
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Black titanium oxide: synthesis, modification, characterization, physiochemical properties, and emerging applications for energy conversion and storage, and environmental sustainability
Xuelan
Hou
 ab,
Yiyang
Li
b,
Hang
Zhang
c,
Peter D.
Lund
c,
James
Kwan
*a and
Shik Chi Edman
Tsang
*b
ab,
Yiyang
Li
b,
Hang
Zhang
c,
Peter D.
Lund
c,
James
Kwan
*a and
Shik Chi Edman
Tsang
*b
aDepartment of Engineering Sciences, University of Oxford, Oxford, OX1 3PJ, UK. E-mail: james.kwan@balliol.ox.ac.uk
bWolfson Catalysis Center, Department of Chemistry, University of Oxford, Oxford, OX1 3QR, UK. E-mail: edman.tsang@chem.ox.ac.uk
cDepartment of Applied Physics, School of Science, Aalto University, P. O. Box 15100, FI-00076 Aalto, Finland
First published on 13th September 2024
Abstract
Since its advent in 2011, black titanium oxide (B-TiOx) has garnered significant attention due to its exceptional optical characteristics, notably its enhanced absorption spectrum ranging from 200 to 2000 nm, in stark contrast to its unmodified counterpart. The escalating urgency to address global climate change has spurred intensified research into this material for sustainable hydrogen production through thermal, photocatalytic, electrocatalytic, or hybrid water-splitting techniques. The rapid advancements in this dynamic field necessitate a comprehensive update. In this review, we endeavor to provide a detailed examination and forward-looking insights into the captivating attributes, synthesis methods, modifications, and characterizations of B-TiOx, as well as a nuanced understanding of its physicochemical properties. We place particular emphasis on the potential integration of B-TiOx into solar and electrochemical energy systems, highlighting its applications in green hydrogen generation, CO2 reduction, and supercapacitor technology, among others. Recent breakthroughs in the structure–property relationship of B-TiOx and its applications, grounded in both theoretical and empirical studies, are underscored. Additionally, we will address the challenges of scaling up B-TiOx production, its long-term stability, and economic viability to align with ambitious future objectives.
 Xuelan Hou | Xuelan Hou is currently an assistant professor at Xi’an Jiaotong University, China. She was a Postdoctoral research assistant at both the Department of Engineering Science and the Department of Chemistry, University of Oxford, UK during the preparation of this review. Her research studies were on enhancing the efficiency and stability of photoelectrochemical cells and solar cells with black color semiconductors. Her research interests are also in black semiconductor synthesis and applications for solar energy systems and electrochemical storage systems. |
Yiyang Li is a Postdoctoral research associate at the Department of Chemistry, University of Oxford, UK. He has been working on the solar-driven photocatalytic overall water splitting for hydrogen evolution, especially the local electric and magnetic field effects in this system. His research interests also include the material design, synthesis, and characterization for other sustainable energy systems. |
Hang Zhang is a Research Fellow at the Department of Applied Physics of Aalto University, Finland. His research covers synthesis, characterization, and self-assembly of nanomaterials, design of hydrogels with novel properties, and bio-inspired soft systems. |
Peter D. Lund is a professor of Engineering Physics and Advanced Energy Systems at the Department of Applied Physics of Aalto University, Finland. He pioneered solar energy and energy storage research in Finland. His specialization areas include both New and renewable energy systems, and Energy and innovation. He is active internationally and holds several positions of trust. |
 James Kwan | James Kwan is an associate professor at the Department of Engineering Science and a Tutorial Fellow at Balliol College, University of Oxford, UK. Before that he obtained a PhD from Columbia University in chemical engineering. Kwan's research interests include the study of ultrasound and cavitation and applying it to address challenges in personal and environmental health. Regarding environmental health, he has a particular interest in reaction engineering and sonocatalysis to improve sonochemical performance and facilitate green chemistry. |
 Shik Chi Edman Tsang | Shik Chi Edman Tsang is a professor of Inorganic Chemistry and Head of Wolfson Catalysis Laboratory at the University of Oxford, UK. He also serves as the catalysis theme coordinator in the department. His main research interests are on nanomaterials and heterogeneous catalysis concerning energy and environment which include developments of catalytic, photocatalytic and electrocatalytic technologies for green chemistry, fine chemicals, cleaner combustion, energy storages, processes and production. Particular expertise is in design and architecture of nanocatalysts and their in situ diffraction and spectroscopic characterization, which can lead to understanding of catalytic surfaces and interfaces. |
1. Introduction
The escalating climate and energy crises have underscored the importance of renewable energy sources such as solar, wind, geothermal, biomass, hydro, and tidal energy. Solar energy, in particular, stands out as the most abundant and clean resource. With an annual solar flux of 3.00 × 1024 Joules (J) per year, reaching the Earth's surface—over 5000 times the global energy consumption in 2021—solar energy is poised to play a pivotal role in the transition to a sustainable society.1,2 However, the full exploitation of solar energy faces several challenges: (i) the development of materials with high solar radiation capture efficiency; (ii) the creation of high-performance, stable devices for solar energy conversion and storage; and (iii) the optimization of energy storage and transportation from an economic and technological standpoint.To capture solar energy sufficiently, ‘black’ semiconductors, such as black titanium oxide (B-TiOx),3–6 black silicon,7–9 black phosphorus,10–12 black bismuth vanadium oxide,13–15 black tungstic oxide,16 and black copper oxide,17 are promising materials with special physicochemical properties for broadened light capture from ultraviolet (UV) to near-infrared (NIR) light, and increased numbers of charge carriers by several magnitudes. Among them, B-TiOx has been regarded as the most promising black semiconductor with low cost and a vast range of applications.
‘Black’ semiconductors, such as B-TiOx, have emerged as promising materials for broad-spectrum light capture, from UV to NIR, and for significantly increasing charge carrier generation. B-TiOx, first synthesized via hydrogenation in 2011, has shown remarkable light absorption in the 360–1300 nm wavelength range and enhanced photocatalytic hydrogen production.3 Its enduring interest stems from its unique physicochemical properties—narrow bandgap, efficient light absorption, rapid electron transport, and high electrical conductivity—coupled with the intrinsic advantages of pristine TiO2, such as non-toxicity, affordability, chemical and mechanical stability, and versatility in shape, size, morphology, and crystalline phase. Efforts to reduce the energy and time costs of B-TiOx production have led to innovations in synthesis, including the manipulation of blackness during fabrication.18–20 For example, one-dimension (1D) B-TiOx nanotubes share the same advantage with both B-TiOx in a low charge carrier recombination rate and with 1D pristine TiO2 nanotubes in a high specific surface area.21–23 However, the energy and time cost of the hydrogenation method to produce B-TiOx were huge. Therefore, tremendous efforts have been devoted to lowering the energy and time costs by manipulating the local degrees of blackness (B°) during synthesis of B-TiOx.24,25
In this context, a lot of progress has been achieved in the synthesis of B-TiOx. Significant progress has been made in optimizing B-TiOx synthesis, with hydrogenation remaining the primary method. Synthesis conditions have been refined, and alternative techniques like plasma treatment and chemical reduction have been explored to lower production costs. Despite these advances, B-TiOx's performance in various applications is hampered by the rapid charge carrier recombination and suboptimal electrical conductivity. Strategies to enhance B-TiOx's industrial-level performance include doping, noble metal nanoparticle decoration, junction construction, and facet engineering, all of which require precise characterization to identify genuine material modifications.26 For instance, the photocatalytic H2 evolution rate of B-TiOx is only 0.388 mmol g−1 h−1 compared to the best recorded value of 21.1 mmol g−1 h−1 in N-TiO2 on MgO (111), which stems from low light utilization and severe charge carrier recombination.3 To optimize B-TiOx towards an industrial-level performance, several strategies have been proposed, such as doping and co-doping elements, decorating noble metal nanoparticles, constructing homo/hetero/tandem junctions, facet engineering, etc.27 At the same time, proper characterizations are necessary to identify the genuine modifications in B-TiOx, resulting from different strategies.28,29 For example, in 2023, Monai et al. used operando scanning transmission electron microscopy and infrared spectroscopy to observe the change of extremely thin top layer of TiOx under hydrogenation reaction conditions.
Concerning the applications of B-TiOx, they are mainly focused on solar energy conversion and storage systems due to their characteristic properties in light absorption, which have been studied both experimentally and theoretically.30 For instance, B-TiOx works as the photocatalyst to split water into H2 and O2 gases by the simulated sunlight source, which has great potential in addressing the challenges in the energy crisis and global warming.31–35 On the other hand, B-TiOx has been investigated as a functional material for efficient and reliable energy storage systems that is driven by its fast electron transport rate, long electron lifetime, relatively high electrical conductivity, vast active sites of Ti3+, and oxygen vacancies (Ov). For example, B-TiOx can serve as a crucial candidate to replace fossil fuels in ion-batteries.36,37 Recently, B-TiOx is renowned for its interaction with metals, known as the strong metal-support interaction, to tune catalytic performance and even product selectivity.
The rapidly increasing amount of literature on B-TiOx calls for a continuous updating of review articles. Among them, a first comprehensive review of its fabrications, physicochemical properties, and applications was published in 2015 by Chen et al.6 To date, several short articles dedicated with special focuses on the potential applications of B-TiOx have also been reported.4,6,30,32–34,38–40 In addition, these reviews have also covered the diversity of synthesis methods and provided understandings of the physicochemical properties of B-TiOx though often in a specific way.4,6,30,32–34,38,39 Meanwhile, continuous studies have deepened our understanding, revealed new properties, enriched designs for devices/installation, and broadened the applications of B-TiOx to address the energy crisis and environmental issues. On the other hand, black semiconductors other than B-TiOx are also gaining increasing attention due to their similar application potential for renewables. With this background, we believe that this comprehensive review serves as a timely update of the recent advances of synthesis, modification, characterization, and potential application of B-TiOx and can also provide a future-oriented perspective on the topic of B-TiOx.
Herein, this review first presents the definition of B-TiOx along with its main advantages in solar light capture in Section 2. Six most important methods for the synthesis of B-TiOx are presented in Section 3 and brief strategies for modification of B-TiOx are outlined in Section 4. In Section 5, characteristic properties of B-TiOx are addressed to understand the material. Representative promising applications of B-TiOx for solar energy utilization, conversion and storage, and electrical energy storage systems are mainly given in Section 6, including water (H2O) splitting into H2, O2, and H2O2 production and supercapacitors. In Section 7, perspectives on key challenges in controlling the local degrees of blackness and reduced depth of B-TiOx, and some new in situ/operando preparation, modification, and characterization techniques are outlined. Large-scale production of B-TiOx and its long-term stability are also considered toward commercialization.
Overall, this review is intended to guide readers through the nuances of cost-effective production methods for B-TiOx and similar black semiconductors, enriching their comprehension of the materials’ distinctive properties and underlying mechanisms. We will specifically highlight recent advancements in synchrotron techniques and machine learning that have been instrumental in tackling the energy and environmental challenges associated with these materials. Furthermore, we will encapsulate the strides made in their potential applications, thereby contributing to a broader technological understanding of B-TiOx.
2. Black titanium oxide
2.1. Definition
Black TiOx is originally named for its ‘black’ appearance, as shown in Fig. 1(a), which contrasts with the white color of the pristine TiO2. In this review, we define a material as black TiOx (B-TiOx), if it satisfies one of the following criteria, or is a modification thereof:41 | ||
| Fig. 1 (a) A photo of unmodified white TiO2 and B-TiOx, and (b) electronic bands of both white TiO2 and B-TiOx: a short-dashed curve is applied to outline a portion of the interface between the crystalline core and the disordered outer layer of B-TiOx. Reprinted with permission from ref. 3. Copyright © 2011, The American Association for the Advancement of Science. | ||
(i) Non-white color of TiO2: the color can be dark, grey, brown, deep blue, etc.
(ii) Narrower bandgap than that of the pristine TiO2 of a strict value of 3.2 eV for anatase TiO2 and 3.0 eV for rutile TiO2. As shown in Fig. 1(b), the bandgap of B-TiO2 is 1.54 eV.
(iii) Light absorption edge longer than 400 nm.
(iv) Structurally disordered surface and subsurface is structurally disordered.
(v) Chemical formula of TiOx, where (0 < x < 2).
(vi) Reductive or inert conditions during synthesis, resulting in names such as ‘reduced TiO2’, ‘defective TiO2’, and ‘hydrogenated TiO2’.
2.2. Potential advantages
(ii) Various shapes, dimensions (D), and sizes: Dimensions (e.g., 0D, 1D, 2D, and 3D), shapes (e.g., nanofiber, nanotube, nanosheet, hierarchical flower-like), and sizes (from nanometers to microns), Fig. 2(a and b).42–53
 | ||
| Fig. 2 (a) Synthesis of the monomicelles as building blocks for mesostructured TiO2 materials and (b) summary of different mesoporous TiO2 structures. Reprinted with permission from ref. 52 Copyright © 2022, Wiley-VCH GmbH. TiO2 films are assembled from (c) 3D printing. Reprinted with permission from ref. 54. Copyright © 2019, Wiley-VCH Verlag GmbH & Co. KGaA, Weinhein. | ||
(iii) Synthesis methods: TiO2 powder is commonly obtained from commercial products such as P25 and from hydrothermal and sol–gel synthesis method using titanium precursors such as titanium tetrachloride.52,53 Films are typically formed by self-assembling methods such as the anodic oxidation to form TiO2 nanotube films on a titanium metal,55–57 hydrothermal method,58–60 atomic layer deposition method,61–65 ink-jet printing method,66etc.; and coating methods (Fig. 2(c)), such as dip-coating, spin-coating, spray-coating,67–69 doctor blade, 3D-printing method,54,70etc.
(iv) Wide range of applications including energy, environment, electronic materials, health and medicine, etc.
(ii) Energy band: narrowed bandgap resulting from shifted valence band locations and thus-formed long band tails and mid-gaps.
(iii) Surface active site: the formation of surface defects of Ti3+ species, –OH, oxygen vacancies (Ov) and surface or near-surface disorders.31
(iv) Interface resistivity: low interface resistivity. (e.g., typical B-TiOx nanotube films are two orders of magnitude lower than anatase TiO2 films71).
(v) Charge carrier: relatively higher charge carrier density, lower charge carrier recombination, and faster charge carriers’ transport rate.72
(vi) Longer lifetime of excited electrons.
These potential advantages enable B-TiOx to form a wide range of functional structures for a wide range of applications, for example as both the anode and cathode electrodes in a fuel cell.3,28,73
3. Synthesis methods
Tremendous effort has been devoted to the synthesis of B-TiOx motivated by its wide-acknowledged advantages in its capability for absorbing the spectrum from UV to NIR range such as oxidation of hydrides.74–76 Recently, the importance of reductive overlayer to boost catalytic reactions have also been emphasized.77–83 Herein, this section will focus on presenting representative synthesis methods: hydrogenation, electrochemical reduction, chemical reduction, solution soaking, plasma, and ultrasonic treatment, along with the costs of economic and energetic considerations. The unique features and parameters of each method to manipulate the degree and depth of blackness (B°) of B-TiOx are highlighted.3.1. Hydrogenation
Hydrogenation is the first reported method to prepare B-TiOx and has also been the most widely used one.3,84 It is typically conducted in a tube furnace (Fig. 3(a)) that enables gas flow to maintain an inert or reductive atmosphere at a temperature range of 200–800 °C for a duration of 0.5–120 h. The gas used in the tube furnace is often a single-component gas such as H2,21,85 N2, CO,86 and NH3,87,88 or a mixture of gases such as H2/N271,89–95 and H2/Ar,96 and vacuum.97 For instance, as shown in Fig. 3(b), the N, C co-doped TiO2 is prepared in 5% H2/Ar at 800 °C using urea as a dopant.98 At lower post-annealing temperature, the time for the synthesis of B-TiOx will increase.99 For example, TiO2 powder has been annealed at the temperature of 200 °C that required 120 h to form B-TiOx.3 In contrast, the time required will be more than 360 h, if the post-annealed temperature is set at room temperature (RT).99 | ||
| Fig. 3 (a) A schematic to show the hydrogeneration method using Ar gas in a tube furnace, and (b) a drawing and photographs concerning the synthesis of N, C-codoped TiO2 samples under H2 gas condition. Reprinted with permission from ref. 98. Copyright © 2020, American Chemical Society. | ||
To manipulate B° of B-TiOx, parameters including atmosphere, post-annealing temperature and duration, and annealing times that can be controlled. As depicted in Fig. 3(b), a group of photographs showing the influence of both annealing temperature and duration on the B° of B-TiOx films along with their bandgaps, and the photoelectrochemical (PEC) water splitting (WS) performance, 21, 33, 38, 38, 36, and 20 μA cm−2, respectively, under the same test conditions, is given. The PECWS performance reveals that the darker of the TiO2 and the narrower of the bandgaps do not necessarily lead to higher activity. Therefore, it is important to control B° of B-TiOx to an optimal range to achieve the highest performance for a specific reaction.
Despite the facile production of B-TiOxvia hydrogenation method, it still needs further consideration for large-scale production of B-TiOx in aspects of energy cost, time, and consumption of high-value gases such as Ar and H2.
3.2. Electrochemical reduction
The electrochemical reduction (ECR) method is carried out in an electrochemical (EC) cell that uses the negative potential with reductive agent(s) to produce B-TiOx.21,100–108In an EC cell, the B-TiOx is often produced at RT for a duration of less than 1 h. For instance, Hou et al. prepared B-TiOx in an EC cell using an organic electrolyte of a mixture of 0.3 wt% ammonium fluoride (NH4F) and 6 vol% deionized H2O in ethylene glycol (EG) under a constant 60 V for 1–3 min, giving a 3 times enhancement in the value of photocurrent density over the pristine TiO2 under the same test conditions.101 To manipulate B° of B-TiOx with the ECR method, parameters including electrolytes (organic or inorganic), applied constant current/potential, and reduction time are to be controlled as listed in Table 1.109–117 From Table 1, there is a reverse relationship between the ECR time and the applied current/potential in both inorganic and organic electrolytes. For example, 30 min is required to produce B-TiOx in 1 M Na2SO4, under a bias of −0.4 VRHE,109 whereas, it only takes 15 s under a bias of −5 V.110,111
| No. | Electrolyte | Bias/Current | Time | Ref. |
|---|---|---|---|---|
| 1 | 1 M Na2SO4 | −0.4 VRHE | 30 min | 109 |
| 2 | 0.5 M Na2SO4 | −5 V | 0.25 min | 110 and 111 |
| 3 | 0.5 M Na2SO4 | −1.0 VSCE | 10 min | 112 |
| −1.2 VSCE | ||||
| −1.4 VSCE | ||||
| −1.6 V SCE | ||||
| 4 | 1 M NaClO4 | −5 mA cm−2 | 10 min | 113 |
| 5 | 0.1 M potassium phosphate buffer solution (KPi) | −5 mA cm−2 | 10 min | 93 |
| 6 | Phosphate buffer solution, pH = 7.2 | −17 mA cm−2 | 1.5 min | 114 |
| 7 | 1 M Li2SO4 aqueous solution | −1.2 VAg/AgCl | 30 min | 115 |
| 8 | EG solution of 0.27 wt% NH4F | −40 V | 3.33 min (200 s) | 100 and 116 |
| 9 | 0.3 wt% NH4F and 6 vol% H2O in EG | −60 V | 1 min | 101 |
| 2 min | ||||
| 3 min | ||||
| 10 | 0.27 wt% NH4F and 5 wt% H2O in EG | −30 V | 15 min | 117 |
| −35 V | 20 min | |||
| −40 V | 30 min | |||
| −45 V | 60 min | |||
| −50 V | 120 min | |||
| 11 | 200 mL EG and 10 mL 1 M of Na2SO4 | −6 V | 0.5 min | 107 |
In comparison to the hydrogenation method, the merits of the ECR method include higher cost-effectiveness and lower energy and time cost. Moreover, the ECR method enables the use of small voltages and currents from renewable electricity to power this process, such as photovoltage from solar cells.118 It should be noted that TiO2 in powder form cannot be employed using this method to prepare the B-TiOx.
3.3. Chemical reduction
The chemical reduction (CR) method involves the use of reductive agents to generate the reductive condition to produce B-TiOx.119–121 There are two kinds of reductive agents commonly used in the CR process: active metals such as Mg122,123 and Al,84,91,124–126 and active hydrogen providing chemicals such as NaBH4,127–135 KBH4,136 and CaH2.137The CR process using active metals such as Al consists of two furnace zones: The high-temperature zone works at the temperature around 800 °C to melt Al to form a reductive condition, and the low-temperature zone is kept at 500 °C to blacken TiO2, which is suitable to prepare both B-TiOx powders, and B-TiOx films.120 In literature, the duration of the CR process is between 0.5–6 h.84,121,125 So far, the two-zone CR method with active metals does not yet provide further information on how to control the B° of B-TiOx.
On the other hand, using active hydrogen providing chemical sources (i.e., KBH4) is another option to produce B-TiOx.127,128,136,138 Here, N-TiO2 and NaBH4 are mixed and grounded for 30 min at RT, which is then post-annealed at 350 °C for 55 min under the Ar gas.136,138 Besides, a liquid-state CR process, viz, a solvent CR process, uses reductant chemicals to produce B-TiOx. To manipulate B° of B-TiOx, the concentration of the chemical, the reaction temperature, and the duration can be adjusted. For example, tuning the weight of KBH4 from 0.025 g, 0.050 g, and 0.100 g, can control the concentration of Ti3+ in B-TiOx.136 The highest performance with KBH4 (0.050 g) gives a photocurrent density of 0.55 mA cm−2, which is 1.11 and 1.43 folds than other treatments: (KBH4, 0.025 g), and (KBH4, 0.100 g), respectively.136
The CR method is a facile and mild method for producing both B-TiOx powders and films. However, further investigations are required to enrich the database.
3.4. Solution soaking
As the name implies, the solution soaking (SS) method synthesizes the B-TiOx by soaking TiO2 in aqueous electrolyte(s). For example, B-TiOx can be produced by soaking TiO2 films in a mixture of EG and NH4F at 60 °C. The B° and the bandgaps of B-TiOx films are controlled by the soaking temperature, the concentration of NH4F, and the soaking time.139 With increasing soaking temperature and the concentration of NH4F, the B° is increased and the bandgap is narrowed. Moreover, the influence of different solutions such as polyol solution and acidic solution along with different fluoride types and different volumes of H2O on the B° of B-TiOx have been studied.140In summary, the SS method is energy and cost-saving, and easy to conduct. At the same time, this method allows large-scale production of B-TiOx powders and films.
3.5. Plasma treatment
The plasma treatment has recently been intensively used to produce B-TiOx.141–150 For instance, Ar/H2 plasma has been utilized to prepare B-TiOx nanosheets by introducing both Ti3+ and Ov species on its surface at a power of 400 W.151 Commonly, the components of the plasma electrode system include a power supply, a gas injection channel, a reactor, and a vessel equipped with a stirring magnetic bar.148 Pylnev et al. controlled the H2 flow rates at 0, 2, 3, 4.5, and 6 scans in a plasma system to manipulate the B° and the bandgaps of corresponding samples145The plasma treatment method is a simple, cost-saving, and energy-saving one in the synthesis of B-TiOx powders and films, which has also been applied to produce other black semiconductors, such as B-BiVO4.13
3.6. Ultrasonic treatment
The ultrasonic method introduces surface defects and disorders to pristine TiO2 by ultrasonication. In addition, ultrasonication is also used to assist the application of B-TiOx in chemical reactions.152,153A scheme in Fig. 4(a) gives an overview of the preparation of Pt single atoms on B-TiOx nanotubes through an ultrasonic method.154 To clearly understand that ultrasonic system and mechanism, a schematic gives in Fig. 4(b) showing the generator of the ultrasound and the depict of mechanism by the generation of acoustic cavitation for introducing –OH to prepare B-TiOx: Acoustic cavitation includes the sequential processes of bubble formation, growth, oscillation, and collapse of bubbles in H2O. The injected –OH forms defects on its surface and gives a 2.33 times improvement in the photocatalysis of acid fuchsin. Fig. 4(c) shows the photos of a series of B-TiOx powders prepared using ultrasonic irradiation duration from 0 to 8 h, where the changes in both B° and bandgap can be observed.155 The B-TiOx powder prepared with ultrasonic method shows the highest enhancement of 22.1 times over the pristine TiO2 powder in photocatalytic degradation of MB. Various parameters can be used to manipulate B° of B-TiOx, including the ultrasonic time, the pH of the suspension, the ratio between the TiO2 (g) and the volume of suspension, the frequency of the ultrasonic wave, the temperature, and the output power density during the ultrasonic irradiation.
 | ||
| Fig. 4 (a) Schematic illustration of the fabrication of Pt single atom/B-TiOx nanotubes. Reprinted with permission from ref. 154. Copyright © 2023 The Authors. Published by American Chemical Society. (b) Schematic illustration of the chemical process of ultrasonic treatment in forming B-TiOx. Reprinted with permission from ref. 153. Copyright © 2017, Elsevier B.V. All rights reserved. (c) Photographs of pristine TiO2 and B-TiOx samples prepared with different ultrasonication duration. Reprinted with permission from ref. 155. Copyright © 2015, The Author(s). | ||
This method to produce B-TiOx is gentle, effective, controllable, and energy-saving, and can enable large-scale production. However, it has not been reported to produce B-TiOx films so far.
4. Modifications
B-TiOx has a wide range of promising applications: as photocatalysts for solar energy conversion and storage into solar fuels, as electronic materials for electrochemical devices, and as biomaterials for health and medicine fields, etc. However, at present, these have not yet been industrialized because the performance and the stability do not meet with the industrial demands. To moderate the demerits of B-TiOx, strategies have been designed to address their limitations.156 For instance, doping strategies are adopted to modify lattice symmetry, electrical conductivity, work function, and semiconductive types of B-TiOx.157–166 In this section, the common modification strategies of B-TiOx will be presented.(i) Loading single atoms (SAs), such as Au, Pt, Ru, Pd, Cu, Nb, Mn.89,167–179
SAs have been new emerging working catalysts for a growing number of applications such as fuel cells because of their maximal atom utilization efficiencies, viz a minimum use of metals, especially noble metals, and simultaneously their contribution in boosting activity, stability, and selectivity of reactions for their unique chemical and physical properties (i.e., distinct as active sites towards reactions). For instance, 4.8% (Pd SA+ NPs) on TiO2, as shown in Fig. 5(a), where the Pd SA is used for the activation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and the Pd NPs for the dissociation of H2, gives a 3.2 times activity of commercial (5.2%) Pd/C benchmark catalyst for hydrogenation of ketone/aldehydes to alcohol under 1
O group and the Pd NPs for the dissociation of H2, gives a 3.2 times activity of commercial (5.2%) Pd/C benchmark catalyst for hydrogenation of ketone/aldehydes to alcohol under 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) atm (H2 pressure) at 25°C.179 Briefly, there are two main types of structures of SAs, as shown in Fig. 5(a): Ru SA substituting a cation at the TiO2 surface, and Ru SA supported on top of TiO2. The two types of SAs generally exhibit different catalytic properties.169
atm (H2 pressure) at 25°C.179 Briefly, there are two main types of structures of SAs, as shown in Fig. 5(a): Ru SA substituting a cation at the TiO2 surface, and Ru SA supported on top of TiO2. The two types of SAs generally exhibit different catalytic properties.169
 | ||
| Fig. 5 (a) The mechanism of the synergistic effect of Pd SA and Pd NPs/TiO2 catalyst for hydrogenation of ketone/aldehydes. Reprinted with permission from ref. 179. Copyright © 2020, The Author(s) (b) Substitutional and supported Rh SAs on the considered TiO2 (110) surfaces, including Ov or adatoms (Red (O), blue (Ti), green (Rh)). Reprinted with permission from ref. 169. Copyright © 2019, The Author(s). | ||
(ii) Doping metal or non-metal elements: Single element doping such as P, N, C, etc., and multi-elements co-doping, such as C–N co-doping, C–N–S co-doping, etc.72,167,180–185
Both self-doping and co-doping of TiO2 can alter the characteristics of its intrinsic properties including optical, electrical, and physical properties (i.e., the n-type semiconductive property), then widening their applications or enhancing performance.186 In Fig. 6(a), the Sc and V co-doping narrow the bandgap of TiO2 from 3.20 eV to 2.72 eV to absorb more sunlight.187 Also, in Fig. 6(b), Cu and V co-doped TiO2 facilitate CO2 reduction reactions into CH4 and CO by introducing Ti3+ to form intermediate band to increase separation of charge carriers, and Ov.188
 | ||
| Fig. 6 (a) The band position and bandgap for pure TiO2 and Sc, V co-doped TiO2 with its relaxed structure of 2 × 2 × 2 supercell. Reprinted with permission from ref. 187. Copyright © 2023 Published by Elsevier B.V. (b) V, Cu co-doped TiO2 for CO2 reduction. Reprinted with permission from ref. 188. Copyright © 2023, The Author(s), under exclusive license to Springer Science Business Media, LLC, part of Springer Nature. | ||
(iii) Decorating with nanoparticles (NPs), such as SiO2 NPs, Au NPs, Ag NPs, Al2O3 NPs, Fe3O4 NPs, etc.176,189–191
(iv) Constructing new junctions: heterojunctions such as Z-scheme heterojunctions and S-scheme heterojunctions, and homojunctions.192–198
(v) Constructing core–shell structures: single-component core–shell structure and multi-component core–shell structure.199,200
(vi) Facet-engineering: expose highly active facets.201–205
(vii) Combination strategy: combining two or multi-strategies of (i)–(vi).196,206
As shown in Fig. 7(a), the Au NPs decorating on the surface of TiO2 (Strategy-iii & iv) is often used to absorb photons because of the localized surface plasmonic resonance derived from the collective oscillation of conductive electrons with its selective injection into TiO2 to facilitate reactions such as HER.207–209 Typically, Au exhibits asymmetrical energy distribution in their plasmonic carriers where h+ are much “hotter” than e−. These hot carriers drive chemical reactions and offer to control selectivity of the reaction, unlike thermal-driven catalysis.209 Other functions from the decorating of small NPs on TiO2 are to increase surface area and surface affinity to anchor dye molecules in the application of solar cells, forming junctions between Fe3O4 and TiO2 to narrow bandgap (Strategy-iv). They also serve as co-catalyst and active sites for accelerating reaction kinetics (in Fig. 7(c)), etc.208
 | ||
| Fig. 7 (a) Schematic of plasmonic metal Au NPs on TiO2 (heterojunctions) with a distribution of energies governed by the Au band structure and the incident photons energy. Reprinted with permission from ref. 207. Copyright © 2018, American Chemical Society. (b) Schematic of Au/TiO2 catalysts: Au NP and TiOx on the facets {001}, {100}, and {101} of TiO2. Reprinted with permission from ref. 210. © 2021 Wiley-VCH GmbH (c) Energy band diagram for bare p-type semiconductor (left) and p-type semiconductor/n-type metal oxide (right) under illumination (the orange sphere denotes a cocatalyst). Reprinted with permission from ref. 208. © 2022 Wiley-VCH GmbH. | ||
Facet controls of B-TiOx (Strategy-vi) show many aspects of interests including the growth rates of facets influence morphology of TiO2: exposure of highly active facets influence catalytic reactions and modify decoration of Au to TiOx, see (Fig. 7(b)). They can then affect light absorption, and charge carriers’ separation and transport.210 Accordingly, facet engineering is a useful strategy to modify B-TiOx towards different applications.211
The constructions of junctions such as n–n, n–p, p–p, and tandem junctions (i.e., n–p–n), (Strategy-iv) are often used to narrow bandgap to capture enough photons, to get a high photovoltage for large open circuit voltage, and to enhance e−/h+ transfer to their surface for a highly active reduction/oxidation reaction, etc.208,212 As shown in Fig. 7(c), the constructed n-p junction shows band bending and a depletion p-type region that allows high collection of photogenerated electrons.208 As a result, CO2 can be reduced with e−. Other functions such as n–n junctions between Fe2O3 and TiO2 are often used to promote water oxidation to O2. In terms of core–shell structure, we will present it in Section 5.3.
5. Characteristic properties
Different synthesis methods and modification strategies of B-TiOx as shown above can result in varied physicochemical properties, which further improve their applications including activity and stability (Table 2).244 This section will elucidate a selection of recent pivotal techniques aimed at acquiring a comprehensive understanding of the properties and structures of B-TiOx and its composites. These techniques serve as a foundation for a rational design approach tailored for specific applications.245 Particularly, the recent advances in synchrotron techniques and density functional theory (DFT) calculations have offered some new structural characterization of modified B-TiOx which has not been made available in the past, i.e., elucidating the effects of different types of oxygen vacancies (Ov) coordination and configuration on oxidation reaction. Emphasizes are placed to new structural refinement from X-ray diffraction and pair distribution function, with atomic precision and high resolution of scanning transmission electron microscopy to show metal single atom dopers. Also, in situ or operando techniques combined with DFT models or machine learning can also deliver new essential information on mechanistic aspects of applications.| No. | Catalyst | Application system | Results | Other important points | Ref. |
|---|---|---|---|---|---|
| Photocatalytic (PC); photoelectrochemical (PEC); hydrogen evolution reaction (HER); oxygen evolution reaction (OER); nitrogen reduction reaction (NRR); solar-to-hydrogen (STH). | |||||
| Strategy (i): modification with single atoms | |||||
| 1 | Cu (0.75 wt%)/B-TiOx NPs | PC H2 evolution | Production rate: | Function: reversible and cooperative photoactivation | 175 |
| Cu/B-TiOx: 16.6 mmol g−1 h−1 | |||||
| Pure TiO2: around 0.488 mmol g−1 h−1 | |||||
| 2 | Pt (0.05 wt%)/2D B-TiOx nanosheets | PC H2 evolution | Production rate: | (i) In situ formation Ti3+ | 50 |
| Pt/B-TiOx: 65 μmol g−1 h−1 (UV light) and 688 μmol g−1 h−1 (1.7 Sun) | (ii) Self-activation and amplification behavior of the catalyst | ||||
| Pure TiO2: 5 μmol g−1 h−1 | (iii) Without sacrificial agents | ||||
| 3 | Pt/B-TiOx nanorod | As photoanode for PEC selective oxidation of glucose to glucaric | Photocurrent density: 1.9 mA cm−2 at 0.6 VRHE, glucaric acid yield 84.3% under simulated sunlight | (i) Optimize oxygen vacancies | 213 |
| (ii) Defects modulate the energy of VB holes that improved charge separation and transportation | |||||
| 4 | Ru (0.29 wt%)/B-TiOx nanosheet | PC H2 evolution | H2 production rate: | Ru atoms serve as charge-trapping sites and facilitate the photo-generated electron separation and transportation | 151 |
| Ru/B-TiOx: 17.81 mmol g−1 h−1 | |||||
| (AQE% of 21.3% at 365 nm) | |||||
| B-TiOx: 0.38 mmol g−1 h−1 | |||||
| 5 | Ru/B-TiOx | PC reduction of N2 to NH3 | NH3 yields rate under mild conditions without any sacrificial agent: | (i) Rational design of catalytic sites for N2 fixation to modulate the local electronic structure | 214 |
| Ru/TiO2: 18.9 μmol g−1 h−1 | (ii) Tune the local atomic structure and accelerate the carriers transfer between Ru single atoms and TiO2 carriers | ||||
| Pristine TiO2: 0 μmol g−1 h−1 | |||||
| 6 | Pd/B-TiOx | Semi-hydrogenation of acetylene | Light irradiation boosts the conversion of acetylene in semi-hydrogenation reaction from 20 to 80% | 215 | |
| The semi-hydrogenation of acetylene can be realized at the temperature of 70 °C | |||||
| 7 | Pd/B-TiOx | PC NOx removal | After 5 h of PC NO removal, NOx removal efficiency is 4–5 times over that of pure TiO2 | The fraction of isolated Pd atoms on TiO2 is quantified by diffuse reflectance infrared Fourier transform spectroscopy using NO as probing the molecule and BaSO4 as the internal standard | 216 |
| 8 | Fe1Sx/B-TiOx sphere | Electrocatalytic NRR | NH3 yields rate: 18.3 μg h−1 mg−1 | (i) FeS2O2 sites are the active centers for electrocatalytic NRR | 217 |
| FE of 17.3% at −0.2 VRHE | (ii) Opened and ordered mesopores can facilitate mass transport and offer an enlarged active surface area for NRR | ||||
| 9 | Au (0.3 wt%)/B-TiOx nanosheets | CO oxidation | 100% conversion at 120 °C with stability for 600 min | (i) The surface defects on B-TiOx can stabilize Au single atomic sites via the Ti–Au–Ti structure | 218 |
| B-TiOx is without CO oxidation before 230 °C | (ii) The Ti–Au–Ti structure promotes the catalytic properties by reducing the energy barrier and relieving the competitive adsorption on isolated Au atomic sites | ||||
| 10 | W, Cu/TiO2-rGO | Electrocatalytic HER, OER, and PC degradation | Overpotential of HER/OER: 121/295 mV @10 mA cm−2 | 219 | |
| Tafel slope of 96/60.3 mV dec−1 in 1 M KOH | |||||
| Photodegradation of ciprofloxacin: | |||||
| W, Cu/TiO2-rGO: 98.5%, | |||||
| P25: 84.2% | |||||
| Strategy (ii): Doping and co-doping | |||||
| 11 | Mn (1 wt%) doping TiO2 NPs | Supercapacitor | The capacitance: 335.52 F g−1, twice of pristine one:132.21 F g−1 | This enhanced electrochemical property is ascribed to the large surface area, mesoporous structure, and appropriate concentration of Mn doping | 220 |
| The charge carrier densities have a double enhancement | |||||
| The anodic and cathodic peak separation is 0.09 V | |||||
| 12 | Co doping TiO2 nanotube | Degradation of organic pollutants | The removal rate (pseudo-first-order rate constant): 0.28 min−1, | (i) A relatively high electronic conductivity associated with the coexistence of Ti4+ and Ti3+ in Co–B-TiOx enables an efficient electron transfer between PMS and the catalyst | 89 |
| Co3O4/rGO: 0.18 min−1 | (ii) A partial reduction of Ti4+ to Ti3+ enhances the degradation of organic chemical contaminants | ||||
| CoFe2O4: 0.17 min−1 | |||||
| Co3O4: 0.13 min−1 | |||||
| Co-TiO2: 0.11 min−1 | |||||
| The arc size in EIS Nyquist plots in the order: bare TiO2 > Co-TiO2 > B-TiOx > Co- B-TiOx | |||||
| 13 | S,N co-doped graphene quantum dots TiO2 NPs | PC production of H2O2 | H2O2 yield rate: 451 μmol L−1 which is 3.2 times higher than that of bare TiO2 under simulated sunlight irradiation | A mechanism of proton-coupled electron transfer to produce H2O2 was proposed for the highly selective two-electron H2O2 production | 221 |
| 14 | N,H co-doped TiO2 | HER | Conductivity: 98.77 μΩ cm−1@300 K | (i) Ov and a new phase TiOxNy incorporation on the film surface | 222 |
| Overpotential (Initial): 0.6 VRHE@10 mA cm−2 | (ii) The cathodic chronoamperometry, generating Ti(III) and Ti(II) facilitate the migration of adsorbed Hads intermediates and perform as active sites that enhanced electrochemical performance | ||||
| Overpotential (After 5 h stability): 0.42 VRHE@10 mA cm−2 | |||||
| Strategy (iii): Decorating with nanoparticles | |||||
| 15 | Au NPs/B-TiOx nanotube | PC degradation | The degradation efficiency of RhB in 60 min: | After loading Au, the electron density increased to 6 times that of B-TiOx | 108 |
| Au/B-TiOx: 82% | Au efficiently transferred electrons and promoted carrier separation | ||||
| B-TiOx: 46% | |||||
| 16 | Au NPs/TiO2 nanorod | PC degradation of RhB | Photodegradation rate under the irradiation of visible light in 80 min: | (i) The hybrid Au–TiO2–C system shows extremely high chemical and mechanical stability | 51 |
| Au/TiO2: >98% | (ii) The enhanced visible-light photocatalysis is caused by the plasmon-induced hot-electrons transfer from Au NPs to neighboring TiO2 | ||||
| Pristine TiO2: 15% | |||||
| 17 | Pt Clusters/B-TiOx | HER | Overpotential of HER: 18 mVRHE@10 mA cm−2 | The created Ov and Pt clusters exhibit synergistic effects for optimizing the reaction kinetics | 223 |
| Tafel slope: 12 mV dec−1 in 0.5 M H2SO4 | |||||
| The current density at 50 mVRHE | |||||
| Pt clusters/B-TiOx: 142 mA cm−2 | |||||
| TiO2: 0.03 mA cm−2 | |||||
| Strategy (iv): Constructing junctions | |||||
| 18 | O-ZnIn2S4 @B-TiOx | PC H2 evolution coupled with oxidative dehydrogenation | H2 and benzaldehyde production rate under visible light O-ZnIn2S4@B-TiOx: 2584.9 and 2880.5 μmol g−1 h−1 | S-scheme heterojunction | 180 |
| Pristine one: 49.24 and 43.38 μmol g−1 h−1 | |||||
| 19 | g-C3N4/TiO2 | PC degradation of X3B dye | The highest degradation rate constant: 0.051 min−1 | Z-scheme Heterojunction | 224 |
| The original one: 0.031 min−1 | |||||
| 20 | TiO2/MoS2/Cu2S | Photothermal-PC fuel production | The PC H2 production rate under visible light | Tandem heterojunctions | 225 |
| TiO2/MoS2/Cu2S: 3376.7 μmol h−1 g−1 | |||||
| B-TiOx: around 211.0 μmol h−1 g−1 | |||||
| 21 | 2D/1D MoS2/TiO2 nanowire | CO2 photoreduction | Under solar irradiation | Heterojunction | 226 |
| CO production rate: 36.3 μmol g−1 h−1 | |||||
| Production rate (TiO2): 6.7 μmol g−1 h−1 (CH4) and 0.9 μmol g−1 h−1 (CO) | |||||
| Under visible light irradiation: | |||||
| CH4 production rate of ca. 81.6 μmol g−1 h−1. | |||||
| Production rate (TiO2): 0 | |||||
| 22 | B-TiOx/WO3 on rGO | PEC WS | Photocurrent density under visible light irradiation: | Multi-composite contributes to efficient photoinduced charge carrier separation and transportation | 227 |
| B-TiOx/WO3/rGO: 0.83 mA cm−2 at 0 V | |||||
| B-TiOx: 0.28 mA cm−2 at 0 V | |||||
| Bandgap of the composites: 1.76 eV | |||||
| 23 | RuTe2/B-TiOx | PC degradation of diclofenac (DCF) | The degradation efficiency of DCF under a light source (250 W xenon lamp) in 120 min: | O2− is the main active substance under irradiation | 228 |
| RuTe2/B-TiOx: 95.2% | |||||
| B-TiOx: around 79.3% | |||||
| 24 | 1T-MoS2@TiO2/Ti | Supercapacitor | The specific capacitance is 428.1 F g−1 at 0.2 A g−1, energy density is 48.2 W h kg−1, power density is 2481.7 W kg−1, and 97% capacitance retention after 10000 cycle |
TiO2 nanotube provides a large surface area, shortens the transfer distance of electrolyte ions, and retards the agglomeration of MoS2 nanosheets, enhancing the electrolyte infiltration; and the direct growth of TiO2 nanotube on Ti foil benefits the electrons transfer in the electrode | 229 |
| 1T-MoS2 nanosheets possess good hydrophilicity and conductivity, which favors the fast transfer of electrolyte ions and electrons | |||||
| Strategy (v): Core–shell structures | |||||
| 25 | Rutile TiO2 (core)-TiO2−x:S (shell) | Water splitting | H2 production rate under simulated solar light: | The core–shell modifications enhanced absorption in visible and near-infrared regions and facilitate charge separation and transport | 124 |
| Core–shell: 0.258 mmol h−1 g−1 | |||||
| (STH: 1.67%) | |||||
| Rutile TiO2: <0.1 mmol h−1 g−1 | |||||
| The photocurrent density of the core–shell is 30-fold higher than the rutile TiO2 under simulated solar light | |||||
| 26 | Ti3+ self-doping B-TiOx nanospheres/g-C3N4 hollow core–shell | PC H2 evolution | H2 production rate: | (i) Heterojunction | 230 |
| Core–shell: ∼808.97 μmol h−1 g−1 | (ii) Ov, Ti3+ self-doping and core–shell nano-heterojunction enhance performance | ||||
| Pristine TiO2: ∼45.04 μmol h−1 g−1 | (iii) Ov improves the visible light response, Ti3+ induces the interface to form a self-hydrogenated shell for reducing the activation barrier of the H2, core shell drives the transfer of photon-generated carriers to promote the photon-generated carrier separation | ||||
| B-TiOx: ∼474.34 μmol h−1 g−1 | |||||
| 27 | Crystal-Amorphous Core–Shell TiO2 NPs | Cancer cells imaging | The limit of detection (LOD) of the 4 NBT molecule on B-TiOx NPs can reach 10−6 M | (i) Heterojunction | 231 |
| (ii) Efficient exciton separation at the crystal–amorphous interface that facilitates charge transfer from the crystal core to the amorphous shell | |||||
| (iii) The Fermi level of the amorphous layer shifts to a low position compared to that of the crystal core, allowing efficient photoinduced charge transfer between the amorphous shell and probe molecules | |||||
| 28 | Crystalline Core with Ti4+ -Amorphous Shell with Ti3+ | PC wastewater treatment | Phenol removal percentage in 180 min under visible light irradiation | Bandgap = 2.96 eV | 232 |
| Core–shell: 76.05% | |||||
| P25: 18.18% | |||||
| The removal rate (pseudo-first-order rate constant): | |||||
| Core–shell: around 0.62 min−1 | |||||
| P25: around 0.13 min−1 | |||||
| 29 | Fe3O4 core @B-TiOx shell | Wideband microwave absorption | Microwave absorption performance is evaluated by reflection loss (RL) | Shell: TiO2–TiO2−x shell | 233 |
| Maximum RL of core–shell: −47.6 dB, the effective absorption bandwidth (RL < −10 dB) spanned 13.0 GHz | |||||
| Others: −14.7 dB of Fe3O4; −27.9 dB of Fe3O4@TiO2 corresponding effective absorption bandwidth of 2.0 GHz and 5.5 GHz, respectively | |||||
| 30 | Cu2S@TiO2 | PC recovery of H2 from H2S | H2 generation rate: | (i) Controlling shell thickness directly influenced the optical and surface-interface properties | 234 |
| Core–shell: 41.6 mmol h−1 g−1, (production efficiency of 10.3%) | (ii) Shell thickness varied from 12.0 to 16.7 nm | ||||
| TiO2: 9.2 mmol h−1 g−1 (production efficiency of 2.27%) | (iii) 3-fold prolonged electron lifetime | ||||
| 31 | Ov amorphous layer (shell) − highly crystalline layer (core) | — | The resistivity of the core shell is 5.9 × 10−3 Ω cm, which is more than five (two) orders of magnitude smaller than that of the crystalline (amorphous) TiO2 | (i) Homojunction | 235 |
| (ii) Metallic conduction is achieved at the crystalline–amorphous homo-interface via electronic interface reconstruction | |||||
| 32 | Nanosized graphene/TiOx (multi-shells) − TiO2 (core) | PC degradation of organic pollutants | The degradation rate constant: | Forming of homo-/hetero-junction on TiO2 | 236 |
| Pristine TiO2: 3.74 × 10−4 min−1 | |||||
| Core–shell: 1.66 × 10−2 min−1 | |||||
| Strategy (vi): Facet engineering | |||||
| 33 | Anatase TiO2 Sheets with dominant {001} facets | PC H2 evolution | H2 evolution rate: | Ov and surface fluorine are subject to obvious surface reconstruction | 237 |
| With Ov: 2332 μmol h−1 m−2 | |||||
| Without Ov: 1333 μmol h−1 m−2 | |||||
| 34 | Au/TiO2 (101) | PC CO2 reduction | CO2 RR to CO and CH4 | (i) Au acts as a cocatalyst to trap the photogenerated electrons from semiconductors for improved charge separation and provide highly active sites for accelerated reaction kinetics | 238 |
| Au/TiO2 (101) and (001) | Au/TiO2 (101): 25.9 and 5.3 μmol g−1 h−1 | (ii) Au serves as light-harvesting antennae to extend the light absorption region based on the injection of plasmonic hot electrons into the semiconductor | |||
| Au/TiO2 (001) | Au/TiO2 (101) and (001): 16.5 and 4.0 μmol g−1 h−1 | ||||
| Au/TiO2 (001): 14.9 and 3.0 μmol g−1 h−1 | |||||
| 35 | TiO2 dominantly exposed of {001} and TiO2 dominantly exposed of {101} facets | Reduction of NO with NH3 | NO conversion (%) | 239 | |
| At 503 K: 10.4% of {001}, 4.3% of {101} | |||||
| At 623 K: 19.6% of {001}, 15.1% of {101} | |||||
| The reaction rate (mol g−1 s−1) | |||||
| At 503 K: 1.71 × 10−6 of {001}, 7.05 × 10−7 of {101} | |||||
| At 623 K: 8.03 × 10−6 of {001}, 6.19 × 10−6 of {101} | |||||
| 36 | TiO2 nanosheets with Ov on {001} facets | PC CO2 reduction to CO | The reduction rate under visible light irradiation: | (i) {101} facets in a small number increase the recombination of photogenerated electrons and holes and inhibit the PC CO2 reduction performance | 240 |
| TiO2 with Ov: 128.5 μmol g−1 h−1 | (ii) The synergistic effect of special unsaturated coordination Ti5c atom and Ov | ||||
| TiO2: 45.2 μmol g−1 h−1 | |||||
| 37 | Graphene quantum dots (GQD) @TiO2 rutile (011) | HER | H2 production rate | The charge transfer from GQD to the TiO2 rutile (011) surface, facilitating electron–hole separation and reducing charge recombination rate | 241 |
| GQD@TiO2 rutile (011): 31063 μmol g−1 h−1 | |||||
| Pristine TiO2 rutile (011): 6931 μmol g−1 h−1 | |||||
| Strategy (vii): multi-strategy | |||||
| 38 | P and C modified Co2P/B-TiOx | PC H2 production | The H2 production rate under simulated solar light: | S-scheme heterojunction | 196 |
| P, C Co2P/B-TiOx: 1.53 mmol g−1 h−1 (normalized rate: 18 μmol h−1 m−2) | |||||
| B-TiOx: 1.3 μmol h−1 m−2 | |||||
| 39 | Fe3O4@SiO2@TiO2/rGO core–shell | PC activation of peroxydisulfate (PDS) towards degradation and mineralization of metronidazole (MNZ) | The removal efficiency of MNZ at the same conditions in 60 min | trapping tests, HO˙1, O2, holes, and SO4˙− species were identified as major reactive species | 242 |
| Under PDS and UV: 94.2 ± 5.36% | |||||
| TiO2 under PDS: 3.1 ± 0.24% | |||||
| TiO2 under UV: 9.6 ± 0.74% | |||||
| 40 | Pt/B-TiOx/CuxO | PC H2 evolution | H2 evolution rate under visible-light excitation (>420 nm) | Ov stabilizes the Pt single atoms deposition that facilitates the separation and transfer of photogenerated charge carriers | 243 |
| Modified catalysts: 26.1 μmol g−1 h−1 | |||||
| TiO2: 0.56 μmol g−1 h−1 | |||||
5.1. Structural aspects
X-ray diffraction (XRD) spectroscopy is a versatile non-destructive analytical technique frequently used for investigating crystalline nanoscale materials to provide structural information such as phase identification, composition, preferred growth orientation, sample purity, crystallite size, etc.246,247 For example, XRD can identify anatase and rutile B-TiOx phases as well as Magnéli phase TiO2 from their characteristic patterns.248,249To confirm the compositions of B-TiOx loaded with Pt on its surface (Fig. 8(a)), the XRD patterns in Fig. 8(b) are used to identify Pt/TiO2–Ov which is composed of anatase TiO2 (PDF# 99-0008) and rutile TiO2 (PDF# 73-1765).250 Further, the observation of the patterns in the 2θ range of 24–26°, confirm indirectly the Ov resulting in weaker peak intensities and broader peaks over others.250 Similarly, XRD can be utilized to confirm multi-composition materials and identify each individual component of B-TiOx prepared by other modification strategies such as doping metal and non-metal elements, constructing Z-Scheme junctions, and core–shell structures.124,197,199,225,227,230,249 The size of the tiny crystalline particles influences the optical and electronic properties of materials.251 XRD can also be used to estimate the average crystallite size of B-TiOx according to Scherrer equation of a peak broadening.90 Chen et al. reported that the average crystallite size of B-TiOx to be 8 nm, which was further corroborated by TEM.3 However, XRD has limited applicability to amorphous materials and nanoparticles (NPs) with small sizes, i.e. 5 nm. Thus, techniques such as pair distribution function (PDF) are needed.
 | ||
| Fig. 8 (a) Diagram of the preparation process of Pt/TiO2–Ov and (b) XRD spectra of TiO2, Pt/TiO2, and Pt/TiO2–Ov. Reprinted with permission from ref. 250. Copyright © 2023. The Authors. Angewandte Chemie published by Wiley-VCH GmbH. | ||
PDF technique can elucidate the atomic arrangement of materials with high spatial resolution (Fig. 9), especially those that do not possess sharp diffraction reflections, such as amorphous materials or small-sized NPs.252–254 This technique is appealing for gaining quantitative insights into information such as the amorphous shell or core in B-TiOx. Particularly, PDF analysis can reveal amorphous, defects and disorder layer of B-TiOx rather than limited information on NP sizes offered by other applications.255–257
 | ||
| Fig. 9 (a) A schematic of the modelling process: spherically averaged distribution of interatomic distances and numbers in a hypothetical square lattice of atoms (points). Reprinted with permission from ref. 254. Copyright © Royal Society. (b) Time-resolved PDF profiles of TiO2 upon annealing in a H2 atmosphere (Black arrows indicate obvious changes in peak position, intensity, and width). Reprinted with permission from ref. 258. Copyright © 2016, American Chemical Society. | ||
Fig. 9(a) presents a schematic illustration of the distribution of atoms and the interatomic distances in a hypothetical lattice of atoms, which can be used for PDFs calculation.254 The atoms at the position r are displayed (r corresponds to the radius of the circle at the intersection point), and upon the intersection of a new atom with the circle, a unit of intensity is incrementally added to the histogram. Thermal motion of materials causes the histogram to broaden into a Gaussians distribution, and the PDF is well represented by a sum of independent Gaussian functions.
Fig. 9(b) displays the in-situ time-resolved PDF profiles of TiO2 annealed under H2 atmosphere and the data is analyzed using Python/PDFgetX3.258 The x-axis is the interatomic distance (r, A) of all pairs of atoms within the sample. The 2 y axes include, one for the temperature, and the other one for the function G(r), viz the probability of finding a pair of atoms at a given interatomic distance r with an integrated intensity depending on the coherence scattering lengths of the elements involved and their multiplicities. The figure shows that the r of Ti–O is1.95/1.98 Å and the r of O–O is 2.53 Å assigned to the four equatorial O ions. When the temperature increases from 25 °C to 600 °C under H2 atmosphere, the interatomic distances of both Ti–Ti, O (3.69 Å) and Ti–Ti (4.74 Å) show obvious shrinkage in both maxima intensity and width, which can be assigned to the removal of oxygen atoms creating the defects.
5.2. Microscopic structures
Microscopic morphologies and structures such as shapes and sizes of B-TiOx, have pronounced effects on the performance of a reaction or in a device.55,82,239,259–264 To characterize morphologies, scanning electron microscope (SEM), transmission electron microscope (TEM), high-resolution TEM (HRTEM), and scanning transmission electron microscopy (STEM) are commonly used.SEM has the potential to provide images of up to 2 million times magnification, which is frequently used to observe multitude of morphologies of B-TiOx (e.g., 1D nanorod, 2D nanosheet, 3D nanoflower, etc.), such as the nano-cubic shape of B-TiOx (Fig. 10(a)).42–51 In addition, sizes can be obtained from SEM images, such as the diameters of nanospheres.265 TEM can achieve high spatial resolution down to 0.1 nm with the possibility to identify the composition and crystal structure of NPs. For instance, it has been used to observe the distribution of Au NPs with an average size of 20 nm deposited on B-TiOx, as shown in Fig. 10(b–d).51 Besides, TEM can provide information on the exposed facets of B-TiOx and the ratio of different facets, along with their modifications.133,201,202,237 For example, B-TiOx in Fig. 10(b) shows a TEM image with an exposed facet of (101). These different exposed facets result in different surface crystallinity influencing the loading amount of Au NPs on each facet, which eventually determines the selectivity and efficiency of the catalyst. For example, in the CO2 reduction to CO and CH4, TiO2 with a dominant exposure of the (101) facet give rates of 25.9 μmol g−1 h−1 for CO and 5.3 μmol g−1 h−1 for CH4, whereas, the TiO2 with the co-exposure of the (101) and (001) facets give rates of 16.5 μmol g−1 h−1 for CO and 4.0 μmol g−1 h−1 for CH4.238 HRTEM can observe features with single atom resolution, i.e., in the Angstrom scale, and provide additional information such as lattice fringe spacing. Fig. 10(e) and (f) presents the HR-TEM images in which the lattice spacings (d) are measured. In Fig. 10(e), there are two d values of 0.250 and 0.234 nm, corresponding to (101) facet of rutile TiO2 and (111) facet of Au NPs, respectively.51 HRTEM has also been utilized to characterize the morphologies and crystallite sizes of NPs, such as core–shell structure and thicknesses. With increasing number of publications using single atoms such as Pt and Au to enhance reactivity, HRTEM has proven to be a fundamental technique in the characterization of B-TiOx.50,151,167–169,175,216–218
 | ||
| Fig. 10 SEM images of B-TiOx (a) template of nanocubes. Reprinted with permission from ref. 44. Copyright © 2018 Elsevier B.V. All rights reserved. TEM images of (b) Au–TiO2 co-exposed (001) and (101). Reprinted with permission from ref. 238. Copyright © 2020, Elsevier B.V. All rights reserved. TEM images of Au–TiO2 (c) scale-bar: 100 nm, (d) scale-bar: 20 nm, and (f) HR-TEM image from the selected area in (e). Reprinted with permission from ref. 51. Copyright © 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinhein. | ||
STEM combined with electron energy loss spectroscopy (EELS) is a powerful technique to characterize thin atomistic layers, based on its extremely high spatial resolution.266–269 In this review, we reveal the use of STEM and EELS to probe details of the interface information between a thin layer of TiOx and their composites under hydrogenation reaction conditions and observe the microstructure change of B-TiOx during the catalytic process.83Fig. 11(a) shows a scheme of an operando electron microscopy setup for the observation of TiOx at both H2 (Fig. 11(b and c)) and CO2:H2 (Fig. 11(d and e)) conditions at 400 °C and atmospheric pressure. The in-situ HAADF-STEM imaging in Fig. 11(b) shows that the TiOx layer encapsulates the Ni (111), and the estimated Ti and Ni atom positions are marked in Fig. 11(c), where the distances of interatomic Ti–Ti and Ni–Ti are around 2.95 ± 0.01 and 2.93 ± 0.09 Å, respectively. When changing gas condition from H2 to CO2 + H2, the interface of Ni–Ti is lost from Fig. 11(d) and results in lower atomic coordination and higher mobility of surface atoms. Meanwhile, as shown in Fig. 11(d), Ni (111) is also lost and only shows some Ni (111). Also, Fig. 11(e) shows a high tensile strain over the entire NP. Fig. 11(f) shows Ni/TiO2 at 600 °C with a completely encapsulated TiOx overlayer. The in-situ EELS analysis confirms the formation of a TiOx layer on Ni, which shows a Ti L2,3 ionization edge signal throughout the entire Ni NP, (Fig. 11(g and h)).
 | ||
| Fig. 11 (a) Schematic of the operando electron microscopy setup and windowed gas cell (climate G +, DENS solutions). (b) Representative atomic-resolution HAADF-STEM image of a Ni NP in a Ni/TiO2 catalyst in H2 at 400 °C. Solid lines indicate TiOx-covered Ni atomic planes, and dashed lines indicate unoccupied facets. Ni (111) and Ni (100) facets are indicated by pink and green, respectively. (c) Estimated positions of Ti (gray) and Ni (color scale) atomic columns. (d) HAADF-STEM image of the same particle as in (b) upon exposure to a CO2:H2 (0.25 bar: 0.75 bar) mixture at 400 °C showing complete re-exposure of Ni and NP restructuring. (e) Estimated atomic column positions of Ni from the same particle shown in (d). Strain maps in the x and y directions resulting from the displacements with respect to the ideal atomic column positions. (f) High-resolution HAADF-STEM image of Ni/TiO2 catalyst in H2 at 600 °C showing a Ni NP encapsulated in a thick TiOx shell. Colored squares correspond to the location where EELS spectra in (g) and (h) were acquired. (g and h) Core loss Ti L2,3 and Ni L2,3 EELS spectra. Reprinted with permission from ref. 83. Copyright © 2023, The American Association for the Advancement of Science. | ||
5.3. Surface and interface disorders
Surface chemistry plays a critical role in determining the applications and performances of B-TiOx catalysis.82,83,268–271 During preparation, modification, and even applications of B-TiOx, surface-disordered layers or defects such as Ov and Ti3+ of B-TiOx are generated that could subtly affect catalytic reactions. For instance, the coordination and configuration of Ov on B-TiOx affect water oxidation reactions.272 According to the study by Yazdanpanah et al. in 2023, they demonstrated that the increase in the concentration of Ov can enlarge acid–base pairs density, which then enhanced the activity and selectivity of ketonization reaction.273The most common surface defects of B-TiOx are Ti3+ species, Ov, –OH, and surface/near-surface disorders that are commonly generated during synthesis and modification processes, which influence their B°, physicochemical property, and performance.89,234,274–276 To identify these different surface states, standard measurements and analysis techniques are needed. As shown in Fig. 12(a), both the binding energy of core-level of Ti 2p and O 1s XPS are changed due to the defects from Ti3+.277 Meanwhile, electron paramagnetic resonance (EPR) or electron spin resonance (ESR) spectra techniques are often used to provide information, i.e. g-tensor values (gx, gy, gz), for defects of B-TiOx.30,91 The EPR signals of gx = 1.9930, gy = 1.9930, and gz = 1.9640 also indicate the presence of defects of B-TiOx from Ti3+ in regular sites of the TiO2 anatase phase, while the g values of gx = 2.0040, gy = 2.0129, and gz = 2.0277 correspond to defects of oxygen ions with trapped holes (cation vacancy or surface exposure).30 The intensity of EPR signal can also reflect the high concentration of surface defects, or alternatively, negligible intensity suggests nonexistent of surface defects.126 For further insights into valence states and coordination environment of Ti on B-TiOx, X-ray absorption fine structure (XAFS) spectroscopy can be invoked.278
 | ||
| Fig. 12 Surface chemical characterization of samples of H–TiO2 (Hydrogenated TiO2), H–TiO2–Ar (post-annealing H–TiO2 in Ar), and H–TiO2–O2 (post-annealing H–TiO2 in O2). (a) XPS spectra of Ti 2p and O 1s. XANES spectra of (b) Ti K-edge, (c) k3-weighted FT-EXAFS spectra, and (d) Ti L-edge XAFS spectra. Reprinted with permission from ref. 277. Copyright © 2021 Elsevier Inc. | ||
The Ti K-edge X-ray absorption near edge structure (XANES) spectra of H–TiO2, H–TiO2–Ar, H–TiO2–O2 samples are shown in Fig. 12(b), the similar spectra indicate a similar local environment. Their Ti pre-edge peaks are located at 4969.4, 4972.0, and 4975.1 eV for the transitions of 1s/3d and 1s/hybridized p–d for the octahedral symmetry.
Among spectra of H–TiO2, H–TiO2–Ar and H–TiO2–O2 samples, H–TiO2–Ar gives the highest pre-edge peak intensity, indicating a decreased Ti oxidation state and strengthened distorted local structures via Ar annealing treatment. This is consistent with the XPS result of Ti3+ on H–TiO2–Ar. The chemical environment of Ti is further examined using extended XAFS (EXAFS) spectroscopy, and the Fourier transform of EXAFS (FT-EXAFS) spectra is displayed in Fig. 12(c). The peak at 1.50 Å is ascribed to Ti–O, while the other one is for Ti–Ti. Ti L-edge spectra in Fig. 12(d) which provides information on the local electronic configuration. The H–TiO2–Ar shows the lowest onset energy position of Ti L-edge from a lower Ti valence state.277
Many recent studies using DFT are employed to provide theoretical understanding on both defects of B-TiOx and reaction pathways.279 Here, we exemplify one of proposed mechanisms to support the importance of B° of B-TiOx on the activity of a chemical reaction.
Fig. 13(a) shows the required energy for the H transfer on both bulk and surface Ov–Ov (0.95 and 0.84 eV) which are lower than that of surface lattice oxygen (Ov–O–Ov, 2.58 eV).280 Indicating H preferentially transfers from the open channel rather than from a blocked one (Fig. 13b), which well supports the importance of controlling B° of B-TiOx.280 Recent observations also support H-transfer pathway over Ov–Ov because H co transfer in certain depth inside a metal oxide.
 | ||
| Fig. 13 (a) Schematic activation energy diagram of by Ov–Ov (left) and Ov–O–Ov (right), and (b) diagram of hydrogen transfer through cascade Ov–Ov (left) and O-obstructed Ov–O–Ov (right) pathways. The structures and activation energy barriers were obtained using DFT. Reprinted with permission from ref. 280. Copyright © 2020, American Chemical Society. (c) Principal Component Analysis (PCA)-decomposed TiOx phase space, with colors representing varying oxygen concentrations. (d) 2D histograms of the PCA-decomposed TiOx phase space, with occurrence rate (colors) overlayed as a third dimension. Occurrence rate is defined as the likelihood of a structure from a given PCA grid occurring in the overall phase space. Dashed lines in (d) provide visual context for the approximate boundary of each phase, where a bin contains greater than 50% of a specific phase. (e)–(g) 2D histograms of the PCA-decomposed TiOx phase space, with coordination number (CN) probabilities (colors) overlayed as a third dimension. CN probabilities represent the likelihood of an atom having the specified CN within a structure that is contained within a given PCA grid for (e) 2F (OTi2), (f) 3F (OTi3), and (g) 4F (OTi4) oxygen CN environments respectively. Reprinted with permission from ref. 281. Copyright © 2023 Royal Society of Chemistry (RSC). | ||
Chapman et al. combines machine learning, DFT, Group theory, and molecular dynamics (MD) to reveal the hydrogen incorporation and transport in TiOx, which can be tailored through compositional engineering.281 For this review, we only discuss on their work using atomic force neural network (AFNN). The AFNN-generated 1944 atom MD trajectories can analyze the local atomic geometries present across the TiOx phase space, which is characterized using the PCA decomposition of the VGOP features. Fig. 13(c) shows TiOx (x = 2, 1.95, 1.9, and 1.85) within this configuration space, which displays a clear separation between each case (TiO2, TiO1.95, TiO1.9, and TiO1.85), but also shows some overlap between them. For example, the TiO2 sub-space shares some overlap with the TiO1.95 sub-space but does not share any portion of the total space with TiO1.9 and TiO1.85. They argued that oscillations provide insight into the size of a given oxygen concentration's portion of the overall phase space, where the circularity of phase space serves as the basis. Fig. 13(d) shows the equilibrium configurations contained within each sub-space, as well as the outlier regions, which provides the probability of each region contained in the phase space. Overall, with the decrease of x from 2 to 1.85, the oscillations about a given chemistry's equilibrium point are reduced and the radius from the center of the approximated clustering is minimized. This seems to indicate that structures can deviate from the equilibrium configuration by a greater extent in TiO2 than in TiO1.85. The effect of oxygen concentration is studied by observing the probability O of 2F (OTi2), 3F (OTi3), and 4F (OTi4) within the system. TiO2 shows a higher rate of the existing of O sites over TiO1.85 in 2F CN environment, where P(TiO2) = 30% and P(TiO1.85) = 23%. It shows a linear trend existing of O for the case of 4F O atoms in Fig. 13(e) and (g), where in Fig. 13(e) the exist rate of TiO2 and TiO1.85 are 9% and 12%, separately. However, the oxygen concentration and the amount of 3F O present within the system is random (Fig. 13(f)).
The core–shell structures are of particular interests and have recently been reported extensively due to their unique electronic and structural interactions of the core component (inner layer) with the shell component (outer layer).28,29 The core–shell structure involves at least one core and one shell layer, enabling the integration of multiple components into a functional system to overcome the demerits of a single component.124,199,200,230,233,234,242 For example, the bandgap of a core–shell structured TiO2 can be excitingly tuned between 1.0 to 2.8 eV rather than 3.0 eV (3.2 eV) of pristine TiO2.
In this review, we discuss on the single-component core–shell structured B-TiOx with disorders.232 One example is shown in Fig. 14(a), where the single component core–shell structured TiO2 is composed of a crystalline core and an amorphous shell, which extends the light absorption edge from around 400 nm to 1300 nm3. The single-component core–shell structured B-TiOx is summarized into four types according to the introduction and distribution of Ti3+ species, as shown in Fig. 14(b): (i) a crystal core with Ti4+ and an amorphous shell with Ti4+; (ii) a crystal core with Ti4+ and an amorphous shell with Ti3+; (iii) a crystal core with Ti3+ and an amorphous shell with Ti4+; and (iv) a crystal core with Ti3+ and an amorphous shell with Ti3+.28,231,232 For instance, the B-TiOx nanotube composes of an anatase core and an amorphous shell with a thickness of 4 nm in Fig. 14(c), which is synthesized by the hydrogenation method under NH3 gas. The nanotube possesses a strong light absorption enhancement in the wavelength range of 300–800 nm.87 The core–shell structures and the thickness of disorder layers can be determined with the HRTEM.73 Furthermore, the simulated density of states (DOS) reveals unprecedented effects of hydrogenation on TiO2. As observed in Fig. 14(d) and (e), the bands of B-TiOx after hydrogenation are severely broadened along with formation of states both below and above the VB in the shell, which are due to the existence of –OH and Ti3+, respectively. The Ti3+ states in the shell of the B-TiOx start just above the VB and fill most of the gap. Meanwhile, the comparison of the DOS of the B-TiOx to the bulk crystalline anatase (Fig. 14(f)) clearly shows the overall upward shift and tailing of the VB.282 The benefits of the core–shell structured TiO2 for applications such as lithium-ion batteries, solar thermal energy, photocatalytic WS, etc., will be addressed in Section 6.
 | ||
| Fig. 14 (a) Schematic illustration of the core–shell structure TiO2. Black dots represent dopants. Reprinted with permission from ref. 3. Copyright © 2011, The American Association for the Advancement of Science. (b) Scheme of four types of destructive core–shell structured TiO2. Reprinted with permission from ref. 28. Copyright © 2019, Elsevier B.V. All rights reserved. (c) Scheme of a core–shell structured B-TiOx nanotube. Reprinted with permission from ref. 87. Copyright © 2016, American Chemical Society. Computed DOS for the core (d), and the shell (e) of TiO2 before and after hydrogenation treatment (f) DOS for the shell and core of B-TiOx are compared to the DOS of bulk crystalline anatase TiO2. Reprinted with permission from ref. 282. Copyright © 2020, Wiley-VCH GmbH. | ||
An interface is a separating layer between two phases including gas–liquid (G–L) interface, gas–solid (G–S) interface, liquid–solid (L–S) interface, solid–solid (S–S) interface, and gas–liquid–solid (G–L–S) interface.234,283–286 These interfaces clearly influence the transport of electrons and ions, the adsorption of reactants, the solubility and diffusion of gas in liquids, etc., and determine the overall cell or chemical reaction performance in the end. Therefore, understanding the interface is important to achieve high stability and efficiency in reaction/device. For instance, the electrode–electrolyte interface is the key factor in influencing the capacity, stability, and performance of a Na-ion battery.46
In terms of B-TiOx, the investigations of interface are accompanied by their studies of synthesis, modifications, and applications. In the synthesis of B-TiOx, there are two types of S–S interface including the phase interface and the interface between the core and shell formed by the same components such as the crystalline-amorphous homo-interface.203,235,287,288 After modifications, the S–S interface is formed between B-TiOx and other materials such as Au NPs and g-C3N4.224 When employing B-TiOx to construct a multi-junction material, a multiple S–S interface might form among different compositions.224,236,241,289,290 In terms of their applications, the interfaces are often discussed among the B-TiOx, the electrolyte (S or L), and the products (S, L, or G), as shown in Fig. 15(a) and (b). In the application of WS to H2 and O2 gases, for example, there will be the S–L–G interface among B-TiOx (S), H2O (L), and O2 (G) and H2 (G); and in the solid oxide fuel cell, the S–S interface is between the B-TiOx electrode and the solid electrolyte.224,241,291–295 To characterize interfacial features, techniques such as XRD, TEM, XPS, etc. are employed. An in-situ TEM images observes and records a reaction taking place on the S–L–G interface, viz, the O2 nanobubbles (G) etching of Au nanorod (S) in aqueous hydrobromic acid (L) with the increasing of the reaction time.294,295 Some cutting-edge TEM techniques reveal that when the distance between G and S is less than 1 nm. In addition, electrochemical impedance spectra (EIS) is used in electrochemical cells such as fuel cells and batteries, to understand the interfaces between the electrode and the electrolyte, and the interfaces of multilayers of the electrode, which will be introduced in Section 5.5.
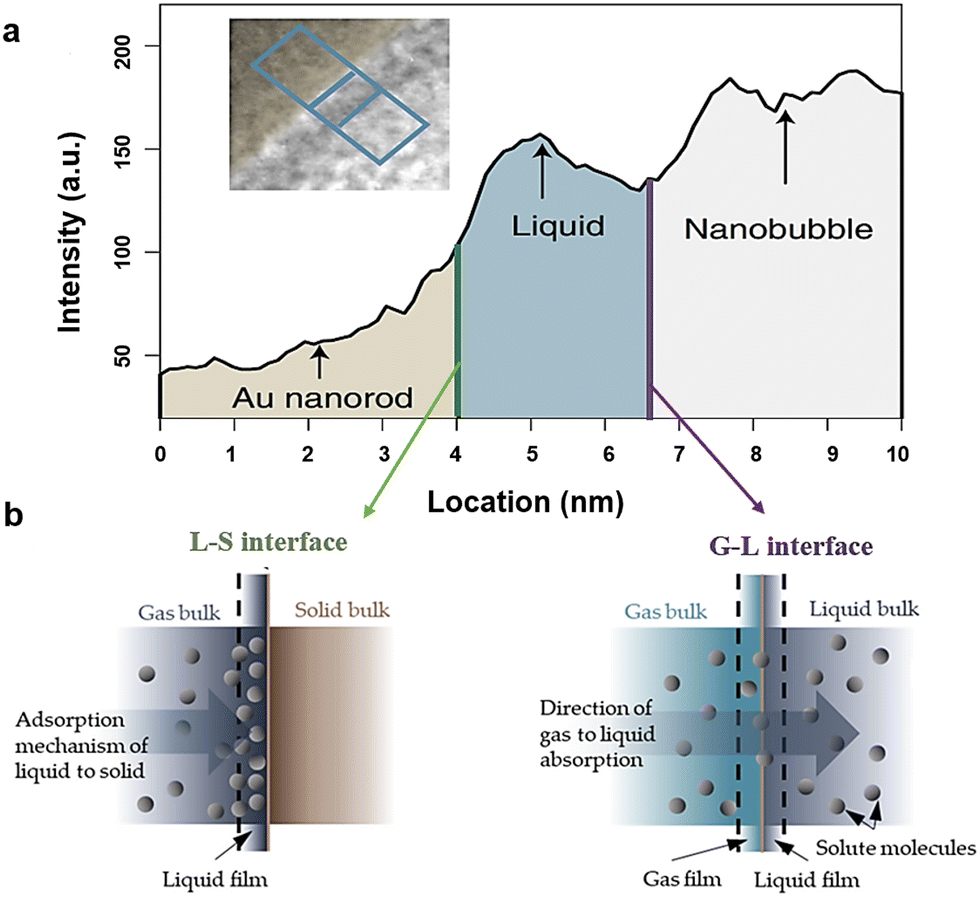 | ||
| Fig. 15 (a) Solid (Au nanorod)–liquid (aqueous hydrobromic acid)–gas (oxygen gas) interface, along with an L–S interface and a G–L in (b). Reprinted with permission from ref. 295. Copyright © 2020, Wiley-VCH GmbH., and Reprinted with permission from ref. 294. | ||
5.4. Energy band structure and optical properties
As stated, both blackening processes and modification strategies can shift the valence band (VB) location of pristine material. Investigations of the band structure and the bandgap energy of semiconductors can therefore help not only in understanding the properties of materials, but also to instruct the future rational design of materials for practical applications. The energy bands of a semiconductor mainly include conduction band (CB), VB, Fermi level (Ef), and bandgap (Eg). The Eg of a semiconductor is defined as the gap between the top of the VB (Ev) and the bottom of the CB (Ec), which determines the solar capture and conversion ability of a semiconductor.236To measure the location of the VB maximum, the X-ray photoelectron spectroscopy (XPS) is a common tool such as the measured VB values of both pristine TiO2 film and B-TiOx film are 3.13 and 1.94 eV, respectively.265 In addition, the ultraviolet photoemission spectroscopy is used to confirm the work function (Φ), which is determined by the vacuum level to find the location of Ef that can be calculated according to eqn (1).
| Φ = hν − Ecut-off | (1) |
The light absorption originated from the electron transmission from VB to CB and then the capacity of light absorption is closely associated with the application potential in solar energy conversion and storage. To measure the light absorption of B-TiOx samples and to compare the optical properties among different samples in parallel, UV-Vis-NIR absorption/reflectance spectroscopy can be a suitable technique. For instance, the UV-Vis-NIR absorption spectra of Pt/TiO2–Ov, and TiO2 are tested in the wavelength range of 200–800 nm and find that that the light absorbance of Pt/TiO2–Ov is stronger than that of pristine TiO2 in the whole wavelength range. If the absorption edge of material is located in a longer wavelength, it will enable a broader light absorption range.120 Additionally, UV-Vis-NIR plots can estimate the bandgap energy using Tauc's relation along with eqn (2)
| Eg (eV) = hυ − 1/β(αhυ)1/n | (2) |
In addition, the incident photon-to-current conversion efficiency (IPCE) recorded with the QE/IPCE measurement system can be used to monitor the ability of the semiconductor converting photons to electrons rather than light absorption – not all absorbed light can be converted into electrons. Typically, a high IPCE value enables a high PEC performance: Ru-doped B-TiOx has a higher IPCE value in the range of 300–600 nm than TiO2 that results in a 40-time higher H2 production rate, 1.91 μmol h−1 cm−2, than that of TiO2.90 If QE/IPCE equipment is absent, eqn (3) can be used to estimate the IPCE value of the photoelectrode at a given wavelength. Meanwhile, the IPCE is also can used to estimate the bandgap energy according to eqn (4)
| IPCE (%) = (Iph ×hν/P × λ) × 100 | (3) |
| Eg (eV) = (IPCE hυ)1/n | (4) |
The apparent QE (%) of the H-atom-involved reactions (H2 evolution reaction and NH3 generation) can be calculated viaeqn (5).296 For example, Li et al. reported the QE of Au-supported Fe3O4/B-TiOx under the photocatalytic WS reaction conditions, and reported a QE of 88.7 ± 2.1% at 437 nm.88 The same group also reported that the QE of a B-TiOx based photocatalyst can reach 97% at 385 nm for the solar-driven seawater splitting.297 Another research group has also previously reported a high QE of 46% at 584 nm, achieved in a solar-driven sacrificial H2 evolution system on a mesoporous B-TiOx.298 Recently, it has been demonstrated that H2 can be generated from methanol on a single-atom-modified TiO2 photocatalyst, which exhibited a high apparent QE of 99.2% at 365 nm with a high-selectivity production of formaldehyde, leading to a nearly zero- carbon-emission process.299
 | (5) |
5.5. Semi-conductivity
Semiconductor type (i.e., n-type and p-type), charge carrier density, and charge carrier transport, separation, and recombination rate of B-TiOx are important for designing and modifying materials towards applications. Surface defects and disorders of B-TiOx show pronounced effect on physicochemical properties, i.e., in facilitating the electrical conductivity and charge transport, efficiently suppressing the recombination of photoinduced charge carriers. Mott–Schottky (M–S), photoluminescence (PL), time-resolved photoluminescence (TRPL), EIS, etc. have been employed to identify such information directly or indirectly.M–S measurement has often been used to identify the type of semiconductor conductivity and the junctions such as n–p–n junctions and to give information on the charge carrier densities. With respect to the semiconductive type: a straight line with a positive slope from M–S plot indicates a n-type characteristic and a negative slope reflects a p-type feature. The different absolute values of both slopes indicate a spectacular disparity in donor densities.300 The carrier densities can be calculated in accordance with the eqn (6). For instance, the calculated charge carrier densities of both pristine TiO2 and B-TiOx according to eqn (6) are 1.24 × 1019 and 5.55 × 1021 cm−3, respectively.120 It can be proven that the blackening treatment increases around 2 magnitude orders of the charge carrier densities of TiO2 which benefits the electrochemical devices.
| Nd = (2/e0εε0) [d(1/C2)/dV] | (6) |
The recombination rate of the free charge carriers, viz, evaluating the separation and utilization efficiency of the photo-stimulated e−/h+ is one of the key parameters in determining their applications.101 Typically, the e−/h+ pair recombination rate of B-TiOx is lower than that of pristine TiO2 that will result in a low peak intensity of the B-TiOx in a PL spectrum.157 To measure the accurate lifetime of the photoexcited e−/h+ pairs, TRPL is needed. For instance, the lifetime of e− in a B-TiOx recorded by TRPL is 2.56 ns, which is longer than that in the pristine TiO2, 1.12 ns.88
The electrical conductivity of a semiconductor relates to the mobility of the charger carriers, which is influenced by the parameters of the blackening process and does also impact the performance of electrical devices and photoelectric cells, i.e., supercapacitors.183,235,301–304 A solid-state conductivity measurement is carried out to measure the resistivities of both pristine TiO2 and B-TiOx, 1170.47 and 15.53 kΩ, respectively, confirming a better electrical conductivity of B-TiOx.71 This higher electrical conductivity of B-TiOx over pristine one attracts rising attention in the fields of electrochemistry and photo-electrochemistry.
The charge transfer resistance among interfaces such as the interface between the electrolyte and the electrode often reflects the charge carrier information of B-TiOx, which is presented in Nyquist plots from EIS tests. In the Nyquist plot, a smaller arc represents a lower resistance at the interface, namely, a higher efficiency of the charge transfer.107,305
Herein, B-TiOx enables a higher charge carrier density, faster separation and utilization efficiency, longer electron lifetime, higher electrical conductivity, and lower interface charge transfer resistance over the pristine one.13
5.6. Other characteristics
In addition to the abovementioned properties of B-TiOx, other information such as chemical structure, phase and polymorph, crystallinity, and molecular interactions with adsorbates can be checked by Raman spectroscopy; the oxidation state and coordination environment of Ti can be determined using XANES technique; the absorption, emission, and photoconductivity of solid, liquid, and gas can be obtained from the Fourier transform infrared spectroscopy. Theoretical understanding are also expected such as the machine learning on PDF analysis for the oxide defect identification through feature extraction and supervised learning.2796. Potential applications
Despite the significant annual market volume of 6 million tonnes for titanium dioxide (TiO2), commercialization of black titanium oxide (B-TiOx) has yet to be realized, with ongoing exploration of its potential market applications. The distinctive properties inherent to B-TiOx underline its recent potential applications in solar energy capture, conversion, and storage devices, presenting promising avenues for utilization in electrical energy storage devices. In this section, the promising applications of B-TiOx on 9 topics towards sustainable future in both energy and environment will be presented: water splitting, carbon dioxide reduction reaction, ammonia synthesis and decomposition, pollutant degradation, solar thermal energy, supercapacitor, ion battery, fuel cell and others (Scheme 1). | ||
| Scheme 1 Potential applications of black titanium oxide under light illumination and bias. PC: Photocatalytic, PEC: Photoelectrocatalytic, EC: Electrocatalytic. | ||
6.1. Water splitting
Water (H2O) needs a Gibbs energy of 237 kJ mol−1 (1.23 V) to be split into H2 and O2, and a Gibbs energy of 354 kJ mol−1 (1.77 V) to be split into H2 and H2O2, as shown in Fig. 18(b).306 The energy can be derived from renewables such as solar energy in Fig. 18(a), to assist water splitting (WS) to form renewable H2 to address the energy crisis.306 The first report on WS to H2 and O2 appeared in 1972 and was tested in a PEC WS cell with a TiO2 anode (active area, 1.0 cm2) and a platinum black cathode in an acid solution (pH = 4.7) under light illumination.307 To the first report on B-TiOx as the photocatalyst for WS dated back to 2011.3 Over more than 10 years of development, B-TiOx and its composites have been studied for WS as photocatalysts, photo-electrocatalysts, and electrocatalysts to produce small molecular chemicals of H2, O2, and H2O2.30,40,76,308–322 The progress and representative reports on these cells will be addressed in the following.Water is required among all WS cells, an aspect that has been seldom considered until recently. The concerns arise from the low conductivity of H2O and the limited amount of fresh H2O.
(i) The conductivity of water:
Pure H2O at the standard STP conditions has very limited ionic conductivity, where the conductivity of ultra-pure water is 5.5 × 10−6 S per m (Simens per meter), drinking water is 0.005–0.05 S per m, and sea water is 5 S per m where equilibrium ionic concentration is extremely low.323 On the other hand, superheated water in sub or supercritical conditions could dramatically increase in ionic concentrations. As a result, WS at high temperature could be useful for exploitation in future since conductivity is important for the performance of both electrolysis and photo-electrolysis WS. The most common way to increase conductivity, is to use inorganic salts (acidic, neutral, and alkaline) such as NaOH/KOH, which are often added into H2O to improve the ionic conductivity of electrolytes and split of H2O into H2/O2 by electrochemical or photochemical means see Fig. 16(a).321 Meanwhile, a question is raised on the both beneficial or undesirable effects (i.e. surface fouling, cost and scaleup issues) of the pH (the concentration of H+/OH−) of the electrolyte on the WS performance. For example, Fig. 16(b) and (c) show the concentration of KOH (1 M, 0.2 M, 0.1 M, and 0.05 M) on both the required electricity to split H2O and the linear sweep voltammetry (LSV) performance.324
 | ||
| Fig. 16 (a) Schematic of a water electrolyser. Reprinted with permission from ref. 321. Copyright © 2022, Springer Nature Limited (b) Long-term stability tests in different KOH electrolyte concentrations (1, 0.2, 0.1, and 0.05 M) for oxygen evolution process, and (c) Polarization curves of samples in 1, 0.2, 0.1, and 0.05 M KOH before and after 10000 s time stability tests. Reprinted with permission from ref. 324. © 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
(ii) The use of seawater instead of freshwater:
Recently, seawater splitting (SWS) has gained increasing attention due to limited availability of fresh water (<1% of earth's water), while sea-H2O makes up a large proportion (around 96.5% of earth's water).320,325,326 While direct electrolysis of seawater has witnessed significant advancements, substantial challenges persist.320,327–329
Seawater contains a high percentage of aggressive chloride (Cl−) ions, which requires the use of robust and efficient catalysts that can withstand Cl− corrosion, especially for the anode. The relationship between the pH, Cl− and the voltage are shown in Fig. 17(b): In acid electrolyte, pH < 7, Cl− would be reduced to Cl2, in base solution pH > 7, Cl− would be oxidized to hypochlorite (HOCl or ClO−).330 In addition to Cl−, other main soluble ions in seawater include sulfate (SO42−), sodium (Na+), magnesium (Mg2+), calcium (Ca2+), bicarbonate (HCO3−), and potassium (K+). The corresponding concentrations are listed in the table in Fig. 17(a).325 These ions might influence the activity and the stability of SWS (Fig. 17d). To solve it, in-directly SWS cells using membrane (Fig. 17(c)) or using distillation method (Fig. 17(e)) to pre-treat seawater would be solutions.331 Despite that additional cost and energy requirement will be unavoidable, the in-direct SWS combing seawater purification technologies is still preferred regarding the activity and stability of WS.332e.g., in Fig. 17(d), both the activity and stability of the WS cell integrated with a seawater purification process are much better than that of the direct SWS.333
 | ||
| Fig. 17 (a) Table of ion concentrations (C) in standard seawater. Reprinted with permission from ref. 325. Copyright © The Royal Society of Chemistry 2021. (b) Pourbaix diagram containing oxygen and Cl− redox reactions. Reprinted with permission from ref. 330. Copyright © 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) Schematic of the integration of the water purification with membranes and water splitting process, and (d) electrolysis durability test at constant current densities of 250mA cm−2 and 400mA cm−2 of 72h. The inset shows the stability test of conventional direct seawater splitting in seawater with commercial electrocatalysts. Reprinted with permission from ref. 333. Copyright © 2022, The Author(s), under exclusive license to Springer Nature Limited. (e) Schematic diagram of saltwater distillation. Reprinted with permission from ref. 331. Copyright © 2023, The Author(s), under exclusive license to Springer Nature Limited. | ||
:1 ratio). The experimentation was conducted under a 1-Sunlight simulator within a Pyrex glass setup, yielding an H2 production rate of 10 mmol h−1 g−1. The methanol in the electrolyte serves as the sacrificial reagent to react with the photogenerated h+.3,122,309 In parallel, PCWS has also been investigated using pure H2O. For example, in Fig. 18(d), N-TiO2/MgO (111) loading by Au yielded an H2 production rate of 20 mmol g−1 h−1.296 At present, the state-of-the-art work in PCWS uses Fe3O4@SiO2/B-TiOx with a loading of 1.0 wt% as photocatalyst, which is carried out under a 1-Sunlight simulator (AM 1.5 G, 100 W cm−2) and an external magnetic field of 180 mT at 270 °C, yielding an H2 production rate of 21.2 ± 0.52 mmol g−1 h−1, which corresponds to an STH of 11.9 ± 0.5%.88
 | ||
| Fig. 18 (a) Light absorption of photocatalysts/photoelectrodes compared with solar spectrum (black) and (b) redox potentials of WS. Reprinted with permission from ref. 306. Copyright © 2022. The Author(s). (c) Comparison of economic benefits from different water oxidation processes. Reprinted with permission from ref. 334. This Copyright © 2019 American Chemical Society. (d) The H2 evolution rates of N-P25 and N-P25/MgO (111). Error bars indicate the standard deviation. Reprinted with permission from ref. 296. Copyright © 2019. The Author(s). (e) The schematic diagram of the flow-by cell assembled with a gas diffusion electrode and a membrane separator for H2O2 production. Reprinted with permission from ref. 335. Copyright © 2021. The Author(s). | ||
Considering the limited freshwater availability (<1% earth's water), PC SWS seems more in line with future goal of green H2 production.336 In the limited scope of PC SWS systems reported so far, the reliance on sacrificial reagents is imperative, giving rise to sustainability concerns and resulting in the generation of carbon-containing by-products. Additionally, the presence of ionic species such as Cl− and Mg2+ in seawater complicates the reaction mechanism, with early attempts revealing adverse effects on photocatalytic performance. Conversely, recent reports highlight that the presence of Cl− ions can enhance reaction kinetics, thereby improving photocatalytic activity.337 Evidently, conflicting perspectives on the impact of seawater prevail over this topic, necessitating a clear unraveling of the role of these ionic species and their interactions with catalyst particles. In 2024, Li et al. reported on a solar-driven PC SWS system for H2 evolution.297 The study highlights that photocatalytic performance can be significantly enhanced in natural seawater due to electrolyte-assisted charge polarization over an N-doped TiO2 photocatalyst. Comprehensive and systematic characterizations, alongside computational studies, indicate that ionic species in seawater can selectively adsorb on photo-polarized facets of opposite charge, which can prolong the charge-carrier lifetime by a factor of five particularly at elevated temperature, leading to an overall energy conversion efficiency of 15.9% at 270 °C.
H2O2 is an important chemical in e.g., medical, and environmental applications, which can be produced by both H2O reduction and oxidation. Recently, PCWS to H2O2 and H2 rather than O2 and H2, has become increasingly attractive due to (i) the value of H2O2 is higher than O2, (ii) the formation of H2O2 is a 2-e− pathway that can moderate the sluggish of the 4-e− pathway for O2 evolution reaction, enabling a high performance of H2 evolution (Fig. 18(c)).221,338 An N, S-co-doped graphene @B-TiOx catalyst is used to produce H2O2 with a rate of 873 μmol L−1 h−1.339 However, the report of B-TiOx in PC H2O2 production is still scarce due to the instability of H2O2 under light irradiation, and the low WS reaction selectivity between O2 and H2O2.335,340
Photoanode and anode. B-TiOx stands as the pioneering and predominantly embraced catalyst for photoanodes in water-splitting (WS) cells. The first report using B-TiOx as a photoanode for water oxidation reaction (WOR) to O2 in a PECWS cell was appeared in 2011.341 In that work, the photoanode was made of the B-TiOx nanowire/FTO glass without loading noble metal with an active area of 0.20–0.25 cm2, which was tested in 1 M NaOH (pH = 13.6) under the 1-Sunlight simulator, AM 1.5 G, 100 mW cm−2. The photocurrent density at 0.4 VRHE (Volt versus the reversible hydrogen electrode) is 1.97 mA cm−2 corresponding to an SHT of 1.63%, and the photocurrent density at 1.23 VRHE is 2.8 mA cm−2. Currently, the state-of-the-art photoanode composed of B-TiOx for WS is B-BiVO4/B-TiOx with an active area of 1.5 cm2. The photocurrent density is 6.12 mA cm−2 at 1.23 VRHE measured in an electrolyte of 0.5 M potassium phosphate buffered to pH = 7 under 1-Sunlight simulator, AM 1.5 G, 100 mW cm−1.215 It is noteworthy that the reporting of the active areas for both photoanodes and photocathodes is crucial, as these two dimensions significantly impact the overall performance.
Very recently, B-TiOx has been used to stabilize the Ru sites through structural-confinement and charge-redistribution for WOR, which extends the catalyst lifetime in a 0.5M H2SO4 solution (pH = 0.3) by 10 orders of magnitude longer than that of commercial (com.) RuO2/TiO2 and better than com. IrO2/TiO2 (Fig. 19a).269 As shown in Fig. 19(b), Ru/TiOx retains stable for a 900-h test at @10 mA cm−2 with an overpotential of 174mV, which is better than reported values shown in Fig. 19(c) in both activity and stability. Meanwhile, the safeguard function from TiOx is further confirmed from comparing the Ru retention rate (%) among Ru/TiOx, RuOx/TiO2, and RuO2/TiO2, 97.3%, 42.2%, and 20.7%, respectively.269 The theoretical calculations are further used to confirm that TiOx stabilizes the Ru active center from the introduction of Ov.
 | ||
| Fig. 19 (a) Chronoamperometric (CP) curves (No iR compensated) of samples of Ru/TiOx, annealed RuOx/TiO2, com. RuO2/TiO2 and com. IrO2/TiO2 for OER at 100mA cm−2 for 50 h, respectively. (b) CP curve of Ru/TiOx for OER at 10 mA cm−2 for 900 h. (c) The overpotentials and durations of stability of Ru/TiOx and state-of-the-art electrocatalysts in for oxygen evolution reaction in acidic media. (d) Inductively coupled plasma-mass spectrometry (ICP-MS) analysis for dissolved Ru ions in electrolyte and Ru mass percentage retained in Ru/TiOx, annealed RuOx/TiO2 and com. RuO2/TiO2 catalyst after the CP test. Reprinted with permission from ref. 269. Copyright © 2023, The Author(s). | ||
Photocathode and cathode. There is a limited number of publications utilizing B-TiOx as a (photo)cathode in water-splitting (WS) cells. This scarcity can be attributed to the inherent instability of metal oxides in the coexisting environment of cathodic potential, initial atomic hydrogen (H*), and gaseous H2.196,222 However, very recently, B-TiOx (photo)cathode has shown remarkable progress in both the activity and the stability in WS cells for the H2 evolution reaction. Hou et al. proposed a multi-step reduction method to form B-TiOx, named R-TNT, which is then directly used as (photo)cathode in a WS cell to produce H2 with a geometric area of 1 cm2.342 The overall current of R-TNT is −221 mA at −1.0 VRHE, which is 17
000-times higher than that of pristine TiO2 (TNT). The long-term stability is carried out at chronopotentiometry (CP) mode of −100, −50, and −10 mA, sequentially, with a decay of 1.3%, 5.2%, and 18.4%.
In terms of the production of H2O2 in PEC/EC cells, both theoretical and experimental investigations have been carried out intensively, also included in recent thorough reviews.335,343 In an electrochemical cell, both WOR and water reduction reaction (WRR) can be carried out to form H2O2, as shown in Fig. 18(e).334,335,344 B-TiOx can work as both the anode catalyst for the WOR to H2O2 and as the cathode catalyst for the WRR to H2O2, which enables high faradaic efficiency of over 100%.193,345,346 For instance, a catalyst of (1T-2H)-MoSe2/B-TiOx is constructed for WOR to produce H2O2 with a rate of 57 μM h−1. A composite of B-TiOx/TiC needs an overpotential of ca.10 mV to form H2O2 with an H2O2 production rate of 7.19 mol g−1 h−1 at 0.3 VRHE and an H2O2 selectivity of 94.1% at 0.5 VRHE.347 In future, bifunctional B-TiOx catalysts are foreseen to be designed to function WOR and WRR simultaneously for H2O2 for a high faradaic efficiency and a high yield. In 2022, one PEC SWS is demonstrated using Au-Gd-Co2B@TiO2 electrocatalyst, which requires an overpotential of 510 mV to reach 1000 mA cm−2 in alkaline seawater. The high activity of Au–Gd–Co2B@TiO2 is assigned to the high concentration of Ov at the interface of Co-Au surface. To date, research reports on B-TiOx for seawater splitting are still limited.348
6.2. Carbon dioxide reduction
The emission of excessive carbon dioxide (CO2) into the atmosphere leads to subtle climate change, manifesting in phenomena including the global warming. Currently, the amount of CO2 released in atmosphere is still increasing: The global energy-related CO2 emissions was 36.8 Gt that grew by 0.9% in 2022, and September 2023 is the warmest September since 1880 so far, which shows a global average surface temperature 1.72 °C above that during the years of 1880–1920.349 Confronting this challenge, extensive research efforts have been devoted to the investigation and development of CO2 capture and conversion techniques for its direct or indirect utilization (Fig. 20(a)).350 Various approaches, including thermochemical, electrochemical, bio-electrochemical, and photocatalytic CO2 reduction reaction (CO2 RR), have been explored to transform CO2 into value-added carbon products such as CO,126,351,352 CH4,65,351,353–355 CH3OH,353,356–360 HCOOH,359,361 C2H5OH,360,361 CH3CHO,362 CH3COOH,359etc. A flowchart in Fig. 20(b) shows the combination of photocatalytic CO2 RR and carbonylation reaction to produce fine chemicals or drugs.363 These different carbon-based chemicals from CO2 RR require different energy inputs corresponding to their thermodynamics (Fig. 20(c)), i.e., which can be provided from batteries or solar cells generated from renewables.364,365 | ||
| Fig. 20 (a) Schematic of direct and indirect utilization of CO. Reprinted with permission from ref. 350. This article is licensed under a Creative Commons Attribution 3.0 License. (b) Schematic illustration of photocatalytic CO2 RR and carbonylation reaction for the utilization of that reduction carbon product. Reprinted with permission from ref. 363. Copyright © 2022, The Author(s). (c) Schematic illustration of required potential energy for photocatalytic CO2 RR for solar fuels and H2O oxidation reaction. Reprinted with permission from ref. 364. Copyright © 2022, The Author(s) under exclusive license to Tianjin University and Springer-Verlag GmbH Germany, part of Springer Nature. (d) and (e) Schematic illustration of the working principles of the electrolysis CO2 conversion cell and the photo-electrolysis CO2 conversion cell. Reprinted with permission from ref. 366. © 2022 The Authors. Advanced Science published by Wiley-VCH GmbH. | ||
The utilization of B-TiOx in CO2 RR has witnessed a significant surge, particularly in photocatalytic (PC) and photo-electrolytic (PEC) processes, which is attributable to its advantageous properties in efficiently capturing solar energy and fine-tuning energy bands.32,34,367–369 An latest review on B-TiOx based materials for photocatalytic CO2 RR was presented in 2022.34
The photocatalytic CO2 RR mechanism displayed in Fig. 20(c) shows the absorption of photons, and the generation of charge carries.364 Similar with PCWS, the migrating e− can reduce the adsorbed CO2 into valuable carbon products, and along with the required potential energies for these redox (Evvs. NHE at pH = 7), while h+ will oxidize H2O to O2. The PEC CO2 RR cell (Fig. 20(e)) produces carbon fuels and O2 in photocathode and photoanode via CO2 RR and HOR, respectively.366 When compared with PC CO2 RR, PEC CO2 RR has more choices in semiconductors in tuning their band position of B-TiOx, which can better meet the requirement of suitable band states and redox potential for CO2 RR with the assistance of an external bias. When compared to EC CO2 RR (Fig. 20(d)), PEC can decrease the overpotentials toward CO2 RR under light irradiation and can provide better control over product selectivity.366 For example, a PEC H-cell is integrated with a TiO2 anode for WOR to O2 and a Pt-reduced graphene oxide cathode for CO2 RR into HCOOH, which uses both solar energy and renewable electricity as the driving force. The photovoltage provided by the anode can compensate and confer cathodic potential for CO2 RR by maintaining the reactions taking place on both electrodes’ surfaces.361 It is worth noting that in both PC and PEC CO2 RR in using B-TiOx, the conduction band (CB) position must be situated below the reduction potential of CO2/carbon products. This arrangement is essential for the activation of CO2 and cleavage of CO bonds. Simultaneously, the valence band (VB) position of B-TiOx should be above the oxidation potential of H2O (refer to Fig. 20(c)). In other words, the B-TiOx and its composites should possess a suitable band structure for both PC and PEC CO2 RR.370
The complex CO2 photoreduction procedures make a substantial difference to their experimental activity and selectivity. In terms of the experimental activity, it is important to design effective B-TiOx based materials combining modification strategies such as doping and defect engineering to simultaneously boost CO2 RR without any sacrificial reagents. Besides, energy inputs (i.e., external potential) should be tuned to realize CO2 RR at reasonable rates. Aside from B-TiOx, the CO2 RR performance is also largely influenced by the concentration of reactants, the adsorption of intermediates, reaction pathways, the electrolyte proton availability, etc. For example, neutral and alkaline pH solutions are preferred to improve the kinetics of CO2 RR and to avoid the HER because of the prevalence of H+ ions in acidic conditions. Catalysts with high overpotential for HER and a contrarily lower overpotential for CO2 RR are desirable. Moreover, there is a wide range of carbon products from CO2 RR, commonly low molecular weight hydrocarbons with 1–4 carbon atoms C1-C4. Meeting market demand, as outlined in Table 3, and concurrently reducing the cost of separating carbon chemicals underscores the importance of achieving high selectivity in CO2 RR towards specific target products.371
| Products | Industrial values | Market prices ($ per kg) | |
|---|---|---|---|
| C1 | Carbon monoxide | Fischer–Tropsch synthesis | 0.06 |
| Methane | Combustion fuel, dry reforming to syngas | 0.18 | |
| Methanol | Gasoline additive, direct methanol fuel cell | 0.58 | |
| Formic acid | Direct formic-acid fuel cell | 0.74 | |
| C2+ | Ethylene | Polyethylene and ethylene glycol production | 1.3 |
| Ethane | Ethylene production | 0.26 | |
| Propane | Domestic combustion fuel | 0.9 | |
| Ethanol | Chemical solvent and fuel, medical use | 1.0 | |
| Ethylene glycol | Poly(ethylene terephthalate) production | 0.75 | |
| Diesel | Aviation fuel | 1.42 | |
| Gasoline | Fuel for internal combustion engines | 1.32 |
For CO2 RR, both the reactant molecule adsorption and the chemical bonds activation are very critical.372–374 As mentioned, B-TiOx has many surface defects such as Ti3+ species, Ov, etc., which can potentially serve as both adsorption and activation sites for the CO2 molecule leading to potential reduction reactions. For instance, Funk et al. reported that CO2 adsorption decreases with increasing defect density of Ov and increases with increasing O2 re-exposure, and also pointed that the defects are the active sites for breaking bonds of reactants of adsorbed O2 and H2O.373 Meanwhile, the adsorption geometry of CO2 and Ov can critically affect the CO2 RR pathway to diverse products. The CO2 adsorbed on Ov sterically inhibits the attack of the proton to the carbon. Therefore, the most feasible reaction pathway for the CO2 undergoing this adsorption mode is the deoxygenation into CO. While, the hydride-like Ti–H–Ti enables TiO2 to act as a hydride reagent during the photocatalytic CO2 RR to preferentially generate formic acid.374
The reaction pathway is contingent upon the catalysts and prevailing reaction conditions, giving rise to notably diverse product distributions. Understanding the generation of these different types of carbonaceous compounds is important for designing catalysts with a high selectivity such as toward the propane rather than ethane.375–377 One example of such a reaction pathway for CO2 RR is shown in Fig. 21 CO2 RR involving e− and H+, and thus the rate-determining steps of these products are governed by the e− or H+ transfer in 9 different ways. CO and HCOOH are produced by the transfer of 2e− and H+, CH3OH and CH4 formations require 6 and 8e− and H+, and the production of CH3CH2OH needs 12e− and H+ for the reaction with CO2˙−intermediate. Besides, the reaction mechanism of multistep intermediates to final products plays a key role in breaking through the kinetics bottleneck. It is generally believed that the pathway and selectivity of the final products are relative to the interaction of surface adsorbates, the replenishment of e− and H+, and the step of hydrogenation and deoxygenation, while product selectivity would decrease dramatically when generating the molecules that require more e−.208 For example, the formation of CH4 (−0.24 V) is thermodynamically more feasible than that of CO (−0.53 V), which means that if sufficient e− and H+ are provided, the formation of CH4 will be preferred. On the other hand, surface structure, morphology, composition, CB location, etc. of B-TiOx catalysts and applied external voltage can affect both the overall selectivity and the activity of the CO2 RR.356,378–380 Another case is the CO2 RR into CO and CH4, where the MoS2/TiO2 composition give production rates of 1.6 μmol g−1 h−1 for CH4 and 3.3 μmol g−1 h−1 for CO; whereas under the same conditions, the pristine TiO2 give 6.7 μmol g−1 h−1 for CH4 and 0.9 μmol g−1 h−1 for CO.226 The effect of the atmosphere (air and inert condition) and the temperature (300 °C, 450 °C and 600 °C) during the post-annealing of TiO2 on the performance of photocatalytic CO2 RR to CO is investigated, giving production rates of 59.54 (air, 450 °C), 172.61 (reductive condition, 300 °C), 179.97 (reductive condition, 450 °C) and 185.39 μmol g−1 h−1 (reductive condition, 600 °C).126
 | ||
| Fig. 21 Possible CO2 reduction reaction (CO2 RR) pathways for the hydrocarbons. Reprinted with permission from ref. 366. © 2022 The Authors. Advanced Science published by Wiley-VCH GmbH. | ||
Recently, TiO2 as a kind of reducible oxide has regained attention from the CO2 hydrogenation pathway. In 2023, Monai et al. presented a study elucidating the reconstruction of Ni/TiOx catalysts. This involved complete removal of TiOx under CO/CO2 hydrogenation conditions at 400 °C (refer to Fig. 22(a)). Interestingly, the process led to the establishment of interfacial sites featuring Ni–TiOx, identified as strong metal-support interaction (SMSI), during the reaction at 600 °C (refer to Fig. 22(c)). This SMSI phenomenon was found to enhance C–C coupling by providing a reservoir for C-species, thereby augmenting C–C coupling activity. The catalytic CO/CO2 hydrogenation performances over 400 °C/600 °C–Ni/TiO2 catalysts were tested at 200–400 °C under 5 bar and shown in Fig. 22(a) and (b). 400-Ni/TiO2 sample gives a higher overall catalytic activity, C1 in Fig. 22(c), C2-C4 in Fig. 22(d), and total yield in Fig. 22(f), but lower C1 (Fig. 22c) and obviously lower C2-C4 (Fig. 22d) stability over 600-Ni/TiO2. Meanwhile, the selectivity of C2+ is 14–20% higher under the CO/CO2 hydrogenation experiments at 200, 250, and 300°C for 600-Ni/TiO2 catalyst because of the formed interface between Ni and TiOx.83
 | ||
| Fig. 22 Model for TiOx overlayer formation after 400 and 600 °C reduction and restructuring under reaction conditions (a) 400 °C reduction, (b) the hydrogenation reaction mechanism on 400-Ni/TiO2, (c) 600 °C reduction, and (d) the hydrogenation reaction mechanism on 600-Ni/TiO2. (e) Catalytic activity for CO2 (dark shade) and CO/CO2 methanation (light shade), (f) C–C coupling activity in the CO/CO2 hydrogenation reaction, and (g) selectivity to hydrocarbons (C1–C4) in the CO/CO2 hydrogenation reaction over 400-Ni/TiO2 and 600-Ni/TiO2. Reprinted with permission from ref. 83. Copyright © 2023, The American Association for the Advancement of Science. | ||
6.3. Ammonia synthesis and decomposition
The atmospheric dinitrogen (N2) can be fixed into ammonia (NH3), nitric oxide, and nitrogen dioxide.381–385 Among them, NH3, as the second most produced chemical in human society, is an important raw material to produce fertilizers, plastics, explosives, nitric acids, and intermediates for dyes and pharmaceuticals. In addition, NH3 is anticipated to be the most promising future energy carrier since it contains no carbon in its molecule with high hydrogen content. To synthesize NH3, conventional Haber–Bosch (H–B) process has been employed under an elevated temperature of around 400 °C and an elevated pressure of around 150 bar, which gives an annual global production of more than 170 Mt (235 Mt in 2021), along with the consumption of about 1.0% of total world energy production and the 1.4% of global CO2 emission. To moderate the energy and environmental issues in the H–B process, new routes have been intensively studied to synthesize green NH3 such as (photo)electrosynthesis of NH3.386–390NH3 has been considered as a H2 carrier for the transportation and storage of H2 at high quantity, which is instead of employing a direct transport and storage of compressed H2 gas or liquid-H2 with inferior hydrogen carrying capacity. There are several advantages to use NH3 as a medium (i) the annual global production of NH3 is large, (ii) liquefication of NH3 requires only a pressure of 8.6 bar at 20 °C, much lower than 700 bar at 25 °C for H2, (iii) NH3 has a high hydrogen capacity, 121 kg H2 m−3 at 10 bar, and a gravimetric hydrogen density of 17.8 wt%, etc.
In this part, we will present the recent progresses in both NH3 synthesis and NH3 decomposition using B-TiOx as the catalyst, with a focus of cycling NH3 for energy transport using renewable energy sources such as solar power.
Even though new pathways in synthesis of NH3 has been intensively studied to lower the energy consumption and reduce CO2 emission, currently, about 90% of NH3 production still relies on the H–B process. The NH3 synthesis via H–B process is understood to follow the dissociative mechanism shown in Fig. 23(a). The first step of the H–B process is the breakage of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond that is also the rate-limiting step of N2 dissociation on the metal sites, followed by subsequent hydrogenation of atomic N to form NH3 before desorbing form the surface.392,393 In contrast, new approaches for the synthesis of NH3 are inspired by the biological enzymes that conduct N2 fixation under mild conditions, which is adopted the associative pathway where the cleavage of N2 is not rate-limiting step. Both experimental studies and DFT calculations show that the fixation of N2 to produce NH3 can proceed via the associative mechanism with the addition of protons proceeds by the distal pathway and the alternating pathway, in Fig. 23(b), and the details has been well discussed by Ye and Tsang in 2023.392
N bond that is also the rate-limiting step of N2 dissociation on the metal sites, followed by subsequent hydrogenation of atomic N to form NH3 before desorbing form the surface.392,393 In contrast, new approaches for the synthesis of NH3 are inspired by the biological enzymes that conduct N2 fixation under mild conditions, which is adopted the associative pathway where the cleavage of N2 is not rate-limiting step. Both experimental studies and DFT calculations show that the fixation of N2 to produce NH3 can proceed via the associative mechanism with the addition of protons proceeds by the distal pathway and the alternating pathway, in Fig. 23(b), and the details has been well discussed by Ye and Tsang in 2023.392
 | ||
| Fig. 23 Mechanisms for ammonia synthesis: (a) dissociation mechanism, and (b) association mechanism, showing the distal pathway (H on the distal N) and alternating pathway (H on both the distal N and the N proximal to the surface site). The reactions initiate at ‘*’. Reprinted with permission from ref. 392. Copyright © 2023, Springer Nature Limited. (c) Single Ti3+ on anatase TiO2 (101) surfaces with Ov for ammonia synthesis. ΔG refers to the free energy, and ΔE refers to the electronic energy. Reprinted with permission from ref. 394. Copyright © 2019, The Author(s). | ||
Both Ti3+ and Ov can serve as active sites (i.e., nitrogen-adsorption sites) to increase the donation of electrons to the anti-bonding π* orbitals of N2, and then to effectively weaken and promote the cleavage of the NN bonds.394,395 For instance, Li et al. reported that the Ov favoring the chemisorption and activation of N2 and the formation of Ti3+. Meanwhile, Cao et al. revealed that adjacent bi-Ti3+ sites on anatase TiO2 (101) can chemisorb and active N2 molecules, but both bi-Ti3+ sites on rutile TiO2 (110) and single and isolated Ti3+ on anatase TiO2 (101) cannot.394 In terms of the reaction mechanism of B-TiOx for NH3 formation, in general, it follows the distal pathway, where hydrogenation takes place on the adsorbed N atoms without previous cleavage of NN but to form *NHx intermediates directly (Fig. 23c).393,394 As a result, the NH3 synthesis can be operated under mild conditions.
In addition, the B-TiOx discussed in this review exhibits promise in both solar energy capture and the regulation of Ti3+ and Ov concentrations through various strategies (see Fig. 24(b)).394 This versatility is advantageous for the photoreduction of N2, even under ambient conditions, with the added benefit of no CO2 emission.107,115,133,181,396–401 For example, a bamboo-like B-TiOx film offers large surface area and Ov. Even without loading of noble-metal co-catalysts, the film can be used for fixing N2 into NH3 under ambient conditions driven by Vis-NIR light, yielding an NH3 generation rate of 48.3 mg m−2 h−1, viz, 178 μmol g−1 h−1.107 As shown in Fig. 24(c), the apparent QE (%) values of B-TiOx are measured at monochromatic wavelengths of 365, 405, 450, 532, 650, and 780 nm, delivering values at 0.39%, 0.12%, 0.11%, 0.15%, 0.24%, and 0.07%, respectively. At the same time, the QE% curve indicates that the synthesis of NH3 can be triggered at visible wavelengths longer than the absorption edge of pristine TiO2 at ca. 400 nm.107 The concept of lattice strain and geometry-modified nanoreactors, B-TiOx nanotubes, is interestingly demonstrated in the synthesis of NH3, which reports a generation rate of 5.50 μg h−1 cm−2 (16.67 μg h−1 mg−1) and a FE(%) of 26% under ambient aqueous conditions.115 In efforts to enhance the yield rate of NH3, reported strategies include the utilization of Ru single atoms to modify B-TiOx, resulting in a notable rate of 22.2 mol g−1 h−1 in the reduction of NO3− to NH3.176 Intriguingly, a dual temperature zone catalyst of TiO2-xHy/Fe (Scheme in Fig. 24(d)), was designed to effectively synthesis of NH3, where Fe as the hot zone to activate N2 and TiO2−xHy as the cooling zone to hydrogenate N to NH3 relying on its Ov.280 Under solar illumination, the apparent temperature of TiO2−xHy/Fe reaches 495 °C but with local temperature difference up to 137 °C between the Fe and TiO2−xHy owing to the plasmonic local heating (Fig. 24(e)). The designed catalysts have surpassed the yields achievable through thermal catalysis in the solar-driven synthesis of NH3.402
 | ||
| Fig. 24 (a) Schematic illustration of required potential energy for photocatalytic nitrogen reduction reactions and a water oxidation reaction. Reprinted with permission from ref. 391. Copyright © 2021 Elsevier Inc. (b) The formation of Ov and Ti3+ sites on the Zr-doping TiO2 for NH3 synthesis. Reprinted with permission from ref. 394. Copyright © 2019, The Author(s). (c) The apparent QE% of NH3 evolution using B-TiOx under the monochromatic lamp irradiation at different wavelengths. Reprinted with permission from ref. 107. Copyright © 2020 Elsevier B.V. All rights reserved. (d) Diagram of N2 and H2 activation and H transfer for NH3 generation over TiO2–xHy/Fe catalyst. Reprinted with permission from ref. 280. Copyright © 2020, American Chemical Society. (e) Local temperature difference of illuminated TiO2-xHy/Fe steady-state non-equilibrium temperature distribution at 726 nm and the proposed dual-temperature-zone NH3 synthesis of TiO2-xHy/Fe. Reprinted with permission from ref. 402. Copyright © 2019 Elsevier Inc. | ||
6.4. Environmental pollutant treatment
The degradation of both inorganic and organic pollutants such as organic dyes and toxic gas of carbon monoxide (CO), holds significant importance in addressing environmental challenges.404–411 Toxic gases purification is becoming a distinctly growing concern. This process often involves gas capture and catalytic degradation, the B-TiOx based materials are unfortunately not suited for gas capture. Thus, we focus on the summary on the degradation of pollutants in liquid solutions that is categorized into advanced oxidation process (AOP) and advanced reduction process (ARP). The AOP has been intensively studied using free radicals to degrade pollutants, meanwhile, ARP has been used as a new approach for removing oxidative pollutants from aqueous solution for water cleaning.412B-TiOx, as an environmentally friendly material, has been an attractive candidate for both AOP and ARP processes,37,113,409–411 and also earns bonuses from its advantages, such as high absorption capacity in a broad wavelength range.
Beyond surface defects, the difference between PEC and PC techniques for the degradation of pollutants, such as MB, RhB, and Cr (VI), using the same B-TiOx catalyst has been discussed. It confirmed that the PEC removal efficiencies are higher than that of PC ones because of the additional power from external voltage.117 With respect to the high performance of the PEC process, the working principle has been addressed and can be divided into two regimes according to the applied potential: (i) at low bias, it worked as the electro-assisted photocatalysis; (ii) at high bias, it worked as a combination of electro-assisted photocatalysis and electrochemical oxidation.117 A PEC degradation mechanism (Fig. 25(a)) has been proposed that Ti3+ serves as the active species for tetracycline (TC) degradation, while, the visible-light light capture can be effectively enhanced from both the unique nanotubes, the narrowed bandgap, and the junctions in enhancement of visible-light harvesting ability.416The proposed TC degradation pathway is displayed in Fig. 25(b), but we would like to stress the importance of ˙O2− and h+ for the oxidation of TC, via continuously attacking TC and intermediate products such as P4 and P5.416
 | ||
| Fig. 25 (a) Proposed mechanism diagrams in Ar–Fe2O3/Ti3+–TiO2-NTs, and (b) Proposed TC pathway by the Ar–Fe2O3/Ti3+–TiO2-NTs photoelectrode. Reprinted with permission from ref. 416. © 2022 Elsevier B.V. All rights reserved. | ||
6.5. Solar thermal energy
Solar thermal energy (STE) involves harnessing solar energy to generate thermal energy, which finds applications in both industrial and residential sectors. Examples include solar cookers and solar-driven interfacial water evaporation systems.419Undoubtedly, a photothermal material with a dark color is of considerable interest as an absorber/collector for STE applications.369,420 B-TiOx has attracted enormous attention in this area due to its significantly extended absorption of solar energy, ranging from UV to NIR.125,147,150,421–426 For instance, illuminated by the simulated solar irradiation (AM 1.5 G, Xe lamp) for 60 s, the temperature of the B-TiOx material increases from RT to 37 °C, while pure TiO2 increases to 28 °C.125 This has been attributed to the significant enhancement of Vis and NIR photo-absorption of B-TiOx originated from the unique oxygen-deficient shells. To further modify B-TiOx, a hierarchical tandem heterojunctions visible-light photocatalyst, B-TiOx/MoS2/Cu2S, was designed and prepared. In this hierarchical structure, the mesoporous B-TiOx can serve as the host to assemble MoS2 and Cu2S. MoS2 acted mainly as the co-catalyst that can effectively transfer and separate photo-generated charge carriers due to suitable band alignments. The introduction of MoS2 and Cu2S extended the photo-response to near infrared region. In addition, both MoS2 and Cu2S with narrow band gap could convert solar light into heat energy. As a result, the light absorption of the B-TiOx/MoS2/Cu2S in the wavelength of around 200–800 nm is stronger than both B-TiOx and B-TiOx/MoS2.225 The temperature change of the B-TiOx, B-TiOx/MoS2, and B-TiOx/MoS2/Cu2S was recorded with an infrared thermograph under light irradiation (λ > 400 nm). After 5 min irradiation, the temperature rose from 25 °C to 49.9 °C (B-TiOx), 53.3 °C (B-TiOx/MoS2), and 58.2 °C (B-TiOx/MoS2/Cu2S), respectively. Furthermore, the photothermal conversion of B-TiOx is often assigned to the high probability of non-radiative relaxation occurring in the Ti 3d orbitals.107 Given the low bandgap energy of B-TiOx, the majority of photons from solar light have significantly possessed higher energy than the bandgap threshold. Consequently, above-bandgap electron–hole pairs are generated. These electron–hole pairs subsequently relax to the band edges, releasing excess of energy in form of heat, likely facilitated through an acoustic-phonon scattering mechanism.427 This is in stark contrast to conventional wide-bandgap semiconductors, where most of absorbed light energy is typically re-emitted as photons following the recombination of electron–hole pairs near the bandgap edge.
Solar steam interfacial evaporation represents a promising strategy for seawater desalination and wastewater purification owing to its environmentally friendly characteristics.123,332 It has been reported recently that metallic λ-Ti3O5 powder, also categorized as B-TiOx, exhibits a high solar absorptivity of 96.4%, which is attributed to Ti–Ti dimer-induced flat-bands around the Fermi level.419 This work has emphasized the critical role of tuning joint densities of states in enhancing solar absorption of photothermal materials, which has been previously overlooked in this field. Combined with the low thermal conductivity of λ-Ti3O5, this novel characteristic ensures efficient solar-to-heat conversion and high thermal localization. As a result, an unprecedentedly high-water evaporation rate of 6.09kg m−2 h−1 (Fig. 26a) was achieved under a 1-Sun irradiation without salt precipitation in the 3D-SSE system (Fig. 26b).419
 | ||
| Fig. 26 (a) Evaporation rates of 3.5wt% saline water as a function of time for the 3D-SSE containing 6wt% λ-Ti3O5 powders under 1sun of irradiation, and photographs of the 3D-SSE at 20 h, 60 h and 100h without salt precipitation. (b) Schematic diagram of 3D-SSE system. Reprinted with permission from ref. 419. Copyright © 2023, The Author(s), under exclusive licence to Springer Nature Limited. Thermal conductivity of anatase single crystals (c) Temperature dependence of the thermal conductivity of three anatase TiO2 single crystals, respectively, from higher to lower κ, test curves and fitted curves (solid lines). Sketches of the difference between heat propagation in (d) pristine single-crystal and in (e) crystal with oxygen vacancies. The color gradients from red to blue illustrate temperature differences across the material. Reprinted with permission from ref. 428. Copyright © 2019, The Author(s). | ||
To some extent, thermal conductivity is related to STE. Fig. 26(c) displays the measurement of the thermal conductivity of TiO2 crystals in the temperature range of 0–300 K using a 4-points measurement method. It has been demonstrated that an increase in the concentration of Ov results in a dramatic decrease in thermal conductivity, leading to high thermal localization. The difference between heat propagation is proposed and sketched in both pristine single-crystal (Fig. 26d) and crystal containing Ov (Fig. 26e).428
6.6. Supercapacitance
Energy storage in supercapacitors is a mature technology, which has unique features in high power density, wide operating temperature range of −40 °C to 220 °C, and long cycle in millions, though further improvements in these aspects are needed.130,286,429,430 Besides, investigations on assembling high-performance supercapacitors with low cost, high surface area, and high electrical conductivity materials have been under continuous investigation.TiO2 has been an obvious candidate for assembling as supercapacitors because of its low cost and chemical stability. However, it is a low electrical conductivity material that hinders its application in supercapacitor. To improve its electrical conductivity for a high-performance supercapacitor, blackening processes have been adapted to modify TiO2.431,432 For instance, reductive chemical of NaBH4 can be used to introduce Ov to form B-TiOx, which exhibits an improved electrical conductivity. Furthermore, B-TiOx can be used to construct supercapacitors with high-power density, a fast charge/discharge rate, long-term life cycles, etc.21,94,110,137,194,291,300,433–437 For example, the supercapacitor assembled with B-TiOx gives a capacitance of 14.3 mF cm−2, which is 14 times higher than that of the supercapacitor assembled with pristine TiO2.110 This low-cost material with high charge capacity value generally serve as a reference for further improvement in superconductors.
6.7. Ion battery
In addition to supercapacitors, an alternative avenue for utilizing renewable energy involves electrochemical storage of chemical energy, subsequently converting it into electrical energy through ion batteries.130,144,264,438–440B-TiOx has gained the unflagging attention from ion batteries such as sodium (Na)-ion batteries, lithium (Li)-ion batteries, magnesium (Mg)-ion batteries, etc. because of its low scalability of the preparation procedure, appreciable capacity, and long-term stability upon reversible operation.130,144,283,441–448 In ion batteries, B-TiOx has been used as both the cathode and the anode materials. Fig. 27(a) shows B-TiOx as the cathode material in a Li–O2 (oxygen) battery, which gives a capacity of 5761 mA h g−1, higher than that of Li–O2 batteries with other two cathodes, i.e., gray TiO2 and carbon nanotube (CNT) with 3620 and 1794 mA h g−1, respectively. The good performance of the Li–O2 battery has been assigned to the surface defects (Ov and Ti3+) of B-TiOx enabling a high electrical conductivity that further promotes the catalytic activity.449 The charge/discharge rate capabilities of B-TiOx assembled Li–O2 batteries have been measured at the current densities of 100, 200, and 500 mA g−1 corresponding to the performances of 5761/4945, 4220/4015 and 2796/2579 mA h g−1, respectively (Fig. 27(b)).449 The long-term stability of these three Li–O2 batteries under the same conditions are shown in Fig. 27(c): 108-cycle-stable for B-TiOx, 40-cycle-stable for gray TiO2, and 10-cycle-stable for CNT.449 In the aspect of modelling, DFT simulations confirmed the function of Ov and Ti3+ of B-TiOx in promoting the reversible capacity and in retaining capacity after long-term cycles of Mg-ion batteries.447 Other important features such as operation at a low temperature of −20 °C has been particularly reported, where the B-TiOx-ion battery retained 84% capacity after 100-cycle charge–discharge tests.450
 | ||
| Fig. 27 (a) The initial discharge/charge profiles of B-TiOx, gray TiO2, and pure CNT cathodes at a current density of 100 mA g−1, (b) the initial discharge/charge profiles of the B-TiOx cathode at different current densities, and (c) cyclability and the terminal voltage of the B-TiOx, gray TiO2, and pure CNT cathodes. Reprinted with permission from ref. 449. Copyright © 2020, Elsevier Ltd and Techna Group S.r.l. | ||
6.8. Fuel cell
Fuel cells operate akin to batteries, where the production of electricity is continuously sustained by chemical consumption. In the case of low-carbon chemicals, such as green H2 and NH3, their utilization in fuel cells further contributes to the advancement of a sustainable society. For example, hydrogen fuel cells use H2 to produce electricity for cars without CO2 emissions.B-TiOx has the major advantages of reasonable electrical conductivity, decreased mass transfer barrier, and large amounts of anchoring sites provided by Ov and –OH groups that arose interests to both the cathode and the anode materials for fuel cells.451–453 For example, a proton exchange membrane (PEM) fuel cell used B-TiOx as both anode (B-TiOx-Anode) and cathode electrode for H2–O2 reaction, which gives a maximum power density of 403 and 52 mW cm−2, respectively.452 For the B-TiOx-Anode, after a 100-h stability test at the constant voltage of 0.45 V, the corresponding current density is still stable at 1500 mA cm−2 without obvious decay, and the polarization curves (before and after 100-h running) are measured to demonstrate their stability results.454 Highly active and durable catalysts for the oxygen reduction reaction (ORR) are important to PEM fuel cells. In 2022, an active protective strategy, using Ta–TiOx nanoparticles as radical scavengers for radicals and H2O2, was demonstrated to protect Fe–N–C catalysts from degradation.455 The cell voltage (Fig. 28a) and power density (Fig. 28b) polarization plots as a function of different current density were shown with (Fe–N–C + 8% Ta–TiOx/KB) and without scavenger (Fe–N–C) under an accelerated durability test (ADT) to confirm the reliability of Ta–TiOx.455 After the ADT, the PEM fuel cell with the Ta–TiOx/KB scavengers showed a current density of 0.63A cm−2 at 0.6V and gave a highest power density of 700mW cm−2, while these of catalyst without scavengers gave 0.39A cm−2 at 0.6V, and 370mW cm−2. Fig. 28(c) compares the current density of the cells with and without the Ta–TiOx/KB scavengers at internal resistance-compensated voltages (ViR-free) of 0.8 ViR-free and 0.9 ViR-free. The cell with Ta–TiOx scavengers at 0.8 ViR-free decreases the decay from 33% to 3%, and at 0.9 ViR-free shows a decrease of decay from 52% to 14%. These findings showed that the Ta–TiOx scavengers play a prominent role in the improvement of the PGM-free cathode durability and demonstrated a new function of B-TiOx to stabilize catalysts. However, at present, the investigations of B-TiOx-based materials in fuel cells are still in infancy stage and further knowledge is being actively pursued.
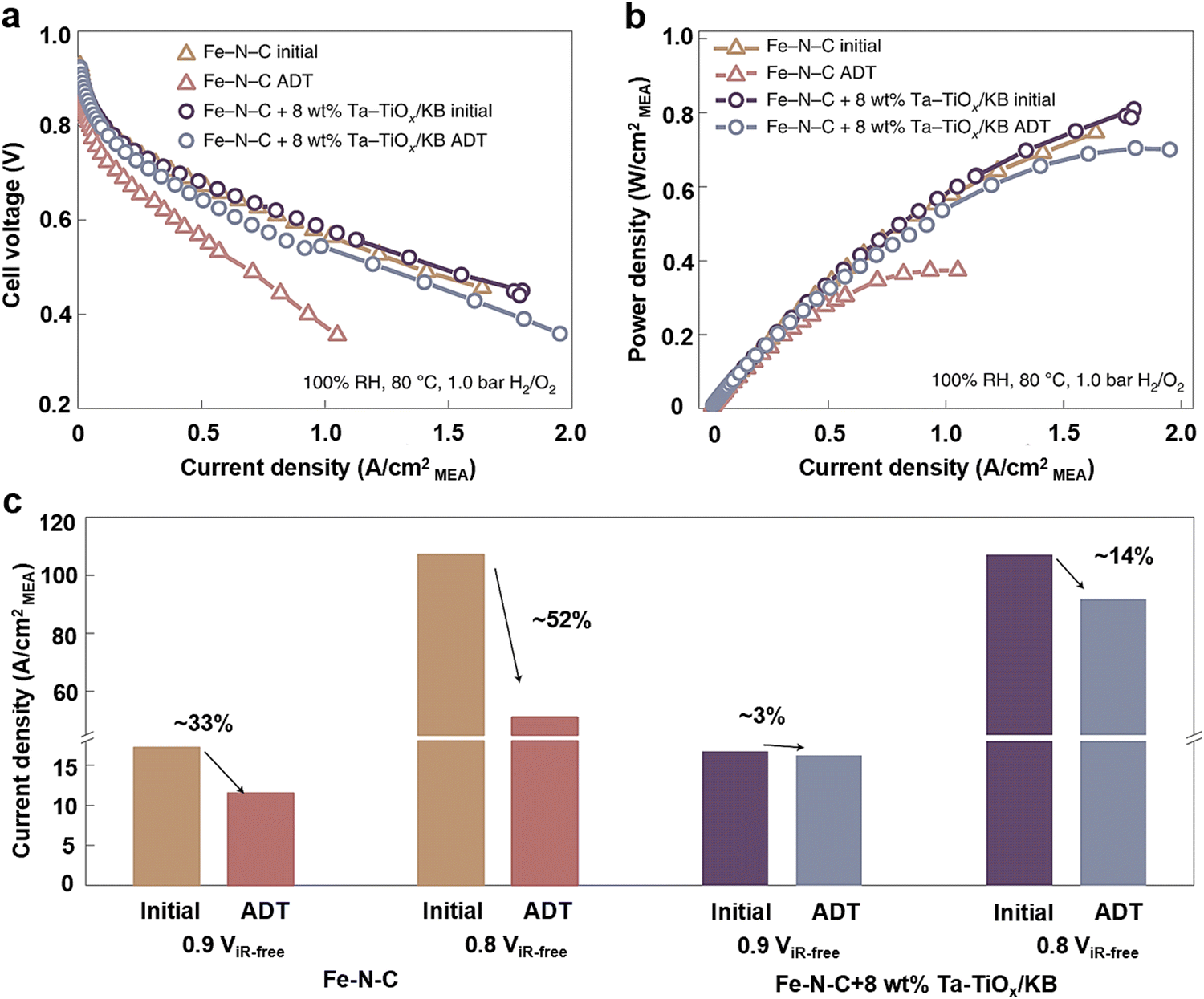 | ||
| Fig. 28 (a) Polarization–discharge voltage curves as a function of current density plots, (b) polarization–discharge power density curves as a function of current density plots for cells before and after the accelerated durability test (ADT). (c) Current density decay comparison for cells with and without Ta–TiOx/KB after the ADT. The y-axis break is used to enhance readability for small current density values. Reprinted with permission from ref. 455. Copyright © 2022, The Author(s), under exclusive license to Springer Nature Limited. | ||
6.9. Other applications
In addition to the aforementioned applications of B-TiOx in energy conversion and storage systems, and waste treatments, other applications such as PEC biomass conversion (Fig. 29),213 solar cells,100,456,457 sensors,458–461 stem cell capture,71 betavoltaics,37,105 smart windows,462 self-cleaning face masks,463,464 and others465,466 have also been investigated. Although these cases are limited in number, their contributions enrich applied fields and research databases, providing valuable insights into the understanding of B-TiOx from diverse perspectives. For instance, dye-719 sensitized solar cells are fabricated with pristine TiO2 and B-TiOx, separately, yielding performances of 2.26% (pristine) and 1.25% (black), which is due to the downshift of the conduction band of B-TiOx during the blackening process.100 | ||
| Fig. 29 B-TiOx is used for PEC oxidation of glucose to glucaric acid (GLA). Reprinted with permission from ref. 213. Copyright © 2023, The Author(s). | ||
7. Conclusions and perspectives
Over the past decade, significant strides have been made in the research of black titanium oxide (B-TiOx), encompassing its synthesis, modification, characterization, and application. Notable achievements include the development of energy and time-efficient synthesis methods for B-TiOx. The focus now shifts towards reducing costs, scaling up production, and refining the controllability of the reduction process. Additionally, addressing the fundamental challenges in understanding the mechanisms by which B-TiOx enhances performance, through in situ or operando characterizations, is an ongoing effort. As highlighted in this review, B-TiOx has demonstrated considerable promise in solar energy utilization, particularly in applications such as green hydrogen production, CO2 reduction, and ammonia synthesis. Despite its potential, B-TiOx remains underexploited in electrochemical cells, where its high charge carrier density, rapid electron transport, and affordability could offer significant advantages. The continued exploration of B-TiOx's applications is expected to play a pivotal role in the development of sustainable energy solutions and environmental remediation. Looking ahead, there is a clear need for in situ techniques that operate across multiple scales to uncover the local structural nuances of B-TiOx during its synthesis, modification, characterization, and application. Advanced diagnostic methods, including cutting-edge synchrotron and high-intensity light sources with superior spatial and temporal resolution, are essential for detailed manipulation of the material's blackness, including the degree and depth, as well as local order–disorder and strain–stress features. These insights are crucial for establishing correlations with catalytic activity, selectivity, and device performance. Furthermore, large-scale demonstrations, supported by predictive models and long-term stability assessments, are vital for transitioning B-TiOx from the laboratory to real-world applications. The successful commercialization of B-TiOx will hinge on these comprehensive evaluations, ensuring that this promising material class can meet the demands of a sustainable future.The remarkable physicochemical attributes of B-TiOx have spurred the advancement of blackening methods, concurrently catalyzing its deployment in renewable energy harnessing, conversion, and both direct and indirect storage into chemical forms or electricity. As laboratory-scale synthesis, modification, and application techniques evolve, the imperative now shifts to scaling up these processes. This escalation is critical to ensure a seamless transition from lab to market, meeting the commercial demand and facilitating widespread adoption.190,467,468 Besides, safety issue, long-term stability, scale-up effect, material cost, and time-cost are needed to be considered and evaluated based on both experiments, theoretical calculations, and models.469 For example, if considering the future industrial issues such as the waste reprocessing costs, manufacturing costs, and the installation, the metal substrates such as titanium metal is highly competitive with the conventional transparent conductive glasses.157,470,471
The blackening of titanium dioxide has gained widespread acceptance for its utility, yet a deeper understanding of B-TiOx remains a necessity. Employing in situ techniques across multiple scales is invaluable for observing, recording, and comprehending the material's formation, modification, and application processes. These techniques are instrumental in elucidating the material's properties, thereby enabling the manipulation of physicochemical characteristics, such as bandgaps, through precise control of the blackening process. Such insights facilitate a balance between cost-efficiency and performance from a commercial standpoint. Additionally, the significance of the reduced metal oxide layers, TiOx, is recognized for their association with highly active catalytic reactions.83 To investigate the formation and influence of thin reducible metal oxide layers on catalytic activity, a combination of in situ and ex-situ analytical tools should be utilized. In situ techniques, including transmission electron microscopy (TEM), X-ray absorption spectroscopy (XAS), and near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS), are essential for real-time observation of the material under reaction conditions. Ex situ methods, such as electron energy loss spectroscopy (EELS) and pair distribution function (PDF) analysis, provide detailed post-reaction insights into the material's structure and electronic properties. Complementing these experimental approaches with density functional theory (DFT) calculations allows for a comprehensive understanding of the material at the atomic level, enabling the correlation of structural features with catalytic performance.472 Thus, further in situ investigations are constantly on call for providing further details in deepening the understanding of B-TiOx. The formation, and the thickness of reducible metal oxide TiO2, viz, B-TiOx, in different reaction conditions is in correlation with the description of B° that can use in situ STEM combined with in situ EELS. Different blackening methods lead to different B° of B-TiOx nanotube/sphere/discs/layers, such as disorder structures and geometric and strain–stress effects, where PDF may be invoked to reveal.
Very recently, Li et al. have pioneered the development of a NAP-XPS technique using trimethylphosphine (TMP) as a surface probe.297 This approach has been employed to demonstrate facet-dependent charge accumulation in a N-doped TiO2 photocatalyst under photoexcitation. The rationale behind this lies in the ability of the nucleophilic TMP molecule to form stable adducts with exposed Lewis acid (LA) sites on the surface, such as the five-coordinated Ti4+. The interaction between TMP and the surface LA sites is very sensitive to the changes in the surface chemical micro-environment and local electron density, resulting in shifts in binding energy on P 2p XPS spectra. It should be emphasized that this technique offers truly surface sensitivity, as TMP molecules can only adsorb on the topmost surface. This contrasts with conventional laboratory based XPS, which provides information on both the surface and subsurface regions. Likewise, it has been demonstrated that the surface features of modified TiO2 materials can also be investigated by the probe-assisted solid state nuclear magnetic resonance technique.473 Foo et al. has recently reported a comprehensive structural study of N-doped TiO2 using variable-temperature synchrotron X-ray powder diffraction.246 Their findings revealed an unusual anisotropic thermal expansion, shedding light on the intricate relationship between subsurface oxygen vacancies, nitrogen doping levels, and photocatalytic activity.
Moreover, the extent of reductive conditions, such as the duration of blackening treatment, can be fine-tuned to regulate the thickness or depth of the blackened layers, thereby affecting their physicochemical properties. To maintain coherence with these alterations, in situ preparation and modification techniques should evolve in tandem with in situ characterization methods. A mechanistic analysis that integrates DFT, molecular dynamics, and machine learning can provide a comprehensive framework from simulation to the calculated preparation and application of materials. This approach not only enhances our understanding of the materials but also accelerates the development process, reducing the time required to transition from laboratory research to market-ready technologies.
Author contributions
Hou X. L.: conceiving the topic, literature search, data collection and analysis, writing – original draft and writing – review & editing. Li Y. Y.: discussion and writing – review & editing. Zhang H.: discussion and writing – review & editing. Lund P. D.: writing – review & editing. Kwan J.: writing – review & editing and supervision. Tsang S. C. E.: conceiving the topic, discussion, writing – review & editing and overall supervision.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
None of the authors has any competing financial or other conflict of interest.Acknowledgements
Financial support by Engineering and Physical Science Research Council (EPSRC), UK (EP/W012316/1) and (EP/X525777/1) is kindly acknowledged. The authors also thank to Miss Qin Yi in providing the image for the TOC.References
- C. Zhang, Y. Xie, H. Zhang, Y. Gu and X. Zhang, Energy, 2023, 262, 125453 CrossRef.
- BP's Statistical Review of World Energy, 2022.
- X. Chen, L. Liu, P. Yu and S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- T. Rajaraman, S. Parikh and V. Gandhi, Chem. Eng. J., 2020, 389, 123918 CrossRef CAS.
- S. Ullattil, S. Narendranath, S. Pillai and P. Periyat, Chem. Eng. J., 2018, 343, 708–736 CrossRef CAS.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- E. Ivanova, J. Hasan, H. Webb, G. Gervinskas, S. Juodkazis, V. Truong, A. Wu, R. Lamb, V. Baulin, G. Watson, J. Watson, D. Mainwaring and R. Crawford, Nat. Commun., 2013, 4, 2838 CrossRef PubMed.
- Y. Yu, Z. Zhang, X. Yin, A. Kvit, Q. Liao, Z. Kang, X. Yan, Y. Zhang and X. Wang, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- J. Lv, T. Zhang, P. Zhang, Y. Zhao and S. Li, Nanoscale Res. Lett., 2018, 13, 1–10 CrossRef PubMed.
- H. Kim, S. Uddin, D.-H. Lien, M. Yeh, N. Azar, S. Balendhran, T. Kim, N. Gupta, Y. Rho, C. Grigoropoulos, K. Crozier and A. Javey, Nature, 2021, 596, 232–237 CrossRef CAS PubMed.
- X. Wang, R. Raghupathy, C. Querebillo, Z. Liao, D. Li, K. Lin, M. Hantusch, Z. Sofer, B. Li, E. Zschech, I. Weidinger, T. Kuhne, H. Mirhosseini, M. Yu and X. Feng, Adv. Mater., 2021, 33, e2008752 CrossRef PubMed.
- H. Shi, S. Fu, Y. Liu, C. Neumann, M. Wang, H. Dong, P. Kot, M. Bonn, H. Wang, A. Turchanin, O. Schmidt, A. Shaygan Nia, S. Yang and X. Feng, Adv. Mater., 2021, 33, e2105694 CrossRef PubMed.
- Z. Tian, P. Zhang, P. Qin, D. Sun, S. Zhang, X. Guo, W. Zhao, D. Zhao and F. Huang, Adv. Energy Mater., 2019, 9, 1901287 CrossRef.
- X. Xu, Y. Xu, F. Xu, G. Jiang, J. Jian, H. Yu, E. Zhang, D. Shchukin, S. Kaskel and H. Wang, J. Mater. Chem. A, 2020, 8, 1636–1645 RSC.
- R. Fernández-Climent, S. Giménez and M. García-Tecedor, Sustainable Energy Fuels, 2020, 4, 5916–5926 RSC.
- G. Wang, Y. Ling, H. Wang, X. Yang, C. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2012, 5, 6180–6187 RSC.
- Y. Zhang, J. Tang, G. Wang, M. Zhang and X. Hu, J. Cryst. Growth, 2006, 294, 278–282 CrossRef CAS.
- X. Chen and S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- X. Chen and A. Selloni, Chem. Rev., 2014, 114, 9281–9282 CrossRef CAS PubMed.
- J. Chen, Z. Xia, H. Li, Q. Li and Y. Zhang, Electrochim. Acta, 2015, 166, 174–182 CrossRef CAS.
- X. Hou, Y. Zhao and Y. Li, Int. J. Hydrogen Energy, 2023, 48, 14279–14286 CrossRef CAS.
- H. Brahmi, R. Neupane, L. Xie, S. Singh, M. Yarali, G. Katwal, S. Chen, M. Paulose, O. Varghese and A. Mavrokefalos, Nanoscale, 2018, 10, 3863–3870 RSC.
- V. Zuñiga-Ibarra, S. Shaji, B. Krishnan, J. Johny, S. Sharma Kanakkillam, D. Avellaneda, J. Martinez, T. Roy and N. Ramos-Delgado, Appl. Surf. Sci., 2019, 483, 156–164 CrossRef.
- N. Rahimi, R. Pax and E. Gray, Prog. Solid State Chem., 2019, 55, 1–19 CrossRef CAS.
- J. Zheng, S. Bao, X. Zhang, H. Wu, R. Chen and P. Jin, Appl. Catal., B, 2016, 183, 69–74 CrossRef CAS.
- W. Fang, M. Xing and J. Zhang, J. Photochem. Photobiol., C, 2017, 32, 21–39 CrossRef CAS.
- H. Hu, Y. Lin and Y. Hu, Chem. Eng. J., 2019, 375, 122029 CrossRef CAS.
- Z. Li, J. Dong, Y. Zhang, T. Zhuang, H. Wang, X. Du, X. Cui and Z. Wang, Compos. Sci. Technol., 2022, 218, 109198 CrossRef CAS.
- A. Naldoni, M. Altomare, G. Zoppellaro, N. Liu, S. Kment, R. Zboril and P. Schmuki, ACS Catal., 2019, 9, 345–364 CrossRef CAS PubMed.
- S. Kim, Y. Cho, R. Rhee and J. Park, Carbon Energy, 2020, 2, 44–53 CrossRef CAS.
- A. Chatzitakis and S. Sartori, Chem. Phys. Chem., 2019, 20, 1272–1281 CrossRef CAS PubMed.
- Z. Li, S. Wang, J. Wu and W. Zhou, Renewable Sustainable Energy Rev., 2022, 156, 111980 CrossRef.
- J. Wang, R. Guo, Z. Bi, X. Chen, X. Hu and W. Pan, Nanoscale, 2022, 14, 11512–11528 RSC.
- Z. Xiu, M. Guo, T. Zhao, K. Pan, Z. Xing, Z. Li and W. Zhou, Chem. Eng. J., 2020, 382, 123011 CrossRef CAS.
- P. Sun, Z. Cao, Y. Zeng, W. Xie, N. Li, D. Luan, S. Yang, L. Yu and D. Lou, Angew. Chem., Int. Ed., 2022, 61, e202115649 CrossRef CAS PubMed.
- L. Placa, P. Vital, L. Gomes, A. Roveda, Jr., D. R. Cardoso, C. Martins and H. Wender, ACS Appl. Mater. Interfaces, 2022, 61, 43259–43271 Search PubMed.
- R. Katal, S. Masudy-Panah, M. Tanhaei, M. Farahani and H. Jiangyong, Chem. Eng. J., 2020, 384, 123384 CrossRef CAS.
- Y. Liu, L. Tian, X. Tan, X. Li and X. Chen, Sci. Bull., 2017, 62, 431–441 CrossRef CAS PubMed.
- Y. Li and E. Tsang, Mater. Today Sustainability, 2020, 9, 100032 CrossRef.
- X. Hou, H. Zhang, R. Raju, Y. Li and P. Lund, J. Power Sources, 2023, 580, 233281 CrossRef CAS.
- R. Vadakkekara and S. Jadkar, ACS Appl. Energy Mater., 2021, 5, 674–684 CrossRef.
- L. Yang, Y. Peng, Y. Yang, J. Liu, Z. Li, Y. Ma, Z. Zhang, Y. Wei, S. Li, Z. Huang and N. Long, ACS Appl. Nano Mater., 2018, 1, 4516–4527 CrossRef CAS.
- A. Ziarati, A. Badiei and R. Luque, Appl. Catal., B, 2018, 238, 177–183 CrossRef CAS.
- O. Secundino-Sánchez, J. Díaz-Reyes, J. Sánchez-Ramírez, J. Arias-Cerón, M. Galván-Arellano and O. Vázquez-Cuchillo, Catal. Today, 2022, 392–393, 13–22 CrossRef.
- K. Lan, Y. Liu, W. Zhang, Y. Liu, A. Elzatahry, R. Wang, Y. Xia, D. Al-Dhayan, N. Zheng and D. Zhao, J. Am. Chem. Soc., 2018, 140, 4135–4143 CrossRef CAS PubMed.
- L. Sheng, T. Liao, L. Kou and Z. Sun, Mater. Today Energy, 2017, 3, 32–39 CrossRef.
- J. Lin, Y. U. Heo, A. Nattestad, Z. Sun, L. Wang, J. Kim and S. Dou, Sci. Rep., 2014, 4, 5769 CrossRef PubMed.
- N.-W. Lee, J.-W. Jung, J.-S. Lee, H.-Y. Jang, I.-D. Kim and W.-H. Ryu, Electrochim. Acta, 2018, 263, 417–425 CrossRef CAS.
- S. Rej, S. Hejazi, Z. Badura, G. Zoppellaro, S. Kalytchuk, Š. Kment, P. Fornasiero and A. Naldoni, ACS Sustainable Chem. Eng., 2022, 10, 17286–17296 CrossRef CAS.
- H. Shi, X. Wang, M. Zheng, X. Wu, Y. Chen, Z. Yang, G. Zhang and H. Duan, Adv. Mater. Interfaces, 2016, 3, 1600588 CrossRef.
- K. Lan, Q. Wei and D. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202200777 CrossRef CAS PubMed.
- Y. Zeng, Y. Wang, S. Zhang and Q. Zhong, Phys. Chem. Chem. Phys., 2018, 20, 22744–22752 RSC.
- C. Xu, T. Liu, W. Guo, Y. Sun, C. Liang, K. Cao, T. Guan, Z. Liang and L. Jiang, Adv. Eng. Mater., 2020, 22, 1901088 CrossRef CAS.
- J. Macak, H. Tsuchiya, A. Ghicov, K. Yasuda, R. Hahn, S. Bauer and P. Schmuki, Curr. Opin. Solid State Mater. Sci., 2007, 11, 3–18 CrossRef CAS.
- J. Macak, H. Tsuchiya and P. Schmuki, Angew. Chem., Int. Ed., 2005, 44, 2100–2102 CrossRef CAS PubMed.
- B. Yin, Q. Qian, Z. Xiong, H. Jiang, Y. Lin and D. Feng, Nanotechnology, 2019, 30, 155702 CrossRef CAS PubMed.
- N. Liu, X. Chen, J. Zhang and J. Schwank, Catal. Today, 2014, 225, 34–51 CrossRef CAS.
- S. Perera, R. Mariano, K. Vu, N. Nour, O. Seitz, Y. Chabal and K. Balkus, ACS Catal., 2012, 2, 949–956 CrossRef CAS.
- G. Yang, B. Yang, T. Xiao and Z. Yan, Appl. Surf. Sci., 2013, 283, 402–410 CrossRef CAS.
- L. Yuan, S. Meng, Y. Zhou and Z. Yue, J. Mater. Chem. A, 2013, 1, 2552–2557 RSC.
- F. Zhuge, J. Qiu, X. Li, X. Gao, X. Gan and W. Yu, Adv. Mater., 2011, 23, 1330–1334 CrossRef CAS PubMed.
- J. Zhang, Y. Kusumawati and T. Pauporté, Electrochim. Acta, 2016, 201, 125–133 CrossRef CAS.
- H. Ali-Loytty, M. Hannula, J. Saari, L. Palmolahti, B. Bhuskute, R. Ulkuniemi, T. Nyyssonen, K. Lahtonen and M. Valden, ACS Appl. Mater. Interfaces, 2019, 11, 2758–2762 CrossRef CAS PubMed.
- M. Liu, L. Zheng, X. Bao, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai, B. Huang and Z. Zheng, Chem. Eng. J., 2021, 405, 126654 CrossRef CAS.
- A. Yakovlev, V. Milichko, E. Pidko, V. Vinogradov and A. Vinogradov, Sci. Rep., 2016, 6, 37090 CrossRef CAS PubMed.
- M. Paik, Y. Lee, H. Yun, S. Lee, S. Hong and S. Seok, Adv. Energy Mater., 2020, 10, 2001799 CrossRef CAS.
- C. Wang, X. Kang, J. Liu, D. Wang, N. Wang, J. Chen, J. Wang, C. Tian and H. Fu, Inorg. Chem. Front., 2023, 10, 1153–1163 RSC.
- J. Kim, D. Suh, C. Kim, Y. Baek, B. Lee, H. Kim, J.-C. Lee and J. Yoon, Desalination, 2016, 397, 157–164 CrossRef CAS.
- C.-Y. Lee, A. Taylor, S. Beirne and G. Wallace, Adv. Energy Mater., 2017, 7, 1701060 CrossRef.
- A. Mazare, J. Park, S. Simons, S. Mohajernia, I. Hwang, J. E. Yoo, H. Schneider, M. Fischer and P. Schmuki, Acta Biomater., 2019, 97, 681–688 CrossRef CAS PubMed.
- H. Pan, M. Sun, X. Wang, M. Zhang, M. Murugananthan and Y. Zhang, Appl. Catal., B, 2022, 307, 121174 CrossRef CAS.
- W. Li, A. Elzatahry, D. Aldhayan and D. Zhao, Chem. Soc. Rev., 2018, 47, 8203–8237 RSC.
- A. Gromov, N. Kouznetsova, S. Yudina and V. Lunin, J. Alloys Compd., 1997, 261, 269–272 CrossRef CAS.
- R. David, Y. Finkelstein, E. Grinberg, S. Samuha, E. Rabkin and D. Cohen, Int. J. Hydrogen Energy, 2020, 45, 25043–25053 CrossRef.
- K. Kato, Y. Xin, J. Hong, K. Katsumata and T. Shirai, Adv. Powder Technol., 2020, 31, 1777–1783 CrossRef CAS.
- J. Gu, J. Aguiar, S. Ferrere, K. Steirer, Y. Yan, C. Xiao, J. Young, M. Al-Jassim, N. Neale and J. Turner, Nat. Energy, 2017, 2(2), 1–8 Search PubMed.
- J. Seo, S. Jeon, S. Lee, D. Lim, J. Kim, J. Kim, S. Ahn and W. Jung, ACS Catal., 2022, 12, 8593–8600 CrossRef CAS.
- J. Huang, S. Yang, X. Tang, L. Yang, W. Chen, Z. Chen, X. Li, Z. Zeng, Z. Tang and X. Gui, Adv. Mater., 2023, e2303737 CrossRef PubMed.
- Y. Xu, S. Chen, X. Chang, X. Wang, G. Sun, Z. Lu, Z. Zhao, C. Pei and J. Gong, ACS Catal., 2023, 13, 6104–6113 CrossRef CAS.
- J. Zhou, G. Li, H. Li, Z. Ma, S. Liu and X. Tang, Fuel, 2023, 343, 128014 CrossRef CAS.
- S. Chen, Y. Xu, X. Chang, Y. Pan, G. Sun, X. Wang, D. Fu, C. Pei, Z. Zhao, D. Su and J. Gong, Science, 2024, 385, 295–300 CrossRef PubMed.
- M. Monai, K. Jenkinson, A. Melcherts, J. Louwen, E. Irmak, S. Van Aert, T. Altantzis, C. Vogt, W. Van der Stam, T. Ducho
![[n with combining breve]](https://www.rsc.org/images/entities/char_006e_0306.gif) , B. Šmíd, E. Groeneveld, P. Berben, S. Bals and B. Weckhuysen, Science, 2023, 380, 644–651 CrossRef CAS PubMed.
, B. Šmíd, E. Groeneveld, P. Berben, S. Bals and B. Weckhuysen, Science, 2023, 380, 644–651 CrossRef CAS PubMed. - J. Li, E.-H. Wu, J. Hou, P. Huang, Z. Xu, Y. Jiang, Q.-S. Liu and Y.-Q. Zhong, RSC Adv., 2020, 10, 34775–34780 RSC.
- N. Denisov, S. Qin, G. Cha, J. Yoo and P. Schmuki, J. Electroanal. Chem., 2020, 872, 114098 CrossRef CAS.
- Y. Tong, S. Fang, C. Wang and Y. Hu, J. Phys. Chem. Solids, 2021, 154, 110053 CrossRef CAS.
- J. Li, C.-H. Liu, X. Li, Z.-Q. Wang, Y.-C. Shao, S.-D. Wang, X.-L. Sun, W.-F. Pong, J.-H. Guo and T.-K. Sham, Chem. Mater., 2016, 28, 4467–4475 CrossRef CAS.
- Y. Li, Z. Wang, Y. Wang, A. Kovács, C. Foo, R. Dunin-Borkowski, Y. Lu, R. Taylor, C. Wu and E. Tsang, Energy Environ. Sci., 2022, 15, 265–277 RSC.
- J. Lim, Y. Yang and M. Hoffmann, Environ. Sci. Technol., 2019, 53, 6972–6980 CrossRef CAS PubMed.
- E. Khorashadizade, S. Mohajernia, S. Hejazi, H. Mehdipour, N. Naseri, O. Moradlou, A. Moshfegh and P. Schmuki, J. Phys. Chem. C, 2021, 125, 6116–6127 CrossRef CAS.
- N. Liu, C. Schneider, D. Freitag, M. Hartmann, U. Venkatesan, J. Muller, E. Spiecker and P. Schmuki, Nano Lett., 2014, 14, 3309–3313 CrossRef CAS PubMed.
- M. Plodinec, I. Grčić, M. Willinger, A. Hammud, X. Huang, I. Panžić and A. Gajović, J. Alloys Compd., 2019, 776, 883–896 CrossRef CAS.
- Y. Yang, L. Kao, Y. Liu, K. Sun, H. Yu, J. Guo, S. Liou and M. Hoffmann, ACS Catal., 2018, 8, 4278–4287 CrossRef CAS PubMed.
- S. Mohajernia, S. Hejazi, A. Mazare, N. Nguyen, I. Hwang, S. Kment, G. Zoppellaro, O. Tomanec, R. Zboril and P. Schmuki, Mater. Today Energy, 2017, 6, 46–52 CrossRef.
- M. Motola, M. Čaplovičová, M. Krbal, H. Sopha, G. Thirunavukkarasu, M. Gregor, G. Plesch and J. Macak, Electrochim. Acta, 2020, 331, 135374 CrossRef CAS.
- H. Brahmi, R. Neupane, L. Xie, S. Singh, M. Yarali, G. Katwal, S. Chen, M. Paulose and A. Mavrokefalos, Nanoscale, 2018, 10, 3863–3870 RSC.
- S. Wang, Z. Zhang, W. Huo, X. Zhang, F. Fang, Z. Xie and J. Jiang, Chem. Eng. J., 2021, 403, 126331 CrossRef CAS.
- E. Lee, C. Park, D. W. Lee, G. Lee, H.-Y. Park, J. Jang, H.-J. Kim, Y.-E. Sung, Y. Tak and S. J. Yoo, ACS Catal., 2020, 10, 12080–12090 CrossRef CAS.
- Y. Xu, M. Melia, L.-K. Tsui, J. Fitz-Gerald and G. Zangari, J. Phys. Chem. C, 2017, 121, 17121–17128 CrossRef CAS.
- H. Li, Z. Chen, C. Tsang, Z. Li, X. Ran, C. Lee, B. Nie, L. Zheng, T. Hung, J. Lu, B. Pan and Y. Li, J. Mater. Chem. A, 2014, 2, 229–236 RSC.
- X. Hou, S. Jiang and Y. Li, Appl. Catal., B, 2019, 258, 117949 CrossRef CAS.
- H. Zhu, M. Zhao, J. Zhou, W. Li, H. Wang, Z. Xu, L. Lu, L. Pei, Z. Shi, S. Yan, Z. Li and Z. Zou, Appl. Catal., B, 2018, 234, 100–108 CrossRef CAS.
- Z. Li, Y. Ding, W. Kang, C. Li, D. Lin, X. Wang, Z. Chen, M. Wu and D. Pan, Electrochim. Acta, 2015, 161, 40–47 CrossRef CAS.
- N. Peighambardoust, S. Khameneh Asl, R. Mohammadpour and S. Asl, Electrochim. Acta, 2018, 270, 245–255 CrossRef CAS.
- Y. Ma, N. Wang, J. Chen, C. Chen, H. San, J. Chen and Z. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 22174–22181 CrossRef CAS PubMed.
- C. Xu, Y. Song, L. Lu, C. Cheng, D. Liu, X. Fang, X. Chen, X. Zhu and D. Li, Nanoscale Res. Lett., 2013, 8, 1–7 CrossRef PubMed.
- Y. Zhang, X. Chen, S. Zhang, L. Yin and Y. Yang, Chem. Eng. J., 2020, 401, 126033 CrossRef CAS.
- Y. Cheng, J. Gao, Q. Shi, Z. Li and W. Huang, J. Alloys Compd., 2022, 901, 163562 CrossRef CAS.
- Z. Zhang, M. Hedhili, H. Zhu and P. Wang, Phys. Chem. Chem. Phys., 2013, 15, 15637 RSC.
- K. Du, G. Liu, M. Li, C. Wu, X. Chen and K. Wang, Electrochim. Acta, 2016, 210, 367–374 CrossRef CAS.
- K. Du, G. Liu, X. Chen and K. Wang, Electrochim. Acta, 2018, 277, 244–254 CrossRef CAS.
- Y. Yang, J. Liao, Y. Li, X. Cao, N. Li, C. Wang and S. Lin, RSC Adv., 2016, 6, 46871–46878 RSC.
- Y. Yang and M. Hoffmann, Environ. Sci. Technol., 2016, 50, 11888–11894 CrossRef CAS PubMed.
- C. Kim, S. Kim, S. Hong, J. Lee and J. Yoon, Phys. Chem. Chem. Phys., 2016, 18, 14370–14375 RSC.
- P. Li, Z. Jin, Z. Fang and G. Yu, Angew. Chem., Int. Ed., 2020, 59, 22610–22616 CrossRef CAS PubMed.
- D. Yu, Y. Zhang, F. Wang and J. Dai, RSC Adv., 2021, 11, 2307–2314 RSC.
- L. Zhu, H. Ma, H. Han, Y. Fu, C. Ma, Z. Yu and X. Dong, RSC Adv., 2018, 8, 18992–19000 RSC.
- X. Hou, L. Fan, Y. Zhao, P. Lund and Y. Li, J. Power Sources, 2022, 524, 231095 CrossRef CAS.
- M. Shang, H. Hu, G. Lu and Y. Bi, J. Mater. Chem. A, 2016, 4, 5849–5853 RSC.
- H. Cui, W. Zhao, C. Yang, H. Yin, T. Lin, Y. Shan, Y. Xie, H. Gu and F. Huang, J. Mater. Chem. A, 2014, 2, 8612–8616 RSC.
- A. Sinhamahapatra, J.-P. Jeon and J.-S. Yu, Energy Environ. Sci., 2015, 8, 3539–3544 RSC.
- L. Hu, Y. Li, W. Zheng, Y.-K. Peng, E. Tsang, L. Lee and K.-Y. Wong, J. Mater. Chem. A, 2020, 8, 22828–22839 RSC.
- M. Ye, J. Jia, Z. Wu, C. Qian, R. Chen, P. O'Brien, W. Sun, Y. Dong and G. Ozin, Adv. Energy Mater., 2017, 7, 1601811 CrossRef.
- C. Yang, Z. Wang, T. Lin, H. Yin, X. Lu, D. Wan, T. Xu, C. Zheng, J. Lin, F. Huang, X. Xie and M. Jiang, J. Am. Chem. Soc., 2013, 135, 17831–17838 CrossRef CAS PubMed.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin, X. Xie and M. Jiang, Energy Environ. Sci., 2013, 6, 3007 RSC.
- J. Gao, Q. Shen, R. Guan, J. Xue, X. Liu, H. Jia, Q. Li and Y. Wu, J. CO2 Util., 2020, 35, 205–215 CrossRef CAS.
- W. Ou, J. Pan, Y. Liu, S. Li, H. Li, W. Zhao, J. Wang, C. Song, Y. Zheng and C. Li, J. Energy Chem., 2020, 43, 188–194 CrossRef.
- Y. Cao, Z. Xing, Z. Li, X. Wu, M. Hu, X. Yan, Q. Zhu, S. Yang and W. Zhou, J. Hazard. Mater., 2018, 343, 181–190 CrossRef CAS PubMed.
- J. Cai, Y. Zhu, D. Liu, M. Meng, Z. Hu and Z. Jiang, ACS Catal., 2015, 5, 1708–1716 CrossRef CAS.
- Z.-W. Zhou, L. Pan, Y.-T. Liu, X.-D. Zhu and X.-M. Xie, Chem. Commun., 2018, 54, 4790–4793 RSC.
- X. Zhang, C. Wang, J. Chen, W. Zhu, A. Liao, Y. Li, J. Wang and L. Ma, ACS Appl. Mater. Interfaces, 2014, 6, 20625–20633 CrossRef CAS PubMed.
- L. Zheng, X. Ye, X. Deng, Y. Wang, Y. Zhao, X. Shi and H. Zheng, ACS Sustainable Chem. Eng., 2020, 8, 15906–15914 CrossRef CAS.
- J. Zhang, Y. Tian, T. Zhang, Z. Li, X. She, Y. Wu, Y. Wang and J. Wu, ChemCatChem, 2020, 12, 2760–2767 CrossRef CAS.
- N. Wan, Z. Xing, J. Kuang, Z. Li, J. Yin, Q. Zhu and W. Zhou, Appl. Surf. Sci., 2018, 457, 287–294 CrossRef CAS.
- X. Yan, Z. Xing, Y. Cao, M. Hu, Z. Li, X. Wu, Q. Zhu, S. Yang and W. Zhou, Appl. Catal., B, 2017, 219, 572–579 CrossRef CAS.
- J. Hou, T. Xu, Y. Ning, B. Huang, Y. Yang and Q. Wang, Spectrochim. Acta, Part A, 2021, 244, 118896 CrossRef CAS PubMed.
- X. Liu, P. Carvalho, M. Getz, T. Norby and A. Chatzitakis, J. Phys. Chem. C, 2019, 123, 21931–21940 CrossRef CAS.
- J. Jiang, Z. Xing, M. Li, Z. Li, X. Wu, M. Hu, J. Wan, N. Wang, A. Besov and W. Zhou, Ind. Eng. Chem. Res., 2017, 56, 7948–7956 CrossRef CAS.
- Z. Li, H. Bian, X. Xiao, J. Shen, C. Zhao, J. Lu and Y. Li, ACS Appl. Nano Mater., 2019, 2, 7372–7378 CrossRef CAS.
- Z. Li, H. Bian, Z. Xu, J. Lu and Y. Li, ACS Appl. Energy Mater., 2020, 3, 6087–6092 CrossRef CAS.
- S. Islam, A. Reed, S. Nagpure, N. Wanninayake, J. Browning, J. Strzalka, D. Kim and S. Rankin, Microporous Mesoporous Mater., 2018, 261, 35–43 CrossRef CAS.
- X. Liu, R. Hua, J. Niu, Z. Zhang and J. Zhang, J. Alloys Compd., 2021, 881, 160509 CrossRef CAS.
- F. Teng, M. Li, C. Gao, G. Zhang, P. Zhang, Y. Wang, L. Chen and E. Xie, Appl. Catal., B, 2014, 148–149, 339–343 CrossRef CAS.
- F. Bella, A. Munoz-Garcia, F. Colo, G. Meligrana, A. Lamberti, M. Destro, M. Pavone and C. Gerbaldi, ACS Omega, 2018, 3, 8440–8450 CrossRef CAS PubMed.
- M. Pylnev, W.-H. Chang and M.-S. Wong, Appl. Surf. Sci., 2018, 462, 285–290 CrossRef CAS.
- M. Pylnev and M.-S. Wong, J. Photochem. Photobiol., A, 2019, 378, 125–130 CrossRef CAS.
- F. Yu, C. Wang, Y. Li, H. Ma, R. Wang, Y. Liu, N. Suzuki, C. Terashima, B. Ohtani, T. Ochiai, A. Fujishima and X. Zhang, Adv. Sci., 2020, 7, 2000204 CrossRef CAS PubMed.
- H.-R. An, S. Park, J. Huh, H. Kim, Y.-C. Lee, Y. Lee, Y. Hong and H. Lee, Appl. Catal., B, 2017, 211, 126–136 CrossRef CAS.
- A. Lepcha, C. Maccato, A. Mettenbörger, T. Andreu, L. Mayrhofer, M. Walter, S. Olthof, T. Ruoko, A. Klein, M. Moseler, K. Meerholz, J. Morante, D. Barreca and S. Mathur, J. Phys. Chem. C, 2015, 119, 18835–18842 CrossRef CAS.
- S. Zhu, Z. Yu, L. Zhang and S. Watanabe, ACS Appl. Nano Mater., 2021, 4, 3940–3948 CrossRef CAS.
- C. Yuan, Y. Shen, C. Zhu, P. Zhu, F. Yang, J. Liu and C. An, ACS Sustainable Chem. Eng., 2022, 10, 10311–10317 CrossRef CAS.
- M. Ji, Y. Choa and Y. Lee, Ultrason. Sonochem., 2021, 74, 105557 CrossRef CAS PubMed.
- C. Fan, X. Fu, L. Shi, S. Yu, G. Qian and Z. Wang, J. Alloys Compd., 2017, 703, 96–102 CrossRef CAS.
- M. Shahrezaei, S. Hejazi, H. Kmentova, V. Sedajova, R. Zboril, A. Naldoni and S. Kment, ACS Appl. Mater. Interfaces, 2023, 15, 37976–37985 CrossRef CAS PubMed.
- C. Fan, C. Chen, J. Wang, X. Fu, Z. Ren, G. Qian and Z. Wang, Sci. Rep., 2015, 5, 11712 CrossRef PubMed.
- Y. Wang, M. Zu, X. Zhou, H. Lin, F. Peng and S. Zhang, Chem. Eng. J., 2020, 381, 122605 CrossRef CAS.
- X. Hou, Z. Li, L. Fan, J. Yuan, P. Lund and Y. Li, Chem. Eng. J., 2021, 425, 131415 CrossRef CAS.
- M. E. Aguirre, R. Zhou, A. Eugene, M. Guzman and M. Grela, Appl. Catal., B, 2017, 217, 485–493 CrossRef CAS.
- Y. Pang, Y. Li, G. Xu, Y. Hu, Z. Kou, Q. Feng, J. Lv, Y. Zhang, J. Wang and Y. Wu, Appl. Catal., B, 2019, 248, 255–263 CrossRef CAS.
- Z. Yan, W. Wang, L. Du, J. Zhu, D. Phillips and J. Xu, Appl. Catal., B, 2020, 275, 119151 CrossRef CAS.
- B. Sun, N. Lu, Y. Su, H. Yu, X. Meng and Z. Gao, Appl. Surf. Sci., 2017, 394, 479–487 CrossRef CAS.
- H. Pan, X. Wang, Z. Xiong, M. Sun, M. Murugananthan and Y. Zhang, Environ. Res., 2021, 198, 111176 CrossRef CAS PubMed.
- X. Hou, K. Aitola and P. Lund, Energy Sci. Eng., 2021, 9, 921–937 CrossRef CAS.
- M. Ge, Q. Li, C. Cao, J. Huang, S. Li, S. Zhang, Z. Chen, K. Zhang, S. Al-Deyab and Y. Lai, Adv. Sci., 2017, 4, 1600152 CrossRef PubMed.
- H. Hosono and M. Kitano, Chem. Rev., 2021, 121, 3121–3185 CrossRef CAS PubMed.
- S. Wu, H. Yu, S. Chen and X. Quan, ACS Catal., 2020, 10, 14380–14389 CrossRef CAS.
- Z. Chen, S. Wu, J. Ma, S. Mine, T. Toyao, M. Matsuoka, L. Wang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 11901–11909 CrossRef CAS PubMed.
- R. Lang, W. Xi, J. Liu, Y. Cui, T. Li, A. Lee, F. Chen, Y. Chen, L. Li, L. Li, J. Lin, S. Miao, X. Liu, A. Wang, X. Wang, J. Luo, B. Qiao, J. Li and T. Zhang, Nat. Commun., 2019, 10, 234 CrossRef PubMed.
- Y. Tang, C. Asokan, M. Xu, G. Graham, X. Pan, P. Christopher, J. Li and P. Sautet, Nat. Commun., 2019, 10, 4488 CrossRef PubMed.
- Z. Xue, M. Yan, Y. Zhang, J. Xu, X. Gao and Y. Wu, Appl. Catal., B, 2023, 325, 122303 CrossRef CAS.
- S. Wu, Z. Chen, W. Yue, S. Mine, T. Toyao, M. Matsuoka, X. Xi, L. Wang and J. Zhang, ACS Catal., 2021, 11, 4362–4371 CrossRef CAS.
- L. Chen, R. Unocic, A. Hoffman, J. Hong, A. Braga, Z. Bao, S. Bare and J. Szanyi, JACS Au, 2021, 1, 977–986 CrossRef CAS PubMed.
- C. Liu, J. Qian, Y. Ye, H. Zhou, C.-J. Sun, C. Sheehan, Z. Zhang, G. Wan, Y.-S. Liu, J. Guo, S. Li, H. Shin, S. Hwang, T. Gunnoe, W. Goddard and S. Zhang, Nat. Catal., 2020, 4, 36–45 CrossRef.
- Y. Zhou, Z. Xie, J. Jiang, J. Wang, X. Song, Q. He, W. Ding and Z. Wei, Nat. Catal., 2020, 3, 454–462 CrossRef CAS.
- B. Lee, S. Park, M. Kim, A. Sinha, S. Lee, E. Jung, W. Chang, K. Lee, J. Kim, S. Cho, H. Kim, K. Nam and T. Hyeon, Nat. Mater., 2019, 18, 620–626 CrossRef CAS PubMed.
- Y. Yao, L. Zhao, J. Dai, J. Wang, C. Fang, G. Zhan, Q. Zheng, W. Hou and L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208215 CrossRef CAS PubMed.
- B. Sarma, F. Maurer, D. Doronkin and J. Grunwaldt, Chem. Rev., 2023, 123, 379–444 CrossRef CAS PubMed.
- F. Abdelghafar, X. Xu, S. Jiang and Z. Shao, Mater. Rep.: Energy, 2022, 2, 100144 CAS.
- L. Kuai, Z. Chen, S. Liu, E. Kan, N. Yu, Y. Ren, C. Fang, X. Li, Y. Li and B. Geng, Nat. Commun., 2020, 11, 48 CrossRef PubMed.
- J. Liu, J. Wan, L. Liu, W. Yang, J. Low, X. Gao and F. Fu, Chem. Eng. J., 2022, 430, 133125 CrossRef CAS.
- Q. Zhang, Y. Li, M. Geng, J. Zhu, H. Sun and B. Jiang, Appl. Catal., B, 2023, 330, 122658 CrossRef CAS.
- M. Ismael, Sol. Energy, 2020, 211, 522–546 CrossRef CAS.
- P. Kodithuwakku, D. Jayasundara, I. Munaweera, R. Jayasinghe, T. Thoradeniya, M. Weerasekera, P. Ajayan and N. Kottegoda, Prog. Solid State Chem., 2022, 67, 100369 CrossRef CAS.
- A. Soleimani-Gorgani, H. Al-Hazmi, A. Esmaeili and S. Habibzadeh, Environ. Res., 2023, 237, 117079 CrossRef CAS PubMed.
- E. Cerrato, E. Gaggero, P. Calza and M. Paganini, Chem. Eng. J. Adv., 2022, 10, 100268 CrossRef CAS.
- M. Dozzi and E. Selli, J. Photochem. Photobiol., C, 2013, 14, 13–28 CrossRef CAS.
- S. Muthukrishnan, R. Vidya and A. Sjåstad, Mater. Chem. Phys., 2023, 299, 127467 CrossRef CAS.
- T. Pawar, D. Contreras López, J. Olivares Romero and J. Vallejo Montesinos, J. Mater. Sci., 2023, 58, 6887–6930 CrossRef CAS.
- M. Coto, G. Divitini, A. Dey, S. Krishnamurthy, N. Ullah, C. Ducati and R. V. Kumar, Mater. Today Chem., 2017, 4, 142–149 CrossRef.
- W. Lee, C. Lee, G. Cha, B. Lee, J. Jeong, H. Park, J. Heo, M. Bootharaju, S. Sunwoo, J. Kim, K. Ahn, D. Kim and T. Hyeon, Nat. Nanotechnol., 2023, 18, 754–762 CrossRef CAS PubMed.
- D. Hasina, M. Saini, M. Kumar, A. Mandal, N. Basu, P. Maiti, S. Srivastava and T. Som, Small, 2023, e2305605 Search PubMed.
- B. He, Z. Wang, P. Xiao, T. Chen, J. Yu and L. Zhang, Adv. Mater., 2022, 34, e2203225 CrossRef PubMed.
- X. Zhang, Y. Zeng, W. Shi, Z. Tao, J. Liao, C. Ai, H. Si, Z. Wang, A. Fisher and S. Lin, Chem. Eng. J., 2022, 429, 131312 CrossRef CAS.
- M. Rajeswari, K. Vanasundari, G. Mahalakshmi, P. Ponnarasi, M. Parthibavarman, M. Shkir and I. Ashraf, Chem. Phys. Lett., 2022, 805, 139936 CrossRef CAS.
- S. Vignesh, S. Chandrasekaran, M. Srinivasan, R. Anbarasan, R. Perumalsamy, E. Arumugam, M. Shkir, H. Algarni and S. AlFaify, Chemosphere, 2022, 288, 132611 CrossRef CAS PubMed.
- L. Chen, X.-L. Song, J.-T. Ren and Z.-Y. Yuan, Appl. Catal., B, 2022, 315, 121546 CrossRef CAS.
- M. Mousavi, M. Soleimani, M. Hamzehloo, A. Badiei and J. Ghasemi, Mater. Chem. Phys., 2021, 258, 123912 CrossRef CAS.
- Y.-P. Zhang, W. Han, Y. Yang, H. Zhang, Y. Wang, L. Wang, X. Sun and F. Zhang, Chem. Eng. J., 2022, 446, 137213 CrossRef CAS.
- T. Nguyen, J. Li, E. Lizundia, M. Niederberger, W. Hamad and M. MacLachlan, Adv. Funct. Mater., 2019, 29, 1904639 CrossRef.
- Y. Xue, F. Wang, H. Luo and J. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 34355–34363 CrossRef CAS PubMed.
- Z. Xiong, Z. Lei, Y. Li, L. Dong, Y. Zhao and J. Zhang, J. Photochem. Photobiol., C, 2018, 36, 24–47 CrossRef CAS.
- Y. Yamazaki, T. Toyonaga, N. Doshita, K. Mori, Y. Kuwahara, S. Yamazaki and H. Yamashita, ACS Appl. Mater. Interfaces, 2022, 14, 2291–2300 CrossRef CAS PubMed.
- P. Bousoulas, P. Asenov, I. Karageorgiou, D. Sakellaropoulos, S. Stathopoulos and D. Tsoukalas, J. Appl. Phys., 2016, 120, 154501 CrossRef.
- C. Wen, H. Jiang, S. Qiao, H. Yang and G. Lu, J. Mater. Chem., 2011, 21, 7052–7061 RSC.
- D. Wang, T. Sheng, J. Chen, H.-F. Wang and P. Hu, Nat. Catal., 2018, 1, 291–299 CrossRef CAS.
- S. Lu, B. Weng, A. Chen, X. Li, H. Huang, X. Sun, W. Feng, Y. Lei, Q. Qian and M. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 13044–13054 CrossRef CAS PubMed.
- J. DuChene, G. Tagliabue, A. Welch, W. Cheng and H. Atwater, Nano Lett., 2018, 18, 2545–2550 CrossRef CAS PubMed.
- L. Putri, B. Ng, W. Ong, S. Chai and A. Mohamed, Adv. Energy Mater., 2022, 12, 2201093 CrossRef CAS.
- X. Du, Y. Huang, X. Pan, B. Han, Y. Su, Q. Jiang, M. Li, H. Tang, G. Li and B. Qiao, Nat. Commun., 2020, 11, 5811 CrossRef CAS PubMed.
- Y. Zhang, J. Liu, K. Qian, A. Jia, D. Li, L. Shi, J. Hu, J. Zhu and W. Huang, Angew. Chem., Int. Ed., 2021, 60, 12074–12081 CrossRef CAS PubMed.
- S. Wang, G. Liu and L. Wang, Chem. Rev., 2019, 119, 5192–5247 CrossRef CAS PubMed.
- M. Malizia, B. Seger, I. Chorkendorff and P. Vesborg, J. Mater. Chem. A, 2014, 2, 6847–6853 RSC.
- Z. Tian, Y. Da, M. Wang, X. Dou, X. Cui, J. Chen, R. Jiang, S. Xi, B. Cui, Y. Luo, H. Yang, Y. Long, Y. Xiao and W. Chen, Nat. Commun., 2023, 14, 142 CrossRef CAS PubMed.
- G. Ren, M. Shi, S. Liu, Z. Li, Z. Zhang and X. Meng, Chem. Eng. J., 2023, 454, 140158 CrossRef CAS.
- Y. Guo, Y. Huang, B. Zeng, B. Han, M. Akri, M. Shi, Y. Zhao, Q. Li, Y. Su, L. Li, Q. Jiang, Y. Cui, L. Li, R. Li, B. Qiao and T. Zhang, Nat. Commun., 2022, 13, 2648 CrossRef CAS PubMed.
- K. Fujiwara and S. Pratsinis, Appl. Catal., B, 2018, 226, 127–134 CrossRef CAS.
- J. Chen, Y. Kang, W. Zhang, Z. Zhang, Y. Chen, Y. Yang, L. Duan, Y. Li and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202203022 CrossRef CAS PubMed.
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1705369 CrossRef PubMed.
- K. Selvakumar, T. Oh, Y. Wang, M. Arunpandian and M. Swaminathan, Chemosphere, 2023, 314, 137694 CrossRef CAS PubMed.
- D. Prashad Ojha, M. Babu Poudel and H. Joo Kim, Mater. Lett., 2020, 264, 127363 CrossRef CAS.
- L. Zheng, H. Su, J. Zhang, L. Walekar, H. Vafaei Molamahmood, B. Zhou, M. Long and Y. Hu, Appl. Catal., B, 2018, 239, 475–484 CrossRef CAS.
- S. Parmar, T. Das, B. Ray, B. Debnath, S. Gosavi, G. Shanker, S. Datar, S. Chakraborty and S. Ogale, Adv. Energy Sustainability Res., 2021, 3, 2100137 CrossRef.
- Z. Wu, P. Yang, Q. Li, W. Xiao, Z. Li, G. Xu, F. Liu, B. Jia, T. Ma, S. Feng and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300406 CrossRef CAS PubMed.
- Z. Huang, Q. Sun, K. Lv, Z. Zhang, M. Li and B. Li, Appl. Catal., B, 2015, 164, 420–427 CrossRef CAS.
- Z. Li, H. Li, S. Wang, F. Yang and W. Zhou, Chem. Eng. J., 2022, 427, 131830 CrossRef CAS.
- A. Hezam, K. Alkanad, M. A. Bajiri, J. Strunk, K. Takahashi, Q. Drmosh, N. Al-Zaqri and L. Krishnappagowda, Small Methods, 2023, 7, e2201103 CrossRef PubMed.
- D.-H. Yoon, M. Biswas and A. Sakthisabarimoorthi, Diamond Relat. Mater., 2022, 129, 109363 CrossRef CAS.
- C. Qiang, N. Li, S. Zuo, Z. Guo, W. Zhan, Z. Li and J. Ma, Sep. Purif. Technol., 2022, 283, 120214 CrossRef CAS.
- J. Zhou, M. Guo, L. Wang, Y. Ding, Z. Zhang, Y. Tang, C. Liu and S. Luo, Chem. Eng. J., 2019, 366, 163–171 CrossRef CAS.
- J. Pan, Z. Dong, B. Wang, Z. Jiang, C. Zhao, J. Wang, C. Song, Y. Zheng and C. Li, Appl. Catal., B, 2019, 242, 92–99 CrossRef CAS.
- J. Lin, W. Ren, A. Li, C. Yao, T. Chen, X. Ma, X. Wang and A. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 4204–4211 CrossRef CAS PubMed.
- R. Nawaz, S. Haider, H. Ullah, M. Akhtar, S. Khan, M. Junaid and N. Khan, J. Environ. Chem. Eng., 2022, 10, 106968 CrossRef CAS.
- X. Shi, Z. Liu, X. Li, W. You, Z. Shao and R. Che, Chem. Eng. J., 2021, 419, 130020 CrossRef CAS.
- V. Navakoteswara Rao, N. Lakshmana Reddy, M. Mamatha Kumari, P. Ravi, M. Sathish, K. Kuruvilla, V. Preethi, K. Reddy, N. Shetti, T. Aminabhavi and M. Shankar, Appl. Catal., B, 2019, 254, 174–185 CrossRef CAS.
- X. Lu, A. Chen, Y. Luo, P. Lu, Y. Dai, E. Enriquez, P. Dowden, H. Xu, P. Kotula, A. Azad, D. Yarotski, R. Prasankumar, A. Taylor, J. Thompson and Q. Jia, Nano Lett., 2016, 16, 5751–5755 CrossRef CAS PubMed.
- K. Kato, Y. Xin, S. Vaucher and T. Shirai, Scr. Mater., 2022, 208, 114358 CrossRef CAS.
- G. Liu, H. Yang, X. Wang, L. Cheng, H. Lu, L. Wang, G. Lu and H. Cheng, J. Phys. Chem. C, 2009, 113, 21784–21788 CrossRef CAS.
- A. Wang, S. Wu, J. Dong, R. Wang, J. Wang, J. Zhang, S. Zhong and S. Bai, Chem. Eng. J., 2021, 404, 127145 CrossRef CAS.
- Q. Shi, Y. Li, Y. Zhou, S. Miao, N. Ta, E. Zhan, J. Liu and W. Shen, J. Mater. Chem. A, 2015, 3, 14409–14415 RSC.
- W. Zhang, J. Xue, Q. Shen, S. Jia, J. Gao, X. Liu and H. Jia, J. Alloys Compd., 2021, 870, 159400 CrossRef CAS.
- F. Ullah, R. Bashiri, N. Muti Mohamed, A. Zaleska-Medynska, C. Kait, U. Ghani, M. Shahid and M. Saheed, Appl. Surf. Sci., 2022, 576, 151871 CrossRef CAS.
- B. Kakavandi, E. Dehghanifard, P. Gholami, M. Noorisepehr and B. MirzaHedayat, Appl. Surf. Sci., 2021, 570, 151145 CrossRef CAS.
- C. Wang, J. Li, E. Paineau, H. Remita and M. Ghazzal, Sol. RRL, 2023, 7, 2200929 CrossRef CAS.
- S. Lee, H. Bae and W. Choi, Acc. Chem. Res., 2023, 56, 867–877 CrossRef CAS PubMed.
- C. Luo, X. Ren, Z. Dai, Y. Zhang, X. Qi and C. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 23265–23286 CrossRef CAS PubMed.
- C. Foo, Y. Li, K. Lebedev, T. Chen, S. Day, C. Tang and E. Tsang, Nat. Commun., 2021, 12, 661 CrossRef CAS PubMed.
- C. Holder and R. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed.
- A. Arif, R. Balgis, T. Ogi, F. Iskandar, A. Kinoshita, K. Nakamura and K. Okuyama, Sci. Rep., 2017, 7, 3646 CrossRef PubMed.
- L. Guo, Y. Jing and B. Chaplin, Environ. Sci. Technol., 2016, 50, 1428–1436 CrossRef CAS PubMed.
- Z. Wu, P. Yang, Q. Li, W. Xiao, Z. Li, G. Xu, F. Liu, B. Jia, T. Ma, S. Feng and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300406 CrossRef CAS PubMed.
- A. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190–200 CrossRef CAS PubMed.
- Z. Chen, S. Mo, H. Lin, Z. Wu, Y. Zhao, X. Hua and P. Zhao, Cell Rep. Phys. Sci., 2023, 101681 CrossRef.
- M. Terban and S. Billinge, Chem. Rev., 2022, 122, 1208–1272 CrossRef CAS PubMed.
- S. Billinge, Philos. Trans. R. Soc., A, 2019, 377, 20180413 CrossRef CAS PubMed.
- S. Zhang, J. Gong, D. Xiao, B. Jayan and A. McGaughey, Comput. Mater. Sci., 2023, 218, 111964 CrossRef CAS.
- X. Hua, Z. Liu, M. Fischer, O. Borkiewicz, P. Chupas, K. Chapman, U. Steiner, P. Bruce and C. Grey, J. Am. Chem. Soc., 2017, 139, 13330–13341 CrossRef CAS PubMed.
- X. Hua, Z. Liu, P. G. Bruce and C. P. Grey, J. Am. Chem. Soc., 2015, 137, 13612–13623 CrossRef CAS PubMed.
- T.-D. Nguyen-Phan, Z. Liu, S. Luo, A. Gamalski, D. Vovchok, W. Xu, E. Stach, D. Polyansky, E. Fujita, J. Rodriguez and S. Senanayake, J. Phys. Chem. C, 2016, 120, 3472–3482 CrossRef CAS.
- F. Haase, A. Bergmann, T. Jones, J. Timoshenko, A. Herzog, H. Jeon, C. Rettenmaier and B. Cuenya, Nat. Energy, 2022, 7, 765–773 CrossRef CAS.
- X. Lv, X. Li, C. Yang, X. Ding, Y. Zhang, Y. Z. Zheng, S. Li, X. Sun and X. Tao, Adv. Funct. Mater., 2020, 30, 1910830 CrossRef CAS.
- Y. Li, S. Wang, Y. Dong, P. Mu, Y. Yang, X. Liu, C. Lin and Q. Huang, Bioact. Mater., 2020, 5, 1062–1070 Search PubMed.
- C. Roy, B. Sebok, S. Scott, E. Fiordaliso, J. Sørensen, A. Bodin, D. Trimarco, C. Damsgaard, P. Vesborg, O. Hansen, I. Stephens, J. Kibsgaard and I. Chorkendorff, Nat. Catal., 2018, 1, 820–829 CrossRef CAS.
- A. Grisafi, J. Nigam and M. Ceriotti, Chem. Sci., 2021, 12, 2078–2090 RSC.
- T.-T. Wei, F.-F. Wang, X.-Z. Li, J.-H. Zhang, Y.-R. Zhu and T.-F. Yi, Sustainable Mater. Technol., 2021, 30, e00355 CrossRef CAS.
- X. Zhang, W. Hu, K. Zhang, J. Wang, B. Sun, H. Li, P. Qiao, L. Wang and W. Zhou, ACS Sustainable Chem. Eng., 2017, 5, 6894–6901 CrossRef CAS.
- N. Dwivedi, R. Yeo, H. Tan, R. Stangl, A. Aberle, C. Bhatia, A. Danner and B. Liao, Adv. Funct. Mater., 2018, 28, 1707018 CrossRef.
- T. Mochizuki, K. Gotoh, Y. Kurokawa, T. Yamamoto and N. Usami, Adv. Mater. Interfaces, 2018, 6, 1801645 CrossRef.
- M. Barzilay, T. Qiu, A. Rappe and Y. Ivry, Adv. Funct. Mater., 2019, 30, 1902549 CrossRef.
- L. Zhou, Y. Shao, F. Yin, J. Li, F. Kang and R. Lv, Nat. Commun., 2023, 14, 7644 CrossRef CAS PubMed.
- M. Yuan, M. Kermanian, T. Agarwal, Z. Yang, S. Yousefiasl, Z. Cheng, P. Ma, J. Lin and A. Maleki, Adv. Mater., 2023, 35, e2304176 CrossRef PubMed.
- C. Christensen, M. Mamakhel, A. Balakrishna, B. Iversen, Y. Chiang and D. Ravnsbaek, Nanoscale, 2019, 11, 12347–12357 RSC.
- J. Gao, J. Xue, S. Jia, Q. Shen, X. Zhang, H. Jia, X. Liu, Q. Li and Y. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 18758–18771 CrossRef CAS PubMed.
- M. Yazdanpanah, M. Fereidooni, V. Marquez, C. Paz, T. Saelee, M. Salazar Villanueva, M. Rittiruam, P. Khajondetchairit, S. Praserthdam and P. Praserthdam, ChemSusChem, 2023, e202301033 Search PubMed.
- H. Chen, J. Li, W. Yang, S. Balaghi, C. Triana, C. Mavrokefalos and G. Patzke, ACS Catal., 2021, 11, 7637–7646 CrossRef CAS.
- J. Kang, Y. Zhang, Z. Chai, X. Qiu, X. Cao, P. Zhang, G. Teobaldi, L. Liu and L. Guo, Adv. Mater., 2021, 33, e2100407 CrossRef PubMed.
- M. Pisarek, M. Krawczyk, M. Holdynski and W. Lisowski, ACS Omega, 2020, 5, 8647–8658 CrossRef CAS PubMed.
- G. Jia, Y. Wang, X. Cui, H. Zhang, J. Zhao, L. H. Li, L. Gu, Q. Zhang, L. Zheng, J. Wu, Q. Wu, D. Singh, W. Li, L. Zhang and W. Zheng, Matter, 2022, 5, 206–218 CrossRef CAS.
- C. Chen, M. Wu, C. Yang, X. Yu, J. Yu, H. Yin, G. Li, G. Su, Z. Hao, M. Song and C. Ma, Cell Rep. Phys. Sci., 2022, 3, 100936 CrossRef CAS.
- S. Zhang, J. Gong, S. Chu, D. Xiao, B. Reeja-Jayan and A. McGaughey, APL Mach. Learn., 2023, 1, 026115 CrossRef.
- C. Mao, J. Wang, Y. Zou, G. Qi, J. Yang Loh, T. Zhang, M. Xia, J. Xu, F. Deng, M. Ghoussoub, N. Kherani, L. Wang, H. Shang, M. Li, J. Li, X. Liu, Z. Ai, G. Ozin, J. Zhao and L. Zhang, J. Am. Chem. Soc., 2020, 142, 17403–17412 CrossRef CAS PubMed.
- J. Chapman, K. Kweon, Y. Zhu, K. Bushick, L. Bayu Aji, C. Colla, H. Mason, N. Goldman, N. Keilbart, S. Qiu, T. Heo, J. Rodriguez and B. Wood, J. Mater. Chem. A, 2023, 11, 8670–8683 RSC.
- S. Selcuk, X. Zhao and A. Selloni, Nat. Mater., 2018, 17, 923–928 CrossRef CAS PubMed.
- J. Bae, D. Kim, H. Yoo, E. Park, Y. Lim, M. Park, Y. Kim and H. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 4541–4547 CrossRef CAS PubMed.
- C. Xue, R. Huang, R. Xue, Q. Chang, N. Li, J. Zhang, S. Hu and J. Yang, J. Alloys Compd., 2022, 909, 164843 CrossRef CAS.
- H. Sugiyama, T. Nakao, M. Miyazaki, H. Abe, Y. Niwa, M. Kitano and H. Hosono, ACS Catal., 2022, 12, 12572–12581 CrossRef CAS.
- R. Lakra, R. Kumar, P. Sahoo, D. Thatoi and A. Soam, Inorg. Chem. Commun., 2021, 133, 108929 CrossRef CAS.
- Y. Zhang, Y. Li, H. Yu, K. Yu and H. Yu, J. Mater. Sci. Technol., 2022, 106, 139–146 CrossRef CAS.
- R. Wang, X. Xue, W. Lu, H. Liu, C. Lai, K. Xi, Y. Che, J. Liu, S. Guo and D. Yang, Nanoscale, 2015, 7, 12833–12838 RSC.
- Y. Yang, P. Gao, Y. Wang, L. Sha, X. Ren, J. Zhang, Y. Chen, T. Wu, P. Yang and X. Li, Nano Energy, 2017, 33, 29–36 CrossRef CAS.
- Y. Yoshida, S. Tokashiki, K. Kubota, R. Shiratuchi, Y. Yamaguchi, M. Kono and S. Hayase, Sol. Energy Mater. Sol. Cells, 2008, 92, 646–650 CrossRef CAS.
- D. Liu, Q. Yu, S. Liu, K. Qian, S. Wang, W. Sun, X.-Q. Yang, F. Kang and B. Li, J. Phys. Chem. C, 2019, 123, 12797–12806 CrossRef CAS.
- G. Cha, S. Mohajernia, N. Nguyen, A. Mazare, N. Denisov, I. Hwang and P. Schmuki, Adv. Energy Mater., 2019, 10, 1903448 CrossRef.
- J. Balajka, M. Hines, W. DeBenedetti, M. Komora, J. Pavelec, M. Schmid and U. Diebold, Science, 2018, 361, 786–789 CrossRef CAS PubMed.
- D. Barauskas, M. Dzikaras, D. Bieliauskas, D. Pelenis, G. Vanagas and D. Viržonis, Sensors, 2020, 20, 3554 CrossRef CAS PubMed.
- W. Wang, T. Xu, J. Chen, J. Shangguan, H. Dong, H. Ma, Q. Zhang, J. Yang, T. Bai, Z. Guo, H. Fang, H. Zheng and L. Sun, Nat. Mater., 2022, 21, 859–863 CrossRef CAS PubMed.
- Y. Li, Y. Peng, L. Hu, J. Zheng, D. Prabhakaran, S. Wu, T. Puchtler, M. Li, K. Wong, R. Taylor and E. Tsang, Nat. Commun., 2019, 10, 4421 CrossRef PubMed.
- Y. Li, H. Zhou, S. Cai, D. Prabhakaran, W. Niu, A. Large, G. Held, R. Taylor, X. Wu and E. Tsang, Nat. Catal., 2024, 7, 77–88 CrossRef CAS.
- M. Xing, J. Zhang, B. Qiu, B. Tian, M. Anpo and M. Che, Small, 2015, 11, 1920–1929 CrossRef CAS PubMed.
- H. Wang, H. Qi, X. Sun, S. Jia, X. Li, T. Miao, L. Xiong, S. Wang, X. Zhang, X. Liu, A. Wang, T. Zhang, W. Huang and J. Tang, Nat. Mater., 2023, 22, 619–626 CrossRef CAS PubMed.
- C. Kim, S. Kim, J. Lee, J. Kim and J. Yoon, ACS Appl. Mater. Interfaces, 2015, 7, 7486–7491 CrossRef CAS PubMed.
- A. Tighineanu, T. Ruff, S. Albu, R. Hahn and P. Schmuki, Chem. Phys. Lett., 2010, 494, 260–263 CrossRef CAS.
- M. Sboui, W. Niu, G. Lu, K. Zhang and J. Pan, Chemosphere, 2023, 310, 136753 CrossRef CAS PubMed.
- J. Liu, J. Yan, Q. Shi, H. Dong, J. Zhang, Z. Wang, W. Huang, B. Chen and H. Zhang, J. Phys. Chem. C, 2019, 123, 4094–4102 CrossRef CAS.
- S. K. Kajli, D. Ray and S. Roy, Nanoscale Adv., 2021, 3, 432–445 RSC.
- H. Huang, X. Hou, J. Xiao, L. Zhao, Q. Huang, H. Chen and Y. Li, Catal. Today, 2019, 330, 189–194 CrossRef CAS.
- X. Guan, S. Shen and S. Mao, Cell Rep. Phys. Sci., 2023, 4, 101211 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Behera, A. Kar and R. Srivastava, Inorg. Chem., 2022, 61, 12781–12796 CrossRef CAS PubMed.
- F. Guzman, S. Chuang and C. Yang, Ind. Eng. Chem. Res., 2013, 52, 61–65 CrossRef CAS.
- Z. Haider and Y. Kang, ACS Appl. Mater. Interfaces, 2014, 6, 10342–10352 CrossRef CAS PubMed.
- R. Shi, H. Ye, F. Liang, Z. Wang, K. Li, Y. Weng, Z. Lin, W. Fu, C. Che and Y. Chen, Adv. Mater., 2018, 30, 1705941 CrossRef PubMed.
- W. Zhou, W. Li, J. Wang, Y. Qu, Y. Yang, Y. Xie, K. Zhang, L. Wang, H. Fu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 9280–9283 CrossRef CAS PubMed.
- B. Sun, W. Zhou, H. Li, L. Ren, P. Qiao, W. Li and H. Fu, Adv. Mater., 2018, 30, 1804282 CrossRef PubMed.
- S. Lin, H. Huang, T. Ma and Y. Zhang, Adv. Sci., 2020, 8, 2002458 CrossRef PubMed.
- N. Liu, S. Mohajernia, N. Nguyen, S. Hejazi, F. Plass, A. Kahnt, T. Yokosawa, A. Osvet, E. Spiecker, D. Guldi and P. Schmuki, ChemSusChem, 2020, 13, 4937–4944 CrossRef CAS.
- A. Juliya, V. Mujeeb, K. Sreenivasan and K. Muraleedharan, J. Photochem. Photobiol., 2021, 8, 100076 CrossRef.
- Q. Liu, D. Ding, C. Ning and X. Wang, Int. J. Hydrogen Energy, 2015, 40, 2107–2114 CrossRef.
- K. Shin and J. Park, ACS Appl. Mater. Interfaces, 2015, 7, 18429–18434 CrossRef CAS PubMed.
- S. Lin, H. Ren, Z. Wu, L. Sun, X.-G. Zhang, Y.-M. Lin, K. H. Zhang, C.-J. Lin, Z.-Q. Tian and J.-F. Li, J. Energy Chem., 2021, 59, 721–729 CrossRef CAS.
- F. Dingenen and S. Verbruggen, Renewable Sustainable Energy Rev., 2021, 142, 110866 CrossRef CAS.
- A. Shih, M. Monteiro, F. Dattila, D. Pavesi, M. Philips, A. da Silva, R. Vos, K. Ojha, S. Park, O. van der Heijden, G. Marcandalli, A. Goyal, M. Villalba, X. Chen, G. Gunasooriya, I. McCrum, R. Mom, N. López and M. Koper, Nat. Rev. Methods Primers, 2022, 2, 84 CrossRef CAS.
- H. Zhao and Z. Yuan, Adv. Energy Mater., 2023, 13, 2300254 CrossRef CAS.
- LENNECH, https://www.lenntech.com/applications, (accessed August 2024).
- X. Ren, J. Zhou, X. Qi, Y. Liu, Z. Huang, Z. Li, Y. Ge, S. Dhanabalan, J. Ponraj, S. Wang, J. Zhong and H. Zhang, Adv. Energy Mater., 2017, 7, 1700396 CrossRef.
- J. Hausmann, R. Schlögl, P. Menezes and M. Driess, Energy Environ. Sci., 2021, 14, 3679–3685 RSC.
- M. Yesupatham, A. Augustin, N. Agamendran, B. Honnappa, M. Shanmugam, P. Sagayaraj, G. Thennarasu, N. Sagaya Selvam and K. Sekar, Sustainable Energy Fuels, 2023, 7, 4727–4757 RSC.
- S. Lv, Y. Wang, Y. Zhou, Q. Liu, C. Song and D. Wang, J. Alloys Compd., 2021, 868, 159144 CrossRef CAS.
- S. Xiao, S. Wu, L. Wu, Y. Dong, J. Liu, L. Wang, X. Chen, Y. Wang, G. Tian, G. Chang, M. Shalom, P. Fornasiero and X. Yang, ACS Nano, 2023, 17, 18217–18226 CrossRef CAS PubMed.
- M. Khan, T. Al-Attas, S. Roy, M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. Ajayan and M. Kibria, Energy Environ. Sci., 2021, 14, 4831–4839 RSC.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- Z. Zhu, H. Zheng, H. Kong, X. Ma and J. Xiong, Nat. Water, 2023, 1, 790–799 CrossRef.
- T. Suguro, F. Kishimoto, N. Kariya, T. Fukui, M. Nakabayashi, N. Shibata, T. Takata, K. Domen and K. Takanabe, Nat. Commun., 2022, 13, 5698 CrossRef CAS PubMed.
- H. Xie, Z. Zhao, T. Liu, Y. Wu, C. Lan, W. Jiang, L. Zhu, Y. Wang, D. Yang and Z. Shao, Nature, 2022, 612, 673–678 CrossRef CAS PubMed.
- J. Liu, Y. Zou, B. Jin, K. Zhang and J. Park, ACS Energy Lett., 2019, 4, 3018–3027 CrossRef CAS.
- J. Tang, T. Zhao, D. Solanki, X. Miao, W. Zhou and S. Hu, Joule, 2021, 5, 1432–1461 CrossRef CAS.
- J. Zhang, W. Hu, S. Cao and L. Piao, Nano Res., 2020, 13, 2313–2322 CrossRef CAS.
- J. Zhang, Y. Lei, S. Cao, W. Hu, L. Piao and X. Chen, Nano Res., 2021, 15, 2013–2022 CrossRef.
- Y. Hong, Y. Cho, E. Go, P. Sharma, H. Cho, B. Lee, S. Lee, S. Park, M. Ko, S. Kwak, C. Yang and J.-W. Jang, Chem. Eng. J., 2021, 418, 129346 CrossRef CAS.
- Y. Yang, B. Zhu, L. Wang, B. Cheng, L. Zhang and J. Yu, Appl. Catal., B, 2022, 317, 121788 CrossRef CAS.
- S. Yu, X. Cheng, Y. Wang, B. Xiao, Y. Xing, J. Ren, Y. Lu, H. Li, C. Zhuang and G. Chen, Nat. Commun., 2022, 13, 4737 CrossRef CAS PubMed.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. Fitzmorris, C. Wang, J. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- X. Hou, K. Aitola, H. Jiang, P. Lund and Y. Li, Catal. Today, 2021, 402, 3–9 CrossRef.
- X. Shi, S. Back, T. Gill, S. Siahrostami and X. Zheng, Chem, 2021, 7, 38–63 CAS.
- Y. Xue, Y. Wang, Z. Pan and K. Sayama, Angew. Chem., Int. Ed., 2021, 60, 10469–10480 CrossRef CAS PubMed.
- S. Xue, L. Tang, Y. Tang, C. Li, M. Li, J. Zhou, W. Chen, F. Zhu and J. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 4423–4431 CrossRef CAS PubMed.
- W. Liu, H. Liu and Z. Ai, J. Hazard. Mater., 2015, 288, 97–103 CrossRef CAS PubMed.
- Z. Xu, J. Liang, Y. Wang, K. Dong, X. Shi, Q. Liu, Y. Luo, T. Li, Y. Jia, A. Asiri, Z. Feng, Y. Wang, D. Ma and X. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 33182–33187 CrossRef CAS PubMed.
- T. u Haq, M. Pasha, Y. Tong, S. Mansour and Y. Haik, Appl. Catal., B, 2022, 301, 120836 CrossRef CAS.
- CO2 earth, https://www.co2.earth, (accessed August 2024).
- R. Sharifian, R. Wagterveld, I. Digdaya, C. Xiang and D. Vermaas, Energy Environ. Sci., 2021, 14, 781–814 RSC.
- Y. Li, Z. Zeng, Y. Zhang, Y. Chen, W. Wang, X. Xu, M. Du, Z. Li and Z. Zou, ACS Sustainable Chem. Eng., 2022, 10, 6382–6388 CrossRef CAS.
- Y. Park, D. Kim, C. Hiragond, J. Lee, J.-W. Jung, C.-H. Cho, I. In and S.-I. In, J. CO2 Util., 2023, 67, 102324 CrossRef CAS.
- J. Low, S. Qiu, D. Xu, C. Jiang and B. Cheng, Appl. Surf. Sci., 2018, 434, 423–432 CrossRef CAS.
- J. Low, L. Zhang, B. Zhu, Z. Liu and J. Yu, ACS Sustainable Chem. Eng., 2018, 6, 15653–15661 CrossRef CAS.
- M. Zubair, H. Kim, A. Razzaq, C. Grimes and S.-I. In, J. CO2 Util., 2018, 26, 70–79 CrossRef CAS.
- J. Liu, H. Shi, Q. Shen, C. Guo and G. Zhao, Appl. Catal., B, 2017, 210, 368–378 CrossRef CAS.
- L. Zhang, H. Cao, Q. Pen, L. Wu, G. Hou, Y. Tang and G. Zheng, Electrochim. Acta, 2018, 283, 1507–1513 CrossRef CAS.
- J. Perini, L. Torquato, K. Irikura and M. Zanoni, J. CO2 Util., 2019, 34, 596–605 CrossRef CAS.
- J. Wu, Y. Feng, B. Logan, C. Dai, X. Han, D. Li and J. Liu, ACS Sustainable Chem. Eng., 2019, 7, 15289–15296 CrossRef CAS.
- M. de Souza, E. Cardoso, G. Fortunato, M. Lanza, C. Nazário, M. Zanoni, G. Maia and J. Cardoso, J. Environ. Chem. Eng., 2021, 9, 105803 CrossRef CAS.
- M. Zhang, J. Cheng, X. Xuan, J. Zhou and K. Cen, ACS Sustainable Chem. Eng., 2016, 4, 6344–6354 CrossRef CAS.
- F. Fu, I. Shown, C. Li, P. Raghunath, T. Lin, T. Billo, H. Wu, C. Wu, P. Chung, M. Lin, L. Chen and K. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 25186–25194 CrossRef CAS PubMed.
- Y. Xia, M. Tang, L. Zhang, J. Liu, C. Jiang, G. Gao, L. Dong, L. Xie and Y. Lan, Nat. Commun., 2022, 13, 2964 CrossRef CAS PubMed.
- M. Li, L. Shen and M.-Q. Yang, Trans. Tianjin Univ., 2022, 28, 506–532 CrossRef CAS.
- V. Kumaravel, J. Bartlett and S. Pillai, ACS Energy Lett., 2020, 5, 486–519 CrossRef CAS.
- W. Zhang, Z. Jin and Z. Chen, Adv. Sci., 2022, 9, e2105204 CrossRef PubMed.
- T. Billo, F. Fu, P. Raghunath, I. Shown, W. Chen, H. Lien, T. Shen, J. Lee, T. Chan, K. Huang, C. Wu, M. Lin, J. Hwang, C. Lee, L. Chen and K. Chen, Small, 2018, 14, 1702928 CrossRef PubMed.
- Y. Hwang, S. Sorcar, J. Lee, J. Jung, C. Cho and S.-I. In, J. Power Sources, 2023, 556, 232430 CrossRef CAS.
- T. Dong, X. Liu, Z. Tang, H. Yuan, D. Jiang, Y. Wang, Z. Liu, X. Zhang, S. Huang, H. Liu, L. Zhao and W. Zhou, Appl. Catal., B, 2023, 326, 122176 CrossRef CAS.
- Y. Liu, M. Xia, D. Ren, S. Nussbaum, J. H. Yum, M. Gratzel, N. Guijarro and K. Sivula, ACS Energy Lett., 2023, 8, 1645–1651 CrossRef CAS PubMed.
- H. Lin, S. Luo, H. Zhang and J. Ye, Joule, 2022, 6, 294–314 CrossRef CAS.
- C. Fu, F. Li, J. Zhang, D. Li, K. Qian, Y. Liu, J. Tang, F. Fan, Q. Zhang, X. Gong and W. Huang, Angew. Chem., Int. Ed., 2021, 60, 6160–6169 CrossRef CAS PubMed.
- S. Funk, B. Hokkanen, E. Johnson and U. Burghaus, Chem. Phys. Lett., 2006, 422, 461–465 CrossRef CAS.
- H. Zhang, Y. Li, J. Wang, N. Wu, H. Sheng, C. Chen and J. Zhao, Appl. Catal., B, 2021, 284, 119692 CrossRef CAS.
- J. Lin, T. Wang, J. Zhou, X. Wu, Z. Ma and S. Liu, Appl. Catal., B, 2023, 324, 122234 CrossRef CAS.
- J. Fan, L. Cheng, J. Fan, Q. Wang, M. Gao, D. Li and J. Feng, J. CO2 Util., 2023, 67, 102333 CrossRef CAS.
- B. Ni, H. Jiang, W. Guo, Q. Xu and Y. Min, Appl. Catal., B, 2022, 307, 121141 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. Scott, X. Liu, A. Engstfeld, S. Horch, B. Seger, I. Stephens, K. Chan, C. Hahn, J. Norskov, T. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- L. Zhang, H. Cao, Y. Lu, H. Zhang, G. Hou, Y. Tang and G. Zheng, J. Colloid Interface Sci., 2020, 568, 198–206 CrossRef CAS PubMed.
- Y. Wang, P. Gao, B. Li, Z. Yin, L. Feng, Y. Liu, Z. Du and L. Zhang, Chemosphere, 2022, 291, 133007 CrossRef CAS PubMed.
- H. Iriawan, S. Andersen, X. Zhang, B. Comer, J. Barrio, P. Chen, A. Medford, I. Stephens, I. Chorkendorff and Y. Shao-Horn, Nat. Rev. Methods Primers, 2021, 1, 56 CrossRef CAS.
- S. Han, H. Li, T. Li, F. Chen, R. Yang, Y. Yu and B. Zhang, Nat. Catal., 2023, 6, 402–414 CrossRef CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- R. Urrego-Ortiz, S. Builes and F. Calle-Vallejo, ACS Catal., 2022, 12, 4784–4791 CrossRef CAS PubMed.
- J. Fu, Y. Zhou, M. Saccoccio, S. Li, R. Sažinas, K. Li, S. Andersen, A. Xu, N. Deissler, J. Mygind, C. Wei, J. Kibsgaard, P. Vesborg, J. Nørskov and I. Chorkendorff, Science, 2023, 379, 707–712 CrossRef PubMed.
- S. Lin, X. Zhang, L. Chen, Q. Zhang, L. Ma and J. Liu, Green Chem., 2022, 24, 9003–9026 RSC.
- J. Z. Bao, H. Maimaiti, X.-W. Zhao, J.-Y. Sun and L.-R. Feng, Appl. Surf. Sci., 2022, 602, 154328 CrossRef CAS.
- Q. Han, C. Wu, H. Jiao, R. Xu, Y. Wang, J. Xie, Q. Guo and J. Tang, Adv. Mater., 2021, 33, e2008180 CrossRef PubMed.
- K. Chen, X. Xu, Q. Mei, J. Huang, G. Yang and Q. Wang, Appl. Catal., B, 2024, 341, 123299 CrossRef CAS.
- X. Huang, F. Dong, G. Zhang, Y. Guo and Z. Tang, Chem. Eng. J., 2021, 419, 129572 CrossRef CAS.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS.
- D. Ye and E. Tsang, Nat. Synth., 2023, 2, 612–623 CrossRef.
- H. Fang, D. Liu, Y. Luo, Y. Zhou, S. Liang, X. Wang, B. Lin and L. Jiang, ACS Catal., 2022, 12, 3938–3954 CrossRef CAS.
- N. Cao, Z. Chen, K. Zang, J. Xu, J. Zhong, J. Luo, X. Xu and G. Zheng, Nat. Commun., 2019, 10, 2877 CrossRef PubMed.
- C. Li, T. Wang, Z. Zhao, W. Yang, J. Li, A. Li, Z. Yang, G. Ozin and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 5278–5282 CrossRef CAS PubMed.
- X. Li, Z. Wu, Y. Zeng, J. Han, S. Zhang and Q. Zhong, Chem. Phys. Lett., 2020, 750, 137494 CrossRef CAS.
- X. Wu, J. Li, R. Zhong, S. Liu, F. Huang, Q. Yang, B. Zhang, X. Wang and F. Zhang, Adv. Funct. Mater., 2024, 2405868 CrossRef.
- A. Mirzaei, M. Eddah, S. Roualdes, D. Ma and M. Chaker, Chem. Eng. J., 2021, 422, 130507 CrossRef CAS.
- Y. Shu, J. Ji, M. Zhou, S. Liang, Q. Xie, S. Li, B. Liu, J. Deng, J. Cao, S. Liu and H. Huang, Appl. Catal., B, 2022, 300, 120688 CrossRef CAS.
- Y. Liu, P. Deng, R. Wu, R. Geioushy, Y. Li, Y. Liu, F. Zhou, H. Li and C. Sun, Front. Phys., 2021, 16, 53503 CrossRef.
- R. Guan, X. Cheng, Y. Chen, Z. Wu, Z. Zhao, Q. Shang, Y. Sun and Z. Sun, Nano Res., 2023, 16, 10770–10778 CrossRef CAS.
- C. Mao, H. Li, H. Gu, J. Wang, Y. Zou, G. Qi, J. Xu, F. Deng, W. Shen, J. Li, S. Liu, J. Zhao and L. Zhang, Chem, 2019, 5, 2702–2717 CAS.
- C. Tang, D. Zhou and Q. Zhang, Mater. Lett., 2012, 79, 42–44 CrossRef CAS.
- M. Trojanowicz, A. Bojanowska-Czajka, I. Bartosiewicz and K. Kulisa, Chem. Eng. J., 2018, 336, 170–199 CrossRef CAS.
- J. Jeong, W. Song, W. Cooper, J. Jung and J. Greaves, Chemosphere, 2010, 78, 533–540 CrossRef CAS PubMed.
- H. Yu, E. Nie, J. Xu, S. Yan, W. Cooper and W. Song, Water Res., 2013, 47, 1909–1918 CrossRef CAS PubMed.
- A. Son, J. Lee, M. Seid, E. Rahman, J. Choe, K. Cho, J. Lee and S. Hong, Appl. Catal., B, 2022, 315, 121543 CrossRef CAS.
- B. Vellanki, B. Batchelor and A. Abdel-Wahab, Environ. Eng. Sci., 2013, 30, 264–271 CrossRef CAS PubMed.
- Y. Liang, G. Huang, X. Xin, Y. Yao, Y. Li, J. Yin, X. Li, Y. Wu and S. Gao, J. Mater. Sci. Technol., 2022, 112, 239–262 CrossRef CAS.
- H. Zhang, Y. Liu, L. Wang, Z. Li, Z. Lu, T. Yang and J. Ma, Environ. Sci. Technol., 2021, 55, 11612–11623 CrossRef CAS PubMed.
- T. Zhang, Y. Liu, Y. Rao, X. Li, D. Yuan, S. Tang and Q. Zhao, Chem. Eng. J., 2020, 384, 123350 CrossRef CAS.
- Q. Li, J. Hu, H. Wang and Z. Wu, Appl. Surf. Sci., 2021, 562, 150250 CrossRef CAS.
- Y.-U. Shin, J. Lim and S. Hong, Desalination, 2022, 538, 115899 CrossRef CAS.
- B. Ou, J. Wang, Y. Wu, S. Zhao and Z. Wang, ACS Omega, 2019, 4, 9664–9672 CrossRef CAS.
- J. Sun, L. Liu and F. Yang, Appl. Catal., B, 2022, 308, 121215 CrossRef CAS.
- M. Jia, Q. Liu, W. Xiong, Z. Yang, C. Zhang, D. Wang, Y. Xiang, H. Peng, J. Tong, J. Cao and H. Xu, Appl. Catal., B, 2022, 310, 121344 CrossRef CAS.
- Y. Xu, Z. He, S. Yu, L. Li, L. Cai and P. Yi, Chem. Eng. J., 2022, 429, 132104 CrossRef CAS.
- X. Liu, G. Liu and S. You, Chemosphere, 2021, 263, 127898 CrossRef CAS PubMed.
- B. Yang, Z. Zhang, P. Liu, X. Fu, J. Wang, Y. Cao, R. Tang, X. Du, W. Chen, S. Li, H. Yan, Z. Li, X. Zhao, G. Qin, X. Chen and L. Zuo, Nature, 2023, 622, 499–506 CrossRef CAS PubMed.
- S. Namboorimadathil Backer, A. Ramachandran, A. Venugopal, A. Mohamed, A. Asok and S. Pillai, ACS Appl. Nano Mater., 2020, 3, 6827–6835 CrossRef CAS.
- L. Ren, W. Zhou, L. Wang, K. Lin, Y. Xu, J. Wu, Y. Xie and H. Fu, Sci. Bull., 2023, 68, 2760–2768 CrossRef CAS PubMed.
- C. Mao, L. Yu, J. Li, J. Zhao and L. Zhang, Appl. Catal., B, 2018, 224, 612–620 CrossRef CAS.
- I. Zada, W. Zhang, P. Sun, M. Imtiaz, N. Iqbal, U. Ghani, R. Naz, Y. Zhang, Y. Li, J. Gu, Q. Liu, D. Pantelić, B. Jelenković and D. Zhang, Appl. Mater. Today, 2020, 20, 100669 CrossRef.
- P. Zheng, J. Tang, Z. Zhou, L. Gong, H. Yang, X. Jia, X. Li, Y. Liu and L. Tan, Surf. Interfaces, 2021, 22, 100901 CrossRef CAS.
- F. Yu, C. Wang, R. Wang, Y. Li, B. Ohtani, A. Fujishima and X. Zhang, Appl. Surf. Sci., 2023, 624, 157119 CrossRef CAS.
- S. Gurbatov, E. Modin, V. Puzikov, P. Tonkaev, D. Storozhenko, A. Sergeev, N. Mintcheva, S. Yamaguchi, N. Tarasenka, A. Chuvilin, S. Makarov, S. Kulinich and A. Kuchmizhak, ACS Appl. Mater. Interfaces, 2021, 13, 6522–6531 CrossRef CAS PubMed.
- J. Umlauff, H. Kalt, W. Langbein, J. Hvam, M. Scholl, J. Söllner, M. Heuken, B. Jobst and D. Hommel, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1390–1393 CrossRef.
- X. Mettan, J. Jaćimović, O. S. Barišić, A. Pisoni, I. Batistić, E. Horváth, S. Brown, L. Rossi, P. Szirmai, B. Farkas, H. Berger and L. Forró, Commun. Phys., 2019, 2, 123 CrossRef.
- Q. Liu, Y. Yang, Y. Ni, Q. Wang, H. Yu, X. Zhu, Z. Ying and Y. Song, Appl. Surf. Sci., 2021, 570, 151175 CrossRef CAS.
- A. Chaichi, G. Venugopalan, R. Devireddy, C. Arges and M. Gartia, ACS Appl. Energy Mater., 2020, 3, 5693–5704 CrossRef CAS.
- X. Lu, G. Wang, T. Zhai, M. Yu, J. Gan, Y. Tong and Y. Li, Nano Lett., 2012, 12, 1690–1696 CrossRef CAS PubMed.
- A. Ramadoss and S. Kim, Carbon, 2013, 63, 434–445 CrossRef CAS.
- J. Zhi, C. Yang, T. Lin, H. Cui, Z. Wang, H. Zhang and F. Huang, Nanoscale, 2016, 8, 4054–4062 RSC.
- S. Yang, Y. Li, J. Sun and B. Cao, J. Power Sources, 2019, 431, 220–225 CrossRef CAS.
- T. Pinto, Y. Núñez-de la Rosa, P. Hammer and J. Aquino, Electrochim. Acta, 2022, 408, 139898 CrossRef CAS.
- R. Huang, J. Zhang, Z. Dong, H. Lin and S. Han, J. Power Sources, 2022, 550, 232169 CrossRef CAS.
- P. Pazhamalai, K. Krishnamoorthy, V. Mariappan and S. Kim, J. Colloid Interface Sci., 2019, 536, 62–70 CrossRef CAS PubMed.
- A. Auer and J. Kunze-Liebhäuser, Small Methods, 2018, 3, 1800385 CrossRef.
- Y. Tang, L. Hong, Q. Wu, J. Li, G. Hou, H. Cao, L. Wu and G. Zheng, Electrochim. Acta, 2016, 195, 27–33 CrossRef CAS.
- J. Wang, W. Liu and C. Wang, J. Alloys Compd., 2022, 921, 166220 CrossRef CAS.
- B. Babu, S. Ullattil, R. Prasannachandran, J. Kavil, P. Periyat and M. Shaijumon, ACS Sustainable Chem. Eng., 2018, 6, 5401–5412 CrossRef CAS.
- J. Kang, J. Kim, S. Lee, S. Wi, C. Kim, S. Hyun, S. Nam, Y. Park and B. Park, Adv. Energy Mater., 2017, 7, 1700814 CrossRef.
- S. Patil, H. Phattepur, B. Kishore, R. Viswanatha and G. Nagaraju, Mater. Renew. Sustainable Energy, 2019, 8, 1–10 CrossRef.
- T. Wu, G. Sun, W. Lu, L. Zhao, A. Mauger, C. Julien, L. Sun, H. Xie and J. Liu, Electrochim. Acta, 2020, 353, 136529 CrossRef CAS.
- F. Zhou, L. Ouyang, J. Liu, X.-S. Yang and M. Zhu, J. Power Sources, 2020, 449, 227549 CrossRef CAS.
- Y. Yang, W. Shi, S. Liao, R. Zhang and S. Leng, J. Alloys Compd., 2018, 746, 619–625 CrossRef CAS.
- Y. Wang, X. Xue, P. Liu, C. Wang, X. Yi, Y. Hu, L. Ma, G. Zhu, R. Chen, T. Chen, J. Ma, J. Liu and Z. Jin, ACS Nano, 2018, 12, 12492–12502 CrossRef CAS PubMed.
- P. Sun, Z. Cao, Y. Zeng, W. Xie, N. Li, D. Luan, S. Yang, L. Yu and D. Lou, Angew. Chem., Int. Ed., 2022, 61, e202115649 CrossRef CAS PubMed.
- J. Ge, G. Du, A. Kalam, X. Bi, S. Ding, Q. Su, B. Xu and A. Al-Sehemi, Ceram. Int., 2021, 47, 6965–6971 CrossRef CAS.
- S.-T. Myung, M. Kikuchi, C. Yoon, H. Yashiro, S.-J. Kim, Y.-K. Sun and B. Scrosati, Energy Environ. Sci., 2013, 6, 2609–2614 RSC.
- C. Zhang, H. Yu, Y. Li, Y. Gao, Y. Zhao, W. Song, Z. Shao and B. Yi, ChemSusChem, 2013, 6, 659–666 CrossRef CAS PubMed.
- C. Zhang, H. Yu, Y. Li, L. Fu, Y. Gao, W. Song, Z. Shao and B. Yi, Nanoscale, 2013, 5, 6834–6841 RSC.
- C. Park, E. Lee, G. Lee and Y. Tak, Appl. Catal., B, 2020, 268, 118414 CrossRef CAS.
- K. Naik, E. Higuchi and H. Inoue, J. Power Sources, 2020, 455, 227972 CrossRef CAS.
- H. Xie, X. Xie, G. Hu, V. Prabhakaran, S. Saha, L. Gonzalez-Lopez, A. Phakatkar, M. Hong, M. Wu, R. Shahbazian-Yassar, V. Ramani, M. Al-Sheikhly, D.-E. Jiang, Y. Shao and L. Hu, Nat. Energy, 2022, 7, 281–289 CrossRef CAS.
- L. Zhang, C. Fu, S. Wang, M. Wang, R. Wang, S. Xiang, Z. Wang, J. Liu, H. Ma, Y. Wang, Y. Yan, M. Chen, L. Shi, Q. Dong, J. Bian and Y. Shi, Adv. Funct. Mater., 2023, 33, 2213961 CrossRef CAS.
- L. Jakob, L. Tutsch, T. Hatt, J. Westraadt, S. Ngongo, M. Glatthaar, M. Bivour and J. Bartsch, Sol. RRL, 2023, 7, 2200929 CrossRef.
- W. Wu and Z. Zhang, J. Mater. Chem. B, 2017, 5, 4883–4889 RSC.
- T. David, K. Gnanasekar, P. Wilson, P. Sagayaraj and T. Mathews, ACS Omega, 2020, 5, 11352–11360 CrossRef CAS PubMed.
- Q. Li, S. Fu, X. Wang, L. Wang, X. Liu, Y. Gao, Q. Li and W. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 57471–57480 CrossRef CAS PubMed.
- L. Duy, R. Yeasmin, S.-I. Han, S. Iqbal, C. Park and H. Seo, Sens. Actuators, B, 2023, 394, 134387 CrossRef CAS.
- S. Zhang, S. Cao, T. Zhang and J. Lee, Adv. Mater., 2020, 32, e2004686 CrossRef PubMed.
- W. Ni, M. Li, J. Cui, Z. Xing, Z. Li, X. Wu, E. Song, M. Gong and W. Zhou, Mater. Sci. Eng., C, 2017, 81, 252–260 CrossRef CAS PubMed.
- M. Zhang, N. Wu, J. Yang and Z. Zhang, ACS Appl. Bio. Mater., 2022, 5, 1341–1347 CrossRef CAS PubMed.
- J. Lawton, S. Thornley, S. Wakeham, M. Thwaites, V. Stolojan and M. Baker, Surf. Coat. Tech., 2024, 476, 130247 CrossRef CAS.
- G. Zhou, B. Sun, X. Hu, L. Sun, Z. Zou, B. Xiao, W. Qiu, B. Wu, J. Li, J. Han, L. Liao, C. Xu, G. Xiao, L. Xiao, J. Cheng, S. Zheng, L. Wang, Q. Song and S. Duan, Adv. Sci., 2021, 8, 2003765 CrossRef CAS.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- Z. Wang, T. Hisatomi, R. Li, K. Sayama, G. Liu, K. Domen, C. Li and L. Wang, Joule, 2021, 5, 344–359 CrossRef CAS.
- H. Asif Javed, A. Qureshi, R. Mehmood, M. Tahir, S. Javed, M. Sarfaraz, M. Javaid, M. Awais and U. Ali, Int. J. Hydrogen Energy, 2021, 46, 14311–14321 CrossRef CAS.
- M. Yang, B. Dong, X. Yang, W. Xiang, Z. Ye, E. Wang, L. Wan, L. Zhao and S. Wang, RSC Adv., 2017, 7, 41738–41744 RSC.
- H. Zhang, L. Xie, C. Huang, Z. Ren, H. Wang, J. Hu, H. Zhang, Z. Jiang and F. Song, Appl. Surf. Sci., 2021, 568, 150933 CrossRef CAS.
- Y. Peng, Y. Hu, H. L. Chou, Y. Fu, I. Teixeira, L. Zhang, H. He and E. Tsang, Nat. Commun., 2017, 8, 675 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |