of lithium siloyl 6.1 with [Cp*HfCl3], resulting in the formation of complex 6.2. This chemistry was extended to the corresponding Zr complex 6.3.97 Subsequently, Sekiguchi demonstrated that trisilacyclopentadiene (6.4) and germadisilacyclopentadiene (6.5) ligands, each incorporating three heavier group 14 atoms within the five-membered ring, could also form complexes with transition metals, again employing a salt-metathesis mechanism. This led to the formation of complexes 6.6 (Fe), 6.7 (Ru), and 6.8 (Rh), where the ‘heavy’ cyclopentadienyl ligands exhibit η5-coordination in all cases (Scheme 45).96,98 Most recently, the group of Roesky demonstrated that the silole dianion 6.9 reacts with [(cot)LaI·(THF)3] (cot = cyclooctatetraene) in the formation of anionic silole complex 6.10 (Scheme 46).99
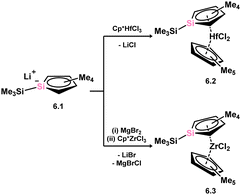 |
| | Scheme 44 Formation of group 4 complexes featuring the sila-cyclopentadienide ligand. | |
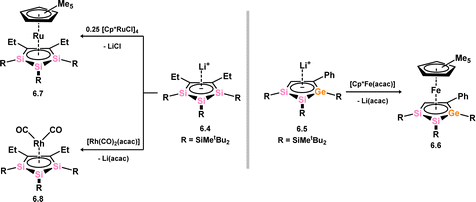 |
| | Scheme 45 Complexes bearing the disilagerma- and trisila-cyclopentadienide ligands. acac = acetylacetonate. | |
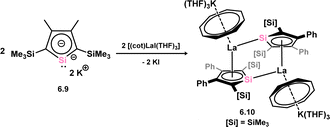 |
| | Scheme 46 Divergent coordination nature of a silole ligand in a lanthanum complex. cot = cyclooctatetraene. | |
Several complexes featuring silicon-containing cyclobutadiene ligands have been synthesised, with the first example being the monosila-cyclobutadiene cobalt complex 6.11. Surprisingly, this complex was accessed through the rearrangement of 4-silatriafulvene 6.12 in the presence of [CpCo(CO)2] after the exchange of both carbonyl ligands (Scheme 47).100 Subsequently, the anionic Cp-free complex 6.13, carrying a tetrasilacyclobutadiene ligand, was obtained through addition of the tetrasilacyclobutadiene dianion [{(tBu2MeSi)Si}4]K2 to [CpCo(CO)2], resulting in the loss of [CpK].101 The [Si4] core in this species demonstrate clearly a delocalised π-system (Fig. 12). Closely related Fe and Ru complexes 6.14 and 6.15 were subsequently prepared through similar synthetic procedures (Scheme 48).102,103
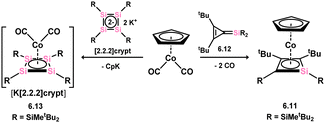 |
| | Scheme 47 Formation of cobalt complexes of the tetrasilacyclobutadidene-diide and silacyclobutadiene-diide ligands via differing routes. | |
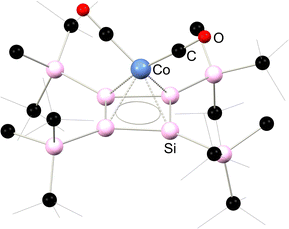 |
| | Fig. 12 Solid-state structure of the anionic part in 6.13. | |
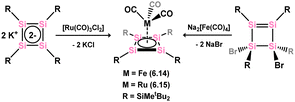 |
| | Scheme 48 Formation of ruthenium and iron complexes of the tetrasilacyclobutadidene-diide ligand. | |
Lastly, two TM-complexes bearing a silabenzene derivative have been documented, both through the direct addition of silabenzene analogue 6.16 to M0 precursors under UV irradiation, so forming 6.17 and 6.18 (Scheme 49). Among these, only the Cr complex 6.17 has been structurally characterised, revealing the silabenzene ligand's η6-coordination with the TM centre.104
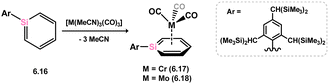 |
| | Scheme 49 Formation of chromium and molybdenum complexes of silabenzene. | |
6.2. Germacycle complexes
The first example of a germoyl-TM complex was published by Tilley and co-workers in 1993, before Si complex 6.2, and represents the first reported example of a heavier Cp-TM complex. 6.19 was synthesised in a similar manner to the described Si system, through the addition of the free lithium germoyl with [(CpRuCl)2] (Scheme 50).105 Some years later, Ge complex 6.20 was reported alongside its Si congener, and was synthesised in the same manner, that is through the addition of lithium-germoyl to [Cp*HfCl3].96 The same group later reported the reaction of the electronically similar lithiogermoyl-hafnocene complex 6.21 with [(dmpe)2MOTf] (M = Rh, Ir), which led to the formation of Ge-metallated germolyl complexes 6.22 and 6.23.106 Müller and co-workers reported closely related dimeric species 6.24 and 6.25, accessed through the addition of dipotassio-germoyl 6.26 with [Cp2MCl2] (M = Ti, Zr), first forming an anionic germylene complex, which dimerises in loss of KCp (Scheme 51).107 The Ti derivative 6.24 undergoes further salt-metathesis reaction with KCp, and ultimately eliminates Cp2, in generating dimeric TiIIIGe-metallated germoyl-Ti dimer 6.27. Complexes similar to dimeric TiIV and ZrIV complexes are also known for yttrium, accessed through the addition of dipotassio-germoyl 6.26 with [Cp*YCl2] and [YCl3·(THF)3], forming 6.28 and 6.29 respectively.108 An η5-coordination mode of the germoyl ring systems is observed in all cases, as is also true for the earlier described siloyl complexes.
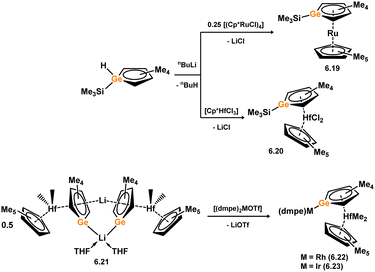 |
| | Scheme 50 Formation of mono- and bi-metallic complexes bearing the geramacyclopentadienide ligand. | |
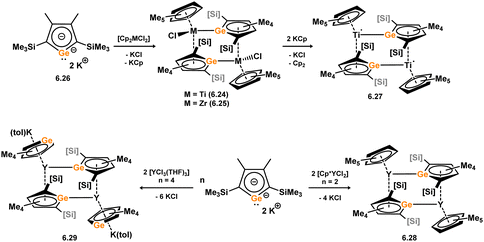 |
| | Scheme 51 Complexation of the germole dianion towards titanium and yttrium, forming dimeric species through Ge–M bonding (M = Ti, Y). | |
Heavier arene complex analogues, that is η6-germabenzene-TM complexes, are also known, albeit rather rare. All such species have been reported by Tokitoh and co-workers, the first in 2003, synthesised through the displacement of nitrile ligands in either [Cr(MeCN)3(CO)3] or [Mo(MeCN)3(CO)3] upon addition of germabenzene analogue 6.30, to [M(CO)3] complexes 6.31 and 6.32, respectively (M = Cr, Mo; Scheme 52).109 This strategy was later extended to the corresponding W complex, 6.33.110 Notably, here it was also shown that addition of germabenzene 6.30 to [(Cp*RuCl)4] did not lead to the desired η6-coordination, but rather to Cl-migration to Ge, forming an η5-ligand bind mode in 6.34.
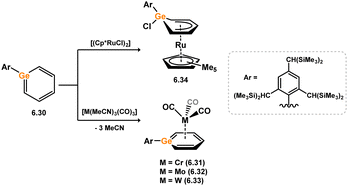 |
| | Scheme 52 Reactivity of germabenzene towards ruthenium(II), and group 6 metal(0) species. | |
6.3. Stannacycle complexes
Though somewhat less common than their Si and Ge counterparts, stannoyl-TM complexes are known. In the single report regarding stable examples of such complexes, Saito and co-workers showed that the anionic heavier ruthenocene analogue 6.35 reacts with a range of carbon and silicon halides through substitution at Sn, generating a family of novel stannoyl complexes, 6.36, 6.37, 6.38, and 6.39 (Scheme 53).111 All systems demonstrate η5-coordination of the stannoyl ring to Ru, though deviations from planarity are observed at the tin centre, the most extreme case being for Cl-substitution, showing a sum of angles at Sn of 306.1°.
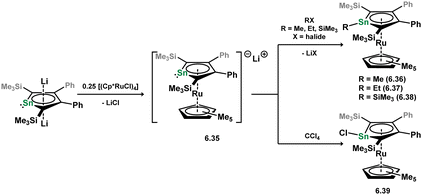 |
| | Scheme 53 Formation of stannacyclopentadienide complexes of ruthenium. | |
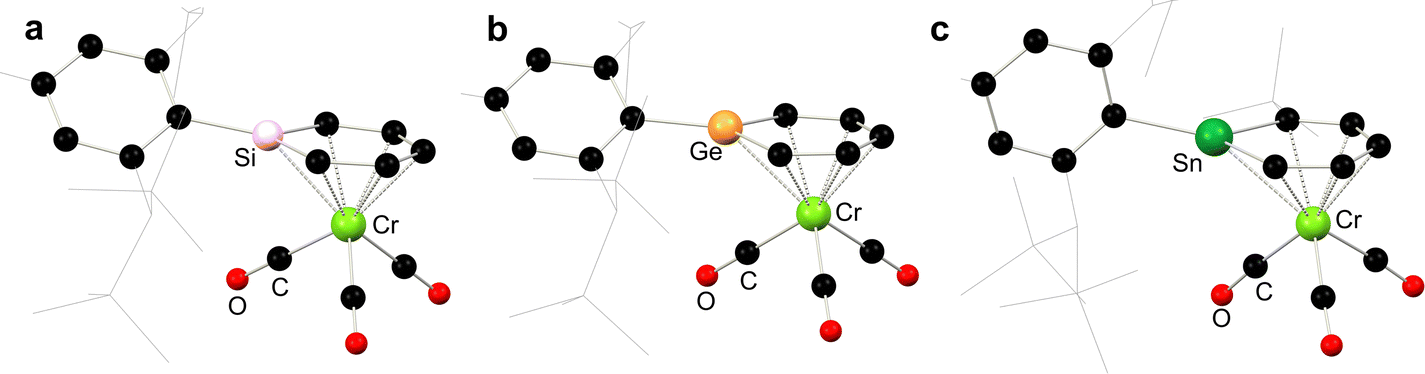
In addition to stannoyl systems described above, stannabenzene derivatives showing η6-coordination are also known. Published alongside the initial synthesis of a stannabenzene compound (vis. 6.40), perhaps better described as a stannanaphthalene, Tokitoh and co-workers showed that this heavier aromatic system readily coordinates the [Cr(CO)3] fragment upon reaction with [Cr(MeCN)3(CO)3], forming 6.41 (Scheme 54).112 It was later demonstrated that a similar reaction is observed for the related [2+2] dimer of stannabenzene 6.42, forming the stannabenezene complex 6.43, giving evidence for a mono-dimer equilibrium for 6.42 species in solution.113 The isolation of this stannabenzene derivative allows for a side-by-side comparison of sila-, germa-, and stannabenzene complexes of [Cr(CO)3] (Fig. 13), whereby all species show a clear planarity in the [EC5] ring, with a notable increase in E–(C5) distance on moving to the heavier elements, presumably due to atomic radius.
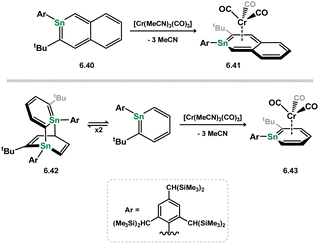 |
| | Scheme 54 Formation of stannabenzene complexes of chromium(0). | |
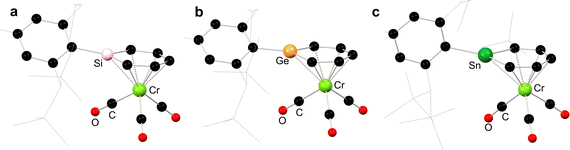 |
| | Fig. 13 Side-by-side comparison of the molecular structures of sila-, germa-, and stannabenzene complexes of [Cr(CO)3]. | |
6.4. Plumbacycle complexes
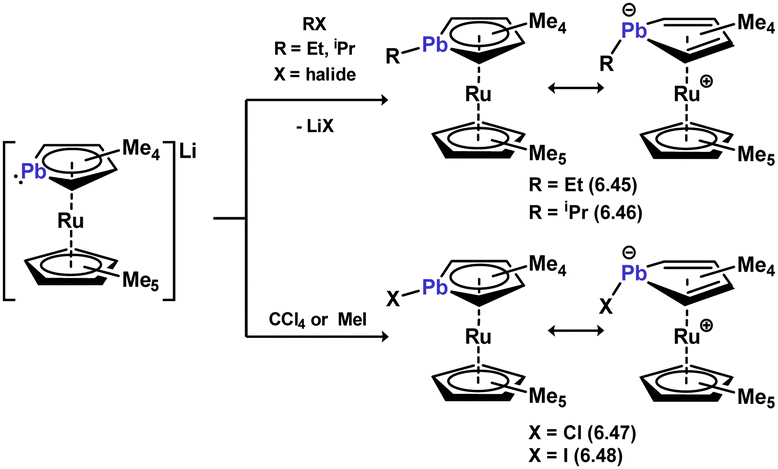
Remarkably, plumboyl-TM complexes are also known, albeit being very rare indeed. As per the described stannoyl systems, these were synthesised through the functionalisation of anionic heavier ruthenocene 6.44 with a range of carbon electrophiles, leading to plumboyl complexes 6.45, 6.46, 6.47, and 6.48 (Scheme 55).114 Unlike the majority of the lighter congeners of such complexes, the [C4Pb] ring in these species deviates significantly from planarity, through pyramidalisation at Pb indicative of a stereoactive lone-pair of electrons. These can therefore be described as η4-systems, reflecting the low inclination for the Pb-based lone electron pairs to partake in bonding interactions.
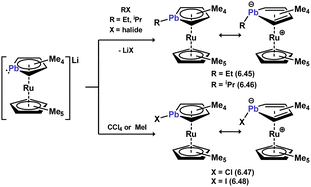 |
| | Scheme 55 Formation of plumacyclopentadienide complexes of ruthenium. | |
7. Summary and outlook
Driven by the discovery and understanding of carbon-centred organometallic π-complexation, research regarding access to and the electronic nature of the heavier derivatives of those systems has been fervent. Since the late 1900s, huge steps forward have been made in accessing heavier group 14 congeners of classical π-complexes, spanning almost the full range of systems known for carbon. This has given further insights into differences between π-bonding in all-carbon systems and those heavier derivatives, whereby deviations away from formal π-complexes, and towards e.g. metallacyclic complexes are observed, which may align with generally weaker π-bonding for Si–Pb. This, however, is largely ligand dependant, allowing for considerable control over the electronic nature and thus reactivity of these systems.
As this review demonstrates, a vast amount of research has been conducted in this field. Still, when one considers the array of compound classes here, and the expanse of the d-block elements, this merely scratches the surface. As our handle on accessing low-oxidation state species for Si–Pb constantly evolves, one can envisage a concomitant and continuous evolution of closely related transition metal chemistry, which, again aligning with related metal-free systems, may lead to a greater focus on further reactivity towards real-world utilisation. For now, there is plenty to explore and discover, and we very much look forward to seeing where this takes this exciting field.
Data availability
No primary research data are included in this review, and as such data sharing is not applicable here.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
TJH thanks the Fonds der Chemischen Industrie (FCI) for generous funding through a Liebig Stipendium, and the ERC for a Starting Grant (Project 101076897 – SINGAMBI). He also thanks Prof. Fässler for his continued support.
Notes and references
- W. C. Zeise, Ann. Phys., 1831, 97, 497–541 CrossRef.
- M. Black, R. H. B. Mais and P. G. Owston, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 1753–1759 CrossRef CAS.
- M. J. S. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C71–C79 Search PubMed.
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- E. O. Fischer and W. Pfab, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1952, 7, 377–379 CrossRef.
- G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125–2126 CrossRef CAS.
- D. M. P. Mingos, J. Organomet. Chem., 2001, 635, 1–8 CrossRef CAS.
- D. V. Banthorpe, Chem. Rev., 1970, 70, 295–322 CrossRef CAS.
-
B. M. Trost and I. Fleming, Comprehensive organic synthesis: selectivity, strategy, and efficiency in modern organic chemistry, Pergamon Press, Oxford, England, 1st edn, 1991 Search PubMed.
- E. O. Fischer and W. Hafner, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1955, 10, 665–668 CrossRef.
- D. Seyferth, Organometallics, 2002, 21, 2800–2820 CrossRef CAS.
- P. P. Power, Chem. Rev., 1999, 99, 3463–3504 CrossRef CAS PubMed.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- D. E. Goldberg, D. H. Harris, M. F. Lappert and K. M. Thomas, J. Chem. Soc., Chem. Commun., 1976, 261–262 RSC.
- P. J. Davidson, D. H. Harris and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1976, 2268–2274 RSC.
- E. A. Carter and W. A. Goddard, J. Phys. Chem., 1986, 90, 998–1001 CrossRef CAS.
- G. Trinquier and J. P. Malrieu, J. Am. Chem. Soc., 1987, 109, 5303–5315 CrossRef CAS.
- M. Lein, A. Krapp and G. Frenking, J. Am. Chem. Soc., 2005, 127, 6290–6299 CrossRef CAS PubMed.
- H. Li, H. Feng, W. Sun, Y. Xie and H. F. Schaefer, Inorg. Chem., 2013, 52, 6849–6859 CrossRef CAS PubMed.
- H. B. Wedler, P. Wendelboe and P. P. Power, Organometallics, 2018, 37, 2929–2936 CrossRef CAS.
- H. B. Wedler, P. Wendelboe, D. J. Tantillo and P. P. Power, Dalton Trans., 2020, 49, 5175–5182 RSC.
- M. Kira, Proc. Jpn. Acad., Ser. B, 2012, 88, 167–191 CrossRef CAS PubMed.
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed.
- M. J. Fink, M. J. Michalczyk, K. J. Haller, R. West and J. Michl, J. Chem. Soc., Chem. Commun., 1983, 1010–1011 RSC.
- E. K. Pham and R. West, J. Am. Chem. Soc., 1989, 111, 7667–7668 CrossRef CAS.
- E. K. Pham and R. West, J. Am. Chem. Soc., 1996, 118, 7871 CrossRef CAS.
- D. H. Berry, J. H. Chey, H. S. Zipin and P. J. Carroll, J. Am. Chem. Soc., 1990, 112, 452–453 CrossRef CAS.
- H. Hashimoto, Y. Sekiguchi, T. Iwamoto, C. Kabuto and M. Kira, Organometallics, 2002, 21, 454–456 CrossRef CAS.
- H. Hashimoto, Y. Sekiguchi, Y. Sekiguchi, T. Iwamoto, C. Kabuto and M. Kira, Can. J. Chem., 2003, 81, 1241–1245 CrossRef CAS.
- M. Kira, Y. Sekiguchi, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 2004, 126, 12778–12779 CrossRef CAS PubMed.
- T. Abe, T. Iwamoto and M. Kira, J. Am. Chem. Soc., 2010, 132, 5008–5009 CrossRef CAS PubMed.
- M. Hartmann, A. Haji-Abdi, K. Abersfelder, P. R. Haycock, A. J. P. White and D. Scheschkewitz, Dalton Trans., 2010, 39, 9288–9295 RSC.
- S. Inoue and C. Eisenhut, J. Am. Chem. Soc., 2013, 135, 18315–18318 CrossRef CAS PubMed.
- R. Fischer, M. Zirngast, M. Flock, J. Baumgartner and C. Marschner, J. Am. Chem. Soc., 2005, 127, 70–71 CrossRef CAS PubMed.
- M. Zirngast, M. Flock, J. Baumgartner and C. Marschner, J. Am. Chem. Soc., 2009, 131, 15952–15962 CrossRef CAS PubMed.
- J. Baumgartner, R. Fischer, J. Fischer, A. Wallner, C. Marschner and U. Flörke, Organometallics, 2005, 24, 6450–6457 CrossRef CAS.
- L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez and D. Polo, Coord. Chem. Rev., 2015, 300, 1–28 CrossRef.
- J. Baumgartner and C. Marschner, Rev. Inorg. Chem., 2014, 34, 119–152 CAS.
- H. Arp, J. Baumgartner, C. Marschner, P. Zark and T. Müller, J. Am. Chem. Soc., 2012, 134, 10864–10875 CrossRef CAS PubMed.
- J. Henning and L. Wesemann, Angew. Chem., Int. Ed., 2012, 51, 12869–12873 CrossRef CAS PubMed.
- C. P. Sindlinger, S. Weiß, H. Schubert and L. Wesemann, Angew. Chem., Int. Ed., 2015, 54, 4087–4091 CrossRef CAS PubMed.
- T. Kuwabara, J. D. Guo, S. Nagase and M. Saito, Angew. Chem., Int. Ed., 2014, 53, 434–438 CrossRef CAS PubMed.
- A. G. Brook, F. Abdesaken, B. Gutekunst, G. Gutekunst and R. K. Kallury, J. Chem. Soc., Chem. Commun., 1981, 191–192 RSC.
- Y. Apeloig, M. Bendikov, M. Yuzefovich, M. Nakash, D. Bravo-Zhivotovskii, D. Bläser and R. Boese, J. Am. Chem. Soc., 1996, 118, 12228–12229 CrossRef CAS.
- C. Couret, J. Escudie, J. Satge and M. Lazraq, J. Am. Chem. Soc., 1987, 109, 4411–4412 CrossRef CAS.
- V. Ya Lee and A. Sekiguchi, Organometallics, 2004, 23, 2822–2834 CrossRef CAS.
- B. K. Campion, R. H. Heyn and T. D. Tilley, J. Am. Chem. Soc., 1988, 110, 7558–7560 CrossRef CAS.
- B. K. Campion, R. H. Heyn, T. D. Tilley and A. L. Rheingold, J. Am. Chem. Soc., 1993, 115, 5527–5537 CrossRef CAS.
- B. K. Campion, R. H. Heyn and T. D. Tilley, J. Am. Chem. Soc., 1990, 112, 4079–4081 CrossRef CAS.
- T. S. Koloski, P. J. Carroll and D. H. Berry, J. Am. Chem. Soc., 1990, 112, 6405–6406 CrossRef CAS.
- S. R. Klei, T. D. Tilley and R. G. Bergman, Organometallics, 2001, 20, 3220–3222 CrossRef CAS.
- D. Bravo-Zhivotovskii, H. Peleg-Vasserman, M. Kosa, G. Molev, M. Botoshanskii and Y. Apeloig, Angew. Chem., Int. Ed., 2004, 43, 745–748 CrossRef CAS PubMed.
- N. Nakata, R. Rodriguez, T. Troadec, N. Saffon-Merceron, J. Sotiropoulos, A. Baceiredo and T. Kato, Angew. Chem., Int. Ed., 2013, 52, 10840–10844 CrossRef CAS PubMed.
- H. Arp, C. Marschner, J. Baumgartner, P. Zark and T. Müller, J. Am. Chem. Soc., 2013, 135, 7949–7959 CrossRef CAS PubMed.
- P. K. Majhi, M. Zimmer, B. Morgenstern and D. Scheschkewitz, J. Am. Chem. Soc., 2021, 143, 8981–8986 CrossRef CAS PubMed.
- N. Wiberg, K. Schurz, G. Reber and G. Müller, J. Chem. Soc., Chem. Commun., 1986, 591–592 RSC.
- A. Meller, G. Ossig, W. Maringgele, D. Stalke, R. Herbst-Irmer, S. Freitag and G. M. Sheldrick, J. Chem. Soc., Chem. Commun., 1991, 1123–1124 RSC.
- M. Draeger, J. Escudie, C. Couret, H. Ranaivonjatovo and J. Satge, Organometallics, 1988, 7, 1010–1013 CrossRef CAS.
- L. Li, T. Fukawa, T. Matsuo, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, Nat. Chem., 2012, 4, 361–365 CrossRef CAS PubMed.
- I. Alvarado-Beltran, A. Rosas-Sánchez, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 10481–10485 CrossRef CAS PubMed.
- D. Wendel, D. Reiter, A. Porzelt, P. J. Altmann, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 17193–17198 CrossRef CAS PubMed.
- L. J. Procopio, P. J. Carroll and D. H. Berry, J. Am. Chem. Soc., 1991, 113, 1870–1872 CrossRef CAS.
- M. Okazaki and S. Ebina, Chem. Lett., 2014, 43, 1089–1091 CrossRef CAS.
- D. A. Straus, S. D. Grumbine and T. D. Tilley, J. Am. Chem. Soc., 1990, 112, 7801–7802 CrossRef CAS.
- T. J. Hadlington, T. Szilvási and M. Driess, J. Am. Chem. Soc., 2019, 141, 3304–3314 CrossRef CAS PubMed.
- B. Minkovich, A. Kaushansky, N. Fridman, D. Bravo-Zhivotovskii and Y. Apeloig, Mendeleev Commun., 2022, 32, 28–32 CrossRef CAS.
- T. Fukuda, H. Hashimoto, S. Sakaki and H. Tobita, Angew. Chem., Int. Ed., 2016, 55, 188–192 CrossRef CAS PubMed.
- T. J. Hadlington, T. Szilvási and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 14282–14286 CrossRef CAS PubMed.
- S. Takahashi, M. A. Ramos-Enríquez, E. Bellan, A. Baceiredo, N. Saffon-Merceron, N. Nakata, D. Hashizume, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2021, 60, 18489–18493 CrossRef CAS PubMed.
- A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2014, 53, 565–570 CrossRef CAS PubMed.
- Z. T. Cygan, J. W. Kampf and M. M. Banaszak Holl, Inorg. Chem., 2003, 42, 7219–7226 CrossRef CAS PubMed.
- T. Matsumoto, Y. Nakaya, N. Itakura and K. Tatsumi, J. Am. Chem. Soc., 2008, 130, 2458–2459 CrossRef CAS PubMed.
- T. Matsumoto, N. Itakura, Y. Nakaya and K. Tatsumi, Chem. Commun., 2011, 47, 1030–1032 RSC.
- L. Pu, B. Twamley and P. P. Power, J. Am. Chem. Soc., 2000, 122, 3524–3525 CrossRef CAS.
- A. Sekiguchi, R. Kinjo and M. Ichinohe, Science, 2004, 305, 1755–1757 CrossRef CAS PubMed.
- N. Wiberg, S. K. Vasisht, G. Fischer and P. Mayer, Z. Anorg. Allg. Chem., 2004, 630, 1823–1828 CrossRef CAS.
- N. Takagi and S. Nagase, Eur. J. Inorg. Chem., 2002, 2775–2778 CrossRef CAS.
- M. Stender, A. D. Phillips, R. J. Wright and P. P. Power, Angew. Chem., Int. Ed., 2002, 41, 1785–1787 CrossRef CAS PubMed.
- A. D. Phillips, R. J. Wright, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2002, 124, 5930–5931 CrossRef CAS PubMed.
- S. Ishida, R. Sugawara, Y. Misawa and T. Iwamoto, Angew. Chem., Int. Ed., 2013, 52, 12869–12873 CrossRef CAS PubMed.
- T. Yamaguchi, A. Sekiguchi and M. Driess, J. Am. Chem. Soc., 2010, 132, 14061–14063 CrossRef CAS PubMed.
- Z. He, L. Liu, F. J. De Zwart, X. Xue, A. W. Ehlers, K. Yan, S. Demeshko, J. I. Van Der Vlugt, B. De Bruin and J. Krogman, Inorg. Chem., 2022, 61, 11725–11733 CrossRef CAS PubMed.
- S. S. Sen, A. Jana, H. W. Roesky and C. Schulzke, Angew. Chem., Int. Ed., 2009, 48, 8536–8538 CrossRef CAS PubMed.
- H. P. Hickox, Y. Wang, Y. Xie, M. Chen, P. Wei, H. F. Schaefer and G. H. Robinson, Angew. Chem., Int. Ed., 2015, 54, 10267–10270 CrossRef CAS PubMed.
- M. Chen, Y. Wang, Y. Xie, P. Wei, R. J. Gilliard, N. A. Schwartz, H. F. Schaefer, P. V. R. Schleyer and G. H. Robinson, Chem. – Eur. J., 2014, 20, 9208–9211 CrossRef CAS PubMed.
- X. Wang, Y. Peng, M. M. Olmstead, H. Hope and P. P. Power, J. Am. Chem. Soc., 2010, 132, 13150–13151 CrossRef CAS PubMed.
- Y. Wang, M. Karni, S. Yao, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 1655–1664 CrossRef CAS PubMed.
- A. Jana, V. Huch, H. S. Rzepa and D. Scheschkewitz, Organometallics, 2015, 34, 2130–2133 CrossRef CAS.
- V. Ya Lee, K. Takanashi, T. Matsuno, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2004, 126, 4758–4759 CrossRef CAS PubMed.
- K. Suzuki, T. Matsuo, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, Science, 2011, 331, 1306–1309 CrossRef CAS PubMed.
- K. Suzuki, Y. Numata, N. Fujita, N. Hayakawa, T. Tanikawa, D. Hashizume, K. Tamao, H. Fueno, K. Tanaka and T. Matsuo, Chem. Commun., 2018, 54, 2200–2203 RSC.
- R. C. Handford, T. A. Wheeler and T. D. Tilley, Chem. – Eur. J., 2020, 26, 6126–6129 CrossRef CAS PubMed.
- W. P. Freeman, T. D. Tilley, L. M. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 1996, 118, 10457–10468 CrossRef CAS.
- K. Wakita, N. Tokitoh, R. Okazaki, N. Takagi and S. Nagase, J. Am. Chem. Soc., 2000, 122, 5648–5649 CrossRef CAS.
- N. Nakata, N. Takeda and N. Tokitoh, J. Am. Chem. Soc., 2002, 124, 6914–6920 CrossRef CAS PubMed.
- J. M. Dysard and T. D. Tilley, J. Am. Chem. Soc., 1998, 120, 8245–8246 CrossRef CAS.
- J. M. Dysard and T. D. Tilley, J. Am. Chem. Soc., 2000, 122, 3097–3105 CrossRef CAS.
- V. Ya Lee, R. Kato, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2005, 127, 13142–13143 CrossRef CAS PubMed.
- X. Sun, L. Münzfeld, D. Jin, A. Hauser and P. W. Roesky, Chem. Commun., 2022, 58, 7976–7979 RSC.
- Y. Kon, K. Sakamoto, C. Kabuto and M. Kira, Organometallics, 2005, 24, 1407–1409 CrossRef CAS.
- K. Takanashi, V. Ya Lee, T. Matsuno, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2005, 127, 5768–5769 CrossRef CAS PubMed.
- K. Takanashi, V. Ya Lee, M. Ichinohe and A. Sekiguchi, Angew. Chem., Int. Ed., 2006, 45, 3269–3272 CrossRef CAS PubMed.
- K. Takanashi, V. Ya Lee and A. Sekiguchi, Organometallics, 2009, 28, 1248–1251 CrossRef CAS.
- A. Shinohara, N. Takeda, T. Sasamori, T. Matsumoto and N. Tokitoh, Organometallics, 2005, 24, 6141–6146 CrossRef CAS.
- W. P. Freeman, T. D. Tilley, A. L. Rheingold and R. L. Ostrander, Angew. Chem., Int. Ed. Engl., 1993, 32, 1744–1745 CrossRef.
- J. M. Dysard and T. D. Tilley, Organometallics, 2000, 19, 2671–2675 CrossRef CAS.
- Z. Dong, O. Janka, J. Kösters, M. Schmidtmann and T. Müller, Angew. Chem., Int. Ed., 2018, 57, 8634–8638 CrossRef CAS PubMed.
- J. Liu, K. Singh, S. Dutta, Z. Feng, D. Koley, G. Tan and X. Wang, Dalton Trans., 2021, 50, 5552–5556 RSC.
- N. Nakata, N. Takeda and N. Tokitoh, Angew. Chem., Int. Ed., 2003, 42, 115–117 CrossRef CAS PubMed.
- N. Tokitoh, N. Nakata, A. Shinohara, N. Takeda and T. Sasamori, Chem. – Eur. J., 2007, 13, 1856–1862 CrossRef CAS PubMed.
- T. Kuwabara, M. Nakada, J. D. Guo, S. Nagase and M. Saito, Dalton Trans., 2015, 44, 16266–16271 RSC.
- Y. Mizuhata, T. Sasamori, N. Takeda and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1050–1051 CrossRef CAS PubMed.
- Y. Mizuhata, S. Fujimori, N. Noda, S. Kanesato and N. Tokitoh, Dalton Trans., 2018, 47, 14436–14444 RSC.
- M. Nakada, T. Kuwabara, S. Furukawa, M. Hada, M. Minoura and M. Saito, Chem. Sci., 2017, 8, 3092–3097 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a