
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in oxidative degradation of plastics
Sewon
Oh
 a and
Erin E.
Stache
*b
a and
Erin E.
Stache
*b
aDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA
bDepartment of Chemistry, Princeton University, Princeton, New Jersey 08544, USA. E-mail: estache@princeton.edu
First published on 17th June 2024
Abstract
Oxidative degradation is a powerful method to degrade plastics into oligomers and small oxidized products. While thermal energy has been conventionally employed as an external stimulus, recent advances in photochemistry have enabled photocatalytic oxidative degradation of polymers under mild conditions. This tutorial review presents an overview of oxidative degradation, from its earliest examples to emerging strategies. This review briefly discusses the motivation and the development of thermal oxidative degradation of polymers with a focus on underlying mechanisms. Then, we will examine modern studies primarily relevant to catalytic thermal oxidative degradation and photocatalytic oxidative degradation. Lastly, we highlight some unique studies using unconventional approaches for oxidative polymer degradation, such as electrochemistry.
 Sewon Oh | Sewon Oh is a PhD student at Cornell University under the supervision of Professor Erin Stache whose research lab is currently at Princeton University. He received his BS degree from Boston College under Professor Jeffery Byers, working towards the synthesis of biodegradable polylactic acid and iron-based catalysts. His current research work is focused on degradation and depolymerization of polymers using photochemistry and development of novel and sustainable materials. |
 Erin E. Stache | Erin Stache earned her BS from the University of Wisconsin-Green Bay and her PhD from Colorado State University in collaboration with Professors Tom Rovis and Abby Doyle. She spent two years as a Cornell Presidential Postdoctoral Fellow in Professor Brett P. Fors's lab at Cornell University before starting her independent career there in 2020. She moved to Princeton University in 2023 as an Assistant Professor of Chemistry. Research in the Stache Lab combines synthetic organic chemistry, photochemistry, materials, and polymer chemistry for synthesis and materials science applications. She also develops degradation and depolymerization strategies for commercial plastics using photothermal conversion. |
Key learning points1. An overview of the oxidative degradation mechanism of plastics.2. Traditional thermal oxidative degradation approaches and limitations. 3. Catalytic oxidative degradation enables the conversion of plastics to value-added feedstocks. 4. Visible light harnessed to promote mild and selective oxidative degradation of plastics. 5. Emerging electrochemical and mechanochemical examples of oxidative degradation. |
1. Introduction
Plastics, also known as polymers, have significantly contributed to our modern society since large-scale production began in the 1950s.1 Consisting of light yet inert carbon-based materials, more than 8 billion metric tons of plastics have been produced in the past 50 years and used in various applications, from packaging materials to automobile parts.2 For several decades, engineers have focused on developing the stability of plastics to ensure consumer confidence. However, this robustness, unluckily, engenders environmental problems because plastics linger in nature without degradation.1,2 These pollutants leak into ecosystems, posing health threats to humans and other living organisms. This tension puts society at a crossroads, where the production of commodity plastics is inexpensive and efficient but engenders a severe environmental concern. The end-of-life fate of these plastics needs to be addressed urgently.3Plastic pollution can be mitigated through a few different types of recycling methods: mechanical recycling, pyrolysis, and incineration (Fig. 1(a)).4–17 For mechanical recycling, plastic waste is collected, ground into polymer pellets, and reused for plastic products.4 Although simple and feasible, this method eventually downgrades the quality of plastics due to polymer chain scission or cross-linking caused by heat, mechanical force, or contaminants like acids.5 Pyrolysis is an alternative approach to chemically convert polymers to monomers (the repeating unit of a polymer) or other small molecule feedstocks under high temperatures in an inert atmosphere.6 This method typically requires high energy inputs (>400 °C); therefore, the operation cost of pyrolysis can be expensive when considering the demand for plastic waste recycling.12 Lastly, incineration is used for energy recovery through the combustion of plastics, yet generates environmentally harmful substances, such as carbon monoxide, ashes, and carcinogenic polyaromatic hydrocarbons (PAHs).15–17 Due to the drawbacks of current recycling techniques, approximately 60% of plastic waste remains unrecycled.3
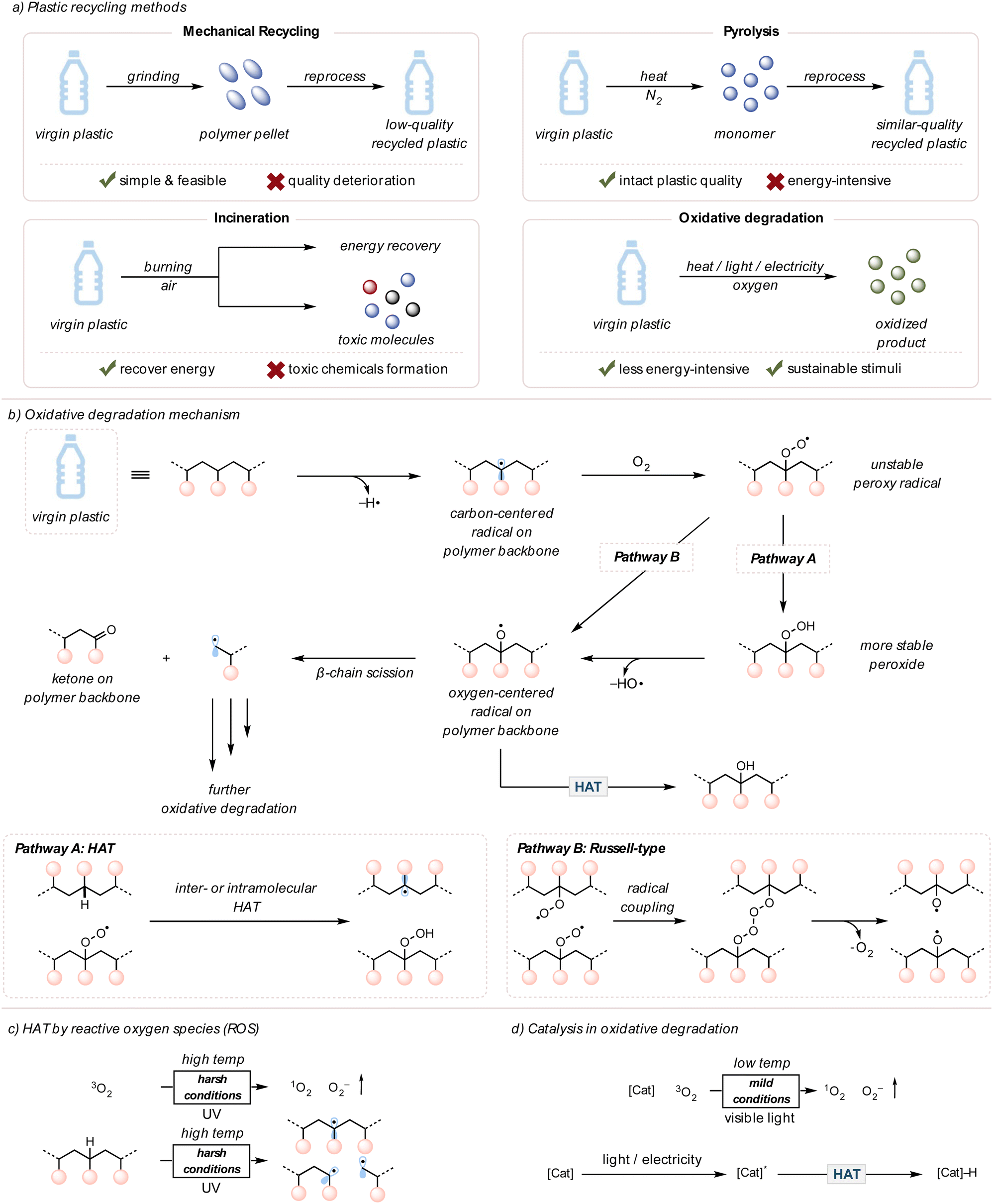 | ||
| Fig. 1 A brief introduction to oxidative degradation. (a) Opportunities and challenges of various recycling methods. (b) Accepted mechanism of oxidative degradation. (c) Uncatalyzed oxidative degradation of polymer under harsh conditions. (d) Catalytic oxidative degradation of polymer under mild conditions. | ||
Chemical upcycling, a conversion of discarded plastics to higher-value materials or feedstocks, is another approach to resolve plastic pollution.18–24 One specific example is oxidative degradation, which uses thermally induced radicals and a cheap oxidant, molecular oxygen (O2), to cleave polymer chains and produce small molecule oxidized compounds.25,26 In the environment, plastics slowly degrade in air under sunlight due to photooxidative degradation, meaning light is involved in the oxidative degradation of plastics.25 Within recent years, photochemistry has been extensively leveraged to chemically upcycle polymers by oxidative degradation under mild conditions with photocatalysts, avoiding the need for high temperatures.27 In addition, light is an environmentally benign stimulus that can be less energy-intensive.28
This tutorial review presents recent advances in thermocatalytic and photocatalytic oxidative degradation of synthetic polymers. To help readers understand this topic, we briefly discuss an overview of oxidative degradation and the mechanisms by which degradation can proceed. Later sections provide more modern examples of oxidative degradation from the literature, including unconventional methods like electrochemical and mechanochemistry. Lastly, we conclude our tutorial review with the challenges for future development of oxidative degradation methods. Photoreforming, biocatalytic degradation, acid/base hydrolysis, and depolymerization are beyond the scope of this work (see these great review articles for more details).29–41
2. An introduction to oxidative degradation
This section aims to present an overview of oxidative degradation of polymers. The degradation mechanism will be reviewed first, followed by the discussions of external stimuli and catalysts that improve the rate of oxidative degradation.2.1 The general mechanism of oxidative degradation of polymers
It has been observed for many years that upon exposure to air, metals naturally corrode, and fresh foods become stale.42,43 Both cases occur due to the oxidation of the compounds by molecular O2 in air. Moreover, the rate of oxidation can be accelerated or decreased depending on the surrounding environments, such as the concentration of O2, temperature, moisture, and pH.44,45Similarly, synthetic plastics in landfills or the environment interact with O2 in air, usually by a radical mechanism.46–48 The resulting post-consumer plastics exhibit yellow coloration and brittle physical properties due to the deterioration of polymer chains.49 The chemical reaction between polymers and O2 promotes chain scissions on the polymer backbone to degrade plastics and install ketone moieties.50 This process is also known as “plastic aging” or “autoxidation,” meaning the spontaneous oxidation of a polymer in the presence of O2.47–49 Previous literature investigated the mechanism of plastics or polymer degradation in the environment through analytical techniques, such as gel permeation chromatography (GPC), mass spectrometry (MS), viscometry, nuclear magnetic resonance (NMR) spectroscopy, and Fourier transform infrared (FTIR) spectroscopy (Fig. 1(b)).46–54
Oxidative degradation is initiated by radical generation on the polymer backbone. This first radical generation step typically requires high energy, such as UV light or high temperatures, to break polymer bonds or species that can abstract hydrogens on the polymer backbone, like singlet O2 or peroxides (later sections will discuss these in further detail). The resulting radicals are quenched by O2 to form unstable peroxy radical intermediates. These peroxy radicals can follow two major pathways, although both routes eventually converge. According to Pathway A, they can perform hydrogen atom transfer (HAT) to abstract from a C–H bond on the polymer backbone, which generates peroxide groups. Upon the O–O bond cleavage, the oxygen-centered radicals are produced.50,55 Pathway B undergoes a Russell-type mechanism in which two peroxy radicals couple to form four consecutive oxygens. Homolytic cleavages regenerate molecular O2 and form two oxygen-centered radicals on polymer backbones.56,57 The oxygen-centered radicals can undergo β-chain scission (beta, meaning two carbons away from oxygen-centered radicals) to yield ketones and alkyl radicals or undergo HAT to form alcohols.
Although the mechanism of oxidative degradation is thoroughly explored, the autoxidation of plastics in nature is slow, resulting in incomplete degradation. The slow autoxidation of polymers is due to their chemical inertness, so the overall concentration of polymeric radicals would be low under mild conditions, such as room temperature. Moreover, hydroperoxyl groups on the polymer backbone do not readily undergo bond cleavage without being in harsh conditions.2 To overcome this kinetic challenge, many researchers utilized external stimuli and catalysts to facilitate efficient oxidative degradation. Both heat and light are important external stimuli that promote the oxidative degradation of polymers.49 In laboratory setups, using high temperatures (>200 °C) or intense UV light (254 nm) improves the polymer degradation rate by increasing the concentration of reactive oxygen species (ROS, such as singlet O2, peroxide, superoxide, etc.) and initial polymeric radicals on the backbone (Fig. 1(c)).49,58–60 However, such intense reaction conditions prevent industry from applying this polymer upcycling strategy due to high energy cost and safety concerns.
2.2 Catalysts
Catalysts are often employed to accelerate the kinetics of chemical reactions under more mild reaction conditions.61 Indeed, academic and industrial efforts conducted pyrolysis at lower temperatures and obtained more selective products after adopting catalysts.6 Likewise, oxidative degradation necessitates using catalysts to speed up polymer degradation under more environmentally friendly conditions and selectively form desired products.49 These catalysts can range from organic compounds to inorganic transition metals and from homogenous to heterogeneous catalysts, depending on the design and purpose of the reactions.61,62 Typically, these catalysts generate ROS or form the excited state species that directly perform HAT on the polymer backbone under mild conditions. This is an invaluable advantage to bypass harsh and unsafe reaction conditions (Fig. 1(d)).63,64 In the case of photo and electrocatalytic systems, specific photocatalysts and electrocatalysts are used and later discussed in this tutorial review.3. Recent advances in thermocatalytic oxidative degradation
This section discusses the oxidative degradation of polymers using heat as an external stimulus. We begin by briefly presenting uncatalyzed thermal oxidative degradation. Effective catalysts are employed to improve the kinetics of degradation and atom economy. The latter part of this section introduces selected examples from the literature to show recent advances and breakthroughs in the thermocatalytic oxidative degradation of polymers.3.1 Thermal oxidative degradation of polymers with molecular oxygen alone
Thermal oxidative degradation of polymers has been extensively investigated in nature and laboratory settings because heat is easily applied to chemical reactions.46 There are many examples in the literature of thermal oxidative degradation without extraneous chemical reagents. Upon exposure to high temperatures in air, these polymers undergo oxidative chain scissions and convert to small molecule oxidized products.55,65 However, there are three significant drawbacks to this method. First, as mentioned in Section 2.2, this technique is inefficient at mild temperatures (for example, at room temperature), so the oxidative degradation must be conducted under harsh conditions and demanding more energy.2,18 Considering the million metric tons of discarded plastics, it would not be economically desirable to build a large reactor that would require high maintenance and operation expenses. Second, the risk of explosion increases with higher temperatures in the presence of O2, making safety another primary concern. Lastly, undesired byproducts are more likely to be formed at extreme temperatures. As the concentration of radicals uncontrollably increases due to numerous C–C bond chain scission events, these radicals undergo side reactions like radical–radical coupling, elimination, and HAT as opposed to being trapped by molecular O2.18 Therefore, to address these problems, it would be more beneficial to decrease temperature while accelerating the efficiency in oxidative degradation.3.2 Thermal oxidative degradation of polymers with oxidants other than oxygen
Nemphos and coworkers showed one of the earliest examples of thermal oxidative degradation of polyethylene (PE) using ozone in 1957 (Fig. 2(a)).66 PE film was placed in an apparatus under an O2 atmosphere with 2% ozone. The oxidation of PE was measured by a change in the concentration of carbonyls on the polymer backbone through IR spectra, with highly oxidized polymers displaying a larger peak in this region. Surprisingly, they observed that PE oxidation started even at room temperature. In contrast, PE oxidation in an atmosphere of pure O2 (with no ozone) did not have comparable results until raising the temperature to 100 °C. Since ozone is a stronger oxidizing agent than O2, oxidative degradation of PE occurred more rapidly. Moreover, they noticed even faster oxidation at higher temperatures when they tested the PE degradation under the same conditions with the temperature range from 25 °C to 137 °C. According to the IR spectrum from Fig. 2(b), the carbonyl stretches (5.6 microns or 1780 cm−1) noticeably increased throughout oxidative degradation. This work inspired many researchers to develop similar strategies to degrade various polymers with ozone oxidants.67,68 | ||
| Fig. 2 (a) PE oxidative degradation in the absence or presence of ozone (2%) in O2. (b) IR spectrum of PE oxidative degradation in ozone (2%) at 137.5 °C for 3 h. Reprinted (adapted) with permission from H. C. Beachell and S. P. Nemphos, Ozone Chem. Technol., 1959, 26, 168–175. Copyright 1959 American Chemical Society. (c) Thermodynamic driving force of hydroxyl radicals to abstract hydrogens from polymers. (d) PKA oxidative degradation in natural garden soil versus H2O2 in water. | ||
Although ozone certainly accelerates the oxidative degradation of polymers, it poses many health risks, making it desirable to use other safer alternatives.69 In fact, other exogenous radical oxidants have commonly been used to decrease the overall degradation temperature while improving the kinetics of oxidation.54,70,71 As such, hydrogen peroxides are reasonable surrogates for ozone. Since the O–O bond of hydrogen peroxide is relatively weak, the homolytic cleavage of hydrogen peroxide yields hydroxyl radicals (˙OH) at low temperatures.72 The hydroxyl radicals have a high bond dissociation energy (ΔHBDE(HO–H) = 119 kcal mol−1, energy required for homolytic cleavage of an O–H bond).73 Conversely, ΔHBDE (C–H) from the polymer backbone is much less than 119 kcal mol−1.74,75 Thus, HAT of the C–H bonds of polymer backbone by hydroxyl radicals is thermodynamically favorable for generating polymeric radicals and water (Fig. 2(c)). The resulting polymeric radicals are then trapped by O2 and undergo oxidative degradation. The initial HAT from the polymer backbone enables facile and effective oxidative degradation of polymers under mild conditions. Gratefully, substoichiometric amounts of peroxide oxidants are typically sufficient to trigger degradation, improving the atom economy of upcycling.
In 2021, Kiatkamjornwong and coworkers compared the biological and chemical degradation of poly(potassium acrylate) and its acrylamide copolymer (Fig. 2(d)).76 Poly(acrylic acid) (PAA), poly(potassium acrylate) (PKA), and its copolymers are known as superabsorbent polymers. Moreover, these polymers have excellent mechanical properties, making them appropriate for diapers or various medical applications. Although PKA undergoes biodegradation in soil, the degradation rate in the natural environment is relatively slow; less than 53 wt% of polymer mass was lost in 10 weeks. However, PKA degraded much more efficiently after PKA was incubated in the presence of hydrogen peroxide at an elevated temperature. Approximately 98.4 wt% of solid PKA turned to liquid (degraded oligomer) after treating the polymer with 12.8% (v/w) of hydrogen peroxide in water at 65 °C in just 7.3 h. As mentioned, hydroxyl radicals from hydrogen peroxide must act as a hydrogen atom abstractor to trigger oxidative degradation. Although the authors did not further identify or characterize liquified products, they showed that the degraded product was innocuous to seed germination, showing that the resulting products were not detrimental to the environment. Besides PKA, there are many similar examples of oxidative degradation of different polymers, including polyolefins and copolymers, with hydrogen peroxide or its analogs.71,77–83
Other reaction conditions can be modified to increase oxidative degradation rates. In 1998, Sen and coworkers oxidatively degraded various polymers in a mixture of high-pressure nitrogen oxides (NO) and O2 at 170 °C for 16 h.84 Although they could quantify the small molecule products, as shown in Table 1, the degradation rate was relatively slow because reactions were conducted in the gas phase. For example, placing 0.26 g of polystyrene (PS) under the above conditions resulted in 0.08 g of benzoic acid and 0.19 g of residual PS and oligomers. To improve upon this degradation method, Zhang and coworkers developed a solution-phase acid-mediated oxidative degradation of PS in 2023 (Fig. 3(a)).85 To activate the benzylic hydrogen of PS, the Zhang group added nitric acid to their degradation conditions as a cheap acid in conjunction with an oxidant. They mixed 20% nitric acid and PS in an autoclave and placed it in an oil bath to heat the system to 180 °C. After 3 h, 90 mol% of PS was converted to benzoic acid. They also observed that nitric acid concentration decreased throughout oxidative degradation because it decomposes to NO2 and O2. Although the exact mechanism is understudied, NO2 has been shown to catalyze the oxidation of alkanes through the formation of various NOx species and superoxides.86,87 Zhang and coworkers proposed that nitric acid catalyzes benzylic radical formation, and O2 acts as a terminal oxidant to cleave the C–C bonds and yield benzoic acid. Moreover, the addition of radical traps, such as 2,2,6,6-tetramethyl-1-piperdinyloxy (TEMPO), suppressed the formation of benzoic acid, which supports that the degradation pathway was through radical chain mechanisms. Lastly, the GPC trace of PS degradation over time points showed that PS was efficiently degraded to styrene oligomers within 2 h (Fig. 3(b)). Although we will not discuss it in detail, there are some examples of the oxidative degradation of oxygen-containing polymers (like lignin and epoxy resins) using different oxidants, such as KMnO4 and acids.88–92
| Polymera | Amount of starting polymer (g) | Amount of nonvolatile products (g) | Products | Amount of products (g) |
|---|---|---|---|---|
| a Standard condition: NO (275 kPa), N2 (3170 kPa), and O2 (690 kPa) at 170 °C for 16 h. b Under the standard condition except using O2 (1035 kPa). | ||||
| Polystyrene | 0.26 | 0.19 | Benzoic acid | 0.08 |
| 4-Nitrobenzoic acid | 0.02 | |||
| 3-Nitrobenzoic acid | 0.02 | |||
| 3,5-Dinitrobenzoic acid | Trace | |||
| High-density polyethyleneb | 0.27 | 0.21 | Succinic acid | 0.08 |
| Glutaric acid | 0.07 | |||
| Adipic acid | 0.02 | |||
| Pimelic acid | 0.03 | |||
| Unknown volatiles | Trace | |||
| Low-density polyethyleneb | 0.26 | 0.22 | Succinic acid | 0.08 |
| Glutaric acid | 0.06 | |||
| Adipic acid | 0.02 | |||
| Pimelic acid | 0.01 | |||
| Unknown volatiles | Trace | |||
| Nylon-6,6 | 0.26 | 0.16 | Succinic acid | 0.01 |
| Glutaric acid | 0.01 | |||
| Adipic acid | 0.06 | |||
| Acetic acid | Trace | |||
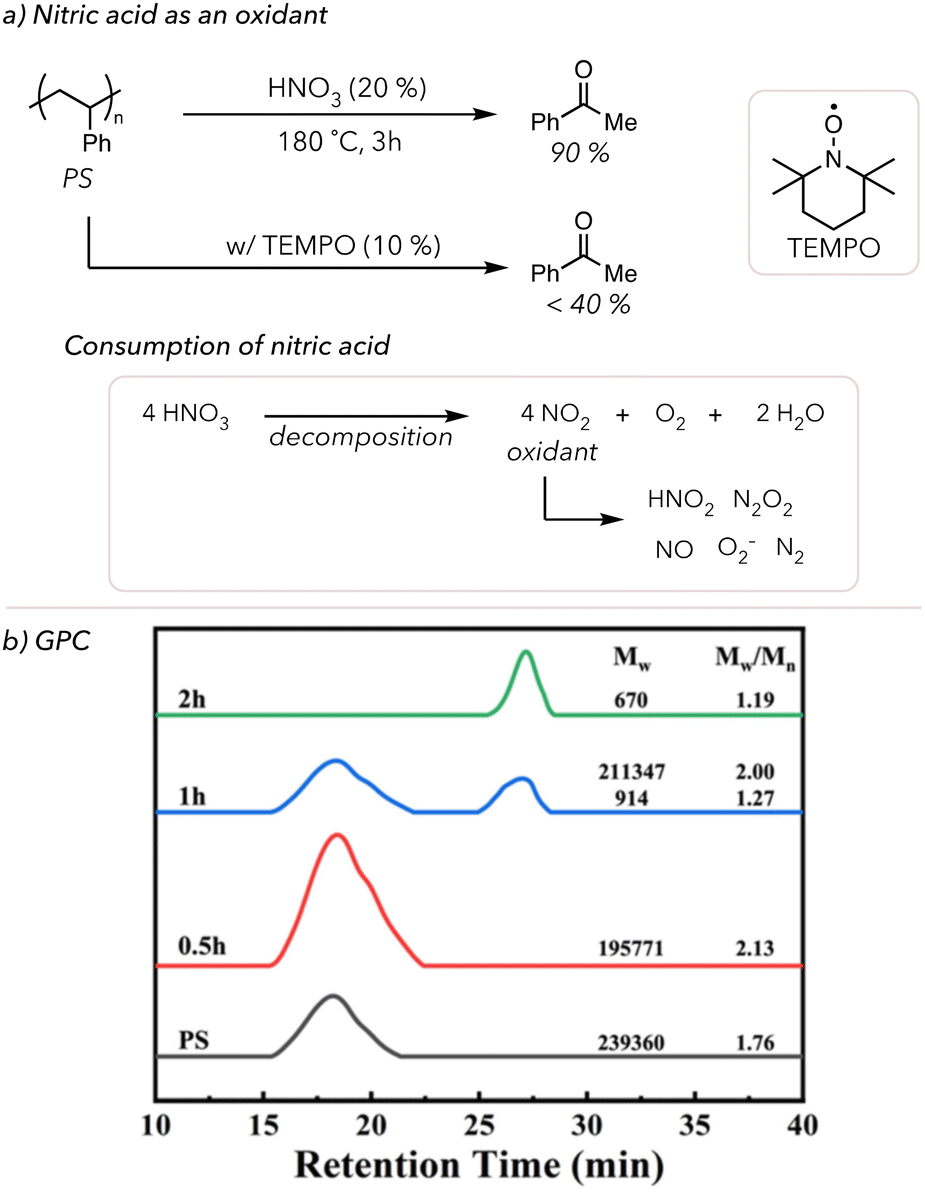 | ||
| Fig. 3 (a) PS oxidative degradation in HNO3 under 180 °C with or with TEMPO. (b) GPC of PS oxidative degradation over the course of 2 h (adapted) with permission from X. Luo, J. Zhan, Q. Mei and S. Zhang, Green Chem., 2023, 25, 6717–6727. Copyright 2023 Green Chemistry (RSC). | ||
3.3 Thermocatalytic oxidative degradation of polymers
Catalysts are crucial for improving the kinetics of reactions while mitigating harsh conditions. Transition metal catalysts adopt different oxidation states, form complexes with various reagents to change their reactivities, and, in some cases, have high turnover numbers under rigorous conditions.93 As such, these catalysts can positively impact the thermal oxidative degradation of polymers. Metal catalysts can directly reduce O2 to promote ROS to trigger HAT from a C–H bond from a polymer backbone under mild conditions. Further, they can behave as Fenton or Fenton-like reagents to rapidly cleave peroxide bonds to generate oxygen-centered radicals on the polymer backbone (Scheme 1).94,95 Another advantage of transition metal catalysts is their recyclability, which makes heterogeneous catalysts particularly suitable for industrial applications.96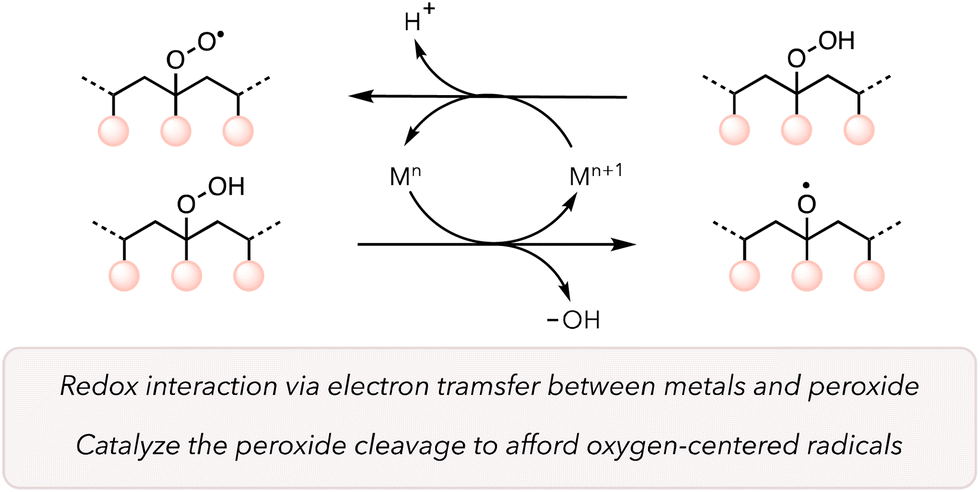 | ||
| Scheme 1 Peroxide cleavage through Fenton chemistry. | ||
The use of transition metals to boost the polymer degradation rate has been investigated since the 1950s.49 In 1993, Rutherford and coworkers screened different metals for polypropylene (PP) degradation at elevated temperatures.97 After casting films with a mixture of PP and metals (150 ppm), they placed those films at 70 and 104 °C to study the embrittlement of the materials (Table 2). They observed that not all transition metals were effective, as the polymer stayed durable even after 2000 h when treated with metals like Ni and Pd. However, in the case of Mn, Co, and Cu at 70 °C and Ce, V, and Fe at 104 °C, PP became brittle over a shorter period, indicating a faster degradation rate. They supported their observations by measuring the molecular weights of various polymer samples. For instance, the molecular weight (Mn) of PP with Mn catalysts reduced from 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1 to 2700 g mol−1 over 2 weeks. They proposed a similar mechanism as presented in Scheme 1. The hydroperoxide groups on polymers are formed through autooxidation due to the interaction between polymers and oxygen. Then transition metals donate an electron to hydroperoxide groups to accelerate the oxidative cleavage of polymer. Moreover, hydroperoxide groups on polymer can reduce metals and form more peroxy radicals, which can induce HAT on the polymer backbone to further oxidize polymers.
500 g mol−1 to 2700 g mol−1 over 2 weeks. They proposed a similar mechanism as presented in Scheme 1. The hydroperoxide groups on polymers are formed through autooxidation due to the interaction between polymers and oxygen. Then transition metals donate an electron to hydroperoxide groups to accelerate the oxidative cleavage of polymer. Moreover, hydroperoxide groups on polymer can reduce metals and form more peroxy radicals, which can induce HAT on the polymer backbone to further oxidize polymers.
| Metal | Time to embrittlement at 104 °C (h) | Time to embrittlement at 70 °C (h) |
|---|---|---|
| Mn(II) | 41 | 541 |
| Co(II) | 41 | 720 |
| Cu(II) | 41 | 723 |
| Ce(II) | 85 | 1610 |
| V(III) | 172 | 2114 |
| Fe(II) | 750 | >3000 |
| Mo(IV) | 1050 | >3000 |
| Zr(IV) | 1400 | >3000 |
| Ni(II) | >2500 | >3000 |
| Ti(II) | >2000 | >2000 |
| Pd(II) | >2000 | >2000 |
| Pt(II) | >2000 | >2000 |
This concept of metal-catalyzed thermal oxidative degradation has been investigated more broadly. In 2003, Partenheimer developed a more complex system in which polymers are treated with Co/Mn/Zr/Br2 catalysts under 70 bar in air at high temperatures (>150 °C) (Fig. 4(a)).98 This system effectively degraded various polymers. Indeed, PS was converted to 88 mol% benzoic acid in 5 h. Their proposed mechanism also relied on the redox chemistry of the metals. They hypothesized two distinct roles of liquid bromine: bromine radicals could abstract C–H bonds on polymer backbone to initiate oxidative degradation, or bromide anion could donate an electron to reduce MnIII to MnII (Fig. 4(b)). Lastly, the role of the Zr catalyst in the reaction mechanism was not extensively studied, but the degradation efficiency slightly improved upon the addition of Zr. While this system rapidly converted polymer to oxidized products, it still relied on high temperatures and pressures. Moreover, this oxidative degradation was conducted in a homogenous solution phase, precluding users from recycling the catalysts.
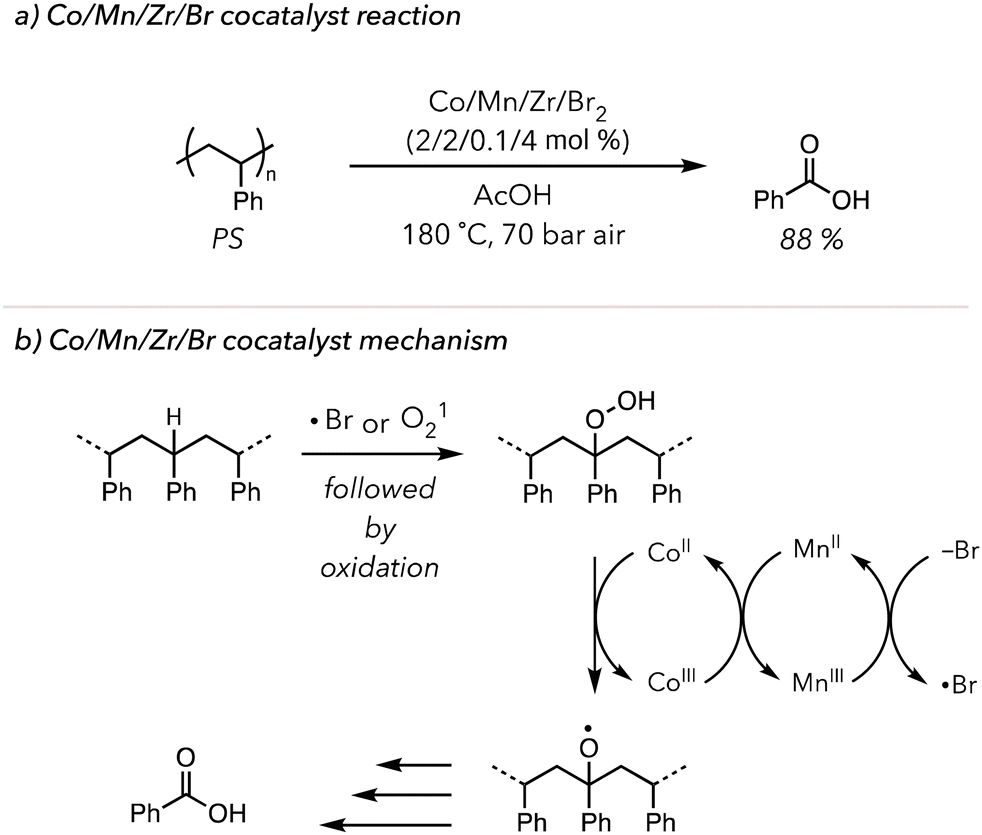 | ||
| Fig. 4 (a) PS oxidative degradation to benzoic acid using Co/Mn/Zr/Br2 as metal co-catalysts. (b) Proposed mechanism of the co-catalysts. | ||
Thermocatalytic oxidative degradation via heterogeneous catalysts has been developed to address the latter issue. In industry, a heterogeneous catalyst is preferred to its homogeneous counterpart because it is generally more stable and can be recycled many times without losing performance. In 1977, Sorvik and coworkers used titanium dioxide (TiO2) as a heterogeneous catalyst to oxidatively degrade low-density PE at 150 °C.99 They witnessed the yellow coloration of polymers, indicating oxidative degradation. Hinge and coworkers published a recent example in 2020, in which they studied the stability of poly(acrylonitrile–butadiene–styrene) (ABS) blended with an Fe or Cu-based pigment at 60 °C for 1440 h.100 The impact toughness of ABS decreased by up to a factor of four compared to the absence of heterogenous pigment. A Fenton-like mechanism pathway was proposed, where peroxides are easily activated through a single electron transfer with metals.
In 2021, Stahl and coworkers fabricated a creative system to degrade biomass in the presence of a Co based heterogeneous catalyst (Scheme 2).101 Acetone was used for the solvent of their degradation system because it solubilizes lignin but not their corresponding oxidized products. Products, consisting of alcohols and carboxylic acids, would crash out in acetone, preventing them from further degradation in O2. They chose Co metal containing polyaniline-doped carbon catalysts (Co-PANI-C) (aniline was polymerized on the carbon support) because this compound has already been shown to catalyze the aerobic oxidation of small molecules. After the oxidative degradation of lignin with 10 wt% catalyst under 35 bar in air with 6% O2 at 190 °C, they recovered 15 wt% of small molecules phenolic products ranging from aldehydes to carboxylic acids in 12 h. Although the yield is not incredibly high, using a one pot system and acetone as a benign solvent is advantageous over traditional methods with multistep degradations and harsher conditions. Apart from transition metal catalysts, nanoparticles, carbon-based materials, and other alternatives can also be used in the thermocatalytic oxidative degradation of polymers.102–105
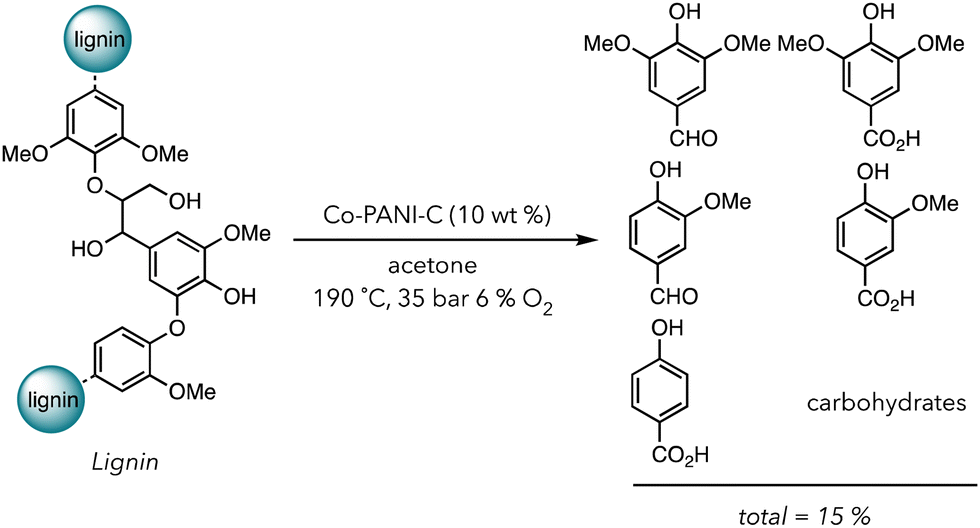 | ||
| Scheme 2 Lignin oxidative degradation with cobalt heterogeneous catalysts. | ||
4. Recent advances in photocatalytic oxidative degradation
This section aims to examine the oxidative degradation of polymers using photochemical approaches. The first part introduces the concept and previous examples of photo-induced oxidative degradation without catalysts. The latter part of this section presents more thorough and recent examples of photocatalytic oxidative degradation for both homogeneous and heterogeneous catalysis.4.1 Photo-induced oxidative degradation of polymers
Photochemistry has enabled new approaches in organic chemistry, as photochemical processes allow us to access novel synthetic routes that traditional thermal chemistry could otherwise not achieve.106 Light is a sustainable energy source that can photo-excite species to perform various mechanistic steps, such as energy transfer, bond scission, single electron transfer, hydrogen atom transfer, and many other chemical transformations.107 In the case of oxidative degradation, direct excitation of polymers, O2, or photocatalysts by light irradiation generates radicals that eventually lead to bond scissions.49,63In nature, the autoxidation rate of plastics increases under sunlight. Particularly, plastics corrode faster when exposed to more intense sunlight under a high concentration of O2. Thus, plastics exposed to air would undergo more rapid oxidative degradation than those in seawater where there is a low concentration of O2, and solar irradiation is less intense.108,109
In the laboratory, many researchers have attempted to initiate photo-induced oxidative degradation by directly irradiating polymers with high-energy photons, such as γ-rays or UV light. Ideally, directly irradiating the polymer would cleave the backbone to yield polymeric radicals. The resultant radicals would be combined to form crosslinks or trapped by O2 to generate oxidized products (Fig. 5(a)).109 Indeed, in 1980, Kuriyama and coworkers demonstrated that after exposing PE to γ-rays, they observed crosslinking under vacuum and oxidative degradation under an O2-rich environment. Therefore, light irradiation and oxygen are necessary to oxidize the polymers, leading to degradation. Other than this previous study, more reports of photo-induced oxidative degradation with other polymers have been published.110–112
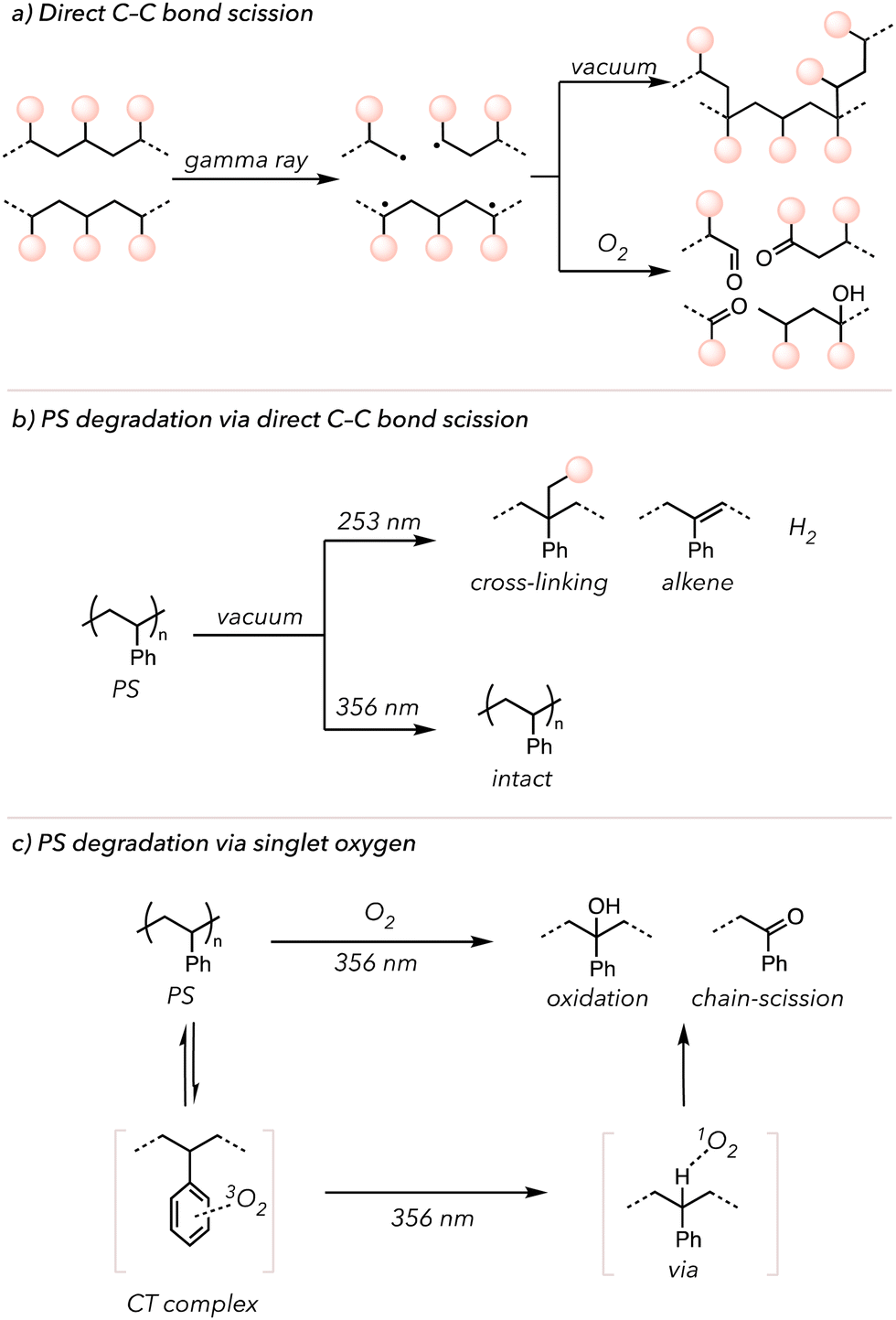 | ||
| Fig. 5 (a) Polymer photodegradation under vacuum vs. O2. (b) PS photodegradation under vacuum with 253 nm vs. 356 nm. (c) PS photo-induced oxidative degradation via PS charge transfer complex with O2. | ||
Singlet O2 can also oxidatively degrade polymers after generation via irradiation. Molecular oxygen exists in its triplet form at the ground state and is excited to a singlet state through photo-excitation or energy transfer from photocatalysts.113 Triplet O2 has two unpaired electrons with the same spins, but singlet O2 undergoes spin inversion to form two unpaired electrons with opposite spins. These opposite spins of electrons (that repel each other) make singlet O2 highly reactive.113 Singlet O2 can serve as an HAT mediator that abstracts C–H bonds from the polymer backbone.114 In 1965, Weir and coworkers irradiated PS with UV light (253 or 356 nm) under vacuum and investigated its effects (Fig. 5(b)).115,116 UV light (253 nm) enabled cleavage of C–H and C–C bonds of PS to generate hydrogen gas and generate alkenes on the backbone. However, no changes were observed when irradiating with 356 nm UV light. Then, they repeated the experiments in the presence of O2. Although relatively slow, PS was oxidized under 356 nm UV light. However, the observation could not be explained through independent activation of PS or O2 by 356 nm UV irradiation. Later, in 1974, Rabek and Ranby explained that the phenyl rings of PS may form a charge transfer (CT) complex with O2, which can then absorb light above 280 nm. Thus, triplet O2 coordinated with PS gets excited to the singlet O2 species that ultimately abstracts hydrogens on the PS backbone to initiate oxidative degradation (Fig. 5(c)).117 Researchers have also incorporated iron-based catalysts into polymers to increase the photo-induced oxidative degradation rate. These catalysts undergo Fenton-like chemistry to accelerate O–O bond scission of the peroxide to oxygen-centered radicals, followed by oxidative polymer chain cleavage.95,118
Although photo-induced oxidative degradation illustrated a potential solution to degrade polymers under more ambient conditions, most early examples use high-energy UV light. Visible light irradiation (>390 nm) would be an ideal alternative.
4.2 Photocatalytic oxidative degradation of polymer with homogeneous catalysts
The past few years have witnessed a renaissance in visible light-mediated oxidative degradation of polymers.119 The use of longer wavelength visible light is beneficial compared to UV light, for it has lower photon energy for a safer alternative.120 However, visible light does not directly excite many commercial polymers (like polyolefins and PS) and cannot generate singlet oxygen. Thus, visible light-absorbing photocatalysts have been pursued to induce photocatalytic oxidative degradation. The products obtained from degradation depend on the polymer microstructure as well as reaction conditions.In 2022, Xiao and coworkers demonstrated the acid-catalyzed photocatalytic oxidative degradation of PS (Fig. 6(a)).121 After irradiating PS with 405 nm light in the presence of 5 mol% p-toluenesulfonic acid monohydrate (pTsOH·H2O) in a mixture of benzene and acetonitrile under 1 bar O2, they recovered approximately 50 mol% of benzoic acid in 15 h. They noticed that adding other strong acids, such as sulfuric acid and triflic acid, also yielded benzoic acid, albeit in more modest yields. In the absence of acid, no degradation was observed. Their mechanistic studies revealed that the strong acid interacts with PS and that this adduct exhibits the characteristics of photosensitizers. According to their UV-vis study, neither PS nor the acids absorb visible light, but the adduct showed a clear absorption peak in the visible light region. They proposed that the photo-excited adduct sensitizes triplet O2 to form singlet O2 that can initiate oxidative degradation of the polymer. Later, in 2024, Zheng and coworkers adopted a similar approach to oxidatively degrade PS using porphyrin-based porous organic polymers (PPOPs).122 They previously developed the catalytic system for PPOPs and discovered that this catalyst serves as a photosensitizer to generate singlet O2. Although they used 370 nm, they conducted their experiment in air and obtained 63 mol% benzoic acid over 16 h. They also extended their study by applying the optimized system to styrene copolymers and still obtained comparable amounts of benzoic acid (>40 mol%).
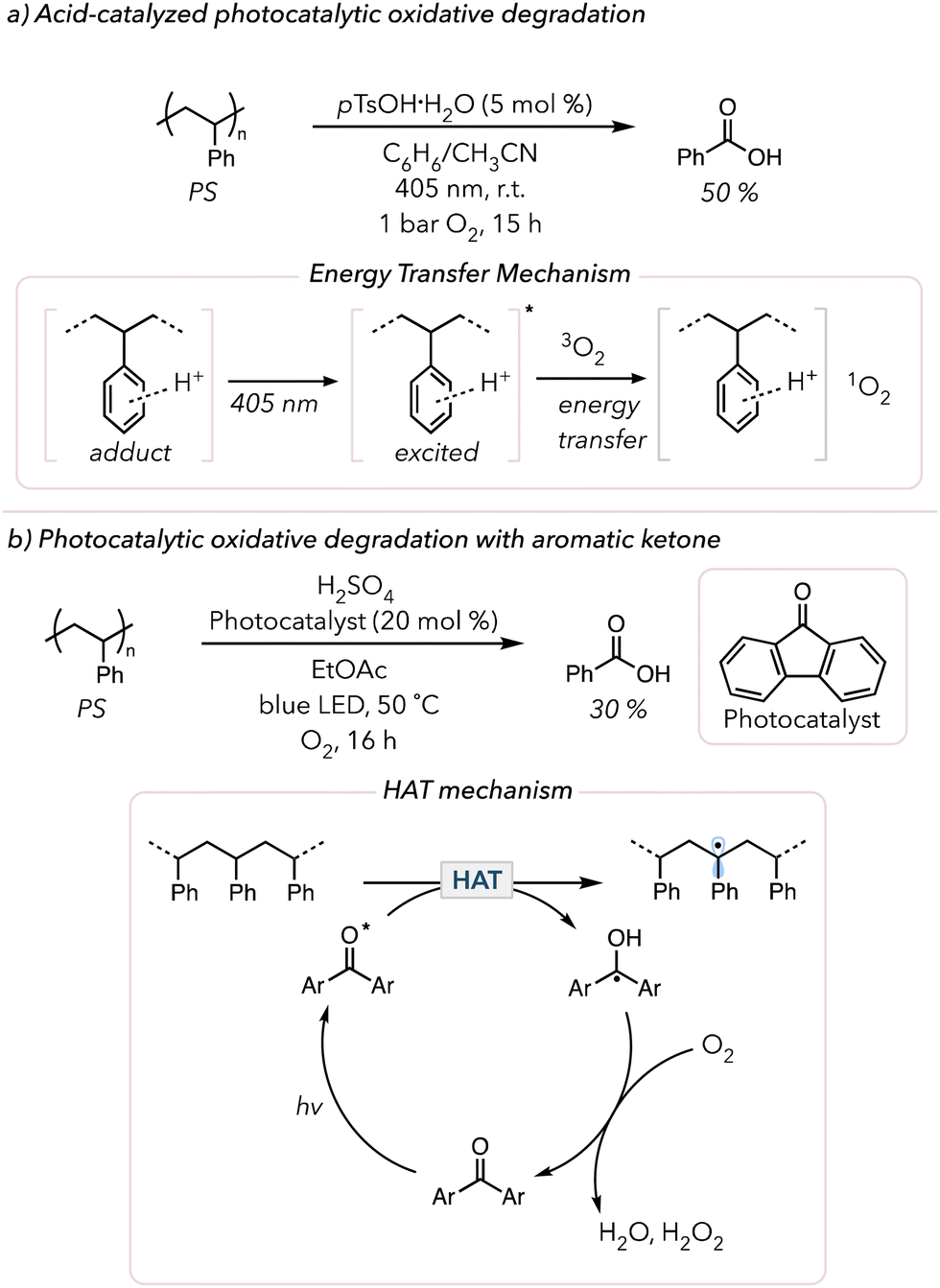 | ||
| Fig. 6 (a) Acid-catalyzed photocatalytic oxidative degradation and mechanism of PS. (b) Photocatalytic oxidative degradation of PS with aromatic ketone as a HAT photocatalyst. | ||
Photocatalysts that enable direct HAT from the polymer backbone can also accelerate oxidative degradation. In 2000, Kaczmarek and coworkers irradiated PS in the presence of benzophenone with UV light and observed that the viscosity of PS quickly dropped.123 They proposed that photoexcited triplet benzophenone abstracts C–H bonds from the PS backbone to trigger oxidative degradation. The resulting ketyl radical on benzophenone is oxidized to the ketone in the presence of O2 to turn over the catalyst. Approximately 20 years later, in 2022, Reisner and coworkers showed the photocatalytic oxidative degradation of PS with fluorenone HAT catalysts under blue LED irradiation.124 Under their optimized conditions, they obtained 30 mol% benzoic acid in 16 h when treating PS with 20 mol% of the HAT catalyst and 1 equiv. of sulfuric acid under an atmosphere of O2 and blue LEDs (Fig. 6(b)). Their mechanistic studies showed no benzoic acid when omitting light or O2. Surprisingly, benzoic acid also did not form without sulfuric acid. They hypothesized that strong acid enabled catalyst turnover. When the ketyl radical on fluorenone was oxidized with O2, the strong acid facilitated the reduction of O2 to water or hydrogen peroxide. Their suggested mechanism was like that proposed by Kaczmarek, in which the HAT catalyst abstracts hydrogens from the PS backbone and the resulting radicals are subsequently quenched by O2 to oxidatively degrade PS to produce benzoic acid. Then, in 2023, Kokotos and coworkers conducted similar studies but attempted to simplify the system further.125 Although they used 390 nm instead of blue LEDs, they successfully removed the need for a strong acid and degraded PS under air using anthraquinone photocatalysts. They oxidatively degraded PS post-consumer products to benzoic acid (25 to 58 mol%). Similarly, uranyl photocatalysts (UO2(NO3)2·6H2O) have been shown to abstract hydrogens from polymer backbone similarly to aromatic ketones.126,127
Aromatic ketones are not the only HAT abstractors employed in photocatalytic oxidative degradation. Halogen radicals (chlorine or bromine radicals) are widely used in organic synthesis for functionalizing C–H bonds.128 Inspired by this technique, researchers used halogen radicals to activate polymer backbones. In 2021, the Zeng and Hu groups separately developed iron chloride (FeClx) catalyzed photocatalytic oxidative degradation of PS. Zeng and coworkers used 10 mol% FeCl3, 10 mol% tetrabutylammonium chloride with 20 mol% trichloroethanol in acetone to degrade PS in 5 days under an O2 balloon to obtain 67% benzoic acid.129 Hu and coworkers modified the system by using 2 mol% FeCl2 in a dichloromethane/acetonitrile solvent mixture irradiated with 400 nm light in an O2 balloon for 66 h and isolated 63 mol% benzoic acid.130 In 2022 and 2023, Stache and coworkers further simplified the degradation conditions by using FeCl3 in acetone irradiated with white LEDs and an O2 balloon to obtain 31 mol% benzoyl products (mainly benzoic acids) in just 20 h (Fig. 7(a)).131,132 The critical step in the mechanism of these photocatalytic oxidative degradation reports is the generation of chlorine radicals. When subjected to polar solvents, FeCl3 is likely to disproportionate, wherein two of the same species react with each other to form two distinct products. When two molecules of FeCl3 react, the resulting products are [FeIIICl2]+ and [FeIIICl4]−. Upon irradiation, [FeIIICl4]− can undergo ligand-to-metal charge transfer (LMCT) (an electron from the ligand is excited to an empty orbital of the metal) to trigger homolytic cleavage of an Fe–Cl bond to result in a chlorine radical and [FeIICl3]−. This chlorine radical abstracts a C–H bond on the PS backbone to initiate oxidative degradation. [FeIICl3]− can now participate in Fenton-like chemistry to cleave peroxide bonds and regenerate [FeIIICl3]. In 2024, Lim and coworkers developed the in situ generation of chlorine radicals from HCl with acridinium photocatalysts to convert PS to benzoic acid.133
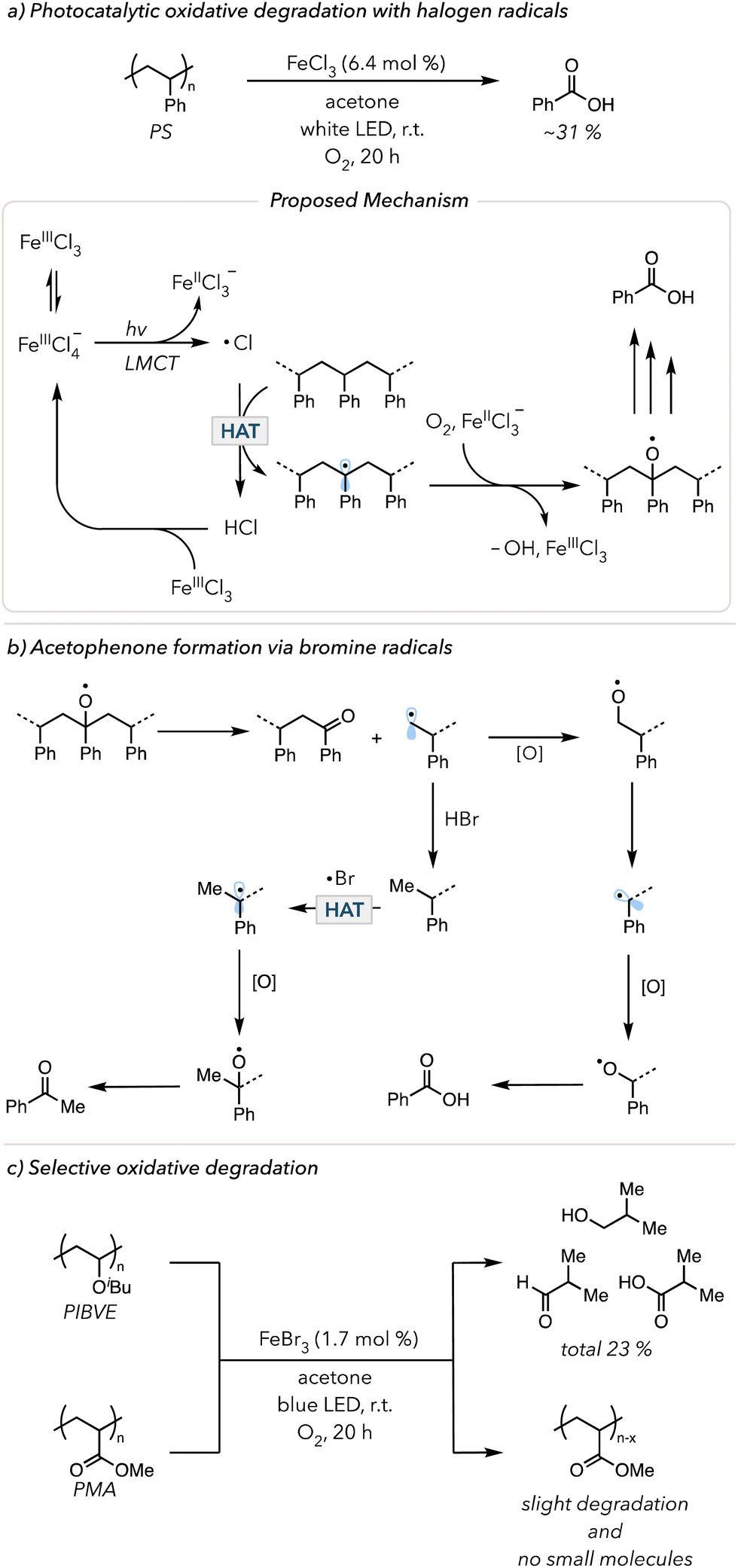 | ||
| Fig. 7 (a) Photocatalytic oxidative degradation of PS using a chlorine radical as a HAT abstractor. (b) Proposed mechanism to form acetophenone and benzoic acid in PS degradation through a bromine radical. (c) Selective photocatalytic oxidative degradation of polymer with electron-rich vs. electron-poor C–H. | ||
In 2023, the Stache group showed oxidative degradation of PS using FeBr3, albeit with lower overall yields of small molecules.132 Interestingly, the product distribution shifted from mostly benzoic acid to an equal amount of benzoic acid and acetophenone. The suggested mechanism is shown in Fig. 7(b). The alkyl radical is oxidized and eventually proceeds toward oxidative degradation to afford benzoic acid. However, H–Br is also known to act as a good hydrogen atom donor for alkyl radicals.134 The primary alkyl radical can easily abstract a hydrogen atom from H–Br. With a methyl group, the subsequent oxidation leads to acetophenone formation. In contrast, H–Cl is a much stronger H–X bond than H–Br; thus, hydrogen atom donation does not readily occur, minimizing the formation of acetophenone. Lastly, the kinetic selectivity of bromine radicals (electrophilic radicals) for electron-rich hydridic C–H bonds enables selective polymer degradation. In 2024, the Stache group demonstrated that bromine radicals could be used to selectively degrade polymers with electron-rich hydrogens (e.g., poly(isobutyl vinyl ethers) (PIBVE)) over polymers with electron-poor hydrogens (e.g., poly(methyl acrylate) (PMA)) (Fig. 7(c)).135
Considering industrial applications, photochemistry has two additional benefits in polymer degradation. First is the potential to use sunlight, an unlimited energy source with a broad wavelength range. While it has already been shown that sunlight can degrade plastics, the degradation is relatively slow.136 In 2024, Su and coworkers used uranyl photocatalysts under sunlight for 3 days in air to convert 20 g of PS to 1.6 g of benzoic acid.127 The second benefit of photochemistry is the ease with which one can transform batch reactions into a photo-flow setup. When using large batch reactors, the light penetration becomes difficult because light is more likely to be absorbed near the surface of the reactors. Photo-flow chemistry is an appropriate choice for scaling because large amounts of samples move through thin tubes with high photon flux.137 Moreover, the product would not be removed from the system in the receiving flask, preventing any side reactions from continuous light irradiation. The Xiao, Stache, and Jiang group scaled their systems and demonstrated the photocatalytic oxidation of PS on a large-scale using photo-flow chemistry.121,126,131
4.3 Photocatalytic oxidative degradation of polymers with heterogeneous catalysts
The works described above are examples of photocatalytic oxidative degradation with homogeneous catalysts. Some recent publications highlight heterogeneous photocatalysis in the oxidative degradation of polymers.28,138 TiO2 is a commonly used heterogeneous photocatalyst due to its outstanding stability in photochemical processes.139 In 2019, Raditoiu and coworkers cast polyolefin films with and without 1 or 2 wt% TiO2 nanoparticles and irradiated them with an Xenon lamp for 192 h (Fig. 8(a) top).140 They observed that while pure polyolefin films did not exhibit any sign of oxidation, films with the photocatalyst began oxidizing almost immediately after the irradiation began. They detected strong carbonyl, lactone, or alcohol peaks in the IR spectrum, which continuously grew with prolonged irradiation. In 2023, Li and coworkers used TiO2 to convert PS to benzoic acid.141 They dissolved PS in DCM, added the photocatalyst, and irradiated the mixture with 370 nm light for 4 h under an O2 atmosphere. Initially, only 8 mol% benzoic acid was recovered. To improve the photocatalytic reactivity, they modified the surface of the photocatalyst with organic substrates, such as potassium stearate. Surprisingly, the surface-modified TiO2 displayed higher photocurrent density and, thus, could improve the conversion of PS to benzoic acid slightly above 40 mol% (Fig. 8(a) bottom). Lastly, they demonstrated the recyclability of the photocatalyst through isolation and resubjecting it to the same conditions. Gratefully, the photocatalyst was recyclable, although its performance decreased by 80% at 6th cycle due to its decomposition.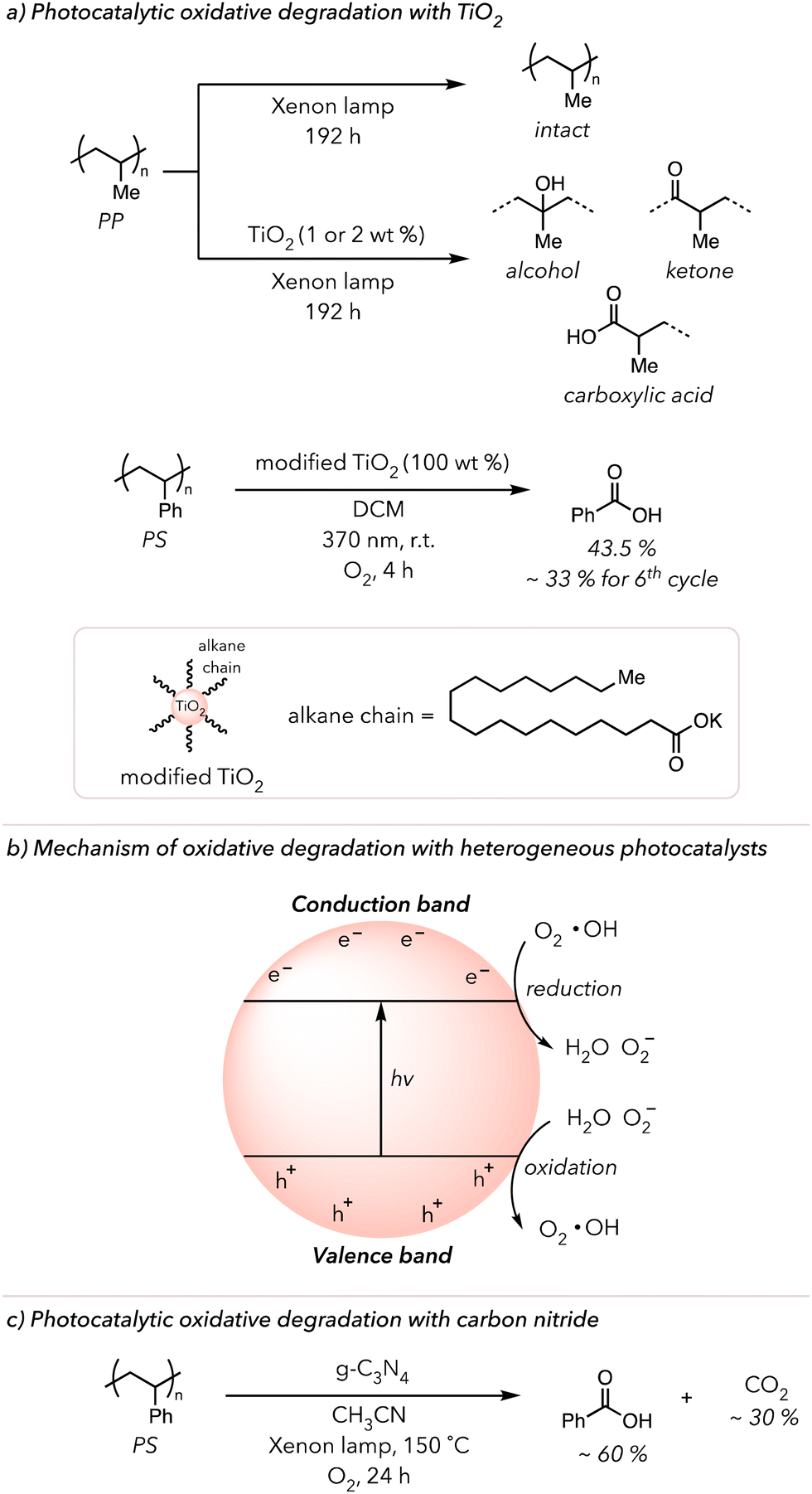 | ||
| Fig. 8 (a) Photocatalytic oxidative degradation of PP or PS using TiO2 as a heterogeneous photocatalyst (b) Proposed general mechanism to photocatalytic oxidative degradation using heterogeneous photocatalysts. (c) Photocatalytic oxidative degradation with graphitic carbon nitride, non-metal photocatalyst. | ||
Besides TiO2 nanoparticles, other heterogeneous photocatalysts, such as Cu, Zn, Nb, and Bi-based nanoparticles, have successfully converted polyolefins to small molecule oxidized products and CO2 or PS to benzoic acids.142–147 According to the suggested mechanism (Fig. 8(b)), the electrons from the valence band are excited to the conduction band to create a hole and electron pairs upon irradiation of nanoparticles.138 These electrons reduce O2 to superoxide radical anions that can react with PS to trigger oxidative degradation. Conversely, the holes oxidize water to O2 or hydroxyl radicals. In either case, ROS generated by these nanoparticles initiates HAT and the subsequent oxidative degradation of polymers.
In 2022, Ma and coworkers utilized graphitic carbon nitride (g-C3N4) catalysts, rather than metal nanoparticles, to oxidatively degrade PS (Fig. 8(c)).148 g-C3N4 catalysts are a graphitic sheet structure with a high surface area that, like metal nanoparticles, generate electron and hole pairs upon irradiation with light. Approximately 90% PS was converted to 60% benzoyl products and 30% CO2 under an O2 atmosphere with xenon light irradiation at 150 °C for 24 h in their optimized system. CO2 was produced because of the decomposition of benzoyl products. The longer the PS sample was irradiated, the more CO2 was generated, while the amounts of benzoyl products decreased. Gratefully, a comparable result was achieved using a broad range of visible light (400–700 nm), so UV light was unnecessary for successful degradation. They postulated that the electrons from excited g-C3N4 reduce O2 to make ROS while the holes oxidized PS. They observed an induction period in which PS was partially oxidized, after which PS became more rapidly oxidized. Lastly, they showcased the recyclability of their heterogeneous photocatalyst by applying their reaction condition 20 times. For the first several rounds of oxidation, catalyst performance remained, but it gradually worsened until the 20th cycle, when the catalyst was no longer very effective in oxidative degradation.
5. Recent advances in other types of oxidative degradation
This section introduces other novel approaches to the oxidative degradation of polymers. The examples in Sections 3 and 4 are thermally or photochemically triggered oxidative degradation, respectively. However, this section aims to highlight the recent advances in photothermal chemistry, electrochemistry, and mechanochemistry in oxidative degradation and raise awareness of these growing fields in chemical upcycling.5.1 Photothermal oxidative degradation of polymers
Photothermal conversion is when light energy is converted to heat energy (Fig. 9(a)).149 Upon light irradiation, photothermal agents emit immense heat, typically through plasmonic localized heating and nonradiative relaxations (see this review paper for a more detailed analysis of these mechanisms).149 Photothermal conversion is particularly attractive for its sustainability and potential spatiotemporal control. A light source allows high heat generation at a desired space and time, avoiding extremely high bulk temperatures for safety. Moreover, strong absorption over a wide range of the electromagnetic spectrum is another asset of photothermal agents, as visible or even IR light can produce heat.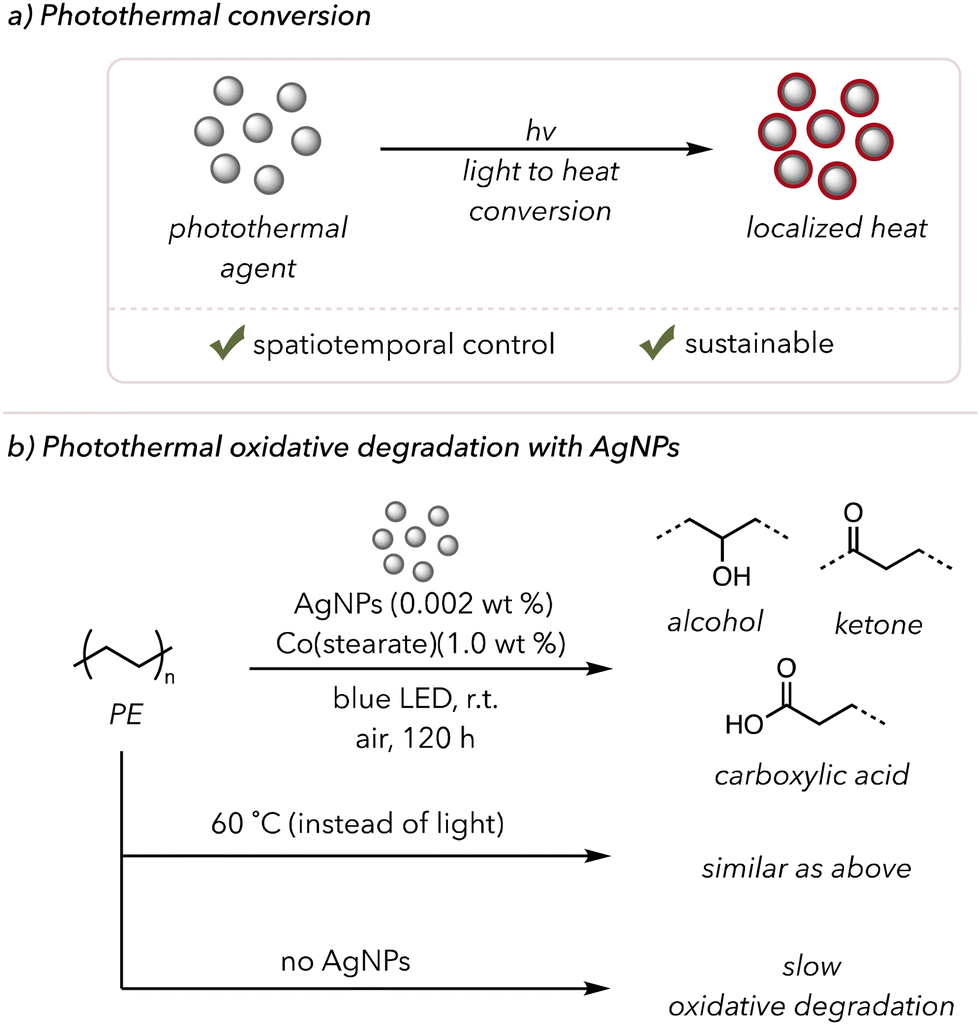 | ||
| Fig. 9 (a) A brief introduction to photothermal conversion. (b) Photothermal oxidative degradation of polymer using AgNPs as a photothermal agent. | ||
The photothermal effect has been widely applied in medical fields, such as cancer therapy, to eradicate toxic cells and bacteria using relatively safe near-IR light without killing essential cells and microorganisms. Recently, organic chemistry has adopted photothermal catalysis to conduct thermodynamically demanding organic synthesis and transformations under mild conditions. Researchers developed photothermal catalytic methods to degrade air pollutants like volatile organic compounds.150,151 Moreover, several recent publications demonstrated the application of photothermal conversion in the chemical recycling of polymers, such as polyolefins, polymethacrylates, and polyesters.152–155
Although photothermal-assisted oxidative degradation has not been extensively studied, a few examples have been found in the literature. In 2019, Clarke and coworkers used Ag-based photothermal nanoparticles (AgNPs) to degrade low-density PE photothermally (Fig. 9(b)).156 As mentioned in previous sections, metals can catalyze the thermal oxidative degradation of polymers at slightly elevated temperatures. Their study used photothermal conversion to generate heat, allowing the metal catalysts to increase degradation rates. AgNPs were chosen as they are efficient photothermal catalysts that absorb blue light. When they irradiated PE film mixed with 0.002 wt% AgNPs and 1.0 wt% cobalt-stearate with blue light, they observed the formation of ketones and alcohols in the sample via UV-vis and FTIR spectroscopy. When irradiating polyethylene films with either AgNPs or cobalt catalysts alone, no significant amount of oxidation occurred over 120 h. PE films with both catalysts or only the cobalt catalyst at 60 °C undergo oxidative degradation, but the film with only AgNPs did not show a sign of polymer degradation. These experiments, overall, emphasized that both a heat source and metal catalysts are necessary and that low incorporation of AgNPs (<0.1 wt%) was enough to heat the polymer to trigger thermocatalytic oxidative degradation.
5.2 Electrocatalytic oxidative degradation of polymer
Applying electrochemistry for organic transformations is another emerging technique in organic chemistry. There has been a boom in electrochemistry in recent years because supplying electrons through externally applied potentials enables diverse chemical reactions that might not be accessible through traditional thermal or photochemistry.157 Further, electricity is considered a clean energy source, bypassing stoichiometric amounts of chemical oxidants or reductants. Other than those listed above, there are many other advantages of using electricity as a stimulus in organic synthesis (see these reviews for more details).157–159 Typical electrochemical reactions consist of two simultaneous redox half-reactions, which are both happening at the electrode surface. Oxidation (removing electrons) occurs at the anode, while reduction (gaining electrons) occurs at the cathode (Fig. 10(a)).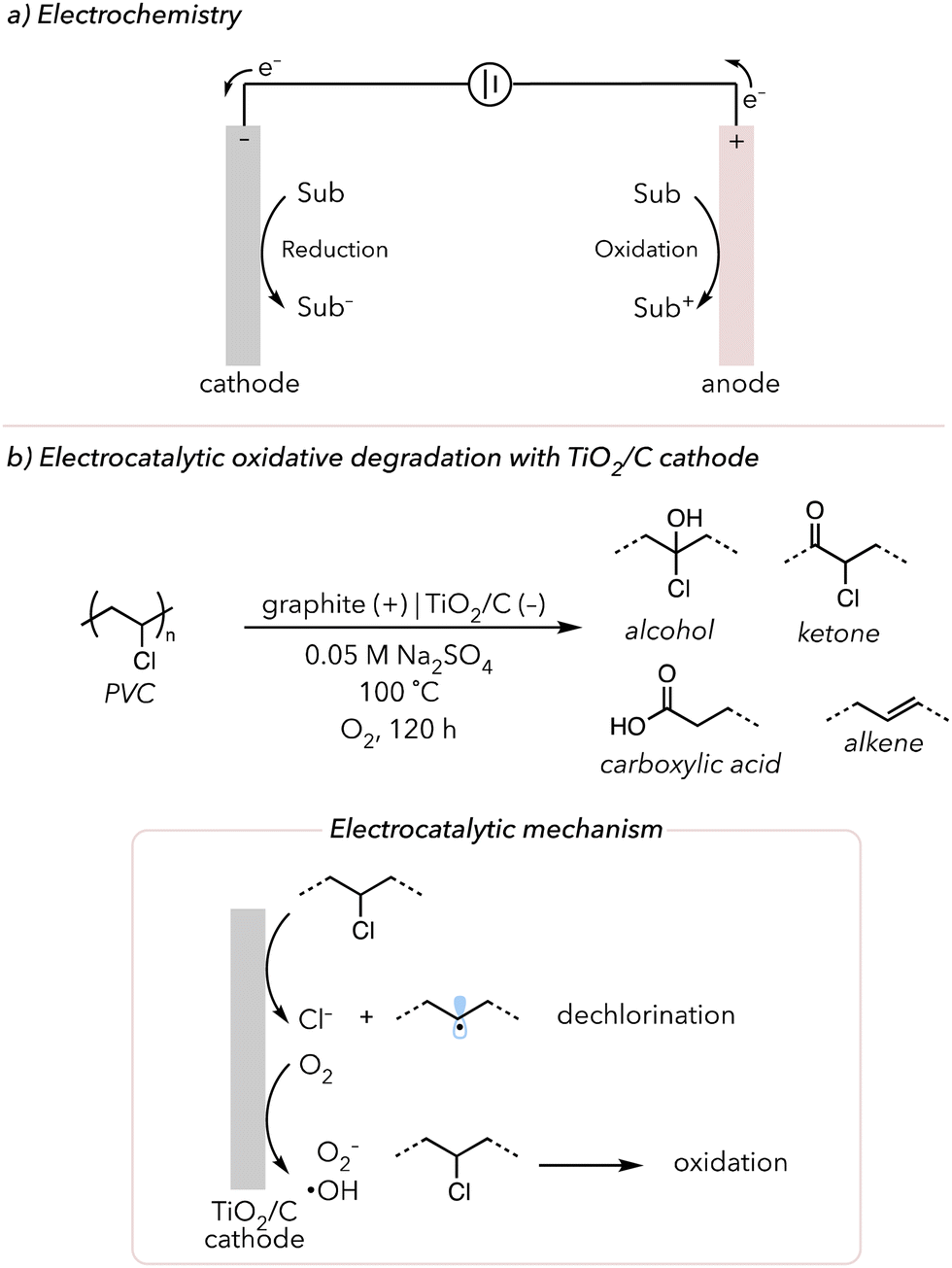 | ||
| Fig. 10 (a) A brief introduction to electrochemistry. (b) Electrocatalytic oxidative degradation of PVC using TiO2/C as cathode. | ||
As with photochemistry, electrochemistry has been used for both the synthesis of compounds under mild conditions and the degradation of toxic small molecules.160,161 In polymer chemistry, various electrochemical processes have been shown to upcycle polymers into more valuable materials or efficiently degrade many different polymers, including polyolefins and biomass.162–171 This section presents a few outstanding examples of electrocatalytic oxidative degradation of polymers with possible mechanisms.
In 2020, Gao and coworkers developed an electro-Fenton-like method using a TiO2/graphite (TiO2/C) cathode to degrade poly(vinyl chloride) (PVC) (Fig. 10(b)).172 Using electrochemistry to generate ROS to degrade organic pollutants and polyolefin microplastics has been studied in the past.173,174 Moreover, the dehalogenation of compounds with chlorines can be conducted electrochemically.175 Thus, using these two concepts, the Gao group attempted to oxidatively degrade PVC and obtain small oxidized products. Their previous work showed that their TiO2/C cathode can reduce O2 to hydroxyl radicals. They then applied their method to oxidatively degrade PVC using their TiO2/C cathode under the O2-rich environment at −0.7 V vs. Ag/AgCl at 100 °C for 6 h. According to their FTIR data, carbonyl and alcohol peaks appeared while the signals for C–Cl and C–H peaks decreased, indicating that the oxidative degradation and dechlorination of PVC are occurring. Approximately 56 wt% of PVC was removed, and 75% of C–Cl bonds were dechlorinated. When assessing the small molecule products remaining after degradation, various oxidized products, like oxalic acids, were observed but eventually decomposed to CO2 or water. Their proposed mechanism indicated that O2 reduction and dehalogenation occur at the TiO2/C cathode. Hydroxyl radicals oxidatively attack saturated PVC and convert polymer to small alcohols or carboxylic acids, which can be further oxidized to gaseous molecules.
In 2022, Román-Leshkov and coworkers demonstrated the use of electrochemically mediated HAT to degrade PS.176 In their system, N-hydroxyphthalimide (NHPI) was used as a mediator, a species used in an electrochemical reaction for facile electron transfer.177,178 NHPI is oxidized to a phthalimide N-oxyl (PINO) radical, abstracting C–H bonds from PS to trigger oxidative degradation. They could degrade PS and identify various products, from benzoyl to pentamer products. In 2024, Fors and coworkers investigated the oxidative degradation of ether-containing polymers such as poly(vinyl ether)s (PVEs), polypropylene oxide (PPO), and polyurethanes (PU).179 In their optimized electrolysis with 0.2 equiv. NHPI, isotatic polypropylene oxide (iPPO) was degraded to acetic acid (14 mol%), formic acid (12 mol%), and acetaldehyde (4 mol%) in 25 h (Fig. 11(a)). Due to the electrophilic nature of N-oxyl radicals, PINO selectively targets electron rich C–H bonds on polymer backbone. Thus, the Fors group also showed that PINO preferentially degraded ether-containing polymers over polymers with electron-poor C–H bonds, such as PMA and poly(vinyl acetate) (PVAc). In their proposed mechanism (Fig. 11(b)), NHIP is converted to PINO through oxidation at the anode, followed by HAT on the polymer backbone to initiate oxidative degradation. Electrons are consumed at the cathode to make hydrogen gas in reaction with protons. The Román-Leshkov and the Fors groups suggested that pyridine and acetic acid are essential in their systems. Pyridine acts as a base to deprotonate NHIP before the oxidation at the anode, while acetic acid acts as a proton source for the sacrificial generation of hydrogen gas.
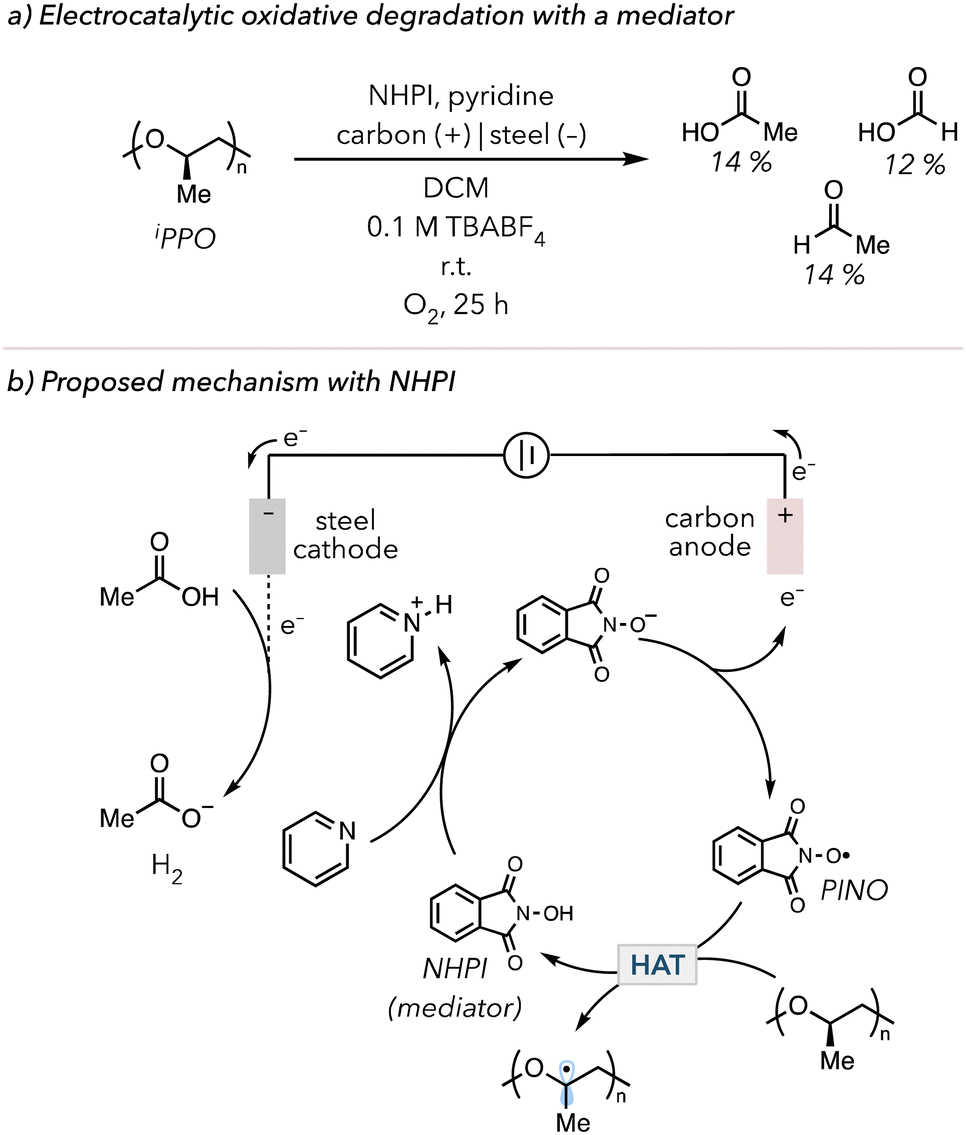 | ||
| Fig. 11 (a) Electrocatalytic oxidative degradation of iPPO with NHPI as a mediator of the electrochemical process. (b) Electrocatalytic oxidative degradation mechanism with NHPI. | ||
5.3 Mechanochemically induced oxidative degradation of polymers
In 2021, Luzinov and coworkers developed mechanochemical processes to oxidatively degrade PS.180 Mechanochemistry uses mechanical force to conduct chemical reactions, replacing other stimuli such as heat or light. It has previously been shown that polymers can be degraded under mechanochemical force, but these studies provide a more in-depth mechanistic understanding.181 In their degradation setup, 1.5 g commercial PS sample was ball-milled for 12 h in air with either hardened steel or tungsten carbide balls. In all cases, the high molecular weight original PS (>80000 g mol−1) was degraded to a much smaller polymer (<8000 g mol−1). In air, approximately 7–8 wt% of volatile organic chemicals were formed primarily styrene. NMR spectra revealed the presence of benzoic acid in the reaction mixture, indicating that PS was oxidatively degraded as well. However, no volatile chemicals were formed when PS was ball-milled under an argon atmosphere. Additionally, no volatile chemicals were detected when PS was ball-milled with a metal-free ball (silicon nitride), even under air. Considering these experiments, they proposed that a combination effect of ball milling and O2 renders efficient degradation of PS. First, ball milling causes C–C bond chain scissions; either O2 or metals trap the resulting carbon radicals. The peroxy radicals lead to oxidative degradation while the metal catalyzes the depolymerization to recover styrene. Although this type of degradation does not yield oxidized products like benzoic acid, O2 is still vital in speeding up the generation of monomers. Even though there are only a few examples of oxidative degradation through mechanochemistry, this field has a potential for further development in future.182,183
Like mechanochemistry, sonochemistry uses ultrasonic sound to trigger either C–C bond scissions or the generation of ROS.184–187 In particular, the rate of polymer degradation increases as more O2 content is present.
6. Conclusion, challenges, and future directions
Oxidative degradation of polymers is an important upcycling method that complements traditional recycling techniques. Despite being studied for many decades, the oxidative degradation of polymers has recently gained more attention, using modern approaches in photochemistry, electrochemistry, and mechanochemistry. In the past, researchers focused on the oxidative degradation of polymers in natural environments (namely autooxidation). Still, more case studies showed creative designs for efficient polymer degradation in the laboratory. To accelerate the rate of oxidative degradation under mild conditions, researchers have used various metal catalysts, photocatalysts, and electrocatalysts that are appropriate for their respective stimuli.Despite the recent advances in this field, there are still many challenges and areas for improvement. The scope of polymers that can be oxidatively degraded needs to be broadened. While PS and polyolefins were thoroughly investigated, other polymers like PVC and polyacrylates are significantly less explored. Additionally, the selectivity of the small molecule products must be improved for commercial implementation. When PS is degraded, for example, over 50 mol% benzoic acid can be recovered and used for other subsequent reactions; however, for the different polymers, it is less likely to achieve specific small molecule products with high yields.188 For instance, PE can degrade to more than 10 small molecules, and overoxidation can cause these molecules to decompose further into CO2. Thus, it is necessary to achieve precise control over the oxidation rate of polyolefins to obtain high yields of specific small molecules while suppressing overoxidation. Lastly, chemists and engineers will need to collaborate to pilot studies on the oxidative degradation of plastics at a large scale. Although some photochemistry and thermal chemistry cases demonstrated the scalability of oxidative degradation, they are not substantial enough to resolve the world's plastic crisis. Moreover, most of the examples (including more contemporary examples) in the literature still use a high concentration of O2 (>1 bar O2 content in air). Such high oxygen concentrations bring about concerns for explosions, so it would be required that large reactors are designed that limit safety concerns but degrade polymers effectively.
Two distinct future directions could mitigate these challenges. The first is called hybrid recycling, in which two different recycling techniques are sequentially used (Fig. 12(a)).189 In 2022, Beckham and coworkers illustrated polymer waste valorization through oxidative degradation and biological funneling.190 Initially, they oxidatively degraded PS, PE, and poly(ethylene terephthalate) (PET) to generate numerous oxidized products, subsequently subjected to microorganisms to selectively produce small molecule products. The oxidized products from the degradation of polymers are more suitable feedstocks for microorganisms than the polymers themselves, as solubility in water increases with higher oxidation and shorter hydrocarbon chains.191 These groups engineered special microorganisms to convert these intermediate chemicals with acetate and carboxylate functionality to polyhydroxyalkanoates or β-ketoadipate. Their robust system allowed the formation of certain specific products from commonly used plastics. In 2023, Williams, and coworkers conducted similar studies in PS where they first converted PS to benzoic acid intermediates and then treated the residues to fungal metabolism to form pharmacologically relevant molecules.192 Therefore, these hybrid recycling systems showed the potential to upcycle waste plastics to valuable materials by altering the mechanistic pathways of bacteria or fungi.
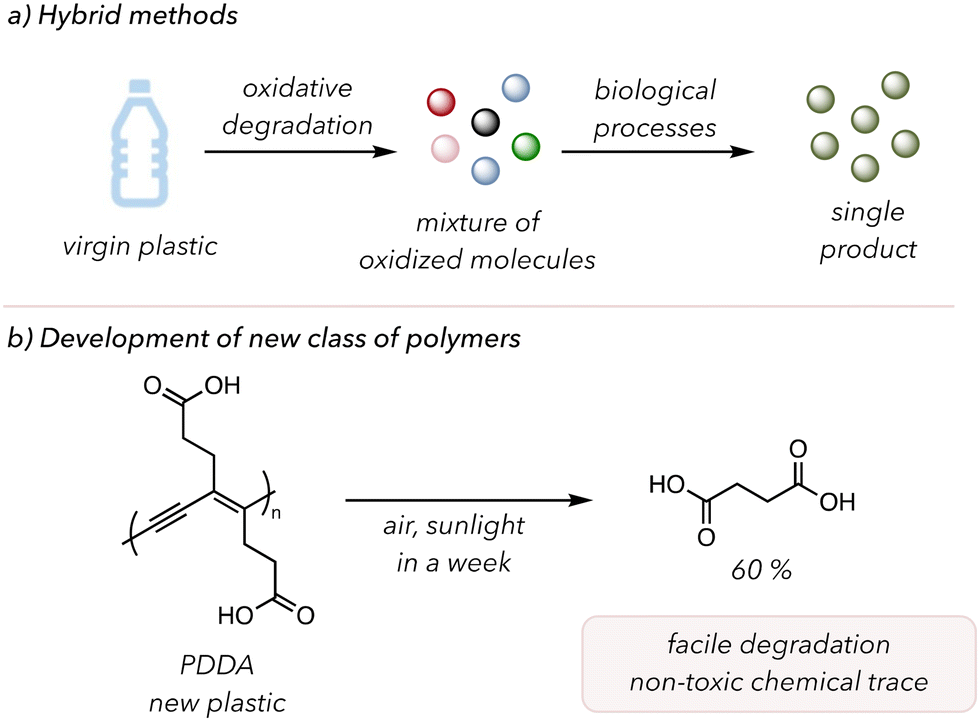 | ||
| Fig. 12 Potential future directions. (a) A hybrid between oxidative degradation and biological processes. (b) Development of new polymers that are functional and easily degradable. | ||
Another field of interest is the invention of a new class of polymers with comparable mechanical properties to those commonly used in our lives but are more prone to oxidative degradation. In 2021, Luo and coworkers synthesized a conjugated polymer susceptible to oxidative degradation after its service life.193 Conjugated polymers are often designed for electronic devices due to their delocalized π-systems. They polymerized deca-4,6-diynedioic acid to make highly conjugated poly(deca-4,6-diynedioic acid) (PDDA) (Fig. 12(b)). Under dark or nitrogen conditions, PDDA showed excellent stability as a functional material. However, upon exposure to sunlight and air in a week, PDDA completely degraded to succinic acids as a major product (60%). The highly conjugated PDDA was sufficiently reactive with O2 to result in efficient degradation, and no photocatalysts or oxidants were required. Moreover, the complete degradation of PDDA would not leave residual microplastics, and succinic acids, naturally occurring chemicals in plant or animal tissues, could be used as a monomer for synthesizing other polymers. Therefore, further developing more materials like PDDA would allow for polymer degradation under mild conditions and low concentrations of O2.
In conclusion, the future of oxidative degradation of polymers should be focused on improving product selectivity while increasing the degradation rate under mild settings close to environmental conditions. Although hybrid methods and the development of new materials are potential solutions, it would be even better to repurpose commercial plastics from waste streams and chemically upcycle them into a desired product in a single step.
Author contributions
S. O. and E. E. S. contributed to writing, reviewing, and editing the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by Cornell University and Princeton University. This research is supported in part by the Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science Early Career Award (DE-SC0024412).References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- P. F. Britt, G. W. Coates, K. I. Winey, J. Byers, E. Chen, B. Coughlin, C. Ellison, J. Garcia, A. Goldman, J. Guzman, J. Hartwig, B. Helms, G. Huber, C. Jenks, J. Martin, M. McCann, S. Miller, H. O’Neill, A. Sadow, S. Scott, L. Sita, D. Vlachos and R. Waymouth, Report of the Basic Energy Sciences Roundtable on Chemical Upcycling of Polymers, USDOE Office of Science (SC), United States, 2019 Search PubMed.
- A. O. C. Iroegbu, S. S. Ray, V. Mbarane, J. C. Bordado and J. P. Sardinha, ACS Omega, 2021, 6, 19343–19355 CrossRef CAS PubMed.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- D. V. A. Ceretti, M. Edeleva, L. Cardon and D. R. D’hooge, Molecules, 2023, 28, 2344 CrossRef CAS PubMed.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed.
- N. Singh, D. Hui, R. Singh, I. P. S. Ahuja, L. Feo and F. Fraternali, Composites, Part B, 2017, 115, 409–422 CrossRef CAS.
- J. Vohlídal, Chem. Teach. Int., 2021, 3, 213–220 CrossRef.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659–4679 CrossRef CAS.
- V. Bellotti, K. Parkatzidis, H. S. Wang, N. De Alwis Watuthanthrige, M. Orfano, A. Monguzzi, N. P. Truong, R. Simonutti and A. Anastasaki, Polym. Chem., 2023, 14, 253–258 RSC.
- T. M. McGuire, A. Buchard and C. Williams, J. Am. Chem. Soc., 2023, 145, 19840–19848 CrossRef CAS PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- B. A. Abel, R. L. Snyder and G. W. Coates, Science, 2021, 373, 783–789 CrossRef CAS PubMed.
- H. Li, J. Wu, Z. Jiang, J. Ma, V. M. Zavala, C. R. Landis, M. Mavrikakis and G. W. Huber, Science, 2023, 381, 660–666 CrossRef CAS PubMed.
- B. Chen, P. Perumal, M. Illikainen and G. Ye, J. Build. Eng., 2023, 71, 106386 CrossRef.
- L. Makarichi, W. Jutidamrongphan and K. Techato, Renewable Sustainable Energy Rev., 2018, 91, 812–821 CrossRef CAS.
- P. Singh, A. Boora and A. Kumar Gupta, IOP Conf. Ser.: Earth Environ. Sci., 2023, 1110, 012042 CrossRef.
- F. Zhang, F. Wang, X. Wei, Y. Yang, S. Xu, D. Deng and Y.-Z. Wang, J. Energy Chem., 2022, 69, 369–388 CrossRef CAS.
- X. Chen, Y. Wang and L. Zhang, ChemSusChem, 2021, 14, 4137–4151 CrossRef CAS PubMed.
- H. Chen, K. Wan, Y. Zhang and Y. Wang, ChemSusChem, 2021, 14, 4123–4136 CrossRef CAS PubMed.
- M. R. Karimi Estahbanati, X. Y. Kong, A. Eslami and H. S. Soo, ChemSusChem, 2021, 14, 4152–4166 CrossRef CAS PubMed.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- B. M. Stadler and J. G. de Vries, Philos. Trans. R. Soc., A, 2021, 379, 20200341 CrossRef CAS PubMed.
- H. Lv, F. Huang and F. Zhang, Langmuir, 2024, 40, 5077–5089 CrossRef CAS PubMed.
- E. Chiellini, A. Corti, S. D’Antone and R. Baciu, Polym. Degrad. Stab., 2006, 91, 2739–2747 CrossRef CAS.
- A. Nasir, T. Yasin and A. Islam, J. Appl. Polym. Sci., 2011, 119, 3315–3320 CrossRef CAS.
- F. Eisenreich, Angew. Chem., Int. Ed., 2023, 62, e202301303 Search PubMed.
- W. Li, W. Zhao, H. Zhu, Z.-J. Li and W. Wang, J. Mater. Chem. A, 2023, 11, 2503–2527 RSC.
- H. Zhou, Y. Wang, Y. Ren, Z. Li, X. Kong, M. Shao and H. Duan, ACS Catal., 2022, 12, 9307–9324 CrossRef CAS.
- Y. Zhang, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2023, 13, 3575–3590 CrossRef CAS.
- H. Abedsoltan, Polym. Eng. Sci., 2023, 63, 2651–2674 CrossRef CAS.
- D. Hetemi and J. Pinson, in Surface Modification of Polymers, ed. J. Pinson and D. Thiry, Wiley, 1st edn, 2019, pp. 211–240 Search PubMed.
- Z. Qin, J. Mou, C. Y. H. Chao, S. S. Chopra, W. Daoud, S. Leu, Z. Ning, C. Y. Tso, C. K. Chan, S. Tang, Z. J. Hathi, M. A. Haque, X. Wang and C. S. K. Lin, ChemSusChem, 2021, 14, 4103–4114 CrossRef CAS PubMed.
- A. Rovaletti, L. De Gioia, P. Fantucci, C. Greco, J. Vertemara, G. Zampella, F. Arrigoni and L. Bertini, Int. J. Mater. Sci., 2023, 24, 6368 CAS.
- C.-C. Chen, L. Dai, L. Ma and R.-T. Guo, Nat. Rev. Chem., 2020, 4, 114–126 CrossRef PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- R. A. Clark and M. P. Shaver, Chem. Rev., 2024, 124, 2617–2650 CrossRef CAS PubMed.
- V. Lohmann, G. R. Jones, N. P. Truong and A. Anastasaki, Chem. Sci., 2024, 15, 832–853 RSC.
- G. R. Jones, H. S. Wang, K. Parkatzidis, R. Whitefield, N. P. Truong and A. Anastasaki, J. Am. Chem. Soc., 2023, 145, 9898–9915 CrossRef CAS PubMed.
- Y. Miao, A. Von Jouanne and A. Yokochi, Polymers, 2021, 13, 449 CrossRef CAS PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- H. Taube, J. Gen. Physiol., 1965, 49, 29–50 CrossRef CAS PubMed.
- S. A. Cichello, J. Food Sci. Technol., 2015, 52, 1889–1895 CrossRef CAS PubMed.
- H. K. Mohammed, S. A. Jafar, J. I. Humadi, S. Sehgal, K. K. Saxena, G. H. Abdullah, L. I. Saeed, M. S. Salman and W. S. Abdullah, Mater. Today: Proc., 2023, S2214785323018643 Search PubMed.
- J. Aromaa, M. Kekkonen, M. Mousapour, A. Jokilaakso and M. Lundström, Corros. Mater. Degrad., 2021, 2, 625–640 CrossRef.
- J. D. Peterson, S. Vyazovkin and C. A. Wight, Macromol. Chem. Phys., 2001, 202, 775–784 CrossRef CAS.
- Z. Cao, J. Yi, Y. Ding, G. Sun and J. Yu, Constr. Build. Mater., 2023, 391, 131808 CrossRef CAS.
- A. V. Tobolsky, Discuss. Faraday Soc., 1947, 2, 384 RSC.
- J. F. Rabek, Comprehensive Chemical Kinetics, Elsevier, 1975, vol. 14, pp.425–538 Search PubMed.
- H. Zweifel, Stabilization of Polymeric Materials, Springer Berlin Heidelberg, Berlin, Heidelberg, 1998, pp.1–40 Search PubMed.
- R. Yang, J. Zhao and Y. Liu, Polym. Degrad. Stab., 2013, 98, 2466–2472 CrossRef CAS.
- M. E. Payne, O. O. Kareem, K. Williams-Pavlantos, C. Wesdemiotis and S. M. Grayson, Polym. Degrad. Stab., 2021, 183, 109388 CrossRef CAS.
- M. C. Celina, E. Linde and E. Martinez, Polym. Degrad. Stab., 2021, 188, 109550 CrossRef CAS.
- L. M. Smith, H. M. Aitken and M. L. Coote, Acc. Chem. Res., 2018, 51, 2006–2013 CrossRef CAS PubMed.
- S. Al-Malaika, Int. Mater. Rev., 2003, 48, 165–185 CrossRef CAS.
- J. A. Howard and K. U. Ingold, J. Am. Chem. Soc., 1968, 90, 1056–1058 CrossRef CAS.
- P. Gijsman, e-Polymers, 2008, 65, 1–34 Search PubMed.
- Q. Liu, J. Li, C. Cong, H. Cui, L. Xu, Y. Zhang, X. Meng and Q. Zhou, Polym. Degrad. Stab., 2020, 177, 109180 CrossRef CAS.
- T. Kashiwagi, A. Inaba, J. E. Brown, K. Hatada, T. Kitayama and E. Masuda, Macromolecules, 1986, 19, 2160–2168 CrossRef CAS.
- M. Doğan, Microsc. Res. Tech., 2021, 84, 2774–2783 CrossRef PubMed.
- E. Roduner, Chem. Soc. Rev., 2014, 43, 8226–8239 RSC.
- B. List, Chem. Rev., 2007, 107, 5413–5415 CrossRef CAS.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- A. M. Kulisiewicz, S. J. Garibay, G. R. Pozza, M. A. Browe, O. Sparr, S. Singh, L. A. Kelly and J. B. DeCoste, ACS Appl. Mater. Interfaces, 2023, 15, 40727–40734 CrossRef CAS PubMed.
- W. L. Hawkins, Polym. Eng. Sci., 1964, 4, 187–192 CrossRef CAS.
- H. C. Beachell and S. P. Nemphos, Ozone Chem. Technol., 1959, 26, 168–175 Search PubMed.
- F. Cataldo, Polym. Degrad. Stab., 2002, 75, 93–98 CrossRef CAS.
- J. Middleton, B. Burks, T. Wells, A. M. Setters, I. Jasiuk, P. Predecki, J. Hoffman and M. Kumosa, Polym. Degrad. Stab., 2013, 98, 436–445 CrossRef CAS.
- J. (Jim) Zhang, Y. Wei and Z. Fang, Front. Immunol., 2019, 10, 2518 CrossRef PubMed.
- C. De Gracia Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758–15764 CrossRef CAS PubMed.
- K. Wisetkhamsai, W. Patthaveekongka and W. Arayapranee, Polymers, 2023, 15, 1031 CrossRef CAS PubMed.
- R. D. Bach and H. B. Schlegel, J. Phys. Chem. A, 2020, 124, 4742–4751 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- E. Giannetti, J. Fluorine Chem., 2005, 126, 623–630 CrossRef.
- J. Zuquan, J. Mol. Struct., 1985, 123, 443–455 CrossRef.
- W. Bankeeree, C. Samathayanon, S. Prasongsuk, P. Lotrakul and S. Kiatkamjornwong, J. Polym. Environ., 2021, 29, 3964–3976 CrossRef CAS.
- Z. Zhang, J. Li, C. Wan, Y. Zhang and S. Wang, ACS Sustainable Chem. Eng., 2021, 9, 2378–2387 CrossRef CAS.
- E. Niki, C. Decker and F. R. Mayo, J. Polym. Sci., Polym. Chem. Ed., 1973, 11, 2813–2845 CrossRef CAS.
- B. Lv, G. Zhao, D. Li and C. Liang, Polym. Degrad. Stab., 2009, 94, 1047–1052 CrossRef CAS.
- H. Su, T. Li, L. Zhu and S. Wang, ChemSusChem, 2021, 14, 4270–4279 CrossRef CAS PubMed.
- X. Wei, Q. Zhang, C. Shen, X. Zhao, F. Zhang, X. Liu, G. Wu, S. Xu and Y.-Z. Wang, Mater. Horiz., 2023, 10, 3694–3701 RSC.
- C. M. Pichler, S. Bhattacharjee, M. Rahaman, T. Uekert and E. Reisner, ACS Catal., 2021, 11, 9159–9167 CrossRef CAS PubMed.
- V. V. Zefirov, I. V. Elmanovich, A. I. Stakhanov, A. A. Pavlov, S. V. Stakhanova, E. P. Kharitonova and M. O. Gallyamov, Polymers, 2022, 14, 744 CrossRef CAS PubMed.
- A. Pifer and A. Sen, Angew. Chem., Int. Ed., 1998, 37, 3306–3308 CrossRef CAS PubMed.
- X. Luo, J. Zhan, Q. Mei and S. Zhang, Green Chem., 2023, 25, 6717–6727 RSC.
- V. N. Parmon, G. I. Panov, A. Uriarte and A. S. Noskov, Catal. Today, 2005, 100, 115–131 CrossRef CAS.
- C. Aellig, C. Girard and I. Hermans, Angew. Chem., Int. Ed., 2011, 50, 12355–12360 Search PubMed.
- K. Yamamoto, T. Hosoya, K. Yoshioka, H. Miyafuji, H. Ohno and T. Yamada, ACS Sustainable Chem. Eng., 2017, 5, 10111–10115 CrossRef CAS.
- M. Das, R. Chacko and S. Varughese, ACS Sustainable Chem. Eng., 2018, 6, 1564–1571 CrossRef CAS.
- T. Hosoya, K. Yamamoto, H. Miyafuji and T. Yamada, RSC Adv., 2020, 10, 19199–19210 RSC.
- M. Zirbes, T. Graßl, R. Neuber and S. R. Waldvogel, Angew. Chem., Int. Ed., 2023, 62, e202219217 Search PubMed.
- D. H. Kim, A. Yu and M. Goh, J. Ind. Eng. Chem., 2021, 96, 76–81 CrossRef CAS.
- H. Yorimitsu, M. Kotora and N. T. Patil, Chem. Rec., 2021, 21, 3335–3337 CrossRef CAS PubMed.
- Z. Osawa, Polym. Degrad. Stab., 1988, 20, 203–236 CrossRef CAS.
- H.-M. Feng, J.-C. Zheng, N.-Y. Lei, L. Yu, K. H.-K. Kong, H.-Q. Yu, T.-C. Lau and M. H. W. Lam, Environ. Sci. Technol., 2011, 45, 744–750 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- A. J. Sipinen and D. R. Rutherford, J. Environ. Polym. Degrad., 1993, 1, 193–202 CrossRef CAS.
- W. Partenheimer, Catal. Today, 2003, 81, 117–135 CrossRef CAS.
- A. Holmström, A. Andersson and E. M. Sörvik, Eur. Polym. J., 1977, 13, 483–487 CrossRef.
- E. Andersen, L. H. Bertelsen, M. Salomonsen, M. Kristensen, P. Kybelund, M. B. Sørensen and M. Hinge, Polym. Degrad. Stab., 2020, 178, 109183 CrossRef CAS.
- H. Luo, E. P. Weeda, M. Alherech, C. W. Anson, S. D. Karlen, Y. Cui, C. E. Foster and S. S. Stahl, J. Am. Chem. Soc., 2021, 143, 15462–15470 CrossRef CAS PubMed.
- Y. T. Al-Majali, S. Forshey and J. P. Trembly, Thermochim. Acta, 2022, 713, 179226 CrossRef CAS.
- R. Aravind, A. K. Sahu, G. S. Brahma and T. Swain, ACS Omega, 2021, 6, 29869–29881 CrossRef CAS PubMed.
- R. V. Jagadeesh, H. Junge, M.-M. Pohl, J. Radnik, A. Brückner and M. Beller, J. Am. Chem. Soc., 2013, 135, 10776–10782 CrossRef CAS PubMed.
- E. O. Fomin, E. S. Trofimchuk, M. A. Moskvina and N. I. Nikonorova, Polym. Sci., Ser. B, 2022, 64, 657–669 CrossRef CAS.
- S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- A. L. Andrady, P. W. Barnes, J. F. Bornman, T. Gouin, S. Madronich, C. C. White, R. G. Zepp and M. A. K. Jansen, Sci. Total Environ., 2022, 851, 158022 CrossRef CAS PubMed.
- T. Seguchi, S. Hashimoto, K. Arakawa, N. Hayakawa, W. Kawakami and I. Kuriyama, Radiat. Phys. Chem., 1981, 17, 195–201 CrossRef CAS.
- O. Chiantore, L. Trossarelli and M. Lazzari, Polymer, 2000, 41, 1657–1668 CrossRef CAS.
- E. Niki, Y. Yamamoto and Y. Kamiya, Stabilization and Degradation of Polymers, 1978, vol. 7, pp.78–95 Search PubMed.
- A. A. Dalinkevich, I. M. Piskarev, L. V. Fomin and T. A. Nenasheva, High Energy Chem., 2023, 57, 252–257 CrossRef CAS.
- E. Yousif and R. Haddad, SpringerPlus, 2013, 2, 398 CrossRef PubMed.
- Q. Y. Lee and H. Li, Micromachines, 2021, 12, 907 CrossRef PubMed.
- N. Grassie and N. A. Weir, J. Appl. Polym. Sci., 1965, 9, 975–986 CrossRef CAS.
- N. Grassie and N. A. Weir, J. Appl. Polym. Sci., 1965, 9, 987–998 CrossRef.
- J. F. Rabek and B. Ranby, Polym. Eng. Sci., 1975, 15, 40–43 CrossRef CAS.
- N. N. H. Azhar, A. Cheng, S. Y. Lee and D. T. C. Ang, J. Polym. Res., 2022, 29, 476 CrossRef CAS.
- K. Parkatzidis, H. S. Wang and A. Anastasaki, Angew. Chem., Int. Ed., 2024, e202402436 Search PubMed.
- N. A. Wong and H. Bahmani, Heliyon, 2022, 8, e10282 CrossRef PubMed.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. Vega Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. McInnes, X. Qi and J. Xiao, J. Am. Chem. Soc., 2022, 144, 6532–6542 CrossRef CAS PubMed.
- S. Xu, S. Liu, W. Song and N. Zheng, Green Chem., 2024, 26, 1363–1369 RSC.
- H. Kaczmarek, A. Kamińska, M. Świątek and S. Sanyal, Eur. Polym. J., 2000, 36, 1167–1173 CrossRef CAS.
- T. Li, A. Vijeta, C. Casadevall, A. S. Gentleman, T. Euser and E. Reisner, ACS Catal., 2022, 12, 8155–8163 CrossRef CAS PubMed.
- N. F. Nikitas, E. Skolia, P. L. Gkizis, I. Triandafillidi and C. G. Kokotos, Green Chem., 2023, 25, 4750–4759 RSC.
- J. Meng, Y. Zhou, D. Li and X. Jiang, Sci. Bull., 2023, 68, 1522–1530 CrossRef CAS PubMed.
- S.-B. Tang, Y.-X. Jiang, K. Li, Z.-X. Wang and J. Su, Ind. Eng. Chem. Res., 2024, 63, 4817–4824 CrossRef CAS.
- S. Bonciolini, T. Noël and L. Capaldo, Eur. J. Org. Chem., 2022, e202200417 CrossRef CAS.
- G. Zhang, Z. Zhang and R. Zeng, Chin. J. Chem., 2021, 39, 3225–3230 CrossRef CAS.
- M. Wang, J. Wen, Y. Huang and P. Hu, ChemSusChem, 2021, 14, 5049–5056 CrossRef CAS PubMed.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- S. Oh and E. E. Stache, ACS Catal., 2023, 13, 10968–10975 CrossRef CAS.
- A. Ong, Z. C. Wong, K. L. O. Chin, W. W. Loh, M. H. Chua, S. J. Ang and J. Y. C. Lim, Chem. Sci., 2024, 15, 1061–1067 RSC.
- C. Walling, M. S. Kharasch and F. R. Mayo, J. Am. Chem. Soc., 1939, 61(10), 2693–2696 CrossRef CAS.
- D. L. Langer, S. Oh and E. E. Stache, Chem. Sci., 2024, 15, 1840–1845 RSC.
- C. P. Ward, C. J. Armstrong, A. N. Walsh, J. H. Jackson and C. M. Reddy, Environ. Sci. Technol. Lett., 2019, 6, 669–674 CrossRef CAS.
- K. C. Harper, E. G. Moschetta, S. V. Bordawekar and S. J. Wittenberger, ACS Cent. Sci., 2019, 5, 109–115 CrossRef CAS PubMed.
- J. Ran, A. Talebian-Kiakalaieh, S. Zhang, E. M. Hashem, M. Guo and S.-Z. Qiao, Chem. Sci., 2024, 15, 1611–1637 RSC.
- W. Fa, J. Wang, S. Ge and C. Chao, Polym. Bull., 2020, 77, 1417–1432 CrossRef CAS.
- V. Raditoiu, A. Raditoiu, M. F. Raduly and L. E. Wagner, Mater. Plast., 2019, 56, 92–96 CrossRef.
- Z. Peng, R. Chen and H. Li, ACS Sustainable Chem. Eng., 2023, 11, 10688–10697 CrossRef CAS.
- T. F. Nelson, C. M. Reddy and C. P. Ward, Environ. Sci. Technol., 2021, 55, 8898–8907 CrossRef CAS PubMed.
- X. Jiao, K. Zheng, Q. Chen, X. Li, Y. Li, W. Shao, J. Xu, J. Zhu, Y. Pan, Y. Sun and Y. Xie, Angew. Chem., Int. Ed., 2020, 59, 15497–15501 Search PubMed.
- P. Kamalian, S. N. Khorasani, A. Abdolmaleki, M. Karevan, S. Khalili, M. Shirani and R. E. Neisiany, J. Polym. Eng., 2020, 40, 181–191 CAS.
- J. D. Acuña-Bedoya, E. Luévano-Hipólito, E. I. Cedillo-González, L. P. Domínguez-Jaimes, A. M. Hurtado and J. M. Hernández-López, J. Environ. Chem. Eng., 2021, 9, 106208 CrossRef.
- V. H. Pino-Ramos, E. Bucio and D. Díaz, Polym. Degrad. Stab., 2021, 190, 109648 CrossRef CAS.
- P. Amato, M. Fantauzzi, F. Sannino, I. Ritacco, G. Santoriello, M. Farnesi Camellone, C. Imparato, A. Bifulco, G. Vitiello, L. Caporaso, A. Rossi and A. Aronne, J. Hazard. Mater., 2024, 463, 132907 CrossRef CAS PubMed.
- R. Cao, M.-Q. Zhang, C. Hu, D. Xiao, M. Wang and D. Ma, Nat. Commun., 2022, 13, 4809 CrossRef CAS PubMed.
- X. Cui, Q. Ruan, X. Zhuo, X. Xia, J. Hu, R. Fu, Y. Li, J. Wang and H. Xu, Chem. Rev., 2023, 123, 6891–6952 CrossRef CAS PubMed.
- M. Zhang, S. Cai, J. Li, E. A. Elimian, J. Chen and H. Jia, J. Hazard. Mater., 2021, 412, 125266 CrossRef CAS PubMed.
- E. Agbovhimen Elimian, M. Zhang, Y. Sun, J. He and H. Jia, Sol. RRL, 2023, 7, 2300238 CrossRef CAS.
- Y. Liu, Q. Zhong, P. Xu, H. Huang, F. Yang, M. Cao, L. He, Q. Zhang and J. Chen, Matter, 2022, 5, 1305–1317 CrossRef CAS.
- C. Xing, H. Cai, D. Kang and W. Sun, Adv. Energy Sustainability Res., 2023, 4, 2300015 CrossRef CAS.
- L. H. Kugelmass, C. Tagnon and E. E. Stache, J. Am. Chem. Soc., 2023, 145, 16090–16097 CrossRef CAS PubMed.
- Y. Miao, Y. Zhao, G. I. N. Waterhouse, R. Shi, L.-Z. Wu and T. Zhang, Nat. Commun., 2023, 14, 4242 CrossRef CAS PubMed.
- G. Firestone, H. Huang, J. R. Bochinski and L. I. Clarke, Nanotechnology, 2019, 30, 475706 CrossRef CAS PubMed.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- N. E. S. Tay, D. Lehnherr and T. Rovis, Chem. Rev., 2022, 122, 2487–2649 CrossRef CAS PubMed.
- J. Rein, S. B. Zacate, K. Mao and S. Lin, Chem. Soc. Rev., 2023, 52, 8106–8125 RSC.
- K. Yang, M. Ji, B. Liang, Y. Zhao, S. Zhai, Z. Ma and Z. Yang, J. Hazard. Mater., 2020, 398, 122892 CrossRef PubMed.
- X. Long, D. Li, R. Huang, S. Huang and S. Zhang, ACS Sustainable Chem. Eng., 2024, 12, 5023–5035 CrossRef CAS.
- M. Kiendrebeogo, M. R. Karimi Estahbanati, Y. Ouarda, P. Drogui and R. D. Tyagi, Sci. Total Environ., 2022, 808, 151897 CrossRef CAS PubMed.
- K. Fukatsu and S. Kokot, Polym. Degrad. Stab., 2001, 72, 353–359 CrossRef CAS.
- H. Du, Q. Wang, G. Chen and J. Wang, TrAC, Trends Anal. Chem., 2022, 157, 116815 CrossRef CAS.
- Y. Han, S. Zhang, X. Zhang and J. Chen, Int. J. Electrochem. Sci., 2020, 15, 3382–3399 CrossRef CAS.
- Y. Zhao, M. Sun, Y. Zhang, Y. Zhao and H. Ge, J. Environ. Chem. Eng., 2022, 10, 107795 CrossRef CAS.
- D. Rauber, T. K. F. Dier, D. A. Volmer and R. Hempelmann, Z. Phys. Chem., 2018, 232, 189–208 CrossRef CAS.
- S. Behera, S. Dinda, R. Saha and B. Mondal, ACS Catal., 2023, 13, 469–474 CrossRef CAS.
- R. S. Weber and K. K. Ramasamy, ACS Omega, 2020, 5, 27735–27740 CrossRef CAS PubMed.
- I. Maksso, R. C. Samanta, Y. Zhan, K. Zhang, S. Warratz and L. Ackermann, Chem. Sci., 2023, 14, 8109–8118 RSC.
- Q. Dong, A. D. Lele, X. Zhao, S. Li, S. Cheng, Y. Wang, M. Cui, M. Guo, A. H. Brozena, Y. Lin, T. Li, L. Xu, A. Qi, I. G. Kevrekidis, J. Mei, X. Pan, D. Liu, Y. Ju and L. Hu, Nature, 2023, 616, 488–494 CrossRef CAS PubMed.
- F. Miao, Y. Liu, M. Gao, X. Yu, P. Xiao, M. Wang, S. Wang and X. Wang, J. Hazard. Mater., 2020, 399, 123023 CrossRef CAS PubMed.
- F. Miao, M. Gao, X. Yu, P. Xiao, M. Wang, Y. Wang, S. Wang and X. Wang, Electrochem. Commun., 2020, 113, 106687 CrossRef CAS.
- M. A. Oturan and J.-J. Aaron, Crit. Rev. Environ. Sci. Technol., 2014, 44, 2577–2641 CrossRef CAS.
- Y. Liu, R. Mao, Y. Tong, H. Lan, G. Zhang, H. Liu and J. Qu, Electrochim. Acta, 2017, 232, 13–21 CrossRef CAS.
- B. Yan, C. Shi, G. T. Beckham, E. Y.-X. Chen and Y. Román-Leshkov, ChemSusChem, 2022, 15, e202102317 CrossRef CAS PubMed.
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS PubMed.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- J. H. Hsu, T. E. Ball, S. Oh, E. E. Stache and B. P. Fors, Angew. Chem., Int. Ed., 2024, 63, e202316578 Search PubMed.
- V. P. Balema, I. Z. Hlova, S. L. Carnahan, M. Seyedi, O. Dolotko, A. J. Rossini and I. Luzinov, New J. Chem., 2021, 45, 2935–2938 RSC.
- J. Sohma, Prog. Polym. Sci., 1989, 14, 451–596 CrossRef CAS.
- V. S. Nguyen, Y. Chang, E. V. Phillips, J. A. Devitt and C. Sievers, ACS Sustainable Chem. Eng., 2023, 11, 7617–7623 CrossRef CAS.
- Y. Chang, S. J. Blanton, R. Andraos, V. S. Nguyen, C. L. Liotta, F. J. Schork and C. Sievers, ACS Sustainable Chem. Eng., 2024, 12, 178–191 CrossRef CAS PubMed.
- A. V. Mohod and P. R. Gogate, Ultrason. Sonochem., 2011, 18, 727–734 CrossRef CAS PubMed.
- E. Bäckström, K. Odelius and M. Hakkarainen, Ind. Eng. Chem. Res., 2017, 56, 14814–14821 CrossRef.
- R. Vinu and G. Madras, Ultrason. Sonochem., 2011, 18, 608–616 CrossRef CAS PubMed.
- M. Das and S. Varughese, ACS Sustainable Chem. Eng., 2016, 4, 2080–2087 CrossRef CAS.
- Y. Qin, T. Zhang, H. Y. V. Ching, G. S. Raman and S. Das, Chem, 2022, 8, 2472–2484 CAS.
- N. Yan, Science, 2022, 378, 132–133 CrossRef CAS PubMed.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl and G. T. Beckham, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- J. Savoldelli, D. Tomback and H. Savoldelli, Waste Manage., 2017, 60, 123–126 CrossRef CAS PubMed.
- C. Rabot, Y. Chen, S.-Y. Lin, B. Miller, Y.-M. Chiang, C. E. Oakley, B. R. Oakley, C. C. C. Wang and T. J. Williams, J. Am. Chem. Soc., 2023, 145, 5222–5230 CrossRef CAS PubMed.
- S. Tian, Q. Yue, C. Liu, M. Li, M. Yin, Y. Gao, F. Meng, B. Z. Tang and L. Luo, J. Am. Chem. Soc., 2021, 143, 10054–10058 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |