
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in peptide macrocyclization strategies
Pengyuan
Fang
a,
Wing-Ka
Pang
b,
Shouhu
Xuan
 c,
Wai-Lun
Chan
*ad and
Ken Cham-Fai
Leung
*b
c,
Wai-Lun
Chan
*ad and
Ken Cham-Fai
Leung
*b
aJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, Fujian, P. R. China. E-mail: kwlchan@nus.edu.sg
bDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Kowloon, Hong Kong SAR, P. R. China. E-mail: cfleung@hkbu.edu.hk
cCAS Key Laboratory of Mechanical Behavior and Design of Materials, Department of Modern Mechanics, University of Science and Technology of China, Hefei, Anhui 230027, P. R. China
dDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore
First published on 19th November 2024
Abstract
Recently, owing to their special spatial structures, peptide-based macrocycles have shown tremendous promise and aroused great interest in multidisciplinary research ranging from potent antibiotics against resistant strains to functional biomaterials with novel properties. Besides traditional monocyclic peptides, many fascinating polycyclic and remarkable higher-order cyclic, spherical and cylindric peptidic systems have come into the limelight owing to breakthroughs in various chemical (e.g., native chemical ligation and transition metal catalysis), biological (e.g., post-translational enzymatic modification and genetic code reprogramming), and supramolecular (e.g., mechanically interlocked, metal-directed folding and self-assembly via noncovalent interactions) macrocyclization strategies developed in recent decades. In this tutorial review, diverse state-of-the-art macrocyclization methodologies and techniques for peptides and peptidomimetics are surveyed and discussed, with insights into their practical advantages and intrinsic limitations. Finally, the synthetic-technical aspects, current unresolved challenges, and outlook of this field are discussed.
 Pengyuan Fang | Pengyuan Fang obtained his Bachelor's degree in Applied Chemistry from Zhengzhou University of Light Industry in 2022. He is currently pursuing his Master's degree in Materials Engineering through the joint program of Fuzhou University and Tianjin University under the guidance of Prof. Yixin Lu and Dr Wai-Lun Chan at the Joint School of National University of Singapore and Tianjin University. |
 Wing-Ka Pang | Wing-Ka Pang obtained his B.Sc. (Hons.) degree in Chemistry from Hong Kong Baptist University and is currently a postgraduate student under the supervision of Professor Ken Cham-Fai Leung in the Department of Chemistry, Hong Kong Baptist University. |
 Shouhu Xuan | Shouhu Xuan was born in Hefei, Anhui, China. He received his B.Sc. degree in Chemistry from Anhui Normal University and PhD degree in Inorganic Chemistry from The University of Science and Technology of China. He worked as a Postdoctoral Fellow at The Chinese University of Hong Kong. He is currently a Professor in the Department of Modern Mechanics, University of Science and Technology of China. His research interests are soft materials, wearable electronic fabrics and intelligent anti-impact materials. |
 Wai-Lun Chan | Wai-Lun Chan received his BSc (Hons.) in Chemistry (2014) and a MA in Communication (2015) from Hong Kong Baptist University, followed by a PhD in Chemistry (2019) from the National University of Singapore under the supervision of Prof. Yixin Lu. After a postdoctoral visit (2020) with Prof. Markus Zweckstetter at the German Center for Neurodegenerative Diseases and Max Planck Institute for Biophysical Chemistry, he was appointed as a Research Assistant Professor (2021) with Prof. Wing-Tak Wong at the Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, and then a Research Associate Professor (2023) with Prof. Yixin Lu at the Advanced Manufacturing Centre, Joint School of National University of Singapore and Tianjin University. His current research interests include asymmetric organocatalysis, natural product synthesis, biomolecule modification, and targeted molecular imaging and phototherapy against cancer and neurodegeneration. |
 Ken Cham-Fai Leung | Ken Cham-Fai Leung is an Associate Professor in the Department of Chemistry at Hong Kong Baptist University (HKBU), Hong Kong SAR, P. R. China. He concurrently holds an Honorary Associate Professorship in the Faculty of Dentistry at The University of Hong Kong (HKU). He is a Fellow of the Royal Society of Chemistry (FRSC, United Kingdom). He was elected as a Founding Member of The Hong Kong Young Academy of Sciences and named as a Fellow of the International Association of Advanced Materials (FIAAM, Sweden). He gained both his B.Sc. and PhD degrees in Chemistry from The Chinese University of Hong Kong, and then conducted three-year postdoctoral research with Professor Sir Fraser Stoddart at The California NanoSystems Institute and Department of Chemistry and Biochemistry, The University of California at Los Angeles, USA. His research interests are in the fields of nanomaterials chemistry, supramolecular polymer materials, catalysis, sensors, and nanomedicine. |
Key learning points(1) Macrocyclic peptides and their derivatives demonstrating different structures and diverse functionalities have become multidisciplinary research megatrends across chemistry, biomedicine, and materials science.(2) Their de novo design, efficient synthesis and precision modification have always been highly challenging but desirable in both academic and industrial settings. (3) Various new macrocyclization strategies for peptides and peptidomimetics have been developed over the recent decades via chemical, biological and supramolecular approaches. (4) Long-standing challenges in macrocyclic peptide synthesis in terms of scope, selectivity, sustainability, and scalability leave much room for further exploration and advancements. |
1. Introduction
Between 1899 and 1908, Emil Fischer (Nobel Prize winner in Chemistry 1902) investigated proteins and first proposed the concept of a “peptide bond”, which preluded and emblematized humankind's odyssey through the vibrant and dynamic research area of “peptide science”. In 1959, Robert Bruce Merrifield (Nobel Prize winner in Chemistry 1984) pioneered solid-phase peptide synthesis (SPPS), a revolutionary breakthrough in the production of synthetic peptides, which is now a well-established common practice in peptide-based drug discovery and development in both academic and industrial settings. Owing to the efforts of scientists in this field globally, since the beginning of the 21st century, there have been many new peptides documented due to serendipitous discoveries from nature, tailored/de novo designs inspired by nature, or just simply curiosity-driven pursuit by humans. It is with such an immensely diverse pool of new peptides of unprecedented forms and functions, as well as the inexhaustible creativities and unlimited perseverance of humankind, that peptide science transcends the traditional knowledge boundaries of organic chemistry and biochemistry, becoming deeply integrated with medicinal chemistry, chemical biology, synthetic biology, inorganic chemistry, supramolecular chemistry, materials science, biomedical science, food science, cosmetic science, and even computational science (Fig. 1).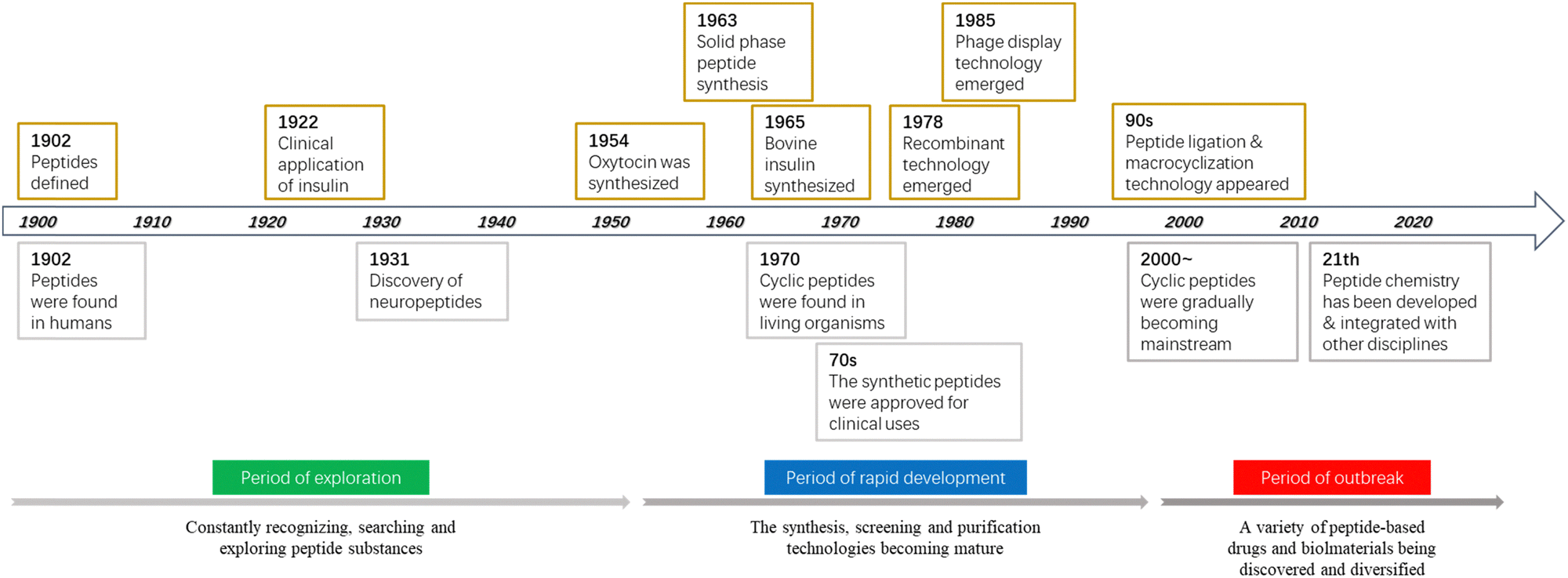 | ||
| Fig. 1 Selected historical development of peptide science. | ||
Peptides are essential components in living organisms and exist as hormones, neurotransmitters, growth factors, and ion channel receptors to, for instance, regulate various physiological activities.1 They have molecular weights between those of small organic molecules and proteins (usually composed of 2–50 amino acids) and are special substances that can be facilely synthesized and modified. Peptide-based drugs are usually favorably endowed with high bioactivity, high specificity, and low toxicity, which surpass small-molecule drugs.2,3 Furthermore, because of their unique spatial configuration, peptides are ideal modalities to modulate protein–protein interactions (PPIs).4 This has aroused a great deal of interest in the medicinal chemistry of therapeutic peptides. In contrast to the intrinsic metabolic instability of linear peptides,5 the constrained flexibility of their cyclic counterparts results in much higher resistance to proteolytic enzymes, with their more rigid conformation manifesting much better specificity when interacting with protein targets.6,7 In addition, the specific, fixed conformation of cyclic peptides reduces the entropic cost of binding to receptors, and thus increases the binding affinity.7,8 According to the “chameleon hypothesis”, cyclic peptides exhibiting smart conformational dynamics (i.e., shielding or exposing their polar functionalities in response to the environment) are found to possess enhanced cell membrane permeability due to their transiently reduced surface polarity and area.6,7,9
In essence, many bioactive natural products are cyclic peptides, such as cyclopeptide alkaloids and cyclotides, with most of them being cyclized precisely through the formation of lactam, lactone, or in the form of disulfide bonds.10 However, this seemingly simple way of ring creation in nature is rather difficult to accomplish by purely traditional chemical means. Firstly, peptide macrocyclization is an entropy-unfavorable process, where intermolecular oligomerization reactions always compete with intramolecular ring-closing reactions; therefore, it must be carried out under either high-dilution conditions or compartmentalization.11 Secondly, the imperfect chemo/stereo/site-selectivity at the points of cyclization, as well as inevitable epimerization of the product due to strong acids/bases or reagents will adversely affect the purity, and hence the bioactivities of cyclic peptides.12 Due to contemporary advances in various novel and efficient macrocyclization strategies and techniques,4 not only the above-mentioned naturally occurring cyclopeptides can be synthesized, but also more structurally complex variants, with the introduction of unnatural amino acids and other non-peptide components, can be accessed with unparalleled properties. Thus, currently, the strategic designs and efficient preparation of macrocyclic peptides and peptidomimetics have become key research hotspots and megatrends in both academia and industry.
Given the multidisciplinary nature of this topic, large knowledge gaps exist for the researchers from different backgrounds (e.g., chemists, biologists, and materials scientists). Thus, in this tutorial review, we aim to provide a panoramic view of the whole research landscape and a reader-friendly, up-to-date catalogue for the synthetic breakthroughs over the recent decades (Fig. 2). In general, peptide macrocyclization strategies can be categorized into three approaches, as follows: (a) chemical, both non-catalytic and catalytic, (b) biological, and (c) supramolecular approaches. Non-catalytic chemical methods include chemoselective ligation, nucleophilic (aromatic) substitution, (cyclo)addition, condensation, and multicomponent reactions between unnatural moieties/external components and amino acid residues; metal-catalyzed chemical methods include carbon–carbon/heteroatom bond formation via click, cross-coupling, C–H activation and olefin metathesis reactions, in addition to a few cases of photochemical examples. These distinctive, diverse chemical approaches, together with the underlying chemical principles, are the focal point of this tutorial. Examples are selected based on their novelty (e.g., new chemistry with reagents/reactions/protocols, compatibility and orthogonality, and creation of new rigid/flexible linkages or functionalities), selectivity (e.g., workable on specific moieties and amino acid residues) and practicality (e.g., reaction cleanness and efficiency, i.e., yield and rate, and operation and subsequent purification simplicity), where they can offer potential novel strategic solutions or alternatives to current macrocyclic peptide synthetic challenges compared with well-established or well-known methods to satisfy new and future demands.
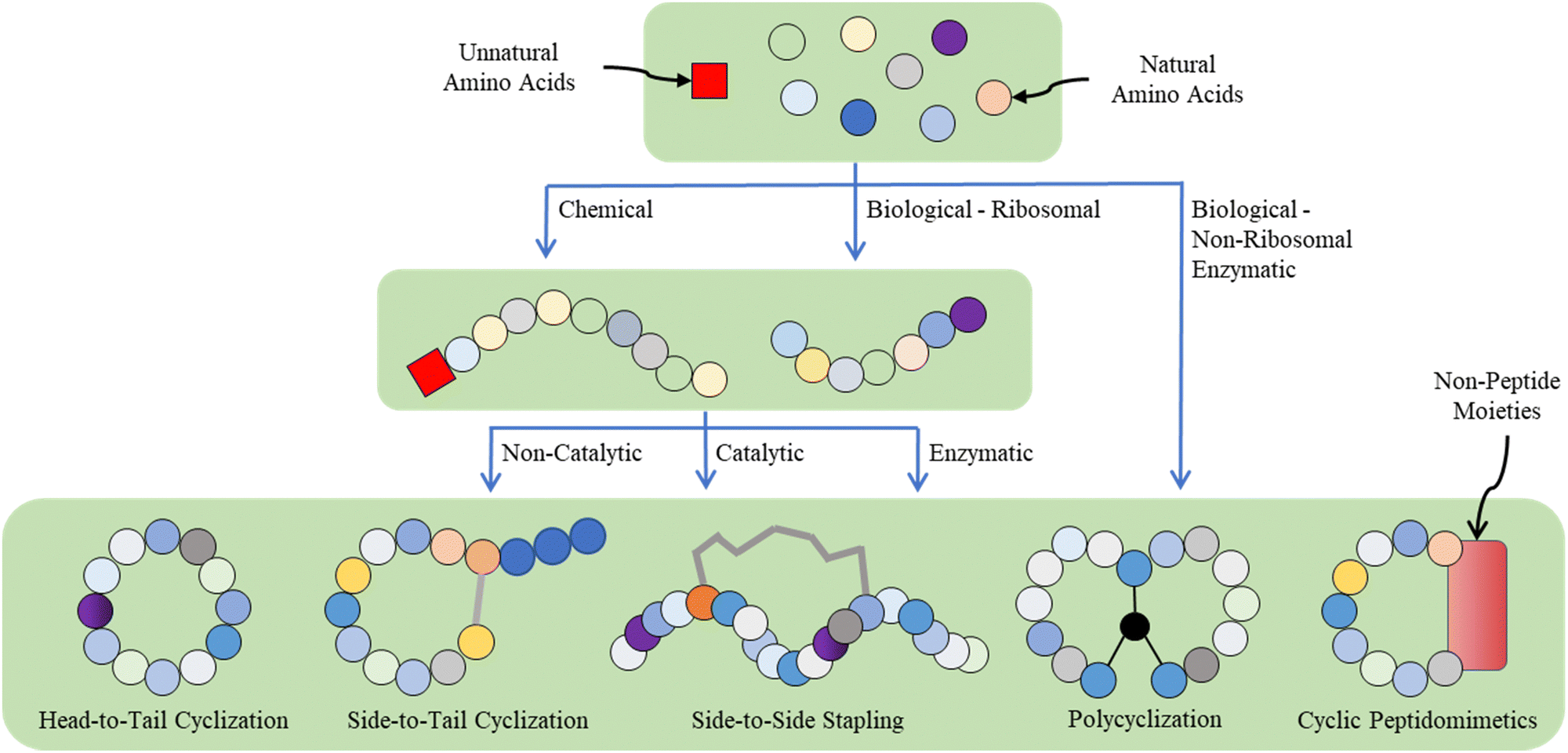 | ||
| Fig. 2 Contemporary synthetic strategies for different categories of peptidic macrocycles. | ||
Owing to the continuous quantum leap in advanced genetic engineering (e.g., genetic code reprogramming) and enzyme engineering (e.g., directed evolution and post-translational enzymatic editing), biosynthetic approaches have recently gained increasing momentum and popularity. From a supramolecular chemistry perspective, many higher-order cyclic peptide-based nano-structures can be obtained by mechanical interlocking and metal/noncovalent interaction-directed self-assembly. Nevertheless, given that they are beyond the scope of this tutorial, the biological and supramolecular approaches are only briefly introduced herein.
Finally, the outstanding features, advantages and limitations of each strategy are highlighted and analyzed, and the general synthetic-technical aspects (e.g., liquid/solid-PPS), and unresolved challenges (e.g., scope, chemo/stereo/site-selectivity, conformational control/stability, sustainability, and scalability) are highlighted and discussed, and subsequently we share our insights into the outlook of this field. We hope that a large multidisciplinary audience can quickly grasp the details and trends in macrocyclic peptide research, thereby contributing to and benefiting from this fundamental applied science and technology for the future betterment of humankind.
2. Non-catalytic chemical macrocyclization strategies
The most common macrocyclization of peptides can be achieved via the straightforward condensation of their terminal amino and carboxyl groups (lactamization). In addition, the thiol group of the cysteine sidechain has inherent nucleophilicity to react with electrophiles; the 1,2 aminothiol moiety of N-terminal cysteine also plays a bifunctional role to undergo different acyl transfers; and the amino group of the terminal sidechain of lysine can be easily subjected to aldehyde condensation to form active imine structures. All these have been reported for the ring formation reactions of peptides. In brief, non-catalytic chemical approaches are largely dependent on the typical functional group transformations of the natural amino acid residues. These methods are usually smartly and carefully designed and performed under relatively optimally mild conditions, with good selectivity and tolerance to other amino acid side chains in most cases.2.1. Ligation-mediated intramolecular cyclization
Direct macrocyclization in the form of amide linkage is the simplest way to synthesize cyclic peptides. However, this amide bond formation usually suffers from very poor chemical selectivity, resulting in isomerization and oligomerization problems. Thus, to circumvent these disadvantages, most amide-connected macrocyclic peptides require the installation of auxiliary groups, the modification of terminal groups, or the mediation of coupling agents. In this regard, native chemical ligation reactions have emerged as a powerful routine strategy for peptide elongation and protein synthesis, and importantly, peptide macrocyclization.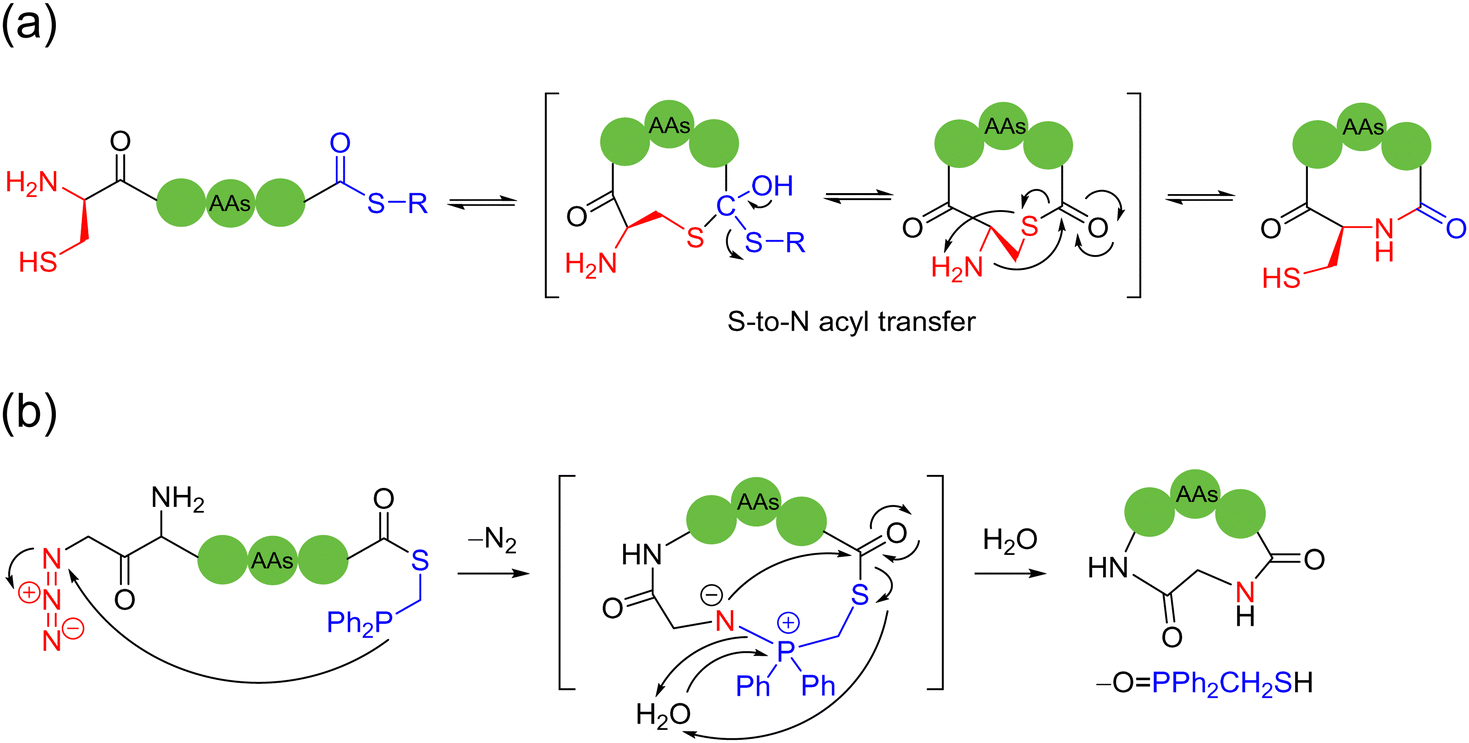 | ||
| Scheme 1 Selected representative ligation-mediated peptide macrocyclization strategies. (a) 1,2-Aminothiol and thioester ligation and S-to-N acyl transfer (native chemical ligation, NCL) and (b) azide and phosphine-linked thioester Staudinger ligation. | ||
The development of the NCL strategy and its extended methods was groundbreaking in the field of cyclic peptide synthesis. Presently, application-wise, NCL methods have progressed substantially in various aspects. For example, solid-phase resins have been employed for the pseudo-dilution effect, which can greatly suppress the side-oligomerization of peptide chains and promote the sought-after intramolecular cyclization.14–16 The incorporation of unnatural amino acids/moieties containing thiols, as well as the application of their desulfurization has also increased the number of reaction sites on peptides, where NCL and other addition reactions can take place beyond cysteines.17,18 Selenocysteine-based NCL can accelerate the rate of the ligation reaction and minimize side-reactions, such as thioester hydrolysis. The seleno-alcohol group can be further treated by alkylation, oxidation, and reduction, which is very helpful for the further derivatization and functionalization of the resulting cyclic peptides.19,20 NCL is a powerful and practical cyclization method with good chemoselectivity, which is appropriate for the preparation of cyclic peptides of various sizes and folding and integrating with continuous-flow and microfluidic technologies.21
Currently, NCL macrocyclization methods are one of the most commonly used strategies and most likely strategies to be translated to industrial production. This is because they showcase a few advantages, as follows: (i) simple preparation of linear peptide precursors and simple operation of the reaction; (ii) no epimerization or racemization of the cyclic peptide products or at the site of ligation; (iii) high water compatibility at neutral pH; and (iv) high reaction specificity in the presence of other internal cysteines, and other unprotected amino acids would not affect the reaction. However, their limitations are also conspicuous, as follows: (i) the presence of a C-terminal thioester and N-terminal cysteine is a required (although the auxiliary-mediated ligation strategy is available) and (ii) only in the case of peptide ring formation, without possibility for further functionalization or addition of new moieties for conformational control. Accordingly, various novel peptide macrocyclization strategies have been developed in the recent decade around the world, as shown herein, to complement and surpass NCL.
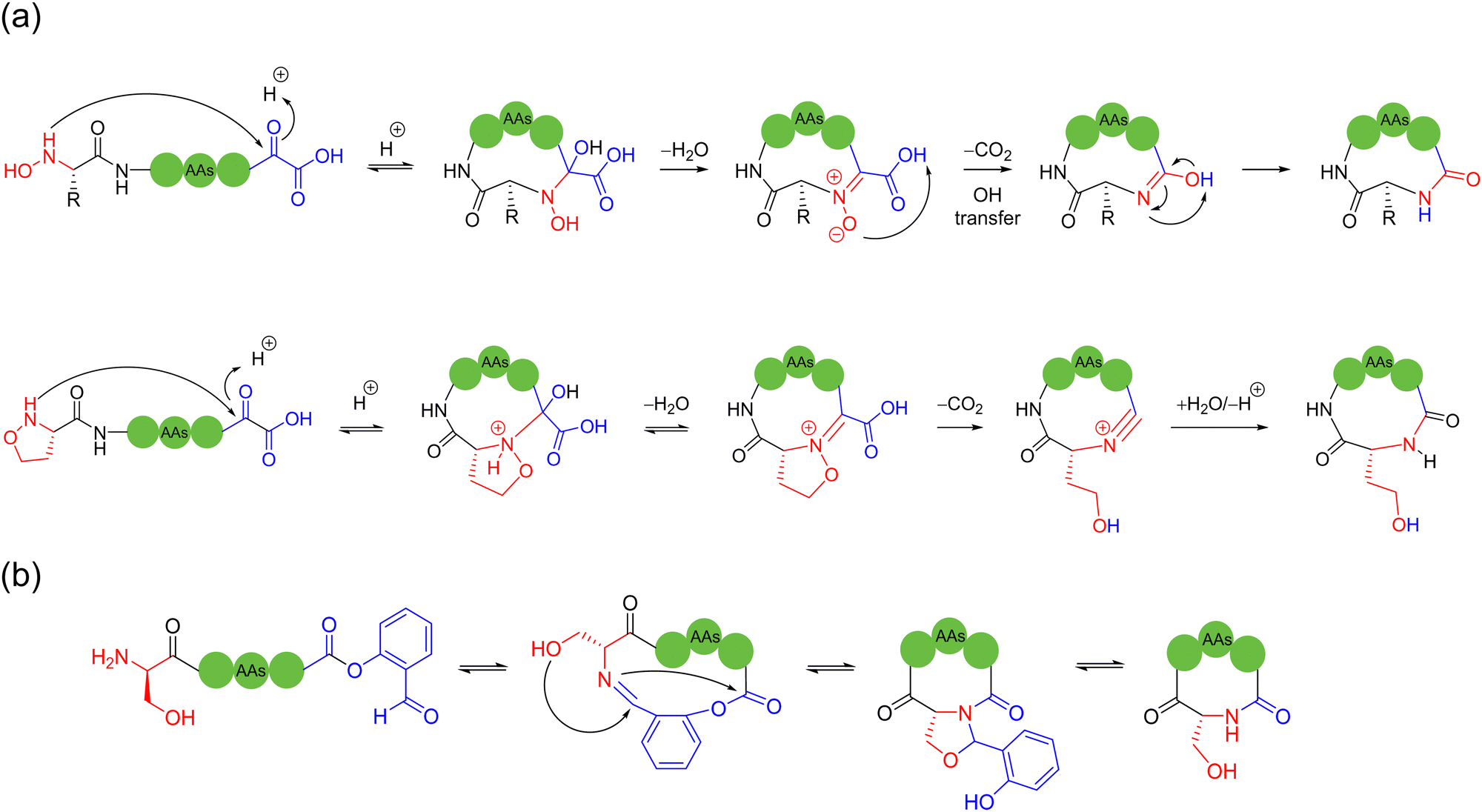 | ||
| Scheme 2 Selected representative ligation-mediated peptide macrocyclization strategies. (a) α-Ketoacid and hydroxylamine (type-1) and 5-oxaproline (type-II) KAHA ligation; (b) serine and salicylaldehyde ester ligation (serine/threonine ligation, STL). | ||
2.2. Thiol-based intramolecular cyclization
In 2020, Hayashi and coworkers developed a disulfide-driven cyclic peptide synthesis (DdCPS) strategy and successfully synthesized some cyclic peptide natural products.35,36 Firstly, on Npys-Cl resin, they prepared a disulfide crosslink between two peptides with two different sulfur-containing peptide fragments, and then cyclized the disulfide-bridged peptides using either thioester ligation or conventional amide coupling methods. Asymmetric disulfide peptides with high purity could be obtained, but this method required over ten steps of solid-phase reaction on the resin, leading to very low product yields. In fact, the disulfide bonds were not stable in most cases.37 Regarding the production of synthetic cyclic peptides and proteins, it is usually preferred that disulfide bonds are reduced to the more stable thioacetal38 or replaced with lactams, thioethers, etc.
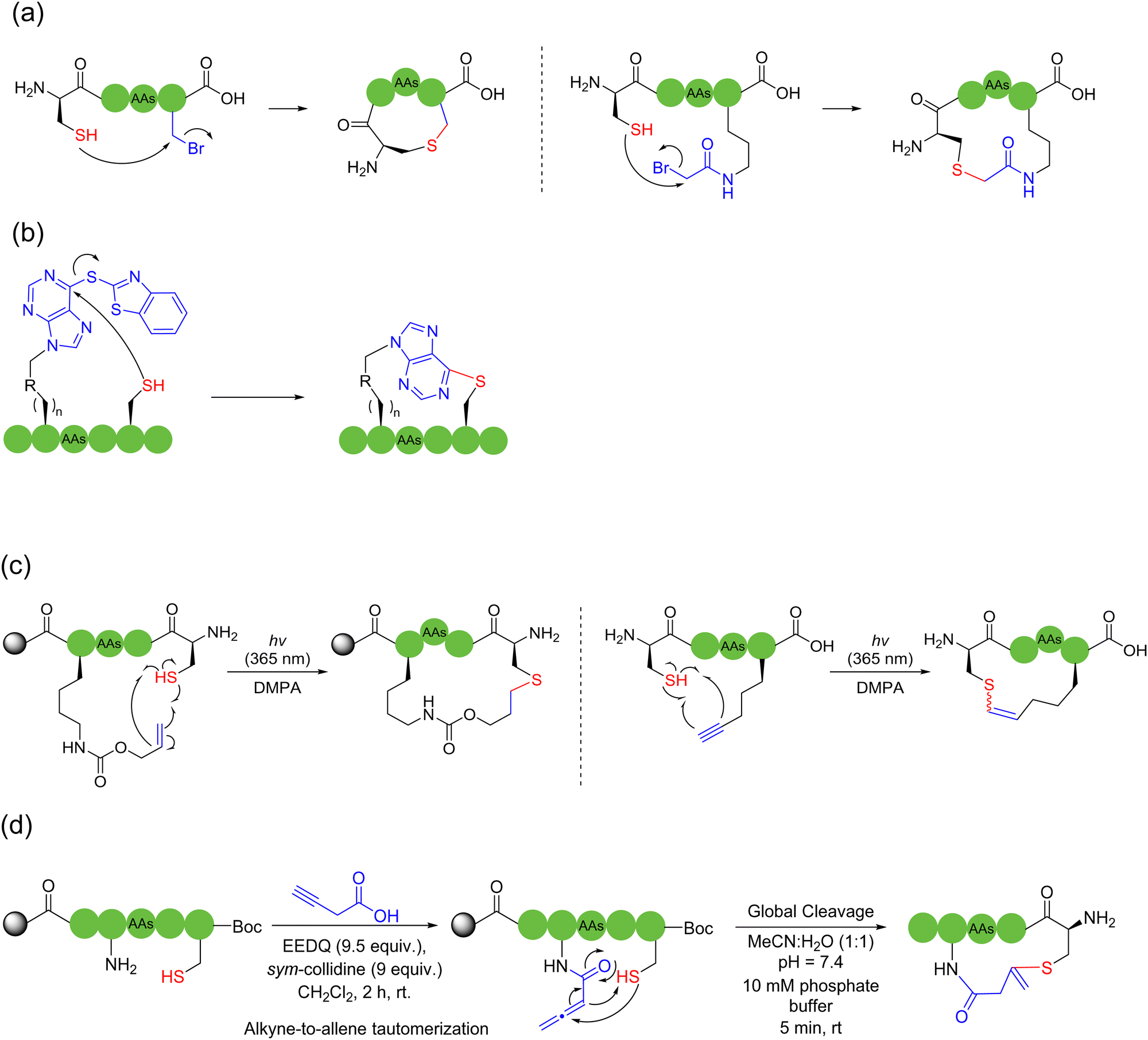 | ||
| Scheme 3 Selected representative strategies of thioether-based peptide macrocyclization with cysteine's thiols. (a) Nucleophilic substitution (SN2); (b) nucleophilic aromatic substitution (SNAr); (c) solid-phase photo-initiated thiol–ene click reaction; (d) on-resin installation of an allenamidyl group for thia-Michael-type click addition. | ||
In 2010, an efficient, photo-initiated, free radical-mediated thiol–ene addition was used for cyclizing peptides. Anseth and coworkers, on Arg-Gly-Asp (RGD) model peptides, exploited cysteine as the thiol source to cyclize with allyloxy carbon (Alloc)-protected lysine containing different active olefin moieties. Thioether bridges were generated on-resin by ultraviolet light and free radical initiators.42 An intermolecular version was carried out by Li and coworkers using the amino acid Ac-Cys-OH.43 Similarly, Li and coworkers further actualized a thiol–alkyne variant by clicking cysteine and alkyne-containing unnatural amino acids,44 bit it delivered a mixture of E-and Z-isomers (Scheme 3c).
Another strategy to employ thiols for solid-phase peptide macrocyclization involves the thia-Michael addition, which occurs between thiols and electron-deficient alkenes. Thioether bonds are formed in the presence of a base or a nucleophilic catalyst. In 2018, Kloxin and coworkers used the commercially available 2-chloroethanesulfonyl chloride to mount the vinyl sulfonamide group, a Michael acceptor, on primary or secondary amines to cyclize with thiol, a Michael donor, on resin; high conversion rates were obtained in a very short time.37 In 2020, Brimble and coworkers successfully documented a solid-phase peptide macrocyclization strategy via the on-resin installation of allenamide on peptides for the chemoselective conjugate addition of the allene with thiols.45 Mechanistically, tautomerization via base-catalyzed alkyne-to-allene isomerization occurs spontaneously and irreversibly at near neutral pH (Scheme 3d). The length of the bridging unit can be adjusted by using a lysine isobody, such as ornithine, or by adding an additional aminoalkyl chain, such as 6-aminocaproic acid.
2.3. Imine-based intramolecular cyclization
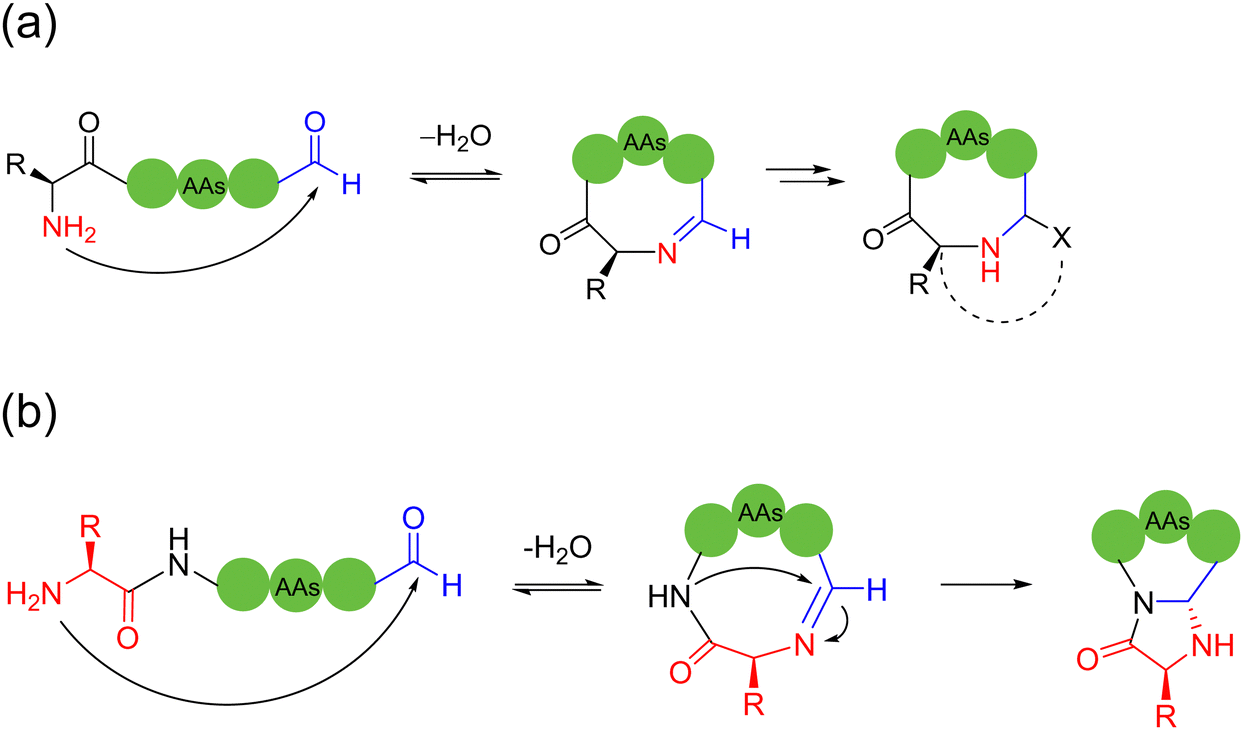 | ||
| Scheme 4 Selected representative strategies of imine-based peptide macrocyclization. (a) Aldehyde and terminal amino group condensation; (b) aldehyde and terminal amino group condensation, and then nucleophilic imine addition cascade. | ||
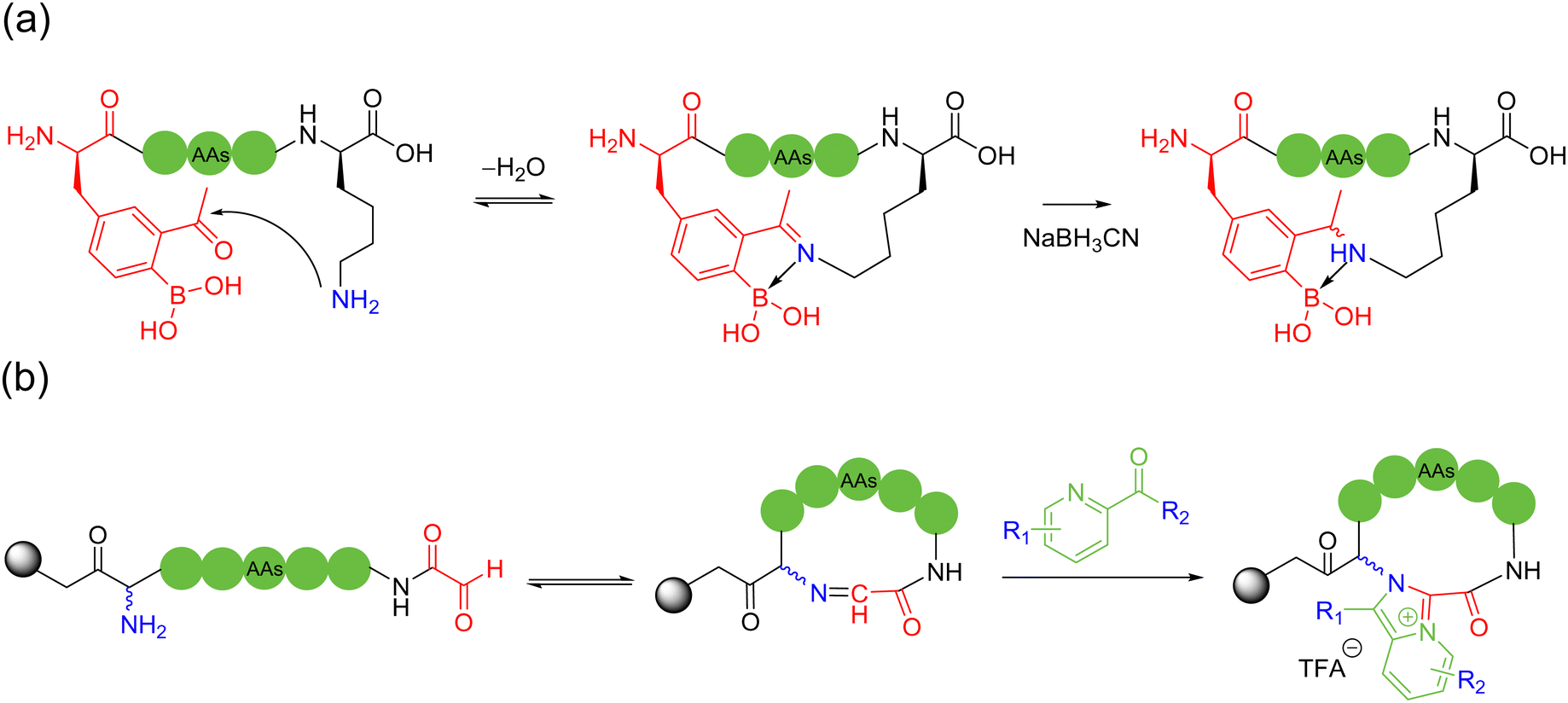 | ||
| Scheme 5 Selected representative strategies for imine-based peptide macrocyclization: (a) 2-acetylphenylboronic acid and lysine amino group condensation, then reduction; (b) oxoaldehyde and lysine amino group condensation, followed by imidazopyridinium grafting. | ||
Most literature-reported imine-mediated intramolecular cyclization methods for peptides tolerate various amino acid sidechains and undergo few oligomerizations based on the reversible imine formation mechanism. These linear peptide precursors containing C-terminal aldehydes can be readily prepared by Fmoc-SPPS, and the internal aldimines formed are capable of introducing a variety of modifications for conformational and structural regulation of the resulting cyclic peptides.
2.4. Multicomponent intermolecular stapling
Multicomponent intermolecular peptide stapling strategies furnish new modes and choices of reaction sites (e.g., in-between) for macrocyclization other than only at both termini in NCL; more importantly, the introduction of new linkages of rigidity and new functional handles creates new opportunities in this field.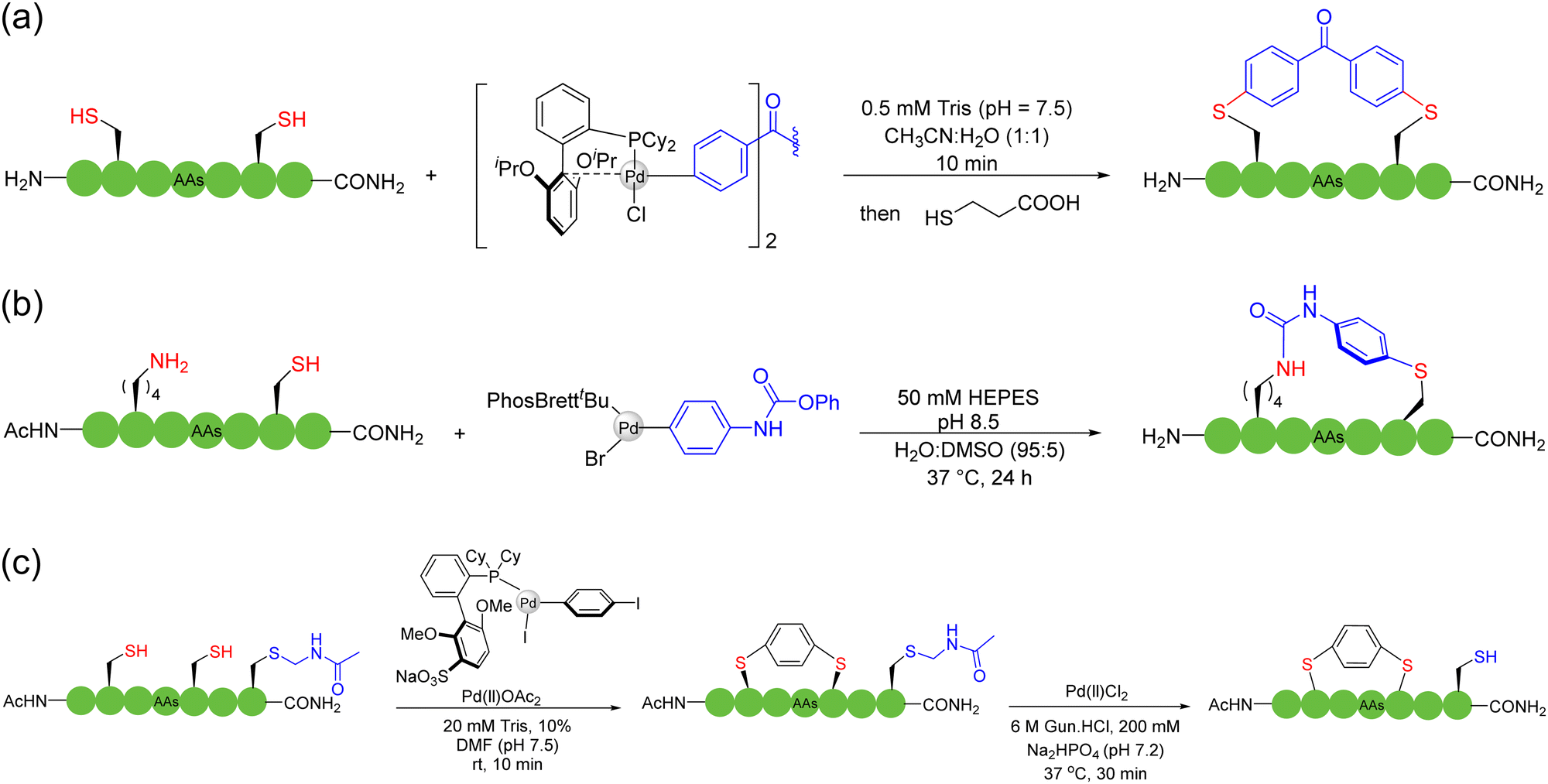 | ||
| Scheme 6 Selected representative strategies of multicomponent peptide stapling between natural amino acid residues with divalent organometallic reagents. (a) Between two cysteines; (b) between a lysine and a cysteine; (c) among multiple cysteines in a stepwise, site-selective manner. | ||
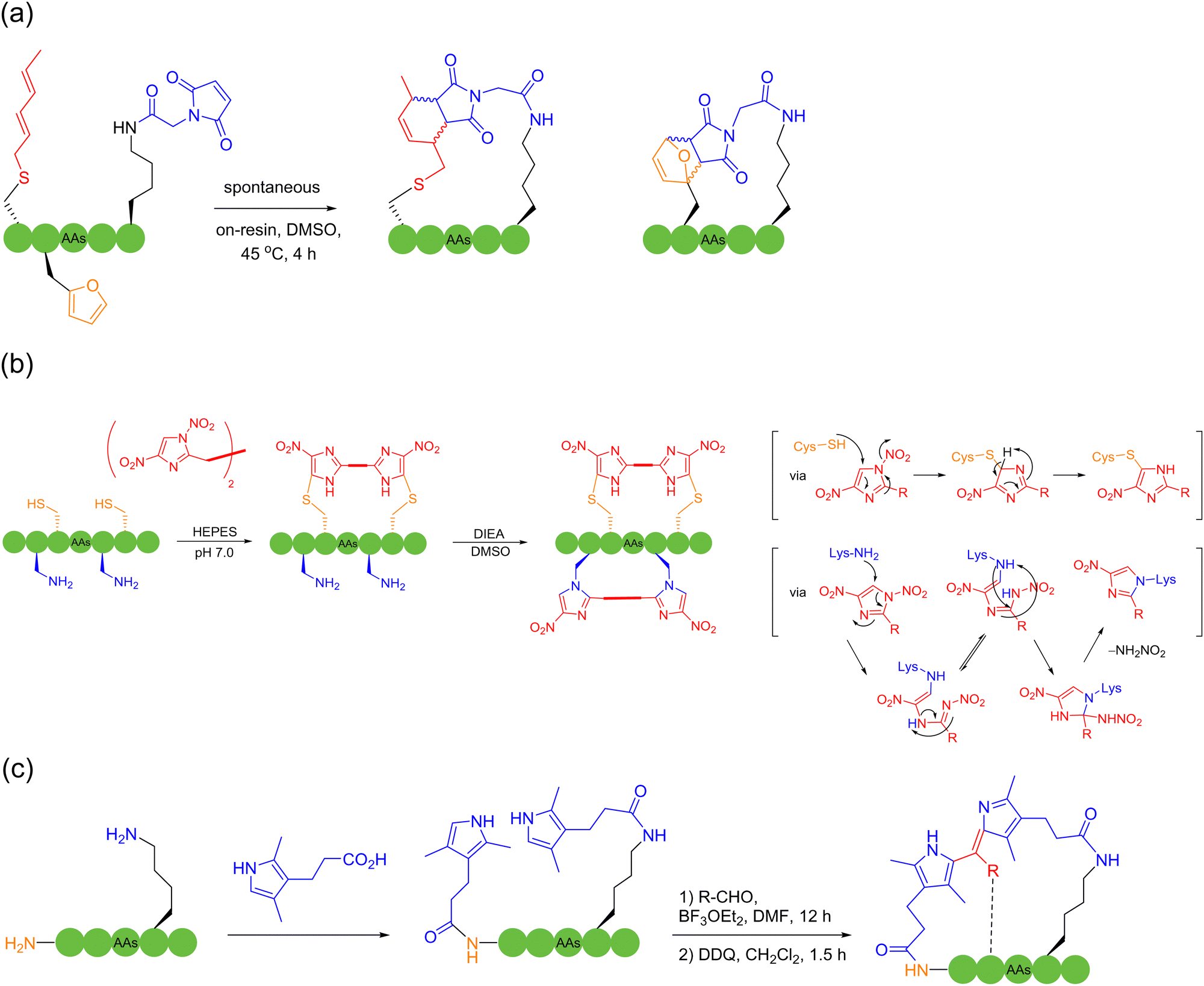
In 2019, Li and coworkers56 and Perrin and coworkers57 independently reported the peptide macrocyclization strategy based on isopeptide bond and isoindole formation. Both strategies used phthalaldehyde (OPA) to react with a lysine and cysteine within an unprotected peptide sequence, which could be saliently adoptable for bicyclic peptide synthesis under mild and metal-free conditions (Scheme 7a). Li and coworkers investigated the further modification of the cyclic peptide products by using dimethylacetamide (DMAC) or N-maleimide derivatives to construct new structures and attach new functional groups. Alternatively, in the study by Perrin and coworkers, different OPA analogues were evaluated and utilized to produce a few bicyclic peptides with different fluorescence properties.
 | ||
| Scheme 7 Selected representative strategies of multicomponent peptide stapling between natural amino acid residues with divalent aldehyde-based linkers. (a) Phthalaldehyde between a cysteine and a lysine; (b) formaldehyde between a lysine and a tyrosine/arginine; (c) phthalaldehyde and (d) α-ketoaldehyde between two primary amines (lysines or lysine with the N-terminus). | ||
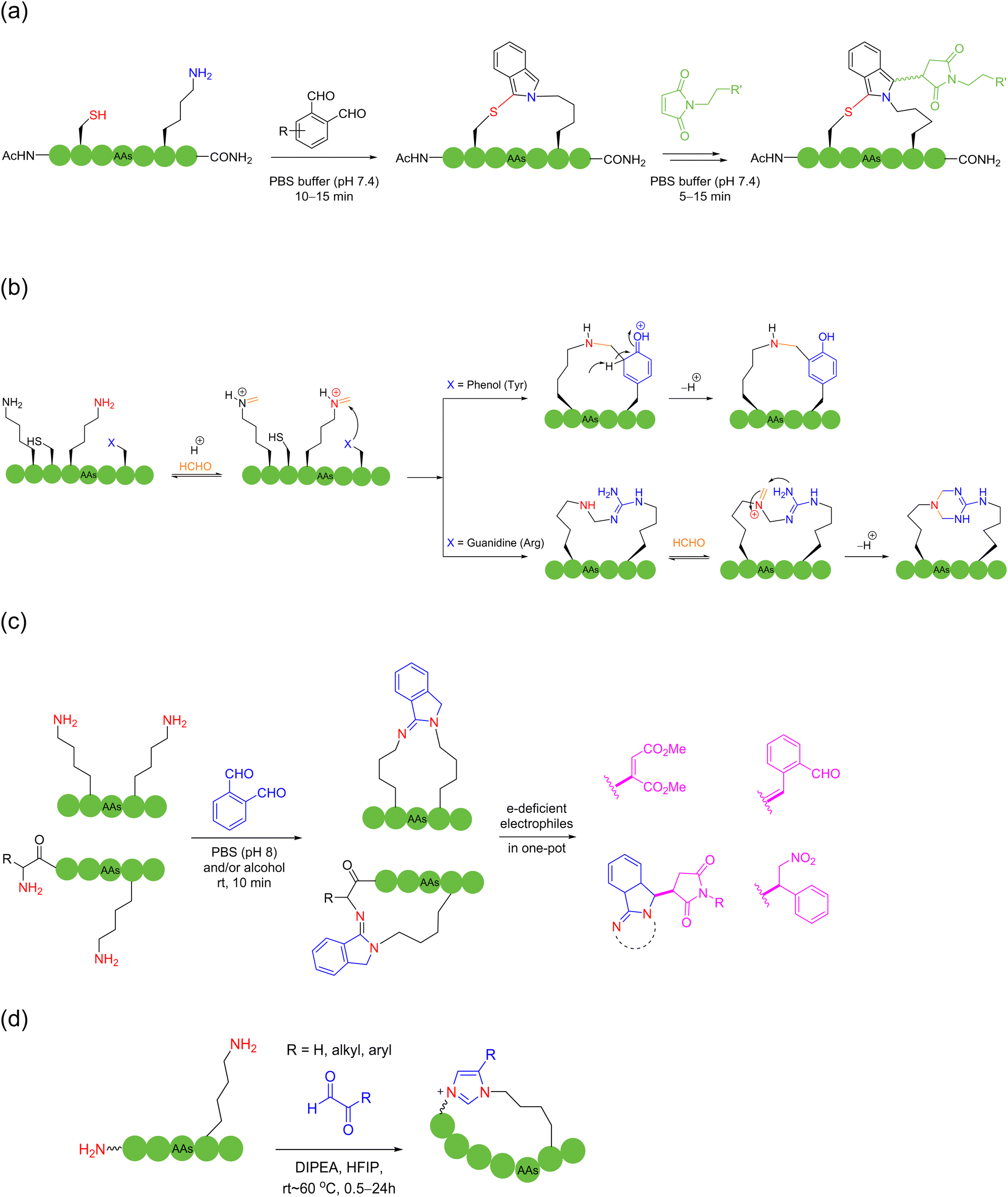
With the help of the anchoring effect of aldehyde-based “glue” molecules on the ε-amino group of lysine, in 2021, Chen and coworkers devised a chemoselective cooperative stapling strategy for the natural peptides lysine and tyrosine/arginine using formaldehyde as the linchpin under mild conditions58 (Scheme 7b). In this reaction, after the reversible condensation of the ε-amino group of lysine with formaldehyde to form imine, it could be crosslinked to the adjacent tyrosine or arginine residues. In the case of tyrosine, the nucleophilic ortho-carbon of the sidechain phenol triggers a Mannich-type cyclization, followed by rearomatization to form a stable benzylamine crosslinked product. Regarding arginine, after nucleophilic attack of the sidechain guanidine group on the imine, the secondary amine of the intermediate forms the imine with formaldehyde, resulting in after another nucleophilic attack, a stable tetrahydrotriazine-stapled product, as well as its exo-isomer.
In 2022, Chen and coworkers reported that OPA could be easily condensed with two primary amines of peptides under mild water conditions in the absence of thiols. They devised different tail-to-side and side-to-side OPA-based macrocyclization of unprotected peptides by suturing two amino groups59 (Scheme 7c). This method was ultrafast and highly compatible with a wide range of peptide substrates, where the isoindolinimine linchpin could be further functionalized with various electron-deficient electrophiles. In 2024, the authors further used different α-ketoaldehyde reagents to crosslink two amino groups, grafting a unique imidazolium moiety into the macrocyclic peptide products. Here, the use of hexafluoroisopropanol (HFIP) solvent was the key to achieving high reactivity, distinctive chemoselectivity for lysine over arginine, and reaction cleanness60 (Scheme 7d).
Besides the well-explored cysteine and lysine residues, in 2023, Sun and coworkers described a chemically selective cross-linking method for tryptophan residues, which was achieved by using dimethyl sulfoxide and visible light.61 The C–S structure was formed by irradiating a blue light emitting diode with thiophenol at room temperature, cyclizing unprotected peptides with high compatibility with most amino acid sidechains. Although the reaction suffered from low product yields, it provided a new perspective. In 2023, Yu, Li, Jiang and coworkers utilized bifunctional triazine moieties to react and staple the phenolic hydroxyl group of two tyrosines of the unprotected peptide62 (Scheme 8).
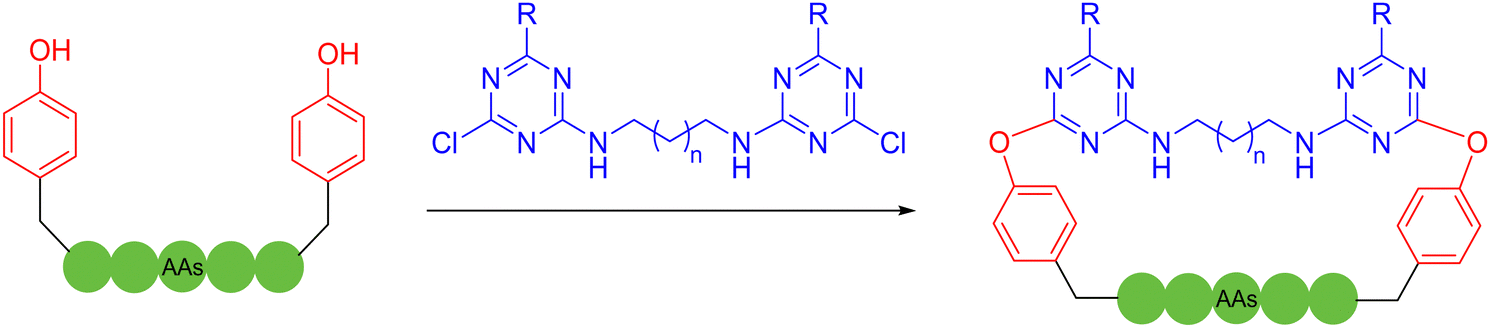 | ||
| Scheme 8 Multicomponent peptide stapling between natural amino acid residues with divalent triazine linkers. | ||
 | ||
| Scheme 9 Selected representative strategies of multicomponent peptide stapling between modified amino acid residues. (a) Diels−Alder [4+2] cycloaddition; (b) 1,4-dinitroimidazole-based skeletal recasting; (c) on-resin dipyrrin construction. | ||
In 2019, Wang and coworkers innovatively employed 1,4-dinitroimidazole, a bifunctional biological coupling reagent, for protein functionalization and peptide macrocyclization.64 In neutral aqueous solutions, 1,4-dinitroimidazole (1,4DNIm) rapidly underwent a substitution reaction with cysteine thiol; in contrast, in the absence of cysteine, 1,4DNIm could react with, under base catalysis, the amino group of lysine with high selectivity through ring-opening and ring-closing mechanisms to form stable peptide conjugates. Cyclization and double cyclization with unprotected peptides were accomplished by using double 1,4DNIm crosslinkers (Scheme 9b). In 2021, Chan, Long, Wong and coworkers disclosed a solid-phase peptide macrocyclization strategy via in situ dipyrrin construction, where dipyrrin was utilized as a multifunctional staple linker.65 The authors first attached commercially available pyrrole moieties onto both N-terminal and amino groups of lysine of resin-bound peptides, and then coupled the two pyrrole moieties with various aldehydes (Scheme 9c). Various alkyl and aromatic groups, as well as different functionalities, could be installed onto the meso-position of dipyrrin (e.g., for fluorescence imaging and sensing), which could also be used as a further reaction site to culminate in highly challenging but sought-after bicyclization.
In recent years, many novel structural and functional staple crosslinkers have been developed for the more precise and efficient construction of complex polycyclic peptide structures, which will be described in the Section 5 of peptide polycyclization.
2.5. Multicomponent reaction-mediated cyclization
Different from NCL, the application of multicomponent reactions (MCRs) in peptide macrocyclization (similar to the previous session) allows not only modification of cyclic peptide backbones, but also the introduction of new linkages and functionalities.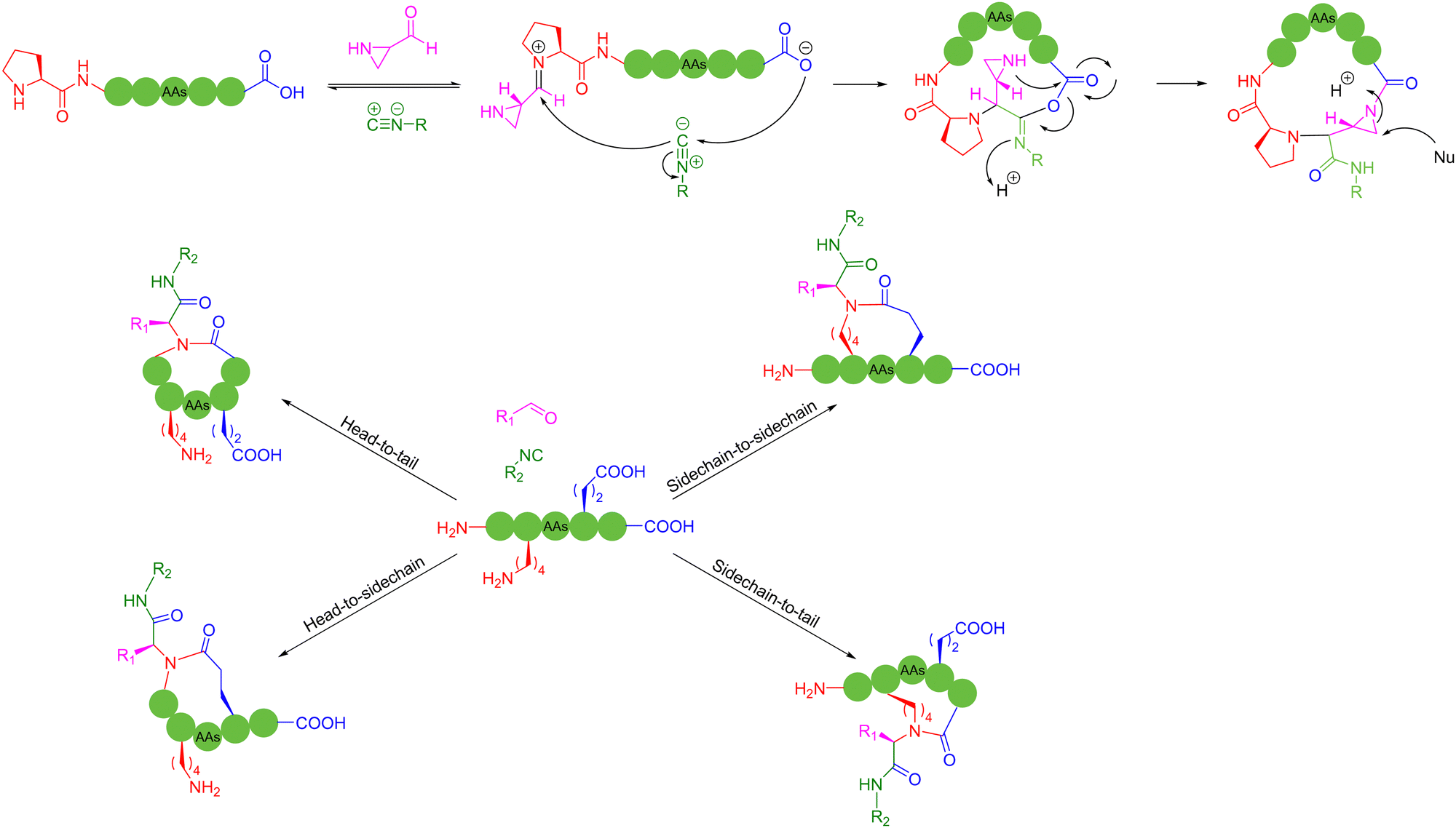 | ||
| Scheme 10 Selected representative Ugi reaction-based peptide macrocyclization strategies. | ||
 | ||
| Scheme 11 Selected representative Petasis reaction-based peptide macrocyclization strategies. | ||
 | ||
| Scheme 12 Selected representative other multicomponent reaction-based peptide macrocyclization strategies. | ||
2.6. Other miscellaneous non-catalytic cyclization strategies
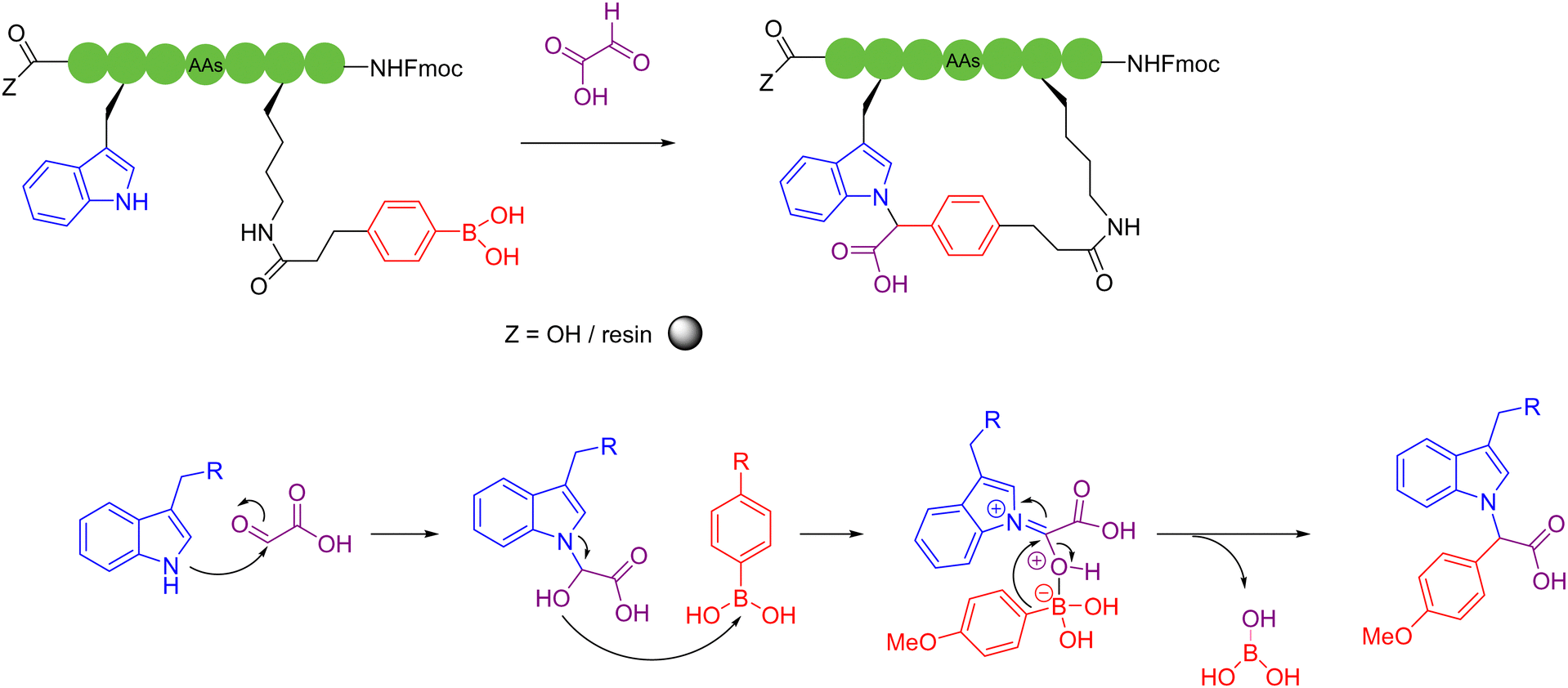
Condensation agents are a class of peptide coupling reagents that are used to promote the direct condensation of carboxylic acids with amines or alcohols to form amide or ester bonds, which play a pivotal role in the synthesis of peptides. In recent years, Zhao and coworkers developed a new type of ynamide coupling reagent, which can be widely used in the synthesis of amides, esters, macrolides, peptides and thiopeptides.72 In 2018, the authors utilized the ynamide, N-methylynetoluenesulfonamide (MYTsA), and monothiocarboxylic acids as thioacyl donors to efficiently synthesize linear thiopeptides without racemization. During this process, amino acid functional groups such as OH, CONH2, and the NH groups of indoles could be protection free, but a long aminolysis reaction time was required, which limits its practical application potential.73 Based on the previous work, in 2022, the authors successfully extended the applicability of ynamide-mediated peptide bond formation to various peptide fragment ligation, head-to-tail cyclization, and even SPPS conditions.74 This strategy employed N-methylynemethylsulfonamide (MYMsA) to robustly form a stabilized intermediate to promote peptide bond formation in a one-pot, two-step manner. High yields of the cyclic products were obtained with excellent chemoselectivity and no detectable epimerization issues compared with other common peptide coupling reagents (Scheme 13a).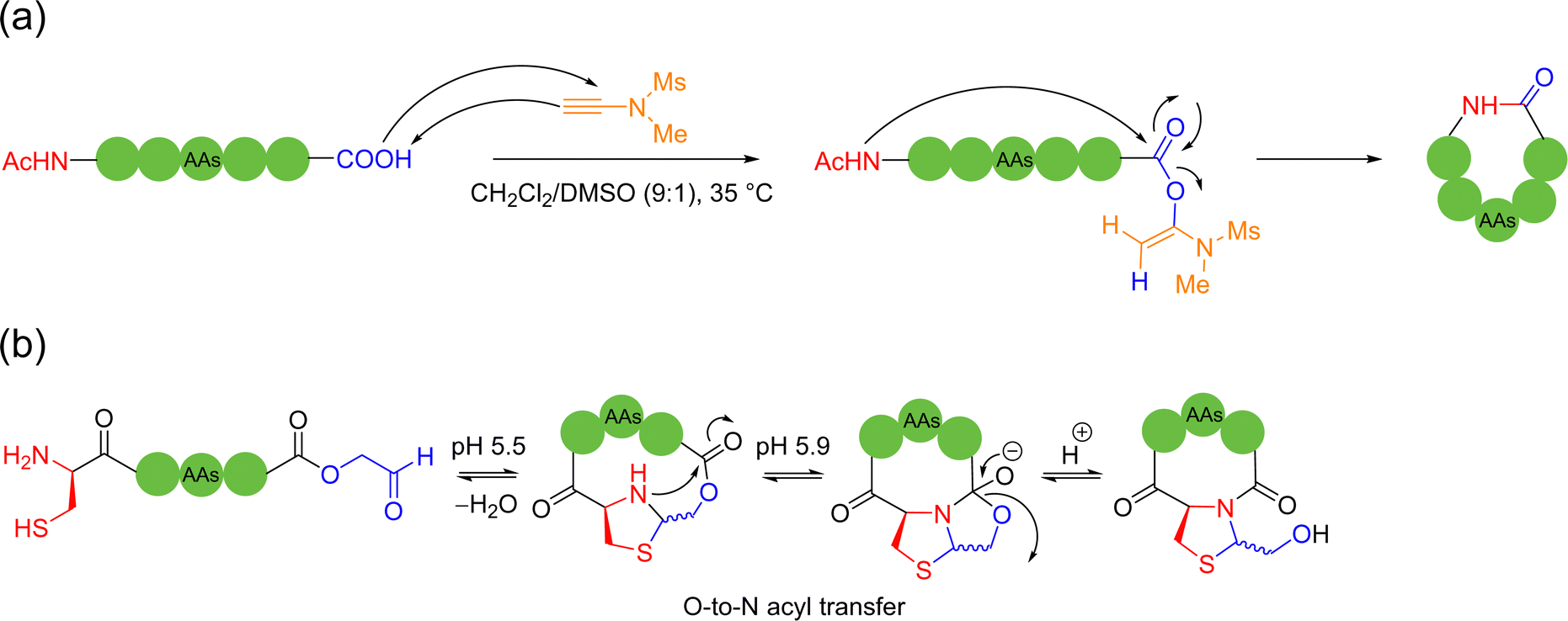 | ||
| Scheme 13 Selected miscellaneous non-catalytic peptide macrocyclization strategies: (a) with ynamide; (b) via thiazolidine grafting. | ||
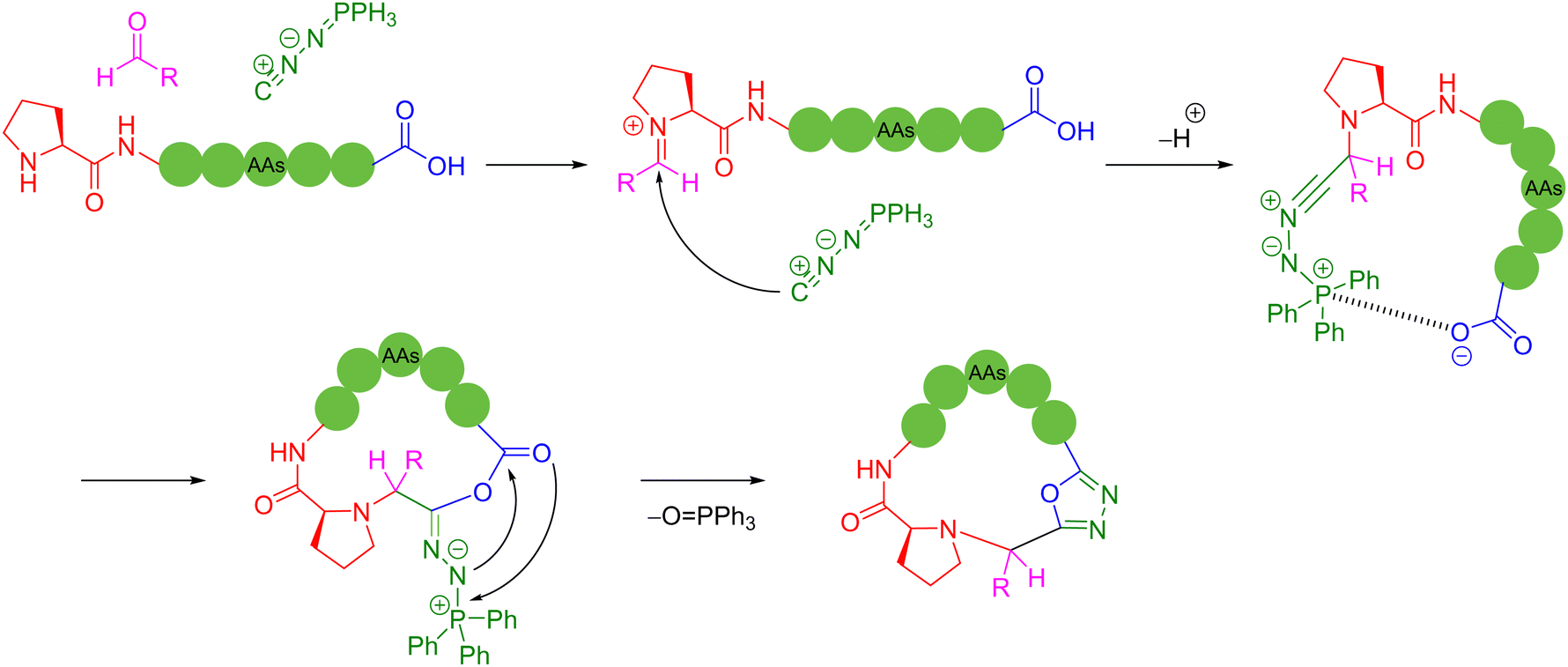
The 1,2-aminothiol of the N-terminal cysteine could easily condense with aldehydes to form thiazolidines, in the protocol first reported by Tam and coworkers to prepare cyclic peptides in 1996.75 In their method, the starting C-terminal ethanolaldehyde ester could first be obtained by the oxidation of glycerol ester under solid-phase peptide synthesis (SPPS). By adjusting the pH value to 5.5 in aqueous solution, an intramolecular condensation between the N-terminal cysteine and C-terminal ethanolaldehyde ester could be triggered to form a thiazolidine intermediate. Subsequently, raising the pH value to 5.9 could enable another intramolecular rearrangement to achieve O-to-N acyl transfer, thus completing the cyclization (Scheme 13b). Only through tuning the pH environment, this reaction could be very simply, conveniently and widely applicable to unprotected peptide main chains without special sequence requirement, except for the two terminal groups. However, the reaction rate of this method is too slow and the inevitable introduction of a new stereocenter on the C-5 carbon of the thiazolidine ring leads to a pair of undesired diastereomers.
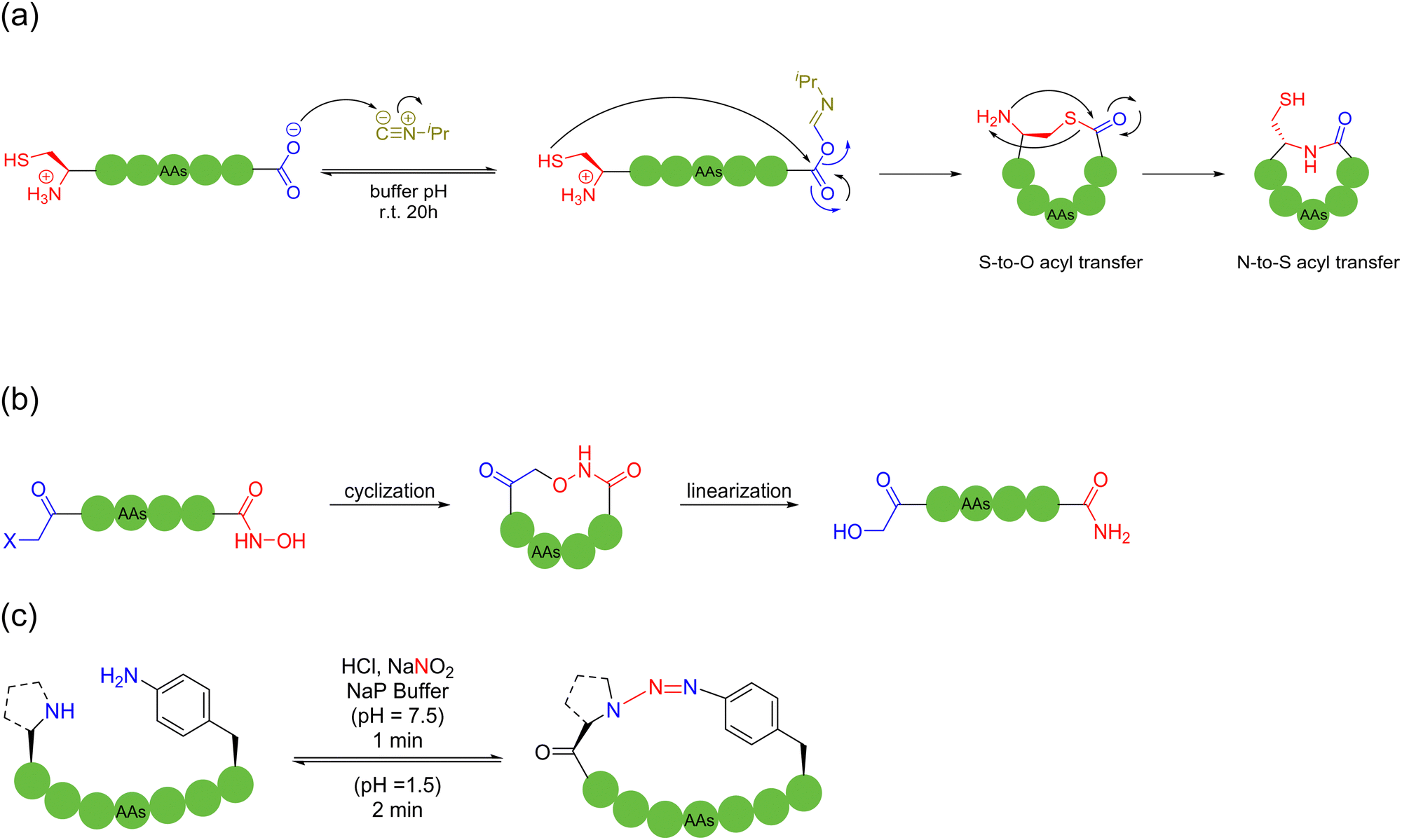
In 2023, Zhang and coworkers reported a chemoselective natural peptide cyclization (NPC) strategy76 (Scheme 14a). In this strategy, the pH of the aqueous solution is adjusted to make the C-terminal carboxylate the major nucleophilic residue to preferentially combine with the isonitrile, thereby forming a mixed-anhydride adduct. Subsequently, the activated C-terminus is captured by the N-terminal cysteine thiol group (SH) via intramolecular thiolysis, leading to the chemically selective formation of thiolactam. Similar to the NCL reactions, the NPC reaction ends by N-to-S acyl transfer. These NPC strategies enabled the intramolecular cyclization of unmodified and protected peptides without the excessive use of additives, organic reagents and solvents. The authors synthesized more than 60, 5- to 20-membered cyclic peptides with simple operation in high efficiency.
 | ||
| Scheme 14 Selected other miscellaneous non-catalytic peptide macrocyclization strategies: (a) with isonitrile; (b) via hydroxamic acid–haloacetamide ring closure; (c) with NaNO2. | ||
In 2024, Kim and coworkers introduced a reversible cyclization methodology for unprotected peptides containing a C-terminal hydroxamic acid and N-terminal electrophile77 (Scheme 14b). In this method, the C-terminal hydroxamic acid and N-terminal haloacetamide were attached via SPPS, followed by the efficient cyclization of unprotected, thiol-free amino acid residue precursor peptides of various lengths and compositions in water under mild conditions. Noticeably, the cyclized peptide could be re-opened by adding excess samarium iodide to reduce the formed hydroxamic linkage in the presence of typical amino acid residues. This hydroxamic acid-driven cyclization strategy embraces certain characteristics, as follows: (i) high compatibility for unprotected peptides, (ii) high stability for the cyclic products, and (iii) high cyclization propensity for low molecular-weight short peptides, rather than undesired oligomerization.
In 2022, Raj and coworkers developed a novel and expeditious arene triazene strategy, which was chemoselective for secondary amines and p-amino phenylalanine on unprotected peptides, with the use of NaNO2, for macrocyclizing peptides, while generating inbuilt triazene chromophores of distinct UV absorbance at the site of cyclization. Saliently, the resulting triazene cyclic peptide products were formed within a minute and found to be highly stable at neutral pH and under harsh conditions. Furthermore, they could quickly respond to various external stimuli, e.g., UV radiation and acidic conditions, leading to reversible ring-opening. In particular, a very broad substrate scope was reported, including different amino acid compositions with all L-amino acids without any turn inducers, various secondary amines and sequences with multiple arginine residues; and different chain lengths (4–20 amino acids)78 (Scheme 14c).
3. Catalytic chemical macrocyclization strategies
Transition metal catalysis have significantly advanced and revolutionized modern synthetic organic chemistry in recent decades. From copper-catalyzed alkyne–azide click cycloaddition to ruthenium-catalyzed ring-closing olefin metathesis, transition metal-based catalytic strategies for peptide macrocyclization have been proven to be highly feasible, efficient, and attractive. As a few examples, photoredox catalytic reactions, cross-coupling reactions, C–H activation, aryl/alkylation of heteroatoms, and many others have been strategically developed. In general, there are two types of mechanistic pathways, where in the first, the metal catalyst activates one of the peptide functional groups for further reacting with another functionalized sidechain or backbone. In the second, the metal catalyst binds two reactive functional groups, and thus the C–C or C–heteroatom bonds can be eventually formed for ring closure. Both pathways depend on the final reductive elimination step in the catalytic cycle. All these strategies provide a completely new and significant synthetic paradigm and platform to access macrocyclic peptides beyond the well-known lactamization and NCL.3.1. Azide–alkyne click cycloaddition-mediated cyclization
In 2001, Kolb, Finn and Sharpless established the featured “click chemistry”.79 In 2002, Meldal and coworkers applied the copper(I)-catalyzed azide–alkyne cycloaddition click reaction (CuAAC) for the ligation of peptides.80 In the presence of unprotected sidechains, this reaction could effectively connect two peptide fragments with a triazole. In 2005, Burgess and coworkers documented the first synthesis of a head-to-tail cyclic peptide by CuAAC, in either organic solvents or water under mild conditions, forming 1,4-disubstituted 1,2,3-triazole-embedded cyclopeptides81 (Scheme 15a). These seminal works laid the theoretical foundation for the applications of CuAAC in peptide chemistry, given its simple operation, high chemoselectivity, and feasibility in the peptide synthesis (the reaction conditions are fully compatible with unprotected sidechains). In particular, it was found that both the synthetic 1,4-disubstituted 1,2,3-triazole and the natural trans-amide bond would have similar spatial structures, which could be used as a β-turn component.80,81 All these advantages are important factors for the wide applications of CuAAC in bioconjugation and biomaterials science.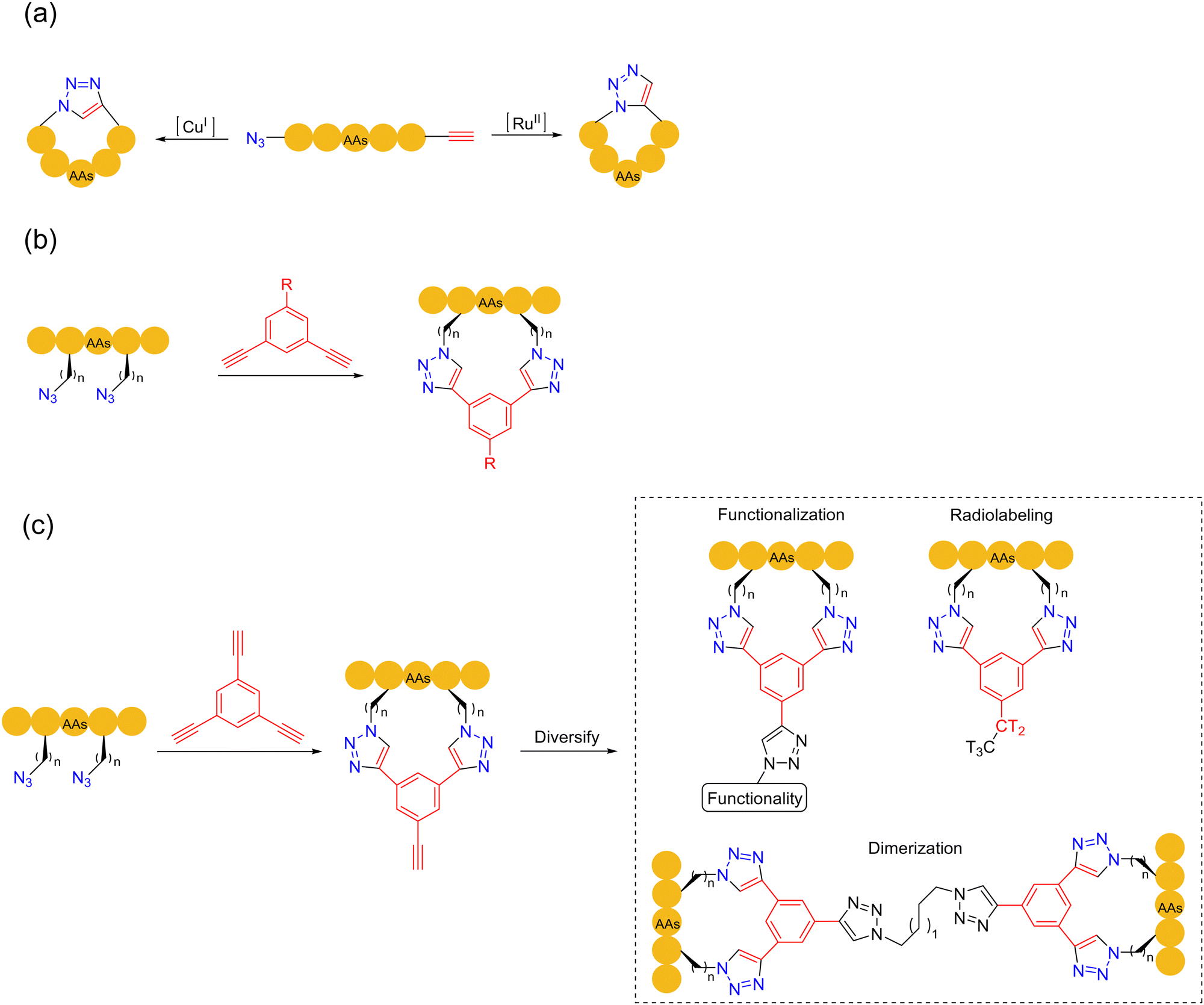 | ||
| Scheme 15 Azide–alkyne click cycloaddition-based peptide macrocyclization strategies. (a) CuAAC and RuAAC; (b) and (c) click-stapling. | ||
The synthesis of cyclic peptide precursors by CuAAC entails the pre-installation of both azide and alkyne moieties on peptides. The azide group can be transferred to the terminal or primary amine in the lysine sidechain by diazo transfer, while the alkyne group can be introduced with unnatural amino acids or chemical modifications of natural amino acid sidechains. The introduction of C-terminal alkynes can be achieved by the Fmoc-SPPS silyl-based alkyne modifying (SAM) linker method.82 CuAAC-based peptide macrocyclizations have been implemented in the form of head-to-tail, sidechain-to-sidechain and sidechain-to-head/tail fashions, which can be carried out in both solid/liquid phases. The double CuAAC-based macrocyclization method was reported by Meldal and coworkers in 2011,83 which played an important role in stabilizing the secondary peptide structure by the rigid triazole moieties instead of flexible disulfide bridges. A second approach, which involves the stapling of tow unnatural/modified azide sidechains with a dialkynyl external component, was developed by Spring and coworkers in 201484 (Scheme 15b). This bioorthogonal two-component stapling method can also be utilized to conjugate other peptides, fluorophores, and functional handles to diversify the functions of the stapled peptides. In 2017, Pedersen and coworkers developed the third generation of the CuAAC strategy. Two amino acid residues bearing azides were combined with 1,3,5-triethynylbenzene to form a staple, with the third free alkyne group being further facilely functionalized or dimerized to obtain more complex and diversified cyclic peptides85 (Scheme 15c).
Although click chemistry is a powerful tool for bioconjugation, especially for proteins and peptides, and even macrocyclization for peptides with rigid nitrogen-based heterocyclic linkages, the CuAAC reaction has certain limitations. Firstly, monitoring the intramolecular CuAAC reaction can be a great challenge because the starting material and the product have the same molecular weight. In addition, a major disadvantage of using the CuAAC reaction in biological systems is that copper species are prone to induce the formation of reactive oxygen species (ROS);86 copper(I) is not stable enough in the physiological environment, which can be oxidized or chelated with other biomolecules. In the worst-case scenario, it may lead to the oxidative degradation of proteins and peptides. Thus, to reduce the biotoxicity of copper ions, the use of tris(triazolylmethyl)amine-based ligands, such as tris(3-hydroxypropyl methyl)amine (THTPA) or tris(1-benzyl-1H-1,2,3-triazolyl-4-yl)methylamine (TBTA), can significantly reduce this oxidative damage. To avoid oxidation completely, the reaction should be carried out under anaerobic conditions. To this end, strain-promoted alkyne-azide cycloaddition (SPAAC), a copper-free click reaction, can serve as a complementary reaction and resolution. A variety of cyclooctyne reagents has been developed for this purpose, and SPAAC has been widely used in biomedicine.87,88
3.2. Ring-closing olefin and alkyne metathesis-mediated cyclization
Compared with the establishment of click reaction, the development of alkene metathesis reaction underwent a much longer process. It took 30–40 years from the discovery of the olefin metathesis reaction in the 1960s to its contemporary wide applications. With the development of the first and second ruthenium-based Grubbs catalysts for olefin metathesis in the 1990s,89 the olefin metathesis reaction gradually became popular. In 2000, Verdine and coworkers synthesized full hydrocarbon bridging a-helical peptides using ring-closing metathesis (RCM)90 (Scheme 16a). In this study, the concept of peptide stapling was proposed for the first time. It demonstrated the ability of RCM reaction to control the helical structure of protein by stapling. The RCM reaction is most commonly used in intramolecular reactions to generate cyclic systems, with its high tolerance to most functional groups, and its usually high product yield makes it highly suitable for modifying relatively large and functionally diverse molecules, e.g., peptides and proteins. RCM-based peptide macrocyclization can be carried out in either the solid or liquid phase, and the hydrocarbon linkage has the advantage of mimicking the disulfide bond to manipulate the configurations of the resulting peptides and proteins. Various creative strategies have been developed for peptide macrocyclization using RCM reactions with relatively more flexible alkene linkages.91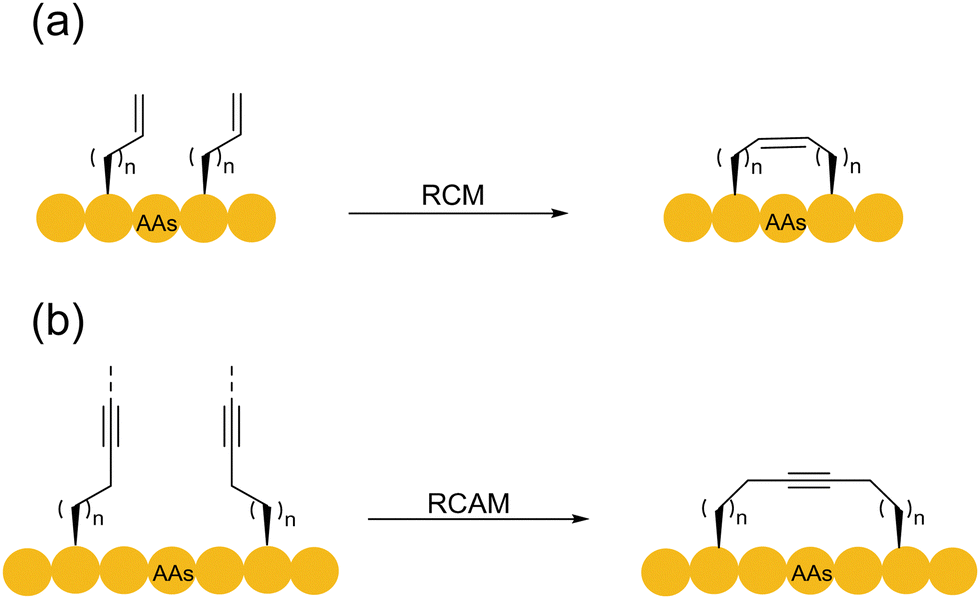 | ||
| Scheme 16 Ring-closing metathesis-based peptide macrocyclization strategies. (a) RCM; (b) RCAM. | ||
Given that each step in the RCM reaction mechanism is a reversible process, and two alkenes are to be released, the overall dynamic equilibrium of the reaction usually shifts to the product side. In the case of complex biomolecular structures such as peptides, the RCM process usually produces a mixture of Z/E-configuration alkenes. However, control of the olefin configuration for peptide synthesis remains challenging. In addition, the most common catalyst for the RCM reaction is the ruthenium-based Grubbs catalyst. However, this class of ruthenium reagents usually induces the decomposition of DNA, proteins, and other substances. To prevent the destruction of peptides by ruthenium, a large amount of Mg2+ ions92,93 is usually added for substrate protection. Yoshiya and coworkers actualized the ring-closing olefin metathesis of unprotected peptides in water by employing a water-soluble ruthenium catalyst, a large excess of MgCl2, and/or acidic conditions, thereby generating three octreotide analogues.93 However, another type of common RCM molybdenum-based catalyst was rarely used due to its lower activity.94
Similarly, alkynes can also undergo metathesis reactions with other alkynes. The ring-closing alkyne metathesis (RCAM) necessitates catalytic high-valence tungsten and molybdenum-based complexes (Scheme 16b). In 2005, Liskamp and coworkers documented the first ring-closing alkyne metathesis for the synthesis of the alkyne mimics of thioether A, B, C, and (D)E ring systems of the lantibiotic nisin Z (with rigid rod-like alkyne linkages).95 The authors used tungsten (W) complex-catalyzed RCAM to synthesize alkyne-bridged cyclic peptides containing 4–7 amino acid residues. Notably, the (D)E ring system of the nisin Z antibiotic was constructed by a combination of RCM and RCAM. The combined use of RCM and RCAM in bicyclic peptide synthesis could solve the selectivity problem of using only a single approach. In 2016, Furstner and coworkers developed an orthogonal RCM–RCAM method to achieve double peptide stapling.96 In this method, the chosen Mo complex-catalyzed RCAM was chemically orthogonal to the Ru-catalyzed RCM, and thus peptides containing two alkyne and two alkene functionalities could be cycled sequentially respectively, in high precision.
RCM and RCAM are very important in the synthesis of all-hydrocarbon stapled peptides. In 2023, Hu and coworkers introduced restrictive anchoring residues containing cyclobutene and synthesized a series of all-hydrocarbon stapled peptides containing different combinations of cyclobutanes using the RCM-based SPPS. Among them, the E7–E7 geometry-specific stapled peptide exhibited higher a-helicity and was found to be more biologically active than its typical hydrocarbon counterparts.97 Because of the linear configuration of the alkynyl groups, it is often necessary to have suitable preorganization for the cyclization of shorter peptide chains, where terminal alkynes with longer alkyl chain linkages can promote the reactions. In general, RCAM is less widely used in peptides than RCM.96 However, control of the Z/E configuration of the double bonds can be achieved by the addition reaction of alkynes.
3.3. Photochemical reaction-mediated cyclization
Photochemical reactions have engendered many new and powerful chemical transformations over the past two decades. With the advances in the field of photochemistry and free radical chemistry, photocatalytic reactions have been extended for the macrocyclization of peptides. In addition to nonmetal catalyzed thiol–ene radical-type photochemical reactions, light-mediated peptide macrocyclization can also be performed using photoredox reactions catalyzed by photoactive transition metals, e.g., iridium and nickel. Relatively flexible hydrocarbon, phenyl ether and thioether/thioamide linkages can be created in the macrocyclic peptide products.In 2014, MacMillan and coworkers developed the Giese reaction for peptide ring formation.98 In this reaction, iridium(III) catalyzed the oxidative decarboxylation of carboxylic acid to form free radicals under excitation by light, which then underwent 1,4-conjugate addition to the Michael receptor. The authors found that this oxidative decarboxylative process also applied to a variety of α-amino acids. In 2017, the authors proposed a decarboxylative macrocyclization method for peptides containing N-terminal Michael receptors99 (Scheme 17a). In this method, upon light irradiation, the Ir(III) catalyst promoted C-terminal decarboxylation to form a-amino radicals. Subsequently, these radicals attacked the N-terminal Michael receptor for 1,4-addition, followed by the critical C–C bond formation to obtain the desired macrocycle, while providing electrophilic α-acyl radicals. The final association with electron-deficient radicals ended the photoredox catalytic cycle. This synthetic method tolerated most natural and non-proteinogenic amino acid sidechains (under protection, except cysteine). It was successfully applied to efficiently cyclize linear precursor peptides comprised of 3 to 15 amino acids.
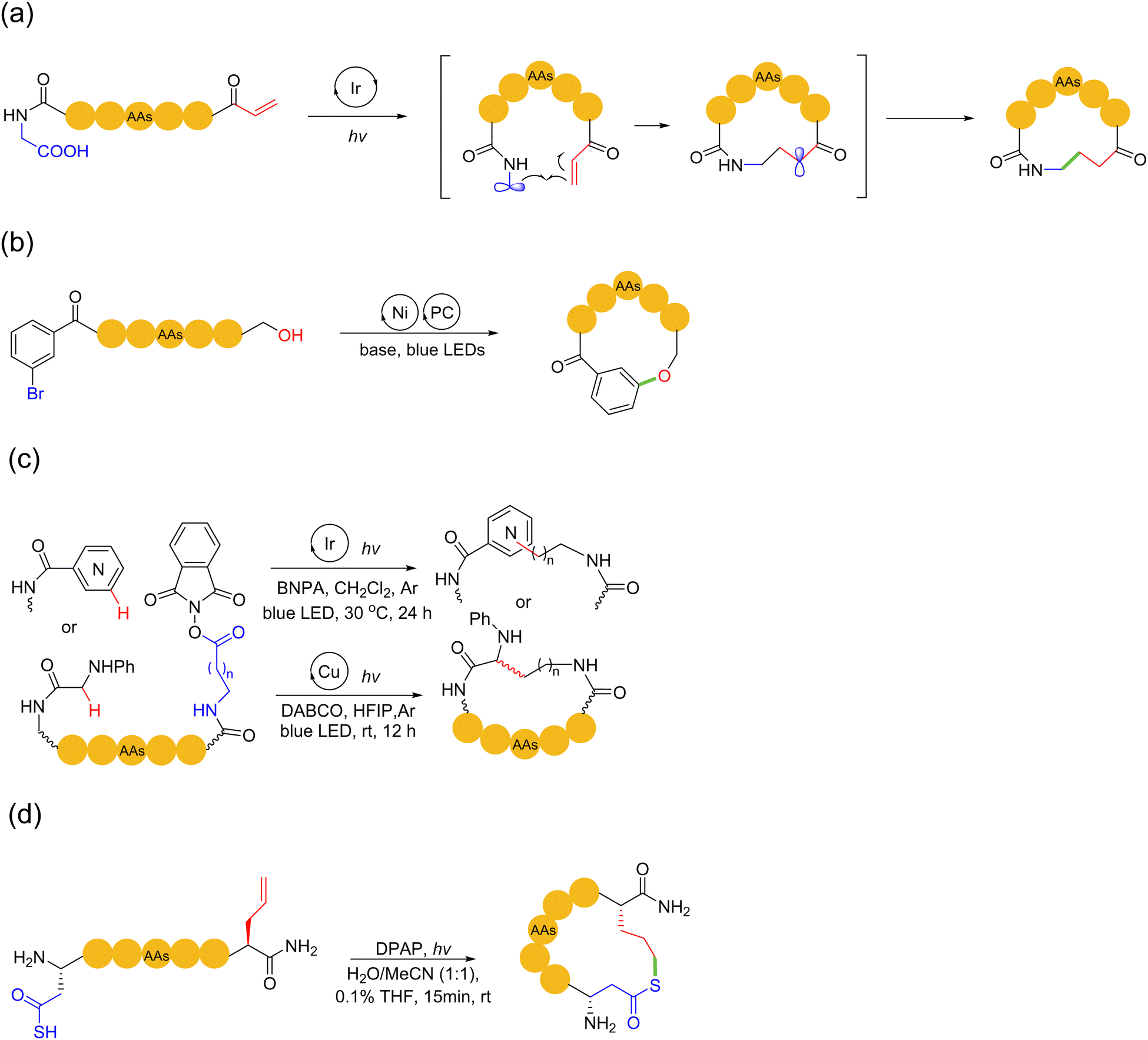 | ||
| Scheme 17 Selected examples of photochemical peptide macrocyclization strategies. (a) Photoredox-mediated decarboxylative radical conjugate addition; (b) Ni/photoredox-promoted C(sp2)–O cross-coupling; (c) photoredox-triggered Minisci reaction; (d) photochemical thio-ene reaction. | ||
Similarly, in 2016, Baran and coworkers disclosed a method for the macrocyclization of peptides based on the nickel-catalyzed Barton decarboxylation/Giese reaction with N-hydroxyphthalimide (NHPI) redox-active esters.100 It should be noted that this method did not rely on the photochemical reactivity of the active ester. In 2019, Sciammetta and coworkers developed a photoredox Ni-catalyzed peptide C(sp2)–O cross-coupling approach101 (Scheme 17b). This work was inspired by the pioneering work of MacMillan and coworker, who realized C(sp2)–O coupling by photoredox nickel catalysis.102 They mounted a bromobenzoyl group at the N-terminus to perform Ni/photoredox catalytic reactions with the hydroxyl of serine and phenol sidechains of tyrosine. The successful formation of ether bonds between the two residues enabled the peptide to achieve sidechain-to-tail macrocyclization with high chemical selectivity in good yield.
In 2021, Chen and coworkers reported two methods for effectively constructing peptide macrocycles through a free radical-mediated intramolecular (Minisci) C–H alkylation reactions of glycines and N-heteroarenes, respectively, under iridium/copper-based photoredox catalysis103 (Scheme 17c). In this work, linear peptide precursors were first equipped with a C-terminal N-(acyloxy)phthalimide ester, which then cyclized with the α C–H bond of the N-terminal glycine or aryl C–H bond of N-heteroarene capping units under mild conditions, enabling the incorporation of lysine, homolysine, and various heteroarene-derived amino acid linchpins into peptide macrocycles. In 2024, Scanlan and coworkers achieved a rapid metal-free photochemical acyl thiol–ene cyclisation strategy in SPPS for fully unprotected peptides bearing a thioacid and alkene to deliver macrocyclic thiolactone peptides104 (Scheme 17d). Concerning the underlying chemistry, γ-thioaspartic acid bearing the required thioacid on the side chain of aspartic acid (Asp) and allylglycine residues were tactically employed to avoid the unfavoured dethiocarboxylation side-reaction under the radical environment.
3.4. Cross-coupling reaction-mediated cyclization
C(sp2)–C(sp2) bonds can be formed very efficiently and reliably via traditional transition metal-catalyzed cross-coupling reactions. Among them, Pd-catalyzed coupling reactions are the most typical and classical examples. Mechanistically, the transition metal is first inserted into the carbon-(pseudo)-halide bond by oxidative addition, then the two organic fragments are bound and joined by an organometallic intermediate (via migratory insertion, β-elimination, isomerization or trans-metallation, etc.), and finally the metal catalyst is regenerated by reductive elimination with the formation of a new carbon–carbon bond. Theoretically and ideally, most of the coupling reactions can be applied to the macrocyclization of peptides, provided that two suitable reactive groups are introduced in the peptides. For example, reactions such as Suzuki–Miyaura, Tsuji–Trost, Sonogashira–Hagihara, and Mizoroki–Heck can be in principle applied. Some examples show that the conditions of palladium-catalyzed cross-coupling reactions have a minimal effect on the enantiomeric purity of the resulting products; among which, Suzuki coupling, as a powerful palladium-catalyzed technique for modifying different carbon–(pseudo) halide bonds, has been shown to have good biocompatibility and bioorthogonality.105 In fact, in reality, the application of traditional transition metal-catalyzed cross-coupling reactions in biomolecules is limited. Most of these reactions can only couple aryl groups or aryl groups with (sp2) alkyl groups. In addition, the need for accurate pre-functionalization of peptides also imposes significant synthetic challenges.In contrast to conventional cross-coupling reactions, C–H activation strategies enable the direct alkylation/arylation of natural amino acid side chains, promisingly avoiding the aforementioned issues. In recent years, there have been increasing examples of peptide modification and cyclization through C–H activation. The metals that can be used for C–H activation include palladium, manganese, and rhodium. The conjugated C–H can be derived from the C atoms of sp-, sp2-, or sp3- hybridization. Among the transition metal-catalyzed peptide macrocyclization strategies, C–H activation has become one of the best methodologies (possibly without pre-functionalization and workable on amino acid residues other than cysteine), in line with NCL, which has been considered an important tool for the late-stage functionalization of biomolecules.106
Pd-catalyzed C(sp2)–H activation was first applied to the macrocyclization of peptides. In 2015, based on the previous first-time direct arylation107 and macrocyclization108 of indole, Albericio, Lavilla, and coworkers reported, for the first time, Pd(OAc)2-catalyzed C(sp2)–H activation for C–C bond formation and peptide stapling between the C2 site of tryptophan and iodinated phenylalanine or tyrosine residues109 (Scheme 18a). They investigated the effect of the distance between the two reaction moieties on the conversion efficiency and showed that both the ring size and the conformation of the peptide were critical for the success of macrocyclization. Based on C(sp2)–H activation and olefination catalyzed by Pd(OAc)2, Wang and coworker developed a series of macrocyclization methods targeting the C-2 position of phenylalanine in the sidechain,110ortho-phenyl position of the terminal methylbenzylacetamide,111ortho-phenyl position of N-terminal sulfonamide,112 indolyl C2/C4 position of tryptophan,113 and N-terminal oxazole motif.114 In their works, the AgOAc incorporated therein acted as an external oxidant to regenerate Pd(II) and restarted the catalytic cycle. These oxidative cross-coupling examples, which included a C–H activation step, avoided the need of pre-functionalization of amino acids. In 2020, Chen and coworkers published a versatile method of Pd-catalyzed picolinamide-directed intramolecular arylation of aryl and alkenyl C–H bonds of amino acid iodinated sidechains for the construction of C(sp2)-linked cyclophane (i.e., biaryl-bridged and aryl–alkene-braced) peptide macrocycles.115 The N-terminal C(sp2) could be derived from either terminal alkenes or aromatic rings.
 | ||
| Scheme 18 Selected peptide macrocyclization strategies via C–H activation on phenylalanine, tyrosine and tryptophan moieties. (a) & (c) C(sp2)–H at tryptophan indole’s C2 position with aryl halide and terminal alkyne, respectively; (b) C(sp3)–H at alanine with aryl halide; (d) C(sp2)–H (d) C(sp2)–H at tryptophan indole’s C7 position with maleimide. | ||
In effect, palladium-catalyzed C(sp3)–H activation in peptide chemistry is of utmost importance, given that it creates the possibility to exploit the peptide conformational constraints of the sidechains of native aliphatic amino acids. Inspired by the intermolecular arylation of C(sp3)–H bond of N-terminal amino acid catalyzed by Pd by Yu and coworkers,116 in 2016, Noisier, Albericio and coworkers reported the Pd-catalyzed late-stage C(sp3)–H activation for peptide stapling117 (Scheme 18b). Coincidentally, Wang and coworkers used almost the same conditions to link a peptide chain with an N-Phth-Ala sequence and an iodine-phenylalanine (Phe) in the form of a C(sp3)–C(sp2) bond.118 This method could be performed in the solution or solid phase, without the need for external directing groups, demonstrating good compatibility with the sidechains of most amino acids. Given the rigidity of the aryl group at the junction, this method was found to be suitable for the cyclization of long-chain peptides. In 2018, Chen and coworkers used the iodobenzene or p-iodo-phenylalanine aryl sidechains of unnatural amino acids and removable the 8-aminoquinoline (AQ) auxiliary groups on the alkyl tail on either C/N-terminus to selectively cyclize linear peptide precursors of different designs to form a variety of cyclic peptides of medium and higher sizes.119 It is worth noting that this method could obtain a 37-membered macrocyclic peptide, which is very rare in the literature.
Strategies of C–H activation catalyzed by other transition metals have also been extensively applied to peptide chemistry. A representative of which is the manganese-catalyzed strategy pioneered by Ackermann and coworkers. As early as 2017, they reported an example of alkynylation based on Mn-catalyzed C–H activation and its successful application to peptide macrocyclization.120 In 2019, they achieved the use of manganese-catalyzed hydroallylation for the modification of peptides, nucleotides, and drug molecules, as well as the macrocyclization of peptides, under mild and heteromerization-free reaction conditions.121 In 2021, they succeeded to use the propargyl ester reagent to install propiolate at multiple sites of the peptide more conveniently by using manganese catalytic activation and realized a variety of cyclized forms of the peptide122 (Scheme 18c). Moreover, C–H alkylation yielded an α,β-unsaturated ester, which could be subjected to further derivations. In 2023, Zheng and coworkers reported a new rhodium-catalyzed macrocyclization strategy via late-stage maleimidation on C(7)–H tryptophan123 (Scheme 18d). Notably, this catalytic system was not limited by the position of tryptophan in the polypeptide, while the maleimide introduced in the cyclic product could also be used as an effective click functional group to further modify the products.
3.5. Heteroatom arylation/alkylation-mediated cyclization
Although heteroatoms (e.g., N, O, and S), other than the amide bonds, do not exist in a large proportion in peptides, they often play critical roles in peptide chemistry and biochemistry. For example, disulfide bonds and thioether bonds can stabilize and regulate the secondary structure of peptides,31,51 and the role of diaryl ether facilitates the synthesis of many natural product peptides such as vancomycin.124 The special nature of heteroatoms gives a special status to the natural amino acids containing these heteroatoms. In 2013, Harran and coworkers developed a method for peptide macrocyclization based on the Tsuji–Trost reaction.125 A common cinnamyl carbonate was attached to the peptide, and then Pd(PPh3)4 was used as a catalyst to selectively react it with the phenol sidechain of tyrosine on the unprotected peptide to obtain a cyclic peptide with cinnamyl ether bond (as cycloetherification or cinnamylation). In 2023, Zhao and coworkers developed the copper-catalyzed Shono-type oxidation of proline residues as a peptide macrocyclization strategy. This strategy used the intramolecular hydroxyl group as a nucleophile to synthesize challenging small-size cyclic peptides with high selectivity under mild reaction conditions; the protocol was easy to operate and well-tolerated different amino acids126 (Scheme 19a). In 2021, Chen and coworkers obtained various medium to large-sized cyclic peptides linked in the form of biaryl thioethers via the chemoselective intramolecular S-arylation of thiols and iodobenzene catalyzed by palladium127 (Scheme 19b). In 2024, Arisawa and coworkers reported a new strategy for the rhodium-catalyzed oxidation of methanol to formaldehyde, converting an unprotected peptide containing two cysteine residues to the cyclic peptide methylene dithioacetals128 (Scheme 19c). Many functional groups in this strategy were well tolerated, and the side-reactions could be suppressed by controlling the concentration of formaldehyde to keep it at a low level during the cyclization step.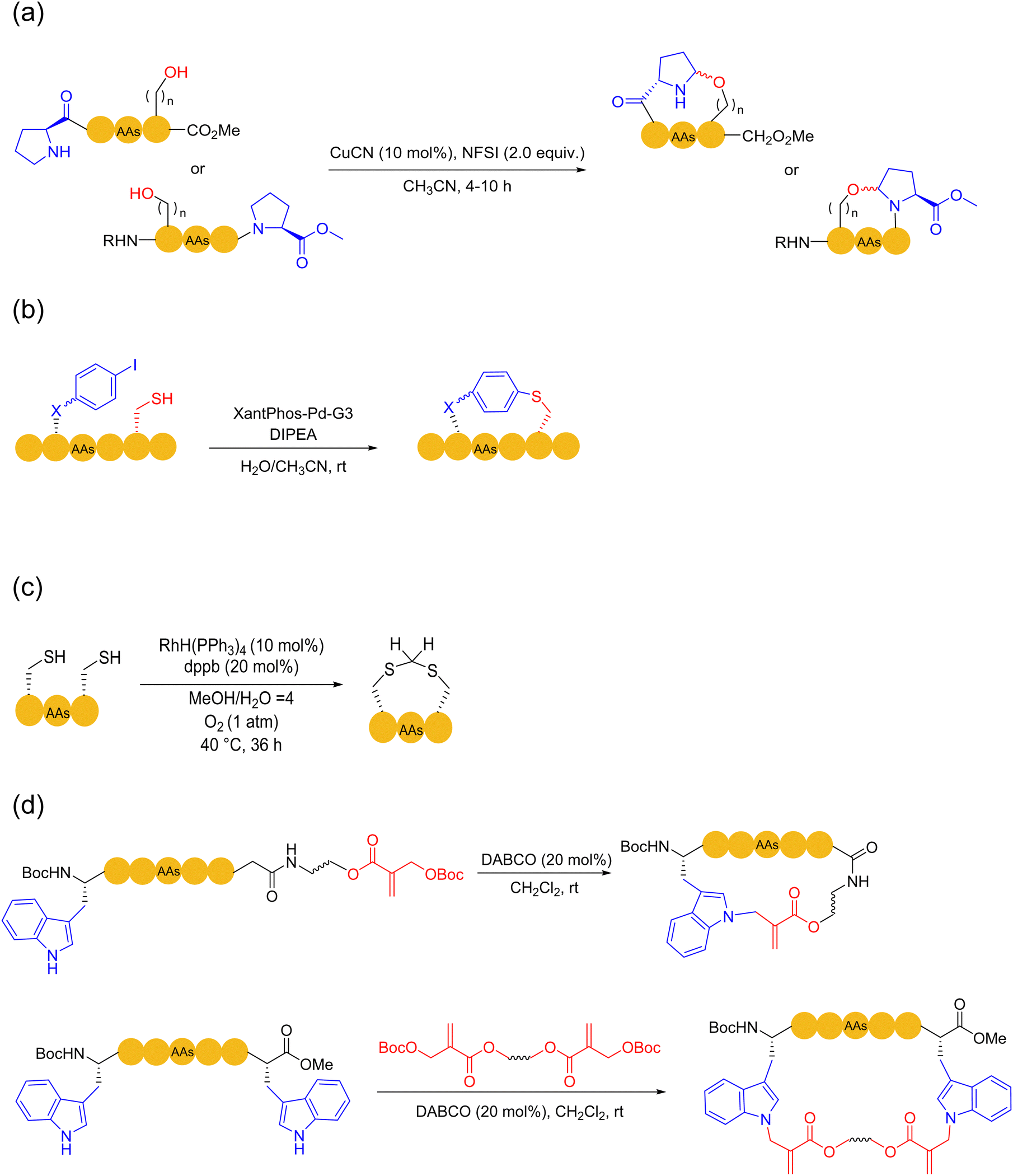 | ||
| Scheme 19 Selected peptide macrocyclization strategies via heteroatom arylations/alkylations: formations of (a) hemiaminal ethers; (b) biaryl thioethers; and (c) dithioacetal; (d) tryptophan indole’s N1 allylation. | ||
The relatively poor nucleophilicity of amino groups compared to O- and S-groups also gives rise to less arylation or alkylation of amino groups in most previously mentioned strategies. In 2023, Chen, Yudin and coworkers disclosed a widely applicable strategy based on the Ulman-type reaction catalyzed by copper.129 The Cu-catalyzed formation of C(aryl)–O or C(aryl)–N bonds between aryl iodide and natural amino acids such as Trp, His, and Tyr residues in various combinations provided a variety of unique tension-bearable multi-joint scaffolds to modulate the backbone conformation of peptides in an unprecedented manner. In the same year, Xie and coworkers combined the use of gold catalysis and silver salts to accomplish the chemo/site-selective arylation of relatively inert N-terminal primary amines (in the presence of other amino acid residues with nucleophilic sidechains).130 DFT calculations provided insight that the silver cation can serve as a transient coordination mask of the more reactive reaction sites. In the case of macrocyclization, two bifunctional linkers, 2-iodobenzoyl chloride and 2-iodobenzaldehyde, were used in their work to form covalent bonds between the amide and amine sites of unprotected peptides, producing cyclic peptides with new aromatic backbone linkage. This strategy showed good biocompatibility and can be further extended to the field of peptide labeling and peptide stapling. In 2024, Xu, Wang, Sun and coworkers reported a rapid late-stage organocatalytic N–H allylation strategy for the indole moieties of tryptophan and Morita–Baylis–Hillman (MBH) carbonates under mild conditions131 (Scheme 19d). The reaction was amenable to both LPPS/SPPS and a new handle was installed for further bioconjugation. Through these methods, various linkers of different rigidity and functionalities can now be installed into macrocyclic peptides by design.
4. Biological macrocyclization strategies
Macrocyclic peptides were first serendipitously discovered in living organisms, where there must be sophisticated biosynthetic pathways for their productions for highly specific biofunctions. Given the recent advances and further understandings in life science and biotechnology (e.g., genetic engineering, enzymatic engineering, and biochemical engineering), scientists at the biology and chemistry interface can now know more about the mysterious macrocyclic peptide synthetic mechanisms, master the exquisite biosynthetic skills, and even manipulate whole microorganisms to generate unnatural peptide macrocycles. To date, there are two basic protocols to form macrocyclic peptides in nature, as follows: (a) via the ribosomal synthesis of mRNA for translation to afford linear peptide precursors for further modifications to cyclic peptide products by different enzymes and (b) via the actions of non-ribosomal peptide synthetase to form cyclic peptide products directly.132 The rational use of genetically modified and highly specific enzyme-catalyzed reactions is powerful strategies for cyclopeptide syntheses (despite unresolved limitations, e.g., low sequence specificity). Judicious employment of thermodynamically less stable substrates, co-solvents and additives, and alteration of the reaction pH/temperature can favor a shift in the reversible reaction equilibrium to the right product side. Recently, a wide variety of biosynthetic techniques has been developed, e.g., genetic codon reprogramming, display technology, and in vitro translation in genetically programmed cells, which not merely speed up the whole de novo design and macrocyclization processes, but also develop novel macrocyclic peptide scaffolds not easily or even impossibly secured by other approaches. Biosynthetic/biocatalytic approaches will emerge as powerful and frontier research in peptide macrocyclization in the years to come. A few typical literature examples are selected and featured below.4.1. Ribosomal synthesis and post-translational enzymatic cyclization
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are named for a class of structurally complex peptidic natural products based on their structural and biosynthetic similarities. Their biosynthetic process has been described in detail by a long name, commencing the synthesis of a biosynthesis gene cluster (BGC) code, translating the gene code within the ribosome, and then synthesizing a linear precursor peptide for post-translational enzymatic modifications. In general, the precursor peptide contains three key regions, as follows: (i) the leader sequence (at the N-terminus, for recognition by post-translationally modifying enzymes, and later removed by proteolysis), (ii) the core sequence (the site to be targeted and modified to access the mature product), and (iii) follower sequence (sometimes additional recognition sequence exists at the C-terminus and will also be proteolytically removed upon RiPP maturation). The leader/follower signal sequences direct the entire precursor peptide to a specific intracellular compartment and the core sequence will be subjected to a series of modifications by post-translationally modifying enzymes. A considerable number of these classes of biologically active peptide natural products is macrocyclic structures.Although precursor peptides contain only proteinogenic amino acids, some unnatural amino acids or even non-amino acid structures can be incorporated during the enzymatic modification steps. One example is the lanthipeptide family, a class of highly active antibacterial agents, where dehydrogenases are typically used to introduce dehydroalanine/dehydrobutyric acid (Dha/Dhb) in their structure. It can be further modified to form lanthionine (Lan), methyllanthionine (MeLan), lemon protein (Lab) and methyl lemon protein (MelAB) motifs. As a hot research topic, thiopeptides can also be modified to form these motifs. In 2021, Wang and coworkers obtained cyclic peptides of 2-aminovinyl-cysteine (AviCys) and 2-aminovinyl-3-methylcysteine (AviMeCys) motifs by using a variety of enzymatic modifications.133 In 2022, Mitchell and coworkers prepared diverse pyridine-based macrocyclic peptides (pyritides), with variations in their macrocycle sequence and size, from linear precursor peptides via a two-site recognition pathway and a key macrocyclase catalyzed [4+2] annulation.134 Inspired by a biosynthetic strategy, in 2014, Fasan and coworkers used chemically selective reactions between cysteine-reactive unnatural amino acids (O-(2-bromoethyl)-tyrosine) and thiols, in combination with split intein-catalyzed protein splicing (for circular ligation), to secure spontaneously and post-translationally modified thioether-linked ribosomally synthesized cyclic peptides in a sidechain-to-sidechain manner in bacterial cells.135
In the process of RiPP natural and artificial biosynthesis, a wide variety of enzymatic macrocyclization reactions has been discovered and strategically employed, including disulfide S–S bond formation, head-to-tail (or sidechain) C–O/N lactonization/lactamization, and C–C/S bond formation,136,137 leading to cyclic peptides of significant structural and functional diversities.
4.2. Non-ribosomal synthesis and enzymatic cyclization
Non-ribosomal peptide synthetases (NRPS) are a large class of multifunctional enzymes that can produce peptides without the cellular ribosomal machinery and messenger RNA. Since Walsh and coworkers first demonstrated the significant potential of NRP cyclase, enzymatic catalysis for accessing cyclic peptide natural products and their related analogues has been greatly developed; the reaction types and mechanisms have been surveyed and discussed in detail in other reviews (Scheme 20).132,138 Previous cyclase catalysis necessitated a minimum peptide length (less than 10 amino acids). Recently, in 2023, Wakimoto and coworkers disclosed a streamlined chemoenzymatic method on a solid support,139 where linear peptides were first linked to the resin through a C-terminal diol ester, and then upon cleavage, the diol ester group underwent head-to-tail cyclization by SurE, a representative penicillin-binding protein-type thioesterase (PBP-type TE). Excellent conversion rates were achieved in cyclizing small size-peptides without epimerization.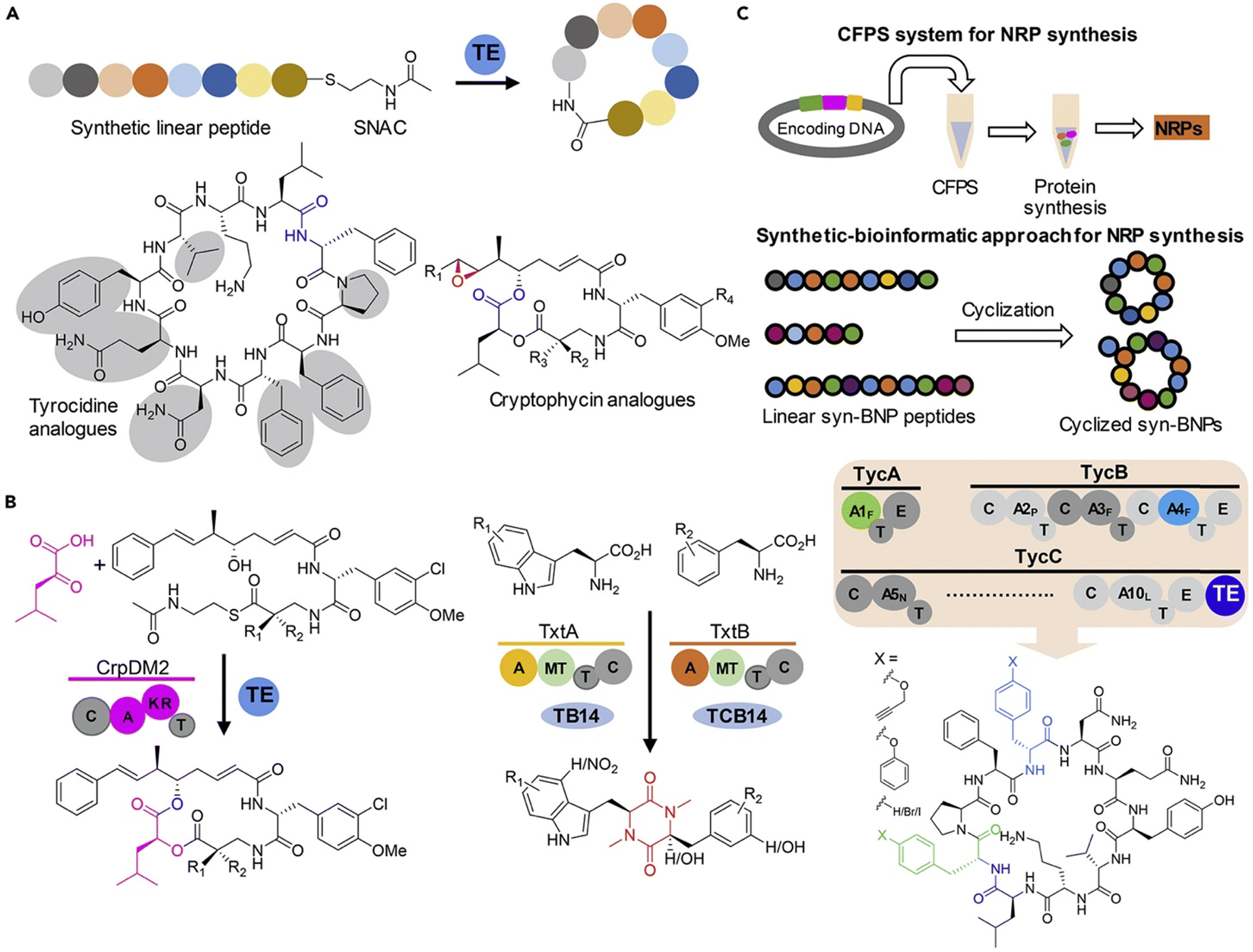 | ||
| Scheme 20 Production of cyclic NRP analogues with different bio-strategies. (a) Chemoenzymatic synthesis of NRPs where recombinant TE alone or with other enzymes is commonly used to catalyze the regiospecific macrocyclization of synthetic linear peptide precursors activated by an SNAC moiety. (b) Biocatalytic synthesis of NRP analogues from synthetic linear peptide precursors or simple building blocks using multifunctional NRPS modules alone or with other enzymes. (c) Cell-free protein synthesis (CFPS) systems and synthetic-bioinformatic natural products (syn-BNPs) approach for in vitro synthesis of NRP analogues. Illustration is adapted with permission from ref. 132. | ||
4.3. Genetic programming-mediated cyclization
In 2009, Suga and coworkers published a pioneering work,140 whose theoretical sources were reported by Kawakami and Aimoto in 2007,141 to apply reprogrammed genetic coding to express and integrate glycolic acid (HOG) into, through ribosomal synthesis, a peptide chain containing Cys-Pro-glycolic acid (C-P-HOG). Recombinant peptide deformylase (PDF) was used to remove the N-terminal formyl group, and methionine aminopeptidase (MAP) was used to cleave methionine to produce the free N-terminus of any desired amino acid. Subsequently, spontaneous rearrangement could generate the corresponding C-terminal diketopiperadine-thioester with the removal of HOG. The subsequent N-to-S acyl transfer of the free amine group with the thioester non-enzymatically formed a backbone cyclized peptide in the form of an amide bond (Scheme 21). This pioneering work allows the incorporation of unnatural amino acids into the expressed peptide to spontaneously generate macrocyclic peptides with different sequences, greatly expanding the applications of this powerful genetic code reprogramming.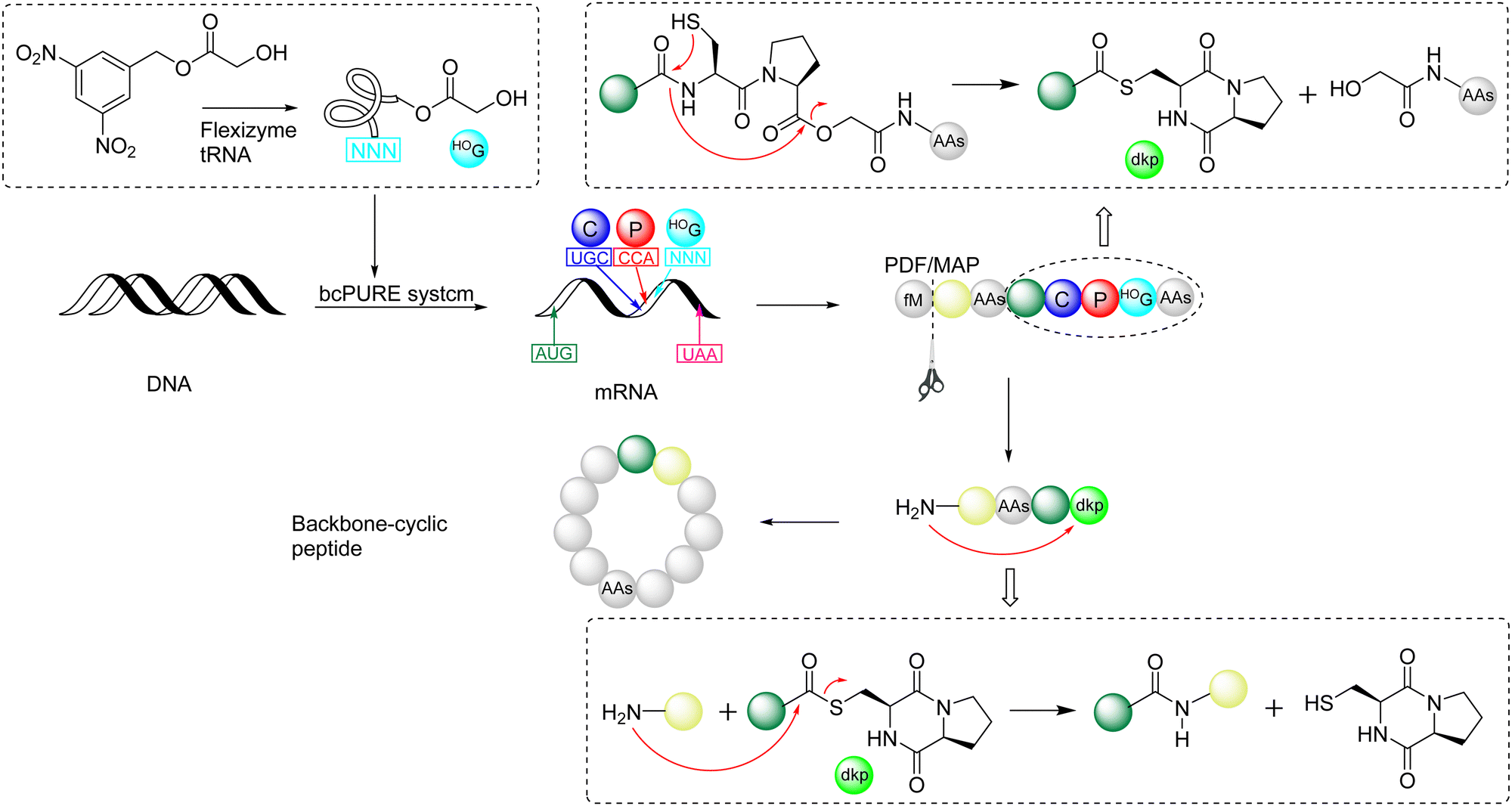 | ||
| Scheme 21 Suga's pioneering work on reprogrammed genetic coding to prepare cyclopeptides.140 | ||
Although the last two decades have witnessed great research progress in the biosynthesis of both genetically encoded and non-natural cyclic peptides, the conventional RiPP and NRP systems can hardly produce desired amino acid sequences. Given that intracellular translation or in vitro translation techniques based on genetic codes can produce various amino acid sequences on demand (as shown in the work by Chin and coworkers in 2022),142 unnatural amino acids can possibly be introduced into the final peptide product by genetic programming techniques, which will certainly open new biosynthetic avenues for macrocyclic peptides.
5. Macrocyclization strategies for polycyclic, interlocked, self-assembled and directed folded peptides
Macrocyclic peptides, which are comprised of two or more ring structures, are known as polycyclic peptides. Compared with their monocyclic counterparts, they demonstrate rigidity and well-defined conformation for better ligand preorganization, thereby exhibiting stronger target binding affinity and higher metabolic stability, where usually the larger their size, the more outstanding these advantages. Polycyclization can greatly improve the cell membrane penetration ability and oral administration performance of peptides, which has become one of the important directions in the development of cyclic peptide therapeutics, as well as imaging agents and diagnostics.143,144 However, polycyclization reactions of peptides/peptidomimetics are far more complex transformations than the monocyclization versions, where the commonplace latter methodologies are usually not translationally suitable/compatible for the former. The significant challenges include achieving precise control of the reaction site-selectivity, extending the substrate scope and functional group tolerance, and realizing the desired conformation control of the resulting polycyclic peptides. At present, the preparation of most bicyclic and polycyclic systems depend on (a) the use of multifunctional handles to react with multiple natural/modified/unnatural amino acid side chains via the so-called stapling reactions, in both solution and solid phases and (b) several intramolecular orthogonal cascade reactions. Elegant and strategic literature works are exemplified and detailed in the following section. Here, the mutual reaction orthogonality of the reported polycyclization methods, paired with NCL cannot be overemphasized.Alternatively, unlike most of the familiar molecular monocyclic and above-mentioned polycyclic peptides, a few novel higher-order peptidic macrocyclic structures are held together by mechanical bonds and weak non-covalent interactions, e.g., (a) by intermolecular interlocking to form peptide (pseudo)-rotaxanes and catenanes and (b) by coordinating with metal ions to fold and form macrocyclic cages and self-assembles. They furnish novel definitions and examples for supramolecular higher-order macrocyclic peptide architectures beyond NCL. Typical examples are also selected and briefly described below.
5.1. Cascade/sequential reaction-mediated polycyclization
Successive multistep modification is a feasible and dominant strategy to obtain polycyclic peptides. A method had been employed by forming a thioether bond between cysteine and oxidized tryptophan residues to construct the first ring in amatoxin derivatives, and then successfully access bicyclic heptapeptides and decapeptides through the end-to-end connection of the peptide chain. Disulfide bonds, as easily accessible functional groups available for transformations, broadly exist indispensably in various functional proteins in nature.32 In 2004, Leatherbarrow and coworkers designed and synthesized a novel protease inhibitor.145 The enzyme inhibitor is a butterfly-type bicyclic peptide containing disulfide bonds, which was synthesized by the mainchain cyclization of the protected precursor peptide, followed by oxidation of the two cysteine side chains within the cyclic peptide to form disulfide bonds after deprotection, resulting in bicyclization. Unlike previous enzyme inhibitors, this bicyclic peptidase inhibitor retained the biological properties of natural proteins well, while achieving a significant reduction in size. As mentioned in Section 2, in 2019, Li and coworkers used NCL to first form a backbone-based macrocyclic peptide, and then OPA to connect amino groups at different positions with the thiol group at the junction of monocyclic ring to achieve bicyclization.56 Perrin and coworkers synthesized an eight-membered monocyclic peptide with an amide linkage by conventional methods, and then modified it with OPA to create a bicyclic peptide scaffold57 (Scheme 22a). With the report of an increasing number of macrocyclization strategies, the synergistic applications of different methods to produce polycyclic peptides start to become prevalent.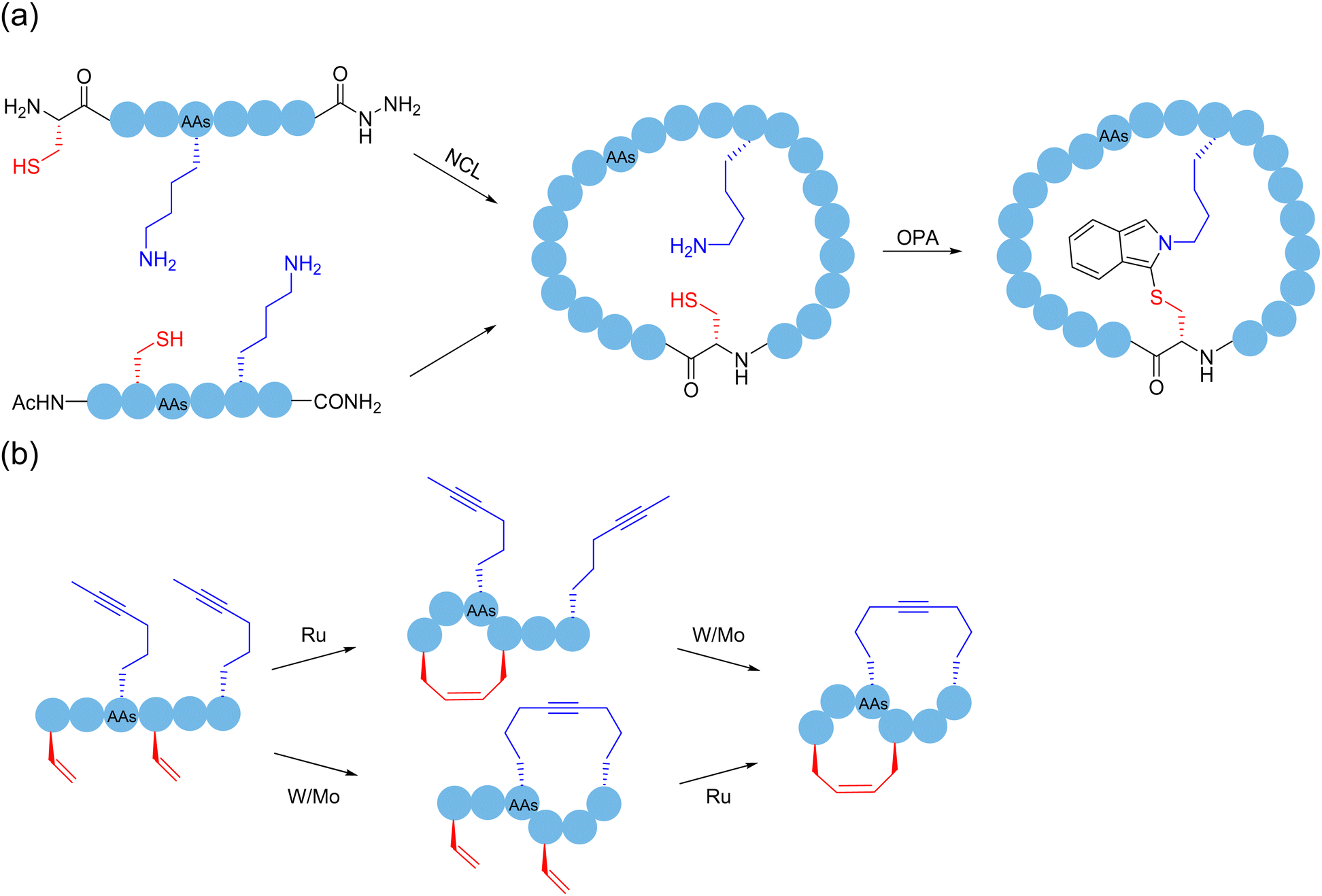 | ||
| Scheme 22 Selected peptide polymacrocyclization strategies via cascade/sequential reactions. (a) NCL with OPA; (b) RCM and RCAM. | ||
Regarding the sidechain-based bicyclization strategies, ruthenium-catalyzed ring-closing olefin metathesis (RCM) and tungsten-catalyzed ring-closing alkyne metathesis (RCAM) have been reported with certain orthogonality and can be cooperatively used to produce bicyclic peptides.95 In 2016, Waldmann and coworkers chemically and regioselectively synthesized sidechain-to-sidechain bicyclic peptides using the orthogonal Mo-catalyzed ring-closing alkyne metathesis (RCAM) and Ru-catalyzed ring-closing olefin metathesis (RCM) systems, and the bicyclic peptide ligands modified by this strategy showed high affinity to the activated Rab GTPase96 (Scheme 22b).
Alternatively, although the use of protective groups in the formation of disulfide bonds on cysteine-rich peptide chains can avoid the side-reactions of thiols, it is still difficult to precisely control the reaction site after removal of the protecting groups, especially when more than one disulfide bond needs to be formed. These unprotected thiols may lead to mispairing during folding. In 2023, Li and coworkers employed a photocleavable protection group to avoid the side reactions of the C-terminal cysteine and continuously form disulfide bonds in a one-pot manner146 (Scheme 23a). Through this sidechain anchoring strategy, they successfully prepared the bicyclopeptide α-symbiont toxin Vc1.1 containing two pairs of disulfide bonds. Given that the biological activities of peptides are chiefly determined by their secondary structures, the formation of polycyclic peptides using the sidechain-to-sidechain stapling approach has high potential to target currently intractable proteins.
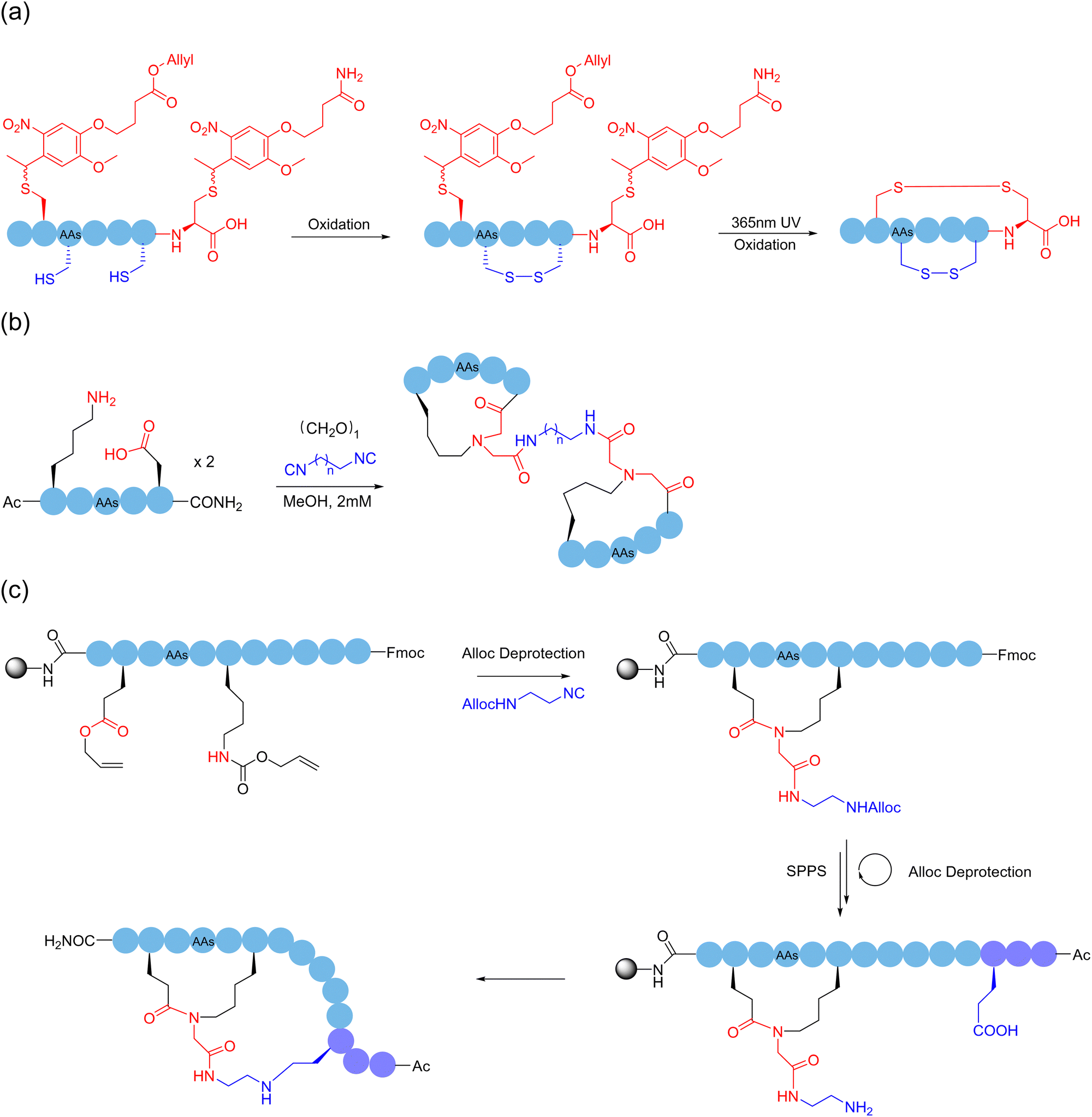 | ||
| Scheme 23 Selected peptide polymacrocyclization strategies through: (a) stepwise site-selective disulfide bridging with photocleavable protective group of cysteine; and (b) and (c) Ugi-type peptide dimerization and double stapling. | ||
Sequential reactions of the same mechanistic type can also be adopted to accomplish ordered polycyclization by some coincidence on a support. In 2022, Huc and coworkers reacted thiol and chloroacetamide to continuously and selectively form a polycyclic structure on the surface of an aromatic polyamide oligomeric helix with a fixed structure.147 The selectivity of the reaction can be related to the ring size, where initially the nearest tryptophan binds to the surface reaction site of the aromatic polyamide oligomeric helix, followed by the formation of the next smallest ring; up to four cyclic peptides of different sizes and shapes could be formed sequentially following the same reaction type. In addition, based on their previous work, Chen and coworkers took advantage of the anchoring effect of formaldehyde on amino groups, and used the three-component condensation reaction of primary amine, formaldehyde, and guanidine, under different anchoring combinations and spatial arrangement in the reaction sequence, to obtain different polycyclic peptide structures cross-linked by the six-membered tetrahydro-1,3 and 5-triazine (4-triazine or THT) linker.148 This reaction used simple reagents to polycyclically modify peptides under relatively mild conditions, which had the advantages of high efficiency and versatility. In 2023, Wessjohann, Rivera and coworkers reported a strategy to construct bicyclic peptides using MCRs.149 The first approach of multicomponent macrocyclization-dimerization involved the assembly of two interlinked β-rotors, where a double Ugi reaction took place for the binding (Scheme 23b). The second approach exploits the efficacy of Ugi macrocyclization on the resin to introduce functional groups, which are subsequently used for macrolactonization, leading to a fused doubly-stapled topology (Scheme 23c).
5.2. Multifunctional/multivalent crosslinker-mediated polycyclization
Among the 20 naturally occurring amino acids, the thiol group in the cysteine sidechain is one of the few ideal reaction sites in which the macrocyclization strategy is not limited to the formation of a monocyclic ring. Due to the alkylation reaction of thiols, cross-linking agents of 1,3,5-tris-(bromomethyl) benzene (TBMB) skeleton were first applied to the synthesis of polycyclic peptides,150–152 to avoid the non-specific modifications caused by excessive reagents. By tuning the electrophilicity, chemists continued to improve different thiol-reactive linkers, and several “trivalent-acceptors” have been developed, including 1,3,5-tris(bromomethyl)benzene (TBMB), N,N′,N′′-(benzene-1,3,5-triyl)-tris(2-bromoacetamide) (TBAB), N,N′,N′′-benzene-1,3,5-triyltrisprop-2-enamide (TAAB), 2,4,6-tris(bromomethyl)-1,3,5-triazine(TBMT), 1,1′,1′′-(1,3,5-triazinane-1,3,5-triyl)tris(2-bromoethan-1-one)(TATB), and 1,3,5-triacryloyl-1,3,5-triazinane (TATA)153,154 (Scheme 24a). By combining them with a mature phage display platform, researchers can take advantage of their high diversity for screening to more quickly identify the molecules that interact with the desired ligand. In 2018, Boeckler and coworkers developed a new thiol-specific coupling agent 1,3,5-tris((pyridin-2-yldisulfanyl)methyl)benzene (TPSMB)155 from 1,3,5-tribromomethyl benzene. Unlike the classical trivalent linker, the bicyclic peptide produced could not only be linked by three disulfide bonds, but also be reduced and re-linearized under physiological conditions. In 2023, Jiang and coworkers innovated a reversible and exchangeable sidechain click stapling strategy in the form of disulfide bonds156 (Scheme 24b). This strategy was realized first via the addition of phthalimide sulfonyl chloride with various commercially available diacetylene to yield a range of double/triple-site electrophiles in varying length and rigidity. Then, expeditious cross-linking happened between the cysteines of the unprotected S-terminal peptide and the triadduct electrophiles in an ambient atmosphere, with removal of the phthalimide. Eventually, a variety of stapled peptides of medium to large sizes was formed in the exchange of disulfide bonds. This novel crosslinking agent of alkyl backbone is easy to obtain, where its length and angle are adjustable, thereby possessing the very advantages of simplicity, rapidity, and flexibility.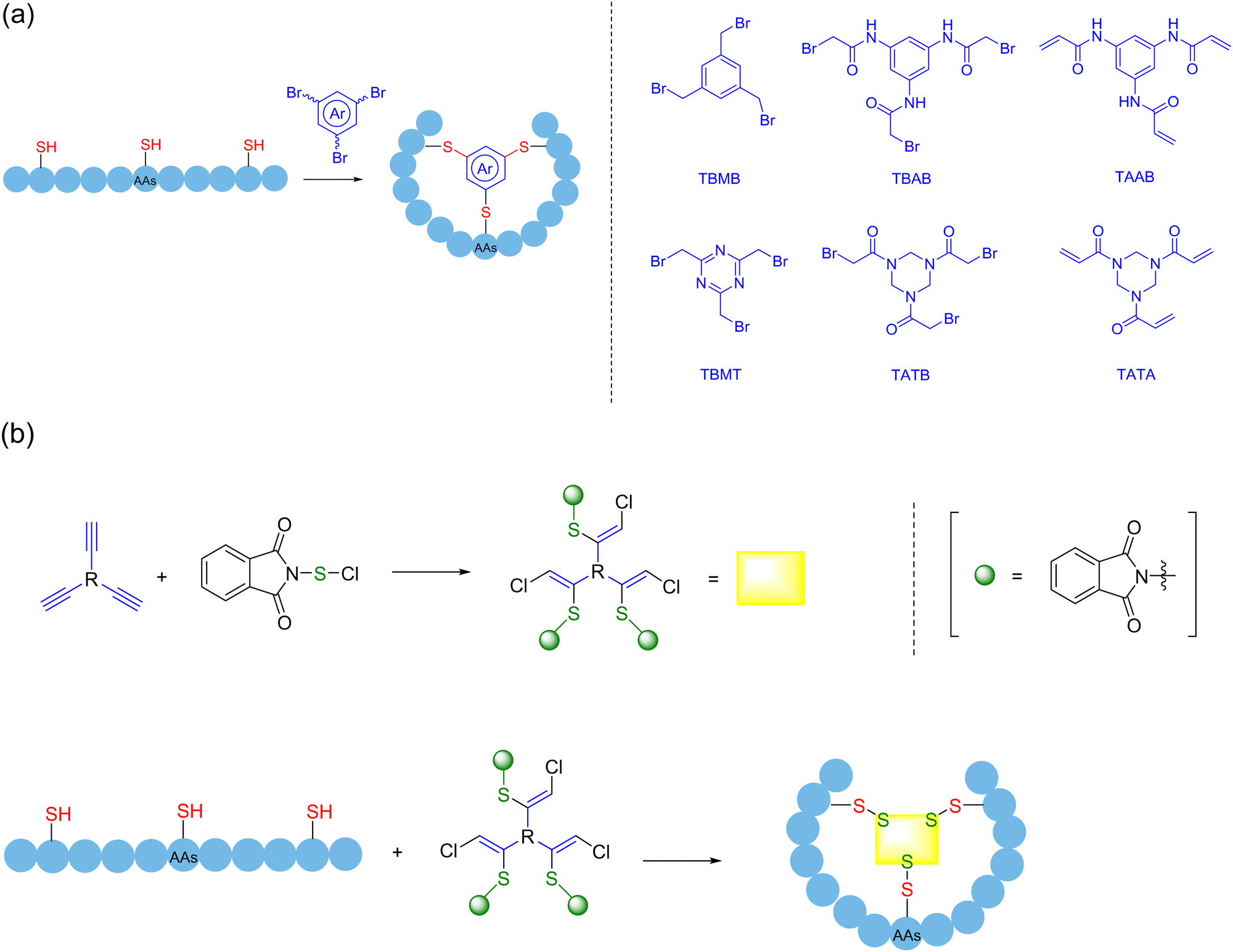 | ||
| Scheme 24 Selected peptide polymacrocyclization strategies with multivalent crosslinkers via nucleophilic substitution reactions. (a) Based on 2-bromoacetamides and acrylamides; (b) based on disulfide clickable electrophiles. | ||
Chemical modification approaches through arylation by introducing new moieties to regulate the structures and functions of different biomolecules have been being extensively explored.157 In 2013, Pentelute and coworkers discovered the facile, room-temperature nucleophilic aromatic substitution chemistry between perfluoroaromatic molecules and the thiolate of cysteine as new NCL and even peptide stapling tactics.158 Since then, a variety of multifunctional crosslinkers with high efficiency and precise control of the reaction sites has been developed based on thiol arylations, in particular in the field of peptide poly-macrocyclization.
In 2017, upon detailed investigation of 2,3,5,6-tetrafluoroterephthalonitrile (4F-2CN) to selectively react with four thiolates of sequentially varied reactivity under mild aqueous conditions through nucleophilic aromatic substitution, Wu and coworkers strategically developed the precisely regulated and efficient locking (PROP-locking) tactic and transformed an unprotected linear peptide containing two cysteine (one from the N-terminal cysteine) and two penicillamine motifs (of different thiol nucleophilicity) into a tricyclic topologies through a one-pot double stapling protocol according to the kinetic sequence of nucleophilic attack159 (Scheme 25a). Notably, the order of aryl C–S and C–N bond formation could be manipulated by the selective substitution of two non-N-terminal thiols of penicillamine.160 In 2018, Pentelute and coworkers reported two methods of cysteine perfluoroarylation chemistry for the synthesis of arginine-rich bicyclic peptides, i.e., by double stapling the thiols in the cysteine sidechain with a decafluorodiphenyl moiety and cross-linking three decafluoroaryl groups with one end already attached to the thiol of a peptide chain with the trivalent 1,3,5-benzenetrithiol161 (Scheme 25b). In 2023, Giuntini, Coxon and coworkers introduced (5,10,15,20-tetrakis(pentafluorophenyl)porphyrin as a platform crosslinker for the synthesis of polycyclic peptides162 (Scheme 25c). This crosslinker allowed the direct modification of unprotected linear peptides containing multiple cysteines under physiological conditions, with the peptides containing two cysteine residues readily stitching by sidechains; or stepwise modifications by subsequently introducing a second peptide in the same manner to obtain bicyclic structures. Similarly, long peptides with more than two cysteine residues could furnish polycyclic products containing up to three peptide “loops”. In 2024, Xu and coworkers presented the first site-selective unsymmetric perfluoroaryl stapling protocol for Ser and Cys residues in unprotected peptides via visible-light-induced radical defluorinative perfluoroarylation, and then SNAr under mild conditions with a high reaction rate; further late-stage functionalization of the resulting stapled peptides was feasible, paving new ways for potential polycyclization.163
 | ||
| Scheme 25 Selected peptide polycyclization strategies with multivalent crosslinkers via nucleophilic aromatic substitution reactions. (a) Between 2,3,5,6-tetrafluoroterephthalonitrile and cysteines/penicillamines; (b) between decafluorodiphenyl moieties and cysteines; (c) between (5,10,15,20-tetrakis(pentafluorophenyl)porphyrins and cysteines. Illustration of (c) is adapted with permission from ref. 162. | ||
In addition to polyfluoroaryl multifunctional crosslinkers, the strategy of using iodoaryl reagents for peptide polycyclization was reported by Chen and coworkers in 2021164 (Scheme 26a). They demonstrated great versatility and efficiency for the selective macrocyclization and polycyclization of native unprotected peptides under mild conditions using a variety of readily available diiodine and triiodoaromatic reagents with different aromatic and heteroaromatic cores by the palladium(II)-catalyzed arylation of cysteine residues. In 2021, Spokoyny and coworkers reported the use of trivalent gold(III)-based reagents for transition metal-mediated arylation reactions to produce bicyclopeptides165 (Scheme 26a). The metal complexes used in this strategy were easy to synthesize, while the products did not require purification, even allowing rapid reactions with peptides under phage-compatible conditions. In 2022, Mudd and coworkers synthesized and explored the scope and generality of four new trivalent gold(III) complexes in the construction of bicyclic peptides166 (Scheme 26a).
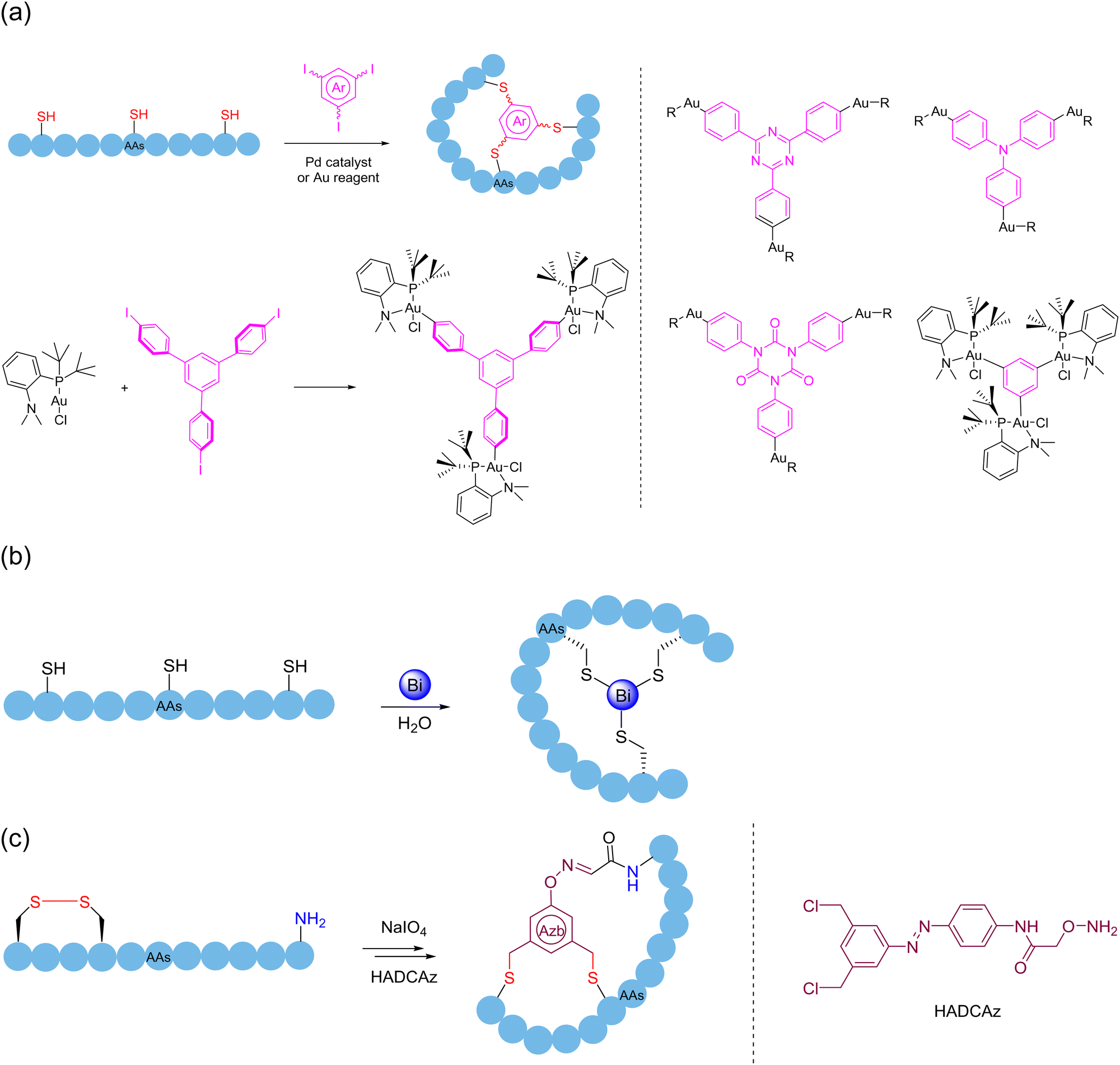 | ||
| Scheme 26 Selected peptide polymacrocyclization strategies: (a) via organometallic catalysts/reagents; (b) through metal coordination; (c) with multifunctional crosslinkers containing a photoswitch moiety. | ||
Bismuth metal is a special element with low biological toxicity, which can also bind with the cysteine residues in glutathione, peptides and proteins.167,168 Inspired by this, Nitsche and coworkers pioneered a novel metal coordination strategy to generate a bicyclic peptide employing a linear precursor peptide with three cysteine residues and a bismuth(III) salt169 (Scheme 26b). Under physiological conditions, a high concentration of bismuth tribromide in DMSO solution was added directly to the aqueous peptide solution, paired with an excess of thiol-free reducing agent tris(2-carboxyethyl) phosphate (TCEP) during the process for ensuring complete reduction of all the cysteine residues. Due to the sulfurophilic property of the trivalent bismuth, the reaction was highly selective and fast to produce stable bicyclic Bi(III)-stapled peptides, which exhibited superior bioactivities, e.g., entering cells through endocytosis in most cases and low toxicity. These bismuth bicyclopeptides offer new blueprints for the future development of cell-penetrating cyclic peptides and bismuth-containing therapeutics.170
Since crosslinkers with two orthogonal reactive functional groups have been successfully designed and synthesized, Derda and coworkers introduced tridentate C2-symmetric light-responsive hydroxyl amine and di-chlorobenzene containing azobenzene (HADCAz) linkers for broad-scope peptide poly-macrocyclization171 (Scheme 26c). These linkers could be linked to the N-terminal serine and two cysteine sidechains of the linear unprotected peptide in one-pot to form a “double-loop” structure. Interestingly, the linker could also be photo-regulated by reversibly converting between the trans and the cis form under blue light (365 nm) irradiation.
A general method for tetracyclic peptide synthesis through the CEPS/CLIPS/CuAAC (“triple-C”) locking methodology was reported by Nuijens, Timmerman and coworkers172 (Scheme 27a). The specific approach was to first use the peptidase variant omniligase-1 (CEPS). The precursor peptide containing two cysteines and two azidohomoalanines (Aha) was initially subjected to head-to-tail enzymatic cyclization, followed by intramolecular polycyclization using a tetravalent crosslinker containing two bromomethyl groups and two alkyne functions by stepwise CLIPS bicyclization (double nucleophilic substitutions) and CuAAC (double click reactions). Later, Maarseveen and coworkers further modified a tetravalent crosslinker scaffold to embrace two active primary bromides with an aldehyde or an aminooxy group,173 using a combination of CEPS, CLIPS, and oxime ligation to generate a tetracyclic peptide from similar unprotected precursors (Scheme 27b). In 2023, Gao and coworkers reported two classes of multifunctional chloroxime-based crosslinkers for their cysteine-directed proximity-driven strategy (i.e., first rapid cysteine labeling, and then proximity-driven amine-selective cyclization) to construct bicyclic peptides.174 This crosslinker enabled different combinations of thiol and amino ligation, including terminal amino-Cys–Cys, Cys–Lys–Cys, and Lys–Cys–Lys. Another chloroxime-based, boronic acid-containing DCA-RMR1 crosslinker triggered the bicyclization of natural peptides through N-terminal amino-Cys–Cys double stapling175 (Scheme 27b).
 | ||
| Scheme 27 Selected peptide polymacrocyclization strategies with (a) stepwise multimodal transformations and (b) multifunctional chloroxime-based crosslinkers. | ||
5.3. Unnatural/modified amino acid-mediated polycyclization
Multifunctional coupling approaches for sidechain stapling, such as the 1,4-dinitroimidazole reagent developed by Wang and coworkers,64 and the on-resin dipyrrin construction by Chan, Long, Wong and coworkers,65 upon modifying the sidechains of amino acids, furnished novel bicyclic peptide structures (Scheme 9c). Albericio, Lavilla, and coworkers exploited diiodo-tyrosine in the peptide sequences and cyclized them with two tryptophans through C–H activation stapling to obtain doubly-stapled peptides109 (Scheme 28a). In 2022, Nitsche and coworkers developed a general bicyclization strategy based on the condensation reaction between two 1,2-aminothiols (to be installed at both C/N-termini) and an unnatural amino acid containing a 2,6-dicyanopyridine (DCP) sidechain (in the middle of the peptide sequence)176 (Scheme 28b). Complex and rigid bicyclopeptides were synthesized selectively and biocompatibly. In 2024, Waser and coworkers developed a novel bivalent hypervalent iodine reagent, ethynylbenziodoxoles (EBxs), which could serve as a rigid staple to modify the amino sidechain of Lys/Orn/Dap, and then react with Cys/Trp/indole-attached Lys residues for peptide macrocyclization; this protocol was also found to be SPPS-compatible, and even orthogonal to HATU- lactamization to accomplish bicyclization in one-pot177 (Scheme 28c). | ||
| Scheme 28 Selected representative peptide bicyclization strategies using unnatural/modified amino acids. (a) Between diiodo-tryosine and tryptophan indoles' C2 position; (b) between 2,6-dicyanopyridine and terminal 1,2-aminothiols; (c) between Cys/Trp/indole-attached Lysine and Lys/Orn/Dap. | ||
5.4. Mechanically interlocked cyclization
With the continuous development of supramolecular chemistry, mechanically interlocked molecules such as rotaxanes and catenanes are widely used in materials science, catalysis, biology, and many other fields.178–181 In fact, mechanical interlocking occurs naturally in DNA and protein molecules, such as lasso structures182 and molecular knots.183 Despite the many difficulties in their synthesis, purification, and characterization, mechanical interlocking peptides can still be synthesized by researchers. The first synthesis of benzamide peptide rotaxanes was reported by Leigh and coworkers in 1999.184 This strategy required the pre-design and synthesis of an axis derived from a short peptide, followed by the formation of a ring around the axis relying on intermolecular hydrogen bond orientation in a low-polar solvent. Since then, different peptidorotaxanes had been successively reported using similar strategies.185–188 In 2006, Leigh and coworkers studied the strategy of re-encapsulation of the axis through the peptide ring to synthesize peptidorotaxanes.189 In 2007, Dawson and coworkers selectively and efficiently formed peptidorotaxanes by crossing linear thioester peptides through a cyclic peptide, which could subsequently be cyclized using NCL ring-closure to afford bicyclic interlocked2 heterocycles190 (Scheme 29a). Early synthetic strategies demonstrated the synthetic accessibility of mechanically bonded interlocked peptides as well as their unique functions and properties. Although amino acid sequences have been introduced into these mechanically interlocked structural molecules for a long period, the key macrocyclization steps are still accomplished by the reactions of non-peptide prefunctionalized components,191 and mechanically interlocked holopeptide structures are still difficult to obtain by purely chemical synthesis. In 2024, Numata and coworkers disclosed the first synthesis of an all-peptide-based rotaxane from a proline-containing cyclic peptide192 (Scheme 29b).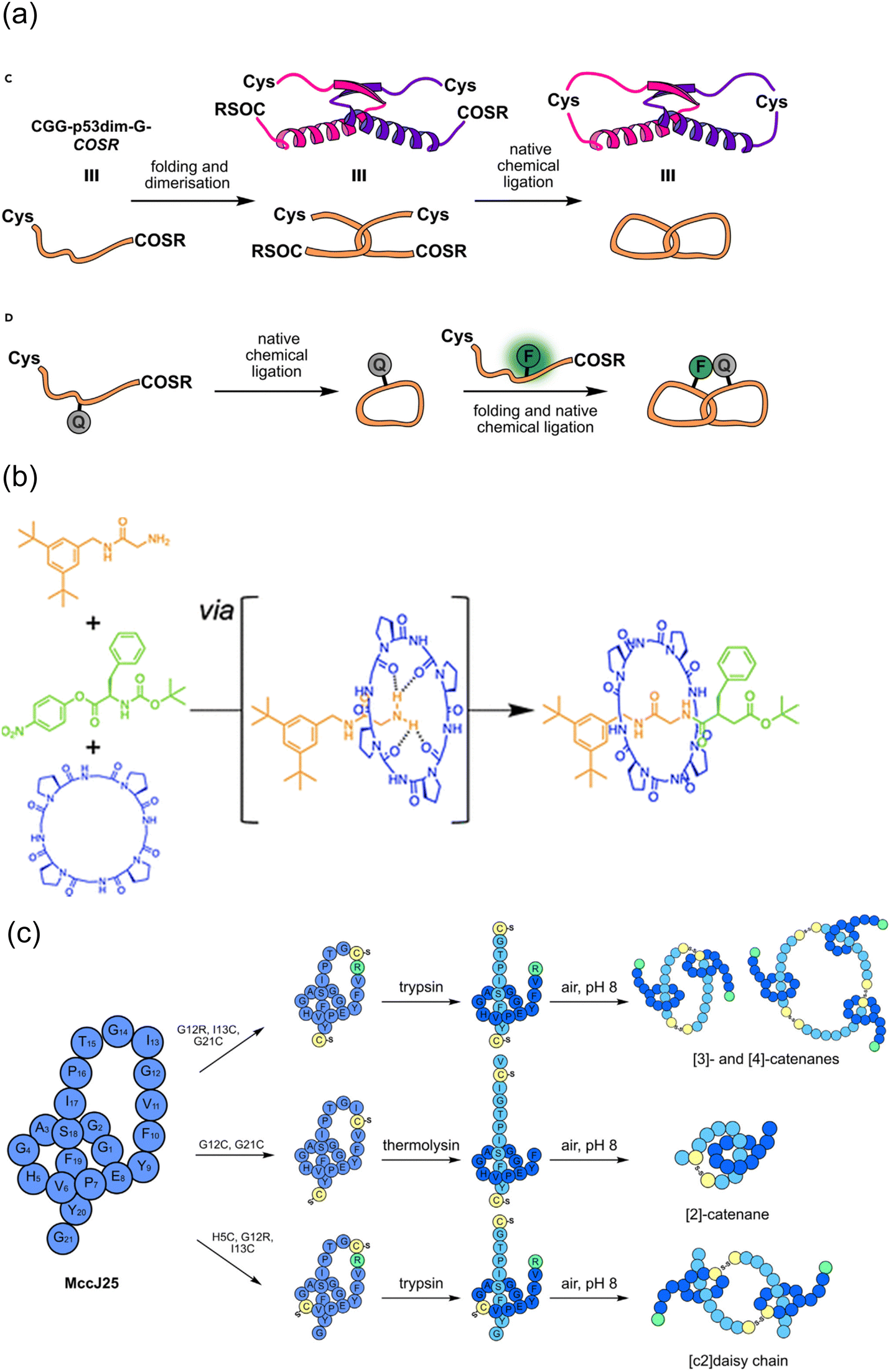 | ||
| Scheme 29 Selected natural and synthetic examples of peptide-based (a) & (c) catenanes and (b) rotaxanes. The Illustrations of (a) and (c) are adapted from ref. 190,193, respectively, and (b) ref. 192, with permission. | ||
Lariat peptides are a class of naturally occurring mechanical interlocking peptides, which are attractive for next-generation antibiotic development. Currently, the combination of gene mutation technology and site-selective chemical modification is the best strategy for the rational design and synthesis of lasso peptides193 (Scheme 29c). Several investigators have provided some excellent strategies in the chemical synthesis of lariat peptides.194,195 Indeed, the full chemical synthesis of natural lasso peptides remains an elusive goal. However, these topologically extraordinary building blocks can be employed to construct a range of interlocked molecular structures. In 2016, Link and coworkers reasoned the use of loop-cleaved lasso peptides as building blocks to create peptidic catenanes. After the mutagenesis of the lasso peptide microcin J25 (MccJ25) with two cysteine residues followed by trypsin cleavage, the [2]rotaxane structure was obtained, where it could further self-assemble into a [3]catenane and [4]catenanes by multimerization through the disulfide linkage at room temperature in aqueous solution.196 Inspired by this surprising discovery, in 2021, they used genetic recombination technology to change the quantity and position of cysteine in the molecular structure of MccJ25, predetermining the building block structure produced after enzymatic cleavage. A range of different mechanical interlocking peptides was generated in water via the covalent self-assembly of different building blocks under mild conditions, including rotaxanes, daisy chains, catenanes, and double-lasso macrocycles.197 In 2021, Ma and coworkers successfully expressed the lariat peptide biosynthesis gene cluster stla with systematic site-directed mutagenesis on the precursor peptide-encoding gene based on a marine Streptomyces heterologous expression system, generating 17 stlassin derivatives and expanding the diversity of lariat peptides.
5.5. Metal-directed stapling, folding and self-assembly
In addition to covalent bonding, the metal ion coordination approach can also provide alternative strategies for accessing structural restricted macrocyclic peptides. In 2014, Fei and coworkers published a peptide stapling strategy based on iridium-(III)-bis(histidine) chelation (Ir-HH cyclization).198 By reacting the Ir(III) complex bis(2-phenylpyridine-C2,N)-bis(aquo)iridium(III) trifluoromethanesulfonate with different model peptide ligands containing two unprotected histidine residues at different positions, various structurally constrained stapled metallopeptides were formed with excellent luminescence imaging capabilities (Scheme 30a). In 2023, similar cyclic ruthenium–peptide conjugates were reported by Sun, Bonnet and coworkers194 (Scheme 30b). By direct coordination coupling of the chiral ruthenium anticancer warhead Λ/Δ-[Ru(Ph2phen)2(OH2)2)2+ to histidine and methionine residues containing the RGD peptide sequence, Ac-MRGDH-NH2, two dimeric isomers of the cyclic ruthenium–peptide complexes, Λ-[1]Cl2 and Δ-[1]Cl2 were created, respectively, which could function as potential brain tumor-targeted phototherapeutic prodrugs. In 2023, Arndt and coworkers took advantage of the surprising affinity of methanobactin OB3b (Mbn-OB3b) for copper ions (Ka ≈ Cu(I) 1034) to generate a conjugated heterocyclic system through a cyclic dehydration-sulfurization procedure.195 As a templated total synthesis example, Cu(I)-bound methanobactin OB3b with a caged structure was obtained, which is another important application of metal ion coordination in peptide chemistry.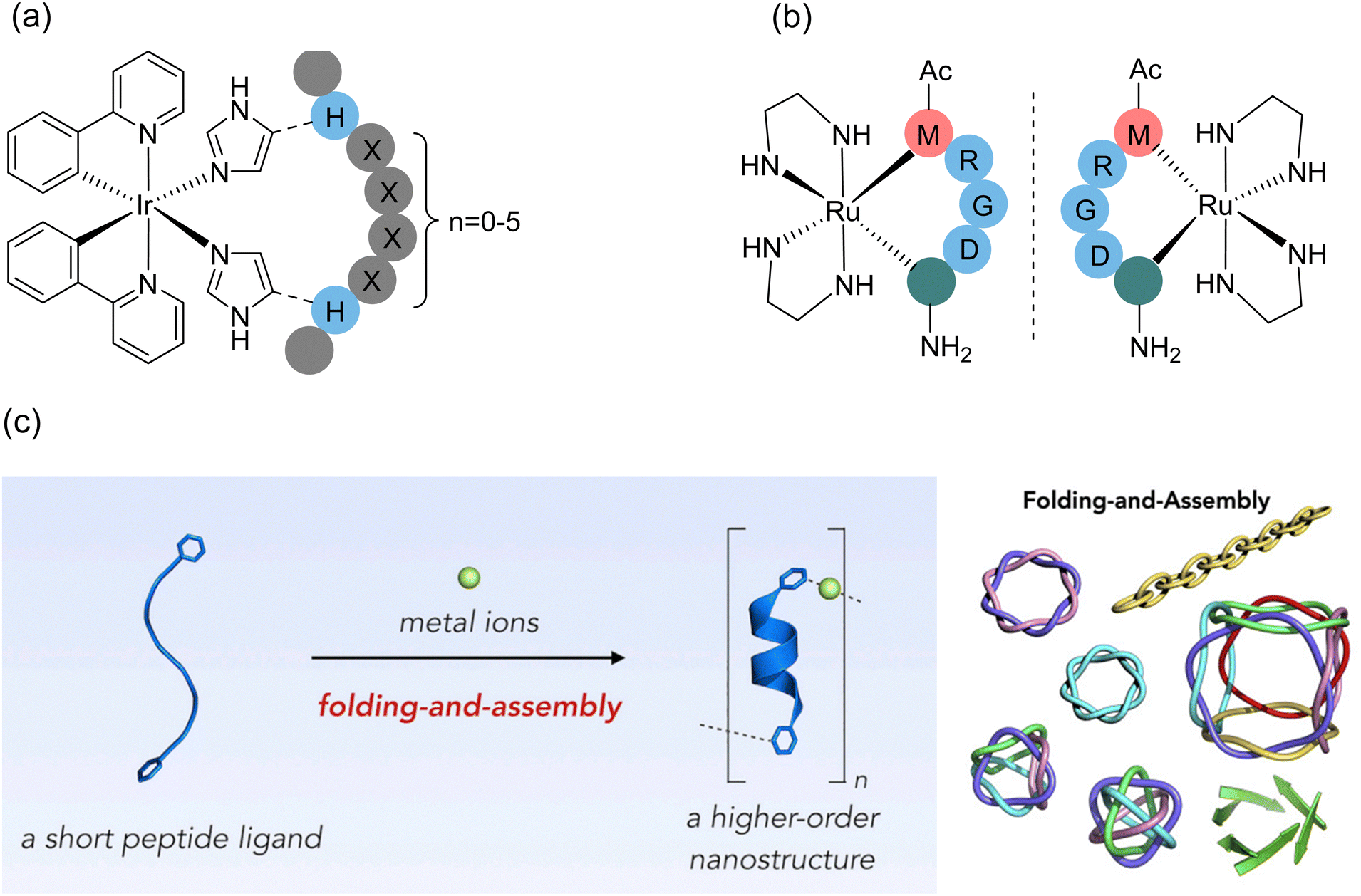 | ||
| Scheme 30 Selected representative cyclic metal–peptide complexes and higher-order self-assembled cyclic metal–peptide nanostructures. (a) & (b) Iridium and ruthenium-chelated stapled peptides; (c) metal ions-directed folding and assembly of short peptide ligands. Illustration of (c) is adapted with permission from ref. 200. | ||
Since short helical peptides were shown to be outstanding building blocks for diverse supramolecular nanostructures, in 2014, Sawada, Fujita, and coworkers proposed a new artificial nano-assembly method for silver(I) and short peptide helices, Gly-Pro-Pro, to obtain a nanosized channel,199 upon the introduction of two terminal pyridines for Ag(I) binding. An ethanolic solution of this modified peptide linker was slowly diffused into an aqueous solution of AgBF4 (56 mm) at 108 °C for one week. The transient folding conformation during the metal-driven assembly process stabilized the peptide-folded porous crystal. Generally, this is the first folding and self-assembly (F&A) strategy for the construction of well-defined peptide-based nanostructures. Later, these authors employed different sequences of short peptides with pyridine–silver ion binding, simultaneously carrying out orthogonal folding and self-assembly processes to obtain a variety of highly entangled metal–peptide frameworks, as well as catenanes200 (Scheme 30c).
6. Technical aspects of peptide macrocyclization strategies
6.1. Comparisons of peptide macrocyclization strategies
Over the past two decades, extensive painstaking efforts have been devoted to exploring the synthesis and applications of macrocyclic peptide compounds, and a variety of strategies has been developed for the preparation of peptidic macrocycles. Ideally, their synthetic protocols should be simple and efficient, highly specific, and probably “traceless” and fully compatible for any amino acid sequence. However, due to the intrinsic differences in the properties of each amino acid, being natural or non-canonical, and the limitations of the current level of science and technology, achieving the ideal macrocyclization reaction for peptide and peptidomimetics is difficult and there is still a long way to go. Despite the dramatic recent advancements, many reported macrocyclization methods still suffer from side-reactions (e.g., isomerization, oligomerization, and epimerization), limited substrate scope, and low yields. In addition, there are many synthetic difficulties, high costs, and environmental issues, i.e., non-sustainability.201 Here, we summarize the different strategies for peptide macrocyclization (Table 1), each of which has significant advantages and obvious limitations. The key to choosing the best macrocyclization strategy is based on the targeted components and applications of the cyclic peptide products.| Chemoselectivity | Regioselectivity | Advantages | Limitations | |
|---|---|---|---|---|
| Non-catalytic chemical strategies | Based on the intrinsic/special reactivity of amino acid sidechains | Based on selective deprotection; the distance between reaction sites | The reaction conditions are mild; natural peptides can be modified | Only limited natural amino acids can serve as reaction sites; use of unnatural amino acids |
| Catalytic chemical strategies | By selectively activating the organic fragments involved in the reaction by the catalyst in use | By controlling the number of amino acids and structures that can participate in the reaction | High cyclization efficiency; good versatility; no excessive requirements in amino acid sequence and composition | Metal ion leaching from the catalyst is costly/difficult to remove; high-loading metal catalysts are expensive |
| Biological strategies | Based on enzymatic recognition of amino acid sequence; expression of genetic factors | Good selectivity and biocompatibility | The design and acquisition of artificial enzymes are difficult and costly | |
| Polycyclic strategies | Based on orthogonality of various methods; special reactivity of amino acid side chain | By orthogonal protection; limiting the number of reaction sites; pre-functionalizing reaction handles | The molecular configuration & conformation can be more restricted & tailored | The threshold is higher and limited methods available |
| Supramolecular strategies | Based on mechanical bonds; metal coordination; intermolecular noncovalent interactions | Moving from the chaos to order increases the application potential of peptides, which can be controlled by fine-tuning the structures | At present, it is still necessary to complete the key macrocyclization steps by introducing a large number of non-peptide structures | |
6.2. Comparisons and remarks for macrocyclic peptide synthesis methods
To date, peptide synthesis technology can be categorized as (i) natural extraction, (ii) chemical synthesis, and (iii) biosynthesis. Chemical synthesis can be further divided into liquid-phase and solid-phase synthesis, where the main difference is whether a solid resin is used. Natural extraction can be regarded as the early form of biosynthesis. In recent years, with the development of biotechnology and genetic engineering, artificially controlled biosynthetic methods have been developed. Biological enzyme catalysis and gene recombination techniques have also been used to synthesize both linear and cyclic peptide products.Although the synthesis and modification of long-chain peptides have been gradually replaced by the solid-phase method, many peptide macrocyclization reactions are still carried out in the liquid phase. Given that the ends of the chain-like precursor peptide in the liquid phase are unconstrained and maintain a high degree of conformational freedom, it has certain advantages in backbone cyclization and sidechain stapling with external components. Regarding the selection of solvents, ranging from their compatibility with physiological conditions to the aspects of cost and environmental protection, water is the best priority choice. For instance, NCL11 and the D–A64 strategies for peptide macrocyclization had been conducted in aqueous media. However, many hydrophobic amino acids (isoleucine, leucine, valine, etc.) or polypeptides with a large molecular weight will suffer from poor solubility or even insolubility in water. Therefore, it is necessary to use mixed or organic solvents to enhance the solubility of the precursor peptides for reacting with other reaction components. In addition, it is necessary to consider whether the polarity of the solvent matches, which will lead to side-reactions and the practical feasibility of the operation. The common solvents for LPPS include DMF, DMSO, water, and mixed THF/MeCN/MeOH solvent system.
In short, the liquid-phase synthesis of cyclic peptides usually requires mild conditions to avoid damaging their sensitive functional groups. Besides, the macrocyclization reaction in the liquid phase usually needs to be carried out at a low concentration (otherwise called high dilution) to reduce the occurrence of oligomerization, isomerization, and many other side-reactions, which will indirectly lead to a reduction in the reaction efficiency, the difficulty of purification, and solvent waste.
The problems that should be given attention when using the solid-phase synthesis method are as follows: (i) the selection of a protective base, (ii) the type of resin, and (iii) the condition of peptide cleavage. In the selection of protecting groups, the Fmoc protective group is preferred in conventional synthesis. Fmoc is relatively easy to remove under alkaline conditions and is relatively stable under acidic conditions. It also has a certain orthogonality with most other amino acid-protecting groups under acidic conditions. Hence, the use of Fmoc protection is more flexible in the choice of synthetic conditions. Another common protecting group, Boc, typically requires deprotection under conditions of strong acids such as TFA and is not suitable for scenarios where reuse is required. It is usually used as a supplement to FMOC protection. Most of the other amino acids also have their own featured protecting groups. In fact, the choice of resin is more diverse, not only the resin materials are different, but also the linkers used to attach amino acids to the resin beads are different. In recent years, polystyrene core resin linkers have been most widely used. As shown in Fig. 3, resins with different linkers have specific cleavage conditions, and different amino acid residues are obtained from the synthetic peptide after separation.202
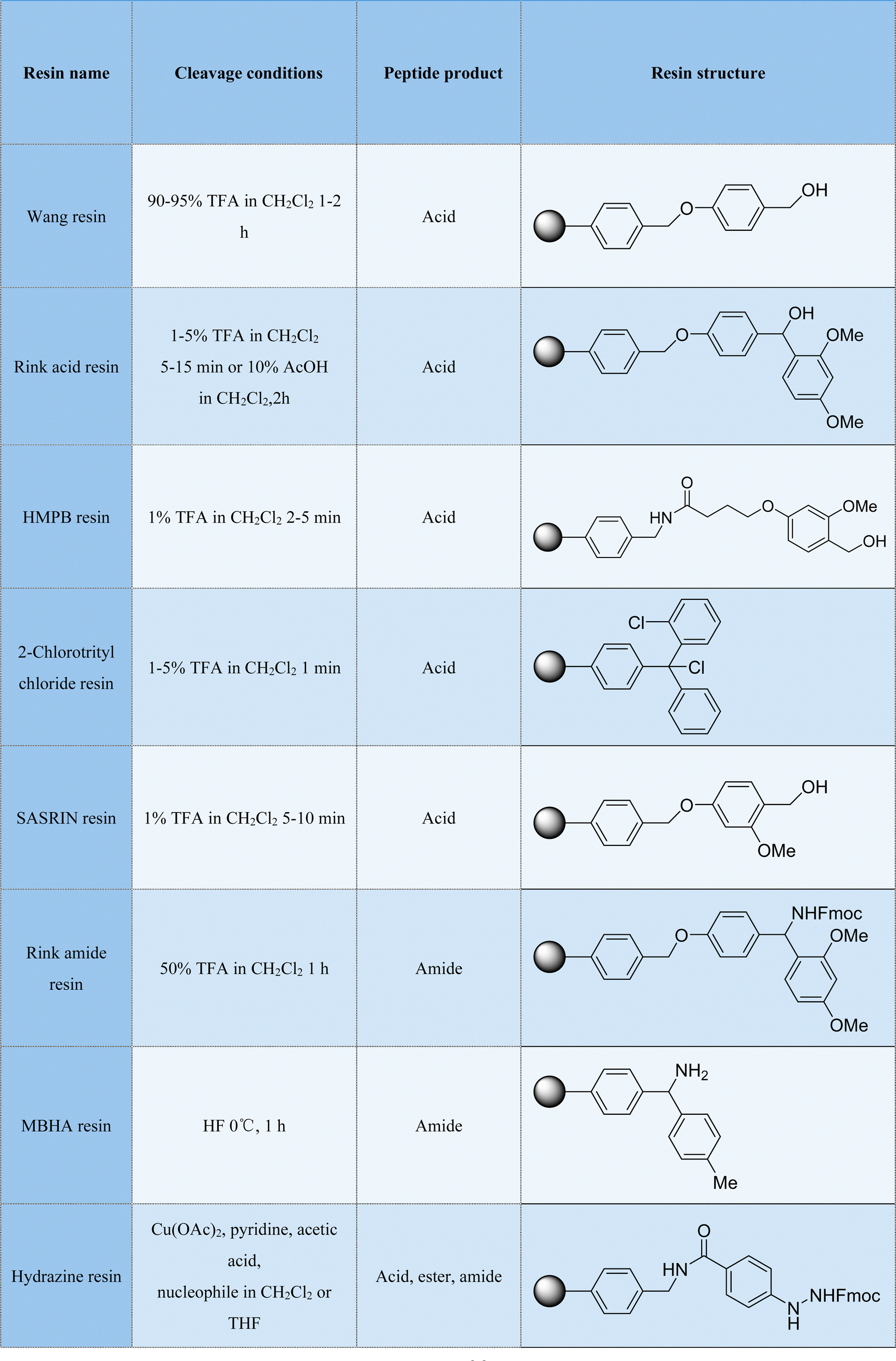 | ||
| Fig. 3 Summary of representative resin structures with their corresponding cleavage conditions and cleavage products. | ||
Based on the pseudo-dilution effect in solid-phase synthesis, the solid-phase synthesis method is very beneficial for intramolecular cyclization. There have been many examples of cyclization on resins, such as macrocyclization of thiol–ene reactions,42 click reactions,203 and ring-closing olefin metathesis.110 In addition, macrocyclization between molecules can also be achieved by adding excess external components such as in glacier coupling,204 Ugi reaction,70 and other cross-coupling strategies.205 Compared with the liquid-phase synthesis method, the solid-phase synthesis method largely avoids the problem of oligomerization of peptides due to the combination of the precursor peptides and resin. Moreover, the solid-phase synthesis method is easy to apply (i.e., high yield and high purity), and has developed into an important tool for the chemical synthesis of peptides. Unfortunately, solid-phase synthesis is generally less economical and requires further improvements in efficiency and sustainability for industrial mass production.
6.3. Characterization and computational analysis
For the development of macrocyclic peptides, monitoring and characterization are the key to their successful synthesis. Therefore, the processes of elongation, modification, and cyclization of polypeptide chains are examined by a variety of methods. Peptide synthesis is usually achieved by solid-phase synthesis. The detection of reaction completeness of peptide chain extension can be performed by detecting the primary and secondary amines by the Kaiser test (Ninhydrin test) and chloranil test (acetaldehyde test), respectively. This detection of incomplete precursor peptides requires separation from the resin by reversed-phase high-performance liquid chromatography (HPLC) to calculate the yield, and mass spectrometry (ESI) to determine the molecular weight to assist qualitative analysis. In the case of shorter peptide chains, proton and carbon nuclear magnetic resonance spectroscopy (1H and 13C-NMR) can also be used to detect the type and number of hydrogen atoms and carbon atoms in different chemical environments for qualitative analysis, respectively. Generally, completion of the cyclization step is accompanied by a change in molecular weight, and the detection of this process can also be preliminarily judged using a combination of HPLC separation and mass spectrometry detection. Regarding some macrocyclization methods, when the cyclic peptide products are not suitable for mass spectrometry analysis or have no molecular weight difference from the precursor peptide (such as azide–alkyne cycloaddition), Fourier transform infrared spectroscopy (FTIR) can also be used to detect the appearance or disappearance of the characteristic peaks of different functional groups.In the case of more complex structures of cyclic peptides, X-ray crystallography can provide more information on their molecular structure including atomic arrangement and spatial structure. However, there are many challenges associated with the X-ray crystallography of cyclic peptides, especially the challenging crystallization of cyclic peptides with larger molecular weights and conformational flexibility. Simultaneously, more difficulties will be encountered in the resolution of their structure because of the complexity of their molecular structure. More often, researchers choose to use other means of representation. For example, the secondary structure of cyclic peptide products (α-helix, β-fold, random coil, etc.) can usually be analyzed by circular dichroism (CD) spectroscopy. CD spectroscopy has high sensitivity in the detection of molecular chiral structure and structural changes. Two-dimensional (2D) NMR can also be widely applied in the analysis of cyclic peptide structures, including NOESY spectrum (detecting the spatial relationship between different residues in the ring); COSY spectrum (the interaction between adjacent amino acid residues); TOCSY spectrum (establishing the interaction network between amino acid residues in the whole molecule to resolve the overall structure); and HSQC spectrum (directly detects the interaction between hydrogen atoms and neighboring nuclei, assisting in determining the carbon spectrum information). The characterization of the tertiary structure of cyclic peptides, including hydrogen bonds, ionic bonds, and hydrophobic interactions, requires a combination of X-ray crystallography, high-resolution mass spectrometry, high-throughput sequencing, and advanced structural biology techniques.
Various peptide-based frameworks/polymers formed by self-assembly of short peptides with metals and cyclic peptide-based nanostructures held through intermolecular forces usually possess three-dimensional regularly arranged crystal structures. Therefore, for the characterization and analysis of these compounds, both solution/solid-state nuclear magnetic resonance spectroscopy can be used to detect their structural information and X-ray diffraction can be used to obtain their detailed crystal structure information. Simultaneously, the morphology and microstructure of the materials can be observed and characterized by scanning electron microscopy (SEM). In the case of more complex peptidic macrocycles, different levels of detection methods are needed to complement each other and comprehensively analyze their compositions and structures.
The three-dimensional conformation of cyclic peptides is closely related to their molecular properties. However, the network of interacting structural elements in macrocyclic peptides generates conformational organization that is challenging to predict and characterize. As the ring size increases, the conformational-activity relationship of the ring molecule becomes complicated. Indeed, cyclic peptide molecules still exhibit some flexibility, with multiple conformations prevalent in solution. These metastable conformations are usually difficult to observe and isolate directly by existing means, which was named by Yudin and coworkers “dark conformational space”.206 Although the presence of the chameleon effect is thought to be one of the reasons for the improved membrane permeability, little is known about the transition between these conformations in reality. Therefore, it is necessary to improve our structural understanding of cyclic peptides to determine or predict their molecular structure and properties. In recent years, conformational studies of a variety of cyclic peptides have been reported. Lokey and coworkers developed a method to elucidate the conformation of cyclic peptides in solution using NMR in combination with density functional theory (DFT) and molecular dynamics (MD) calculations.207 The nearly exhaustive backbone sampling, sequence design, and energy landscape calculation by Hosseinzadeh, Bhardwaj, Mulligan, and coworkers enumerated stable structures that could be adopted by macrocyclic peptides composed of L-and D-amino acids. Over 200 possible structures were computationally designed and predicted to fold into a single stable structure.208 Lin and coworkers, using MD simulation results, trained machine learning models to predict the structure of cyclic peptides.209 In 2023, Ovchinnikov, Bhardwaj and coworkers reported the use of AlphaFold and RoseTTAFold for the structure prediction and design of cyclic peptides.210 Deep learning techniques based on huge databases provide considerable opportunities for the structure prediction and design of biomolecules such as cyclic peptides. Establishing a huge database to analyze and predict the structure of cyclic peptide molecules, as well as probably various peptide-based metal frameworks/polymers/supramolecules/nanomaterials through deep learning, holds tremendous promise in the coming future and is a milestone in this field. However, at present, the comprehensive simulation of cyclic peptides is still in the initial stage of development, and the establishment of each data library is only available for the algorithm prediction of a single specific structure or type of cyclic peptide.211,212 Thus, there is still a large number of tasks to be completed to realize huge and comprehensive knowledge networks.
7. Summary and outlook
The structural diversity, stability, and modularity of cyclic peptides (Fig. 4) make them excellent compounds for generating self-assembling materials with exceptional properties for use as therapeutic drugs. In the last decade, bioactive and functional cyclic peptides have been continuously reported, and peptide macrocyclization strategies, other than lactamization and NCL, have been greatly enriched. However, most of them are limited by the presence of certain natural/unnatural amino acids with specific functional groups and specific sequences. The ultimate goal of precise macrocyclization with arbitrary amino acids or arbitrary amino acid sequences is still a long way off. In essence, there are very few literature-reported methods that are expected to achieve potential translational, industrial-scale production, given that most either incur significant economic costs or are environmentally unfriendly. Therefore, more green, efficient, sustainable, and controllable macrocyclization strategies need to be further designed and optimized.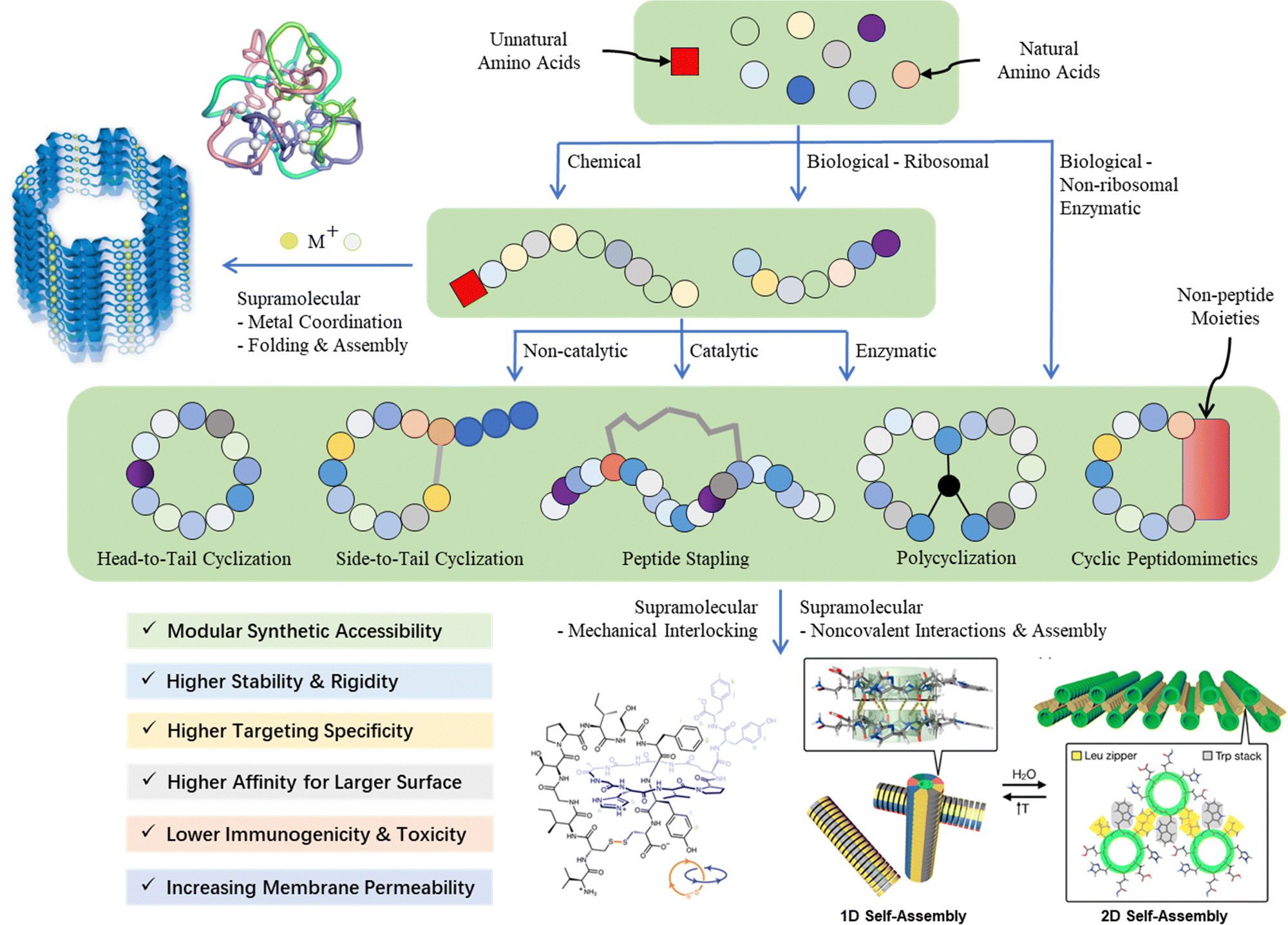 | ||
| Fig. 4 Overview of synthetic strategies for different categories of peptidic macrocycles and their salient advantages compared with those of their simple linear counterparts. Illustrations of selected examples are adapted with permission from ref. 197, 199 and 213. | ||
Alternatively, how can the “right structure” be obtained and stabilized at the theoretical level? This is the most intuitive problem faced by the de novo design of cyclic peptides. In fact, secondary structures such as hydrophobic cores or helices and β-sheets other than the β-hairpin are difficult to obtain for small-sized cyclic peptides, and cyclic peptides with larger molecular weights have a variety of conformations in solution. Designing peptide macrocycles with rigidity, high tension, and complex topology remains an unsolved but pressing fundamental challenge. At present, we have an insufficient understanding of the correlations at the theoretical level such as “the correlation of chemical structure-spatial conformation-macroscopic properties”, and less structural information can be obtained, which makes it difficult to accurately predict the structure of macrocyclic peptides. Briefly, the current limited development of cyclic peptides is due to the inadequacy of macrocyclization methods and insufficient diversity of cyclic peptides.
Future exploration of non-ribosomal peptide synthetases (NRPS), a class of multifunctional enzymes, will be definitely highly promising. These enzymes have modular domains containing characteristic motifs, which include (i) the adenylation (A)-domain for substrate recognition and activation, (ii) peptidyl carrier protein PCP-domain (also known as thiolation (T) domain) for cellular transport to their respective catalytic locations, (iii) the condensation (C)-domain, which catalyzes the formation of peptide bonds, and (iv) thioesterase (TE)-domain (releasing the growing peptide through hydrolysis, oligomerization or cyclization) (Scheme 31). These enzymes can synthesize nonribosomal peptides by condensing amino acid sequences (transfer of the aminoacyl or peptidyl group), where this mechanism is very common in microorganisms.214 Further evolution or engineering of these enzymes will adamantly give rise to unprecedented and unexpected results.
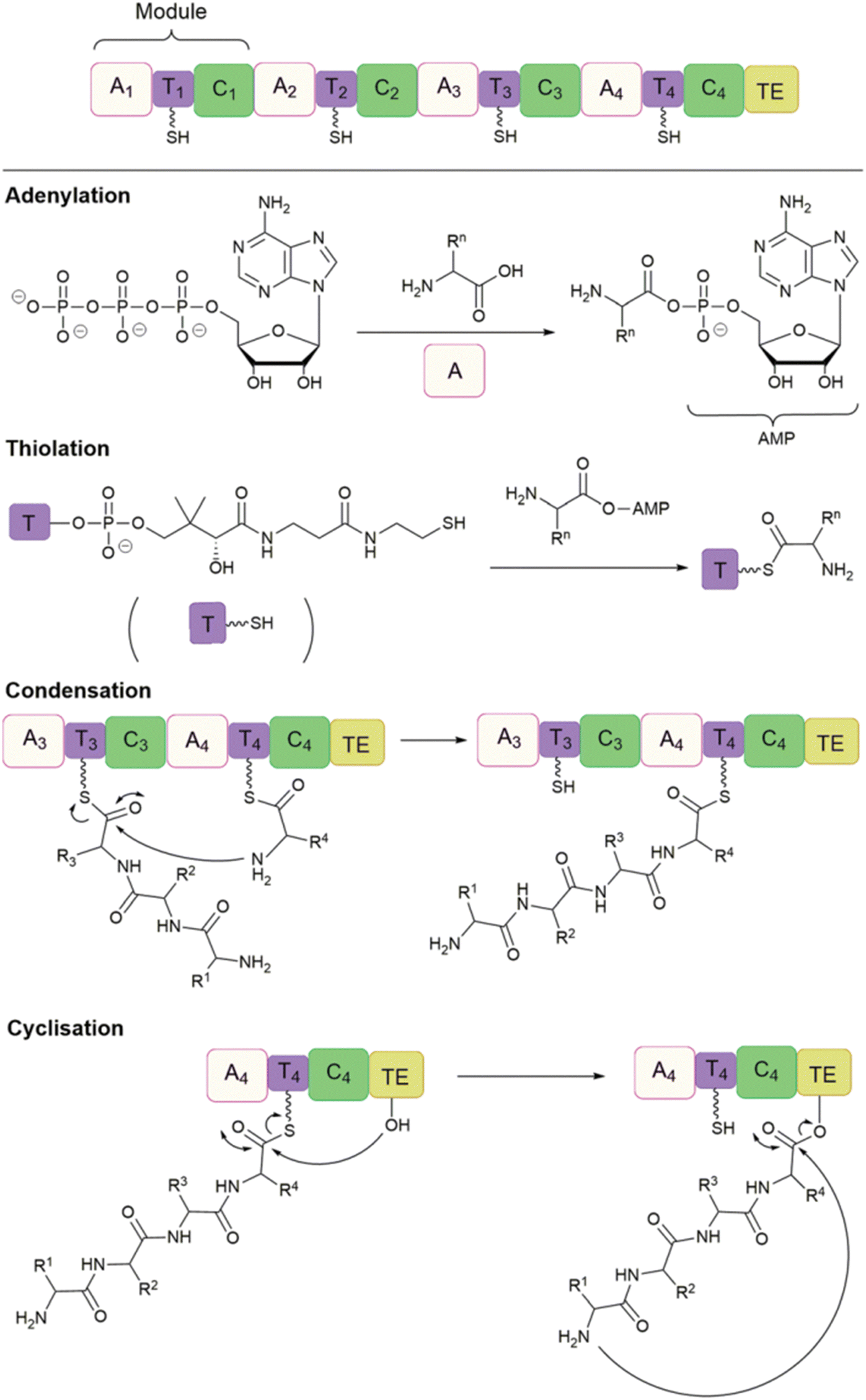 | ||
| Scheme 31 Non-ribosomal peptide synthesis (NRPS) modules composed of adenylation (A), thiolation (T) and condensation (C) domains with a terminal thioesterase (TE) domain. Illustration is adapted with permission from ref. 214. | ||
Regarding another interesting aspect, in 2022, Carreira and coworkers documented the first total syntheses of tricyclic mutanobactins A, B and D by employing a novel thiazepanone amino acid building block.215 In 2024, Perrin and coworkers presented an interesting indole-based peptide stapling tactic by reacting 5-hydroxypyrroloindoline with either a cysteine-thiol or tryptophan-indole to yield the corresponding tryptathionine or 2,2′-bisindole-stapled peptides.216
It is expected that with the progress of basic research on ring-closing metathesis217,218 and traditional strategies on complex macrocyclic suprastructure formation,219–221 more researchers will enter the field of peptide macrocyclization.222 Automated continuous-flow synthesis has recently been reported for growing long-chained peptides, peptide-nucleic acid conjugates, and traditional cyclic peptides; alternatively, cyclic peptide structures start to be predictable and optimizable by deep learning. In the case of the structure and conformation prediction of new macrocyclic peptide scaffolds, more advanced programs/software with excellent and specific chemo/bio-informatics (such as AlphaFold and RoseTTAFold for proteins) need to be invented. It is expected that these technologies will provide theoretical and technical support for ideal drug discovery and the development of “accurate prediction, efficient synthesis, and rapid screening” for cyclic peptides, paired with the development of novel and practical macrocyclization strategies for their synthesis, and hopefully manufacturing. With global concerted research efforts from multidisciplinary experts, as well as the new blood in different areas, this highly dynamic, multidisciplinary and evolving research field of the synthesis of peptidic macrocycles and beyond (i.e., their derivatives/hybrids/conjugates/variants/frameworks/self-assemblies/polymers), which is of paramount importance, will and must be taken to next strata, with new drugs, new probes, new materials, and new tools, to resolve contemporary global scientific and societal challenges and the betterment of humankind.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.Conflicts of interest
There is no competing interest to declare.Acknowledgements
This work is partially supported by Hong Kong Baptist University (RMGS-2022-13-07). P. Fang and W.-L. Chan are grateful for Prof Yixin Lu's academic support from National University of Singapore.References
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- H. Y. Zhang and S. Y. Chen, RSC Chem. Biol., 2022, 3, 18–31 RSC.
- C. Bechtler and C. Lamers, RSC Med. Chem., 2021, 12, 1325–1351 RSC.
- T. A. F. Cardote and A. Ciulli, ChemMedChem, 2016, 11, 787–794 CrossRef CAS PubMed.
- P. G. Dougherty, A. Sahni and D. Pei, Chem. Rev., 2019, 119, 10241–10287 CrossRef CAS PubMed.
- H. Shao, V. Adebomi, A. Bruce, M. Raj and K. N. Houk, Angew. Chem., Int. Ed., 2023, 62, e202307210 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Z. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- L. K. Buckton, M. N. Rahimi and S. R. McAlpine, Chem. – Eur. J., 2021, 27, 1487–1513 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- L. Zhang and J. P. Tam, J. Am. Chem. Soc., 1997, 119, 2363–2370 CrossRef CAS.
- J. P. Tam, Y. Lu and Q. Yu, J. Am. Chem. Soc., 1999, 121, 4316–4324 CrossRef CAS.
- V. Agouridas, O. E. Mahdi, V. Diemer, M. Cargoet, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS.
- J. A. Camarero, G. J. Cotton, A. Adeva and T. W. Muir, J. Pept. Res., 1998, 51, 303–316 CrossRef CAS.
- J. Tulla-Puche and G. Barany, J. Org. Chem., 2004, 69, 4101–4107 CrossRef CAS PubMed.
- B. H. Gless and C. A. Olsen, J. Org. Chem., 2018, 83, 10525–10534 CrossRef CAS PubMed.
- L. R. Malins and R. J. Payne, Aust. J. Chem., 2015, 68, 521–537 CrossRef CAS.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- R. Quaderer and D. Hilvert, Chem. Commun., 2002, 2620–2621 RSC.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- N. Ollivier, T. Toupy, R. C. Hartkoorn, R. Desmet, J. M. Monbaliu and O. Melnyk, Nat. Commun., 2018, 9, 2847 CrossRef PubMed.
- R. Kleineweischede and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2008, 47, 5984–5988 CrossRef PubMed.
- J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef PubMed.
- T. Fukuzumi, L. Ju and J. W. Bode, Org. Biomol. Chem., 2012, 10, 5837–5844 RSC.
- V. R. Pattabiraman, A. O. Ogunkoya and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5114–5118 CrossRef PubMed.
- X. Li, H. Y. Lam, Y. Zhang and C. K. Chan, Org. Lett., 2010, 12, 1724–1727 CrossRef PubMed.
- H. Y. Lam and X. Li, Synlett, 2014, 1339–1344 Search PubMed.
- C. L. Lee, H. Y. Lam and X. Li, Nat. Prod. Rep., 2015, 32, 1274–1279 RSC.
- C. T. T. Wong, H. Y. Lam, T. Song, G. Chen and X. Li, Angew. Chem., Int. Ed., 2013, 52, 10212–10215 CrossRef PubMed.
- C. Xu, J. Xu, H. Liu and X. Li, Chin. Chem. Lett., 2018, 29, 1119–1122 CrossRef.
- I. Bosnjak, V. Bojovic, T. Segvic-Bubic and A. Bielen, Protein Eng., Des. Sel., 2014, 27, 65–72 CrossRef PubMed.
- B. K. W. Chunga and A. K. Yudina, Org. Biomol. Chem., 2015, 13, 8768–8779 RSC.
- Y. Zheng, L. Zhai, Y. Zhao and C. Wu, J. Am. Chem. Soc., 2015, 137, 15094–15097 CrossRef PubMed.
- Y. Zheng, Z. Li, J. Ren, W. Liu, Y. Wu, Y. Zhao and C. Wu, Chem. Sci., 2017, 8, 2547–2552 RSC.
- A. Taguchi, K. Kobayashi, Y. Cui, K. Takayama, A. Taniguchi and Y. Hayashi, J. Org. Chem., 2020, 85, 1495–1503 CrossRef PubMed.
- H. Shida, A. Taguchi, S. Konno, K. Takayama, A. Taniguchi and Y. Hayashi, Chem. Pharm. Bull., 2023, 71, 435–440 CrossRef PubMed.
- B. P. Sutherland, B. M. El-Zaatari, N. I. Halaszynski, J. M. French, S. Bai and C. J. Kloxin, Bioconjugate Chem., 2018, 29, 3987–3992 CrossRef PubMed.
- R. Mousa, S. Lansky, G. Shoham and N. Metanis, Chem. Sci., 2018, 9, 4814–4820 RSC.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- F. M. Brunel and P. E. Dawson, Chem. Commun., 2005, 2552–2554 RSC.
- J. Li, W. Lai, A. Pang, L. Liu, L. Ye and X. Xiong, Org. Lett., 2022, 24, 1673–1677 CrossRef.
- A. A. Aimetti, R. K. Shoemaker, C. C. Lin and K. S. Anseth, Chem. Commun., 2010, 46, 4061–4063 RSC.
- Y. Tian, D. Yang, X. Ye and Z. Li, Chem. Rec., 2017, 17, 874–885 CrossRef.
- Y. Tian, J. Li, H. Zhao, X. Zeng, D. Wang, Q. Liu, X. Niu, X. Huang, N. Xu and Z. Li, Chem. Sci., 2016, 7, 3325–3330 RSC.
- A. J. Cameron, P. W. R. Harris and M. A. Brimble, Angew. Chem., Int. Ed., 2020, 59, 18054–18061 CrossRef.
- L. R. Malins, J. N. Degruyter, K. J. Robbins, P. M. Scola, M. D. Eastgate, M. R. Ghadiri and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 5233–5241 CrossRef.
- V. Adebomi, R. D. Cohen, R. Wills, H. A. H. Chavers, G. E. Martin and M. Raj, Angew. Chem., Int. Ed., 2019, 131, 19249–19256 CrossRef.
- A. Bandyopadhyay and J. Gao, J. Am. Chem. Soc., 2016, 138, 2098–2101 CrossRef PubMed.
- B. Li, J. Parker, J. Tong and T. Kodadek, J. Am. Chem. Soc., 2024, 146, 14633–14644 CrossRef.
- Y. Wang and D. H.-C. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931–10934 CrossRef PubMed.
- D. P. Fairlie and A. D. de Araujo, Pept. Sci., 2016, 106, 843–852 CrossRef PubMed.
- Q. Zuo, Y. Li, X. Lai, G. Bao, L. Chen, Z. He, X. Song, R. E. P. Wang, Y. Shi, H. Luo, W. Sun and R. Wang, Adv. Sci., 2024, 2308491 CrossRef PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef PubMed.
- K. Kubota, P. Dai, B. L. Pentelute and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 3128–3133 CrossRef PubMed.
- X. Lin, O. Harel and M. Jbara, Angew. Chem., Int. Ed., 2024, 63, e202317511 CrossRef PubMed.
- Y. Zhang, Q. Zhang, C. T. T. Wong and X. Li, J. Am. Chem. Soc., 2019, 141, 12274–12279 CrossRef PubMed.
- M. Todorovic, K. D. Schwab, J. Zeisler, C. C. Zhang, F. Benard and D. M. Perrin, Angew. Chem., Int. Ed., 2019, 58, 14120–14124 CrossRef PubMed.
- B. Li, H. Tang, A. Turlik, Z. Wan, X.-S. Xue, L. Li, X. Yang, J. Li, G. He, K. N. Houk and G. Chen, Angew. Chem., Int. Ed., 2021, 60, 6646–6652 CrossRef.
- B. Li, L. Wang, X. Chen, X. Chu, H. Tang, J. Zhang, G. He, L. Li and G. Chen, Nat. Commun., 2022, 13, 311 CrossRef.
- P. Guo, X. Chu, C. Wu, T. Qiao, W. Guan, C. Zhou, T. Wang, C. Tian, G. He and G. Chen, Angew. Chem., Int. Ed., 2024, e202318893 Search PubMed.
- G. Bao, P. Wang, J. Li, X. Song, T. Yu, J. Zhang, Y. Li, Z. He, E. Ruiyao, X. Miao, J. Xie, J. Ni, R. Wang and W. Sun, CCS Chem., 2023, 1547–1556 Search PubMed.
- Y. Zhang, R. Yin, H. Jiang, C. Wang, X. Wang, D. Wang, K. Zhang, R. Yu, X. Li and T. Jiang, Org. Lett., 2023, 25, 2248–2252 CrossRef PubMed.
- J. E. Montgomery, J. A. Donnelly, S. W. Fanning, T. E. Speltz, X. H. Shangguan, J. S. Coukos, G. L. Greene and R. E. Moellering, J. Am. Chem. Soc., 2019, 141, 16374–16381 CrossRef PubMed.
- Q. Luo, Y. Tao, W. Sheng, J. Lu and H. Wang, Nat. Commun., 2019, 10, 142 CrossRef PubMed.
- Y. Wu, H. Chau, W. Thor, K. H. Y. Chan, X. Ma, W. Chan, N. Long and K. Wong, Angew. Chem., Int. Ed., 2021, 133, 20463–20469 CrossRef.
- R. Hili, V. Rai and A. K. Yudin, J. Am. Chem. Soc., 2010, 132, 2889–2891 CrossRef.
- A. V. Vasco, C. S. Pérez, F. E. Morales, H. E. Garay, D. Vasilev, J. A. Gavín, L. A. Wessjohann and D. G. Rivera, J. Org. Chem., 2015, 80, 6697–6707 CrossRef.
- A. Doemling, M. Ricardo, A. Ali, J. Plewka, E. Surmiak, B. Labuzek, C. Neochoritis, J. Atmaj, L. Skalniak, R. Zhang, T. Holak, M. Groves and D. Rivera, Angew. Chem., Int. Ed., 2020, 59, 5235–5241 CrossRef PubMed.
- A. Yamaguchi, S. J. Kaldas, S. D. Appavoo, D. B. Diaz and A. K. Yudin, Chem. Commun., 2019, 55, 10567–10570 RSC.
- S. Krajcovicova and D. R. Spring, Angew. Chem., Int. Ed., 2023, 62, e202307782 CrossRef PubMed.
- J. R. Frost, C. C. G. Scully and A. K. Yudin, Nat. Chem., 2016, 8, 1105–1111 CrossRef PubMed.
- T. Liu, S. Xu and J. Zhao, Chin. J. Org. Chem., 2021, 41, 873–887 CrossRef.
- J. Yang, C. Wang, S. Xu and J. Zhao, Angew. Chem., Int. Ed., 2019, 58, 1382–1386 CrossRef PubMed.
- S. Xu, D. Jiang, Z. Peng, L. Hu, T. Liu, L. Zhao and J. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202212247 CrossRef.
- P. Botti, T. D. Pallin and J. P. Tam, J. Am. Chem. Soc., 1996, 118, 10018–10024 CrossRef.
- H. Chen and Q. Zhang, J. Am. Chem. Soc., 2023, 145, 27218–27224 CrossRef.
- E. Fowler and J. Kim, Chem, 2024, 10, 730–745 Search PubMed.
- O. Nwajiobi, A. K. Verma and M. Raj, J. Am. Chem. Soc., 2022, 144, 4633–4641 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- Y. Angell and K. Burgess, J. Org. Chem., 2005, 70, 9595–9598 CrossRef CAS PubMed.
- M. Strack, É. Billard, D. Chatenet and W. D. Lubell, Bioorg. Med. Chem. Lett., 2017, 27, 3412–3416 CrossRef PubMed.
- K. Holland-Nell and M. Meldal, Angew. Chem., Int. Ed., 2011, 50, 5204–5206 CrossRef PubMed.
- Y. H. Lau, P. de Andrade, S.-T. Quah, M. Rossmann, L. Laraia, N. Skold, T. J. Sum, P. J. E. Rowling, T. L. Joseph, C. Verma, M. Hyvonen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC.
- T. P. Thu, C. O. Larsen, T. Rondbjerg, M. De Foresta, M. B. A. Kunze, A. Marek, J. H. Loper, L.-E. Boyhus, A. Knuhtsen, K. Lindorff-Larsen and D. S. Pedersen, Chem. – Eur. J., 2017, 23, 3490–3495 CrossRef.
- S. Li, H. Cai, J. He, H. Chen, S. Lam, T. Cai, Z. Zhu, S. J. Bark and C. Cai, Bioconjugate Chem., 2016, 27, 2315–2322 CrossRef.
- Y. Wang and Q. Hu, Adv. NanoBiomed Res., 2023, 3, 2200128 CrossRef.
- Y. H. Lau, Y. Wu, M. Rossmann, B. X. Tan, P. de Andrade, Y. S. Tan, C. Verma, G. J. McKenzie, A. R. Venkitaraman, M. Hyvçnen and D. R. Spring, Angew. Chem., Int. Ed., 2015, 54, 15410–15413 CrossRef.
- D. Astruc, New J. Chem., 2005, 29, 42–56 RSC.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef.
- E. C. Gleeson, W. R. Jackson and A. J. Robinson, Tetrahedron Lett., 2016, 57, 4325–4333 CrossRef CAS.
- X. Lu, L. Fan, C. B. Phelps, C. P. Davie and C. P. Donahue, Bioconjugate Chem., 2017, 28, 1625–1629 CrossRef CAS PubMed.
- S. Masuda, S. Tsuda and T. Yoshiya, Org. Biomol. Chem., 2018, 16, 9364–9367 RSC.
- A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef PubMed.
- N. Ghalit, A. J. Poot, A. Fürstner, D. T. S. Rijkers and R. M. J. Liskamp, Org. Lett., 2005, 7, 2961–2964 CrossRef PubMed.
- P. M. Cromm, S. Schaubach, J. Spiegel, A. Fürstner, T. N. Grossmann and H. Waldmann, Nat. Commun., 2016, 7, 11300 CrossRef PubMed.
- B. Chen, C. Liu, W. Cong, F. Gao, Y. Zou, L. Su, L. Liu, A. Hillisch, L. Lehmann, D. Bierer, X. Li and H. Hu, Chem. Sci., 2023, 14, 11499–11506 RSC.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef PubMed.
- S. J. McCarver, J. Qiao, J. Carpenter, R. M. Borzilleri, M. A. Poss, M. D. Eastgate, M. M. Miller and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2017, 56, 728–732 CrossRef PubMed.
- T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260–265 CrossRef PubMed.
- H. Lee, N. C. Boyer, Q. Deng, H. Y. Kim, T. K. Sawyer and N. Sciammentta, Chem. Sci., 2019, 10, 5073–5078 RSC.
- J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330–334 CrossRef PubMed.
- X. Hu, X. Chen, B. Li, G. He and G. Chen, Org. Lett., 2021, 23, 716–721 CrossRef CAS PubMed.
- A. Benny and E. M. Scanlan, Chem. Commun., 2024, 60, 7950–7953 RSC.
- H. Gruß and N. Sewald, Chem. Eur. J., 2020, 26, 5328–5340 CrossRef PubMed.
- W. Wang, M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700–14717 CrossRef PubMed.
- J. Ruiz-Rodríguez, F. Albericio and R. Lavilla, Chem. – Eur. J., 2010, 16, 1124–1127 CrossRef PubMed.
- H. Dong, C. Limberakis, S. Liras, D. Priceb and K. James, Chem. Commun., 2012, 48, 11644–11646 RSC.
- L. Mendive-Tapia, S. Preciado, J. Garcıa, R. Ramon, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed.
- Z. Bai, C. Cai, Z. Yu and H. Wang, Angew. Chem., Int. Ed., 2018, 130, 14108–14112 CrossRef.
- J. Tan, J. Wu, S. Liu, H. Yao and H. Wang, Sci. Adv., 2019, 5, eaaw0323 CrossRef.
- J. Tang, H. Chen, Y. He, W. Sheng, Q. Bai and H. Wang, Nat. Commun., 2018, 9, 3383 CrossRef.
- Z. Bai, C. Cai, W. Sheng, Y. Ren and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 14686–14692 CrossRef.
- S. Liu, Ch Cai, Z. Bai, W. Sheng, J. Tan and H. Wang, Org. Lett., 2021, 23, 2933–2937 CrossRef.
- B. Han, B. Li, L. Qi, P. Yang, G. He and G. Chen, Org. Lett., 2020, 22, 6879–6883 CrossRef.
- W. Gong, G. Zhang, T. Liu, R. Giri and J. Yu, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef PubMed.
- A. F. M. Noisier, J. Garc, I. A. Ionut and F. Albericio, Angew. Chem., Int. Ed., 2016, 128, 314–318 Search PubMed.
- J. Tang, Y. He, H. Chen, W. Sheng and H. Wang, Chem. Sci., 2017, 8, 4565–4570 RSC.
- X. Zhang, G. Lu, M. Sun, M. Mahankali, Y. Ma, M. Zhang, W. Hua, Y. Hu, Q. Wang, J. Chen, G. He, X. Qi, W. Shen, P. Liu and G. Chen, Nat. Chem., 2018, 10, 540–548 CrossRef PubMed.
- Z. Ruan, N. Sauermann, E. Manoni and L. Ackermann, Angew. Chem., Int. Ed., 2017, 129, 3172–3176 CrossRef PubMed.
- N. Kaplaneris, T. Rogge, R. Yin, H. Wang, G. Sirvinskaite and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 3476–3480 CrossRef PubMed.
- N. Kaplaneris, F. Kaltenhauser, G. Sirvinskaite, S. Fan, T. De Oliveira, L. C. Conradi and L. Ackermann, Sci. Adv., 2021, 7, eabe6202 CrossRef PubMed.
- P. Wang, J. Liu, X. Zhu, Kenry, Z. Yan, J. Yan, J. Jiang, M. Fu, J. Ge, Q. Zhu and Y. Zheng, Nat. Commun., 2023, 14, 3973 CrossRef PubMed.
- S. Shabani and C. A. Hutton, Org. Lett., 2020, 22, 4557–4561 CrossRef PubMed.
- K. V. Lawson, T. E. Rose and P. G. Harran, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3753–E3760 Search PubMed.
- Z. Chang, S. Wang, J. Huang, G. Chen, Z. Tang, R. Wang and D. Zhao, Sci. Adv., 2023, 9, eadj3090 CrossRef PubMed.
- P. Yang, X. Wang, B. Li, Y. Yang, J. Yue, Y. Suo, H. Tong, G. He, X. Lu and G. Chen, Chem. Sci., 2021, 12, 5804–5810 RSC.
- K. Fukumoto, M. Yazaki and M. Arisawa, Org. Lett., 2024, 26, 221–224 CrossRef PubMed.
- P. Yang, M. J. Širvinskas, B. Li, N. W. Heller, H. Rong, G. He, A. K. Yudin and G. Chen, J. Am. Chem. Soc., 2023, 145, 13968–13978 CrossRef.
- W. Li, Y. Chen, Y. Chen, S. Xia, W. Chang, Ch Zhu, K. N. Houk, Y. Liang and J. Xie, J. Am. Chem. Soc., 2023, 145, 14865–14873 CrossRef.
- Y. Liu, G. Li, W. Ma, G. Bao, Y. Li, Z. He, Z. Xu, R. Wang and W. Sun, Chem. Sci., 2024, 15, 11099–11107 RSC.
- D. Liu, G. M. Rubin, D. Dhakal, M. Chen and Y. Ding, iScience, 2021, 24, 102512 CrossRef PubMed.
- J. Lu, Y. Wu, Y. Li and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 1951–1958 CrossRef PubMed.
- D. T. Nguyen, T. T. Le, A. J. Rice, G. A. Hudson, W. A. van der Donk and D. A. Mitchell, J. Am. Chem. Soc., 2022, 144, 11263–11269 CrossRef PubMed.
- N. Bionda, A. L. Cryan and R. Fasan, ACS Chem. Biol., 2014, 9, 2008–2013 CrossRef PubMed.
- J. Lu, Y. Li, Z. Bai, H. Lv and H. Wang, Nat. Prod. Rep., 2021, 38, 981–992 RSC.
- Z. L. Budimir, R. S. Patel, A. Eggly, C. N. Evans, H. M. Rondon-Cordero, J. J. Adams, C. Das and E. I. Parkinson, Nat. Chem. Biol., 2024, 20, 120–128 CrossRef PubMed.
- M. L. Adrover-Castellano, J. J. Schmidt and D. H. Sherman, ChemCatChem, 2021, 13, 2095–2116 CrossRef PubMed.
- M. Kobayashi, K. Fujita, K. Matsuda and T. Wakimoto, J. Am. Chem. Soc., 2023, 145, 3270–3275 CrossRef PubMed.
- T. Kawakami, A. Ohta, M. Ohuchi, H. Ashigai, H. Murakami and H. Suga, Nat. Chem. Biol., 2009, 5, 888–890 CrossRef PubMed.
- T. Kawakami and S. Aimoto, Chem. Lett., 2007, 36, 76–77 CrossRef.
- M. Spinck, C. Piedrafita, W. E. Robertson, T. S. Elliott, D. Cervettini, D. de la Torre and J. W. Chin, Nat. Chem., 2023, 15, 61–69 CrossRef PubMed.
- V. Baeriswyl and C. Heinis, ChemMedChem, 2013, 8, 377–384 CrossRef PubMed.
- D. Feng, L. Liu, Y. Shi, P. Du, S. Xu, Z. Zhu, J. Xu and H. Yao, Chin. Chem. Lett., 2023, 34, 108026 CrossRef.
- A. M. Jaulent and R. J. Leatherbarrow, Protein Eng., Des. Sel., 2004, 17, 681–687 CrossRef PubMed.
- J. Luo, Y. Gao, R. Zhao, J. Shi and Y. Li, Org. Biomol. Chem., 2023, 21, 8863–8867 RSC.
- S. Dengler, C. Douat and I. Huc, Angew. Chem., Int. Ed., 2022, 61, e202211138 CrossRef PubMed.
- B. Li, Z. Wan, H. Zheng, S. Cai, H. Tian, H. Tang, X. Chu, G. He, D. Guo, X. Xue and G. Chen, J. Am. Chem. Soc., 2022, 144, 10080–10090 CrossRef PubMed.
- A. V. Vasco, Y. Méndez, C. González, C. S. Pérez, L. Reguera, L. A. Wessjohann and D. G. Rivera, ChemBioChem, 2023, 24, e202300229 CrossRef PubMed.
- D. S. Kemp and P. E. McNamara, J. Org. Chem., 1985, 50, 5834–5838 CrossRef.
- P. Timmerman, J. Beld, W. C. Puijk and R. H. Meloen, ChemBioChem, 2005, 6, 821–824 CrossRef PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef PubMed.
- S. Chen, J. Morales-Sanfrutos, A. Angelini, B. Cutting and C. Heinis, ChemBioChem, 2012, 13, 1032–1038 CrossRef PubMed.
- K. Deyle, X. Kong and C. Heinis, Acc. Chem. Res., 2017, 50, 1866–1874 CrossRef PubMed.
- C. Ernst, J. Heidrich, C. Sessler, J. Sindlinger, D. Schwarzer, P. Koch and F. M. Boeckler, Front. Chem., 2018, 6, 484 CrossRef PubMed.
- Q. Yu, L. Bai and X. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202314379 CrossRef PubMed.
- C. Zhang, E. V. Vinogradova, A. M. Spokoyny, S. L. Buchwald and B. L. Pentelute, Angew. Chem., Int. Ed., 2019, 58, 4810–4839 CrossRef PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef PubMed.
- W. Liu, Y. Zheng, X. Kong, C. Heinis, Y. Zhao and C. Wu, Angew. Chem., Int. Ed., 2017, 56, 4458–4463 CrossRef PubMed.
- J. Wang, M. Zha, Q. Fei, W. Liu, Y. Zhao and C. Wu, Chem. – Eur. J., 2017, 23, 15150–15155 CrossRef PubMed.
- J. M. Wolfe, C. M. Fadzen, R. L. Holden, M. Yao, G. J. Hanson and B. L. Pentelute, Angew. Chem., Int. Ed., 2018, 57, 4756–4759 CrossRef PubMed.
- P. Dognini, T. Chaudhry, G. Scagnetti, M. Assante, G. S. M. Hanson, K. Ross, F. Giuntini and C. R. Coxon, Chem. – Eur. J., 2023, 29, e202301410 CrossRef PubMed.
- M. Wang, D. Pan, Q. Zhang, Y. Lei, C. Wang, H. Jia, L. Mou, X. Miao, X. Ren and Z. Xu, J. Am. Chem. Soc., 2024, 146, 6675–6685 CrossRef PubMed.
- X. Chu, L. Shen, B. Li, P. Yang, C. Du, X. Wang, G. He, S. Messaoudi and G. Chen, Org. Lett., 2021, 23, 8001–8006 CrossRef PubMed.
- J. M. Stauber, A. L. Rheingold and A. M. Spokoyny, Inorg. Chem., 2021, 60, 5054–5062 CrossRef PubMed.
- G. E. Mudd, S. J. Stanway, D. R. Witty, A. Thomas, S. Baldo, A. D. Bond, P. Beswick and A. Highton, Bioconjugate Chem., 2022, 33, 1441–1445 CrossRef PubMed.
- M. Matzapetakis, D. Ghosh, T. Weng, J. E. Penner-Hahn and V. L. Pecoraro, J. Biol. Inorg. Chem., 2006, 11, 876–890 CrossRef PubMed.
- C. Nitsche, M. C. Mahawaththa, W. Becker, T. Huber and G. Otting, Chem. Commun., 2017, 53, 10894–10897 RSC.
- S. Voss, J. Rademann and C. Nitsche, Angew. Chem., Int. Ed., 2022, 61, e202113857 CrossRef PubMed.
- S. Voss, L. Adair, K. Achazi, H. Kim, S. Bergemann, R. Bartenschlager, E. New, J. Rademann and C. Nitsche, Angew. Chem., Int. Ed., 2023, 63, e202318615 CrossRef PubMed.
- M. R. Jafari, H. Yu, J. M. Wickware, Y. Lin and R. Derda, Org. Biomol. Chem., 2018, 16, 7588–7594 RSC.
- G. Richelle, M. Schmidt, H. Ippel, T. Hackeng, J. van Maarseveen, T. Nuijens and P. Timmerman, ChemBioChem, 2018, 19, 1934–1938 CrossRef PubMed.
- D. E. Streefkerk, M. Schmidt, J. H. Ippel, T. M. Hackeng, T. Nuijens, P. Timmerman and J. H. van Maarseveen, Org. Lett., 2019, 21, 2095–2100 CrossRef PubMed.
- F. Chen, N. Pinnette, F. Yang and J. Gao, Angew. Chem., Int. Ed., 2023, 62, e202306813 CrossRef PubMed.
- R. M. Reja, B. Chau and J. Gao, Org. Lett., 2023, 25, 4489–4492 CrossRef CAS PubMed.
- S. Ullrich, J. George, A. E. Coram, R. Morewood and C. Nitsche, Angew. Chem., Int. Ed., 2022, 61, e202208400 CrossRef CAS PubMed.
- X. Liu, O. Mykhailenko, A. Faraone and J. Waser, Angew. Chem., Int. Ed., 2024, e202404747 CAS.
- X. Wang, X. Bao, M. McFarland-Mancini, I. Isaacsohn, A. F. Drew and D. B. Smithrud, J. Am. Chem. Soc., 2007, 129, 7284–7293 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef PubMed.
- A. W. Heard, J. M. Suárez and S. M. Goldup, Nat. Rev. Chem., 2022, 6, 182–196 CrossRef PubMed.
- N. Pairault, R. Barat, I. Tranoy-Opalinski, B. Renoux, M. Thomas and S. Papot, C. R. Chimie, 2016, 19, 103–112 CrossRef.
- M. O. Maksimov, S. Pan and A. J. Link, Nat. Prod. Rep., 2012, 29, 996–1006 RSC.
- N. C. H. Lim and S. E. Jackson, J. Phys.: Condens. Matter, 2015, 27, 354101 CrossRef PubMed.
- W. Clegg, C. Gimenez-Saiz, D. A. Leigh, A. Murphy, A. M. Z. Slawin and S. J. Teat, J. Am. Chem. Soc., 1999, 121, 4124–4129 CrossRef.
- M. Asakawa, G. Brancato, M. Fanti, D. A. Leigh, T. Shimizu, A. M. Z. Slawin, J. K. Y. Wong, F. Zerbetto and S. Zhang, J. Am. Chem. Soc., 2002, 9, 2939–2950 CrossRef PubMed.
- H. Onagi and J. Rebek. Jr, Chem. Commun., 2005, 4604–4606 RSC.
- A. Moretto, I. Menegazzo, M. Crisma, E. J. Shotton, H. Nowell, S. Mammi and C. Toniolo, Angew. Chem., Int. Ed., 2009, 48, 8986–8989 CrossRef PubMed.
- A. Fernandes, A. Viterisi, F. Coutrot, S. Potok, D. A. Leigh, V. Aucagne and S. Papot, Angew. Chem., Int. Ed., 2009, 121, 6565–6569 CrossRef.
- V. Aucagne, D. A. Leigh, J. S. Lock and A. R. Thomson, J. Am. Chem. Soc., 2006, 128, 1784–1785 CrossRef CAS PubMed.
- J. W. Blankenship and P. E. Dawson, Protein Sci., 2007, 16, 1249–1256 CrossRef CAS PubMed.
- S. Pilon, S. I. Jørgensen and J. H. van Maarseveen, ACS Org. Inorg. Au, 2021, 1, 37–42 CrossRef CAS PubMed.
- T. Kurita, M. Higashi, J. Gimenez-Dejoz, S. Fujita, H. Uji, H. Sato and K. Numata, Biomacromolecules, 2024, 25, 3661–3670 CrossRef CAS PubMed.
- T. Liu, X. Ma, J. Yu, W. Yang, G. Wang, Z. Wang, Y. Ge, J. Song, H. Han, W. Zhang, D. Yang, X. Liu and M. Ma, Chem. Sci., 2021, 12, 12353–12364 RSC.
- L. Zhang, P. Wang, X. Zhou, L. Bretin, X. Zeng, Y. Husiev, E. A. Polanco, G. Zhao, L. S. Wijaya, T. Biver, S. E. Le Dévédec, W. Sun and S. Bonnet, J. Am. Chem. Soc., 2023, 145, 14963–14980 CrossRef CAS PubMed.
- E. Cupioli, F. J. M. Gaigne, A. Sachse, P. Buday, W. Weigand, P. Liebing and H. D. Arndt, Angew. Chem., Int. Ed., 2023, 62, e202304901 CrossRef CAS PubMed.
- C. D. Allen and A. J. Link, J. Am. Chem. Soc., 2016, 138, 14214–14217 CrossRef CAS PubMed.
- H. V. Schröder, Y. Zhang and A. J. Link, Nat. Chem., 2021, 13, 850–857 CrossRef PubMed.
- X. Ma, J. Jia, R. Cao, X. Wang and H. Fei, J. Am. Chem. Soc., 2014, 136, 17734–17737 CrossRef CAS PubMed.
- T. Sawada, A. Matsumoto and M. Fujita, Chem, 2014, 126, 7356–7360 Search PubMed.
- T. Sawada and M. Fujita, Chem, 2020, 6, 1861–1876 CAS.
- L. Ferrazzano, M. Catani, A. Cavazzini, G. Martelli, D. Corbisiero, P. Cantelmi, T. Fantoni, A. Mattellone, C. De Luca, S. Felletti, W. Cabri and A. Tolomelli, Green Chem., 2022, 24, 975–1020 RSC.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 RSC.
- S. Ingale and P. E. Dawson, Org. Lett., 2011, 13, 2822–2825 CrossRef PubMed.
- P. A. Cistrone, A. P. Silvestri, J. C. J. Hintzen and P. E. Dawson, ChemBioChem, 2018, 19, 1031–1035 CrossRef PubMed.
- H. Asfaw, T. Wetzlar, M. S. Martinez-Martinez and P. Imming, Bioorg. Med. Chem. Lett., 2018, 28, 2899–2905 CrossRef PubMed.
- D. B. Diaz, S. D. Appavoo, A. F. Bogdanchikova, Y. Lebedev, T. J. McTiernan, G. D. P. Gomes and A. K. Yudin, Nat. Chem., 2021, 13, 218–225 CrossRef PubMed.
- Q. Nhu, N. Nguyen, J. Schwochert, D. J. Tantillo and R. S. Lokey, Phys. Chem. Chem. Phys., 2018, 20, 14003–14012 RSC.
- P. Hosseinzadeh, G. Bhardwaj, V. K. Mulligan, M. D. Shortridge, T. W. Craven, F. Pardo-Avila, S. A. Rettie, D. E. Kim, D.-A. Silva, Y. M. Ibrahim, I. K. Webb, J. R. Cort, J. N. Adkins, G. Varani and D. Baker, Science, 2017, 358, 1461–1466 CrossRef PubMed.
- J. Miao, M. L. Descoteaux and Y. Lin, Chem. Sci., 2021, 12, 14927–14936 RSC.
- S. A. Rettie, K. V. Campbell, A. K. Bera, A. Kang, S. Kozlov, J. De La Cruz, V. Adebomi, G. Zhou, F. DiMaio, S. Ovchinnikov and G. Bhardwaj, bioRxiv, 2023, 529956 Search PubMed.
- R. Thakkar, D. Upreti, S. Ishiguro, M. Tamura and J. Comer, RSC Med. Chem., 2023, 14, 658–670 RSC.
- A. Ohta, M. Tanada, S. Shinohara, Y. Morita, K. Nakano, Y. Yamagishi, R. Takano, S. Kariyuki, T. Iida, A. Matsuo, K. Ozeki, T. Emura, Y. Sakurai, K. Takano, A. Higashida, M. Kojima, T. Muraoka, R. Takeyama, T. Kato, K. Kimura, K. Ogawa, K. Ohara, S. Tanaka, Y. Kikuchi, N. Hisada, R. Hayashi, Y. Nishimura, K. Nomura, T. Tachibana, M. Irie, H. Kawada, T. Torizawa, N. Murao, T. Kotake, M. Tanaka, S. Ishikawa, T. Miyake, M. Tamiya, M. Arai, A. Chiyoda, S. Akai, H. Sase, S. Kuramoto, T. Ito, T. Shiraishi, T. Kojima and H. Iikura, J. Am. Chem. Soc., 2023, 145, 24035–24051 CrossRef CAS PubMed.
- Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Chem. Rev., 2021, 121, 13936–13995 CrossRef CAS PubMed.
- H. C. Hayes, L. Y. P. Luk and Y. H. Tsai, Org. Biomol. Chem., 2021, 19, 3983–4001 RSC.
- M. E. Hansen, S. O. Yasmin, S. Wolfrum and E. M. Carreira, Angew. Chem., Int. Ed., 2022, 61, e202203051 CrossRef CAS PubMed.
- M. Todorovic, A. Blanc, Z. Wang, J. Lozada, J. Froelich, J. Zeisler, C. Zhang, H. Merkens, F. Benard and D. M. Perrin, Chem. – Eur. J., 2024, 30, e202304270 CrossRef CAS PubMed.
- S. J. Cantrill, R. H. Grubbs, D. Lanari, K. C.-F. Leung, A. Nelson, K. G. Poulin-Kerstien, S. P. Smidt, J. F. Stoddart and D. A. Tirrell, Org. Lett., 2005, 7, 4213–4216 CrossRef CAS PubMed.
- H. Hou, K. C.-F. Leung, D. Lanari, A. Nelson, J. F. Stoddart and R. H. Grubbs, J. Am. Chem. Soc., 2006, 128, 15358–15359 CrossRef CAS PubMed.
- C.-S. Kwan, R. Zhao, M. A. Van Hove, Z. Cai and K. C.-F. Leung, Nat. Commun., 2018, 9, 497 CrossRef PubMed.
- D. N. Tritton, F.-K. Tang, G. B. Bodedla, F.-W. Lee, C.-S. Kwan, K. C.-F. Leung, X. Zhu and W.-Y. Wong, Coord. Chem. Rev., 2022, 459, 214390 CrossRef.
- W.-L. Chan, C. Xie, W.-S. Lo, J.-C. G. Bünzli, W.-K. Wong and K.-L. Wong, Chem. Soc. Rev., 2021, 50, 12189–12257 RSC.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |