
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defect-enabling zirconium-based metal–organic frameworks for energy and environmental remediation applications
Saba
Daliran
 *a,
Ali Reza
Oveisi
*b,
Chung-Wei
Kung
*c,
Unal
Sen
d,
Amarajothi
Dhakshinamoorthy
ef,
Cheng-Hsun
Chuang
c,
Mostafa
Khajeh
b,
Mustafa
Erkartal
g and
Joseph T.
Hupp
*h
*a,
Ali Reza
Oveisi
*b,
Chung-Wei
Kung
*c,
Unal
Sen
d,
Amarajothi
Dhakshinamoorthy
ef,
Cheng-Hsun
Chuang
c,
Mostafa
Khajeh
b,
Mustafa
Erkartal
g and
Joseph T.
Hupp
*h
aDepartment of Organic Chemistry, Faculty of Chemistry, Lorestan University, Khorramabad 68151-44316, Iran. E-mail: daliran.s@lu.ac.ir
bDepartment of Chemistry, University of Zabol, P.O. Box: 98615-538, Zabol, Iran. E-mail: aroveisi@uoz.ac.ir; alir.oveisi@gmail.com
cDepartment of Chemical Engineering, National Cheng Kung University, 1 University Road, Tainan City 70101, Taiwan. E-mail: cwkung@mail.ncku.edu.tw
dDepartment of Materials Science and Engineering, Faculty of Engineering, Eskisehir Technical University, Eskisehir 26555, Turkey
eDepartamento de Quimica, Universitat Politècnica de València, Av. De los Naranjos s/n, 46022 Valencia, Spain
fSchool of Chemistry, Madurai Kamaraj University, Madurai 625021, India
gDepartment of Basic Sciences, Faculty of Engineering, Architecture and Design, Bartin University, Bartin 74110, Turkey
hDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA. E-mail: j-hupp@northwestern.edu
First published on 14th May 2024
Abstract
This comprehensive review explores the diverse applications of defective zirconium-based metal–organic frameworks (Zr-MOFs) in energy and environmental remediation. Zr-MOFs have gained significant attention due to their unique properties, and deliberate introduction of defects further enhances their functionality. The review encompasses several areas where defective Zr-MOFs exhibit promise, including environmental remediation, detoxification of chemical warfare agents, photocatalytic energy conversions, and electrochemical applications. Defects play a pivotal role by creating open sites within the framework, facilitating effective adsorption and remediation of pollutants. They also contribute to the catalytic activity of Zr-MOFs, enabling efficient energy conversion processes such as hydrogen production and CO2 reduction. The review underscores the importance of defect manipulation, including control over their distribution and type, to optimize the performance of Zr-MOFs. Through tailored defect engineering and precise selection of functional groups, researchers can enhance the selectivity and efficiency of Zr-MOFs for specific applications. Additionally, pore size manipulation influences the adsorption capacity and transport properties of Zr-MOFs, further expanding their potential in environmental remediation and energy conversion. Defective Zr-MOFs exhibit remarkable stability and synthetic versatility, making them suitable for diverse environmental conditions and allowing for the introduction of missing linkers, cluster defects, or post-synthetic modifications to precisely tailor their properties. Overall, this review highlights the promising prospects of defective Zr-MOFs in addressing energy and environmental challenges, positioning them as versatile tools for sustainable solutions and paving the way for advancements in various sectors toward a cleaner and more sustainable future.
 Saba Daliran | Saba Daliran studied Pure Chemistry at the Lorestan University (LU) and conducted her Organic Chemistry MSc and PhD at the Bu-Ali Sina University. She spent 9 months as a visiting scholar at the Universitat Politècnica de València (UPV), working in the group of Prof. Hermenegildo Garcia. She did two postdoctoral research, awarded by the Iran National Science Foundation (INSF) and the Iran Science Elites Federation (ISEF). Dr Daliran has served as a Guest Associate Editor for Frontiers Journal. Now, she is an Assistant Professor at the LU where focuses her research on synthesis and applications of (nano)porous materials. |
 Ali Reza Oveisi | Ali Reza Oveisi is an Associate Professor at the University of Zabol (UOZ) (PhD in 2015 from the Bu-Ali Sina University) with research interests focusing on MOFs, COFs, and POPs. He was a visiting scholar at the Northwestern University, in USA, joining the group of Prof. J. T. Hupp and Prof. O. K. Farha. Dr Oveisi has received numerous awards including “Young Scientific Elites” by the Iran Science Elites Federation and “110 TOP Scientists in Iran” by the Iran National Science Foundation, etc. He has also acted as a guest editor for Frontiers in Catalysis and Frontiers in Chemical Engineering. |
 Chung-Wei Kung | Chung-Wei Kung earned his BS and PhD degrees from Department of Chemical Engineering, National Taiwan University, Taiwan in 2011 and 2015, respectively. He joined The Hupp and Farha Groups in Northwestern University, United States as a visiting scholar and postdoctoral researcher during 2013–2014 and 2016–2018, respectively. He joined Department of Chemical Engineering, National Cheng Kung University, Taiwan as an assistant professor to launch his independent research group in August 2018, and became associate professor in 2021. His research focuses on the design and synthesis of metal–organic frameworks (MOFs), MOF-based nanocomposites and relevant porous materials for various electrochemical applications. |
 Unal Sen | Unal Sen received his BSc in Chemistry from Bilkent University and his PhD in engineering from Tohoku University. He conducted postdoctoral research at Northwestern University under the supervision of Prof. Hupp and Prof. Farha. He currently works as a faculty member at Eskisehir Technical University, Department of Materials Science and Engineering. Dr Sen's research focuses on MOFs, MOF-derived carbon-based materials, and MOF-polymer hybrid materials with applications in advanced ceramics, energy storage and conversion, gas storage and separation, and environmental remediation. He was recognized for his outstanding work with the Japan Society for the Promotion of Science RONPAKU Fellowship. |
 Amarajothi Dhakshinamoorthy | Amarajothi Dhakshinamoorthy received his PhD degree in 2009 from Madurai Kamaraj University, India. Later, he worked as a postdoctoral researcher with Prof. Hermenegildo Garcia at Technical University of Valencia for four years. Currently, he is serving as UGC-Assistant Professor at School of Chemistry, Madurai Kamaraj University. His research interests include catalytic applications of metal–organic frameworks and related materials. He received Young Scientist Award 2014 for Chemical Sciences from The Academy of Sciences, India. He has co-authored over 200 publications, six book chapters and one patent. He is also an Early Carrier Advisory Board member for Molecular Catalysis, Elsevier. |
 Joseph T. Hupp | Joseph T. Hupp is the Morrison Professor of Chemistry at Northwestern University. He was appointed as chair of the Editorial Board of Energy & Environmental Science (2018–2022) and was an associate editor of Journal of the American Chemical Society (2003–2014). He has been designated by Clarivate Analytics/Thomson-Reuters as “Highly Cited Researcher” in 2014–2023. He was elected to the American Academy of Arts and Sciences in 2021. His group synthesizes and investigates molecular materials, supramolecular assemblies, and reticular molecules (MOFs, defective MOFs, HOFs. etc.), aiming to solve problems involving solar energy conversion, chemical fuel storage, sensing, separations, or selective catalysis. |
1. Introduction
According to the Oxford Dictionary, a “defect” is a fault in something or in the way it has been made, which means that it is not perfect. Although this idea may not be attractive in everyday life, it is interesting to note that researchers from different disciplines are actively researching the deliberate creation of defects in solid-state materials.1–3 Rather than striving for perfection, scientists take advantage of the potential of imperfections to provide properties that would otherwise be impossible to achieve. A prime example of this is in semiconductor materials, where the addition of dopants allows for the precise control of electrical conductivity, thus expanding their range of uses.4 Furthermore, in the realm of low-dimensional materials such as graphene, careful manipulation of defects can enhance the sensing, electrical, and mechanical properties, thus opening the door to technological advancement.5–7 This growing recognition of the significance of defects contrasts with the traditional view of perfection in materials science. This underscores the importance of customizing materials to meet specific needs by exploiting and manipulating defects. By taking advantage of the unique characteristics and capabilities that arise from these imperfections, researchers are pushing the boundaries of materials engineering and fostering innovation in various fields.Metal–organic frameworks (MOFs) have been the subject of much attention in many fields because of their remarkable characteristics, such as high surface adsorption capacity, tunable pore size, and catalytically active sites.8–11 These qualities make them particularly attractive for gas storage purposes, especially for adsorbing gases, such as hydrogen, methane, and carbon dioxide.12 The essence of MOF design is the self-assembly of metal ions and organic linkers, which enables the alteration of the pore size, hydrothermal stability, and thermal stability by incorporating various metal centers and acid binders to obtain the desired characteristics. Recent studies have explored the possibility of using MOFs as photocatalysts for various applications, including hydrogen production, carbon dioxide reduction, organic pollutant degradation, and heavy metal removal by combining metal ions with photosensitive organic ligands.13,14 It is important to recognize that applications of many discoveries have yet to move substantially beyond the concept demonstration stage, mainly because of the reliance on idealized experimental conditions and the financial obstacles associated with large-scale implementation. These factors can hinder the practical application of MOFs in real-world scenarios.15 Nevertheless, examples of successful, large-scale commercialization exist – most notably, MOF-based storage and controlled release of “electronic gases” used for phosphorous and arsenic doping of semiconductors during manufacturing.16
MOFs possess unique characteristics that distinguish them from other porous materials, emphasizing their design. This allows researchers to manipulate the composition of MOFs to introduce heterogeneity and incorporate disorder into their structures. Yaghi et al. have presented various approaches for achieving “heterogeneity within order”.17 In this context, heterogeneity refers not only to incorporating different building blocks within the framework and pores of MOFs, but also includes deviations from perfect regularity or multiple pore sizes in an otherwise highly ordered system.18 Examples include surface blending with other substances as well as the integration of nanoscale inorganic components with MOF materials. In addition, the properties of MOFs can be altered by incorporating diverse linkers and combining multiple metal-containing secondary building units or different types of metal ions with similar topological functions.19–22 This approach is considered to be an attractive method for modifying the properties of MOFs. Defects within MOFs can be viewed as a specific form of heterogeneity, in which certain structural components are selectively removed in a non-periodic manner. Furthermore, when there are variations in the composition without any elements removed, these deviations may also be classified as defects.18 Defect engineering in MOFs has gained significant attention because of its potential to enhance the structural and surface properties of these materials, leading to improved adsorption processes.23,24
Defects can include missing clusters, missing linkers, or structural rearrangements that create dynamic behavior within the material related to the metal-linker bonding.25 Dynamic changes could occur in the coordination number of metal-based clusters26,27 and could involve the rearrangement of inorganic building units, decoordination of linkers, or dynamic capping/decapping of defective sites.28 Defects in MOFs could act as active sites for dynamic interactions with the targeted compounds, and could also participate in exchange processes with guest molecules. Dynamic behaviours of defects in MOFs in aqueous media have been reported in several studies. For example, MFM-300(Sc), a Sc(III)-based MOF, has been reported to show the switch-on/switch-off dynamic behavior of its coordinatively unsaturated metal sites in water.29 In another work, first-principle molecular dynamics simulations have revealed that the presence of protic solvents such as water or methanol can induce the dynamic structural rearrangements in UiO-66, a Zr(IV)-based MOF, leading to the formation of defects with both undercoordinated and overcoordinated metal-based nodes.30 In addition, MFM-300(Sc) was also reported to exhibit a reversible dynamic change in its structure upon the adsorption of ammonia.31
To gain a comprehensive understanding of these imperfections and their importance in MOFs, it is essential to provide a comprehensive overview of the most common defects encountered in MOFs. These defects can be classified as point defects, which include (i) missing-linker defects, (ii) missing-node defects, (iii) functionalized node defects, and (iv) functionalized linker defects. Fig. 1 displays various types of defects in MOFs, with one of the most commonly reported MOFs with defects, Zr(IV)-based UiO-66, as an example. Investigating and studying these issues in depth are essential for increasing our understanding of MOFs and exploring their wide range of potential uses.
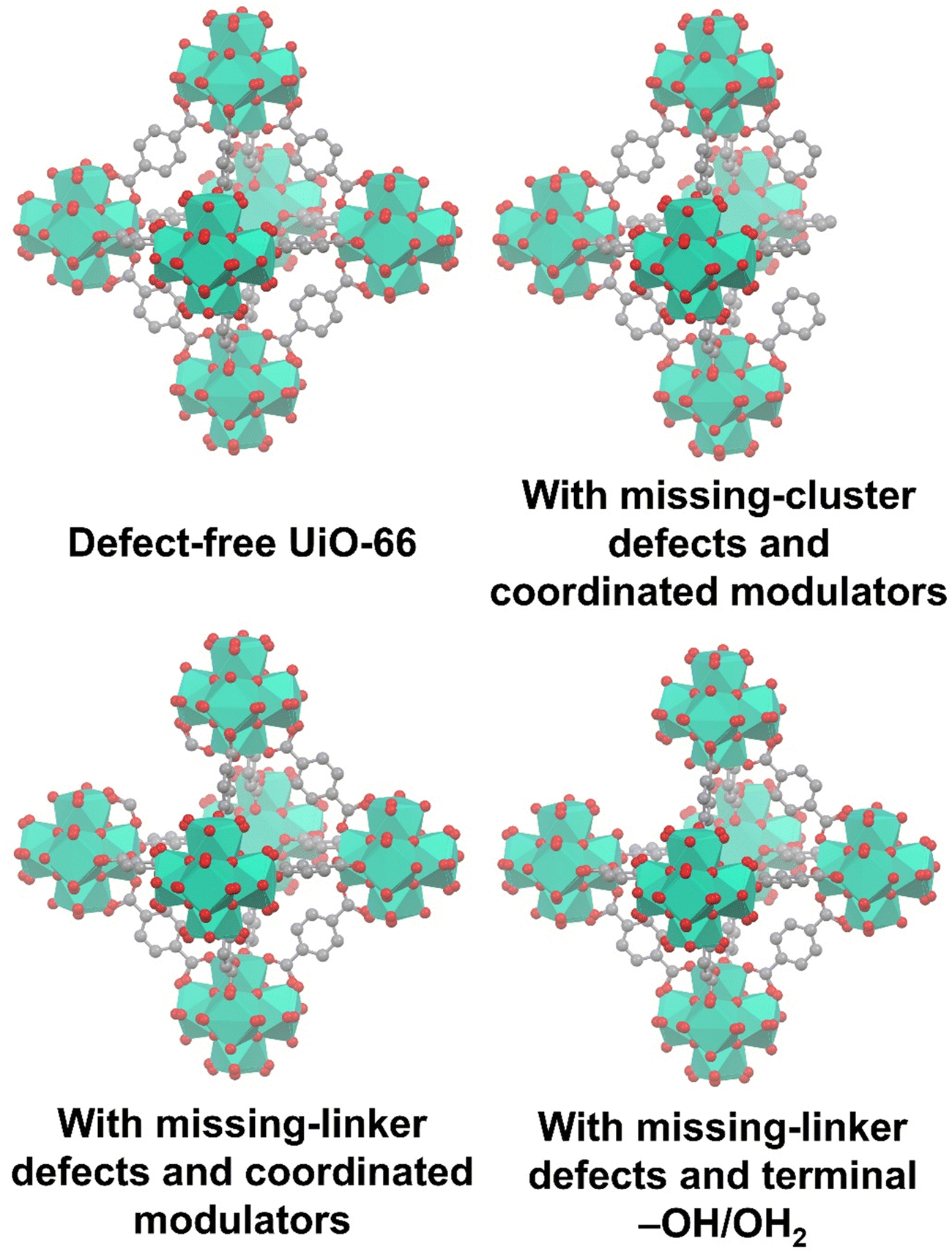 | ||
| Fig. 1 Common defects in a zirconium-based MOF, UiO-66. Hydrogen atoms are not shown in all structures. | ||
The occurrence of linker defects could be attributed to the absence of individual linker molecules in the arrangement. When enough defects are present in a critical concentration and arrangement, they form missing-node (missing-cluster) defects. In principle, missing-node effects could consist of sets of linkers that surround and point to a void space. Or, they could be defined by modulators that surround a void space and lack the ability to fill the void via further coordination bond formation. In practice, missing-node defects appear to be mainly the second type – consistent with high-concentration modulators (monotopic ligands) outcompeting low-concentration linkers for capping a node, in the absence of additional node-forming units.32–36
Understanding and characterizing the defects in MOFs is crucial for tailoring their properties and optimizing their performance for diverse applications. The use of various analytical techniques enables the discernment and quantification of defects within MOF structures, which is essential for enhancing their efficacy and versatility. Thermogravimetric analysis (TGA) is an example of a methodology that is effective for determining defect densities but is generally ineffective for distinguishing between missing-node and missing-linker defects. Nevertheless, for solvothermally synthesized UiO-type MOFs, the identification of missing-node defects is aided by the tendency of these defects to form in a correlated fashion, such that they are periodically arranged in small domains.35,37 Within these domains, nominally forbidden reflections can yield new, low-angle peaks in powder X-ray diffraction (PXRD) patterns. If the peaks are sufficiently prominent, the size of the corresponding domain can be estimated from the peak widths using the Scherrer equation. Correlated missing-node domains generally comprise 8-connected, rather than 12-connected, nodes that are organized in a reo topology rather than the fcu topology of idealized, defect-free frameworks. These observations provide a rationale for the apparent ceiling of four missing-linkers per node for defective UiO-type frameworks. Further studies show that UiO-type frameworks featuring, on average, more than 8 but fewer than 12 linker connections per node (i.e., on average, less than 4 but more than 0 missing-linker defects per node), consist, in some instances, of co-crystallized reo and fcu domains.32
For the UiO-type framework domains characterized by reo topology, the maximum pore diameter is roughly three times that found in the fcu domains. As such, pore-size distributions from N2 isotherms can reveal the presence of missing-node defects. Other indirect indicators include subtle shifts in Raman and IR vibrational frequencies.32 Direct observation and mapping of neighboring reo and fcu domains has been demonstrated for the hafnium version of UiO-66 via scanning electron diffraction (SED);37 see also the work by Zheng, et al.38 It is important to note that in the MOF field, any changes to the linker or node that are not the same as those in the original framework are considered defects.39–41 Potentiometric acid–base titration performed with the suspension of accurately weighted MOF crystals may provide quantitative information regarding the amounts of dissociated ligands and protons, which are highly relevant to the concentration of defects in the zirconium-based MOF (Zr-MOF). Farha and coworkers pioneered the use of this technique to determine the number of defect sites in UiO-66 and quantify the amount of titratable protons present in each hexa-zirconium node. The titration curve of UiO-66 revealed three pKa values of 3.52, 6.79, and 8.30, associated with the μ3-OH, Zr-OH2, and Zr-OH groups present in the nodes, respectively.42 By calculating the difference in the amount of NaOH consumed between two equivalence points, the number of defects composed of terminal aqua/hydroxo pairs and the amount of modulator coordinated on the node can be obtained.42,43 Subsequent studies by Lang et al. revealed that titration could also change the structure of UiO-66 and cause the continuous release of terephthalate linkers from the MOF during the measurement when the pH was higher than 5.44 Consequently, information from the titration curve is also associated with the dynamic formation of new defects through linker dissociation during the titration process; coupling with another technique such as high-performance liquid chromatography is thus required to accurately quantify the amounts of released linkers and modulators.44 Solution-phase NMR and infrared (IR) spectroscopic methodologies are pivotal in discerning imperfections within Zr-MOFs. Utilizing 1H NMR, modulator-induced monocarboxylates were identified.45 Additionally, IR spectroscopy, particularly diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), accurately identifies hydroxyl groups present in the nodes of Zr-MOFs.46 This comprehensive analysis provides crucial insights into the structural integrity of defective Zr-MOFs.
Advances in MOF-applicable structural characterization techniques will likely continue to enhance our understanding of defect structures, defect siting, and defect evolution,47 including real-time observation of evolution. Foremost among these are low-dose transmission electron microscopy (TEM), SED, and atomic-force microscopy (AFM) with sub-nanometer resolution.48–50 Of course, AFM reports on the external surface of the structure. The application of electron diffraction to ever smaller MOF crystallites or to domains within larger crystallites,37 may reveal defect structures, including internal defects that are difficult to observe via PXRD, where information is averaged over many crystallites, or via synchrotron-enabled single-crystal X-ray diffraction, where individual nanocrystallites can indeed be examined, but not down to the size that can be interrogated via electron diffraction. The de Broglie wavelength of electrons is sufficiently small such that resolution constraints, as evidenced by the Scherrer broadening of diffraction peaks in X-ray measurements, are absent. As with PXRD measurements,35,51 particularly instructive may be the observation of low-angle electron-diffraction peaks that reveal the existence of periodicity for structural defects. The application of PDF (pair-distribution function) analysis to data52,53 from total X-ray scattering measurements may provide information about node restructuring, and associated node-localized defect formation, together with other irregular structural features,54,55 that cannot easily be accessed via conventional X-ray crystallographic methods.56 Lastly, Ameloot, et al. have demonstrated how confocal microscopy can be used to visualize planar defects in the interiors of MOF crystallites.57
Much of the above discussion concerns periodically ordered missing-node defects. For some applications, randomly distributed missing-linker defects might prove desirable. Taking Zr-based UiO-66 as an example, an approach that likely provides the latter is exemplified by including trans-1,4-cyclohexanedicarboxylic acid in an otherwise standard mixture for solvothermal synthesis of the MOF.58 The resulting material contains some number of trans-1,4-cyclohexanedicarboxylate (CDC) ions in place of 1,4-benzenedicarboxylate (BDC) ions as linkers. The flexibility of CDC facilitates size-matching and, therefore, the realization of crystallinity in the mixed-linker product (see also, work demonstrated by Bueken, et al.59). Notably, CDC thermally degrades at temperatures below the decomposition temperature for the overall framework. Heating, therefore, allows missing-linker defects to be formed wherever CDC is found. A related approach, described by Zhou and co-workers, entails selective chemical decomposition and, therefore, linker labialization of just one type of linker within a mixed-linker MOF.60 The authors showed how the approach could be used to obtain randomly distributed pores of varying size from a material initially characterized by uniform porosity. The Zhou group then showed that thermal labialization and mesopore formation could be engendered in spatially selective fashion, with microscopic rather than nanoscopic resolution, by employing a laser to heat a material.61
The deliberate introduction of defects in MOFs and their subsequent utilization as heterogeneous catalysts represent a prominent and actively pursued research area. This approach involves intentionally incorporating missing-linker defects, which introduces Lewis acidic sites and missing-node defects that enhance the diffusion of products and reactants within the MOF structure. In addition, a noteworthy number of dynamic Brønsted sites can be formed as the result of defective structures.26,28,62–64 These defect-engineering strategies offer intriguing possibilities for catalytic applications. Several catalytic reactions, including alkene dimerization,65,66 benzaldehyde cyanosilylation,67 methanol dehydration,68 and isomerization of α-pinene oxide62 have been successfully demonstrated using defective MOFs.18,69 Despite these achievements, it is important to acknowledge that defective MOFs are not always more advantageous compared to the ideal “perfect” MOFs in terms of overall catalytic activity and cost-effectiveness. Nevertheless, defective MOFs are valuable platforms for exploring and optimizing novel heterogeneous catalysts with a highly tunable structure-to-property relationship. They provide insights into the effects of defects on the catalytic performance and offer opportunities for further investigation and advancement in bridging the performance gap between traditional catalysts and MOF-based systems.70
The potential applications of imperfect MOFs are primarily centered on chemical catalysis, prompting extensive research on further chemical modifications of defective frameworks. Strategies, such as incorporating different linker functionalities or modifying the metal node, have been pursued to manipulate properties such as framework hydrophilicity, accessibility of reactive sites, and extended coordination space near reactive centers.71 The involvement of computational scientists has also played a significant role in advancing this field; however, conventional simulation techniques face challenges in explicitly addressing defect sites and their distribution owing to periodic boundary conditions. To overcome these limitations, scale-bridging techniques have emerged as a viable solution for incorporating defects into models and calculating properties such as gas sorption and mechanical behavior.60,72,73 The field of defect chemistry in MOFs continues to garner increasing attention, as evidenced by the growing number of publications, reflecting their potential to drive advances in catalysis, materials design, and the broader scope of MOF applications.
Zr-MOFs exhibit remarkable stability compared with many other types of MOFs constructed from various metal ions or clusters.11,51,74–92 Their stability is evident in multiple aspects: they can withstand high temperatures up to 450 °C without undergoing significant degradation, they can be immersed in various organic solvents and acidic aqueous solutions without experiencing notable damage, and they can maintain their crystal structure even under high-pressure conditions.93 The exceptional stability of Zr-MOFs can be attributed to the robustness of the Zr-carboxylate bond and the high level of connectivity among the metal clusters.
The superior stability of Zr-MOFs opens up a wide range of potential applications. One prominent application is in gas and/or vapor separations, where Zr-MOFs can efficiently adsorb and separate various gases and vapor owing to their highly porous and robust structures.94–114 Additionally, Zr-MOFs have shown promising activities in heterogeneous catalysis/photocatalysis, enabling efficient chemical reactions with excellent catalyst stability and reusability.74,112,115–138 With the exceptional chemical stability in water, Zr-MOFs can also be utilized in both the adsorption of ions/dyes/contaminants and catalysis in aqueous media.134,139–150 Another exciting application is in the field of drug delivery,151–154 where Zr-MOFs can serve as carriers for therapeutic agents and provide controlled and targeted release.155 Furthermore, Zr-MOFs exhibit the potential for sensing applications, enabling the detection of specific molecules or ions with high sensitivity and selectivity.156–163 Finally, Zr-MOFs are useful in electrochemistry, offering opportunities for energy storage and conversion devices with enhanced stability and performance.164–170
The incorporation of defects into Zr-MOFs provides an intriguing avenue to augment the functionality and application potential of these materials. The introduction of defects such as missing clusters or missing linkers into the structure of Zr-MOFs significantly enhances their chemical activity and functional diversity. This heightened chemical activity and functional diversity enable the customized modification of Zr-MOFs to meet specific application requirements. For instance, defective Zr-MOFs with vacant sites on Zr centers can be employed in environmental remediation processes, where these open sites serve as active binding sites for pollutants.171 Consequently, defective Zr-MOFs effectively adsorb and eliminate contaminants from water or air.172 Moreover, defective Zr-MOFs are useful for energy-conversion applications. These defects can serve as active sites or be utilized to incorporate catalysts for photocatalytic reactions such as hydrogen evolution and CO2 reduction. Furthermore, post-synthetic methods allow for the further modification of defective Zr-MOFs, introducing additional functional groups or catalytically active sites within the porous framework or fine-tuning the pore size. This inclusion of functional groups and pore size manipulation enhanced the selectivity of defective Zr-MOFs towards different substrates.173 Defects in Zr-MOFs can also play important roles in inducing redox conductivity for electrons in the framework and modulating the microenvironment near the electrode surface upon the electrochemical energy conversion and storage. Overall, the introduction of defects into Zr-MOFs broadens their possibilities and expands their application potential in environmental remediation and energy-related purposes.
The creation of defects in Zr-MOFs has been a topic of interest among numerous researchers, who have presented various strategies for achieving this goal.174–176 Methods can be categorized into two primary types: de novo synthesis and post-synthetic treatment. The de novo synthesis method incorporates a modulator and mixed-linker approach. In contrast, post-synthetic modification involves post-synthetic exchange and etching. The synthesis of Zr-MOFs can be influenced by various factors, such as the choice of solvent, Zr-based precursor, modulator nature, modulator concentration, linker concentration, temperature, and crystal growth rate, leading to changes in the generation of defects.177–180 Monoprotic acids are typically employed as modulators to generate defects during the cultivation of Zr-MOFs. Some frequently employed monoprotic acid modulators for synthesizing defective Zr-MOFs are hydrochloric acid (HCl, pKa = −7.00),181,182 and monocarboxylic acids such as formic acid (HFo, pKa = 3.77),183,184 acetic acid (HAc, pKa = 4.76),43,183,184 benzoic acid (BA, pKa = 4.20),184 trifluoroacetic acid (TFA, pKa = 0.23),34,185–188 amino acids (AAs),189 dichloroacetic acid (DCA, pKa = 1.30),154 trichloroacetic acid (TCA, pKa = 0.66),190,191 difluoroacetic acid (DIF, pKa = 1.24),34,192 mercaptoacetic acid/thioglycolic acid (MA, pKa = 3.83),193,194 dodecanoic acid (DDA, pKa = 5.3),195 and 2-thiophenecarboxylic acids (pKa = 3.49).196 Surfactants such as cetyltrimethylammonium bromide (CTAB) can also induce the defects in Zr-MOFs.197,198 In one of the key studies, Shearer and coworkers have observed a good correlation between the defect concentration and the Brønsted acidity of the modulator employed to create defects.34 Further, concentration of missing-linker or missing-cluster defects has also been controlled by altering the acidity of the modulator or the concentration of the modulator.199,200 Milner and coworkers showed that the variation in the structure of modulator can also lead to differences in both the defect level and particle size of Zr-MOFs.196
Besides these facts, in the presence of the same modulator, the synthetic temperature for growing Zr-MOFs is also the key factor that determines the number of defective sites in MOFs.43 On the other hand, another strategy to generate defects during the synthesis of Zr-MOFs is the mixed-linker approach mentioned previously,62 in where the labile linker may be removed afterword by post-synthetic treatments to create the defective cavity.58,60 In addition to the “de novo” synthesis, another category of approaches to create defects in Zr-MOFs is post-synthetic exchange or etching, which can further create defects in as-synthesized Zr-MOFs. Among these post-synthetic methods, post-synthetic ligand exchange, which involves the exchange of linker in the as-synthesized Zr-MOF by another ligand, has been widely demonstrated to create defects in Zr-MOFs.201,202 On the other hand, unlike other MOFs, the post-synthetic exchange of metal ions in Zr-MOFs is relatively challenging and may result in the deposition of metal oxides in the Zr-MOF.203 Post-synthetic treatments can also be used for defect healing to reduce the amount of defects in Zr-MOFs.181 The process of creating defects in Zr-MOFs via synthetic methods has been thoroughly documented in review articles that have been published.18,21,186,204
In this review, we summarize the environmental remediation and energy-related applications of defective Zr-MOFs. We have discussed how the introduction of defects in Zr-MOFs generates open sites that can be utilized for the adsorption and remediation of pollutants as well as for catalyzing photochemical and electrochemical reactions in energy conversion and storage processes. We emphasized the significance of functional groups and pore size manipulation in enhancing the selectivity and efficiency of defective Zr-MOFs. By tailoring the defects and controlling their distribution within the framework, researchers can further optimize the performance of defective Zr-MOFs for environmental remediation and energy-conversion applications. Furthermore, we highlight the advantages of defective Zr-MOFs over other materials in terms of stability and tunability. These materials exhibit high stability and synthetic versatility, enabling the introduction of missing-linkers or -cluster defects, and post-synthetic modifications. These defects create “open sites” on the Zr centers, augmenting the adsorption capacity and catalytic activity of defective Zr-MOFs.
2. Applications
In the following sections, the use of Zr-MOFs with defects in energy-related and environmental applications demonstrated in previous studies will be highlighted. These applications include electrocatalytic energy conversions, electrochemical energy storage, electrochemical sensing, proton-conducting materials, photocatalytic energy conversions, remediation of organic and inorganic pollutants in water, and the detoxification of chemical warfare agents. The critical role of defects in Zr-MOFs in these applications, either for incorporating active sites or providing distinct chemical functionalities to enhance the performance will be discussed. It should be noticed that herein we only cover the Zr-MOFs containing the “defects” that do not possess obvious long-range order and are randomly distributed within the framework structure. Those Zr-MOFs with well-defined eight-connected, six-connected, or four-connected clusters in their structures, which were sometimes defined as the Zr-MOFs with “inherent defects”,18,205 are not included in the following sections since such inherent defects in fact have the well-defined long-range order.2.1. Environmental remediation
Defect engineering has been widely used for improving the adsorption capacity and selectivity of Zr-MOFs and related materials, which are highly appealing for environmental remediation. Strategies that can induce defect formation, which have been discussed above, include modulated synthesis, mixed-linker approach, post-synthetic exchange and post-synthetic etching. By employing these approaches, the defect density, particle size, porosity as well as specific surface area,189,206 hydrophobicity,207,208 surface charge,7 and adsorption properties of Zr-MOFs can be modulated and improved. The missing-linker and/or missing-cluster defects can lead to the coordination vacancy, i.e., open metal sites or coordinatively unsaturated sites (CUSs) on the hexa-zirconium nodes, which are largely saturated with hydroxyl/aqua (–OH/H2O) ligands or other occupying ligands, depending on the synthetic condition and activation process.63,74,209–213 Such defects in Zr-MOFs can provide the following benefits for environmental applications: (a) additional or larger pore apertures and higher porosity,33,34,43,76,214 allowing the additional accessibility and faster diffusion of the targeted molecule to internal active sites; (b) more Zr(IV) CUSs as Lewis-acid sites and/or more active Zr-OH/Zr-H2O binding sites (note that at pH = 7, terminal aqua ligands are generally changed to hydroxo ligands);215–217 (c) the increased hydrophilicity in the pore to facilitate the penetration of aqueous media.207,208,218 Furthermore, the particle size and surface charge of MOFs are other important factors for the adsorption and removal of contaminates, which can also be tuned by the modulator-based synthesis.146,206,219–221 Overall, an increase in the defect level of Zr-MOFs usually leads to an increase in the number of active sites, porosity, and therefore, the resulting overall adsorption capacity for environmental remediation. However, a synergistic effect of defect density, porosity, stability, particle size, hydrophilicity, and the resulting performance of Zr-MOFs should be considered during the design of Zr-MOFs aiming for the removal of pollutants and catalysis applications.| Defective Zr-MOF | Strategy for defect formation | BET surface areaa (m2 g−1) | Pore size (nm)/pore volumea (cm3 g−1) | Pollutant (or analyte)/adsorption capacity (mg g−1) | Selectivity | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| a Dash indicates that values were not reported. | |||||||||
| Organic dyes/pigments | |||||||||
| UiO-66-15 | BA-modulated synthesis and post-treatment with HCl | 1890 | 1.5/0.88 | Safranine T (ST)/370 | ST over crystal violet (CV) | 220 | |||
| N-heterocycle-doped UiO-66 | N-doping strategy by using nitrogen-based compounds and mechanochemistry | 1550 | 1.24/0.5 | Rhodamine B (RhB)/385 | RhB over ST | 7 | |||
| W-UiO-66-12 | Long-term aging in water (water-induced dangling linker) | 1000 | 2.9/0.73 | Methyl red (MR)/55; methyl orange (MO)/59; malachite green (MG)/9 | MR and MO over MG | 222 | |||
| UiO-66-Y (AA-doped UiO-66) | AA modulated synthesis | 1100 | 1.6–2.3/0.79 | Sunset yellow FCF (SY)/353 | SY over MB, RhB, congo red (CR), and Coomassie brilliant blue R-250 (CBB) | 189 | |||
| (BDC)100−x(NDC)x-60 | Mixed linker with BA-modulated synthesis | 1190–1440 | 0.6–1.6/— | MB/18–62% | — | 223 | |||
| MOF-808-OH | Mixed linker with HFo-modulated synthesis | 2611 | 1.7/0.87 | SY/612; MO/659; quinoline yellow (QY)/701; MG/284; MB/218; C(VI)/164 | SY, MO, and QY over Cr(VI), MB, and MG | 224 | |||
| MOF-808-NH2 | 3085 | 1.7/0.95 | SY/626; MO/690; QY/702; MG/273; MB/208; Cr(VI)/153 | ||||||
| Herbicides and agrochemicals | |||||||||
| UiO-67 | HCl- and HAc-modulated synthesis | 2172 | 1.17 and 1.61/1.12 | Glyphosate (GP)/540; glufosinate (GF)360 | — | 46 | |||
| UiO-67 (100–200 nm) | Modulated synthesis with both HFo and HCl | — | ∼1.4 and 1.6/— | GP/1335 (7.90 mmol g−1) | — | 206 | |||
| UiO-67 (250–350 nm) | TFA-modulated synthesis | GP/1.034 (6.12 mmol g−1) | |||||||
| UiO-67 (500–700 nm) | HCl-modulated synthesis | GP/919 (5.44 mmol g−1) | |||||||
| UiO-67 (1000–2000 nm) | HFo-modulated synthesis | GP/542 (3.21 mmol g−1) | |||||||
| UiO-67 | HCl-modulated synthesis | 2345 | 1.25/0.93 | Atrazine/26 | — | 208 | |||
| UiO-66 | HCl-modulated synthesis | 730 | 1.92/0.046 | 2,4-Dichlorophenoxyacetic acid (2,4-D)/370; ciprofloxacin/112; naproxen (NPX)/44 | — | 225 | |||
| UiO-66 | HFo-modulated synthesis | — | — | Fungicides/— | — | 226 | |||
| UiO-66 | TFA-modulated synthesis | 1405 | — | Fungicides/— | — | 227 | |||
| Pharmaceuticals | |||||||||
| UiO-66 | HCl-modulated synthesis | 1640 | ∼1.0–2.1/0.66 | Carbamazepine (CBZ)/427 (for UiO-67) | — | 228 | |||
| UiO-67 | HCl-modulated synthesis | 2344 | >2/1.07 | ||||||
| UiO-66-NH2 | HCl-modulated synthesis | 1110 | 0.4/0.51 | CBZ/32 | — | 229 | |||
| UiO-67-BA(10) | BA-modulated synthesis | 2900 | —/1.23 | CBZ/294; ibuprofen (IBU)/213 | — | 230 | |||
| UiO-66 | Zr-L1 nanoparticles (NPs) | BA- and HCl-modulated synthesis | 1160 | ∼0.6–1.5/— | Calcein/4.9 (wt%); α-cyano-4-hydroxycinnamic acid (α-CHC)/31.0 (wt%) | — | 231 | ||
| Zr-L2 | HCl-modulated synthesis (BA and HCl for Zr-L2 NPs) | 607 | Calcein/1.0 (wt%); α-CHC/3.1 (wt%)[α-CHC/1 (wt%) for Zr-L2 NPs] | ||||||
| Zr-L3 | HCl-modulated synthesis (BA and HCl for Zr-L3 NPs) | 794 | Calcein/1.0 (wt%); α-CHC/5.0 (wt%)[α-CHC/2 (wt%) for Zr-L3 NPs] | ||||||
| Zr-L4 | HCl-modulated synthesis (BA and HCl for Zr-L4 NPs) | 1031 | Calcein/1.2 (wt%); α-CHC/7.2 (wt%)[α-CHC/4 (wt%) for Zr-L4 NPs] | ||||||
| Zr-L5 | L-Proline- and HCl- modulated synthesis | 1295 | Calcein/2.1 (wt%); α-CHC/14.8 (wt%)[α-CHC/12 (wt%) for Zr-L5 NPs] | ||||||
| Zr-L6 | L-Proline- and HCl- modulated synthesis | 2483 | Calcein/15.2 (wt%); α-CHC/20.3 (wt%)[α-CHC/20 (wt%) for Zr-L6 NPs] | ||||||
| Mesoporous DCA-loaded UiO-66 | DCA@Zr-L1small | DCA-modulated synthesis | 890 | —/0.87 | 5-Fluorouracil (5-FU)/1.9 (wt%) | — | 154 | ||
| DCA@Zr-L2small | 640 | —/0.81 | 5-FU/3.8 (wt%) | ||||||
| DCA@Zr-L3small | 900 | —/1.12 | 5-FU/4.3 (wt%) | ||||||
| DCA@Zr-L4small | 990 | —/1.21 | 5-FU/2.4 (wt%) | ||||||
| MVTM MOF | α-CHC/DCA@UiO-66 | Multivariate-modulation strategy (MTVM) | ∼1500 | 0.8 and 1.1/0.77 | Anticancer cytotoxicity treatment/— | MCF-7 breast cancer over HEK293 kidney cells | 155 | ||
| AL/DCA@UiO-66 | 370 | —/0.35 | |||||||
| α-CHC/AL/DCA@UiO-66 | 635 | 0.8 and 1.1/0.42 | |||||||
| UiO-66 | HCl-modulated synthesis | 1510 | 1.0 and 1.5/0.63 | Nonsteroidal anti-inflammatory drugs (NSAIDs)/overall up to 3 (mmol g−1); IBU/3; ketoprofen/2.5; NPX/2.2; indomethacin/1.74; furosemide/1.71; salicylic acid/1.13 | Drugs over amitriptyline | 232 | |||
| H-UiO-66 | In situ self-assembly template strategy | 1170 | 17.3/1.06 | Tetracycline/670 | — | 233 | |||
| UiO-66-7.8 | Lauric acid-modulated synthesis | 1410 | 0.57–8.50/1.53 | PPCPs like IBU/80 | Extracting pharmaceuticals in complicated wastewater | 234 | |||
| UiO-66 | HAc-modulated synthesis | 1030 | ∼1–2.2/— | Diclofenac (DCF)/320 | — | 235 | |||
| d-MOF-801 | HFo-modulated synthesis in aqueous media | 1070 | 1.13/0.37 | DCF/680 | — | 236 | |||
| Hf-UiO-66 | Post-synthetic metal incorporation | 780 | ∼0.65/0.50 | Cefoperanoz/346 | — | 237 | |||
| Mesoporous UiO-66 (12A-high) | Acid-like DDA-modulated synthesis | 1974 | 0.77–8.26/— | 17 Pharmaceuticals/80 for IBU | Pharmaceuticals in humic acid, bovine serum albumin, and bovine hemoglobin | 195 | |||
| Organoarsenic compounds | |||||||||
| UiO-66-20 | BA-modulated synthesis | 1570 | 1.7 and 2.3/0.87 | Roxarsone (ROX)/730 | — | 212 | |||
| UiO-66-D-NH2 | Post treatment with HCl | 975 | 0.5/— | p-Arsenilic acid (p-ASA)/98; ROX/91 | — | 238 | |||
| UiO-66-D | 1370 | 0.9/— | p-ASA/68; ROX/70 | ||||||
| Other organic pollutants | |||||||||
| UiO-66-10 | HCl-modulated synthesis | 1420 | 0.8, 1.1, 1.6, 2.0/0.72 | Perfluorooctanesulfonate (PFOS)/1.24 (mmol g−1); perfluorobutanesulfonate (PFBS)/1.74 (mmol g−1) | — | 239 | |||
| UiO-66-25 | 1400 | 0.8, 1.1, 1.6, 2.0/0.72 | PFOS/0.82 (mmol g−1); PFBS/— | ||||||
| hcp UiO-66-50 | 690 | 0.8, 1.1, and ∼2.0–10/0.32 | PFOS/0.19 (mmol g−1) | ||||||
| UA-1 | Mixed-linker synthesis | 960 | 5.3/0/11 | Formaldehyde/49 | — | 240 | |||
| D200 | TFA-modulated synthesis and postsynthetic heat treatment | 1800 | 1.3, 1.6, and 2.0/0.84 | SO2, cyclohexane, and benzene/7.20–10.0 (mmol cm−3) | 241 | ||||
| CTAB-U-x | CTAB-U-0.3 | Surfactant-modified synthesis | 1103 | 1.0–1.5/0.49 | Toluene/180 | — | 197 | ||
| CTAB-U-0.5 | 885 | 1.0–1.7/0.47 | Toluene/275 | ||||||
| CTAB-U-1 | 962 | 1.0–1.6/0.55 | Toluene/200 | ||||||
| C-U-N-X | C-U-N-0.3 | Surfactant-modified synthesis | 490 | 1.06–2.1/0.26 | Toluene/230 (for C-U-N-0.5) | — | 198 | ||
| C-U-N-0.5 | 795 | 1.06–2.2/0.39 | |||||||
| C-U-N-1 | 590 | 1.06–2.1/0.34 | |||||||
| UiO-66/MOF-5 (M-U-0.01) | Template strategy (MOF as template) | 1170 | 0.9–2.2/0.95 | Toluene/257 | — | 242 | |||
| UiO-67 | UiO-67-HFo-40 | Acid-modulated synthesis and microwave-assisted approach | 1900 | ∼0.9 and ∼1.9/0.95 | Toluene/320 | — | 243 | ||
| UiO-67-HFo-50 | 2450 | ∼0.9 and ∼1.9/1.30 | Toluene/370 | ||||||
| UiO-67-HFo-60 | 2500 | ∼0.9 and ∼1.9/1.46 | Toluene/470 | ||||||
| UiO-67-HFo-80 | 2050 | ∼0.9 and ∼1.9/1.22 | Toluene/425 | ||||||
| UiO-67-HAc | 2450 | ∼0.9 and ∼1.9/1.33 | Toluene/420 | ||||||
| UiO-67-HPr | 2380 | ∼0.9 and ∼1.9/1.30 | Toluene/4040 | ||||||
| UiO-67-BA | BA-modulated synthesis | 1768 | 1.0–3.0/0.75 | Toluene/480 | — | 244 | |||
| UiO-67-HFo | HFo-modulated synthesis | 1770 | 1.0–3.1/0.79 | Toluene/390 | |||||
| UiO-67-HAc | HAc-modulated synthesis | 1956 | 1.0–4.1/0.79 | Toluene/330 | |||||
| UiO-67 | 67-ben-3 | BA-modulated synthesis | 1345 | 1.0–2.9/0.63 | Toluene/296 | — | 245 | ||
| 67-ben-6 | 1690 | 1.0–2.9/0.73 | Toluene/330 | ||||||
| 67-ben-10 | 1770 | 1.0–3.0/0.75 | Toluene/480; benzene/156; o-xylene/382; m-xylene/180; p-xylene/340; acetone/78 | ||||||
| 67-ben-14 | 1250 | 1.0–3.1/0.58 | Toluene/247 | ||||||
| UiO-67-xBA | UiO-67-3BA | HCl- and BA-modulated synthesis | 2645 | ∼0.8/1.16 | Dimethyl phthalate (DMP)/∼190; phthalic acid (PA)/∼380 | — | 246 | ||
| UiO-67-10BA | 2705 | ∼0.8/1.21 | DMP/∼200; PA/∼400 | ||||||
| UiO-67-30BA | 3040 | ∼0.9/1.44 | DMP/230; PA/430 | ||||||
| UiO-66 | UiO-66-1.6HAc | HAc-modulated synthesis | 1057 | ∼5.0–15/0.793 | DMP/90; PA/205 | 190 | |||
| UiO-66-0.2TCA | TCA-modulated synthesis | 1420 | Up to 3.9/0.784 | DMP/∼75; PA/∼220 | |||||
| Zr-fumarate-MOF | HFo-modulated synthesis | 960 | 0.5–0.9 and 1–2/0.38 | C3 hydrocarbons: propylene (1.63 mmol g−1); propane (1.27 mmol g−1) | Propylene over propane | 247 | |||
2.1.1.1. Organic dyes/pigments removal. Dyes, as soluble organic compounds, are extensively used in the manufacturing of paper printing, textiles, food, leather, plastics, pharmaceuticals, etc.248 Accordingly, there have been growing worries about the contamination of water resources by dyes as a result of their obvious colors, high stability, and potential toxicity. Thus, it is urgent to remove dyes from the contaminated wastewater before entering the environment. Up to now, Zr-based MOFs possessing excellent chemical and thermal stabilities have been developed for the removal of dye molecules from wastewater.249–251 In addition, the research of defect-engineered Zr-MOFs to adsorb and remove dyeing wastewater to reduce their environmental issue has attracted increasing interest.252
For instance, Wang and coworkers have studied the affinity of defective UiO-66 for safranine T (ST) and crystal violet (CV), two cationic dyes with different sizes (ST, about 11.4 × 9.2 Å in size; CV, about 13.9 × 12.3 Å in size).220 They modulated the defect density with various adding BA as a modulator and post-synthetic treatment with 1 M HCl to yield a UiO-66-X series (X = 0, 5, 10, 15, and 20, X represents the equivalent of BA with respect to the BDC linker). BA could contribute to the MOF formation, removing through the post-synthetic acid treatment. The approach successfully increased the surface areas and total pore volume of the UiO-66. Among the synthesized UiO-66-X solids, UiO-66-15 exhibited the largest BET surface area and pore volume, which were higher than those of non-defective UiO-66 (1200 m2 g−1 and 0.49 cm3 g−1) and the other UiO-66s prepared with modulators such as HAc, HCl, and TFA. In addition, the defective UiO-66 showed a large pore of about 1.5 nm in addition to the observed pore of 1.15 nm for the non-defective UiO-66. Notably, the defective UiO-66 retained its chemical stability as compared with its non-defective analog. Thanks to the defects in the UiO-66-15 and the size-exclusion effect, the MOF could selectively adsorb ST from the mixture of ST and CV. The defective UiO-66 exhibited a notable maximum adsorption capacity for ST dye adsorption (adsorption conditions: T = 25 °C, adsorbent amount: 10 mg, concentration = 200 ppm for adsorption kinetics experiment, contact time = 24 h) as compared with that of the defect-free UiO-66 (39 mg g−1).
Hu and collaborators reported the synthesis of defective UiO-66 using alkaline nitrogen heterocycles (dopamine, pyrrole, and 2-methylimidazole) as modulators through an in situ one-pot mechanochemical approach.7 The monodentate N-heterocycles were successfully attached to the Zr6 nodes by partial BDC linker substitution, which resulting in the missing linker defect in the MOF skeleton. This strategy significantly increased the surface area and pore volume of the N-coordinated UiO-66 MOFs compared to the parent UiO-66. Among these MOFs, pyrrole-doped UiO-66, namely Np-UiO-66, displayed the highest surface area and pore volume along with typical mesoporous behavior. The defective MOF was then assessed for highly selective adsorption of two cationic dyes, rhodamine B (RhB) and ST, which have almost the same molecular sizes (RhB; 14.1 × 9.8 Å in size; ST, 12.9 × 9.6 Å in size) but are different in acid–base groups. The respective MOF was found to have two times higher adsorption capacity for RhB (384 mg g−1) (adsorption conditions: T = 303 K, pH = 7.0, adsorbent amount: 10 mg, concentration = 4–130 ppm, contact time = 24 h) and 223 times higher selectively towards RhB over ST (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) than the original UiO-66. This enhanced adsorption performance was ascribed to the N-modified surface and defective sites in UiO-66 pores. In addition, the Np-UiO-66 displayed a better resistance to alkaline corrosion than pristine UiO-66.
:1 ratio) than the original UiO-66. This enhanced adsorption performance was ascribed to the N-modified surface and defective sites in UiO-66 pores. In addition, the Np-UiO-66 displayed a better resistance to alkaline corrosion than pristine UiO-66.
In a related study, the UiO-66 nanoparticles (NPs) aged in water for a long time of 12 months (W-UiO-66-12) showed a higher affinity to adsorb anionic methyl red (MR) and methyl orange (MO) dyes than to the cationic malachite green (MG) compared to the pristine MOF, mostly due to electrostatic forces between the anionic dyes and the free carboxylic acid functionalities created during the aging process.222 The long-term aging procedure could result in the partial hydrolysis of the framework and, thereby, lead to the bond cleavage between the BDC linkers and Zr6 nodes, creating some defect sites on the surface (dangling linker defect). The BET surface area decreased from 1280 m2 g−1 for UiO-66 to 1,000 m2 g−1 for W-UiO-66-12, but the microporosity (1.7 nm) of the pristine MOF was changed to mesoporosity (2.9 nm).
Very recently, Wang and coworkers designed and synthesized pH-switchable UiO-66s through in situ AAs (valine (V, pKa values of 2.32 and 9.62), threonine (T, pKa values of 2.09 and 9.10), and tyrosine (Y, pKa values of 2.20 and 9.11)) doping method for selective adsorption of anionic dyes (sunset yellow FCF (SY) and acid orange II (AO)).189 They used AAs as modulators during the MOF assembly to generate defective UiO-66s (UiO-66-V, UiO-66-T, and UiO-66-Y) not only with larger pore sizes but also with the incorporation of amino-functionality into the MOF. The carboxylate functionality of AA can compete with those of the BDC ligand for coordination with Zr6 nodes. Notably, amino groups of AA-doped UiO-66s play a substantial pH-responsive feature for the selectivity of anionic/cationic dyes with changing pH (pH on–off switchable property). The rigid Y was competitively coordinated with Zr nodes rather than the two AAs, and caused to the formation of more defects with a larger pore size and a higher fraction of mesoporosity/microporosity in UiO-66-Y (a hybrid type I/IV isotherm) with irregular cubical shape particles (in sizes of 150–250 nm) compared to the parent UiO-66 (BET surface area = 1220 m2 g−1, pore volume = 0.62 cm3 g−1, and a type I isotherm) with octahedral shape particles (in average size of 150 nm). As a result, UiO-66-Y improved adsorption capacity and diffusion rate (168 × 10−3 min−1) for anionic SY, respectively, ∼1.9 and 5.9 folds higher than of those obtained with parent UiO-66 (conditions: T = 298 K, pH = 6.0, dye concentration = 0.1 mmol L−1, V = 50 mL, adsorbate dosage = 10 mg). Interestingly, it presented a high selectivity for anionic dyes over cationic MB and RhB dyes with the same molecular dimensions (adsorption uptake for SY was about 10-fold observed for MB and RhB). In addition, UiO-66-Y revealed a higher affinity for SY than AO, due to the presence of more sulfonic acids on SY, which caused a higher anionic property than the AO at the studied condition. The observed decrease in adsorption uptakes of other anionic dyes such as Coomassie brilliant blue R-250 CBB, (27.3 × 17.0 × 9.1 Å) and congo red (CR, 26.2 × 8.1 × 5.0 Å) rather than SY (11.9 × 8.2 × 2.4 Å) are attributed to larger dynamic diameters of CBB and CR molecules than that of SY molecule. Further, the defective UiO-66s showed stable self-adsorption/desorption and regenerated via pH-switching for six subsequent uses.
In another work, UiO-66 MOFs (BDCNDC-60 series) were synthesized through using BDC and 1,4-naphthalenedicarboxylic acid (NDC) as linkers with different proportions and using BA as the modulator (Fig. 2) and the activity of these solids was tested in the adsorption of MB and its photocatalytic degradation.223 The experimental evidence indicates that active zirconium sites can also participate in the degradation process and catalyze oxidative demethylation of MB in addition to the linker modification with naphthalene rings (π–π stacking between NDC-linker and MB and bandgap reduction).
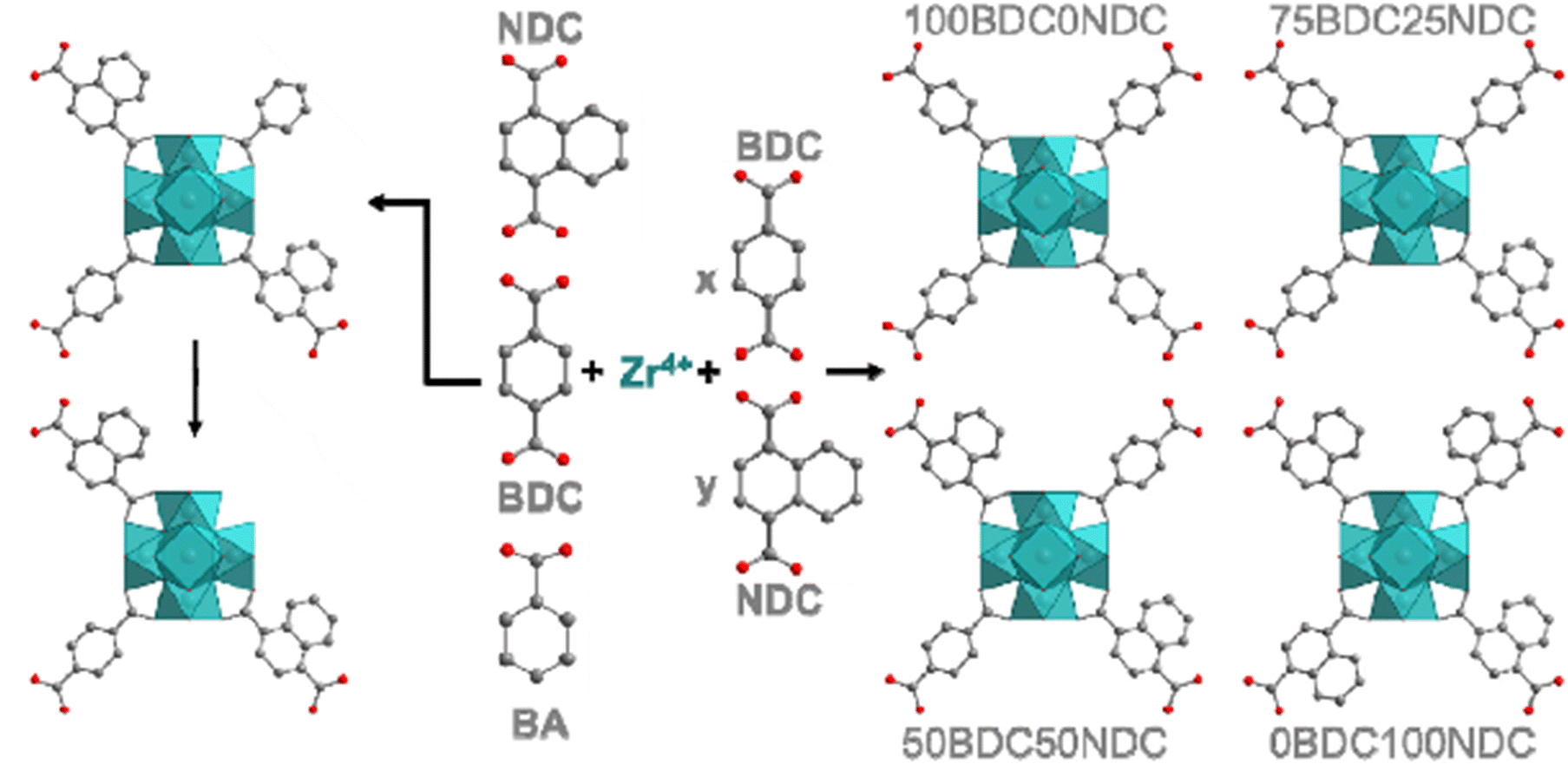 | ||
| Fig. 2 Schematic synthetic plan towards formation of the UiO-66 MOFs. The left one displays the usage of BA for defect formation. Reprinted from ref. 223 Copyright 2021 Elsevier. | ||
Phan and co-workers have reported the synthesis of two defective MOFs, MOF-808-OH and MOF-808-NH2 by partially replacing 1,3,5-benzenetricarboxylate (BTC) linker with 5-hydroxyisophthalate and 5-aminoisophthalate, respectively (Fig. 3).224 The number of defect sites was approximately 7 and 3 mol% for MOF-808-OH and MOF-808-NH2, respectively, as evidenced by 1H NMR spectroscopy. In this way, these defective sites enhanced the acidic centers around Zr-clusters due to the missing-linker defect, thus exhibiting higher adsorption capacity towards various anionic organic dyes and Cr(VI) species. The experimental results showed that the adsorption capacity of defective MOFs (MOF-808-OH and MOF-808-NH2) was increased by 30–60% for anionic contaminants including SY, quinoline yellow (QY), MO, and potassium dichromate compared to MOF-808 without defective sites. On the other hand, the defective MOFs showed 5–15% better performance in the removal of cationic dyes (MG and MB) than the MOF-808. It is worth noting that published examples of defective MOF-808 are relatively rare compared to those of defective UiO-66. Since the perfect MOF-808 is already constructed from six-connected hexa-zirconium clusters that can allow further coordination of additional carboxylate-based ligands, advanced characterizations are usually required to verify the presence of missing-linker defects caused by the second ligand rather than the direct coordination of the second ligand onto the node without replacing the linker.
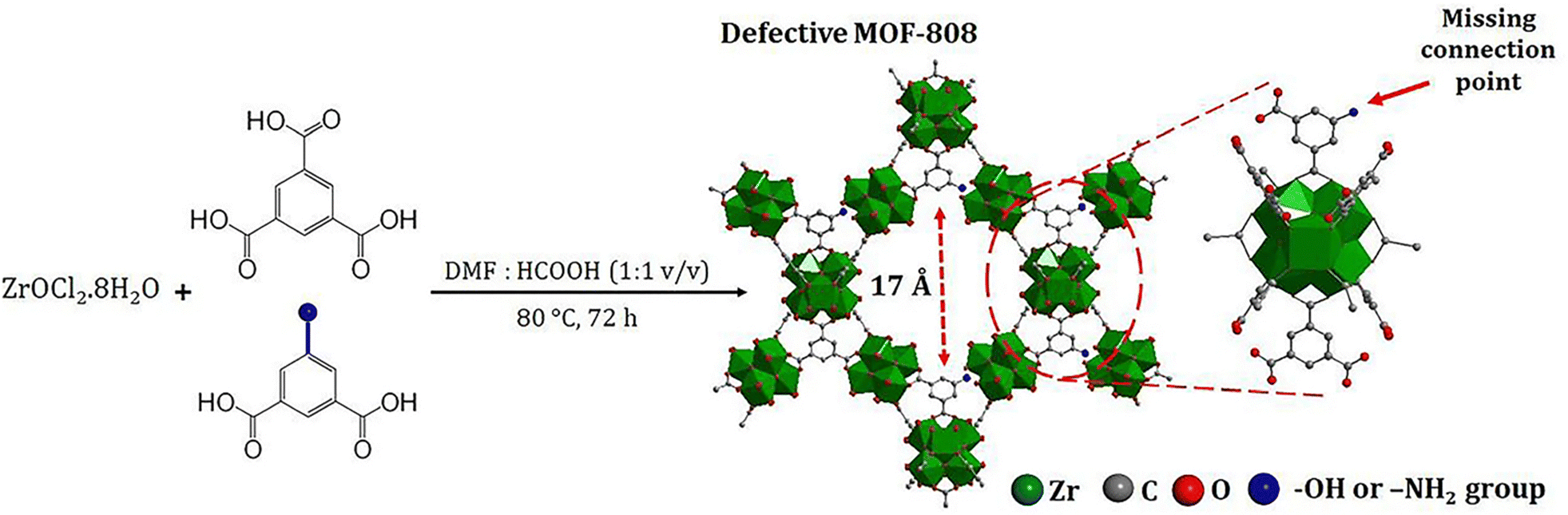 | ||
| Fig. 3 Defect-engineered MOF-808-OH and MOF-808-NH2. Reprinted from ref. 224 Copyright 2023 Royal Society of Chemistry. | ||
2.1.1.2. Herbicides and agrochemicals removal. Not to be confused with organophosphorous insecticides,253 glyphosate (GP) and glufosinate (GF) are widely used as herbicides, specifically for broad leaf weeds. They function by inhibiting plant enzymes (Scheme 1). At low exposures, whether acute or chronic, they appear to pose little threat to humans or to aquatic fauna. Nevertheless, the environmental consequences of GP and GF accumulation in soil and water are not fully understood. GF is a racemic mixture. In water at pH 4 (see Scheme 1), the dominant forms of both are negatively charged zwitterions.
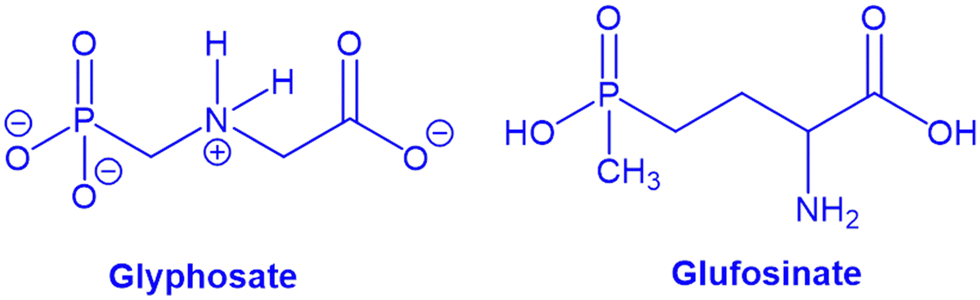 | ||
| Scheme 1 Structures of GP and GF as herbicides. | ||
Zhu et al. looked at the ability of defective UiO-67 to adsorb GP and GF from water at pH 4.46 The MOF was synthesized by using HCl and HAc as acidic modulators which engender missing-linker defects. At this pH, acetate is lost from the MOF node, and each defect site presents an aqua and a terminal-hydroxo ligand. Given a pKa value around 3.3,42,254 the four μ3-hydroxos of the node core should be largely deprotonated, such that the overall framework carries a net negative charge. One might anticipate that anionic guests like GP and GF would be excluded from the pores of UiO-67. Zhu et al., however, observed sizable adsorption capacities, i.e. 540 (3.20 mmol) and 360 (1.86 mmol) mg g−1 for GP and GF, respectively (adsorption condition: T = 25 °C, pH 4, adsorbent concentration = 0.03 g L−1, contact time = 5 h). The prolific uptake reflects, at least in part, the ability of oxy-Zr(IV) species, including Zr-MOFs,255–257 to bind phosphonates tenaciously via zirconium-oxygen bond formation.258 The anticipated stoichiometry is one phosphonate per missing-linker defect. The above capacities translate to about 3 GP per node and 2 GF per node, where the defect density is 1.2 per node. Thus, additional factors must be involved.
In a related study, Pankajakshan, et al. have used HFo, HCl, TFA, and a mixture of HFo and HCl (1:1) as modifiers to tune both the density of missing-linker defects in UiO-67 and the size of the MOF crystallites from micrometer to nanometer (UiO-67: 1–2 μm, HFo; 500–700 nm, HCl; 250–350 nm, TFA; HFo:HCl (1:1), 100–200 nm).206 Notably, the smallest crystallites featured the largest fraction of missing linkers. These samples were evaluated for the adsorptive removal of GP from water. Qualitatively consistent with exposure of the largest proportion of Lewis acidic Zr sites, the HFo:HCl form of UiO-67 displayed by far the greatest uptake, as well as the fastest uptake. The latter finding is consistent with rate control via intracrystalline diffusion of GP. Ignoring, for the moment, differences in defect density and, therefore, average aperture size, the rate of diffusion-limited uptake should scale inversely as the square of the length of the crystallite.
Atrazine (6-chloro-N-ethyl-N′-(1-methylethyl)-1,3,5-triazine-2,4-diamine, molecular dimensions: 0.94 × 0.84 × 0.32 nm) is a widely used synthetic herbicide in agriculture and is a potent endocrine disrupting compound.259 The adsorptive removal of atrazine from water was studied by three MOFs – ZIF-8, UiO-66, and UiO-67.208 Interestingly, UiO-67 showed an adsorption efficiency of 98% in 2 min, whereas ZIF-8 reached the same efficiency only after 50 min. To some extent, it is surprising that UiO-66 was poor for the removal of the hydrophobic atrazine because of the small pores and hydrophilic character of the framework. As a result of the missing linker defects utilizing HCl, UiO-67 showed the presence of both micropores and mesopores (>2.0 nm), capable of faster transport adsorption of the micro-pollutant into the MOF with a maximum adsorption capacity of 26 mg g−1. In another work, Akpinar and coworkers clarified the mechanism of atrazine adsorption which was in line with adjusting the size of linker π-systems in the MOF; a larger π-system offers more π(MOF)–π(atrazine) interactions, thereby engendering more adsorption capacity,260 at least on a “per pore” basis.
Bayazit et al. studied an HCl-modulated UiO-66 with missing-linker defects for the adsorption of a synthetic auxin, namely 2,4-dichlorophenoxyacetic acid (2,4-D), which is a toxic herbicide sometimes found in water supplies.225 The nominal MOF showed a maximum adsorption capacity of ∼370 mg g−1 for 2,4-D (adsorption condition: T = 25 °C, pH 5.5, contact time = 2 h, adsorbate solution = 50 mg L−1). In another study, a defective MOF was obtained by using HFo modulator.226 In this study, a highly crystalline defective UiO-66 was evaluated as an adsorbent for dispersive micro-solid-phase extraction coupled to liquid chromatography/mass spectrometry (d-μ-SPE-LC/MS) for the determination of fungicides in aqueous samples with satisfactory recoveries (>80%). In addition, Wang and co-workers demonstrated the efficacy of defective UiO-66 as the solid-phase microextraction material for selective extraction of the fungicides, such as chlorothalonil, vinclozolin, dimethachlon, procymidone and iprodione, from fruit samples.227
2.1.1.3. Pharmaceuticals adsorptive removal. Pharmaceuticals and personal care products (PPCPs) are among a group of emerging organic pollutants, which can cause a series of severe health problems for humankind.261
In 2017, Akpinar et al. reported the application of defective UiO-66 and UiO-67 MOFs for the adsorption of carbamazepine (CBZ, as an anticonvulsant drug, kinetic diameter of 0.74 nm) from water.228 They used HCl as a modulator during the assembly of the MOFs, which resulted in highly porous, quasi-spherical shape and small MOF particle sizes including more open active Zr sites and more numbers of –OH groups on nodes, comparing to those of the defect-free UiO-66 and UiO-67. While UiO-66 showed a poor performance of around 35% efficiency in removing CBZ, UiO-67 exhibited an effective removal of 95% in only 2 min (100 mg L−1 aqueous CBZ solution, adsorbent concentration 1.2 mg mL−1, pH 5). The superior performance of UiO-67 is due to the impact of missing-linker defects. Physisorption processes, the hydrophobic and π–π interactions are ruled out in the adsorption mechanism. In another work, a defective UiO-66-NH2 was considered as adsorbent for the extraction and determination of CBZ in water and urine samples (extraction conditions: pH 7.5, adsorbent amount 5 mg, sample volume 100 μL, total time <12 min).229 The missing-linker defects and the amino functionality dominated the increase of CBZ adsorption. On the other hand, series of defective UiO-67 frameworks were fabricated with BA as the modulator in another study, and the performances of these solids in the adsorption of CBZ and ibuprofen (IBU) from water were studied. Among the various defective MOFs, UiO-67-BA(10) (10 stands for the molar ratio (%) of BA/ZrCl4) exhibited significant adsorption capacities for CBZ and IBU with 294 and 213 mg g−1, respectively. The adsorption capacity of UiO-67(Zr)-BA(10) for CBZ was relatively higher compared to the most reported adsorbents.230 The adsorption performance of UiO-67(Zr)-BA(10) for CBZ was ∼4.5 times higher than the commercial activated carbon. This superior performance of UiO-67(Zr)-BA(10) is attributed to many factors like synergism between porosity and defect sites, π–π interaction, H-bonding, and van der Waals interactions. Additionally, the adsorption performance was retained without any decrease for at least four cycles.230
Forgan and Fairen-Jimenez groups have introduced the adsorption of a series of anti-cancer drugs, carboxylate and phosphate-containing moieties by Zr-MOFs utilizing a defect-modulated MOF synthetic approach, that they termed defect drug loading.154,155,231 Note that the broader goal was not to remove the targeted compounds from solution, but, instead, to employ MOF nanocrystallites as vectors for delivery and controlled release of guest species, including delivery to the interiors of HELA cells. They examined eight linkers, of varying size and functionality, and along with several modulators (HCl, HAc, L-proline, BA) in preparing a series of defective Zr-MOFs (Zr-L1 to Zr-L8, Scheme 2).231 Consistent with the presence of missing-linker defects, all samples (e.g. Zr-L1, Zr-L2, Zr-L4; L1 is BDC and L2-L4 are functionalized BDC) yielded type II nitrogen (77 K) isotherms. When BDC linker and a mixture of BA and HCl modulators were used, a defective and mesoporous UiO-66, termed Zr-L1 was formed. Notably, higher loadings of calecin (4.9 wt%)262 as a fluorescent molecule similar to doxorubicin drug and α-CHC (31.0 wt%, as an anti-cancer drug, α-cyano-4-hydroxycinnamic acid, 10.6 × 5.9 Å in size) were found for the unfunctionalized version of UiO-66 (Zr-L1), compared to those for other synthesized UiO-6X compounds (except Zr-L6 for calecin: 15.2 wt%). This behavior was attributed to the presence of the missing-linker defects associated with a favorable balance between MOF pore size and the size of the guest molecule. While no doubt true for α-CHC, a possible alternative interpretation of the 4.9 wt% uptake of calecin is that adsorbate is size excluded from the UiO-66 crystallite interior, but faces little steric impediment to carboxylate-based grafting to Zr(IV) at defects sites on the exterior surfaces of crystallites. Regardless, all MOF samples proved capable of delivering guest molecules to the interiors of HELA cells.
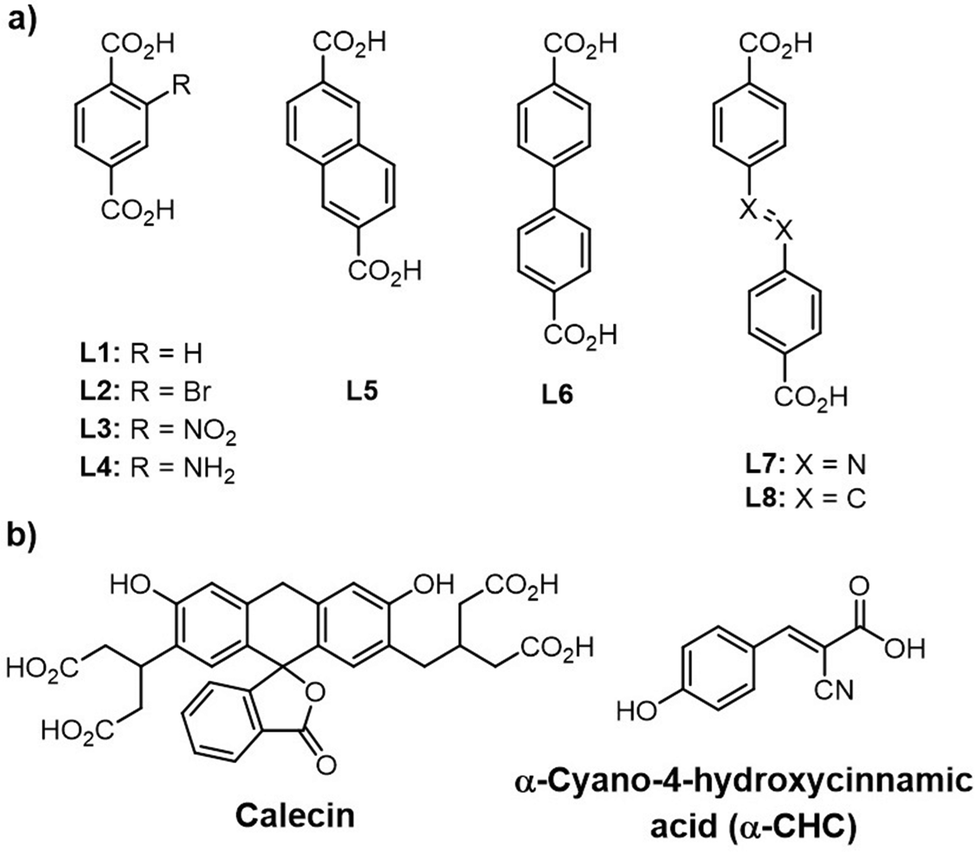 | ||
| Scheme 2 (a) Structures of organic linkers employed for the synthesis of various Zr-based MOFs (Zr-L1 to Zr-L8) and (b) structures of candidate guest molecules.231 | ||
In the next work, they used DCA, an anti-cancer probe molecule, as a modulator during the solvothermal synthesis of UiO-66s, resulting in a very small (10–30 nm) and highly defective DCA-loaded MOF nanaoparticles (DCA@Zr-L1-4small).154 DCA serves as a size and defect control molecule as connected to the Zr6 nodes in place of linkers. Although the BET surface areas were lower than defect-free UiO-66 (1200 m2 g−1), but, the defects induced mesoporosity in the MOF with exceptionally high pore volumes (∼0.8–1.2 cm3 g−1), capable of effective adsorption of 5-fluorouracil (5-FU, as an anticancer drug) about 1.9–4.3 wt% and demonstrating enhanced therapeutic effects compared to the free drugs.
Moreover, Forgan and co-workers have reported a new drug-delivery strategy, so-called multivariate modulation (MTVM) strategy,263 wherein three delivery candidates/linkers are incorporated within the pores of UiO-66 during solvothermal formation of the MOF itself (Scheme 3).155 They are uniformly distributed via coordination to Zr(IV) at missing-linker defect sites. Later, a fourth drug is loaded post-synthetically into the MVT MOFs to afford nanoparticles loaded with cocktails of drugs. The activity of these solids was tested in vitro for selective anticancer cytotoxicity against MCF-7 breast cancer cells. For example, the anticancer activity of the double drug combinations (5-FU@DCA@UiO-66 and α-CHC/DCA@UiO-66) towards MCF-7 breast cancer cells is significantly increased compared to the free drugs. Interestingly, a very sizable increase in selectivity towards cancer cells is achieved by 5-FU@α-CHC/AL/DCA@UiO-66.
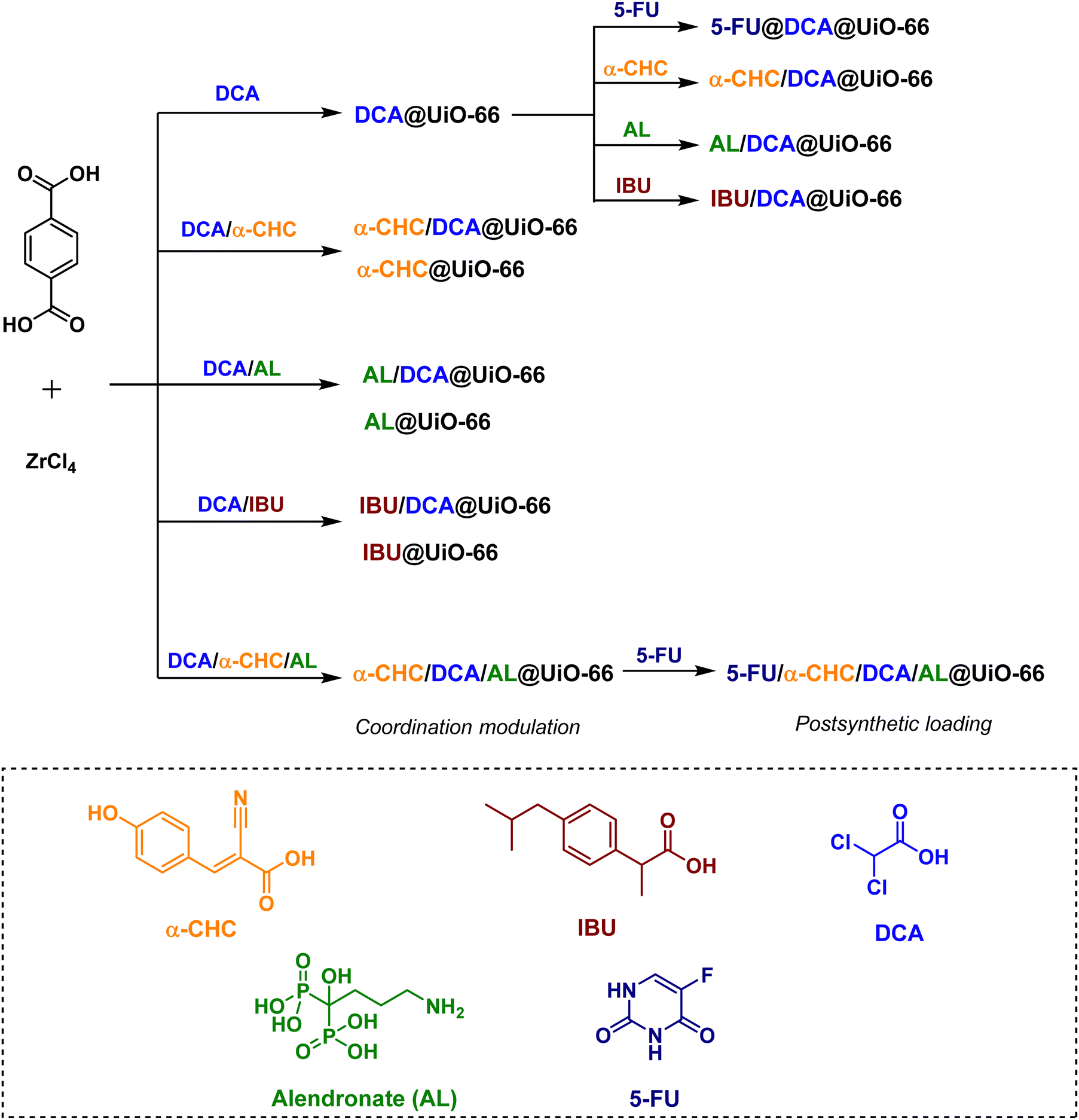 | ||
| Scheme 3 Drug-modulated UiO-66s and their post-synthetic drug loading.155 | ||
Nonsteroidal anti-inflammatory drugs (NSAIDs) are mainly anionic aromatic compounds found in ground water and surface water may cause harm to the environment and human health.264 Lin et al. applied three Zr-MOFs, defective UiO-66 [Zr6O4(μ3-OH)4(BDC)5], MOF-808 [Zr6O4(μ3-OH)4(BTC)2], and MOF-802 [Zr6O4(μ3-OH)4(PZDC)5, PZDC = 3,5-pyrazoledicarboxylic acid], respectively, with 10, 6, and 10-connected Zr clusters for the adsorptive removal of a serious of NSAIDs from water.232 Defective UiO-66, containing 1.0 and 1.5 nm micropores, achieved maximum adsorption capacities up to ∼3 mmol g−1 (amount of adsorbent = 6.0 mg, pharmaceutical solution = 1.0 mmol L−1, pH = ∼6.0), substantially higher than those of MOF-808 and MOF-802. Acid-treatment resulted in the formation of defect sites. In the respective MOF, the presence of two free terminal –OH/–OH2 groups on the unsaturated-coordinated Zr clusters and the larger number of BDC linkers showed high adsorption affinity toward anionic pharmaceuticals, carboxylic acid-containing drugs, such as IBU, ketoprofen, naproxen (NPX, a typical NSAID), indomethacin, and furosemide via a synergistic effect of chemical adsorption and π–π interactions. The adsorption of the other anion drug, salicylic acid, was strongly due to chemical adsorption without a noticeable π–π interaction. Also, acetophenone (min-width, 0.59 nm) with no carboxyl groups, as a neutral pharmaceutical compound, adsorbed by defective UiO-66 because of only π–π interactions between the benzene rings of pharmaceuticals and the MOF linkers. Besides, the adsorption of amitriptyline in the MOFs was very low due to the electrostatic repulsion that occurred at the studied pH (pH ∼ 6) between the cationic sites (Zr-OH2+) and the protonated tertiary amine groups (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH+–) of the drug (the pKa value of amitriptyline is 8.8). The work showed the importance of terminal –OH/–OH2 groups on the node, large porosity, and high uptake affinity of defective MOFs for effective pharmaceuticals adsorption.
NH+–) of the drug (the pKa value of amitriptyline is 8.8). The work showed the importance of terminal –OH/–OH2 groups on the node, large porosity, and high uptake affinity of defective MOFs for effective pharmaceuticals adsorption.
In another work reported by Bayazit et al. mentioned previously, they also investigated the adsorptive removal of ciprofloxacin, a typical fluoroquinolone antibiotic and NPX by a defective UiO-66.225 HCl treatment led to the formation of defective sites in the framework. The particle sizes were about 0.28–0.30 μm. Remarkably, it exhibited maximum adsorption capacities of 112 and 44 mg g−1, for CPX and NPX, respectively (adsorption conditions: T = 25 °C, pH 5.5, contact time = 2 h, adsorbate solutions = 10 mg L−1).
One of the strategies to prepare hierarchically porous MOFs is utilizing the self-assembly template,265 in which the less stable metal–organic assemblies such as nanosized MOF-5 are used as the template during the construction of another more stable MOF.265 After the synthesis of the targeted MOF, the template was removed to create mesopores in the hierarchical MOF. Zhang et al. reported this strategy for the synthesis of a hierarchical-pore variant of UiO-66 with different mesopore sizes, H-UiO-66-3.8 nm and H-UiO-66-17.3 nm, for the adsorptive removal of antibiotic tetracycline (TC) in wastewater.233 The H-UiO-66s were obtained through an in situ self-assembly template strategy by using Zn(NO3)2·6H2O and BA. H-UiO-66-17.3 nm exhibited a type-IV curve (mesoporous) and enhanced pore volume of compared to pristine UiO-66. Accordingly, H-UiO-66-17.3 nm showed enhanced removal efficiency of >99% for TC and a maximum adsorption uptake of 670 mg g−1 (adsorption conditions: T = 30 °C, pH 5, time = 12 h, adsorbate solutions = 38 mg L−1), and the material performs greater than the pristine UiO-66 (125.72 mg g−1), H-UiO-66-3.8 (490 mg g−1), MIL-101(Cr)-NH2 (14 mg g−1),266 MIL-101(Cr) (39.88 mg g−1),249 NiFeC nanocomposites (592 mg g−1),267 and carbon disulfide-modified magnetic ion-imprinted chitosan-Fe(III) (520 mg g−1).268 The interaction between the Zr6 cluster of H-UiO-66 and the O atoms of the amide group and a neighboring hydroxyl group (at C-3) in TC and π–π stacking between the H-UiO-66 linker and TC associated with the mesoporosity of 17.3 nm, played a vital role for the high adsorption of the analyte.
Other studies also reported the use of defective Zr-MOFs for the removal of pharmaceutical pollutants from aqueous solutions. For example, defect-rich MOFs were prepared by tuning the node connectivity ranging from 7.8 to 9.2 and their activities for the removal of IBU were evaluated. The initial rates of defective UiO-66-7.8 was 60 times higher compared to conventional UiO-66 materials (UiO-66-11.5). In addition, the maximum IBU equilibrium capacity was gradually enhanced with increasing the defective density and decreasing the particle size from UiO-66-11.5 to UiO-66-7.8. The as-prepared materials were proved to be highly efficient in removing these detected ones within 3 min (below the detection limits).234 Another study showed that among various controls tested, a higher degree of structural damage within the framework of UiO-66 exhibited 320 mg g−1 of diclofenac sodium (DCF) adsorption capacity due to the presence of a high population of defective (active) sites. This value was much higher compared to perfect UiO-66 solid (54 mg g−1).235
In addition to defective UiO-type MOFs, Li and Prasetya synthesized the defective MOF-801 NPs (d-MOF-801, a defective Zr-fumarate MOF) at room temperature by using water as a medium rather than dimethylformamide (Fig. 4).236 The performance of the defective MOF-801 was tested in the adsorption of DCF from water streams. A series of defective MOF-801 were prepared by varying the concentration of the modulator (HFo) relative to the ZrCl4. Among the various solids screened, the highest adsorption capacity for the defective MOF-801 (d-MOF-801(35) with 35 equivalent of HFo) was 680 mg g−1, which is nearly 4 times higher compared to non-defective MOF-801. This superior performance of the defective MOF-801 was due to its pore enlargement thus offering a facile pathway to access the adsorption sites.
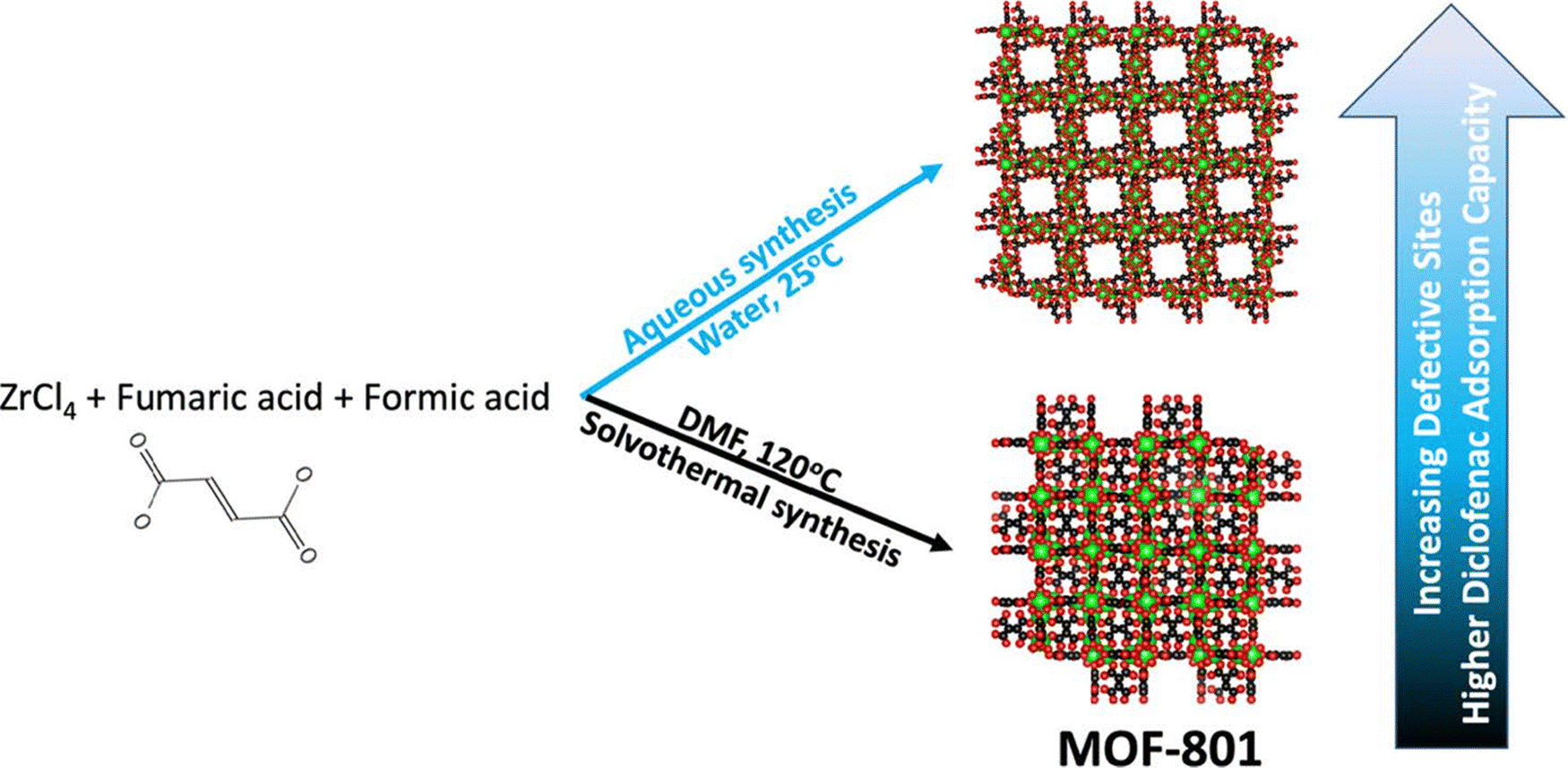 | ||
| Fig. 4 Schematic synthesis of defective MOF-801 for DCF adsorption. Reprinted from ref. 236 Copyright 2022 Elsevier. | ||
Bimetallic MOFs, Hf-UiO-66 and Ti-UiO-66 have been synthesized through the post-synthetic modification of UiO-66 and the activity of these solids was studied in the adsorption cefoperazone.237 Among the various solids screened for this adsorption, Hf-UiO-66 showed 346 mg g−1 adsorption capacity of cefoperazone which is higher than that of UiO-66. This enhanced activity of Hf-UiO-66 is due to the introduction of Hf(IV) by changing the coordination environment in UiO-66 solid, thus creating defective sites by changing the surface charges and enlarged pores. A series of analysis (FT-IR, XPS, zeta potential) has confirmed various driving forces like electrostatic attraction, π–π stacking, hydrogen bonding and metal complexation that are responsible for high adsorption capacity of the bimetallic MOFs. In addition, Hf-UiO-66 retained its activity up to five reuses. As an aside, Hf(IV) and Ti(IV) were initially thought to incorporate via exchange with node Zr(IV) ions – and, indeed, many papers posit this mechanism. Subsequent works by Cohen and co-workers and Gascon and co-workers have demonstrated that exchange does not occur.136,203 Instead, incorporation is better described as grafting or deposition. The absence of exchange-type incorporation of Ti(IV) can be understood both in terms of the resistance of node-site zirconium ions to substitution and to the inability of Ti(IV) to coordinate more than six atoms. Zr(IV), in contrast, can accommodate eight.
In another precedent, a set of controllable hierarchically porous Zr-based MOFs were designed by using series of monocarboxylic acids possessing different kinetic diameters like HAc (2 Å), pentanoic acid (5 Å), n-caprylic acid (8 Å), n-dodecanoic acid (DDA, 12 Å), n-myristic acid (14 Å), n-pentacyclic acid (15 Å) and n-palmitic acid (16 Å) as modulators. These selected carboxylic acids exhibit higher pKa values ranging between 4.74–4.89 compared to BDC (pKa = 3.51), thus they could competitively coordinate with the hexa-zirconium clusters and be completely expelled later to create hierarchical pores. Thus, fifteen isostructural MOFs were obtained with different surface areas and pore sizes. The highest surface area was attained using pentanoic acid as the modulator, while the use of DDA introduced a large number of mesoporous than the others. The adsorption behaviour of these solids was measured with seventeen pharmaceuticals. This study has shown that the influence of the mesoporous formation process can overcome the disadvantages of competitive interaction for active sites in the adsorption process. The adsorption rates were found to in the order of UiO-66 (12A-high) > UiO-67 > MIL-101. Furthermore, complicated co-existed constitutes with large molecular sizes could not hinder the adsorption process due to the strong size exclusion effect.195
2.1.1.4. Organoarsenic compound removal. The increasing use of organoarsenics, such as 4-hydroxy-3-nitrobenzenearsenic acid (Roxarsone, ROX) and 4-aminobenzenearsenic acid (p-arsenilic acid, p-ASA) as feed additives has resulted in a high discharge of organic wastes into the environment. Moreover, these compounds can degrade into toxic inorganic arsenic species.269
Li and coworkers presented defect engineering strategy in UiO-66 using variable amounts of BA as a modulator during the framework construction, resulting in highly porous MOFs, named as UiO-66-2, UiO-66-10, and UiO-66-20 (2, 10, 20 are the molar equivalents of BA used in the synthetic procedure), with tunable pore size and linker vacancy defects.212 Remarkably, it was observed that the increase of BA not only enhanced BET surface area and pore volume of MOFs, but also resulted in gradual deviation of the isotherm types from type I (microporous structure) to type IV (mesoporous structure). Among the synthesized UiO-66s, UiO-66-20 revealed the highest BET surface area and additional pore volume from the mesopore sizes of 1.7 and 2.3 nm, and thereby, exhibited the highest adsorption capacity of 730 mg g−1 for removal of ROX (adsorption conditions: T = 25 °C, pH 4, time = 30 min, initial adsorbate solution = 0.1 g L−1). The defects in UiO-66 not only led to an improved porosity, but also increased the number of Zr-OH groups which are mainly involved as active adsorption sites for chemical bonding and efficient capture of ROX.
Lin et al. in 2019 described a defective amino-functionalized UiO-66 (UiO-66-D-NH2) for the removal of p-ASA and ROX as emerging pollutants in the environment.238 The research work indicated maximum adsorption capacities in trend of UiO-66-D-NH2 > UiO-66-D > UiO-66 > ZrO2 for both p-ASA and ROX. The combination effects of defective sites (more Zr-OH sites) and amino functionary are worked together for the enhanced adsorption efficiency of UiO-66-D-NH2 (Fig. 5).
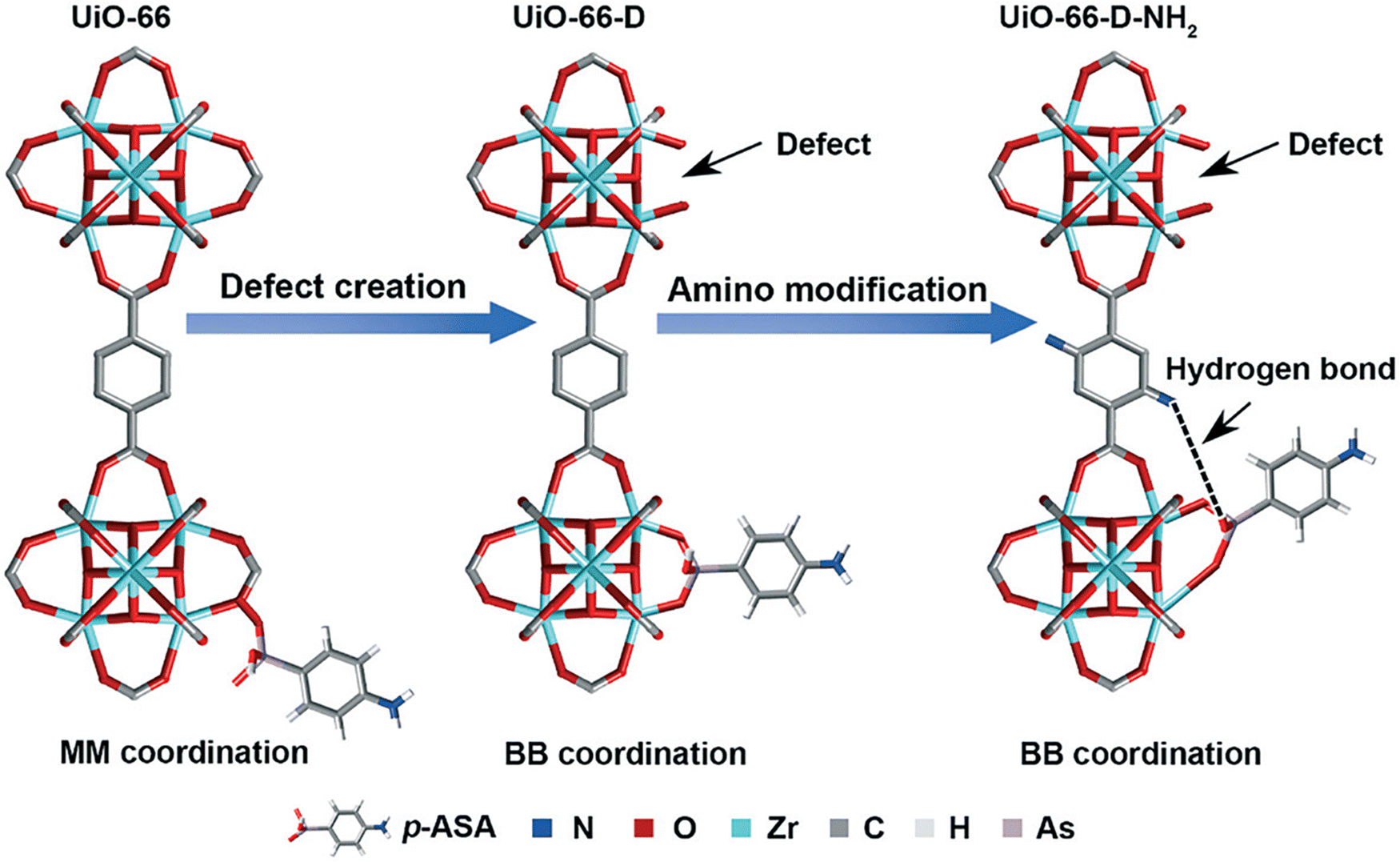 | ||
| Fig. 5 Synergistic mechanism of UiO-66-D-NH2 for adsorption of organoarsenic (MM = monodentate mononuclear As–Zr complex, BB = bidentate binuclear As–Zr complex). Reprinted from ref. 238 Copyright 2019 Royal Society of Chemistry. | ||
Recently, several water-stable Zr-MOFs have been studied for the adsorptive removal of p-ASA and ROX from water.270 Among the considered MOFs, MOF-808F (Zr6(μ3-O)4(μ3-OH)4(Formate)6(BTC)2) with pores of 1.0 and 1.8 nm showed a remarkable maximum adsorption of capacity of 710 mg g−1 for ROX (adsorption conditions: T = 25 °C, pH 4, time = 4 h, initial adsorbate solution = 25 mg L−1), which is slightly lower than that of the defective UiO-66 as mentioned before (UiO-66-20, 730 mg g−1).212 Zr-MOFs such as MOF-808, defective UiO-66 and NU-1000 containing Zr(IV) CUSs with a large number of Zr-OH(H2O) groups demonstrated strong coordination interactions between the MOF and p-ASA. While the synergistic effect of electrostatic, hydrogen bonding and π–π stacking were also found for p-ASA adsorption over MOF-808F, defect-free UiO-67, DUT-67, and NU-1000B (NU-1000 containing benzoate ligands) without or with very few Zr-OH(H2O) groups.270
2.1.1.5. Other organic pollutants removal. Perfluoroalkyl sulfonates (PFASs) such as perfluorooctanesulfonate (PFOS) and perfluorobutanesulfonate (PFBS), a group of emerging organic pollutants (EOCs), are found in wastewater released from their use in commercial and industrial applications. They are persistent in the environment and can prompt health problems such as disruption of the immune system. Also, it was concluded that there is a probable link between exposure to PFAS and cancers.271 Therefore, there is a need to examine porous MOFs as adsorbents for fast and high adsorption capacities of PFASs.
Clark et al. studied the removal of PFOS and PFBS in simulated wastewater by defective UiO-66-X materials.239 Different amounts of defects were presented in the UiO-66 framework (UiO-66-X materials, X symbolizes the vol% of conc. HCl; 10, 25, or 50) by varying amounts of concentrated HCl during the solvothermal synthesis of MOF. Defective UiO-66 (namely, UiO-66-10, UiO-66-25, and UiO-66-50) exhibited remarkable adsorption uptakes and fast kinetics for PFASs (diagonal dimension of PFOS as larger analogue is ∼1.6 nm) than the defective-free structure, DF-UiO-66, due to the introduction of larger pores (∼2.0 nm), the enlarged internal surface area and more concentration of Zr(IV) CUSs that can interact with the sulfonate moiety of PFASs. UiO-66-10 presented expressively a higher adsorption uptake than to UiO-66-25 (with the same surface area and pore structure of UiO-66-10) and UiO-66-50. The maximum adsorption capacities of 1.24 mmol g−1 and 1.74 mmol g−1 were found, respectively, for PFOS and PFBS (Condition: initial concentration = 500 mg L−1, sorbent loading = 0.5 g L−1, pH = 5, time = 60 min). The presence of co-existing ions such as chloride showed a slight effect on PFOS adsorption, whereas anionic sulfate and chromate competed for the adsorption sites in the MOF. By using 50 vol% of HCl (a high ZrCl4:HCl molar ratio, ∼1:180), an unexpected UiO-66-50 with hcp crystalline phase (versus common fcu-phase MOF) and a low BET surface area was formed. This is due to the presence of a denser framework caused by condensation of the Zr6 cluster to Zr12 double cluster during the MOF assembly. The lower adsorption capacity of UiO-66-25 than UiO-66-10 is due to both the electrostatic repulsion between ionized PFOS (pKa = −3.27) and increased amounts of terminal chloride on nodes (at defect sites) and decreased framework hydrophobicity as a result of more missing-linker defects in the structure. The present work exhibited not only the effect of structural MOF defects on surface charge and hydrophobicity of framework in the adsorption of PFASs, but also highlighted that a good balance between increased amount of Zr(IV) CUSs and decreased hydrophobicity caused by framework defects is needed for an effective adsorption process.
Volatile organic compounds (VOCs) are an important class of indoor air pollutants that can cause serious environmental and health problems.272,273 Formaldehyde (HCHO), benzene, and toluene are representative VOCs. In 2019, Ezugwu et al. used defective UiO-66-NH2 nano-octahedra (UA-x, where x (0.25, 0.5, or 1) is mole ratio of NH2-BDC to total BDC linkers (NH2-BDC plus BDC), NH2-BDC = 2-aminoterephthalic acid) for efficient HCHO capture and its regenerative conversion into less toxic molecules of HFo and methanol via Cannizzaro-like disproportionation reaction (Fig. 6).240 The MOF (UA-1) revealed a superior HCHO uptake of 49 mg g−1 in about 10 minutes at room temperature. The remarkable performance is mostly ascribed to the well-defined MOF structure, large porosity, and the abundant availability basic amino groups, which are in close proximity to a large number of Lewis acidic Zr-sites in the framework.
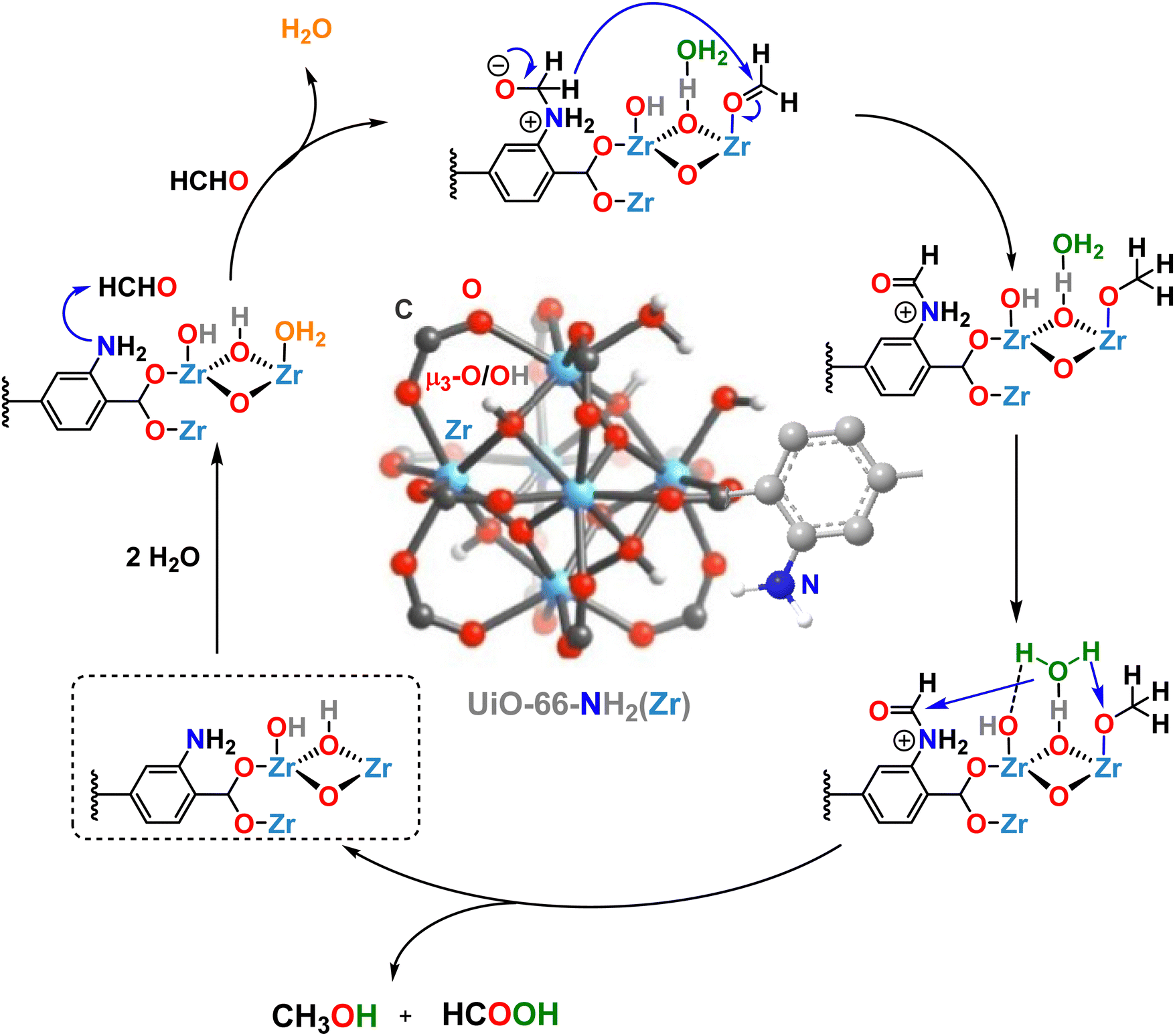 | ||
| Fig. 6 Suggested adsorption and Cannizzaro-type conversion mechanism of HCHO over defective UiO-66(Zr)-NH2.240 | ||
Jiao and coworkers extended the concept of defect to synthesize two defective UiO-66 (denoted here D) materials, with and without TFA compensating groups, so-called D200 and D320 (the number refers to the heating temperature), by varying the concentration of TFA modulator in the MOF synthesis followed by post-synthetic heat treatment, respectively, at 200 °C and 320 °C under reduced pressure (Fig. 7).241 The BET surface area of the pristine UiO-66 was 1180 m2 g−1 (pore volume = 0.56 cm3 g−1). For D200, the addition of TFA (20 equivalent) to the synthesis mixture and heating the as-prepared solid at 200 °C caused a higher BET surface area of 1800 m2 g−1 with a pore volume of 0.84 cm3 g−1. From the pore-size distributions, D200 showed supermicropores compared to the parent UiO-66 (ca. 0.6 nm). These higher values are related to the formation of missing cluster defects in the frameworks. The obtained solid at 320 °C under vacuum, D320 had a much lower BET surface area of 850 m2 g−1 and pore volume of 0.69 cm3 g−1 than the parent MOF. This is due to the total removal of coordinated TFA groups to Zr sites and consequently partial conversion to amorphous phase of the defective UiO-66 structure and the formation of ZrO2 nanorods. As a result of the removal of coordinated TFA, however, D320 exhibited larger pore sizes of 5 nm and 9 nm, typical mesoporous materials. Then, they investigated the defective UiO-66 for adsorption of SO2 (kinetic diameter = 0.41 nm), benzene (0.58 nm), and cyclohexane (∼0.61 nm). In comparison to the pristine UiO-66, the defective UiO-66, D200, could adsorb significantly more of each guest, with the most striking enhancement being found for the largest guest, cyclohexane. Further, the incorporation of trifluoroacetate groups in defective UiO-66, D200, not only resulted in an increase in the MOF pore size but also enhanced chemical stability against water and acidic media.
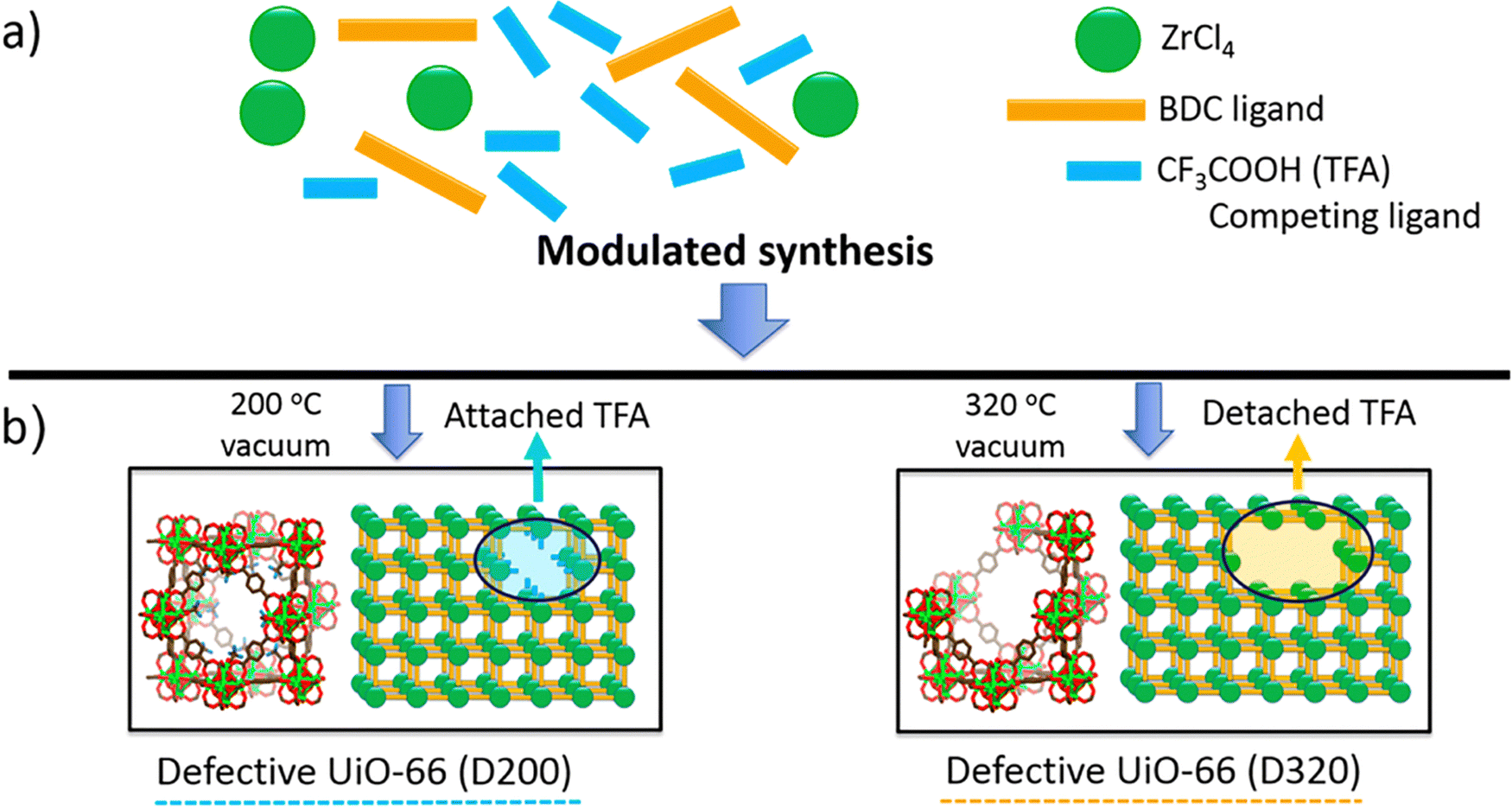 | ||
| Fig. 7 (a) Illustration of defective UiO-66 with TFA and (b) proposed structural conversions after post-synthetic heat reaction. Reprinted with permission from ref. 241 Copyright 2017 American Chemical Society. | ||
In another work, defective UiO-66 materials modified with CTAB surfactant were considered as adsorbents for removal of toluene.197 The MOFs are called CTAB-U-x (x, which is the molar ratio of CTAB:Zr(IV), is 0.3, 0.5, or 1). CTAB considerably could affect porosity, surface area, and morphology of the framework. As a result, the BET surface area and total pore volume of CTAB-U-0.5 were lower than to UiO-66 (1335 m2 g−1 and pore volume of 0.83 cm3 g−1) and the others. In addition, compared to UiO-66 with agglomerated small particles ranging from 150 to 200 nm, CTAB-U-0.5 showed agglomerated small spherical particles of 200–300 nm. Interestingly, CTAB-U-0.5 exhibited a greater adsorption capacity of 275 mg g−1 than to UiO-66 (150 mg g−1), CTAB-U-0.3 (180 mg g−1), and CTAB-U-1 (200 mg g−1), more likely due to the being of missing-linker defect sites and stronger π–π interactions. Similarly, defective UiO-66-NH2 using CTAB as a template was prepared and examined for toluene capture.198 Among the synthesized C-U-N-X MOFs (C, U-N, and X signify, respectively, CTAB, UiO-66-NH2 and the molar ratio of CTAB/Zr(IV): 0.3, 0.5 and 1), C-U-N-0.5 revealed the highest toluene uptake of 230 mg g−1, which is 1.4-fold higher than to defect-free UiO-66-NH2 (568 m2 g−1 and pore volume of 0.36 cm3 g−1), because of the synergistic effects of the π–π, –NH2, OH–CH3 and OH–π hydrogen bonding interactions. Another noticeable example for toluene adsorption was reported by Lu and coworkers, demonstrating the design of a modified defective UiO-66, UiO-66/MOF-5 (M-U-0.01, where 0.01 is the ratio of Zr(IV)/MOF-5), by using MOF-5 as a template during the synthesis of UiO-66.242 The results showed that the defective UiO-66/MOF-5 had the higher toluene sorption property than to UiO-66 (1.7 times)197 and displayed good thermal stability and reproducibility.
In 2020, some defective Zr-based biphenyl dicarboxylate MOFs, defective UiO-67s, were prepared under microwave-assisted continuous flow by using various monocarboxylic acids (HFo, HAc, propionic acid (HPr), and BA) as modulators. Notably, the synthesis time was only 10 min.243 The effects of both the modulator acidity and concentration on the formation of missing-linker and missing-cluster defects in UiO-67 were investigated. The pore structure, pore size distributions, surface area, and morphology of UiO-67 were controlled by the modulator. After that, the adsorption of toluene by the defective UiO-67 samples was explored. In the presence of BA at the short time of 10 min, no solid crystal was obtained. However, the use of HFo as the modulator with a HFo/Zr(IV) ratio of 40:1 equiv. resulted in the formation of UiO-67-HFo-40 in 76% yield. Particles of UiO-67-HFo-40 were characterized by unusually small, irregular morphology, and by low crystallinity. Large particles/crystallites were obtained by increasing the number of equivalents of HFo to 60. A further increase, to 80 equivalents, engendered the formation of crystallites of octahedral morphology. Defect-free UiO-67 contains 6 biphenyl dicarboxylate (BPDC) linkers per Zr6-node. Consistent with missing-linker formation, UiO-67-HFo-40, UiO-67-HFo-50, UiO-67-HFo-60, UiO-67-HFo-80, UiO-67-HAc, and UiO-67-HPr were found to contain, respectively, 4.98, 4.32, 4.08, 3.82, 4.23, and 4.36 linkers per node. The porosities were also assessed via analysis of N2 adsorption isotherms collected at 77 K. The following trend was observed: UiO-67-HFo-60 > UiO-67-HAc > UiO-67-HPr ≈ UiO-67-HFo-50 > UiO-67-HFo-80 > UiO-67-HFo-40. Note that pore volume is correlated with BET surface area (as expected), and that BET surface areas for defective UiO-type MOFs can exceed the theoretical maxima for perfectly crystalline, defect-free MOFs. Indeed, the experimental observation of surface areas that exceed those obtained computationally for perfect UiO-type MOFs is a good indication of the presence of missing-linker and missing-node defects. The pore sizes in defect-free UiO-67 are 1.2 and 1.6 nm, while defective MOF samples showed sizes at about 0.9 and 1.9 nm. The modulator can deprotonate in solution, and consequently, can compete with the linker for coordination of Zr(IV) during the MOF synthesis. HFo (pKa = 3.77) is stronger acid than both HAc (pKa = 4.74) and HPr (pKa = 4.87), and thus, should more effectively compete with linkers for node coordination. In turn, higher defect densities and higher VOC capacities might be expected. Consistent with these simple ideas, defective UiO-67-HFo exhibited a higher toluene capacity (470 mg g−1) than UiO-67-HAc (420 mg g−1), UiO-67-HPr (400 mg g−1), and previously reported MOF materials such as UiO-66 (130 mg g−1),274 UiO-66-NH2 (250 mg g−1),275 and the CTAB-modified UiO-66 (275 mg g−1).197
Liu et al. studied experimentally and theoretically that the degree of deficiency in UiO-67 can be controlled by acid-modulator strategy (HAc, HFo and BA) for exceptional toluene uptake, albeit in the presence of water vapors.244 The numbers of missing-linker and compensating-linker defects in UiO-67 materials were estimated by TGA and 1H NMR, respectively. The highest toluene capture (480 mg g−1) was found with the UiO-67-BA due to its highest missing linker defects (∼3.4) and higher compensating linker defects, which displayed 2-folds higher than to conventional UiO-67 (240 mg g−1). Adsorption mechanisms via density functional theory (DFT) calculations were proved that the cooperation effects between π–π, OH–CH3, and OH–π hydrogen bonding improved toluene adsorption, and OH–π was the most substantial interaction.
In another related precedent, Zhang and coworkers have evaluated the effects of the molar ratio of Zr6 node:BPDC:BA modulator (1:1:X (X = 0, 3, 6, 10, and 14)) on defect density within UiO-67, named as 67-ben-X.245 They then examined the effects of defects on VOC uptake capacity and kinetics. The VOCs examined included such as toluene, acetone, benzene, and ortho-, meta-, and para-xylene, and it was found that UiO-67-BA-10 (67-ben-10) achieved the highest adsorption capacities. Theory-based comparisons between uptakes of various VOCs yielded results qualitatively consistent with experiments, and in line with expectations based on differences in the polarizability and molecular dimensions of VOCs.
Phthalic acid esters are a class of endocrine-disrupting chemicals (EDCs), which have received growing attention in recent years due to their high demand in plastic products.276–278 It is crucial to eliminate such EDCs from water because of their harmful effects on humans and aquatic life through contaminated potable water. In this context, Liu and coworkers have reported the use of HCl and different amounts of BA as modulators to adjust the quantity of missing-linker defects in UiO-67, as denoted UiO-67-xBA (x represented the molar ratio of BA to ligands).246 Then, the defective UiO-67 structures were tested for the adsorptive removal of dimethyl phthalate (DMP, the molecular size is 7.8 × 6 Å) and phthalic acid (PA, the molecular size of PA is about 6 × 6 Å) in water. UiO-67-30BA [calculated molecular formula: Zr6O4(OH)4(BPDC)4.0(H5C6OO)1.6Cl0.4(OH)2.0] revealed the highest surface area as compared to those of UiO-67-3BA, UiO-67-10BA, and UiO-67 (BET surface area = 1810 m2 g−1, pore volume 0.62 cm3 g−1). The use of BA not only enhanced the crystallinity and the porosity of the UiO-67 but also could maintain almost the quantity of benzene rings in UiO-67 and π–π interactions as the result of the BPDC linker replacement with BA during formation of defects in MOFs. Interestingly, UiO-67-30BA with the highest defects indicated high PA and DMP adsorption uptake in comparison to that of the others (equilibrium times for uptake of PA and DMP on the MOF were about 1 and 1.6 h, respectively). In addition, diffusion and access of molecules to pores can affect the kinetics and efficiency of the adsorption process. It was found that electrostatic and π–π interactions were enhanced between PA and UiO-67, while in the adsorption of DMP, π–π interactions were important.
A facile etching method with monocarboxylic acids (HAc, TFA and TCA) was reported for the induction of defective sites into UiO-66 for the efficient removal of PA and DMP from aqueous solutions.190 Compared to UiO-66, defective UiO-66 solids showed increased surface area and pore volume, due to the introduction of the mesopores to the framework, and thereby, enhanced the adsorption capacity for PA and DMP (18.05% up of PA and 41.59% up of DMP to UiO-66). Among the prepared MOF materials, UiO-66-1.6HAc (1.6 refers to concentration of HAc of 1.6 mol L−1) and UiO-66-0.2TCA with mediate defect exhibited, respectively, the largest pore volume and the highest specific surface area.
Iacomi and coworkers reported a combined computational and experimental study on the adsorption of C3 hydrocarbons (propane and propylene), in a defective Zr-fumarate MOF (MOF-801).247 Defective Zr-fumarate-MOF was obtained by adding HFo (100 equiv.) as a modulator to the MOF precursors under a hydrothermal condition. The defective sample showed the appearance of new pores (∼1–2 nm) larger than those of tetrahedral and octahedral cages (0.5–1.0 nm). In this work, the presence of missing-linker defects indicated a slight effect on the equilibrium adsorption behavior of C3 hydrocarbons. This is more likely the result of the small size of the fumarate linker which after its removal or its replacement with formate cannot create significantly larger pores in the framework. Dramatically, the experiments were simulated by a model of 1/8 of missing-clusters in the MOF (Fig. 8). It is interesting to note that TGA and 1H NMR proved ineffective for distinguishing between missing-linker and missing-cluster defects. However, the XRD patterns supported the presence of missing-cluster defects. The thermodynamic propane-propylene separation data indicated that the existence of defects, mostly, resulted in an unexpected change in the selectivity from propylene to propane, when compared with that of observed in the pristine structure. The pristine structure was selective for propylene, based on the diffusion and size exclusion (kinetic diameter of propylene and propane are 0.40 nm and 0.43 nm, respectively).
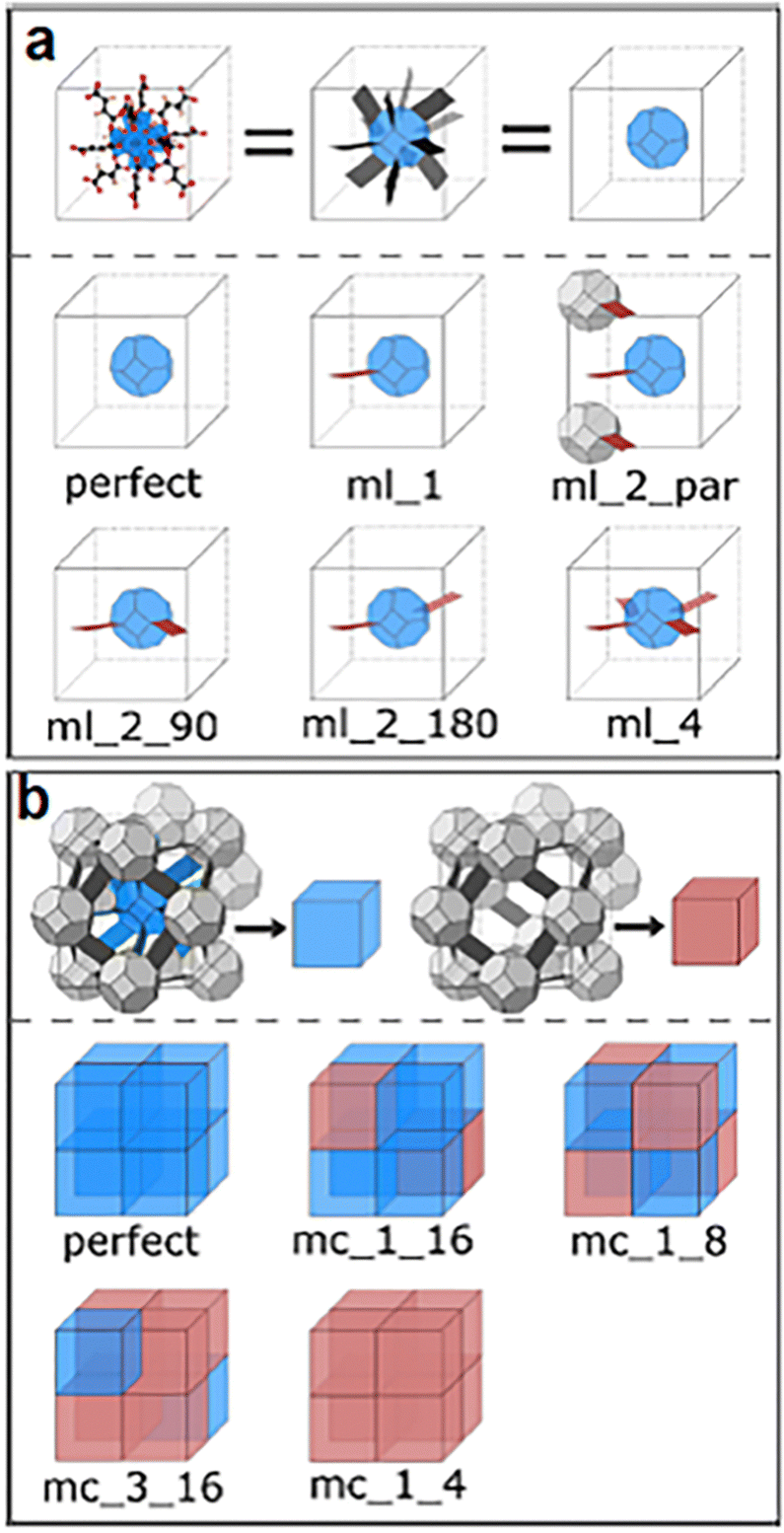 | ||
| Fig. 8 Depiction of (a) missing-linker defects (ml is referred to missing linker, followed by the number and, where applicable, the relative positions where linkers were detached) and (b) missing-cluster defects and relating supercells (mc_1_4, mc_3_16, mc_1_8, and mc_1_16 (mc = missing cluster) stand for defects with 1/4, 3/16, 1/8, and 1/16 of the total number of clusters removed). Reprinted with permission from ref. 247 Copyright 2019 American Chemical Society. | ||
| Defective Zr-MOF | Strategy for defect formation | BET surface areaa (m2 g−1) | Pore size (nm)/pore volumea (cm3 g−1) | Pollutant (or analyte)/adsorption capacity (mg g−1) | Selectivity | Ref. |
|---|---|---|---|---|---|---|
| a Dash indicates that values were not reported. | ||||||
| UiO-66-1 | HAc-modulated synthesis | 750 | 1.41/0.42 | Phosphate/415 | Phosphate over interfering ions/molecules | 279 |
| UiO-66-2 | 860 | 1.47/0.46 | Phosphate/326 | |||
| UiO-66-3 | 1600 | 1.54/0.51 | Phosphate/286 | |||
| HCl-UiO-66 | HCl-modulated synthesis | 1554 | <2 nm/— | Phosphate/1.60 (mmol g−1) | Phosphate over interfering ions/molecules | 280 |
| HCl-UiO-66-NH2 | 1255 | <2 nm/— | Phosphate/0.70 (mmol g−1) | |||
| HP-UiO-OA | Template-assisted strategy | 650 | 2.5–16/0.51 | Phosphate/190 | Phosphate over competitive anions/molecules | 281 |
| HP-UiO-BA | Butyric acid-modulated synthesis | 950 | <∼8/0.42 | Phosphate/80 | ||
| HCl-UiO-66 | HCl-modulated synthesis | 1650 | 1.2–1.6/0.68 | Arsenate (As(V))/∼100 (3 As(V) per Zr6 node) | Anionic As(V) oxyanions over As(III) | 282 |
| HCl-UiO-66(SH)2 | HCl-modulated synthesis | 750 | 1.2–1.6/0.32 | As(III)/10 (0.26 As(III) per Zr6 node); As(V)/40 (1 As(V) per Zr6 node) | — | |
| UiO-66-36TFA | TFA-modulated synthesis | 1690 | 1.5 and 2/0.52 | As(V)/200 | As(V) over competitive anions | 186 |
| HP-UiO-66-40% | Mixed linker and post-thermolysis | 550 | <10/0.40 | As(V)/250 | As(V) over interfering anions | 283 |
| HP-UiO-66-30% | 715 | <10/0.50 | As(V)/210 | — | ||
| HP-UiO-66-0% | 830 | <2/0.44 | As(V)/84 | — | ||
| Fe-UiO-66-M | BA and post-synesthetic Fe incorporation | 980 | 5.35/0.72 | As(V)/340 | — | 284 |
| UiO-66-HCl | HCl-modulated synthesis | 1500 | 0.8 and 1.2/0.75 | Se(VI) oxyanions/87 | — | 192 |
| UiO-66-12TFA | TFA-modulated synthesis | 1530 | 0.8 and 1.2/0.61 | Se(VI) oxyanions/63 | ||
| UiO-66-36TFA | TFA-modulated synthesis | 1255 | 1.0 and 1.2/0.64 | Se(VI) oxyanions/55 | ||
| UiO-66-Ac | HAc-modulated synthesis | 1320 | 0.8 and 1.8/0.50 | Se(VI) oxyanions/46 | ||
| PCN-134 | Mixed linker and BA-modulated synthesis | 1950 | — | Cr(VI)/0.19 (mmol g−1) | Cr(VI) over interfering anions | 285 |
| UiO-66-def | HCl-modulated synthesis | 1165 | — | Cr(VI)/22 | — | 286 |
| UiO-66-(OH)2-def | — | — | Cr(VI)/75 | — | ||
| U-OCl-Ti | ZrOCl2·8H2O as the precursor and post incorporation of titanium | 460 | >2.0/— | Cr(V)/21 | — | 287 |
| UiO-66-20D | BA-modulated synthesis | 1730 | ∼1.0–2.1/0.84 | U(VI)/350 | U(VI) over competing metal ions | 288 |
| HP-UiO-66-35 | DDA-modulated synthesis | 705 | 15.2/— | U(VI)/120 | 289 | |
| HP-UiO-66-15 | 780 | 11.5/— | U(VI)/1200 | |||
| M808-4 | Mixed linker and HFo-modulated synthesis | 770 | ∼3.29/0.45 | U(VI)/418 | U(VI) over competing metal ions | 290 |
| M808-1 | 875 | ∼2.55/0.53 | U(VI)/318 | — | ||
| Zr-MSA-MA | MA-modulated synthesis | 175 | 3.8/0.93 | Hg(II)/715 | Hg(II) over competing metal ions | 193 |
| UiO-66-SH | BA-modulated synthesis and post treatment with HCl and mercaptoisobutyric acid | 1570 | 0.5–2.0/— | Hg(II)/785 | Hg(II) over competing metal ions | 291 |
| UiO-66(A) | HCl-modulated synthesis | 1050 | ∼1.0 and 1.5/0.75 | Pt(IV)/144 | — | 292 |
| UiO-66(B) | HCl-modulated synthesis | 680 | ∼1.0 and 1.5/0.82 | Pt(IV)/100 | — | |
| UiO-66 | HCl-modulated synthesis | 1510 | ∼1.1 and 1.6/0.63 | Pd(II)/105 | Pd(II) over competing metal ions | 219 |
| UiO-66-TFA | TFA-modulated synthesis | — | — | Sb(V)/380 | — | 293 |
| UiO-66-BA | BA-modulated synthesis | — | — | Sb(V)/306 | ||
| UiO-66-HAc | HAc-modulated synthesis | — | — | Sb(V)/197 | ||
| UiO-66-1:1–120 |
Altering the concentration of the linker and synthetic temperature | 1147 | —/0.31 | Pb(II)/125 | Pb(II) over competing metal ions | 178 |
| UiO-66-1:1–120 |
1060 | —/0.28 | Pb(II)/110 | |||
| UiO-66-1:2–120 |
1456 | —/0.39 | Pb(II)/175 | |||
| UiO-66-1:2–150 |
1240 | —/0.53 | Pb(II)/150 | |||
| SS-NH2-UiO-66-5 | Seignette salt (SS)-modulated synthesis | 125 | 26.0–50.0 and 50.0–120/0.094 | Pb(II)/186 | Pb(II) over competing metal ions | 294 |
Due to the serious problems of inorganic pollutants consisting of metal and non-metal species that could affect human health and the environment, removing them from contaminated water is required.295–299
Gu and coworkers synthesized UiO-66 materials with different sizes and morphologies by changing both the ratio of HAc as a modulator and reaction time toward a highly efficient adsorbent for the removal of phosphate from water.279 When the amount of HAc and reaction time are increased, the particle size, BET surface area, and pore size of UiO-66 were notably increased, which is caused by the missing-linker defects. It was indicated that UiO-66 (UiO-66-1) with the smallest size and lowest surface area exhibited the fastest uptake rate and highest adsorption capacity towards phosphate capture. This indicated that the exchange and replacement of BDC linkers with phosphate on Zr6-nodes is more important than the interaction between defect sites, from larger specific surface area, and phosphate. It was also concluded that HAc, contributing to the framework formation, is not able to create tremendously extra adsorption sites for coordination as increased further.
In another similar work, the adsorptive removal of phosphate from water by using high-defect-density (prepared with HCl as a modulator) versus low-defect-density (prepared with HAc as a modulator) versions of UiO-66 and UiO-66-NH2 was explored.280 As one might anticipate, the affinity of HCl-UiO-66 for phosphate exceeded that of HAc-UiO-66, and similarly the affinity of HCl-UiO-66-NH2 for phosphate exceeded that of HAc-UiO-66-NH2. In water at pH 7, the dominant form of phosphate is H2PO41−. The authors find that phosphate adsorbs at missing-linker defect sites via displacement of node aqua (and terminal hydroxo) ligands. Once these sites are filled, however, phosphate displaces the MOF linkers (Fig. 9), as evidenced, in part, by leaching of linkers into solution. The enormous affinity of phosphate (and to a lesser extent, carbonate and hydroxide) for oxy-zirconium nodes, and the ensuing framework degradation, is an Achilles’ heel for otherwise robust Zr-MOFs. Notably, Lin and co-workers showed that phosphate displacement of organic linkers (via a process they termed “topotactic ligand extraction”) can be used as an advantage to form a porous crystalline oxy-zirconium-phosphate solid displaying admirable affinity for actinide ions in aqueous solutions.300
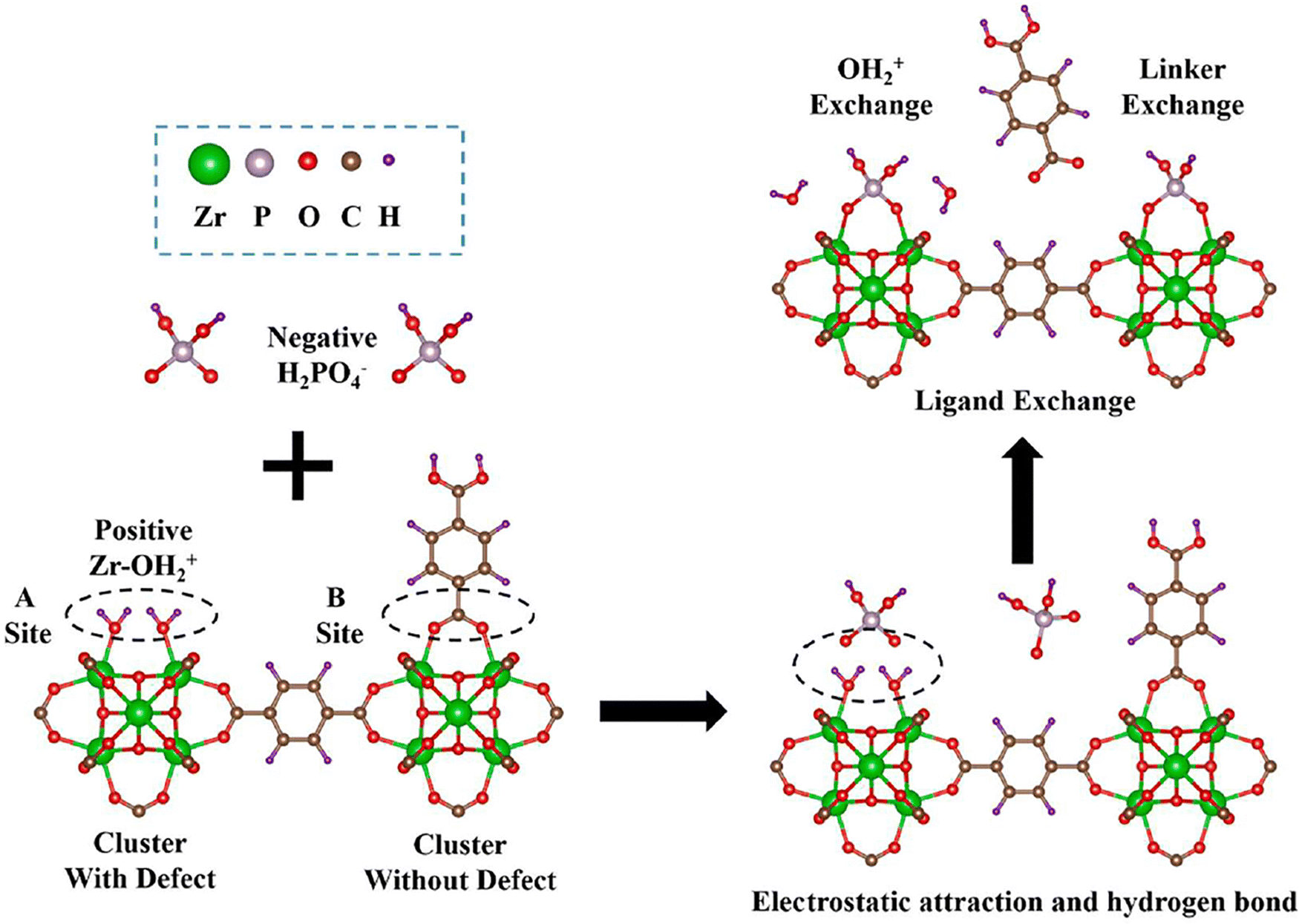 | ||
| Fig. 9 Phosphate adsorption on nodes of linker-defective UiO-66. Binding at site A entails displacement of node aqua and terminal-hydroxo ligands (rather than pairs of aqua ligands, as sketched). At site B, phosphate binds via displacement of carboxylate-terminated linkers. Reprinted with permission from ref. 280 Copyright 2021 Elsevier. | ||
UiO-type MOFs displaying hierarchical porosity (HP-MOFs) are inherently defective, especially with regard to missing clusters.281 In this aspect, a template-assisted strategy was employed for HP-MOFs by ordering the type and number of defects by utilizing monocarboxylic acids with varying chain lengths as modulators (Fig. 10). The introduced aliphatic acids with various chain lengths (C1–C8) and higher pKa values behave as capping agents to pre-coordinate on the hexa-zirconium clusters. The increased alkyl chain length provides a strong steric encumbrance and limits the replacement of such monocarboxylic acids by the BDC linker, resulting in the generation of larger pores with abundant defective sites upon the subsequent activation process. Further, as the chain length is increased, the defect density is also increased. HP-UiO-66(Zr) showed higher uptake capacity and faster sorption kinetics than the pristine UiO-66(Zr). Among various HP-MOFs tested, the HP-UiO-66-OA (OA = octanoic acid) afforded an adsorption capacity of 190 mg phosphate per g, which is 4.8 and ∼2 times greater than those found for pristine UiO-66 and UiO-67.46
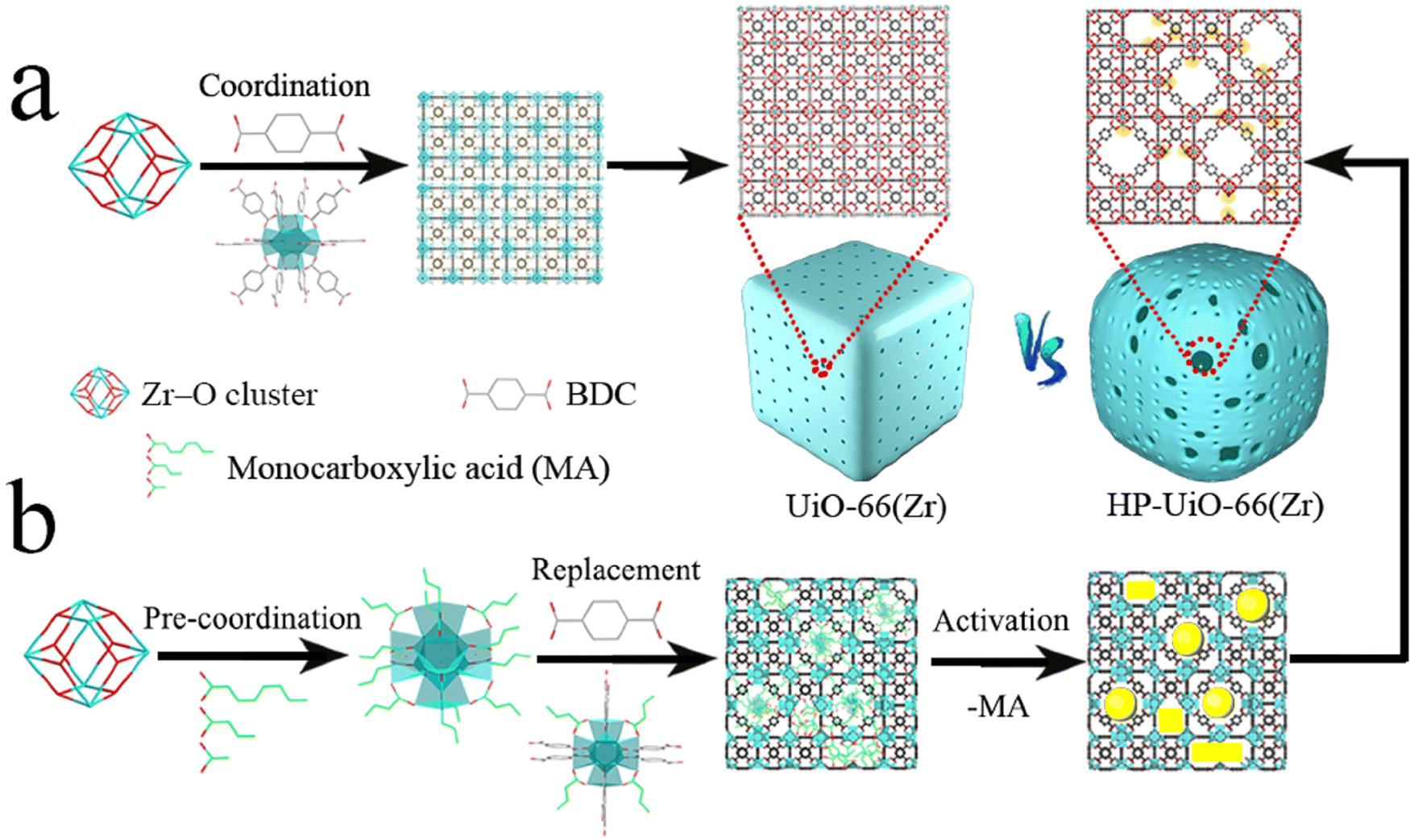 | ||
| Fig. 10 Schematic of stages in the formation of (a) pristine UiO-66(Zr) and (b) defective HP-UiO-66-MA. Reprinted with permission from ref. 281 Copyright 2021 American Chemical Society. | ||
DFT was used to predict and interpret the influence of defects and node-metal-ion identity (Hf, Zr, or Ce) on phosphate binding. Defect-containing versions of UiO-66 were found to be superior to idealized defect-free versions. Binding strength was calculated to vary in the order Hf > Zr > Ce.301
In a related study, Audu et al. described an HCl-modulator approach to the synthesis of a series of UiO-66 samples of tunable crystallite size and missing-linker defect density,282 such that defect densities a large as 3 per node could be obtained (versus ∼1 per node for samples synthesized by using HAc as a modulator, HAc-UiO-66|11/12). The higher defect densities for HCl-UiO-66 samples translated to larger pores, as well as greater numbers of potential binding sites for oxy-As(V) species. As a result, the HCl-UiO-66 showed higher uptake and faster adsorption of As(V) than the largely defect-free HAc-UiO-66. The findings suggest that both the particle size and defect density of Zr-MOFs can play key roles in As(V) adsorption. The missing-linker sites of Zr6 nodes are known to adsorb the As(V) oxyanions over the formation of Zr–O–As bonds (Fig. 11). For capture of oxy-As(III) species, an HCl-UiO-66 derivative featuring thiolated linkers proved effective. Notably, the persistence of missing-linker defects rendered the derivative functional for simultaneous capture of As(III) and As(V) species.
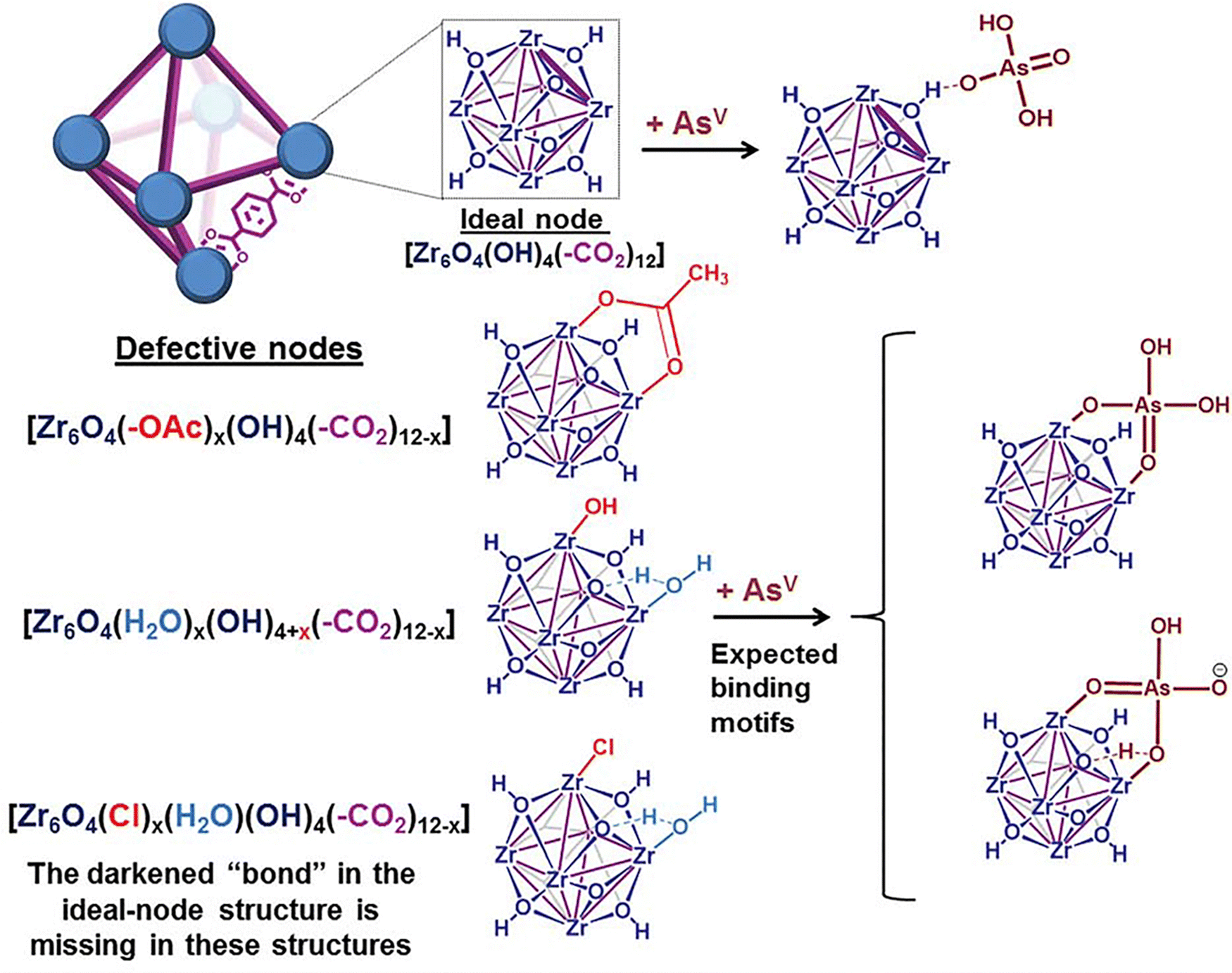 | ||
| Fig. 11 Structural scheme of ideal and missing-linker defective UiO-66, with either 12-coordinated ideal [Zr6O4(OH)4(–CO2)12] or defective [Zr6O4(OH)X(–CO2)y] nodes. Reprinted with permission from ref. 282 Copyright 2016 Royal Society of Chemistry. | ||
Assaad and coworkers described the suitability of defect-engineering of UiO-66 for arsenate (AsO34−, As(V)) removal from aqueous media.186 UiO-66 MOFs with different densities of defects were synthesized solvothermally by varying the type and the amount of the modulator (HAc or TFA). The density of defect, surface area and porosity of UiO-66 were increased as the acidity and amount of the modulator were increased. As a result, UiO-66-36TFA (36 means 36 equiv. of TFA) with the highest free adsorption/Lewis acid sites presented the highest adsorption uptake about 200 mg g−1 at pH 7, when compared to that of other samples reported in this work synthesized with less equivalents of TFA.
In another study, Xu et al. synthesized a hierarchically porous UiO-66 (HP-UiO-66) with tunable oxygen vacancies/defects and mesopores via facile selective ligand thermolysis of a series of UiO-66-X% (X%, ratio by mass of NH2-BDC:total ligand, X = 0–40).283 NH2-BDC as thermolabile ligands were selectively decomposed by adjusting the temperature, leading to generation of a large number of oxygen vacancies and abundant zirconium CUS sites in the defective MOF. As a result, the mesopores HP-UiO-66 (mesopore value = 61%) was obtained, reaching an exceptional As(V) capture capacity of up to 250 mg g−1 under neutral pH at a fast rate, and culminating in the formation of Zr–O–As bonds. Similarly, a mesoporous MOF, Fe-UiO-66-M (M stand for mesoporous) was synthesized with the high surface area of 980 m2 g−1, medium pore size of ∼5.35 nm and particle size of ∼25 nm.284 The adsorption capacity of Fe-UiO-66-M was examined in the removal of As(V) under a wide pH ranges between 1–11. The experimental outcomes exhibited that As(V) adsorption uptake reaches to 340 mg g−1 with the extraction efficiency of 94% at pH 3 under the optimized conditions. The adsorption capacity of As(V) with Fe-UiO-66-M MOF outperformed the reported adsorbents in the literature like Fe/Mg-MIL-88B,302 Fe3O4@UiO-66,303 and MIL-100 (Fe).304 The enhanced behavior of Fe-UiO-66-M MOF is evidently a result of the mesostructured crystalline structure which facilitates diffusive transportation. Furthermore, defective sites favored the electrostatic interaction and the formation of As–O–Zr and Fe–O–As bonds within the framework.
It is worth mentioning that the abundant CUS sites in defective Zr-MOFs and relevant materials play an important role in the adsorption of As(V) and As(III), and this effect is crucial even after converting the defective Zr-MOF into MOF-derived tetragonal zirconium oxide.305 Such displaceable and terminal –OH/H2O pairs on the nodes of Zr-MOFs are also capable of adsorbing oxyanions (selenite (SeO32−) and selenate (SeO42−)), as demonstrated by Howarth et al.306
Li et al. synthesized different types of Zr-MOFs using the modulator-assisted strategy to clarify the effect of different defects and “capping” ligands on the adsorptive removal of Se(VI) oxyanions.192 The use of HAc and HCl as modulators resulted in the production of Zr-MOFs with missing-linker defects including UiO-66-HAc, UiO-66-HCl and UiO-67-HCl. Whereas, DIF and TFA (sometimes also abbreviated as Trif) gave Zr-MOFs with missing-cluster defects including UiO-66-36DIF, UiO-66-12TFA (the amount of added TFA; 12 equiv.) and UiO-66-36TFA (36 equiv. TFA used) (Fig. 12).34,192 Impressively, UiO-66-HCl exhibited a superior Se(VI) adsorption uptake of 87 mg g−1 than NU-1000 (79 mg g−1, including 8-connected Zr6 nodes and pore diameter of 1.2 and 3.1 nm), MOF-808 (60 mg g−1 with 6-connected Zr6 nodes and pore diameter of 1.84 nm), UiO-66-12TFA (63 mg g−1), UiO-66-36TFA (55 mg g−1), UiO-66-HAc (46 mg g−1), and defect-free UiO-66 (34 mg g−1 with pore diameter of ∼0.7 and ∼0.8 nm), indicating that the modulator-assisted strategy can be a vital tool to create proper defects in Zr-MOFs and expand their aperture sizes to form extra binding sites for oxyanions.192 The increase in defect concentration and pore spacing values associated with the decrease in the particle sizes are responsible for the high performance of the HCl-modulated MOF. In addition, smaller capping ligands (HO−, Cl−, or H2O) on defect sites of Zr6-nodes in HCl-UiO-66 could more easily be exchanged by Se(VI) oxyanions in contrary to the monocarboxylic acid ligands.
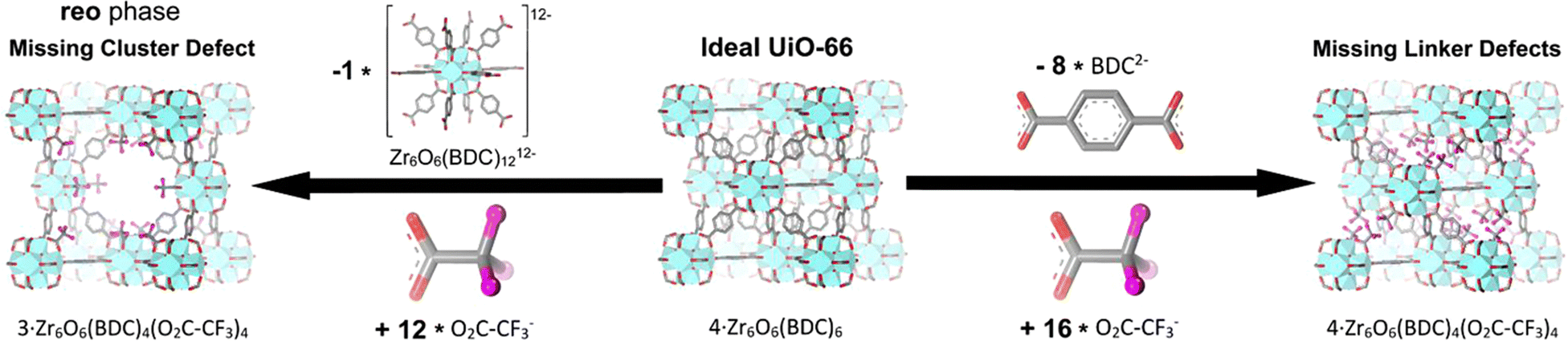 | ||
| Fig. 12 Structural scheme and compositional differences between the ideal and defective UiO-66 samples. Reprinted with permission from ref. 34 Copyright 2016 American Chemical Society. | ||
Zhou and coworkers have reported thermodynamically favored construction of mixed-linker Zr-MOFs, PCN-133 (Zr6O4[OH]4[BTB]2[DCDPS]3, BTB = benzenetribenzoate, DCDPS = 4,4′-dicarboxydiphenyl sulfone) and PCN-134 (Zr6O4[OH]6[H2O]2[BTB]2[TCPP], TCPP = tetrakis(4-carboxyphenyl)porphyrin), in which the density of structural defects and adsorption performance of MOFs could be considerably adjusted by tailoring missing auxiliary linkers (TCPP and DCDPS) and BA modulating reagent during the MOF assembly.285 For example, PCN-134 with a TCPP ratio of 25%, PCN-134-25% TCPP, displayed the highest N2 uptake, while PCN-134-22% TCPP indicated the highest adsorption of Cr2O72− (0.19 mmol g−1 within 10 min) comparing with those of the other PCN-134 MOF. The superior performance of PCN-134-22% TCPP was a result of a strong correlation between stability, porosity, and defects. The terminal hydroxyl or aqua groups on missing-linker defect sites acted as coordinating sites for dichromate capture (Scheme 4).
 | ||
| Scheme 4 Proposed adsorption pathway of dichromate onto PCN-134.285 | ||
In 2020, Saiz et al. clarified that the introduction of hydroxyl and amine functionalities on organic linkers could act synergistically with the missing-linker defects in UiO-66 derivatives to accelerate both adsorption and reduction of Cr(VI) species in aqueous solutions.286 The defective Zr-MOFs (UiO-66, UiO-66-(OH)2 and UiO-66-NH2) were prepared through adding concentrated HCl as a modulator during the synthetic process.
Feng et al. demonstrated that defect density, surface area, pore size, particle size and adsorption performance of UiO-66-NH2 towards Cr(VI) can be precisely tuned, in part by using ZrOCl2·8H2O in place of ZrCl4 as synthetic reagent and the post-synthetically incorporated Ti(IV).287 The use of ZrOCl2·8H2O as the precursor could generate hexa-zirconium nodes in the MOF that are not fully coordinated by twelve linkers. Although described as Ti(IV) substitution for Zr(IV), incorporation of Ti(IV) is better characterized as grafting to intact hexa-zirconium-oxy nodes.136,203 Under optimal synthetic and post-synthetic conditions, the obtained UiO-66-NH2(Zr/Ti) (U-OCl-Ti) material possessed smaller particle size, higher defect density, and greater Cr(VI) uptake compared to the conventionally synthesized UiO-66-NH2. XPS measurements appear to implicate both the amino substituent and the node-grafted titanium ion in binding Cr(VI). Curiously, irradiation with blue light engenders reduction of the notoriously carcinogenic Cr(VI) to much less dangerous Cr(III). The basis for light absorption is linker-localized electronic excitation of the yellow-appearing NH2-BDC component of the MOF. A plausible chromium reduction scheme is electron-transfer from the photo-excited linker to the grafted Ti(IV) ion to yield Ti(III),307 which in turn, reduces the adsorbed chromium ion by one electron. Prolonged irradiation can lead to the repetitive electron transfer to Ti(IV) and then to chromium until Cr(III) is obtained. Somewhat puzzling is the absence of reversal of the reduction process via back electron transfer to the light-absorbing linker. Presumably residual benzoate (modulator), methanol (from sample rinsing), or some other species is functioning as a sacrificial oxidant in the photo-driven scheme of chromium reduction.
Yuan and et al. described the relationship between defect engineering in UiO-66 MOF and adsorption of U(VI) ions in acidic media.288 Missing-linker defects, the number of active Zr-OH sites, BET surface area, pore volume and sorption capacity of MOFs (denoted as UiO-66-XD, XD = different equivalents of BA with respect to BDC linker) were greater than before with increasing BA concentration. Of the various examples examined, UiO-66-20D (20 equivalents of BA with respect to BDC linker) yielded the largest BET surface area and pore volume as well as the highest capacity for the uptake of U(VI).
Yin and collaborators solvothermally synthesized a hierarchically mesoporous UiO-66 (HP-UiO-66) by employing DDA as a synthetic modulator.289 Compared to the conventionally synthesized UiO-66, HP-UiO-66-35 (obtained by using a 35:1 molar ratio of DDA:ZrCl4) offers more defects and significant numbers of mesopores. HP-UiO-66-35 displayed a maximum U(VI) adsorption capacity of around 1220 mg g−1 and a rapid approach to adsorption equilibrium (∼2 min). At pH = 8 (roughly the pH of seawater), with an initial solution U(VI) concentration of 100 mg L−1 (i.e. 100 ppm (parts per million)) and a modest excess sorbent, the authors found that 87% of the dissolved U(VI) could be captured by HP-UiO-66-35 (and therefore leaving behind 13 ppm). The nominal concentration of U(VI) in seawater is considerably lower, ∼3.3 ppb (parts per billion)308 – underscoring the magnitude of the challenge of direct extraction from seawater. Notably, at pH = 8, in the absence of dissolved CO2/HCO3−, the dominant forms of dissolved U(VI) are (UO2)3(OH)5+ and (UO2)4(OH)7+ (i.e. cationic clusters), and the MOF is anticipated to carry a net negative charge. Thus, the clusters would be similar in size to the apertures of defect-free UiO-66, resulting in either size exclusion from, or very slow transport through, the MOF interior. One might expect the introduction of hierarchical mesoporosity, to greatly enhance the kinetics of U(VI) uptake, and indeed, this is the case.
In another work, a series of MOF-808 solids were prepared with adjustable missing-linker defects via the manipulation of the concentration of the carboxylic acids of BTC and BDC.290 Among these solids tested for the adsorption capacity, MOF-808 with the highest defect density, MOF-808-4, exhibited selective extraction of U(VI) in the presence of coexisting competitive metal ions in seawater. Interestingly, MOF-808-4 showed remarkable U(VI) extraction activity in real seawater with an uptake value of 5.8 mg g−1 in 28 days, which is ∼10 times higher than that found for vanadium (Fig. 13). These adsorption experiments indicate the existence of an abundance of labile node coordination sites formed during the creation of defects. The solid was used for five cycles with no significant loss of adsorption activity.
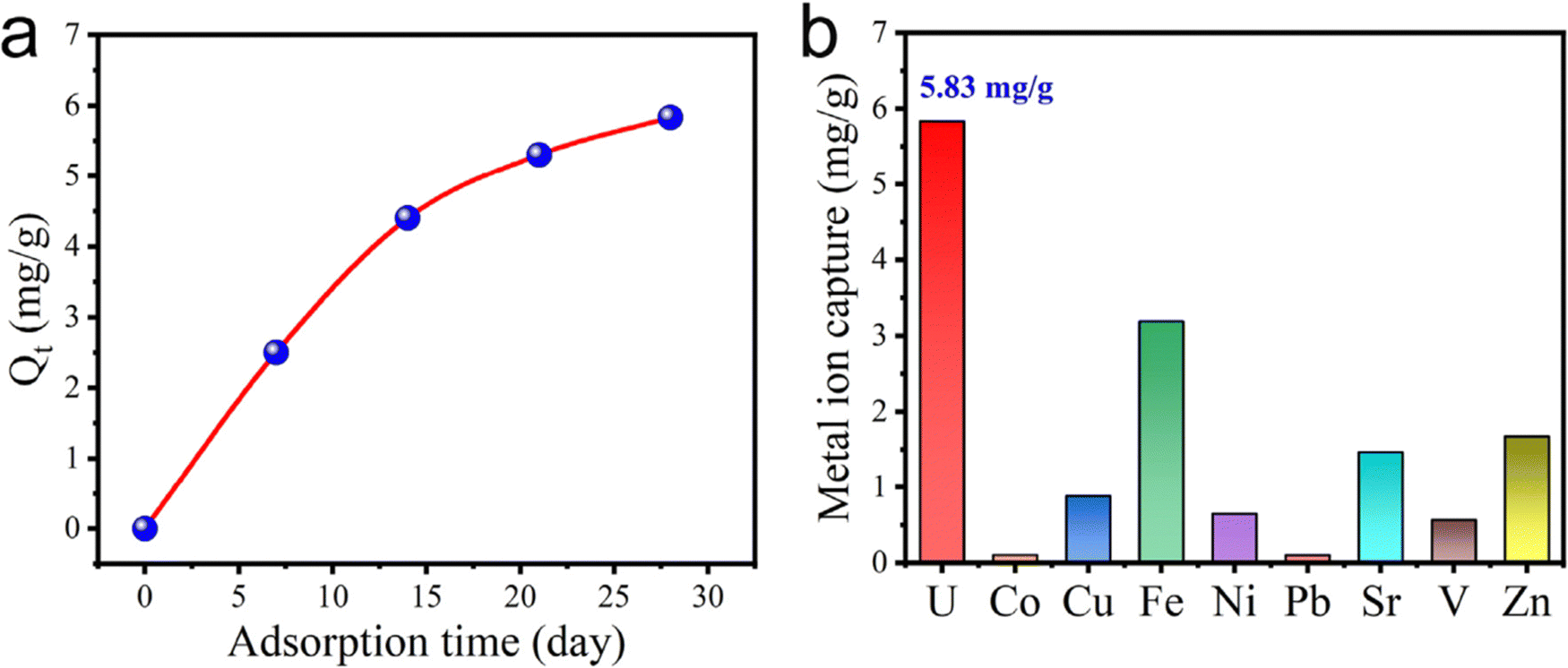 | ||
| Fig. 13 (a) Kinetics and (b) adsorption capacity of U(VI) over coexisting ions (Co(II), Cu(II), Fe(III), Ni(II), Pb(II), Sr(II), V(V), and Zn(II)) on MOF-808-4 in real seawater. Reprinted with permission from ref. 290 Copyright 2022 Elsevier. | ||
In a recent study, Zr-MSA-MA (MSA = mercaptosuccinic acid) was synthesized by using MA as the modulator (Fig. 14).193 It should be noticed that the topology and diffraction patterns of Zr-MSA MOFs are similar to those of UiO-66. One of the advantages of using MA as the modulator compared to HAc and HFo is that the former provides a high density of binding sites along with the creation of defective sites. Under the optimized reaction conditions, Zr-MSA-MA-1 showed a high adsorption capacity of 715 mg g−1 for Hg(II), which is superior to other MOF-based adsorbents such as thiol-functionalized NU-1100 (322 mg g−1),309 SH-Fe3O4@SiO2@UiO-66 (282 mg g−1),310 and UiO-66(SH)2 (236 mg g−1).311 This superior performance of this MOF is due to strong chemical bonding between Hg2+ and –SH groups.
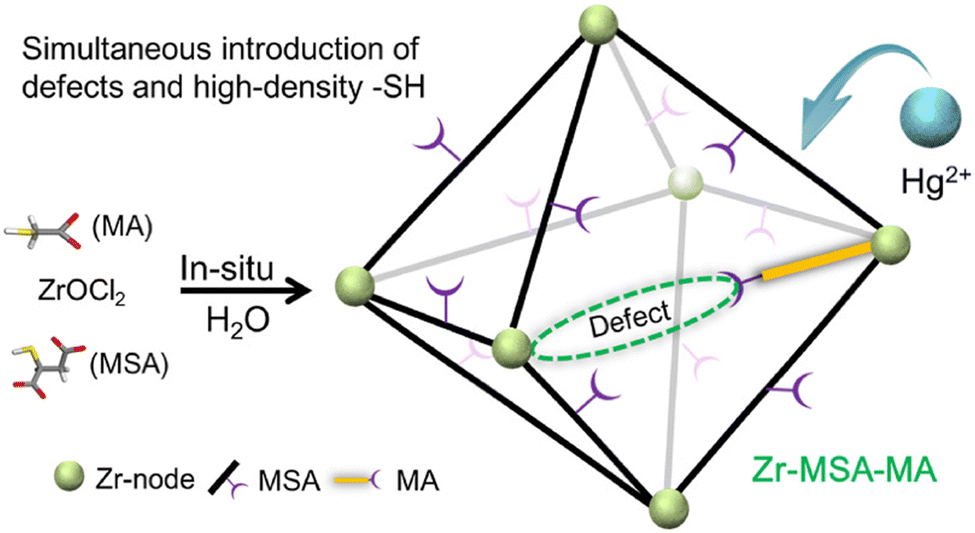 | ||
| Fig. 14 Schematic presentation of the Zr-MSA-MA framework for the extraction of mercury ions. Reprinted with permission from ref. 193 Copyright 2023 Elsevier. | ||
In another effort, at first, a defective UiO-66, was achieved by using a modulator synthetic strategy with BA (as a crystal growth modulator) and HCl treatment, then post-modified with 3-mercaptoisobutyric acid to give thiol-functionalized UiO-66, UiO-66-SH, through reaction between the formed defect sites and carboxylic group of MIA (Fig. 15).291 Comparing to the pristine UiO-66, the obtained UiO-66-SH showed a high BET surface area of 1570 m2 g−1 and was applied for fast (20 min) and selective adsorption of Hg(II) ions from aqueous media, presenting a high adsorption capacity of 785 mg g−1 at pH 4. In addition, the respective MOF showed a high adsorption toward organic mercury species (methylmercury, ethylmercury, and phenylmercury).
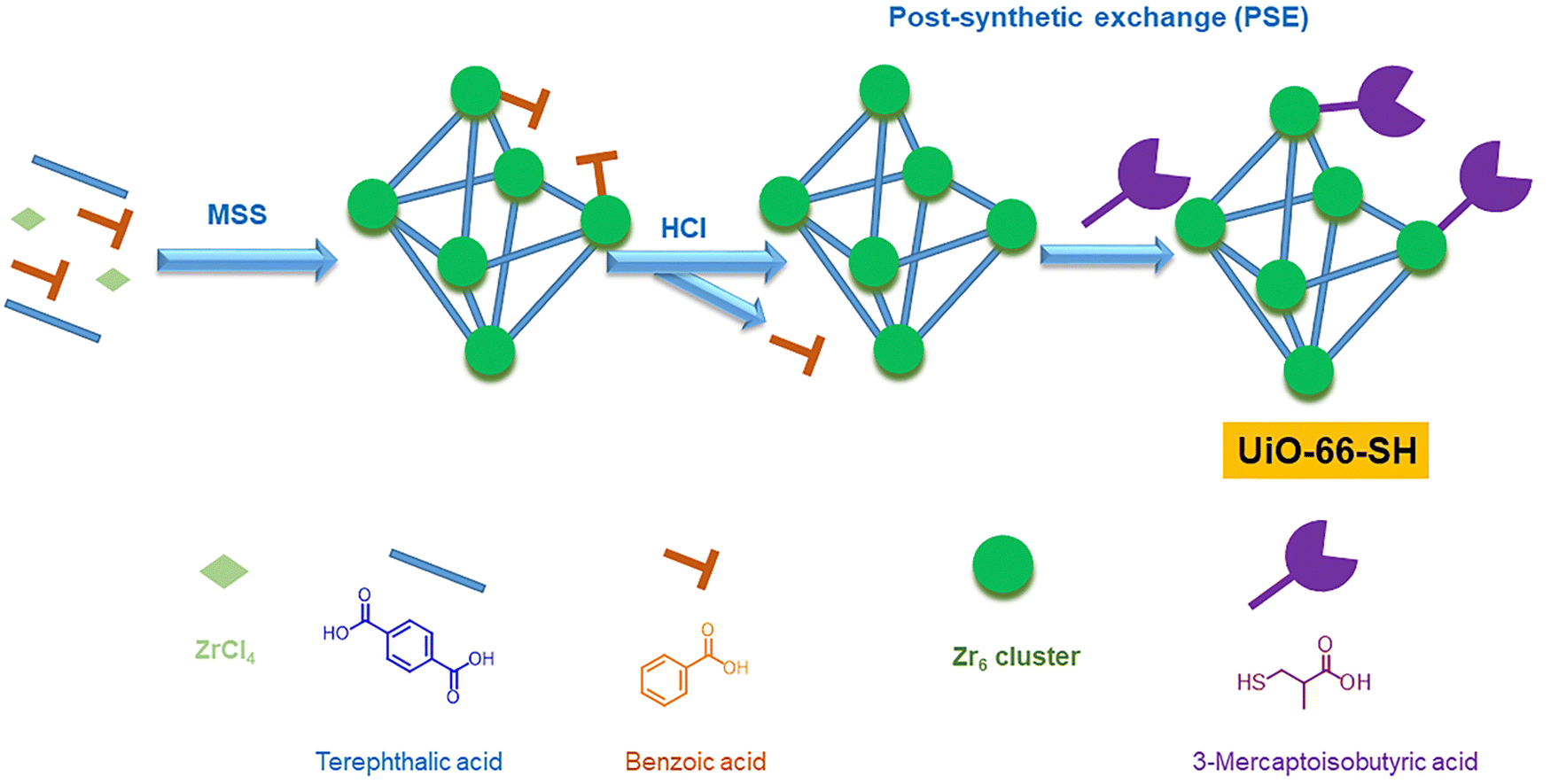 | ||
| Fig. 15 Illustration of the synthetic scheme for the preparation of UiO-66-SH.291 | ||
In 2019, Lin and coworkers synthesized a defective UiO-66 with 10-connected Zr nodes and a correspondingly large number of potentially reactive Zr sites (Zr-OH/H2O) using a modulator synthetic approach by varying amount of concentrated HCl and then applied it for adsorption and recovery of Pt(IV) anions from acidic media (pH = 1).292 The optimized UiO-66 (UiO-66(A)) was shown to have the most rapid uptake and highest Pt(IV) adsorption capacity (∼144 mg g−1). The favorable behavior was attributed to either local electrostatic attraction (PtCl62−Zr4+) or to node-based substitution of nonstructural, terminal-hydroxo ligands by PtCl62−. Although the pKb of the terminal hydroxo ligand has not been reported, available results for other Zr-MOFs suggest that at pH = 1,74 a significant fraction of the hydroxos may convert to aqua ligands, leaving the framework with a net positive charge and an ability to bring in charge-compensating anions (such as PtCl62−) via ion exchange.
Similarly, a defect-engineering of UiO-66 structure has exhibited superb adsorptive affinities for Pd(II) (105 mg g−1) from acidic aqueous solutions, which is lower than that of MOF-808 containing more terminal OH/H2O ligands or other (formate/chloride) occupying ligands on the Zr6 clusters.219 In another similar work, a series of UiO-66-X (X: HAc, BA, TFA) solids (Fig. 16) were obtained by using HAc, BA, and TFA as modulators, and their adsorption behavior is tested with Sb(V) removal.293 A battery of characterization techniques like XRD, TGA and EPR indicated the existence of defective sites in UiO-66-X solids. The experimental results indicated that the adsorption of Sb(V) with UiO-66-HAc (or here as UiO-66-AA), UiO-66-BA and UiO-66-TFA increased by 54%, 140% and 198% compared to the pristine UiO-66. The superior activity afforded by UiO-66-TFA is ascribed due to the presence of most defective sites and lower adsorption energy and higher charge transfer as evidenced by DFT calculations.
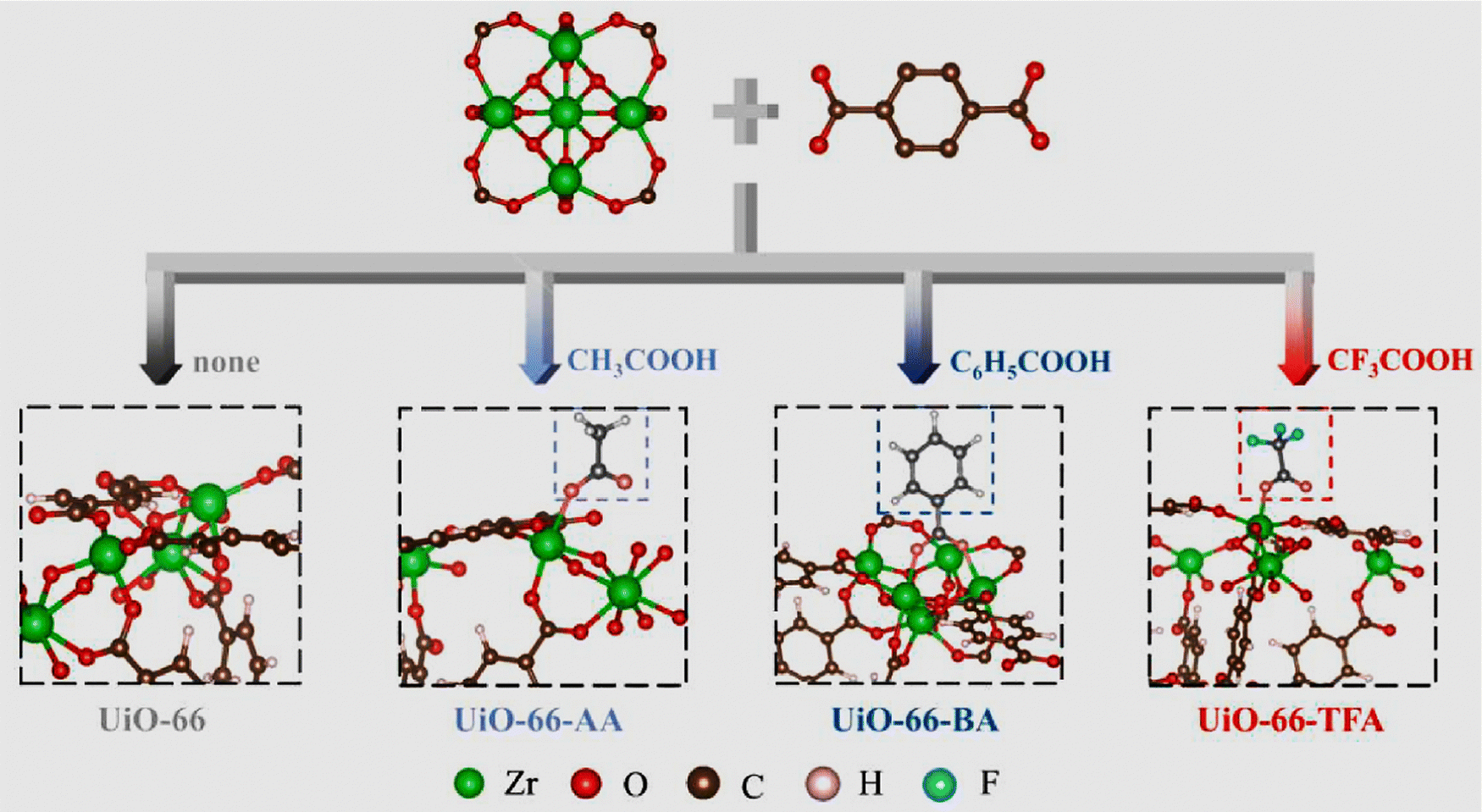 | ||
| Fig. 16 Synthetic design of the UiO-66-X with different modulators. Reprinted with permission from ref. 293 Copyright 2022 Elsevier. | ||
Wang and co-workers have reported the synthesis of two series of defective UiO-66 MOFs by varying the concentration of linker and adjusting the temperature of the reaction.178 The synthesized samples were designated as UiO-66-M/L-x, where M/L represents the Zr/BDC ratio and x stands for the temperature during the synthesis. Results summarized in Fig. 17 point to modest increases in Pb(II) sorption capacity and in Pb(II) binding constant with increasing fraction of defects. The enhancements might reflect an increased capacity of the MOF to engage in cation-exchange, or the enhancements might be due to grafting of lead ions to nodes at defect sites. Also aiming for removing Pb(II) from wastewater, in another recent study, SS-NH2-UiO-66-X materials (SS: Seignette salt/potassium sodium tartrate) were synthesized by using different concentrations of SS (X: SS mmol per 100 mg of NH2-UiO-66) to alter the porosity and manipulate the adsorption capacity of UiO-66-NH2. The primary structural consequences of SS introduction appear to be: (a) MOF etching to create mesopores as large as 50 nm, and (b) an increase in the number of missing-linker defects. Notably, MOF crystallinity was retained. Among the obtained materials, SS-NH2-UiO-66-5 exhibited the highest adsorption capacity and fastest kinetics for Pb(II) uptake. The beneficial effects were attributed to increases in the relative number pore dimension and to chelation of Pb(II) by a linker-pendant amine and a node-sited terminal (hydr)oxo ligand. Interestingly, the possibility of coordination of Pb(II) onto oxygen-rich, node-grafted tartrate ions, perhaps in tandem with a linker amino substituent, does not seem to have been considered. The maximum adsorption uptake of SS-NH2-UiO-66-5 was found to be 186 mg g−1 at pH = 5.46, which was ∼34-fold higher than that of ideal NH2-UiO-66.294
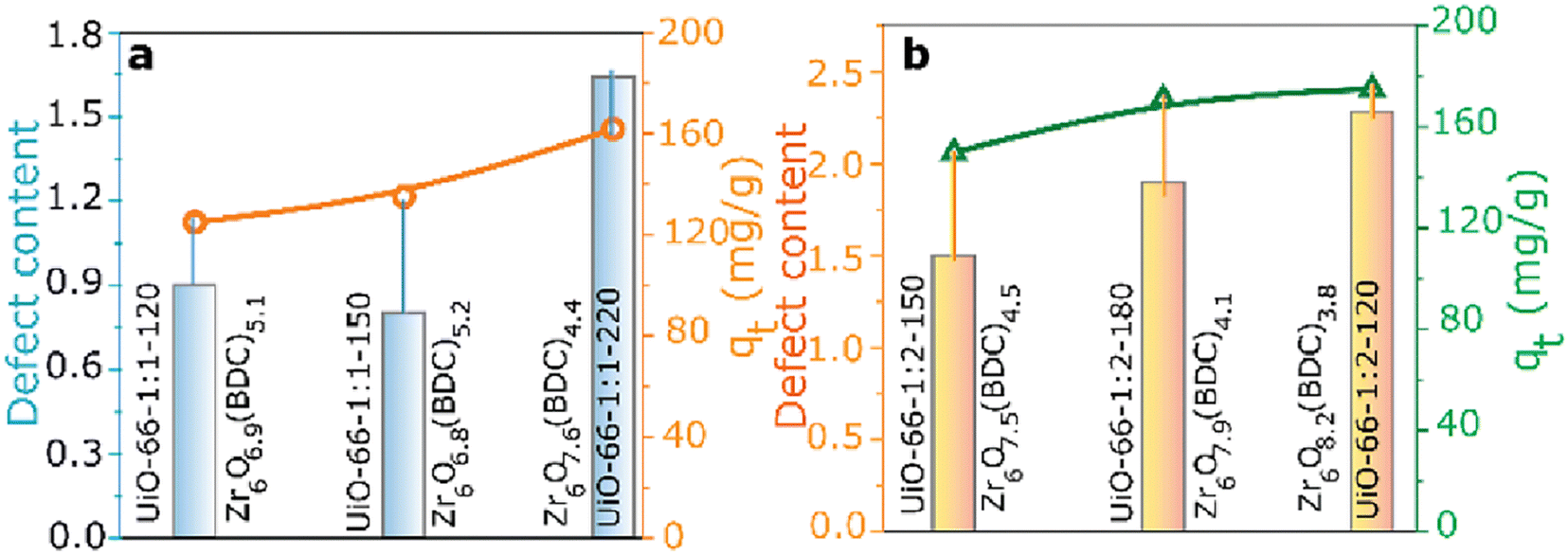 | ||
| Fig. 17 The relationship between the defect density and Pb2+ removal (a) and (b). Reprinted with permission from ref. 178 Copyright 2021 American Chemical Society. | ||
Among the most toxic CWAs, organophosphorous nerve agents receive considerable attention. They can irreversibly inhibit acetylcholinesterase (AChE), an enzyme that accelerates hydrolysis of acetylcholine in the nervous system. AChE inhibition results in buildup of acetylcholine, leading to an inability to relax contracted muscles, including pulmonary muscles, and, consequently, rapid and severe incapacitation or death.312,313Scheme 5 shows examples of agents, as well as simulants, i.e. much lower toxicity analogues that display degradation kinetics and mechanistic pathways similar to real agents when treated with degradation catalysts. Thus, as a leaving group during hydrolysis (i.e., degradation) the nitrophenoxide group in DMNP and DENP is a remarkably good kinetic mimic for fluoride in GB and GD. While exposure to DENP in small quantities is relatively safe for humans, the compound is an effective and widely used agricultural insecticide, where it is known as paraoxon. (“Oxon” signifies that the compound contains a PO bond). Agricultural runoff can lead to surface and ground water contamination with paraoxon, and a need for remediation.314
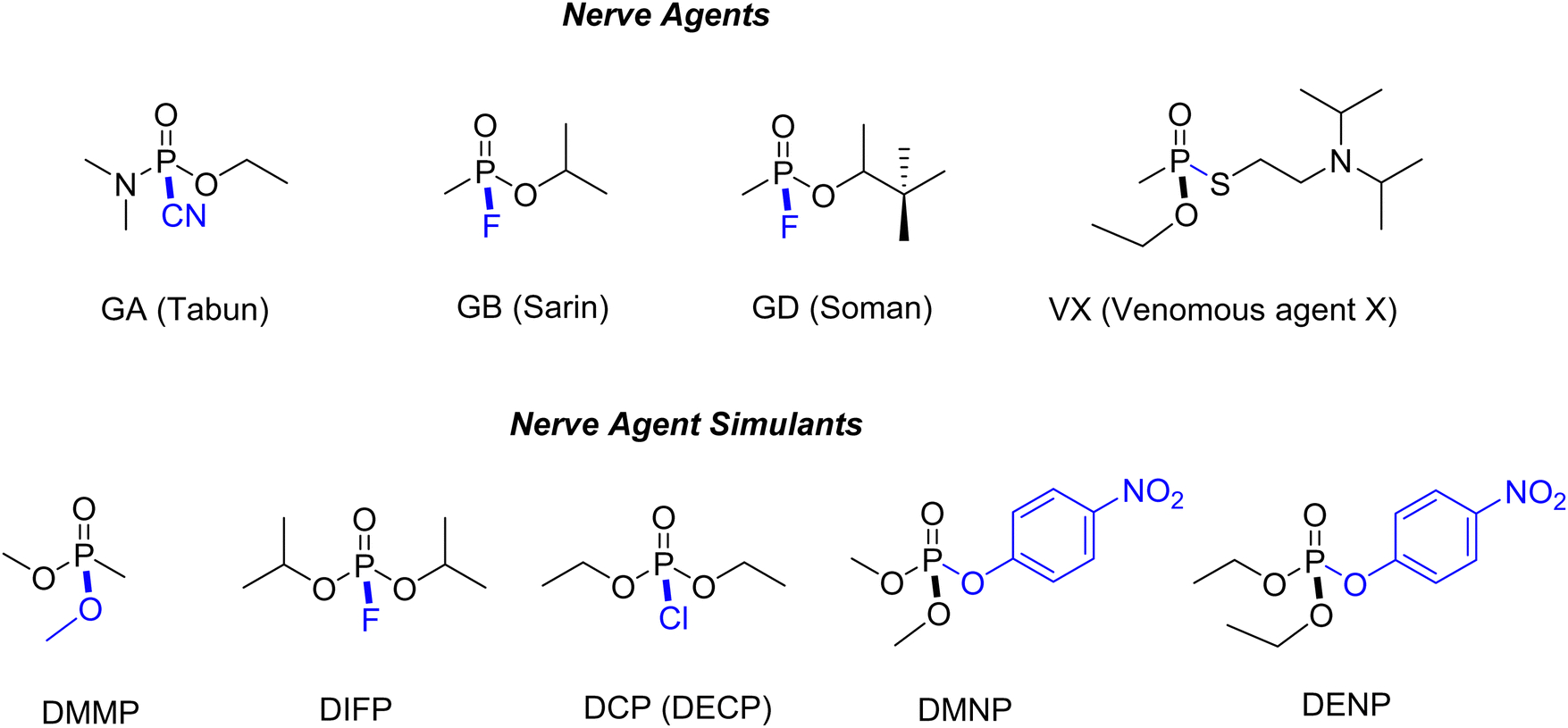 | ||
| Scheme 5 Examples of nerve agent-type CWAs and their simulants: Tabun (military designation: GA; ethyl dimethylamidocyanophosphate), sarin (military designation: GB; Trilone 46, O-isopropyl methylphosphonofluoridate), soman (military designation: GD; pinacolyl methylfluorophosphonate), venomous agent X (VX; O-ethyl S-[(2-diisopropylamino)ethyl] methylphosphonothioate), DMMP (dimethyl methylphosphonate), DIFP (diisopropyl-fluorophosphate), DCP (DECP, diethylchlorophosphate), DMNP (methylparaoxon, dimethyl-4-nitrophenylphosphate), and DENP (paraoxon, diethyl-4-nitrophenylphosphate). | ||
Catalytic degradation of nerve agents and simulants by Zr(IV)-containing MOFs312,315–320 is centered almost entirely on Lewis-acid activation (i.e. weakening) of P–X bonds, where X− is a leaving group, and activation is accomplished via the intermediacy of a Zr(IV)⋯OP interaction (i.e., via simulant (or agent) sorption or weak coordination); see step 1 below. Beyond this, however, there is considerable confusion in the extent literature regarding mechanistic details and the identity and nature of rate-determining steps. Hydrolysis occurs via nucleophilic attack of the P–X bond, where the nucleophile in aqueous solutions is either hydroxide ion or water; see steps 2A & B. Solvent-assisted product desorption then resets the system and enables catalytic cycling; see steps 3A & B in the overall scheme below (Scheme 6). For simplicity, the scheme omits diffusive transport of reactants to active sites on or within the MOF crystallite – a step that, in principle, could be rate-limiting,321,322 especially for larger MOFs crystallites and/or for catalytic MOFs featuring comparatively small apertures and channels.
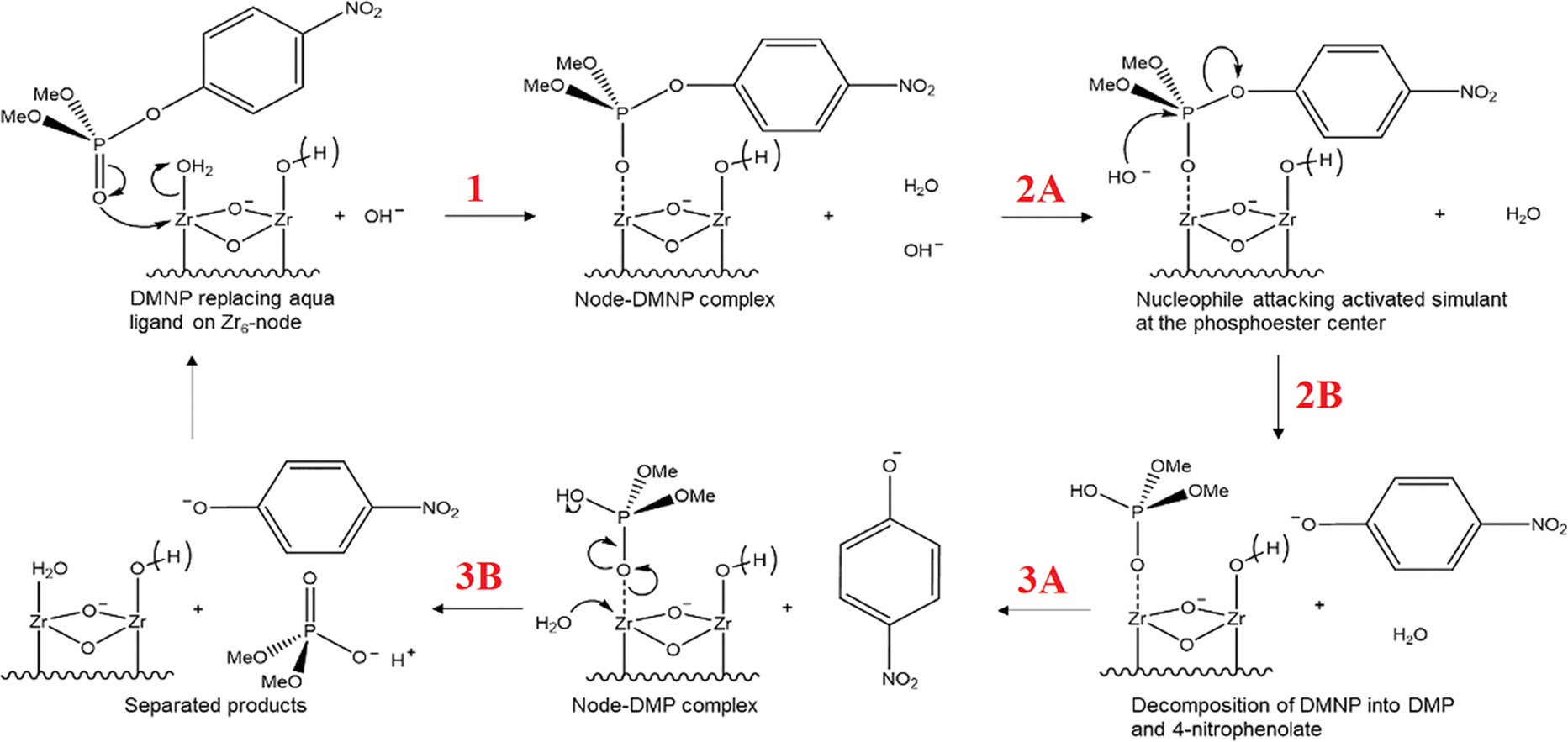 | ||
| Scheme 6 Degradation pathways for nerve agents or simulants by Zr(IV)-containing MOFs.320 | ||
Notably, because it would lack open coordination sites, a hypothetical, defect-free 12-connected Zr-MOFs, such as UiO-66 or one of its many congeners and derivatives, would be incapable of functioning as an array of Lewis acids and catalyzing hydrolytic degradation of simulants or agents. Further, UiO-66, with its comparatively small apertures, lacks the ability sterically to admit DENP and DEMP to the MOF interior, although reactants such as GB likely can permeate the MOF. To engender or enhance permeability toward reactants, missing-linker or missing-node defects are required. To engender catalytic activity, missing-liker defects are required, as these provide a basis for reactant to access to the Lewis acid sites (Zr(IV) sites). In general, in the case of UiO-66s, the adsorption and degradation performance can be tuned by controlling the defect density in the structure.
An important question is “what is present when linkers are missing?” In dry samples, coordination sites could be occupied by modulators, or, if modulators are removed by conventional treatment with hot DMF + aqueous HCl, they will be largely supplanted by ligated formate, generated via the decomposition of DMF.74 Soaking MOFs for several hours in water often is sufficient to remove formate and replace it, at least nominally, with a terminal aqua, hydroxo ligand pair. In practice, most aqua ligands are deprotonated at pH 7 and the framework acquires an overall negative charge. Heating dry samples in vacuum can remove formate, or modulators such as trifluoroacetate,187 as the corresponding conjugate acid, where the needed proton for molecular charge neutralization and volatilization can be recruited from μ3-OH moieties on the node. Depending on the temperature, however, heating can engender node restructuring, as shown by Chapman, et al.56,323
Lewis acid catalysis of agent or simulant hydrolysis starts, as shown in Scheme 7, by displacement of an aqua ligand from the node; linkers and terminal hydroxo ligands are effectively substitution inert (although terminal hydroxo ligands can be transiently protonated to yield substitution-labile aqua ligands). As the solution pH increases, the availability of displaceable aqua ligands systematically decreases. At pH 5.5 roughly half the expected aqua ligands are present (with the other half deprotonated and, therefore, refractory toward displacement). At pH 10, however, only about 1 node in 60,000 presents a displaceable aqua ligand; see Scheme 7.324 Consistent with this picture, Katz and co-workers observed that the rate of hydrolysis of DMNP by defective UiO-66 systematically decreases as the pH increased from 8.8 to 10.2; see Fig. 18.324 From the rate modulation they concluded that step 1 in the mechanistic Scheme 6 is rate-determining.
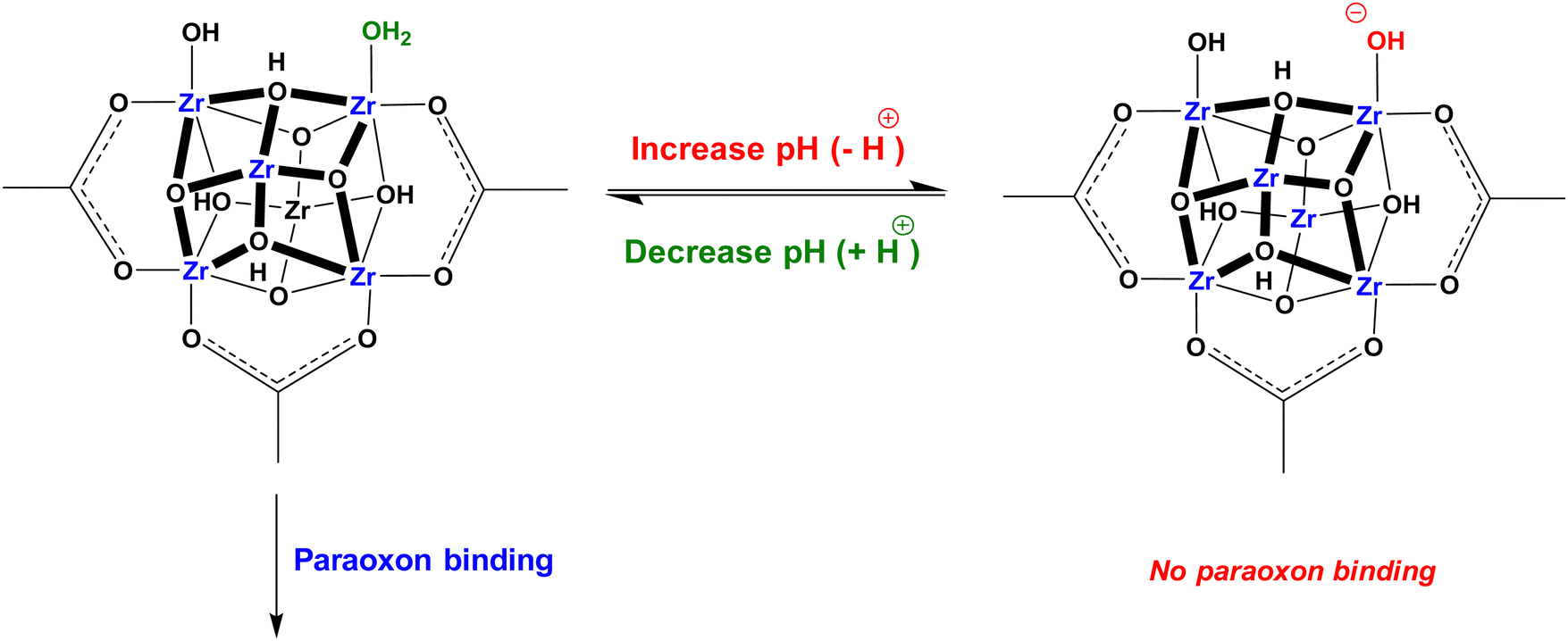 | ||
| Scheme 7 Illustration of pH dependence of composition of node ligands at missing-linker site. DMNP can bind to Lewis-acidic Zr(IV) only at aqua coordination sites. Although not significant here, the four bridging hydroxos should be shown as deprotonated at pHs where catalysis can be observed.324 | ||
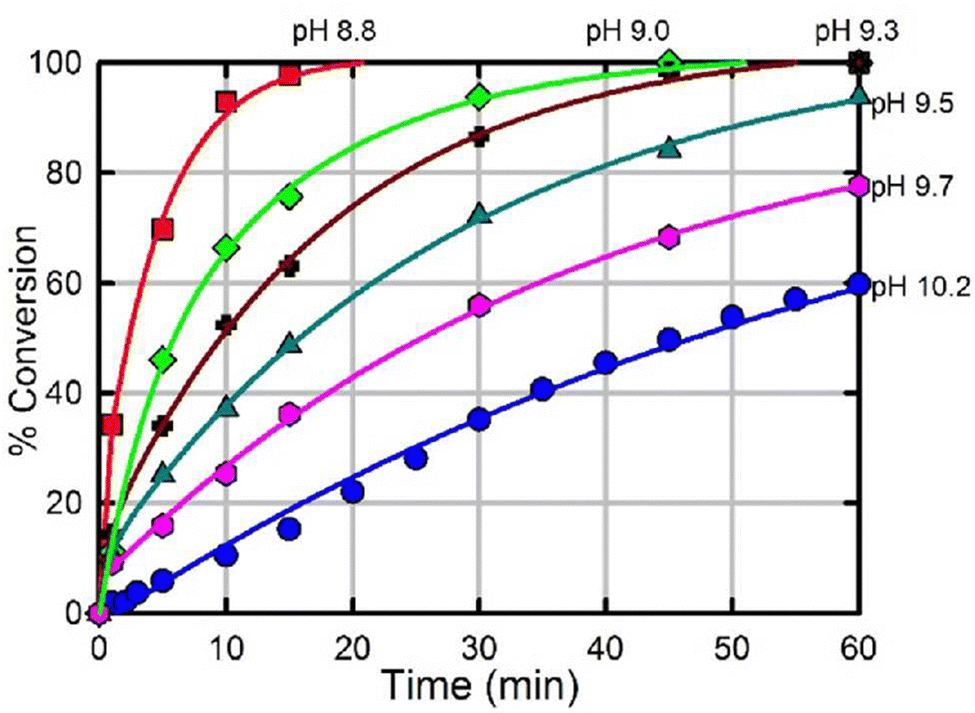 | ||
| Fig. 18 Hydrolysis rate of DMNP, as a function of time and pH for defective UiO-66 as the catalyst. Reprinted with permission from ref. 324 Copyright 2015 American Chemical Society. | ||
When step 1 is fast, step 2 can influence the rate. In isolation, the rate depends directly on the concentration of the nucleophile. If water were the nucleophile, the concentration would be unchanged with pH. For solution-phase hydroxide as the nucleophile, however, the nucleophile concentration (activity) will increase ten-fold for each unit of increase of pH. Counterbalancing this contribution is a ten-fold decrease in available aqua sites per unit increase of pH. If no other pH-influenced effects are important, the observed rate should be nearly pH independent when step 2 is rate-determining. For NU-1000 as the catalyst, the observed rate is indeed nearly independent of pH. From this finding and others, Liao, et al. concluded that the nucleophile for Zr-MOF-catalyzed hydrolysis is solution-phase hydroxide and that, for NU-1000, step 2 is rate determining.254
Finally, Liao and co-workers observed that successive half-lives for simulant hydrolysis are successively longer.325 For a pseudo-1st-order reaction, successive half-lives should be identical. By closely examining initial rates, the authors were able to establish that the cause of the progressively increasing half-lives is product inhibition. The oxy, methoxy phosphorous product is anionic and displays a greater affinity for the node than does simulant. Even so, they were able to observe tens to hundreds of turnovers, establishing that hydrolysis of DMNP in aqueous solution is catalytic.
In contrast, experiments run in high-vacuum have typically yielded only a single hydrolytic turnover – consistent with the absence of a reservoir water, and therefore hydroxide.326,327 Interestingly, measurements made in humid atmospheres, sufficient to fill MOF pores with water, similarly have yielded only about one turnover. This peculiar result can be understood, at least qualitatively, by considering concentrations of relevant species. An upper limit for catalytic sites is one per missing-linker – roughly 4 per node for a UiO-type MOF having a high density of defects. This density of active sites in the candidate catalyst translates to a concentration of roughly 2 M – meaning that a single catalytic turnover would yield roughly 2 M of catalyst-inhibiting, anionic product. From the few available measures, binding constants for DMNP and Sarin hydrolysis products on Zr-MOF nodes range from about 10 to 140 M−1.325 On this basis, a 2-M solution of pore-confined hydrolysis product, with no external reservoir for product dilution, can be expected to severely inhibit the catalyst and nearly completely arrest reaction progress after 1 turnover.
Katz and co-workers214 showed that by including HCl in solvothermal syntheses, the density of missing-linker defects for UiO-66, UiO-67, and various derivatives, could be reliably increased to ∼2 per node, corresponding to an average connectivity of 8; see also, earlier work by Voertmele, et al.,187 specifically for UiO-66 (Note that one missing linker opens potential sorption or reaction sites on two nodes). Conversely, Shearer, et al. showed that missing-linker defects for UiO-66 could be systematically decreased by raising the synthesis temperature and increasing the ratio of linker to Zr.76 A salutary outcome is a very sizable increase in thermal stability.
Capitalizing on these observations, Peterson et al. evaluated the influence of missing-linker defects within UiO-66-NH2 for capture and/or detoxification of the following CWAs and simulants: chlorine gas, 2-chloroethyl ethylsulfide (2-CEES or CEES; a simulant of sulfur mustard, yperite or HD agent) vapor, Soman, DMNP, and VX.328 They examined samples of UiO-66-NH2 offering “low,” “medium,” or “high” densities of missing-linker defects. Results exhibited that different amounts of HCl modulator and synthesis temperature resulted in differences in particle size and defects density, thus affecting the porosity, surface area, and the density of amino groups, giving low, moderate, and high-defect density MOFs. It was shown that the presence of more defects in UiO-66-NH2 resulted in more active sites and larger pores for accommodation and hydrolysis of nerve agents, but an optimum defect is needed for chemicals requiring amino-linker functionality for their removal. Consistent with Table 3, they found that the extent of uptake of targets featuring small kinetic diameters is little affected by varying the density of missing linkers. For targets subjected to hydrolytic degradation, a figure-of-merit is the time required to degrade half the amount initially present. For both DMNP and GD (see structures above), changing the density of defects from “low” to “medium” has an enormous effect, accelerating the degradation rate by ∼15 to 20-fold; but, further increasing the defect density elicits only marginal additional acceleration. DMNP and GD are size excluded by MOF apertures lacking defects. Consequently, at low defect density, catalysis is largely restricted to the outermost portions of each MOF crystallite. Slightly more sterically demanding than DMNP and GD is VX. Substantial rate acceleration for VX is attained only for samples offering “large” densities of defects (defined as roughly 4 defects per node). Evidently, with “medium” levels of defect density, rapid admission of VX to the MOF interior and its bounty of active sites does not occur.
In 2018, Bůžek and coworkers proposed that the in situ formation of defects, not formerly existent in the UiO-66 structure, can form in water through the cleavage of Zr-carboxylate bonds with linker/modulator, and thereby, new catalytic sites on Zr-nodes are formed. These sites are posited to be effective for degradation of DMNP.44 Arguably, this important point, i.e. the creation of catalytically active sites via water-assisted displacement of carboxylate-anchored modulators, is implicit in the majority of studies of simulant hydrolysis in liquid-water microenvironments.
Momeni and Cramer studied computationally the effects number and morphology of defect sites on the performance of various Zr6 and Zr12 MOFs (UiO-66, and also MOF-808 and NU-1000) for detoxification of sarin nerve agent.210 They found that increasing the number of missing-linker defect sites enhances overall catalytic hydrolytic activity. In addition to the obvious consequence of boosting the number of active sites, the presence of increasing numbers of defects engendered favorable hydrogen-bonding interactions and favorable reactant orientations relative to active sites.
In related work, Harvey et al. investigated computationally the impact of missing-linker defects in UiO-66s on binding of CWA simulants DECP, DIFP, and GB and inferred that Zr-OH and μ3-OH acted as contributing binding sites for the chemicals.329 While this inference may well be correct in the absence of significant amounts of adsorbed water or methanol, the four μ3-OH moieties are deprotonated and, therefore, unavailable in the presence of water if the pH is above ∼4 (i.e. pKa = ∼3.3).254 The pKa of aqua ligands, however, is around 5.5 – meaning that these will have been largely converted to additional terminal hydroxo ligands at pH 7.
Hwang et al. synthesized and investigated, for adsorption of DMMP, a set of zirconium-BTC-based MOF-808 samples featuring formate (F), propionate (P), and valerate (V) as charge-balancing ligands (CBLs) (Fig. 19).146 Breakthrough experiments showed that despite the lower porosities of MOF-808-P (R = C2H5; BET surface area 1525 m2 g−1, pore volume 0.62 cm3 g−1, pore size <1.6 nm) and MOF-808-V (R = C4H9; BET surface area 1260 m2 g−1, pore volume 0.72, pore size <1.7 nm), relative to MOF-808-F (R = H; BET surface area 1800 m2 g−1, pore volume 0.76, pore size ∼1.4–2.2 nm), the capacities of the former for uptake of DMMP are ∼2.6-fold larger. The authors concluded that open Zr Lewis acid sites at defect sites play a crucial role in the adsorption of DMMP. Note that if CBLs are fully retained as secondary ligands on the node, Lewis acidic Zr(IV) sites will be exposed to DMMP only if a bidentate CBL (or linker) carboxylate protonates and becomes monodentate. The available evidence strongly suggests that this indeed does happen with propionate and valerate CBLs, as sketched in Fig. 19; also see the work by Rayder, et al.330 The authors also inferred that Brønsted acid sites (μ3-OH, OH) may facilitate DMMP sorption.42,146 For solution-phase studies, Brønsted sites may well be important at some pHs, the bridging hydroxo units should be largely deprotonated at pH 7. The circumstances are more complex, however, for vapor-phase uptake, especially if the vapor includes water.146 Notably, the authors found that DMMP capacity loss due to competitive adsorption of water at 15% humidity was relatively unimportant. In a related study, albeit with NU-1008 rather than MOF-808-X, Wang, et al. found that co-adsorption of water considerably accelerates diffusive transport of water – a desirable outcome for practical applications.331
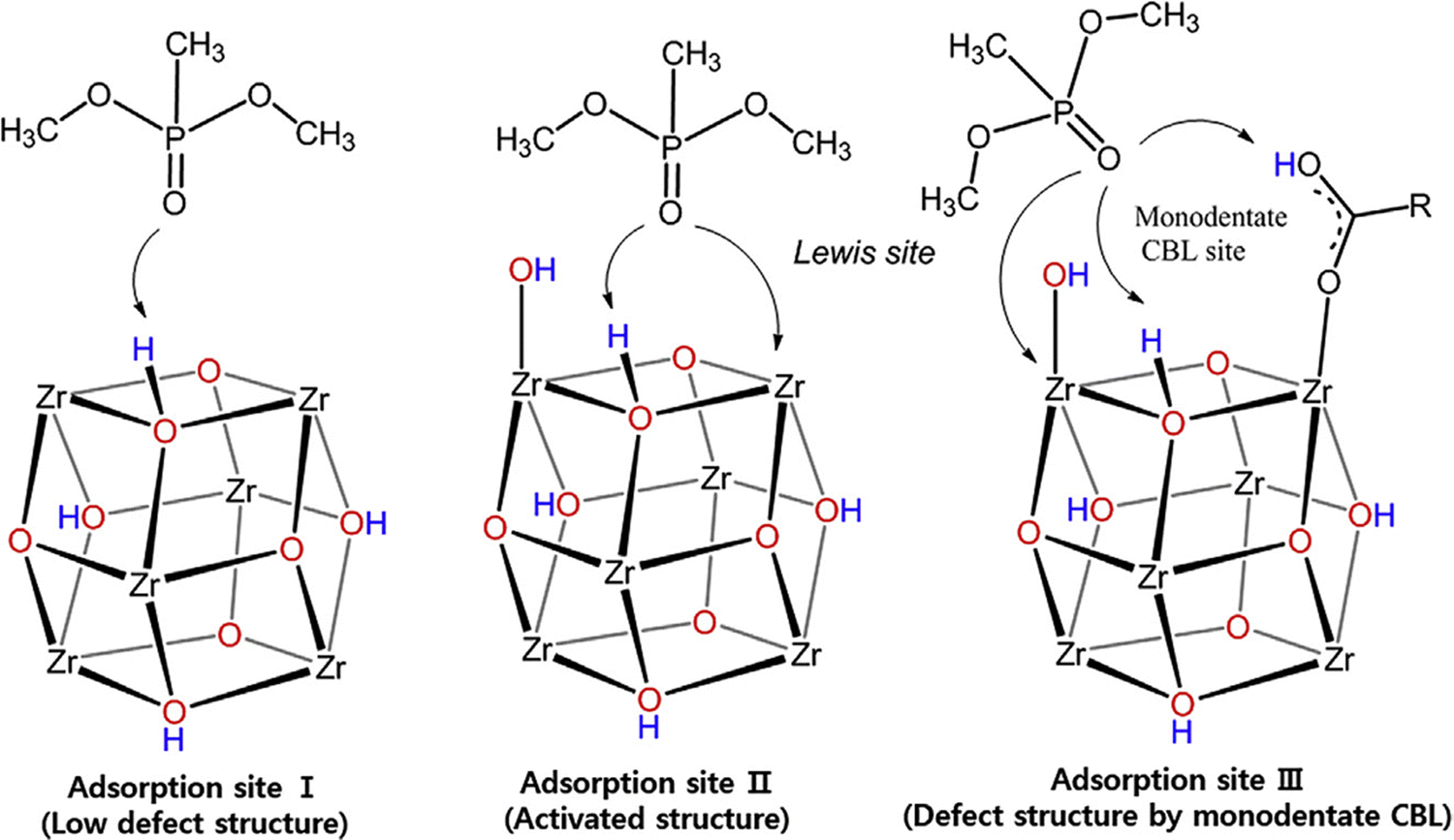 | ||
| Fig. 19 Candidate adsorption sites for vapor-phase DMMP on MOF-808-X (X = F, P, and V). The drawings are simplified to omit coordinated linkers and bidentate CBLs. Reprinted with permission from ref. 146 Copyright 2018 Elsevier. | ||
Cho et al. suggested that the number of defects and particle size of UiO-66 can be altered by altering the concentrations of synthesis reagents.221 The use of HCl modulator during MOF synthesis and the lowest DMF solvent level afforded the smallest spherical particle size (100 nm) and highest defect density (1.8 missing-linker per cluster) and pore volume (0.54 cm3 g−1), namely UiO-66S, which in combination with 4-ethylmorpholine as an organic base showed >2-fold higher catalytic activity (t1/2 = 0.3 min, turnover frequency (TOF) = 0.5 s−1) for hydrolytic degradation of DMNP. Note that while reaction half-lives, t1/2, are useful for comparing catalysts within a given study, comparisons between studies can be complicated by the anticipated dependence of t1/2 on the amount of catalyst present. Indeed, increasing or decreasing the amount of catalyst present is a convenient way of shifting the reaction time into a regime where it is easily measured. Comparisons of turnover frequencies (TOFs) on a per-active-site or per-node basis, i.e. normalized comparisons, are typically more useful.
The Bandosz group observed that when CEES is delivered via the vapor phase, dehydrohalogenation and partial oxidation are the main mechanisms for detoxification over defective UiO-66, whereas hydrolysis is the most important pathway when liquid CEES is degraded (Scheme 8).332
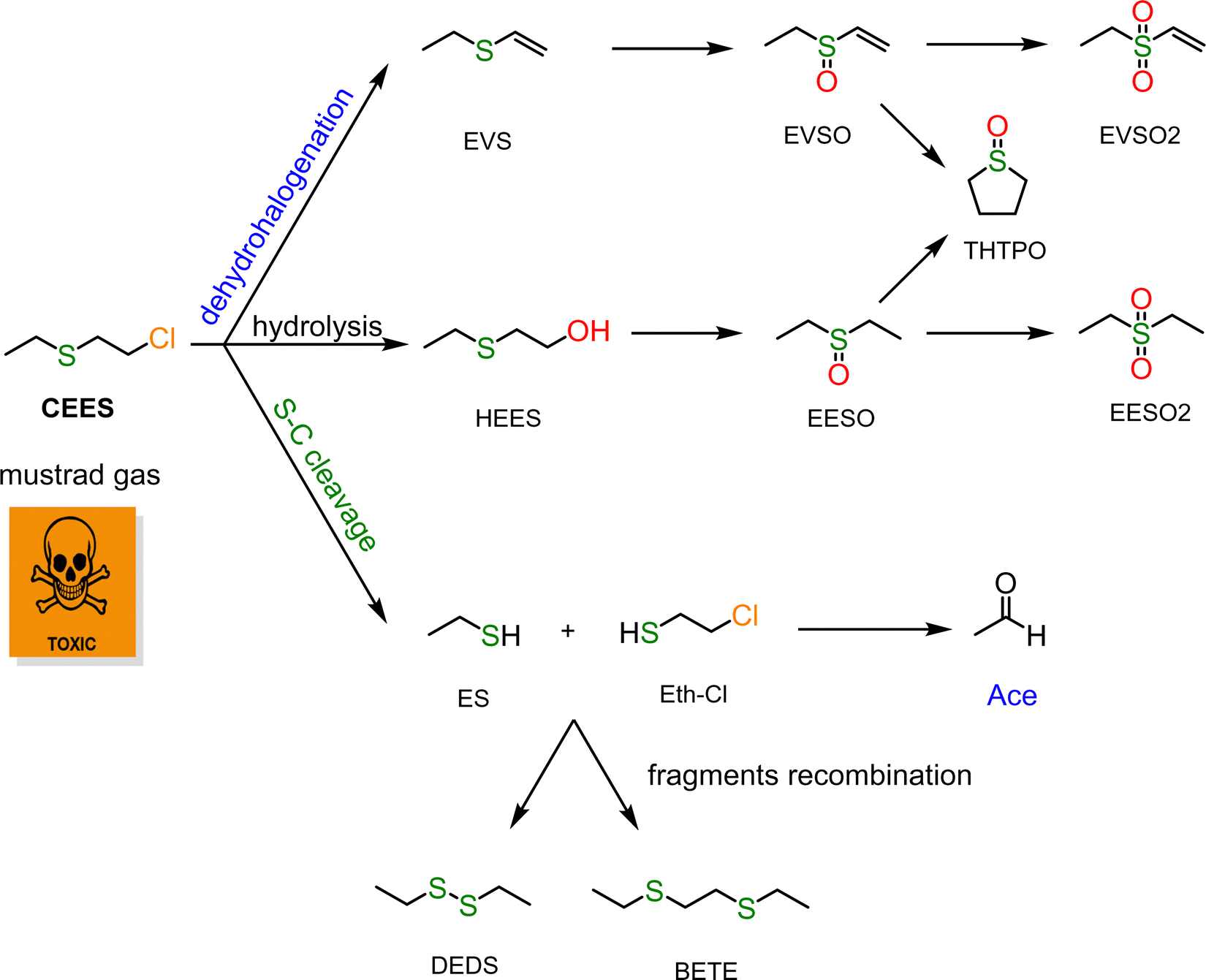 | ||
| Scheme 8 Conceivable mechanisms of detoxification of CEES.332 | ||
Mondloch and co-workers described, for thermally treated and fully dehydrated NU-1000, a distinctly different type of effect that, nonetheless, is relevant to both catalytic and stoichiometric inactivation of chemical threats.319 Further structurally assessed by Chapman and co-workers,56 the thermally generated defect is neither a missing-linker nor a missing-node. Instead, it is a symmetry-lowering distortion of the dehydrated Zr6(μ3-O)88+ core of the MOF node to yield Zr6(μ3-O)7O8+. Notably, the distortion persists even after MOF immersion in water and node rehydration. As illustrated in Fig. 20, the distorted site, with its structural void,56,211superficially resembles an oxygen vacancy of the sort that can be generated on or in zirconia via heating at very high temperature in a reducing atmosphere.333 The thermally disordered MOF node, however, retains a full complement of oxygen atoms and displays no evidence of vacancy-initiated reduction of Zr(IV) to Zr(III) or Zr(II). A further consequence of irreversible node distortion (defect formation) in NU-1000 is a ca.10-fold enhancement in the rate of degradative, catalytic hydrolysis of the agent simulant, DMNP.319 The chemical basis for the rate enhancement has yet to be established. Distortion driven enhancement of active-site Lewis acidity, however, is a probable contributor.
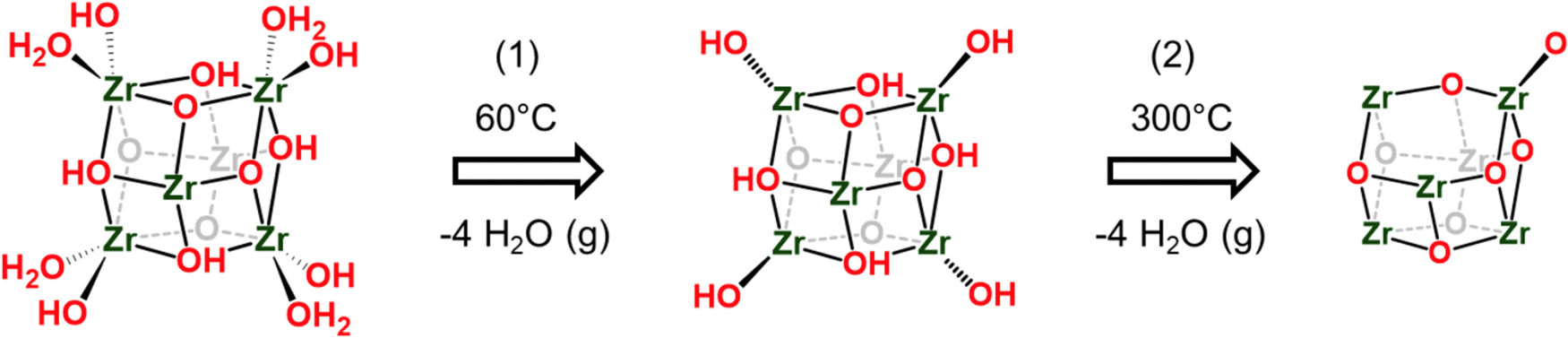 | ||
| Fig. 20 Stepwise thermal dehydration and symmetry-lowering distortion of the node of modulator-free NU-1000. | ||
The irreversible node distortion culminates in the presentation of highly Lewis acidic Zr(IV) sites and a highly Lewis basic site in the form of a terminal oxo ligand. These sites are proximal and can act in concert to accomplish irreversible capture of ammonia, a potential industrial chemical threat, by splitting it into NH2− and H+, with the former bridging a pair of Zr(IV) ions and the latter converting the terminal oxo ligand to a terminal hydroxo.334 It is reasonable to expect that disordered nodes in Zr-MOFs will prove capable of immobilizing and degrading other chemical threats via similar heterolytic bond-splitting.
2.2. Energy-related applications
For example, Gascon et al. reported that after installing reducible Ti(IV) on defect sites in NH2-UiO-66, the obtained functionalized Zr-MOF exhibited photocatalytic activity for evolution of molecular hydrogen (Fig. 21(a)).136 With 30 mg of the Ti-installed NH2-UiO-66 dispersed in the mixture of acetonitrile, water and triethylamine, a hydrogen production rate of 0.192 μmol h−1 was achieved under illumination, and the Zr-MOF could preserve its crystallinity after the photocatalysis. A systematic experimental study was demonstrated by Jiang, Zhang and coworkers to probe the effect of structural defect present in NH2-UiO-66 incorporated with Pt NPs on the resulting photocatalytic activity for hydrogen evolution reaction (HER) (Fig. 21(b)).199 These findings suggested that even though more defects present in the Zr-MOF result in a higher specific surface area, there is an optimal degree of defects to achieve the highest photocatalytic activity after incorporating Pt NPs. Owing to its fastest relaxation kinetics and highest charge-separation efficiency, the optimal MOF-based photocatalyst with moderate structural defects, Pt@UiO-66-NH2-100, could achieve a hydrogen production rate of 381.2 μmol g−1 h−1 under illumination in the presence of triethanolamine (TEOA) as the sacrificial agent. The Pt@UiO-66-NH2-100 was reported to achieve almost the same hydrogen production rate after ten cycles of two-hour photocatalytic experiments, and the crystallinity of the MOF was unchanged after 48 h of the long-term reaction. On the other hand, the Ti-loaded NH2-UiO-66 was also used as the photocatalyst to reduce Cr(IV) to Cr(III) under visible light, as reported by Yao et al.287
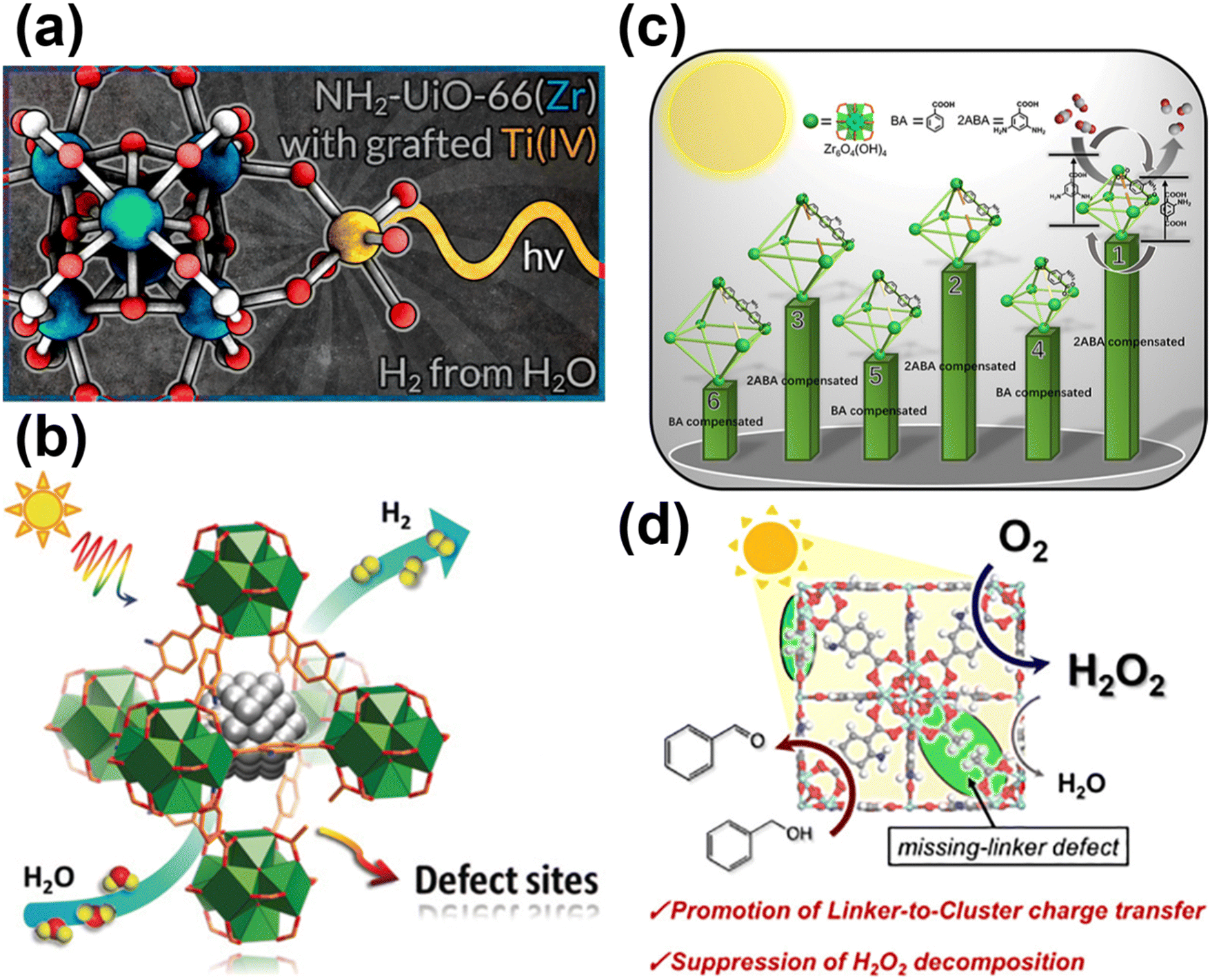 | ||
| Fig. 21 Schematic representations of the (a) defective NH2-UiO-66 installed with Ti(IV) for photocatalytic HER, reprinted with permission from ref. 136 Copyright 2017 American Chemical Society, (b) Pt NPs confined in the defective NH2-UiO-66 for photocatalytic HER, reprinted with permission from ref. 199 Copyright 2019 John Wiley & Sons, (c) defective Zr-MOFs for modulating the activity toward photocatalytic CO2 reduction, reprinted with permission from ref. 341 Copyright 2021 American Chemical Society and (d) missing-linker defects in NH2-UiO-66 for boosting the photocatalytic activity to produce hydrogen peroxide from oxygen gas, reprinted with permission from ref. 342 Copyright 2021 American Chemical Society. | ||
In addition to HER, the conversion of CO2 into valuable chemicals and fuels by using solar energy is another crucial solar-to-fuel photocatalytic route which is promising to address both the global warming and increasing energy demand.336 Recently, MOFs have received great attention as photocatalysts for CO2 reduction due to their electronic structures, excellent CO2 adsorption capacities (at least in the gas phase), and tailorable light absorption capabilities.343 The chemically robust Zr-MOFs thus also became attractive candidates for such a photocatalytic process, and the defect engineering in UiO-66 and its derivatives may also play an important role to turn the resulting photocatalytic activity. For example, Zhu, Fan and coworkers created defects in NH2-UiO-66 by tuning the crystallization temperature in the presence of concentrated HCl as the modulator.344 As a result, the defective NH2-UiO-66 synthesized at 473 K could convert CO2 into formate with a production rate of 129.8 μmol g−1 h−1 under illumination in the presence of TEOA in CO2-purged acetonitrile, which is much higher than that achieved by using the defect-free MOF (24.6 μmol g−1 h−1). The defective NH2-UiO-66 was also recycled and reused for five cycles of the photocatalytic experiments, and it was found that the production rate of formate is almost the same during the five cycles of experiments, and the crystallinity of the MOF could be well preserved. Recently, Sun, Ma and coworkers further performed a systematic study to investigate the effects of modulator used to create defects and the length of linkers on the resulting photocatalytic activity for CO2 reduction of the UiO-based Zr-MOF series.341 Both the type of defects and the length of linkers were found to remarkably change the absorption energy of the Zr-MOFs, which results in the distinct photocatalytic activity of these defective Zr-MOFs for CO2 reduction. In the solvent-free system under illumination with the addition of TEOA, the UiO-66-NH2-2ABA synthesized by using 3,5-diamino-benzoate as the modulator was found to be capable of converting CO2 into CO with a production rate of 17.5 μmol g−1 h−1, which is higher than those achieved by the UiO-66-NH2 synthesized with BA as well as both defective UiO-67 and UiO-68 with larger pore sizes (Fig. 21(c)). The UiO-66-NH2-2ABA was subjected to six cycles of the five-hour photocatalytic tests, and it was found that the CO production rate is almost the same for every cycle. The crystallinity of the MOF was still observable after the solvent-free photocatalytic reaction. Another study from the same research group showed that in the solvent-free system with TEOA, UiO-66-NH2 samples featuring ligand vacancies could achieve a much higher photocatalytic activity for converting CO2 into CO (30.5 μmol g−1 h−1) compared to those with modulator-capped defective sites and missing-cluster defects.345
In addition to photocatalytic hydrogen evolution and CO2 reduction, another notable example is the use of defective Zr-MOFs for the photocatalytic production of hydrogen peroxide from the oxygen gas, demonstrated by Yamashita et al.342 The authors found that when more missing-linker defects are present in NH2-UiO-66, the decomposition of hydrogen peroxide could be suppressed, resulting in the increase in the net production rate of hydrogen peroxide during the photocatalytic process (Fig. 21(d)). With 1 mg mL−1 of the optimal defective NH2-UiO-66 dispersed in the mixture of acetonitrile and TEOA, a H2O2 production rate of 120.7 μmol h−1 L−1 could be achieved, which is 2.3 times higher than that achieved by the pristine NH2-UiO-66. Both the crystallinity and morphology of the defective NH2-UiO-66 could be well preserved after the photocatalysis. This work points to the importance of defect engineering in Zr-MOFs for such an eco-friendly and safe approach to produce hydrogen peroxide.
Finally, while several studies have posited linker-to-cluster (Zr) charge transfer as the basis for light absorption and/or light-initiated charge separation, most computational studies have concluded that the empty 4d orbitals of Zr(IV) are too high in energy to be populated either directly or indirectly via absorption of visible light.346–348 Instead, absorption is ascribed computationally to linker-localized transitions, most notably with amino-functionalized linkers (blue-light absorbers that render MOFs yellow). Computations appear to support this picture, even when defects shift the energies of empty states.
Seemingly at odds are EPR signals that conceivably could be assigned to formation of Zr(III). Remarkably similar signals have been observed, however, for O2− when the ion is adsorbed/ligated either to exposed Zr(IV) coordination sites on MOF nodes, or on similar sites on zirconia.349,350 Electron-transfer from photo-excited linkers to even trace amounts of O2 might be expected to yield easily observable EPR signals due to adsorbed superoxide anion. Note that exposure of Zr(IV) sites to ligation of O2− becomes increasingly more likely as the density of missing-linker defects increases. Finally, in interpreting photocatalytic responses from Zr-MOFs, it may be important to consider the possibility of hole or electron transfer from photoexcited linkers to putative charge scavengers (ascorbate, triethanolamine, triethylamine, formate, etc.135), rather than directly to intended reactants. Regardless of how scavengers capture charges, a not uncommon occurrence is for the fragmenting scavenger to yield a high-energy radical that can transfer an additional charge – effectively yielding downstream a two-electron (or hole) conversion of a target such as CO2, based on absorption of one photon.351–353 Related thermally driven electron-transfer reactions involving often overlooked species like node-ligated formate have been shown to initiate otherwise puzzling “autocatalytic” metal-ion to zero-valent metal cluster reduction reactions.354
Owing to their exceptional chemical stabilities in aqueous solutions over a wide range of pH,361–364 (albeit, not highly alkaline solutions), Zr-MOFs have been considered as attractive candidate materials for various electrochemical applications that require aqueous media such as electrocatalysis, electroanalytical systems and supercapacitors.365–367 However, the electrically insulating nature of most MOFs significantly limits their direct use for electrochemical and electronic applications.368,369 Thus, to utilize Zr-MOFs as the active materials located near the electrode surface for certain electrochemical applications, long-range charge-transport pathways during the electrochemical processes are necessary; “redox hopping” thus has become the most common charge-transport pathway in Zr-MOF thin films.365,370–373 As the hexa-zirconium nodes are redox-innocent, to render the electronic conduction via redox hopping in Zr-MOFs, the redox-active moieties need to be either present in the organic linker or installed on the node.371 Thus, for UiO-66 and its derivatives, which are commonly reported Zr-MOFs but constructed from redox-inactive organic linkers, their missing-linker or missing-cluster defects become advantageous for immobilizing the redox-active species in order to facilitate the charge hopping in MOF which is required for electrochemical processes.
For example, Hupp et al. reported the installation of a redox-active ferrocene derivative on the missing-linker defects of UiO-66, and the resulting functionalized UiO-66 could show electrochemical activity relying on the redox hopping between the coordinated ferrocene moieties.374 In addition, Tang and coworkers demonstrated the incorporation of polysulfides on the defect sites of UiO-66.375 The authors further installed polysulfides within defective UiO-66 crystals interconnected by electrically conductive carbon nanotubes, and the obtained redox-active sulfur-loaded MOF-based composites could be applied for lithium–sulfur batteries to achieve an initial capacity of 925 mA h g−1 at 0.5 A g−1 and a very low fading rate of 0.071% per cycle at 1 A g−1 over 800 cycles.375 Similarly, the defective UiO-66 was also applied for lithium–sulfur batteries by coordinating the lithium thiophosphate on the terminal –OH/H2O sites presented in the framework, as demonstrated by Thoi and coworkers.376 The same group has also reported the lithiation of defective sites in UiO-66 for the application in lithium–sulfur batteries (Fig. 22(a)).377 With the lithiated defective UiO-66 as the additive in the cathodes for lithium–sulfur batteries, an average maximum capacity of 1272 mA h g−1 was achieved, which is higher than those achieved by the batteries with the defective UiO-66 (918 mA h g−1) and lithiated less defective UiO-66 (1050 mA h g−1) as additives. Hod et al. showed that the defective sites of UiO-66 could be used to immobilize redox-active Hemin, and the redox-hopping rate was found to be highly tunable by changing the density of defects in the parent UiO-66 framework.378 As Hemin can catalyze oxygen reduction, the defective UiO-66 functionalized with Hemin was applied for electrocatalytic oxygen reduction (Fig. 22(b)). Further axial coordination of the electron-donating 2-methylimidazole on the Hemin immobilized in UiO-66 was found to accelerate the charge hopping rate and thus enhance the electrocatalytic activity for oxygen reduction, as demonstrated in another study published by the same group.379 The study also reveals that the Hemin-functionalized defective UiO-66 could exhibit a stable electrocatalytic activity for oxygen reduction in the acetonitrile-water cosolvent electrolyte for four hours, and the morphology of the Zr-MOF was found to be barely altered.
 | ||
| Fig. 22 Schematic representations of the (a) lithiation of defective UiO-66 for lithium–sulfur batteries, reprinted with permission from ref. 377 Copyright 2019 American Chemical Society, (b) installation of electrocatalytic Hemin in defective UiO-66 for oxygen reduction, reprinted with permission from ref. 378 Copyright 2019 American Chemical Society, (c) installation of electrocatalytic Ir(III)/Ir(IV) sites on the missing-linker defects of UiO-66 for electrocatalytic water oxidation, reprinted with permission from ref. 380 Copyright 2019 Elsevier and (d) use of defective UiO-66 membrane deposited on the silver electrode to modulate the selectivity toward electrochemical CO2 reduction, reprinted with permission from ref. 168 Copyright 2019 John Wiley and Sons. | ||
In addition to the installations of organic moieties and polysulfides, the redox-active metal ions could also be immobilized on the missing-linker defect sites of Zr-MOFs. For example, Kung et al. demonstrated the solution-phase post-synthetic decoration of redox-active Mn(II) ions on the terminal –OH/H2O sites presented in defective UiO-66 nanocrystals to engender node-to-node redox hopping.381 Since redox-active manganese species are widely utilized as pseudocapacitive materials, such Mn-decorated UiO-66 nanocrystals could be applied for aqueous-based supercapacitors after being composited with electronically conductive nanocarbon.381 A similar approach was used to immobilize redox-active Ir(III)/Ir(IV) ions in defective UiO-66,382 and the resulting functionalized Zr-MOF could be utilized in the electrochemical detection of nitrite in aqueous solutions,380 as demonstrated by the same research group. With the iridium-functionalized defective UiO-66 as the electrocatalyst, a sensitivity of 168.8 μA mM−1 cm−2 and a limit of detection of 0.41 μM were achieved for detecting nitrite in a weakly acidic HClO4/NaClO4 aqueous electrolyte (pH = 5). Since such iridium-based species are capable of electrocatalyzing the oxidation of water to molecular oxygen, the important half reaction of water splitting, in acidic electrolytes, and Zr-MOFs are chemically stable in strong acids, UiO-66 nanocrystals grown on conductive carbon nanotubes with the iridium sites coordinated on the missing-linker defects were applied for the electrochemical conversion of water into oxygen gas (Fig. 22(c)).380,383 The optimal nanocomposite with iridium-functionalized defective UiO-66 and carbon nanotubes could achieve a current density of 10 mA cm−2 for oxygen evolution with an overpotential of 430 mV. The study also found that both the crystallinity and morphology of the defective UiO-66 are unchanged after the electrocatalytic oxygen evolution in the aqueous electrolyte containing 0.1 M of HClO4. It should be noted that in addition to the defect sites in UiO-66, the defective cavities caused during the activation of other Zr-MOFs may also play a role in confining the electrochemically active metallic NPs inside. For example, the mesoporous cavities in a porphyrinic Zr-MOF, NU-902, were utilized to confine silver NPs with the size of around 3 nm that are capable of electrocatalytically oxidizing nitrite ions.384
Apart from the immobilization of redox-active or catalytically active sites on the defects of Zr-MOFs aiming for electrochemical applications, recently, a few studies have attempted to utilize the defects in MOFs to directly adjust the chemical environments near the electrode surface to further modulate the selectivity of complicated electrochemical reactions occurring on the underlying electrode. Since the defective MOF here only plays the role similar to a porous “sieve” without providing any active site for electrochemical reactions to occur, the electrical conductivity of MOF is no longer necessary in such systems.385 The first example of such concept was demonstrated by Hod and coworkers.168 In this work, the porous and continuous UiO-66 membrane with enriched missing-cluster defects was grown on the surface of silver electrode that is electrocatalytic for converting CO2 into CO, and the authors found that with the help of the electrically insulating defective UiO-66 coating, the selectivity toward CO2 reduction against the competing hydrogen evolution occurring on the silver surface can be largely improved owing to a second-sphere effect (Fig. 22(d)). With the optimal defective UiO-66 membrane, the selectivity toward CO could reach 79% at −0.8 V vs. reversible hydrogen electrode (RHE), much higher than that achieved by the bare silver electrode (43%). The authors then further post-synthetically coordinated the positively charged 3(carboxypropyl)-trimethyl-ammonium in the defective UiO-66 to provide the electrostatic repulsion to protons, the reactant of hydrogen evolution; the selectivity of CO2 reduction toward the formation of CO could thus be further enhanced to 89% at −0.8 V vs. RHE. A similar concept was utilized in electrochemical sensors; the missing-linker defects of UiO-66 were used to selectively adsorb the targeted analytes, dopamine (DA), in order to boost the selectivity of the DA sensor against other interferents, as reported in a recent work by Kung et al.386 Very recently, the same concept of utilizing defective Zr-MOF to modulate the microenvironment near the electrode/catalyst surface was also applied to electrocatalytic reduction of nitrate to generate ammonia, as demonstrated by Yan and coworkers. To electrochemically convert nitrate ions present in wastewater into ammonia, an eight-electron process with nine involving protons is required. It was found that with the defective UiO-66 coated on the surface of a CuZn-based electrocatalyst, selectivity toward producing ammonia against the formation of unwanted nitrite can be facilitated with the help of terminal carboxylic acid groups present in the defective MOF to provide sufficient protons to the catalyst surface.387 At an applied potential of −1.0 V vs. RHE, the defective UiO-66-coated CuZn could achieve a faradaic efficiency of 91.4% for converting nitrate into ammonia, which is obviously higher than that achieved by the bare CuZn (30.6%). The selectivity of the reaction was found unchanged after ten cycles of the electrocatalysis, and both the crystallinity and morphology of the defective UiO-66 could be preserved after the reaction.
Another noticeable electrochemical application of Zr-MOFs is the use of such materials as proton conductors, which can be further served as solid-state electrolytes of fuel cells. Compared to the commonly used Nafion and polymer-based proton conductors, proton-conducting MOFs are more structurally characterizable and have the opportunity to conduct protons at a low relative humidity (RH).256 The porous and water-stable characteristics of Zr-MOFs allow them to be deployed as robust proton-conducting materials.256,388 Thus, several researchers have focused on imparting acidity to the pores of Zr-MOFs by using linkers possessing pendant moieties such as nonstructural carboxylates, amines, phosphonates, or sulfonates, in order to design a range of highly proton-conducting Zr-MOFs.389 However, the effect of defects present in Zr-MOFs on the resulting proton conductivity have only rarely to be investigated. Kitagawa et al. prepared various UiO-66 materials with different degrees of missing-linker defects to investigate the effect of defect density on proton conductivity.390 The proton conductivity was reported to increase by nearly three orders of magnitude to 7 × 10−3 S cm−1 at 65 °C and 95% RH. The crystallinity of the defective UiO-66 could be preserved after both the pelletizing process and the impedance measurements operated at high RH. The Lewis acid sites provide binding sites for water molecules which facilitates the proton transport together with the increase in proton mobility due to increased pore volume. Another noticeable example was recently reported by Qu and coworkers, who synthesized the xerogels containing a defective Zr-MOF, MOF-801, for developing anhydrous proton conductors.391 The findings suggested that with defects present in MOF-801, the proton conductivity of the resulting xerogel could be enhanced owing to the structural changes of water clusters; an anhydrous proton conductivity of 3.6 × 10−3 S cm−1 at 40 °C was achieved with the use of defective MOF-801 xerogel.391 Another recent example demonstrated by Matoga and coworkers investigated the effect of missing-linker defects present in a sulfonate-containing two-dimensional (2D) Zr-MOF, JUK-14, on the resulting proton conductivity.392 The findings suggested that with missing-linker defects in the 2D Zr-MOF, higher proton conductivity values can be achieved at 60% and 75% RH compared to those achieved by the defect-free 2D MOF. The 2D Zr-MOFs reported here were also found to keep their structural integrity after the impedance measurements under a wide range of RH between 30% and 90%.
2.3. Other relevant applications
Membranes are valuable candidates for sustainable energy and environment and hold great promise for clean energy technologies. This section documents some applications of defective Zr-MOFs in developing membranes and optical sensors. Composite membranes with defective MOFs are one of the emerging fields of MOF modification for diverse applications, however, so far this field has not been extensively investigated. Lyu et al. computed potential utility of defective UiO-66 comprising different types (missing-linker or missing-cluster), density and unsaturated Zr sites compensated by modulators during the MOF synthesis such as chloride, hydroxide or monocarboxylates including formate, acetate, and trifluoroacetate (Fig. 23).393 | ||
| Fig. 23 Depiction of (a) defect-free and (b) and (c) defective UiO-66 frameworks, having two missing linkers and one missing cluster, respectively. (d)–(h) Representation for compensating the unsaturated metal sites created by the defects, one defect center (i.e., two unsaturated Zr atoms) coordinated with various ligands. Color code: carbon, gray; hydrogen, white; oxygen, red; zirconium, cyan; chlorine, green and fluorine, magenta. (i) Water permeability and salt rejection of both defect-free and defective membranes. The concentration of BDC vacancies is displayed in the figure, changing from 1/8 to 1/4 to 1/2 (i.e., missing-cluster structures), which are related to 3, 6, and 12 linkers missing per unit cell, respectively. Reprinted with permission from ref. 393 Copyright 2019 American Chemical Society. | ||
The studies show that UiO-66-containing membranes with high performance, in terms of water permeability and salt rejection, can be obtained by adjusting pore size and water affinity. Compared with a nominally defect-free membrane, the missing-cluster membrane obtained via trifluoroacetate was shown to have a higher water permeability (800 L m−2 h−1) with salt rejection of >99%. In addition, desirable water intrusion behavior of defective samples can be improved by presenting hydrophilic compensating ligands. This work opens the possibility of MOF defect engineering as emerging reverse osmosis membranes in desalination, lowering energy and operating costs.
Zhang et al. investigated effects of crystal size and missing-linker defects on optical sensing performance for chemical vapors and found that the small particle size with a high loading of defects increased the sensitivity and decreased the response time and recovery time of the MOF-based sensor.394
In a recent study, Liu and co-workers showed that robust membranes for dye-exclusion from permeating water could be fabricated via seeded growth of polycrystalline MOF-801 on the exterior of a porous alumina tube (Fig. 24).395 The membrane exhibited admirable rejection of a molecular probe dye (rejection rate >99.50%), while achieving admirable water flux >31 L m−2 h−1 bar−1. The authors reasoned that the flux could be boosted by increasing the density of missing-linker defects (by 2.6-fold) through varying the concentration of HFo used for synthesizing the MOF. A ca. 20% increase in flux was indeed found; but, it was accompanied by a ca. 3-fold increase in amount of dye transmitted. Clearly, however, the integrated assembly approach highlighted in Fig. 24 shows promise.
 | ||
| Fig. 24 Schematic diagram of the synthesis of MOF-801 membranes by epitaxial growth. Reproduced with permission from ref. 395 Copyright 2023 John Wiley and Sons. | ||
3. Conclusions and outlook
With their exceptional chemical and thermal stability, Zr-MOFs have been widely examined for various energy-related and environmental applications requiring aqueous media or high-temperature conditions. Although there is already a high structural diversity for Zr-MOFs, the introduction of modulable defects on the hexa-zirconium clusters of the MOFs further increases the structural flexibility and chemical functionality of Zr-MOFs. Taking the most commonly reported defective Zr-MOF, UiO-66, as an example, the proportion of defects can be broadly tuned without altering its main crystalline structure. These sites can serve as the active sites to adsorb organic pollutants or inorganic targeted ions, which could be utilized in removing pollutants from environmental samples. By exposing Zr(IV) sites to candidate reactants, missing-linker defects enable node-based Lewis-acid catalysis – most notably for detoxification of chemical warfare agents, as well as for chemically related pesticides in water. In addition, defects can be used as grafting sites for post-synthetic installation of photosensitizing moieties, specific molecular catalysts, single-metal-atom catalysts, or other desirable chemical moieties. A broad and well-established, positive feature of post-synthetic modification is that it allows framework tailoring to be accomplished under chemical conditions that differ from those of the parent synthesis. Post-synthetic installation of redox-active sites at defect sites can be exploited to engender charge-hopping-based, electrical conductivity, that, in turn, enables otherwise-insulating Zr-MOFs and their composites, to be deployed in electrochemical energy storage and conversion schemes. An arguably underexplored notion, that can be useful for engendering greater overall electrical conductivity, is to integrate frameworks with electronic conductors such as carbon nanotubes. An open challenge or opportunity here is to achieve precise chemical and geometric control over integration and co-location of defect-containing Zr-MOFs and electronic conductors. Finally, by supporting nonstructural, Brønsted-acidic oxy-ligands, defect sites on nodes can enhance proton conduction, and more generally, ion conduction, by frameworks.Missing-node and missing-linker defects, especially if periodically sited, can provide MOF-interior access to molecules that would otherwise be sterically excluded. Conversely, access can be restricted by grafting to missing-linker defects, species that exclude specific guest molecules sterically or that select (or exclude) guests based on favorable (or unfavorable) aperture/guest or pore/guest hydrophobic, hydrophilic, London dispersion or coulombic interactions, including pH-tunable interactions.
Chemically intriguing, but, thus far, under-explored are framework-topology-specific, node-distortion defects. The few available studies point to irreversibly enhanced Lewis acidity (and basicity), and, in turn, enhanced condensed-phase catalytic activity for hydrolysis reactions. In the absence of solvent, effect formation is accompanied by symmetry-lowering decreases in Zr(IV) coordination number, and sufficient enhancement of Lewis acidity and basicity to engender heterolytic cleavage of N–H, S–H, and other covalent bonds, concomitant with grafting of the resulting fragments to the distorted node. Thus, defects formed by node distortion can serve to capture and degrade unwanted volatile chemicals. While little explored, they also should, via the same chemistry, be capable of enabling persistent functionalization of Zr-MOF nodes via otherwise weak sulfur-zirconium and nitrogen-zirconium bond formation. In turn, this chemistry may open up new several potential applications.
Certain desirable properties scale with the density of structural defects; the density of defects, however, has an upper limit. Clearly useful strategy would be synthetic methods that permit defect densities to be tightly controlled and that permit defect densities to be increased, without sacrificing MOF porosity or crystallinity. Conceivably, the use of larger clusters as node cores – for example, Zr12O22 in place of Zr6O8 – could be turned to advantage in controlling or enhancing the density and/or catalytic activity of missing-linker defects.126,396 Alternatively, repetitively grafting additional, non-structural Zr(IV) ions or entire pre-formed clusters, onto defect-presenting nodes might well prove effective for boosting the density of desired missing-linker-like sites.
The chemical-stability limitations of carboxylate-based Zr-MOFs themselves should be considered. In highly alkaline aqueous solutions and in solutions containing strongly coordinating ions (e.g., phosphate, carbonate and their conjugate acids), these materials are susceptible to degradation, including loss of crystallinity, loss of porosity, and even partial dissolution and loss of linkers. Degradation is driven by substitutional displacement of linker termini (carboxylates) from Zr-nodes. In certain contexts – most notably, intentional post-synthetic alteration of framework composition via solvent-assisted linker exchange – the susceptibility of carboxylates to displacement is a functional asset. Curiously, a closely related problem has been encountered with liquid-junction type, dye-sensitized solar cells (DSCs), where dyes are anchored via carboxylate termini to materials such as TiO2. For DSCs, dye loss has been suppressed by: (a) atomic-layer-deposition (ALD) of alumina or titania such that the added inert metal-oxide enshrouds carboxylates, thereby physically preventing their displacement from the underlying TiO2 surface,397,398 (b) similar blocking and encapsulation via self-limiting, conformal growth of sub-nanometer silica coatings,399 and (c) use of much more strongly adhering phosphonate groups in place of carboxylates as dye anchors.400 To our knowledge, neither node-centric ALD of protective metal oxides nor self-limiting growth of protective silica has been used to suppress chemical degradation of Zr-MOFs, although either or both would appear to be feasible if they can be implemented without too great loss in framework porosity. A handful of examples of phosphonate-based Zr-MOFs has been described.401–403 These materials indeed do tend to display much greater chemical stability when challenged by strongly coordinating oxy-anions. Unfortunately, the apparent irreversibility of node-linker bond formation (with phosphonates) tends to yield amorphous frameworks, typically of low porosity. A general solution to the chemical degradation problem, whether via an advance in phosphonate-enabled synthesis of porous, crystalline MOFs, or via another approach, would substantially broaden the application of defect-containing Zr-MOFs – especially in the realm of electrochemically driven energy conversion where, for example, hydroxide and bicarbonate may feature prominently in aqueous electrolytes for oxygen evolution and carbon dioxide reduction. Regardless, we suggest that increased understanding of the chemical nature and promise of defects, together with enhanced control over their density and siting, will translate to increasingly sophisticated and/or useful demonstrations of applications, especially applications directed toward challenging energy- and environment-related problems.
Finally, advances in the techniques that are capable of characterizing defects in MOFs, including the real-time observation of defect formation, will further enhance our understanding of defect structure, defect siting, and defect evolution.47,404 Overall, we believe that more and more opportunities in utilizing defective Zr-MOFs and related materials in energy-related and environmental applications will emerge, and these materials will be integrated into practical devices or units in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. T. H. gratefully thanks the support from U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (grant number: DE-FG02-87ER-13808). We thank the support from National Science and Technology Council of Taiwan (grant numbers: 112-2223-E-006-003-MY3, 112-2218-E006-022 and 113-2923-E-006-006-MY2) and Ministry of Education, Taiwan (Yushan Young Scholar Program and Higher Education Sprout Project). A. D. is beneficiary of a grant María Zambrano in Universitat Politècnica de València within the framework of the grants for the retraining in the Spanish university system (Spanish Ministry of Universities, financed by the European Union, NextGeneration EU). A. R. O. and M. K. acknowledge financial support from the University of Zabol (grant numbers: IR-UOZ-GR-9381 and IR-UOZ-GR-8175) and Iran National Science Foundation (INSF, grant number 4025637). S. D. also thank the Lorestan University for the grant support. U. S. and M. E. extend their acknowledgment to The Scientific and Technological Research Council of Turkey (TUBITAK) for providing funding under grant numbers 120M698 and 122M312. We thank Dr. Boris Kramar for constructing key figures.References
- D. Chen, H. Zhuang, M. Chen, P. Y. Huang, V. Vlcek and Y. Jiao, Appl. Phys. Rev., 2023, 10, 021310 CAS.
- T. Vojta, Annu. Rev. Condens. Matter Phys., 2019, 10, 233–252 CrossRef.
- G. Wolfowicz, F. J. Heremans, C. P. Anderson, S. Kanai, H. Seo, A. Gali, G. Galli and D. D. Awschalom, Nat. Rev. Mater., 2021, 6, 906–925 CrossRef CAS.
- E. G. Seebauer and M. C. Kratzer, Mater. Sci. Eng., R, 2006, 55, 57–149 CrossRef.
- T. Tang, Z. Wang and J. Guan, Chin. J. Catal., 2022, 43, 636–678 CrossRef CAS.
- S. Chandrasekaran, D. Ma, Y. Ge, L. Deng, C. Bowen, J. Roscow, Y. Zhang, Z. Lin, R. D. K. Misra, J. Li, P. Zhang and H. Zhang, Nano Energy, 2020, 77, 105080 CrossRef CAS.
- P. Hu, Z. Zhao, X. Sun, Y. Muhammad, J. Li, S. Chen, C. Pang, T. Liao and Z. Zhao, Chem. Eng. J., 2019, 356, 329–340 CrossRef CAS.
- G. Cai, P. Yan, L. Zhang, H. C. Zhou and H. L. Jiang, Chem. Rev., 2021, 121, 12278–12326 CrossRef CAS PubMed.
- S. Daliran, A. R. Oveisi, Y. Peng, A. López-Magano, M. Khajeh, R. Mas-Ballesté, J. Alemán, R. Luque and H. Garcia, Chem. Soc. Rev., 2022, 51, 7810–7882 RSC.
- K. O. Kirlikovali, S. L. Hanna, F. A. Son and O. K. Farha, ACS Nanosci. Au, 2023, 3, 37–45 CrossRef CAS PubMed.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- W. Fan, X. Zhang, Z. Kang, X. Liu and D. Sun, Coord. Chem. Rev., 2021, 443, 213968 CrossRef CAS.
- J. Chen, R. Abazari, K. A. Adegoke, N. W. Maxakato, O. S. Bello, M. Tahir, S. Tasleem, S. Sanati, A. M. Kirillov and Y. Zhou, Coord. Chem. Rev., 2022, 469, 214664 CrossRef CAS.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2023, 123, 445–490 CrossRef PubMed.
- Z. Wang, L. Liu, Z. Li, N. Goyal, T. Du, J. He and G. K. Li, Energy Fuels, 2022, 36, 2927–2944 CrossRef CAS.
- Z. Chen, M. C. Wasson, R. J. Drout, L. Robison, K. B. Idrees, J. G. Knapp, F. A. Son, X. Zhang, W. Hierse, C. Kühn, S. Marx, B. Hernandez and O. K. Farha, Faraday Discuss., 2021, 225, 9–69 RSC.
- H. Furukawa, U. Müller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417–3430 CrossRef CAS PubMed.
- S. Dissegna, K. Epp, W. R. Heinz, G. Kieslich and R. A. Fischer, Adv. Mater., 2018, 30, 1704501 CrossRef PubMed.
- I. E. Khalil, C. Xue, W. Liu, X. Li, Y. Shen, S. Li, W. Zhang and F. Huo, Adv. Funct. Mater., 2021, 31, 2010052 CrossRef CAS.
- X. Feng, H. S. Jena, C. Krishnaraj, K. Leus, G. Wang, H. Chen, C. Jia and P. Van Der Voort, ACS Appl. Mater. Interfaces, 2021, 13, 60715–60735 CrossRef CAS PubMed.
- W. Xiang, Y. Zhang, Y. Chen, C. J. Liu and X. Tu, J. Mater. Chem. A, 2020, 8, 21526–21546 RSC.
- R. S. Forgan, Chem. Sci., 2020, 11, 4546–4562 RSC.
- X. Hou, J. Wang, B. Mousavi, N. Klomkliang and S. Chaemchuen, Dalton Trans., 2022, 51, 8133–8159 RSC.
- Z. Yu, S. Jamdade, X. Yu, X. Cai and D. S. Sholl, J. Phys. Chem. Lett., 2023, 14, 6658–6665 CrossRef CAS PubMed.
- A. W. Thornton, R. Babarao, A. Jain, F. Trousselet and F. X. Coudert, Dalton Trans., 2016, 45, 4352–4359 RSC.
- J. Hajek, C. Caratelli, R. Demuynck, K. De Wispelaere, L. Vanduyfhuys, M. Waroquier and V. Van Speybroeck, Chem. Sci., 2018, 9, 2723–2732 RSC.
- C. K. Brozek, V. K. Michaelis, T.-C. Ong, L. Bellarosa, N. López, R. G. Griffin and M. Dincă, ACS Cent. Sci., 2015, 1, 252–260 CrossRef CAS PubMed.
- S. Ling and B. Slater, Chem. Sci., 2016, 7, 4706–4712 RSC.
- R. A. Peralta, P. Lyu, A. López-Olvera, J. L. Obeso, C. Leyva, N. C. Jeong, I. A. Ibarra and G. Maurin, Angew. Chem., Int. Ed., 2022, 61, e202210857 CrossRef CAS PubMed.
- C. Caratelli, J. Hajek, E. J. Meijer, M. Waroquier and V. Van Speybroeck, Chem. – Eur. J., 2019, 25, 15315–15325 CrossRef CAS PubMed.
- P. Lyu, A. M. Wright, A. López-Olvera, P. G. M. Mileo, J. A. Zárate, E. Martínez-Ahumada, V. Martis, D. R. Williams, M. Dincă, I. A. Ibarra and G. Maurin, Chem. Mater., 2021, 33, 6186–6192 CrossRef CAS.
- B. T. Yost, B. Gibbons, A. Wilson, A. J. Morris and L. E. McNeil, RSC Adv., 2022, 12, 22440–22447 RSC.
- B. Shan, S. M. McIntyre, M. R. Armstrong, Y. Shen and B. Mu, Ind. Eng. Chem. Res., 2018, 57, 14233–14241 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CrossRef CAS PubMed.
- X. Feng, H. S. Jena, C. Krishnaraj, D. Arenas-Esteban, K. Leus, G. Wang, J. Sun, M. Rüscher, J. Timoshenko, B. Roldan Cuenya, S. Bals and P. V. D. Voort, J. Am. Chem. Soc., 2021, 143, 21511–21518 CrossRef CAS PubMed.
- D. N. Johnstone, F. C. N. Firth, C. P. Grey, P. A. Midgley, M. J. Cliffe and S. M. Collins, J. Am. Chem. Soc., 2020, 142, 13081–13089 CrossRef CAS PubMed.
- A. Zheng, K. Yin, R. Pan, M. Zhu, Y. Xiong and L. Sun, Nanomaterials, 2023, 13, 1742 CrossRef CAS PubMed.
- J. N. Hall and P. Bollini, React. Chem. Eng., 2019, 4, 207–222 RSC.
- Y. Shan, G. Zhang, Y. Shi and H. Pang, Cell Rep. Phys. Sci., 2023, 4, 101301 CrossRef CAS.
- X. Yan, L. Zhuang, Z. Zhu and X. Yao, Nanoscale, 2021, 13, 3327–3345 RSC.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC.
- M. R. DeStefano, T. Islamoglu, S. J. Garibay, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 1357–1361 CrossRef CAS.
- D. Bůžek, J. Demel and K. Lang, Inorg. Chem., 2018, 57, 14290–14297 CrossRef PubMed.
- C. Atzori, G. C. Shearer, L. Maschio, B. Civalleri, F. Bonino, C. Lamberti, S. Svelle, K. P. Lillerud and S. Bordiga, J. Phys. Chem. C, 2017, 121, 9312–9324 CrossRef CAS.
- X. Zhu, B. Li, J. Yang, Y. Li, W. Zhao, J. Shi and J. Gu, ACS Appl. Mater. Interfaces, 2015, 7, 223–231 CrossRef CAS PubMed.
- S. Tatay, S. Martínez-Giménez, A. Rubio-Gaspar, J. Castells-Gil, Z. Dong, A. Mayoral, N. Almora-Barrios, N. M. Padial and C. Martí-Gastaldo, PREPRINT (Version 1) available at Research Square, 2023 DOI:10.21203/rs.3.rs-2374720/v1.
- N. Hosono, A. Terashima, S. Kusaka, R. Matsuda and S. Kitagawa, Nat. Chem., 2019, 11, 109–116 CrossRef CAS PubMed.
- L. Liu, Z. Chen, J. Wang, D. Zhang, Y. Zhu, S. Ling, K.-W. Huang, Y. Belmabkhout, K. Adil, Y. Zhang, B. Slater, M. Eddaoudi and Y. Han, Nat. Chem., 2019, 11, 622–628 CrossRef CAS PubMed.
- M.-S. Yao, K.-i Otake and S. Kitagawa, Trends Chem., 2023, 5, 588–604 CrossRef CAS.
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Cryst. Growth Des., 2014, 14, 5370–5372 CrossRef.
- D. A. Keen and A. L. Goodwin, Nature, 2015, 521, 303–309 CrossRef CAS PubMed.
- X. Yang, P. Juhas, C. L. Farrow and S. J. Billinge, arXiv, 2014, preprint, arXiv.1402.3163, DOI:10.48550/arXiv.1402.3163.
- A. F. Sapnik, I. Bechis, A. M. Bumstead, T. Johnson, P. A. Chater, D. A. Keen, K. E. Jelfs and T. D. Bennett, Nat. Commun., 2022, 13, 2173 CrossRef CAS PubMed.
- C. Castillo-Blas, J. M. Moreno, I. Romero-Muñiz and A. E. Platero-Prats, Nanoscale, 2020, 12, 15577–15587 RSC.
- Z. Chen, G. D. Stroscio, J. Liu, Z. Lu, J. T. Hupp, L. Gagliardi and K. W. Chapman, J. Am. Chem. Soc., 2023, 145, 268–276 CrossRef CAS PubMed.
- R. Ameloot, F. Vermoortele, J. Hofkens, F. C. De Schryver, D. E. De Vos and M. B. J. Roeffaers, Angew. Chem., Int. Ed., 2013, 52, 401–405 CrossRef CAS PubMed.
- B. Bueken, N. Van Velthoven, A. Krajnc, S. Smolders, F. Taulelle, C. Mellot-Draznieks, G. Mali, T. D. Bennett and D. De Vos, Chem. Mater., 2017, 29, 10478–10486 CrossRef CAS.
- B. Bueken, F. Vermoortele, M. J. Cliffe, M. T. Wharmby, D. Foucher, J. Wieme, L. Vanduyfhuys, C. Martineau, N. Stock, F. Taulelle, V. Van Speybroeck, A. L. Goodwin and D. De Vos, Chem. – Eur. J., 2016, 22, 3264–3267 CrossRef CAS PubMed.
- S. Yuan, L. Zou, J. S. Qin, J. Li, L. Huang, L. Feng, X. Wang, M. Bosch, A. Alsalme, T. Cagin and H. C. Zhou, Nat. Commun., 2017, 8, 15356 CrossRef CAS PubMed.
- K.-Y. Wang, L. Feng, T.-H. Yan, S. Wu, E. A. Joseph and H.-C. Zhou, Angew. Chem., Int. Ed., 2020, 59, 11349–11354 CrossRef CAS PubMed.
- X. Feng, J. Hajek, H. S. Jena, G. Wang, S. K. P. Veerapandian, R. Morent, N. De Geyter, K. Leyssens, A. E. J. Hoffman, V. Meynen, C. Marquez, D. E. De Vos, V. Van Speybroeck, K. Leus and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 3174–3183 CrossRef CAS PubMed.
- C. Caratelli, J. Hajek, S. M. J. Rogge, S. Vandenbrande, E. J. Meijer, M. Waroquier and V. Van Speybroeck, ChemPhysChem, 2018, 19, 420–429 CrossRef CAS PubMed.
- F. G. Cirujano and F. X. Llabrés i Xamena, J. Phys. Chem. Lett., 2020, 11, 4879–4890 CrossRef CAS PubMed.
- L. Löbbert, S. Chheda, J. Zheng, N. Khetrapal, J. Schmid, R. Zhao, C. A. Gaggioli, D. M. Camaioni, R. Bermejo-Deval, O. Y. Gutiérrez, Y. Liu, J. I. Siepmann, M. Neurock, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2023, 145, 1407–1422 CrossRef PubMed.
- W. Zhang, M. Kauer, O. Halbherr, K. Epp, P. Guo, M. I. Gonzalez, D. J. Xiao, C. Wiktor, F. X. Llabrés i Xamena, C. Wöll, Y. Wang, M. Muhler and R. A. Fischer, Chem. – Eur. J., 2016, 22, 14297–14307 CrossRef CAS PubMed.
- S. Dissegna, R. Hardian, K. Epp, G. Kieslich, M.-V. Coulet, P. Llewellyn and R. A. Fischer, CrystEngComm, 2017, 19, 4137–4141 RSC.
- Y. Xiao, M. Zhang, D. Yang, L. Zhang, S. Zhuang, J. Tang, Z. Zhang and X. Qiao, ACS Appl. Mater. Interfaces, 2023, 15, 34675–34681 CrossRef CAS PubMed.
- V. Pascanu, G. González Miera, A. K. Inge and B. Martín-Matute, J. Am. Chem. Soc., 2019, 141, 7223–7234 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. Santiago-Portillo, A. M. Asiri and H. Garcia, ChemCatChem, 2019, 11, 899–923 CrossRef CAS.
- A. Jrad, B. J. Abu Tarboush, M. Hmadeh and M. Ahmad, Appl. Catal., A, 2019, 570, 31–41 CrossRef CAS.
- Z. Fang, J. P. Dürholt, M. Kauer, W. Zhang, C. Lochenie, B. Jee, B. Albada, N. Metzler-Nolte, A. Pöppl, B. Weber, M. Muhler, Y. Wang, R. Schmid and R. A. Fischer, J. Am. Chem. Soc., 2014, 136, 9627–9636 CrossRef CAS PubMed.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37, 1248–1257 CrossRef CAS.
- Z. Lu, J. Liu, X. Zhang, Y. Liao, R. Wang, K. Zhang, J. Lyu, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2020, 142, 21110–21121 CrossRef CAS PubMed.
- L. Feng, G. S. Day, K.-Y. Wang, S. Yuan and H.-C. Zhou, Chem, 2020, 6, 2902–2923 CAS.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- G. Kaur, S. Øien-Ødegaard, A. Lazzarini, S. M. Chavan, S. Bordiga, K. P. Lillerud and U. Olsbye, Cryst. Growth Des., 2019, 19, 4246–4251 CrossRef CAS.
- H. L. B. Boström, S. Emmerling, F. Heck, C. Koschnick, A. J. Jones, M. J. Cliffe, R. Al Natour, M. Bonneau, V. Guillerm, O. Shekhah, M. Eddaoudi, J. Lopez-Cabrelles, S. Furukawa, M. Romero-Angel, C. Martí-Gastaldo, M. Yan, A. J. Morris, I. Romero-Muñiz, Y. Xiong, A. E. Platero-Prats, J. Roth, W. L. Queen, K. S. Mertin, D. E. Schier, N. R. Champness, H. H.-M. Yeung and B. V. Lotsch, Adv. Mater., 2023, 2304832, DOI:10.1002/adma.202304832.
- P. Gimeno-Fonquernie, W. Liang, J. Albalad, A. Kuznicki, J. R. Price, E. D. Bloch, C. J. Doonan and C. J. Sumby, Chem. Commun., 2022, 58, 957–960 RSC.
- Y. Zhao, S. Qi, Z. Niu, Y. Peng, C. Shan, G. Verma, L. Wojtas, Z. Zhang, B. Zhang, Y. Feng, Y.-S. Chen and S. Ma, J. Am. Chem. Soc., 2019, 141, 14443–14450 CrossRef CAS PubMed.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- S. Leubner, V. E. G. Bengtsson, K. Synnatschke, J. Gosch, A. Koch, H. Reinsch, H. Xu, C. Backes, X. Zou and N. Stock, J. Am. Chem. Soc., 2020, 142, 15995–16000 CrossRef CAS PubMed.
- S. Leubner, H. Zhao, N. Van Velthoven, M. Henrion, H. Reinsch, D. E. De Vos, U. Kolb and N. Stock, Angew. Chem., Int. Ed., 2019, 58, 10995–11000 CrossRef CAS PubMed.
- R. Dai, F. Peng, P. Ji, K. Lu, C. Wang, J. Sun and W. Lin, Inorg. Chem., 2017, 56, 8128–8134 CrossRef CAS PubMed.
- K. A. Milakin, S. Gupta, L. Kobera, A. Mahun, M. Konefał, O. Kočková, O. Taboubi, Z. Morávková, J. M. Chin, K. Allahyarli and P. Bober, ACS Appl. Mater. Interfaces, 2023, 15, 23813–23823 CrossRef CAS PubMed.
- C. Koschnick, M. W. Terban, R. Frison, M. Etter, F. A. Böhm, D. M. Proserpio, S. Krause, R. E. Dinnebier, S. Canossa and B. V. Lotsch, J. Am. Chem. Soc., 2023, 145, 10051–10060 CrossRef CAS PubMed.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- A. Torres-Huerta, D. Galicia-Badillo, A. Aguilar-Granda, J. T. Bryant, F. J. Uribe-Romo and B. Rodríguez-Molina, Chem. Sci., 2020, 11, 11579–11583 RSC.
- N. Alsadun, G. Mouchaham, V. Guillerm, J. Czaban-Jóźwiak, A. Shkurenko, H. Jiang, P. M. Bhatt, P. Parvatkar and M. Eddaoudi, J. Am. Chem. Soc., 2020, 142, 20547–20553 CrossRef CAS PubMed.
- C. Koschnick, R. Stäglich, T. Scholz, M. W. Terban, A. von Mankowski, G. Savasci, F. Binder, A. Schökel, M. Etter, J. Nuss, R. Siegel, L. S. Germann, C. Ochsenfeld, R. E. Dinnebier, J. Senker and B. V. Lotsch, Nat. Commun., 2021, 12, 3099 CrossRef CAS PubMed.
- D. Azarifar, R. Ghorbani-Vaghei, S. Daliran and A. R. Oveisi, ChemCatChem, 2017, 9, 1992–2000 CrossRef CAS.
- S. Karamzadeh, E. Sanchooli, A. R. Oveisi, S. Daliran and R. Luque, Appl. Catal., B, 2022, 303, 120815 CrossRef CAS.
- Y. Bai, Y. Dou, L. H. Xie, W. Rutledge, J. R. Li and H. C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- H. R. Abid, H. Tian, H. M. Ang, M. O. Tade, C. E. Buckley and S. Wang, Chem. Eng. J., 2012, 187, 415–420 CrossRef CAS.
- O. G. Nik, X. Y. Chen and S. Kaliaguine, J. Membr. Sci., 2012, 413–414, 48–61 CrossRef CAS.
- Q. Yang, A. D. Wiersum, P. L. Llewellyn, V. Guillerm, C. Serre and G. Maurin, Chem. Commun., 2011, 47, 9603–9605 RSC.
- J. Zhu, P. M. Usov, W. Xu, P. J. Celis-Salazar, S. Lin, M. C. Kessinger, C. Landaverde-Alvarado, M. Cai, A. M. May, C. Slebodnick, D. Zhu, S. D. Senanayake and A. J. Morris, J. Am. Chem. Soc., 2018, 140, 993–1003 CrossRef CAS PubMed.
- B. Ghalei, K. Wakimoto, C. Y. Wu, A. P. Isfahani, T. Yamamoto, K. Sakurai, M. Higuchi, B. K. Chang, S. Kitagawa and E. Sivaniah, Angew. Chem., Int. Ed., 2019, 58, 19034–19040 CrossRef CAS PubMed.
- B. Bueken, H. Reinsch, N. Reimer, I. Stassen, F. Vermoortele, R. Ameloot, N. Stock, C. E. A. Kirschhock and D. De Vos, Chem. Commun., 2014, 50, 10055–10058 RSC.
- W. Gong, Y. Xie, X. Wang, K. O. Kirlikovali, K. B. Idrees, F. Sha, H. Xie, Y. Liu, B. Chen, Y. Cui and O. K. Farha, J. Am. Chem. Soc., 2023, 145, 2679–2689 CrossRef CAS PubMed.
- Y. Shi, Y. Xie, T. Alshahrani and B. Chen, CrystEngComm, 2023, 25, 1643–1647 RSC.
- Y. Zhang, Y. Liu, Y. Hang, G. Liu, B. Mo, J. Li, W. Ji, G. Liu and W. Jin, J. Membr. Sci., 2024, 689, 122171 CrossRef CAS.
- J. Shen, G. Liu, K. Huang, Q. Li, K. Guan, Y. Li and W. Jin, J. Membr. Sci., 2016, 513, 155–165 CrossRef CAS.
- S. Wang, N. Xhaferaj, M. Wahiduzzaman, K. Oyekan, X. Li, K. Wei, B. Zheng, A. Tissot, J. Marrot, W. Shepard, C. Martineau-Corcos, Y. Filinchuk, K. Tan, G. Maurin and C. Serre, J. Am. Chem. Soc., 2019, 141, 17207–17216 CrossRef CAS PubMed.
- M. P. M. Poschmann, Ö. Alan, S. Ito, C. Näther, G. Friedrichs and N. Stock, Inorg. Chem., 2023, 62, 12252–12259 CrossRef CAS PubMed.
- H. Wang, X. Dong, J. Lin, S. J. Teat, S. Jensen, J. Cure, E. V. Alexandrov, Q. Xia, K. Tan, Q. Wang, D. H. Olson, D. M. Proserpio, Y. J. Chabal, T. Thonhauser, J. Sun, Y. Han and J. Li, Nat. Commun., 2018, 9, 1745 CrossRef PubMed.
- W.-Y. Gao, T. Thiounn, L. Wojtas, Y.-S. Chen and S. Ma, Sci. China: Chem., 2016, 59, 980–983 CrossRef CAS.
- G. Zhang, F. Xie, T. M. Osborn Popp, A. Patel, E. M. Cedeño Morales, K. Tan, R. Crichton, G. Hall, J. Zhang, A. J. Nieuwkoop and J. Li, CrystEngComm, 2023, 25, 1067–1075 RSC.
- R. K. Mah, B. S. Gelfand, J. M. Taylor and G. K. H. Shimizu, Inorg. Chem. Front., 2015, 2, 273–277 RSC.
- J. Kim, D. Nam, H. Kitagawa, D.-W. Lim and W. Choe, Nano Res., 2021, 14, 392–397 CrossRef CAS.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- G. Liu, Z. Wang, C. Chen, J. Li, G. Zhou, W. Ji, G. Liu and W. Jin, Chem. Commun., 2023, 59, 8075–8078 RSC.
- Z. Lu, J. Duan, L. Du, Q. Liu, N. M. Schweitzer and J. T. Hupp, J. Mater. Chem. A, 2022, 10, 6442–6447 RSC.
- W. Gong, X. Chen, K. M. Fahy, J. Dong, Y. Liu, O. K. Farha and Y. Cui, J. Am. Chem. Soc., 2023, 145, 13869–13878 CrossRef CAS PubMed.
- Y. He, C. Li, X. B. Chen, Z. Shi and S. Feng, ACS Appl. Mater. Interfaces, 2022, 14, 28977–28984 CrossRef CAS PubMed.
- M. Pander, M. Janeta and W. Bury, ACS Appl. Mater. Interfaces, 2021, 13, 8344–8352 CrossRef CAS PubMed.
- B. Villoria-del-Álamo, S. Rojas-Buzo, P. García-García and A. Corma, Chem. – Eur. J., 2021, 27, 4588–4598 CrossRef PubMed.
- A. Das, N. Anbu, M. Sk, A. Dhakshinamoorthy and S. Biswas, Inorg. Chem., 2019, 58, 5163–5172 CrossRef CAS PubMed.
- K. D. Nguyen, C. Kutzscher, F. Drache, I. Senkovska and S. Kaskel, Inorg. Chem., 2018, 57, 1483–1489 CrossRef CAS PubMed.
- C. A. Trickett, T. M. Osborn Popp, J. Su, C. Yan, J. Weisberg, A. Huq, P. Urban, J. Jiang, M. J. Kalmutzki, Q. Liu, J. Baek, M. P. Head-Gordon, G. A. Somorjai, J. A. Reimer and O. M. Yaghi, Nat. Chem., 2019, 11, 170–176 CrossRef CAS PubMed.
- M. D. Korzyński, D. F. Consoli, S. Zhang, Y. Román-Leshkov and M. Dincă, J. Am. Chem. Soc., 2018, 140, 6956–6960 CrossRef PubMed.
- S. Yuan, L. Zou, H. Li, Y.-P. Chen, J. Qin, Q. Zhang, W. Lu, M. B. Hall and H.-C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 10776–10780 CrossRef CAS PubMed.
- J. Li, J.-Y. Huang, Y.-X. Meng, L. Li, L.-L. Zhang and H.-L. Jiang, Chem. Commun., 2023, 59, 2541–2559 RSC.
- D. Yang and B. C. Gates, Acc. Chem. Res., 2021, 54, 1982–1991 CrossRef CAS PubMed.
- S. Das, D. Yang, E. T. Conley and B. C. Gates, ACS Catal., 2023, 13, 14173–14188 CrossRef CAS.
- X. Chen, Y. Lyu, Z. Wang, X. Qiao, B. C. Gates and D. Yang, ACS Catal., 2020, 10, 2906–2914 CrossRef CAS.
- Y. Zhu, J. Zheng, J. Ye, Y. Cui, K. Koh, L. Kovarik, D. M. Camaioni, J. L. Fulton, D. G. Truhlar, M. Neurock, C. J. Cramer, O. Y. Gutiérrez and J. A. Lercher, Nat. Commun., 2020, 11, 5849 CrossRef CAS PubMed.
- J. Zheng, L. Löbbert, S. Chheda, N. Khetrapal, J. Schmid, C. A. Gaggioli, B. Yeh, R. Bermejo-Deval, R. K. Motkuri, M. Balasubramanian, J. L. Fulton, O. Y. Gutiérrez, J. I. Siepmann, M. Neurock, L. Gagliardi and J. A. Lercher, J. Catal., 2022, 413, 176–183 CrossRef CAS.
- Z. Sharifzadeh, S. A. A. Razavi and A. Morsali, Green Chem., 2023, 25, 8661–8678 RSC.
- P. Ji, X. Feng, S. S. Veroneau, Y. Song and W. Lin, J. Am. Chem. Soc., 2017, 139, 15600–15603 CrossRef CAS PubMed.
- J. Zhang, B. An, Z. Li, Y. Cao, Y. Dai, W. Wang, L. Zeng, W. Lin and C. Wang, J. Am. Chem. Soc., 2021, 143, 8829–8837 CrossRef CAS PubMed.
- B. R. Reiner, N. T. Mucha, A. Rothstein, J. S. Temme, P. Duan, K. Schmidt-Rohr, B. M. Foxman and C. R. Wade, Inorg. Chem., 2018, 57, 2663–2672 CrossRef CAS PubMed.
- A. M. Rasero-Almansa, A. Corma, M. Iglesias and F. Sánchez, ChemCatChem, 2014, 6, 3426–3433 CrossRef CAS.
- W. Gong, M. Kazem-Rostami, F. A. Son, S. Su, K. M. Fahy, H. Xie, T. Islamoglu, Y. Liu, J. F. Stoddart, Y. Cui and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 22574–22581 CrossRef CAS PubMed.
- Y. Benseghir, A. Solé-Daura, D. R. Cairnie, A. L. Robinson, M. Duguet, P. Mialane, P. Gairola, M. Gomez-Mingot, M. Fontecave, D. Iovan, B. Bonnett, A. J. Morris, A. Dolbecq and C. Mellot-Draznieks, J. Mater. Chem. A, 2022, 10, 18103–18115 RSC.
- J. G. Santaclara, A. I. Olivos-Suarez, A. Gonzalez-Nelson, D. Osadchii, M. A. Nasalevich, M. A. van der Veen, F. Kapteijn, A. M. Sheveleva, S. L. Veber, M. V. Fedin, A. T. Murray, C. H. Hendon, A. Walsh and J. Gascon, Chem. Mater., 2017, 29, 8963–8967 CrossRef CAS.
- H.-H. He, J.-P. Yuan, P.-Y. Cai, K.-Y. Wang, L. Feng, A. Kirchon, J. Li, L.-L. Zhang, H.-C. Zhou and Y. Fang, J. Am. Chem. Soc., 2023, 145, 17164–17175 CrossRef CAS PubMed.
- S. Daliran, M. Khajeh, A. R. Oveisi, J. Albero and H. García, ACS Appl. Mater. Interfaces, 2022, 14, 36515–36526 CrossRef CAS PubMed.
- D. Banerjee, W. Xu, Z. Nie, L. E. V. Johnson, C. Coghlan, M. L. Sushko, D. Kim, M. J. Schweiger, A. A. Kruger, C. J. Doonan and P. K. Thallapally, Inorg. Chem., 2016, 55, 8241–8243 CrossRef CAS PubMed.
- T. He, Y.-Z. Zhang, X.-J. Kong, J. Yu, X.-L. Lv, Y. Wu, Z.-J. Guo and J.-R. Li, ACS Appl. Mater. Interfaces, 2018, 10, 16650–16659 CrossRef CAS PubMed.
- D. Li, M. Kassymova, X. Cai, S.-Q. Zang and H.-L. Jiang, Coord. Chem. Rev., 2020, 412, 213262 CrossRef CAS.
- A. Gutiérrez-Serpa, T. Kundu, J. Pasán, A. I. Jiménez-Abizanda, S. Kaskel, I. Senkovska and V. Pino, ACS Appl. Mater. Interfaces, 2022, 14, 4510–4521 CrossRef PubMed.
- J. Hou, H. Wang and H. Zhang, Ind. Eng. Chem. Res., 2020, 59, 12907–12923 CrossRef CAS.
- J. Hou, H. Zhang, H. Wang, A. W. Thornton and K. Konstas, J. Mater. Chem. A, 2023, 11, 13223–13230 RSC.
- J. Hou, H. Zhang, J. Lu, X. Li, C. Zhao, H. Wang, A. W. Thornton and K. Konstas, J. Membr. Sci., 2023, 674, 121511 CrossRef CAS.
- K. H. Cho, S. K. Chitale, S.-J. Kim, G.-Y. Cha, D.-Y. Hong, S. G. Ryu, J.-S. Chang and Y. K. Hwang, Microporous Mesoporous Mater., 2019, 285, 61–69 CrossRef CAS.
- S. Nazri, M. Khajeh, A. R. Oveisi, R. Luque, E. Rodríguez-Castellón and M. Ghaffari-Moghaddam, Sep. Purif. Technol., 2021, 259, 118197 CrossRef CAS.
- S. Daliran, M. Ghazagh-Miri, A. R. Oveisi, M. Khajeh, S. Navalón, M. Âlvaro, M. Ghaffari-Moghaddam, H. Samareh Delarami and H. García, ACS Appl. Mater. Interfaces, 2020, 12, 25221–25232 CrossRef CAS PubMed.
- R. Oktavian, R. Schireman, L. T. Glasby, G. Huang, F. Zanca, D. Fairen-Jimenez, M. T. Ruggiero and P. Z. Moghadam, ACS Appl. Mater. Interfaces, 2022, 14, 56938–56947 CrossRef CAS PubMed.
- Z. Chen, P. Li, X. Wang, K.-I. Otake, X. Zhang, L. Robison, A. Atilgan, T. Islamoglu, M. G. Hall, G. W. Peterson, J. F. Stoddart and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 12229–12235 CrossRef CAS PubMed.
- K. E. deKrafft, W. S. Boyle, L. M. Burk, O. Z. Zhou and W. Lin, J. Mater. Chem., 2012, 22, 18139–18144 RSC.
- X. Chen, B. B. Mendes, Y. Zhuang, J. Conniot, S. Mercado Argandona, F. Melle, D. P. Sousa, D. Perl, A. Chivu, H. K. Patra, W. Shepard, J. Conde and D. Fairen-Jimenez, J. Am. Chem. Soc., 2024, 146, 1644–1656 CrossRef CAS PubMed.
- K. Ma, Y. H. Cheung, H. Xie, X. Wang, M. Evangelopoulos, K. O. Kirlikovali, S. Su, X. Wang, C. A. Mirkin, J. H. Xin and O. K. Farha, Chem. Mater., 2023, 35, 2342–2352 CrossRef CAS.
- I. Abánades Lázaro, S. Abánades Lázaro and R. S. Forgan, Chem. Commun., 2018, 54, 2792–2795 RSC.
- I. Abánades Lázaro, C. J. R. Wells and R. S. Forgan, Angew. Chem., Int. Ed., 2020, 59, 5211–5217 CrossRef PubMed.
- C. Jia, T. He and G. M. Wang, Coord. Chem. Rev., 2023, 476, 214930 CrossRef CAS.
- F. Drache, V. Bon, I. Senkovska, M. Adam, A. Eychmüller and S. Kaskel, Eur. J. Inorg. Chem., 2016, 4483–4489 CrossRef CAS.
- S. Ghosh, J. Krishnan, V. Karthik, A. Dhakshinamoorthy and S. Biswas, Inorg. Chem., 2023, 62, 8605–8614 CrossRef CAS PubMed.
- S. Ghosh, J. Krishnan, S. S. Hossain, A. Dhakshinamoorthy and S. Biswas, ACS Appl. Mater. Interfaces, 2023, 15, 26843–26851 CrossRef CAS PubMed.
- G. Nickerl, I. Senkovska and S. Kaskel, Chem. Commun., 2015, 51, 2280–2282 RSC.
- H.-L. Jiang, D. Feng, K. Wang, Z.-Y. Gu, Z. Wei, Y.-P. Chen and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 13934–13938 CrossRef CAS PubMed.
- B. Wang, P. Wang, L.-H. Xie, R.-B. Lin, J. Lv, J.-R. Li and B. Chen, Nat. Commun., 2019, 10, 3861 CrossRef PubMed.
- B. Wang, Q. Yang, C. Guo, Y. Sun, L.-H. Xie and J.-R. Li, ACS Appl. Mater. Interfaces, 2017, 9, 10286–10295 CrossRef CAS PubMed.
- A. E. Baumann, X. Han, M. M. Butala and V. S. Thoi, J. Am. Chem. Soc., 2019, 141, 17891–17899 CrossRef CAS PubMed.
- H. Fei, S. Pullen, A. Wagner, S. Ott and S. M. Cohen, Chem. Commun., 2015, 51, 66–69 RSC.
- F. Su, S. Zhang, H. Ji, H. Zhao, J. Y. Tian, C. S. Liu, Z. Zhang, S. Fang, X. Zhu and M. Du, ACS Sens., 2017, 2, 998–1005 CrossRef CAS PubMed.
- W. A. Maza, A. J. Haring, S. R. Ahrenholtz, C. C. Epley, S. Y. Lin and A. J. Morris, Chem. Sci., 2016, 7, 719–727 RSC.
- S. Mukhopadhyay, R. Shimoni, I. Liberman, R. Ifraemov, I. Rozenberg and I. Hod, Angew. Chem., Int. Ed., 2021, 60, 13423–13429 CrossRef CAS PubMed.
- M. O. Cichocka, Z. Liang, D. Feng, S. Back, S. Siahrostami, X. Wang, L. Samperisi, Y. Sun, H. Xu, N. Hedin, H. Zheng, X. Zou, H.-C. Zhou and Z. Huang, J. Am. Chem. Soc., 2020, 142, 15386–15395 CrossRef CAS PubMed.
- A. Van Wyk, T. Smith, J. Park and P. Deria, J. Am. Chem. Soc., 2018, 140, 2756–2760 CrossRef CAS PubMed.
- H. Liu, M. Cheng, Y. Liu, G. Zhang, L. Li, L. Du, B. Li, S. Xiao, G. Wang and X. Yang, Coord. Chem. Rev., 2022, 458, 214428 CrossRef CAS.
- M. Fu, X. Deng, S. Q. Wang, F. Yang, L. C. Lin, M. J. Zaworotko and Y. Dong, Sep. Purif. Technol., 2022, 288, 120620 CrossRef CAS.
- J. Yan, T. Ji, Y. Sun, S. Meng, C. Wang and Y. Liu, J. Membr. Sci., 2022, 661, 120959 CrossRef CAS.
- Y. Feng, Q. Chen, M. Jiang and J. Yao, Ind. Eng. Chem. Res., 2019, 58, 17646–17659 CrossRef CAS.
- J. Ren, M. Ledwaba, N. M. Musyoka, H. W. Langmi, M. Mathe, S. Liao and W. Pang, Coord. Chem. Rev., 2017, 349, 169–197 CrossRef CAS.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- S. Chaemchuen, Z. Luo, K. Zhou, B. Mousavi, S. Phatanasri, M. Jaroniec and F. Verpoort, J. Catal., 2017, 354, 84–91 CrossRef CAS.
- S. Ali, Z. Zuhra, Y. Abbas, Y. Shu, M. Ahmad and Z. Wang, Langmuir, 2021, 37, 13602–13609 CrossRef CAS PubMed.
- N. Precisvalle, P. H. Ho, T. Cacciaguerra, S. Deabate, L. Girard, G. Toquer, K. D. Nguyen, H. V. Le, F. Di Renzo, A. Martucci and P. Trens, Microporous Mesoporous Mater., 2021, 324, 111293 CrossRef CAS.
- O. V. Gutov, M. G. Hevia, E. C. Escudero-Adán and A. Shafir, Inorg. Chem., 2015, 54, 8396–8400 CrossRef CAS PubMed.
- J. Duncan, D. Sengupta, S. Bose, K. O. Kirlikovali and O. K. Farha, Sustainable Chem. Environ., 2023, 3, 100032 CrossRef.
- F. Ragon, P. Horcajada, H. Chevreau, Y. K. Hwang, U. H. Lee, S. R. Miller, T. Devic, J.-S. Chang and C. Serre, Inorg. Chem., 2014, 53, 2491–2500 CrossRef CAS PubMed.
- D. Yang, M. A. Ortuño, V. Bernales, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2018, 140, 3751–3759 CrossRef CAS PubMed.
- R. Wei, C. A. Gaggioli, G. Li, T. Islamoglu, Z. Zhang, P. Yu, O. K. Farha, C. J. Cramer, L. Gagliardi, D. Yang and B. C. Gates, Chem. Mater., 2019, 31, 1655–1663 CrossRef CAS.
- Z. Hu, I. Castano, S. Wang, Y. Wang, Y. Peng, Y. Qian, C. Chi, X. Wang and D. Zhao, Cryst. Growth Des., 2016, 16, 2295–2301 CrossRef CAS.
- N. Assaad, G. Sabeh and M. Hmadeh, ACS Appl. Nano Mater., 2020, 3, 8997–9008 CrossRef CAS.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- B. Van de Voorde, I. Stassen, B. Bueken, F. Vermoortele, D. De Vos, R. Ameloot, J.-C. Tan and T. D. Bennett, J. Mater. Chem. A, 2015, 3, 1737–1742 RSC.
- R. Wang, L. Liu, S. Subhan, Y. Muhammad, Y. Hu, M. Huang, Y. Peng, Z. Zhao and Z. Zhao, Chem. Eng. J., 2020, 395, 124958 CrossRef CAS.
- P. Wang, L. Sun, J. Ye, Q. Liu, Z. Fei, X. Chen, Z. Zhang, J. Tang, M. Cui and X. Qiao, Microporous Mesoporous Mater., 2021, 312, 110778 CrossRef CAS.
- E. Tervola, K.-N. Truong, J. S. Ward, A. Priimagi and K. Rissanen, RSC Adv., 2020, 10, 29385–29393 RSC.
- J. Li, Y. Liu, X. Wang, G. Zhao, Y. Ai, B. Han, T. Wen, T. Hayat, A. Alsaedi and X. Wang, Chem. Eng. J., 2017, 330, 1012–1021 CrossRef CAS.
- X. Gao, B. Liu and X. Zhao, Chemosphere, 2023, 317, 137891 CrossRef CAS PubMed.
- R. Rippel, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 DOI:10.1002/14356007.a16_265.
- Y. Cao, X. Li, G. Yu and B. Wang, J. Hazard. Mater., 2023, 442, 130025 CrossRef CAS PubMed.
- F. E. Chen, T. A. Pitt, D. J. Okong’o, L. G. Wetherbee, J. J. Fuentes-Rivera and P. J. Milner, Chem. Mater., 2022, 34, 3383–3394 CrossRef CAS PubMed.
- X. Zhang, Y. Yang, L. Song, J. Chen, Y. Yang and Y. Wang, J. Hazard. Mater., 2019, 365, 597–605 CrossRef CAS PubMed.
- X. Shi, X. Zhang, F. Bi, Z. Zheng, L. Sheng, J. Xu, Z. Wang and Y. Yang, J. Mol. Liq., 2020, 316, 113812 CrossRef CAS.
- X. Ma, L. Wang, Q. Zhang and H.-L. Jiang, Angew. Chem., Int. Ed., 2019, 58, 12175–12179 CrossRef CAS PubMed.
- Z. Xue, K. Liu, Q. Liu, Y. Li, M. Li, C.-Y. Su, N. Ogiwara, H. Kobayashi, H. Kitagawa, M. Liu and G. Li, Nat. Commun., 2019, 10, 5048 CrossRef PubMed.
- M. Taddei, R. J. Wakeham, A. Koutsianos, E. Andreoli and A. R. Barron, Angew. Chem., Int. Ed., 2018, 57, 11706–11710 CrossRef CAS PubMed.
- Z. Hu, S. Faucher, Y. Zhuo, Y. Sun, S. Wang and D. Zhao, Chem. – Eur. J., 2015, 21, 17246–17255 CrossRef CAS PubMed.
- M. S. Denny, Jr., L. R. Parent, J. P. Patterson, S. K. Meena, H. Pham, P. Abellan, Q. M. Ramasse, F. Paesani, N. C. Gianneschi and S. M. Cohen, J. Am. Chem. Soc., 2018, 140, 1348–1357 CrossRef PubMed.
- Y. Wang, C. Peng, T. Jiang and X. Li, Front. Energy, 2021, 15, 656–666 CrossRef.
- Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Chem. Commun., 2016, 52, 7806–7809 RSC.
- A. Pankajakshan, M. Sinha, A. A. Ojha and S. Mandal, ACS Omega, 2018, 3, 7832–7839 CrossRef CAS PubMed.
- Y. Huang, Y. Jiao, T. Chen, Y. Gong, S. Wang, Y. Liu, D. S. Sholl and K. S. Walton, ACS Appl. Mater. Interfaces, 2020, 12, 34413–34422 CrossRef CAS PubMed.
- I. Akpinar and A. O. Yazaydin, J. Chem. Eng. Data, 2018, 63, 2368–2375 CrossRef CAS.
- Ü. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- M. R. Momeni and C. J. Cramer, J. Phys. Chem. C, 2019, 123, 15157–15165 CrossRef CAS.
- J. Liu, J. L. Prelesnik, R. Patel, B. V. Kramar, R. Wang, C. D. Malliakas, L. X. Chen, J. I. Siepmann and J. T. Hupp, J. Am. Chem. Soc., 2023, 145, 27975–27983 CrossRef CAS PubMed.
- B. Li, X. Zhu, K. Hu, Y. Li, J. Feng, J. Shi and J. Gu, J. Hazard. Mater., 2016, 302, 57–64 CrossRef CAS PubMed.
- M. Erkartal and U. Sen, ACS Appl. Mater. Interfaces, 2018, 10, 787–795 CrossRef CAS PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- I. Ahmed, M. M. H. Mondol, M. J. Jung, G. H. Lee and S. H. Jhung, Coord. Chem. Rev., 2023, 475, 214912 CrossRef CAS.
- D. Yang and B. C. Gates, Adv. Mater., 2024, 36, 2305611 CrossRef CAS PubMed.
- R. S. H. Khoo, C. Fiankor, S. Yang, W. Hu, C. Yang, J. Lu, M. D. Morton, X. Zhang, Y. Liu, J. Huang and J. Zhang, J. Am. Chem. Soc., 2023, 145, 24052–24060 CrossRef CAS PubMed.
- G. Jajko, S. Calero, P. Kozyra, W. Makowski, A. Sławek, B. Gil and J. J. Gutiérrez-Sevillano, Commun. Chem., 2022, 5, 120 CrossRef CAS PubMed.
- S. Lin, Y. Zhao, J. K. Bediako, C.-W. Cho, A. K. Sarkar, C.-R. Lim and Y.-S. Yun, Chem. Eng. J., 2019, 362, 280–286 CrossRef CAS.
- K. Wang, C. Li, Y. Liang, T. Han, H. Huang, Q. Yang, D. Liu and C. Zhong, Chem. Eng. J., 2016, 289, 486–493 CrossRef CAS.
- K. Y. Cho, J. Y. Seo, H.-J. Kim, S. J. Pai, X. H. Do, H. G. Yoon, S. S. Hwang, S. S. Han and K.-Y. Baek, Appl. Catal., B, 2019, 245, 635–647 CrossRef CAS.
- F. Ahmadijokani, R. Mohammadkhani, S. Ahmadipouya, A. Shokrgozar, M. Rezakazemi, H. Molavi, T. M. Aminabhavi and M. Arjmand, Chem. Eng. J., 2020, 399, 125346 CrossRef CAS.
- V. V. Butova, A. M. Aboraia, M. Solayman, I. S. Yahia, H. Y. Zahran, A. F. Abd El-Rehim, H. Algarni, G. Khabiri and A. V. Soldatov, Microporous Mesoporous Mater., 2021, 325, 111314 CrossRef CAS.
- K. D. Nguyen, N. T. Vo, K. T. M. Le, K. V. Ho, N. T. S. Phan, P. H. Ho and H. V. Le, New J. Chem., 2023, 47, 6433–6447 RSC.
- Ş. S. Bayazit and S. Şahin, J. Environ. Chem. Eng., 2020, 8, 103901 CrossRef.
- S. Zhang, Z. Hua, H. Zhao, W. Yao, Y. Wu, D. Fu and J. Sun, J. Sep. Sci., 2021, 44, 2113–2120 CrossRef CAS PubMed.
- Y. Li, X. Zang, Y. Li, S. Zhang, C. Wang and Z. Wang, Microchem. J., 2023, 190, 108608 CrossRef CAS.
- I. Akpinar and A. O. Yazaydin, Ind. Eng. Chem. Res., 2017, 56, 15122–15130 CrossRef CAS.
- M. R. Rezaei Kahkha, A. R. Oveisi, M. Kaykhaii and B. Rezaei Kahkha, Chem. Cent. J., 2018, 12, 77 CrossRef PubMed.
- M. M. H. Mondol, D. K. Yoo and S. H. Jhung, J. Environ. Chem. Eng., 2022, 10, 108560 CrossRef CAS.
- C. Orellana-Tavra, R. J. Marshall, E. F. Baxter, I. A. Lázaro, A. Tao, A. K. Cheetham, R. S. Forgan and D. Fairen-Jimenez, J. Mater. Chem. B, 2016, 4, 7697–7707 RSC.
- S. Lin, Y. Zhao and Y.-S. Yun, ACS Appl. Mater. Interfaces, 2018, 10, 28076–28085 CrossRef CAS PubMed.
- Y. Zhang, Q. Ruan, Y. Peng, G. Han, H. Huang and C. Zhong, J. Colloid Interface Sci., 2018, 525, 39–47 CrossRef CAS PubMed.
- Y. Cao, X. Li and B. Wang, J. Cleaner Prod., 2022, 380, 135060 CrossRef CAS.
- S. Zhuang and J. Wang, Chemosphere, 2021, 281, 130997 CrossRef CAS PubMed.
- N. Prasetya and K. Li, Sep. Purif. Technol., 2022, 301, 122024 CrossRef CAS.
- M. Qin, Y. Shi, D. Lu, J. Deng, G. Shi and T. Zhou, Appl. Surf. Sci., 2022, 595, 153494 CrossRef CAS.
- Y. Xu, J. Lv, Y. Song, X. Zhou, C. Tian, X. Hong, Y. Cai, C. Zhao and Z. Lin, Environ. Sci.: Nano, 2019, 6, 3590–3600 RSC.
- C. A. Clark, K. N. Heck, C. D. Powell and M. S. Wong, ACS Sustainable Chem. Eng., 2019, 7, 6619–6628 CrossRef CAS.
- C. I. Ezugwu, S. Zhang, S. Li, S. Shi, C. Li, F. Verpoort, J. Yu and S. Liu, Environ. Sci.: Nano, 2019, 6, 2931–2936 RSC.
- Y. Jiao, Y. Liu, G. Zhu, J. T. Hungerford, S. Bhattacharyya, R. P. Lively, D. S. Sholl and K. S. Walton, J. Phys. Chem. C, 2017, 121, 23471–23479 CrossRef CAS.
- X. Zhang, X. Shi, J. Chen, Y. Yang and G. Lu, J. Environ. Chem. Eng., 2019, 7, 103405 CrossRef CAS.
- T. K. Vo, V. N. Le, D. T. Quang, M. Song, D. Kim and J. Kim, Microporous Mesoporous Mater., 2020, 306, 110405 CrossRef CAS.
- X. Zhang, X. Shi, Q. Zhao, Y. Li, J. Wang, Y. Yang, F. Bi, J. Xu and N. Liu, Chem. Eng. J., 2022, 427, 131573 CrossRef CAS.
- Q. Zhao, Q. Du, Y. Yang, Z. Zhao, J. Cheng, F. Bi, X. Shi, J. Xu and X. Zhang, Chem. Eng. J., 2022, 433, 134510 CrossRef CAS.
- Q. Liu, J. Ye, Y. Han, P. Wang, Z. Fei, X. Chen, Z. Zhang, J. Tang, M. Cui and X. Qiao, J. Mol. Liq., 2021, 321, 114477 CrossRef CAS.
- P. Iacomi, F. Formalik, J. Marreiros, J. Shang, J. Rogacka, A. Mohmeyer, P. Behrens, R. Ameloot, B. Kuchta and P. L. Llewellyn, Chem. Mater., 2019, 31, 8413–8423 CrossRef CAS.
- B. Lellis, C. Z. Fávaro-Polonio, J. A. Pamphile and J. C. Polonio, Biotechnol. Res. Innov., 2019, 3, 275–290 CrossRef.
- J. Qiu, Y. Feng, X. Zhang, M. Jia and J. Yao, J. Colloid Interface Sci., 2017, 499, 151–158 CrossRef CAS PubMed.
- K.-D. Zhang, F.-C. Tsai, N. Ma, Y. Xia, H.-L. Liu, X.-Q. Zhan, X.-Y. Yu, X.-Z. Zeng, T. Jiang, D. Shi and C.-J. Chang, Materials, 2017, 10, 205 CrossRef PubMed.
- R. J. Drout, L. Robison, Z. Chen, T. Islamoglu and O. K. Farha, Trends Chem., 2019, 1, 304–317 CrossRef CAS.
- X.-Q. Zhan, F.-C. Tsai, L. Xie, K.-D. Zhang, H.-L. Liu, N. Ma, D. Shi and T. Jiang, Nanomaterials, 2018, 8, 655 CrossRef PubMed.
- S. A. Saghir, K. Husain and R. A. Ansari, in Patty's Toxicology, ed. E. Bingham, B. Cohrssen and C. H. Powell, 2024, pp. 1–162 DOI:10.1002/0471125474.tox095.pub3.
- Y. Liao, T. R. Sheridan, J. Liu, Z. Lu, K. Ma, H. Yang, O. K. Farha and J. T. Hupp, ACS Catal., 2024, 14, 437–448 CrossRef CAS.
- P. Deria, W. Bury, I. Hod, C.-W. Kung, O. Karagiaridi, J. T. Hupp and O. K. Farha, Inorg. Chem., 2015, 54, 2185–2192 CrossRef CAS PubMed.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- K. J. Gagnon, H. P. Perry and A. Clearfield, Chem. Rev., 2012, 112, 1034–1054 CrossRef CAS PubMed.
- T. B. Hayes, A. Collins, M. Lee, M. Mendoza, N. Noriega, A. A. Stuart and A. Vonk, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5476–5480 CrossRef CAS PubMed.
- I. Akpinar, R. J. Drout, T. Islamoglu, S. Kato, J. Lyu and O. K. Farha, ACS Appl. Mater. Interfaces, 2019, 11, 6097–6103 CrossRef CAS PubMed.
- A. J. Ebele, M. Abou-Elwafa Abdallah and S. Harrad, Emerging Contam., 2017, 3, 1–16 CrossRef.
- C. Orellana-Tavra, E. F. Baxter, T. Tian, T. D. Bennett, N. K. H. Slater, A. K. Cheetham and D. Fairen-Jimenez, Chem. Commun., 2015, 51, 13878–13881 RSC.
- X.-W. Yan, Z. Sharifzadeh, S.-J. Wang and A. Morsali, J. Cleaner Prod., 2024, 443, 140704 CrossRef CAS.
- Y. Zhou, D. M. Boudreau and A. N. Freedman, Pharmacoepidemiol. Drug Saf., 2014, 23, 43–50 CrossRef CAS PubMed.
- H. Huang, J.-R. Li, K. Wang, T. Han, M. Tong, L. Li, Y. Xie, Q. Yang, D. Liu and C. Zhong, Nat. Commun., 2015, 6, 8847 CrossRef CAS PubMed.
- N. Tian, Q. Jia, H. Su, Y. Zhi, A. Ma, J. Wu and S. Shan, J. Porous Mater., 2016, 23, 1269–1278 CrossRef CAS.
- F. Liu, W. Zhang, W. Chen, J. Wang, Q. Yang, W. Zhu and J. Wang, Chem. Eng. J., 2017, 310, 187–196 CrossRef CAS.
- A. Chen, C. Shang, J. Shao, Y. Lin, S. Luo, J. Zhang, H. Huang, M. Lei and Q. Zeng, Carbohydr. Polym., 2017, 155, 19–27 CrossRef CAS PubMed.
- R. Sierra-Alvarez, I. Cortinas and J. A. Field, J. Hazard. Mater., 2010, 175, 352–358 CrossRef CAS PubMed.
- Z.-J. Lin, H.-Q. Zheng, Y.-N. Zeng, Y.-L. Wang, J. Chen, G.-J. Cao, J.-F. Gu and B. Chen, Chem. Eng. J., 2019, 378, 122196 CrossRef CAS.
- C. Lau, K. Anitole, C. Hodes, D. Lai, A. Pfahles-Hutchens and J. Seed, Toxicol. Sci, 2007, 99, 366–394 CrossRef CAS PubMed.
- C. Jiang, D. Li, P. Zhang, J. Li, J. Wang and J. Yu, Build Environ., 2017, 117, 118–126 CrossRef.
- Y. Lu, J. Liu, B. Lu, A. Jiang and C. Wan, J. Hazard. Mater., 2010, 182, 204–209 CrossRef CAS PubMed.
- T. K. Vo, V. N. Le, K. S. Yoo, M. Song, D. Kim and J. Kim, Cryst. Growth Des., 2019, 19, 4949–4956 CrossRef CAS.
- K. Vellingiri, P. Kumar, A. Deep and K.-H. Kim, Chem. Eng. J., 2017, 307, 1116–1126 CrossRef CAS.
- H. Sun, R. Ma, Y. Nan and R. Feng, Ecotoxicol. Environ. Saf., 2019, 169, 353–360 CrossRef CAS PubMed.
- D.-W. Gao and Z.-D. Wen, Sci. Total Environ., 2016, 541, 986–1001 CrossRef CAS PubMed.
- L. P. Padhye, H. Yao, F. T. Kung'u and C.-H. Huang, Water Res., 2014, 51, 266–276 CrossRef CAS PubMed.
- Y. Gu, D. Xie, Y. Ma, W. Qin, H. Zhang, G. Wang, Y. Zhang and H. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 32151–32160 CrossRef CAS PubMed.
- L. Wang, X. Wen, J. Li, P. Zeng, Y. Song and H. Yu, Chem. Eng. J., 2021, 405, 126681 CrossRef CAS.
- M. Li, Y. Liu, F. Li, C. Shen, Y. V. Kaneti, Y. Yamauchi, B. Yuliarto, B. Chen and C.-C. Wang, Environ. Sci. Technol., 2021, 55, 13209–13218 CAS.
- C. O. Audu, H. G. T. Nguyen, C.-Y. Chang, M. J. Katz, L. Mao, O. K. Farha, J. T. Hupp and S. T. Nguyen, Chem. Sci., 2016, 7, 6492–6498 RSC.
- R. Xu, Q. Ji, P. Zhao, M. Jian, C. Xiang, C. Hu, G. Zhang, C. Tang, R. Liu, X. Zhang and J. Qu, J. Mater. Chem. A, 2020, 8, 7870–7879 RSC.
- N. D. Hai, M. B. Nguyen, V. M. Tan, N. T. Huu, L. B. Phuong, P. T. M. Huong and T. D. Nguyen, Int. J. Environ. Sci. Technol., 2022, 20, 10075–10088 CrossRef.
- S. Yuan, J.-S. Qin, L. Zou, Y.-P. Chen, X. Wang, Q. Zhang and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6636–6642 CrossRef CAS PubMed.
- P. G. Saiz, N. Iglesias, B. González Navarrete, M. Rosales, Y. M. Quintero, A. Reizabal, J. Orive, A. Fidalgo Marijuan, E. S. Larrea, A. C. Lopes, L. Lezama, A. García, S. Lanceros-Mendez, M. I. Arriortua and R. Fernández de Luis, Chem. – Eur. J., 2020, 26, 13861–13872 CrossRef CAS PubMed.
- Y. Feng, Q. Chen, M. Cao, N. Ling and J. Yao, ACS Appl. Nano Mater., 2019, 2, 5973–5980 CrossRef CAS.
- L. Yuan, M. Tian, J. Lan, X. Cao, X. Wang, Z. Chai, J. K. Gibson and W. Shi, Chem. Commun., 2018, 54, 370–373 RSC.
- C. Yin, Q. Liu, R. Chen, J. Liu, J. Yu, D. Song and J. Wang, Ind. Eng. Chem. Res., 2019, 58, 1159–1166 CrossRef CAS.
- Z. Zhao, R. Lei, Y. Zhang, T. Cai and B. Han, J. Mol. Liq., 2022, 367, 120514 CrossRef CAS.
- J. Li, Y. Liu, Y. Ai, A. Alsaedi, T. Hayat and X. Wang, Chem. Eng. J., 2018, 354, 790–801 CrossRef CAS.
- S. Lin, J. K. Bediako, M.-H. Song, J.-A. Kim, C.-W. Cho, Y. Zhao, J.-W. Choi and Y.-S. Yun, ACS Sustainable Chem. Eng., 2019, 7, 7510–7518 CrossRef CAS.
- M. Peng, D. You, H. Shi, P. Shao, W. Ren, L. Yang, X. Sheng, J. Shao, X. Ding, L. Ding, H. Wang, K. Yu and X. Luo, Chem. Eng. J., 2022, 448, 137612 CrossRef CAS.
- Y.-H. Li, C.-C. Wang, X. Zeng, X.-Z. Sun, C. Zhao, H. Fu and P. Wang, Chem. Eng. J., 2022, 442, 136276 CrossRef CAS.
- M. Vhahangwele and L. M. Khathutshelo, in Heavy Metals, ed. M. S. Hosam El-Din and F. A. Refaat, IntechOpen, Rijeka, 2018, ch. Environmental Contamination by Heavy Metals DOI:10.5772/intechopen.76082.
- A. J. Howarth, Y. Liu, J. T. Hupp and O. K. Farha, CrystEngComm, 2015, 17, 7245–7253 RSC.
- Z. S. Moghaddam, M. Kaykhaii, M. Khajeh and A. R. Oveisi, Spectrochim. Acta, Part A, 2018, 194, 76–82 CrossRef CAS PubMed.
- S. Daliran, A. R. Oveisi, M. Khajeh, A. Barkhordar and A. Dhakshinamoorthy, Polyhedron, 2023, 237, 116392 CrossRef CAS.
- S. Nohtani, M. Khajeh, A. R. Oveisi, M. G. Moghaddam and M. Rakhshanipour, J. Mol. Liq., 2023, 385, 122422 CrossRef CAS.
- C. W. Abney, K. M. L. Taylor-Pashow, S. R. Russell, Y. Chen, R. Samantaray, J. V. Lockard and W. Lin, Chem. Mater., 2014, 26, 5231–5243 CrossRef CAS.
- R. Stanton and D. J. Trivedi, Chem. Mater., 2021, 33, 5730–5737 CrossRef CAS.
- Y. Gu, D. Xie, Y. Wang, W. Qin, H. Zhang, G. Wang, Y. Zhang and H. Zhao, Chem. Eng. J., 2019, 357, 579–588 CrossRef CAS.
- J.-B. Huo, L. Xu, X. Chen, Y. Zhang, J.-C. E. Yang, B. Yuan and M.-L. Fu, Microporous Mesoporous Mater., 2019, 276, 68–75 CrossRef CAS.
- E. Berardozzi, J. S. Tuninetti, F. S. G. Einschlag, O. Azzaroni, M. Ceolín and M. Rafti, J. Inorg. Organomet. Polym. Mater., 2021, 31, 1185–1194 CrossRef CAS.
- G. Qu, P. Jia, T. Zhang, Z. Li, C. Chen and Y. Zhao, Chemosphere, 2022, 288, 132594 CrossRef CAS PubMed.
- A. J. Howarth, M. J. Katz, T. C. Wang, A. E. Platero-Prats, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 7488–7494 CrossRef CAS PubMed.
- B. V. Kramar, B. T. Phelan, E. A. Sprague-Klein, B. T. Diroll, S. Lee, K.-I. Otake, R. Palmer, M. W. Mara, O. K. Farha, J. T. Hupp and L. X. Chen, Energy Fuels, 2021, 35, 19081–19095 CrossRef CAS.
- K. Oguma, T. Suzuki and K. Saito, Talanta, 2011, 84, 1209–1214 CrossRef CAS PubMed.
- Y. He, Y.-L. Hou, Y.-L. Wong, R. Xiao, M.-Q. Li, Z. Hao, J. Huang, L. Wang, M. Zeller, J. He and Z. Xu, J. Mater. Chem. A, 2018, 6, 1648–1654 RSC.
- L. Huang, M. He, B. Chen and B. Hu, J. Mater. Chem. A, 2016, 4, 5159–5166 RSC.
- K. Leus, J. P. H. Perez, K. Folens, M. Meledina, G. Van Tendeloo, G. Du Laing and P. Van Der Voort, Faraday Discuss., 2017, 201, 145–161 RSC.
- T. Islamoglu, Z. Chen, M. C. Wasson, C. T. Buru, K. O. Kirlikovali, U. Afrin, M. R. Mian and O. K. Farha, Chem. Rev., 2020, 120, 8130–8160 CrossRef CAS PubMed.
- Y. J. Jang, K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2015, 115, PR1–PR76 CrossRef CAS PubMed.
- J. Kaushal, M. Khatri and S. K. Arya, Ecotoxicol. Environ. Saf., 2021, 207, 111483 CrossRef CAS PubMed.
- M. J. Katz, J. E. Mondloch, R. K. Totten, J. K. Park, S. T. Nguyen, O. K. Farha and J. T. Hupp, Angew. Chem., Int. Ed., 2014, 53, 497–501 CrossRef CAS PubMed.
- Y. Liu, A. J. Howarth, N. A. Vermeulen, S.-Y. Moon, J. T. Hupp and O. K. Farha, Coord. Chem. Rev., 2017, 346, 101–111 CrossRef CAS.
- Y. Liu, S.-Y. Moon, J. T. Hupp and O. K. Farha, ACS Nano, 2015, 9, 12358–12364 CrossRef CAS PubMed.
- Y. Chen, X. Zhang, M. R. Mian, F. A. Son, K. Zhang, R. Cao, Z. Chen, S.-J. Lee, K. B. Idrees, T. A. Goetjen, J. Lyu, P. Li, Q. Xia, Z. Li, J. T. Hupp, T. Islamoglu, A. Napolitano, G. W. Peterson and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 21428–21438 CrossRef CAS PubMed.
- J. E. Mondloch, M. J. Katz, W. C. Isley Iii, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512–516 CrossRef CAS PubMed.
- H. Chen, P. Liao, M. L. Mendonca and R. Q. Snurr, J. Phys. Chem. C, 2018, 122, 12362–12368 CrossRef CAS.
- P. Li, R. C. Klet, S.-Y. Moon, T. C. Wang, P. Deria, A. W. Peters, B. M. Klahr, H.-J. Park, S. S. Al-Juaid, J. T. Hupp and O. K. Farha, Chem. Commun., 2015, 51, 10925–10928 RSC.
- P. Li, S.-Y. Moon, M. A. Guelta, L. Lin, D. A. Gómez-Gualdrón, R. Q. Snurr, S. P. Harvey, J. T. Hupp and O. K. Farha, ACS Nano, 2016, 10, 9174–9182 CrossRef CAS PubMed.
- A. E. Platero-Prats, A. Mavrandonakis, L. C. Gallington, Y. Liu, J. T. Hupp, O. K. Farha, C. J. Cramer and K. W. Chapman, J. Am. Chem. Soc., 2016, 138, 4178–4185 CrossRef CAS PubMed.
- M. J. Katz, R. C. Klet, S.-Y. Moon, J. E. Mondloch, J. T. Hupp and O. K. Farha, ACS Catal., 2015, 5, 4637–4642 CrossRef CAS.
- Y. Liao, T. Sheridan, J. Liu, O. Farha and J. Hupp, ACS Appl. Mater. Interfaces, 2021, 13, 30565–30575 CrossRef CAS PubMed.
- D. M. Driscoll, D. Troya, P. M. Usov, A. J. Maynes, A. J. Morris and J. R. Morris, J. Phys. Chem. C, 2018, 122, 14582–14589 CrossRef CAS.
- A. M. Plonka, Q. Wang, W. O. Gordon, A. Balboa, D. Troya, W. Guo, C. H. Sharp, S. D. Senanayake, J. R. Morris, C. L. Hill and A. I. Frenkel, J. Am. Chem. Soc., 2017, 139, 599–602 CrossRef CAS PubMed.
- G. W. Peterson, M. R. Destefano, S. J. Garibay, A. Ploskonka, M. McEntee, M. Hall, C. J. Karwacki, J. T. Hupp and O. K. Farha, Chem. – Eur. J., 2017, 23, 15913–15916 CrossRef CAS PubMed.
- J. A. Harvey, J. A. Greathouse and D. F. Sava Gallis, J. Phys. Chem. C, 2018, 122, 26889–26896 CrossRef CAS.
- T. M. Rayder, F. Formalik, S. M. Vornholt, H. Frank, S. Lee, M. Alzayer, Z. Chen, D. Sengupta, T. Islamoglu, F. Paesani, K. W. Chapman, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2023, 145, 11195–11205 CrossRef CAS PubMed.
- R. Wang, K. Shi, J. Liu, R. Q. Snurr and J. T. Hupp, J. Am. Chem. Soc., 2023, 145, 13979–13988 CrossRef CAS PubMed.
- D. A. Giannakoudakis and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2020, 12, 14678–14689 CrossRef CAS PubMed.
- D. Eder and R. Kramer, Phys. Chem. Chem. Phys., 2002, 4, 795–801 RSC.
- J. Liu, Z. Chen, R. Wang, S. Alayoglu, T. Islamoglu, S.-J. Lee, T. R. Sheridan, H. Chen, R. Q. Snurr, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2021, 13, 22485–22494 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- Z. Liang, C. Qu, D. Xia, R. Zou and Q. Xu, Angew. Chem., Int. Ed., 2018, 57, 9604–9633 CrossRef CAS PubMed.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- A. De Vos, K. Hendrickx, P. Van Der Voort, V. Van Speybroeck and K. Lejaeghere, Chem. Mater., 2017, 29, 3006–3019 CrossRef CAS PubMed.
- M. Taddei, G. M. Schukraft, M. E. A. Warwick, D. Tiana, M. J. McPherson, D. R. Jones and C. Petit, J. Mater. Chem. A, 2019, 7, 23781–23786 RSC.
- S.-Q. Wang, X. Wang, X.-Y. Zhang, X.-M. Cheng, J. Ma and W.-Y. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 61578–61586 CrossRef CAS PubMed.
- Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, J. Phys. Chem. C, 2021, 125, 27909–27918 CrossRef CAS.
- R. Li, W. Zhang and K. Zhou, Adv. Mater., 2018, 30, 1705512 CrossRef PubMed.
- Y. Fu, J. Wu, R. Du, K. Guo, R. Ma, F. Zhang, W. Zhu and M. Fan, RSC Adv., 2019, 9, 37733–37738 RSC.
- S.-Q. Wang, X. Gu, X. Wang, X.-Y. Zhang, X.-Y. Dao, X.-M. Cheng, J. Ma and W.-Y. Sun, Chem. Eng. J., 2022, 429, 132157 CrossRef CAS.
- X.-P. Wu, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2018, 140, 7904–7912 CrossRef CAS PubMed.
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed.
- J. L. Mancuso, A. M. Mroz, K. N. Le and C. H. Hendon, Chem. Rev., 2020, 120, 8641–8715 CrossRef CAS PubMed.
- E. Giamello, M. Volante, B. Fubini, F. Geobaldo and C. Morterra, Mater. Chem. Phys., 1991, 29, 379–386 CrossRef CAS.
- P. Ji, T. Drake, A. Murakami, P. Oliveres, J. H. Skone and W. Lin, J. Am. Chem. Soc., 2018, 140, 10553–10561 CrossRef CAS PubMed.
- B. Probst, A. Rodenberg, M. Guttentag, P. Hamm and R. Alberto, Inorg. Chem., 2010, 49, 6453–6460 CrossRef CAS PubMed.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726–7727 CrossRef CAS PubMed.
- M. Bonchio, J. Bonin, O. Ishitani, T.-B. Lu, T. Morikawa, A. J. Morris, E. Reisner, D. Sarkar, F. M. Toma and M. Robert, Nat. Catal., 2023, 6, 657–665 CrossRef.
- Y. Yang, X. Zhang, S. Kanchanakungwankul, Z. Lu, H. Noh, Z. H. Syed, O. K. Farha, D. G. Truhlar and J. T. Hupp, J. Am. Chem. Soc., 2020, 142, 21169–21177 CrossRef CAS PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- D.-W. Hwang, S. Lee, M. Seo and T. D. Chung, Anal. Chim. Acta, 2018, 1033, 1–34 CrossRef CAS PubMed.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- S. Yuan, J.-S. Qin, C. T. Lollar and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 440–450 CrossRef CAS PubMed.
- J. E. Mondloch, M. J. Katz, N. Planas, D. Semrouni, L. Gagliardi, J. T. Hupp and O. K. Farha, Chem. Commun., 2014, 50, 8944–8946 RSC.
- J.-H. Li, Y.-S. Wang, Y.-C. Chen and C.-W. Kung, Appl. Sci., 2019, 9, 2427 CrossRef CAS.
- C.-H. Chuang and C.-W. Kung, Electroanalysis, 2020, 32, 1885–1895 CrossRef CAS.
- K. M. Choi, H. M. Jeong, J. H. Park, Y.-B. Zhang, J. K. Kang and O. M. Yaghi, ACS Nano, 2014, 8, 7451–7457 CrossRef CAS PubMed.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- S. Lin, P. M. Usov and A. J. Morris, Chem. Commun., 2018, 54, 6965–6974 RSC.
- C.-W. Kung, S. Goswami, I. Hod, T. C. Wang, J. Duan, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2020, 53, 1187–1195 CrossRef CAS PubMed.
- A. T. Castner, H. Su, E. Svensson Grape, A. K. Inge, B. A. Johnson, M. S. G. Ahlquist and S. Ott, J. Am. Chem. Soc., 2022, 144, 5910–5920 CrossRef CAS PubMed.
- K. Maindan, X. Li, J. Yu and P. Deria, J. Phys. Chem. B, 2019, 123, 8814–8822 CrossRef CAS PubMed.
- R. H. Palmer, J. Liu, C.-W. Kung, I. Hod, O. K. Farha and J. T. Hupp, Langmuir, 2018, 34, 4707–4714 CrossRef CAS PubMed.
- Y. Pu, W. Wu, J. Liu, T. Liu, F. Ding, J. Zhang and Z. Tang, RSC Adv., 2018, 8, 18604–18612 RSC.
- A. E. Baumann, X. Han, M. M. Butala and V. S. Thoi, J. Am. Chem. Soc., 2019, 141, 17891–17899 CrossRef CAS PubMed.
- A. E. Baumann, D. A. Burns, J. C. Díaz and V. S. Thoi, ACS Appl. Mater. Interfaces, 2019, 11, 2159–2167 CrossRef CAS PubMed.
- R. Shimoni, W. He, I. Liberman and I. Hod, J. Phys. Chem. C, 2019, 123, 5531–5539 CrossRef CAS.
- I. Liberman, R. Shimoni, R. Ifraemov, I. Rozenberg, C. Singh and I. Hod, J. Am. Chem. Soc., 2020, 142, 1933–1940 CrossRef CAS PubMed.
- T.-E. Chang, C.-H. Chuang and C.-W. Kung, Electrochem. Commun., 2021, 122, 106899 CrossRef CAS.
- Y. S. Wang, Y. C. Chen, J. H. Li and C. W. Kung, Eur. J. Inorg. Chem., 2019, 3036–3044 CrossRef CAS.
- C.-H. Chuang, J.-H. Li, Y.-C. Chen, Y.-S. Wang and C.-W. Kung, J. Phys. Chem. C, 2020, 124, 20854–20863 CrossRef CAS.
- T.-E. Chang, C.-H. Chuang, Y.-H. Chen, Y.-C. Wang, Y.-J. Gu and C.-W. Kung, ChemCatChem, 2022, 14, e202200199 CrossRef CAS.
- Y.-C. Wang, Y.-C. Chen, W.-S. Chuang, J.-H. Li, Y.-S. Wang, C.-H. Chuang, C.-Y. Chen and C.-W. Kung, ACS Appl. Nano Mater., 2020, 3, 9440–9448 CrossRef CAS.
- C.-H. Shen, Y.-N. Chang, Y.-L. Chen and C.-W. Kung, ACS Mater. Lett., 2023, 5, 1938–1943 CrossRef CAS.
- Y.-N. Chang, C.-H. Shen, C.-W. Huang, M.-D. Tsai and C.-W. Kung, ACS Appl. Nano Mater., 2023, 6, 3675–3684 CrossRef CAS.
- Z. Wang, S. Liu, M. Wang, L. Zhang, Y. Jiang, T. Qian, J. Xiong, C. Yang and C. Yan, ACS Catal., 2023, 13, 9125–9135 CrossRef CAS.
- L. Feng, H.-B. Hou and H. Zhou, Dalton Trans., 2020, 49, 17130–17139 RSC.
- X. Chen and G. Li, Inorg. Chem. Front., 2020, 7, 3765–3784 RSC.
- J. M. Taylor, S. Dekura, R. Ikeda and H. Kitagawa, Chem. Mater., 2015, 27, 2286–2289 CrossRef CAS.
- J. Tang, F. Zhang, X. Liang, G. Dai and F. Qu, J. Colloid Interface Sci., 2022, 607, 181–191 CrossRef CAS PubMed.
- M. Szufla, J. A. R. Navarro, K. Góra-Marek and D. Matoga, ACS Appl. Mater. Interfaces, 2023, 15, 28184–28192 CrossRef CAS PubMed.
- Q. Lyu, X. Deng, S. Hu, L.-C. Lin and W. S. W. Ho, J. Phys. Chem. C, 2019, 123, 16118–16126 CrossRef.
- R. Zhang, D. Zhang, Y. Yao, Q. Zhang, Y. Xu, Y. Wu, H. Yu and G. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 21010–21017 CrossRef CAS PubMed.
- F. Yan, J. Yan, Y. Sun, T. Ji, Y. He, X. Guo and Y. Liu, Eur. J. Inorg. Chem., 2023, e202200679 CrossRef CAS.
- M. J. Cliffe, E. Castillo-Martínez, Y. Wu, J. Lee, A. C. Forse, F. C. N. Firth, P. Z. Moghadam, D. Fairen-Jimenez, M. W. Gaultois, J. A. Hill, O. V. Magdysyuk, B. Slater, A. L. Goodwin and C. P. Grey, J. Am. Chem. Soc., 2017, 139, 5397–5404 CrossRef CAS PubMed.
- H.-J. Son, C. Prasittichai, J. E. Mondloch, L. Luo, J. Wu, D. W. Kim, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 11529–11532 CrossRef CAS PubMed.
- W. L. Hoffeditz, H.-J. Son, M. J. Pellin, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2016, 8, 34863–34869 CrossRef CAS PubMed.
- H.-J. Son, X. Wang, C. Prasittichai, N. C. Jeong, T. Aaltonen, R. G. Gordon and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 9537–9540 CrossRef CAS PubMed.
- P. Péchy, F. P. Rotzinger, M. K. Nazeeruddin, O. Kohle, S. M. Zakeeruddin, R. Humphry-Baker and M. Grätzel, J. Chem. Soc., Chem. Commun., 1995, 65–66, 10.1039/C39950000065.
- M. Taddei, F. Costantino, F. Marmottini, A. Comotti, P. Sozzani and R. Vivani, Chem. Commun., 2014, 50, 14831–14834 RSC.
- M. Taddei, F. Costantino, R. Vivani, S. Sabatini, S.-H. Lim and S. M. Cohen, Chem. Commun., 2014, 50, 5737–5740 RSC.
- T. Zheng, Z. Yang, D. Gui, Z. Liu, X. Wang, X. Dai, S. Liu, L. Zhang, Y. Gao, L. Chen, D. Sheng, Y. Wang, J. Diwu, J. Wang, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017, 8, 15369 CrossRef CAS PubMed.
- F. Sha, H. Xie, K. O. Kirlikovali, W. Gong, Y. Chen and O. K. Farha, ACS Mater. Lett., 2024, 1396–1403, DOI:10.1021/acsmaterialslett.4c00199.
| This journal is © The Royal Society of Chemistry 2024 |