
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactive capture and electrochemical conversion of CO2 with ionic liquids and deep eutectic solvents
Saudagar
Dongare
 a,
Muhammad
Zeeshan
a,
Ahmet Safa
Aydogdu
bc,
Ruth
Dikki
a,
Samira F.
Kurtoğlu-Öztulum
bcd,
Oguz Kagan
Coskun
a,
Miguel
Muñoz
a,
Avishek
Banerjee
e,
Manu
Gautam
f,
R. Dominic
Ross
g,
Jared S.
Stanley
h,
Rowan S.
Brower
i,
Baleeswaraiah
Muchharla
j,
Robert L.
Sacci
k,
Jesús M.
Velázquez
i,
Bijandra
Kumar
j,
Jenny Y.
Yang
h,
Christopher
Hahn
g,
Seda
Keskin
bc,
Carlos G.
Morales-Guio
e,
Alper
Uzun
bcl,
Joshua M.
Spurgeon
f and
Burcu
Gurkan
*a
a,
Muhammad
Zeeshan
a,
Ahmet Safa
Aydogdu
bc,
Ruth
Dikki
a,
Samira F.
Kurtoğlu-Öztulum
bcd,
Oguz Kagan
Coskun
a,
Miguel
Muñoz
a,
Avishek
Banerjee
e,
Manu
Gautam
f,
R. Dominic
Ross
g,
Jared S.
Stanley
h,
Rowan S.
Brower
i,
Baleeswaraiah
Muchharla
j,
Robert L.
Sacci
k,
Jesús M.
Velázquez
i,
Bijandra
Kumar
j,
Jenny Y.
Yang
h,
Christopher
Hahn
g,
Seda
Keskin
bc,
Carlos G.
Morales-Guio
e,
Alper
Uzun
bcl,
Joshua M.
Spurgeon
f and
Burcu
Gurkan
*a
aChemical and Biomolecular Engineering, Case Western Reserve University, Cleveland, OH 44106, USA. E-mail: beg23@case.edu
bDepartment of Chemical and Biological Engineering, Koç University, Rumelifeneri Yolu, Sariyer, 34450 Istanbul, Turkey
cKoç University TÜPRAŞ Energy Center (KUTEM), Koç University, Rumelifeneri Yolu, Sariyer, 34450 Istanbul, Turkey
dDepartment of Materials Science and Technology, Faculty of Science, Turkish-German University, Sahinkaya Cad., Beykoz, 34820 Istanbul, Turkey
eDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, CA 90095, USA
fConn Center for Renewable Energy Research, University of Louisville, Louisville, KY 40292, USA
gMaterials Science Division, Lawrence Livermore National Laboratory, Livermore, CA 94550, USA
hDepartment of Chemistry, University of California, Irvine, Irvine, CA 92697, USA
iDepartment of Chemistry, University of California, Davis, Davis, CA 95616, USA
jDepartment of Mathematics, Computer Science, & Engineering Technology, Elizabeth City State University, 1704 Weeksville Road, Elizabeth City, NC 27909, USA
kChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37830, USA
lKoç University Surface Science and Technology Center (KUYTAM), Koç University, Rumelifeneri Yolu, Sariyer, 34450 Istanbul, Turkey
First published on 24th June 2024
Abstract
Ionic liquids (ILs) and deep eutectic solvents (DESs) have tremendous potential for reactive capture and conversion (RCC) of CO2 due to their wide electrochemical stability window, low volatility, and high CO2 solubility. There is environmental and economic interest in the direct utilization of the captured CO2 using electrified and modular processes that forgo the thermal- or pressure-swing regeneration steps to concentrate CO2, eliminating the need to compress, transport, or store the gas. The conventional electrochemical conversion of CO2 with aqueous electrolytes presents limited CO2 solubility and high energy requirement to achieve industrially relevant products. Additionally, aqueous systems have competitive hydrogen evolution. In the past decade, there has been significant progress toward the design of ILs and DESs, and their composites to separate CO2 from dilute streams. In parallel, but not necessarily in synergy, there have been studies focused on a few select ILs and DESs for electrochemical reduction of CO2, often diluting them with aqueous or non-aqueous solvents. The resulting electrode–electrolyte interfaces present a complex speciation for RCC. In this review, we describe how the ILs and DESs are tuned for RCC and specifically address the CO2 chemisorption and electroreduction mechanisms. Critical bulk and interfacial properties of ILs and DESs are discussed in the context of RCC, and the potential of these electrolytes are presented through a techno-economic evaluation.
 Saudagar Dongare | Saudagar Dongare is a Postdoctoral Researcher at Case Western Reserve University in the Gurkan group. He is a member of the 4C EFRC Energy Early Career Network. He received his PhD in Chemical Engineering from Thapar Institute of Engineering and Technology, India. His research interests include environmental electrocatalysis, developing electrode materials and electrolytes for electrochemical CO2 reduction, and studying the electrode–electrolyte interfaces with in situ spectroscopy. |
 Left to Right (members of the Gurkan group at CWRU): Muhammad Zeeshan; Ruth Dikki; Miguel Muñoz; Oguz Kagan Coskun. | Dr Zeeshan is a postdoctoral researcher; he earned his PhD degree in Chemical Engineering from Koç University, Türkiye; his expertise is the development of composites for CO2 separations. Ruth Dikki is a PhD student; her thesis involves the synthesis and characterization of CO2 capture solvents. Miguel Muñoz is a PhD student and a member of the Student Research Organization of BEES2 EFRC; his thesis involves the understanding of transport and electron transfer reactions in structured electrolytes; he earned his MS degree in Chemical Science and Technology from the Autonomous University of Zacatecas, Mexico. Oguz Kagan Coskun is a PhD student; his thesis involves the probing of interfaces in electrocatalysis through electroanalytical techniques; he earned his MS degree in Metallurgical and Materials Engineering from Istanbul Technical University, Türkiye. |
 Left to Right (from Koç University): Ahmet Safa Aydogdu; Samira F. Kurtoğlu-Öztulum; Seda Kesin, Alper Uzun. | Ahmet Safa Aydogdu is a PhD student, co-advised by Prof. Seda Keskin and Prof. Alper Uzun in Chemical and Biological Engineering Department (CheBioEng) at Koç University (KU), Türkiye; his thesis focuses on ionic liquids and MOFs for CO2 capture and separation. Samira F. Kurtoğlu-Öztulum is an Assistant Professor at Turkish-German University; she got her PhD in CheBioEng from KU; her research focuses on catalysis for energy-related applications. Seda Keskin is a Professor and the Director of the Nanomaterials, Energy, and Molecular Modeling Research Group; she completed her PhD at Georgia Institute of Technology; her group currently focuses on MOFs for energy applications. Alper Uzun is a Professor and the Director of the UzunLab Materials for Energy Research Group at KU; previously he worked as an Associate Research Engineer at ConocoPhillips/Phillips66 Bartlesville Technology Center and completed postdoctoral training at the University of California, Berkeley after receiving his PhD from University of California, Davis. |
 Left to Right (members of 4C EFRC): Manu Gautam; Joshua M. Spurgeon; Avishek Banerjee; Carlos G. Morales-Guio. | Manu Gautam is a postdoctoral researcher in the Spurgeon group at the University of Louisville; his research involves the examination of contaminants in electrochemical CO2 reduction. Joshua M. Spurgeon is the Theme Leader for Solar Fuels research at the Conn Center; he was previously the Project Lead for the Interface group and Processing, Materials, and Integration Team at JCAP (2013). Avishek Banarjee is a PhD student in the Morales-Guio group at the University of California, Los Angeles working on CO2 electrolyzes; he earned his MS at McGill University. Carlos G. Morales-Guio is an Assistant Professor at the UCLA Samueli School of Engineering; he completed his postdoctoral training at Stanford University with a fellowship from the Swiss National Science Foundation; his group specializes in electrochemical engineering for catalytic processes. |
 Left to Right (members of 4C EFRC). Top row: Baleeswaraiah Muchharla; Bijandra Kumar; Jared S. Stanley; Jenny Y. Yang. Bottom Row: Rowan S. Brower; Jesús M. Velázquez; Robert L. Sacci; R. Dominic Ross; Christopher Hahn. | Baleeswaraiah Muchharla is a postdoctoral researcher in the Kumar group; Bijandra Kumar is an Associate Professor at the Elizabeth City State University researching CO2 reduction, energy storage, and sensors. Jared S. Stanley is a PhD student in the Yang group developing molecular catalysts; Jenny Yang is a Professor and Chancellor's Fellow at the University of California, Irvine; she is the Director of 4C EFRC. Rowan S. Brower is a PhD student in the Velázquez group developing catalysts; Jesús M. Velázquez is an Associate Professor at the University of California, Davis. Robert L. Sacci is the Interim Group Leader of the Energy Storage & Conversion Group at Oak Ridge National Laboratory; his group images solid–liquid interfaces. R. Dominic Ross is a postdoctoral researcher under the supervision of Christopher Hahn who is the Deputy Group Leader of the materials for Energy and Climate Security at Lawrence Livermore National Laboratory and the Deputy Director of 4C EFRC. |
 Burcu Gurkan | Burcu (Eksioglu) Gurkan is a Professor at Case Western Reserve University; she is the Associate Editor for ACS Applied Engineering Materials; she serves as the Deputy Director of BEES2 (Breakthrough Electrolytes for Energy Storage Systems) – a Department of Energy Frontier Research Center (EFRC) where she leads the effort in developing structured electrolytes for flow batteries and a Key Researcher in 4C (Center for Closing the Carbon Cycle) EFRC where her team develops functional electrolytes for capture and conversion of CO2. Her team synthesizes and investigates deep eutectic solvents and ionic liquids for solving problems in separations, electrocatalysis, energy storage and conversion. |
1. Introduction
The current carbon economy relies on fossil fuels for energy generation and the production of chemicals, resulting in record-high CO2 emissions.1,2 CO2 is a greenhouse gas and therefore, it is environmentally desirable to capture CO2 from the atmosphere and emission sources.1 CO2 capture from post-combustion flue gas and the atmosphere is critical for net-zero and net-negative emissions, respectively. To prevent further deleterious effects of CO2 in the atmosphere on climate change, approximately 10 gigatons of CO2/year needs to be removed to limit global warming to a maximum of 1.5 °C by 2050.3–6 This goal is ambitious and requires a cooperative effort to simultaneously implement emission mitigation and negative emission technologies in parallel to decarbonization efforts toward a more sustainable future. To incentivize these implementations, technologies for capturing and converting CO2 into higher value products need to become more energy efficient.2Currently, chemical separations make up 10–15% of all the energy consumed in the USA, according to the 2019 report by the National Academies of Sciences, Engineering and Medicine: “A Research Agenda for Transforming Separation Science”.3 In negative emission technologies,4 such as CO2 separation from direct air, achieving favorable energetics and practical gas–volume processability depends on our ability to design highly selective materials and interfaces.5,6 Further, the reduction of the captured CO2 to produce other carbon products such as fuels and commodity chemicals via chemical, thermal, biochemical, photochemical, and electrochemical reactions are possible.7–9 However, the critical challenges are the requirement of substantial energy to activate CO2, which is otherwise a very stable molecule, and the lack of highly selective catalysts. Therefore, materials that can multi-function for selective absorption and conversion with lower energy requirements have transformative potential for emission mitigation and negative emissions science, which are pressing societal needs. Another challenge when converting CO2 to fuels and feedstocks is the availability of CO2. While CO2 is present in many industrial waste streams, its concentration is typically low (Table 1), therefore it is necessary to concentrate CO2 from these dilute sources.
| Stream | Contents (vol%) | |||||
|---|---|---|---|---|---|---|
| CO2 | N2 | O2 | H2O | Other major gases | Trace contaminants | |
| Biogas | 30–50 | 0–1 | 0–1 | 15 | CH4 | |
| Cement Plant | 18 | 56.5 | 7.5 | 18.2 | — | CO, NOx, SO2, HCl |
| Flue gas (coal) | 11 | 76 | 6 | 10–18 | Ar | NOx, SO2, Hg, Cd, other heavy metals |
| Flue gas (natural gas) | 3 | 76 | 14 | 6 | Ar | NOx |
| Air | 0.04 | 78 | 21 | 1 | Ar | — |
Traditionally, CO2 separation and concentration are achieved by thermal, moisture, or pressure-swing processes utilizing sorbents and have substantial energy requirements;13 current capture approaches yield CO2 at a cost of up to $200 per ton. The energetic and capital costs are largely due to low CO2 concentration and sorbent regeneration. With decreasing partial pressure of CO2 in the available stream, sorbents with higher capacity and selectivity are desired. For example, the direct air capture (DAC) of CO2 is a critical technology platform for CO2 removal (carbon dioxide removal, CDR) to achieve negative emissions technologies.7,8 This goal necessitates the use of sorbents with high binding constants (i.e., enthalpy) for highly selective CO2 absorption at ambient temperature and pressures. Consequently, the CO2 release in the concentration step becomes more energy intensive. Further, the reactive contaminants in the CO2 streams, if not removed, can degrade the sorbent in the capture process and poison the catalyst in the post CO2 conversion process. Therefore, discovery of sorbents with high CO2 capacity and selectivity, stability (e.g., against humidity, thermal-swing, feed contaminants) and regenerability with minimal energy input remain a critical challenge.
The theoretical minimum energy cost for CO2 capture and concentration (CCC) from flue gas (∼5–15% CO2 concentration) using the aqueous amine (i.e., monoethanolamine, MEA) solvent, the benchmark, is calculated as 8.4 kJ mol−1.14 This requirement increases to 20 kJ mol−1 for DAC by earlier estimates and further increases to 30 kJ mol−1 when taking into account the irreversibility of practical sorbents.15 While the CCC from flue gas and DAC processes are not alternatives to each other, the energetics of various processes are often compared to provide a perspective of the needs within a portfolio of solutions. Most common sorbents require thermal swing operation for regeneration and this step is the most energy intensive step in the processes. In addition, sorbents exposed to high temperatures during thermal swing undergo thermal degradation, thus necessitating material or solvent replacement. Pressure- and thermal-swings also require significant infrastructure, reducing their applicability with smaller point sources. For example, regeneration of the sorbent in DAC is currently estimated to consume 95% of the total energy consumption and requires 75% of the capital cost.16,17 Combined capture and conversion bypasses the costly step of regeneration of the captured CO2 from sorbents. It also provides for direct, low temperature utilization that minimizes sorbent degradation. By eliminating the biggest energetic and capital cost from CCC, direct reduction of captured CO2 would make the largest difference in improving the overall efficiency from CO2-source-to-products.18 While pure CO2 from current DAC methods are expected to cost more than $200 per ton of CO2, combined carbon capture and utilization (CCU) is estimated to achieve carbon negative processes at $20–25 per ton of CO2.19
The potential economic and energy intensity advantages have led to broad interest in CCU strategies. However, most research and development has focused on integration and intensification of CCU at the systems and process level. There has not been as significant focus on tailoring the integration of CCU at the molecular scale for efficient and selective capture with direct conversion of captured CO2, or reactive capture of CO2 (RCC). While there has been substantial research in the two separate areas – electrochemical CO2 capture and concentration (eCCC)20 and catalysts for electrolytic CO2 reduction reactions (CO2RR), these two disciplines have often not been discussed together. While the eCCC community has focused on selectivity, stability, and efficiency at scale, efforts in CO2RR have primarily used pure CO2 streams to improve rate, selectivity, and durability. The unique metrics defined for successful eCCC and CO2RR do not necessarily translate to RCC. Fig. 1 illustrates the concepts of eCCC, CO2RR, and RCC.
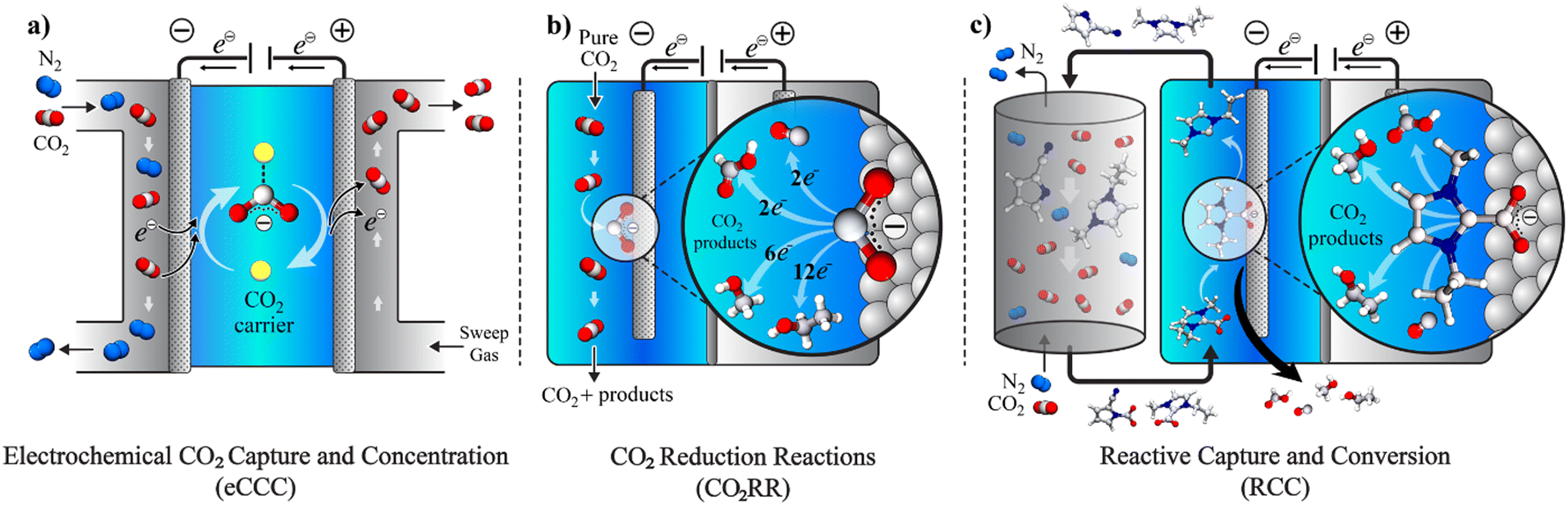 | ||
| Fig. 1 Schematic illustration of eCCC, CO2RR, and RCC processes. (a) A redox active carrier (represented by yellow sphere) binds with CO2 upon reduction (generating a nucleophile) at the cathode, which is a gas diffusion electrode in contact with feed gas, and the carrier releases the CO2 upon oxidation at the anode, thereby pumping CO2 from the feed to the effluent side in an eCCC process. (b) The CO2 that is physisorbed by the electrolyte is reduced to CO2˙− at the cathode (also the catalyst) in a CO2RR process and this intermediate then undergoes hydrogenation in the availability of a proton source, or C–C coupling at the cathode/catalyst surface, thus achieving heterogenous electrocatalytic conversion of CO2. (c) The electron is injected from the electrode to the chemisorbed CO2 either through a molecular catalyst or a functionalized electrolyte component, thus RCC achieves direct conversion of the chemisorbed CO2. | ||
In this review, we focus on materials and electrolytes that have the potential for the advancements in RCC. Specifically, the recent developments, key considerations, and current specific challenges in RCC with multifunctional solvents such as ionic liquids (ILs), deep eutectic solvents (DESs), and more generally electrolytes with IL and DES components are presented.
ILs are salts that melt below 100 °C, an arbitrary temperature chosen to differentiate ILs from molten salts.21 ILs are typically based on an organic bulky cation and an organic or inorganic anion. Since the review article22 by Welton published in 1999, interest in ILs has grown tremendously and they have been proposed for numerous applications such as catalysis, separations, energy storage, sensors, and drug delivery.23–28 Their versatility is associated with their tunability for specific applications through the immense structural diversity and combination of ions. ILs present lack of flammability, negligible vapor pressures, and generally high thermal stability, hence they are regarded as green solvents. The specific relation of ILs to RCC is their multifunctionality in synergizing the capture and conversion processes. More specifically, ILs have high CO2 absorption capacity and selectivity, with tunable absorption reaction enthalpy. Further, ILs are ionically conductive and can serve as the electrolyte in electrochemical conversion reactions in addition to serving as the liquid sorbent for the capture process. Since the functionalized ILs complex with CO2, they provide large availability of the reactants for the electrocatalytic conversion reaction (as long as the chemisorption complex is electrochemically active), thus enhancing their utility for RCC.
As alternatives to ILs, DESs are easier to obtain as they are mixtures of two or more components where the resulting liquid demonstrates significantly depressed melting point compared to the parent compounds (lower than the ideal liquid mixture).29 There are different classes of DESs as first introduced by Abbott.30,31 However, DESs that have been studied for CO2 capture and conversion are mostly those formed from mixtures of hydrogen bond acceptor (HBA) such as a halide salt and hydrogen bond donor (HBD) compounds such as urea and glycerol.32 DESs present similar advantages as ILs including low vapor pressures, high CO2 solubilities, high ionic conductivities, and generally tunable physical properties. As emphasized by Coutinho and colleagues,33 ‘deep eutectic’ behavior of these solvents is what distinguishes them from simple mixtures or eutectic mixtures. However, as DESs are adapted and further tuned for specific applications, the confirmation of the phase behavior gets overlooked and many simple mixtures are referred as DESs.33 As is the case, for DES applications in both CO2 capture and electrochemical conversion where the formulations for tuning viscosity, conductivity, solubility, polarity, and electrochemical stability for RCC yield mixtures that loose this classification identity. Further, it is often difficult to measure the melting point of these mixtures and often only a glass transition temperature is seen. Therefore, these solvents are also referred as low-transition-temperature mixtures (LTTMs).34 Nevertheless, these mixtures still offer some of the advantageous properties such as low volatility and high solubilities.35 Example structures for ILs and DESs developed or utilized for CO2 capture and electrochemical conversion are shown in Fig. 2.
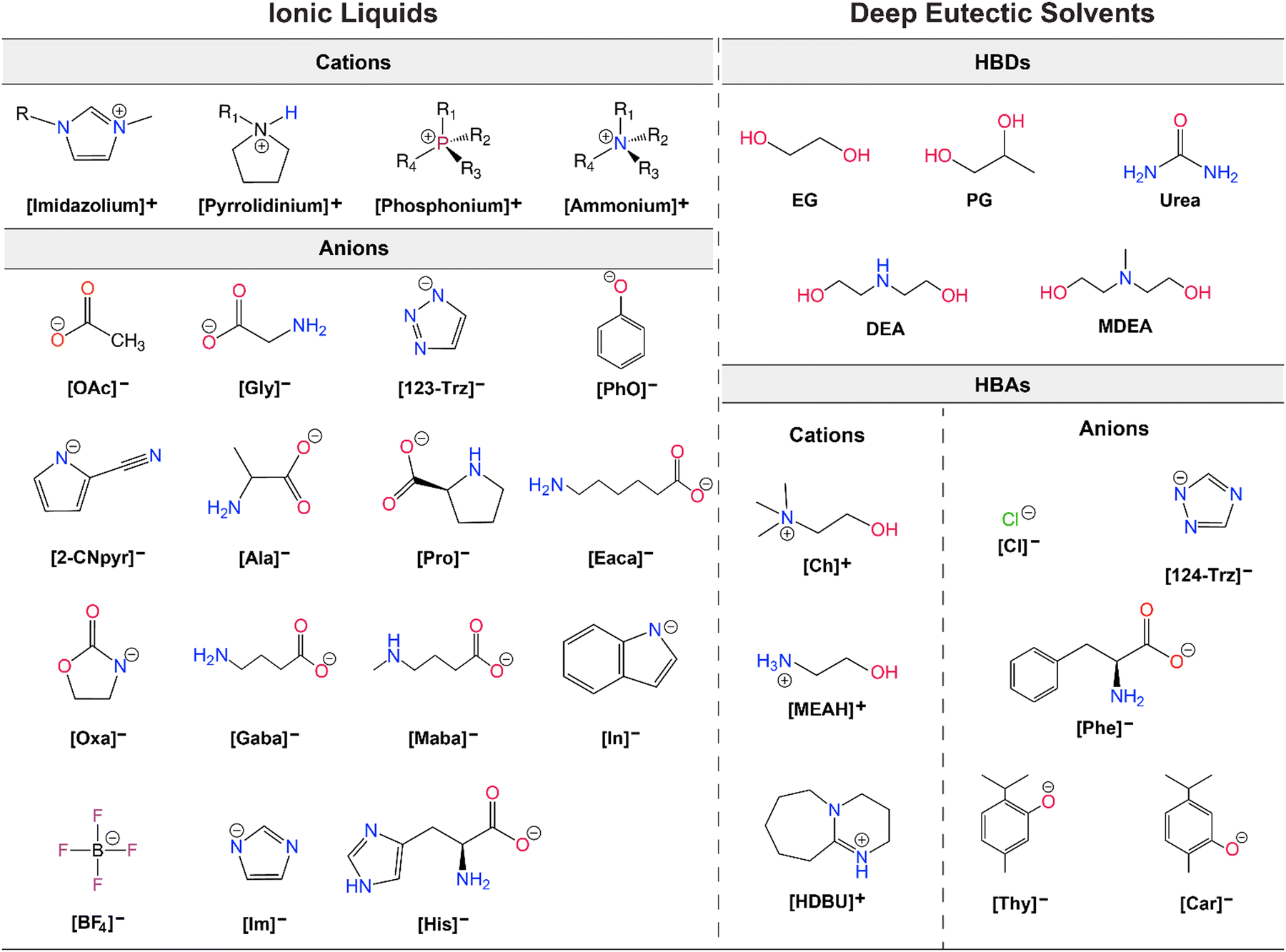 | ||
| Fig. 2 Example structures of ILs (left) and DESs (right) studied for CO2 capture and electrochemical conversion. | ||
While the focus of this review is RCC with electrolytes based on ILs and DESs, state-of-the-art solvents, electrolytes, and materials are introduced to provide a background and a perspective of the field. In particular ILs, DESs, and the related composite materials for CO2 capture are reviewed with critical evaluations of their adaptability in electrochemical conversion approaches of CO2. As a more conventional approach, efforts on the thermal conversion of CO2 involving ILs are also summarized. A focused review on the mechanisms, thermodynamics, and kinetics of CO2RR and RCC, as governed by the microenvironment created by ILs and DESs is presented with an emphasis on the topics of debate and recent findings. Further, a list of critical properties and key factors specific to IL and DES electrolytes for the combined capture and conversion processes are provided with the aim of better integration of the two fields toward the advancement of IL/DES systems for electrocatalysis in general. Specific takeaways from the techno-economic analysis of CO2 electrolyzers are summarized as they relate to RCC with ILs. Finally, conclusion with a brief outlook are presented.
2. Functional solvents as capture media
2.1 Overview of sorption
Significant advances have been made in the development of sorbents both for CO2 capture from point source of emissions (e.g., flue gas streams) and DAC of CO2 (at concentrations as low as 0.4 vol% of CO2). These sorbents are categorized as liquid solvents (e.g., amines, ILs, DESs, and alkaline solutions) and solid sorbents (e.g., activated carbons, metal organic frameworks (MOFs), covalent organic frameworks (COFs), zeolites, silicas, porous organic polymers, carbon nanotubes, and particularly amine-modified porous supports). CO2 is typically captured by liquid solvents via chemisorption, whereas in solid sorbents, CO2 can either be chemisorbed or physisorbed onto the sorbent surface. The weak, reversible interactions of the physisorption process enable efficient desorption at moderate temperatures (40–60 °C), promoting energy conservation during regeneration. Moreover, rapid sorption and desorption kinetics associated with the physisorption contribute to fast sorbent cycling. However, the limited CO2 capacity and low capture efficiency of such sorbents specifically for the capture from dilute streams pose challenges for RCC.Among the carbon capture technologies, benchmark of industrial processes for CO2 capture is absorption by aqueous alkanolamines and aqueous hydroxide solutions. Amines have many advantages such as excellent CO2 capacity, high efficiency, and they are relatively inexpensive. However, CO2 binds chemically with amines, hence, considerable amount of energy (−60 to −100 kJ mol−1 or 4–6 GJ per ton CO2) is required to regenerate the solvent for the next cycle, which significantly lowers the efficacy of the overall process.36,37 Furthermore, regeneration of sorbents at relatively high temperatures (e.g., CaCO3 requires higher than 800 °C for regeneration during DAC process) results in evaporation and susceptibility to thermal degradation of solvents. Other problems include corrosivity, toxicity, and oxidative degradation of the amine. Compared to liquid solvents, solid-based sorbents have shown promising potential to capture CO2 from flue gas in terms of selective sorption capacity and lower energy for regeneration. Particularly, MOFs have received a lot of attention for CO2 capture owing to their structural tunability,38,39 as well as their high surface areas, high CO2 capacities, and lower isosteric heat of adsorption (−20 to −30 kJ mol−1) than the traditional sorbents such as zeolites,40 activated carbons,41 and silicas (−30 to −40 kJ mol−1).42
Once CO2 is captured from a gas mixture, it can be released either by an increase in temperature (temperature-swing) or through a reduction in pressure (pressure-swing). In temperature-swing adsorption/absorption technique, the temperature of the sorbent is increased to release the weakly bound CO2, driven by the increasing kinetic energy of the molecules. High temperatures directly break the chemical bonds between CO2 and functional groups of the sorbent, releasing the captured gas. Although this approach is effective, it is energy-intensive, and the sorbents are prone to degradation at elevated temperatures. Alternatively, pressure-swing adsorption/absorption lowers the pressure to release the captured CO2. In the last decade, researchers have reported moisture-swing,43–45 electro-swing,46–48 and microwave-assisted (dielectric heating)49–51 regeneration methods to desorb CO2, which are comparatively more effective than conventional thermal heating owing to targeted heating, rapid desorption, and suppression of sorbent degradation. As a follow-up to these CCC processes where CO2 is released during regeneration, conversion of the concentrated CO2via CO2RR provides higher-value products, such as fuels and commodity chemicals. Better yet, if the CCC process is replaced by employing an RCC approach where the captured CO2 is directly utilized for fuel production, it would, in theory, be a cost-effective solution. Chemisorption, characterized by chemical bond formation between the solvent components and CO2, facilitates higher CO2 capacities particularly at low partial pressures, making chemisorbing solvents interesting for achieving large-scale CO2 conversion, specifically directly from CO2 emission sources. However, not all of the capture sorbents are adaptable for RCC. Here, we present the insights to each of the capture media and provide a perspective on their adaptability for RCC.
2.2 Solvent types
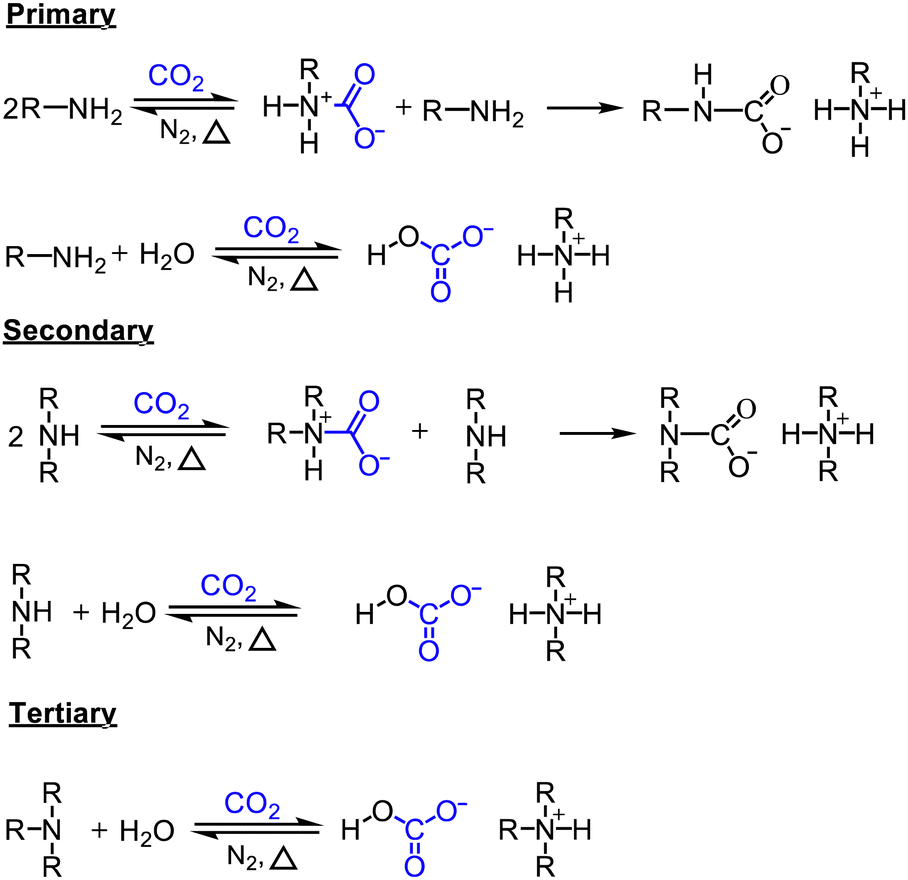 | ||
| Scheme 1 CO2 absorption by aqueous amines. | ||
Because CO2 is an acidic gas, a key feature in an organic sorbent is its basicity. Aqueous amines and aqueous alkali bases have been studied for CO2 capture for decades in pressure-swing and temperature-swing absorption–desorption processes for point sources.60–62 The CO2 cyclic absorption capacities are found to be enhanced by increased basicity and the heat of absorption was reported to follow an order of primary/secondary>tertiary, with few exceptions as seen in Fig. 3.63 Regeneration of amines, is an energy-intensive process because of high binding energy of amine with CO2, in the order of −80 kJ mol−1.64 Thus, innovative heat integration schemes have been carried out to reduce the parasitic energy consumption of CO2 capture technologies. Another challenge is fugitive emission of the solvent vapor, which must be re-condensed to prevent loss of active material, leading to an even greater energy penalty.65 Nevertheless, these materials have been employed for post-combustion flue gas and direct air capture owing to their availability.1
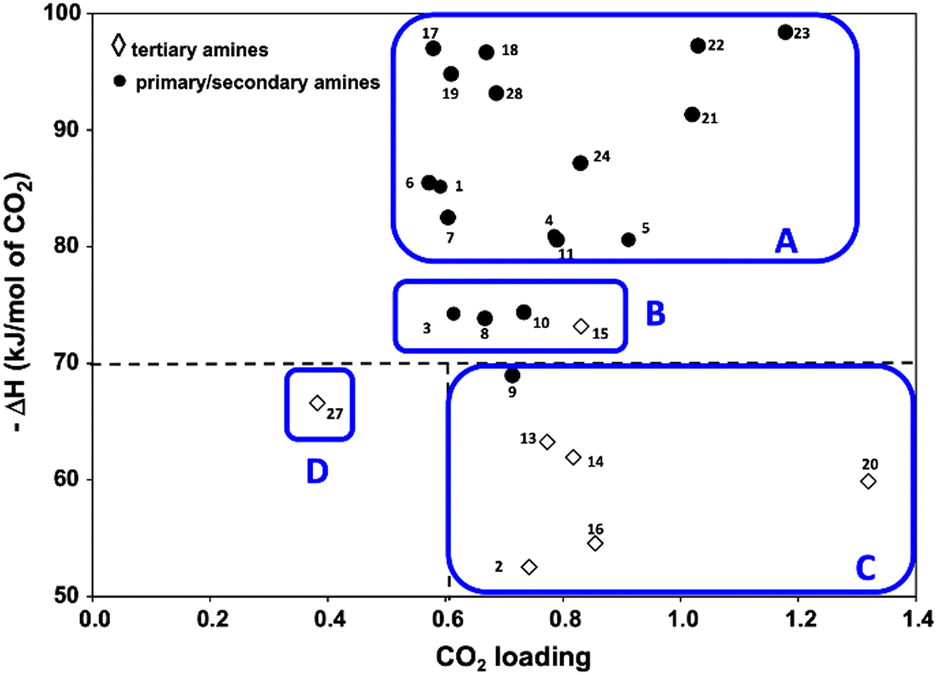 | ||
| Fig. 3 Heat of absorption of aqueous amine solutions (primary, secondary, and tertiary amines). Classes A, B, and C are organized as high, moderate, and low ΔH. Class D (tertiary) is also with low ΔH but has very low CO2 capacity. Reprinted with permission from ref. 63 Copyright 2017, Elsevier. | ||
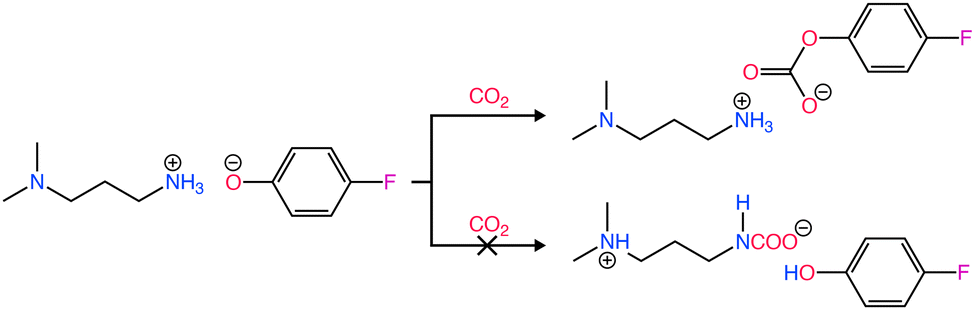
Recently, there has been interesting studies utilizing amines in RCC with the aim of replacing the conventional amine recovery unit in commercially available amine-based CO2 capture processes with CO2 electrolysis.66–68 This approach can substitute the most energy-intensive step in industrial amine scrubbing processes while concurrently providing a pathway for CO2 utilization.69 As depicted in Scheme 1, the carbamate and bicarbonate are the primary CO2 species and the concentration of free dissolved CO2 is negligible. However, during the CO2 electrolysis in amine solutions, there has not been direct evidence showing the conversion of the amine complexed CO2 (i.e., carbamate and bicarbonate species) on the electrode surface. For instance, despite the high CO2 capture capacity of MEA, Chen et al.70 observed dominance of the hydrogen evolution reaction (HER) (>80% faradaic efficiency, FE) and negligible CO2 conversion rates and CO selectivities over smooth surfaces of Bi, Pb, Pd, Ag, Cu, and Zn in CO2-saturated 30% (w/w) aqueous MEA solution containing 0.1% (w/w) cetrimonium bromide. These findings suggest that free dissolved CO2 primarily serves as the active species during CO2 electrolysis. Subsequently, Gallent et al.66 proposed a process wherein CO2 must be liberated from amine solutions before undergoing reduction to form various products. In this method, a CO2-rich propylene carbonate solvent with 1 M 2-amino-2-methyl-1-propanol (AMP) and 0.7 M tetraethylammonium chloride is introduced into the cathodic side of an electrochemical reactor operated at approximately 75 °C. Here, the captured CO2 is desorbed in close proximity to the cathode electrode surface and subsequently electrochemically converted into formate (giving up to 50% FE) and some oxalate and glycolate over a Pb surface. Further, other studies also highlighted the issues related to electrochemical instability, high volatility, and the corrosive nature of the amine towards the metal electrodes commonly used in RCC such as copper.71,72 Achieving long-term stability in RCC requires selecting the right electrode material, electrolyte composition, and understanding the fundamental processes both in the bulk and at the electrode–electrolyte interfaces so that bottom-up approaches can be developed. Functional ILs are considered as alternatives to aqueous amines for RCC due to their corrosion-resistant properties and higher CO2 capacities.73,74
The aqueous ammonia-based CO2 capture yields ammonium bicarbonate (NH4HCO3) as a product. There has been reports of NH4HCO3 electrolysis for utilization of the captured CO2 for integrated processes where the on-site generated NH3 from nitrate can be used for RCC, thus generating formate.80 Syngas production has also been reported for the direct electroreduction of NH4HCO3 on bromine-modified Ag electrocatalyst.81 Separately, electrochemically driven C–N bond formation from NH3 and CO2 over a copper catalyst toward formamide and acetamide products is documented.82 However, the desired product selectivities for these processes have heretofore remained very low.
CO2-reactive ILs typically contain a nucleophilic functional group such as hydroxyl (–OH), amine (–NH2) or amino acids, that can participate in hydrogen bonding with CO2, via a nucleophilic attack mechanism similar to amine-based sorbents. Initially, this functionalization comes in the form of alkyl chains containing a primary amine, usually attached to the cation. However, ILs can be functionalized by introducing strong nucleophilic alcohol and amine moieties that allows for the chemisorption of CO2 through a Lewis type acid–base reaction. Functionalized ILs offer impressive CO2 capacity particularly at low partial pressures through the formation of carbonates and carbamates. The study by Bates et al. in 2002 was the first to report amine functional ILs for CO2 capture, namely the task specific ILs, where they were able to attain almost 0.5 mol of chemisorbed CO2 per mol of IL at 1 bar CO2 and room temperature, according to the scheme shown in Scheme 2.90 Following studies demonstrated improved CO2 capacities, as high as 2 mol CO2 per mol IL for the ILs utilizing biofriendly, cheap, and readily available amino acids with nucleophilic amine groups for CO2 chemisorption.91,92 Similarly, Wang et al.,93 investigated Lewis type acid–base interactions observed between CO2 and the ILs, demonstrating the tunability of different aprotic trihexyl(tetradecyl)phosphonium cation-based ILs with varied anions. By varying the basicity of the anions in these solvents, the absorption enthalpy of CO2 was varied, making it possible to tune the energy requirement for CO2 desorption. For instance, the highly basic anion containing solvents, such as those containing the imidazolate anion, form high CO2 capacity ILs with high CO2–IL absorption enthalpies, making it harder to regenerate at elevated temperatures compared to the ILs containing the less basic triazolide anions.
 | ||
| Scheme 2 The CO2 chemisorption of a task specific IL where the imidazolium cation is functionalized with –NH2 moiety. Adapted with permission from ref. 90 Copyright 2002, American Chemical Society. | ||
Functionalized ILs demonstrate an increase in viscosity upon CO2 complexation, associated with formation of intermolecular H-bonding.94 For example, the viscosity of trihexyl(tetradecyl)-phosphonium isoleucinate ([P66614][Iso]) increases over 200-fold after CO2 absorption.91 This limitation makes them less suitable for industrial CO2 capture due to slow sorption rates driven by limitations in diffusion and mass transfer. Besides the ionic associations and H-bonding, this is in part due to the high molecular weight of the IL, which further translates to low CO2 gravimetric capacities (<0.1 g CO2 per g IL).95 Therefore, the follow-up research focused on eliminating the increased H-bonding and reducing molecular weight as discussed later.96
Aside from aprotic ILs, protic ionic liquids (P-ILs), such as those formed from the combination of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) superbase with weak acids such as imidazole, have shown great promise in the field of CO2 separations.97 CO2 chemisorption in P-ILs, as shown in Scheme 3, is generally first induced by a proton transfer reaction from imidazole type molecules to the superbase, creating the electron rich imidazolate anion which is attractive for CO2 chemisorption. However, the proton activities in P-ILs typically result in a decrease in their molar capacity in comparison to aprotic ILs, which potentially results from a decrease in the Coulombic interactions that aids in the absorption of CO2. Additionally, to attain higher gravimetric capacities with ILs, Noorani and Mehrdad98 studied the use of choline cation which has a relatively lower molecular mass when paired with different amino acids. However, choline–amino acid based ILs are known to be extremely viscous and typically require dilutants to improve their CO2 absorption rates. The following sections provide an overview of different classes of functionalized ILs and discusses each type relevant to CO2 capture with a perspective on their adaptability for RCC.
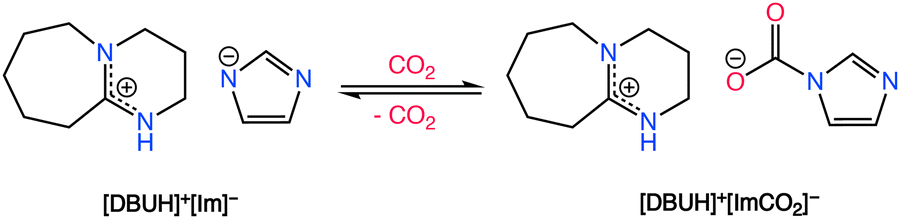 | ||
| Scheme 3 CO2 chemisorption by a DBU-based PIL. Adapted with permission from ref. 97 Copyright 2017, American Chemical Society. | ||
2.3 Functionalized ILs
ILs are classified into several categories based on the combination of cations and anions, each exhibiting unique biological, physical, chemical, and thermal properties. Fig. 4 summarizes the category of functionalized ILs as discussed in this section. Task-specific ionic liquids (TS-ILs) are functionalized ILs that have received attention for their tunable properties achieved by altering cation–anion combinations, making them versatile for applications in organic synthesis, catalysis, and chiral compound synthesis.99 Chiral ILs are useful for asymmetric inductions, having use in the liquid chiral chromatography, stereoselective polymerization, and chiral compound synthesis.99 Switchable Polarity ILs (SPS-ILs) undergo polarity changes upon activation,100 and bio-based ILs address the toxicity concerns by utilizing sustainable bio-precursors.101 Polymerized ILs (poly-ILs) form repeating motifs and are utilized in diverse applications, including polymer electrolytes and sensors.102 Energetic ILs offer advantages over conventional energetic compounds, such as 2,4,6-trinitrotoluene (TNT), 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (HNIW), because of their higher density and thermal stability,103 while neutral ILs exhibit weak electrostatic interactions and are used as inert solvents.104 Protic ILs have redeemable Brønsted acidic protons,105 and metallic ILs involve metal salts with specific counter anions,106 showcasing a wide array of applications across various scientific and industrial domains.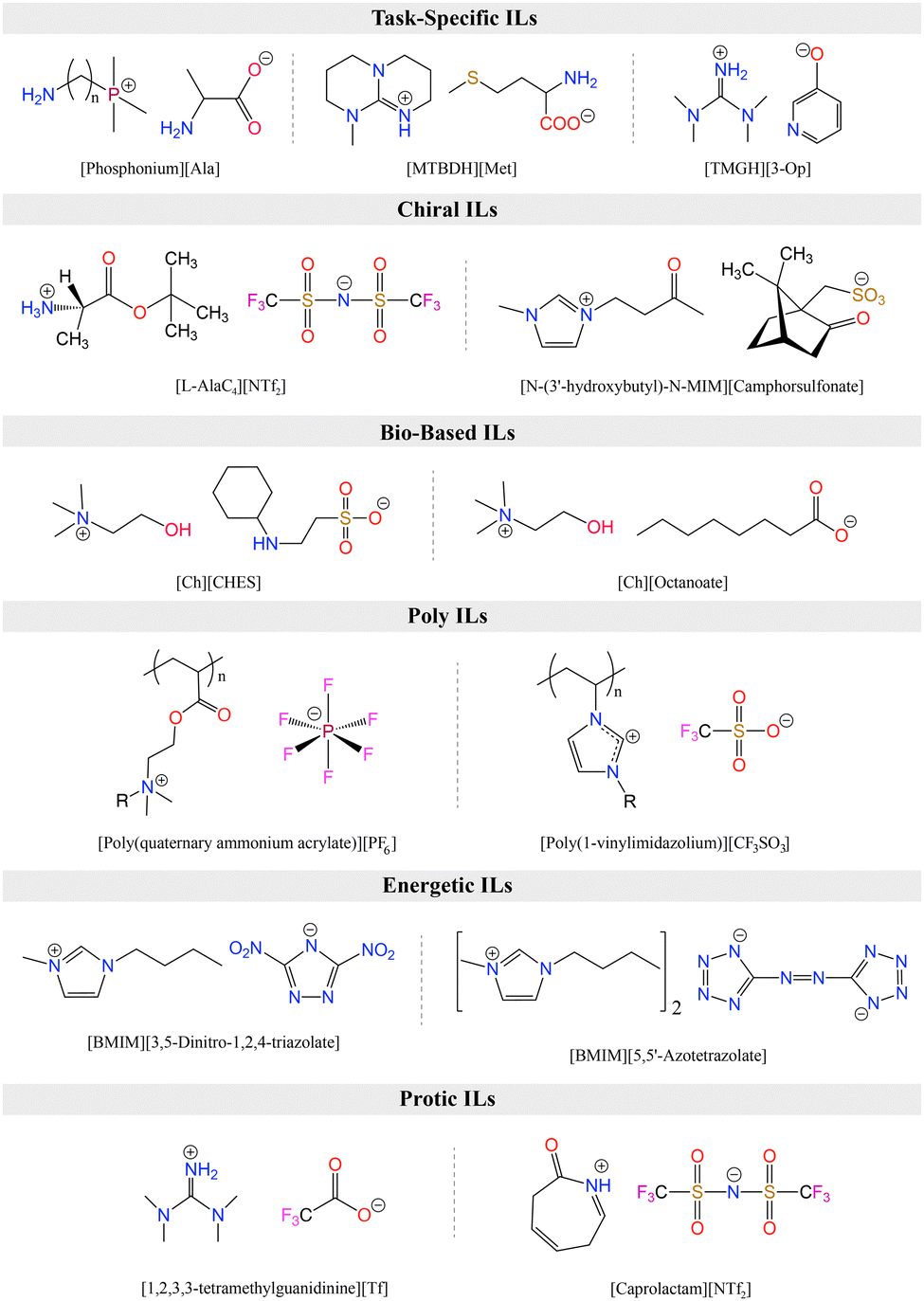 | ||
| Fig. 4 Classes of ILs with representative structures. | ||
Among different types of ILs, TS-ILs received a considerable amount of attention for CO2 capture.107 Such ILs are modified by the incorporation of various functional groups, such as amino,54 carboxyl,108 imidazolyl,109 pyridyl,110 alkoxyl,111 and hydroxyl,112 into their molecular structure. Introduction of these functional groups alters the physical and chemical properties of the ILs, enabling them to create additional or enhanced chemical interactions with CO2.113 This chemical reactivity significantly boosts the CO2 absorption capacity, making TS-ILs crucial for achieving high absorption efficiency. Notably, as mentioned before, TS-ILs have demonstrated the ability to absorb equimolar or multi-molar amounts of CO2, addressing the limitations of conventional ILs and offering a more effective solution for carbon capture in scenarios with low CO2 partial pressures, such as post-combustion flue gas.114
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 carbamate mechanism (2 mol of amino: 1 mol of CO2) at 1 bar, as in Scheme 2. This carbon capture capacity surpassed the performance of conventional alkanolamine aqueous solutions. However, a substantial increase in the viscosity upon complexation with CO2 was observed. Similarly, Mehrdad et al. reported the impact of hydrogen bonding in their investigation of three amino acid ILs ([BMIM][Gly], [BMIM][Ala], and [BMIM][Val]) for CO2 capture.115 Their study explored both physical and chemical sorption mechanisms, revealing that the hydrogen bonding in these ILs led to elevated viscosity. Further exploration led to the synthesis of tetraalkylammonium amino acid ILs (AAILs) by Wu et al., demonstrating low viscosity and high capture capacities approaching 0.5 mole of CO2 per mole of IL for [N2222][Ala], [N2222][β-Ala], and [N2224][Ala] at 1 bar and 40 °C.116,117 Hence, these investigations highlight the nuanced approaches in tailoring amino-functionalized ILs for enhanced CO2 absorption.
:1 carbamate mechanism (2 mol of amino: 1 mol of CO2) at 1 bar, as in Scheme 2. This carbon capture capacity surpassed the performance of conventional alkanolamine aqueous solutions. However, a substantial increase in the viscosity upon complexation with CO2 was observed. Similarly, Mehrdad et al. reported the impact of hydrogen bonding in their investigation of three amino acid ILs ([BMIM][Gly], [BMIM][Ala], and [BMIM][Val]) for CO2 capture.115 Their study explored both physical and chemical sorption mechanisms, revealing that the hydrogen bonding in these ILs led to elevated viscosity. Further exploration led to the synthesis of tetraalkylammonium amino acid ILs (AAILs) by Wu et al., demonstrating low viscosity and high capture capacities approaching 0.5 mole of CO2 per mole of IL for [N2222][Ala], [N2222][β-Ala], and [N2224][Ala] at 1 bar and 40 °C.116,117 Hence, these investigations highlight the nuanced approaches in tailoring amino-functionalized ILs for enhanced CO2 absorption.
 | ||
| Scheme 4 Chemical reaction of CO2 with carboxylate-functionalized ILs. Adapted with permission from ref. 125 Copyright 2022, Elsevier. | ||
Effect of the basicity of the anion is further analyzed in various studies demonstrating different basicity on azolate anions can induce the extraction of an active hydrogen atom from the cation, leading to the formation of carbene or ylide compounds that subsequently interact with CO2 to create carbene–CO2 or ylide–CO2 complexes,132–135 as in Scheme 5. The reversibility of CO2 absorption on AHA ILs was further investigated with [P2228][2-CNpyr] and [P2222][BnIm], both in the absence and presence of water.136 However, it is worth noting that the CO2 absorption mechanism in moist conditions is different, thus often enhancing CO2 absorption capacity.136 However, the presence of co-adsorbed moisture necessitates increased thermal energy to regenerate the solvent due to the high heat capacity of water (4.2 J g−1 K−1).137 Bonilla et al.,136 showed that in the presence of water, CO2 capture mechanism includes an additional reaction pathway in AHA ILs. One pathway leads to the carbamate formation (expected reaction of the anion with CO2) and the other pathway leads to the bicarbonate formation. Accordingly, the anion is protonated by water and CO2 reacts with the hydroxide to form bicarbonate.
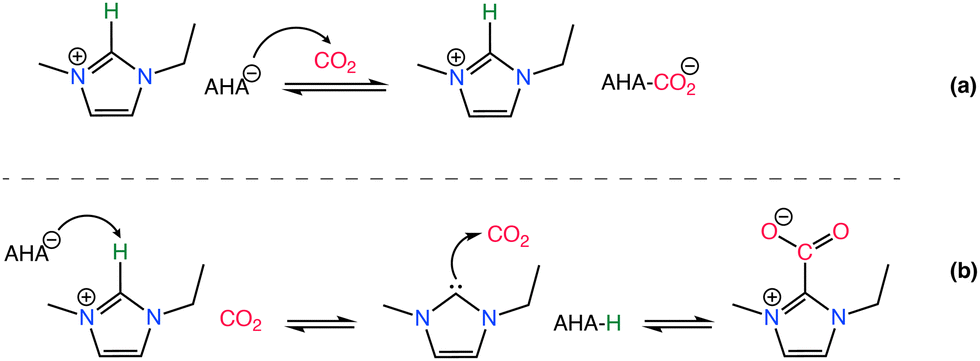 | ||
| Scheme 5 CO2 chemisorption by an AHA IL where the CO2 binds to the nucleophilic anion (a) and to the imidazolium cation through the C2 position (b). Adapted with permission from ref. 132 Copyright 2019, American Chemical Society. | ||
The versatility of the azolate ILs is underscored by their maintained viscosities without further increase, ease of regeneration (reaction enthalpies in the order of −50 kJ mol−1),138 and the potential formation of carbene and ylide complexes,135,139,140 providing a rich landscape for designing tailored ILs for enhanced CO2 capture efficiency. Further, some of the earlier works with ILs in CO2RR and RCC are based on azolate ILs as discussed later.
Similarly, phenol can be used to synthesize phenoxide based-functionalized ILs via the deprotonation of hydrogen atom from the –OH group of phenol to form phenolate anion. For example, a study investigated various phenolate ILs by modifying the phenolate anion structure with different substituents.143 Performance measurements demonstrated that absorption capacities of these ILs varied with carbonyl-substituted phenolates.144 The findings indicate that incorporation of a CHO group into the anion resulted in a notable increase in CO2 capacity. Specifically, a molar increase of 0.2 was observed when comparing [P66614][PhO] and [P66614][4-CHO-PhO], with the CO2 capacity rising from 0.81 to 1.01 mol CO2 per mol IL.
Fluorinated phenolate ILs were also examined to have high CO2 capacities. Specifically, a notable increase of approximately 20% in CO2 capacity was observed for [DMAPAH][4F-PhO] and [P4444][4-F-PhO] in comparison to their respective counterparts [DMAPAH][2F-PhO] and [P4444][PhO].145,146 The chemisorption mechanism for [DMAPAH][4F-PhO] is shown in Scheme 6. Additionally, phenoxides have no direct bond between the O atoms and the hydrogen atoms, hence, the absence of any intermolecular hydrogen bond interactions results in reduced viscosity.144 Overall, the tunability of these functional groups highlights the potential of alkoxide and phenolate functionalized ILs in capturing CO2, showcasing their role in environmentally relevant processes.
 | ||
| Scheme 6 CO2 chemisorption by a phenoxide functionalized IL; CO2 binding the hydroxy in the fluorine-substituted phenolic anion was favored. Adapted with permission from 146 Copyright 2018, Elsevier. | ||
| Ionic liquids | T A (°C) | Capacity (mol CO2 per kg solvent) | T D (°C) | Viscosity at TA (mPa s) |
|---|---|---|---|---|
| [Ch][Gly]149 | 25 | 2.69 | — | — |
| [Ch][βAla]149 | 2.65 | — | — | |
| [Ch][Pro]149 | 1.92 | 50 | — | |
| [Ch][Phe]149 | 0.78 | — | — | |
| [HDBU][Im]150 | 40.0 | 4.41 | — | 19 |
| [HDBU][Ind]150 | 3.02 | — | — | |
| [HDBU][124-Trz]150 | 1.73 | — | 307 | |
| [HDBN][Im]151 | 40.2 | 3.38 | ||
| 1.61 (0.1 bar) | 80.2 | 8.3 | ||
| [HDBN][Pyr]151 | 3.38 | |||
| 1.30 (0.1 bar) | — | 5.1 | ||
| [HDBU][Pyr]151 | 3.04 | |||
| 1.41 (0.1 bar) | — | 9.7 | ||
| [EMIM][OAc]152 | 40.2 | 1.65 | — | — |
| [EMIM][Ala]152 | 1.89 | — | 34.5 | |
| [EMIM][Gly]152 | 2.32 | — | 39.4 | |
| [P66614][3HMPz]153 | 25.0 | 1.65 | 60.0 | 922.6 |
| [P66614][5HMPz]153 | 0.74 | — | — | |
| [P66614][1HDMPz]153 | 1.16 | — | — | |
| [P66614][1HMPz]153 | 1.43 | — | — | |
| [N1111][Eaca]154 | 40.2 | 4.02 | ||
| 2.84 (0.1 bar) | 80.2 | 85.5 | ||
| [N1111][Maba]154 | 4.05 | |||
| 2.79 (0.1 bar) | — | 116.3 | ||
| [N1111][Gaba]154 | 3.29 | |||
| 2.55 (0.1 bar) | — | 140.5 | ||
| [P66614][MN]155 | 25 | 1.53 (1 bar) | 1235 |
2.4 Deep eutectic solvents (DESs)
In the context of CO2 capture, where it is essential to prioritize high selectivity, absorption capacity, low desorption energy, and stability, DESs stand out as effective contenders as capture media. Common examples of DESs for CO2 capture include HBAs that are often ammonium or phosphonium salts and HBDs such as ethylene glycol, urea, or other organic compounds with functional groups such as the carboxylic acid, amide, alcohol, and thiol groups. A common characteristic of DESs is the significant depression in the melting point of the liquid at the deep eutectic composition compared to the individual constituents. Although earlier reports revealed advantageous properties, such as the formation of low viscous solvents with the highest conductivity at the eutectic composition, eutectic solvents formed at different HBA:HBD compositions in the vicinity of the eutectic point have also been of interest35 because of the similar desirable properties they present for application in biotechnology, catalysis, and gas separations and conversion. Given the various degree of melting point depression obtained in different HBA:HBD compositions discussed here, these mixtures will be referred to as eutectic solvents for simplicity.A common example of eutectic solvents includes reline, formed by mixing solid choline chloride salt ([Ch][Cl]) and urea (in a 1:2 molar ratio) which result in the formation of a room temperature liquid. Other examples of eutectic solvents include ethaline and glycine, both of which contain [Ch][Cl] as HBA, with ethylene glycol (EG) and glycerol (Gly) as HBDs, respectively. Ethaline contains about 1:4 molar ratio of [Ch][Cl]:EG, whereas glycine contains about 1:2 molar ratio of [Ch][Cl]:Gly, both of which have been studied for CO2 capture and separation applications.156,157 In these solvents, CO2 absorption is mainly dominated by the physical absorption of CO2 through the occupation of the entropic voids that are maximized at the deep eutectic composition.32 While physisorbing eutectic solvents show appreciable CO2 capacities at elevated pressures, they are less effective for low partial pressure CO2 separations due to their low CO2 selectivity and uptake. Through precise adjustment of the CO2 affinities of the functional groups present in the HBAs and HBDs, eutectic solvents can be tuned to both physisorb and chemisorb CO2. As such, introducing CO2 reactive functional groups into eutectic solvent results in the formation of more suitable solvents for applications such as DAC, where CO2 is captured from air with ultra-low CO2 concentration (i.e., 410 ppm).158 Increasing the CO2 affinity of the HBAs/HBDs typically involves incorporation of the nucleophilic moieties that allows formation of CO2 reactive eutectic solvents with appreciable CO2 capacity and selectivity even under CO2 concentrations as low as 0.5% of the feed.150,159,160 Commonly utilized eutectic solvents for the process of chemical absorption consist of ILs, amines, and alkanol amines via the creation of N–C, O–C, and C–C bonds.125,161
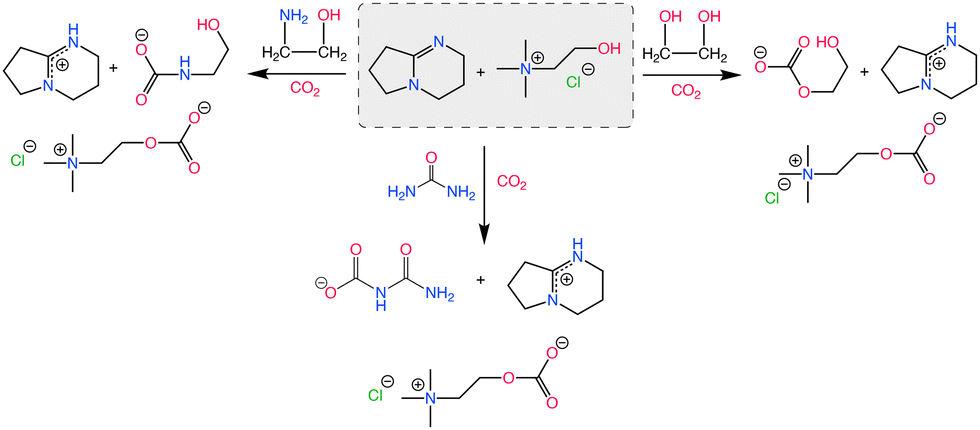
Earlier examples of DESs with CO2 chemisorption include the addition of superbases to ethaline such as the one reported by Bhawna et al.,162 where 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) were the active components. Superbases participates in strong proton sharing interactions with ethaline, inducing the formation of electron rich (nucleophilic) moieties with high CO2 affinity, which facilitates chemisorption as shown in Scheme 7.
 | ||
| Scheme 7 CO2 chemisorption in superbase-added [Ch][Cl] based eutectic solvent. Adapted with permission from ref. 162 Copyright 2017, John Wiley and Sons. | ||
The strong proton sharing induced by the superbase results from their high proton affinity as strong Brønsted-Lowry bases. Several studies have further shown how the proton activities, influenced by the presence of strong Brønsted–Lowry bases, affect the CO2 chemisorption in eutectic solvents. For instance, Cui et al.159 showed the impact of phosphonium and ammonium based HBAs with imidazolate and triazolate conjugate base anions, both of which have high HBA ability, on the chemisorption of CO2 to EG. The EG chemisorption was initiated by an induced proton sharing interaction due to the presence of the strong conjugate bases. These tetraethylphosphonium ([P222]+) and tetraethylammonium ([N222]+) based eutectic solvents, capable of absorbing almost 1 mol CO2 per mol solvent at 1 bar CO2 and at 25 °C were easily regenerable at 70 °C; with the [P222]+ and [N222]+ counterions showing very similar absorption behaviors. Similarly, Wang et al.163 studied the impact of HBDs on CO2 where both EG and triazole HBDs were used. While EG HBD systems were observed to show appreciable CO2 capacities, the use of triazole HBDs in place of EG resulted in a sufficient decrease in the CO2 chemisorbing ability of the eutectic solvents despite the buildup of electron density resulting from the proton sharing interactions, highlighting the complex interplay between components in eutectic solvents. Wang et al.164 also observed the reactivity of EG in bio-phenol based eutectic solvents; however, changes in the HBD from EG to 4-methylimidazole was accompanied by a change in the chemisorption sites and CO2 absorption mechanism. While eutectic solvents employing EG as the HBD exhibited primary chemisorption by EG itself, replacing EG with 4-methylimidazole HBD resulted in a shift, with bio-phenol derivatives becoming the predominant chemisorption sites. This potentially results from a combination of the changes in the proton activities present within these solvents, as well as the differences in the localization of electron density in the various binding sites.
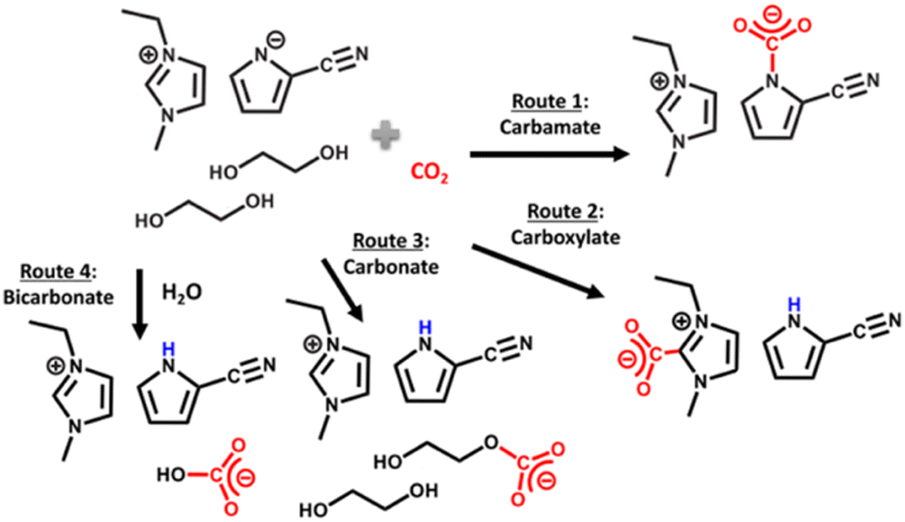
Lee et al.160 reported on the eutectic formation when [EMIM][2-CNpyr] IL was mixed with the common HBD ethylene glycol. Similar to previous reports, in the presence of a basic anion, EG was deprotonated neutralizing the pyrrolide that enabled CO2 binding to EG, thus forming carbonate species in addition to the known CO2 binding mechanism to the IL component itself. Scheme 8 illustrates the CO2 absorption reaction by this reported eutectic solvent. Further, from the transition state calculations performed at the density functional theory (DFT) level, the authors showed the significantly reduced reaction energies for CO2 binding to both the IL anion and the cation in the presence of EG due to the hydrogen bonding interactions. These interactions resulted in lowered energy requirements for the regeneration of the solvent where regeneration temperatures as low as 40 °C was possible for a cyclable working capacity of CO2.
 | ||
| Scheme 8 CO2 chemisorption in DES composed of [EMIM][2-CNpyr] and EG. Reprinted with permission from ref. 160 under CC-BY-NC-ND license. | ||
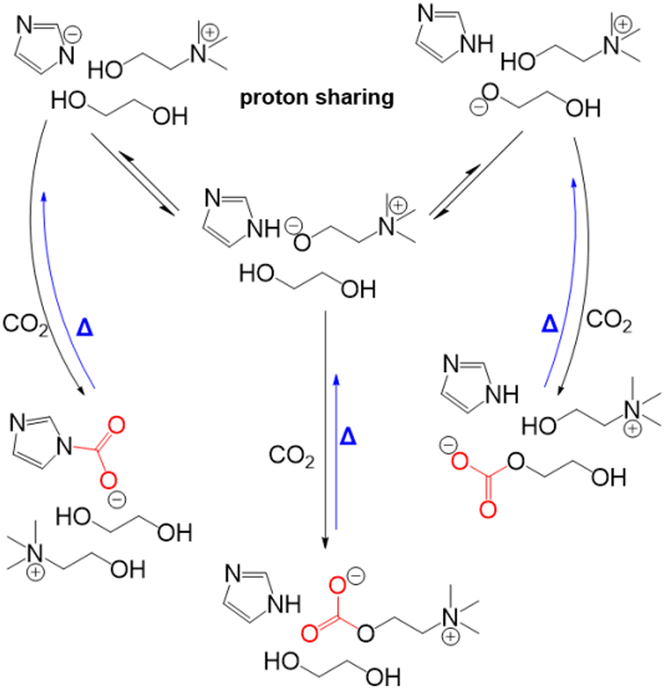
Dikki et al.165 further demonstrated a very similar EG-CO2 chemisorption behaviors in choline based eutectic solvents with EG as the HBD. By tuning the proton affinity of the conjugate base anions in the different eutectic solvent systems, they observed a change in the concentration of CO2 chemisorbed by EG. For instance, the concentration of CO2 bound to the eutectics was greatest in the imidazolate containing solvents with the highest proton affinity (pKb of imidazolate in H2O = −0.52), followed by those with triazolate anion-based solvent (pKb of triazolate in H2O = 4.03). Notably, EG-CO2 chemisorption was non-detectable in the chloride-based eutectic solvent (pKb of chloride in H2O = ∼20), thus demonstrating the tunability of CO2 capacity through modifying the proton activity.
Scheme 9 shows the representative proton sharing and the CO2 binding in this reactive eutectic solvent. It was further observed that anions such as imidazolate, triazolate, and phenolate systems with electron density delocalization by resonance were less likely to interact with CO2 compared to EG with a more localized electron density. Aside from its ability to participate in the CO2 chemisorption process, studies have shown that EG can modulate proton activity in these mixtures.
 | ||
| Scheme 9 CO2 chemisorption in a eutectic solvent composed of choline imidazole and ethylene glycol. Reprinted from ref. 165 with permission from the Royal Society of Chemistry. Copyright 2024. | ||
As reported by Klemm et al.,149 mixing EG with choline amino acid based salts (i.e., [Ch][Pro]) resulted in an increase in the chemisorption of CO2 to –NH2 group present in [Pro]− in the eutectic solvents when compared to the neat [Ch][Pro]. Through a combination of experimental NMR spectroscopy and computational methods including molecular dynamics (MD) simulations and DFT calculations, authors elucidated CO2 absorption mechanism. Analysis demonstrated that hydrogen bonding interactions facilitated by EG play a crucial role in preventing proton transfer between [Pro]–[Pro] species upon CO2 binding to [Pro]−. With the inclusion of EG, deactivation of CO2 binding sites resulting from intermolecular proton transfer was prevented and higher CO2 capacity was obtained with the eutectic solvent compared to neat [Ch][Pro].
While the chemical makeup of the eutectic mixtures can be tuned for CO2 absorption, significant changes are also noted in physical properties including the basicity, polarity, thermal stability, density, and viscosity.166 These properties are important when regenerating these solvents at elevated temperatures. Like ILs, different functional groups present in the HBAs and the HBDs influence the binding energies of the CO2, which further translate to large variations in the amount of energy required to desorb the captured CO2. For instance, among the studied choline-based eutectic solvents with imidazolate, triazolate, 2-cyanopyrilide, and phenolate, Dikki et al.,165 found that the triazolate solvents were easily regenerated at 50 °C while the imidazolate eutectics required elevation up to 70 °C for regeneration for the same working capacity post absorption at 25 °C. This difference was in spite of having similar CO2 binding sites in both systems, thus showing the tunability of the energy required to desorb the chemisorbed CO2 in these solvents through the proton activity and the electrostatics induced by the hydrogen bonding network. Since it is desired to minimize water in these liquids to maintain a low energy requirement for regeneration, there has been some examples of hydrophobic DESs as well.167Table 3 summarizes the gravimetric capacities of different CO2 reactive ILs and eutectic solvent systems reported in literature, along with the changes in viscosities post CO2 saturation. Considering the target CO2 loading quoted by DAC companies (e.g., Climeworks) is 1 mmol CO2 per g of sorbent, these results are very promising.
| Eutectic solvents | T A (°C) | Capacity (mol CO2 per kg) | T D (°C) | Viscosity at TA (mPa s) | Viscosity with CO2 at TA (mPa s) |
|---|---|---|---|---|---|
| Ch±ImH:EG (1:2)165 |
25 | 3.25 | 70 | 200 | 191 |
| Ch±PhOH:EG (1:2)165 |
2.49 | 50 | 356 | 185 | |
| [Ch][2-CNpyr]:EG (1:2)165 |
2.41 | 95 | 178 | ||
| [Ch][124-Trz]:EG (1:2)165 |
2.36 | 171 | 157 | ||
| Ch±ImH:PG (1:2)166 |
25 | 2.75 | 70 | 369 | 430 |
| Ch±PhOH:PG (1:2)166 |
2.35 | 5 | 644 | 409 | |
| [Ch][2-CNpyr]:PG (1:2)166 |
1.67 | 170 | 340 | ||
| [Ch][124-Trz]:PG (1:2)166 |
2.03 | 335 | 349 | ||
| Ch±ImH:MEA (1:2)166 |
4.60 | — | 128 | — | |
| Ch±PhOH:MEA (1:2)166 |
3.13 | — | 182 | — | |
| [Ch][124-Trz]:MEA (1:2)166 |
4.92 | 60 | 121 | — | |
| [N2222][123-Trz]::EG (1:2)163 |
25 | 2.48 | 60 | — | — |
| [P2222][123-Trz]:EG (1:2)159 |
25 | 2.68 | 70 | — | — |
| [P2222][Im]:EG (1:2)159 |
2.69 | — | — | — | |
| [EMIM][2-CNpyr]:EG (1:2)160 |
25 | 2.59 | 40 | 45 | — |
| [Ch][Pro]:EG (1:2)149 |
25 | 2.10 | 50 | — | — |
| [Ch][βAla]:EG (1:2)149 |
1.83 | — | — | ||
| [Ch][Gly]:EG (1:2)149 |
1.69 | — | — | ||
| [Ch][Phe]:EG (1:2)149 |
0.45 | — | — | ||
| [HBDU][Im]:EG (7:3)150 |
40 | 3.20 | 70 | 31.48 | 166.51 |
| [HBDU][Ind]:EG (7:3)150 |
2.66 | — | 36.99 | 114.18 | |
| [HBDU][123-Trz]:EG (7:3)150 |
2.45 | 197.72 | 205.26 | ||
| [HBDU][Im]:EG EG (6:4)150 |
2.68 | — | — | ||
| [HBDU][Im]:EG EG (5:5)150 |
2.48 | — | — | ||
| [HBDU][Im]:EG EG (4:6)150 |
1.86 | — | — | ||
| [Ch][Cl]:MEA (1:5)168 |
30 | 6.18 | — | ∼25 | — |
| [Ch][Cl]:DEA (1:6)168 |
3.89 | ∼75 | — | ||
| [Ch][Cl]:MDEA (1:7)168 |
1.39 | ∼350 | — | ||
| [HDBU][Car]:EG (1:2)169 |
25 | 2.27 | 70 | 389 | — |
| [HDBU][Car]:EG (1:3)169 |
2.03 | — | 228 | — | |
| [HDBU][Car]:EG (1:4)169 |
1.80 | 166 | — | ||
| [HDBU][Thy]:EG (1:2)169 |
2.27 | 70 | 477 | — | |
| [HDBU][Thy]:EG (1:3)169 |
2.03 | — | 263 | — | |
| [HDBU][Thy]:EG (1:4)169 |
1.82 | 179 | — | ||
| [MEAH][Cl]:MDEA (1:3)170 |
25 | 2.64 | 80 | 262 | ∼1500 |
| [DEAH][Cl]:MDEA (1:3)170 |
2.46 | — | ∼260 | ∼1450 | |
| [MDEAH][Cl]:MDEA (1:3)170 |
1.34 | ∼250 | ∼420 | ||
| [DBUH][4-F-PhO]:EG (1:5)171 |
25 | 1.74 | 75 | 87 | — |
| [N2222][4-PyO]:DMSO (1:4)172 |
40 | 2.06 | — | 6.5 | — |
| [N2222][CH(CN)2]:Eim (1:1)173 |
30 | 2.8 | 120 | 12.7 | 115 |
From Table 3, it can be gathered that the viscosity of the HBDs component primarily governs the viscosity of the resulting liquid mixture. It is also noticed that by replacing the hydroxyl functionalized HBAs with those with amine functionalized HBAs, viscosity of the eutectic solvent is reduced. However, the viscosities of these solvents are generally increased with CO2 saturation and the starting viscosity range is one to two orders of magnitude higher compared to aqueous amines. Therefore, the advantages from their lowered regeneration energy and the disadvantage from their high viscosities should be analysed in a coupled manner to further improve the CO2 capture and separation performance of DES sorbents.
2.5 Composite materials with ILs/DESs
Although ILs provide several advantages as sorbents, such as low volatility and high thermal stability at CO2 capture conditions, they exhibit quite high viscosities, which can increase even more upon CO2 saturation, reducing the mass transfer.174 However, having a low viscosity is another prerequisite for a good sorbent.126 To mitigate the negative effects arising from the high viscosities of ILs, they can be immobilized on porous materials. An increased contact area of the IL with the CO2 gas increases the sorption rate.175 Furthermore, this is a cost-efficient solution when considering the costs of the ILs.176For CO2 capture, several composites (Table 4) were considered by depositing ILs on high-surface-area supports, such as metal organic frameworks (MOFs),175,177–181 covalent organic frameworks (COFs),179,182,183 carbonaceous materials,175,181,184–188 zeolites,189,190 synthetic and natural clay minerals,191–193 mesoporous and microporous silica and alumina,175,194–196 and polymers.197,198 ILs immobilized within the pores of these materials, improved several aspects of the CO2 capture process, owing to the increased gas–liquid interface.199 For example, Arellano et al. demonstrated a sevenfold higher CO2 uptake for [EMIM][NTf2]-zincate/SBA15 composite compared to the bulk IL.200 Likewise, [BMIM][NTf2]/MOF-808, with an IL thickness layer of 1.7 nm on the external MOF, provided a normalized CO2 uptake of 38.7 mmol g−1 at 100 kPa and 25 °C; 1000 times higher than the bulk IL.201
| IL/porous composites | P (bar) | T A (°C) | T D (°C) | CO2 uptake (mmol g−1) |
|---|---|---|---|---|
| [HEMIM][DCA]/ZIF-8202 | 1 | 25 | 125 | 0.42 |
| [BMIM][SCN]/ZIF-8203 | 1 | 25 | 125 | 0.33 |
| [BMIM][BF4]/ZIF-8177 | 1 | 25 | 125 | 0.27 |
| [BMIM][PF6]/ZIF-8204 | 1 | 25 | 125 | 0.39 |
| [BMIM][OAc]/ZIF-8205 | 1 | 30 | 80 | 1.03 |
| [BMIM][MeSO3]/ZIF-8206 | 1 | 25 | 125 | 0.39 |
| [BMIM][CF3SO3]/ZIF-8206 | 0.37 | |||
| [BMIM][MeSO4]/ZIF-8206 | 0.41 | |||
| [EMIM][OAc]/ZIF-8207 | 1 | 30 | 100 | 0.59 |
| [EMIM][NTf2]/ZIF-8207 | 0.43 | |||
| [C2OHMIM][NTf2]/ZIF-8207 | 0.41 | |||
| [C10MIM][NTf2]/ZIF-8207 | 0.45 | |||
| [P66614][NTf2]/ZIF-8208 | 1 | 30 | 100 | 0.42 |
| [C6MIM][DCA]/ZIF-8208 | 0.43 | |||
| [C6MIM][C(CN)3]/ZIF-8208 | 0.45 | |||
| [C6MIM][Cl]/ZIF-8208 | 0.43 | |||
| [BMIM][FeCl4]/ZIF-8208 | 0.41 | |||
| [BMIM][OAc]/CuBTC209 | 1 | 30 | 80 | 1.90 |
| [BMIM][NTf2]/CuBTC209 | 2.1 | |||
| PIL/CuBTC210 | 0.2 | 25 | 200 | 0.8 |
| [EMIM][NTf2]/MOF-808201 | 1 | 25 | 60 | 3.0 |
| [DETA][OAc]/MIL-101(Cr)211 | 1 | 25 | 100 | 2.35 |
| BUCT-C19212 | 1 | 25 | 150 | 1.88 |
| AFIL-ZIF-8213 | 1 | 25 | 150 | 2.38 |
| Ni-MOF/IL-3214 | 1 | 25 | 80 | 4.34 |
| [BMIM][Cl]/SBA-15215 | 0.4 | 25 | 65 | 2.40 |
| [BMIM][NTf2]/alumina216 | 0.4 | 25 | 65 | 1.30 |
| [C4MIM][NTf2]/Silica216 | 1.27 | |||
| [BMIM][MeSO4]/MIL-53(Al)217 | 1 | 25 | 110 | 0.89 |
| [TETA][NO3]/SBA-15218 | 0.15 | 40 | 150 | 2.12 |
| [BMIM][OAc]/SBA-15219 | 0.04 | 25 | 120 | 0.93 |
| [EMIM][OAc]SBA-15220 | 1 | 30 | 110 | 2.20 |
| [EMIM][OAc]/MOF-177221 | 1 | 30 | 120 | 0.4 |
| [EMIM][Gly]/activated carbon222 | 0.04 | 50 | 100 | 0.58 |
| [EMIM][Gly]/PMMA198 | 0.023 | 30 | 150 | 0.88 |
| [EMIM][Lys]/PMMA198 | 1.05 | |||
| [N1111][Gly]/PMMA223 | 0.023 | 30 | 100 | 2.14 |
| [EMIM][Gly]/PMMA224 | 0.023 | 30 | 100 | 0.90 |
| [EMIM][Lys]/MCM-41225 | 0.15 | 30 | 100 | 1.50 |
| [N1111][Gly]/silica136 | 1.5 | 30 | 80 | 0.65 |
| [Vbtma][Gly]/activated carbon187 | 1 | 25 | 110 | 1.51 |
| [TEA][Tau]/SBA-15226 | 0.1 | 25 | 110 | 1 |
| [P4444][2-Op]/silica227 | 0.15 | 50 | 120 | 1.49 |
| [P66614][124-Trz]/SBA-15228 | 1 | 20 | 50 | 0.70 |
| [P4443][Cl]/silica gel229 | 1 | 0 | 150 | 1.07 |
| [P4443][BF4]/silica gel229 | 1.30 | |||
| [(EtO)3SiP8883][NTf2]/activated carbon188 | 2 | 25 | 80 | 0.87 |
| [EMIM][Lys]/silica230 | 1 | 25 | 105 | 0.61 |
| [DAMT][BF4]/graphene231 | 1 | 25 | 120 | 0.88 |
| [NAIm][Br]/ZIF-67232 | 0.15 | 25 | 200 | 0.68 |
| [Et4NBr]/Py-COF233 | 1 | 25 | 100 | 1.97 |
Similar to IL-based hybrid composites, impregnation of DESs into porous materials has recently emerged as a promising strategy to design hybrid materials for CO2 capture and separation. This approach aims to increase the affinity of the hybrid sorbents towards CO2 for enhanced selectivities and to increase the CO2-DES contact area for improved sorption rates. Furthermore, tunability of DES composition allows to tailor CO2 interaction strength with the binding sites. Impregnation of porous materials including activated carbon,234 silica,235 and MOFs236 with DESs also showed to be effective in terms of increasing the additional CO2 binding sites in the composite, compared to the parent compounds.
Such composites of ILs or DESs with porous materials are synthesized by a variety of methods, either through in situ methods, such as ionothermal synthesis, or by post synthesis routes, like wet impregnation, incipient wetness impregnation, capillary action, ship-in-a-bottle, and grafting.237 Wet impregnation, one of the most frequently used routes, results in a noncovalent interaction between the IL/DES and the surface of the porous material. On the other hand, methods, such as grafting, involve a covalent bond formation between the liquid and the surface,229 which can offer advantages, such as maintaining the porosity of the host material and higher stability, in the expense of more complicated synthesis procedure. In this section, we focus on physically impregnated porous materials; more details on the CO2 uptake and separation performance of composites obtained by grafting ILs on supports can be found in the review article by Zheng et al.175
One of the crucial properties of IL/porous material composites determining their applicability is their thermal stability limits. The thermogravimetric analysis data of a composite typically presents a two-step decomposition, associated with the decomposition of the IL and the porous material separately.238,239 However, if the porous material is highly stable, as in the case of metal oxides or zeolites, typically only one decomposition step, corresponding to the decomposition of the IL only, is observed.240,241
Interestingly, thermal stability limits of these composites are generally reported to be largely different than those of their individual components (the IL and the porous material),238,240 representing the formation of a new hybrid material having different characteristics. These characteristics are mostly set by the molecular interactions between the IL molecules and the surface of the porous material. These interactions also play a major role in determining the gas sorption properties of the composite. For instance, Kinik et al. presented the results of a detailed infrared (IR) spectroscopy-based analysis by comparing the IR spectrum of [BMIM][PF6] in the bulk state with that of the composite prepared by incorporating the same IL into the cages of ZIF-8.204 Data showed that the IR bands belonging to the imidazolium ring of the IL blue-shifted when the IL was immobilized on the MOF, while the features associated with the [PF6]− anion were red-shifting. The weakening of the P–F bond upon immobilizing the IL on ZIF-8 demonstrated an electron-sharing between the anion and the ZIF-8, which evidenced a direct interaction between MOF and the anion of the IL. The extend of such interactions, and their consequences on the gas sorption properties, depends significantly on the individual components of a composite, and hence different combinations of an IL with different porous materials offer significantly different performance measures.237 The research has been directed towards tuning these interactions towards improved ability for separating CO2 from different gas mixtures.
In one of the earliest reports introducing the potential of IL incorporated MOFs for CO2 separation, Sezginel et al. immobilized [BMIM][BF4] into Cu-BTC at IL loadings of 5, 20, and 30 wt% by impregnation.242 Data showed that the ideal sorption selectivities, calculated as the ratio of adsorbed amounts of single-component gases, exhibited significant changes. For instance, the CO2/H2 selectivity was almost 1.5-times higher compared to that of the pristine CuBTC (Fig. 5a).242 Similarly, when [BMIM][PF6] was incorporated into ZIF-8, the CO2/CH4 and CO2/N2 selectivities were more than two-times higher than for that of pristine ZIF-8 at low pressures (up to 0.4 bar), because of the high CO2 affinity of [PF6]−.204 Even though up to 0.4 bar the CO2 uptake in the composite increased in comparison to the pristine ZIF-8 owing to the newly created adsorption sites, CO2 uptake measurements showed that the composite underperformed compared to pristine ZIF-8 above 0.4 bar.204 This result was as expected, because the majority of the pore volume of the ZIF-8 was occupied by the IL molecules in the composite, largely reducing the available pore space for gas storage at relatively high pressures.
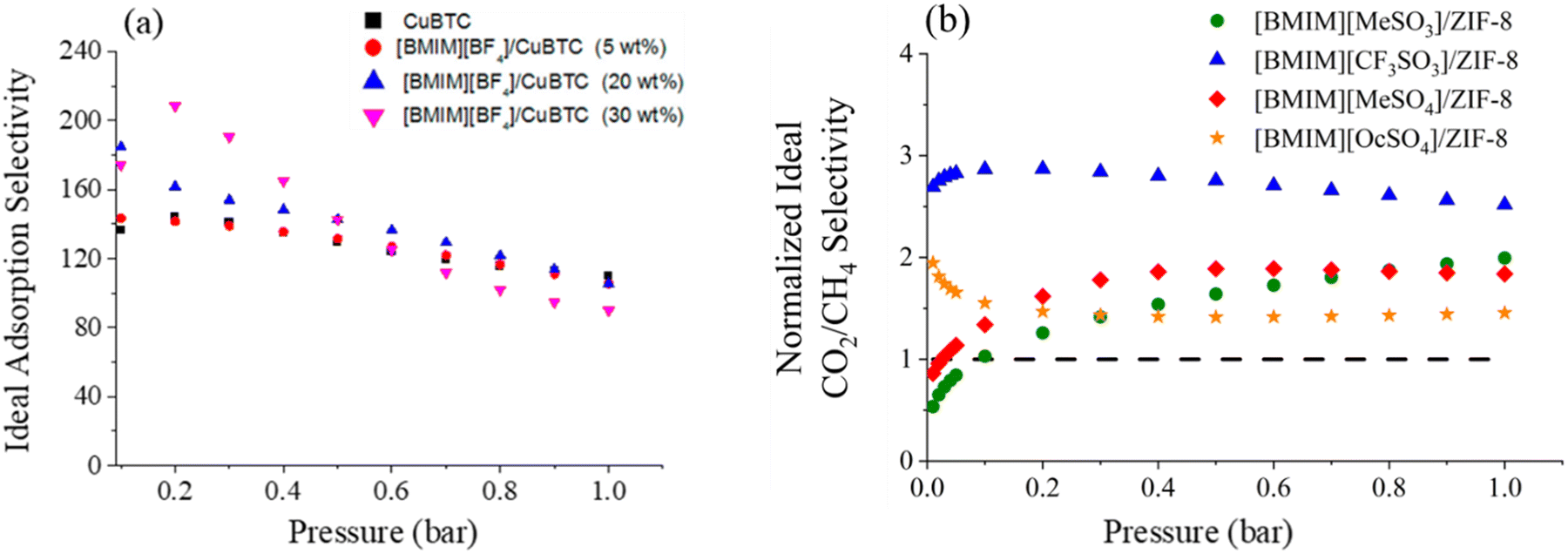 | ||
| Fig. 5 Ideal adsorption selectivities of (a) CO2/H2 for IL-based CuBTC composite with various IL loadings. Reprinted with permission from ref. 242 Copyright 2016, American Chemical Society. (b) CO2/CH4 selectivity for IL-based ZIF-8 composite. Reprinted with permission from ref. 206 Copyright 2020, Elsevier. | ||
Investigating the structural factors controlling the separation performance of such composites, Zeeshan et al. demonstrated a direct influence of the size and electronic structure of the anion of the IL.206 The composite formed by using the IL with a fluorinated anion ([BMIM][CF3SO3]/ZIF-8) provided a three-fold increase in CO2/CH4 selectivity compared to the composite formed with the non-fluorinated IL ([BMIM][MeSO3]/ZIF-8) at 1 bar (Fig. 5b). On the other hand, the composite having an IL with a small-sized anion ([BMIM][MeSO4]/ZIF-8) exhibited a CO2/CH4 selectivity 3.3-times higher at 1 bar compared to the selectivity of a similar composite having an IL with a bulkier anion ([BMIM][OcSO4]/ZIF-8). Even though all composites led to decreased CO2 capacities compared to the pristine ZIF-8, the composites were superior in terms of CO2/CH4 selectivity, controlled by the choice of the anion. Similarly, Nozari et al.243 investigated that the methylation at the C2 position on the imidazolium ring ([BMMIM][PF6] vs. [BMIM][PF6]) effects the interaction between the IL and CuBTC in composites which had a direct influence on the CO2 separation performance. The composite prepared by non-methylated IL ([BMIM][PF6]/CuBTC) provided a higher CO2/N2 selectivity compared to the selectivity of the composite prepared by the methylated IL ([BMMIM][PF6]/CuBTC) up to 200 mbar.243
The effect of IL structure on the CO2 storage and separation performance of the composite was investigated by considering metal oxides as supports as well. Mirzaei et al. immobilized various imidazolium ILs ([BMIM][NO3], [BMIM][SCN], [BMIM][HSO4], and [BMIM][DCA]) on silica and investigated the CO2 capacity of both bulk ILs and silica-supported ILs.244 Among the bulk ILs, [BMIM][DCA] and among supported ILs, [BMIM][HSO4]/SiO2 provided the highest CO2 capacities (0.42 and 0.53 mmol g−1, respectively, at 25 °C and 1 bar). The CO2 capacity of the composites was higher compared to the corresponding capacity of the bulk IL, which was attributed to the synergistic effects of silica and IL.
To investigate the extend of such synergistic effects between the IL molecules and the surface, pyridinium ILs with opposite hydrophobic characters (hydrophilic [BMPyr][DCA] and hydrophobic [BMPyr][PF6]) were immobilized on the surface of reduced graphene aerogel (rGA), whose surface characteristics were tuned by simple heat treatments at different temperatures prior to deposition of the ILs.184 Detailed characterization indicated that the heat treatment of GAs facilitates the partial removal of surface oxygen-containing groups and tunes the surface area by the formation of micropores.186,245 Even though the CO2 uptakes of the IL/rGA composites were outperformed by pristine rGA, a remarkable CO2/CH4 selectivity of 450.9 was obtained at 1 mbar and 25 °C for [BMPyr][PF6] immobilized on GA reduced at 300 °C. Similar findings regarding the decreased uptake of CO2 along with an increased CO2 selectivity over other gases, such as N2 and CH4, with the immobilization of an IL to pristine porous materials were also observed for [BMIM][Cl]/SBA-15,215 [BMIM][MeSO4]/MIL-53(Al),217 [BMIM][PF6]/MIL-53(Al),246 [BMIM][CF3SO3]/MIL-53(Al),246 [BMIM][SbF6]/MIL-53(Al),246 [BMIM][NTf2]/MIL-53(Al),246 [BMIM][BF4]/MIL-53(Al),246 [BMIM][SCN]/ZIF-8,203 1,3-bis (3-trimethoxysilylpropyl) imidazolium chloride/MCM-41,247 [BMIM][PF6]/Cu-BTC,248 [MPPyr][DCA]/MIL-101(Cr),249 [EMIM][DEP]/Cu-BTC,250 [BMIM][PF6]/rGA,186 and [BMIM][NTf2]/CuBTC.248 However, it is worth noting that several composites prepared by using conventional ILs, such as [BMIM][Cl]/montmorillonite,193 [BMIM][Cl]/α-zirconium phosphate,193 and [BMIM][Cl]/LAPONITE®,192 can provide an enhanced CO2 uptake compared to the pristine host material. This result was observed mostly when the porous material has very limited or no CO2 uptake.193
Amino- and amino acid-functionalized ILs were also used to prepare composites with porous materials187,225 to obtain higher CO2 capacities, owing to the chemisorption by the amine moiety.74,90 For example, a composite obtained by immobilizing a polyamine-based P-IL on SBA-15 ([TEPA][NO3]/SBA-15) provided a 7.8-fold CO2 uptake enhancement upon IL incorporation into the mesoporous silica (Fig. 6a).252 IR spectroscopy measurements performed on the composite after CO2 uptake experiments confirmed that CO2 is captured through the amine groups as new IR features associated with the carbamate moieties formed. The chemisorption of CO2 was further confirmed by isosteric heat of adsorption measurements and DFT calculations.
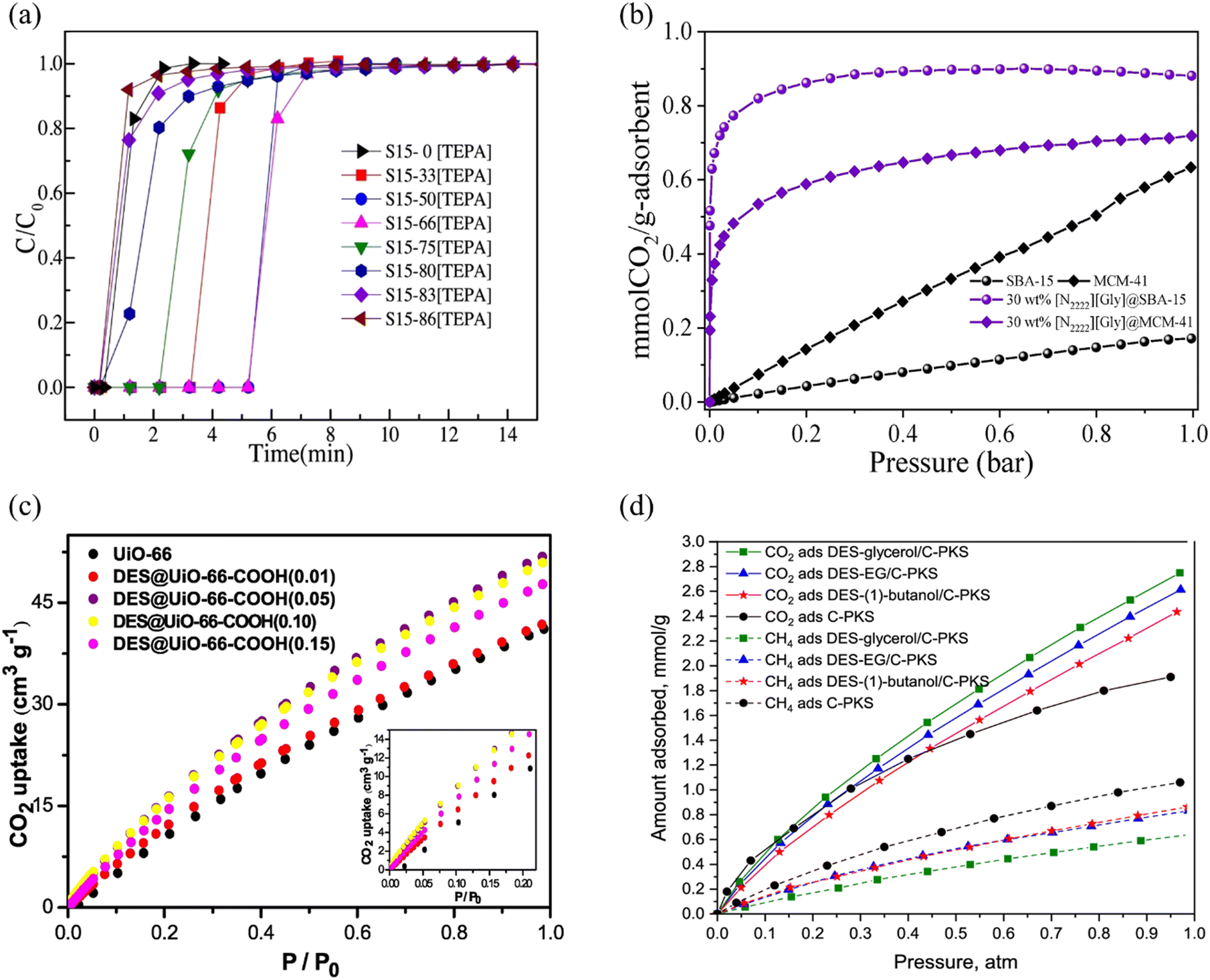 | ||
| Fig. 6 (a) CO2 breakthrough curves of SBA-15 and TEPA immobilized SBA-15 at 40 °C and 0.15 bar. Reprinted with permission from ref. 252 Copyright 2019, Elsevier. (b) CO2 uptake of amino acid-functionalized IL composites with MCM-41 and SBA-15 at 40 °C. Adapted with permission from ref. 253 Copyright 2023, Elsevier. (c) CO2 adsorption isotherms of DES@UiO-66 MOFs at 25 °C. Reprinted with permission from ref. 254 Copyright 2019, Elsevier. (d) Adsorption isotherms of CO2 at 30 °C for DES-impregnated C-PKS. Reprinted with permission from ref. 255 under CC-BY-NC-ND 4.0 license. | ||
Uehara et al. investigated the CO2 uptake of various ILs having [EMIM]+ and [P4444]+ cations, and amino acid based anions (glycine, L-lysine, L-histidine, L-alanine, β-alanine, L-arginine) immobilized on PMMA microspheres by impregnation.198 Overall, the amino acid based ILs with [P4444]+ cation provided better CO2 capacities. All composites reached their maximum uptake at an IL loading of 50 wt%, while PMMA itself provided almost no CO2 capacity. Furthermore, the CO2 adsorption on these amino acid-based IL/PMMA composites were determined as chemisorption. A comparison among anions showed that glycine and L-lysine anions having linear side chains with only one primary amino group provided higher CO2 capacities, while the L-histidine and L-arginine having secondary or tertiary amino groups provided lower CO2 capture although they have more amine groups. This difference was attributed to the presence of multiple amino groups blocking the pores of the supports and hindering the interaction with CO2.187,198 Similarly, Huang et al. immobilized an IL, 1-aminopropyl-3-methylimidazolium lysine ([APMIM][Lys]), having amino functional groups on both the cation and the anion, on PMMA and on pore expanded-SBA-15.256 All prepared composites having varying IL loadings provided significantly higher CO2 capacity than the pristine supports. Wu et al. showed that [C2OHmim][Lys] incorporation on a porous polymer, (GDX-103 (Poly divinylbenzene porous beads)) provides six-times more CO2 capacity than the pristine GDX-103 by using an IL loading of 60 wt%, at 1 bar and 40 °C.197
One challenge in the field of CO2 capture and separation is the removal of trace amount of CO2. To mitigate this challenge, Zheng and coworkers immobilized an amino acid-functionalized IL, [N2222][Gly], by impregnation at different loadings on SBA-15 and MCM-41, where both a higher CO2 capacity and a higher CO2 selectivity were achieved compared to host materials.253 [N2222][Gly]/SBA-15 composite at an IL loading of 60 wt% provided an CO2/N2 selectivity of 11545 at 0.005 bar and 15 °C, 288-times higher than that of the pristine SBA-15. Moreover, at 0.005 bar and 40 °C, 2200-times higher CO2 capacity was achieved upon IL incorporation (Fig. 6b). This outstanding performance was associated with the chemisorption of CO2 by the ILs and the newly formed ultra-micropores (smaller than 0.65 nm), especially with 60 wt% IL loading, which were not present in pristine SBA-15 (having a pore size distribution between 4.5 and 9.5 nm). As provided by these examples, amine functionalization is a promising approach to enhance the CO2 uptake of IL/porous composites.
As an alternative to carboxyl-, amine-, and amino acid-functionalized ILs, Arellano et al. demonstrated the efficiency of impregnating a zinc-functionalized IL on microporous alumino-silicate,257 non-porous nano-silica,257 non-ordered mesoporous silica,257 bio-templated mesoporous silica beads,258 and ordered mesoporous silica.200 It was shown that the zinc functionalized IL having a high viscosity could be effectively immobilized as a thin layer on the supports to make use of its interaction sites with CO2 and to overcome the mass transport challenge due to high viscosity. The efficient wetting of the zinc functionalized IL ([EMIM][NTf2] zincate) provided access to the pores of the porous host material. It has been shown that the zinc functionalized bulk IL, provided superior CO2 uptake compared to its non-functionalized analogue ([EMIM][NTf2]), owing to the additional sites for chemisorption.257
Similar to the IL/MOF composites, studies with DESs have also reported enhanced CO2 capacity. Li et al.254 showed that incorporation of reline into a UiO-66 framework not only increased the CO2 capture capacity but also preserved the open metal sites within the MOF, offering additional binding sites for CO2. Results showed that DES-incorporated UiO-66 had a CO2 uptake of 2.35 mmol g−1 compared to 1.85 mmol g−1 of pristine UiO-66 at 25 °C and 1 bar, such increase in uptake capacity was attributed to the combination of physical and chemical adsorption via –NH2 and –OH groups (Fig. 6c). Similarly, Mehrdad and co-workers236 prepared NH2-MIL-53(Al) incorporated DESs ([Ch][Cl] mixed with different amines) and reported two-fold improvement in the CO2 uptake compared to the pristine MOF at 25 °C and 5 bar. This improvement in CO2 capacity was ascribed to the dual-mode CO2 capture mechanism within DES-impregnated NH2-MIL-53(Al): it binds to the DES monolayer formed on the pore surfaces via hydrogen bonding, and secondly, it interacts with the residual DES confined within the pores. Additionally, the amine groups within the MOF structure also contributed to the overall CO2 uptake.
Composites made up of DES-impregnated porous carbon and silica have also been demonstrated. For instance, Mukti et al.259 extended this concept to modify porous carbon surface by impregnating with DES ([Ch][Cl]:Gly). Gas adsorption experiments showed that CO2 uptake was increased from 1.85 mmol g−1 (pristine Carbon)202 to 2.85 mmol g−1 (DES-modified carbon) at 30 °C and 1 bar. In another example, Hussin et al.260 functionalized palm shell based activated carbon with 14 different combinations of DESs. Among the studied samples, DES-functionalized (choline hydroxide + urea, 1:05 mole ratio) activated carbon achieved the highest CO2 uptake (0.85 mmol g−1) compared to pristine carbon (0.15 mmol g−1) at 25 °C and feed concentration of 10 vol% CO2 in He. The same group studied three types of DES-functionalized activated carbon and reported that the combination of tetrabutylammonium bromide (TBAB) and triethylene glycol (TEG) (mole ratio 1:2) is the most effective, achieving a maximum CO2 adsorption capacity of 0.37 mmol g−1 in the composite at 25 °C and 0.1 bar. Ariyanto et al.255 explored the potential of porous carbon derived from the palm kernel shell (C-PKS) impregnated with a DESs, which were composed of a ChCl and the HBDs of 1-butanol, ethylene glycol, and glycerol. Adsorption isotherm revealed that CO2 adsorption capacity in DES ([Ch][Cl]:glycerol)/C-PKS increased to 2.75 mmol g−1 in comparison to pristine C-PKS, which had a CO2 adsorption capacity of 1.70 mmol g−1 at 30 °C and 1 bar (Fig. 6d). Ghazali et al.235 investigated modification of mesoporous silica-gel with a mixture of [Ch][Cl], urea, and polyethyleneimine to prepare a hybrid material and reported a CO2 capacity of 1.15 mmol g−1 at 25 °C and 1 bar for DES incorporated silica gel composite, which was 2.5 times higher than the CO2 uptake of pristine silica gel.
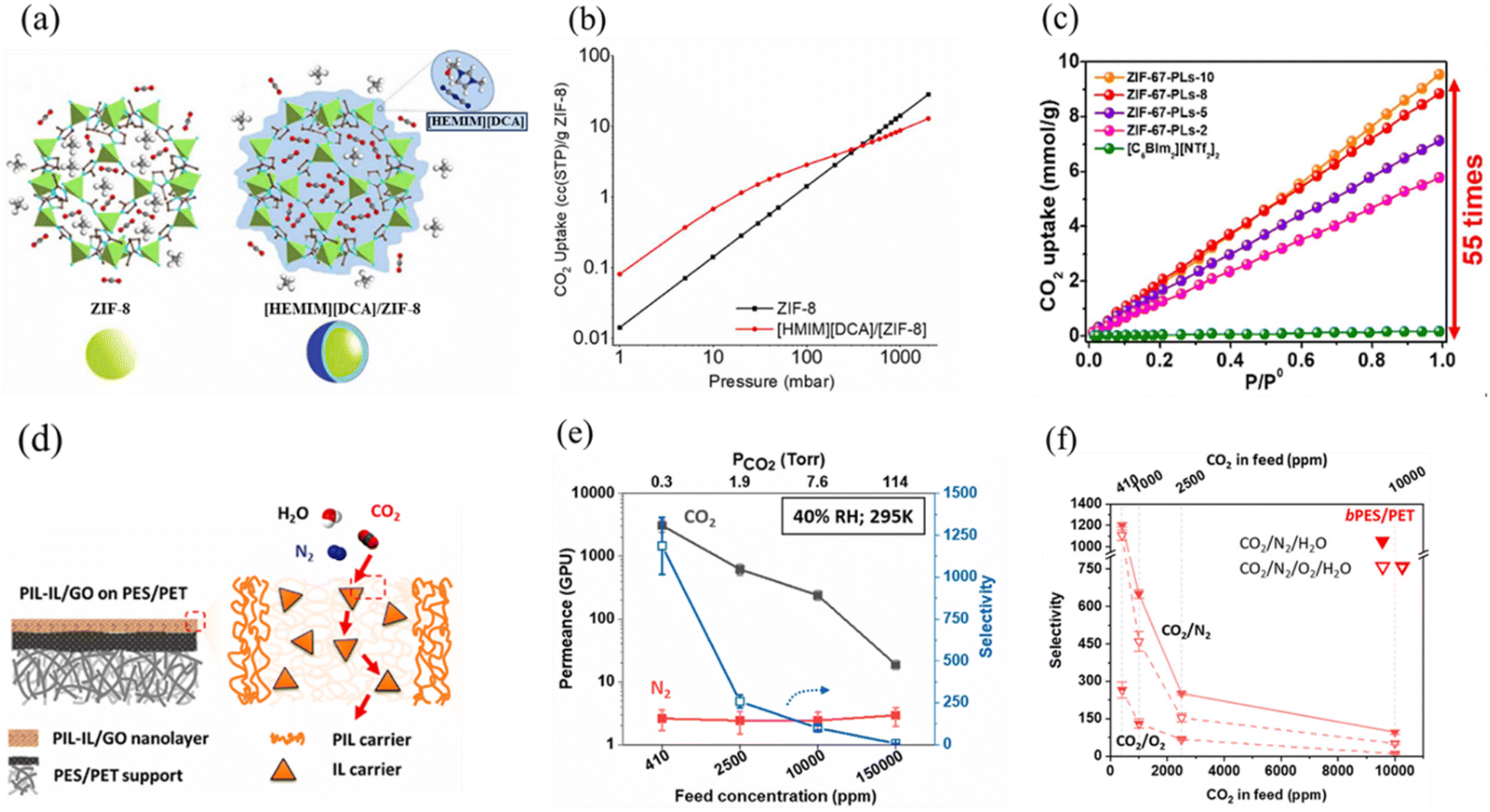 | ||
| Fig. 7 (a) Schematic representation of the proposed core–shell type IL/MOF structure; (b) CO2 uptake of ZIF-8 and IL/ZIF-8 composite at 25 °C. Reprinted with permission from ref. 202 Copyright 2018, American Chemical Society. (c) CO2 adsorption isotherms of bulk IL and ZIF-67-PLs with different loadings at 25 °C. Reprinted with permission from ref. 262 Copyright 2021, Elsevier. (d) Schematics of the fabricated PIL-IL/GO membrane on PES/PET substrate; (e) Measured CO2 and N2 permeances PIL-IL/GO thin film composite membrane (right) as a function of CO2 concentration in the feed at 22 °C and 40% relative humidity (RH). Reprinted with permission from ref. 263 Copyright 2021, Elsevier. (f) CO2/N2 and CO2/O2 selectivities of PIL–IL/GO on bPES/PET substrate at 22 °C and 40% RH. Reprinted from ref. 264 with permission from the Royal Society of Chemistry. Copyright 2022. | ||
For the core–shell type composites mentioned so far, IL loadings are restricted under the incipient wetness limit so that the composite remains in its powder form. When the incipient wetness is exceeded by a large extend, however, porous liquids (PLs) are obtained. The PLs are another novel concept that combines the intrinsic characteristics of porous materials with liquid solvents.265 A recent review paper provides a comprehensive categorization of three distinct types of PLs based on their structures,266 where type I presents intrinsic rigid porosity, type II is for porous materials dissolved in a sterically hindered solvent, and type III is where the porous material such as a MOF is dispersed in the solvent. Among these, type III PLs are synthesized by employing an excess amount of dispersive solvent that fails to permeate the pores of porous materials, hence preserving their liquid form.267 The unique combination of high ionic conductivity, very low volatility, and high solubilities of ILs makes them promising candidates for the synthesis of type III PLs.268 Commonly utilized porous materials for PL synthesis encompass zeolites, mesoporous silica, COFs, and MOFs. Among these, MOFs emerge as particularly promising porous materials for integration with ILs, facilitating the synthesis of highly stable porous ionic liquids (PoILs).269 Additionally, PoILs exhibit significant potential for CO2 capture, owing to the easily tunable structure of ILs.
Some examples of PoILs include [P4444][Lev] and [C6(BIm)2][NTf2] with ZIF-8 and ZIF-67, respectively.262,270 When 5 wt% ZIF-8 was dispersed in [P4444][Lev], the resulting PoIL exhibited a CO2 sorption capacity of 1.5 mmol CO2 per g PoILs, approximately 20% higher than that of the bulk IL, at 30 °C and 2 bar. Moreover, 10 wt% ZIF-67 dispersed in [C6(BIm)2][NTf2] displayed a remarkable CO2 capacity of 9.54 mmol CO2 per g PoILs (Fig. 7c). This result represents a 55-fold increase compared to the bulk IL, observed at 25 °C and 1 bar. Various porous materials, including silicalite-1 (a type of ZSM-5) and ZIF-8, were also employed for the synthesis of PoILs dispersed in [DBU-PEG][NTf2].271 The results revealed that the PoIL consists of 30 wt% ZIF-8 and remaining as [DBU-PEG][NTf2] exhibited superior performance, approximately 4.7-times higher than the bulk IL. However, the effective application of type III PoILs for CO2 capture is significantly constrained due to the requirement of bulky ILs (low gravimetric uptake) and porous materials with pore apertures sufficiently small to prevent the permeation of ILs into their pores.
As evidenced from these above-mentioned studies, hybrid materials, such as those obtained by confining ILs or functionalized ILs in the pores of porous supports, core–shell type composites, ENILs or PoILs provide opportunities towards reaching high CO2 uptake and particularly high CO2 selectivity over other gases, such as CH4, N2, C2H2, H2. Hence, such composites can be used as fillers in polymer matrix to form mixed matrix membranes (MMMs) to boost the CO2 separation performance. Alternatively, the ILs can be used in membrane-based gas separation in the form of supported ionic liquid membranes (SILMs),272 where the IL is directly impregnated into the pores of the membrane. However, they have certain drawbacks such as swelling and IL-leaching.273 IL polymer membranes (ILPMs), which are obtained by physically mixing the IL and polymer can be a potential solution to this problem, however, they lack mechanical strength, particularly when the IL loading is high.237 Therefore, combining ILs with inorganic fillers in the polymer matrix to obtain MMMs is a promising approach. In this way, high selectivity obtained by IL/porous material samples can be used and the IL can offer a better compatibility between the inorganic filler and the polymer by acting as a wetting agent.
Several ILs have been tested to enhance the performance of MOFs and zeolites as fillers in MMMs.274 For example, Habib et al. fabricated an IL/MOF/polymer MMM by using [MPPyr][DCA]/MIL-101(Cr) composite as a filler in Pebax.275 [MPPyr][DCA]/MIL-101(Cr)/Pebax performed 45-times and 10-times higher CO2/N2 and CO2/CH4 selectivities, than pristine Pebax, respectively. Similarly, Mahboubi et al. synthesized [BMIM][OAc]/Al2O3 nanoparticles/Pebax-1657 MMM and demonstrated that the CO2 permeability and selectivity of CO2 over CH4 improved compared to the Pebax membrane.276
Similar examples of confined ILs in membrane separations have been demonstrated. For example, Lee and Gurkan263 fabricated a composite membrane where a functionalized IL was impregnated into a graphene oxide nanoframework that served as a micron-thick selective layer supported on an ultrafiltration membrane for separation of CO2 from air (Fig. 7d). This study demonstrated the first facilitated transport membranes with a functionalized IL for separating CO2 from air. Accordingly, a CO2 permeance of 3090 GPU (1 GPU = 3.348 × 10−10 mol m−2 s−1 Pa−1) and a CO2/N2 selectivity of 1180 were demonstrated at conditions relevant to direct air capture (295 K and 40% RH) (Fig. 7e). Later, they also demonstrated a CO2/O2 selectivity of 300 with a 410 ppm of CO2 at 1 atm feed gas with helium sweeping on the permeate side (Fig. 7f).264 They noted a significant reduction in the separation performance under higher transmembrane pressures. Thin membranes based on graphene oxide layers offer a very low resistance; however, they are also prone to defect formation under high transmembrane pressures. To enhance their durability and mechanical stability, reinforcement by poly ionic liquids and increased membrane thicknesses are viable strategies as demonstrated in the discussed examples but these types of membranes are currently difficult to adapt for roll-to-roll fabrication to assemble membrane modules.
457 based on our search of this data center using Conquest software.278 Other host materials used, such as zeolites, mesoporous silica, or carbonaceous materials, also provide a substantial flexibility. In addition to this, when considering that the possible anion/cation combination for ILs is almost unlimited number, hence, it is impossible to synthesize every possible IL/porous material composite and test them for CO2 capture and separation. Similarly, there is virtually unlimited possibilities of DES compositions with varying HBAs and HBDs. There has been a lot of progress in the prediction of ILs and DES bulk properties using machine learning (ML). For example, interpretable and explainable ML models to predict the solubility of CO2 in chemically reactive DESs were demonstrated by integrating the quantum chemistry-derived Sigma σ-profiles.279 Sigma profile is used to quantify the relative probability of a molecular surface segment having a screening charge density. By utilizing σ-profiles as inputs, the developed models capture the molecular interactions.280 Among the investigated ML models, feed forward neural network (FFNN) showed the optimal performance on the CO2 solubility predictions including DESs with chemisorption capability.279 Therefore, molecular simulations and ML approaches are crucial to screen a large number of ILs/DESs and porous material combinations to determine the best candidates.
In this regard, for instance, Polat et al. used Grand Canonical Monte Carlo (GCMC) simulations to predict the CO2 uptakes and selectivities over CH4 and N2 for various IL/CuBTC composites that have ILs with the same [BMIM]+ cation but different anions, and compared their computational results with experimental measurements.281 It was demonstrated that all of the IL/CuBTC composites yielded higher CO2 selectivities than the pristine CuBTC and the predicted values agreed well with the experimental results. In another study,282 high throughput molecular simulations were combined with experiments to analyze the CO2/N2 separation performances of IL/MOF composites. Experimentally, three IL/MOF composites, [BMIM][BF4]/UiO-66, [BMIM][BF4]/ZIF-8, and [BMIM][BF4]/CuBTC, were synthesized and tested for CO2 and N2 uptakes and their CO2/N2 selectivities were determined to compare the experimental results with simulations to confirm the accuracy of the methodology. Thereafter, CO2/N2 selectivities of 1085 composites were predicted by using a single type of IL ([BMIM][BF4]) in combination with various MOFs. Accordingly, 97.6% of the studied MOFs provided higher CO2/N2 selectivity and 82.8% of them provided an increased CO2 working capacity.
Gulbalkan et al. employed a computational screening strategy by integrating conductor-like screening model for realistic solvents (COSMO-RS) calculations, GCMC and MD simulations, and DFT calculations to explore the capabilities of different IL/COF composites for CO2/N2 selectivity by considering adsorption and membrane-based separation methods.283 The CO2/N2 selectivity of TpPa-1-F2 increased from 12 to 26 upon [BMPyrr][DCA] incorporation at VSA conditions. Yan et al. investigated the optimal IL loading in IL/COF composites by high-throughput computational screening and ML.284 Instead of the frequently used gravimetric loading ratio (wt%), they proposed a new “volumetric loading (vol%)” descriptor, which was defined as the ratio of inserted IL molecules into the COF over the maximum number of ILs that can be loaded into the pore of the COF. Among 15410 screened IL/COF composites, the highest CO2/N2 selectivity was reached when the volumetric IL loading was 35 vol%, showing that computational studies can also provide insights on the optimum IL loading for synthesis to achieve the best gas separation performance.
By combining various computational tools, including quantum chemical calculations (COSMO-RS- and DFT) and molecular simulations (GCMC), Zeeshan et al.285 identified [BMPyrr][DCA] as the best pyrrolidinium IL based candidate to be combined with UiO-66 for obtaining superior CO2 separation performance. The experimental data obtained on this composite demonstrated that the [BMPyrr][DCA]/UiO-66 offers practically infinite CO2/N2 selectivity owing to extremely poor solubility of N2 and superior solubility of CO2 in [BMPyrr][DCA]. These results demonstrated the great potential of computational efforts in the way of designing composites with exceptional CO2 separation performance, especially when they are integrated in a hierarchical way.
3. CO2 thermal conversion
Reduction of CO2via chemical, thermal, biochemical, photochemical, and electrochemical reactions have been reported.7–9 The critical challenges are the requirement of substantial energy to activate CO2 and the lack of highly selective catalysts. The conventional CO2 conversion is thermal with an appropriate catalyst. Typical catalysts for the conversion of CO2 are very broad; ranging from noble and non-noble metal-based homogeneous or heterogeneous catalysts to metal-free ones. Although noble metals, such as Pd, Pt, Ir, Rh, Ru, or Au, provide several advantages, including high CO2 conversion performance, they are scarce and costly.286 Thus, cost-effective non-noble metal-based catalysts such as Cu-, Ni-, and Fe-based ones, metal oxides, zeolites or ordered mesoporous silica,287,288 and in particular metal-free catalysts, such as graphene and carbon nanotubes289 or porous organic polymers, have gained significant attention for thermal, as well as photo- and electro-catalytic CO2 conversion. This section provides an overview of the thermal catalysis of CO2 as a point of reference for conversion processes utilizing ILs (Fig. 8). | ||
| Fig. 8 Overview of CO2 thermal catalysis by IL in a continuous flow reactor mostly used for hydrogenation of CO2 (left) and a batch reactor mostly used of epoxidation of CO2 (right). The zoomed-in panels for the flow reactor represent the possible configurations where an IL layer itself or with metal NPs serves as the catalyst surface (top) or the IL-coated metal NPs are decorated over the porous support (bottom). The zoomed-in panels for the batch reactor represent the cases of IL acting as the homogenous catalyst as well as the reaction media (top) and the NPs dispersed in the IL media (bottom). | ||
ILs offer several advantages for their utilization in thermocatalytic conversion of CO2, since they can be used as both the sorption media and catalytically active phase itself, enabling an integrated capture and conversion technology consisting of a metal-free catalyst.290 Furthermore, they can also act as solvents for the reaction, where they can host externally added metal-based homogeneous or heterogeneous catalysts, or they can be themselves supported by high-surface area materials (supported ionic liquid phase catalysts, SILPs).290 Advantages which further foster their utilization in thermal conversion of CO2 are their relatively high stability at CO2 conversion temperatures and pressures, and their low vapor pressures and low melting points. By pairing of specific anions and cations or by attaching functional groups depending on the active sites needed for a specific product by CO2 conversion, ILs can be used in a very flexible way for CO2 conversion.
The thermal conversion of CO2 with systems involving ILs comprises mostly the following reactions: (i) cycloaddition of CO2 to epoxides to produce cyclic carbonates291 and the reaction of CO2 and methanol to produce linear carbonates,292 (ii) the hydrogenation of CO2 to products, such as methane, methanol, formic acid, methyl formate, or long-chain hydrocarbons, (iii) the synthesis of nitrogen containing compounds, such as quinazoline-2,4(1H,3H)-dione,293 and (iv) the synthesis of sulfur containing compounds, such as benzothiazole.294 Detailed discussion on the latter two routes regarding the production of N- and S-containing compounds can be found elsewhere.286 For the cycloaddition reaction of CO2 to epoxides, ILs can act as the catalysts themselves without the addition of any co-catalyst or solvent, while the hydrogenation of CO2 to products, such as methanol, methane, or long chain hydrocarbons, mostly necessitates the presence of other active sites of noble or non-noble metals. In the following sections, a summary of different kinds of approaches to convert CO2 to valuable products in IL-containing systems is provided with a specific focus on the CO2 cycloaddition to epoxides and CO2 hydrogenation.
3.1 CO2 conversion to cyclic carbonates
Cycloaddition of CO2 to epoxides is one of the most widely studied routes for the thermal conversion of CO2. Several ILs, SILPs, and ILs hosting metals have been considered for this reaction. For the cycloaddition reaction, the catalyst should involve multiple active sites which are crucial for the ring opening, CO2 insertion, and ring closure steps.295 A Lewis acid site or HBD is needed to polarize the epoxide.237,295 This active site can be obtained by metal sites in conventional catalysts.237 However, in IL-catalyzed systems, the cation, which provides hydrogen bonds, can act as a Lewis acid site.290 The anion, on the other hand, acts as a nucleophile to induce the ring opening of the epoxide.296 Thus, the selection of an IL is quite important as the cation and anion are the active sites for this reaction. Furthermore, the interaction of ILs dictates how available the anion and cation are for this reaction.297There are several studies reporting the use of different classes of neat ILs for the reactions involving the cycloaddition reaction of CO2 to epoxides to obtain cyclic carbonates. The very first demonstration of using ILs for solvent-free and metal-free cycloaddition reaction was by Peng et al.298 A series of imidazolium and pyridinium ILs were used as catalysts in a batch reactor to produce propylene carbonate from propylene oxide and CO2 at 2 MPa CO2 pressure, where [BMIM][BF4] was found as the best performing IL.298 Then, it has been shown that the use of supercritical CO2 is more effective for such reaction systems.299 Several ILs,299–302 P-ILs,303–306 poly-ILs,296,307–310 and dicationic ILs311 have been further investigated as catalyst for the cycloaddition reaction. Scheme 10 illustrates a proposed reaction mechanism of the cycloaddition of CO2 with epoxides.300 Accordingly, the anion plays a crucial role in the nucleophilic ring opening of the epoxide, which is the rate-determining step for this reaction. For this reaction, P-ILs are specifically advantageous since it is easy to synthesize them at relatively low-cost.303 Besides, poly-ILs can be also used as heterogeneous catalysts for the cycloaddition reaction. These ILs offer advantages in terms of ease of separation from the reaction medium, as opposed to ILs which are homogeneous catalysts.102 Other catalysts reported were amino acid-based ILs,312,313 and ILs having metals (metal ILs)314 for the cycloaddition of CO2 to epoxides.
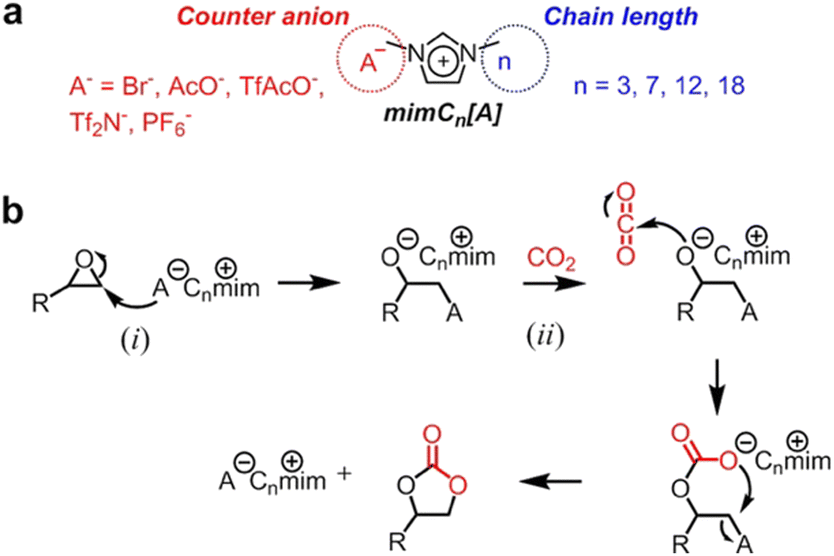 | ||
| Scheme 10 Proposed mechanism for CO2 cycloaddition reaction of CO2 to epoxides to cyclic carbonates by imidazolium ILs. A− is representing the anion and n in the cation (mimCn+) is representing the chain length on the imidazolium. Reprinted with permission from ref. 300. Copyright 2018, John Wiley and Sons. | ||
To further improve the catalytic performance, several novel functionalized ILs,315–320 functionalized poly-ILs,292,307,321,322 and functionalized P-ILs323 were tested for the cycloaddition reaction. Functionalization leads to an even higher flexibility of ILs and a control of active sites for the reaction allowing the synthesis of sites with desired nucleophilic and electrophilic character.324 For example, hydroxyl (–OH) functionalization on imidazolium-based ILs offers an eased activation of the epoxide thanks to the hydrogen bonding with the oxygen atom on the epoxide.319 In addition to –OH, other HBD moieties have been incorporated to ILs, such as –NH– and –COOH groups.295 Although the introduction of such groups can polarize the epoxide and accelerate the slow step of the cycloaddition reaction, it can also result in an intramolecular hydrogen bonding between the HBD functional group and the anion of the IL itself (typically halides), which can cause a lowered catalytic activity.325 Thus, controlling the position and number of HBD groups is crucial. To overcome this issue, Zhang et al.325 synthesized a novel family of imidazolium based ILs with multiple weak HBD groups (e.g., N–H and C–H groups) and obtained a high performance for the reaction between epichlorohydrin and CO2. In another example demonstrating the use of functionalized ILs, the synergistic effect of pyridine and bromide anion sites in mesoporous pyridine-functionalized binuclear poly(ionic liquid) showed an excellent yield of 95% at 2 MPa and 100 °C for the cycloaddition of low-concentration CO2 (15% CO2, balance N2) and epichlorohydrin without a co-solvent or co-catalyst.322
Multiple active sites for cycloaddition reaction can be obtained by synthesizing composites of ILs with porous materials, such as MOFs and COFs as heterogeneous catalysts.237 These composites provide substantial flexibility, because both the IL structure and the porous material can be functionalized and combined to reach a high performance. IL/MOF,326,327 IL/COF, IL/metal oxide,328 IL/carbon,329 IL/zeolite, IL/mesoporous silica330,331 composites have been considered widely for the cycloaddition reaction.237 For instance, a bi-functionalized IL ([HeMOEim][Br]) was impregnated on a MOF (MIL-53(Al)) and it reached a yield of 95.7% cyclopropyl carbonate for the low-CO2 concentration cycloaddition reaction at 130 °C, 0.1 MPa, and using a catalyst dosage of 0.2 mol% without the addition of any solvent or co-catalyst.332 The synergistic effect of the hydroxyl group of the IL and the Al sites of the MOF acted as Lewis acid sites to activate the epoxide, while the halide anion served as the nucleophile. Multiple functional sites could be obtained also by immobilizing a dicationic IL, having two imidazolium rings with COOH groups and two Br− anions, on SBA-15.333 A high yield (97.4%) and high selectivity (99.1%) of cyclic carbonates at a reaction temperature of 130 °C and at a CO2 pressure of 2 MPa (50 mg catalyst) were achieved thanks to the presence of multiple active sites and the successful dispersion of the IL on the support.
Another CO2 fixation approach through thermal catalysis is the reaction of CO2 and alcohols to form linear carbonates.334 For example, He et al.292 demonstrated that ternary copolymerized bifunctional poly-ILs having hydroxyls, halogen anions, and tertiary N sites are effective catalysts for the solvent- and co-catalyst-free dimethyl carbonate production from CO2 and methanol. In another example, Ding and coworkers335 synthesized porous sulfonyl binuclear carbonate poly(ionic liquid)s for utilization as a catalyst in dimethyl carbonate production where a yield of 80% was achieved without any co-catalyst from the reaction between methanol and the low concentration CO2, representing flue gas (15% CO2 + 85% N2).
3.2 CO2 hydrogenation
CO2 can be hydrogenated into a variety of products, such as formic acid, formaldehyde, methanol, methane, or long-chain hydrocarbons, by thermal catalysis. The most direct CO2 conversion route is the CO2 conversion to methanol, which can then be used as a platform molecule to produce several other chemicals.336 In another route, CO2 can be converted to CO by the reverse water gas shift reaction (RWGS). The produced syngas can further be converted to methanol, methane, and formaldehyde, or to long-chain hydrocarbons by Fischer–Tropsch synthesis. Another approach is the direct synthesis of hydrocarbons by Fischer–Tropsch from CO2 and H2, where RWGS reaction and the Fischer–Tropsch synthesis is performed simultaneously in a single-pass reactor, which needs sophisticated multifunctional catalyst design. In such a system, to shift the equilibrium to CO formation by the RWGS reaction (newly formed CO then enters the Fischer–Tropsch path to produce long-chain hydrocarbons), the co-formed water should be constantly removed.290 This could be achieved by introducing a hydrophobic IL nearby the active site, to reduce the concentration of H2O formed during the reaction.337 In addition, when ILs cover the active sites, they can also control the effective CO2 concentration. Choosing an IL having high CO2 solubility can significantly enhance the performance. Therefore, the use of ILs has several advantageous functions in hydrogenation of CO2 by thermal catalysis, when used as modifiers for the main active site or as solvents. These main active sites of the thermal CO2 hydrogenation are often provided by another active phase of noble or non-noble metal catalyst.338Melo et al.339 used in situ prepared Ru nanoparticle catalysts in an imidazolium IL ([C8MIM][NTf2]) for CO2 hydrogenation to methane. A methane yield of 69% using 0.24 mol% catalyst at 40 bar and 150 °C was achieved through the ability of [C8MIM][NTf2] to stabilize Ru nanoparticles (2.5 nm).339 The yield could be improved further to 84% by conducting a systematic study revealing [C8mim][NfO] is a better IL to host the Ru nanoparticles.340 Wang et al., on the other hand, encapsulated Ru complexes into poly(ionic liquid)s and reached almost 100% CH4 selectivity at 160 °C in a batch reactor by feeding H2 at a pressure of 3 MPa and CO2 at a pressure of 2 MPa with this catalyst.341
CO2 can be thermally hydrogenated to obtain long-chain hydrocarbons as well. Qadir et al.342 used RuFe alloy nanoparticles in two different ILs (in the hydrophobic [BMIM][NTf2] and the basic [BMIM][OAc]) for CO2 hydrogenation. The reaction path depended on the IL structure: RuFe nanoparticles in [BMIM][NTf2] favored the long-chain hydrocarbon formation path, while RuFe nanoparticles in [BMIM][OAc] led to the production of formic acid. Hydrophobic [BMIM][NTf2] controlled the H2O concentration near the active sites, shifting the equilibrium to the products. RuFe nanoparticles in [BMIM][NTf2] provided 12% CO2 conversion and 10% selectivity towards the long-chain hydrocarbons (C7–C21) at a relatively mild temperature of 150 °C and reaction pressure of 8.5 bar (CO2:H2, 1:4). Qadir et al.343 also investigated Ru/Ni core shell nanoparticles in the same hydrophobic IL, [BMIM][NTf2], and reached 30% conversion at 150 °C and at 8.5 bar (CO2:H2, 1:4) with 5% CH4, 79% C2+ alkane, and 16% olefin selectivity, respectively. Further details regarding the CO2 conversion in IL mediated catalytic systems can be found in a recent review article.344
Formic acid production by CO2 hydrogenation is one of the most studied reactions in IL containing systems. Typically, bases are added to the reaction environment to consume the produced formic acid, so that the thermodynamic equilibrium shifts to the products.345 This approach results in extra cost, as the produced waste (the formic acid salts produced because of the acid–base reaction) should be purified from the pure formic acid.345 Several reports showed that using basic ILs as buffering agents is a promising approach to shift the equilibrium to the products. For example, Weilhard and coworkers346 used a basic IL as a buffer ([BMMIM][OAc]) together with a Ru-based homogeneous catalyst (Ru pincer N-heterocyclic carbenes) and a Lewis acid catalyst to obtain comparable CO2 hydrogenation performance to formic acid to base-containing systems. In several other reports, homogeneous catalysts consisting of metal complexes were used together with ILs and with or without a co-solvent to produce formic acid as an alternative to a reaction medium containing bases.347–350
Using ploy-ILs as support materials for noble metals, such as Pd, to obtain heterogeneous catalysts for formic acid production by CO2 hydrogenation is another approach.351,352 IL-mediated heterogeneous catalysts for formic acid production can also be obtained by SILP systems.353 For example, Anandaraj and coworkers synthesized SILPs on SiO2 using various imidazolium ILs with different anions, followed by the immobilization of Ru nanoparticles by impregnation on the SILP materials.353 All Ru immobilized SILP catalysts with a high Ru dispersion (ranging between 0.8 ± 0.3 and 2.9 ± 0.8 nm) provided up to ten-times more catalytic performance towards formic acid production by CO2 hydrogenation than a reference Ru/SiO2 catalyst (1.5 ± 0.5 nm) despite their similar dispersions, emphasizing the importance of having ILs in close proximity to Ru as modifiers.
Few studies were reported involving the usage of ILs for the methanol synthesis by CO2 and H2. Generally, high pressures exceeding 2 MPa and temperatures above 200 °C are needed for the direct synthesis of methanol from CO2 and H2. The industrial methanol synthesis catalyst is Cu/ZnO/Al2O3 which remains superior to alternatives reported to date.354 Several attempts were made to enhance the methanol synthesis performance of Cu/ZnO/Al2O3 by adding noble metals such as Pt, Au, Rh, and Pd or promoters such as Zr, Ga, and F.354 Although these approaches increase the catalytic performance, they are not feasible from an economic point of view. In contrast, ILs can be used as cost-effective and metal-free modifiers for Cu/ZnO/Al2O3. They can act as sorbents for methanol, which eventually can shift the rection equilibrium to the products. For example, Zebarjad et al.355 used [EMIM][BF4], in a membrane reactor. A CO2 conversion of 42% was obtained at a temperature of 220 °C and pressure of 24 bar by using a commercial Cu-based methanol synthesis catalysts where a feed consisting of a mixture of CO2, CO, H2 and N2, and a sweep liquid consisting of [EMIM][BF4] with a flow rate of 1 cc min−1 were the testing conditions. The obtained conversion value of 42% was even higher than the equilibrium conversion (35.5%) at that condition. It remained higher when compared to the conventional organic solvent tetraethylene glycol dimethyl ether (TGDE), where a conversion of 38.4% was obtained under the identical conditions (220 °C and 24 bar). The constant in situ removal of methanol from the system by [EMIM][BF4] was proven to be an efficient approach to shift the equilibrium towards the products. Therefore, Zebarjad and coworkers further investigated the CO2 and methanol solubilities of [EMIM][BF4] at high pressure and temperature.356 Their findings demonstrate that CO2 has negligible solubility in [EMIM][BF4] under high temperature and pressure conditions. However, it was demonstrated that the solubility of methanol in [EMIM][BF4] was quite high, which is a desired property to obtain a high performing sweep liquid in processes involving methanol production. It was also found that the solubility of methanol in [EMIM][BF4] was higher than in a petroleum-based solvent, TGDE. Similarly, Reichert et al.357 showed that adding a solution of Li[NTf2] doped [P1444][NTf2] as a sorbent to the methanol synthesis reaction medium provided a yield of 63.3%. Using solely [P1444][NTf2] resulted in no observed enhancement in product yield, indicating that addition of an alkali salt (Li[NTf2]) is crucial because of its high-water sorption capacity which shifts the equilibrium drastically toward the products.
Other IL-modified CO2 reduction routes include methyl formate production from CO2 hydrogenation in the presence of alcohols, the hydroformylation of olefins with CO2 and H2 to yield aldehydes, and the alkoxycarbonylation of olefins with CO2 and methanol to yield carboxylic esters. Further details can be found in related review articles.286,290
For most of the above mentioned examples comprising the IL-mediated CO2 hydrogenation reactions, temperatures exceeding 150 °C are needed. This need comes with a challenge related with the limited thermal stabilities of ILs, which can vary significantly depending on the anion/cation pair.240 These stability limits should be high enough to ensure stable operation at the desired conditions. Even if a certain IL has a sufficiently high thermal stability in the bulk state, its stability limits can deviate substantially when they are immobilized on high-surface area supports, such as on metal oxides240,241 or MOFs.238 This phenomenon should be taken into account when using the SILP phase for CO2 conversion. In addition, it should also be noted that any co-catalysts present with ILs, such as metal nanoparticles, can also accelerate the catalytic decomposition of ILs at elevated temperatures. Besides, for the catalyst systems having ILs in direct contact with metal complexes or nanoparticles, ILs can donate or withdraw electrons to/from the metal, and the presence of such electronic interactions can potentially influence the electronic structure of the active sites.358–360 This possibility should be taken into consideration when designing such catalysts; the structure–performance relationships should be investigated focusing particularly on characterizing these electronic interactions, and their consequences on the catalytic properties.
In the light of the temperature, pressure, and catalyst requirements of thermal conversions, electrochemical approaches provide a more benign alternative where the reactions can be driven by electricity under ambient conditions. ILs maintain their importance in electrochemical conversions as they discussed in the next section.
4. CO2 electrocatalytic conversion
Electrochemical reduction of CO2 is a 154-year-old, still unresolved challenge, which was first studied in 1870 by M. E. Royer361 in Paris. Various CO2 reaction pathways have been suggested based on the physicochemical features of the metal catalyst.362–368 Hori et al.369 showed conversion of CO2 to other chemicals beyond CO, including gaseous C2 products, and even tri-carbons can be achieved, specifically with a copper catalyst in aqueous electrolytes. Among the major challenges associated with CO2RR are the large energy requirements, low efficiencies due to competition with the hydrogen evolution reaction (HER), and limited solubility of CO2 in the liquid. On the other hand, electroreduction of CO2 offers the potential to achieve higher conversion efficiency and product selectivity compared to other conversion methods.370 Theoretically, the overall potentials of +0.2–0.6 V (vs. Normal Hydrogen Electrode, SHE) are sufficient to reduce CO2 to CO, formic acid, methane, and other products in an aqueous solution (1 M electrolyte, pH = 7, 25 °C, 1 atm CO2) as seen in Table 5. However, the thermodynamic potential values in nonaqueous electrolytes can be different.371 Further, the overpotentials required for the formation of CO2 reduction products are independent of the bulk electrolyte pH on an absolute potential scale.372,373 In practice, very negative potentials must be applied to reduce CO2 to intermediates. The first step in CO2RR is the anion radical formation, CO2−, by one electron transfer from the electrode to the CO2 on the electrode surface. This initial step involves an energetically unfavorable intermediate, which requires a potential of −1.9 V (vs. SHE).| Half reactions of electrochemical CO2 reduction | Electrode potentials (V vs. SHE) at pH 7 |
|---|---|
| CO2(g) + e− → CO2˙− | −1.90 |
| CO2(g) + 2H+ + 2e− → HCOOH(l) | −0.61 |
| CO2(g) + H2O(l) + 2e− → HCOO(aq)− + OH− | −0.43 |
| CO2(g) + 2H+ + 2e− → CO(g) + H2O(l) | −0.53 |
| CO2(g) + 2H2O(l) + 2e− → CO(g) + OH− | −0.52 |
| CO2(g) + 4H+ + 2e− → HCHO(l) + H2O(l) | −0.48 |
| CO2(g) + 3H2O(l) + 4e− → HCHO(l) + 4OH− | −0.89 |
| CO2(g) + 6H+ + 6e− → CH3OH(l) + H2O(l) | −0.38 |
| CO2(g) + 5H2O(l) + 6e− → CH3OH(g) + 6OH− | −0.81 |
| CO2(g) + 8H+ + 8e− → CH4(g) + 2H2O(l) | −0.24 |
| CO2(g) + 6H2O(l) + 8e− → CH4(g) + 8OH− | −0.25 |
| 2CO2(g) + 12H+ + 12e− → C2H4(g) + 4H2O(l) | 0.06 |
| 2CO2(g) + 8 H2O + 12e− → C2H4(g) + 12OH− | −0.34 |
| 2CO2(g) + 12H+ + 12e− → CH3CH2OH(l) + 3H2O(l) | 0.08 |
| 2CO2(g) + 9H2O + 12e− → CH3CH2OH(l) + 12OH− | −0.33 |
ILs are shown to lower the activation energy of CO2− formation371 by co-catalyzing the reaction at the metal electrode surface, suppressing HER,375,376 and increasing CO2 solubility377,378 with control over the reaction pathway.379 An overview of the discussed reactions in the literature, as interpreted by us, are represented in Fig. 9. However, the exact mechanism by which ILs lower the overpotential (difference between the thermodynamic and experimental reduction potential) is unclear. Whether an inner sphere electron transfer380 for surface adsorbed CO2 or a carboxylated [EMIM]+ promoted route371 takes place is debated.
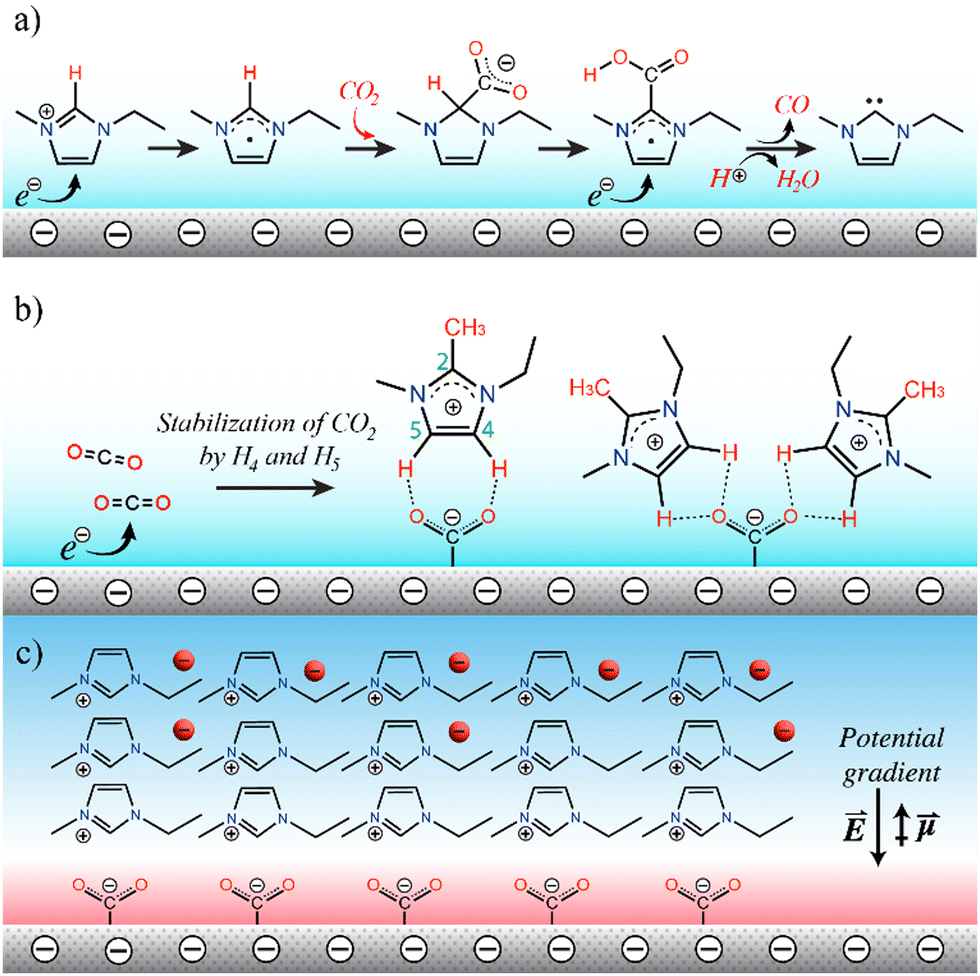 | ||
| Fig. 9 Overview of the discussed reaction routes in the literature describing the co-catalytic role of [EMIM]+ during CO2 electrolysis. | ||
Briefly, Han-Scherer et al.381 and Neyrizi et al.382 reported the formation of an intermediate [EMIM]+-CO2−, providing a low-energy pathway toward the conversion of CO2 to CO on Ag surfaces, as illustrated in Fig. 9a. Stabilization of the high-energy CO2− intermediate by hydrogen bonding through the C4–H or C5–H functionality of C2-substituted imidazolium cations are reported by Lau et al.,383 as represented in Fig. 9b. Similarly to this study, Min et al.384 also showed C4-H and C5-H promoted CO generation; however this was in parallel to C2-H promoted HER as the C2 was not substituted. Liu et al.385 discussed the ionic screening effects on acceleration the CO generation through the first steps of CO2RR. Recently, Noh et al.386 reported that the imidazolium cations form an ordered structure at the electrode surface under negative applied potential, decreasing the entropy of activation, as represented in Fig. 9c. There are also few reports showing the importance of anion identity in tunning product selectivity.387
DES electrolytes are believed to play a similar role to ILs where increased Faradaic efficiencies towards CO production on Ag were reported,388,389 compared to aqueous systems. Although studies with DESs are more scarce, the main challenge for both electrolytes for CO2RR is their high viscosity and the related mass transport limitations that ultimately result in low current densities. Therefore, these solvents are often diluted with an appropriate solvent to tune viscosity and conductivity. In 2016, Kenis, Masel and coworkers reported390 on the successful strategy of adding additional supporting salt (e.g., KCl) in imidazolium chloride and DES based electrolytes in order to improve conductivity and therefore the current density. With these enhanced formulations, the question of course is whether the IL or the DES components function in the same way as they would without additional diluents or supporting salts. It is therefore important to develop a fundamental understanding of the effects of solvation, electric field, and surface adsorbed species on CO2RR. While the physical models of the electrode–electrolyte interfaces with IL and DES based electrolytes are being developed to shed light to the CO2RR mechanism, the reactive systems (e.g., CO2 chemisorbing ILs and DESs) bring more complexity to these models as the electron transfer reaction is accompanied by chemical reactions, and therefore present multiple scientific questions that are new and inherent to RCC. Before delving into the specific challenges of RCC with reactive electrolytes, we must first revisit CO2RR with more traditional systems and outline how incorporation of ILs alters CO2RR thermodynamics and kinetics. We can then begin to understand RCC with functionalized ILs and DESs.
4.1 State-of-art electrochemical CO2 conversion
Since Hori's work,369 published in the 1990s utilizing different metal electrodes for CO2RR, considerable efforts have been dedicated to enhancing the performance of both metal and alloy electrodes. Achieving high selectivity in CO2RR remains primary research focus due to close standard reduction potentials from CO and formic acid to multicarbon chemicals (C2+). In 2017, Rossmeisl and co-workers391 subsequently categorized metal electrodes into four groups: (i) Ti, Fe, Ni, Pd, Ga, and Pt electrodes primarily undergo the HER; (ii) Ag, Au, and Zn electrodes predominantly yield CO as the main reduction product; (iii) Cd, Hg, In, Tl, Sn, and Pb electrodes primarily generate formic acid/formate, and (iv) Cu which forms multicarbon products.Cu-based materials demonstrate remarkable selectivity in catalyzing CO2RR yielding a range of hydrocarbons and alcohols. Over the years, numerous strategies have been investigated to enhance the selectivity of hydrocarbons and alcohols over copper. These approaches include adjusting surface morphology, surface alloying, controlling oxidation states, and achieving atomic dispersion. These efforts have primarily focused on modulating binding energies of reaction intermediates and the electrode surface. For example, Gu et al.392 reported 70% FE for ethanol and n-propanol over defect-site-rich Cu surfaces which enhances local CO production and, thereby, increase the surface *CO coverage and CO2-to-alcohol selectivity in aqueous 0.1 M KHCO3 electrolyte. Despite these advancements, the challenge persists in managing hydrogen production, which often competes with or exceeds the desired product yield. This is due to the aqueous nature of electrolytes, which necessitates precise control in adsorption of CO2 or CO2 intermediates on the catalyst active sites.393
Further, there are several material classes beyond transition metal monometallics that exhibit promise in CO2 electrocatalysis, including metal alloys, carbon-based materials, transition metal chalcogenides, nanostructured materials, and single-atom catalysts. Despite the success demonstrated by a diverse range of material compositions and structures in aqueous CO2RR conditions, their performance in IL electrolytes, which present unique advantages such as a broader electrochemical window and improved thermal stability, remains largely unexplored. Although these electrode materials have proven effective in aqueous CO2RR, they still face limitations that is discussed herein.
4.2 IL and DES electrolytes
Acknowledging the limitations of conventional aqueous electrolytes and saturation of the electrode materials research, recent efforts in CO2RR has focused on electrolyte effects.424–426 In particular, the challenges raised by the low solubility of CO2, excessive HER, and the instability of metal and metal oxide catalysts in aqueous environments are the main motivators for electrolytes discovery and formulations. Due to the high CO2 capacity of ILs and DESs, electrolytes based on these solvents have gained interest in CO2RR and more recently in RCC. For example, electrode materials encountering low catalytic activity, ILs and DESs can alleviate these issues by providing a conductive environment and increasing CO2 concentration at the electrode surface, enabling enhanced mass transport of reactants and intermediates to active sites within these electrode materials. Moreover, tuning solvent–solute interactions in ILs can stabilize catalytic active sites and inhibit undesirable side reactions, enhancing catalyst efficiency and selectivity. Overall, harnessing these benefits of IL and DES environments in CO2RR and RCC holds promise in overcoming challenges such as stability, selectivity, and mass transport limitations.ILs as electrolytes present wide electrochemical stability, high CO2 solubility, negligible vapor pressures, and tunable physical properties.381,427–429 The ionic DESs share similar properties to ILs and have been studied for CO2RR in recent years. ILs are reported to activate CO2, otherwise thermodynamically very stable, and accelerate its transport to the reaction interface, acting as “co-catalysts”.371 Analogous arguments can be envisioned for DESs; however, there are far fewer CO2RR studies with both solvents, than for CO2 capture. Furthermore, when it comes to RCC, there is scarcity in the literature.
Both CO2RR and RCC studies to date including ILs have mostly involved the imidazolium cation and a variety of anions. While neat ILs demonstrate high CO2 solubilities, they also present mass transfer limitations due to their high viscosities (i.e., >15 cP at 25 °C). Functionalized ILs generally present even higher viscosities and may further increase in viscosity after saturation with CO2 due to the increased hydrogen bonding as elaborated earlier. DESs are also viscous in nature as they are concentrated and involve an extensive hydrogen bonding network.430 Therefore, CO2RR and RCC studies to date examined ILs and DESs mostly in the presence of co-solvents such as water, acetonitrile, and dimethyl sulfoxide.
A review article published in 2024 summarizes the use of ILs and DESs for CO2RR.435 Accordingly, various heterogeneous metallic electrodes have been used in the presence of ILs in a variety of cell configurations for the production of many different CO2 reduction products (e.g., CO, HCOOH, CH3OH, oxalate, polyethylene, etc.) as summarized in Table 6. The most relevant electrocatalytic performance metrics, including selectivity, current density and the main product faradaic efficiency, are included. Despite the controversy surrounding the hydrolytic decomposition of [EMIM][BF4] through [BF4]− hydrolysis to HF in the presence of water,436 this IL has become famous for CO2RR studies. In many cases, the introduction of an imidazolium-based IL, both in organic and aqueous solutions, has been found to significantly enhance catalytic activity and selectivity, promoting the reduction of CO2 to CO and other valuable products while suppressing the HER. Apart from high selectivity for CO and HCOOH in IL based electrolytes on Ag and Pb, respectively, another interesting C1 product, methanol, was reported on atomically dispersed Sn catalysts on a defective CuO support.437 This electrocatalyst showed remarkable selectivity of 88.6% for methanol production at a current density of 67.0 mA cm−2. This study highlights the synergistic effect of catalyst design and the IL based electrolytes in achieving high selectivity and efficiency for methanol production.
| IL | WE[Ref] | Catholyte | Anolyte | CO2RR onset | Type of cell | E ve (V) | CDtotal (mA cm−2) | FE (%) |
|---|---|---|---|---|---|---|---|---|
| a The bulk electrolysis was performed at constant current density at −10 mA cm−2 for 4.5 h. | ||||||||
| [EMIM][BF4] | Ag NPs/GDE376 | 10.5 mol% IL in water | 0.1 M H2SO4 | −0.5 V vs. RHE | Flow | −0.7 V vs. SHE | — | ∼100% (CO) |
| MoS2438 | 4 mol% IL in water | 0.1 M H2SO4 | −0.164 V vs. RHE | Two-compartment | −0.764 V vs. RHE | 65 | 98% (CO) | |
| Ag385 | 0.9 M IL in acetonitrile with 0.1 M [TBA][BF4] | — | −2.01 V vs. Ag/Ag+ | Two-compartment | −2.5 V vs. Ag/Ag+ | 78.5 | ∼100% (CO) | |
| Ag-coated Al foam439 | 8 mol% IL in water | 0.5 M H2SO4 | −1.1 V vs. Pt | Flow | −1.8 V vs. Pt | 36.6 | 75% (CO) | |
| Nanostructured Cu440 | 2–8 mol% IL in water | 2–8 mol% IL in water | −1.2 V vs. Fc/Fc+ | Single-compartment | −1.55 V vs. Fc/Fc+ | — | 83% (HCOOH) | |
| Ag441 | 0.7 M IL in acetonitrile | — | −1.9 V vs. Ag/Ag+ | Two-compartment | −2.5 V vs. Ag/Ag+ | ∼40 | 80% (CO) | |
| [EMIM][NTf2] | Pb379 | 0.1 M IL in acetonitrile with 0.1 M TEAP | 0.1 M IL in acetonitrile with 0.1 M TEAP | −2.12 V vs. Ag/Ag+ | Two-compartment | −2.40 V vs. Ag/Ag+ | — | 40% (CO), 60% (carboxylate) |
| [BMIM][BF4] | Sn dispersed CuO437 | 25 mol% IL in water | −1.9 V vs. Ag/Ag+ | Two-compartment | −2.0 V vs. Ag/Ag+ | 67.0 | 88.6% (CH3OH) | |
| [BMIM][BF4] [BMIM][OAc] [BMIM][CF3SO3] | Ag442 | 0.3 M IL in acetonitrile | 0.1 M KOH | −2.0 to −1.88 V vs. Ag/Ag+ depends on IL | Two-compartment cell | −1.65 V vs. Ag/Ag+ | −20 | 20–97% (CO) depending on the IL |
| [BMIM][Cl] | Ag443 | IL with 20 wt% water | 0.1 M H2SO4 | −1.5 V vs. SCE | Two-compartment | — | −2.4 | 99% (CO) |
| [DMIM][BF4] | Ag444 | 2 mM IL in acetonitrile with 0.1 M Bu4NPF6 | 2 mM IL in acetonitrile with 0.1 M Bu4NPF6 | −2.28 V vs. Fc/Fc+ | Two-compartment | −2.3 V vs. Fc/Fc+ | — | 45.1% (CO), 53.4% (C2O42−), 0.4% (HCO2−) |
| [MM][NTf2] | Au382 | 0.5 mol% IL in acetonitrile | 0.5 mol% IL in acetonitrile | −0.8 V vs. Ag/Ag+ | Single compartment | 10 | 100% (CO) | |
Combining the non-functionalized ILs (i.e., 0.5 M [BMIM][PF6]/acetonitrile) with N,P-co-doped carbon aerogel (NPCA) electrolytes, one of the highest current densities in CO2RR for CO generation was achieved with 99.1% FE and 143.6 mA cm−2 current density.445 This performance was attributed to the synergy between the electrode and the electrolyte, where the large electrochemically active surface area of the NPCA electrode and the improved electronic conductivity achieved by the N-/P-doping were complemented by high CO2 availability at the electrode surface by the IL as well as the stabilization of the [BMIM-CO2](ad) complex that has a lower reaction barrier.
Apart from the bulk metals examined with IL electrolytes, electrocatalysis with MOFs also have interesting behavior in the presence of ILs. For example, Kang et al.434 performed CO2 electroreduction using a Zn-based MOF (Zn-1,3,5-benzenetricarboxylic acid metal–organic frameworks (Zn-BTC MOFs) deposited on carbon paper (CP)) in the neat imidazolium IL ([BMIM][BF4]). They reported the adsorption of CO intermediate on the catalyst framework, stable enough to accommodate up to 6-electron transfers and the generation of CH4 (80.1% FE at −2.2 V vs. Ag/Ag+). The stabilization of the intermediate was believed to be due to the surface adsorbed layer of imidazolium cations on the surface after the Zn-BTC/CP cathode immersed in the IL electrolyte. The authors also performed CO2, CO, and CH4 adsorption experiments at 1 atm and 298 K on the Zn-BTC/CP. The obtained adsorption capacities showed stronger binding of CO2 and CO over CH4 on Zn-BTC/CP. Furthermore, electrolysis with bulk metal foils (Ag, Au, Fe, Pt, Cu, and Zn) produced very low CH4 formation rates in comparison to Zn-BTC/CP under similar conditions, confirming the synergistic effects of the IL electrolyte and the MOF.
Similarly, Zhu et al.446 reported an in situ electrosynthesized hollow copper metal–organic framework (Cu-MOF) that selectively produces formate (FEformate = 98.2%) with a very high current density of 102.1 mA cm−2 in a 0.5 M [BMIM][BF4]/acetonitrile electrolyte containing 1.0 M water. To the best of our knowledge, this is the first report of such high current densities in a simple H-cell configuration. The authors ascribed this to the availability of abundant exposed edges, which offer more active sites for catalytic reactions, and the unique morphology facilitating direct contact between the catalyst and the substrate, favoring electron transfer. This facilitates efficient electron transfer from the inner metallic Cu-gauze to the outer shell of the Cu layer, and subsequently to the CO2 substrate.
In the summary, significant progress has been observed in CO2RR utilizing MOF-related electrocatalysts. However, achieving high selectivity and high current densities requires the dilution of IL with a suitable solvent, as it allows better transport of reactant to the electrode surface. The utilization of MOF structures in neat IL electrolyte may pose mass transfer limitations during CO2RR, giving low current densities.434 Therefore, a preferable strategy involves immobilizing IL on MOFs to serve as an electrocatalyst directly. This approach facilitates the study of functional IL, enabling selective CO2 capture from dilute streams, followed by conversion.
From the studies thus far involving the imidazolium IL, it is inferred that the complex formation between the cation and CO2 plays an important role in improving the CO2RR efficiency and the thermodynamics.447 While the formation of the imidazolium-CO2 complex does not occur in the absence of the electric field with non-functionalized ILs (e.g., [EMIM][BF4] or [BMIM][PF6] do not spontaneously chemisorb CO2), it does with functionalized ILs involving basic anions. Therefore, exploring functional ILs with CO2 chemisorption is gaining interest for RCC.
| ILs | WE[Ref] | Catholyte | Anolyte | CO2RR onset | Type of cell | E ve (V) | CDtotal (mA cm−2) | FE (%) |
|---|---|---|---|---|---|---|---|---|
| [P66614][124-Trz] | Ag377 | 0.1 M IL in acetonitrile w/0.7 M H2O | 0.1 M IL in acetonitrile w/0.7 M H2O | −0.90 V Ag/Ag+ | Two compartment cell | −0.7 V vs. Ag/Ag+ | — | 95% (HCOO−) |
| [BMIM][124-Trz] | Pb448 | 0.7 M IL in acetonitrile w/5 wt% H2O | 0.1 M H2SO4 | −1.78 V Ag/Ag+ | Two compartment cell | −2.1 V vs. Ag/Ag+ | 24.5 | 95.2% (HCOOH) |
| [P4444][4-MF-PhO] | Pb449 | 0.5 M IL in acetonitrile | 0.1 M H2SO4 | −2.42 V Ag/Ag+ | Two compartment cell | −2.6 V vs. Ag/Ag+ | 12.6 | 93.8% (C2O42−) |
| [EMIM][2-CNpyr] | Ag450 | 0.1 M IL in acetonitrile w/0.1 M TEAP | 0.1 M IL in acetonitrile w/0.1 M TEAP | −1.92 V Ag/Ag+ | Single compartment cell | −2.1 V vs. Ag/Ag+ | 6 | 94% (CO) |
| [EMIM][2-CNpyr] | Cu451 | 0.5 M IL in acetonitrile w/0.1 M TEAP | 0.1 M H2SO4 | −1.90 V Ag/Ag+ | Two compartment cell | −2.1 V vs. Ag/Ag+ | 7.2 | 11% (Succinate) |
To the best of our knowledge, the first report utilizing a functional IL for RCC was published in 2009 by Compton et al.452 They examined neat [BMIM][OAc] with a CO2 solubility of 1520 mM (at 1 bar and 25 °C) and reported reduction at −1.3 V vs. Ag wire on a Pt electrode at room temperature. While this study did not explain the reduction mechanism or report any specific products, they noted the irreversible nature of the reaction and that CO2 complexes with the anion.
Later, Hollingsworth et al.377 highlighted the superbasic tetraalkyl phosphonium IL, [P66614][124-Trz], for RCC where CO2 binds to the anion [124-Trz]−. This interaction is said to enable the reduction of the chemisorbed CO2 at −0.7 V vs. Ag/Ag+ on Ag. This potential is corresponding to applied overpotentials as low as 0.17 V, yielding formate as the primary product. The control experiments of the IL with a non-reactive anion ([NTf2]−) showed a selectivity towards CO production. This study points to the significance of reactive anions in providing alternative pathways with reduced activation energy for RCC. Nonetheless, the current density was limited, below 1 mA cm−2, also pointing to the necessity for further tuning electrolyte formulations. Feng et al.448 combined the favored imidazolium cation with the superbasic anion, 1-butyl-3-methylimidazolium 1,2,4-triazolide ([BMIM][124-Trz]), to further enhance the RCC performance. The FE and current density for formic acid, the only observed reaction product, reached 95.2% and 24.5 mA·cm−2, respectively, on a Pb surface. The main explanation offered was that the CO2 bound on the anion interacts closely with the C2 hydrogen of the imidazolium, thus lowering the energy barrier for generating formic acid. This was further supported by DFT calculations where the CO2 in [124-Trz]-CO2− is mostly in an sp2 hybridized state and more representative of that in formic acid. On the other hand, they also discussed the possible mechanism of imidazolium reduction followed by the electron transfer to CO2 facilitated by this imidazolium radical, as with previous studies, where oxalate formation is prevented. However, the [124-Trz]-CO2− complex formation offers a lower-energy barrier for generating formic acid, and thus serves as the main mechanism for CO2 reduction. These findings once again confirmed the importance of the ionic microhabitat in enhancing CO2 reduction.
Another example of aprotic IL with a reactive anion, 4-(methoxycarbonyl) phenol, and phosphonium cation was reported to produce oxalate on a Pb electrode.449 The IL, [P4444][4-MF-PhO], was diluted in acetonitrile (0.5 M) and oxalate was generated with 93.8% FE at −2.6 V vs. Ag/Ag+. They attributed this outstanding selectivity to the effectiveness of [P4444][4-MF-PhO] in activating the CO2 molecule through ester and phenoxy double active sites. Furthermore, the IL was discussed to tune the local electronic structure of Pb to facilitate CO2 activation and dimerization of the surface adsorbed CO2˙− to form *C2O42−.
Recently, Gurkan and co-workers450,451 have reported another functional IL with AHA and imidazolium cation, [EMIM][2-CNpyr], where they discussed the utilization of the CO2 complexes with both the anion and the cation through the proposed mechanism shown in Fig. 10. They performed a systematic study where the anion and the cation were varied to investigate the effective role of the IL components. Accordingly, the pre-activation of CO2 was achieved chemically through the carboxylate formation via the carbene intermediate of the [EMIM]+ cation and the carbamate formation via binding to the nucleophilic [2-CNpyr]− anion. The carboxylic acid intermediate was confirmed by in situ surface enhanced Raman spectroscopy (SERS) and the organic radical formation, attributed to imidazolium, was further confirmed by electron paramagnetic resonance (EPR) spectroscopy. Specifically, the onset potential for RCC was observed to positively shifted by 240 mV when the IL was introduced to the acetonitrile-based electrolyte. The main product was CO, as seen on the surface by SERS and in the gas phase by (GC) with >94% FE at −2.1 V vs. Ag/Ag+ over a Ag electrode. In their follow up work, they reported the same IL on Cu where various C2+ products (∼20% FE) including succinate were obtained.451 This study specifically examined the electrode–electrolyte interface, highlighting the role of the HBD (protonated anion) that is generated in situ from CO2 absorption for controlling the product selectivity. Interestingly, the ethane and ethylene formation were enhanced with an intentional increase in HBD concentration. This study suggests that the hydrogen bonding network offered by ILs and DESs within the interfacial region helps regulate RCC product selectivity and energetics in aprotic solvents.
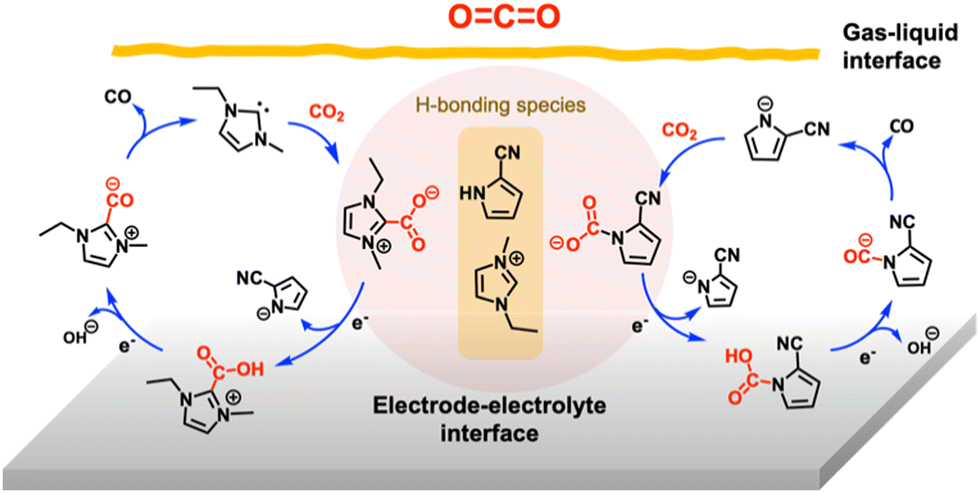 | ||
| Fig. 10 The proposed CO2 conversion reaction routes on Ag facilitated by the functionalized IL, [EMIM][2-CNpyr]. Reprinted with permission from ref. 450 Copyright 2023, American Chemical Society. | ||
From the above discussed studies, it can be concluded that CO2 reduction onset potential shift positively with both the functionalized and non-functionalized ILs and produce similar products on the same electrodes; however, the mechanisms by which the electron transfer and the proton transfer differ. The main underlying reason is that the non-functional ILs are entirely composed of ions even after CO2 exposure, while functional ILs contain an important proportion of neutral molecules (≥50 mol%) (potentially serving as native HBDs),138,453 which directly impact the distribution of charged species and oriented dipoles at the electrode/solution interface. Therefore, it is critical to examine the electrode–electrolyte interfaces by in situ spectroscopy techniques complemented by first principle calculations to unravel roles of the individual components in facilitating RCC and governing selectivity.
:urea (1:2) DES (a.k.a, reline) for selective CO formation with Ag nanoparticles supported on a gas diffusion electrode.390 The electrolyte composition was systematically varied from conventional (i.e., KOH, KHCO3, and KCl) to reline in an aqueous solution. The highest selectivity for CO (FECO = 94.1%) was observed with 2 M [Ch][Cl]:urea/water at a total current density of 11.6 mA cm−2. The authors explained the possibility of greater stabilization of the CO2− intermediate by the ammonium species, enabling high selectivity for CO compared to that of other conventional electrolytes. Furthermore, they highlighted the limitations of the DES electrolyte in achieving high current density due to its high viscosity and low conductivity. To improve the conductivity of the electrolyte, they introduced 1.5 M KCl to the 0.5 M DES/water mixture, which improved the electrolyte conductivity from 34.5 mS cm−1 to 131.4 mS cm−1, thus achieving a two-fold increase in total current density (22.1 mA cm−2) without appreciable change in CO selectivity at the same cell potential.
In a subsequent study in 2019, Garg et al.388 conducted concentration-dependent CO selectivity investigations on Ag by varying the reline concentration from 50 to 70 wt% in water. At all tested reline concentrations, more than 95% FECO was observed at −0.884 V vs. the reversible hydrogen electrode (RHE). The authors attributed this high selectivity to reline-mediated interactions with the Ag surface, resulting in the restructuring of Ag with low-coordinated atoms and facilitating CO2 reduction through choline and urea functionalities. Briefly, the elevated reline concentration was found to dissolve native Ag-oxide layers on the Ag foil, with the dissolved Ag-oxides subsequently being electrodeposited back onto the Ag foil during constant potential CO2 electrolysis experiments (−1.084 V vs. RHE) as confirmed by the morphology analysis shown in Fig. 11a. This dissolution/deposition process resulted in low-coordinated silver atoms on the cathode surface, thereby increasing the surface area and promoting the adsorption of urea and choline cations. Consequently, this led to a significant reduction in HER and enhanced CO2RR efficiency.
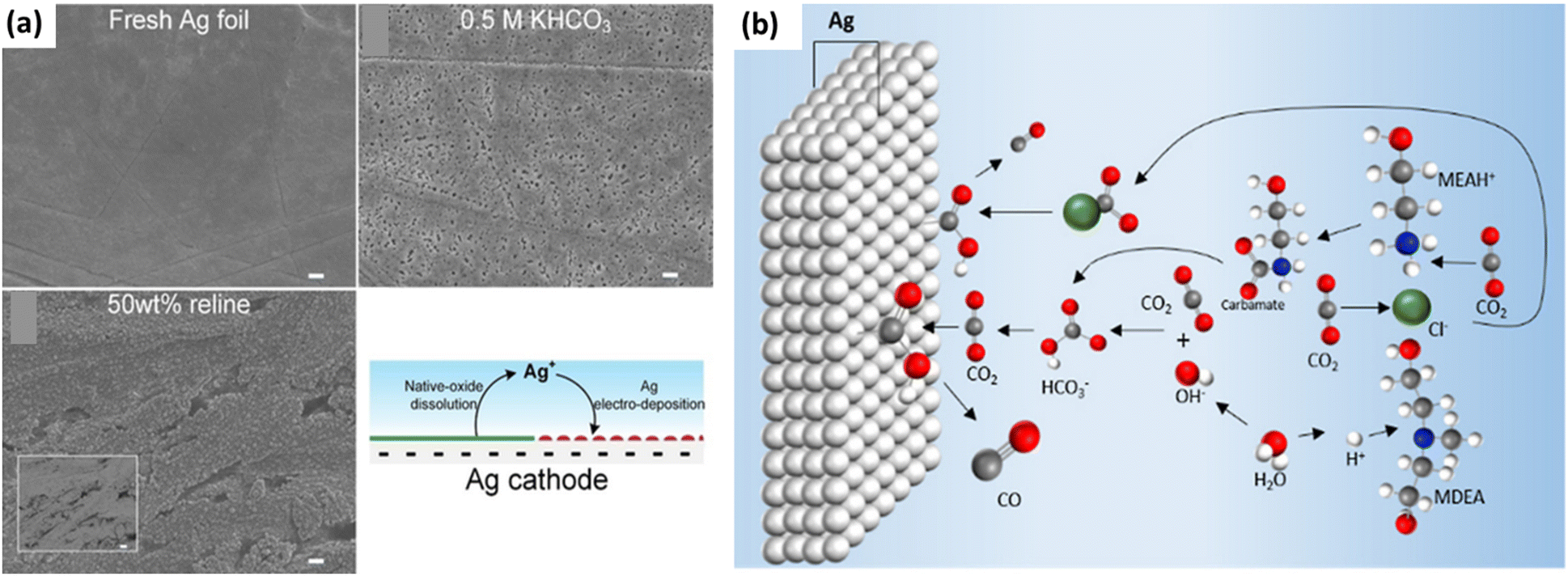 | ||
| Fig. 11 (a) SEM images of fresh Ag foil and Ag foils after CO2 electrolysis in 0.5 M KHCO3 and 50 wt% reline; scale bar: 1 μm. (Top) Stability test over Ag foil in CO2-saturated 50 wt% reline and 0.5 M KHCO3 for 8 h at −0.984 V vs. RHE (bottom). Reprinted with permission from ref. 388. Copyright 2019, John Wiley and Sons. (b) Schematic representation of [MEAH][Cl]:MDEA DES mediated CO2 reduction to CO on Ag surface. Reprinted with permission from ref. 455. Copyright 2021, Elsevier. | ||
Further examples of DESs studied for CO2RR and RCC are summarized in Table 8 where the reported CO2 reduction onset potential, product selectivity, and the main experimental conditions are summarized. Most of the DESs reported present very low total current densities (in the range of 0.1 to 1 mA cm−2). With the high viscosity of these electrolytes, the presence of a large hydrodynamic boundary layer at the catalyst surface is very likely. Therefore, it is difficult for CO2 to reach the electrode surface, thus yielding low current density. Therefore, most studies have included the addition of co-solvents to facilitate the CO2 species transport to the electrode surface. Importantly, the nature of the solvent largely affects the electrolysis performance as recently highlighted by the study of Vasilyev et al.389 where mixtures of [Ch][Cl] and ethylene glycol (1:2) DES (a.k.a, ethaline) with water, acetonitrile, propylene carbonate, and 2-(2-ethoxyethoxy)ethanol co-solvents were examined for CO2RR on Ag electrodes. In particular, the viscosity was reduced from 1244 cP to 1.5 cP with the addition of water to DES (0.1 M DES in water), which led to an increas in total current density. However, the CO selectivity was only 23% since water encouraged the production of mainly H2 at −1.6 V vs. Ag/AgCl. Further improvements in CO selectivity (>90%) and total current densities were observed by performing electrolysis in other aprotic solvents.
| DES | WE | Catholyte | Anolyte | CO2RR onset | Type of cell | E ve (V) | CDtotal (mA cm−2) | FECO (%) |
|---|---|---|---|---|---|---|---|---|
| [Ch][Cl]:EG (1:2) |
Au metal sheet456 | Neat DES | 1 M KOH | − 0.70 V vs. Ag/Ag+ | Two compartment | −1.60 V vs. Ag/Ag+ | 1.1 | 81.8 |
| [Ch][Cl]:urea (1:2) |
Ag foil389 | Neat DES | 0.5 M H2SO4 | −1.40 V vs. Ag/AgCl | Two compartment | −1.40 V vs. Ag/AgCl | 0.1 | 15.8 |
| [Ch][Cl]:EG (1:2) |
Neat DES (1:2) |
−1.43 V vs. Ag/AgCl | −1.43 V vs. Ag/AgCl | 0.4 | 78 | |||
| [Ch][Cl]:EG (1:2) |
1 M DES in acetonitrile | −1.52 V vs. Ag/AgCl | −1.7 V vs. Ag/AgCl | 20 | 98.8 | |||
| [Ch][Cl]:urea (1:2) |
Ag NPs/GDE390 | 2 M DES in H2O | — | ∼0.70 V vs. RHE | Flow | −0.83 V vs. RHE | 11.6 | 94.1 |
| [Ch][Cl]:EG (1:2) |
Cu0.3Zn9.7 deposited on Cu foam457 | 1 M DES in propylene carbonate | 0.1 M H2SO4 | — | Two compartment | −1 V vs. RHE | ∼16 | 94.9 |
| [Ch][Cl]:urea (1:2) |
Roughened Ag foil388 | 50 wt% DES in water | 0.5 M H2SO4 | — | Two compartment | −0.884 V vs. RHE | ∼4.8 | 96 |
| [MEAH][Cl]:MDEA (1:4) |
Ag foil455 | 0.4 M DES in H2O | 0.1 M H2SO4 | −0.80 V vs. RHE | Two compartment | −1.1 V vs. RHE | 10.5 | 71 |
Amine-based DESs have been extensively studied for CO2 capture owing to their remarkable CO2 uptake capacities. However, their application in RCC is limited. To date, only one study, conducted by Ahmad et al.,455 utilized an amine-based DES composed of monoethanolamine hydrochloride ([MEAH][Cl]) and methyldiethanolamine (MDEA) on Ag, Cu, and Zn surfaces. Consistent with previous research utilizing a 50 wt% reline/water electrolyte on Ag surfaces,388 nano-sized agglomerates were observed with [MEAH][Cl]:MDEA (1:4) DES. Due to the enhanced surface area, remarkably high CO selectivity was obtained; 71% at −1.1 V vs. RHE in the aqueous solutions containing 0.4 M DES. The authors explained the stabilization of CO2 species in the form of *COOH or *CHOO in the presence of protic species to selectively produce CO as shown in Fig. 11b. However, changing the molar ratio of [MEAH][Cl]:MDEA DES from 1:4 to 1:3, and 1:2 led to a decline in CO selectivity, from 71%, 43%, to 5%, respectively, alongside an increase in HER. This was attributed to the increased presence of protic species near the electrode while decreasing the HBD ratio, subsequently promoting greater H2 production. Additionally, the authors mentioned the narrower potential windows accessible with the DES on Zn and Cu, compared to Ag. Notably, during electrolysis experiments with Cu surfaces, a color change was observed in the electrolyte, further confirming the instability of Cu in the presence of amine-based DESs.
Both IL and DES electrolytes are composed of a variety of charged species and display a complex electrical double-layer structure,458 as evidenced by numerous probing techniques including advanced spectroscopy459 and simulations.460,461 Therefore, understanding the molecular-level RCC mechanism is crucial for enhancing the catalytic activity. The following sections summarize experimental and computational studies on the aspects of the surface species, double-layer structure, potential-dependent behavior of the electrode–electrolyte interface that controls the reaction energetics, kinetics, and product selectivity.
| ΔG = −nFE | (1) |
485 C mol−1) and E is the potential. The discrepancy between the actual potential applied (E) and the theoretical thermodynamic potential needed (E0) for the electrochemical reaction is termed overpotential (η = E − E0), indicating how far a reaction is from its equilibrium state on the Gibbs energy scale.
Experimentally observed overpotentials can arise from various distinct physical phenomena,462 including activation overpotential (also called the charge-transfer overpotential, ηactivation), concentration overpotential (ηmass-transport), and resistance overpotential, ηohmic-drop, which can be significant for viscous electrolytes such as ILs and DESs. Accordingly, the overall overpotential is the sum of all:
| η = ηactivation + ηmass-transport + ηohmic-drop | (2) |
In the context of electrochemical CO2 reduction, due to the high stability and inert nature of CO2 molecules, the process of converting CO2 requires a significant overpotential. From a thermodynamic perspective, multi-electron transfer reactions to yield products are more favorable than the direct one-electron reduction of CO2 to form the radical anion,463 CO2−, as shown in Table 5. Consequently, the primary factor in the total overpotential required for CO2 conversion is the activation overpotential, ηactivation.464 Thus, the stabilization of the intermediate radical anion, which forms upon the initial electron transfer to CO2, becomes a critical step especially in nonaqueous electrolytes.
The use of ILs as solvents or as electrolytes have generally led to anodic shifts in potential for non-catalytic and catalytic reductions versus the electrochemical reference. These milder potentials are often directly correlated to reductions in overpotential under equivalent conditions without ILs. However, ILs can modify the standard potential or the CO2 reduction reaction. As a result, the magnitude of anodic shifts may not directly correlate to the magnitude in the overpotential reduction. Matsubara, Grills, and coworkers have provided valuable experimental data and analysis regarding the overpotential of solutions containing imidazolium cations as solvent or electrolyte in CH3CN, particularly in the presence of CO2 and with H2O.465,466 In presence of larger amount of water in an organic solvent and under CO2, bicarbonate can form. Bicarbonate has a sufficiently similar pKa to imidazolium that it can from an equilibrium between imidazolium carboxylate and water; effectively, the imidazolium can behave as a proton source during catalysis.
When ILs are used as electrolytes in organic or aqueous solvents, the catalytic activity can generally be determined using standard methods as the viscosity of the solution does not change significantly.467 However, the diffusion constants of both the catalyst and substrates is expected to be much lower due to the high viscosity, which leads to lower current responses. In order to more accurately determine rates, some studies have measured the diffusion constants, and determined that despite the lower catalytic currents, the second order rate constants for catalysis was improved by up to an order of magnitude.468
Overpotential, along with current density and FE represent the key metrics for evaluating the performance of an electrocatalytic process. These indices are closely linked to the kinetics of catalytic reactions, with current density being directly associated with the applied overpotential as expressed through the Butler–Volmer expression (eqn (3)):
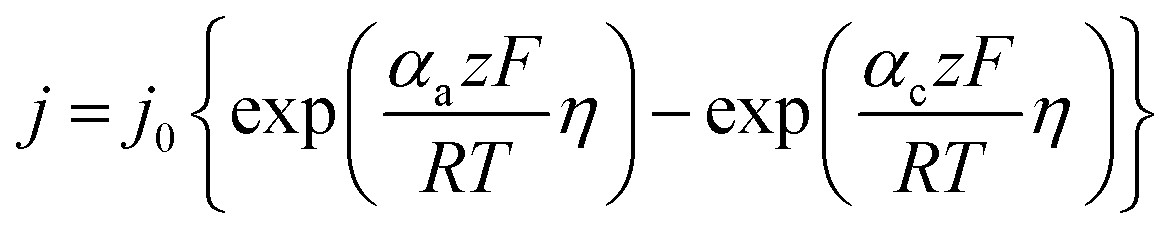 | (3) |
ILs have been said to co-catalyze CO2RR since they helps overcome the activation overpotential by stabilizing the radical anion intermediate. Decreased overpotentials imply that the electrochemical reactions can proceed with less additional voltage, enhancing the energy efficiency and performance of CO2 electrolyzers. Furthermore, ILs enhance CO2 solubility, ensuring higher concentrations of CO2 at the electrode surface and reducing concentration overpotentials. On the other hand, due to the high viscosity of ILs and DESs, they present significant ohmic overpotentials. Therefore, designing an electrochemical cell to minimize overpotentials due to ohmic drops and using a stable, well-defined reference electrode are crucial for consistent and comparative measurements in RCC.467
To quantitively compare CO2 reduction in ILs and DESs vs. conventional aqueous and nonaqueous media, average turnover frequencies (TOF) are determined from the reported performances as shown in Fig. 12. TOF analysis provides an estimate of intrinsic reaction rates across different catalysts, reaction media, and products. This analysis provides a quantitative comparison of activity on a per active site, per time basis, which allows for a fair comparison of reaction kinetics between catalysts with different surface areas and structures. By plotting against overpotential, it becomes more facile to compare catalysts that may be measured with different electrode morphologies (e.g. metal foils, nanoparticles), reference electrodes, electrolyte environments, and apparent current densities.
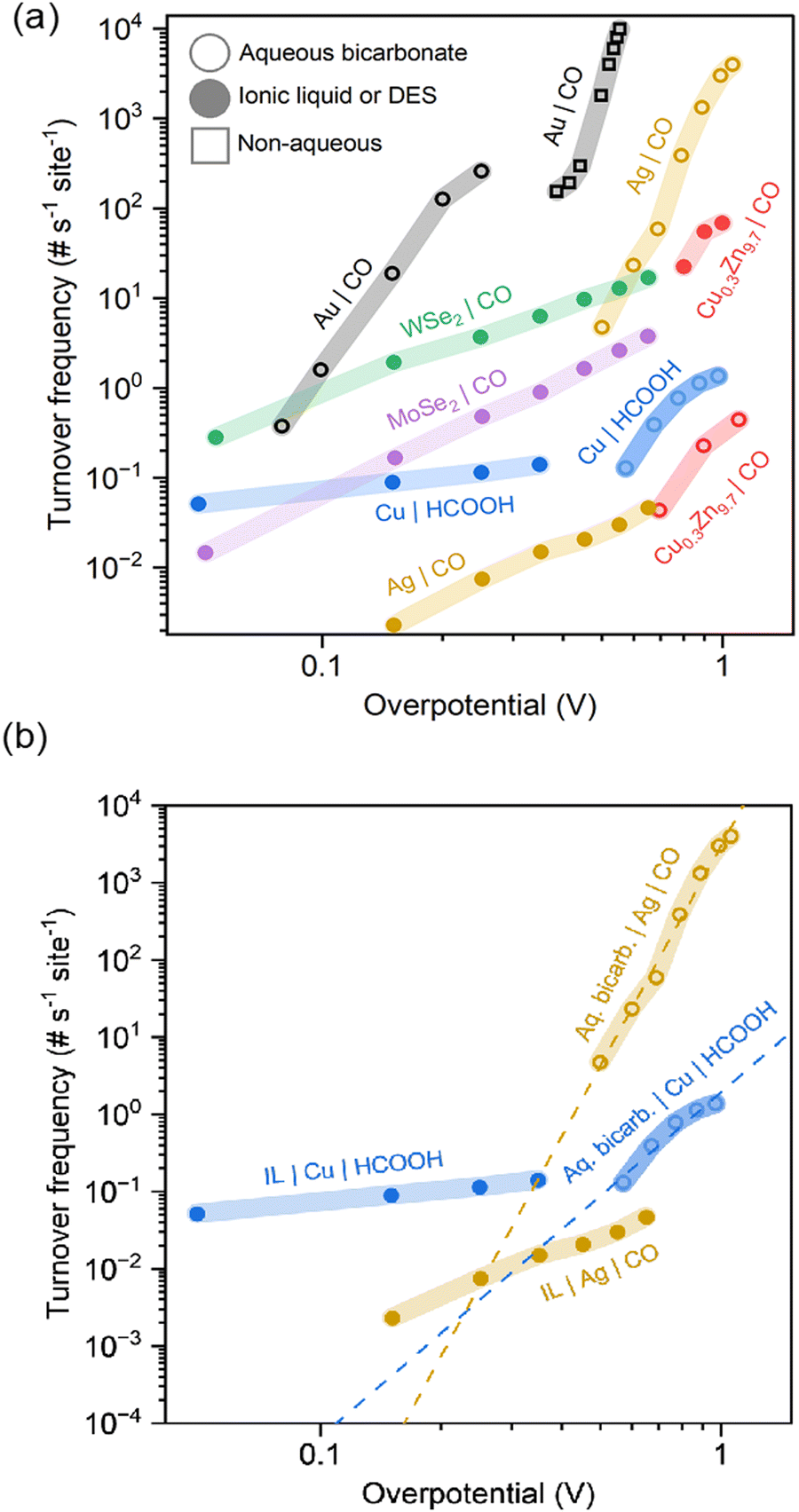 | ||
| Fig. 12 (a) Calculated turnover frequency (TOF) of various catalysts in aqueous media,457,477 non-aqueous media,478 and ILs414,440,457 plotted against the overpotential. The thermodynamic limiting potentials were taken as the standard values from aqueous media (−0.12 V vs. RHE for CO2 to HCOOH and −0.10 V vs. RHE for CO2 to CO) except in the case of Cu (in IL) where the value was taken as −1.32 V vs. Fc/Fc+.440 For WSe2, MoSe2, and Ag in IL, TOFs were previously reported based on roughness factors (determined by CV measurements) relative to reference samples. All other surface areas were converted to active site densities on an assumption of (111) facets, except for Cu0.3Zn9.7 which was converted assuming Zn(0001). For Cu in IL, the surface area was previously reported based on a measurement of ferricyanide reduction and use of Randles–Sevcik equation. For examples where ECSA was not previously reported, reported capacitance values were converted using a standard specific capacitance of 10 μF cm−2,479 and 29 μF cm−2 for Cu.480 (b) TOF of CO2 to CO on Ag and CO2 to HCOOH on Cu in ILs414 and aqueous media481vs. potential replotted to highlight differences in slope and onset overpotential. | ||
TOF comparisons draw attention to the fact that transition metal dichalcogenides (TMDs, i.e. WSe2 and MoSe2) are typically highly active compared to pure metals in solutions containing ILs as shown in Fig. 12a. The large TOFs of WSe2 and MoSe2 at low overpotentials are especially notable considering that these materials are typically highly HER active and thus usually completely inactive for CO2RR.482,483 The compositional and faceting design space of TMDs could be especially promising for realizing high activity RCC at low overpotentials. Due to drastically differing active site densities, the apparent current densities observed in typical electrochemical scans comparing metals and TMDs is vastly different, and thus benefits from this careful TOF analysis.
These comparisons also make clear that onset overpotentials for CO2 reduction in both dilute and concentrated ILs are typically on the order of ∼50–150 mV, much lower than similar catalysts measured in typical aqueous bicarbonate electrolytes. For example, CO2 to HCOOH on Cu in [EMIM][BF4]:H2O (92:8 v-v) reaches a similar TOF (∼10−1 s−1 site−1) at a ∼270 mV lower overpotential than Cu measured in typical aqueous conditions as illustrated in Fig. 12b. Importantly, at similar TOFs, HCOOH represents over 60% of the FE over Cu in IL, whereas it represents ∼20% of the FE of Cu measured under conventional aqueous conditions, which is suggestive of the mechanistic changes between aqueous conditions and ILs. Due to the high intrinsic activity, using ILs could allow for low overpotential production of HCOOH without byproducts. This is also similar to the trend of CO production on Ag. This analysis again illustrates the role of the ILs in RCC.
For heterogeneous catalysts an estimation of the electrochemically active surface area (ECSA) and the density of active sites on the catalyst surface is required. Double layer capacitance measurements are some of the most broadly used methods for assessing catalyst ECSAs as the capacitance is proportional to surface area. However, knowledge of the specific capacitance, an intrinsic material parameter, is needed for a quantitative conversion of the double layer capacitance to a catalyst surface area. Additionally, an assumption of the catalyst faceting (or characterization to elucidate the exposed facets) is necessary to estimate the density of exposed active sites, which is complex for typical polycrystalline catalysts. Since certain sites may be more active than others and even exhibit ensemble effects, normalizing to the total surface area provides a conservative lower bound for comparing the average TOFs of heterogeneous catalysts. Due to the high viscosity of ILs limiting mass transport, these measurements may reflect a mixed regime of mass transport and kinetic contributions, whereas many of the examples under aqueous conditions are more representative of intrinsic kinetics.
In general, catalysts measured in ILs seem to have larger Tafel slopes than catalysts measured in aqueous electrolytes. Since ILs are typically measured in small static electrochemical cells with poor convection, this could be reflective of mixed kinetic and mass transport contributions. Using forced convection strategies to alleviate mass transport limitations along with careful consideration of ECSA could be critical strategies to better understanding RCC in ILs. Alternatively, well-defined mass transport environments (i.e. rotating disk electrodes) could be used to better quantitatively account for these mass transport effects. Characterization of the catalyst faceting by imaging or diffraction could also allow for better active site quantification and may be aided by in situ studies to understand dynamic changes over catalysts in ILs that are distinct from aqueous environments. Specific capacitances for ECSA determination could also be determined under more uniform conditions, such as in a defined aprotic electrolyte.
Recently, Coskun et al.451 studied the interfacial behavior of reactive IL ([EMIM][2-CNpyr]) on a Cu surface, where the IL can chemisorbed CO2 and yield carboxylate ([EMIM]+-CO2−) and carbamate ([2-CNpyr-COO]) adducts. The complex interfacial structure is probed with SERS as shown in Fig. 13a and b under N2 and CO2, respectively. Upon negative polarization of the electrode, there is increased interactions between [EMIM]+ and the surface as evident from the increased intensity of 1347, 1380, and 1423 cm−1 peaks. With CO2, these peaks experience a blue shift. Additionally, peaks at 1116, 1342, and 1380 cm−1 associated with C4 and C5 of [EMIM]+ and N–CH2 moiety of the ethyl group exhibit a relatively higher enhancement in intensity with polarization compared to other peaks, suggesting an orientation change of the cation evidenced at −1.5 V vs. Ag/Ag+, as illustrated in Fig. 13c. This orientation was further supported by theoretical calculations as depicted in Fig. 13d. Simultaneously, the stretching of the [2-CNpyr]− ring at 1398 cm−1, faintly observed under N2, intensifies and shifts to 1405 cm−1 due to the closer proximity of the neutral 2-CNpyrH species formed via protonation of the anion upon CO2 saturation. The presence of 2-CNpyrH upon exposure to CO2 on the electrode surface is also confirmed by the shift in the –CN stretching vibration from 2189 cm−1 under N2 to 2223 cm−1 under CO2. Based on the findings from SERS analysis, the interfacial configuration was described as a layer rich in cations infiltrated with anions under N2. However, under CO2, the surface was predominantly characterized by [EMIM]+, [EMIM]+-CO2−, 2-CNpyrH, and fewer [2-CNpyr]− species. Notably, the presence of pyrrole species contributed to the creation of a distinct microenvironment by the reactive IL for RCC, as the anion forms complexes with CO2 to generate carbamate adducts and can transport CO2 to the electrode surface. Furthermore, the protonated anion, 2-CNpyrH, exhibits hydrogen bond donor properties which might also be the reason for the lowered overpotential. These unique interfacial configurations resulting from the reorientation of IL cations can influence the catalytic efficacy of electrochemical CO2 utilization by modulating the electric field at the interface and facilitating CO2 mass transport from the bulk to the electrode interface.
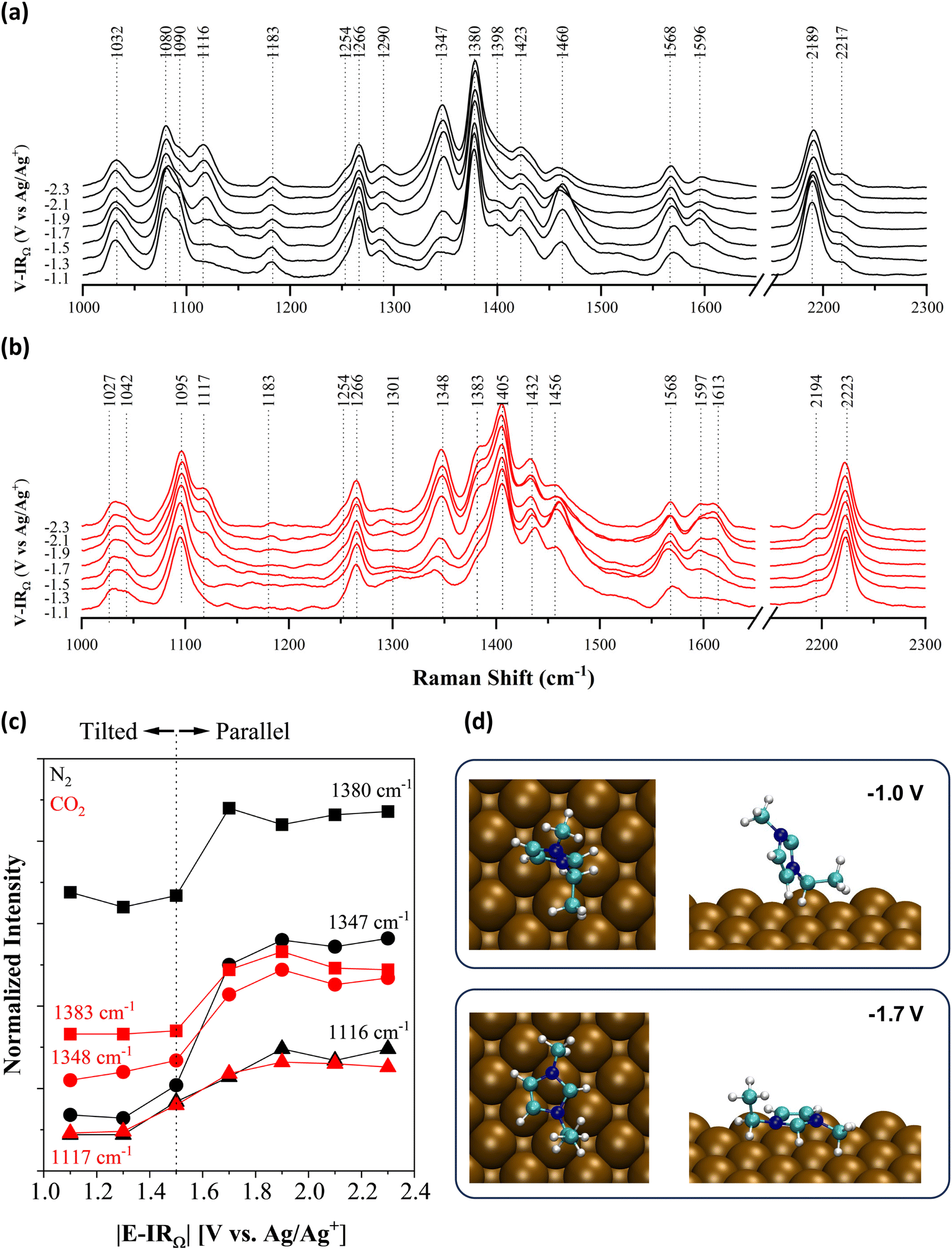 | ||
| Fig. 13 Potential dependent SERS spectra of [EMIM][2-CNpyr] under (a) N2 and (b) CO2, (c) SERS peak intensity as a function of absolute applied potential (E-IRΩ). The dotted line marks the potential of the change in the orientation of imidazolium species under N2 (black) and CO2 (red). Peaks: 1116 cm−1 (triangles) for δ(C4C5-H); 1347 cm−1 for υ(Im-ring) + υ(CH2(N)) (circle); 1380 cm−1 for υ(Im-ring) + υ(CH2(N)) + υ(CH3) (square). (d) Lowest energy geometries calculated for [EMIM]+ at −1.0 and −1.7 V on Cu(100) (top and side views) indicating the preference of the parallel orientation at a more negative potential. Atom color code: blue = N; cyan = C; white = H. Reprinted with permission from ref. 451. Copyright 2023, John Wiley and Sons. | ||
The extent of charge density and the hydrogen bonding between the ions impact how they structure near the electrode surface.490 The interfacial structure in ILs are believed to be heterogenous and extend a few nanometers away from the electrode surface as examined by various techniques. Even when ILs are diluted with other solvents, they tend to crowd the surface with applied polarization. Therefore, their electric field effects can be significant. In the case of solutes undergoing electron transfer reactions in the presence of the IL or DES components, the reaction intermediates can present a varied levels of interaction with these components. Specifically, as the oxidation state changes, the solvent rearrangement may be significant thus impacting the reaction energy and the kinetics.
The earlier work with physisorbing imidazolium ILs reported decreased overpotentials for CO2RR with the speculation that electrogenerated imidazolium carbene complexes with CO2 at the C2-position.371 Although this complex formation has been reported also by others,379,491 it was suggested that it served as a byproduct rather than an intermediate leading to the reduction of CO2. The viewpoints presented in other studies381 propose an alternative intermediate featuring a tetrahedral carbon at the C2-position, while C2–H is still present, resulting from coupling between the electrogenerated EMIM radical (as opposed to the carbene) and CO2−. By employing operando spectroscopy, Kemna et al.459 reported the formation of the C2-COOH (carboxylic acid) adduct, via the transient carbene intermediate, that is key to CO2 conversion to CO and formate on Pt electrodes with the same [EMIM][BF4] IL as with the previous studies. Other researchers suggest that the interaction of the CO2− radical at the C2-position of the imidazolium ring might not be the sole reason for the enhancements in the overpotentials reported. Lau et al.383 investigated the structure–activity relationships of imidazolium salts featuring various substituents to probe the role of C4,5-protons in co-catalyzing CO2 reduction. They proposed that hydrogen bonding between these protons and adsorbed CO2− stabilizes the latter, potentially explaining the reduced overpotential. Tanner et al.380 further claimed that the formation of the imidazolium–CO2 complex is improbable to have a substantial impact on the overall reaction and the enhancement of CO2RR facilitated by imidazolium cations is most pronounced for Ag, indicating a synergy between the electrode material and the co-catalyst. The suggested mechanism by the authors involves an initial chemical step, the desorption of IL cation, before an electron is transferred to CO2. This proposed mechanism indicates that CO2 directly receives the initial electron rather than the electron first being accepted by the [EMIM]+ molecule to generate a cation radical that would then assist the bending of CO2 and the stabilization of the radical anion of CO2.
It should be noted that studies to date utilized different heterogenous catalysts (e.g., working electrode), different reference electrodes, varying water content, and different supporting salts, which makes it difficult to develop a uniform understanding or make meaningful comparisons. Furthermore, it has been difficult to probe the dynamic electrode–electrolyte interfaces with evolving gases and speciation at different timescales. Sum frequency generation (SFG) is a useful method in probing surfaces and interfaces,492 and it has been shown to complement surface enhanced spectroscopy techniques for CO2RR mechanistic studies. Braunschweig et al.493 provided evidence for an interfacial imidazolium-CO2 adduct by utilizing SFG where a vibrational mode attributed to this adduct was observed at 2355 cm−1 (non-centrosymmetric CO2 mode) at notably more positive potentials. Thus, they explained this complex lowering the potential barrier for CO2 reduction in [EMIM][BF4] on Pt. Garcia Rey and Dlott494 also utilized SFG to note a structural transition within the IL on Ag with [EMIM][BF4] (0.3 to 77 mol% water). A dip in the non-resonant SFG signal precisely at the onset potential, exhibiting distinct curvatures on either side, was evaluated by the authors as an indication of interfacial structure change. Furthermore, this dip coincided with a Stark shift of the CO product, hinting at a change in the interfacial electric field. Intriguingly, this structural transition occurred irrespective of CO2 presence, suggesting an inherent property of the IL itself. Moreover, the same researchers observed a shift in the non-resonant SFG dip and the threshold potential for CO2RR towards less negative potentials when the amount of water in the electrolyte was increased. In contrast to the mentioned SFG studies, Papasizza and Cuesta495 could not observe any band attributable to the interfacial CO2 or the imidazolium-CO2 adduct by using an attenuated total reflection (ATR) surface-enhanced infrared absorption (SEIRA) on Au with [EMIM][BF4] (18 mol% in water). They revealed CO as the primary adsorbed product of CO2RR under these conditions. The integrated absorbance of the band corresponding to solvated CO2 mirrored the current.
Returning to the effect of water, numerous studies agree on the enhancement of CO2RR with the presence of water in IL-based electrolytes.376,496 This outcome is expected as CO2RR necessitates both proton and electron transfers, and water serves as an effective proton donor. However, the notable drawback of introducing water into IL systems is the enhancement of HER. Nonetheless, some reports propose that imidazolium cations could mitigate HER, even at water concentrations as high as 90 mol%.376 Conversely, in their ATR-SEIRAS study, Papasizza and Cuesta495 documented significant HER with an 82 mol% water and 18 mol% [EMIM][BF4] mixture. Feaster et al.497 observed that incorporating 0.1 M [EMIM][Cl] into acidic aqueous electrolyte curbed HER activity by 10–75% on transition metal catalysts, while no such suppression occurred in basic electrolyte. They argue that this phenomenon arises because [EMIM]+ displaces interfacial H3O+, but not interfacial H2O. The augmentation in CO2RR may also stem from alterations in the product distribution with imidazolium reduction. For instance, it is reported498 that the introduction of protons to pure [BMIM][NTf2] skewed the equilibrium between the imidazolium radical and the carbene towards the former, potentially influencing the CO2RR mechanism. Furthermore, Fortunati et al.442 discusses that carbene can bind to active sites on the electrode and the basicity of the anion determines the equilibrium between [EMIM]+ and its radical form.
Aside from experimental investigations, theoretical computations have sought to identify the pivotal intermediate involved in the CO2RR within IL-based systems. Wang et al.499 directed their attention to the electrochemical reduction of CO2 in aqueous electrolytes of [EMIM][BF4]. Employing the DFT method, they meticulously optimized all conceivable intermediate structures of [EMIM]+-CO2− complexes at the C2-position in the aqueous solutions, with the solvation environment addressed through an implicit water solvation model. Subsequently, they computed the Gibbs free energies of all intermediates and identified a thermodynamically favorable reaction pathway. In this pathway, [EMIM]+ undergoes reduction before the formation of [EMIM]+-CO2− complexes, with hydrogen at the C2-position acting as the proton donor to yield the key intermediate, [EMIM]+–COOH. However, despite the calculated redox potentials aligning with the thermodynamic pathways, achieving all steps in an exergonic manner necessitates an applied potential of −2.4 V vs. SHE, notably more negative than the experimental onset potentials (−0.25 V vs. SHE).375 The authors attributed this disparity to the absence of the electrode surface in their calculations. In addition to the interactions between [EMIM]+ and CO2−, the impacts of double layer effects on the CO2RR are also noteworthy and can be explored through computational methodologies. Nørskov and co-workers500,501 utilized DFT to examine the influence of the electric field on the CO2RR at the interface. Their findings revealed that cations proximate to CO2 exert a notable stabilizing influence on adsorbed CO2 on weakly adsorbing metals like Ag, with this effect attributed not to specific chemical bonding but to the interfacial electric field.
Implicit solvent models may be insufficient to capture the non-covalent interactions present in the microenvironment formed by ILs/DESs. In an effort to explicitly model double layer effects, Lim et al.502 devised a multiscale approach to focus on determining the free energies of adsorbed intermediates *CO2−, *COOH, and *CO in both aqueous solution and [EMIM][BF4]–water mixtures. The results indicated a decrease in the overpotential for the CO2 to CO conversion by 310 mV in the latter compared to the former. The primary distinction between the aqueous solution and the IL-containing solution is the robust electrostatic attraction between cations and anions, a microenvironment formed at the interface through non-covalent interactions among water, [BF4]− and [EMIM]+. This microenvironment facilitated stabilization of *COOH.
The mechanisms discussed above primarily apply to conventional ILs, where the CO2 is only physically dissolved in the absence of an electric field, and the constructed electrochemical interface is simpler compared to CO2-reactive ILs in RCC. For instance, one of the earlier examples of RCC utilized [P66614][124-Trz]377 where basicity of the [124-Trz]− anion prompts its direct binding with CO2 ([124-Trz]-CO2−) resulting in the formation of an additional CO2-carrying species within the interface.503 Thus, an alternative low-energy pathway for CO2 conversion to formate was achieved. This phenomenon underscores the role of the CO2-reactive, functionalized ILs, in lowering the reaction energy barrier in RCC. When such highly basic anions are combined with a cation like [EMIM]+, a more complicated interplay emerges as discussed by the studies of Gurkan and co-workers.450,451
4.3 Electrolyzers for RCC
The electrolyzer design and cell configuration are important considerations in RCC. The electrolyzer architecture and the operating conditions (e.g., temperature, pressure, applied potential, current) influence the transport of the active species and products between the bulk of the solvent and the electrode surface, as well as the temporal speciation of the capture agent and CO2 adducts. The reactor and catalyst design also determines the rate of equilibration between the species in the gas and liquid phases. Recent studies on RCC have been carried out in different reactor designs which have different mass transport characteristics. The commonly used electrochemical reactors, the H-cell and the flow cell configuration, have also been used extensively for CO2RR in the past and recent work has shown that hydrodynamics in the cell indeed determine the product distribution obtained.473,504 To date, very few works have been reported for RCC, but it can be anticipated that the type of reactors used will also impact the product distribution. In some of the RCC systems discussed in this section, the membrane separating the cathode and anode compartments is used to control the generation and transport of protons that shift the equilibrium and release CO2 in the proximity of the catalyst. Electrolyzer design is thus tightly tied to the performance of RCC systems and needs to be carefully studied.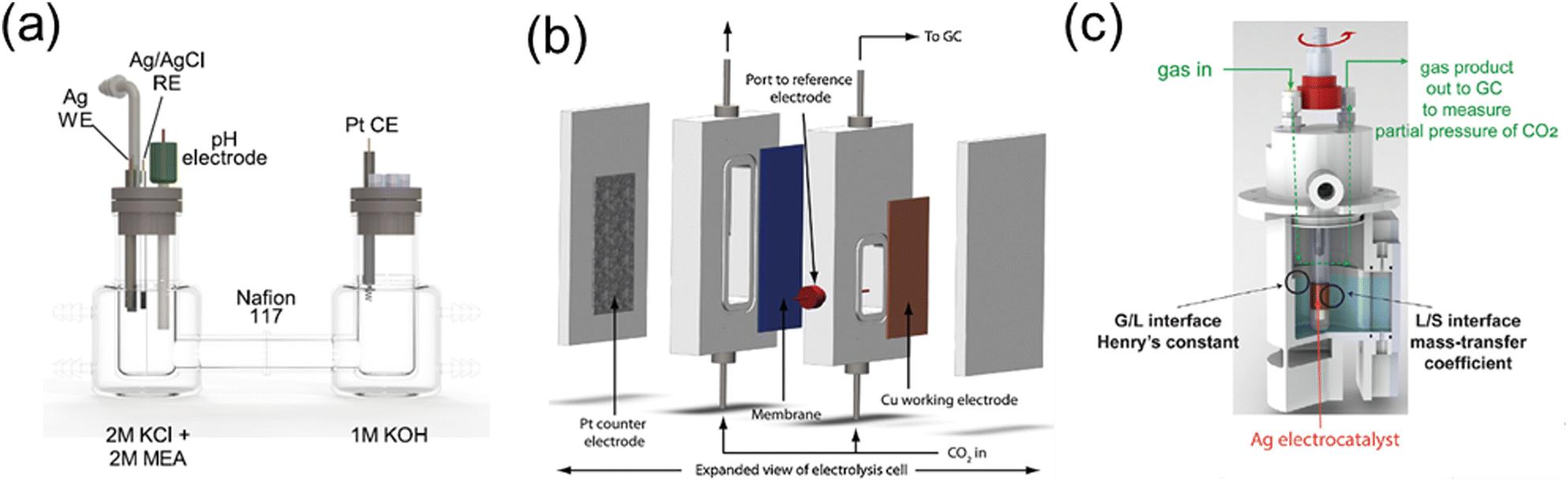 | ||
| Fig. 14 Examples of electrolyzer designs for activity benchmarking. (a) The conventional H-type cell. Reprinted with permission from ref. 505. Copyright 2023, American Chemical Society. (b) The sandwich-type compression cell. Reprinted from ref. 506 with permission from the Royal Society of Chemistry. Copyright 2012. (c) The gas-tight rotating cylinder electrode cell. Reprinted with permission from ref. 507. Copyright 2023, Elsevier. | ||
Alternatively, a sandwich-type compression cell has advantages for activity benchmarking, including lower cell resistance with a short cathode to anode separation, and typically large electrode areas relative to a small volume of electrolyte to aid in the sensitivity of product detection (Fig. 14b).506 Safipour et al.508 investigated CO2RR in the presence of MEA in a compression cell configuration and observed a maximum partial current density for CO of 5 mA cm−2, which decreased with increasing MEA concentration. Compression cells are to a first approximation amenable to the modeling of transport in and out of the surface of the catalysts under the assumption of the absence of primary and secondary current distributions. With support of a 1D continuum reaction-transport model, Safipour et al.508 concluded that the addition of MEA to the KHCO3 electrolyte does not change HCO3− concentration, however, concentration of monoethanolammonium ([MEAH]+) increases which acts as a proton source for H2 and reduces the local CO2 concentration, in turn decreasing the CO partial current density, with no evidence of any direct reduction of the carbamate.
RCE cells are particularly useful for benchmarking electrocatalytic activity because they can control mass transport by varying the rotation speed of the electrode, thus permitting a better analysis of the inherent kinetics at the WE. For instance, Shen et al. used an RCE cell to investigate RCC in bicarbonate and amine-based CO2 capture solutions on Ag (Fig. 14c).507 They studied different mass transport regimes by changing the rotation speed and observed different faradaic efficiencies and partial current densities for CO, ultimately concluding that the produced CO was primarily derived from the unbound dissolved CO2 within the system while the bound CO2-absorber species contributed only at highly negative potential.
The RCE cell system is unique in that it measures the partial pressure of CO2 in the headspace of the cell, which is in a pseudo-equilibrium with the solution. The partial pressure of CO2 can be used to estimate the amount of free and dissolved CO2 in the bulk of the electrolyte and facilitates the discrimination between different carbon sources during RCC. Transport of mass, heat, and charge affect kinetics in RCC and thus needs to be considered while designing electrolyzers for this reaction. A cell design with a thick hydrodynamic layer, like the H-cell, could slow down the local mass transfer of the species resulting in lower faradaic efficiency.509 Furthermore, cell resistance and uniform current distribution are some other factors that can potentially affect product distributions in electrochemical systems, especially for organic solvents with lower conductivity. The RCE cell provides an optimal balance for these concerns and is thus a state-of-the-art electrolyzer design for RCC activity benchmarking.
In any electrochemical CO2 conversion reactor, some key criteria should be met for high performance: (1) a high mass flux of the reactant (i.e., CO2 or CO2-adduct) to the cathode catalyst to ensure high product selectivity uninhibited by mass transfer limitations, (2) high catalyst activity and surface area to minimize the activation overpotential, (3) low ohmic losses in the form of solution, membrane, and electrode series resistance to keep the operating cell voltage low, (4) effective membrane separation of the cathode and anode to maintain low product crossover and back reactions,510 and (5) stable system components under operating conditions to promote long-term device stability.511,512 For electrolyzers under either gas-phase or liquid-phase cathode operation, flow cell design architectures have demonstrated the greatest success at achieving high-current–density, high-selectivity electrochemical CO2 reduction.513–515 In the best aqueous-based flow electrolyzers, current densities >1 A cm−2 have been achieved with sufficient CO2 mass flux to even reach 90% FE for multicarbon C2+ products.516–519 Similar current densities and FEs have been achieved in state-of-the-art flow electrolyzers designed for liquid-product formic acid production.520–522 Several possible designs for different reactor types for integrated CO2 capture and conversion are shown in Fig. 15, which will be referred to when discussing literature reports on electrolyzers for captured CO2 conversion.
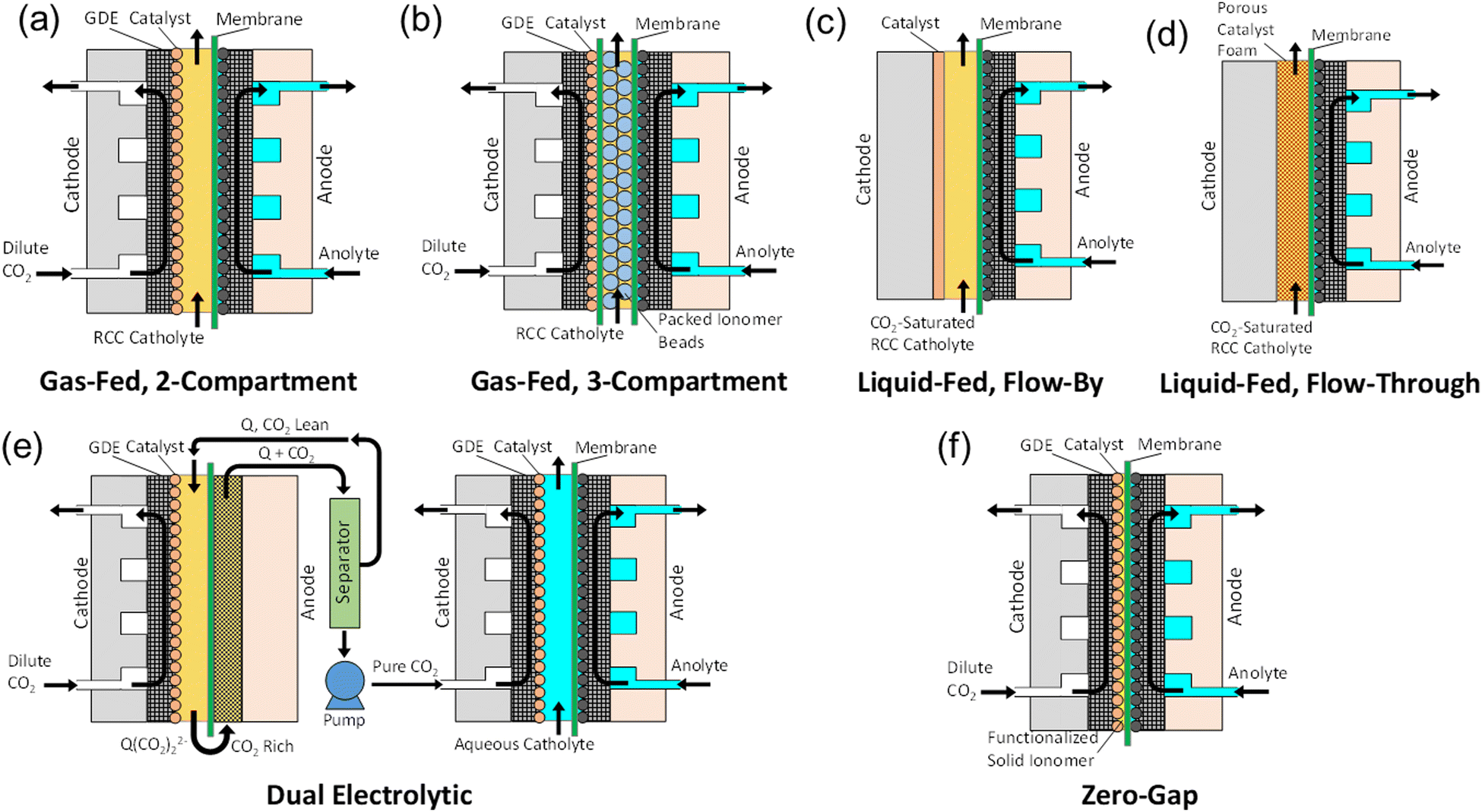 | ||
| Fig. 15 Schematics of electrolyzer design concepts for high-performance integrated CO2 capture and conversion. (a) Gas-fed, two-compartment GDE flow electrolyzer. (b) Gas-fed, three-compartment GDE flow electrolyzer. (c) Liquid-fed, flow-by electrolyzer. (d) Liquid-fed, flow-through electrolyzer. (e) Dual electrolytic cells. Q represents a generic CO2 binding species, such as an organic quinone redox species. (f) Gas-fed, zero-gap flow electrolyzer. | ||
To achieve high CO2 mass flux to the cathode, gas-fed flow electrolyzers typically rely on a gas diffusion electrode (GDE) with a conductive microporous structure to diffusively disperse reactant uniformly across the active catalytic area (Fig. 15a).523,524 Moreover, the GDE interface is designed to be hydrophobic to prevent catholyte wetting of the pores, thus maintaining a high-surface–area catalyst/electrolyte/CO2 triple-phase boundary for high CO2 mass flux despite the relatively low solubility of CO2 in water.513 At present, it is unclear how effective the GDE reactor architecture could be for RCC systems. Functional solvents for carbon capture, including ILs and other non-aqueous media, complicate the interfacial interactions at the GDE. Organic solvents, in particular, are generally much lower in surface tension than water and can readily wet and subsequently flood most GDEs, thus impeding gaseous CO2 mass transfer. Additionally, for an RCC mechanism involving the direct reduction of a CO2-adduct, such as a bound CO2–IL, liquid-phase mass transfer of the adduct species to the catalyst would still be a concern. Thus, liquid-phase reactor designs with sufficient sorbent species concentration to prevent mass transfer limitations of the CO2-adduct to the cathode from inhibiting selectivity at high currents may be an important feature for RCC flow electrolyzer design. For instance, microfluidic electrolyzer designs have been demonstrated for aqueous CO2RR in which the cathode and anode are separated by thin (<1 mm) electrolyte flow channels,525–527 with one such microfluidic reactor reported to achieve >1 A cm−2 for CO2RR.518 These types of narrow-dimension liquid-phase catholyte flow reactors may be a suitable approach for RCC in functional solvents.
We must note here that the discussion in this section is descriptive of the state-of-the art in CO2RR and RCC but that many knowledge gaps remain ahead in the path to understanding how to translate bench-scale catalysts to industrial scale RCC electrolyzers. For instance, we have indicated that using of organic solvents in GDEs is problematic because of flooding in CO2RR applications. We have also indicated that ILs change in viscosity as these are loaded with CO2. However, there could be a dynamic range where organic liquids and ILs mixtures can result in desirable properties for dynamic transport of CO2 species within a pore while changing reaction energetics at the surface of the catalyst while preventing flooding. RCC systems are nonlinear systems and will require large experimental datasets that could eventually capture the full complexity of this reaction. As we develop a better understanding of the variables relevant to RCC performance, we anticipate that new electrolyzer concepts will be developed.
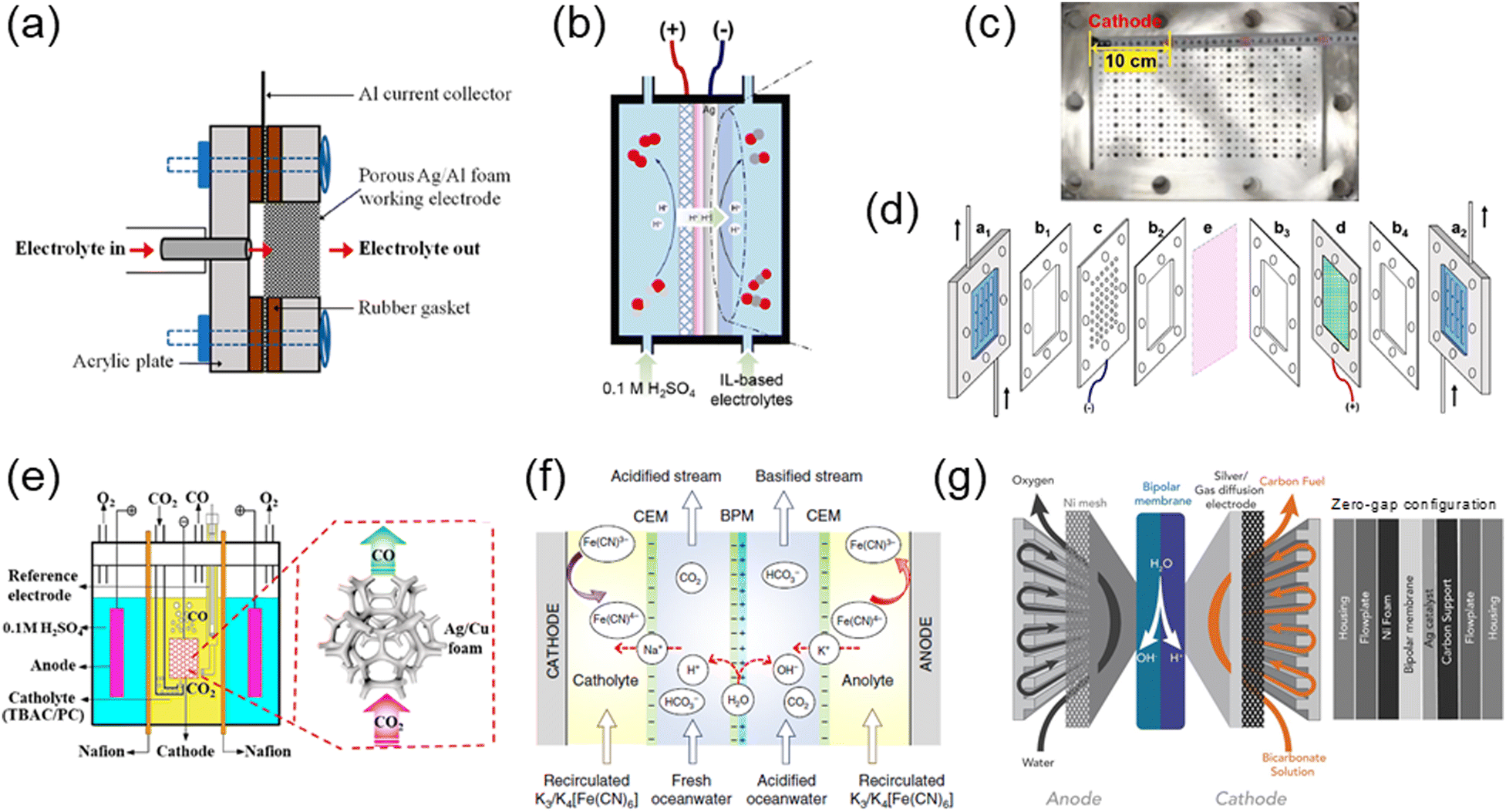 | ||
| Fig. 16 Examples of electrolyzer device design for high performance RCC. (a) The cathode chamber of a CO2-presaturated IL flow electrolyzer with a liquid-fed, flow-through porous cathode. Reprinted with permission from ref. 439 Copyright 2019, American Chemical Society. (b) A liquid-fed, flow-by IL electrolyzer and corresponding (c) image of a large perforated Ag plate cathode and (d) exploded view full cell schematic. Reprinted with permission from ref. 534 Copyright 2022, Elsevier. (e) A non-aqueous solvent porous flow-through cathode electrolyzer. Reprinted with permission from ref. 535 Copyright 2022, American Chemical Society. (f) A bipolar membrane electrodialysis cell for CO2 separation from oceanwater and subsequent coupling in a dual electrolytic cell. Reprinted with permission from ref. 536 under CC-BY license. (g) Flow cell electrolyzer with a bipolar membrane in a zero-gap configuration for bicarbonate reduction. Adapted with permission from ref. 537 Copyright 2019, Elsevier. | ||
There have also been a couple reports of investigations into larger size IL-based flow electrolyzers for CO2 conversion. For instance, a 30 cm2 Zn foil cathode was used with an ion exchange membrane in a pressurized flow cell using 1-ethyl-3-methylimidazolium trifluoromethanesulfonate ([EMIM][TfO]) in 90 wt% water acidified with H2SO4.538 At a pressure of 30 bar, a CO selectivity of 82% FE was achieved at an applied current density of 3 mA cm−2. Additionally, Yuan et al. reported an effort to scale up an IL-based liquid-fed, flow-by electrolyzer (Fig. 15c) for electrochemical CO2RR.534 This flow cell used CO2-saturated [BMIM][BF4] at an optimal concentration of 0.1 M in acetonitrile with 5 wt% water as catholyte separated by a Nafion membrane from aqueous 0.1 M H2SO4 anolyte. Acetonitrile solvent was used to reduce the IL viscosity, as the observed decline in CO2RR performance at higher IL concentrations and flow rates were attributed to viscosity-related mass transfer limitations. A small amount of water was added as a proton source, which was also reported to enhance stability. The sizable electrolyzer used a perforated Ag plate cathode of 495 cm2 (Fig. 16b–d). Under the optimal operating conditions, this large IL-based flow electrolyzer achieved a total current of 6.32 A (or 12.8 mA cm−2) at 4.0 V with 83.9% FE for CO and <2% FE for H2. Furthermore, it was demonstrated to be stable over 10 h and outperform an analogous aqueous 0.1 KHCO3 flow electrolyzer in both stability and CO selectivity.534 While better mass transfer and innovative flow-through cathode structuring may be needed to improve the current density, the strong performance of this scaled-up IL-based electrolyzer is encouraging for the prospects of commercially feasible reactive carbon capture.
Few dual electrolytic systems have yet been reported for integrated CO2 capture and conversion. Notably, Digdaya et al. demonstrated a directly coupled dual electrolytic system using a bipolar membrane electrodialysis cell and a vapor-fed CO2RR cell to capture and convert CO2 from ocean water (Fig. 16f).536 The bipolar membrane cell used a reversible ferro/ferricyanide redox couple separated from the ocean water by cation exchange membranes to drive CO2 release via the acidification of bicarbonate. The outlet CO2 stream was then sent through tandem vapor-fed cells, first a pre-electrolysis oxygen reduction reaction cell to remove residual O2 that would parasitically hurt CO2RR performance, and then a subsequent GDE CO2RR flow cell catalyzed by either Cu or Ag. The combined system led to a CO2 capture efficiency of 71% with up to 95% FE to CO on Ag.
Although not many dual electrolytic cells have been characterized for integrated CO2 capture and conversion in the literature, there are a several reported electrochemical CO2 separation techniques that might be suitable for incorporation into a dual electrolytic system.545,546 For instance, electrochemical cycling of organic quinone redox species in the presence of dilute CO2 sources can selectively bind CO2 during reduction and then oxidatively release it in a purified stream (Fig. 15e),547 which was effectively demonstrated for 1,4-naphthoquinone in the ionic liquid [EMIM] tricyanomethanide, [TCM].89 In another case, a liquid naphthoquinone sorbent was coupled to a ferrocene-derived counter electrolyte in a continuous electrochemical CO2 capture and release system.548 Quinones for CO2 separation were also employed in a novel faradaic electro-swing reactive adsorption process in which a solid-state electrochemical cell with a polyanthraquinone–carbon nanotube electrode captured CO2 upon reduction and subsequently released it during the anodic swing.47 The electro-swing process was effectively demonstrated with CO2 concentrations as low as 6000 ppm and at typical flue gas concentrations (∼10%) with a CO2 separation faradaic efficiency >90%. Electrochemically mediated amine regeneration (EMAR) is another approach to CO2 separation that may be amenable to dual electrolytic cell operation. In EMAR, CO2 binds to an amine sorbent to form a carbamate, which is then electrochemically oxidized to release the CO2 and subsequently reduced to regenerate the amine.545 In one example of continuous EMAR, ethylene diamine sorbent was used in a Cu ion-mediated process to achieve a CO2 separation efficiency >80% from flue gas.549 In another, ethoxyethylamine in dimethyl sulfoxide was used in an electrochemical cation-swing process in which different Lewis acid cations entered the electrolyte depending on the direction of current flow, and the cation identity led to a reversible carbamic acid-to-carbamate conversion that released purified CO2.550 These kinds of electrochemical CO2 separation techniques could be designed for efficient coupling to optimized gas-fed CO2RR reactors for a dual electrolytic integrated conversion of dilute CO2 streams.
As reported previously for IL-based CO2RR, complexing of the imidazolium cations with CO2 led to lower onset potentials and suppressed side reactions.371 Researchers have sought to gain some of the benefits of functional solvent ILs while bypassing issues arising from the use of liquid electrolyte by incorporating imidazolium groups into solid polymer electrolyte membranes for use in zero-gap membrane electrode assembly electrolyzers. With a methylimidazolium-substituted styrene copolymer anion exchange membrane, a gas-fed CO2RR electrolyzer was operated as high as 200 mA cm−2 at 3 V for 1000 h with >95% CO FE.551,552 These imidazolium-functionalized membranes were trademarked as SustainionTM and incorporated some of the functional characteristics of imidazolium-based ILs. More recently, benzimidazolium cations grafted onto alkaline polyelectrolytes enabled efficient CO2RR conversion to ethylene over Cu catalyst at up to 331 mA cm−2.553 Similarly, EMIM-functionalized Mo3P nanoparticles coated in anion exchange ionomer were reported to produce propane, a C3 product, at 91% FE and a current density of 395 mA cm−2.554 In another, IL-MOF hybrid catalyst which immobilized 1-butyl-3-methylimidazolium hexafluorophosphate, [BMIM][PF6], within the porous MOF achieved CO2RR at >65% FE for CH4.555 Each of these examples highlights the feasibility of immobilizing functional IL or other sorbent groups at the ionomer and/or catalyst interface to enable some of the benefits of functional solvents in a zero-gap electrolyzer architecture.
4.4 Homogeneous CO2 electrocatalysis
There is an extensive literature on the study of heterogeneous catalysts for CO2RR in IL containing electrolytes. By comparison, there are far fewer studies that have examined the impact of IL salts as solvents or electrolyte for homogeneous catalysts. These studies have investigated catalysts that are already known for their selectivity towards CO2 reduction in conventional solvents. They include catalysts selective towards CO production such as Fe(TPP)Cl,556,557fac-Re(bpy)(CO)3Cl,465,468,558 or Ni(cyclam)Cl2,559 and towards HCOO− production such as (Rh(bpy)(Cp*)Cl).560 The molecular structures are shown in at the top of Fig. 17. The bottom shows catalysts that have incorporated imidazolium type functionalities in their secondary coordination sphere to enhance catalytic activity. A brief summary of electrocatalytic activity of the homogeneous catalyst for CO2RR is given in Table 9.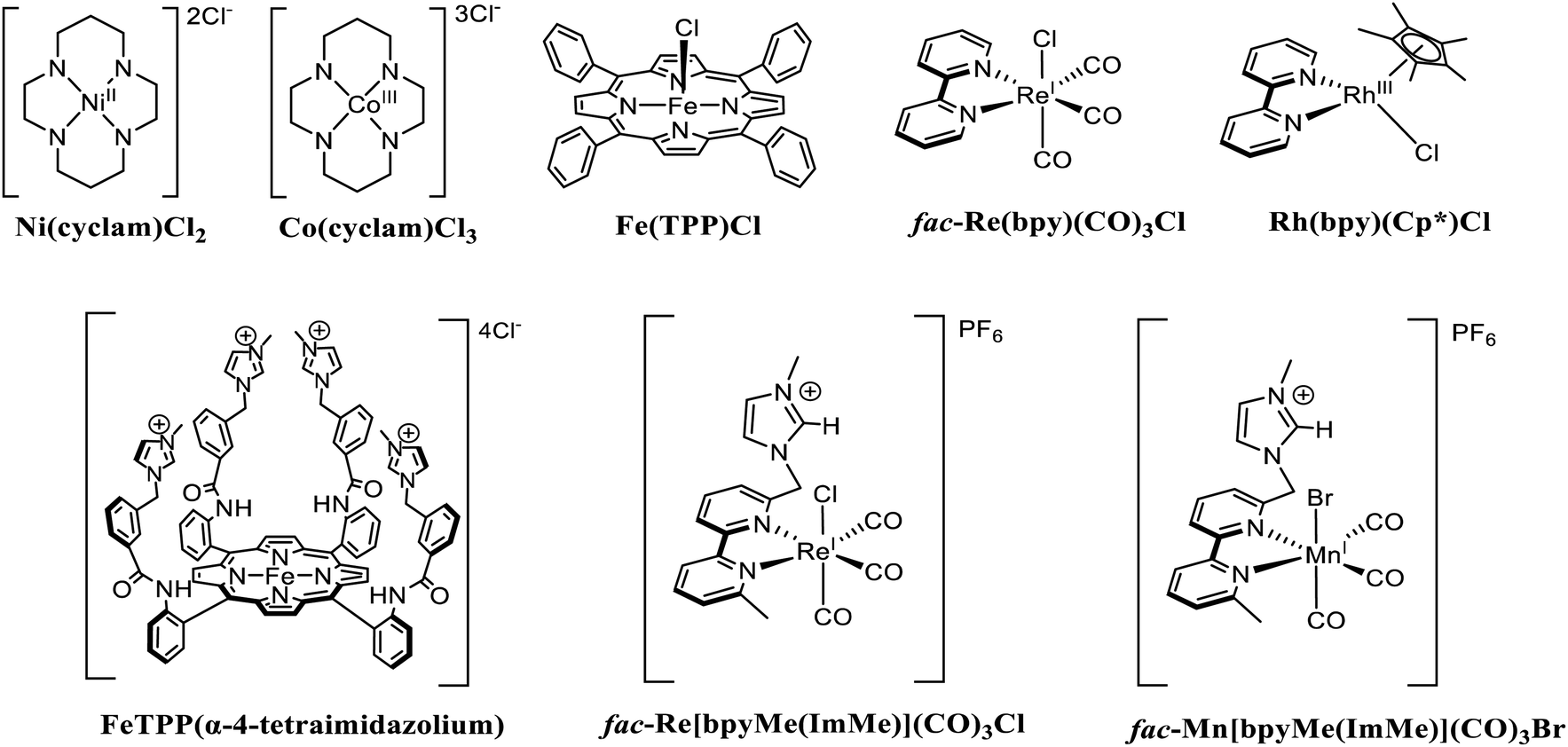 | ||
| Fig. 17 Example structures of homogeneous electrocatalysts studied for CO2RR in IL media. | ||
| Homogeneous catalyst[Ref] (concentration) | Catholyte | Proton source | E ve (V) | FE % (product) | TOFproduct | TONproduct |
|---|---|---|---|---|---|---|
| [Ni(cyclam)Cl2]559 (80 mM) | Neat [BMIM][BF4] | None added | −1.4 V vs. Ag/AgCl | 95.2% (CO) | 0.73 h−1 | 2.19 |
| Neat [BMIM][NTf2] | None added | 75.1% (CO) | 0.06 h−1 | 0.17 | ||
| [Co(cyclam)Cl3]559 (80 mM) | Neat [BMIM][BF4] | None added | 84.9% (CO) | 0.08 h−1 | 0.22 | |
| Neat [BMIM][NTf2] | None added | 62% (CO) | 0.04 h−1 | 0.11 | ||
| FeTPPCl556 (0.5 mM) | 0.3 M [BMIM][BF4] in DMF with 0.1 M [TBA][PF6] | 1 M TFE | −1.56 V vs. NHE | 93% (CO) | 190 s−1 | 27.4 × 105 |
| [Rh(bpy)(Cp*)Cl]Cl560 (1 mM) | 0.5 M [EMIM][PF6] in acetonitrile | 5% v/v H2O | −1.89 V vs. Fc+/Fc | 90% (HCOO−), 9% (H2) | — | — |
| −1.83 V vs. Fc+/Fc | 91% (HCOO−), 9% (H2) | — | — | |||
| 0.1 M [EMIM][BF6] in acetonitrile | 0.1 M CH3COOH | −0.9 V vs. Ag/AgCl | 46% (HCOO−), 40% (H2) | — | — | |
| Re(bpy)(CO)3Cl468 (0.5 mM) | Neat [EMIM][TCB] | 50 mM H2O | −1.8 V vs. Fc+/Fc | 88% (CO) | — | — |
| Re(bpy)(CO)3Cl558 (1 mM) | 0.5 M [EMIM][PF6] in acetonitrile | 1.5 M TFE | −1.75 V vs. Fc+/Fc | 96% (CO), 1% (H2) | — | — |
| −2.05 V vs. Fc+/Fc | 77% (CO), 12% (H2) | — | — | |||
| 0.5 M [BMPyrr][PF6] in acetonitrile | 80% (CO), 10% (H2) | — | — | |||
| 0.5 M [EMIM][BF4] in acetonitrile | 71% (CO), 18% (H2) | — | — | |||
| Homogeneous catalyst incorporated with imidazolium in the secondary coordination sphere | ||||||
| FeTPP(α-4-tetraimidazolium)561 (0.5 mM) | 0.1 M KCl (aq.) | H2O | −0.948 V vs. NHE | 91% (CO) | 14986 s−1 |
1.08 × 108 |
| {Re[bpyMe(ImMe)](CO)3Cl}PF6562 (1 mM) | 0.1 M [TBA][PF6] in acetonitrile | 9.4 M H2O | −1.74 V vs. Fc+/Fc | 73% (CO) | — | — |
| {Mn[bpyMe(ImMe)](CO)3Br}PF6563 (1 mM) | 0.1 M [TBA][PF6] in acetonitrile | 9.25 M H2O | −1.56 V vs. Fc+/Fc | 77.7% (CO) | — | — |
| −1.82 V vs. Fc+/Fc | 70.3% (CO) | — | — | |||
| Catalysts immobilized onto electrode surfaces | ||||||
| Re(bpy)(CO)3Cl@HPC564 | [EMIM][BF4] with H2O (5% v/v) | H2O | −1.75 V vs. Fc+/Fc | 79% (CO) | 0.3 s−1 | 260 |
| −2.05 V vs. Fc+/Fc | 76% (HCOO−) | 2.2 s−1 | 1950 | |||
| ZnTAPP@ITO565 | Neat [BMIM][BF4] | None added | −0.8 V vs. Ag/AgCl | 14.5% (CO) | — | 2.74 |
| FeTAPP@ITO565 | 79.6% (CO) | — | 9.18 | |||
These catalysts have been studied using ILs either as the primary solvent or as the electrolyte in organic/aqueous solvents. Additionally, there are two examples of catalysts that have been immobilized onto surfaces,557,564 as well as modified molecular catalysts that incorporate IL cations in the secondary coordination sphere.561–563 In all cases, the presence of small quantity (0.5–80 mM) of these homogeneous catalysts in neat IL or IL diluted in solvent result in anodic shifts of CO2RR onset potential. These anodic potential shifts are generally larger than those observed in organic solvents saturated with non-IL electrolytes, indicating these changes are not solely due to the greater ionic strength of the ILs.468,556 Studies that explored the effect of [BF4]−versus [PF6]− found minimal effects on the electrochemical and electrocatalytic response of fac-Re(bpy)(CO)3Cl558 and [Rh(bpy)(Cp*)Cl].560 [TCB]− and [NTF2]− were both used as anions with [EMIM]+ to study the electrocatalytic activity of fac-ReCl(bpy)(CO)3 and exhibited no significant differences.468 However, [NTF2]− was found to have an inhibitory effect on catalysis compared to [BF4]− with [Ni(cyclam)Cl].559
Similarly, few studies have focused on the effect of different IL cations, either imidazolium ([BMIM]+, [EMIM]+) or pyrrolidiniums ([BMPym]+, [BMPyrr]+). One study with fac-Re(bpy)(CO)3Cl used the protic imidazolium cation [DiMIM]+, but catalysis under CO2 reduction conditions only resulted in hydrogen due to the acidity of the cation.558 Under non-protic conditions with fac-ReCl(bpy)(CO)3Cl the imidazolium cations [BMIM]+ and [EMIM]+ led to greater anodic shifts than the pyrrolidinium cation,558 [BMPym]+ relative to ferrocene. The first reduction of fac-ReCl(bpy)(CO)3Cl is metal-based and leads to loss of the chloride ligand and is shifted anodically by ∼80 mV. The second reduction is centered on the bpy ligand to form the anionic compound [fac-Re(bpy)(CO)3]− and corresponds to the onset of electrocatalysis. The second reduction exhibits the largest anodic change (up to 450 mV) in potential under aprotic conditions. However, these changes to the reduction potential were minimized when the acid source trifluoroethanol was added to the IL. A computational study on this complex in ILs indicates the imidazolium cation stabilizes this anionic species through an electrostatic π+–π interaction.465 Adding of an external acid source, such as trifluoroethanol, is expected to minimize this electrostatic stabilization by protonating the reduced metal complex.558 Despite the muted effect that the addition of an acid source had on the reduction potential, in all cases, the catalytic current with the imidazolium and pyrrolidinium cations was increased, indicating a beneficial effect on the catalytic rate. The rate enhancement is attributed to the imidazolium facilitating C–OH bond cleavage through a hydrogen-bonding interaction with CO2 bound to the catalyst, which is believed to be the rate-determining step in catalysis.465
The impact of imidazolium-based and pyrrolidinium-based cations as electrolyte were also investigated with [Rh(bpy)(Cp*)Cl]Cl, which catalyzes the reduction of CO2 to HCOO−.560 In a mixed 95:5 acetonitrile:water solution, the presence of the IL cations results in a minimal change to the first reduction, which is metal-based. Like the prior example, [fac-Re(bpy)(CO)3]−, the second reduction occurs on the bpy ligand to generate the active catalyst and leads to the onset of catalysis. This reduction occurs at a more anodic potential (<100 mV) for both IL cations, with EMIM providing a larger effect. The EMIM cation also gives a greater current density than TBA, whereas BMIM results in a similar current density, and [BMPyrr] results in a lower current density. When acetic acid was used as an acid source, [EMIM][BF4] resulted in almost double the faradaic efficiency (up to 66%) for formate compared to the non-IL electrolyte [TBA][BF4]. The catalyst was also tested under aqueous conditions, where it displayed greater selectivity and more anodic catalytic onset potentials when [EMIM][BF4] was the electrolyte compared to [TBA][PF6]. Again, similar to [fac-Re(bpy)(CO)3]−, DFT calculations indicate that [EMIM]+ stabilized the reduced species through π+–π interactions, resulting in more anodic potentials. Furthermore, the computational studies suggest cations associated with the catalyst inhibit the HER via electrostatic interactions, resulting in improved selectivity for HCO2−.
As diffusion can slow the overall rate of catalysis due to the high viscosity of neat ILs, efforts have been made to immobilize molecular catalysts onto electrodes. These ‘heterogenized’ catalysts only require consideration of substrate diffusion to the electrode. Fac-Re(bpy)(CO)3Cl catalyst was immobilized onto a conductive carbon porous material.564 Interestingly, the catalyst selectivity changed with the applied catalytic potential. At the most anodic potentials, CO was the dominant product; however, at more cathodic potentials, formic acid was observed. The latter is not normally a product of CO2 reduction with this catalyst when it is not immobilized and may be due to the local hydrophobic nature of the catalyst support. Formate was also observed as a product in another study under aqueous (non-IL) conditions where this catalyst was immobilized in Nafion.557 The FE of H2 also increases with greater concentrations of water. However, smaller amounts of water, down to 1%, led to lower current densities due to the higher viscosity of the solution. Thus, the ideal water content (5%) provided a good optimization between catalyst selectivity (75% FE for CO) and activity. Porphyrin-based catalysts containing Fe and Zn were immobilized onto indium tin oxide electrodes using in a conducting polymer.565 With BMImBF4 as the solvent under aprotic conditions, the Fe derivative demonstrated the highest faradaic efficiency for CO, with 79.6% and almost 10 TON, which are both higher than freely diffusing catalysts under the same conditions.
Another strategy to impart the positive local impact of imidazolium is to modify the ligand to contain these functionalities in the secondary coordination sphere. In the first study of this type, the bpy ligand in the selective CO2 reduction catalysts [fac-Re(bpy)(CO)3]− was modified to contain an imidazolium functionality near the bpy ligand.562 Similar to these catalysts in IL solvents or when IL electrolytes are used, this complex demonstrated an increased catalytic activity and selectivity at more anodic potentials than the unfunctionalized complex. Furthermore, the synthesis of a variant with a methylated imidazolium complex did not demonstrate similar improvements in activity, indicating the acidic C–H is critical to the improved catalysis. DFT studies indicate the imidazolium facilitates the loss of Cl− required to generate the active catalyst while not impacting the reductive properties of the bpy ligand, as well as facilitating the cleavage of the C–O bond to Re-bound CO2. These studies were pursued further using imidazolium functionalized fac-Mn(bpy)(CO)3 catalysts.563 Similar to the Re analog, they found that the imidazolium functionalities enhance catalytic activity, resulting in the reduction of CO2 to CO in the presence of H2O at much more anodic potentials than the unfunctionalized congener. DFT studies indicated a synergistic interaction between the imidazolium and water molecules, likely due to the acidity of the C–H proton, which can participate in hydrogen-bonding interactions.
In another study, Fe(porphyrin) catalysts were modified to contain four pendant imidazolium cations.561 In DMF:H2O 9:1 mixture, the first reduction potential that corresponds to the Fe(III/II) reduction event is not impacted, indicating there is minimal inductive effect with the imidazolium functionalities. However, the next two reductive events, which have generally been assigned as ligand-based reductions,566 exhibit anodic shifts of about 120 and 415 mV, respectively, with the larger shift associated with the third reduction corresponding to the catalytic wave. As a result, the catalytic wave was 375 mV more positive compared to unfunctionalized Fe(TPP)Cl. The placement of the imidazolium fragments on the ligand framework is also important; the addition of four 1-benzyl-3-methyl-imidazolium chlorides with Fe(TPP) exhibits no enhanced catalytic ability. In aqueous phosphate buffer, a higher current is observed despite the fact that CO2 is only about one-third as soluble compared to DMF. Thus, pre-organizing the IL environment via incorporation into the second-coordination sphere leads to the same beneficial anodic shifts and enhanced catalytic activity.
Although studies with molecular catalysts in ILs are still nascent, the mechanisms by which these cations improve catalysis are likely different. For heterogeneous catalysts, imidazolium cations are believed to stabilize the formation of the CO2 radical anion. However, the catalytic potentials for most homogeneous catalysts are positive of that required to form the radical. Instead, the cations are believed to stabilize the reduced forms of the catalysts through electrostatic interactions.
These stabilizing effects appear to be more pronounced at ligand-based reductions, which can result in π+–π interactions. Enhanced catalytic rates have been attributed to hydrogen-bonding interactions between the imidazolium C–H and catalyst-carboxylate intermediates to facilitate bond cleavage or otherwise lower transition state structures. As the catalysts that have been studied typically have selectivity for CO2 reduction in organic solvents, the IL cation's ability to suppress the hydrogen evolution has not been discussed extensively. However, the electrostatic impact of a closely associated imidazolium cation to the catalyst has been suggested as important to suppressing proton approach for hydrogen evolution. Homogeneous catalysts have also been immobilized onto electrode surfaces for use with ILs, although their resulting catalytic behavior may be due to both the IL and the local environment of the catalyst support. Additionally, studies using imidazolium functionalities incorporated into the secondary coordination sphere of molecular catalysts through ligand design demonstrate that the beneficial impacts on catalysis can be replicated through intramolecular interactions.
4.5 Technoeconomic analysis
| System metric | Key performance parameters | Current state-of-the-art |
|---|---|---|
| Current density | >0.2 A cm−2 | 0.5–1.8 A cm−2 |
| Energy efficiency | >50% | ca. 40% |
| Faradaic efficiency | >80% | 80–90% |
| Full cell potential @ 300 mA cm−2 | <3.0 V, better <2.5 V, ideally <2.0 V | 2.7 V |
| Single pass conversion | >50% | 30–70% |
| System temperature | 60–90 °C | 40–80 °C |
| Additional notes and challenges: | ||
| Stability | <10 μV h−1 | |
| Cost of electricity | <0.04 USD kW h−1 | |
Jouny et al.570 used a TEA study to calculate the end-of-life net present value (NPV) of a generalized CO2 electrolyzer system for the production of 100 tons/day of various CO2 reduction products. The authors suggested that current density has a minimum impact on NPV after a threshold value of 200–400 mA cm−2 and that the required maximum cell voltage for profitable conditions can vary based on the desired product. In their review, Somoza-Tornos et al.571 emphasized concerns related to the high variability in the CO2RR product costs estimated via different TEA studies. This might be due to the variable input parameters (e.g., price of CO2 and electricity) and assumptions, and market values of different products during the period of studies.
Besides the economic feasibility of CO and HCOOH products, Shin et al.572 and Gao et al.,573 provided the roadmap for C2 chemical production by a detailed TEA study. To achieve economic viability for these products, the cell performance required significant improvements to achieve targeted price of $0.53 per kg. This assumed a further reduction in the electricity price to $0.01 kW h−1 and improvements in the electrolyzer cost in addition to selling the hydrogen byproduct and using carbon credits. Considering the importance of energy consumption, Leonzio et al.574 assessed that eliminating CO2 crossover alone could drastically impact these estimations.
The CO2RR can be coupled with numerous other organic oxidation reactions (OORs). Na et al.576 performed a thorough study encompassing 132768 process calculations to evaluate the economic feasibility of 295 electrochemical coproduction processes. The study indicates that coupling CO2RR with OORs, mainly 2,5-furandicarboxylic acid (FDCA) and 2-furoic acid, results in making the production of every CO2RR candidate economically feasible. It should be noted that the increased complexity of the OOR systems, particularly in comparison to the OER, requires intricate multistep separation processes and continuous monitoring of both anodic and cathodic products.577
Sisler et al.578 reported a comparative TEA study for C2H4 production via a single-step CO2 reduction in an alkaline flow cell, neutral MEA electrolyzers, and a CO2–CO–C2H4 tandem electrolysis process. The TEA study suggested that the CO2–CO–C2H4 tandem process required a significantly lower electrical energy efficiency (52% less) in comparison to the single-step process, and thus it was a more economically viable approach for C2H4 production. Similar to the use of multiple electrolyzers in tandem, TEA modeling has indicated that the use of stacked electrolyzers can be beneficial in comparison to a single chamber (SC) electrolyzer. For example, it was reported that an electro-stack electrolyzer achieved CO production at a significantly lower cost of $0.37 per kg compared to $0.41 per kg for the analogous SC electrolyzer.579 The costs associated with MEA replacement and capital expenses were modestly lower for the stacked electrolyzer system.
In the TEA study conducted by Li et al.,584 they aimed to gain insights into the energy dynamics of both sequential gas feed and integrated CO2 capture and electrochemical conversion processes. According to their findings, in the base scenario (with CO FE of 70% and cell voltage of 4 V), assuming a 50% single-pass conversion efficiency, the integrated process shows marginal benefits over the sequential process due to the substantial energy demand for CO2 electrolysis (Fig. 18a). However, under optimistic conditions (with CO FE of 90% and cell voltage of 3 V), and assuming similar CO2 electrolysis efficiency in both scenarios, the integrated route demonstrates a significant 44% reduction in energy consumption compared to the sequential process. This reduction is primarily attributed to the decreased requirement for thermal energy associated with amine regeneration, as well as the reduced electricity needed for CO2 compression and product purification (Fig. 18b). The integrated route presents a compelling advantage with a 22% reduction in energy costs compared to the sequential route (Fig. 18c). This significant decrease in energy consumption enhances the attractiveness of the integrated approach. The authors also compared the energy consumption considering 100% single-pass conversion efficiency for sequential process and optimistic conditions for the integrated process (Fig. 18). Even in this scenario, the integrated approach retains a maximum overall energy advantage of 26% and an energy cost benefit of 11% (Fig. 18).
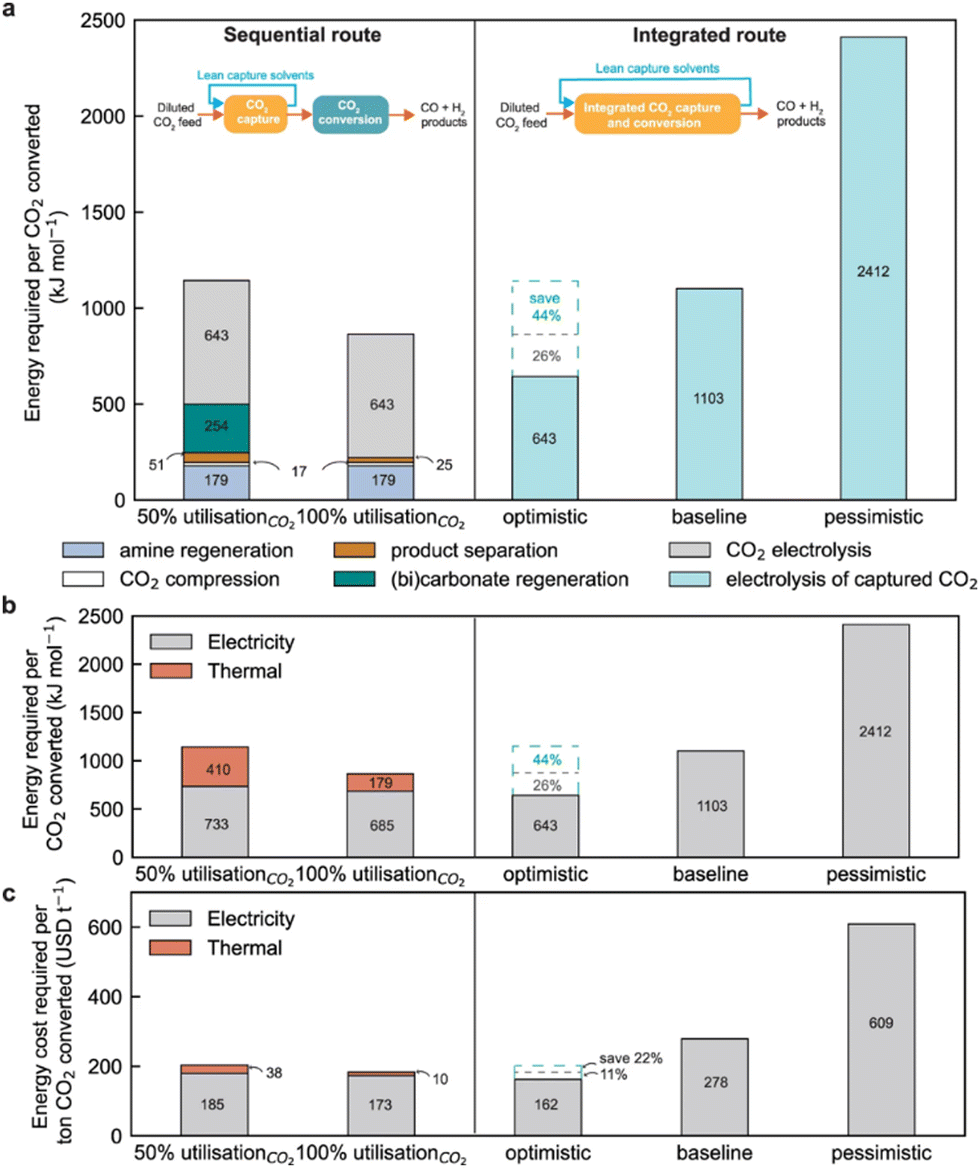 | ||
| Fig. 18 Energy analysis for sequential and integrated CO2RR: (a) overall energy consumption and energy requirements for various subprocesses, (b) the breakdown of thermal and electricity consumption, and (c) cost analysis of energy consumption in different scenarios. This data has been adapted with permission from ref. 584 under CC-BY 4.0 licence. | ||
5. Critical properties for reactive capture and electrochemical conversion of CO2
This section summarizes the IL/DES properties of interest for CCC, CO2RR, and RCC, that are discussed at length in previous sections to provide a closure on the main discussions. Fig. 19 captures the critical points highlighted in this section.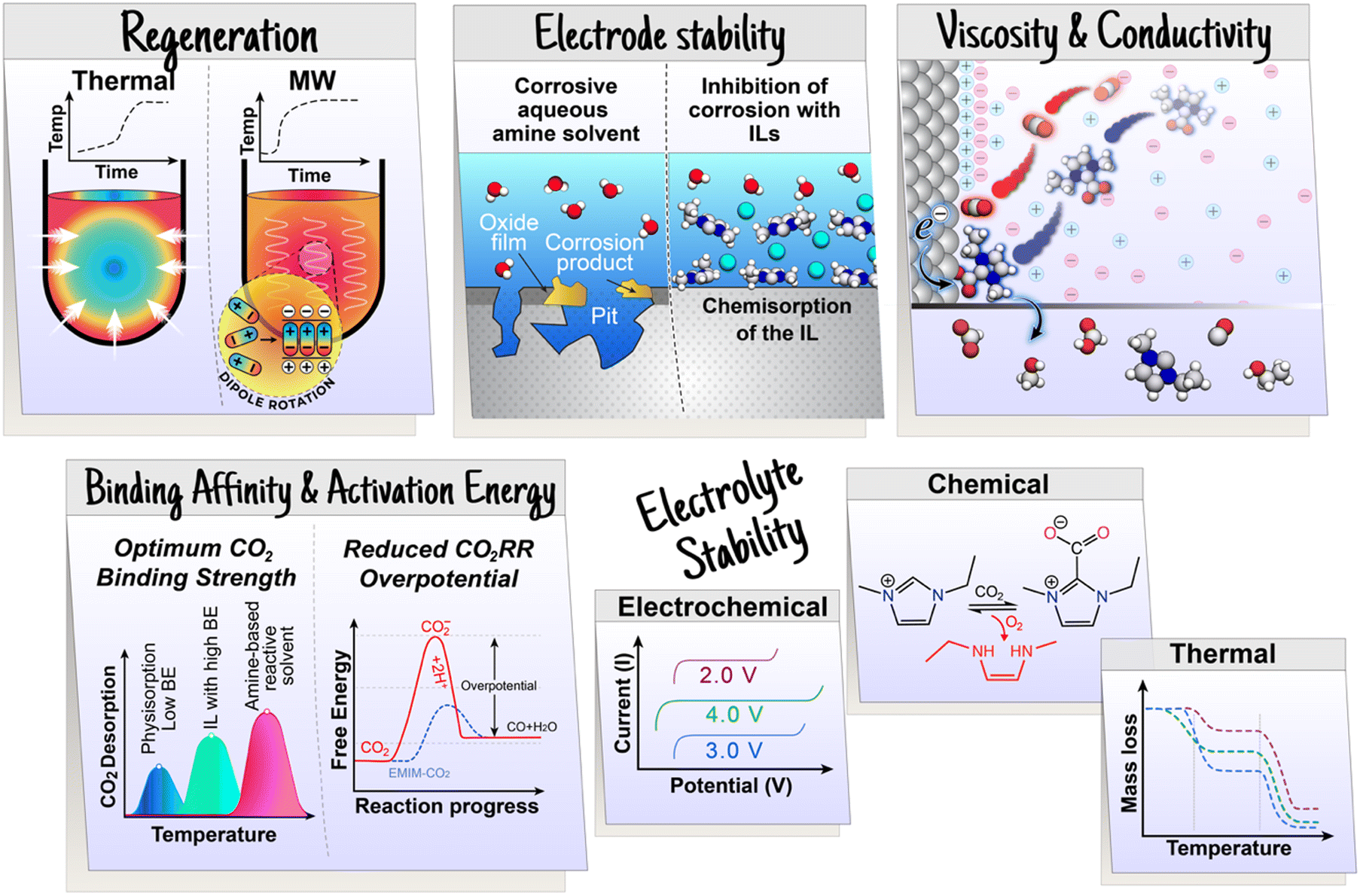 | ||
| Fig. 19 Schematic illustration of critical properties for RCC. | ||
5.1 Viscosity and conductivity
The high viscosity of ILs/DESs can hinder mass transfer, potentially limiting CO2 uptake.585 The high viscosity of IL/DES contributes to ohmic resistance and, therefore, can limit electrocatalytic CO2 conversion. Such high viscosity (>100 cP at 25 °C) limits their realization as neat electrolytes for effective RCC.91,586,587 In comparison, the viscosity of water is 1 cP.Amine functionalized ILs demonstrate great absorption capacities compared to conventional ILs; however, the bulk viscosities of these ILs dramatically increase upon CO2 absorption, resulting in a gel-like structure.90,587,588 The reason behind the significant increase in the viscosity of these ILs following reaction with CO2 has been extensively studied.586,589 A detailed molecular dynamic simulation reveals that the substantial rise in viscosity is attributed to the establishment of robust and densely interconnected hydrogen-bonded networks between the zwitterion and di-cation species formed due to the CO2 reaction with the amino-tailored cation.94
The experimental assessment of viscosity changes in amino-functionalized ILs, namely [aP4443][Gly], [aP4443][Ala], [aP4443][Val], and [aP4443][Leu], reveal a diminished CO2 absorption for all of these ILs (less than 0.2 mol CO2 per mol IL).587 This outcome is attributed to an increase in viscosity, which notably reduces CO2 diffusivity. The [Ala]− and [Gly]− anions have been further investigated in a different study paired with the phosphonium cation ([P66614]+), focusing on variations in viscosity following CO2 absorption.91 The findings indicated a substantial 48-fold and 117-fold escalation in viscosity at room temperature upon CO2 absorption for [P66614][Gly] and [P66614][Ala], respectively. This phenomenon is ascribed to the development of hydrogen bond networks within the CO2-complexed ILs.
Several studies attempted to reduce the viscosity of amino-functionalized ILs by substituting the cation with phosphonium and ammonium cations.116,590 Despite observing a reduction in the bulk phase viscosity of the IL, the significant increase in viscosity upon CO2 absorption remains unresolved. In other research, the pronounced increase in viscosity was addressed by strategically eliminating the hydrogen on the amine through anion-functionalization in AHA ILs.126 This approach prevented the generation of an acidic proton during the CO2 and IL reaction, thereby avoiding the formation of intermolecular hydrogen bonds. Despite the high viscosities of the demonstrated AHA ILs (e.g., 370–500 cP at 25 °C), they did not exhibit further increase with CO2 absorption. Solvents that have lower viscosity tend to be more favorable for CO2 capture and utilization, as the mass transfer rate is generally higher and pumping costs are lower. In efforts to further reduce the viscosity and increase the gravimetric capacity, AHAs were later paired with smaller phosphonium and ammonium cations,96 and the imidazolium cation.453,591 Further, when imidazolium AHA IL was diluted with EG (IL:EG, 1:2 molar ratio), thus forming DES, as demonstrated by Lee et al.,160 the viscosity was lowered from 70 to about 45 cP at 25 °C.
Despite these efforts, the viscosities of functionalized ILs and DESs remain high for RCC applications where the transport of CO2-limiting conditions. Such viscosity-controlled energy losses due to mass transport overpotential and ohmic losses are not overcome by improved CO2 concentration. Therefore, all of the electrolysis studies within RCC with ILs and DESs to date involve a co-solvent so that appreciable amounts of conversion products can be generated and quantified for reporting product distribution and faradaic efficiencies.
5.2 CO2 binding affinity
Amine solutions, such as MEA, are used for industrial carbon capture, however, the regeneration of captured CO2 requires a significant amount of energy input. The considerable energy demand associated with MEA can be largely ascribed to the substantial heat of reaction, reaching up to −85 kJ mol−1 of CO2 at 40 °C.64 Such high heat of reaction, along with heat losses from water vaporization in the stripper renders them interchangeable. Additional challenges include issues, such as corrosiveness, amine volatilization, and oxidative degradation of the amine.592 In this regard, the potential of ILs emerges due to their elevated CO2 capacity and lower enthalpies in comparison to the amine solvents commonly employed in industrial processes for CO2 capture. The enthalpy associated with the physical absorption of CO2 by ILs is approximately −20 kJ mol−1. This energy suggests that the energy needed to release CO2 physically absorbed by ILs during the regeneration step is only a quarter of that required in the amine solution method. Despite this efficiency, the CO2 absorption capacity of these ILs is limited to around 3 mol% under atmospheric pressure.593 Functionalizing ILs with CO2-reactive chemisorption sites enhances the capture capacity of ILs, addressing limitations seen in the physical absorption process, as discussed earlier. Thus, TS-ILs are tailored with specific chemical properties to enhance the interaction and capture of CO2 molecules, thereby increasing the affinity between the sorbent and CO2. However, the functionalization of ILs with chemisorption sites significantly increases the heat of sorption (ΔH), resulting in higher energy consumption during regeneration phase.For instance, the ΔH of two amino functionalized IL, [P66614][Pro] and [P66614][Met], were compared by using the same phosphonium cation.74 The enthalpies of sorption between amino-functionalized ILs and CO2 were experimentally measured to be −80 kJ mol−1 and −64 kJ mol−1 of CO2, respectively, at 25 °C and 2–3 bar pressure. These results exhibit a noteworthy alignment with the calculated reaction energies for the gas-phase anions, demonstrating that the phosphonium cation has an insignificant effect on ΔH of CO2 and the absorption of CO2 is dominated by the anion of the ILs. Another study analyzed the enthalpy of carbamate formation on amino functionalized [Pro]− and [Ala]− anions with a different phosphonium cation upon CO2 absorption.594 The ΔH value for [P4444][Pro] was −52.8 kJ mol−1 compared to [P4444][Ala] at −75.3 kJ mol−1. This difference suggests that the carbamic acid formation reaction is more favorable with [Ala]−, despite [Pro]−-based ILs exhibiting a higher CO2 uptake. Carbamic acids displayed strong intramolecular hydrogen bonding, contributing to their stability. Additionally, intermolecular hydrogen bonds likely exist between carbamic acids, further enhancing their overall stability. The enthalpy associated with CO2 absorption in carboxylate functionalized [P66614][OAc] was also determined in the same study through the analysis of the temperature-dependent variation in the natural logarithm of the equilibrium constant. The calculated enthalpy value is −37.6 kJ mol−1 between 20–40 °C under 5–30 kPa, indicating a chemical absorption process, yet the heat of absorption is significantly lower than amino functionalized ILs.
A similar calculation of CO2 absorption enthalpy is conducted on an azolate functionalized IL, [P66614][2-CNpyr], over the temperature range of 22–55 °C via the temperature dependence of equilibrium constants and differential calorimeter.126 The results were further verified by a first principles G3 calculation. The experimental reaction enthalpy is determined as −53 ± 5 kJ mol−1, with −10 kJ mol−1 attributed to the enthalpy of physisorption, aligning with the enthalpy of non-functionalized ILs. The remaining enthalpy is associated with chemisorption facilitated by the functional group of the anion. The results further exhibited an excellent concordance with the G3-computed reaction energy of −49 kJ mol−1 for gas phase anion at 25 °C. A different approach is considered by investigating the CO2 absorption enthalpy of another azolate functionalized IL, 1,1,3,3-tetramethylguanidinium imidazole ([TMG][Im]), at a CO2 concentration of 15% to mimic the industrial process conditions.128 The enthalpy of the reaction was determined by exploiting the temperature dependence of equilibrium constants within the range of 30 to 50 °C, yielding a value of −30.3 kJ mol−1. Consequently, the captured CO2 exhibits greater ease of release. A more comprehensive investigation is undertaken to explore the impact of various AHA anions, specifically indazolide, imidazolide, pyrrolide, pyrazolide, and triazolide, in ILs featuring the phosphonium cation [P66614], on the enthalpy of CO2 absorption.138 The reaction enthalpy of the [Inda]−, [BnIm]−, [6-BrBnIm]−, [2-SCH3BnIm]−, [2-CNPyr]−, [3-CF3Pyra]−, [3-CH3-5-CF3Pyra]−, [123-Triz]−, and [124-Trz]− anions were experimentally determined as −54, −52, −48, −41, −45, −44, −41, −37, and −42 kJ mol−1 CO2 at 22 °C, respectively. It is found that while [123-Trz]− has the lowest reaction enthalpy, [Inda]− has the highest reaction enthalpy among all AHA anions. However, all the AHA ILs demonstrated a significantly lower absorption enthalpy compared to conventional amine solvents, such as MEA, and AEEA, reaching enthalpies of −80 kJ mol−1 CO2 at 40 °C,64 hence having a potential for CO2 capture utilization with significantly lower regeneration energy requirement.
The enthalpy of the reaction between the IL and the CO2 is the main driving force for the capture of CO2. As a result, higher reaction enthalpies typically result in higher absorption. However, high absorption enthalpy results in challenging desorption along with elevated energy requirements for the regeneration step. Therefore, reducing the enthalpy of reactions on chemisorption dominated absorption is crucial to overcome these issues.
The effects of different substituents on the reaction enthalpy of CO2 with the ILs having phenolic anions were investigated in a computational study.143 The geometry and energy optimization of each anion, anion–CO2 complexes, and gas CO2 is calculated at the B3LYP/6-31++G(p,d) level. The results demonstrated that the addition of electron-withdrawing groups, such as Cl, to the structure of the phenolate anion decreased the reaction enthalpy. In contrast, addition of the electron-donating groups, such as Me, resulted in an increase in the reaction enthalpy. Hence, the reaction enthalpy of the [P66614][3-Cl-PhO] is calculated as −31.4 kJ mol−1. Conversely, [P66614][4-MeO-PhO] had an enthalpy of −51.4 kJ mol−1. The calculation of Mulliken atomic charge of the oxygen atom further revealed that as the substituent atoms gets more electron-withdrawing, the weaker the charge on the oxygen atom gets. In contrary, increase in the electron-donating ability of the groups leads to an increase in the charge of the oxygen atom of the phenolic anion. The comparison of the [4-Cl-PhO]−, and [2,4,6-Cl-PhO]− anions show a significant change on the charge of the oxygen atom with the addition of Cl groups, from −0.6833 to −0.6270, respectively. The results observed on the change of reaction enthalpy and charge of oxygen atom showcased the roughly linear correlation between the Mulliken charge of the oxygen atom and enthalpy of CO2 absorption. Among 13 ILs that were studied, [P66614][4-MeO-PhO] had the highest enthalpy of −51.4 kJ mol−1 along with highest Mulliken charge of −0.7062 on the oxygen atom, whereas [P66614][4-NO2-PhO] had the lowest oxygen charge of −0.6187, therefore, the lowest reaction enthalpy with −17.1 kJ mol−1. Hence, the effect of substituents plays a crucial role in designing ILs having low energy requirement for desorption.
A distinct methodology for fine-tuning the enthalpy and capacity of CO2 absorption is proposed, involving the modulation of the basicity of ILs.93 This approach integrates experimental and computational tools to systematically determine the optimum between absorption enthalpy and improved CO2 capacity. ILs with various functionalization, utilizing the [P66614] cation paired with eight different anions characterized by basicity between 8.2–19.8 pKa, were investigated. An observable and noteworthy reduction in CO2 capacity is noted, shifting from 1.02 mol CO2 per mol IL in [P66614][Pyr] to 0.08 mol CO2 per mol IL in [P66614][Tetz], correlated with a decline in the pKa values of the respective weak proton donors in DMSO, dropping from 19.8 to 8.2, respectively. The effect of basicity on the CO2 absorption enthalpy of ILs is further investigated by calculation of gas-phase reaction energetics at the B3LYP/6-31++G(p,d) level. Comparison of absorption enthalpy between the most and least basic ILs demonstrated a significant difference, with [P66614][Pyr] having −91.0 kJ mol−1, whereas [P66614][Tetz] having −19.1 kJ mol−1, which is in the range of physisorption.593 However, a decrease in pKa from 18.6 for [P66614][Im] to 13.9 for [P66614][134-Trz] resulted in only a 5% decrease in CO2 capacity preserving almost equimolar absorption ability, although a substantial decrease in enthalpy of CO2 absorption was observed from −89.9 kJ mol−1 to −56.4 kJ mol−1. Therefore, the enthalpy of CO2 absorption in these basic ILs can be systematically adjusted by manipulating their basicity while preserving their absorption capacity. However, steric effects can also be important impacting binding affinity.
A study underscored the significance of considering entropy variations arising from structural differences in anion functionalized ILs.595 Methylbenzolate-based ILs were synthesized having amino groups both in ortho- and para-positions. The results indicated that [P66614][p-AA] exhibited a CO2 absorption capacity of 0.94 mole of CO2 per mole of IL, while [P66614][o-AA] showed a lower capacity of 0.60 mole of CO2 per mole of IL at 30 °C. Notably, the Mulliken charge analysis of the N atom in these ILs revealed that [o-AA] had a stronger interaction with CO2 (Mulliken charge: −0.584) compared to [p-AA] (Mulliken charge: −0.529), suggesting higher absorption enthalpy. However, the unexpected increase in CO2 absorption capacity from [P66614][o-AA] to [P66614][p-AA] was attributed to the entropic effect, challenging the conventional enthalpy-driven perspective in CO2 capture. The computation of the enthalpy of CO2 absorption in these ILs indicated that the IL with the amino group in the para-position exhibited an enthalpy of −41 kJ mol−1. This value marked a notable decrease from −56 kJ mol−1 observed when the amino group was positioned ortho-, all while maintaining its equimolar CO2 absorption capacity. Therefore, the higher CO2 capacity and the lower enthalpy of the [P66614][p-AA] is attributed to the difference in the formation of intermolecular hydrogen bonding as a result of entropic differences. These differences emphasize the importance of considering both enthalpy and entropy in the Gibbs free energy calculation for a more comprehensive understanding of CO2 capture mechanisms.
The importance of hydrogen bonding and the proton activity in DESs (Section 2.4) was also highlighted in a series of publications by the Gurkan group.149,160,165,166 Similar to ILs, basicity and steric effects are both at play in governing the CO2 absorption strength in DESs, which ultimately impact the regeneration energy in CO2 capture. This, again, is an important concept for RCC since the CO2 bound on an IL ion or DES component in the electrolyte needs to be released at the electrode surface for hydrogenation and C–C coupling without the consumption of the IL/DES.
In summary, although functionalized ILs offers significant enhancements to the CO2 absorption capacity compared to the conventional ILs, the increase in enthalpy of the CO2 absorption can be very drastic due to the high exothermic effect of chemisorption of CO2 on the reactive functionalized sites, reaching enthalpies as high as −80 to 90 kJ mol−1.74 Modifications such as addition of substituent groups, changing the basicity, and consideration of entropic effects, are pivotal in fine-tuning the enthalpy of CO2 absorption when designing high capacity ILs/DESs for CO2 absorption, facilitating the ease of regeneration as well as recovery in RCC.
5.3 Regeneration energy and other factors impacting cyclability
The primary determinants of the effectiveness of absorbents include the CO2 capacities and regenerability. An absorbent exhibiting a substantial heat of reaction implies a correspondingly significant energy requirement for its decomplexation during regeneration. However, the heat of reaction, or enthalpy of reaction, is linked to CO2 capacity, as indicated by the equation ln(K) = −ΔG/RT, where K represents the equilibrium constant for the complexation reaction, and ΔG denotes the Gibbs free energy change. The relationship is further delineated by ΔG = ΔH − TΔS where ΔH is the CO2 absorption enthalpy, ΔS is the change in entropy associated with CO2 absorption, and T is the temperature. Consequently, a trade-off arises between the desire for enhanced CO2 capacity at a specific temperature and the preference to minimize the heat required for reversing the reaction. Hence, evaluation of regeneration performance of functionalized ILs is critical, given their higher reaction enthalpies compared to conventional ILs. The same thermodynamic principles apply to DESs. In both cases, functionalization leads to a higher thermal energy requirement for regeneration of the sorbents.The regeneration of an amino-functionalized IL, [BMIM][Gly], is assessed through the measurement of CO2 sorption at 25 °C and 0.9 bar, followed by vacuum treatment for 1 hour at 25 °C in each of 5 consecutive cycles.115 The results indicated that [BMIM][Gly] could be desorbed effectively within 1 hour under the conditions tested and the CO2 absorption capacity exhibited no significant variations across five consecutive cycles (retained capacity of 95.4% after the fifth cycle). The slight decrease in the capacity was attributed to the marginally imperfect desorption of CO2 under the tested conditions. In another study, the regeneration performance of the amino-functionalized IL, 1-aminoethyl-2,3-dimethylimidazolium taurine ([AEMMIM][Tau]) was assessed over six consecutive absorption–desorption cycles.596 The findings revealed an approximate 5% reduction in the absorption capacity of the IL when desorption occurred at 80 °C under 20 Pa, indicating the regenerability of the IL, particularly under slightly elevated temperatures.
A more comprehensive investigation of the effect of temperature on the regeneration on amino functionalized [APMIM][Lys] was conducted by changing the regeneration temperature between 90 and 120 °C.597 It was found that the reabsorption loading increased with higher regeneration temperatures. The regeneration efficiency fluctuated between 79.0 and 84.4% as the temperature was increased from 90 to 110 °C, suggesting incomplete regeneration. However, [APmim][Lys] achieved a regeneration efficiency of 99.6% at 120 °C, establishing it as the optimal regeneration temperature. Subsequently, the results of five absorption–desorption cycle at 120 °C illustrated the regeneration efficiency of CO2 in the saturated [APMIM][Lys] solution, which ranged between 99.1 and 100%. The regeneration performance of [APMIM][Lys] is compared with the widely used amine solvent MEA over 5 cycles. MEA, in contrast, could only preserve 81% of its initial absorption capacity (0.46 mol of CO2 per mol of MEA),598 highlighting the potential of [APMIM][Lys] as an absorbent for repetitive cycles.
The regeneration of carboxylate-functionalized ILs was investigated utilizing a 1:1 [P4444][HCO2]/H2O and [P2228][CH3CO2]/H2O mixture as CO2 capture media.599 The IL/H2O mixtures underwent regeneration at 70 °C by flowing dry N2 or air through a stirred liquid mixture for 15 minutes. The results demonstrated complete regeneration (100%) for the [P4444][HCO2] system and 90% regeneration for the [P2228][CH3CO2] system after two absorption–desorption cycles.
A comprehensive analysis of the regeneration process for an azolate-functionalized ionic liquid, [TMG][Im], was conducted using IR spectroscopy.128 IR spectra were obtained for CO2-flowed [TMG][Im] at 35 °C for 40 minutes, and N2-bubbled [TMG][Im] at 65 °C for 30 minutes following absorption. The CO2-free [TMG][Im] and regenerated [TMG][Im] did not exhibit any differences in their IR spectra, while additional carbonyl peaks were observed in the CO2-absorbed [TMG][Im]. Furthermore, a five-cycle absorption–desorption process resulted in a ±5% change in the CO2 capture capacity of [TMG][Im], indicating the successful regeneration of the IL.
Co-absorption of water and other impurities from CO2 sources such as oxygen in DAC applications, can alter the reaction mechanism in functionalized ILs and DESs, and hence the regeneration energy and absorption–desorption cyclability. In fact, there are very few studies that demonstrate cycling beyond 5 to 10. Further, the side reactions leading to thermal and oxidative degradation in ILs/DESs are not well understood. The side reactions can be accelerated during regeneration with the elevation of temperature and co-adsorption of water, O2, and other gases. Oxidative degradation has been reported for amine functionalized adsorbents, such as aminosilane.600 Further, poly(ethylenimine) is reported to readily oxidize with increased temperature due to the presence of secondary amines.601 Recently, the role of water on the oxidative degradation of solid amine sorbents including supported poly(ethylenimine) has recently been reported by Jones and colleagues.602 They report a radical mediated autooxidative degradation mechanism that is accelerated by water. DES and IL solvents share common structural motifs to these previously studied amines and therefore similar oxidation mechanisms may occur. Lee et al. reported radical and SN2 side reactions that occur in functionalized IL [EMIM][2-CNpyr], due primarily to the nucleophilic anion, that degrade the sorbent to lose 50% of its CO2 capacity after exposure to oxygen at 80 °C continuously for a week.51 Despite the reported loss in capacity, the functionalized IL demonstrate superior stability compared to the solid amines where CO2 capacity loss in the orders of 80% and 61% have been reported under dry air at 120 °C[71] and 80 °C72 for 7 days, respectively.
Most DESs and ILs are hygroscopic, similar to salts. While the absorbed water improves transport and CO2 uptake capacity, it is not desirable from the point of view of thermal regeneration and the energy cost. For instance, absorbed water reacts with CO2 in the presence of a base such as in the functionalized systems, thus forming bicarbonate, which is energetically less favorable to release CO2 compared to CO2-IL complexes. Lee et al.263 measured the CO2 capacity of the reactive IL, [EMIM][2-CNpyr], as a function of CO2 feed and relative humidity. While the viscosity was seen to decrease, CO2 capacity increased slightly with increasing relative humidity hence the increasing water content. The increase in capacity is due to the additional CO2 absorbed by the water. Similar chemistry occurs in an amine functionalized DESs.160 The common DES ethaline absorbs about 6 wt% water when exposed to air603 and maintains its H-bonding network up to 30 wt% water,604 beyond which it becomes a simple solution. The reactive IL, [EMIM][2-CNpyr], was mixed with ethylene glycol to achieve a functionalized DES, in another study by Lee et al.160 The authors reported induced electrostatics as a result of H-bonding, as demonstrates by DFT calculations of the reaction pathways where the CO2 binding energies were reduced, compared to the IL itself, thereby enabling regeneration at 40 °C after absorption at 25 °C. On the other hand, these types of DESs present lower thermal stabilities, compared to ILs, as the volatile component evaporates at elevated temperatures. Further, the amount of sorbed water can influence the phase behavior in addition to the transport properties and the CO2 capacities.
Considering the high heat capacity of water, it is desirable to eliminate its co-absorption with CO2 sorbents through the inclusion of hydrophobic moieties in reactive solvents and hydrophobic film barriers or supports in composites for RCC. In fact, the presence of water is detrimental for most solid sorbents such as zeolites and MOFs, thus necessitating a pre-absorption column to first remove water from the gas mixture. On the other hand, water is tolerable with ILs, DESs, and their composites where the reaction mechanism, absorption capacity, transport properties, and regeneration temperature may be affected without loss of performance. Lee et al.190 demonstrated a one-to-one comparison of the breakthrough performance of an encapsulated reactive IL (an example of an ENIL) and zeolite 13× where the zeolite lost its CO2 selectivity completely with co-absorption of water and the ENIL demonstrated maintained selectivity accompanied with improved CO2 capacity. However, the mass transfer limitation in ENIL breakthrough curves was apparent in the presence of water in addition to the need for increasing the regeneration temperature from 40 to 90 °C.
In contrast to the notion of molecular and composite designs for increased hydrophobicity to suppress water effects, Lackner and colleagues13,43,44 demonstrated a moisture-swing regeneration that is based on quaternary ammonium hydroxide resins where the CO2 is absorbed under water-lean conditions and released under water-rich conditions. Further, they coupled this approach with the conventional thermal-swing regeneration in an effort to minimize the regeneration energetics and improve the CO2 desorption rates.
The issue of high energy consumption during thermal solvent regeneration may also be circumvented by dielectric heating by irradiation instead of conventional thermal heating by convection. A faster CO2 release was reported in aqueous amines by the use of microwaves.49 Microwave regeneration of dry alkanolamines is reported to decrease the energy requirement by 10-fold.50 Similarly, the dielectric response was increased with the number of amine functional groups in mesoporous silica which resulted in 4-fold increase in adsorbent regeneration rate.605 The proof of concept on the utility of dielectric heating for solvent regeneration was recently demonstrated for functionalized ILs and DESs.51,165 In particular, the absorption–desorption cyclability of [EMIM][2-CNpyr] up to 10 cycles was possible with microwaves.51 The authors reported improved cycling efficiency in comparison to conventional thermal heating. They also noted the prospects of improved stability since the exposure to elevated temperatures that accelerate degradation kinetics are suppressed with the dielectric heating approach.
In summary, functionalized ILs and DESs demand more energy-intensive regeneration processes, primarily attributed to their high level of chemisorption from reactive functional groups, as compared to conventional ILs and DESs. However, in contrast to conventional amine solvents like MEA, functionalized ILs/DESs exhibit superior regenerability across repetitive absorption–desorption cycles. Nevertheless, it is noteworthy that amine-functionalized ILs/DESs necessitate more energy-intensive regeneration steps compared to carboxylate and azolate functionalized ones, primarily due to their higher enthalpy of reaction. Therefore, there is a tradeoff between high CO2 capacity/selectivity and the required regeneration energy. As a result, sorbents for applications such as DAC where very high CO2 capacities and selectivities are needed. Key performance indicators will be the ease of regeneration with a maintained working capacity, stability, and cyclability. The sorbent regeneration step contributes the most to the operating cost of CO2 removal processes, and thus further optimization of the capture and regeneration energetics of the chemically and thermally stable ILs and DESs must be pursued.
5.4 Stability of functional electrolytes during RCC
The stability of the functional electrolyte in RCC signifies that performance indicators (e.g., CO2 uptake, solvent regeneration, mass transfer, faradaic efficiency, current density, conversion, and energy efficiency) must be preserved during long-term operation without significant decay. Better stability corresponds to lower maintenance and replacement costs associated with the consumption of catalysts, electrodes, exchange membranes, and electrolytes. In this subsection, the stability issues of functional solvents/electrolytes, both chemical and electrochemical, are reviewed under RCC conditions.The main sources of industrial CO2, such as power plants and petroleum industries, contain impurities. The flue gas from typical coal-fired power plants consists of 13% CO2, 68% N2, 16% water, 3% O2, and other residuals such as hydrocarbons, NOx, SOx, H2S, particulates, mercury, and chlorides.606 The exposure of small amount of O2, water, NOx, SOx, and H2S has significant impact on the stability of functional CO2 capture solvents. Some of these impurities strongly bind with the functional solvents and poison the catalyst with preferential adsorption. ILs with fluorine-containing anions (e.g., [BF4]−, [PF6]−) have been very popular for CO2RR studies in the last few years; however, such anions may undergo hydrolysis, forming hydrofluoric acid,607 which is highly corrosive. This reactivity is particularly inconvenient for treating exhaust gases arising from combustion processes, which can contain as much as 10% water. As a result, utilizing anions that are hydrogen-bond acceptors thus strongly interacting with water can result in weakened cation–anion interactions in imidazolium-based ILs.132 These intermolecular changes influence the RCC reaction mechanism, CO2 uptake capacity, and CO2 binding mechanism and strength. In addition, higher amounts of water in ILs also favor the formation of H2 over the reduction of CO2 species, highlighting the multiple effects of water on the stability and performance of ILs for RCC.
Imidazolium based ILs are considered co-catalysts for CO2 electrolysis, however, reactions involving imidazolium cations can impede RCC. Under moderately basic conditions, imidazolium can undergo deprotonation at the weakly acidic C2 position and the generated nucleophilic N-heterocyclic carbenes can react with aldehydes, sulfur, H2S, and CO2.608 This reactivity is also true under the negative applied potentials since imidazolium is reduced to form a carbene with a an electron transfer reaction, which then combines with CO2 available in the bulk and generates [EMIM]+-CO2− complex. This complex is reported as an inactive CO2RR byproduct due to the strong energy requirements to break [EMIM]+-CO2− bonds. However, the presence of some water can reactivate [EMIM]+-CO2− complexes during RCC to dramatically increase CO2 reduction rates.432
Further, the presence of sulfur impurities in the CO2-stream has the capacity to chemically interact with the n-heterocyclic carbene generated by a single electron reduction of imidazolium and form imidazole-2-thiones.609 The sulfur-containing complexes have been shown to cause significant metal surface poisoning which would negatively impact on the RCC performance. However, most of the existing RCC studies is centered on CO2 capture from the pure CO2 stream or CO2 diluted in inert gas and their subsequent conversion, and almost no studies have considered SO2 or similar acidic gases potentially present in real flue gas. Therefore, additional studies are required to evaluate the potential effects of these impurities during the electrolysis step.
Aside from the chemical side reactions, the electrochemical stability window (the difference in voltage between the onset potentials of oxidation and reduction reactions of the electrolyte itself) is a critical property. The electrochemical stability window of ILs depends not only on the chemical structure, but also on the electrode materials. The exact determination of the stability window is also dependent on the sweep rate of the potential, temperature, atmosphere, and impurities. In general, ILs are reported to have high electrochemical stability compared to that of conventional solvents. However, the presence of trace amounts of unavoidable impurities limit their electrochemical windows.610 Most of the literature reports IL stability using an inert glassy carbon electrode. However, for electrocatalysis, active metal or metal oxide electrodes are more relevant to consider. For instance, Dongare et al.467 observed earlier cathodic degradation of [EMIM][2-CNpyr] on Ag (−2.3 V vs. Ag/Ag+), Au (−2.1 V vs. Ag/Ag+), Sn (−2.05 V vs. Ag/Ag+), in comparison to glassy carbon (−2.45 V vs. Ag/Ag+). The anodic degradation was observed to start from 0.5 V vs. Ag/Ag+ on an Ag surface. Additionally, they confirmed the anodic degradation of [EMIM][2-CNpyr] on Pt and carbon rod, noting a color change of IL from light yellow to dark brown during the bulk electrolysis experiment conducted at −2.1 V vs. Ag/Ag+. Such degradation products could crossover during the RCC; hence, ILs with nucleophilic anions should be avoided in the anolyte. Furthermore, authors also reported increased HER when the aqueous anolyte was used with the non-aqueous catholyte containing the CO2-reactive IL. The increase in HER current was attributed to the crossover of H+ from the anolyte to the catholyte. To address these issues, the strategy of integrated electrolysis (coupled/paired electrolysis or co-electrolysis) has emerged in recent years, where the aqueous electrolytes at the anode side is replaced with other oxidation reactions such as electrooxidation of glycerol611 or methanol.612
Further, CO2 electroreduction takes place at potentials that are very close to the cathodic limit of most ILs. Therefore, qualitative and quantitative in-depth investigations of the cathodic stability of ILs are necessary. Recently, Michez et al.491 reported the stability of [BMIM][NTf2] by studying its reductive decomposition on Au electrodes. Using NMR and gas chromatography, they confirmed the formation of the neutral dimers by recombination of two imidazole-2-yl radicals (generated by 1e− transfer to imidazolium cation) with >80% FE at −2.5 V vs. Fc+/Fc. Despite the general recognition of ILs as highly stable electrolytes, the cathodic limit is relevant to examine as it can significantly impact the performance of CO2 electrolysis.
5.5. Electrode stability
Various materials ranging from pure metals to SACs are being explored for their potential use in RCC systems, each offering distinct advantages. However, their long term thermal and electrochemical stability must also be considered for practical implementation. Electrodes used in CO2RR undergo various stability issues including corrosion, surface passivation, and catalyst deactivation due to chemical reactions with CO2, water, and electrolyte salts, which degrade the electrode surface and diminish performance overtime. In the context of stability challenges present in aqueous electrolytes, ILs offer improved durability and composition control by mitigating surface restructuring and corrosion that would occur at more negative potentials and higher temperatures. For example, the work by Parada et al., demonstrated corrosion resistance properties of imidazolium based ILs through a process called ‘solid catalyst ionic liquid layer’ that shielded electrodes from degradation by forming stable passivation layers on the surface.613 For electrode materials encountering low catalytic activity, ILs can alleviate these issues by providing a conductive environment and increasing CO2 concentration at the electrode surface. Moreover, tailored solvent–solute interactions in ILs can stabilize catalytic active sites and inhibit undesirable side reactions, enhancing catalyst efficiency and selectivity.6. Conclusions and outlook
This review summarizes the recent advancements and offers a perspective in the reactive capture and thermal and electrochemical conversion of CO2, with a specific focus on the roles of the functional ILs and DESs as the capture agents as well as the catalysts and the electrolyte media. Specific advantages of these solvents according to their molecular design are discussed in the context of the state-of-the-art CO2 capture figures of merit. While there has been significant progress in improving the CO2 capacity of ILs and DESs for separations and conversion by thermal catalysis, utilization of these solvents for RCC is relatively recent. Therefore, there is still a need to pursue research on the specific reaction mechanisms and the compatibility of ILs and DESs with the electrode materials in terms of understanding stability, interfacial structure, and surface adsorption as they relate to the thermodynamics and kinetics of RCC. These studies will assist in advancing functional electrolytes and electrolyzer design simultaneously.As most ILs and DESs demonstrate increased viscosity with CO2 saturation, thus limiting the mass transfer processes, they are commonly supported through porous materials to increase gas–liquid surface area and improve transport properties without compromising the CO2 capacities. Among the various support structures investigated for CO2 capture with ILs, MOFs have been of interest also as an active material for tuning the CO2RR selectivity with high current densities. To date, a few of the MOFs, mostly those with Cu and Zn,446,614 have been investigated due to the availability of multiple active sites exposed for CO2 binding. Furthermore, when these MOFs are wetted by IL electrolytes, there is believed to be increased availability of CO2 and longer residence time for reaction intermediates on the surface, thus enabling multi electron transfer reactions. Therefore, these types of materials could be interesting to further explore as catalysts in RCC.
Despite advancement in ILs, DESs, and composites based on these liquids with porous supports, the current state of the CO2 capture field necessitates improved solvents/sorbents for simultaneous achievement of high CO2 capacities, fast transport, non-thermal or low energy for regeneration, and long-term stability under high oxygen and humidity conditions, as underscored by the 2019 NASEM report on NET and Reliable Sequestration.615 To advance the discoveries toward this goal, besides the simulations and ML approaches, the characterization of the gas–liquid, gas–solid, and liquid–solid interfaces as well as temporal changes in these interfaces are quite important. For the composites, the interactions taking place between the IL molecules and the surface of porous material are critical factors governing affinity-based capture and CO2 transport. The presence of the IL can alter several characteristics of the support material, such as morphology, surface area, pore volume, and thermal stability. Concomitantly, when the IL coating on the porous material is thin, the properties of the liquid layer might be significantly different compared to the bulk. The structural changes in the IL associated with the presence of a surface and surface-specific interactions can further result in kinetic effects. For example, the confinement of ILs,616 has been discussed to impact liquid structure and dynamics where deviations from the bulk have been reported, such as faster molecular motion near walls.617 Usually, confinement is defined by the relative size of the confining structure with respect to the molecular dimension of the liquid. ILs with approximately 1 nm ion size are said to be confined when they are in a 10 nm-sized pore. The examination of such buried interfaces in these length-scales with nm-scale resolution is often difficult, especially as they evolve during absorption–desorption events, thus necessitating the utilization of advanced in situ characterization techniques. Furthermore, most experimental research on IL/DES based hybrid materials has been directed towards enhancing CO2 capture and separation performance in pure gas composition under dry conditions; however, practical applications involve gas mixtures with N2, O2, and moisture, which significantly impact the material's overall performance and stability. Future efforts need to prioritize testing with multi-component gas mixtures, reflecting real-world conditions, and incorporate moisture to understand how water impacts both CO2 capture efficiency, potential regeneration processes, and the corresponding lifetime of the material under operation conditions. These assessments are also relevant for RCC processes as direct utilization of CO2 from the capture media present the possibility of co-absorbed impurities from the CO2 source and their side reactions.
Regarding the use of ILs and DESs for the thermal conversion of CO2 into valuable products, determination of the structural factors controlling the thermal stability limits is crucial. These limits can largely deviate from those of the bulk liquids when there are direct interactions with the surface of the support. Concomitantly, these interactions become even more significant in the presence of a metal complex or a metal nanoparticle in the catalyst structure. In such cases, ILs and DESs can substantially alter the electronic structure on the active metal centers. Hence, the research should be directed towards elucidating these interactions and their consequences on the catalytic properties.
In review of the electrocatalytic conversion of CO2 using functional ILs and DESs, the most critical challenge is the high viscosity of these liquids as electrolytes. While high faradaic efficiencies and lower overpotentials were possible under controlled experimental conditions, the obtained current densities have been typically very low (<25 mA cm−2), therefore, the overall performance still needs improvements to be industrially relevant. The high viscosities impede mass transport of CO2 or the CO2 carrying specie to the electrode surface. ILs, even in dilution, present interfacial structuring that increases resistance. Therefore, there is an interplay between the increased IL concentration for increased CO2 availability and the increased interfacial resistance due to IL ion accumulation in controlling the current density and CO2 conversion rates. Additionally, in cases where CO2 is captured in carboxylate form, it is not electrochemically active, hence the utilization of the captured CO2 remains limited. As a result, further studies are needed to investigate the behavior of carboxylate adducts at the interface and develop solutions to unlock their electrochemical utilization. Some of the transport challenges can also be addressed through the RCC electrolyzer design, aiming for high liquid-phase CO2-adduct species mass flux to the active catalyst by forced convection through highly porous foam cathodes to enable operation at simultaneously high current density and selectivity. The electrolyte stability, evolution in interfacial resistance, and the cross-over through the separator are critical parameters to examine further for practical RCC electrolyzers.
Returning back to the fundamental aspects, answers to several scientific questions remain incomplete: How do ILs and DESs alter the interfacial microenvironment? What are the critical elements of the microenvironment that enables the capture CO2 to be reduced at potentials and rates that are feasible for scalable RCC? How does the hydrogen bonding in the electrolyte affect the CO2 reduction energetics and mechanisms? How can the change in the interfacial pH in the presence of ILs and DESs be assessed? How do ILs/DESs influence the electric field on the electrode surface? The answers to these questions will help advance the state of functional electrolytes for RCC.
Author contributions
S. D. and B. G. created the outline of this review, wrote the original draft, and edited; B. G. managed the project. A. S. A., S. F. K. O., S. K., and A. U., contributed to Sections 2.1–2.3 (CO2 capture) and led Section 3 (thermal conversion). M. Z. wrote Section 2.5 (composites), contributed to Section 5 (sorbent properties), and oversaw the discussions and editing in Section 2 (CO2 capture). R. D. wrote Sections 2.4 and 2.5 and constructed tables (CO2 capture with DESs); O. K. C wrote Section 4.2.4 (thermodynamics & kinetics) and 4.2.5 (microenvironments); M. M. contributed to Section 5 (electrolyte properties) and created all of the original graphics. A. B., M. G. C. G. M.-G., and J. M. S. wrote Section 4.3 (electrolyzers). R. D. R. and C. H. contributed to Section 4.2.4 (Tafel slope and turn over frequencies); J. S. S. and J. Y. Y. wrote Section 4.4 (homogenous catalysts); R. S. B. and J. M. V. wrote Section 4.1 (electrodes) and 5.5 (electrode stability); R. L. S. contributed to editing of the manuscript. B. M. and B. K. wrote Section 4.5 (technoeconomic analysis) with contributions from J. M. S. All authors discussed, edited, and reviewed the manuscript before submission.List of abbreviations
| AFIL | 1-(3-Aminopropl)-3-methylimidazolium chloride |
| Ag NPs | Silver nanoparticles |
| Ag/Ag+ | Silver/silver nitrate reference electrode |
| AHA | Aprotic heterocyclic anions |
| AMP | 2-Amino-2-methyl-1-propanol |
| CCC | CO2 capture and concentration |
| CD | Current density |
| CDR | CO2 removal |
| *C2O42− | Adsorbed oxalate |
| *CO | Adsorbed CO |
| CO2˙− | CO2 anion radical |
| CO2RR | Electrolytic CO2 reduction reactions |
| CO32− | Carbonate anion |
| COFs | Covalent organic frameworks |
| CP | Carbon paper |
| CuO | Cupric oxide |
| DAC | Direct air capture |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DES | Deep eutectic solvents |
| DETA | Diethylenetriamine |
| DFT | Density functional theory |
| DMSO | Dimethyl sulphoxide |
| eCCC | Electrochemical CO2 capture and concentration |
| ECSA | Electrochemical active surface area |
| EDA | Electron donor–acceptor |
| EG | Ethylene glycol |
| Eim | 1-Ethylimidazole |
| ENILs | Encapsulated ionic liquids |
| E ve | Electrolysis potential/actual potential applied |
| E 0ve | Theoretical thermodynamic potential |
| Fc/Fc+ | Ferrocenium/ferrocene reference potential |
| FE | Faradaic efficiency |
| FTIR | Fourier-transform infrared spectroscopy |
| GCMC | Grand Canonical Monte Carlo |
| GDE | Gas diffusion electrode |
| HBA | Hydrogen bond acceptor |
| HBD | Hydrogen bond donor |
| HCO3− | Bicarbonate anion |
| HCOO− | Formate |
| HCOOH | Formic acid |
| HER | Hydrogen evaluation reaction |
| HNIW | 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane |
| HRMAS NMR | High resolution magic angle spinning nuclear magnetic resonance |
| ILPMs | Ionic liquid polymer membranes |
| ILs | Ionic liquids |
| KCl | Potassium chloride |
| KHCO3 | Potassium bicarbonate |
| KOH | Potassium hydroxide |
| LTTMs | Low-transition-temperature mixtures |
| MEA | Monoethanolamine |
| MMMs | Mixed matrix membranes |
| MOFs | Metal–organic frameworks |
| MoS2 | Molybdenum disulfide |
| NH3 | Ammonia |
| NH4HCO3 | Ammonium bicarbonate |
| NHE | Normal hydrogen electrode |
| NPCA | N,P-Co-doped carbon aerogel |
| OH− | Hydroxide ions |
| PEI | Poly(ethylenimine) |
| P-ILs | Protic ionic liquids |
| PMMA | Poly(methyl methacrylate) |
| PSA | Pressure-swing adsorption/absorption |
| RCC | Reactive capture and conversion |
| RCE | Rotating cylinder electrode |
| reline | DES formed by choline chloride and urea |
| rGA | Reduced graphene aerogel |
| RWGS | Rreverse water gas shift reaction |
| SFG | Sum frequency generation |
| SHE | Standard hydrogen electrode |
| SILMs | Supported ionic liquid membranes |
| SILP | Supported ionic liquid phase |
| SPS-IL | Switchable polarity ionic liquids |
| TEAP | Tetraethylammonium perchlorate |
| TFE | Trifluoroethanol |
| TNT | 2,4,6-Trinitrotoluene |
| TOF | Turnover frequencies |
| TSA | Temperature-swing adsorption/absorption |
| TS-IL | Task-specific ionic liquid |
| VFT | Vogel–Fulcher–Tammann |
| WE | Working electrode |
| ZIF | Zeolitic imidazolate frameworks |
| Zn–BTC | Zn-1,3,5-benzenetricarboxylic acid |
| [124-Trz]-CO2− | CO2 bind to 124-triazole anion to form carbamate |
| [123-Trz]− | 123-Triazolate |
| [2-CNpyr]− | 2-Cyanopyrrolide anion |
| [Ala]− | Alaninate |
| [AEMMIM][Tau] | 1-Aminoethyl-2, 3-dimethylimidazolium taurine |
| [aP4443][Ala] | (3-Aminopropyl)tributylphosphonium l-α-aminopropionic acid |
| [aP4443][Gly] | (3-Aminopropyl)tributylphosphonium aminoethanoic acid |
| [aP4443][Leu] | (3-Aminopropyl)tributylphosphonium l-α-amino-4-methylvaleric acid |
| [aP4443][Val] | (3-Aminopropyl)tributylphosphonium l-α-aminoisovaleric acid |
| [APBIM][BF4] | 1-Propylamide-3-butyl imidazolium tetrafluoroborate |
| [APMIM][Lys] | 1-Aminopropyl-3-methylimidazolium lysine |
| [BF4]− | Tetrafluoroborate |
| [BMIM][124-Trz] | 1-Butyl-3-methylimidazolium 1,2,4-triazolide |
| [BMIM][Ala] | 1-Butyl-3-methylimidazolium alaninate |
| [BMIM][BF4] | 1-n-Butyl-3-methylimidazolium tetrafluoroborate |
| [BMIM][CF3SO3] | 1-n-Butyl-3-methylimidazolium trifluoromethanesulfonate |
| [BMIM][Cl] | 1-n-Butyl-3-methylimidazolium chloride |
| [BMIM][DCA] | 1-n-Butyl-3-methylimidazolium dicyanamide |
| [BMIM][FeCl4] | 1-Butyl-3-methylimidazolium tetrachloroferrate |
| [BMIM][Gly] | 1-Butyl-3-methylimidazolium glycinate |
| [BMIM][HSO4] | 1-n-butyl-3-methylimidazolium hydrogen sulfate |
| [BMIM][MeSO3] | 1-n-Butyl-3-methylimidazolium methanesulfonate |
| [BMIM][NO3] | 1-n-Butyl-3-methylimidazolium nitrate |
| [BMIM][NTf2] | 1-Butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide |
| [BMIM][OAc] | 1-Butyl-3-methylimidazolium acetate |
| [BMIM][OcSO4] | 1-n-Butyl-3-methylimidazolium octyl sulfate |
| [BMIM][PF6] | 1-n-Butyl-3-methylimidazolium hexafluorophosphate |
| [BMIM][SbF6] | 1-n-Butyl-3-methylimidazolium hexafluoroantimonate |
| [BMIM][SCN] | 1-n-Butyl-3-methylimidazolium thiocyanate |
| [BMIM][Val] | 1-Butyl-3-methylimidazolium valinate |
| [BMIM]+ | 1-n-Butyl-3-methylimidazolium |
| [BMMIM][OAc] | 1,2-Dimethyl-3-butylimidazolium acetate |
| [BMMIM][PF6] | 1-n-Butyl-2,3-dimethylimidazolium hexafluorophosphate |
| [BMPyr][DCA] | 1-n-Butyl-1-methylpyrrolidinium dicyanamide |
| [BMPyr][PF6] | 1-n-Butyl-1-methylpyrrolidinium hexafluorophosphate |
| [C10MIM][NTf2] | 1-Decyl-3-methylimidazolium bis(trifluoromethyl)sulfonylimide |
| [C2OHMIM][Lys] | 1-Hydroxyethyl-3-methylimidazolium lysine |
| [C2OHMIM][NTf2] | 1-(2-Hydroxyethyl)-3-methylimidazolium bis(trifluoromethyl)sulfonylimide |
| [C6MIM][C(CN)3] | 1-Hexyl-3-methylimidazolium tricyanomethanide |
| [C6MIM][Cl] | 1-Hexyl-3-methylimidazolium chloride |
| [C6MIM][DCA] | 1-Hexyl-3-methylimidazolium tricyanomethanide |
| [C8MIM][NfO] | 1-Octyl-3-methylimidazolim perfluorobutanesulfonate |
| [C8MIM][NTf2] | 1-Octyl-3-methylimidazolium bistrifluoromethanesulfonylimide |
| [Car]− | Carvacrol |
| [Ch]+ | Choline |
| [Ch][Cl] | Choline chloride |
| [Cl]− | Chloride |
| [DAMT][BF4] | 3,5-Diamino-1-methyl-1,2,4-triazolium tetrafluoroborate |
| [DBUH][4-F-PhO] | IL Formed by 1,8-diazabicyclo[5.4.0]undecane-7-ene (DBU) and 4-fluorophenol (4-F-PhOH) |
| [DCA]− | Dicyanamide |
| [DETA][OAc] | Diethylenetriamine acetate |
| [DMIM][BF4] | 1-Decyl-3-methylimidazolium tetrafluoroborate |
| [Eaca]− | 6-Aminocaproicate |
| [EMIM][2-CNpyr] | 1-Ethyl-3-methylimidazolium 2-cyanopyrrolide |
| [EMIM][BF4] | 1-Ethyl-3-methylimidazolium tetrafluoroborate |
| [EMIM][Cl] | 1-Ethyl-3-methylimidazolium chloride |
| [EMIM][DEP] | 1-Ethyl-3-methylimidazolium diethyl phosphate |
| [EMIM][NTf2] | 1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide |
| [EMIM][OAc] | 1-Ethyl-3-methylimidazolium acetate |
| [EMIM][TfO] | 1-Ethyl-3-methylimidazolium trifluoromethanesulfonate |
| [EMIM]+ | 1-Ethyl-3-methylimidazolium |
| [EMIM]+-CO2− | 1-Ethyl-3-methylimidazolium carboxylate |
| [Gaba]− | γ-Aminobutyricate |
| [Gly]− | Glycinate |
| [HDBU]+ | 1,8-Diazabicyclo[5,4,0]undec-7-ene imidazole |
| [HEMIM][DCA] | 1-(2-Hydroxyethyl)-3-methylimidazolium dicyanamide |
| [HeMOEim][Br] | 1-(2-Hydroxyl-ethyl)-3-methoxyethyl-imidazolium bromide |
| [His]− | Histidine |
| [In]− | Indole |
| [Im]− | Imidazole |
| [Maba]− | n-Methyl-4-aminobutyricate |
| [MEAH]+ | Monoethanolamine |
| [MEAH][Cl]:MDEA | DES formed with monoethanolamine hydrochloride ([meah][cl]) and methyldiethanolamine (mdea) |
| [MM][ NTf2] | 1,3-Dimethylimidazolium |
| [MPPyr][DCA] | 1-Methyl-1-propyl pyrrolidinium dicyanamide |
| [N1111]− | Tetramethylammonium anion |
| [N2222][Ala] | Tetraethylammonium α-alaninate |
| [N2222][4-PyO] | Tetraethylamminonium 4-hydroxypyridine |
| [N2222][CH(CN)2] | Tetraethylamminonium malononitrile |
| [N2222][Gly] | Tetraethylammonium glycinate |
| [N2222][β-Ala] | Tetraethylammonium β-alaninate |
| [N2224][Ala] | Triethylbutylammonium α-alaninate |
| [NAIm][Br] | 1-(3-Aminopropyl)-3-(2-bromoethyl)imidazole |
| [NTf2]− | Bis(trifluoromethylsulfonyl) imide |
| [NTf2]− | Bis(trifluoromethylsulfonyl) imide |
| [OAc]− | Acetate |
| [Oxa]− | Oxazole |
| [P1444][NTf2] | Tributylmethyl-phosphonium bis(trifuoromethylsulfonyl)imid |
| [P2222][BnIm] | Triethylphosphonium benzimidazolate |
| [P2228][2-CNPyr] | Triethyloctylphosphonium 2-cyanopyrrolide |
| [P4442][DAA] | Tributylethylphosphonium diacetamide |
| [P4442][Suc] | Tributylethylphosphonium succinimide |
| [P4444][2-Op] | Tetrabutylphosphonium 2-hydroxylpyridium |
| [P4444][4-MF-PhO] | Tetrabutylphosphonium 4-(methoxycarbonyl) phenol |
| [P4444][Ala] | Tetrabutylphosphonium α-alaninate |
| [P4444][Gly] | Ttetrabutylphosphonium glycinate |
| [P4444][HCO2] | Tetrabutylphosphonium methanoate |
| [P4444][Lys] | Tetrabutylphosphonium lysinate |
| [P4444][OAc] | Tetrabutylphosphonium acetate |
| [P4444][Pro] | Tetrabutylphosphonium prolinate |
| [P4444][Ser] | Tetrabutylphosphonium serinate |
| [P4444][β-Ala] | Tetrabutylphosphonium β-alaninate |
| [P4444]+ | Tetrabutylphosphonium |
| [P66614][123-Trz] | Trihetyl(tetradecyl)phosphonium 1,2,3-triazolide |
| [P66614][124-Trz] | Trihexyltetradecylphosphonium 1,2,4-triazolide |
| [P66614][134-Trz] | Trihexy(tetradecyl)phosphonium 1,3,4-trizolate |
| [P66614][2-CNpyr] | Trihetyl(tetradecyl)phosphonium 2-cyanopyrrolide |
| [P66614][2-Op] | Trihetyl(tetradecyl)phosphonium 2-hydroxylpyridium |
| [P66614][2-SCH3BnIm] | Trihetyl(tetradecyl)phosphonium 2-methlythio-benzimidazolide |
| [P66614][3-CF3Pyra] | Trihetyl(tetradecyl)phosphonium 3-trifluoromethyl-pyrazolide |
| [P66614][3-CH3-5-CF3Pyra] | Trihetyl(tetradecyl)phosphonium 3-methly-5-trifluoromethyl-pyrazolide |
| [P66614][3-Cl-PhO] | Trihexyl(tetradecyl)phosphonium o-chlorophenolate |
| [P66614][3-Op] | Trihetyl(tetradecyl)phosphonium 3-hydroxylpyridium |
| [P66614][4-MeO-PhO] | Trihexyl(tetradecyl)phosphonium p-methoxyphenolate |
| [P66614][4-NO2-PhO] | Trihexyl(tetradecyl)phosphonium p-nitrophenolate |
| [P66614][4-Op] | Trihetyl(tetradecyl)phosphonium 4-hydroxylpyridium |
| [P66614][6-BrBnIm] | Trihetyl(tetradecyl)phosphonium 6-bromo-benzimidazolide |
| [P66614][Ala] | Trihetyl(tetradecyl)phosphonium α-alaninate |
| [P66614][BnIm] | Trihetyl(tetradecyl)phosphonium benzimidazolide |
| [P66614][Gly] | Trihetyl(tetradecyl)phosphonium glycinate |
| [P66614][Im] | Trihexyl(tetradecyl)phosphonium imidazole |
| [P66614][Inda] | Trihetyl(tetradecyl)phosphonium indazolide |
| [P66614][Iso] | Trihexyl(tetradecyl)-phosphonium isoleucinate |
| [P66614][Met] | Trihetyl(tetradecyl)phosphonium methioninate |
| [P66614][MN] | Trihetyl(tetradecyl)phosphonium malononitrile |
| [P66614][NTf2] | Trihexyl(tetradecyl)phosphonium bis(trifluoromethyl)sulfonylimide |
| [P66614][OAc] | Trihetyl(tetradecyl)phosphonium acetate |
| [P66614][Pro] | Trihetyl(tetradecyl)phosphonium prolinate |
| [P66614][Pyr] | Trihexyl(tetradecyl)phosphonium pyrazole |
| [P66614][Tetz] | Trihexyl(tetradecyl)phosphonium tetrazole |
| [PF6]− | Hexafluorophosphate |
| [Phe]− | Phenylalaninate |
| [PhO]− | Phenolate |
| [Pro]− | Prolinate |
| [PMIM][NTf2] | 1-Propyl-3-methylimidazoliumbis(trifluoro-methylsulfonyl)imide |
| [Tea][Tau] | Tetra-ethylalkylammonium taurinate |
| [TEPA][NO3] | Tetraethylenepentammonium nitrate |
| [TETA][NO3] | Triethylenetetrammonium nitrate |
| [TfO]− | Trifluoromethylsulfonate anion |
| [Thy]− | Thymol |
| [TMG][Im] | 1,1,3,3-Tetramethylguanidinium imidazole |
| [Vbtma][gly] | Dimethylethylenediammonium propanoate |
| Fe(TPP)Cl | Iron tetraphenylporphyrin chloride |
| fac-Re(bpy)(CO)3Cl | Dicationic Re bipyridine-type complex, fac-Re(6,6′-(2-((trimethylammonio)-methyl)phenyl)-2,2′-bipyridine) (CO)3Cl hexafluorophosphate (12+) |
| Ni(cyclam)Cl2 | Ni-Cyclam complex |
| Rh(bpy)(Cp*)Cl | [Rh(bpy)(Cp*)Cl]Cl complex where bpy = 2,2′-bipyridine and Cp* = pentamethylcyclopentadienyl |
| Zn(TPP)Cl | Zinc tetraphenylporphyrin chloride |
| Co(cyclam)Cl3 | Co-Cyclam complex |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Center for Closing the Carbon Cycle, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under Award Number DE-SC0023427 (electrochemical conversion; S. D., A. B., M. G., R. D. R., J. S., R. S. B., J. M. V., J. Y. Y., C. H., C. G. M.-G., J. M. S., B. G.), Basic Energy Sciences, award # DE-SC0022214 (sorbents for CO2 capture; M. Z., R. D., B. G.), Breakthrough Electrolytes for Energy Storage (BEES2) under award # DE-SC0019409 (electrolyte properties; M. M., B. G.), and National Science Foundation Career award # 2045111 from the Division of Chemical, Bioengineering, Environmental and Transport Systems (interfacial analysis of ionic liquid electrolytes; O. K. C., B. G.). A. U. thanks the Fulbright Türkiye's Visiting Scholar Program, the Koç University Visiting Scholar Program, and the Scientific and Technological Research Council of Türkiye (TUBITAK) 2219 Program. B. K.'s efforts were supported by the Department of Energy, National Nuclear Security Administration (NNSA) grants (DE-NA0004112 and DE-NA0004007).References
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- W. Wang, S. P. Wang, X. B. Ma and J. L. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- National Academies of Sciences Engineering and Medicine (U.S.). Committee on a Research Agenda for a New Era in Separation Science, A research agenda for transforming separation science, 2019.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- Y. S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed.
- Z.-Z. Yang, Y.-N. Zhao and L.-N. He, RSC Adv., 2011, 1, 545–567 RSC.
- C. Finn, S. Schnittger, L. J. Yellowlees and J. B. Love, Chem. Commun., 2012, 48, 1392–1399 RSC.
- H. R. Jhong, S. C. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- A. Calbry-Muzyka, H. Madi, F. Rüsch-Pfund, M. Gandiglio and S. Biollaz, Renewable Energy, 2022, 181, 1000–1007 CrossRef CAS.
- R. W. Baker, B. Freeman, J. Kniep, Y. I. Huang and T. C. Merkel, Ind. Eng. Chem. Res., 2018, 57, 15963–15970 CrossRef CAS.
- D. O. E. DOE/NETL Advanced Carbon Dioxide Capture R&D Program: Technology Update, NETL, Eds., Department of Energy: Morgantown, WV, 2010.
- X. Y. Shi, H. Xiao, H. Azarabadi, J. Z. Song, X. L. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed.
- F. Zeman, Environ. Sci. Technol., 2007, 41, 7558–7563 CrossRef CAS PubMed.
- R. Baciocchi, G. Storti and M. Mazzotti, Chem. Eng. Process., 2006, 45, 1047–1058 CrossRef CAS.
- S. Chatterjee and K.-W. Huang, Nat. Commun., 2020, 11, 3287 CrossRef CAS PubMed.
- G. Realmonte, L. Drouet, A. Gambhir, J. Glynn, A. Hawkes, A. C. Köberle and M. Tavoni, Nat. Commun., 2019, 10, 3277 CrossRef CAS PubMed.
- M. C. Freyman, Z. Huang, D. Ravikumar, E. B. Duoss, Y. Li, S. E. Baker, S. H. Pang and J. A. Schaidle, Joule, 2023, 7, 631–651 CrossRef CAS.
- D. J. Heldebrant, J. Kothandaraman, N. M. Dowell and L. Brickett, Chem. Sci., 2022, 13, 6445–6456 RSC.
- A. M. Zito, L. E. Clarke, J. M. Barlow, D. Bím, Z. Zhang, K. M. Ripley, C. J. Li, A. Kummeth, M. E. Leonard, A. N. Alexandrova, F. R. Brushett and J. Y. Yang, Chem. Rev., 2023, 123, 8069–8098 CrossRef CAS PubMed.
- J. S. Wilkes, Green Chem., 2002, 4, 73–80 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- R. Sheldon, Chem. Commun., 2001, 2399–2407, 10.1039/b107270f.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- K. S. Egorova, E. G. Gordeev and V. P. Ananikov, Chem. Rev., 2017, 117, 7132–7189 CrossRef CAS PubMed.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- G. García, S. Aparicio, R. Ullah and M. Atilhan, Energy Fuels, 2015, 29, 2616–2644 CrossRef.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef CAS.
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- R. Ghahremani, R. F. Savinell and B. Gurkan, J. Electrochem. Soc., 2022, 169, 030520 CrossRef CAS.
- X. Li, S. Wang and C. Chen, Energy Procedia, 2013, 37, 1836–1843 CrossRef CAS.
- R.-T. Guo, G.-Y. Li, Y. Liu and W.-G. Pan, Energy Fuels, 2023, 37, 15429–15452 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- S. Mahajan and M. Lahtinen, J. Environ. Chem. Eng., 2022, 10, 108930 CrossRef CAS.
- D. G. Boer, J. Langerak and P. P. Pescarmona, ACS Appl. Mater. Interfaces, 2023, 6, 2634–2656 CAS.
- A. E. Creamer and B. Gao, Environ. Sci. Technol., 2016, 50, 7276–7289 CrossRef CAS PubMed.
- M. Pardakhti, T. Jafari, Z. Tobin, B. Dutta, E. Moharreri, N. S. Shemshaki, S. Suib and R. Srivastava, ACS Appl. Mater. Interfaces, 2019, 11, 34533–34559 CrossRef CAS PubMed.
- K. S. Lackner, Eur. Phys. J.: Spec. Top., 2009, 176, 93–106 Search PubMed.
- T. Wang, K. S. Lackner and A. Wright, Environ. Sci. Technol., 2011, 45, 6670–6675 CrossRef CAS PubMed.
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed.
- M. C. Stern and T. A. Hatton, RSC Adv., 2014, 4, 5906–5914 RSC.
- S. Voskian and T. A. Hatton, Energy Environ. Sci., 2019, 12, 3530–3547 RSC.
- M. Rahimi, G. Catalini, S. Hariharan, M. Wang, M. Puccini and T. A. Hatton, Cell Rep. Phys. Sci., 2020, 1, 100033 CrossRef.
- S. Tsubaki, K. Furusawa, H. Yamada, T. Kato, T. Higashii, S. Fujii and Y. Wada, ACS Sustainable Chem. Eng., 2020, 8, 13593–13599 CrossRef CAS.
- J. Yang, H. Y. Tan, Q. X. Low, B. P. Binks and J. M. Chin, J. Mater. Chem. A, 2015, 3, 6440–6446 RSC.
- Y.-Y. Lee, E. Cagli, A. Klemm, Y. Park, R. Dikki, M. K. Kidder and B. Gurkan, ChemSusChem, 2023, 16, e202300118 CrossRef CAS PubMed.
- S.-M. Kim, K.-M. Kim, B.-K. Choi, J.-H. Mun, B.-J. Shin, U. Lee, C.-H. Shin, J. Choi, B.-M. Min and U. Lee, Chem. Eng. J., 2022, 428, 132044 CrossRef CAS.
- S. Chen, X. Han, X. Sun, X. Luo and Z. Liang, Chem. Eng. J., 2020, 386, 121295 CrossRef CAS.
- R. Zhang, X. He, T. Liu, C. E. Li, M. Xiao, H. Ling, X. Hu, X. Zhang, F. Tang and H. A. Luo, Sep. Purif. Technol., 2022, 295, 121292 CrossRef CAS.
- M. Xiao, D. Cui, Q. Yang, Z. Liang, G. Puxty, H. Yu, L. Li, W. Conway and P. Feron, Chem. Eng. Sci., 2021, 229, 116009 CrossRef CAS.
- N. E. L. Hadri, D. V. Quang and M. R. M. Abu-Zahra, Energy Proc., 2015, 75, 2268–2286 CrossRef CAS.
- A. C. Forse and P. J. Milner, Chem. Sci., 2021, 12, 508–516 RSC.
- J. Xiang, D. Wei, W. Mao, T. Liu, Q. Luo, Y. Huang, Z. Liang and X. Luo, Sep. Purif. Technol., 2024, 330, 125310 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- G. F. Versteeg, L. A. J. Van Dijck and W. P. M. Van Swaaij, Chem. Eng. Commun., 1996, 144, 113–158 CrossRef CAS.
- C. H. Yu, C. H. Huang and C. S. Tan, Aerosol Air Qual. Res., 2012, 12, 745–769 CrossRef CAS.
- P. Singh, D. W. F. Brilman and M. J. Groeneveld, Int. J. Greenhouse Gas Control, 2011, 5, 61–68 CrossRef CAS.
- N. El Hadri, D. V. Quang, E. L. V. Goetheer and M. R. M. Abu Zahra, Appl. Energy, 2017, 185, 1433–1449 CrossRef CAS.
- I. Kim and H. F. Svendsen, Ind. Eng. Chem. Res., 2007, 46, 5803–5809 CrossRef CAS.
- T. Nguyen, M. Hilliard and G. T. Rochelle, Int. J. Greenhouse Gas Control, 2010, 4, 707–715 CrossRef CAS.
- E. Pérez-Gallent, C. Vankani, C. Sánchez-Martínez, A. Anastasopol and E. Goetheer, Ind. Eng. Chem. Res., 2021, 60, 4269–4278 CrossRef.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- M. Abdinejad, Z. Mirza, X.-A. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 1715–1720 CrossRef CAS.
- M. Li, K. Yang, M. Abdinejad, C. Zhao and T. Burdyny, Nanoscale, 2022, 14, 11892–11908 RSC.
- L. Chen, F. Li, Y. Zhang, C. L. Bentley, M. Horne, A. M. Bond and J. Zhang, ChemSusChem, 2017, 10, 4109–4118 CrossRef CAS PubMed.
- D. Filotás, T. Nagy, L. Nagy, P. Mizsey and G. Nagy, Electroanalysis, 2018, 30, 690–697 CrossRef.
- J. C. S. Choi, A. Banerjee, R. L. Sacci, G. M. Veith and C. Stieber, et al., ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-5xsp5 This content is a preprint and has not been peer-reviewed.
- Y. Wu, J. Xu, K. Mumford, G. W. Stevens, W. Fei and Y. Wang, Green Chem. Eng., 2020, 1, 16–32 CrossRef.
- B. E. Gurkan, J. C. de la Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. A. Price, W. F. Schneider and J. F. Brennecke, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS PubMed.
- J.-F. Pérez-Calvo, D. Sutter, M. Gazzani and M. Mazzotti, Sep. Purif. Technol., 2021, 274, 118959 CrossRef.
- F. O. Ochedi, J. Yu, H. Yu, Y. Liu and A. Hussain, Environ. Chem. Lett., 2021, 19, 77–109 CrossRef CAS.
- F. Wang, J. Zhao, H. Miao, J. Zhao, H. Zhang, J. Yuan and J. Yan, Appl. Energy, 2018, 230, 734–749 CrossRef CAS.
- K. Jiao, X. Chen, X. Bie, D. Liu, M. Qiu and S. Ma, Sci. Rep., 2021, 11, 10457 CrossRef CAS PubMed.
- D. Bonalumi and A. Giuffrida, Energy, 2016, 117, 439–449 CrossRef CAS.
- H. Liu, Y. Chen, J. Lee, S. Gu and W. Li, ACS Energy Lett., 2022, 7, 4483–4489 CrossRef CAS.
- H. Li, J. Gao, Q. Du, J. Shan, Y. Zhang, S. Wu and Z. Wang, Energy, 2021, 216, 119250 CrossRef CAS.
- J. Li and N. Kornienko, Chem. Sci., 2022, 13, 3957–3964 RSC.
- U. K. Ghosh, S. E. Kentish and G. W. Stevens, Energy Proc., 2009, 1, 1075–1081 CrossRef CAS.
- R. Ramezani and R. Di Felice, Green Energy Environ., 2021, 6, 83–90 CrossRef CAS.
- V. Sang Sefidi and P. Luis, Ind. Eng. Chem. Res., 2019, 58, 20181–20194 CrossRef CAS.
- U. E. Aronu, A. F. Ciftja, I. Kim and A. Hartono, Energy Proc., 2013, 37, 233–240 CrossRef CAS.
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. X. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 CrossRef CAS.
- M. Ramdin, T. W. de Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS.
- B. Gurkan, F. Simeon and T. A. Hatton, ACS Sustainable Chem. Eng., 2015, 3, 1394–1405 CrossRef CAS.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- B. F. Goodrich, J. C. De La Fuente, B. E. Gurkan, D. J. Zadigian, E. A. Price, Y. Huang and J. F. Brennecke, Ind. Eng. Chem. Res., 2011, 50, 111–118 CrossRef CAS.
- H. Ohno and K. Fukumoto, Acc. Chem. Res., 2007, 40, 1122–1129 CrossRef CAS PubMed.
- C. Wang, X. Luo, H. Luo, D.-E. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 4918–4922 CrossRef CAS PubMed.
- K. E. Gutowski and E. J. Maginn, J. Am. Chem. Soc., 2008, 130, 14690–14704 CrossRef CAS PubMed.
- X. Zhang, X. Zhang, H. Dong, Z. Zhao, S. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668–6681 RSC.
- P. Brown, B. E. Gurkan and T. A. Hatton, AIChE J., 2015, 61, 2280–2285 CrossRef CAS.
- X. Zhu, M. Song and Y. Xu, ACS Sustainable Chem. Eng., 2017, 5, 8192–8198 CrossRef CAS.
- N. Noorani and A. Mehrdad, J. Mol. Liq., 2022, 357, 119078 CrossRef CAS.
- R. Giernoth, Angew. Chem., Int. Ed., 2010, 49, 2834–2839 CrossRef CAS PubMed.
- Z. M. Memon, E. Yilmaz and M. Soylak, J. Mol. Liq., 2017, 229, 459–464 CrossRef CAS.
- A. Brzęczek-Szafran, P. Więcek, M. Guzik and A. Chrobok, RSC Adv., 2020, 10, 18355–18359 RSC.
- W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC.
- Q. Zhang and J. n M. Shreeve, Chem. Rev., 2014, 114, 10527–10574 CrossRef CAS PubMed.
- S. K. Singh and A. W. Savoy, J. Mol. Liq., 2020, 297, 112038 CrossRef CAS.
- S. K. Singh and P. L. Dhepe, Green Chem., 2016, 18, 4098–4108 RSC.
- I. J. Lin and C. S. Vasam, J. Organomet. Chem., 2005, 690, 3498–3512 CrossRef CAS.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- Y. Wang, C. Zheng, Y. Wang, H. Chen and Y. Xu, J. Environ. Chem. Eng., 2019, 7, 102774 CrossRef CAS.
- S. Kang, Y. G. Chung, J. H. Kang and H. Song, J. Mol. Liq., 2020, 297, 111825 CrossRef CAS.
- X. Luo, Y. Guo, F. Ding, H. Zhao, G. Cui, H. Li and C. Wang, Angew. Chem., Int. Ed., 2014, 126, 7173–7177 CrossRef.
- X. Gao, X. Liu, A. Mariani, G. A. Elia, M. Lechner, C. Streb and S. Passerini, Energy Environ. Sci., 2020, 13, 2559–2569 RSC.
- G. Shi, R. Zhai, H. Li and C. Wang, Green Chem., 2021, 23, 592–596 RSC.
- Q. R. Sheridan, W. F. Schneider and E. J. Maginn, Chem. Rev., 2018, 118, 5242–5260 CrossRef CAS PubMed.
- R. Zhang, Q. Ke, Z. Zhang, B. Zhou, G. Cui and H. Lu, Int. J. Mol. Sci., 2022, 23, 11401 CrossRef CAS PubMed.
- N. Noorani and A. Mehrdad, Fluid Phase Equilib., 2020, 517, 112591 CrossRef CAS.
- Y.-Y. Jiang, G.-N. Wang, Z. Zhou, Y.-T. Wu, J. Geng and Z.-B. Zhang, Chem. Commun., 2008, 505–507 RSC.
- H. Yu, Y.-T. Wu, Y.-Y. Jiang, Z. Zhou and Z.-B. Zhang, New J. Chem., 2009, 33, 2385–2390 RSC.
- M. B. Shiflett, D. J. Kasprzak, C. P. Junk and A. Yokozeki, J. Chem. Thermodyn., 2008, 40, 25–31 CrossRef CAS.
- M. B. Shiflett, B. A. Elliott, S. R. Lustig, S. Sabesan, M. S. Kelkar and A. Yokozeki, ChemPhysChem, 2012, 13, 1806–1817 CrossRef CAS PubMed.
- E. J. Maginn, Design and evaluation of ionic liquids as novel CO2 absorbents, University of Notre Dame (US), 2004.
- G. Gurau, H. Rodríguez, S. P. Kelley, P. Janiczek, R. S. Kalb and R. D. Rogers, Angew. Chem., Int. Ed., 2011, 50, 12024–12026 CrossRef CAS PubMed.
- P. J. Carvalho and J. A. Coutinho, J. Phys. Chem. Lett., 2010, 1, 774–780 CrossRef CAS.
- P. J. Carvalho, V. H. Álvarez, B. Schröder, A. M. Gil, I. M. Marrucho, M. Aznar, L. M. Santos and J. A. Coutinho, J. Phys. Chem. B, 2009, 113, 6803–6812 CrossRef CAS PubMed.
- P. Raveendran and S. L. Wallen, J. Am. Chem. Soc., 2002, 124, 12590–12599 CrossRef PubMed.
- M. Mei, X. Hu, Z. Song, L. Chen, L. Deng and Z. Qi, J. Mol. Liq., 2022, 348, 118036 CrossRef CAS.
- B. Gurkan, B. Goodrich, E. Mindrup, L. Ficke, M. Massel, S. Seo, T. Senftle, H. Wu, M. Glaser and J. Shah, J. Phys. Chem. Lett., 2010, 1, 3494–3499 CrossRef CAS.
- G. Cui, N. Zhao, J. Wang and C. Wang, Chem. – Asian J., 2017, 12, 2863–2872 CrossRef CAS PubMed.
- X. Lei, Y. Xu, L. Zhu and X. Wang, RSC Adv., 2014, 4, 7052–7057 RSC.
- F. Li, Y. Bai, S. Zeng, X. Liang, H. Wang, F. Huo and X. Zhang, Int. J. Greenhouse Gas Control, 2019, 90, 102801 CrossRef CAS.
- X. Zhang, W. Xiong, L. Peng, Y. Wu and X. Hu, AIChE J., 2020, 66, e16936 CrossRef CAS.
- X. Zhu, M. Song, B. Ling, S. Wang and X. Luo, J. Solution Chem., 2020, 49, 257–271 CrossRef CAS.
- S. Oh, O. Morales-Collazo and J. F. Brennecke, J. Phys. Chem. B, 2019, 123, 8274–8284 CrossRef CAS PubMed.
- J. Hu, L. Chen, M. Shi and C. Zhang, Fluid Phase Equilib., 2018, 459, 208–218 CrossRef CAS.
- K. Mei, X. He, K. Chen, X. Zhou, H. Li and C. Wang, Ind. Eng. Chem. Res., 2017, 56, 8066–8072 CrossRef CAS.
- T. R. Gohndrone, T. Song, M. A. DeSilva and J. F. Brennecke, J. Phys. Chem. B, 2021, 125, 6649–6657 CrossRef CAS PubMed.
- G. M. Avelar Bonilla, O. Morales-Collazo and J. F. Brennecke, ACS Sustainable Chem. Eng., 2019, 7, 16858–16869 CrossRef CAS.
- D. V. Quang, A. Dindi, A. V. Rayer, N. E. Hadri, A. Abdulkadir and M. R. M. Abu-Zahra, Greenhouse Gases: Sci. Technol., 2015, 5, 91–101 CrossRef CAS.
- S. Seo, M. Quiroz-Guzman, M. A. DeSilva, T. B. Lee, Y. Huang, B. F. Goodrich, W. F. Schneider and J. F. Brennecke, J. Phys. Chem. B, 2014, 118, 5740–5751 CrossRef CAS PubMed.
- S. Oh, O. Morales-Collazo, A. N. Keller and J. F. Brennecke, J. Phys. Chem. B, 2020, 124, 8877–8887 CrossRef CAS PubMed.
- V. V. Chaban and N. A. Andreeva, J. Ionic Liq., 2023, 3, 100068 CrossRef.
- D. W. Keith, G. Holmes, D. S. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- A. Sodiq, Y. Abdullatif, B. Aissa, A. Ostovar, N. Nassar, M. El-Naas and A. Amhamed, Environ. Technol. Innovation, 2023, 29, 102991 CrossRef CAS.
- C. Wang, H. Luo, H. Li, X. Zhu, B. Yu and S. Dai, Chem. – Eur. J., 2012, 18, 2153–2160 CrossRef CAS PubMed.
- F. Ding, X. He, X. Luo, W. Lin, K. Chen, H. Li and C. Wang, Chem. Commun., 2014, 50, 15041–15044 RSC.
- X.-M. Zhang, K. Huang, S. Xia, Y.-L. Chen, Y.-T. Wu and X.-B. Hu, Chem. Eng. J., 2015, 274, 30–38 CrossRef CAS.
- T. Zhao, X. Zhang, Z. Tu, Y. Wu and X. Hu, J. Mol. Liq., 2018, 268, 617–624 CrossRef CAS.
- Y. Huang, G. Cui, Y. Zhao, H. Wang, Z. Li, S. Dai and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 13293–13297 CrossRef CAS PubMed.
- X. An, X. Du, D. Duan, L. Shi, X. Hao, H. Lu, G. Guan and C. Peng, Phys. Chem. Chem. Phys., 2017, 19, 1134–1142 RSC.
- A. Klemm, S. P. Vicchio, S. Bhattacharjee, E. Cagli, Y. Park, M. Zeeshan, R. Dikki, H. Liu, M. K. Kidder, R. B. Getman and B. Gurkan, ACS Sustainable Chem. Eng., 2023, 11, 3740–3749 CrossRef CAS.
- H. Yan, L. Zhao, Y. Bai, F. Li, H. Dong, H. Wang, X. Zhang and S. Zeng, ACS Sustainable Chem. Eng., 2020, 8, 2523–2530 CrossRef CAS.
- W. Xiong, M. Shi, L. Peng, X. Zhang, X. Hu and Y. Wu, Sep. Purif. Technol., 2021, 263, 118417 CrossRef CAS.
- F.-F. Chen, K. Huang, J.-P. Fan and D.-J. Tao, AIChE J., 2018, 64, 632–639 CrossRef CAS.
- L. Qiu, Y. Fu, Z. Yang, A. C. Johnson, C.-L. Do-Thanh, B. P. Thapaliya, S. M. Mahurin, L.-N. He, D.-E. Jiang and S. Dai, ChemSusChem, 2024, 17, e202301329 CrossRef CAS PubMed.
- S. Wen, T. Wang, X. Zhang, W. Xu, X. Hu and Y. Wu, AIChE J., 2023, 69, e18206 CrossRef CAS.
- X. Suo, Y. Fu, C.-L. Do-Thanh, L.-Q. Qiu, D.-E. Jiang, S. M. Mahurin, Z. Yang and S. Dai, J. Am. Chem. Soc., 2022, 144, 21658–21663 CrossRef CAS PubMed.
- R. B. Leron and M.-H. Li, J. Chem. Thermodyn., 2013, 57, 131–136 CrossRef CAS.
- R. B. Leron and M.-H. Li, Thermochim. Acta, 2013, 551, 14–19 CrossRef CAS.
- V. Alizadeh, L. Esser and B. Kirchner, J. Chem. Phys., 2021, 154, 094503 CrossRef CAS PubMed.
- G. Cui, M. Lv and D. Yang, Chem. Commun., 2019, 55, 1426–1429 RSC.
- Y.-Y. Lee, D. Penley, A. Klemm, W. Dean and B. Gurkan, ACS Sustainable Chem. Eng., 2021, 9, 1090–1098 CrossRef CAS.
- J. Ruan, X. Ye, R. Wang, L. Chen, L. Deng and Z. Qi, Fuel, 2023, 334, 126709 CrossRef CAS.
- Bhawna, A. Pandey and S. Pandey, ChemistrySelect, 2017, 2, 11422–11430 CrossRef CAS.
- Z. Wang, C. Wu, Z. Wang, S. Zhang and D. Yang, Chem. Commun., 2022, 58, 7376–7379 RSC.
- Z. Wang, Z. Wang, J. Chen, C. Wu and D. Yang, Molecules, 2021, 26, 7167 CrossRef CAS PubMed.
- R. Dikki, E. Cagli, D. Penley, M. Karayilan and B. Gurkan, Chem. Commun., 2023, 59, 12027–12030 RSC.
- R. Dikki, V. Khokhar, M. Zeeshan, S. Bhattacharjee, O. K. Coskun, R. Getman and B. Gurkan, Green Chem., 2024, 26, 3441 RSC.
- Y. Gu, Y. Hou, S. Ren, Y. Sun and W. Wu, ACS Omega, 2020, 5, 6809–6816 CrossRef CAS PubMed.
- X. Liu, Q. Ao, S. Shi and S. Li, Mater. Res. Express, 2022, 9, 015504 CrossRef CAS.
- Z. Wang, Z. Wang, X. Huang, D. Yang, C. Wu and J. Chen, Chem. Commun., 2022, 58, 2160–2163 RSC.
- N. Ahmad, X. Lin, X. Wang, J. Xu and X. Xu, Fuel, 2021, 293, 120466 CrossRef CAS.
- Z. Wang, M. Chen, B. Lu, S. Zhang and D. Yang, ACS Sustainable Chem. Eng., 2023, 11, 6272–6279 CrossRef CAS.
- G. Cui, Y. Xu, D. Hu, Y. Zhou, C. Ge, H. Liu, W. Fan, Z. Zhang, B. Chen, Q. Ke, Y. Chen, B. Zhou, W. Zhang, R. Zhang and H. Lu, Chem. Eng. J., 2023, 469, 143991 CrossRef CAS.
- M. Chen, W. Xiong, W. Chen, S. Li, F. Zhang and Y. Wu, AIChE J., 2024, 70, e18319 CrossRef CAS.
- F. F. Chen, K. Huang, Y. Zhou, Z. Q. Tian, X. Zhu, D. J. Tao, D. E. Jiang and S. Dai, Angew. Chem., Int. Ed., 2016, 55, 7166–7170 CrossRef CAS PubMed.
- S. Zheng, S. Zeng, Y. Li, L. Bai, Y. Bai, X. Zhang, X. Liang and S. Zhang, AIChE J., 2021, 68, e17500 CrossRef.
- T. Zhou, C. Gui, L. Sun, Y. Hu, H. Lyu, Z. Wang, Z. Song and G. Yu, Chem. Rev., 2023, 123, 12170–12253 CrossRef CAS PubMed.
- B. Koyuturk, C. Altintas, F. P. Kinik, S. Keskin and A. Uzun, J. Phys. Chem. C, 2017, 121, 10370–10381 CrossRef CAS.
- V. Nozari, S. Keskin and A. Uzun, ACS Omega, 2017, 2, 6613–6618 CrossRef CAS PubMed.
- I. Cota and F. F. Martinez, Coord. Chem. Rev., 2017, 351, 189–204 CrossRef CAS.
- X. Li, K. Chen, R. Guo and Z. Wei, Chem. Rev., 2023, 123, 10432–10467 CrossRef CAS PubMed.
- A. H. Ab Rahim, N. M. Yunus and M. A. Bustam, Molecules, 2023, 28, 7091 CrossRef CAS PubMed.
- Y. Zhang, M. X. Wu, G. Zhou, X. H. Wang and X. Liu, Adv. Funct. Mater., 2021, 31, 2104996 CrossRef CAS.
- M. Yin, L. Wang and S. Tang, ACS Appl. Mater. Interfaces, 2022, 14, 55674–55685 CrossRef CAS PubMed.
- H. P. Caglayan, U. Unal, S. Keskin and A. Uzun, ACS Appl. Nano Mater., 2023, 6, 2203–2217 CrossRef CAS.
- O. Durak, M. Zeeshan, S. Keskin and A. Uzun, Chem. Eng. J., 2022, 437, 135436 CrossRef CAS.
- M. Zeeshan, K. Yalcin, F. E. Sarac Oztuna, U. Unal, S. Keskin and A. Uzun, Carbon, 2021, 171, 79–87 CrossRef CAS.
- M. S. Raja Shahrom, A. R. Nordin and C. D. Wilfred, J. Environ. Chem. Eng., 2019, 7, 103319 CrossRef CAS.
- X. He, J. Zhu, H. Wang, M. Zhou and S. Zhang, Coatings, 2019, 9, 590 CrossRef CAS.
- Y. Yu, J. Mai, L. Huang, L. Wang and X. Li, RSC Adv., 2014, 4, 12756 RSC.
- Y. Y. Lee, K. Edgehouse, A. Klemm, H. Mao, E. Pentzer and B. Gurkan, ACS Appl. Mater. Interfaces, 2020, 12, 19184–19193 CrossRef CAS PubMed.
- S. Chen, Y. Chao, J. Wu, H. Ye, X. Luo and Z. Liang, Ind. Eng. Chem. Res., 2022, 61, 11953–11963 CrossRef CAS.
- Y. Xu, Y. Zhou, J. Liu and L. Sun, J. Energy Chem., 2017, 26, 1026–1029 CrossRef.
- Y. Zhou, J. Liu, M. Xiao, Y. Meng and L. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 5547–5555 CrossRef CAS PubMed.
- M. Mirzaei, B. Mokhtarani, A. Badiei and A. Sharifi, Chem. Eng. Technol., 2018, 41, 1272–1281 CrossRef CAS.
- M. Balsamo, A. Erto, A. Lancia, G. Totarella, F. Montagnaro and R. Turco, Fuel, 2018, 218, 155–161 CrossRef CAS.
- S. Yoshida, K. Takahashi, S. Kudo, S. Iwamura, I. Ogino and S. R. Mukai, Ind. Eng. Chem. Res., 2017, 56, 2834–2839 CrossRef CAS.
- J. Wu, Z. Yang, J. Xie, P. Zhu, J. Wei, R. Jin and H. Yang, Langmuir, 2023, 39, 2729–2738 CrossRef CAS PubMed.
- Y. Uehara, D. Karami and N. Mahinpey, Energy Fuels, 2018, 32, 5345–5354 CrossRef CAS.
- D. N. Lapshin, M. Jorge, E. E. B. Campbell and L. Sarkisov, J. Mater. Chem. A, 2020, 8, 11781–11799 RSC.
- I. H. Arellano, J. Huang and P. Pendleton, Chem. Eng. J., 2015, 281, 119–125 CrossRef CAS.
- M. Zhao, Y. Ban and W. Yang, Chem. Eng. J., 2022, 439, 135650 CrossRef CAS.
- M. Zeeshan, V. Nozari, M. B. Yagci, T. Isık, U. Unal, V. Ortalan, S. Keskin and A. Uzun, J. Am. Chem. Soc., 2018, 140, 10113–10116 CrossRef CAS PubMed.
- M. Zeeshan, S. Keskin and A. Uzun, Polyhedron, 2018, 155, 485–492 CrossRef CAS.
- F. P. Kinik, C. Altintas, V. Balci, B. Koyuturk, A. Uzun and S. Keskin, ACS Appl. Mater. Interfaces, 2016, 8, 30992–31005 CrossRef CAS PubMed.
- M. Mohamedali, H. Ibrahim and A. Henni, Chem. Eng. J., 2018, 334, 817–828 CrossRef CAS.
- M. Zeeshan, H. Kulak, S. Kavak, H. M. Polat, O. Durak, S. Keskin and A. Uzun, Microporous Mesoporous Mater., 2020, 306, 110446 CrossRef CAS.
- T. J. Ferreira, R. P. P. L. Ribeiro, J. P. B. Mota, L. P. N. Rebelo, J. M. S. S. Esperança and I. A. A. C. Esteves, ACS Appl. Nano Mater., 2019, 2, 7933–7950 CrossRef CAS.
- T. J. Ferreira, A. T. Vera, B. A. de Moura, L. M. Esteves, M. Tariq, J. M. S. S. Esperança and I. A. A. C. Esteves, Front. Chem., 2020, 8, 590191 CrossRef CAS PubMed.
- M. Mohamedali, A. Henni and H. Ibrahim, Microporous Mesoporous Mater., 2019, 284, 98–110 CrossRef CAS.
- G. Yang, J. Yu, S. Peng, K. Sheng and H. Zhang, Polymers, 2020, 12, 370 CrossRef CAS PubMed.
- C. Chen, N. Feng, Q. Guo, Z. Li, X. Li, J. Ding, L. Wang, H. Wan and G. Guan, J. Colloid Interface Sci., 2018, 521, 91–101 CrossRef CAS PubMed.
- J. Yang, M. Tong, G. Han, M. Chang, T. Yan, Y. Ying, Q. Yang and D. Liu, Adv. Funct. Mater., 2023, 33, 2213743 CrossRef CAS.
- Y. Zhang, Y. Huang, S. Chen, L. Shi, J. Wang, Q. Yi and F. Pei, Chem. Eng. J., 2023, 471, 144580 CrossRef CAS.
- L. Qiu, L. Peng, D. Moitra, H. Liu, Y. Fu, Z. Dong, W. Hu, M. Lei, D.-E. Jiang, H. Lin, J. Hu, K. A. McGarry, I. Popovs, M. Li, A. S. Ivanov, Z. Yang and S. Dai, Small, 2023, 19, 2370338 CrossRef.
- B. B. Polesso, F. L. Bernard, H. Z. Ferrari, E. A. Duarte, F. D. Vecchia and S. Einloft, Heliyon, 2019, 5, e02183 CrossRef PubMed.
- B. B. Polesso, R. Duczinski, F. L. Bernard, H. Z. Ferrari, M. da Luz, F. Dalla Vecchia, S. M. C. de Menezes and S. Einloft, Am. J. Mater. Sci., 2019, 22, e20180810 CAS.
- H. Kulak, H. M. Polat, S. Kavak, S. Keskin and A. Uzun, Energy Technol., 2019, 7, 1900157 CrossRef PubMed.
- W. Zhang, E. Gao, Y. Li, M. T. Bernards, Y. Li, G. Cao, Y. He and Y. Shi, Energy Fuels, 2019, 33, 8967–8975 CrossRef CAS.
- J.-Z. Yin, M.-Y. Zhen, P. Cai, D. Zhou, Z.-J. Li, H.-Y. Zhu and Q.-Q. Xu, Mater. Res. Express, 2018, 5, 065060 CrossRef.
- F. Liu, K. Huang and L. Jiang, AIChE J., 2018, 64, 3671–3680 CrossRef CAS.
- M. Mohamedali, A. Henni and H. Ibrahim, Adsorption, 2019, 25, 675–692 CrossRef CAS.
- A. Erto, A. Silvestre-Albero, J. Silvestre-Albero, F. Rodríguez-Reinoso, M. Balsamo, A. Lancia and F. Montagnaro, J. Colloid Interface Sci., 2015, 448, 41–50 CrossRef CAS PubMed.
- J. Ren, Z. Li, Y. Chen, Z. Yang and X. Lu, Chin. J. Chem. Eng., 2018, 26, 2377–2384 CrossRef CAS.
- Y. Uehara, D. Karami and N. Mahinpey, Ind. Eng. Chem. Res., 2017, 56, 14316–14323 CrossRef CAS.
- Y. Uehara, D. Karami and N. Mahinpey, Adsorption, 2019, 25, 703–716 CrossRef CAS.
- K. N. Ruckart, R. A. O’Brien, S. M. Woodard, K. N. West and T. G. Glover, J. Phys. Chem. C, 2015, 119, 20681–20697 CrossRef CAS.
- C. Xue, L. Feng, Q. Zhang, R. Huang, Y. Hao, X. Ma, G. Liu, K. Li and X. Hao, Int. J. Greenhouse Gas Control, 2019, 84, 111–120 CrossRef CAS.
- J. Cheng, Y. Li, L. Hu, J. Liu, J. Zhou and K. Cen, Fuel Process. Technol., 2018, 172, 216–224 CrossRef CAS.
- J. M. Zhu, F. Xin, Y. C. Sun and X. C. Dong, Theor. Found. Chem. Eng., 2014, 48, 787–792 CrossRef CAS.
- V. Hiremath, A. H. Jadhav, H. Lee, S. Kwon and J. G. Seo, Chem. Eng. J., 2016, 287, 602–617 CrossRef CAS.
- P. Tamilarasan and S. Ramaprabhu, RSC Adv., 2016, 6, 3032–3040 RSC.
- X. Song, J. Yu, M. Wei, R. Li, X. Pan, G. Yang and H. Tang, Materials, 2019, 12, 2361 CrossRef CAS PubMed.
- B. Dong, L. Wang, S. Zhao, R. Ge, X. Song, Y. Wang and Y. Gao, Chem. Commun., 2016, 52, 7082–7085 RSC.
- N. Z. Zulkurnai, U. F. Md Ali, N. Ibrahim and N. S. Abdul Manan, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 206, 012001 Search PubMed.
- Z. Ghazali, N. Suhaili, M. N. A. Tahari, M. A. Yarmo, N. H. Hassan and R. Othaman, J. Mater. Res. Technol., 2020, 9, 3249–3260 CrossRef CAS.
- N. Noorani and A. Mehrdad, Sci. Rep., 2023, 13, 13012 CrossRef CAS PubMed.
- O. Durak, M. Zeeshan, N. Habib, H. C. Gulbalkan, A. A. A. M. Alsuhile, H. P. Caglayan, S. F. Kurtoğlu-Öztulum, Y. Zhao, Z. P. Haslak, A. Uzun and S. Keskin, Microporous Mesoporous Mater., 2022, 332, 111703 CrossRef CAS.
- M. Zeeshan, V. Nozari, S. Keskin and A. Uzun, Ind. Eng. Chem. Res., 2019, 58, 14124–14138 CrossRef CAS.
- O. Durak, H. Kulak, S. Kavak, H. M. Polat, S. Keskin and A. Uzun, J. Phys.: Condens. Matter, 2020, 32, 484001 CrossRef CAS PubMed.
- S. F. Kurtoğlu-Öztulum, A. Jalal and A. Uzun, J. Mol. Liq., 2022, 363, 119804 CrossRef.
- M. Babucci, A. Akcay, V. Balci and A. Uzun, Langmuir, 2015, 31, 9163–9176 CrossRef CAS PubMed.
- K. B. Sezginel, S. Keskin and A. Uzun, Langmuir, 2016, 32, 1139–1147 CrossRef CAS PubMed.
- V. Nozari, M. Zeeshan, S. Keskin and A. Uzun, CrystEngComm, 2018, 20, 7137–7143 RSC.
- M. Mirzaei, A. R. Badiei, B. Mokhtarani and A. Sharifi, J. Mol. Liq., 2017, 232, 462–470 CrossRef CAS.
- Y. Cheng, S. Zhou, P. Hu, G. Zhao, Y. Li, X. Zhang and W. Han, Sci. Rep., 2017, 7, 1439 CrossRef PubMed.
- S. Kavak, H. M. Polat, H. Kulak, S. Keskin and A. Uzun, Chem. – Asian J., 2019, 14, 3655–3667 CrossRef CAS PubMed.
- R. Duczinski, F. Bernard, M. Rojas, E. Duarte, V. Chaban, F. D. Vecchia, S. Menezes and S. Einloft, J. Nat. Gas Sci. Eng., 2018, 54, 54–64 CrossRef CAS.
- F. W. M. d Silva, G. M. Magalhães, E. O. Jardim, J. Silvestre-Albero, A. Sepúlveda-Escribano, D. C. S. de Azevedo and S. M. P. de Lucena, Adsorpt. Sci. Technol., 2015, 33, 223–242 CrossRef.
- N. Habib, O. Durak, H. C. Gulbalkan, A. S. Aydogdu, S. Keskin and A. Uzun, ACS Appl. Eng. Mater., 2023, 1, 1473–1481 CrossRef CAS PubMed.
- M. Zeeshan, H. C. Gulbalkan, Z. P. Haslak, S. Keskin and A. Uzun, Microporous Mesoporous Mater., 2021, 316, 110947 CrossRef CAS.
- R. Santiago, J. Lemus, D. Hospital-Benito, C. Moya, J. Bedia, N. Alonso-Morales, J. J. Rodriguez and J. Palomar, ACS Sustainable Chem. Eng., 2019, 7, 13089–13097 CrossRef CAS.
- W. Zhang, E. Gao, Y. Li, M. T. Bernards, Y. He and Y. Shi, J. CO2 Util., 2019, 34, 606–615 CrossRef CAS.
- S. Zheng, S. Zeng, G. Li, X. Yao, Z. Li and X. Zhang, Chem. Eng. J., 2023, 451, 138736 CrossRef CAS.
- Z. Li, W. Sun, C. Chen, Q. Guo, X. Li, M. Gu, N. Feng, J. Ding, H. Wan and G. Guan, Appl. Surf. Sci., 2019, 480, 770–778 CrossRef CAS.
- T. Ariyanto, K. Masruroh, G. Y. S. Pambayun, N. I. F. Mukti, R. B. Cahyono, A. Prasetya and I. Prasetyo, ACS Omega, 2021, 6, 19194–19201 CrossRef CAS PubMed.
- Z. Huang, M. Mohamedali, D. Karami and N. Mahinpey, Fuel, 2022, 310, 122284 CrossRef CAS.
- I. H. Arellano, J. Huang and P. Pendleton, RSC Adv., 2015, 5, 65074–65083 RSC.
- I. H. Arellano, S. H. Madani, J. Huang and P. Pendleton, Chem. Eng. J., 2016, 283, 692–702 CrossRef CAS.
- N. I. F. Mukti, T. Ariyanto, W. B. Sediawan and I. Prasetyo, RSC Adv., 2023, 13, 23158–23168 RSC.
- F. Hussin, M. K. Aroua and R. Yusoff, J. Environ. Chem. Eng., 2021, 9, 105333 CrossRef CAS.
- N. Hussain Solangi, F. Hussin, A. Anjum, N. Sabzoi, S. Ali Mazari, N. M. Mubarak, M. Kheireddine Aroua, M. T. H. Siddiqui and S. Saeed Qureshi, J. Mol. Liq., 2023, 374, 121266 CrossRef CAS.
- X. Li, D. Wang, Z. He, F. Su, N. Zhang, Y. Xin, H. Wang, X. Tian, Y. Zheng and D. Yao, Chem. Eng. J., 2021, 417, 129239 CrossRef CAS.
- Y.-Y. Lee and B. Gurkan, J. Membr. Sci., 2021, 638, 119652 CrossRef CAS.
- Y.-Y. Lee, N. P. Wickramasinghe, R. Dikki, D. L. Jan and B. Gurkan, Nanoscale, 2022, 14, 12638–12650 RSC.
- N. O'Reilly, N. Giri and S. L. James, Chem. – Eur. J., 2007, 13, 3020–3025 CrossRef PubMed.
- M. Z. Ahmad and A. Fuoco, Curr. Res. Green Sustainable Chem., 2021, 4, 100070 CrossRef CAS.
- B. D. Egleston, A. Mroz, K. E. Jelfs and R. L. Greenaway, Chem. Sci., 2022, 13, 5042–5054 RSC.
- S. Zhang, K. Dokko and M. Watanabe, Chem. Sci., 2015, 6, 3684–3691 RSC.
- J. Avila, C. Červinka, P. Y. Dugas, A. A. Pádua and M. Costa Gomes, Adv. Mater. Interfaces, 2021, 8, 2001982 CrossRef CAS.
- J. Avila, L. F. Lepre, C. C. Santini, M. Tiano, S. Denis-Quanquin, K. Chung Szeto, A. A. Padua and M. Costa Gomes, Angew. Chem., Int. Ed., 2021, 133, 12986–12992 CrossRef.
- W. Shan, P. F. Fulvio, L. Kong, J. A. Schott, C.-L. Do-Thanh, T. Tian, X. Hu, S. M. Mahurin, H. Xing and S. Dai, ACS Appl. Mater. Interfaces, 2018, 10, 32–36 CrossRef CAS PubMed.
- P. Scovazzo, A. E. Visser, J. H. DavisJr, R. D. Rogers, C. A. Koval, D. L. DuBois and R. D. Noble, Ionic Liquids, American Chemical Society, 2002, ch. 6, vol. 18, pp. 69–87 Search PubMed.
- N. N. R. Ahmad, C. P. Leo, A. W. Mohammad, N. Shaari and W. L. Ang, Int. J. Energy Res., 2021, 45, 9800–9830 CrossRef CAS.
- W. Luo, C. Wang, M. Jin, F. Li, H. Li, Z. Zhang, X. Zhang, Y. Liang, G. Huang and T. Zhou, Sep. Purif. Technol., 2024, 330, 125406 CrossRef CAS.
- N. Habib, Ö. Durak, A. Uzun and S. Keskin, Sep. Purif. Technol., 2023, 312, 123346 CrossRef CAS.
- R. Mahboubi, E. Joudaki, R. M. Behbahani and N. Azizi, Mater. Today Commun., 2023, 36, 106542 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2002, 58, 389–397 CrossRef PubMed.
- M. Mohan, O. Demerdash, B. A. Simmons, J. C. Smith, M. K. Kidder and S. Singh, Green Chem., 2023, 25, 3475–3492 RSC.
- M. Mohan, K. S. Jetti, M. D. Demerdash, M. O. Kidder and J. C. Smith, J. Chem. Theory Comput., 2024, 12(18), 7040–7054 CAS.
- H. M. Polat, M. Zeeshan, A. Uzun and S. Keskin, Chem. Eng. J., 2019, 373, 1179–1189 CrossRef CAS.
- H. M. Polat, S. Kavak, H. Kulak, A. Uzun and S. Keskin, Chem. Eng. J., 2020, 394, 124916 CrossRef CAS.
- H. C. Gulbalkan, Z. P. Haslak, C. Altintas, A. Uzun and S. Keskin, Sep. Purif. Technol., 2022, 287, 120578 CrossRef CAS.
- T. Yan, M. Tong, D. Liu, Q. Yang and C. Zhong, J. Mater. Chem. A, 2023, 11, 14911–14920 RSC.
- M. Zeeshan, H. C. Gulbalkan, O. Durak, Z. P. Haslak, U. Unal, S. Keskin and A. Uzun, Adv. Funct. Mater., 2022, 32, 2204149 CrossRef CAS.
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC.
- A. Velty and A. Corma, Chem. Soc. Rev., 2023, 52, 1773–1946 RSC.
- Z. Xie, M. Zhu, A. Nambo, J. B. Jasinski and M. A. Carreon, Dalton Trans., 2013, 42, 6732–6735 RSC.
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819 CrossRef.
- M. I. Qadir and J. Dupont, Angew. Chem., Int. Ed., 2023, 62, e202301497 CrossRef PubMed.
- Z. J. Li, J. F. Sun, Q. Q. Xu and J. Z. Yin, ChemCatChem, 2021, 13, 1848–1866 CrossRef CAS.
- Y. He, D. Jiang, X. Li, J. Ding, H. Li, H. Wan and G. Guan, J. CO2 Util., 2021, 44, 101427 CrossRef CAS.
- T. Chen, Y. Guo and Y. Xu, Chem. Commun., 2023, 59, 12282–12285 RSC.
- X. Gao, J. Zhao, Y. Gao, Y. Deng, Y. Shi, J. He and Y. Li, New J. Chem., 2023, 47, 17449–17455 RSC.
- M. Liu, X. Wang, Y. Jiang, J. Sun and M. Arai, Catal. Rev., 2018, 61, 214–269 CrossRef.
- Y. Xie, Q. Sun, Y. Fu, L. Song, J. Liang, X. Xu, H. Wang, J. Li, S. Tu, X. Lu and J. Li, J. Mater. Chem. A, 2017, 5, 25594–25600 RSC.
- P. Migowski, P. Lozano and J. Dupont, Green Chem., 2023, 25, 1237–1260 RSC.
- J. Peng and Y. Deng, New J. Chem., 2001, 25, 639–641 RSC.
- H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896–897, 10.1039/b212823c.
- J. Byun and K. A. I. Zhang, ChemCatChem, 2018, 10, 4610–4616 CrossRef CAS.
- Á. Mesías-Salazar, R. S. Rojas, F. Carrillo-Hermosilla, J. Martínez, A. Antiñolo, O. S. Trofymchuk, F. M. Nachtigall, L. S. Santos and C. G. Daniliuc, New J. Chem., 2024, 48, 105–111 RSC.
- F. Norouzi and A. Abdolmaleki, Fuel, 2023, 334 Search PubMed.
- L. Xiao, D. Su, C. Yue and W. Wu, J. CO2 Util., 2014, 6, 1–6 CrossRef CAS.
- Z. Zhang, F. Fan, H. Xing, Q. Yang, Z. Bao and Q. Ren, ACS Sustainable Chem. Eng., 2017, 5, 2841–2846 CrossRef CAS.
- X. Meng, Z. Ju, S. Zhang, X. Liang, N. von Solms, X. Zhang and X. Zhang, Green Chem., 2019, 21, 3456–3463 RSC.
- C. Luo, J. Wang, H. Lu, K. Wu, Y. Liu, Y. Zhu, B. Wang and B. Liang, Green Chem., 2022, 24, 8292–8301 RSC.
- Q. Yi, T. Liu, X. Wang, Y. Shan, X. Li, M. Ding, L. Shi, H. Zeng and Y. Wu, Appl. Catal., B, 2021, 283, 119620 CrossRef CAS.
- Y. Zhang, M. Qiu, J. Li, H. Wu, L. Shi and Q. Yi, Fuel, 2023, 332, 126191 CrossRef CAS.
- B. Chen, S. Zhang and Y. Zhang, Green Chem., 2023, 25, 7743–7755 RSC.
- A. Dani, E. Groppo, C. Barolo, J. G. Vitillo and S. Bordiga, J. Mater. Chem. A, 2015, 3, 8508–8518 RSC.
- J. D. Ndayambaje, I. Shabbir, Q. Zhao, L. Dong, Q. Su and W. Cheng, Catal. Sci. Technol., 2024, 14, 293–305 RSC.
- Y. Qu, Y. Chen and J. Sun, J. CO2 Util., 2022, 56, 101840 CrossRef CAS.
- Y. Qu, J. Lan, Y. Chen and J. Sun, Sustainable Energy Fuels, 2021, 5, 2494–2503 RSC.
- G. Chen, J. Zhang, X. Cheng, X. Tan, J. Shi, D. Tan, B. Zhang, Q. Wan, F. Zhang, L. Liu, B. Han and G. Yang, ChemCatChem, 2020, 12, 1963–1967 CrossRef CAS.
- Y. Toda, Y. Komiyama, A. Kikuchi and H. Suga, ACS Catal., 2016, 6, 6906–6910 CrossRef CAS.
- J. Steinbauer, L. Longwitz, M. Frank, J. Epping, U. Kragl and T. Werner, Green Chem., 2017, 19, 4435–4445 RSC.
- J. Sun, L. Han, W. Cheng, J. Wang, X. Zhang and S. Zhang, ChemSusChem, 2011, 4, 502–507 CrossRef CAS PubMed.
- X. Meng, H. He, Y. Nie, X. Zhang, S. Zhang and J. Wang, ACS Sustainable Chem. Eng., 2017, 5, 3081–3086 CrossRef CAS.
- S. Yue, P. Wang and X. Hao, Fuel, 2019, 251, 233–241 CrossRef CAS.
- Á. F. Arruda da Mata, N. Glanzmann, P. H. Fazza Stroppa, F. Terra Martins, R. P. das Chagas, A. D. da Silva and J. L. S. Milani, New J. Chem., 2022, 46, 12237–12243 RSC.
- Y. Zou, Y. Ge, Q. Zhang, W. Liu, X. Li, G. Cheng and H. Ke, Catal. Sci. Technol., 2022, 12, 273–281 RSC.
- P. Wang, Q. Lv, Y. Tao, L. Cheng, R. Li, Y. Jiao, C. Fang, H. Li, C. Geng, C. Sun, J. Ding, H. Wan and G. Guan, Mol. Catal., 2023, 544, 113157 CrossRef CAS.
- C. Li, F. Liu, T. Zhao, J. Gu, P. Chen and T. Chen, Mol. Catal., 2021, 511, 111756 CrossRef CAS.
- Y. Fu, Y. Xu, Z. Zeng, A.-R. Ibrahim, J. Yang, S. Yang, Y. Xie, Y. Hong, Y. Su, H. Wang, Y. Wang, L. Peng, J. Li and W. L. Queen, Green Energy Environ., 2023, 8, 478–486 CrossRef CAS.
- A. Zhang, C. Chen, C. Zuo, X. Xu, T. Cai, X. Li, Y. Yuan, H. Yang and G. Meng, Green Chem., 2022, 24, 7194–7207 RSC.
- D. Dong, X. Zhao, C. Pu, Y. Yao, B. Zhao, G. Tian, G. Chang and X. Yang, Inorg. Chem., 2023, 62, 20528–20536 CrossRef CAS PubMed.
- Y. Sun, H. Huang, H. Vardhan, B. Aguila, C. Zhong, J. A. Perman, A. M. Al-Enizi, A. Nafady and S. Ma, ACS Appl. Mater. Interfaces, 2018, 10, 27124–27130 CrossRef CAS PubMed.
- V. K. Tomazett, G. Chacon, G. Marin, M. V. Castegnaro, R. P. das Chagas, L. M. Lião, J. Dupont and M. I. Qadir, J. CO2 Util., 2023, 69, 102400 CrossRef CAS.
- N. Wu, Y. Zou, R. Xu, J. Zhong and J. Li, J. CO2 Util., 2021, 52, 101702 CrossRef CAS.
- J. Sun, Z. Li, X. Li, M. Xue and J. Yin, Catal. Lett., 2021, 152, 2669–2677 CrossRef.
- Q. Su, Y. Qi, X. Yao, W. Cheng, L. Dong, S. Chen and S. Zhang, Green Chem., 2018, 20, 3232–3241 RSC.
- L. Sun, M. Yin and S. Tang, J. Environ. Chem. Eng., 2023, 11(5), 110843 CrossRef CAS.
- Z. Shi, Q. Su, T. Ying, X. Tan, L. Deng, L. Dong and W. Cheng, J. CO2 Util., 2020, 39, 101162 CrossRef CAS.
- Y. He, X. Li, W. Cai, H. Lu, J. Ding, H. Li, H. Wan and G. Guan, ACS Sustainable Chem. Eng., 2021, 9, 7074–7085 CrossRef CAS.
- J. Ding, P. Wang, Y. He, L. Cheng, X. Li, C. Fang, H. Li, H. Wan and G. Guan, Appl. Catal., B, 2023, 324, 122278 CrossRef CAS.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- T. Sasaki, Curr. Opin. Green Sustainable Chem., 2022, 36, 100633 CrossRef CAS.
- S. J. Louis Anandaraj, L. Kang, S. DeBeer, A. Bordet and W. Leitner, Small, 2023, 19, 2206806 CrossRef CAS PubMed.
- C. I. Melo, A. Szczepanska, E. Bogel-Lukasik, M. Nunes da Ponte and L. C. Branco, ChemSusChem, 2016, 9, 1081–1084 CrossRef CAS PubMed.
- C. I. Melo, D. Rente, M. Nunes da Ponte, E. Bogel-Łukasik and L. C. Branco, ACS Sustainable Chem. Eng., 2019, 7(14), 11963–11969 CAS.
- R. Wang, Y.-R. Du, G.-R. Ding, R. Zhang, P.-X. Guan and B.-H. Xu, ACS Sustainable Chem. Eng., 2022, 10, 5363–5373 CrossRef CAS.
- M. I. Qadir, A. Weilhard, J. A. Fernandes, I. de Pedro, B. J. C. Vieira, J. C. Waerenborgh and J. Dupont, ACS Catal., 2018, 8, 1621–1627 CrossRef CAS.
- M. I. Qadir, F. Bernardi, J. D. Scholten, D. L. Baptista and J. Dupont, Appl. Catal., B, 2019, 252, 10–17 CrossRef CAS.
- M. F. Santos, M. L. Alcantara, C. A. O. Nascimento, G. S. Bassani and R. M. B. Alves, Clean Technol. Environ. Policy, 2024, 26, 11–29 CrossRef CAS.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed.
- A. Weilhard, S. P. Argent and V. Sans, Nat. Commun., 2021, 12, 231 CrossRef CAS PubMed.
- L. Piccirilli, B. Rabell, R. Padilla, A. Riisager, S. Das and M. Nielsen, J. Am. Chem. Soc., 2023, 145, 5655–5663 CrossRef CAS PubMed.
- W. Ma, J. Hu, L. Zhou, Y. Wu, J. Geng and X. Hu, Green Chem., 2022, 24, 6727–6732 RSC.
- R. Webber, M. I. Qadir, E. Sola, M. Martín, E. Suárez and J. Dupont, Catal. Commun., 2020, 146, 106125 CrossRef CAS.
- R. Kanega, M. Z. Ertem, N. Onishi, D. J. Szalda, E. Fujita and Y. Himeda, Organometallics, 2020, 39, 1519–1531 CrossRef CAS.
- Q. Li, T. Huang, Z. Zhang, M. Xiao, H. Gai, Y. Zhou and H. Song, Mol. Catal., 2021, 509, 111644 CrossRef CAS.
- B. Feng, Z. Zhang, J. Wang, D. Yang, Q. Li, Y. Liu, H. Gai, T. Huang and H. Song, Fuel, 2022, 325, 124853 CrossRef CAS.
- S. J. Louis Anandaraj, L. Kang, S. DeBeer, A. Bordet and W. Leitner, Small, 2023, 19, e2206806 CrossRef PubMed.
- P. Schwiderowski, H. Ruland and M. Muhler, Curr. Opin. Green Sustainable Chem., 2022, 38, 100688 CrossRef CAS.
- F. S. Zebarjad, S. Hu, Z. Li and T. T. Tsotsis, Ind. Eng. Chem. Res., 2019, 58, 11811–11820 CrossRef CAS.
- F. S. Zebarjad, Z. Wang, H. Li, S. Hu, Y. Tang and T. T. Tsotsis, Chem. Eng. Sci., 2021, 242, 116722 CrossRef CAS.
- J. Reichert, S. Maerten, K. Meltzer, A. Tremel, M. Baldauf, P. Wasserscheid and J. Albert, Sustainable Energy Fuels, 2019, 3, 3399–3405 RSC.
- M. Babucci, C.-Y. Fang, A. S. Hoffman, S. R. Bare, B. C. Gates and A. Uzun, ACS Catal., 2017, 7, 6969–6972 CrossRef CAS.
- M. Babucci, C.-Y. Fang, J. E. Perez-Aguilar, A. S. Hoffman, A. Boubnov, E. Guan, S. R. Bare, B. C. Gates and A. Uzun, Chem. Sci., 2019, 10, 2623–2632 RSC.
- A. Jalal and A. Uzun, J. Catal., 2017, 350, 86–96 CrossRef CAS.
- M. E. Royer and C. R. Hebd, Acad. Sci., Ser. D, 1870, 70, 731 Search PubMed.
- P. G. Russell, N. Kovac, S. Srinivasan and M. Steinberg, J. Electrochem. Soc., 1977, 124, 1329–1338 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- Y. Hori, in Modern Aspects of Electrochemistry, ed.C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer, New York, New York, NY, 2008, pp. 89–189 DOI:10.1007/978-0-387-49489-0_3.
- C. Shi, K. Chan, J. S. Yoo and J. K. Nørskov, Org. Process Res. Dev., 2016, 20, 1424–1430 CrossRef CAS.
- S. Ringe, C. G. Morales-Guio, L. D. Chen, M. Fields, T. F. Jaramillo, C. Hahn and K. Chan, Nat. Commun., 2020, 11, 33 CrossRef CAS PubMed.
- C. Hahn and T. F. Jaramillo, Joule, 2020, 4, 292–294 CrossRef.
- J. T. Feaster, C. Shi, E. R. Cave, T. T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Norskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- N. S. Spinner, J. A. Vega and W. E. Mustain, Catal. Sci. Technol., 2012, 2, 19–28 RSC.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- A. Bard, Standard potentials in aqueous solution, Routledge, 2017 Search PubMed.
- B. A. Rosen, J. L. Haan, P. Mukherjee, B. Braunschweig, W. Zhu, A. Salehi-Khojin, D. D. Dlott and R. I. Masel, J. Phys. Chem. C, 2012, 116, 15307–15312 CrossRef CAS.
- B. A. Rosen, W. Zhu, G. Kaul, A. Salehi-Khojin and R. I. Masel, J. Electrochem. Soc., 2013, 160, H138 CrossRef CAS.
- N. Hollingsworth, S. F. R. Taylor, M. T. Galante, J. Jacquemin, C. Longo, K. B. Holt, N. H. de Leeuw and C. Hardacre, Angew. Chem., Int. Ed., 2015, 54, 14164–14168 CrossRef CAS PubMed.
- L. Chen, S.-X. Guo, F. Li, C. Bentley, M. Horne, A. M. Bond and J. Zhang, ChemSusChem, 2016, 9, 1271–1278 CrossRef CAS PubMed.
- L. Sun, G. K. Ramesha, P. V. Kamat and J. F. J. L. Brennecke, Langmuir, 2014, 30, 6302–6308 CrossRef CAS PubMed.
- E. E. L. Tanner, C. Batchelor-McAuley and R. G. Compton, J. Phys. Chem. C, 2016, 120, 26442–26447 CrossRef CAS.
- F. A. Hanc-Scherer, M. A. Montiel, V. Montiel, E. Herrero and C. M. Sanchez-Sanchez, Phys. Chem. Chem. Phys., 2015, 17, 23909–23916 RSC.
- S. Neyrizi, J. Kiewiet, M. A. Hempenius and G. Mul, ACS Energy Lett., 2022, 7, 3439–3446 CrossRef CAS PubMed.
- G. P. S. Lau, M. Schreier, D. Vasilyev, R. Scopelliti, M. Grätzel and P. J. Dyson, J. Am. Chem. Soc., 2016, 138, 7820–7823 CrossRef CAS PubMed.
- Z. Min, B. Chang, C. Shao, X. Su, N. Wang, Z. Li, H. Wang, Y. Zhao, M. Fan and J. Wang, Appl. Catal., B, 2023, 326, 122185 CrossRef CAS.
- B. Liu, W. Guo and M. A. Gebbie, ACS Catal., 2022, 12, 9706–9716 CrossRef CAS.
- S. Noh, Y. J. Cho, G. Zhang and M. Schreier, J. Am. Chem. Soc., 2023, 145, 27657–27663 CrossRef CAS PubMed.
- S. Sharifi Golru and E. J. Biddinger, Electrochim. Acta, 2020, 361, 136787 CrossRef CAS.
- S. Garg, M. Li, T. E. Rufford, L. Ge, V. Rudolph, R. Knibbe, M. Konarova and G. G. X. Wang, ChemSusChem, 2020, 13, 304–311 CrossRef CAS PubMed.
- D. V. Vasilyev, A. V. Rudnev, P. Broekmann and P. J. Dyson, ChemSusChem, 2019, 12, 1635–1639 CrossRef CAS PubMed.
- S. Verma, X. Lu, S. C. Ma, R. I. Masel and P. J. A. Kenis, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084 RSC.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS.
- Z. Lin, C. Han, G. E. P. O’Connell and X. Lu, Angew. Chem., Int. Ed., 2023, 62, e202301435 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- E. L. Clark, R. Nielsen, J. E. Sørensen, J. L. Needham, B. Seger and I. Chorkendorff, ACS Energy Lett., 2023, 8, 4414–4420 CrossRef CAS PubMed.
- W. Zhu, L. Zhang, P. Yang, X. Chang, H. Dong, A. Li, C. Hu, Z. Huang, Z.-J. Zhao and J. Gong, Small, 2018, 14, 1703314 CrossRef PubMed.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- J. Park, C. Jeong, M. Na, Y. Oh, K.-S. Lee, Y. Yang and H. R. Byon, ACS Catal., 2024, 14, 3198–3207 CrossRef CAS.
- D. Bagchi, J. Raj, A. K. Singh, A. Cherevotan, S. Roy, K. S. Manoj, C. P. Vinod and S. C. Peter, Adv. Mater., 2022, 34, 2109426 CrossRef CAS PubMed.
- M. Ding, Z. Chen, C. Liu, Y. Wang, C. Li, X. Li, T. Zheng, Q. Jiang and C. Xia, Mater. Rep.: Energy, 2023, 3, 100175 CAS.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X.-D. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- A. Hasani, M. A. Teklagne, H. H. Do, S. H. Hong, Q. Van Le, S. H. Ahn and S. Y. Kim, Carbon Energy, 2020, 2, 158–175 CrossRef CAS.
- S. Oh, J. R. Gallagher, J. T. Miller and Y. Surendranath, J. Am. Chem. Soc., 2016, 138, 1820–1823 CrossRef CAS PubMed.
- L. Ye, Y. Ying, D. Sun, Z. Zhang, L. Fei, Z. Wen, J. Qiao and H. Huang, Angew. Chem., Int. Ed., 2020, 59, 3244–3251 CrossRef CAS PubMed.
- Y. Zhu, X. Yang, C. Peng, C. Priest, Y. Mei and G. Wu, Small, 2021, 17, 2005148 CrossRef CAS PubMed.
- K. Zhao and X. Quan, ACS Catal., 2021, 11, 2076–2097 CrossRef CAS.
- M. Balamurugan, H.-Y. Jeong, V. S. K. Choutipalli, J. S. Hong, H. Seo, N. Saravanan, J. H. Jang, K.-G. Lee, Y. H. Lee, S. W. Im, V. Subramanian, S. H. Kim and K. T. Nam, Small, 2020, 16, 2000955 CrossRef CAS PubMed.
- L. Wang, Z. Sofer and M. Pumera, ACS Nano, 2020, 14, 21–25 CrossRef CAS PubMed.
- S. Palchoudhury, K. Ramasamy, J. Han, P. Chen and A. Gupta, Nanoscale Adv., 2023, 5, 2724–2742 RSC.
- A. Zahid, A. Shah and I. Shah, Nanomaterials, 2022, 12, 1380 CrossRef CAS PubMed.
- X. Zheng, P. De Luna, F. P. García de Arquer, B. Zhang, N. Becknell, M. B. Ross, Y. Li, M. N. Banis, Y. Li, M. Liu, O. Voznyy, C. T. Dinh, T. Zhuang, P. Stadler, Y. Cui, X. Du, P. Yang and E. H. Sargent, Joule, 2017, 1, 794–805 CrossRef CAS.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- M. Dai and R. Wang, Small, 2021, 17, 2006813 CrossRef CAS PubMed.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- X. Liu, H. Yang, J. He, H. Liu, L. Song, L. Li and J. Luo, Small, 2018, 14, 1704049 CrossRef PubMed.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 677 CrossRef CAS PubMed.
- E. Sedano Varo, R. Egeberg Tankard, J. Kryger-Baggesen, J. Jinschek, S. Helveg, I. Chorkendorff, C. D. Damsgaard and J. Kibsgaard, J. Am. Chem. Soc., 2024, 146, 2015–2023 CrossRef CAS PubMed.
- A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 CrossRef.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8, 3602–3635 CrossRef CAS.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- A. Asserghine, A. Baby, E. Gao, H. Zhao and J. Rodríguez-López, Electrochim. Acta, 2024, 475, 143620 CrossRef CAS.
- L. Liu, M. Li, F. Chen and H. Huang, Small Struct., 2023, 4, 2200188 CrossRef CAS.
- Y. Cheng, S. Yang, S. P. Jiang and S. Wang, Small Methods, 2019, 3, 1800440 CrossRef.
- D. F. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- B. Deng, M. Huang, X. Zhao, S. Mou and F. Dong, ACS Catal., 2022, 12, 331–362 CrossRef CAS.
- G. Marcandalli, M. C. O. Monteiro, A. Goyal and M. T. M. Koper, Acc. Chem. Res., 2022, 55, 1900–1911 CrossRef CAS PubMed.
- Y. Matsubara, D. C. Grills and Y. J. A. C. Kuwahara, Journal, 2015, 5, 6440–6452 CAS.
- X. Tan, X. Sun and B. J. N. S. R. Han, Natl. Sci. Rev., 2022, 9, nwab022 CrossRef PubMed.
- L. M. Welch, M. Vijayaraghavan, F. Greenwell, J. Satherley and A. J. Cowan, Faraday Discuss., 2021, 230, 331–343 RSC.
- S. Spittle, D. Poe, B. Doherty, C. Kolodziej, L. Heroux, M. A. Haque, H. Squire, T. Cosby, Y. Zhang, C. Fraenza, S. Bhattacharyya, M. Tyagi, J. Peng, R. A. Elgammal, T. Zawodzinski, M. Tuckerman, S. Greenbaum, B. Gurkan, C. Burda, M. Dadmun, E. J. Maginn and J. Sangoro, Nat. Commun., 2022, 13, 219 CrossRef CAS PubMed.
- G. Zhao, T. Jiang, B. Han, Z. Li, J. Zhang, Z. Liu, J. He and W. Wu, J. Supercrit. Fluids, 2004, 32, 287–291 CrossRef CAS.
- B. Ratschmeier, A. Kemna and B. Braunschweig, ChemElectroChem, 2020, 7, 1765–1774 CrossRef CAS.
- A. Atifi, D. W. Boyce, J. L. DiMeglio and J. Rosenthal, ACS Catal., 2018, 8, 2857–2863 CrossRef CAS PubMed.
- X. Kang, Q. Zhu, X. Sun, J. Hu, J. Zhang, Z. Liu and B. Han, Chem. Sci., 2016, 7, 266–273 RSC.
- A. Leal-Duaso, Y. Adjez and C. M. Sánchez-Sánchez, ChemElectroChem, 2024, 11, e202300771 CrossRef CAS.
- M. G. Freire, C. M. S. S. Neves, I. M. Marrucho, J. A. P. Coutinho and A. M. Fernandes, J. Phys. Chem. A, 2010, 114, 3744–3749 CrossRef CAS PubMed.
- W. Guo, S. Liu, X. Tan, R. Wu, X. Yan, C. Chen, Q. Zhu, L. Zheng, J. Ma, J. Zhang, Y. Huang, X. Sun and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21979–21987 CrossRef CAS PubMed.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Král, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470 CrossRef CAS PubMed.
- V. Vedharathinam, Z. Qi, C. Horwood, B. Bourcier, M. Stadermann, J. Biener and M. Biener, ACS Catal., 2019, 9, 10605–10611 CrossRef CAS.
- T. N. Huan, P. Simon, G. Rousse, I. Génois, V. Artero and M. Fontecave, Chem. Sci., 2017, 8, 742–747 RSC.
- W. Guo, B. Liu and M. A. Gebbie, J. Phys. Chem. C, 2023, 127, 14243–14254 CrossRef CAS.
- A. Fortunati, F. Risplendi, M. Re Fiorentin, G. Cicero, E. Parisi, M. Castellino, E. Simone, B. Iliev, T. J. S. Schubert, N. Russo and S. Hernández, Commun. Chem., 2023, 6, 84 CrossRef CAS PubMed.
- F. Zhou, S. Liu, B. Yang, P. Wang, A. S. Alshammari and Y. Deng, Electrochem. Commun., 2014, 46, 103–106 CrossRef CAS.
- S.-F. Zhao, M. Horne, A. M. Bond and J. Zhang, J. Phys. Chem. C, 2016, 120, 23989–24001 CrossRef CAS.
- C. Chen, X. Sun, X. Yan, Y. Wu, H. Liu, Q. Zhu, B. B. A. Bediako and B. Han, Angew. Chem., Int. Ed., 2020, 59, 11123–11129 CrossRef CAS PubMed.
- Q. Zhu, D. Yang, H. Liu, X. Sun, C. Chen, J. Bi, J. Liu, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 59, 8896–8901 CrossRef CAS PubMed.
- M. Aghaie, N. Rezaei and S. Zendehboudi, Renewable Sustainable Energy Rev., 2018, 96, 502–525 CrossRef CAS.
- J. Feng, S. Zeng, H. Liu, J. Feng, H. Gao, L. Bai, H. Dong, S. Zhang and X. Zhang, ChemSusChem, 2018, 11, 3191–3197 CrossRef CAS PubMed.
- C. Jiang, S. Zeng, X. Ma, J. Feng, G. Li, L. Bai, F. Li, X. Ji and X. Zhang, AIChE J., 2023, 69, e17859 CrossRef CAS.
- S. Dongare, O. K. Coskun, E. Cagli, K. Y. C. Lee, G. Rao, R. D. Britt, L. A. Berben and B. Gurkan, ACS Catal., 2023, 13, 7812–7821 CrossRef CAS PubMed.
- O. K. Coskun, S. Dongare, B. Doherty, A. Klemm, M. Tuckerman and B. Gurkan, Angew. Chem., Int. Ed., 2024, 63, e202312163 CrossRef CAS PubMed.
- L. E. Barrosse-Antle and R. G. Compton, Chem. Commun., 2009, 3744–3746, 10.1039/B906320J.
- S. Seo, M. A. DeSilva and J. F. Brennecke, J. Phys. Chem. B, 2014, 118, 14870–14879 CrossRef CAS PubMed.
- D. V. Vasilyev and P. J. Dyson, ACS Catal., 2021, 11, 1392–1405 CrossRef CAS.
- N. Ahmad, X. Wang, P. Sun, Y. Chen, F. Rehman, J. Xu and X. Xu, Renewable Energy, 2021, 177, 23–33 CrossRef CAS.
- S. Imteyaz, C. M. Suresh, T. Kausar and P. P. Ingole, J. CO2 Util., 2023, 68, 102349 CrossRef CAS.
- Z. Zhang, S. Li, Y. Rao, L. Yang, W. Yan and H. Xu, Chem. Eng. J., 2024, 479, 147376 CrossRef CAS.
- O. K. Coskun, M. Muñoz, S. Dongare, W. Dean and B. E. Gurkan, Langmuir, 2024, 40, 3283–3300 CAS.
- A. Kemna, N. García Rey and B. Braunschweig, ACS Catal., 2019, 9, 6284–6292 CrossRef CAS.
- D. V. Vasilyev, S. Shyshkanov, E. Shirzadi, S. A. Katsyuba, M. K. Nazeeruddin and P. J. Dyson, ACS Appl. Energy Mater., 2020, 3, 4690–4698 CrossRef CAS.
- G. Li, C. Jiang, S. Zeng, K. Peng, L. Yuan, J. Chu and X. Zhang, J. Mol. Liq., 2023, 375, 121392 CrossRef CAS.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- A. W. B. Aylmer-Kelly, A. Bewick, P. R. Cantrill and A. M. Tuxford, Faraday Discuss. Chem. Soc., 1973, 56, 96–107 RSC.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS PubMed.
- Y. Matsubara, D. C. Grills and Y. Kuwahara, ACS Catal., 2015, 5, 6440–6452 CrossRef CAS.
- Y. Matsubara, D. C. Grills and Y. Koide, ACS Omega, 2016, 1, 1393–1411 CrossRef CAS PubMed.
- S. Dongare, O. K. Coskun, E. Cagli, J. S. Stanley, A. Q. Mir, R. S. Brower, J. M. Velázquez, J. Y. Yang, R. L. Sacci and B. Gurkan, Langmuir, 2024, 40, 9426–9438 CrossRef CAS PubMed.
- D. C. Grills, Y. Matsubara, Y. Kuwahara, S. R. Golisz, D. A. Kurtz and B. A. Mello, J. Phys. Chem. Lett., 2014, 5, 2033–2038 CrossRef CAS PubMed.
- M. Dunwell, W. Luc, Y. Yan, F. Jiao and B. Xu, ACS Catal., 2018, 8, 8121–8129 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- J. Newman and N. P. Balsara, Electrochemical systems, John Wiley & Sons, 2021 Search PubMed.
- N. B. Watkins, Z. J. Schiffer, Y. Lai, C. B. Musgrave, III, H. A. Atwater, W. A. Goddard, III, T. Agapie, J. C. Peters and J. M. Gregoire, ACS Energy Lett., 2023, 8, 2185–2192 CrossRef CAS.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed.
- N. A. Shaheen, M. Ijjada, M. B. Vukmirovic and R. Akolkar, J. Electrochem. Soc., 2020, 167, 143505 CrossRef CAS.
- X. Shen, N. Sinclair, J. Wainright and R. F. Savinell, J. Electrochem. Soc., 2021, 168, 056520 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- A. T. Chu, O. Jung, W. L. Toh and Y. Surendranath, J. Am. Chem. Soc., 2023, 145, 9617–9623 CrossRef CAS PubMed.
- C. Wei, S. Sun, D. Mandler, X. Wang, S. Z. Qiao and Z. J. Xu, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- Z. Zhou, X. Hu, J. Li, H. Xie and L. Wen, Adv. Sci., 2024, 11, 2309963 CrossRef CAS PubMed.
- A. Thevenon, A. Rosas-Hernández, A. M. Fontani Herreros, T. Agapie and J. C. Peters, ACS Catal., 2021, 11, 4530–4537 CrossRef CAS.
- G. Shi, L. Yu, X. Ba, X. Zhang, J. Zhou and Y. Yu, Dalton Trans., 2017, 46, 10569–10577 RSC.
- L. Yu, Y. Xie, J. Zhou, Y. Li, Y. Yu and Z. Ren, J. Mater. Chem. A, 2018, 6, 4706–4713 RSC.
- W. Dean, J. Klein and B. Gurkan, J. Electrochem. Soc., 2021, 168, 026503 CrossRef CAS.
- Q. Zhang, Y. Han, Y. Wang, S. Ye and T. Yan, Electrochem. Commun., 2014, 38, 44–46 CrossRef CAS.
- S. Baldelli, Acc. Chem. Res., 2008, 41, 421–431 CrossRef CAS PubMed.
- V. O. Santos, M. B. Alves, M. S. Carvalho, P. A. Z. Suarez and J. C. Rubim, J. Phys. Chem. B, 2006, 110, 20379–20385 CrossRef CAS PubMed.
- J.-Y. Fu, X.-C. Li, Z. Yu, X.-N. Huang-Fu, J.-A. Fan, Z.-Q. Zhang, S. Huang, J.-F. Zheng, Y.-H. Wang and X.-S. Zhou, Langmuir, 2022, 38, 6209–6216 CrossRef CAS PubMed.
- J. M. Klein, H. Squire and B. Gurkan, J. Phys. Chem. C, 2020, 124, 5613–5623 CrossRef CAS.
- W. Dean, D. Penley, Y.-Y. Lee, R. Ghahremani, S. Dongare and B. Gurkan, J. Phys. Chem. C, 2022, 126, 14598–14610 CrossRef CAS.
- R. Michez, T. Doneux, C. Buess-Herman and M. Luhmer, ChemPhysChem, 2017, 18, 2208–2216 CrossRef CAS PubMed.
- N. G. Rey and D. D. Dlott, J. Electroanal. Chem., 2017, 800, 114–125 CrossRef CAS.
- B. Braunschweig, P. Mukherjee, J. L. Haan and D. D. Dlott, J. Electroanal. Chem., 2017, 800, 144–150 CrossRef CAS.
- N. García Rey and D. D. Dlott, Phys. Chem. Chem. Phys., 2017, 19, 10491–10501 RSC.
- M. Papasizza and A. Cuesta, ACS Catal., 2018, 8, 6345–6352 CrossRef CAS.
- D. Vasilyev, E. Shirzadi, A. V. Rudnev, P. Broekmann and P. J. Dyson, ACS Appl. Energy Mater., 2018, 1, 5124–5128 CAS.
- J. T. Feaster, A. L. Jongerius, X. Liu, M. Urushihara, S. A. Nitopi, C. Hahn, K. Chan, J. K. Nørskov and T. F. Jaramillo, Langmuir, 2017, 33, 9464–9471 CrossRef CAS PubMed.
- R. Michez, J. Vander Steen, T. Doneux, M. Luhmer and C. Buess-Herman, Electrochim. Acta, 2018, 270, 434–439 CrossRef CAS.
- Y. Wang, M. Hatakeyama, K. Ogata, M. Wakabayashi, F. Jin and S. Nakamura, Phys. Chem. Chem. Phys., 2015, 17, 23521–23531 RSC.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- M. Urushihara, K. Chan, C. Shi and J. K. Nørskov, J. Phys. Chem. C, 2015, 119, 20023–20029 CrossRef CAS.
- H.-K. Lim, Y. Kwon, H. S. Kim, J. Jeon, Y.-H. Kim, J.-A. Lim, B.-S. Kim, J. Choi and H. Kim, ACS Catal., 2018, 8, 2420–2427 CrossRef CAS.
- Y. Hu, J. Feng, X. Zhang, H. Gao, S. Jin, L. Liu and W. Shen, ChemistrySelect, 2021, 6, 9873–9879 CrossRef CAS.
- J. Jang, M. Rüscher, M. Winzely and C. G. Morales-Guio, AIChE J., 2022, 68, e17605 CrossRef CAS.
- G. Leverick, E. M. Bernhardt, A. I. Ismail, J. H. Law, A. Arifutzzaman, M. K. Aroua and B. M. Gallant, ACS Catal., 2023, 13, 12322–12337 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- K. Shen, D. Cheng, E. Reyes-Lopez, J. Jang, P. Sautet and C. G. Morales-Guio, Joule, 2023, 7, 1260–1276 CrossRef CAS.
- J. Safipour, A. Z. Weber and A. T. Bell, ACS Energy Lett., 2023, 8, 5012–5017 CrossRef CAS.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
- Q. Xu, S. Liu, F. Longhin, G. Kastlunger, I. Chorkendorff and B. Seger, Adv. Mater., 2024, 36, 2306741 CrossRef CAS PubMed.
- Z. Wang, Y. Zhou, P. Qiu, C. Xia, W. Fang, J. Jin, L. Huang, P. Deng, Y. Su, R. Crespo-Otero, X. Tian, B. You, W. Guo, D. Di Tommaso, Y. Pang, S. Ding and B. Y. Xia, Adv. Mater., 2023, 35, 2303052 CrossRef CAS PubMed.
- H. Shen, Z. Gu and G. Zheng, Sci. Bull., 2019, 64, 1805–1816 CrossRef CAS PubMed.
- D. Ma, T. Jin, K. Xie and H. Huang, J. Mater. Chem. A, 2021, 9, 20897–20918 RSC.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- Q. Chen, X. Wang, Y. Zhou, Y. Tan, H. Li, J. Fu and M. Liu, Adv. Mater., 2024, 36, 2303902 CrossRef CAS PubMed.
- Y. Cao, Z. Chen, P. Li, A. Ozden, P. Ou, W. Ni, J. Abed, E. Shirzadi, J. Zhang, D. Sinton, J. Ge and E. H. Sargent, Nat. Commun., 2023, 14, 2387 CrossRef CAS PubMed.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- K. Fernández-Caso, G. Díaz-Sainz, M. Alvarez-Guerra and A. Irabien, ACS Energy Lett., 2023, 8, 1992–2024 CrossRef.
- T. Fan, W. Ma, M. Xie, H. Liu, J. Zhang, S. Yang, P. Huang, Y. Dong, Z. Chen and X. Yi, Cell Rep. Phys. Sci., 2021, 2, 100353 CrossRef CAS.
- I. Grigioni, L. K. Sagar, Y. C. Li, G. Lee, Y. Yan, K. Bertens, R. K. Miao, X. Wang, J. Abed, D. H. Won, F. P. García de Arquer, A. H. Ip, D. Sinton and E. H. Sargent, ACS Energy Lett., 2021, 6, 79–84 CrossRef CAS.
- Y. Luo, K. Zhang, Y. Li and Y. Wang, InfoMat, 2021, 3, 1313–1332 CrossRef CAS.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2022, 7, 55–64 CrossRef CAS.
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109 CrossRef CAS.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- B. Endrődi, G. Bencsik, F. Darvas, R. Jones, K. Rajeshwar and C. Janáky, Prog. Energy Combust. Sci., 2017, 62, 133–154 CrossRef.
- M. Alvarez-Guerra, J. Albo, E. Alvarez-Guerra and A. Irabien, Energy Environ. Sci., 2015, 8, 2574–2599 RSC.
- M. König, J. Vaes, E. Klemm and D. Pant, iScience, 2019, 19, 135–160 CrossRef PubMed.
- D. Yang, Q. Zhu and B. Han, Innovation, 2020, 1, 100016 CAS.
- A. Salehi-Khojin, H.-R. M. Jhong, B. A. Rosen, W. Zhu, S. Ma, P. J. A. Kenis and R. I. Masel, J. Phys. Chem. C, 2013, 117, 1627–1632 CrossRef CAS.
- S. S. Neubauer, B. Schmid, C. Reller, D. M. Guldi and G. Schmid, ChemElectroChem, 2017, 4, 160–167 CrossRef CAS.
- Y. Miao, N. Siri-Nguan, T. Sornchamni, G. N. Jovanovic and A. F. Yokochi, Chem. Eng. J., 2018, 333, 300–309 CrossRef CAS.
- L. Yuan, L. Zhang, J. Feng, C. Jiang, J. Feng, C. Li, S. Zeng and X. Zhang, Chem. Eng. J., 2022, 450, 138378 CrossRef CAS.
- S. Wu, Y. Hua, J. Zhang, F. Shen, W. Song, X. Zhang, J. Liu, B. Yang, Y. Dai and J. Shi, Energy Fuels, 2022, 36, 3771–3777 CrossRef CAS.
- I. A. Digdaya, I. Sullivan, M. Lin, L. Han, W.-H. Cheng, H. A. Atwater and C. Xiang, Nat. Commun., 2020, 11, 4412 CrossRef CAS PubMed.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- S. Messias, M. M. Sousa, M. Nunes da Ponte, C. M. Rangel, T. Pardal and A. S. Reis Machado, React. Chem. Eng., 2019, 4, 1982–1990 RSC.
- M. Gautam, F. Nkurunziza, M. C. Mulvehill, S. S. Uttarwar, D. T. Hofsommer, C. A. Grapperhaus and J. M. Spurgeon, ChemSusChem, 2023, e202301337 Search PubMed.
- M. König, S.-H. Lin, J. Vaes, D. Pant and E. Klemm, Faraday Discuss., 2021, 230, 360–374 RSC.
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, ECS Trans., 2017, 77, 1425 CrossRef CAS.
- C. Jiang, A. W. Nichols, J. F. Walzer and C. W. Machan, Inorg. Chem., 2020, 59, 1883–1892 CrossRef CAS PubMed.
- Q. Xia, K. Zhang, T. Zheng, L. An, C. Xia and X. Zhang, ACS Energy Lett., 2023, 8, 2840–2857 CrossRef CAS.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS.
- K. M. Diederichsen, R. Sharifian, J. S. Kang, Y. Liu, S. Kim, B. M. Gallant, D. Vermaas and T. A. Hatton, Nat. Rev. Methods Primers, 2022, 2, 68 CrossRef CAS.
- J. H. Rheinhardt, P. Singh, P. Tarakeshwar and D. A. Buttry, ACS Energy Lett., 2017, 2, 454–461 CrossRef CAS.
- F. Simeon, M. C. Stern, K. M. Diederichsen, Y. Liu, H. J. Herzog and T. A. Hatton, J. Phys. Chem. C, 2022, 126, 1389–1399 CrossRef CAS.
- K. M. Diederichsen, Y. Liu, N. Ozbek, H. Seo and T. A. Hatton, Joule, 2022, 6, 221–239 CrossRef CAS.
- M. Wang, M. Rahimi, A. Kumar, S. Hariharan, W. Choi and T. A. Hatton, Appl. Energy, 2019, 255, 113879 CrossRef CAS.
- F.-Y. Kuo, S. E. Jerng and B. M. Gallant, ACS Cent. Sci., 2023, 9, 1750–1757 CrossRef CAS PubMed.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- M. Pellerite, M. Kaplun, C. Hartmann-Thompson, K. A. Lewinski, N. Kunz, T. Gregar, J. Baetzold, D. Lutz, M. Quast, Z. Liu, H. Yang, S. D. Sajjad, Y. Gao and R. Masel, ECS Trans., 2017, 80, 945 CrossRef CAS.
- L. Xue, Z. Gao, T. Ning, W. Li, J. Li, J. Yin, L. Xiao, G. Wang and L. Zhuang, Angew. Chem., Int. Ed., 2023, 62, e202309519 CrossRef CAS PubMed.
- M. Esmaeilirad, Z. Jiang, A. M. Harzandi, A. Kondori, M. Tamadoni Saray, C. U. Segre, R. Shahbazian-Yassar, A. M. Rappe and M. Asadi, Nat. Energy, 2023, 8, 891–900 CrossRef CAS.
- E. P. Delmo, Y. Wang, J. Wang, S. Zhu, T. Li, X. Qin, Y. Tian, Q. Zhao, J. Jang, Y. Wang, M. Gu, L. Zhang and M. Shao, Chin. J. Catal., 2022, 43, 1687–1696 CrossRef CAS.
- J. Choi, T. M. Benedetti, R. Jalili, A. Walker, G. G. Wallace and D. L. Officer, Chem. – Eur. J., 2016, 22, 14158–14161 CrossRef CAS PubMed.
- T. Yoshida, K. Tsutsumida, S. Teratani, K. Yasufuku and M. Kaneko, J. Chem. Soc., Chem. Commun., 1993, 631–633, 10.1039/C39930000631.
- E. Vichou, Y. Li, M. Gomez-Mingot, M. Fontecave and C. M. Sánchez-Sánchez, J. Phys. Chem. C, 2020, 124, 23764–23772 CrossRef CAS.
- J. Honores, D. Quezada, M. García, K. Calfumán, J. P. Muena, M. J. Aguirre, M. C. Arévalo and M. Isaacs, Green Chem., 2017, 19, 1155–1162 RSC.
- E. Vichou, A. Solé-Daura, C. Mellot-Draznieks, Y. Li, M. Gomez-Mingot, M. Fontecave and C. M. Sánchez-Sánchez, ChemSusChem, 2022, 15, e202201566 CrossRef CAS PubMed.
- A. Khadhraoui, P. Gotico, B. Boitrel, W. Leibl, Z. Halime and A. Aukauloo, Chem. Commun., 2018, 54, 11630–11633 RSC.
- S. Sung, D. Kumar, M. Gil-Sepulcre and M. Nippe, J. Am. Chem. Soc., 2017, 139, 13993–13996 CrossRef CAS PubMed.
- S. Sung, X. Li, L. M. Wolf, J. R. Meeder, N. S. Bhuvanesh, K. A. Grice, J. A. Panetier and M. Nippe, J. Am. Chem. Soc., 2019, 141, 6569–6582 CrossRef CAS PubMed.
- D. Grammatico, H. N. Tran, Y. Li, S. Pugliese, L. Billon, B.-L. Su and M. Fontecave, ChemSusChem, 2020, 13, 6418–6425 CrossRef CAS PubMed.
- D. Quezada, J. Honores, M. J. Aguirre and M. Isaacs, J. Coord. Chem., 2014, 67, 4090–4100 CrossRef CAS.
- C. Römelt, J. Song, M. Tarrago, J. A. Rees, M. van Gastel, T. Weyhermüller, S. DeBeer, E. Bill, F. Neese and S. Ye, Inorg. Chem., 2017, 56, 4745–4750 CrossRef PubMed.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti, G. Harrison and E. Tzimas, Int. J. Hydrogen Energy, 2016, 41, 16444–16462 CrossRef.
- D. Segets, C. Andronescu and U.-P. Apfel, Nat. Commun., 2023, 14, 7950 CrossRef CAS PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2020, 59, 8121–8123 CrossRef CAS.
- A. Somoza-Tornos, O. J. Guerra, A. M. Crow, W. A. Smith and B.-M. Hodge, iScience, 2021, 24, 102813 CrossRef CAS PubMed.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustainable, 2021, 4, 911–919 CrossRef.
- T. Gao, B. Xia, K. Yang, D. Li, T. Shao, S. Chen, Q. Li and J. Duan, Energy Fuels, 2023, 37, 17997–18008 CrossRef CAS.
- G. Leonzio, B. Chachuat and N. Shah, Sustain. Prod. Consum., 2023, 43, 124–139 CrossRef.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- Á. Vass, B. Endrődi and C. Janáky, Curr. Opin. Electrochem., 2021, 25, 100621 CrossRef.
- J. Sisler, S. Khan, A. H. Ip, M. W. Schreiber, S. A. Jaffer, E. R. Bobicki, C.-T. Dinh and E. H. Sargent, ACS Energy Lett., 2021, 6, 997–1002 CrossRef CAS.
- J. W. Sun, H. Q. Fu, P. F. Liu, A. Chen, P. Liu, H. G. Yang and H. Zhao, EES Catal., 2023, 1, 934–949 RSC.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- J. M. Spurgeon, N. Theaker, C. A. Phipps, S. S. Uttarwar and C. A. Grapperhaus, ACS Sustainable Chem. Eng., 2022, 10, 12882–12894 CrossRef CAS.
- Y. Li, F. Li, A. Laaksonen, C. Wang, P. Cobden, P. Boden, Y. Liu, X. Zhang and X. Ji, Ind. Chem. Mater., 2023, 1, 410–430 RSC.
- N. Gao, C. Quiroz-Arita, L. A. Diaz and T. E. Lister, J. CO2 Util., 2021, 43, 101365 CrossRef CAS.
- M. Li, E. Irtem, H.-P. Iglesias van Montfort, M. Abdinejad and T. Burdyny, Nat. Commun., 2022, 13, 5398 CrossRef CAS PubMed.
- H. Weingärtner, Angew. Chem., Int. Ed., 2008, 47, 654–670 CrossRef PubMed.
- M. D. Soutullo, C. I. Odom, B. F. Wicker, C. N. Henderson, A. C. Stenson and J. H. Davis, Chem. Mater., 2007, 19, 3581–3583 CrossRef CAS.
- Y. Zhang, S. Zhang, X. Lu, Q. Zhou, W. Fan and X. Zhang, Chem. – Eur. J., 2009, 15, 3003–3011 CrossRef CAS PubMed.
- X. Li, M. Hou, Z. Zhang, B. Han, G. Yang, X. Wang and L. Zou, Green Chem., 2008, 10, 879–884 RSC.
- G. Yu, S. Zhang, G. Zhou, X. Liu and X. Chen, AIChE J., 2007, 53, 3210–3221 CrossRef CAS.
- J. Kagimoto, K. Noguchi, K. Murata, K. Fukumoto, N. Nakamura and H. Ohno, Chem. Lett., 2008, 37, 1026–1027 CrossRef CAS.
- Y.-Y. Lee, K. Edgehouse, A. Klemm, H. Mao, E. Pentzer and B. Gurkan, ACS Appl. Mater. Interfaces, 2020, 12, 19184–19193 CrossRef CAS PubMed.
- R. E. Siegel, S. Pattanayak and L. A. Berben, ACS Catal., 2023, 13, 766–784 CrossRef CAS.
- J. Huang and T. Rüther, Aust. J. Chem., 2009, 62, 298–308 CrossRef CAS.
- Q. Yang, Z. Wang, Z. Bao, Z. Zhang, Y. Yang, Q. Ren, H. Xing and S. Dai, ChemSusChem, 2016, 9, 806–812 CrossRef CAS PubMed.
- X. Y. Luo, F. Ding, W. J. Lin, Y. Q. Qi, H. R. Li and C. M. Wang, J. Phys. Chem. Lett., 2014, 5, 381–386 CrossRef CAS PubMed.
- Z. Xue, Z. Zhang, J. Han, Y. Chen and T. Mu, Int. J. Greenhouse Gas Control, 2011, 5, 628–633 CrossRef CAS.
- Z. Zhou, X. Zhou, G. Jing and B. Lv, Energy Fuels, 2016, 30, 7489–7495 CrossRef CAS.
- B. Lv, Y. Shi, C. Sun, N. Liu, W. Li and S. Li, Chem. Eng. J., 2015, 270, 372–377 CrossRef CAS.
- K. Anderson, M. P. Atkins, J. Estager, Y. Kuah, S. Ng, A. A. Oliferenko, N. V. Plechkova, A. V. Puga, K. R. Seddon and D. F. Wassell, Green Chem., 2015, 17, 4340–4354 RSC.
- C.-J. Yoo, S. J. Park and C. W. Jones, Ind. Eng. Chem. Res., 2020, 59, 7061–7071 CrossRef CAS.
- P. Bollini, S. Choi, J. H. Drese and C. W. Jones, Energy Fuels, 2011, 25, 2416–2425 CrossRef CAS.
- J. S. A. Carneiro, G. Innocenti, H. J. Moon, Y. Guta, L. Proaño, C. Sievers, M. A. Sakwa-Novak, E. W. Ping and C. W. Jones, Angew. Chem., Int. Ed., 2023, 62, e202302887 CrossRef CAS PubMed.
- Y. Chen, D. Yu, W. Chen, L. Fu and T. Mu, Phys. Chem. Chem. Phys., 2019, 21, 2601–2610 RSC.
- T. Zhekenov, N. Toksanbayev, Z. Kazakbayeva, D. Shah and F. S. Mjalli, Fluid Phase Equilib., 2017, 441, 43–48 CrossRef CAS.
- H. Nigar, B. Garcia-Baños, F. L. Peñaranda-Foix, J. M. Catalá-Civera, R. Mallada and J. Santamaría, AIChE J., 2016, 62, 547–555 CrossRef CAS.
- J. F. Brennecke and B. E. Gurkan, J. Phys. Chem. Lett., 2010, 1, 3459–3464 CrossRef CAS.
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC.
- B. Wang, L. Qin, T. Mu, Z. Xue and G. Gao, Chem. Rev., 2017, 117, 7113–7131 CrossRef CAS PubMed.
- M. Feroci, M. Orsini and A. Inesi, Adv. Synth. Catal., 2009, 351, 2067–2070 CrossRef CAS.
- S. Doblinger, T. J. Donati and D. S. Silvester, J. Phys. Chem. C, 2020, 124, 20309–20319 CrossRef CAS.
- M. S. E. Houache, R. Safari, U. O. Nwabara, T. Rafaïdeen, G. A. Botton, P. J. A. Kenis, S. Baranton, C. Coutanceau and E. A. Baranova, ACS Appl. Energy Mater., 2020, 3, 8725–8738 CrossRef CAS.
- X. Wei, Y. Li, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 3148–3155 CrossRef CAS PubMed.
- W. A. Parada, D. V. Vasilyev, K. J. J. Mayrhofer and I. Katsounaros, ACS Appl. Mater. Interfaces, 2022, 14, 14193–14201 CrossRef CAS PubMed.
- J. H. Cho, C. Lee, S. H. Hong, H. Y. Jang, S. Back, M.-G. Seo, M. Lee, H.-K. Min, Y. Choi, Y. J. Jang, S. H. Ahn, H. W. Jang and S. Y. Kim, Adv. Mater., 2023, 35, 2208224 CrossRef CAS PubMed.
- National Academies of Sciences, Division on Earth, Life Studies, Ocean Studies Board, Board on Chemical Sciences, Board on Earth Sciences, Board on Energy et al. Negative emissions technologies and reliable sequestration: a research agenda. (2019).
- S. Perkin, Phys. Chem. Chem. Phys., 2012, 14, 5052–5062 RSC.
- C. Pinilla, M. G. Del Pópolo, R. M. Lynden-Bell and J. Kohanoff, J. Phys. Chem. B, 2005, 109, 17922–17927 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |