
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding photochemical degradation mechanisms in photoactive layer materials for organic solar cells
Jianhua
Han
*ab,
Han
Xu
a,
Sri Harish Kumar
Paleti
ac,
Anirudh
Sharma
 a and
Derya
Baran
*a
a and
Derya
Baran
*a
aMaterials Science and Engineering Program (MSE), Physical Science and Engineering Division (PSE), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: derya.baran@kaust.edu.sa
bInstitut für Anorganische Chemie and Institute for Sustainable Chemistry & Catalysis with Boron (ICB), Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: jianhua.han@uni-wuerzburg.de
cDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 41296, Göteborg, Sweden
First published on 13th June 2024
Abstract
Over the past decades, the field of organic solar cells (OSCs) has witnessed a significant evolution in materials chemistry, which has resulted in a remarkable enhancement of device performance, achieving efficiencies of over 19%. The photoactive layer materials in OSCs play a crucial role in light absorption, charge generation, transport and stability. To facilitate the scale-up of OSCs, it is imperative to address the photostability of these electron acceptor and donor materials, as their photochemical degradation process remains a challenge during the photo-to-electric conversion. In this review, we present an overview of the development of electron acceptor and donor materials, emphasizing the crucial aspects of their chemical stability behavior that are linked to the photostability of OSCs. Throughout each section, we highlight the photochemical degradation pathways for electron acceptor and donor materials, and their link to device degradation. We also discuss the existing interdisciplinary challenges and obstacles that impede the development of photostable materials. Finally, we offer insights into strategies aimed at enhancing photochemical stability and discuss future directions for developing photostable photo-active layers, facilitating the commercialization of OSCs.
 Jianhua Han | Dr. Jianhua Han completed his BSc at East China University of Science and Technology, and PhD at Dalian University of Technology under the supervision of Prof. Jinyan Wang and Xigao Jian. Following his doctoral studies, he conducted postdoctoral research with Prof. Renqiang Yang at Qingdao Institute of Bioenergy and Bioprocess Technology, and then moved to the group of Prof. Derya Baran at KAUST. In 2022, he joined the research group of Prof. Todd B. Marder at Universität Würzburg as an Alexander von Humboldt Postdoctoral Fellow. His research interests cover synthetic chemistry and polymer materials for stable organic electronic devices. |
 Han Xu | Han Xu completed his BSc at Zhejiang University and his MSc at Northeast Normal University. Currently, he is pursuing a PhD degree under the supervision of Prof. Derya Baran at King Abdullah University of Science and Technology. His main research interests lie in developing stable organic solar cells. |
 Sri Harish Kumar Paleti | Dr. Sri Harish Kumar Paleti is currently pursuing his post-doctoral studies in Prof. Christian Müller's group at Chalmers University of Technology. He holds a PhD degree in Materials Science and Engineering from King Abdullah University of Science and Technology (2022) under the supervision of Prof. Derya Baran and an MSc (Hons.) in Advanced Materials and Processes from the Friedrich Alexander University Erlangen Nürnberg (2017). His research interests include the stability of organic photovoltaic devices as well as doping of organic semiconductors. |
 Anirudh Sharma | Dr. Anirudh Sharma earned his PhD from Flinders University, South Australia, specializing in Nanotechnology and Surface Physics. He conducted subsequent research at the University of South Australia and the University of Bordeaux, France. Anirudh was the recipient of the Common Wealth Scientific and Industrial Research Organization (CSIRO) Future Manufacturing Flagship fellowship and has also served as a visiting researcher at the Max Planck Institute for Polymer Research. Currently, he is a research fellow at KAUST in Prof. Baran's research group OMEGALAB. His expertise lies in photovoltaics, surface physics, and the thermomechanical properties of organic semiconductors. |
 Derya Baran | Prof. Derya Baran is an Associate Professor in Materials Science and Engineering Program at KAUST. She has completed her PhD at Friedrich Alexander University Erlangen Nürnberg in 2014, followed by a joint Helmholtz Postdoctoral fellowship between Julich Research Center and Imperial College London. Prof. Baran's research group OMEGALAB focuses on the processable semiconductors for electronic applications and materials for printed electronics. |
1. Introduction
Solution-processed organic solar cells (OSCs) demonstrate great potential for sustainable applications in solar power generation,1–3 offering various advantages including light weight, mechanical flexibility, utilization of low-cost materials, “green-solvent” fabrication processes, and compatibility with the integration with large-area, flat, or curved electronic devices.4–7 In recent years, numerous chemists have made a dedicated effort to develop electron acceptor (A) and donor (D) materials and boost device performances to over 19% efficiency.8–14 For high-performance OSCs, the incorporation of a bulk heterojunction (BHJ) photoactive layer is crucial.10,15,16 The BHJ layer consists of a continuous network of A and D materials, which can facilitate efficient exciton dissociation, charge generation, and transportation processes.15,17–19 The community has witnessed several well-established models and correlations between the molecular structures of photovoltaic (PV) materials and device performance, enabling the continuous improvement of power conversion efficiencies (PCEs) over the past two decades.10,11,20 However, the commercialization of high-performance OSCs with large-scale production and long-term stability remains a challenge in the field.5,21,22 This challenge arises not only from cost-effectiveness concerns but also from the need for enhanced stability.3,23–26 Therefore, to overcome these hurdles and identify stable materials, it is essential to comprehend the degradation mechanisms and establish correlations between chemical structures and device stability. Such understanding can guide material design and minimize the need for optimization via trial-and-error synthetic procedures that are often complex and time-consuming.25–28Several factors that can influence the stability of OSCs as shown in Fig. 1 are mechanical stability (for flexible and stretchable devices), morphological stability under heat stress, solar irradiance, oxygen exposure, and moisture ingress.24,29,30 Generally, OSC devices consist of electrodes, transport layers, and active layers. All these components in devices would suffer from environmental stress, thus impacting the overall stability of devices. It is well known that during the photo-to-electric conversion process, photovoltaic materials are inevitably subjected to illumination, either from indoor LED lighting or outdoor solar irradiance. Therefore, it is crucial to employ a D/A combination with excellent photochemical stability for the successful commercialization of OSCs. Notably, the illumination also increases the temperature of the device during operation, subsequently influencing the thermally triggered morphological stability. It is important to differentiate between photostability and heat-induced morphological stability (thermal stability), following the International Summit on Organic Photovoltaic Stability (ISOS) protocol, which can be assessed through photostability measurements (such as ISOS-L) and thermostability measurements (such as ISOS-D), respectively.31–34 Generally, heat-induced morphological stability (thermal stability) is related to physical thermodynamic behavior (mostly w/o a chemical process).27,35 On the other hand, the photochemical stability of PV materials under illumination plays an important role in the photodegradation of OSC devices. Therefore, the photochemical degradation pathways of these materials are crucial for understanding the photodegradation mechanism of OSCs.
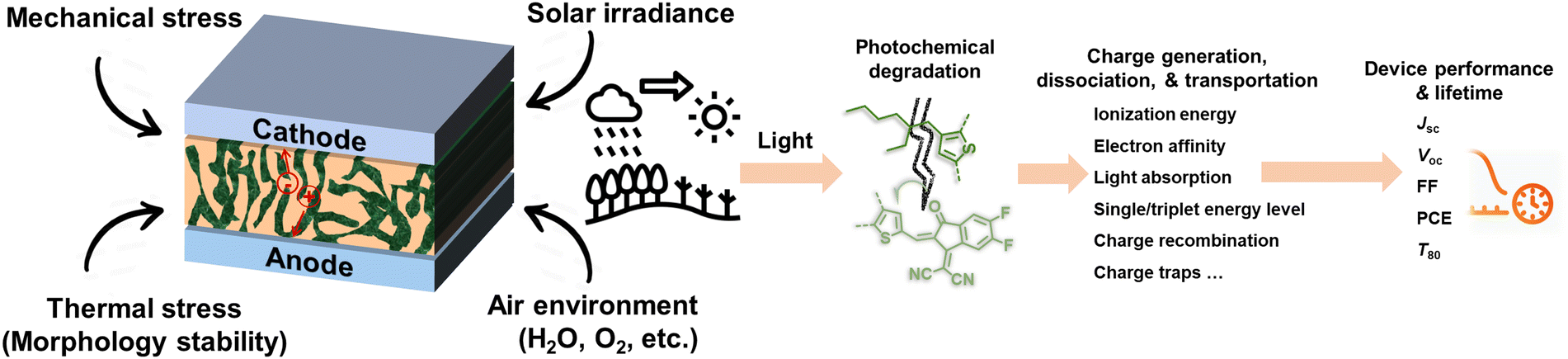 | ||
| Fig. 1 Environmental factors and light-induced degradation of OSCs. | ||
This review primarily focuses on the light-induced photochemical degradation of PV materials, aiming to provide guidance for synthetic chemists and the OSC community in designing stable OSCs and minimizing trial-and-error efforts. The heat-induced morphological changes (thermal stability) that occur during photostability measurements and under outdoor conditions are not covered in this review. Such thermal stability is well reviewed and can be found in the related ref. 27, 35 and 36. Generally, solar irradiance and illumination impact photostability in the following ways: (i) triggering photo-chemical reactions in photovoltaic molecules, (ii) causing changes in their photo-physical properties, and (iii) introducing additional traps and recombination sites, and leading to device degradation. As illustrated in Fig. 1, the photostability of OSCs is governed by the photochemical stability of these photovoltaic materials.29,37–40 The related light-driven nm-scale morphological changes will be discussed in Section 5. It is worth mentioning that the chemical reactions between PV materials and additives can influence the device stability,20 which is also discussed in this review (Section 5.1).
The review begins with an overview of the development of OSCs and the advancements achieved through the design of photovoltaic acceptors and donors, resulting in remarkable PCEs from 2% to over 19%.3,10 We then provide a summary of the chemical stability of electron acceptors (Section 2), including PCBM-, PDI-, IDTBR-, ITIC-, and BTP-family acceptors as shown in Fig. 2. Then, we focus on the photochemical stability of polymer donors in Section 3, covering from polymer donors P3HT, PM6, to D18 (chemical structures detailed in Fig. 2, with corresponding abbreviations provided at the end of this review). We present the photo-degradation mechanisms of these photovoltaic acceptors and donors and discuss how light-induced chemical reactions impact molecular structural changes, leading to device degradation. Furthermore, we highlight the photochemical degradation pathways of small molecular donors, polymer acceptors and single-component materials (Section 4), which have been overlooked within the community. Finally, we discuss strategies to prevent the photochemical degradation of PV materials and the outstanding challenges associated with designing stable materials with future perspectives for OSCs as a green energy source towards commercialization. Although the molecules discussed in this review are mainly used in OSCs, the insights into the relationship between the photochemical stability of organic semiconductors and device stability will also be relevant for a range of other organic semiconductors used in organic photodetectors, organic light-emitting diodes, and integrated organic electronics for light-related device applications.
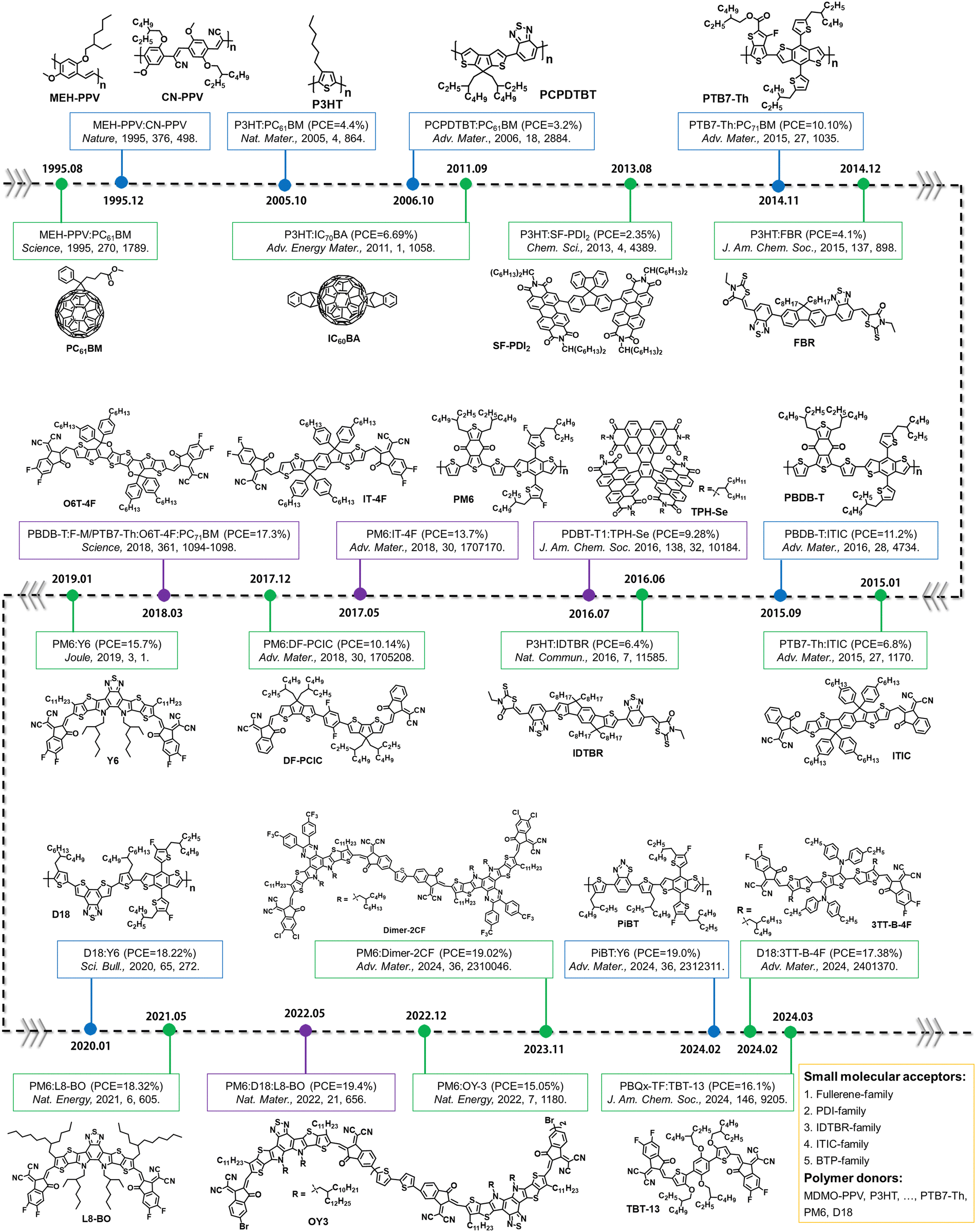 | ||
| Fig. 2 Evolution of electron acceptors and polymer donors for OSCs. Since 1995, the development of OSCs has been driven by small molecular acceptors and polymer donors,8,41,42,43,44–64 and the investigation of precise photochemical degradation pathways was primarily focused on the small molecular acceptors and polymer donors as model systems (see Sections 2 and 3). Other PV materials (small molecular donors, polymer acceptors, and single-component materials) are presented and discussed in Section 4. | ||
2. Chemical stability and degradation mechanisms of electron acceptors
As depicted in Fig. 2, research on OSCs has revolved around emerging acceptors, including the classic PCBM-family, ITIC-family, and the more recent BTP-family acceptors with PCEs over 19%,3,11 along with dimer/multimer acceptors and low-cost non-fused ring acceptors.12,14,16,65–79 It is well known that light can induce photochemical reactions, leading to the formation of photodegradation products.24,37,38,80 Furthermore, the presence of O2 and H2O during illumination can facilitate these photochemical reactions and also result in the generation of additional photodegraded products.29,81 Consequently, the photodegradation mechanisms and photochemical stability of photovoltaic materials may differ when exposed to ambient air or an inert environment. During large-scale manufacturing of OSCs, the photoactive layers will inevitably encounter ambient conditions (O2 and H2O), and a photostable material under ambient air can also reduce the manufacturing costs. For the long-term applications, high-quality encapsulation for OSC devices can efficiently prevent oxygen and moisture from the diffusion into the active layers, and thereby improve device lifetime.24,82,83 Therefore, it is crucial to understand the photodegradation mechanisms of PV materials both with and without O2/H2O environments. Given these considerations, the following section focuses on reviewing the chemical stability and photodegradation mechanisms of the electron acceptors in ambient air and an O2-free inert environments.2.1 Fullerene derivatives
Electron acceptors based on fullerene derivatives, particularly PC61BM and PC71BM (the abbreviations provided at the end of this review), were widely used in the early stages of OSC research, leading to single-junction BHJ OSCs with PCEs exceeding 11%.84,85 As illustrated in Fig. 3(a), in the presence of light and O2, the fullerene-based derivatives are prone to undergo a photo-oxidation process, primarily affecting the fullerene cage. This photodegradation can occur even under low light conditions such as sample preparation and ambient characterization in laboratory environments.38 During the photochemical degradation process under illumination, PCBM derivatives form various oxidative defects within the fullerene cage, such as epoxide, diol, and carbonyl structures, as confirmed experimentally through matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF), ultraviolet photoelectron spectroscopy (UPS), and near-edge X-ray absorption fine structure (NEXAFS) analysis with the theoretical support of density functional theory (DFT) calculations.38,86 As shown in Fig. 3(b) and (c), the presence of oxidized PCBM in the active layers increases energetic disorder. Li et al. have quantitatively correlated the fraction of oxidized fullerene with decreased electron mobility and charge lifetime.38 The increasing number of additional trap states induced by oxidized PCBM contributes to degradation in charge carrier densities, electron transport, and recombination, leading to losses in open-circuit voltage (Voc), short-circuit current (Jsc), and fill factor (FF) of the devices.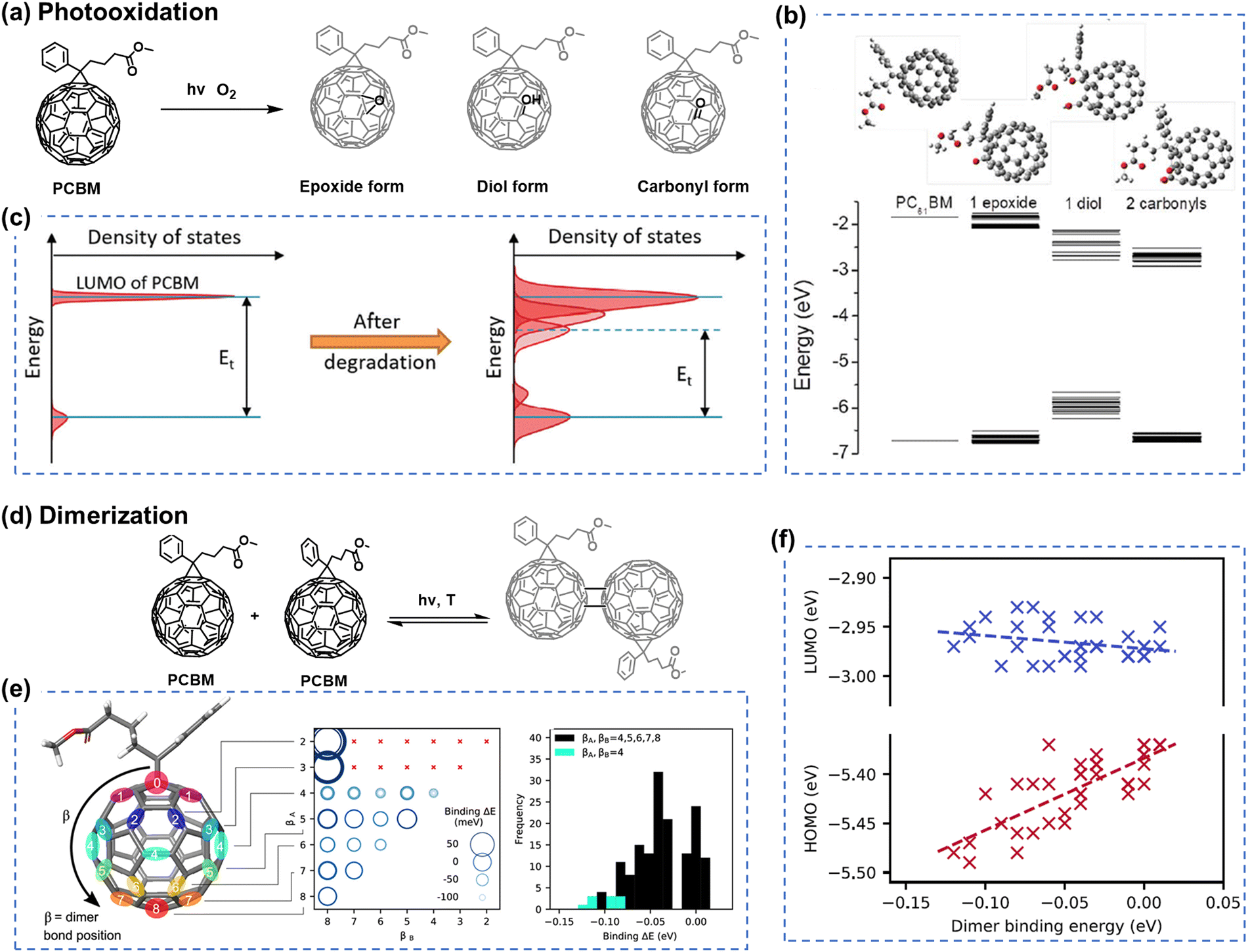 | ||
| Fig. 3 (a) Photooxidation process of PCBM under combined light and oxygen stresses. (b) Energy levels of the HOMO and LUMO for the neat PCBM and various photo-oxidated PCBMs with one epoxide, one diol, or two carbonyl defects, highlighting various positions on the fullerene cage. (c) Schematic illustrating the energetic disorder induced by photodegradation products.38 Reproduced from ref. 38 with permission from RSC Publishing, Copyright 2018. (d) Dimerization of PCBM under light and thermal stresses. (e) DFT calculations of potential PCBM dimer configurations. Reactive sites at double bonds between hexagonal faces, featuring 8 classes with equivalent distances from the PCBM tail unit. (f) Dependency of the PCBM dimer binding energy on each fullerene class presented in Fig. 3(e). These energetic disorders can induce the performance degradation in OSCs.87 Reproduced from ref. 87 with permission from the American Chemical Society, Copyright 2019. | ||
Besides the photochemical reactions of electron acceptors exposed to light and oxygen, Li et al. have demonstrated that the formation of superoxide radical ions in the active layers is another crucial factor that controls the photostability of electron acceptors.88 The correlation is observed between the lowest unoccupied molecular orbital (LUMO) level of electron acceptors and the resulting environmental stability of OSCs, influenced by the formation of superoxide in the blends. Consequently, electron acceptors with deeper LUMOs and suppressed oxidation processes are desired to achieve long-term stability for fullerene-based OSCs in the presence of O2.
When transitioning to an environment without O2, as depicted in Fig. 3(d), fullerenes tend to form dimers under illumination. Interestingly, Durrant et al. have demonstrated that fullerenes can form different types of dimers, ranging from weak to strongly bound, depending on the intensity of the illumination.87 These distinct dimers exhibit different LUMO and highest occupied molecular orbital (HOMO) levels, as shown in Fig. 3(e) and (f). Additionally, it has been proven that this dimerization process is reversible under light and heat stress.89 Therefore, light-induced dimerization can be considered a post-treatment process to optimize device performance, similar to thermal annealing.90 This indicates that although thermal stability is not driven by the same mechanism as photochemical stability, such dimeric chemical structures and reversible process with light/heat stress allow the researchers to build the correlation between morphological stability and photostability in PCBM-based devices when stability tests are conducted in an inert atmosphere (in the absence of O2 and without the formation of oxidation products).91
However, it is worth mentioning, in general, that the photostability of OSCs (chemical structure changes and reactions) is not directly linked to their morphological stability under thermal stress.27 This is particularly applicable in the case of the relatively thermal stable non-fullerene acceptor (NFA) blends under room temperature, such as ITIC- and BTP-family NFAs.36,92,93 A more detailed discussion is given in Section 5.2.
2.2 PDI derivatives
Although fullerene-based OSCs have achieved PCEs of 11%, there still exists an efficiency gap between OSCs and perovskite/Si cells that needs to be addressed for commercialization.21,94–96 To reduce this gap, efforts are being made to develop NFAs that can overcome the limitations of fullerene derivatives, such as difficulties in chemical modification of fullerene cages, weak absorption in the visible (vis) and near-infrared (NIR) range, and variability in band gaps.97,98 As illustrated in Fig. 2, the field of OSCs has experienced rapid advancements with the emergence of NFAs. Perylene diimide (PDI) units, as one of the NFAs, exhibit good light absorption and favorable 3D charge transport channels similar to fullerenes, such as CRP-1 (Fig. 4(a)) with the record PCEs of 11.2% in PDI derivatives so far.64,99,100 While significant attention has been directed towards exploring the efficiency of PDI derivatives. Limited studies have investigated the chemical stability and photodegradation mechanisms of PDI-based OSCs. Min et al. conducted photostability testing (with O2) for five different PDI-based NFAs. As presented in Fig. 4(b)–(d), both of them suffered from UV-vis absorption losses after the photooxidation process.81 It is noteworthy that the PDI moiety itself possesses excellent thermal and photostability (w/o O2), making it suitable for applications as lasers, filter materials, and structural components in different environments.101,102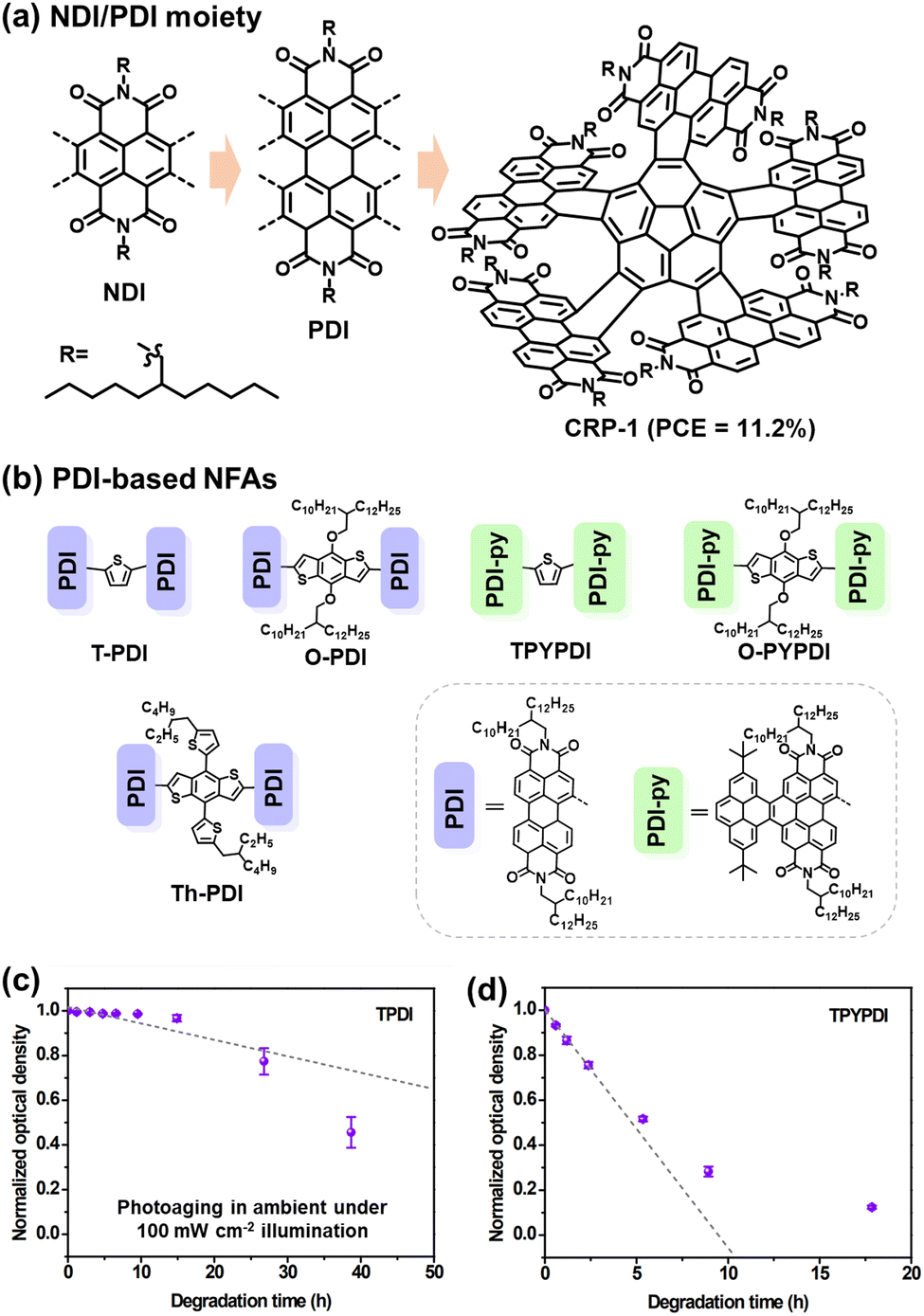 | ||
| Fig. 4 (a) Chemical structures of naphthalene diimide and perylene diimide (NDI/PDI) derivatives and the PDI-based acceptor with the record PCEs obtained in binary OSCs to date.99 (b)–(d) Chemical structures of PDI-based acceptors and their UV-vis absorption loss under illumination (photooxidation), relative to the initial maximum optical density before photoaging.81 Reproduced from ref. 81 with permission from RSC Publishing, Copyright 2019. | ||
In OSC research, some reports have shown that PDI derivatives can serve as stable transport layers, contributing to the achievement of highly stable OSCs.103 Moreover, PDI moieties have demonstrated good photostability even when in contact with photo-catalyst ZnO.104 Therefore, although the efficiency of PDI-family NFAs currently lags far behind that of ITIC- and BTP-family NFAs, PDI derivatives could still be potentially considered as a class of NFAs with promising photostability.
2.3 IDTBR and IDFBR derivatives (A–D–A type NFAs)
The acceptor–donor–acceptor (A–D–A) type small molecule acceptors have emerged as a particularly promising alternative to fullerenes and 3D-type PDIs during the past few years.3 Notably, their capacity to consistently achieve PCEs exceeding 13% marks a significant stride in the field over fullerene counterparts since 2016.57 The classification as A–D–A type acceptors derives from their distinctive molecular architecture: a central electron-rich donor core flanked on either side by electron-deficient acceptor units.The fluorene moiety emerged as one of the pioneering donor units in A–D–A type NFAs, chosen for its facile synthesis, ready availability, and the ease with which solubilizing chains could be incorporated to exert control over structural characteristics.10 In 2015, Holliday et al. synthesized an A–D–A type NFA named IDFBR (Fig. 5), featuring an alkylated fluorene donor at its core, flanked by electron-withdrawing benzothiadiazole and 3-ethylrhodanine units in the periphery.51 Subsequently, the substitution of the fluorene donor unit in IDFBR with an indacenodithiophene (IDT) core resulted in another notable A–D–A type acceptor known as IDTBR (Fig. 5(a)).54 Baran et al. reported that the o-IDTBR, equipped with phenyl side-chains, demonstrated a PCE of 7.8% when paired with polymer donor P3HT.18 Transitioning to EH-IDTBR, featuring branched alkyl side chains, yielded an impressive 11.1% PCE, facilitated by the use of the non-chlorinated processing solvent mesitylene without any additives.105
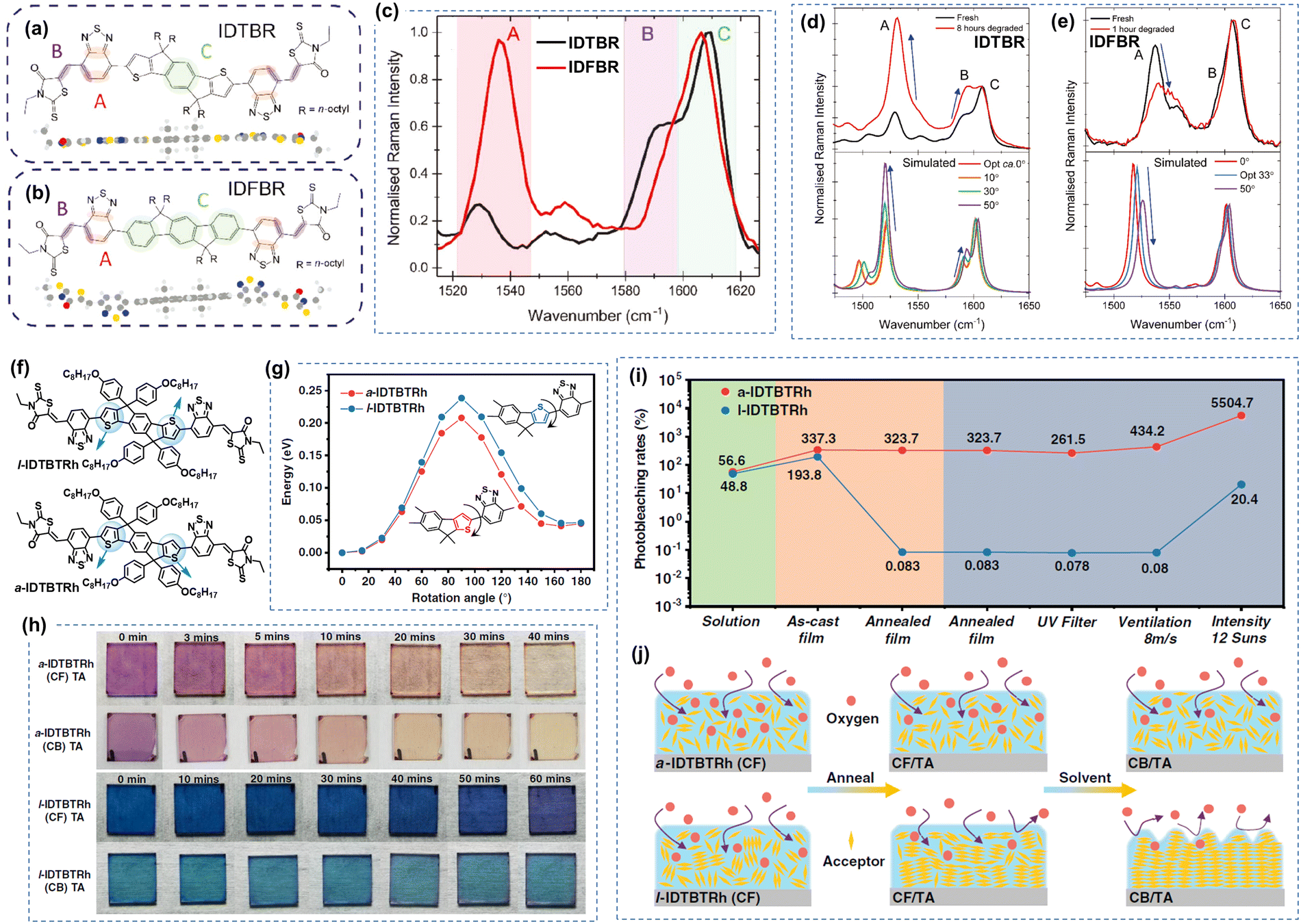 | ||
| Fig. 5 Photochemical stability (with O2) of IDTBR and IDFBR-based acceptors. (a) and (b) Chemical structures of IDTBR and IDFBR. (c) Simulated Raman spectra and the assignment of Raman peaks. (d) and (e) Photodegradation behaviors of IDTBR and IDFBR were investigated by Raman spectroscopy.106 Reproduced from ref. 106 with permission from Wiley-VCH, Copyright 2019. (f) Chemical Structures of a-IDTBTRh and l-IDTBTRh. (g) Energy barriers for the rotation modes. (h) Images of the photobleaching effect of thin films. (i) Photobleaching rates of a-IDTBTRh and l-IDTBTRh. (j) Schematic for the photooxidation process of a-IDTBTRh and l-IDTBTRh-based thin films processed from a different treatment.107 Reproduced from ref. 107 with permission from Wiley-VCH, Copyright 2021. | ||
Despite the structure optimization on IDTBR-type NFAs for achieving high PCEs, the photostability of IDTBR derivatives was mainly investigated under an O2 environment. As illustrated in Fig. 5(a)–(e), Kim et al. have systematically probed the pivotal influence of molecular structure and conformation on the photostability of IDTBR and IDFBR-based NFAs in ambient air using Raman spectroscopy.106 This investigation entails a comparison between the planar IDTBR and the nonplanar IDFBR, which exhibit structural similarities but distinct conformations. The study unveils a tripartite degradation pathway: (1) initiation of photoinduced conformational change: a phase involves the induction of photoinduced conformational changes, specifically torsional rotation related to the dihedral angles between the core and benzothiadiazole units. This conformational modulation is proposed to be instigated by noncovalent interactions with environmental moieties. (2) Subsequent photo-oxidation and fragmentation: such photo-oxidation and fragmentation processes precipitate chromophore bleaching and the formation of degradation byproducts. (3) Final chromophore bleaching: the ultimate phase culminates in complete chromophore bleaching. This comprehensive analysis underscores the pivotal role of the initial conformational transition as a decisive prerequisite for the ensuing stages of degradation.
Min et al. explored the impact of molecular aggregation and ordering on the photostability through a comparative analysis of two A–D–A-type isomeric electron acceptors: a-IDTBTRh and l-IDTBTRh as shown in Fig. 5(f)–(j).107 Despite sharing identical elemental composition, these two isomers exhibit significant differences in molecular conformation and thin film morphology. The study reveals that both molecules display similar photobleaching behaviors in solution with illumination under air. However, a significant contrast emerges in thin-film states: the photobleaching rate difference between a-IDTBTRh (amorphous film) and l-IDTBTRh (crystalline film) is nearly four orders of magnitude, underscoring the dominant influence of molecular aggregation on determining photo-oxidation stability. As presented in Fig. 5(j), such photo-oxidation rate in thin films is mainly affected by the O2 diffusion from the surface into bulk thin films.
Under an inert atmosphere, Kim et al. found that the vinyl group is still the weak link in IDTBR and IDFBR-based NFAs, leading to structural twisting and device degradation.106 It is worth noting that Li et al. proposed a molecular design strategy to improve the intrinsic chemical and photochemical stability of IDTBR-based NFAs.108 The incorporation of ring-locked carbon–carbon double bonds within the D–A conjugation enhances steric hindrance against nucleophilic attacks and promotes the intramolecular C–H/O interactions, leading to improved photostability both under the O2 environment and an inert atmosphere.
2.4 ITIC derivatives (A–D–A type NFAs)
As of now, a diverse range of A–D–A type NFAs has been developed, featuring the linkage of two electron-withdrawing groups through a conjugated core, including 9,9′-dialkylfluorene, spirobifluorene, and dithienosilole moieties.1,10,97,100 This review focus, from the standpoint of photochemical stability, centers on A–D–A-types NFAs with IDT cores as the most prevalent NFAs during the past few years.10 In the last few years, the community has systematically explored and demonstrated the photochemical stability and photodegradation mechanisms of NFAs using these IDT-based NFAs and ITIC derivatives as model systems. Generally, there are three distinct photodegradation pathways for ITIC derivatives and IDT-based NFAs corresponding to different aging conditions: (i) involving an oxidation process; (ii) photoisomerization and electrocyclic reaction (under an inert atmosphere); (iii) the break of A–D–A conjugation effect from the weak vinyl groups in the presence of the photocatalyst ZnO. We summarize these three identified pathways in detail as follows.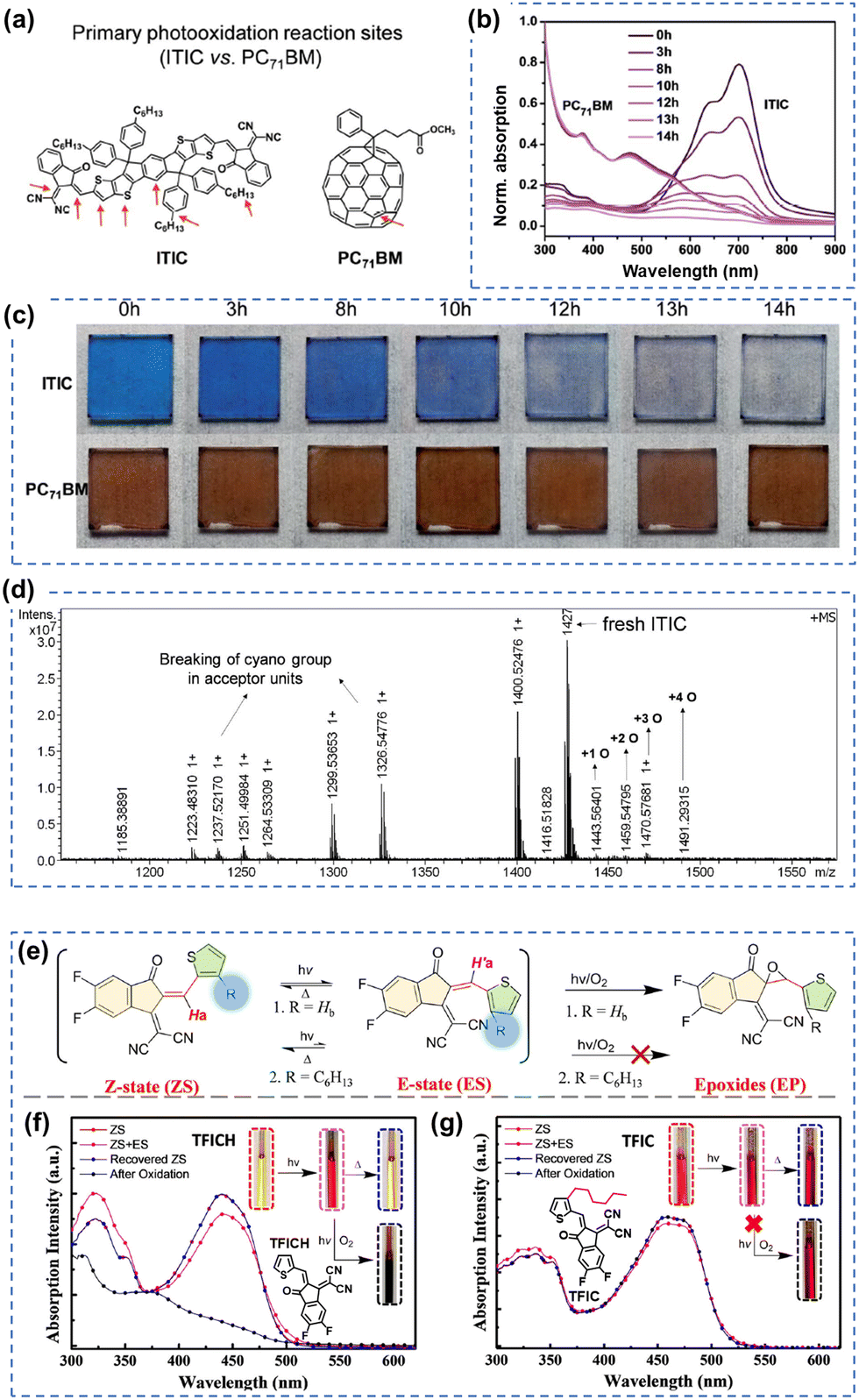 | ||
| Fig. 6 Photochemical stability (with O2) of the A–D–A-type and ITIC-series acceptors. (a) Chemical structures and primary photo-oxidation reaction sites of ITIC and PC71BM. (b) UV-vis spectra and (c) photographs of photo-oxidized ITIC and PC71BM films at different exposure times. (d) MALDI-TOF spectrum of neat ITIC film degraded under one sun for 10 hours (in dry air).81 Reproduced from ref. 81 with permission from RSC Publishing, Copyright 2019. (e) Photoisomerization and photooxidation of TFICH and TFIC. The chemical structures are given in Fig. 6(f) and (g). Photoisomerization of (f) TFICH and (g) TFIC after 10 hours of illumination with ZS-ES reversible process upon heat treatment, and further decomposition (oxidation) of TFICH after 220 hours of illumination.109 Reproduced from ref. 109, CC BY. | ||
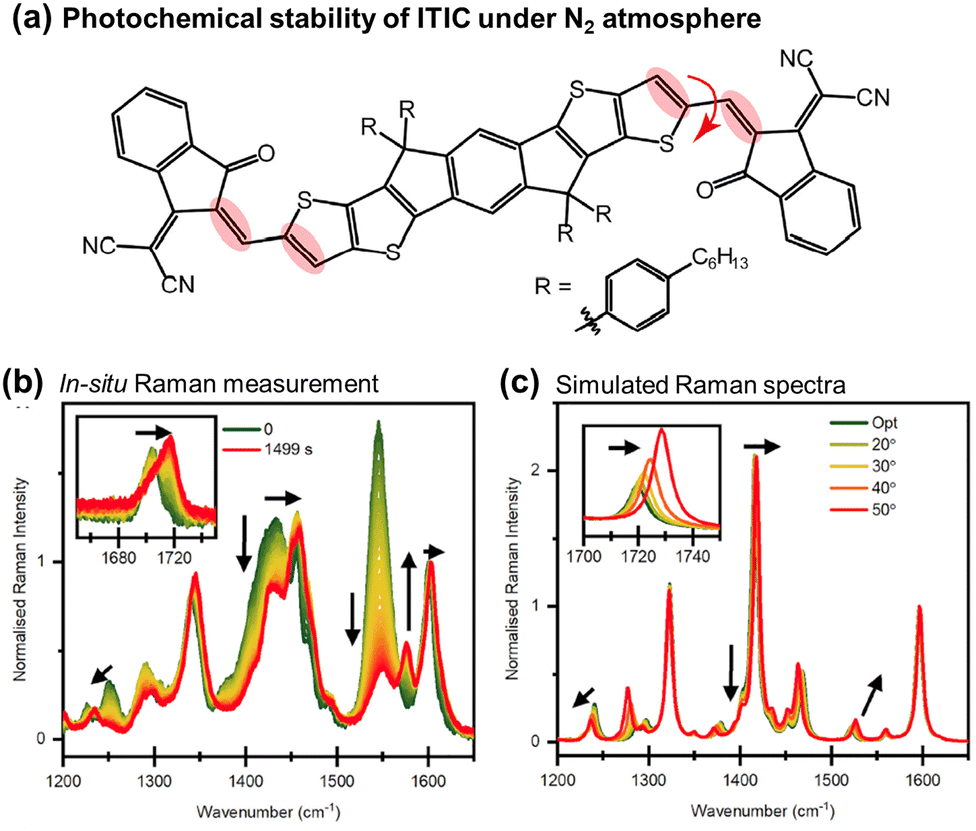 | ||
| Fig. 7 (a) Chemical structure of ITIC with emphasized Raman vibrational modes during photoaging. The red arrow denotes the modified dihedral angle in Fig. 7(c). (b) In situ Raman spectra recorded using 514 nm laser under the N2 atmosphere. Black arrows indicate the primary peak changes during in situ degradation. (c) DFT-simulated Raman spectra with increasing dihedral angles across the C–C bond in the vinylene linkage. Black arrows emphasize peak changes.110 Reproduced from ref. 110, CC BY. | ||
In 2021, for the first time, Perepichka et al. successfully isolated the photodegradation products and identified the photodegradation pathways for ITIC-family NFAs.37 As summarized in Fig. 8(a), they demonstrated that under an inert atmosphere (without a photooxidation process), IT-4F undergoes a photoisomerization process and then a six-electron electrocyclic reaction between the end-group unit (dicyanomethylene moiety) and the thiophene unit (in the backbone and near the dicyanomethylene moiety), followed by a 1,5-sigmatropic hydride shift. The related degradation pathway is confirmed by the isolated photodegradation product P1 with detailed structure characterization studies for the first time, such as NMR spectra, mass spectra (MS), and transition-state DFT calculations. They further examined this by mixing the photodegradation product P1 into fresh BHJ layers and correlated the photochemical stability with device stability.
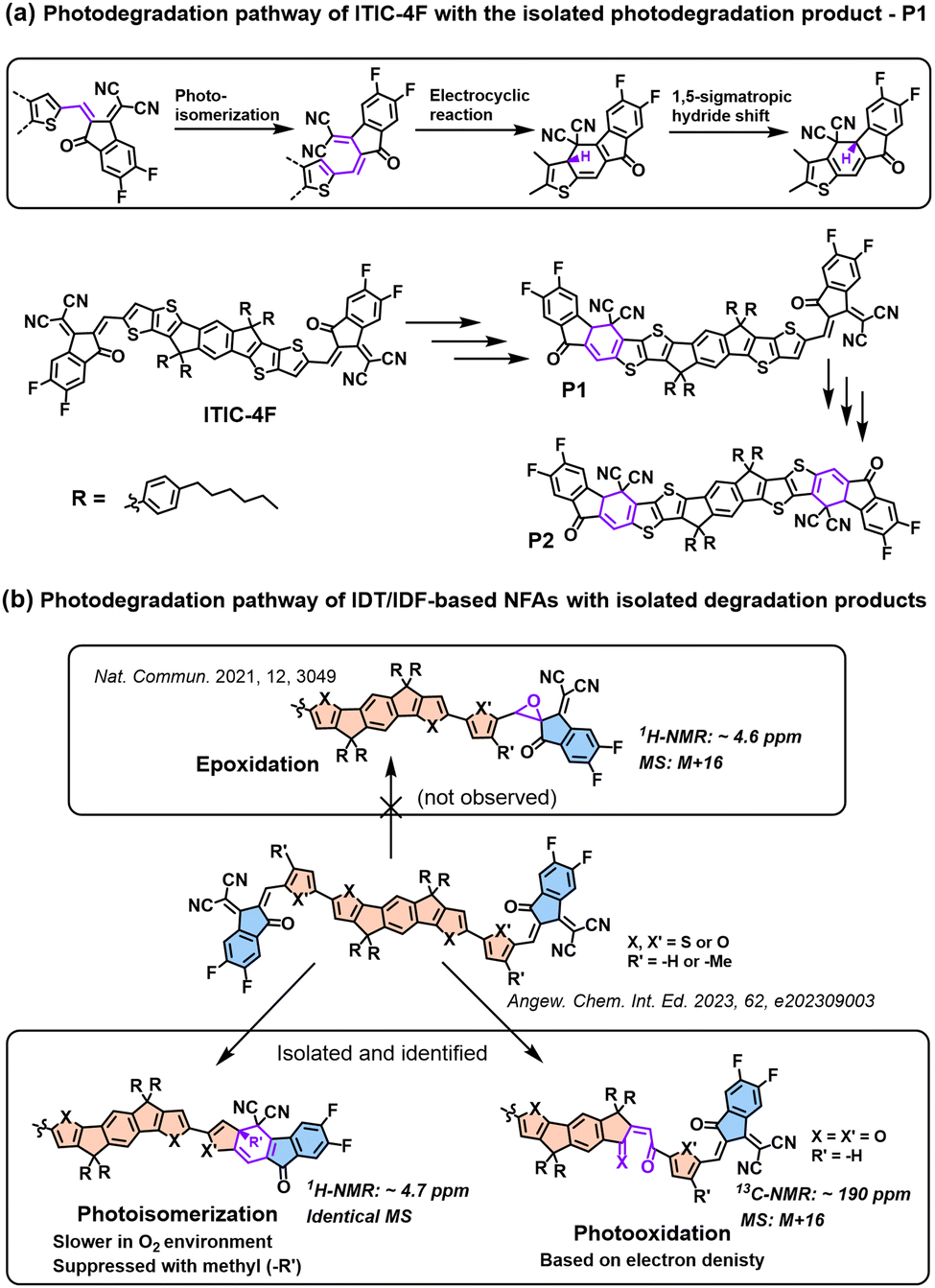 | ||
| Fig. 8 (a) Photodegradation pathways of ITIC-4F. P1 is successfully isolated as one of the photodegradation products.37 (b) Photodegradation pathways of IDT/IDF-based NFAs with isolated and identified photodegradation products.80 | ||
Notably, in 2023, Perepichka et al. systematically investigated the photodegradation mechanisms of ITIC-type and other IDT-based NFAs under both an O2 environment and an inert atmosphere.80 First, Perepichka and co-workers showed that oxygen should selectively attack the electron-rich core rather than the electron-deficient vinylene bridge as reported by Li et al.109 Under an O2 environment, the photooxidation process, such as the formation of 1,4-enedione or ketothione, is a more favorable pathway than the 6-electron electrocyclic photoisomerization. Second, based on the clarified photodegradation pathway, they developed a series of furan-based NFAs and achieved superior photovoltaic performance and photostability compared to thiophene-based analogs. The addition of methyl groups to the furan linkers not only reverses the conformational change but also improves intermolecular electronic coupling. Unlike the electrocyclic photoisomerization observed in many NFAs with thiophene moieties, the photodegradation of furan-based NFAs stems from the [4+2] cycloaddition reaction of 1O2 to the furan core units, resulting in the formation of a 1,4-enedione. Interestingly, the use of methyl-furan as the linker effectively mitigates both of the two degradation pathways, leading to remarkable photostability, which is proven by transition-state DFT calculations.
The complicated structures of ITIC-type NFAs (compared to fullerenes) are one of the reasons, leading to that the understanding of photostability mechanism lags far behind the development of their PCEs. Using the simplified 1,1-dicyanomethylene-3-indanon (INCN) compounds as model systems, Castellano et al. conducted a series of designed experiments and complementary ground- and excited-state computations.111 As shown in Fig. 9, they offer an exhaustive examination of the photochemical characteristics of a series of INCN-based molecules, featuring variations of INCN units in (hetero-)aryl substitution, alkyl group positioning, and halogen substitution. Notably, the photoisomerization reactions are observed across all cases, facilitated by selective wavelengths of excitation spanning the ultraviolet and visible regions. In agreement with the work reported by Perepichka et al.,80 Castellano and coworkers still didn’t observe evidence of the photooxidation on vinylene groups as previously reported by Li et al.109 Intriguingly, under irradiation at 454 nm, one of the eight INCN-based molecules exhibits both Z/E photoisomerization and sequential pericyclic reactions.
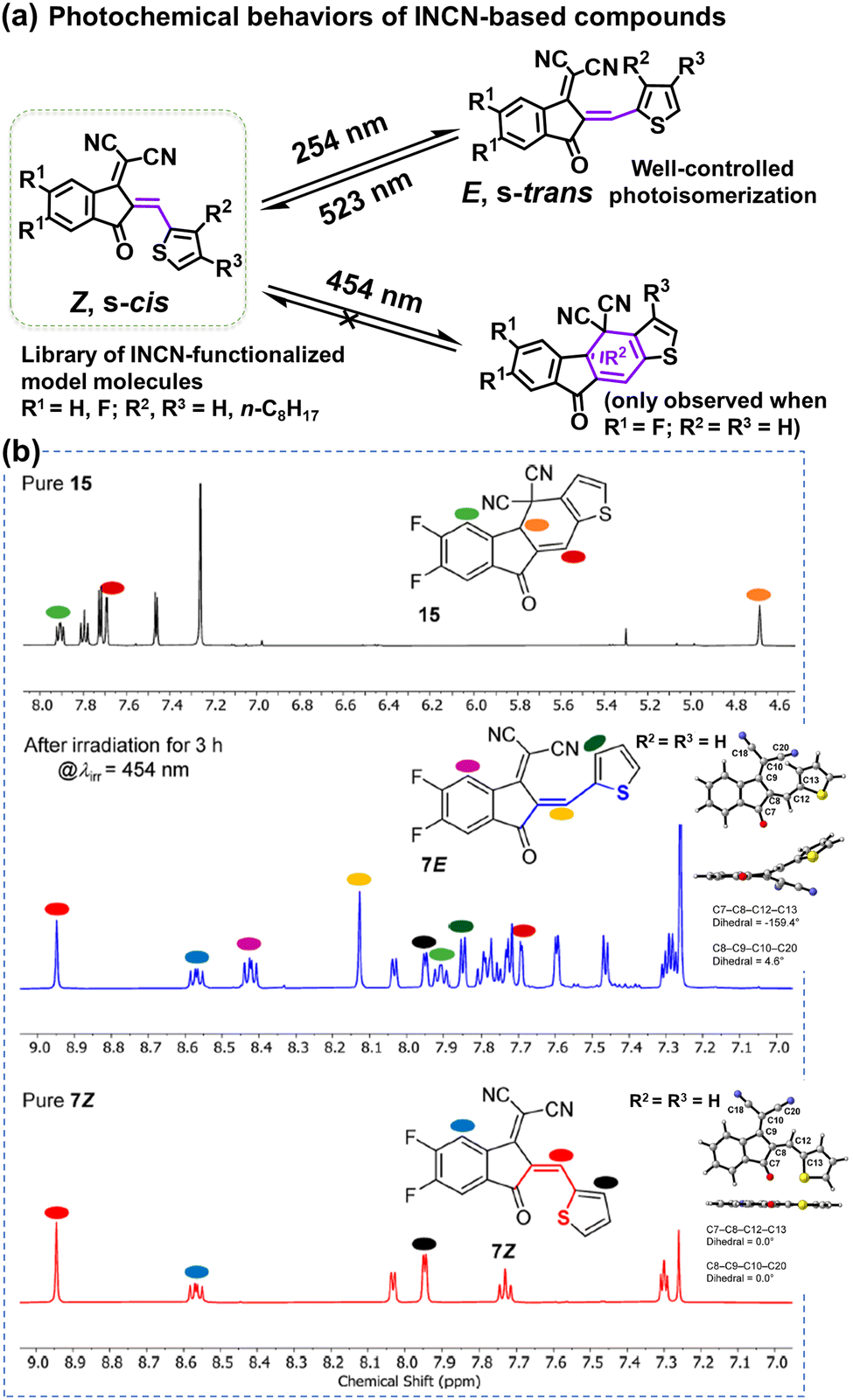
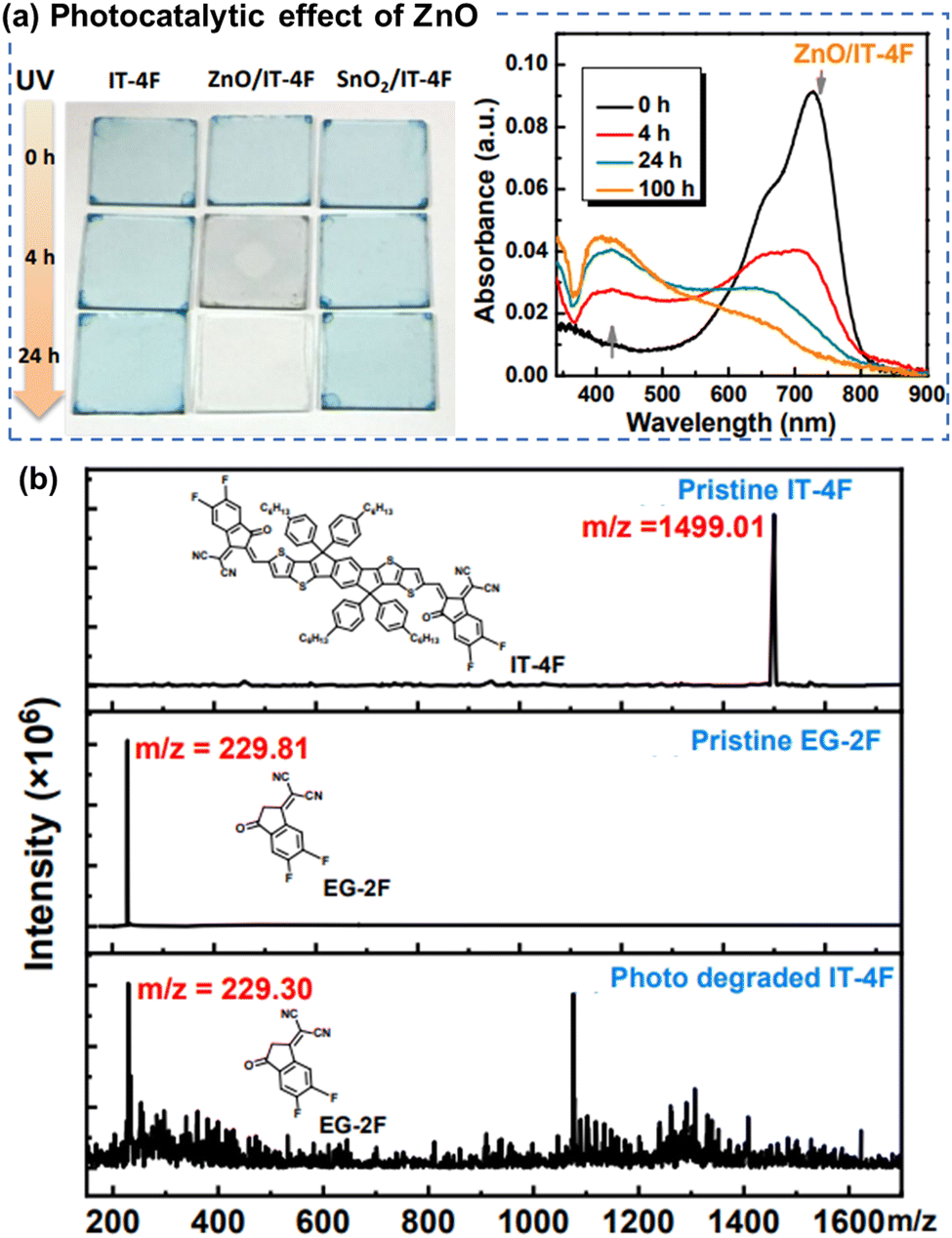 | ||
| Fig. 10 (a) Photographs and UV-vis absorption spectra of the neat ITIC-4F films during photo-aging, showing the photocatalytic effect of ZnO on chemical degradation of ITIC-based NFAs. (b) MALDI-TOF spectra of the fresh and photoaged ITIC-4F-based film.112 Reproduced from ref. 112 with permission from RSC Publishing, Copyright 2019. | ||
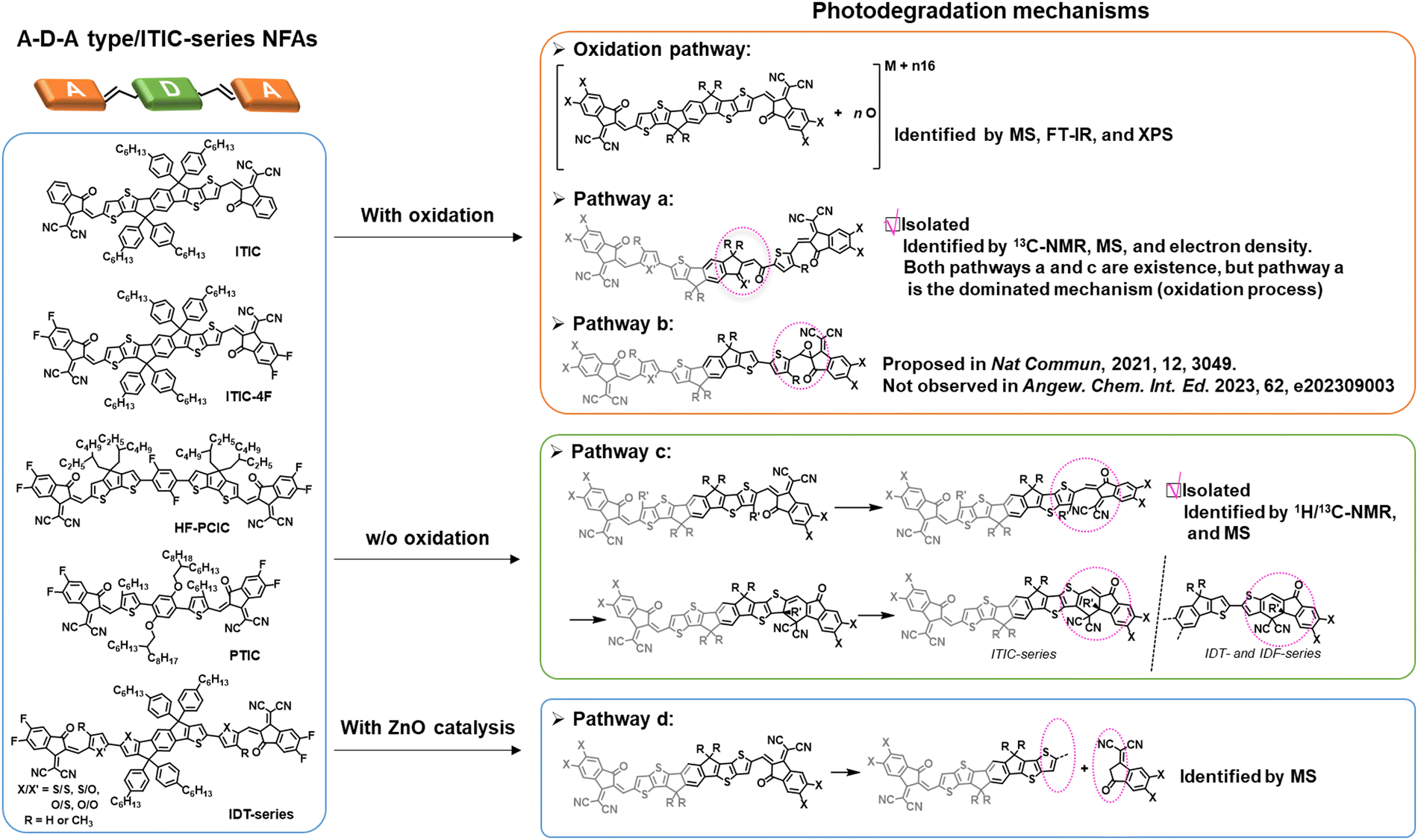 | ||
| Fig. 11 Summary of the photochemical degradation pathways and photodegradation mechanisms for ITIC- and IDT-based NFAs. | ||
2.5 BTP derivatives (A–D–A–D–A type NFAs)
In early 2019, Zou et al. developed an acceptor with central fused dithienothiophen[3.2-b]-pyrrolobenzothiadiazole core, named as BTP-4F (also known as Y6), featuring an A–D–A–D–A type backbone.63 These BTP-series NFAs with narrow bandgap and high electron mobility enable the efficiencies of OSCs to surpass 19%.8,60,117 Consequently, OSC research has entered a new era with BTP-NFAs as the most efficient acceptors to date. Recent reviews and perspectives have summarized and outlined the ongoing enhancements in BTP-NFAs, focusing on refining their optical and electrical properties to achieve further breakthroughs in PCEs.3,9,11,94,96,98,118–121 In the last four years, the photostability of BTP-NFAs has predominantly been explored under controlled atmospheres (w/o O2), while investigations into their oxidation processes remain relatively limited.29,118 Therefore, this review aims to delve into the chemical stability and photodegradation mechanisms of BTP-based NFAs without oxidation reactions.Recently, Kim et al. summarized the key molecular perspectives for high chemical stability of NFAs,25 and demonstrated the increased photostability from ITIC-X (X = –Me, –H, and –F), IEICO-4F, and Y6, using in situ Raman measurements.122 As shown in Fig. 12, owing to the existence of outer side-chains (–C11H23) in Y6 and strong intermolecular interactions in IEICO-4F and Y6, the rotation process of vinyl groups is suppressed from ITIC-X (X = –Me, –H, and –F), IEICO-4F, to Y6, leading to the improved photostability.
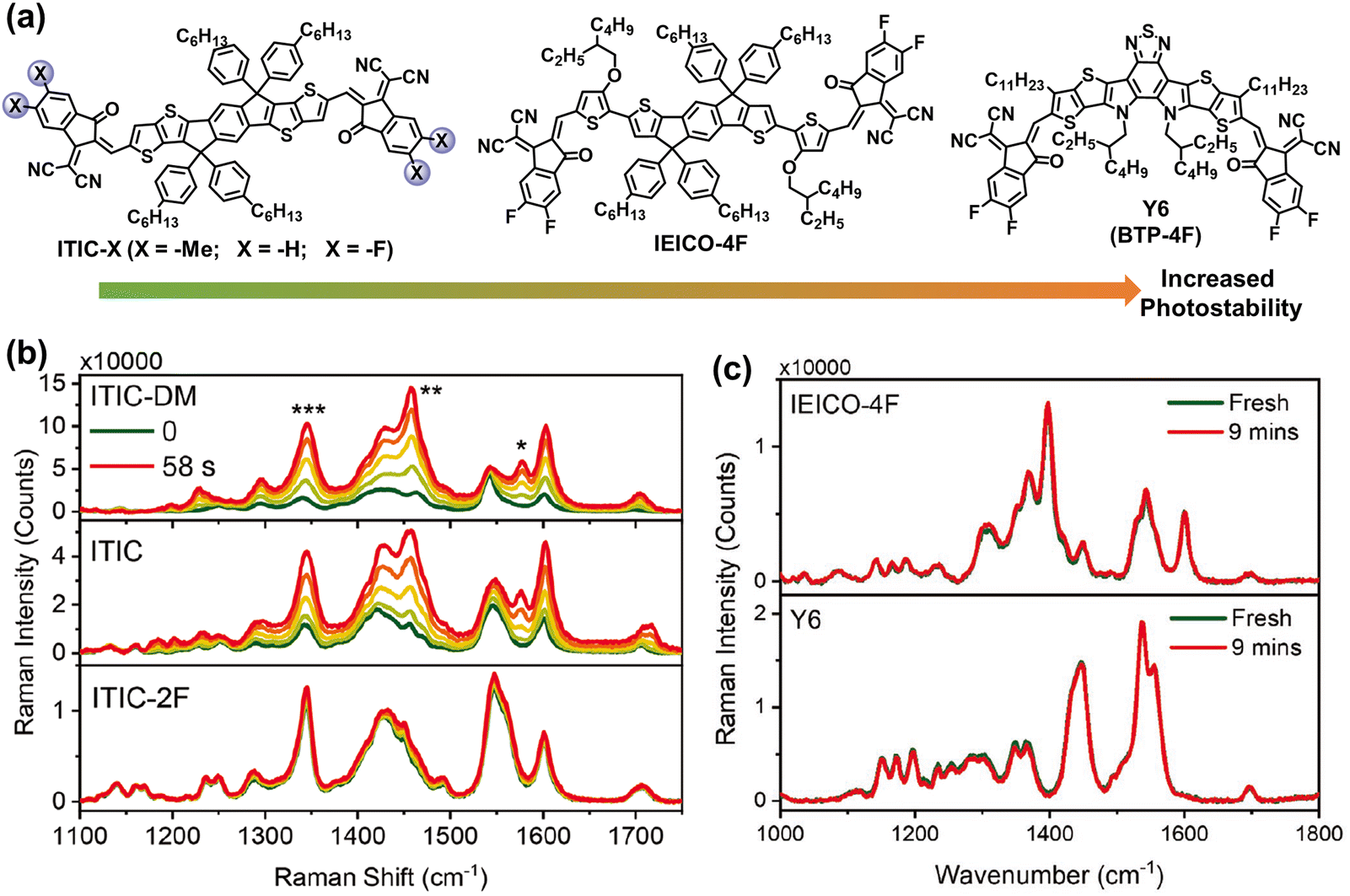 | ||
| Fig. 12 (a) Chemical structures of ITIC-Me, ITIC-H, ITIC-F, IEICO-4F, and BTP-4F (also known as Y6) with increased photostability. (b) In situ Raman spectra (514 nm laser) of the neat ITIC-NFA films. (c) In situ Raman spectra (633 nm laser degradation before and after 9 min) of the IEICO-4F and Y6 neat films.122 Reproduced from ref. 122, CC BY. | ||
Similar to ITIC derivatives, BTP-NFAs also suffered from the conjugation breakage from the vinyl linkage between the end groups and core units. As shown in Fig. 13(a), Wang et al. showed the loss of end-groups and the formation of ketone groups in BTP-NFAs (contacting with ZnO) as the main photodegradation products using MALDI-TOF spectra.123 Loo et al. demonstrated the loss of end-groups in BTP-NFAs after the photoaging process when devices used C60-SAM (self-assembled monolayer) coated ZnO as the transport layer, while the photodegradation products are end-capped with methyl groups, identified by MALDI-TOF spectra (Fig. 13(b)).39
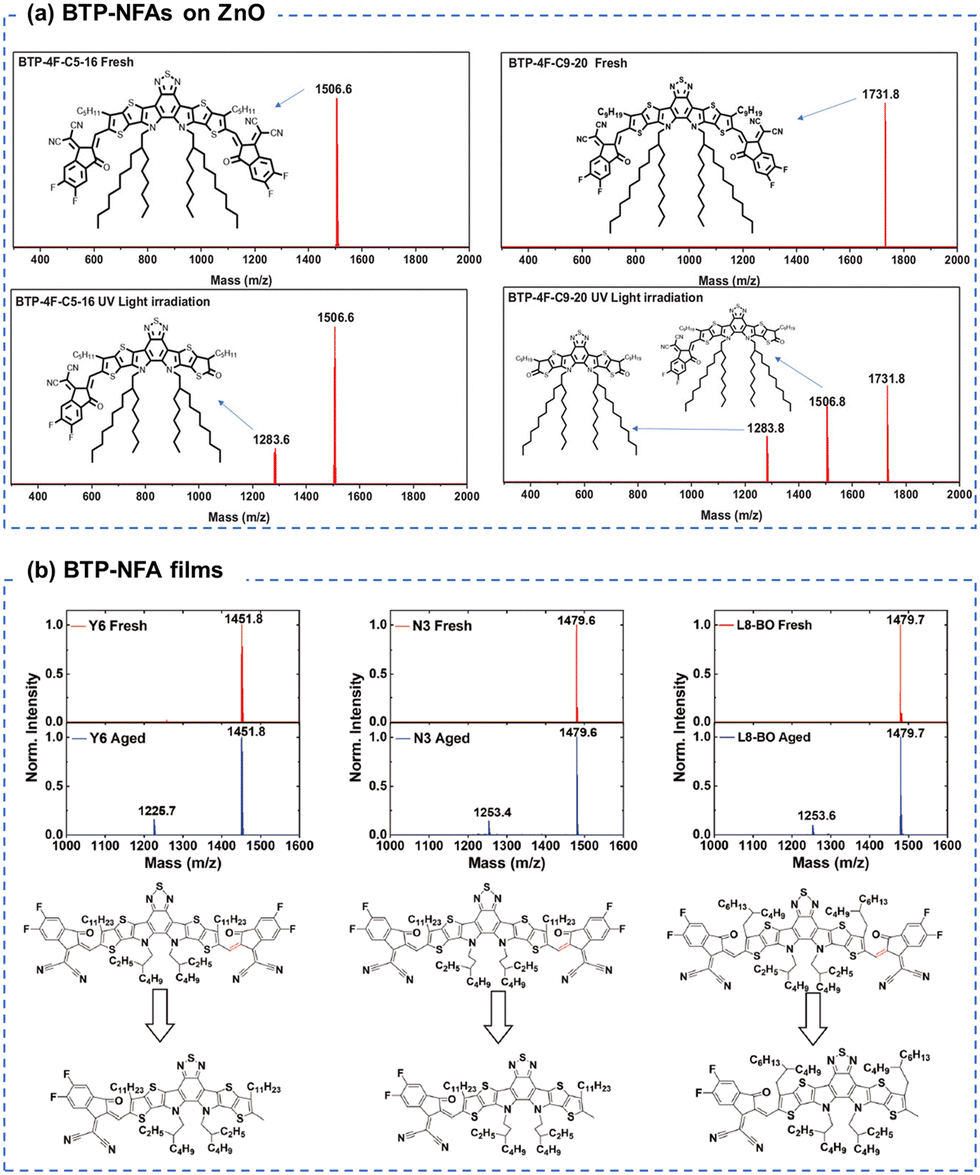 | ||
| Fig. 13 (a) MALDIT-TOF spectra of the BTP-based NFA films before and after photo-aging with ZnO layers.123 Reproduced from ref. 123 with permission from Wiley-VCH, Copyright 2022. (b) MALDIT-TOF spectra of the BTP-based NFA films before and after photo-aging.39 Reproduced from ref. 39 with permission from Wiley-VCH, Copyright 2023. | ||
The rotation of vinyl groups in A–D–A type NFAs is the initial process that induced the photodegradation of NFAs, which is proved by the isolated photodegradation products in ITIC-NFAs.37,80,111 Although the existence of out-side-chains in BTP-NFAs can suppress such rotation modes, the flexible vinyl groups are still able to rotate under illumination. As shown in Fig. 14, Jen et al. synthesized a series of BTP-NFAs with local-isomerized conjugated side-groups, and proved the isomerization on their geometries and torsion angles can module the photostability of OSC devices, which also behaves differently from their thermal stability.124,125
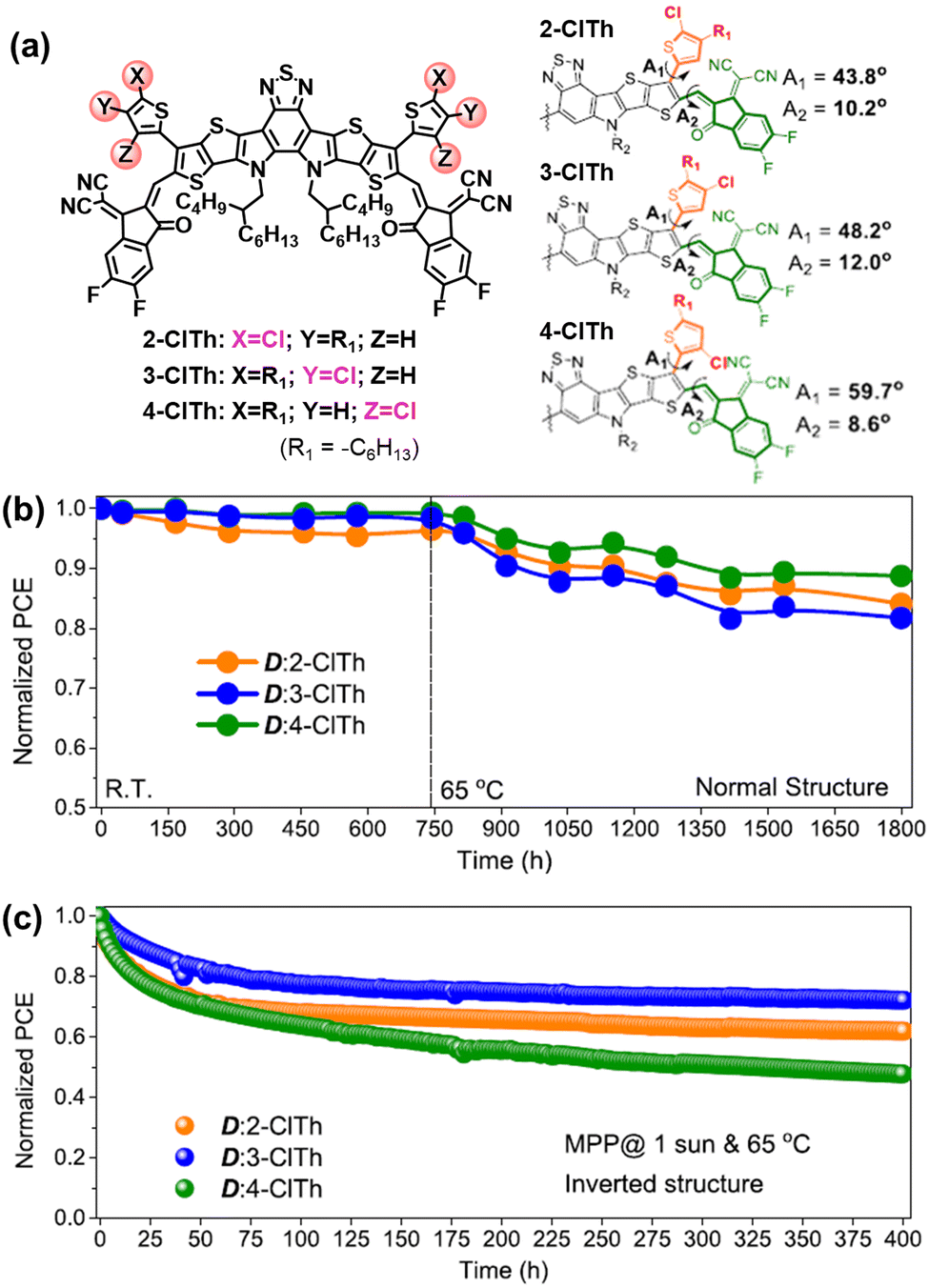 | ||
| Fig. 14 (a) Chemical structures of BTP-based NFAs with locally isomerized conjugated side-groups. (b) Storage (R.T.) and thermal stability (65 °C) of their normal-structure devices. (c) Photostability (maximum-power-point tracking, 65 °C) of the corresponding inverted devices.124 Reproduced from ref. 124 with permission from the American Chemical Society, Copyright 2023. | ||
Baran et al. utilized six different NFAs with Cl/F-endgroups and side-chains as a platform to study how the molecular structures of BTP-NFAs affect the photostability and outdoor stability of OSC devices.83 As summarized in Fig. 15, the increased energy barrier for the rotation of vinyl groups can suppress the photodegradation process of NFAs at the molecular level (steric hindrance from side-chains), and the high quadrupole moments in NFAs (fluorinated end-groups) can further prevent the rotation of vinyl groups in the solid states, which is consistent with the work reported by Kim et al.122 The observed photostability trend among Y6, BTP-eC9, and BTP-BO-4Cl (PEDOT:PSS-based devices, Fig. 15(b)) is also consistent with the one reported by Yu et al.126 These results show that preventing the rotation of vinyl groups is not only the key in ITIC-NFAs, but also important in BTP-NFAs, which is also reported by Du et al.127 This is because vinyl groups exhibit high isomerization reactivity under thermal and light stress, which has been recognized since the discovery of polyacetylene film (Nobel lecture).128 Locking the vinyl groups by rational chemical design has been proven as an effective strategy for improving the photostability of ITIC-based NFAs.108,129 Therefore, this would also be a promising avenue for achieving photostable BTP-type NFAs.130
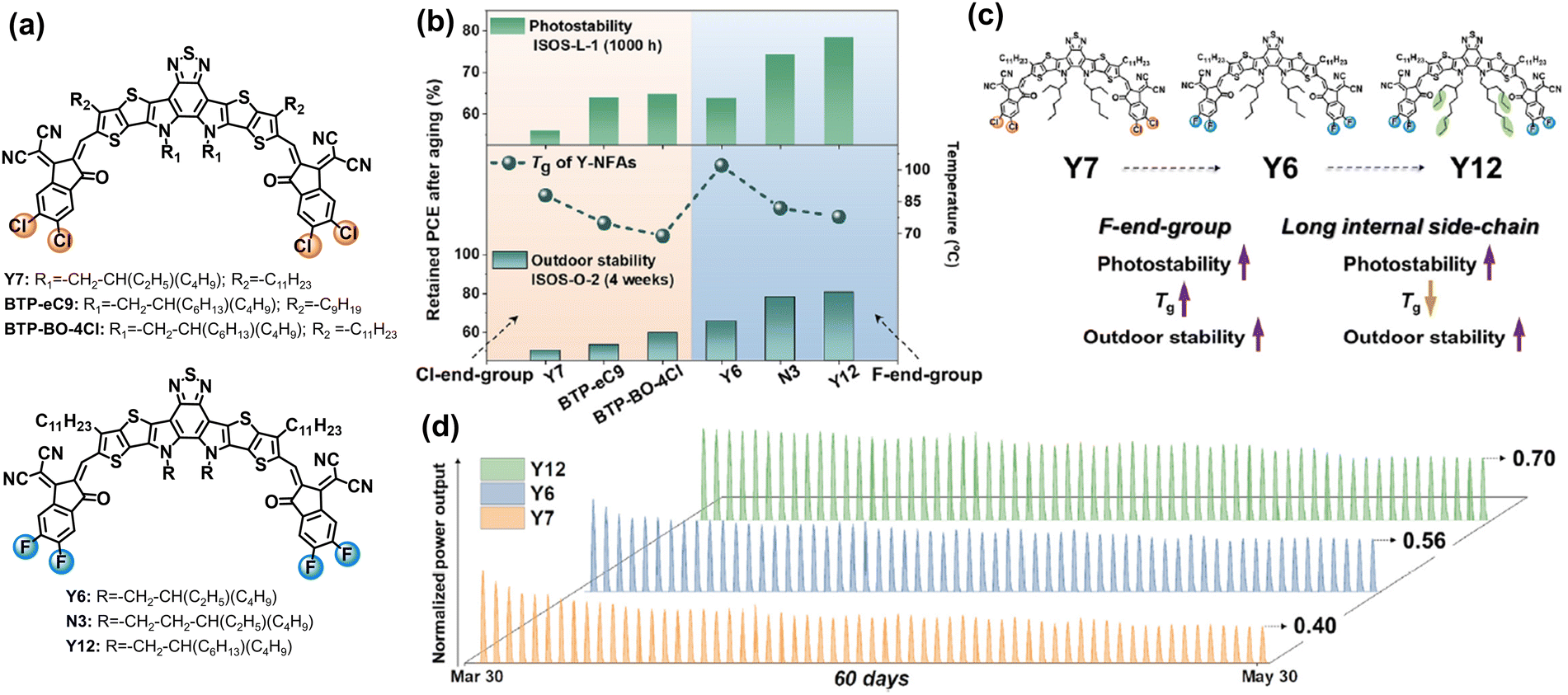 | ||
| Fig. 15 (a) Chemical structures of Cl- and F-endgroup-based BTP-NFAs with different side-chains. (b) Photostability and outdoor stability of the NFA-based devices. (c) Molecular engineering on improving photostability and outdoor stability of the devices. (d) 60-day outdoor operation of the Y7-, Y6-, and Y12-based devices under hot and sunny Saudi climates.83 Reproduced from ref. 83 with permission from Elsevier Publishing, Copyright 2023. | ||
3. Chemical stability and degradation mechanisms of polymer donors
The limited diffusion length of excitons presents a challenge in achieving effective exciton separation upon generation and recombination in most organic semiconductors.131 To address this, a prevailing strategy involves the nanoscale blending of electron acceptor and donor components, forming a BHJ blend.132 This BHJ morphological approach remains a cornerstone in the realm of OSC research.3 Therefore, despite the photostability of electron acceptors discussed in Section 2, the photostability of polymer donors stands as another critical factor governing device stability.Fig. 16 delineates two primary categories of polymer donors so far: D-type and D–A type. Low-cost P3HT and other D-type polymers exhibit relatively wide band gaps. To further mitigate this, the strategy of D–A hybridization has been extensively employed, integrating alternating D and A segments along the polymer backbone. This integration fosters molecular orbital mixing between D and A units, engendering a set of hybridized molecular orbitals with an effective band gap smaller than that of D-type polymer donors.131 Consequently, D–A polymers have dominated OSC research over the past 10 years, and allow for precise control over bandgaps and energy levels via meticulous monomer selection.133 This progress has led to the development of various D–A polymer donors, including PTB7-Th, PM6, D18, and PTQ10.3,134,135
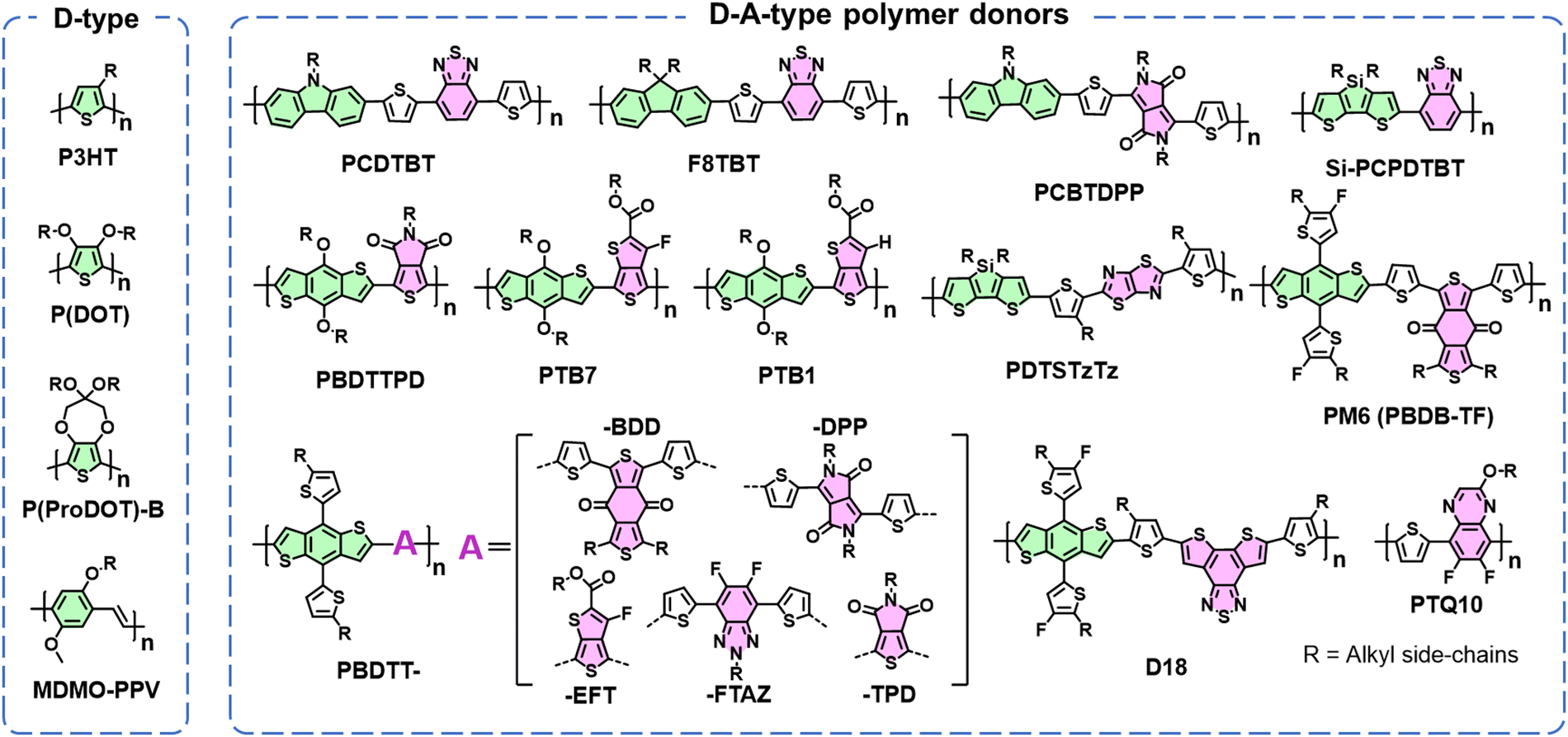 | ||
| Fig. 16 Chemical structures of D-type and D–A-type polymer donors used in OSCs. | ||
In the following section, we will comprehensively summarize the photodegradation mechanisms of both D-type and D–A-type polymer donors, encompassing scenarios involving and excluding oxidation processes. The photostability for small molecular donors, polymer acceptors, and single-component materials will be discussed in Section 4.
3.1 Photodegradation mechanisms of polymer donors involving an oxidation proces
This section focuses on delineating the photochemical degradation pathways for polymer donors, primarily emphasizing the oxidative processes. Among these donors, P3HT, as a star polymer, remains a subject of active investigation within the community owing to its cost-effectiveness in comparison to other D–A-type polymer donors. Over the last two decades, extensive research has elucidated the photodegradation pathways of P3HT as summarized in Fig. 17. Under illumination in an oxygen environment, P3HT undergoes diverse photodegradation pathways, including H-abstraction, β-scission, cage reactions, and oxidation. These reactions generate various photodegradation products, which were identified through MS, FT-IR, and Raman spectra analyses. Consequently, the material experiences conjugation loss, alterations in HOMO/LUMO levels, and a decline in UV-vis-NIR absorbance, impacting its inherent photophysical properties and consequently leading to device degradation.136–141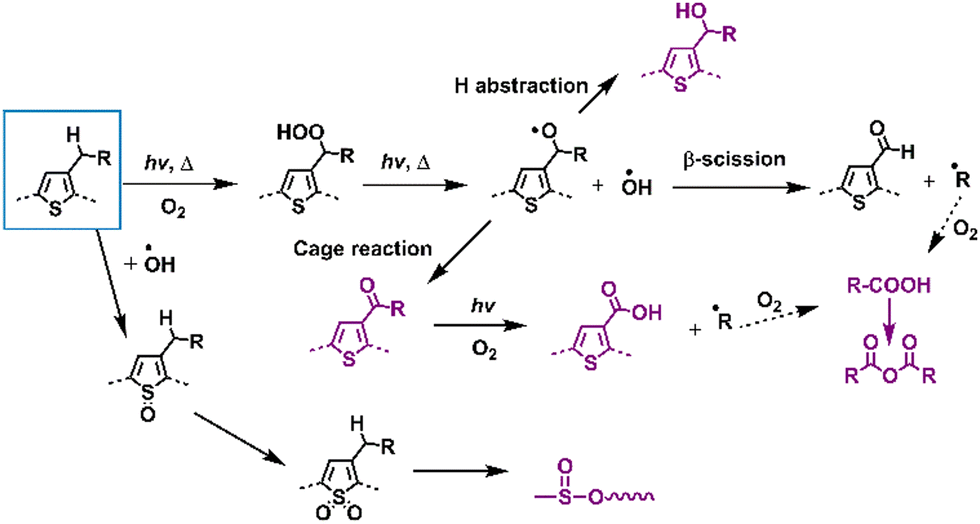 | ||
| Fig. 17 Photodegradation mechanisms (with O2) of classic D-type polymer donors – poly(thiophene) derivatives.136–139,141,142 | ||
Generally, D–A-type polymer donors share the following design concept: conjugated backbones serve as charge transport channels, and alkyl side-chains contribute to their favorable solution processability. The conjugated backbones of these polymers are susceptible to oxidation and ring-opening reactions under illumination in the presence of oxygen. The side-chains suffered from H-abstraction and scission reactions. While exploring D–A-type polymer donors, Troshin et al. investigated photooxidation across 16 conjugated polymers using electron spin resonance (ESR) spectroscopy. They observed that even trace amounts of oxygen (as low as 900 ppm) can facilitate and trigger photooxidation in all studied polymers. Notably, polymers with lower-lying HOMO energy levels, reflecting higher oxidation potentials, demonstrated better resistance to photooxidation. This emphasizes the role of energy levels in determining the polymer susceptibility to oxidation under light.143
Despite the oxidation sites on polymer backbones, the alkyl side-chains are proved to be weak links in polymer donors, along the photochemical degradation pathways, such as H-abstraction and β-scission. Tournebize et al. have highlighted the significance of side-chain degradation behaviors in polymer donors, positing them as a hindrance to the commercial viability of these materials.144,145
Recently, Ratcliff et al. investigated the ambient-induced photodegradation pathways of five donor polymers, featuring the same alkyl thienyl-substituted-benzodithiophene (BDTT also known as BDT) unit but with different A units.146 Through controlled photobleaching in air and simultaneous absorptance monitoring, as shown in Fig. 18, they identified three different pathways. By tracking the photoaging process with XPS, they assessed near-surface chemical changes, particularly oxygen additions to sulfur and nitrogen on the BDTT or distinct A units. The results show that alkyl chain oxygen addition typically acted as the initial site of attack, followed by direct oxidation on the sulfur atom in the conjugated backbone.
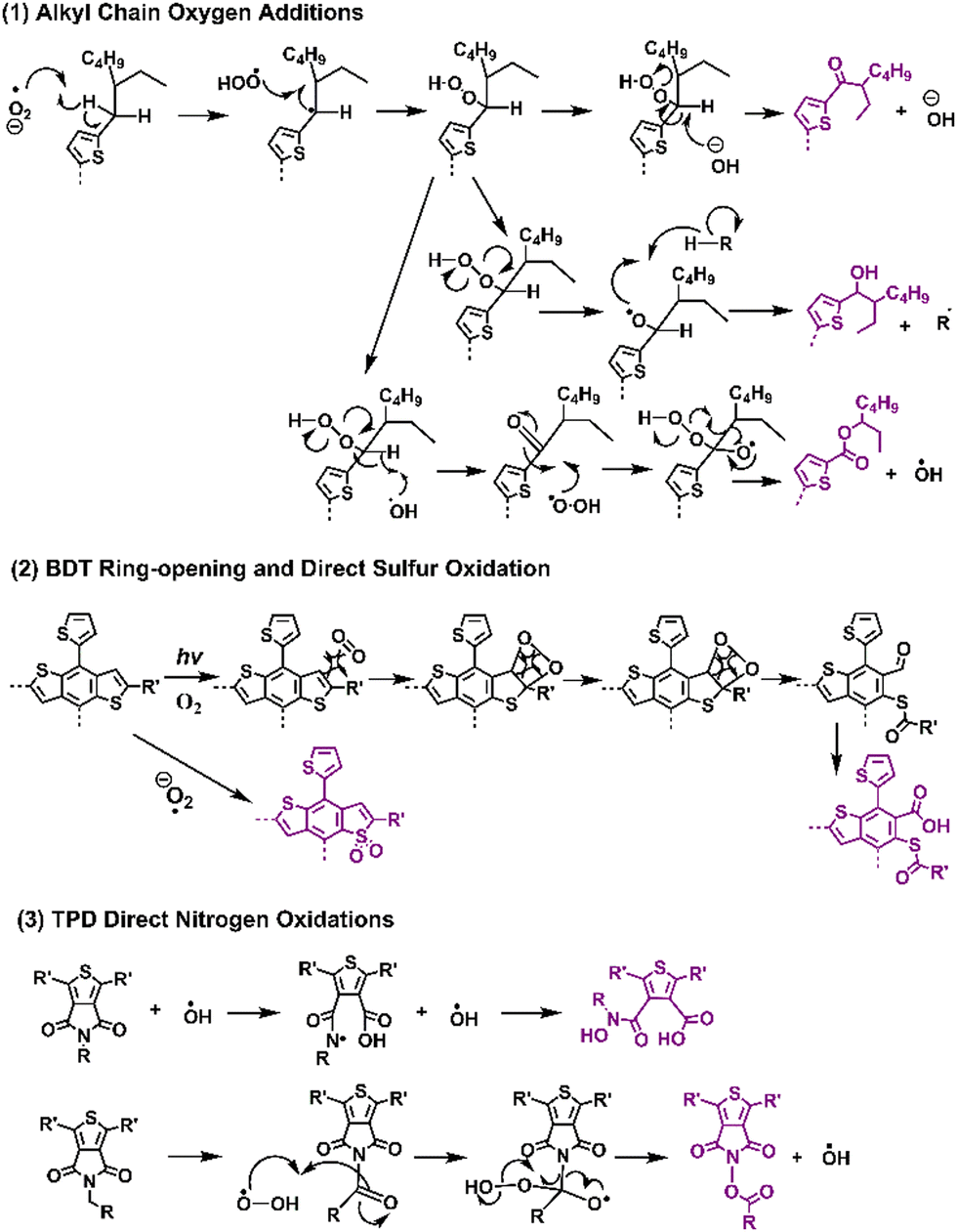 | ||
| Fig. 18 Photodegradation mechanisms (with O2) and reaction pathways for the classic donor and acceptor block units in D–A type polymer donors.144,146 | ||
In Fig. 19, the proposed photodegradation products for five classic D–A polymers were established through XPS analysis. Ratcliff et al. revealed that sulfur oxidation typically commenced after the saturation of alkyl chain oxygenation.146 Notably, this result aligns with the findings of Manceau et al.,136 indicating that direct sulfur oxidation is initiated by hydroxy radicals generated from alkyl chain oxygenation, serving as the key step. Meanwhile, ring opening reactions of thiophene/furan rings in NFAs (Fig. 8(b) and the pathway a in Fig. 10) and PBDTT-EFT (also known as PTB7-Th in Fig. 19) are induced by Diels–Alder addition of singlet oxygen.80,136,146 The photochemical degradation mechanism illustrated in Fig. 19 can extend beyond D–A polymer architectures, proving relevant for a wider range of organic semiconductors. This significance also arises from the common structural template shared by most polymer donors, featuring alkyl chains on a conjugated backbone, which is consistent with the degradation pathways reported by Tournebize et al.144,145 As alkyl side-chains in polymer donors are essential for ensuring the solution-processability, this contradictory behavior still presents a considerable challenge in developing stable OSCs when the device operates under an O2 environment.
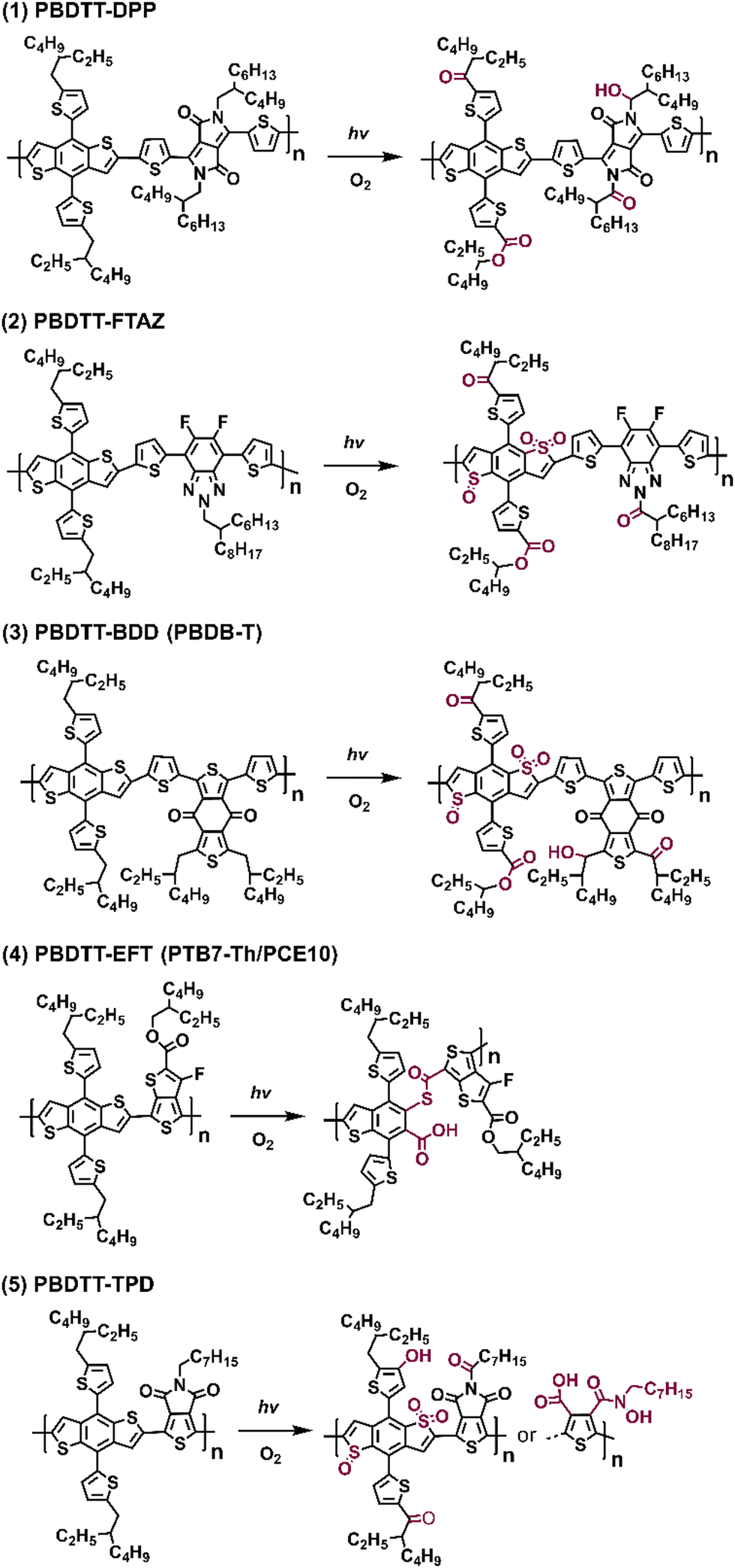 | ||
| Fig. 19 Photodegradation products (with O2) of five different D–A type polymer donors.146 | ||
3.2 Photodegradation mechanisms of polymer donors without involving an oxidation process
With the help of high-quality encapsulation, OSC can operate with the exclusion of ambient air and prevent the ingress of oxygen and water into the BHJ layers.82,83 Therefore, understanding the photodegradation mechanisms of polymer donors without involving an oxidation process is also important for the commercialization of OSCs. Taking P3HT as an example, researchers have shown that H-abstraction and β-scission are two main degradation pathways followed by radical recombination with the formation of cross-linking products.137 Such cross-linking reactions can be observed in a wide range of D–A polymers when exposed to light (without O2).40,147 In 2019, Troshin et al. discovered a commonality by investigating a library of over 20 conjugated polymers and diverse structural small molecules, showing that all these materials readily undergo light-induced crosslinking. They proposed that this phenomenon likely involves light-driven [2+2] cycloaddition and radical addition photochemical reactions.40 Recently, Troshin et al. found that such photodegradation pathways come from side-chain breakage.147In 2022, Reynolds et al. conducted a comprehensive study on the photostability of D-type polymer donors under oxygen-free conditions, comprising a family of dialkoxy-functionalized thiophene polymers.148 Employing UV-vis-NIR spectroscopy, XPS, gel permeation chromatography (GPC), NMR, and cyclic voltammetry (CV) measurements, they explored the photochemical degradation pathways, such as crosslinking, chain scission, and chemical alterations affecting the conjugated backbones (Fig. 20). The findings highlighted that crosslinking through alkyl side chains emerged as the primary degradation pathway under oxygen-free conditions. Based on these insights, they suggested promising strategies to enhance photostability in conjugated polymers, including moving away from tertiary carbon branching to either quaternary carbon branching or linear counterpart (without branching points) in side-chains, or shifting the side-chain branching point further from the main backbone. These modifications not only hold promises for improving the photostability of D-type polymers but also would be informative for achieving photostable D–A-type polymer donors used in OSCs.
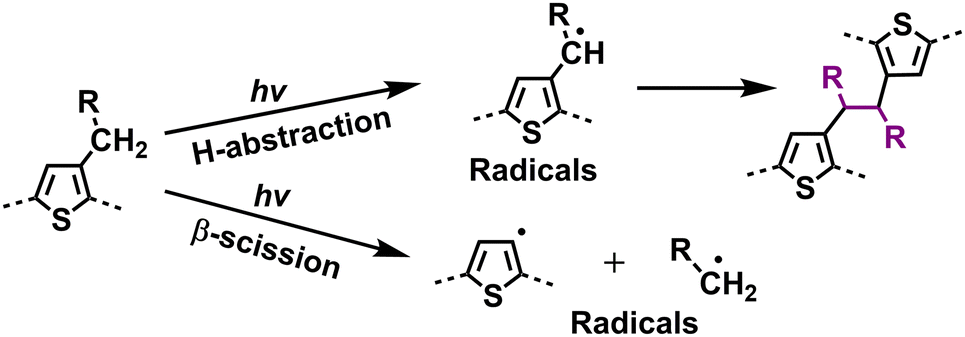 | ||
| Fig. 20 Photodegradation mechanism (without O2) for polymer donors: identified side-chain cross-linking pathways.144,145,148 | ||
3.3 The role of photodegradation of polymer donors in the state-of-the-art NFA OSCs
As summarized in the last two sections, it is well-known both polymer donors and electron acceptors are subjected to photodegradation. Therefore, it is important to understand the role and contribution of polymer donors and NFAs to the photodegradation effect in state-of-the-art OSCs.Gillett et al. investigated the photodegradation effect of PM6 and Y6 on the device stability (ZnO-based devices w/o UV, aging under white LED and in air). They investigated the photostability of three classic donor polymers – PM6, D18, and PTQ10 when paired with Y6. Upon subjecting PM6 and D18 to light exposure in ambient air, they observed a notable increase in the conversion of singlet excitons into trapped interchain polaron pairs within a timeframe of less than 100 fs. This phenomenon surpassed electron transfer to Y6, leading to a considerable reduction in the overall charge generation yield. As shown in Fig. 21, using in situ Raman measurements, they found that a twisting tendency within the BDT-thiophene motif, shared by PM6 and D18, is the main reason. As such, compared to Y6, the polymer donors (PM6 and D18) emerge as a weak link impacting overall OSC stability.29 Loo et al. also investigated the photodegradation effect of PM6 and Y6 on the device stability (C60-SAM/ZnO-based devices, aging under AM 1.5G illumination in an N2 atmosphere).39 They found that photochemically, PM6 is relatively stable after photo-aging for 200 h. In contrast, photochemical decomposition of Y-NFAs is responsible for the degradation of high-efficiency OSCs.
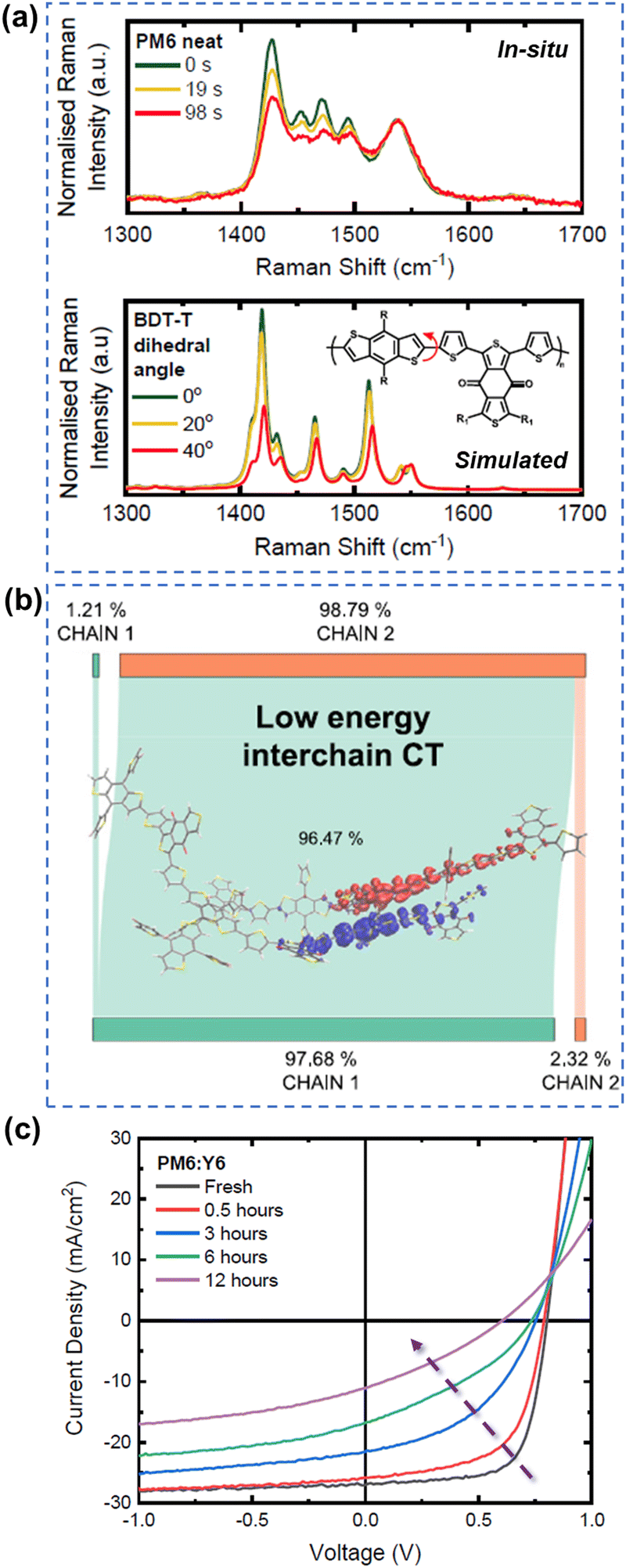 | ||
| Fig. 21 (a) In situ Raman measurements and simulated Raman spectra for understanding photodegradation (with O2) of PM6. (b) Structure twisting effect of PM6 induced low energy interchain charge transfer states. (c) J–V curves of PM6:Y6-based devices during the photoaging process (with O2).29 Reproduced from ref. 29, CC BY. | ||
To unravel the photo-degradation mechanism within PM6:Y6-based OSCs, Yu et al. employed various device fabrication methods and photoaging procedures, strategically isolating the individual contributions of PM6 and Y6.126 As depicted in Fig. 22, the comparison between devices exposed solely to the donor PM6, solely to the acceptor Y6, and those exposed as a blend strongly suggests that the degradation of PM6 predominantly governs the photodegradation process in PM6:Y6-based devices. This research presents a novel approach to investigating limitations hampering device photostability, highlighting the pivotal role of enhancing the photostability of polymer donors as a straightforward solution to improving overall device stability. Interestingly, this finding aligns with conclusions drawn from studies on perovskite-organic tandem solar cells as reported by Riedl et al.149
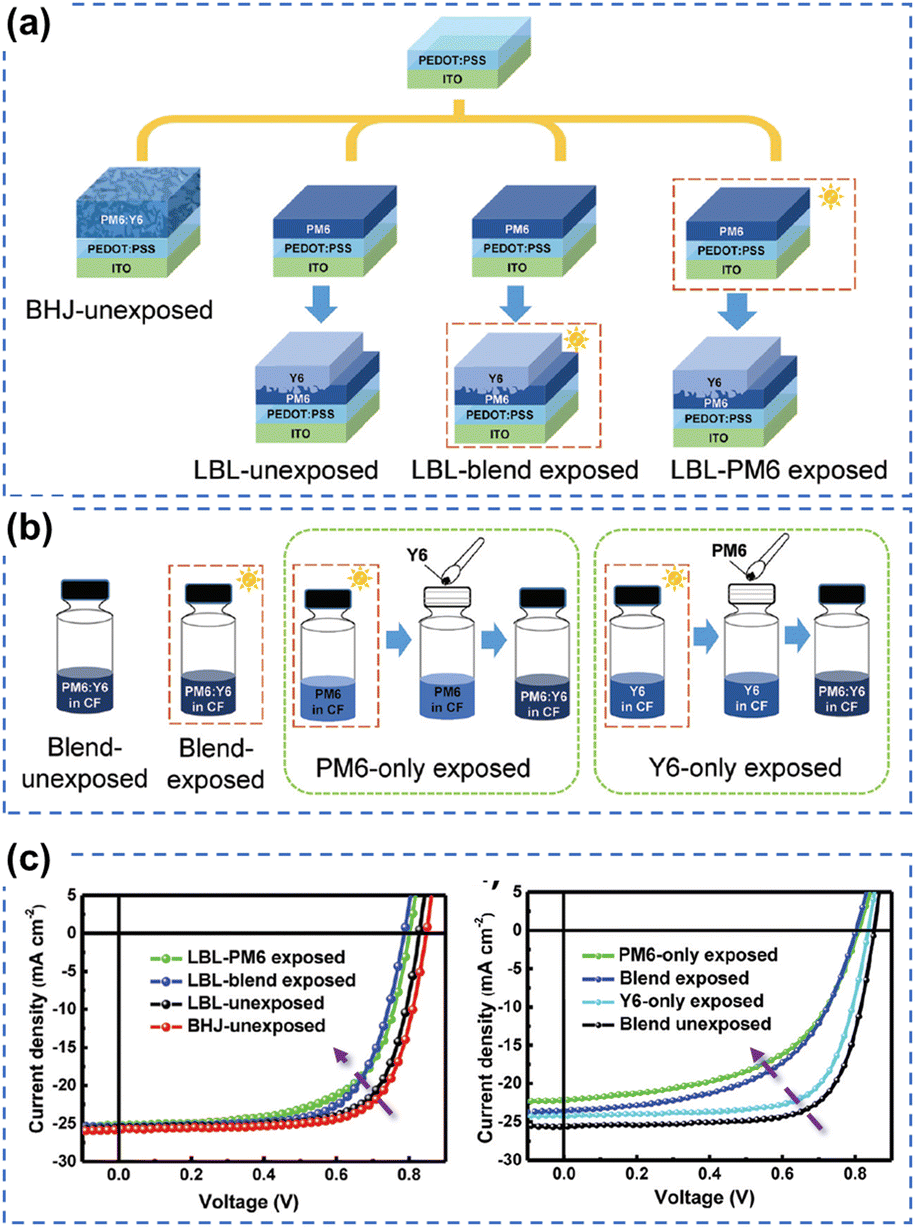 | ||
| Fig. 22 (a) and (b) Device fabrication process for understanding the role of photodegradation (without O2) of PM6 and Y6 in OSCs. (c) J–V curves of PM6:Y6-based devices using different device fabrication and photoaging processes (without O2).126 Reproduced from ref. 126 with permission from RSC Publishing, Copyright 2021. | ||
4. Photochemical stability of small molecular donors, polymer acceptors, and single component materials
Given the performance of BHJ-type OSCs leading the development in this field,2,3,21,25,117,118 extensive investigations have centered around small molecular acceptors and polymer donors, serving as models to unravel the photodegradation mechanisms of PV materials. However, as summarized in Fig. 23, there still exist other PV materials, including small molecular donors, polymer acceptors, and single-component materials.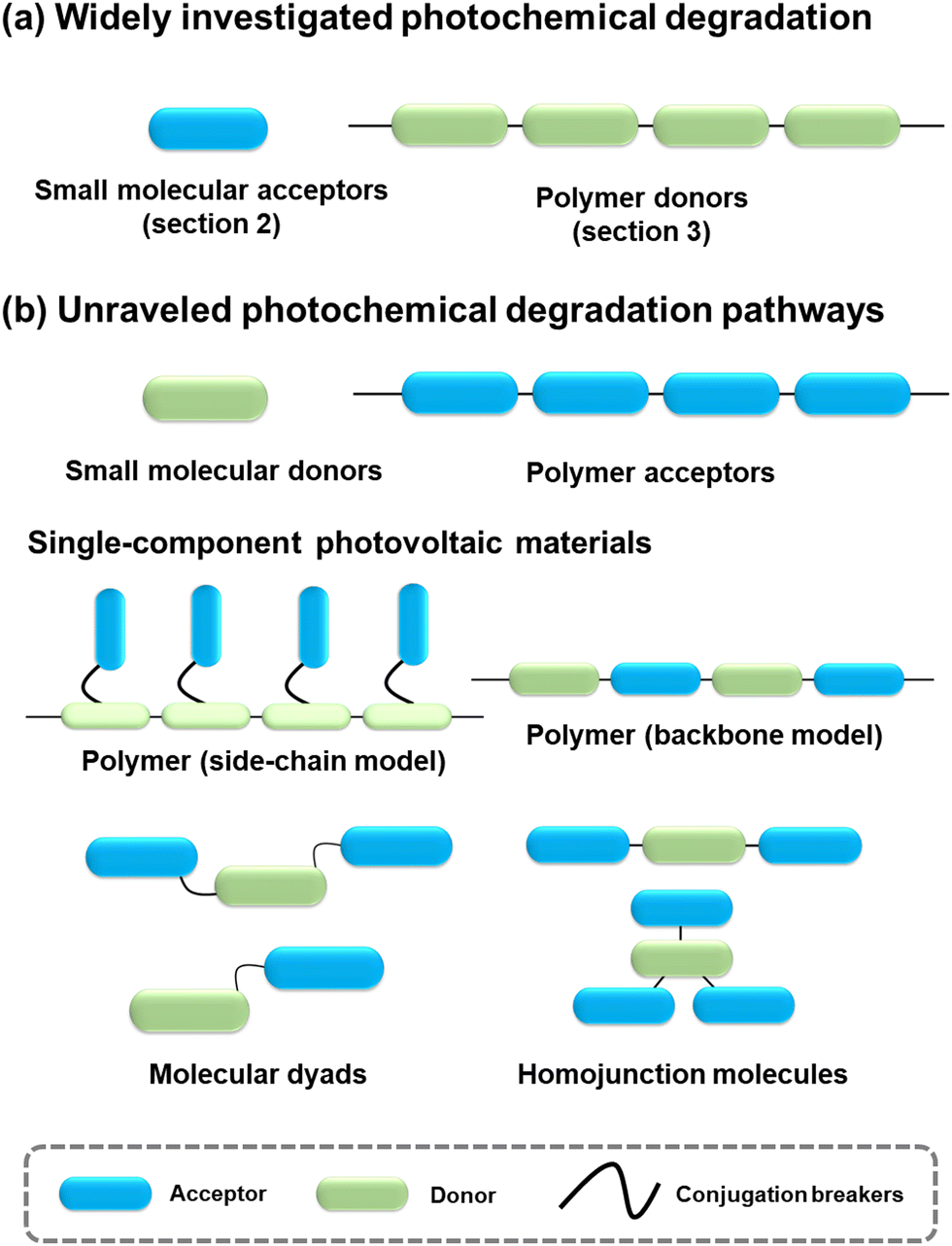 | ||
| Fig. 23 Schematic diagrams of the photovoltaic materials: (a) widely investigated photochemical degradation mechanism for small molecular acceptors and polymer donors, and (b) limited studies on photochemical pathways for the other PV materials. | ||
All small-molecular OSCs, consisting of both small molecular donors (Fig. 24(a)) and acceptors, offer distinct advantages such as well-defined chemical structures, minimal batch-to-batch variations, straightforward synthesis and purification methods, and easily adaptable properties. The development of small-molecule donors and NFAs has boosted PCEs of the devices exceeding 18%, rivalling those of their polymer-based counterparts.150–164 The recently reported all-small-molecule ternary solar cells have delivered PCEs of 18.1% with promising operational stability.160 Despite the reported photostability data of all-small-molecule-based devices,160,165,166 the precise photochemical degradation pathways for these small molecular donors remain unclear.
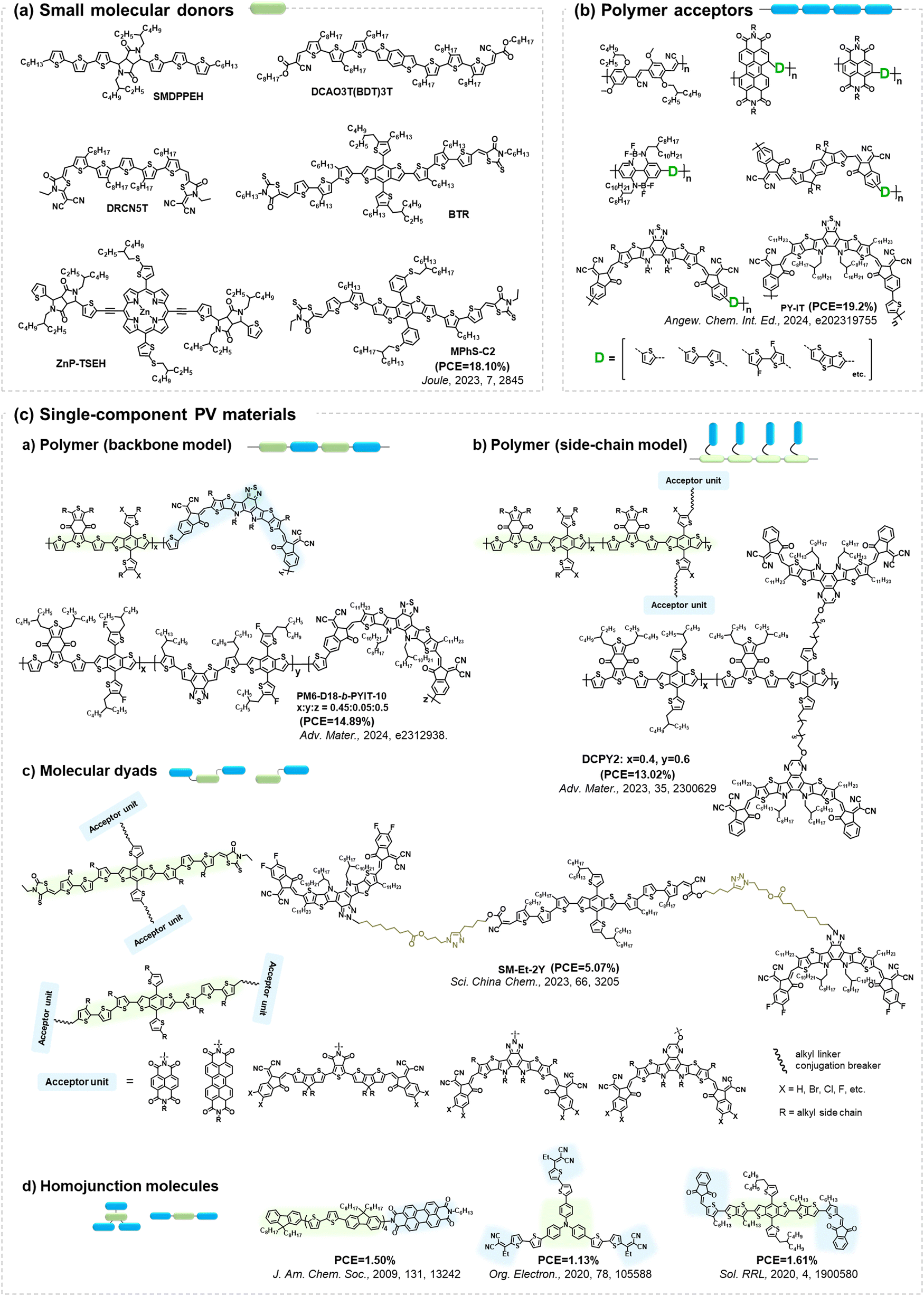 | ||
| Fig. 24 Molecular architectures of representative PV materials in Fig. 23 (Section 4). Despite advancements in PCEs and reporting device stability/lifetime, studies on the precise photochemical degradation pathways of these PV materials remain limited. | ||
A similar scenario applies to all-polymer solar cells, comprising polymer donors and polymer acceptors, where their efficiency has increased by over 18%.167–172 In the molecular architectures of polymer acceptors depicted in Fig. 24(b), the primary advantage of all-polymer solar cells is their enhanced thermal stability in comparison to other OSCs comprising small molecular PV materials. The related thermal degradation mechanisms have been elucidated, revealing that polymerized acceptors tend to display low thermal diffusion coefficients and maintain stable morphology under thermal stress.167 Many researchers have also conducted the photostability measurements on these devices.173–177 However, little attention has been given to investigating the precise photochemical degradation pathways for these polymer acceptors. Unlike small molecular acceptors (fullerenes and ITIC-/BTP-NFAs as discussed in Section 2), analysing the photochemical reactions of polymers and isolating photodegraded polymeric products are challenging due to limited characterization methods and the intrinsic complexity of polymers. To achieve photostable all-polymer solar cells, a deeper understanding of the photochemical degradation pathways of polymer acceptors is imperative and significant for ensuring their photostability and minimizing trial-and-error efforts.
In addition to BHJ-type active layers comprising electron donor and acceptor materials, PV materials based on covalently linked electron donors and acceptors (referred to as “single-component materials”) have also been developed as photoactive layers in OSCs for the past two decades.72,178–193 As depicted in Fig. 24(c), recent advancements in single-component material OPVs have marked a significant stride towards achieving competitive PCEs exceeding 14%.192 The advantages of single-component materials over other donor and acceptor materials in BHJ blends include the simplification of device fabrication process and the covalently stabilized D–A units, which facilitate thermally stable morphology and excellent device stability.193 Researchers have also presented photostability results on single-component material OPVs, indicating promising device performance even under harsh environmental conditions such as high temperature and continuous illumination.179,181,184–186,188,189,191,194 However, the understanding of photochemical degradation mechanisms in these systems remains unexplored. Questions persist regarding whether the pendent acceptor units would follow the same photochemical degradation pathway as discussed in Section 2, or if the donor units undergo oxidation under illumination in the presence of O2, and how the molecular structures of single-component materials influence the reaction rates of photochemical degradation pathways.
As summarized from Sections 2–4, current investigations into photodegradation mechanisms and photochemical degradation pathways predominantly focus on small molecular acceptors and polymer donors. Rather than solely reporting photostability results, comprehending the photodegradation pathways can effectively bridge the gap between molecular design efforts within the chemistry community and the ultimate stability performance within the device community. This understanding can help avoid reliance on trial-and-error approaches for materials design and device engineering towards enhancing overall device stability in PV technology development.
5. Chemical reactions involving additives and chemical stability of photovoltaic materials with light-driven morphological changes
Over the past decades, the exploration of photochemical pathways in OSCs has predominantly centred around small molecular acceptors and polymer donors as discussed in the above Sections 2 and 3. Furthermore, there are still other factors related to the photostability of OSCs, which will be discussed in the following sections.5.1 The side reactions between PV materials and additives
The intrinsic photochemical stability of PV molecules holds significant importance in their photostability, and it is also essential to note the existence of side reactions between PV materials and additives. For instance, in the case of the classic additive 1,8-di-iodooctane (DIO), Jacobs et al. discovered that residual DIO can persist within the BHJ layers after the device fabrication and related post-treatment process.195 Specifically, the weak C–I bond in DIO exhibits susceptibility to dissociation with the formation of radicals upon exposure to illumination. As depicted in Fig. 25, Brabec et al. demonstrated the capacity for both PCBM and ITIC to engage in radical reactions with DIO, forming adducts, a finding confirmed through high-performance liquid chromatography–mass spectrometry (HPLC-MS) analysis.196 This photochemical reaction further accelerates the photodegradation process in OSCs. Meanwhile, other research groups have also reported on this similar additive-accelerated photodegradation process in OSCs.197–199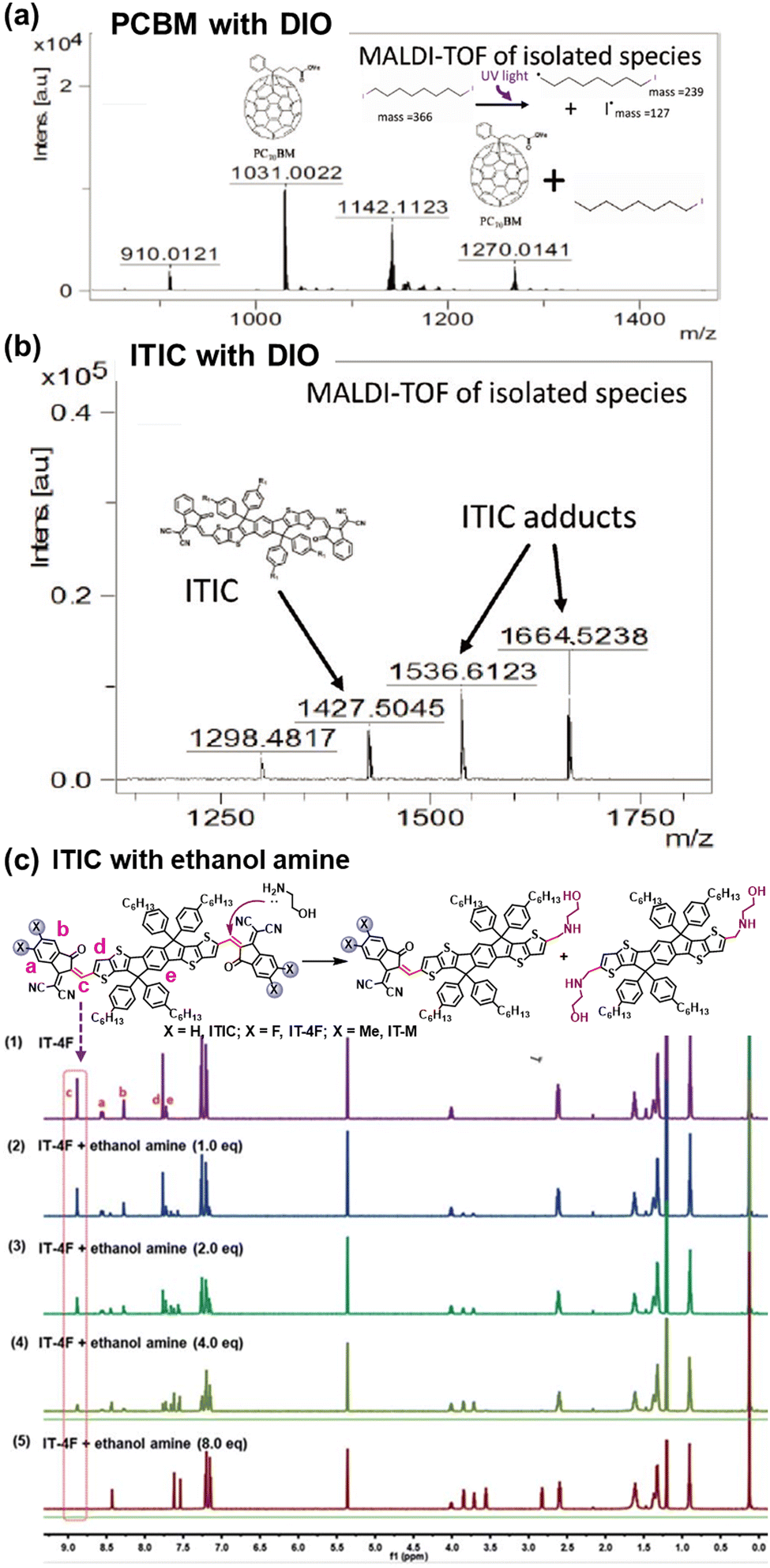 | ||
| Fig. 25 Chemical reactions between photovoltaic materials and additives: (a) and (b) DIO and (c) PEI.196,200 Reproduced from ref. 196 with permission from Wiley-VCH, Copyright 2019. Reproduced from ref. 200, CC BY. | ||
In addition to DIO, amine-containing materials, such as polyethylenimine (PEI) and ethoxylated PEI (PEIE), are also reactive compounds with NFAs. For fullerene-based OSCs, PEI/PEIE is a widely used material for doping or interface treatment to improve device performance.201–203 However, when PEI/PEIE meets NFAs, a voltage drop occurs, leading to a typical “S” shape current–voltage (J–V) curves.23,200,204 It was found that there are “toxic” chemical reactions between PEI/PEIE and NFAs.200 As shown in Fig. 25(c), generally, the amine group acts as a nucleophile, engaging in an addition reaction with the vinyl group in NFAs. This reaction ultimately disrupts the double bond (vinyl linker) and the extensive conjugated structure of the molecule, leading to the loss of device performance.23 Such “toxic” chemical reactions between amine-containing materials and NFAs can be efficiently suppressed by reducing the chemical reactivity of –NH groups, achieved through methods like metal ion-chelated PEI,200,205 chelation with small molecules,206,207 modification of the amine groups,208,209 and protonation of the amino groups.210
Zhou et al. observed that NFAs also react chemically with bases.211 Through the introduction of sodium hydroxide into the ITIC solution, OH anions function as nucleophiles under basic conditions, targeting the vinyl group and producing ITIC-OH (ITIC after OH− addition). The colour of the solution transitions from blue to orange upon mixing ITIC with PEIE. Upon adding acetic acid to the ITIC-OH solution, ITIC-OH reverts to ITIC. This interconversion between ITIC and ITIC-OH is reversible under both acidic and basic conditions.
These existing chemical reactions between PV materials and additives suggest that the chemical stability of OSCs not only depends on the intrinsic chemical stability of PV molecules but is also influenced by the reactive additives utilized during the device fabrication process.
5.2 Photochemical degradation mechanisms and the light-induced morphological changes in OSCs
Thermally induced morphological stability, driven by thermal diffusion and crystallization processes, is a phenomenon related to heat stress.27,35,36,212,213 In contrast, photostability is linked to light stress. Generally, light can trigger photochemical reactions, yielding photodegraded products that result in device degradation.24 This process involves a photochemical reaction inducing molecular structural changes at the sub-nm scale.30,37,80,81,107,109,111,112,143,144,146,148,214,215 Under illumination, morphological changes at nm-scale also occur, either due to molecular alterations or light-induced molecular movements at the nm-scale.29,216–220 As depicted in Fig. 26, conducting thermal stability measurements at temperatures close to those during photostability measurements can aid in understanding thermally induced morphological effects. However, comprehending the light-induced changes at the sub-nm and nm scales requires further analysis to elucidate the photostability of OSCs.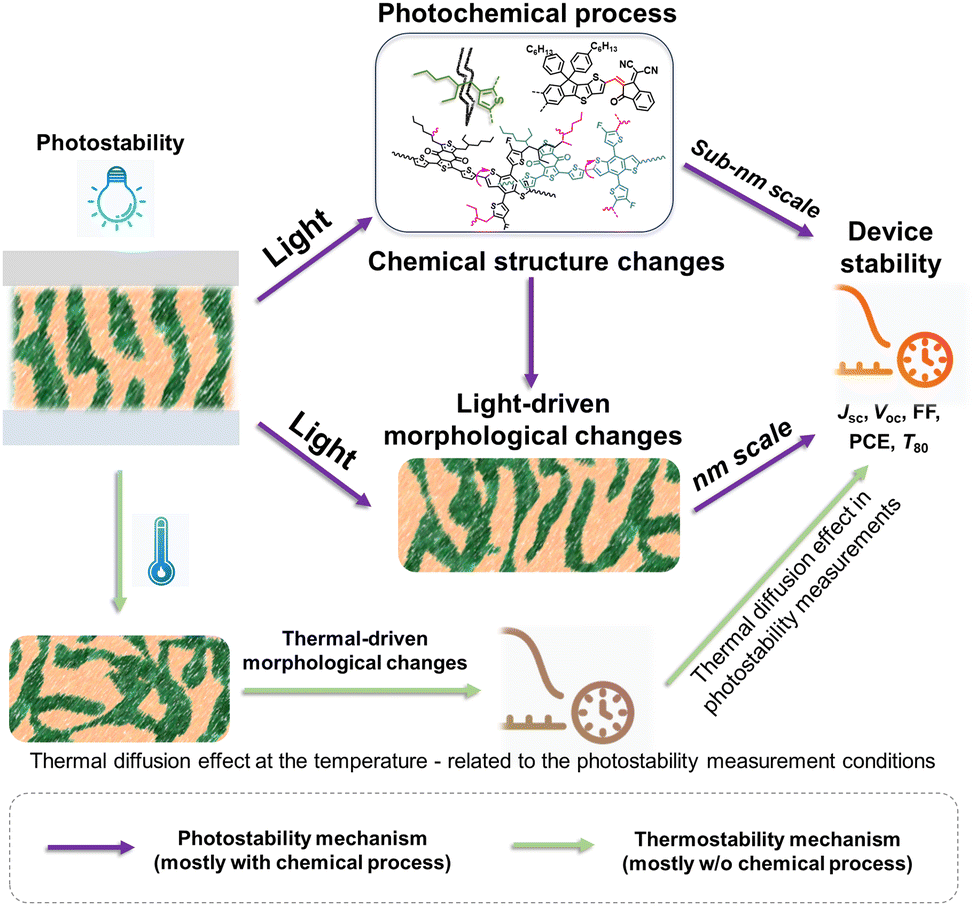 | ||
| Fig. 26 The distinction and connection between the light-induced molecular structural changes (sub-nm scale) and light-driven morphological changes (nm scale), and the mechanism differences between photostability and thermal stability. | ||
In the era dominated by fullerene-based materials, the study of photostability is linked to the photochemical reactions and the related light-induced morphological changes. This connection arises from the reversible light-induced dimerization process experienced by fullerenes, which can be correlated with their morphological characteristics.89,90 However, in the era of NFAs, the complexity of chemical reactions would complicate the light-induced morphological changes.80 It is also reasonable that light-induced molecular changes (chemical degradation) at the sub-nm scale could trigger the morphological changes at the nm-scale.29,216,220,221
It is noteworthy that several research groups observed no clear or minor morphological changes in NFA-based OSCs during photostability measurements through techniques such as atomic force microscopy (AFM), grazing-incidence wide-angle X-ray scattering (GIWAXS), and grazing-incidence small-angle X-ray scattering (GISAXS).39,114,149,217 Detecting mixed D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A domains in BHJ layers also remains a persistent challenge within the research community. These findings underscore the limitations of conventional morphological techniques in tracking the photodegradation behaviors in NFA-based devices. In a recent study, Müller-Buschbaum et al. employed AFM, GIWAXS, and GISAXS to investigate four distinct PM6-based devices incorporating Y6, ITIC-4F, ITIC-4Cl, and PC71BM. While AFM and GIWAXS exhibited only minor changes during photoaging, notably, a clear trend of light-induced nm scale morphological changes emerged from operando-GISAXS measurements.216 This highlights the necessity of addressing the photochemically induced molecular changes at the sub-nanometer scale in conjunction with the light-induced nano-scale morphological changes.
:A domains in BHJ layers also remains a persistent challenge within the research community. These findings underscore the limitations of conventional morphological techniques in tracking the photodegradation behaviors in NFA-based devices. In a recent study, Müller-Buschbaum et al. employed AFM, GIWAXS, and GISAXS to investigate four distinct PM6-based devices incorporating Y6, ITIC-4F, ITIC-4Cl, and PC71BM. While AFM and GIWAXS exhibited only minor changes during photoaging, notably, a clear trend of light-induced nm scale morphological changes emerged from operando-GISAXS measurements.216 This highlights the necessity of addressing the photochemically induced molecular changes at the sub-nanometer scale in conjunction with the light-induced nano-scale morphological changes.
6. Conclusions and outlooks
This review delves into the photodegradation mechanisms of electron acceptors and donors as photo-active layers in OSCs. As illustrated in Fig. 27, unlike thermal-induced morphological changes (mostly w/o a chemical process), the photostability of PV materials primarily encompasses photochemically induced molecular alterations at the sub-nanometer scale and the light-induced nano-scale morphological changes. Over the past decades, investigations into photochemical degradation pathways have predominantly employed polymer donors and NFAs as the model systems. Herein, we highlight the advancements in strategies aimed at enhancing the photochemical stability of OSCs from a chemical perspective, as depicted in Fig. 27.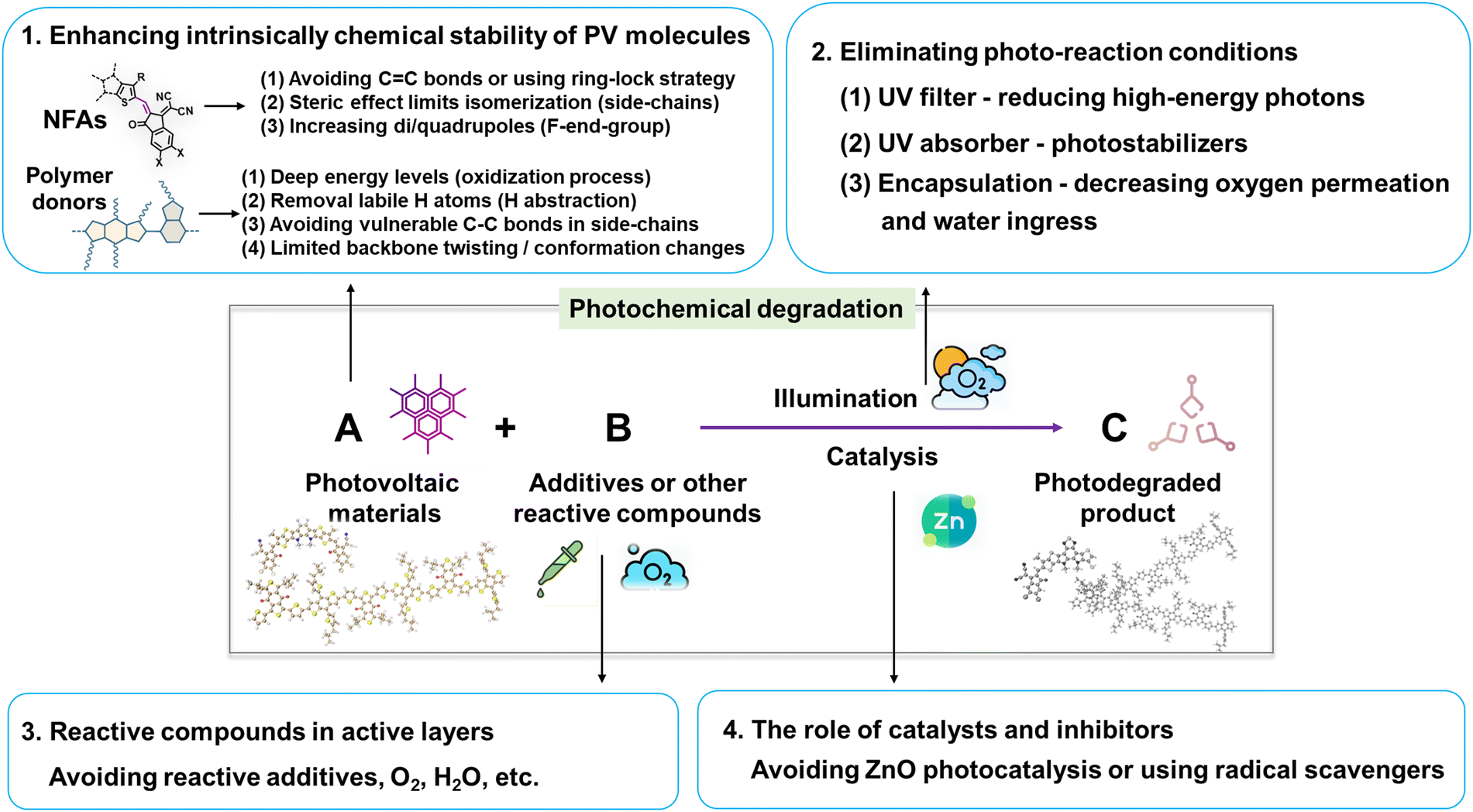 | ||
| Fig. 27 Strategies for enhancing photostability of PV materials and the OSC devices. | ||
(1) Enhancing the intrinsic photochemical stability of PV materials: (i) NFAs: Since the discoveries in organic semiconductors (Nobel lecture),128 it has been well-established that vinyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds) exhibit considerable flexibility and susceptibility to isomerization reactions. Numerous studies have confirmed the vulnerability of the vinyl group as the weak link in NFAs as summarized in Section 2. Therefore, one effective strategy to enhance photostability involves circumventing the use of vinyl groups and employing approaches like the ring-lock strategy.108,129,130 However, this necessitates a different molecular design concept for NFAs, particularly in the linking unit between core and end-group moieties. Another approach involves reinforcing resistance to isomerization reactions, achieved through the use of steric side-chains (demonstrating improved photostability from ITIC to Y-NFAs via outer side-chains) and high di/quadrupoles in NFAs (enhancing stability through fluorinated end-groups).25,106,122
C bonds) exhibit considerable flexibility and susceptibility to isomerization reactions. Numerous studies have confirmed the vulnerability of the vinyl group as the weak link in NFAs as summarized in Section 2. Therefore, one effective strategy to enhance photostability involves circumventing the use of vinyl groups and employing approaches like the ring-lock strategy.108,129,130 However, this necessitates a different molecular design concept for NFAs, particularly in the linking unit between core and end-group moieties. Another approach involves reinforcing resistance to isomerization reactions, achieved through the use of steric side-chains (demonstrating improved photostability from ITIC to Y-NFAs via outer side-chains) and high di/quadrupoles in NFAs (enhancing stability through fluorinated end-groups).25,106,122
(ii) Polymer donors: under O2 conditions, the deep energy levels of donors benefit in suppressing the oxidation process.143 Notably, avoiding the use of BDT-thiophene units is also an effective strategy for limiting the backbone twisting effect in polymer donors (associated with device degradation).29 Even in the absence of O2 and H2O, polymer donors are still susceptible to photodegradation through H-abstraction and homolytic scissions in side-chains, along with backbone twisting as summarized in Section 3. Hence, the necessity to avoid using labile H and vulnerable single chemical bonds (C–C/X, X as Cl, Br, etc) in side-chains persists in ensuring a restrained backbone conformation.
Other strategies are (2) Elimination of photoaging conditions: this involves the use of UV filters and UV absorber/stabilizers,196,222 and coupled with high-quality encapsulation to prevent the ingress of O2 and H2O.24,82 (3) Avoidance of reactive compounds: this includes refraining from using reactive components like DIO and reactive amines. (4) Catalysis and inhibitors: this entails refraining from the use of photocatalytic ZnO with UV light.112 Additionally, incorporating radical scavengers is also an effective strategy in suppressing the photodegradation pathways in NFAs and polymer donors.81,223
In summary, while significant progress has been made in developing high-performance OSCs, achieving stable OSCs with high PCEs remains challenging.224,225 Addressing these challenges requires enhancing device photostability while concurrently reducing the cost of PV materials. Future research endeavors should prioritize a deeper exploration of photochemical degradation pathways and the development of photostable and high-performance PV materials that are cost-effective and conducive to large-scale synthesis and green-solvent processability, thereby facilitating industrial-level scale-up and commercialization of OSCs.
Abbreviations
| OSCs | Organic solar cells |
| D/A | Donor/acceptor |
| BHJ | Bulk heterojunction |
| PV | Photovoltaics |
| PCEs | Power conversion efficiencies |
| ISOS | International summit on organic photovoltaics |
| PCBM | Phenyl-C61-butyric acid methyl ester or phenyl-C71-butyric acid methyl ester, also known as PC61BM or PC71BM |
| PDI | Perylenediimide |
| IDTBR | (5Z)-3-Ethyl-2-sulfanylidene-5-[[4-[9,9,18,18-tetrakis(R)-15-[7-[(Z)-(3-ethyl-4-oxo-2-sulfanylidene-1,3-thiazolidin-5-ylidene)methyl]-2,1,3-benzothiadiazol-4-yl]-5,14-dithiapentacyclo[10.6.0.0.3,1004,8.013,17]octadeca-1(12),2,4(8),6,10,13(17),15-heptaen-6-yl]-2,1,3-benzothiadiazol-7-yl]methylidene]-1,3-thiazolidin-4-one; herein, R = n-octyl for O-IDTBR and R = 2-ethylhexyl for EH-IDTBR |
| ITIC | 3,9-bis(2-Methylene-(3-(1,1-dicyanomethylene)-indanone)-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b]-dithiophene), also known as ITIC-H |
| BTP | Dithienothiophen[3.2-b]pyrrolobenzothiadiazole |
| BTP-4F | (2,20-((2Z,20Z)-((12,13-bis(2-Ethylhexyl)-3,9-diundecyl-12,13-dihydro-[1,2,5]thiadiazolo[3,4-e] thieno[2'′′,30′′:4′,50] thieno[20,30:4,5]pyrrolo[3,2-g]thieno[20,30:4,5] thieno[3,2-b]indole-2,10-diyl)bis(methanylylidene))bis(5,6-difluoro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene)) dimalononitrile), also known as Y6 |
| MDMO-PPV | Poly[2-methoxy-5-(3′,7′-dimethyloctyloxy)-p-phenylene vinylene] |
| P3HT | Poly(3-hexylthiophene) |
| PM6 | Poly[(2,6-(4,8-bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl) benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione))] |
| D18 | Poly[(2,6-(4,8-bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-5,5′-(5,8-bis(4-(2-butyloctyl)thiophen-2-yl)dithieno[3′,2′:3,4;2′′,3′′:5,6]benzo[1,2-c][1,2,5] thiadiazole)] |
| MALDI-TOF | Matrix-assisted laser desorption/ionization – time of flight |
| UPS | Ultraviolet photoelectron spectroscopy |
| NEXAFS | Near-edge X-ray absorption fine structure |
| V OC | Open-circuit voltage |
| J SC | Short-circuit current density |
| FF | Fill factor |
| LUMO | Lowest unoccupied molecular orbital |
| HOMO | Highest occupied molecular orbital |
| Vis-NIR | Visible-near-infrared |
| NDI | Naphthalene diimide |
| IDT | Indacenodithiophene |
| FBR | 5,5′-[(9,9-Dioctyl-9H-fluorene-2,7-diyl)bis(2,1,3-benzothiadiazole-7,4-diylmethylidyne)]bis[3-ethyl-2-thioxo-4-thiazolidinone] |
| IDFBR | (5Z)-3-Ethyl-5-[[4-[2-[7-[(Z)-(3-ethyl-4-oxo-2-sulfanylidene-1,3-thiazolidin-5-ylidene)methyl]-2,1,3-benzothiadiazol-4-yl]-6,6,12,12-tetraoctylindeno[1,2-b]fluoren-8-yl]-2,1,3-benzothiadiazol-7-yl]methylidene]-2-sulfanylidene-1,3-thiazolidin-4-one |
| NFAs | Non-fullerene acceptors |
| FT-IR | Fourier transform infrared absorption spectra |
| XPS | X-ray photoelectron spectroscopy |
| NMR | Nuclear magnetic resonance |
| ITIC-4F | 3,9-bis(2-Methylene-((3-(1,1-dicyanomethylene)-6,7-difluoro)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene, also known as IT-4F |
| ITIC-M | 2-[(2Z)-2-[[20-[(E)-[1-(Dicyanomethylidene)-5,6-dimethyl-3-oxoinden-2-ylidene]methyl]-12,12,24,24-tetrakis(4-hexylphenyl)-5,9,17,21-tetrathiaheptacyclo[13.9.0.0.3,130.4,1106,10.0.16,23018,22]tetracosa-1(15),2,4(11),6(10), 7,13,16(23),18(22),19-nonaen-8-yl]methylidene]-5,6-dimethyl-3-oxoinden-1-ylidene]propanedinitrile |
| ITIC-4Cl | 3,9-bis(2-Methylene-((3-(1,1-dicyanomethylene)-chloro)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno [1,2-b:5,6-b′]dithiophene |
| DFT | Density functional theory |
| MS | Mass spectrometry |
| INCN | 1,1-Dicyanomethylene-3-indanon |
| ETL | Electron transport layer |
| IEICO-4F | 2,2′-[[4,4,9,9-tetrakis(4-hexylphenyl)-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene-2,7-diyl]bis[[4-[(2-ethylhexyl)oxy]-5,2-thiophenediyl]methylidyne(5,6-difluoro-3-oxo-1H-indene-2,1(3H)-diylidene)]]bis [propanedinitrile] |
| SAM | Self-assembled monolayer |
| PEDOT:PSS | Poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate). |
| PTB7-Th | Poly([2,6′-4,8-di(5-ethylhexylthienyl)benzo[1,2-b;3,3-b]dithiophene]) |
| PTQ10 | Poly[(thiophene)-alt-(6,7-difluoro-2-(2-hexyldecyloxy)quinoxaline)] |
| ESR | Electron spin resonance |
| BDT | Thienyl-substituted-benzodithiophene |
| GPC | Gel permeation chromatography |
| CV | Cyclic voltammetry |
| DIO | 1,8-Di-iodooctane |
| HPLC–MS | High performance liquid chromatography–mass spectrometry |
| PEI | Polyethylenimine |
| PEIE | Ethoxylated polyethylenimine |
| J–V | Current–voltage |
| AFM | Atomic force microscopy |
| GIWAXS | Grazing-incidence wide-angle X-ray scattering |
| GISAXS | Grazing-incidence small-angle X-ray scattering |
Author contributions
J. H. prepared the first draft of the manuscript. S. K. P., H. X., A. S., and D. B. reviewed and revised the manuscript. D. B. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under award no: OSR-2019-CARF/CCF-3079, and KAUST Solar Center. J. H. expresses his gratitude to the Alexander von Humboldt Foundation and the support during his stay in Todd B. Marder's group at Julius-Maximilians-Universität Würzburg. Icons in Fig. 1, 26, and 27 were sourced from https://www.flaticon.com.Notes and references
- H. Yu, J. Wang, Q. Zhou, J. Qin, Y. Wang, X. Lu and P. Cheng, Chem. Soc. Rev., 2023, 52, 4132–4148 RSC.
- Y. Li, X. Huang, H. K. M. Sheriff and S. R. Forrest, Nat. Rev. Mater., 2022, 8, 186–201 CrossRef.
- G. Zhang, F. R. Lin, F. Qi, T. Heumuller, A. Distler, H. J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Jen and H. L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed.
- D. Corzo, D. Rosas-Villalva, A. C, G. Tostado-Blázquez, E. B. Alexandre, L. H. Hernandez, J. Han, H. Xu, M. Babics, S. De Wolf and D. Baran, Nat. Energy, 2022, 8, 62–73 CrossRef.
- I. McCulloch, M. Chabinyc, C. Brabec, C. B. Nielsen and S. E. Watkins, Nat. Mater., 2023, 22, 1304–1310 CrossRef CAS PubMed.
- J. W. Lee, C. Sun, S. W. Lee, G. U. Kim, S. Li, C. Wang, T. S. Kim, Y. H. Kim and B. J. Kim, Energy Environ. Sci., 2022, 15, 4672–4685 RSC.
- J.-W. Lee, H.-G. Lee, E. S. Oh, S.-W. Lee, T. N.-L. Phan, S. Li, T.-S. Kim and B. J. Kim, Joule, 2024, 8, 204–223 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C. C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- Z. Zheng, J. Wang, P. Bi, J. Ren, Y. Wang, Y. Yang, X. Liu, S. Zhang and J. Hou, Joule, 2022, 6, 171–184 CrossRef CAS.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- D. Meng, R. Zheng, Y. Zhao, E. Zhang, L. Dou and Y. Yang, Adv. Mater., 2022, 34, e2107330 CrossRef PubMed.
- J. W. Lee, C. Sun, T. N. L. Phan, D. C. Lee, Z. Tan, H. Jeon, S. Cho, S. K. Kwon, Y. H. Kim and B. Kim, Energy Environ. Sci., 2023, 16, 3339–3349 RSC.
- Y. Zhang, W. Deng, C. E. Petoukhoff, X. Xia, Y. Lang, H. Xia, H. Tang, H. T. Chandran, S. Mahadevan, K. Liu, P. W. K. Fong, Y. Luo, J. Wu, S.-W. Tsang, F. Laquai, H. Wu, X. Lu, Y. Yang and G. Li, Joule, 2024, 8, 509–526 CrossRef CAS.
- J. W. Lee, J. S. Park, H. Jeon, S. Lee, D. Jeong, C. Lee, Y. H. Kim and B. J. Kim, Chem. Soc. Rev., 2024, 53, 4674–4706 RSC.
- S. Karuthedath, J. Gorenflot, Y. Firdaus, N. Chaturvedi, C. S. P. De Castro, G. T. Harrison, J. I. Khan, A. Markina, A. H. Balawi, T. A. D. Pena, W. Liu, R. Z. Liang, A. Sharma, S. H. K. Paleti, W. Zhang, Y. Lin, E. Alarousu, S. Lopatin, D. H. Anjum, P. M. Beaujuge, S. De Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, D. Andrienko and F. Laquai, Nat. Mater., 2022, 21, 378 CrossRef CAS PubMed.
- J. Yi, G. Zhang, H. Yu and H. Yan, Nat. Rev. Mater., 2023, 9, 46–62 CrossRef.
- N. Gasparini, A. Salleo, I. McCulloch and D. Baran, Nat. Rev. Mater., 2019, 4, 229–242 CrossRef.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Rohr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2017, 16, 363–369 CrossRef CAS PubMed.
- L. Wang, C. Chen, Y. W. Fu, C. H. Guo, D. H. Li, J. C. Cheng, W. Sun, Z. R. Gan, Y. D. Sun, B. J. Zhou, C. H. Liu, D. Liu, W. Li and T. Wang, Nat. Energy, 2024, 9, 208–218 CrossRef CAS.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- M. Riede, D. Spoltore and K. Leo, Adv. Energy Mater., 2020, 11, 2002653 CrossRef.
- B. Azzopardi, C. J. M. Emmott, A. Urbina, F. C. Krebs, J. Mutale and J. Nelson, Energy Environ. Sci., 2011, 4, 3741–3753 RSC.
- P. Jiang, L. Hu, L. Sun, Z. Li, H. Han and Y. Zhou, Chem. Sci., 2022, 13, 4714–4739 RSC.
- P. Cheng and X. Zhan, Chem. Soc. Rev., 2016, 45, 2544–2582 RSC.
- J. Luke, E. J. Yang, C. Labanti, S. Y. Park and J. S. Kim, Nat. Rev. Mater., 2023, 8, 839–852 CrossRef CAS.
- P. Ding, D. Yang, S. Yang and Z. Ge, Chem. Soc. Rev., 2024, 53, 2350–2387 RSC.
- Z. X. Peng, N. Stingelin, H. Ade and J. J. Michels, Nat. Rev. Mater., 2023, 8, 439–455 CrossRef.
- L. Lüer, I. M. Peters, A. S. Smith, E. Dorschky, B. M. Eskofier, F. Liers, J. Franke, M. Sjarov, M. Brossog, D. M. Guldi, A. Maier and C. J. Brabec, Joule, 2024, 8, 295–311 CrossRef.
- Y. W. Wang, J. Luke, A. Privitera, N. Rolland, C. Labanti, G. Londi, V. Lemaur, D. T. W. Toolan, A. J. Sneyd, S. Jeong, D. P. Qian, Y. Olivier, L. Sorace, J. S. Kim, D. Beljonne, Z. Li and A. J. Gillett, Joule, 2023, 7, 810–829 CrossRef CAS.
- Y. W. Li, T. F. Li and Y. Z. Lin, Mater. Chem. Front., 2021, 5, 2907–2930 RSC.
- M. O. Reese, S. A. Gevorgyan, M. Jorgensen, E. Bundgaard, S. R. Kurtz, D. S. Ginley, D. C. Olson, M. T. Lloyd, P. Moryillo, E. A. Katz, A. Elschner, O. Haillant, T. R. Currier, V. Shrotriya, M. Hermenau, M. Riede, K. R. Kirov, G. Trimmel, T. Rath, O. Inganäs, F. L. Zhang, M. Andersson, K. Tvingstedt, M. Lira-Cantu, D. Laird, C. McGuiness, S. Gowrisanker, M. Pannone, M. Xiao, J. Hauch, R. Steim, D. M. DeLongchamp, R. Rösch, H. Hoppe, N. Espinosa, A. Urbina, G. Yaman-Uzunoglu, J. B. Bonekamp, A. J. J. M. van Breemen, C. Girotto, E. Voroshazi and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2011, 95, 1253–1267 CrossRef CAS.
- Y. Zhang, I. D. W. Samuel, T. Wang and D. G. Lidzey, Adv. Sci., 2018, 5, 1800434 CrossRef PubMed.
- L. Duan and A. Uddin, Adv. Sci., 2020, 7, 1903259 CrossRef CAS PubMed.
- G. Tregnago, Nat. Energy, 2022, 7, 567 CrossRef.
- M. Ghasemi, N. Balar, Z. Peng, H. Hu, Y. Qin, T. Kim, J. J. Rech, M. Bidwell, W. Mask, I. McCulloch, W. You, A. Amassian, C. Risko, B. T. O’Connor and H. Ade, Nat. Mater., 2021, 20, 525–532 CrossRef CAS PubMed.
- S. Siddika, Z. X. Peng, N. Balar, X. Y. Dong, X. W. Zhong, W. You, H. Ade and B. T. O’Connor, Joule, 2023, 7, 1593–1608 CrossRef CAS.
- Y. Che, M. R. Niazi, R. Izquierdo and D. F. Perepichka, Angew. Chem., Int. Ed., 2021, 60, 24833–24837 CrossRef CAS PubMed.
- H. K. H. Lee, A. M. Telford, J. A. Röhr, M. F. Wyatt, B. Rice, J. Y. Wu, A. D. Maciel, S. M. Tuladhar, E. Speller, J. McGettrick, J. R. Searle, S. Pont, T. Watson, T. Kirchartz, J. R. Durrant, W. C. Tsoi, J. Nelson and Z. Li, Energy Environ. Sci., 2018, 11, 417–428 RSC.
- T. R. Liu, Q. C. Burlingame, M. R. Ivancevic, X. Liu, J. N. Hu, B. P. Rand and Y. L. Loo, Adv. Energy Mater., 2023, 13, 2300046 CrossRef CAS.
- O. R. Yamilova, I. V. Martynov, A. S. Brandvold, I. V. Klimovich, A. H. Balzer, A. V. Akkuratov, I. E. Kusnetsov, N. Stingelin and P. A. Troshin, Adv. Energy Mater., 2020, 10, 1903163 CrossRef CAS.
- N. Yang, Y. Cui, T. Zhang, C. An, Z. Chen, Y. Xiao, Y. Yu, Y. Wang, X.-T. Hao and J. Hou, J. Am. Chem. Soc., 2024, 146, 9205–9215 CrossRef CAS PubMed.
- Y. Liang, D. Zhang, Z. Wu, T. Jia, L. Lüer, H. Tang, L. Hong, J. Zhang, K. Zhang, C. J. Brabec, N. Li and F. Huang, Nat. Energy, 2022, 7, 1180–1190 CrossRef CAS.
- X. Gu, R. Zeng, T. He, G. Zhou, C. Li, N. Yu, F. Han, Y. Hou, J. Lv, M. Zhang, J. Zhang, Z. Wei, Z. Tang, H. Zhu, Y. Cai, G. Long, F. Liu, X. Zhang and H. Huang, Adv. Mater., 2024, e2401370 CrossRef PubMed.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- G. Li, V. Shrotriya, J. S. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- D. Mühlbacher, M. Scharber, M. Morana, C. Brabec, Z. Zhu, D. Waller and R. Gaudiana, Adv. Mater., 2006, 18, 2884–2889 CrossRef.
- Y. Sun, C. Cui, H. Wang and Y. Li, Adv. Energy Mater., 2011, 1, 1058–1061 CrossRef CAS.
- Q. F. Yan, Y. Zhou, Y. Q. Zheng, J. Pei and D. H. Zhao, Chem. Sci., 2013, 4, 4389–4394 RSC.
- J. D. Chen, C. Cui, Y. Q. Li, L. Zhou, Q. D. Ou, C. Li, Y. Li and J. X. Tang, Adv. Mater., 2015, 27, 1035–1041 CrossRef CAS PubMed.
- S. Holliday, R. S. Ashraf, C. B. Nielsen, M. Kirkus, J. A. Rohr, C. H. Tan, E. Collado-Fregoso, A. C. Knall, J. R. Durrant, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 898–904 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- D. Baran, T. Kirchartz, S. Wheeler, S. Dimitrov, M. Abdelsamie, J. Gorman, R. S. Ashraf, S. Holliday, A. Wadsworth and N. Gasparini, Energy Environ. Sci., 2016, 9, 3783–3793 RSC.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C. H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganas, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS PubMed.
- S. Li, L. Zhan, F. Liu, J. Ren, M. Shi, C. Z. Li, T. P. Russell and H. Chen, Adv. Mater., 2018, 30, 1705208 CrossRef PubMed.
- W. Li, L. Ye, S. Li, H. Yao, H. Ade and J. Hou, Adv. Mater., 2018, 30, 1707170 CrossRef PubMed.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H. L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- T. Lin, Y. Hai, Y. Luo, L. Feng, T. Jia, J. Wu, R. Ma, T. A. Dela Pena, Y. Li, Z. Xing, M. Li, M. Wang, B. Xiao, K. S. Wong, S. Liu and G. Li, Adv. Mater., 2024, e2312311 CrossRef PubMed.
- M. Lv, Q. Wang, J. Zhang, Y. Wang, Z. G. Zhang, T. Wang, H. Zhang, K. Lu, Z. Wei and D. Deng, Adv. Mater., 2024, 36, e2310046 CrossRef PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- D. Meng, H. T. Fu, C. Y. Xiao, X. Y. Meng, T. Winands, W. Ma, W. Wei, B. B. Fan, L. J. Huo, N. L. Doltsinis, Y. Li, Y. M. Sun and Z. H. Wang, J. Am. Chem. Soc., 2016, 138, 10184–10190 CrossRef CAS PubMed.
- L. Ma, S. Zhang, J. Zhu, J. Wang, J. Ren, J. Zhang and J. Hou, Nat. Commun., 2021, 12, 5093 CrossRef CAS PubMed.
- A. Mishra and G. D. Sharma, Angew. Chem., Int. Ed., 2023, 62, e202219245 CrossRef CAS PubMed.
- X. Zhang, X. Gu and H. Huang, Acc. Chem. Res., 2024, 57, 981–991 CrossRef CAS PubMed.
- D. Luo, C. J. Brabec and A. K. K. Kyaw, Nano Energy, 2023, 114, 108661 CrossRef CAS.
- Z. P. Yu, Z. X. Liu, F. X. Chen, R. Qin, T. K. Lau, J. L. Yin, X. Kong, X. Lu, M. Shi, C. Z. Li and H. Chen, Nat. Commun., 2019, 10, 2152 CrossRef PubMed.
- D. L. Ma, Q. Q. Zhang and C. Z. Li, Angew. Chem., Int. Ed., 2023, 62, e202214931 CrossRef CAS PubMed.
- J. Wang, Q. Luan, P. Wang, C. Han, F. Bi, C. Yang, Y. Li and X. Bao, Adv. Funct. Mater., 2023, 33, 2301575 CrossRef CAS.
- L. J. Bu, X. Y. Guo, B. Yu, Y. Qu, Z. Y. Xie, D. H. Yan, Y. H. Geng and F. S. Wang, J. Am. Chem. Soc., 2009, 131, 13242–13243 CrossRef CAS PubMed.
- H. Bai, Q. Fan, R. Ma, X. Guo, W. Ma and M. Zhang, Chin. J. Chem., 2024, 42, 1307–1318 CrossRef CAS.
- F. Qi, Y. Li, F. R. Lin and A. K. Jen, ChemSusChem, 2024, e202301559 CrossRef PubMed.
- C. Sun, J.-W. Lee, C. Lee, D. Lee, S. Cho, S.-K. Kwon, B. J. Kim and Y.-H. Kim, Joule, 2023, 7, 416–430 CrossRef CAS.
- C. Sun, J. W. Lee, Z. Tan, T. N. L. Phan, D. Han, H. G. Lee, S. Lee, S. K. Kwon, B. J. Kim and Y. H. Kim, Adv. Energy Mater., 2023, 13, 2301283 CrossRef CAS.
- J. W. Lee, C. Sun, J. Lee, D. J. Kim, W. J. Kang, S. Lee, D. Kim, J. Park, T. N. L. Phan, Z. Tan, F. S. Kim, J. Y. Lee, X. Bao, T. S. Kim, Y. H. Kim and B. J. Kim, Adv. Energy Mater., 2024, 14, 2303872 CrossRef CAS.
- D. W. Li, H. R. Zhang, X. Y. Cui, Y. N. Chen, N. Wei, G. L. Ran, H. Lu, S. H. Chen, W. K. Zhang, C. H. Li, Y. H. Liu, Y. Q. Liu and Z. S. Bo, Adv. Mater., 2024, 36, 2310362 CrossRef CAS PubMed.
- N. Yang, Y. Cui, Y. Xiao, Z. Chen, T. Zhang, Y. Yu, J. Ren, W. Wang, L. Ma and J. Hou, Angew. Chem., Int. Ed., 2024, e202403753 CAS.
- Y. Che, M. R. Niazi, Q. Chan, P. Ghamari, T. Yu, C. Ruchlin, H. Yu, H. Yan, D. Ma, S. S. Xiao, R. Izquierdo and D. F. Perepichka, Angew. Chem., Int. Ed., 2023, 62, e202309003 CrossRef CAS PubMed.
- J. Guo, Y. Wu, R. Sun, W. Wang, J. Guo, Q. Wu, X. F. Tang, C. K. Sun, Z. H. Luo, K. Chang, Z. H. Zhang, J. Yuan, T. F. Li, W. H. Tang, E. J. Zhou, Z. Xiao, L. M. Ding, Y. P. Zou, X. W. Zhan, C. L. Yang, Z. Li, C. J. Brabec, Y. F. Li and J. Min, J. Mater. Chem. A, 2019, 7, 25088–25101 RSC.
- R. Azmi, E. Ugur, A. Seitkhan, F. Aljamaan, A. S. Subbiah, J. Liu, G. T. Harrison, M. I. Nugraha, M. K. Eswaran, M. Babics, Y. Chen, F. Xu, T. G. Allen, A. U. Rehman, C. L. Wang, T. D. Anthopoulos, U. Schwingenschlogl, M. De Bastiani, E. Aydin and S. De Wolf, Science, 2022, 376, 73–77 CrossRef CAS PubMed.
- H. Xu, J. H. Han, S. Chen, Y. Liu, L. H. Hernandez, J. Bertrandie, M. Babics, S. Alam, D. R. Villalva, S. H. K. Paleti, J. Gorenflot, C. Herok, N. Ramos, J. Troughton, A. Sharma, T. B. Marder, B. Engels, J. Martin, S. De Wolf, F. Laquai and D. Baran, Joule, 2023, 7, 2135–2151 CrossRef CAS.
- F. L. Zhang, O. Inganäs, Y. H. Zhou and K. Vandewal, Nat. Sci. Rev., 2016, 3, 222–239 CrossRef CAS.
- J. B. Zhao, Y. K. Li, G. F. Yang, K. Jiang, H. R. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- A. S. Anselmo, A. Dzwilewski, K. Svensson and E. Moons, Chem. Phys. Lett., 2016, 652, 220–224 CrossRef CAS.
- S. Pont, S. Osella, A. Smith, A. V. Marsh, Z. Li, D. Beljonne, J. T. Cabral and J. R. Durrant, Chem. Mater., 2019, 31, 6076–6083 CrossRef CAS.
- E. M. Speller, A. J. Clarke, N. Aristidou, M. F. Wyatt, L. Francas, G. Fish, H. Cha, H. K. H. Lee, J. Luke, A. Wadsworth, A. D. Evans, I. McCulloch, J. S. Kim, S. A. Haque, J. R. Durrant, S. D. Dimitrov, W. C. Tsoi and Z. Li, ACS Energy Lett., 2019, 4, 846–852 CrossRef CAS PubMed.
- S. Pont, J. R. Durrant and J. T. Cabral, Adv. Energy Mater., 2019, 9, 1803948 CrossRef.
- H. C. Wong, Z. Li, C. H. Tan, H. Zhong, Z. Huang, H. Bronstein, I. McCulloch, J. T. Cabral and J. R. Durrant, ACS Nano, 2014, 8, 1297–1308 CrossRef CAS PubMed.
- T. Heumueller, W. R. Mateker, A. Distler, U. F. Fritze, R. Cheacharoen, W. H. Nguyen, M. Biele, M. Salvador, M. von Delius, H. J. Egelhaaf, M. D. McGehee and C. J. Brabec, Energy Environ. Sci., 2016, 9, 247–256 RSC.
- D. Lee, Y. Jang, J. Kim, S. Y. Jeong, H. Y. Woo, D. G. Lee, J. Kim, Y. Lee, C. Lee and W. Lee, J. Mater. Chem. A, 2023, 11, 3008–3017 RSC.
- L. Yu, D. Qian, S. Marina, F. A. A. Nugroho, A. Sharma, S. Hultmark, A. I. Hofmann, R. Kroon, J. Benduhn, D. M. Smilgies, K. Vandewal, M. R. Andersson, C. Langhammer, J. Martin, F. Gao and C. Muller, ACS Appl. Mater. Interfaces, 2019, 11, 21766–21774 CrossRef CAS PubMed.
- O. Almora, D. Baran, G. C. Bazan, C. I. Cabrera, S. Erten-Ela, K. Forberich, F. Guo, J. Hauch, A. W. Y. Ho-Baillie, T. J. Jacobsson, R. A. J. Janssen, T. Kirchartz, N. Kopidakis, M. A. Loi, R. R. Lunt, X. Mathew, M. D. McGehee, J. Min, D. B. Mitzi, M. K. Nazeeruddin, J. Nelson, A. F. Nogueira, U. W. Paetzold, B. P. Rand, U. Rau, H. J. Snaith, E. Unger, L. Vaillant-Roca, C. Yang, H. L. Yip and C. J. Brabec, Adv. Energy Mater., 2023, 13, 2203313 CrossRef CAS.
- O. Almora, D. Baran, G. C. Bazan, C. Berger, C. Cabrera, K. R. Catchpole, S. Erten-Ela, F. Guo, J. Hauch, A. W. Y. Ho-Baillie, T. J. Jacobsson, R. A. J. Janssen, T. Kirchartz, N. Kopidakis, Y. F. Li, M. A. Loi, R. R. Lunt, X. Mathew, M. D. McGehee, J. Min, D. B. Mitzi, M. K. Nazeeruddin, J. Nelson, A. F. Nogueira, U. W. Paetzold, N. G. Park, B. P. Rand, U. Rau, H. J. Snaith, E. Unger, L. Vaillant-Roca, H. L. Yip and C. J. Brabec, Adv. Energy Mater., 2021, 11, 2102526 CrossRef CAS.
- O. Almora, D. Baran, G. C. Bazan, C. Berger, C. I. Cabrera, K. R. Catchpole, S. Erten-Ela, F. Guo, J. Hauch, A. W. Y. Ho-Baillie, T. J. Jacobsson, R. A. J. Janssen, T. Kirchartz, N. Kopidakis, Y. Li, M. A. Loi, R. R. Lunt, X. Mathew, M. D. McGehee, J. Min, D. B. Mitzi, M. K. Nazeeruddin, J. Nelson, A. F. Nogueira, U. W. Paetzold, N. G. Park, B. P. Rand, U. Rau, H. J. Snaith, E. Unger, L. Vaillant-Roca, H. L. Yip and C. J. Brabec, Adv. Energy Mater., 2020, 11, 2002774 CrossRef.
- Y. Z. Lin and X. W. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC.
- J. Wang and X. Zhan, Acc. Chem. Res., 2021, 54, 132–143 CrossRef CAS PubMed.
- S. Chen, D. Meng, J. Huang, N. Liang, Y. Li, F. Liu, H. Yan and Z. Wang, CCS Chem., 2021, 3, 78–84 CrossRef CAS.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- S. Kumar, J. Shukla, Y. Kumar and P. Mukhopadhyay, Org. Chem. Front., 2018, 5, 2254–2276 RSC.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- J. Yao, Q. Chen, C. Zhang, Z. G. Zhang and Y. F. Li, Susmat, 2022, 2, 243–263 CrossRef CAS.
- M. Sadeghianlemraski, C. R. Harding, G. C. Welch and H. Aziz, ACS Appl. Energy Mater., 2020, 3, 11655–11665 CrossRef CAS.
- A. Wadsworth, R. S. Ashraf, M. Abdelsamie, S. Pont, M. Little, M. Moser, Z. Hamid, M. Neophytou, W. M. Zhang, A. Amassian, J. R. Durrant, D. Baran and I. McCulloch, ACS Energy Lett., 2017, 2, 1494–1500 CrossRef CAS.
- J. Luke, E. M. Speller, A. Wadsworth, M. F. Wyatt, S. Dimitrov, H. K. H. Lee, Z. Li, W. C. Tsoi, I. McCulloch, D. Bagnis, J. R. Durrant and J. S. Kim, Adv. Energy Mater., 2019, 9, 1803755 CrossRef.
- J. Guo, Y. Wu, R. Sun, W. Wang, J. F. Li, E. J. Zhou, J. Guo, T. Wang, Q. Wu, Z. H. Luo, W. Gao, Y. M. Pan, C. L. Yang and J. Min, Sol. RRL, 2021, 5, 2000704 CrossRef CAS.
- H. T. Liu, W. Wang, Y. H. Zhou and Z. A. Li, J. Mater. Chem. A, 2021, 9, 1080–1088 RSC.
- Z. X. Liu, Z. P. Yu, Z. Shen, C. He, T. K. Lau, Z. Chen, H. Zhu, X. Lu, Z. Xie, H. Chen and C. Z. Li, Nat. Commun., 2021, 12, 3049 CrossRef CAS PubMed.
- A. J. Clarke, J. Luke, R. Meitzner, J. Y. Wu, Y. M. Wang, H. K. H. Lee, E. M. Speller, H. Bristow, Y. J. Cha, M. J. Newman, K. Hooper, A. Evans, F. Gao, H. Hoppe, I. McCulloch, U. S. Schubert, T. M. Watson, J. R. Durrant, W. C. Tsoi, J. S. Kim and Z. Li, Cell Rep. Phys. Sci., 2021, 2, 100498 CrossRef CAS.
- P. Das, C. T. Kornman, I. Ghiviriga, K. A. Abboud and R. K. Castellano, Chem. Mater., 2023, 35, 8122–8134 CrossRef CAS.
- Y. Y. Jiang, L. L. Sun, F. Y. Jiang, C. Xie, L. Hu, X. Y. Dong, F. Qin, T. F. Liu, L. Hu, X. S. Jiang and Y. H. Zhou, Mater. Horiz., 2019, 6, 1438–1443 RSC.
- S. Park and H. J. Son, J. Mater. Chem. A, 2019, 7, 25830–25837 RSC.
- M. Gunther, D. Blatte, A. L. Oechsle, S. S. Rivas, A. A. Yousefi Amin, P. Muller-Buschbaum, T. Bein and T. Ameri, ACS Appl. Mater. Interfaces, 2021, 13, 19072–19084 CrossRef PubMed.
- L. Y. Su, H. H. Huang, C. E. Tsai, C. H. Hou, J. J. Shyue, C. H. Lu, C. W. Pao, M. H. Yu, L. Wang and C. C. Chueh, Small, 2022, 18, e2107834 CrossRef PubMed.
- S. Balasubramanian, M. A. Leon-Luna, B. Romero, M. Madsen and V. Turkovic, ACS Appl. Mater. Interfaces, 2023, 15, 39647–39656 CrossRef CAS PubMed.
- S. Guan, Y. Li, C. Xu, N. Yin, C. Xu, C. Wang, M. Wang, Y. Xu, Q. Chen, D. Wang, L. Zuo and H. Chen, Adv. Mater., 2024, e2400342 CrossRef PubMed.
- S. Shoaee, H. M. Luong, J. Song, Y. Zou, T. Q. Nguyen and D. Neher, Adv. Mater., 2023, e2302005 Search PubMed.
- S. X. Li, C. Z. Li, M. M. Shi and H. Z. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS.
- Q. Y. Wei, W. Liu, M. Leclerc, J. Yuan, H. G. Chen and Y. P. Zou, Sci. China: Chem., 2020, 63, 1352–1366 CrossRef CAS.
- J. Wang, Z. Zheng, Y. Zu, Y. Wang, X. Liu, S. Zhang, M. Zhang and J. Hou, Adv. Mater., 2021, 33, e2102787 CrossRef PubMed.
- J. Luke, E. J. Yang, Y. C. Chin, Y. X. Che, L. Winkler, D. Whatling, C. Labanti, S. Y. Park and J. S. Kim, Adv. Energy Mater., 2022, 12, 2201267 CrossRef CAS.
- B. J. Zhou, L. Wang, Y. Liu, C. H. Guo, D. H. Li, J. L. Cai, Y. W. Fu, C. Chen, D. Liu, Y. H. Zhou, W. Li and T. Wang, Adv. Funct. Mater., 2022, 32, 2206042 CrossRef CAS.
- B. Fan, W. Gao, R. Zhang, W. Kaminsky, F. R. Lin, X. Xia, Q. Fan, Y. Li, Y. An, Y. Wu, M. Liu, X. Lu, W. J. Li, H. L. Yip, F. Gao and A. K. Jen, J. Am. Chem. Soc., 2023, 145, 5909–5919 CrossRef CAS PubMed.
- B. Fan, W. Gao, X. Wu, X. Xia, Y. Wu, F. R. Lin, Q. Fan, X. Lu, W. J. Li, W. Ma and A. K. Jen, Nat. Commun., 2022, 13, 5946 CrossRef CAS PubMed.
- Y. S. Zhao, Z. N. Wu, X. Liu, Z. P. Zhong, R. H. Zhu and J. S. Yu, J. Mater. Chem. C, 2021, 9, 13972–13980 RSC.
- W. Q. Zhang, X. Y. Du, Y. L. Ma, J. W. Qiao, Q. D. Zheng and X. T. Hao, Adv. Funct. Mater., 2024, 34, 2308591 CrossRef CAS.
- H. Shirakawa, Angew. Chem., Int. Ed., 2001, 40, 2574–2580 CrossRef PubMed.
- X. Zhu, S. Liu, Q. Yue, W. Liu, S. Sun and S. Xu, CCS Chem., 2021, 3, 1070–1080 CrossRef CAS.
- W. Liu, S. Xu, H. Lai, W. Liu, F. He and X. Zhu, CCS Chem., 2023, 5, 654–668 CrossRef CAS.
- S. Holliday, Y. L. Li and C. K. Luscombe, Prog. Polym. Sci., 2017, 70, 34–51 CrossRef CAS.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CrossRef CAS PubMed.
- Z. Zheng, H. Yao, L. Ye, Y. Xu, S. Zhang and J. Hou, Mater. Today, 2020, 35, 115–130 CrossRef CAS.
- C. An and J. Hou, Acc. Mater. Res., 2022, 3, 540–551 CrossRef CAS.
- Z. Wang, Z. Peng, Z. Xiao, D. Seyitliyev, K. Gundogdu, L. Ding and H. Ade, Adv. Mater., 2020, 32, e2005386 CrossRef PubMed.
- M. Manceau, A. Rivaton, J. L. Gardette, S. Guillerez and N. Lemaître, Polym. Degrad. Stab., 2009, 94, 898–907 CrossRef CAS.
- H. Hintz, H. J. Egelhaaf, L. Lüer, J. Hauch, H. Peisert and T. Chassé, Chem. Mater., 2010, 23, 145–154 CrossRef.
- M. Manceau, S. Chambon, A. Rivaton, J. L. Gardette, S. Guillerez and N. Lemaître, Sol. Energy Mater. Sol. Cells, 2010, 94, 1572–1577 CrossRef CAS.
- R. A. Street, J. E. Northrup and B. S. Krusor, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 205211 CrossRef.
- A. Tournebize, P. O. Bussière, P. Wong-Wah-Chung, S. Thérias, A. Rivaton, J. L. Gardette, S. Beaupré and M. Leclerc, Adv. Energy Mater., 2013, 3, 478–487 CrossRef CAS.
- R. A. Street, Y. Yang, B. C. Thompson and I. McCulloch, J. Phys. Chem. C, 2016, 120, 22169–22178 CrossRef CAS.
- N. Sai, K. Leung, J. Zador and G. Henkelman, Phys. Chem. Chem. Phys., 2014, 16, 8092–8099 RSC.
- I. V. Martynov, L. N. Inasaridze and P. A. Troshin, ChemSusChem, 2022, 15, e202101336 CrossRef CAS PubMed.
- A. Tournebize, J.-L. Gardette, C. Taviot-Guého, D. Bégué, M. A. Arnaud, C. Dagron-Lartigau, H. Medlej, R. C. Hiorns, S. Beaupré, M. Leclerc and A. Rivaton, Polym. Degrad. Stab., 2015, 112, 175–184 CrossRef CAS.
- A. Tournebize, I. F. Domínguez, G. E. Morse, C. Taviot-Guého, A. Rivaton, H. Peisert and T. Chassé, Polym. Degrad. Stab., 2017, 146, 155–160 CrossRef CAS.
- M. A. Anderson, A. Hamstra, B. W. Larson and E. L. Ratcliff, J. Mater. Chem. A, 2023, 11, 17858–17871 RSC.
- P. M. Kuznetsov, I. V. Martynov, I. S. Zhidkov, L. G. Gutsev, E. A. Komissarova, A. V. Maskaev, A. I. Kukharenko, F. A. Prudnov and P. A. Troshin, J. Phys. Chem. B, 2023, 127, 6432–6439 CrossRef CAS PubMed.
- D. E. Shen, A. W. Lang, G. S. Collier, A. M. Österholm, E. M. Smith, A. L. Tomlinson and J. R. Reynolds, Chem. Mater., 2022, 34, 1041–1051 CrossRef CAS.
- K. O. Brinkmann, T. Becker, F. Zimmermann, C. Kreusel, T. Gahlmann, M. Theisen, T. Haeger, S. Olthof, C. Tuckmantel, M. Gunster, T. Maschwitz, F. Gobelsmann, C. Koch, D. Hertel, P. Caprioglio, F. Pena-Camargo, L. Perdigon-Toro, A. Al-Ashouri, L. Merten, A. Hinderhofer, L. Gomell, S. Zhang, F. Schreiber, S. Albrecht, K. Meerholz, D. Neher, M. Stolterfoht and T. Riedl, Nature, 2022, 604, 280–286 CrossRef CAS PubMed.
- H. Gao, Y. Sun, L. Meng, C. Han, X. Wan and Y. Chen, Small, 2023, 19, e2205594 CrossRef PubMed.
- W. R. Xu, Y. L. Chang, X. W. Zhu, Z. H. Wei, X. L. Zhang, X. N. Sun, K. Lu and Z. X. Wei, Chin. Chem. Lett., 2022, 33, 123–132 CrossRef CAS.
- C. Y. Xu, Z. J. Zhao, K. X. Yang, L. B. Niu, X. L. Ma, Z. J. Zhou, X. L. Zhang and F. J. Zhang, J. Mater. Chem. A, 2022, 10, 6291–6329 RSC.
- Z. Zhang, Y. Wang, C. Sun, Z. Liu, H. Wang, L. Xue and Z. G. Zhang, Nano Select, 2021, 3, 233–247 CrossRef.
- H. Tang, C. Q. Yan, J. M. Huang, Z. P. Kan, Z. Y. Xiao, K. Sun, G. Li and S. R. Lu, Matter, 2020, 3, 1403–1432 CrossRef.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- X. Sun, J. Lv, C. Zhang, K. Wang, C. Yang, H. Hu and X. Ouyang, Sol. RRL, 2023, 7, 2300332 CrossRef CAS.
- H. Tang, C. Yan, J. Huang, Z. Kan, Z. Xiao, K. Sun, G. Li and S. Lu, Matter, 2020, 3, 1403–1432 CrossRef.
- C. Xu, Z. Zhao, K. Yang, L. Niu, X. Ma, Z. Zhou, X. Zhang and F. Zhang, J. Mater. Chem. A, 2022, 10, 6291–6329 RSC.
- Y. Sun, L. Nian, Y. Kan, Y. Ren, Z. Chen, L. Zhu, M. Zhang, H. Yin, H. Xu, J. Li, X. Hao, F. Liu, K. Gao and Y. Li, Joule, 2022, 6, 2835–2848 CrossRef CAS.
- Y. Gao, X. Yang, R. Sun, L.-Y. Xu, Z. Chen, M. Zhang, H. Zhu and J. Min, Joule, 2023, 7, 2845–2858 CrossRef CAS.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886–3893 CrossRef CAS PubMed.
- W. Xu, Y. Chang, X. Zhu, Z. Wei, X. Zhang, X. Sun, K. Lu and Z. Wei, Chin. Chem. Lett., 2022, 33, 123–132 CrossRef CAS.
- A. B. Tamayo, X.-D. Dang, B. Walker, J. Seo, T. Kent and T.-Q. Nguyen, Appl. Phys. Lett., 2009, 94, 103301 CrossRef.
- B. Walker, A. B. Tamayo, X. D. Dang, P. Zalar, J. H. Seo, A. Garcia, M. Tantiwiwat and T. Q. Nguyen, Adv. Funct. Mater., 2009, 19, 3063–3069 CrossRef CAS.
- R. Sun, Y. Wu, J. Guo, Y. H. Wang, F. Qin, B. X. Shen, D. H. Li, T. Wang, Y. W. Li, Y. H. Zhou, G. H. Lu, Y. F. Li and J. Min, Energy Environ. Sci., 2021, 14, 3174–3183 RSC.
- R. Cheacharoen, W. R. Mateker, Q. Zhang, B. Kan, D. Sarkisian, X. F. Liu, J. A. Love, X. J. Wan, Y. S. Chen, T. Q. Nguyen, G. C. Bazan and M. D. McGehee, Sol. Energy Mater. Sol. Cells, 2017, 161, 368–376 CrossRef CAS.
- K. K. Zhou, K. H. Xian and L. Ye, InfoMat, 2022, 4, e12270 CrossRef CAS.
- M. Kataria, H. D. Chau, N. Y. Kwon, S. H. Park, M. J. Cho and D. H. Choi, ACS Energy Lett., 2022, 7, 3835–3854 CrossRef CAS.
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- T. Yang and C. L. Zhan, Sci. China: Chem., 2023, 66, 2513–2531 CrossRef CAS.
- Z. Wang, X. Wang, L. Tu, H. Wang, M. Du, T. Dai, Q. Guo, Y. Shi and E. Zhou, Angew. Chem., Int. Ed., 2024, e202319755 CAS.
- B. Liu, W. Xu, R. Ma, J. W. Lee, T. A. Dela Pena, W. Yang, B. Li, M. Li, J. Wu, Y. Wang, C. Zhang, J. Yang, J. Wang, S. Ning, Z. Wang, J. Li, H. Wang, G. Li, B. J. Kim, L. Niu, X. Guo and H. Sun, Adv. Mater., 2023, 35, e2308334 CrossRef PubMed.
- R. Sun, W. Wang, H. Yu, Z. Chen, X. Xia, H. Shen, J. Guo, M. Shi, Y. Zheng, Y. Wu, W. Yang, T. Wang, Q. Wu, Y. Yang, X. Lu, J. Xia, C. J. Brabec, H. Yan, Y. Li and J. Min, Joule, 2021, 5, 1548–1565 CrossRef CAS.
- W. Y. Yang, W. Wang, Y. H. Wang, R. Sun, J. Guo, H. N. Li, M. M. Shi, J. Guo, Y. Wu, T. Wang, G. H. Lu, C. J. Brabec, Y. F. Li and J. Min, Joule, 2021, 5, 1209–1230 CrossRef.
- J. L. Song, L. L. Ye, C. H. Liu, Y. H. Cai, C. Zhang, G. C. Yue, Y. Li, M. H. Jee, Y. Zhao, D. H. Wei, H. Y. Woo and Y. M. Sun, Energy Environ. Sci., 2023, 16, 5371–5380 RSC.
- J. Guo, T. Wang, Y. Wu, R. Sun, Q. Wu, W. Wang, H. Wang, X. Xia, X. Lu, T. Wang and J. Min, J. Mater. Chem. C, 2022, 10, 1246–1258 RSC.
- Q. Wu, W. Wang, Z. Chen, X. Xia, M. Gao, H. Shen, H. Zhu, X. Lu, L. Ye, J. Xia and J. Min, J. Mater. Chem. C, 2022, 10, 1850–1861 RSC.
- S. Liang, C. Xiao, C. Xie, B. Liu, H. Fang and W. Li, Adv. Mater., 2023, 35, e2300629 CrossRef PubMed.
- J. Guo, Y. Wu, W. Wang, T. Wang and J. Min, Sol. RRL, 2022, 6, 2101024 CrossRef CAS.
- X. Yang, M. Chen, S. Wang, Y. Gao, Y. Shao, L.-Y. Xu, Y. Wu, Y. Wang, R. S. Ashraf, S. Ponomarenko, Y. Luponosov and J. Min, Giant, 2023, 16, 100191 CrossRef CAS.
- S. Liang, X. Jiang, C. Xiao, C. Li, Q. Chen and W. Li, Acc. Chem. Res., 2021, 54, 2227–2237 CrossRef CAS PubMed.
- S. Liang, J. Wang, Y. Ouyang, W. L. Tan, C. R. McNeill, Q. Chen, Z. Tang and W. Li, Macromolecules, 2022, 55, 2517–2523 CrossRef CAS.
- D. Xia, S. Zhou, W. L. Tan, S. Karuthedath, C. Xiao, C. Zhao, F. Laquai, C. R. McNeill and W. Li, Aggregate, 2022, 4, e279 CrossRef.
- Y. K. He, N. Li, T. Heumüller, J. Wortmann, B. Hanisch, A. Aubele, S. Lucas, G. T. Feng, X. D. Jiang, W. W. Li, P. Bäuerle and C. J. Brabec, Joule, 2022, 6, 1160–1171 CrossRef CAS.
- W. Wei, Y. Gao, Y. Wu, X. Yang, Z. Chen, Z. Chen, T. Wang, R. Sun, Q. Wu, X. Hao, H. Zhu, S. Ponomarenko, Y. Luponosov and J. Min, J. Mater. Chem. A, 2022, 10, 18753–18761 RSC.
- Y. Gao, B. Xiao, S. Wang, M. Chen, W. Wang, X. Yang, Y. Shao, R. Sun and J. Min, Sci. China: Chem., 2023, 66, 3205–3212 CrossRef CAS.
- X. Yang, Y. Wu, Y. Gao, J. Guo, W. Wang and J. Min, Adv. Funct. Mater., 2022, 33, 2208412 CrossRef.
- Y. Cheng, Q. Mao, C. Zhou, X. Huang, J. Liu, J. Deng, Z. Sun, S. Jeong, Y. Cho, Y. Zhang, B. Huang, F. Wu, C. Yang and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202308267 CrossRef CAS PubMed.
- Y. He, N. Li and C. J. Brabec, Org. Mater., 2021, 03, 228–244 CrossRef CAS.
- J. Roncali, Adv. Energy Mater., 2021, 11, 2102987 CrossRef CAS.
- B. Li, X. Yang, S. Li and J. Yuan, Energy Environ. Sci., 2023, 16, 723–744 RSC.
- Y. Cheng, B. Huang, Q. Mao, X. Huang, J. Liu, C. Zhou, W. Zhou, X. Ren, S. Kim, W. Kim, Z. Sun, F. Wu, C. Yang and L. Chen, Adv. Mater., 2024, e2312938 CrossRef PubMed.
- Y. He, B. Wang, L. Lüer, G. Feng, A. Osvet, T. Heumüller, C. Liu, W. Li, D. M. Guldi, N. Li and C. J. Brabec, Adv. Energy Mater., 2021, 12, 2103406 CrossRef.
- Y. Wu, J. Guo, W. Wang, Z. Chen, Z. Chen, R. Sun, Q. Wu, T. Wang, X. Hao, H. Zhu and J. Min, Joule, 2021, 5, 1800–1815 CrossRef CAS.
- I. E. Jacobs, F. Wang, Z. I. B. Valdez, A. N. A. Oviedo, D. J. Bilsky and A. J. Moulé, J. Mater. Chem. C, 2018, 6, 219–225 RSC.
- A. Classen, T. Heumueller, I. Wabra, J. Gerner, Y. K. He, L. Einsiedler, N. Li, G. J. Matt, A. Osvet, X. Y. Du, A. Hirsch and C. J. Brabec, Adv. Energy Mater., 2019, 9, 1902124 CrossRef CAS.
- X. Xu, J. Xiao, G. Zhang, L. Wei, X. Jiao, H. L. Yip and Y. Cao, Sci. Bull., 2020, 65, 208–216 CrossRef CAS PubMed.
- D. Yang, F. C. Löhrer, V. Körstgens, A. Schreiber, S. Bernstorff, J. M. Buriak and P. Müller-Buschbaum, ACS Energy Lett., 2019, 4, 464–470 CrossRef CAS.
- H. T. Liu, Y. B. Li, S. H. Xu, Y. H. Zhou and Z. A. Li, Adv. Funct. Mater., 2021, 31, 2106735 CrossRef CAS.
- F. Qin, W. Wang, L. Sun, X. Jiang, L. Hu, S. Xiong, T. Liu, X. Dong, J. Li, Y. Jiang, J. Hou, K. Fukuda, T. Someya and Y. Zhou, Nat. Commun., 2020, 11, 4508 CrossRef CAS PubMed.
- B. W. Xu and J. H. Hou, Adv. Energy Mater., 2018, 8, 1800022 CrossRef.
- D. Baran, N. Gasparini, A. Wadsworth, C. H. Tan, N. Wehbe, X. Song, Z. Hamid, W. Zhang, M. Neophytou, T. Kirchartz, C. J. Brabec, J. R. Durrant and I. McCulloch, Nat. Commun., 2018, 9, 2059 CrossRef PubMed.
- F. Qin, L. Sun, H. Chen, Y. Liu, X. Lu, W. Wang, T. Liu, X. Dong, P. Jiang, Y. Jiang, L. Wang and Y. Zhou, Adv. Mater., 2021, 33, e2103017 CrossRef PubMed.
- L. Hu, Y. Liu, L. Mao, S. X. Xiong, L. L. Sun, N. Zhao, F. Qin, Y. Y. Jiang and Y. H. Zhou, J. Mater. Chem. A, 2018, 6, 2273–2278 RSC.
- W. Zeng, X. Zhou, B. Du, L. Hu, C. Xie, W. Wang, Y. Jiang, T. Wang and Y. Zhou, Adv. Energy Sustainability Res., 2021, 2, 2000094 CrossRef CAS.
- A. Prasetio, M. Jahandar, S. Kim, J. Heo, Y. H. Kim and D. C. Lim, Adv. Sci., 2021, 8, 2100865 CrossRef CAS PubMed.
- Z. Q. Zhang, Z. L. Zhang, Y. F. Yu, B. Zhao, S. Li, J. Zhang and S. T. Tan, J. Energy Chem., 2020, 47, 196–202 CrossRef.
- Y. Li, T. Li, J. Wang, X. Zhan and Y. Lin, Sci. Bull., 2022, 67, 171–177 CrossRef CAS PubMed.
- M. Kyeong, J. Lee, M. Daboczi, K. Stewart, H. F. Yao, H. J. Cha, J. Luke, K. Lee, J. R. Durrant, J. S. Kim and S. Hong, J. Mater. Chem. A, 2021, 9, 13506–13514 RSC.
- S. Xiong, L. Hu, L. Hu, L. Sun, F. Qin, X. Liu, M. Fahlman and Y. Zhou, Adv. Mater., 2019, 31, e1806616 CrossRef PubMed.
- X. Y. Zhu, L. Hu, W. Wang, X. S. Jiang, L. Hu and Y. H. Zhou, ACS Appl. Energy Mater., 2019, 2, 7602–7608 CrossRef CAS.
- Y. P. Qin, N. Balar, Z. X. Peng, A. Gadisa, I. Angunawela, A. Bagui, S. Kashani, J. H. Hou and H. Ade, Joule, 2021, 5, 2129–2147 CrossRef CAS.
- S. H. K. Paleti, S. Hultmark, J. Han, Y. Wen, H. Xu, S. Chen, E. Jarsvall, I. Jalan, D. R. Villalva, A. Sharma, J. I. Khan, E. Moons, R. Li, L. Yu, J. Gorenflot, F. Laquai, C. Muller and D. Baran, Nat. Commun., 2023, 14, 4608 CrossRef CAS PubMed.
- J. Xiao, N. Li, Q. Yin, Y. Min and H. L. Yip, Adv. Opt. Mater., 2024, 12, 2302202 CrossRef CAS.
- Z. Xing, X. Wu, T. Chen, S. Ye, S. Wang, Y. Pan, S. Li, M. Shi and H. Chen, J. Mater. Chem. A, 2024, 12, 11286–11294 RSC.
- X. Y. Jiang, A. J. Gillett, T. L. Zheng, X. Song, J. E. Heger, K. Sun, L. V. Spanier, R. J. Guo, S. Z. Liang, S. Bernstorff and P. Müller-Buschbaum, Energy Environ. Sci., 2023, 16, 5970–5981 RSC.
- D. F. Zhang, C. Liu, K. C. Zhang, Y. H. Jia, W. K. Zhong, W. D. Qiu, Y. F. Li, T. Heumueller, K. Forberich, V. M. Le Corre, L. Lüer, N. Li, F. Huang, C. J. Brabec and L. Ying, Energy Environ. Sci., 2023, 16, 5970–5981 RSC.
- Y. Li, X. Huang, A. R. Mencke, S. K. Kandappa, T. Wang, K. Ding, Z. Q. Jiang, A. Amassian, L. S. Liao, M. E. Thompson and S. R. Forrest, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2301118120 CrossRef CAS PubMed.
- J. Y. Xiao, M. R. Ren, G. C. Zhang, J. B. Wang, D. L. Zhang, L. L. Liu, N. Li, C. J. Brabec, H. L. Yip and Y. Cao, Sol. RRL, 2019, 3, 1900077 CrossRef.
- N. Gasparini, H. K. Paleti, J. Bertrandie, G. L. Cai, G. C. Zhang, A. Wadsworth, X. H. Lu, H. L. Yip, I. McCulloch and D. Baran, ACS Energy Lett., 2020, 5, 1371–1379 CrossRef CAS.
- S. H. K. Paleti, S. Hultmark, N. Ramos, N. Gasparini, A. H. Emwas, J. Martin, C. Müller and D. Baran, Sol. RRL, 2022, 6, 2200436 CrossRef CAS.
- Y. J. Cui, Z. Chen, P. P. Zhu, W. Ma, H. M. Zhu, X. F. Liao and Y. W. Chen, Sci. China: Chem., 2023, 66, 1179–1189 CrossRef CAS.
- M. Salvador, N. Gasparini, J. D. Perea, H. Paleti, A. Distler, L. N. Inasaridze, P. A. Troshin, L. Lüer, H. J. Egelhaaf and C. Brabec, Energy Environ. Sci., 2017, 10, 2005–2016 RSC.
- A. Karki, A. J. Gillett, R. H. Friend and T. Q. Nguyen, Adv. Energy Mater., 2020, 11, 2003441 CrossRef.
- Q. Burlingame, M. Ball and Y. L. Loo, Nat. Energy, 2020, 5, 947–949 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |