
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSERS in 3D cell models: a powerful tool in cancer research
Lara
Troncoso-Afonso
 ab,
Gail A.
Vinnacombe-Willson
a,
Clara
García-Astrain
ac and
Luis M.
Liz-Márzan
*acd
ab,
Gail A.
Vinnacombe-Willson
a,
Clara
García-Astrain
ac and
Luis M.
Liz-Márzan
*acd
aBioNanoPlasmonics Laboratory, CIC biomaGUNE, Basque Research and Technology Alliance (BRTA), 20014 Donostia-San Sebastián, Spain. E-mail: llizmarzan@cicbiomagune.es
bDepartment of Applied Chemistry, University of the Basque Country, 20018 Donostia-San Sebastián, Gipuzkoa, Spain
cCentro de Investigación Biomédica en Red de Bioingeniería Biomateriales, y Nanomedicina (CIBER-BBN), Paseo de Miramón 182, 20014 Donostia-San Sebastián, Spain
dIkerbasque Basque Foundation for Science, 48013 Bilbao, Spain
First published on 12th April 2024
Abstract
Unraveling the cellular and molecular mechanisms underlying tumoral processes is fundamental for the diagnosis and treatment of cancer. In this regard, three-dimensional (3D) cancer cell models more realistically mimic tumors compared to conventional 2D cell cultures and are more attractive for performing such studies. Nonetheless, the analysis of such architectures is challenging because most available techniques are destructive, resulting in the loss of biochemical information. On the contrary, surface-enhanced Raman spectroscopy (SERS) is a non-invasive analytical tool that can record the structural fingerprint of molecules present in complex biological environments. The implementation of SERS in 3D cancer models can be leveraged to track therapeutics, the production of cancer-related metabolites, different signaling and communication pathways, and to image the different cellular components and structural features. In this review, we highlight recent progress in the use of SERS for the evaluation of cancer diagnosis and therapy in 3D tumoral models. We outline strategies for the delivery and design of SERS tags and shed light on the possibilities this technique offers for studying different cellular processes, through either biosensing or bioimaging modalities. Finally, we address current challenges and future directions, such as overcoming the limitations of SERS and the need for the development of user-friendly and robust data analysis methods. Continued development of SERS 3D bioimaging and biosensing systems, techniques, and analytical strategies, can provide significant contributions for early disease detection, novel cancer therapies, and the realization of patient-tailored medicine.
 Lara Troncoso-Afonso | Lara Troncoso-Afonso received her B.Sc in Chemistry in 2021 and her M.Sc. in Chemistry at the interface with Biology and Materials Science in 2023, both from the University of Santiago de Compostela. In 2023 she obtained a ‘’FPU’’ fellowship from the Ministry of Education and Science of Spain, to pursue her PhD and she joined BioNanoPlasmonics group led by Luis Liz-Marzán at CIC biomaGUNE, in Donostia-San Sebastián, where she is involved in the ERC project 4DbioSERS. Her current research is focused on the use of SERS to detect cancer-related biomarkers in 3D cell models. |
 Gail A. Vinnacombe-Willson | Dr Gail A. Vinnacombe-Willson received her B.Sc. in Chemistry from the University of San Francisco. She graduated with her PhD in Materials Chemistry from the University of California, Los Angeles, with her thesis on gold nanoparticle patterning and substrate growth. In 2022, she was awarded with the Marie Skłodowska-Curie postdoctoral fellowship for project PLASMOSTEMFATE. She is currently a postdoctoral researcher in the BioNanoPlasmonics group at CIC biomaGUNE in Donostia-San Sebastián, Spain, where she is involved in the European project 4D bioSERS. Her current research includes the development of SERS substrates based on hydrogels for biomedical applications. |
 Clara García-Astrain | Clara García-Astrain is a Juan de la Cierva Incorporación Fellow in the Bionanoplasmoncis Lab lead by Prof. Luis M. Liz-Marzán (CICbiomaGUNE, Donostia-San Sebastián, Spain). In 2015 she obtained her PhD from the University of the Basque Country (Donostia-San Sebastián, Spain), after which she completed postdocs at the Institute of Chemistry and Processes for Energy, Environment and Health (ICPEES) from the University of Strasbourg/CNRS (Strasbourg, France) and the Basque Centre for Materials, Applications and Nanostructures (BCMaterials, Leioa, Spain). In 2018, she moved to CICbiomaGUNE where she focuses on the development of polymer nanocomposites for 3D sensing and imaging applications. |
 Luis M. Liz-Márzan | Luis Liz-Marzán is an Ikerbasque Professor at CIC biomaGUNE, in San Sebastián (Spain), where he served as the Scientific Director from 2012 to 2021. He obtained a PhD in chemistry from the University of Santiago de Compostela (1992), and after a postdoctoral stay at Utrecht University, he became Professor at the University of Vigo (1995–2012), where he currently holds a part-time professorship. Liz-Marzán has contributed to develop the colloidal synthesis and self-assembly of metal nanocrystals, as well as the characterization and application of their plasmonic properties. His most recent research focuses on various biomedical applications of plasmonic nanostructures. |
1. Introduction
Cancer is caused by the uncontrolled proliferation and migration of abnormal cells, resulting in the formation of tumors and spreading to other parts of the body in what we know as metastasis.1 An ordinary cellular cycle includes growth, mitosis, and apoptosis, but if this cycle is disrupted, cells may start dividing rapidly, eventually leading to tumor formation. Cancer affects millions of people worldwide; in 2020, 18.1 million people were diagnosed, and 10 million people died of this disease.2 Standard clinical approaches for cancer treatment are surgery, radiotherapy, and chemotherapy. However, surgery is not always feasible, and radiotherapy and chemotherapy are not cell-specific, usually causing unwanted damage to healthy cells and tissues.3,4 Moreover, the discovery of chemotherapeutic agents takes 10–13 years on average, with only 5% of potential candidates reaching the market.5Tumors are not only composed of tumoral cells but also contain cancer associated fibroblasts (CAFs), endothelial cells, adipocytes, and immune cells (natural killer cells, T cells, B cells, macrophages, etc.).6 All these cellular components are present in the tumor microenvironment and constantly interact with tumor cells, influencing their proliferation, differentiation, migration, decision making, and response to therapeutics.7 Studying both cell–cell and cell-environment signaling and communication is fundamental in cancer research.8 Although creating an in vitro cell model that considers all the aspects of the tumor microenvironment is difficult, reproducing tumor ecosystems with only a few of the main components may be enough to build more robust models than conventional two-dimensional (2D) mono-cultures of tumor cells.9 The limitations of standard 2D cell cultures to reproduce tumoral physiological features are well-known contributors to the high drug development attrition rates.10 Indeed, to narrow down the translational gap between 2D cellular models and patients, researchers have placed efforts towards developing reliable three-dimensional (3D) cell models where cancer cells can be cultured in a spatially relevant fashion, together with the components of the tumor microenvironment, to mimic native cell–cell and cell–matrix interactions, as well as their physicochemical and mechanical properties.11
Nonetheless, 3D cancer models need to be combined with robust analytical tools capable of imaging these biological constructs and detecting different biomarkers that would regulate cellular behavior, communication, and proliferation.12 Conventional techniques include flow cytometry, colorimetry, bright field, dark field, or differential interference contrast optical microscopies, as well as scanning or transmission electron microscopies (SEM and TEM, respectively). However, most of them require fixation, sectioning, or destruction of the sample, thereby resulting in loss of biochemical information and presenting a barrier for time-dependent studies.13 The most intensely used techniques for imaging in biological settings are those based on fluorescence,14 from conventional wide field and confocal laser scanning fluorescence microscopy, to nonlinear microscopy based on two- or multi-photon absorption, and super resolution techniques such as stimulated emission depletion (STED), stochastic optical reconstruction microscopy (STORM), photo-activated localization microscopy (PALM), etc.15 Despite the usefulness of these techniques for cell imaging, fluorescence-based microscopy is limited by its dependency on either exogenous (chromophores, dyes, quantum dots, etc.) or endogenous (cellular autofluorescence) fluorophores, preventing non-fluorescent or weakly fluorescent molecular species to be probed.16 Moreover, in biological settings some fluorophores might be quenched and suffer from photobleaching.17 Another issue when using fluorescence in 3D cell models is the limited penetration depth due to poor light transmission through non-transparent matrices, such as those present in biological settings.18
Thus, studying the components of complex 3D biological models still poses significant challenges and there is a need for continued development of advanced analytical tools that can probe these systems. In this context, Raman spectroscopy is a non-destructive, non-invasive, and label-free technique of growing interest in biophysical and biochemical research.19 Raman spectroscopy measures (inelastic) light scattering from molecules upon excitation with a highly intense light source. Light scattering is caused by the polarization of the molecular electron cloud under the oscillating electromagnetic field of incident photons, which leaves the molecule in a higher energy state. This process can be explained by considering the formation of a short-lived complex between the photon and the molecule, which is called the virtual state of the molecule. This virtual state is not stable, and the photon is quickly re-emitted as scattered light. In case the molecule retains the same energy and thus the incident and scattered photons have the same frequency, i.e., light is elastically scattered, the phenomenon is known as Rayleigh scattering (Fig. 1A). However, the molecule may also gain or lose energy, so that the scattered photon will have a lower (Stokes) or higher (anti-Stokes) frequency compared to the incident one, so that light is inelastically scattered, which is the basis of Raman scattering (Fig. 1A). The frequency shift of Raman scattered photons can be correlated with the excitation of different molecular vibrational modes, thereby providing information about the molecule's chemical structure. Therefore, Raman scattering is complementary to IR absorption spectroscopy, as both techniques provide a vibrational chemical fingerprint of molecules. However, Raman spectroscopy is of greater interest for biomedical applications due to the high IR absorption but weak scattering by water.20 However, the main limitation of Raman spectroscopy is that only one of every 106–108 photons is inelastically scattered, thus requiring longer acquisition times than the dynamic processes occurring inside in vivo samples, with their components in constant motion.21 Moreover, the scattering cross-sections for many metabolites are extremely low, preventing biomarker detection at low concentrations. Fifty years ago, it was discovered that the intensity in Raman measurements can be largely increased when the molecules are in contact with a metallic surface,22–24 giving rise to a new technique that we know as surface-enhanced Raman Spectroscopy (SERS).25 This technique has attracted much attention ever since and has been extensively applied in biomedical research.26
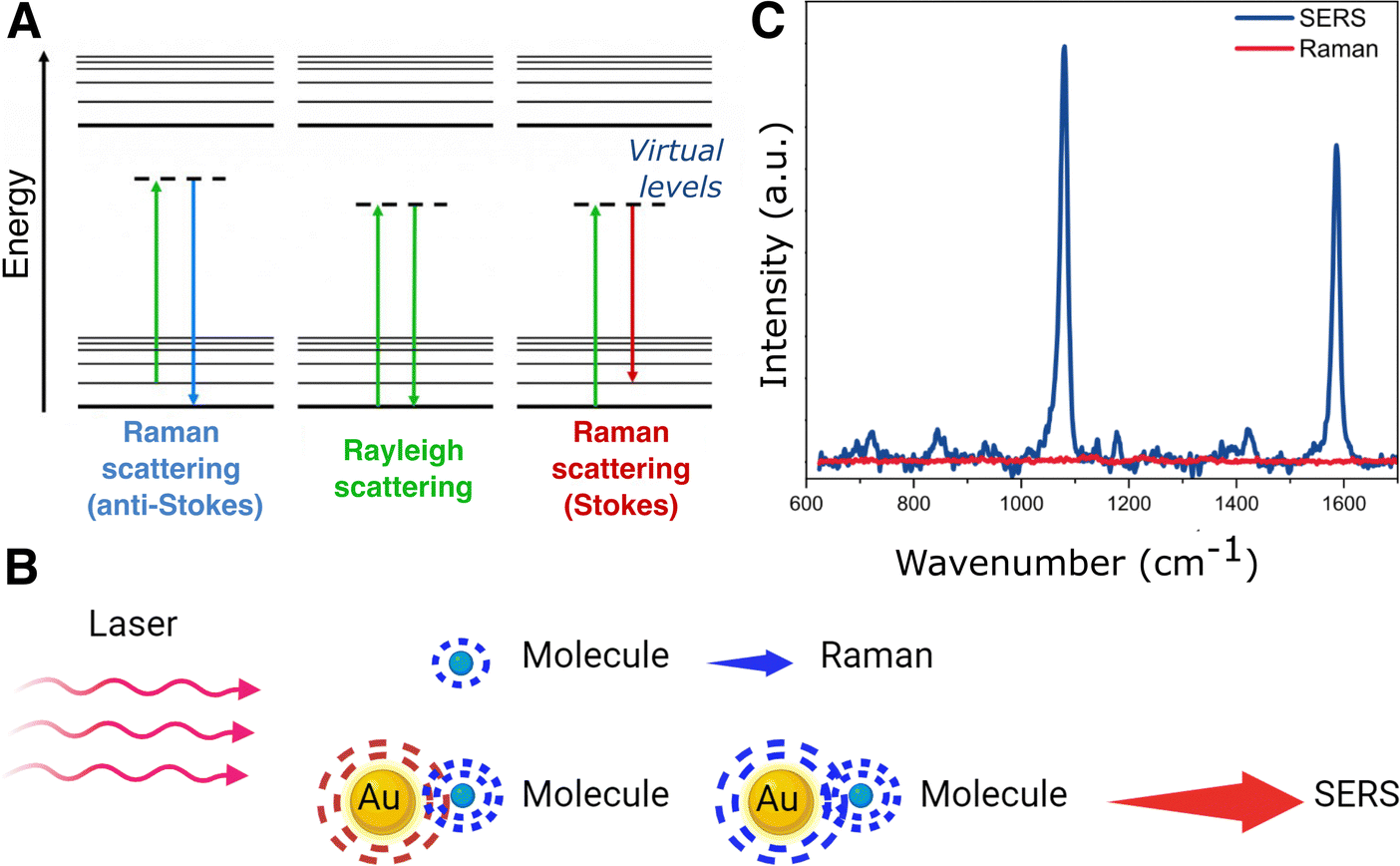 | ||
| Fig. 1 (A) Jablonski diagram for scattering processes. (B) Schematic of the surface-enhanced Raman scattering (SERS) principle: a metal nanoparticle is excited with laser irradiation matching the particle's localized surface plasmon resonance. For molecules close to the surface of the nanoparticle, the Raman signal is significantly enhanced. (C) Comparison of SERS and Raman spectra for 4-mercaptobenzoic acid. | ||
The surface-enhanced Raman effect occurs when molecules are in close proximity to nanostructured metal surfaces, leading to a significant increase in their Raman scattering intensity, thereby revealing molecular vibrational fingerprints and enabling detection at low concentrations (Fig. 1B and C). This drastic enhancement of the Raman signal stems from a combination of plasmon-assisted amplification of the electromagnetic field (electromagnetic mechanism) and the resonant transfer of electrons between the analyte and the metal (chemical mechanism).27,28 Plasmonic enhancement of the local electric field is observed in metal particles that have smaller spatial dimensions than the wavelength of incoming light. The interaction of light with metal nanoparticles (NPs) leads to a collective oscillation of the conduction electrons in the metal, which can be resonantly amplified by the incident electromagnetic field, the so-called localized surface plasmon resonance (LSPR).29,30 In short, the LSPR effect causes an enhancement in the local electric field, altering the polarizability of adsorbed molecules, thereby increasing the efficiency of inelastic scattering events.31,32 The electromagnetic field can be further enhanced when confined within short interparticle distances or at sharp NP features, creating regions called “hotspots”.33–35 The size, morphology, composition and spatial arrangement of the metallic nanostructures are crucial parameters to fine-tune the LSPR and achieve the desired enhancement, as further discussed in Section 3 (Fig. 2). Thus, the pursuit of methods for the preparation of different metallic nanostructures has become a key element to achieve highly reproducible and precise (bio)sensors. Two main fabrication methods are usually distinguished: top-down and bottom-up approaches. Concerning the former, nanolithography is arguably the most popular technique, whereas the latter is usually based on chemical methods for the synthesis of colloidal NPs and their self-assembly into nanostructured substrates. By carefully designing these nanostructures, the local field enhancement can then be leveraged to boost the Raman scattering signal of nearby molecules by up to a factor of ∼1014 (Fig. 1C).36–39 SERS can therefore be employed to detect analytes, such as pesticides, cancer therapeutics, and biomarkers, at extremely low concentrations, even down to the single-molecule level.40 The incorporation of such plasmonic NPs into 3D cell models has been envisaged to allow for the in situ detection of biomolecules in the tumoral environment, allowing for real-time, non-invasive monitoring of tumor activity. The combination of SERS and 3D cell models may thus result in a powerful tool for the detection of tumor biomarkers at low concentrations, eventually contributing to the development of more precise and personalized medicine.41 It is also worth mentioning that, besides cancer research, this technique can be applied to numerous applications in the biomedical field, such as virus42 and bacteria43 detection, monitoring of cardiovascular diseases,44 study of neurodegenerative diseases,45 monitoring drug delivery,46 as well as for the development of point-of-care devices for rapid detection of a wide range of biomarkers.47
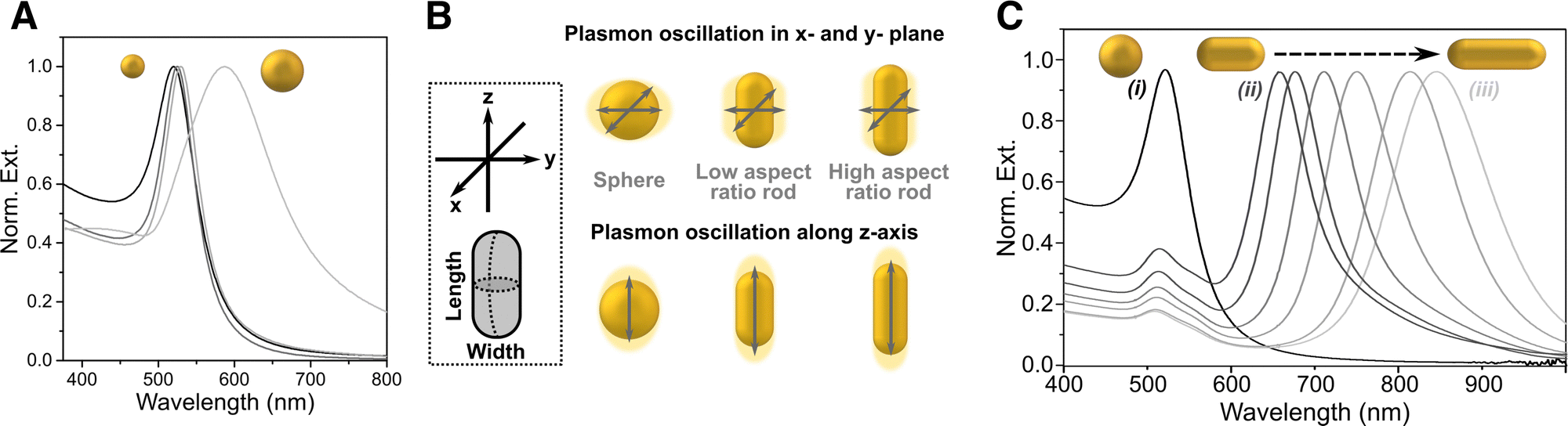 | ||
| Fig. 2 (A) Extinction spectra for spheres of different sizes. (B) Schematic showing the different plasmon oscillations of an isotropic sphere vs. an anisotropic nanorod. (C) Extinction spectra of gold nanorods with changing aspect ratio. [Adapted from ref. 48]. | ||
The goal of this review is to discuss the use of SERS in combination with different 3D cell models, to boost cancer diagnosis and treatment. Multicomponent plasmonic systems referred to as “SERS tags” are often applied for both biosensing and bioimaging in 3D. SERS tags (also termed nanotags) are composed of a plasmonic nanoparticle (core), labelled with so-called “Raman reporters” (RaRs), molecules featuring high Raman scattering cross-sections and affinity for the metal NP surface. Such tags may be subsequently coated with macromolecules such as polymers, lipids, peptides, proteins, or with biorecognition motifs like antibodies or aptamers, to promote different functionalities or target specific biological interactions, leading to a wide variety of configurations depending on the particular application of SERS measurements. In this review, biosensing refers to cases in which the chemical information provided by or encoded to SERS tags is used to detect and/or identify analytes. Additionally, “bare” nanoparticles without RaRs can also provide chemical information via “label-free” sensing. Bioimaging refers to instances in which SERS tags are tracked within or around cancer cells and at tumor sites in vivo. We thus begin by noting the differences between in vitro and in vivo 3D cell models, with examples of the various types of models that have been explored. We then cover design rules and describe SERS tags that have been used to interrogate such 3D models. Next, we address how SERS bioimaging can be combined with other techniques to obtain complementary information and higher imaging resolution (multimodal imaging). We also describe the use of “deep” SERS imaging techniques to overcome the limitations of light penetration through opaque materials or those containing many different interfaces/materials, such as tissues. Finally, we discuss the use of SERS for cancer biomarker detection, drug testing, and pH sensing in tumoral environments.
2. Three-dimensional cell models
In this section we introduce the main categories of 3D cell models that have been developed so far, aiming to better recapitulate human tumoral environments, compared to 2D cell cultures.2.1. Spheroids
Spheroids result from the self-assembly of substrate-seeded cells, and their formation is driven by cell–cell adhesion forces that are stronger than cell–surface interactions.49 These 3D structures were first obtained by Moscona et al. from organ rudiments of early chick embryo.50 Nowadays, plenty of methods are available for spheroid formation from single or co-cultures, such as hanging drop51 or rotating culture,52 and more sophisticated methods like microfluidics or 3D bioprinting.53 Spheroids enable studies regarding cell–cell signaling, cell–cell and cell–ECM physical interactions, growth kinetics, gene expression, and drug resistance phenomena.54 In fact, it has been demonstrated that spheroids are able to mimic the original tissue, for example building hepatocyte spheroids with liver-like functions.55 In cancer research, spheroids have been extensively used because they can mimic in vivo solid tumors, in terms of structural organization and gradients of nutrients, oxygen, and pH, which are established within three-dimensional structures. Multicellular tumor spheroids can be structurally divided into an external proliferating zone, an internal quiescent zone, and a necrotic core (Fig. 3A).56 Despite the advantages of spheroids, cellular aggregates greater than 1–2 mm in thickness show a limited exchange of nutrients, oxygen, and waste metabolites, resulting in cell death.57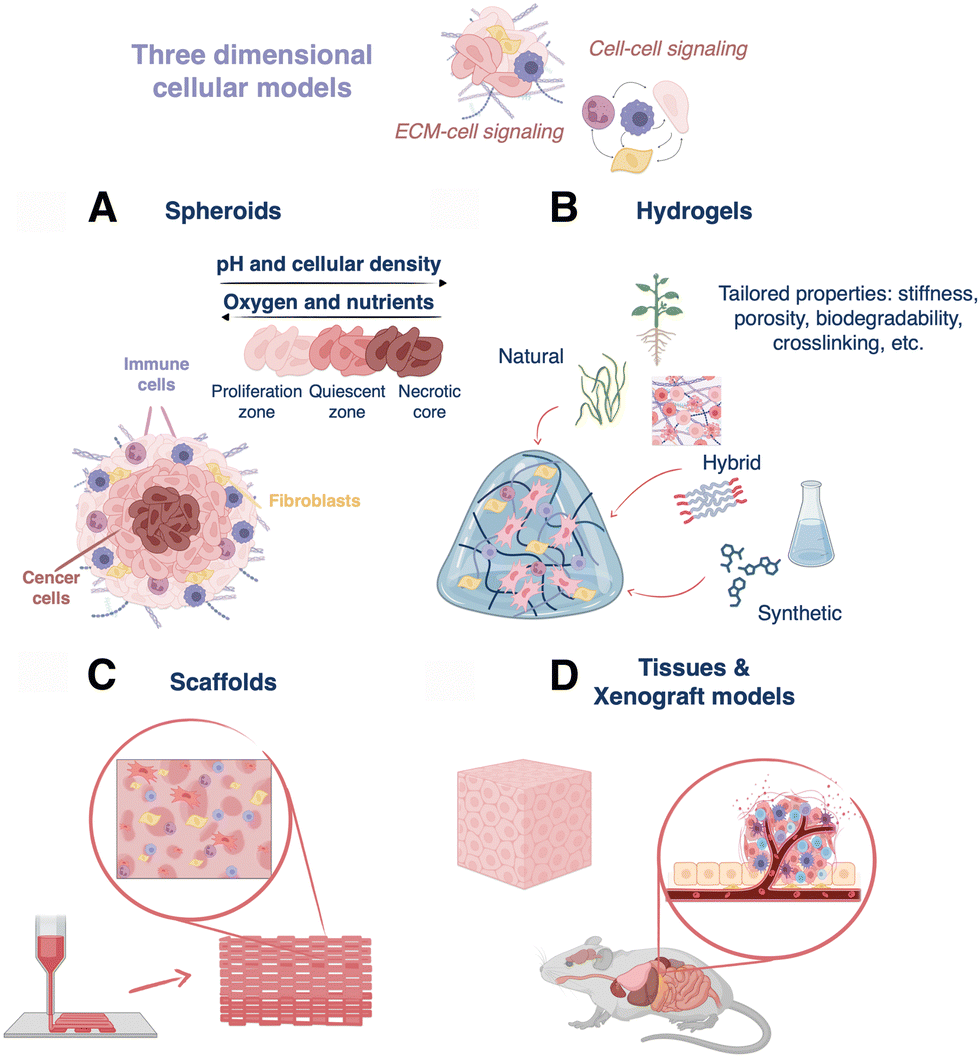 | ||
| Fig. 3 Diagram describing different 3D cell models: (A) 3D spheroids, (B) hydrogels for cell culture, (C) hydrogel-based 3D printed scaffolds, and (D) tissues and xenograft models. | ||
2.2. Bulk hydrogels
Hydrogels are hydrophilic polymer networks that display great biocompatibility and ability to retain water.58 These matrices allow the flux of oxygen and nutrients, have similar mechanical properties as those of many soft tissues, and are able to support cell adhesion and protein sequestration.58 They can be crosslinked via chemical (click chemistry, radical reactions, Michael-additions, etc.) or physical (thermal, hydrogen bonding, ionic interactions, etc.) mechanisms. Moreover, hydrogels can be chemically modified to display particular stiffness, porosity, ease of functionalization, crosslinking type, biodegradability, and cell compatibility, which are important features to build in vitro tissue-like architectures (Fig. 3B).59 Hydrogel-based 3D cell cultures can be obtained by encapsulating cells within the hydrogel prior to crosslinking or by seeding cells after crosslinking.Hydrogels can be classified depending on their polymeric origin as natural, synthetic, or hybrid (semi-synthetic). On one hand, natural hydrogels include polysaccharides (chitosan, dextran, etc.), proteins/peptides (gelatin, elastin, Matrigel, etc.), and decellularized extracellular matrix-based polymeric networks. They are extracted from different biological sources, for instance, collagen, fibrin, and hyaluronic acid are proteins of native ECM, alginate and agarose derive from marine algae, and cellulose is a component of the vegetal cellular wall. In the case of dECM-based hydrogels, they are obtained from decellularized tissue, and thus retain proteins (collagen, fibronectin, laminin, etc.), as well as some growth factors and polysaccharides (proteoglycans, glycosaminoglycans, etc.).60 As a result, they reproduce the microstructure, biochemical, and biophysical features of the native tissue, and are able to assist growth and tissue reconstruction.61 In general, natural hydrogels are biocompatible (low toxicity), bioactive (promote cell activity), and readily available. Nonetheless, their extraction and purification can be expensive, and as natural products, batch-to-batch variability is high, leading to poor reproducibility between different cultures.62 On the other hand, the group of synthetic hydrogels includes poly(ethylene glycol) (PEG), poly(vinyl alcohol) (PVA), poly(2-hydroxy ethyl methacrylate), poly(glycolic acid), and poly(lactic acid), among others. These polymers display well-defined structures and can be uniformly produced, but they may also restrict cellular functions.58 Finally, hybrid materials obtained through the combination of synthetic and natural polymers are a third option.63 These composites combine the properties of both polymer types and can be designed to mimic the biophysical and biochemical features of the natural extracellular matrix.64 In any case, the choice of one or another possibility finally relies on the particularities of the cell culture and the type of targeted tissue or tumor. Different combinations of hydrogels for cell culture are subsequently addressed in Section 3.3.
2.3. Scaffolds
Scaffolds are constructs designed to support cell growth outside of the body, trying to reproduce the extracellular microenvironment. These 3D structures should display biocompatibility, reproducibility, high porosity, pre-designed biodegradability, as well as suitable mechanical and biochemical properties to promote cell attachment, proliferation, and migration.65 Over the past decade, many materials such as metals (orthopedics), ceramics (implants), and polymers (tissue engineering), have been used to prepare scaffolds for biomedical applications.52 Focusing here on polymers, hydrogel-based scaffolds are of particular interest because of the properties described in Section 2.2 above. Different methods can be used to build up hydrogel-based scaffolds, ranging from classical templating to more sophisticated 3D printing,66 which enables the construction of multiscale architectures by precise deposition of materials in x, y, and z directions. Three primary 3D printing techniques can be mentioned: extrusion, laser-assisted, and inkjet printing.67 Hydrogels can be used as inks because they are viscoelastic polymers with non-Newtonian properties, allowing for extrusion from a nozzle to yield a controlled deposition of filaments.68 In addition, low-viscosity hydrogels can be combined with cells, resulting in specialized bioinks that can be used to accurately position cells within the scaffold.69 Otherwise, cells can be seeded and cultured directly on top of the hydrogel-based scaffold post-printing, allowing them to grow and proliferate with the scaffold support.70–72 In short, these complex structures display high resolution in all spatial dimensions and tunable biochemical, biophysical, and mechanical properties for the reliable and reproducible production of 3D cell models (Fig. 3C).2.4. Xenograft models and tissues
Tissues represent another common 3D model, which can be studied in vivo in live animals and/or ex vivo as cultured or resected tissue (Fig. 3D). Although human tissues can be cultured ex vivo, they require the use of dynamic bioreactor setups and culture over several days (even up to 60 days).73 Thus, as an alternative for ex vivo studies, porcine tissues and organs are relatively easy to obtain, while being compositionally similar to human tissues.74In vivo, immunocompromised or humanized mice and rats implanted with human cancer cells are the most common models. In general, the design of biologically accurate cancer models is challenging because oncogenesis results from genetic and epigenetic anomalies that arise and evolve uniquely patient-to-patient, and tissue inhomogeneities exist, even within the same tumor.75,76 This is why, although many studies are based on implementing tumors from immortal cancer cell lines in xenograft models (especially because they are relatively easy to engraft/obtain), patient-derived xenografts (PDX) are also gaining traction as 3D in vivo models.75,77 However, PDX models also have their limitations, for example, the surrounding ECM, cells, vasculature, etc. – the so-called “tumor stroma” – in animal models is not the same as that of the primary tumor, and accurately replicating the stroma is challenging.78,79 Moreover, the body size, anatomy, physiology and pathophysiology of rodents differs significantly from those in humans, and when immunocompromised animals are used, limitations arise in the conclusions that can be drawn from drug resistance studies.5,80,81 With PDX models, one also faces the challenge that engraftment success rate can vary wildly, often leading to the loss of difficult-to-obtain patient-derived samples.82 Ultimately, these drawbacks have promoted research interest in human tissue engineering.83 However, until now, owing to their high engraftment success rates with commercial cell lines and their ability to replicate tumor microenvironments with good fidelity, xenograft models have enabled significant developments in the design and implementation of SERS probes in 3D, which will be discussed further in Sections 3 and 4.3. Designing SERS tags for three-dimensional cell models
Naturally, the composition, concentration, and geometry of the plasmonic NP units plays a significant role in determining the biocompatibility, scattering intensity, and measurement conditions. Metals with plasmonic responses in the ultraviolet (UV)-visible-near infrared (NIR) spectral range include gold, silver, copper, and aluminum but, to date, primarily silver and gold have found applications in biological environments.84 Gold and silver NPs (AuNPs and AgNPs, respectively) display large optical cross sections, can be readily bio-functionalized, and their optical response can be tailored – through NP size and shape – across the visible and NIR regions of the electromagnetic spectrum.85 The ability to tune the LSPR wavelength in the NIR is key for matching the “biological transparency windows” where there is maximum penetration of light and minimum tissue autofluorescence and photodamage. Even though AgNPs provide higher field enhancements and stronger SERS activity, they can be cytotoxic and are prone to degradation via oxidation and sulfidation.86–88 However, these negative properties can be mitigated in certain configurations, and relevant strategies will be discussed in the following section. Compared to AgNPs, AuNPs have higher biocompatibility and stability against oxidation, rendering them the preferred choice for incorporation in 3D cell models.89 Gold nanostars (AuNSts), nanoshells, nanoprisms, and nanorods (AuNRs) can exhibit LSPR modes in the NIR.90–93 Even though AuNPs are particularly stable compared to other plasmonic metals, some precautions still need to be taken to prevent reshaping when using geometries with sharp features.94Beyond the SERS tags themselves, measurement parameters such as acquisition time, laser wavelength, power density, etc., must be carefully optimized for biological studies to ensure cell viability. Tuning measurement conditions is important because the common geometries used in SERS (nanostars, nanoshells, and nanorods) can efficiently generate localized heat in response to NIR irradiation,95–97 which may damage or alter the behavior of cells or biological systems under study.98 Cellular homeostasis takes place between 37 and 41 °C, and higher temperatures closer to ∼48 °C cause the tertiary structure of proteins to be disrupted, a process that becomes irreversible above ∼50 °C.99 Depending on the size, shape, and configuration of the metallic nanostructures and the irradiation conditions, it is possible to reach temperatures higher than 100 °C at the nanoscale.95,97 Therefore, the laser energy delivered to NP-labelled cells must be controlled to avoid laser-induced (photo)toxicity while providing a reliable SERS signal.98 Therefore, the optimization of several imaging parameters is required to maintain cell viability in 3D systems. Irradiation time and repeated exposure of cells to the laser beam are important factors to bear in mind, low laser power and/or fast acquisition times being optimal. Alternatively, off-resonance Raman, whereby laser irradiation wavelengths are mismatched with the LSPR, can also prevent photothermal damage, which has been applied for SERS studies on tissues.100
Altogether, careful control over spectral acquisition parameters is essential, and a number of considerations are to be made in the design of the tag itself, as will be further discussed in Subsection 3.1. In the following subsections (3.2–3.4), we will discuss strategies for incorporating plasmonic nanotags into different types of 3D models.
3.1. SERS labelling for sensing and imaging
Generally, to effectively monitor the spatial and temporal distribution of distinct cell populations, it is necessary to employ labelling strategies, such as fluorescence or radioactivity, among other examples.101 In the case of SERS imaging, decoration of the surface of plasmonic NPs with different RaRs results in a variety of SERS tags that can be used to track cells upon internalization (Fig. 4). The purpose of this section is to provide a concise overview of various SERS labelling options; for more comprehensive information regarding SERS nanotag configuration, readers are referred to recently published reviews.102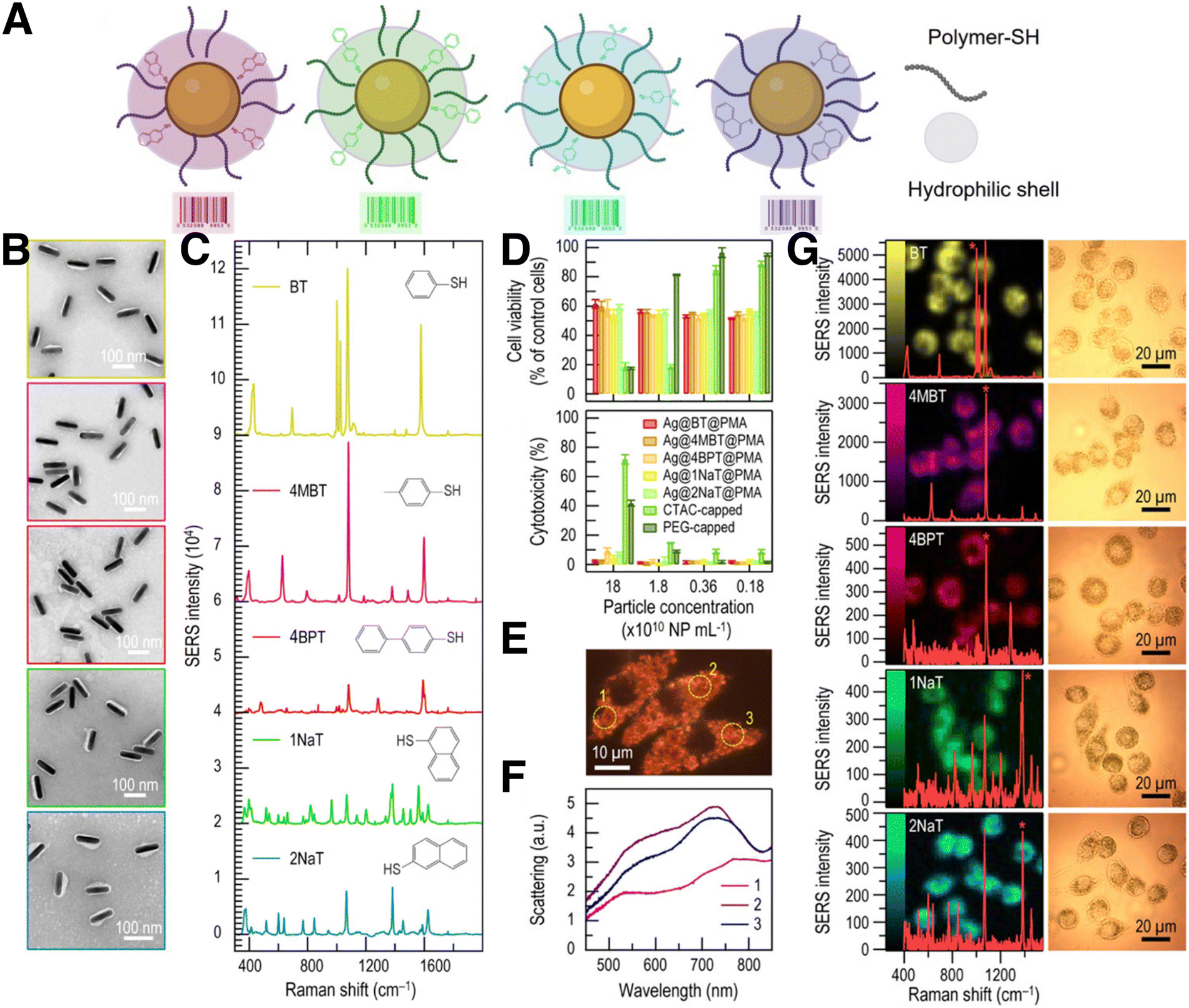 | ||
| Fig. 4 (A) Schematic view of SERS tags comprising a metal NP core functionalized with the selected Raman reporter and a thiol-binding polymer, and further stabilized with an amphiphilic polymeric shell. (B) TEM images of silver nanorods (AgNRs) coated with PMA and functionalized with five different RaRs. From top to bottom: benzenethiol (BT), 4-methyl benzenethiol (4-MBT), 4-BPT, 1-NAT, 2-NAT. (C) Corresponding SERS spectra of the samples in B. (D) Cell viability and cytotoxicity results of AgNRs incubated with J774 macrophage cells, from MTT and LDH viability assays, respectively. (E) Real-color dark-field scattering image and (F) dark-field scattering spectra measured from three individual J774 cells containing AgNRs functionalized with BT. (G) SERS images measured from J774 cells containing AuNRs with different RaRs and their corresponding spectra (left) and bright field images of the cells (right). [Adapted with permission from ref. 110 Copyright (2020) American Chemical Society] | ||
Overall, the designed SERS tags must be biocompatible and stable under biologically relevant conditions.102 Therefore, after RaR adsorption, an external coating is usually deposited to improve their stability and biocompatibility, and to modulate intracellular uptake (Fig. 4A). The application of this coating holds significance in preventing RaR leakage, safeguarding against SERS signal contamination from other interfering molecules, mitigating the potential toxicity of NPs, and minimizing unintended intensity fluctuations resulting from plasmon coupling induced by particle–particle interactions. Commonly employed encapsulation strategies include different types of coatings made of peptides or proteins,103,104 liposomes,105 silica,106 or polymers.107,108 The choice of coating should be determined by the final application, considering factors such as biodegradability, long-term stability, agglomeration, binding competition, etc. As an example, coatings based on amphiphilic polymers, such as dodecylamine-modified polyisobutylene-alt-maleic polymer (PMA), have been demonstrated to improve the stability of SERS tags over extended periods of time.108 In this system, NP internalization can be enhanced through an additional coating step with a cationic polymer such as poly-L-arginine hydrochloride (PA), because cellular uptake is known to be enhanced for positively charged NPs.107
Whereas most NPs used for SERS cell labelling relate to anisotropic structures featuring plasmon resonances in the NIR region, synthetic strategies can also be implemented to fine-tune the aggregation of spherical AuNPs and redshift their plasmon resonances through plasmon coupling. To this end, the ligand ratio between RaRs and stabilizing polymer layer can be tuned to control NP aggregation. For example, the use of poly(isoprene)-diethylenetriamine (PI-DETA) as NP ligand and poly(isoprene)-block-poly(ethylene glycol) (PI-b-PEG) as a biocompatible micelle builder, combined with a solvent-induced self-assembly process, has been reported to encapsulate AuNP clusters. The number of encapsulated particles can be adjusted by varying the PI-b-PEG to PI-DETA ratio, and the particles can be labelled with RaR molecules.109 Pre-labelling of individual NPs with RaRs appears to be more effective than RaR incorporation into the assemblies, to ultimately introduce them into the resulting hotspots. In this case, PI-DETA was partially replaced by RaRs, resulting in a less dense ligand shell with NPs in closer proximity. Therefore, the type of RaR and the number of incorporated molecules will influence NP aggregation and should be addressed carefully. Enhancement factors of 104–105 were reported for these systems, with tunable LSPR depending on parameters such as incubation time and RaR content.109
Although most reported systems for SERS bioimaging rely on AuNPs, due to their well-known biocompatibility, silver-containing NPs can also be protected from external conditions by means of appropriate synthetic protocols, to prevent oxidation. Zhuo et al. reported the codification of monodisperse Ag nanorods (AgNRs) with up to five different RaRs, for intracellular SERS imaging (Fig. 4B–G). As mentioned above, the unique dielectric properties of Ag are hindered by its high cytotoxicity and poor stability in biological settings. However, as previously reported for AuNRs, PMA can be also used to coat and stabilize AgNPs. PMA-coated Au@Ag nanorods derived from Au bipyramids were reported to be stable, non-cytotoxic, and biocompatible, as well as suitable for SERS imaging of J774 macrophages, upon labelling with various RaRs.110
Another interesting feature of SERS tags is related to multiplexing, owing to narrow SERS peaks and the possibility of pre-labelling different cell populations with different SERS tags and relocating the tagged cells in a mixed co-culture via simultaneous excitation with a single laser source.98 Multiplex imaging based on SERS tags conjugated to antibodies or aptamers designed for active cell targeting can also be useful for imaging distinct cell types within mixed cultures (Fig. 5). In one example, Nima et al. employed up to four different RaR molecules; 4-mercaptobenzoic acid (4-MBA), 4-aminothiophenol (4-ATP), 4-nitrothiophenol (4-NTP) and 4-(methylsulfanyl) thiophenol (4-MSTP), linked to four breast cancer markers (anti-epithelial cell adhesion molecule, anti-CD44, anti-keratin, and anti-insulin-like growth factor antigen) for the detection of circulating tumor cells.111 With this approach, the specificity of the detection of a particular cell type can be enhanced, reducing the number of false readings. The identification of human epidermal growth factor receptor 2 (HER2)-positive breast cancer cells has also been possible by SERS imaging.112 Anti-HER2 antibody-conjugated AuNPs were prepared to target breast cancer cells using 4-MBA as both RaR and conjugation site for attaching antibodies.112 Following this strategy, it was possible to distinguish between HER2-positive and HER2-negative expressing cells, according to differences in SERS signal intensity. SERS tag labelling can also be used to distinguish between cancerous and healthy cell lines. In an example, Rodal-Cedeira et al. reported the bioconjugation of nanocapsules with antibodies against three different cell surface receptors epidermal growth factor receptor (EGFR), epithelial cell adhesion molecule (EpCAM), and CD44.113 SERS detection was carried out simultaneously in a co-culture of human epithelial carcinoma A431 cell line and nontumoral murine fibroblasts 3T3 2.2. cells. The results showed that 3T3 2.2 cell line only expressed CD44, whereas A431 cells expressed all three antibodies, demonstrating the ability of the SERS tags to distinguish between both cell populations.
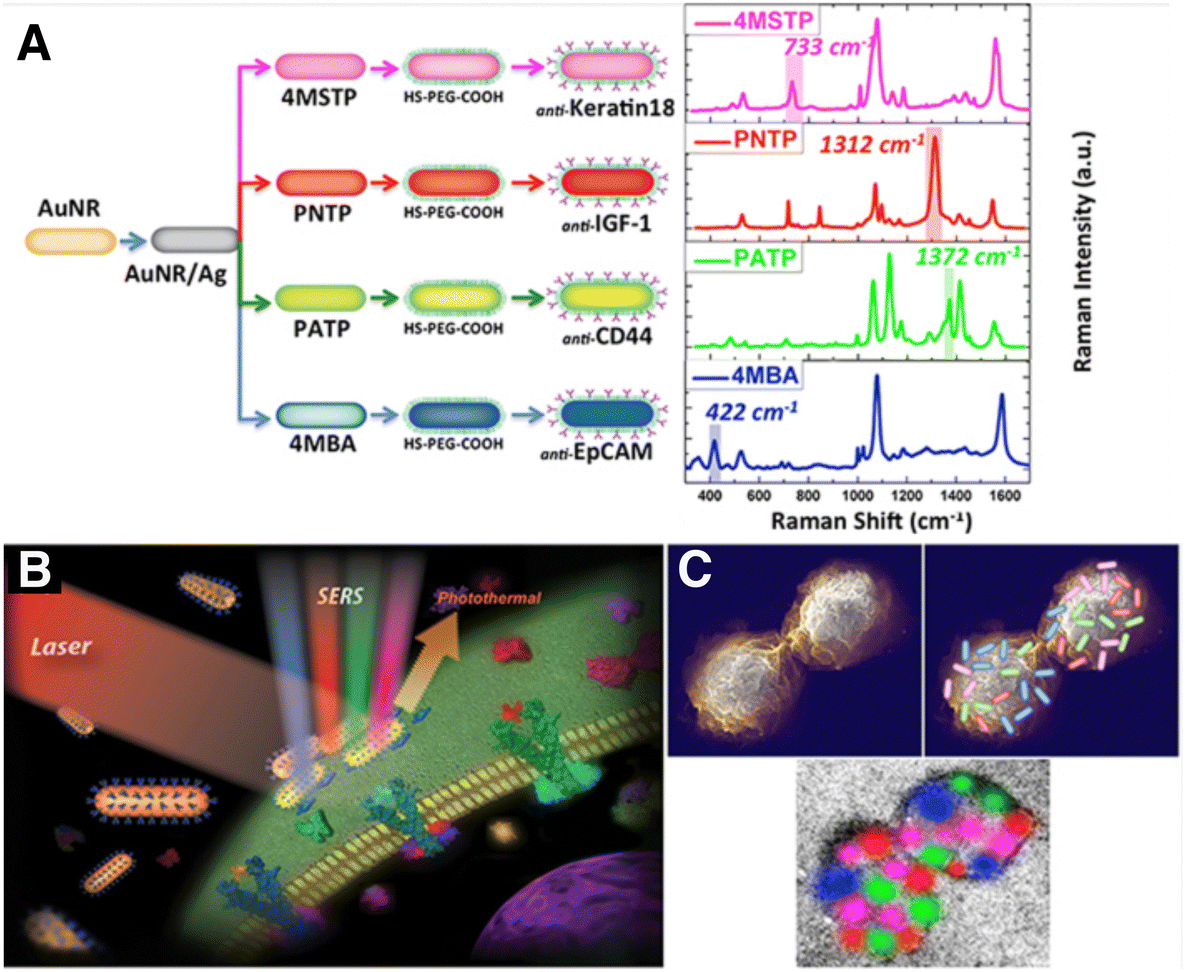 | ||
| Fig. 5 (A) Scheme showing four types of nanotags functionalized with different antibodies and their corresponding SERS spectra. Non-overlapping peaks from each SERS spectrum were assigned a different color, as indicated: 4MBA/anti-EpCAM (blue), 4-NTP/anti-IGF-1 (red), 4-ATP/anti-CD44 (green), 4-MSTP/anti-Keratin18 (magenta). Schemes showing proposed (B) breast cancer cell surface targeting by SERS tags and (C) multi-color 2D spatial distribution of SERS intensities, showing how the tags can assist in mapping the cell surface. [Reproduced from ref. 111 with permission from Springer Nature]. | ||
3.2. Implementing SERS tags in spheroids
Incorporation of SERS tags in spheroids can be accomplished through two main methods: prior internalization into individual cells or post penetration from the periphery into the spheroid core. Cell internalization is determined by the ability of a SERS tag or biofunctionalized NPs (without RaRs) to travel through the plasma membrane, whereas post-penetration depends on the system's diffusion through interstitial spaces, with size, geometry, and surface charge being the most relevant parameters in both cases.114–116 Sujai and co-workers compared the internalization efficiency of neutral, positively and negatively charged AuNPs, functionalized with 2-(4-methylthiobenzylidenamlononitrile), within 2D HeLa cell cultures and multi-layered HeLa spheroids.117 Their results demonstrated greater internalization of positively charged NPs in the 2D HeLa cell cultures, where negatively charged particles had a higher penetration into the core of the spheroids. Similarly, McCabe et al. compared NP localization in 2D glioblastoma U87-MG cell cultures and spheroids.118 Gold NPs functionalized with (4-1H-pyrazol-4-yl) pyridine were encapsulated in a silica shell, then tenascin-C antibody was immobilized on the tags to target U87-MG glioblastoma cells which overexpress tenascin-C protein. Although the antibody labelled-NPs were found throughout the whole cell in U87-MG monolayer culture, they were shown to accumulate in the spheroid periphery. The authors identified the larger size of the particle, with a diameter >30 nm being optimal for spheroid penetration, and the blocking of active transport processes due to a lack of nutrients and oxygen in the spheroid core, as the cause of this result.119Aside from the size and charge of the SERS tags, incubation method and time can also affect their final localization in the spheroid structure. Jamieson et al. fabricated multicellular tumor spheroids, then performed incubations with SERS tags either in 2D cell culture before spheroid formation or with RaR-labelled nanoshells at different time points during spheroid formation (Fig. 6).120 This approach resulted in spheroids with SERS tags either in the core (Fig. 6A), intermediate area (Fig. 6B), outer zones (Fig. 6C), or throughout the spheroid (Fig. 6D). Drug treatment of MCF-7 and PC3 spheroids was studied by monitoring the SERS spectra of the RaRs, 4-MBA, naphthoquinone, and methylnaphthoquinone, present on the tags, from which pH and redox potential gradients could also be determined. In summary, the type of cells involved, the spheroid building method and the morphological (size, charge, and coating) characteristics of AuNPs are of great importance for SERS sensing and imaging in these 3D models.121 Thus, all parameters must be carefully chosen to maintain viability and achieve efficient internalization and sensing/imaging throughout spheroids based on different cell lines.122
 | ||
| Fig. 6 Photothermal optical coherence tomographs showing SERS nanosensors in the core, intermediate, and outer zones of a spheroid. Optical coherence tomograph (left column), photothermal image (central left column), overlay of both (central right column), and schematic representation (right column) of (A) gold nanoshells in the interior, (B) intermediate part, and (C) outer part, and (D) throughout the spheroid. [Reproduced from ref. 120 with permission from the Royal Society of Chemistry]. | ||
3.3. Fabrication of plasmonic-hydrogel hybrid materials
Bulk hydrogels can be combined with plasmonic NPs to turn the resulting composite into a SERS substrate for biodetection. The incorporation of either SERS tags or bare metal NPs (either to be subsequently functionalized with RaRs or applied for label-free sensing) within hydrogel-based matrices has been reported for various metals, resulting in nanocomposites with applications in drug delivery, as bioactive implants, and stimuli-responsive materials for tissue engineering.123 In fact, bulk hydrogels containing plasmonic NPs can serve both as SERS substrates and as plasmonic inks to 3D print scaffolds in which cells can be cultured and monitored.Among the various synthesis protocols for hydrogel-NP composites we highlight the following common approaches: (a) hydrogel formation in NP suspension; (b) NP embedding into the hydrogels after gelation; (c) in situ formation of NPs within the gel; and (d) the use of NPs as crosslinking agents.124 In general, the rheological (yield stress, shear stress, storage and loss modulus, etc.) and physicochemical (response to temperature, swelling, hydrophobicity, etc.) properties of the hydrogel, as well as the optical (absorption, scattering, reflection, emission, etc.) properties of the NPs, may differ compared to those of the resulting composite.125,126 The study carried out by García-Astrain et al. assessed shear thinning, yield point, and viscoelastic modulus for a mixture of HAMA and κ-carrageenan polymers, containing either bare or PEG-coated AuNRs or AuNSts.71 The effect of NP concentration was interrogated, which was found not to affect the shear thinning, but was determined to alter the storage modulus (G′), which was shown to increase with the addition of up to 0.5 mM of [Au0], then decreasing for higher concentrations. Despite these changes to the physical properties of the gel, the resulting composite was successfully used as an ink for printing high resolution scaffolds (as described in Section 2.3) with 250 μm interlinear spacing, which could be applied for biosensing (see Section 5.1). In summary, the viscoelastic properties of plasmonic hydrogels can be tailored by selection of the hydrogel component, NP type, concentration, and functionalization.
Most often, plasmonic NPs are mixed with the hydrogel prior to constructing the scaffold as in the previous example (Fig. 7A), but the formation of NPs in situ, i.e., within the gel, has also been explored (Fig. 7B). As another example of the former, Plou et al. described the fabrication of SERS active 3D printed scaffolds using a bioink containing gelatin, alginate and AuNRs.72 Regarding the latter approach, Lehman and colleagues built plasmonic Au/pHEMA composite scaffolds by including gold NP precursor (Au3+) in a pHEMA matrix and then incubating in sodium ascorbate solution, after the polymer was cured with UV-light, to form NPs in situ. By following this protocol, the authors aimed to prevent AuNP aggregation during processing and 3D printing.127 However, the chemicals used for gold reduction can also react with the hydrogel and the resulting material might contain potentially cytotoxic unreacted chemical species or reaction by-products, which would hinder the use of these scaffolds for 3D cell culture.128
 | ||
| Fig. 7 Scheme for 3D printing of plasmonic scaffolds, using (A) hybrid bioinks comprising NP colloids and hydrogels, or (B) hybrid bioinks made from chemical precursors and hydrogels. | ||
3.4. Integrating SERS tags in tissues
SERS tags can be incorporated into tissues ex vivo by either direct injection,129 placing a tissue slice on a SERS substrate,130 or by enclosing a chamber containing SERS tags within the tissue.131,132 For in vivo 3D SERS studies related to cancer, rodent xenograft models are common, as described in Section 2.4. While the earliest example of in vivo SERS biosensing from the group of Van Duyne applied a plasmonic substrate implanted in rats, the use of colloidal NPs is currently seen as a requirement to extend the imaging/sensing capabilities beyond 2D.41,102,133 In some studies, colloidal nanotags are delivered by direct subcutaneous, muscular, or (∼1 cm) deep muscular injection,134 but it is also possible to “passively” deliver the SERS tags to tumor sites via intravenous injection, thanks to the so-called enhanced permeation and retention (EPR) effect, i.e., the tendency of NPs to accumulate in tissues with high vascular permeability (e.g., tumors).135,136 In this sense, NPs with sizes of 5–200 nm have been reported for “passive’’ tumor targeting because both smaller and larger NPs are cleared through either the kidney or the mononuclear phagocyte system.137 Alternatively, “active” targeting using antibodies, aptamers, or other ligands, may enhance the accumulation of plasmonic tags introduced intravenously at selected tumor regions, which can be helpful both as a diagnostic tool and for further increasing the SERS signal at the target site.102,103,109,138–1404. SERS bioimaging in 3D cell models
McAughtrie et al. reported for the first time the combination of Raman and SERS for 3D cell imaging, with the aim of simultaneously performing multiple component detection and confirming NP internalization, without requiring additional intracellular imaging techniques, such as TEM.141 3D cell mapping with Raman spectroscopy was combined with SERS mapping to confirm the localization of the SERS tags in the interior of Chinese hamster ovarian cells. Silver citrate-capped NPs were coated with four different RaRs, 4-mercaptopyridine (4-Mpy), 5′5-dithiobis(2-nitrobenzoic acid) (DTNB), 4-nitrobenzenethiol (NBT) and 2-napthalenethiol (2-NAT), then NP aggregation was induced by adding 1,6-hexamethylenediamine to produce hotspots. Finally, poly(vinylpyrrolidone) (PVP) was added as quencher for hot spot stabilization. The SERS tags were later delivered to the cells and volume mapping was performed by collection of Raman and SERS images with 0.5 mm resolution in x- and y-directions, and 1 mm resolution in the z-direction. The authors were able to identify three out of the four tags intracellularly. A certain degree of colocalization was reported due to their simultaneous administration to the cells, but the tags did not contain any functionalities to target specific subcellular regions.The uptake and distribution of NPs within cells might also change over time. Here again, SERS imaging can be used as a non-invasive tool, to estimate in situ the number of particles remaining in cells over time, through a correlation between signal intensity and number of SERS tags. Lenzi et al. evaluated the average SERS intensity per NP by using correlated TEM and SERS.142 AuNSts and AuNRs with four different RaRs (4-methyl benzenethiol (4-MBT), benzenethiol (BT), 2-NAT and 4-bisphenilthiol (4-BPT)) were applied to develop an application software (SERSTEM App), that can be used for the correlation.
One of the advantages that SERS offers over fluorescence microscopy is the possibility to perform simultaneous SERS imaging and sensing. In this way, 3D cell maps of SERS tags and cell biomarkers can be achieved simultaneously to provide information regarding their spatiotemporal distribution. Chen et al. reported the use of labelled and label-free SERS for 3D HeLa cell imaging, using spherical gold NPs labelled with 4-MBA, crystal violet, and crystal violet acetate. For the incorporation of these three dyes, a poly-allylamine (PAH) shell was incorporated onto the NPs to reduce aggregation. The PAH shell also introduced –NH2 groups on the NP surface that could be used for subsequent functionalization.143
4.1. In vitro SERS bioimaging
Although only few studies have reported the use of SERS imaging in 3D, this field is undergoing significant expansion. SERS imaging has been used to study the differences of NP uptake between 2D cell monolayers and 3D cell models, which eventually provides information about the possible mechanisms behind NP internalization and diffusion, which depend on the RaR composition/charge and NP size and geometry, as discussed in Section 3.2.117,118 In one example, Liu et al. designed SERS tags with different sizes, each coated with a particular RaR (34 nm – Au@MBA@Ag; 60 nm – Au@4-mercaptonitrobenzoic acid (MNBA)@Ag; 108 nm – Au@2,3,5,6-tetrafluoro-4-mercaptobenzoic acid (TFMBA)@Ag; 147 nm – Au@4-mercaptopyridine(MPy)@Ag) to compare their accumulation in T47D human breast cancer multicellular spheroids (Fig. 8A).144 The highest degree of internalization and diffusion across interstitial spaces was ultimately achieved for the 60 nm Au@MNBA@Ag NPs (Fig. 8B and C).145,146 This result is consistent with other uptake studies (albeit not performed in spheroids), which found that NPs with dimensions close to 50 nm more efficiently penetrate into cells by endocytosis. It has been suggested that the binding of single nanoparticles with diameters much smaller than 50 nm is not energetically favorable and that the membrane encapsulation process for particles much larger than 50 nm is kinetically disfavored.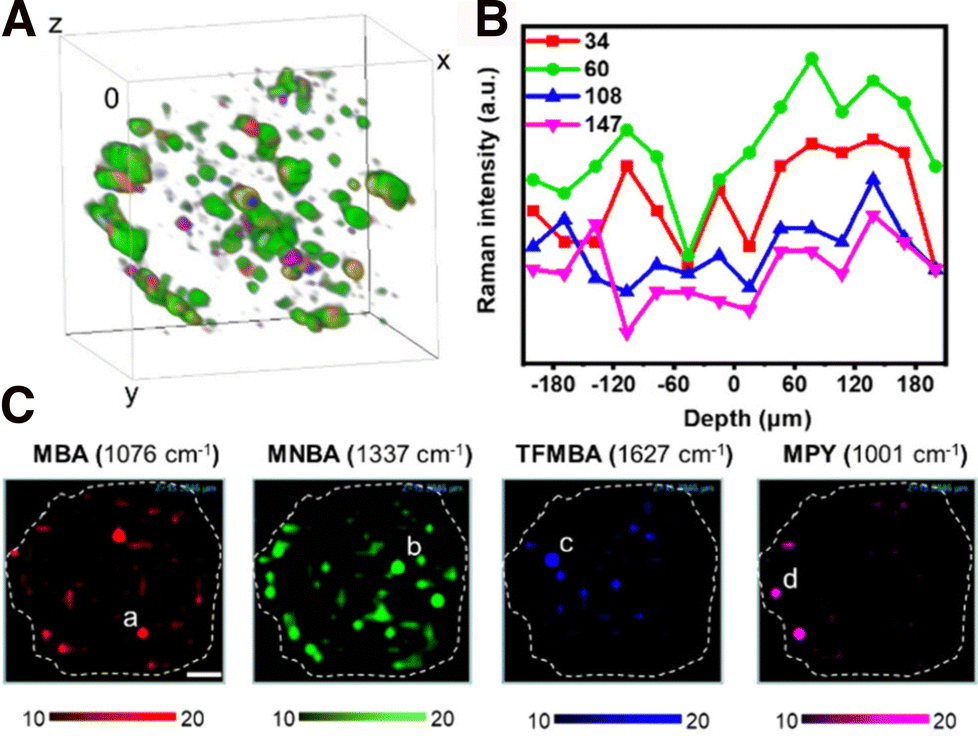 | ||
| Fig. 8 (A) Reconstruction of a T47D spheroid using the SERS intensities from four SERS tags (color code provided in C). (B) Average SERS intensity from 34, 60, 108, and 147 nm NPs in each layer. (C) Individual SERS mapping channels of the spheroid with RaRs [Adapted with permission from ref. 144 Copyright (2023) American Chemical Society]. | ||
Instead of using labels, the spectral bands associated with different biomolecules ubiquitously present in cells can be used to image nucleic acid, lipid, and protein-rich regions. Pan and co-workers reported the synthesis of silver nanowires at the tip of a carbon nanoelectrode, for recording spectral data at various penetration depths in MCF7 breast cancer spheroids via direct insertion.147 The different spectral bands were attributed to lipids, proteins, phospholipids, and nucleic acids, and their distribution was imaged at different regions of the spheroid (cellular wall, interstitial spaces, etc.). More recently, the same group reported the use of this setup to determine intracellular and extracellular pH.148
Besides single cells or cell clusters, SERS imaging has been recently implemented in 3D substrates, such as polymers or tissues, thereby expanding the applications of SERS bioimaging to more complex 3D cell models. Vantasin et al. reported the incorporation of octahedral silver hexapod microparticles labelled with the RaR 4-ATP, into layered matrices containing different polymer components.149 The microparticles were mixed into blended and layered polymer systems, and inhomogeneities in the materials were identified with 3D SERS mapping. Overall, these results support the importance of polymer composition, assembly, and transparency for designing complex 3D scaffolds for SERS imaging.
Another example of 3D SERS imaging was reported by Jimenez de Aberasturi et al., where a 3D cell culture model was used, comprising unlabeled and SERS tag-labeled fibroblasts, in a layer-by-layer system.107 In this work, AuNSts and AuNRs were encoded with 4-BPT and 2-NAT, respectively, or labelled with fluorescent dyes. Signal overlap was avoided by using layers of unlabeled cells and Pearson's correlation coefficients were employed for linear correlation with the corresponding reference spectrum. This complex 3D sample was imaged with 5 μm resolution in x, y, and z, and a well-defined layered structure could be observed within an area of 104 mm2 in x and y. Due to the instrument configuration, the measurements were carried out from the top to the bottom layers, resulting in a lower definition of the 3D map toward the bottom of the sample and the need for data processing to enhance the signal-to-noise ratio. Therefore, the visualization of living organisms relies on the development of compatible 3D model configurations that improve the sensitivity and specificity of the technique. Measurement setups and acquisition times should also be compatible with biological samples. Additionally, there is a growing need for data analysis techniques to extract valuable insights from complex biological data, as will be discussed in Section 6.
4.2. In vivo SERS bioimaging
Noninvasive imaging methods like positron emission tomography/computerized tomography (PET/CT) and magnetic resonance imaging (MRI) are commonly used for clinical staging, to determine the extent of the disease in cancer patients. However, these techniques are susceptible to errors from potential artifacts, and techniques like PET can undesirably damage nearby tissue because of the use of high-energy radiation. In this context, SERS requires the application of a comparatively milder irradiation source,150 has lower interference with tissue autofluorescence when using NIR SERS tags, and opens the door to multiplexed in vivo bioimaging.151 In one example, Nie and co-workers showed the implementation of SERS imaging in a clinically practical setup, for the precise indication of tumor margins using a handheld pen (Fig. 9A).152 The “Spectro Pen” incorporates a diode laser with 785 nm irradiation, in a compact head unit that acts as the light source and collection probe, connected via an optical fiber to a spectrometer for recording fluorescence and Raman signals. The collection of Stokes-shifted light is facilitated by the attenuation of Rayleigh scattered light using a dichroic mirror and long-pass filter, as well as by minimization of the silica signals from the fiber via physical filtering in the excitation and emission pathways.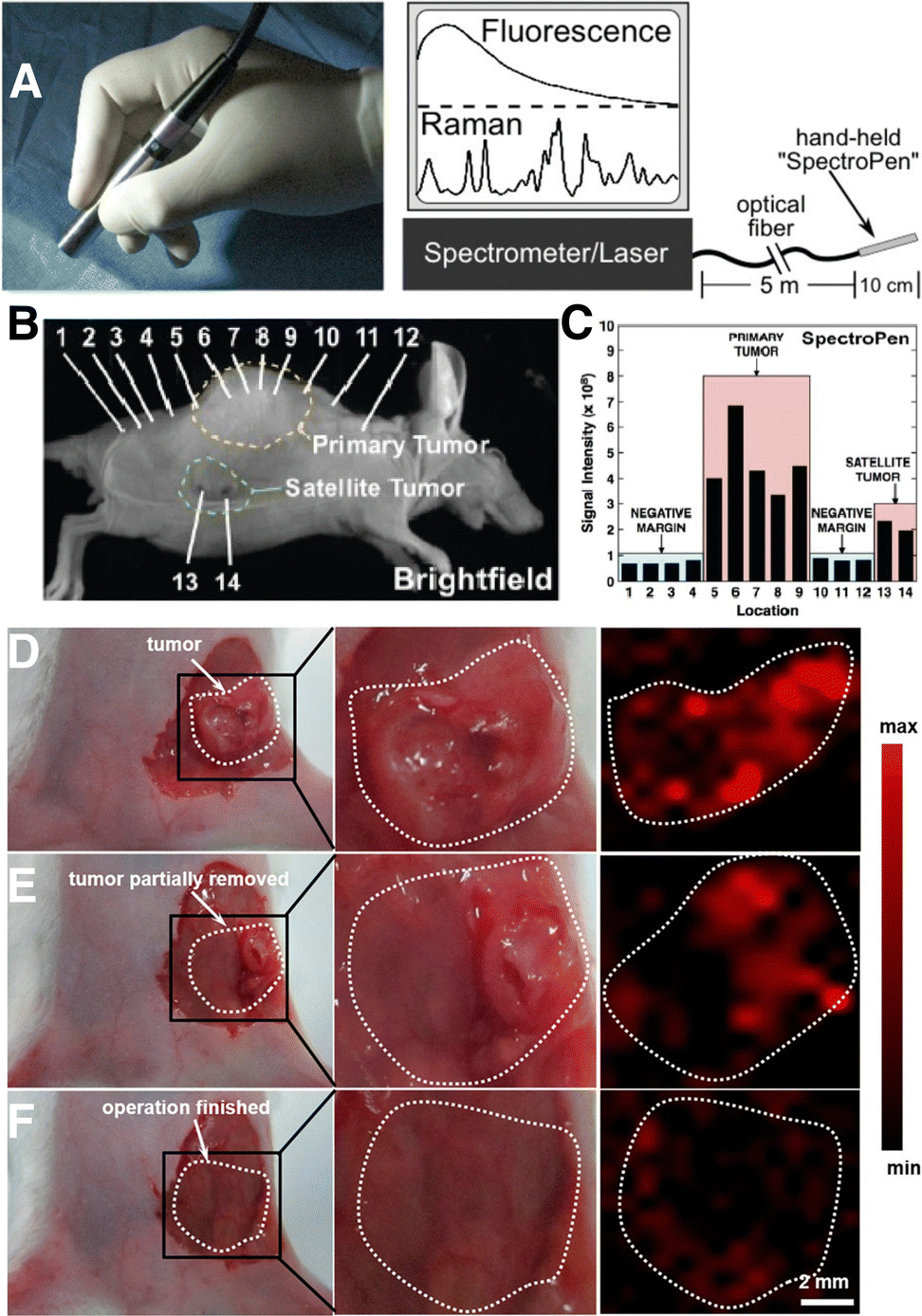 | ||
| Fig. 9 (A) Photograph (left) of a Raman imaging pen (“SpectroPen”) and schematic showing the measurement configuration. (B) Brightfield image of a mouse xenograft model with a 4T1 breast cancer tumor. (C) Results for mapping of primary and satellite tumor boundaries with the SpectroPen setup. [Adapted with permission from ref. 152 Copyright (2023) American Chemical Society] (D)–(F) digital photographs (left and center) and corresponding SERS images (right) of a 4T1 tumor-bearing mouse (D) prior to resection, (E) following partial removal of the tumor, and (F) after complete resection of the primary tumor. The bright regions in panel F correspond to residual microtumors. [Adapted with permission from ref. 156 Copyright (2021) American Chemical Society]. | ||
More recently, tumor margin identification has been demonstrated for breast,153 ovarian,151,154 and brain155 cancer xenograft models. The high imaging resolution provided by SERS is especially useful for identifying micrometastases or microtumors, which are microscopic collections of cancer cells that are often missed by standard imaging tests.153 In the work of Wen and colleagues, SERS tags based on AuNSts were coated with the RaR 4-NTP, encapsulated in silica, functionalized with PEG-silane, and intravenously delivered into mice bearing 4T1 breast cancer tumors.156 SERS imaging specifically indicated the tumor boundaries pre- and intra-operatively (Fig. 9D and E). Following resection of the primary tumor, SERS imaging of the surgical bed led to identification of multiple residual tumor foci and satellite microtumors (Fig. 9F). This work showed that SERS can be useful for ensuring complete removal of cancer cells, following image-guided resection.
Whereas tags built from NPs with LSPR in the first NIR biological window (∼700–950 nm) are the most common ones, scattering is further diminished at longer wavelengths, and therefore recent works have explored the second biological window (NIR-II) at 1000–1700 nm for further extending the limits of resolution.154,157,158 SERS tags targeting the NIR-II often include plasmonic cores comprising gold stars, cages, or nanorods with increased anisotropy (aspect rato) or hollow structures.158 In one example, Li and co-workers prepared porous AgAu nanocubes with LSPR in the NIR-II, capped with the dye IR 1061 as RaR and a polyethyleneimine/hyaluronic acid stabilizing shell. These tags were incubated in multicellular 4T1 breast cancer tumor spheroids for 12 h. Successful imaging of the spheroids following subcutaneous injection in animal models provided proof-of-concept demonstration that NIR-II tags could be used for imaging microtumors. In the same work, tags were also injected into the tail vein of solid tumor-bearing mice, which demonstrated nonspecific accumulation at the tumoral site due to the EPR effect. Overall, in vivo imaging requires both high resolution and high throughput, with multimodal imaging offering an opportunity to improve both aspects, which we discuss in the following section.
4.3. Multimodal bioimaging
A wide variety of bioimaging modalities have been reported and implemented at different extent, each of them showing certain advantages and drawbacks. Therefore, numerous attempts have also been directed to the combination of technologies, which is usually known as multimodal bioimaging. Same as other modalities, SERS can also be combined with other techniques, in a way that advantage can be taken of mutual benefits for imaging biological models with high fidelity, resolution, and overall greater reliability and speed.159 Multimodality can be achieved e.g., by integrating plasmonic tags with magnetic components, for combined SERS, CT and MRI.160 The combination of plasmonic and magnetic NPs can also be useful for therapeutic purposes, and gold coated-iron oxide NPs have demonstrated excellent performance in multimodal imaging, hyperthermia treatments, and drug delivery.161 SERS can also be readily combined with photoacoustic imaging, which has been shown for anisotropic NPs like AuNRs and AuNSts.151 Yet another possibility is the correlation of SERS imaging with nanothermometry, which has been applied to monitor temperature at the nanoscale and to manage photothermal heating effects during SERS measurements.162,163 As an example, rare earth-doped calcium fluoride NPs have been combined with AuNSts and AuNRs to produce heater/thermometer nanotags that could be excited at the same wavelength within the biological transparency window, to heat and measure temperature within 3D tumor models.148,164Overall, arguably the most common multimodal imaging combination for 3D cell models is dual SERS-fluorescence imaging, whereby NPs are functionalized with both RaRs and fluorescent tags, to create dual-mode tags. Although we mainly focus on SERS-fluorescence multimodal imaging in this section, we direct the reader to other in-depth reviews on multimodal imaging, for extended information on other methods.159,165
In one example of SERS-fluorescence multimodal imaging, AuNRs coated with mesoporous silica shells were labelled with the RaR DTNB and the outer silica shell was doped with fluorescent Rhodamine B isothiocyanate (Fig. 10).166 These dual tags were additionally conjugated with folic acid as a target ligand for HeLa cells. By tuning the wavelength of the excitation source, fluorescence (543 nm) and SERS signals (633 nm) could be generated separately. This type of dual nanotags has been employed to label MDA-MB-231 triple-negative breast cancer cells. In other examples, AuNPs were labelled with Malachite green isothiocyanate (MGITC) and tris(2,2′-bipyridyl)ruthenium(II) chloride hexahydrate (Ru(bpy)3) as RaRs, protected with an inner silica shell, and MGITC-modified fluorophores incorporated prior to an additional silica coating step. The intermediate coating with silica was intended to prevent the leaching of RaRs, as well as fluorescence quenching from the metal particle cores.167 SERS tags can also be used in combination with fluorescently labelled polymer beads. For instance, AuNSts adsorbed on fluorescent polystyrene beads can be tuned to improve the SERS signal, which has been used for multimodal imaging of Human lung epithelial cancer cells (adenocarcinomic human alveolar basal epithelial cells A549), MCF7 breast cancer cells, and murine J774 macrophages.168
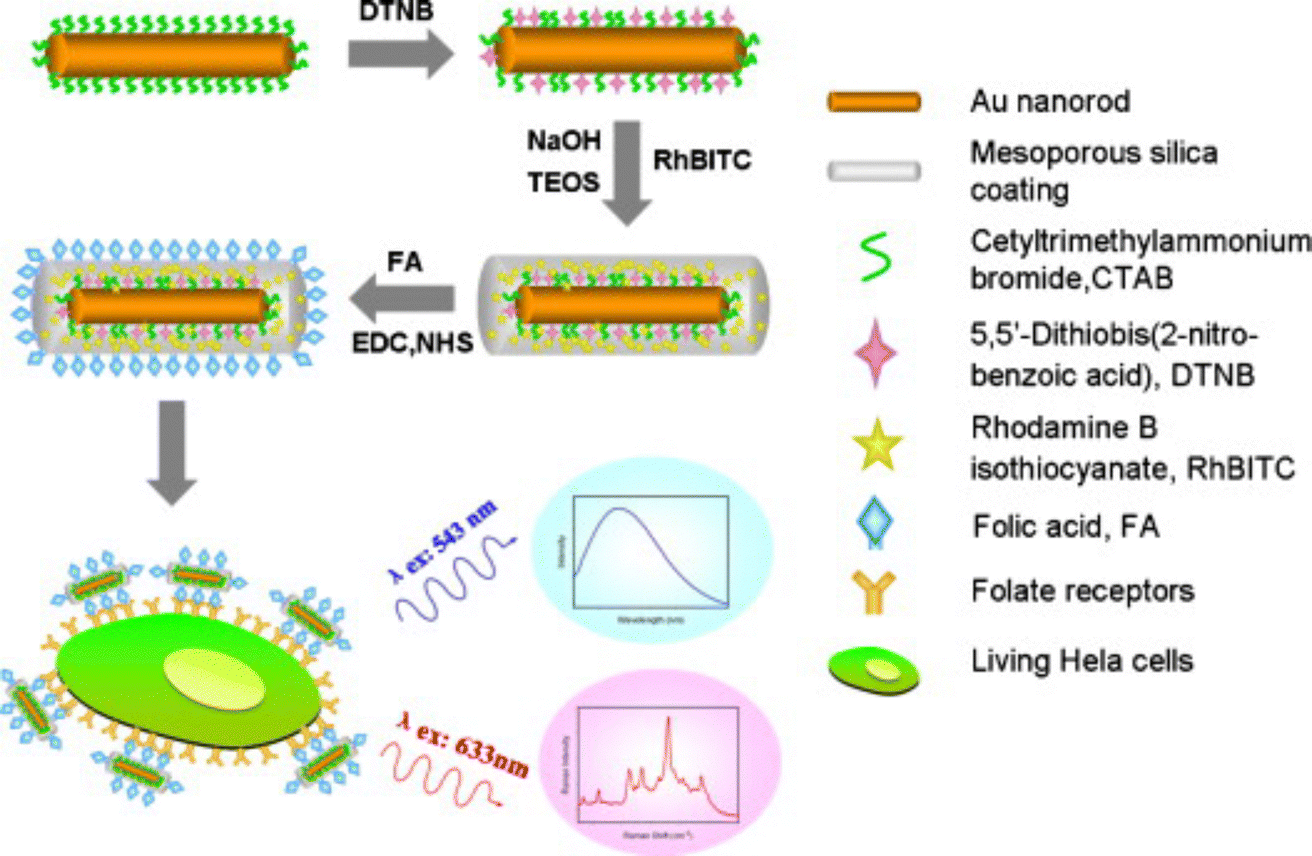 | ||
| Fig. 10 Schematic view of the fabrication of SERS-fluorescence nanotags for targeting cancer cells, using mesoporous silica-coated AuNRs. Three main steps were followed for the fabrication of the tag. First, the surface of AuNRs is labeled with DTNB as RaR. Then, an outer shell of mesoporous silica doped with Rhodamine B isothiocyanate is coated on the labelled AuNRs for fluorescence imaging. Finally, folic acid is conjugated on the surface of the particles as targeting ligand. [Adapted from ref. 166 with permission from Elsevier]. | ||
The concept of multimodal imaging can be similarly applied to the visualization of scaffolds, as well as 3D models. Polymers, for example, can incorporate either dyes or SERS-labelled NPs, for combined use of both imaging techniques on a single material. Strozyk et al. proposed the use of electrohydrodynamic co-jetting to prepare poly(lactic-co-glycolic acid) (PLGA) compartmentalized fibers, with dual fluorescence and SERS activity.169 One of the polymer inks contained nanostars labelled with 4-BPT and a green dye, whereas the other was labelled with nanostars carrying 2-NAT and a blue dye. The absorption and emission profiles of the fluorescent dyes were selected to be far away from the excitation wavelength of the plasmonic cores in the SERS tags. 3D SERS and confocal fluorescence imaging were used to study the distribution of all labels inside the matrix, clearly demonstrating the compartmentalization of the fibers. In another work by the same authors, human dermal fibroblasts (HDF) were labelled with AuNSts and AuNRs functionalized with 1-NAT, 2-NAT, and 4-MBT, to prove the multiplexing ability of the technique toward imaging cell populations grown inside a scaffold (Fig. 11A and B).170 When comparing the stability of the SERS tags with respect to the embedded dyes, under UV irradiation, photobleaching of the dyes was clearly observed whereas the SERS tags were not altered (Fig. 11C and D). The SERS signal intensity remained stable up to 4 days of incubation in vitro, but was reduced after 8 days. When incorporated into the scaffolds, it was possible to image SERS-labelled HDFs up to 25 days in vitro, with a homogeneous distribution, also in the z-direction (100 μm). It is worth noting that complex data analysis, such as multiple linear regression analysis or true component analysis, are required for data postprocessing. The authors demonstrated that SERS imaging offers advantages for repetitive sample measurements with stable signals, an important aspect when monitoring cell models over prolonged periods.
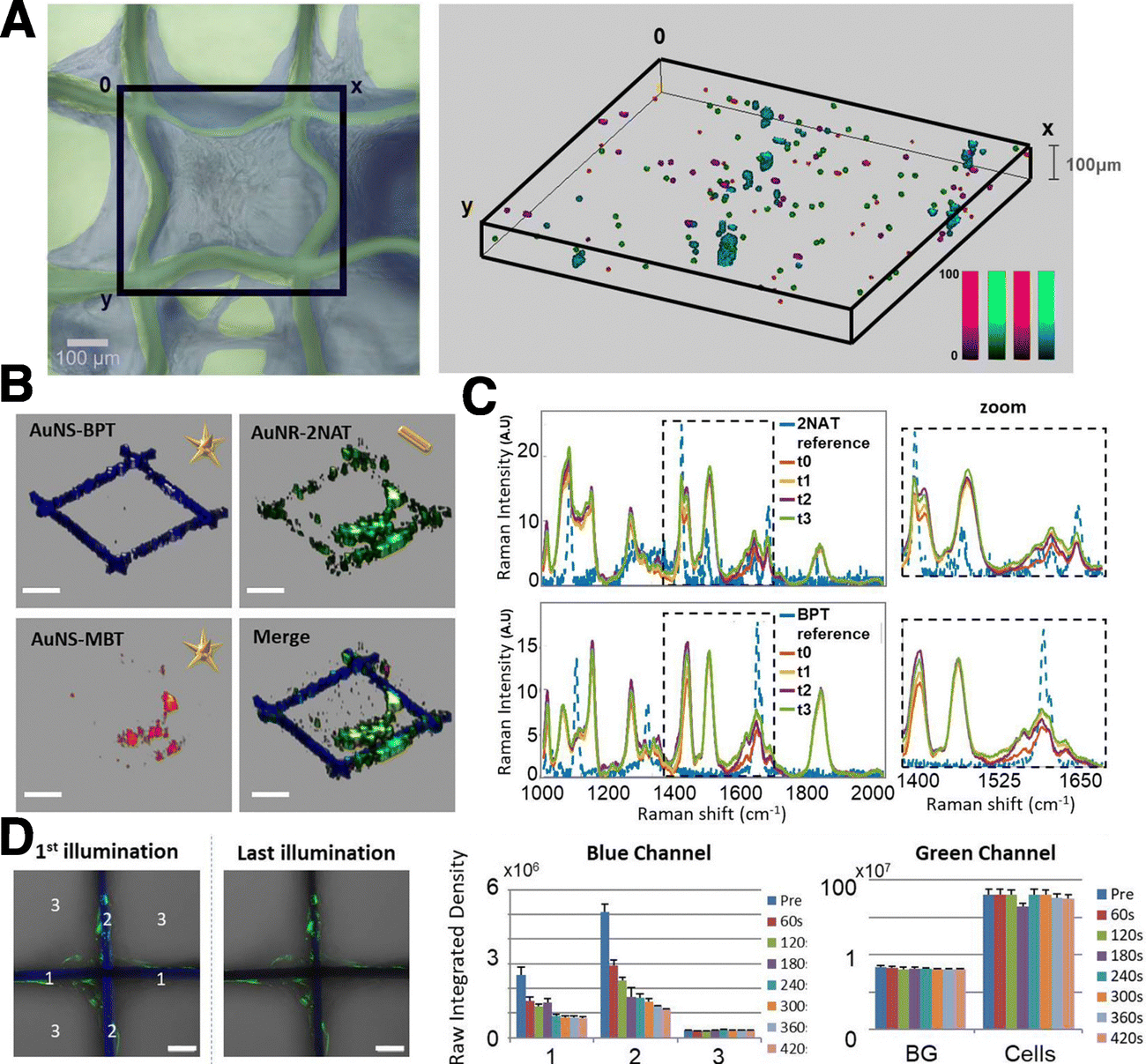 | ||
| Fig. 11 Comparison of SERS and fluorescence stability in multimodal scaffolds. (A) 3D SERS mapping of HDF cells after 25 DIV as viewed in bright field microscopy (left) and 3D SERS imaging (right). AuNSts labelled with 1-NAT (magenta), 2-NAT (green) and 4-MBT (red). Cells containing the three tags are shown in cyan. (B) Individual and merged 3D reconstructions of four different layers from different z-stack measurements. AuNSts labelled with 4-bisphenylthiol (4-BPT) (scaffold) appear in blue, AuNRs labelled with 2-NAT (HDF) are shown in green, and AuNSt labelled with 4-MBT (HDF) are shown in red (scale bars = 200 μm). (C) Photobleaching test using cell-internalized AuNRs labelled with 2-NAT and AuNSts labelled with BPT inside PLGA scaffolds. The area was repeatedly illuminated 4× for 1 h, with a 785 nm laser. For fluorescence imaging, a different area was irradiated 7× for 60 s, using a 405 nm laser. Images show the first and last illumination SERS maps, as well as the corresponding average spectra. Scale bars: 50 μm (D) fluorescence imaging bleaching tests. Left images show the blue and green channels overlaid with the corresponding optical images for the first and last illuminations. Scale bars: 50 μm. The bar graphs show the evolution of the fluorescence intensity for both fluorophores. [Adapted with permission from ref. 170 Copyright (2022) American Chemical Society]. | ||
SERS and fluorescence multimodal imaging also offers the possibility to perform in vivo deep tissue imaging. Among various options, NIR fluorescence-SERS tags are ideal candidates due to their high contrast and deeper detection ability, related to lower absorption and scattering of NIR light by biological tissue. The combination of both imaging techniques is particularly useful because fluorescence imaging can provide fast and wide-area detection to identify the target spot, whereas SERS offers high specificity and contrast at the nanotag location. For example, the distribution and excretion of intravenously injected AuNRs in deep tissues of live mice has been reported to assess their toxicity and potential application.171 Nanorods were coated with thiolated PEG and diethylthiatricarbocyanine, both as fluorescent dye and RaR. The LSPR of AuNRs was tuned to avoid overlap with the fluorescence emission band of the RaR-dye, thereby preventing quenching. When the rods were subcutaneously injected to a mouse, real-time imaging was carried out by monitoring fluorescence, and SERS spectra were then recorded from the fluorescent site. When intravenously administered, AuNRs accumulated mostly in the liver of the mouse and partly in the tail. The intensity of the fluorescence signal was weaker than the one detected subcutaneously due to the location of the liver in the mouse body. However, the SERS spectra still had a high contrast in deep tissues and could be differentiated from the background. Sentinel lymph node mapping, as well as in vivo tumor targeting, were also explored following the same strategy, with bright fluorescent and SERS signals being obtained after 2 min and 24 h, respectively. Histological analysis revealed that a high amount of intravenously injected NPs did not induce any tissue/organ/nerve toxicity to mice. Along with multimodal imaging, these types of tags can be also employed for photodynamic therapy (PDT). As an example, silica-coated AuNRs were doped with protoporphyrin IX as a photosensitizer, to have them accumulate in the tumor site and ultimately combine multimodal imaging with PDT.172 In short, SERS can be complemented with multiple techniques to investigate biological settings, fluorescence having been the most exploited modality so far.
4.4. “Deep” SERS bioimaging techniques
Although we have seen significant developments based on traditional SERS setups, conventional measurement configurations are still limited by the penetration depth of light, which is only tens of mm at its best, even in the biological transparency window. For standard SERS measurements, optical clearing agents can be used to increase light penetration by reducing refractive index changes throughout the tissue (i.e., via dehydration).173 However, these techniques can be destructive to the sample, and are therefore primarily used for ex vivo analyses. Surface-enhanced resonance Raman spectroscopy (SERRS), a method pioneered by Stacy and Van Duyne for the analysis of tissues, is capable of accessing signal intensities that are orders of magnitude higher than those for standard SERS.174 In SERRS, a laser line with a frequency closely matching an electronic excitation in the RaR is used to amplify the Raman scattering signal (Fig. 12A). The RaRs used for SERRS are usually dyes, selected to absorb light at wavelengths matching standard continuous-wave laser lines (532, 633, 785, 808 nm, etc.).174 A number of studies have validated SERRS for 3D bioimaging,175,176 as well as for sensing pH, oligonucleotides, thrombin, and other biologically relevant molecules.177–180 Although SERRS gives comparable sensitivities to fluorescence-based approaches,181 it can also provide information regarding the local microenvironment, while avoiding challenges related to photobleaching and the need for specific biological labels. Despite these benefits, SERRS tags must contain a chromophore at the illumination wavelength (preferably in the NIR) and provide a configurational response to the desired analytes or environmental changes, which is challenging.182 This barrier may inhibit in vivo applications of SERRS. For instance, pH-sensitive SERRS tags have been reported to exhibit narrower sensitivity ranges (∼2–4 pH units), compared to standard RaRs (reaching ∼6 or more pH units, required for pH sensing in organelles characterized by pH ∼4–8).183–186 Surface-enhanced hyper-Raman scattering (SEHRS) in part offers a solution by performing the electronic excitation via a non-linear two-photon illumination process (at tunable wavelength), enabling resonance Raman for a wider breadth of RaRs (Fig. 12B). However, SEHRS requires a pulsed laser, whereas SERRS can be performed with more standard setups.187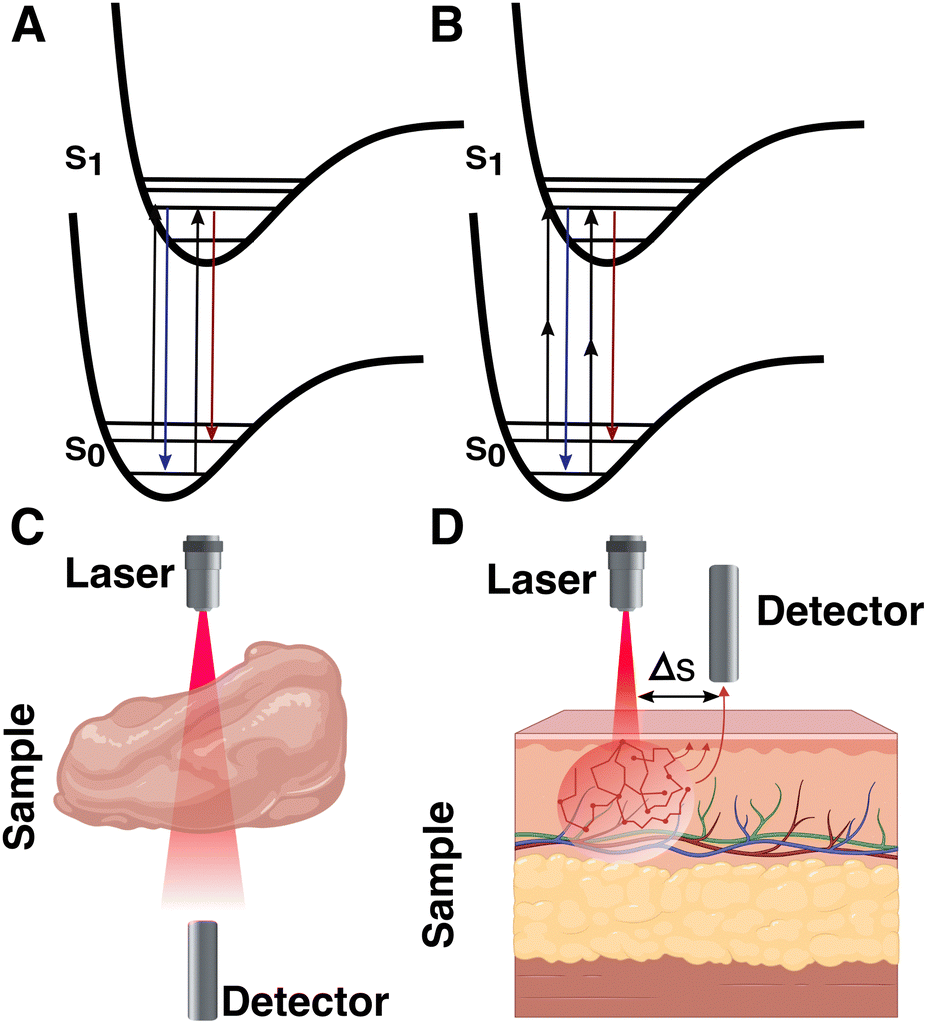 | ||
| Fig. 12 Jablonski diagrams of (A) surface-enhanced resonance Raman spectroscopy (SERRS) and (B) surface-enhanced hyper-Raman scattering (SEHRS). Schematics showing the measurement configuration for (C) transmission Raman spectroscopy (TRS) and (D) basic spatially offset Raman spectroscopy (SORS). | ||
There are many relevant cases where spectroscopic measurements should be made at depths beyond a few mm, e.g., for deep-seated “phantom” and intracranial tumors.40 Such extreme systems generally require alternative measurement setups whereby the configuration or position of the laser and/or detector are modified. Various “deep Raman imaging” techniques have been developed for this purpose, including transmission Raman spectroscopy (TRS), and (surface-enhanced) spatially offset Raman spectroscopy ((SE)SORS). Both TRS and SORS/SESORS improve the signal of scattered photons coming from deeper within a target material, by changing the spatial configuration of the source and detector. For the former, the detector is placed opposite the source. The latter method is more general, with the detector separated from the tag laser by certain distance (Δs) and/or at a different angle (Fig. 12C and D).131,188
Early demonstrations by Matousek's team showed that the spectra corresponding to SERS tags embedded 50 mm deep in porcine muscle tissue could be detected with TRS ex vivo.131,189,190 More recently, Zhang et al. applied TRS for 3D deep-seated tumor sensing in vivo, in nude mice.132 While these works provide substantial and impressive improvement in 3D bioimaging/sensing compared to standard SERS, measurement in transmission requires the probe light to traverse completely through the sample. Therefore, this setup is not compatible with certain sample configurations or materials of widths greater than tens of mm, thereby limiting the in vivo applicability of the technique to (mainly) certain physical configurations in live mouse (or even smaller) models.
Compared to TRS, SESORS offers a more general solution for samples that are incompatible with transmission measurements.191 The implementation of SESORS alone enables imaging in up to ∼6 mm deep tissue192 and ∼3–5 mm of bone, for detecting RaRs193 and biomolecules,194,195 respectively. In one example, Moody et al. showed that SESORS (with 1 mm offset) could be applied to detect melatonin, epinephrine, and serotonin (distinguished using principal component analysis; PCA) in a synthetic brain model, through a 2 mm thick cat skull (Fig. 13A and B).194 However, the limit of detection was only 100 μM, a couple orders of magnitude higher than physiologically relevant concentrations. Later, Vo-Dinh and co-workers demonstrated SESORS detection of glioblastoma (but not 3D bioimaging) through a 5 mm monkey skull, which is comparable to human skulls with average bone thicknesses of 3–14 mm.195 The sensing and imaging depth accessible by SESORS can be further extended through combination with SERRS, which is often referred to as SESORRS. This technique was applied to probing multicellular tumor spheroid breast cancer models, at depths up to 15 mm, and in porcine muscle tissues at 25 mm.196
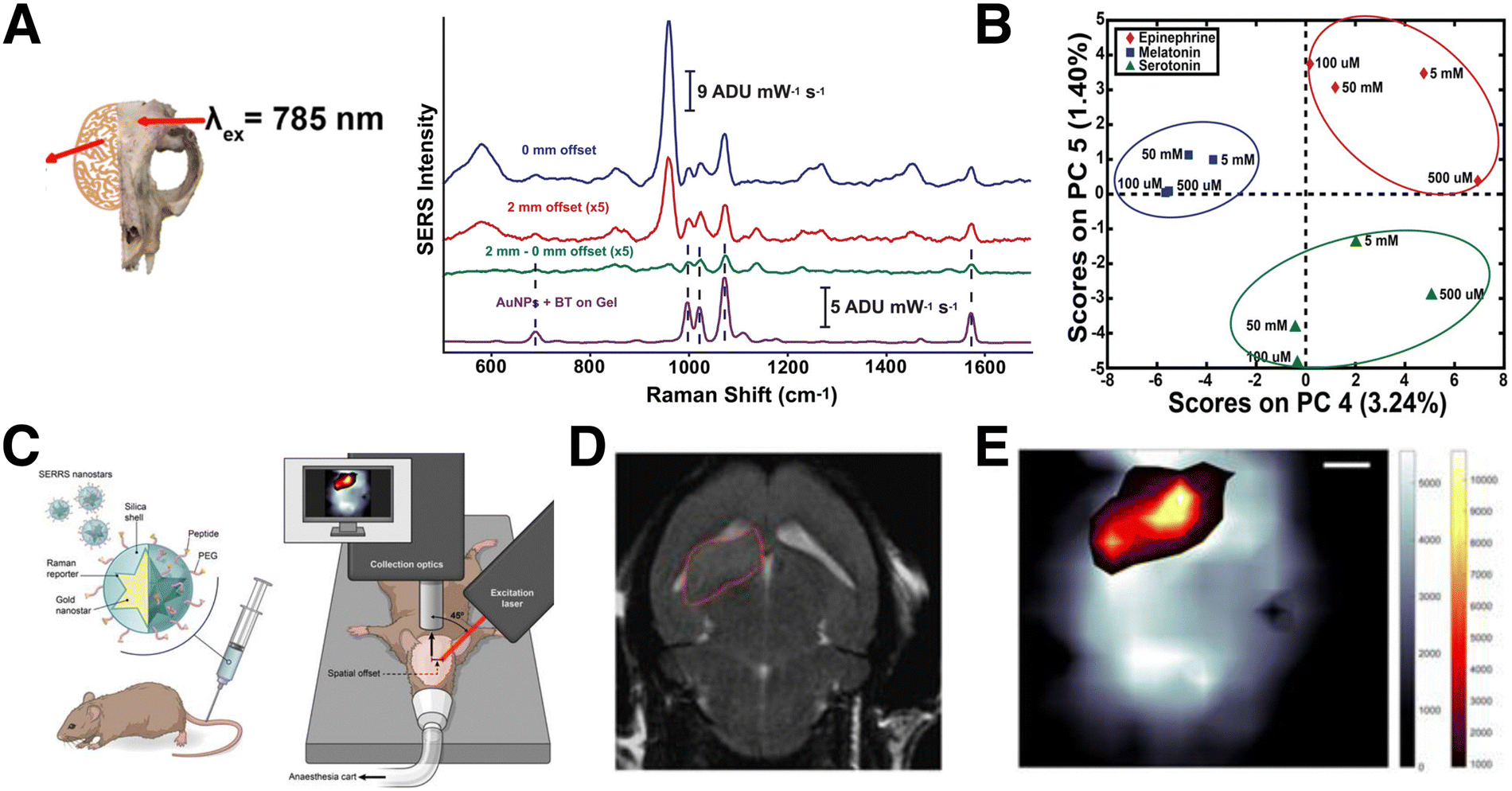 | ||
| Fig. 13 (A) Spectra obtained from a polymer brain model, through a cat skull (inset), without the addition of NPs or serotonin analyte (blue), after the addition of 100 μM serotonin and NP tags with 0 offset (pink), after the addition of 100 μM serotonin and NP tags with 1 mm offset (green), and of 100 μM serotonin drop-cast on NP tags outside the mouse skull model on plain gel (purple). [Adapted with permission from ref. 194 Copyright (2017) American Chemical Society]. (B) Schematic view of a setup for SESORRS live mouse brain imaging. (C) 2D axial T2-weighted MRI measured 4 weeks following injection of fibroblasts in the region outlined in red, and (D) the corresponding SERS map, showing the overlay of the SERS tag signal (orange/yellow) and that corresponding to skull/bone (grayscale). [Adapted from ref. 197 with permissions from Ivyspring International Publisher] | ||
For bioimaging through bone, Nicolson and co-workers carried out a notable study showing that SESORRS facilitates 3D bioimaging of mouse brains in vivo in live mice (Fig. 13C–E).197 The SESORRS maps showed good agreement with MRI imaging of the tumor model (Fig. 13D and E). Measurement depth can be further maximized by combining resonance Raman with other imaging techniques, e.g., in the work of Neuschmelting et al., where image-guided brain tumor resection was achieved by combining SERRS with multispectral optoacoustic tomography (MSOT).176 Overall, these alternative SERS setups continue to push the limits of 3D spectroscopic analysis in tissue sections and in vivo environments. While an intensive account of alternative SERS techniques is beyond the scope of this review, we encourage those readers interested in learning about these methods to other comprehensive reviews on SERRS,182,198 TRS/SESORS,188,199 and SEHRS.187
5. SERS biosensing in 3D
SERS is a powerful technique for interrogating cancer microenvironments, through the monitoring of biomarkers (such as growth factors, nucleotides, receptors, etc.), cell viability, or local pH, among other possibilities, and at different levels of complexity, from serum samples and 3D cell cultures through 3D constructs.200 The evaluation of both the tumor microenvironment and cancer-related cellular markers enables precise characterization of the tumor, which may yield diagnostic information and inform therapeutic strategies, because the overexpression of certain biomarkers is characteristic of disease evolution.201,202 Additionally, SERS can also be used for combined theranostic studies.84,136,139,203–2055.1. Sensing cancer-related biomarkers
Uncontrolled cell proliferation is mediated by many different signaling pathways, giving rise to a wide variety of valuable biomarkers, which can be more accurately studied in a 3D environment. For instance, biomarkers related to communication phenomena between tumor cells and the tumor microenvironment are often lacking in 2D models, but present in 3D.206 Qian et al. studied the production of several biomarkers in 3D cultures by SERS. As an example, the interactions between tumoral and endothelial cells can induce angiogenesis, in turn promoting tumor growth and metastasis.207,208 The production of the angiogenic cytokine vascular endothelial growth factor (VEGF) was monitored through the design of a multifunctional microfluidic platform combining the 3D cell culture unit with a SERS detection unit (Fig. 14). In the first unit, they co-cultured MCF-7 and human umbilical vein endothelial cells in a 3D collagen matrix (Fig. 14A). After a few days, culture medium was guided inside the SERS detection unit and the secretion of VEGF could be determined with a detection limit of 100 pg mL−1 (Fig. 14B). The applied SERS tags were composed of AuNRs functionalized with DTNB and an outer Ag layer for improved SERS enhancement (Au@DTNB@AgNRs), as well as aptamers for the selective detection of VEGF. The use of a SERS microfluidic chip was validated for in situ interrogation of the tumor cell-endothelial cell interaction. Apart from angiogenesis inducers, adenosine also plays an important role in the development of tumors and metastases. García-Astrain et al. described the design of a scaffold for 3D cell culture and adenosine detection (down to 10 μM), using 3D printed AuNR-containing scaffolds (see Section 3.3). Moreover, the scaffolds were shown to be fully biocompatible for HeLa cells.71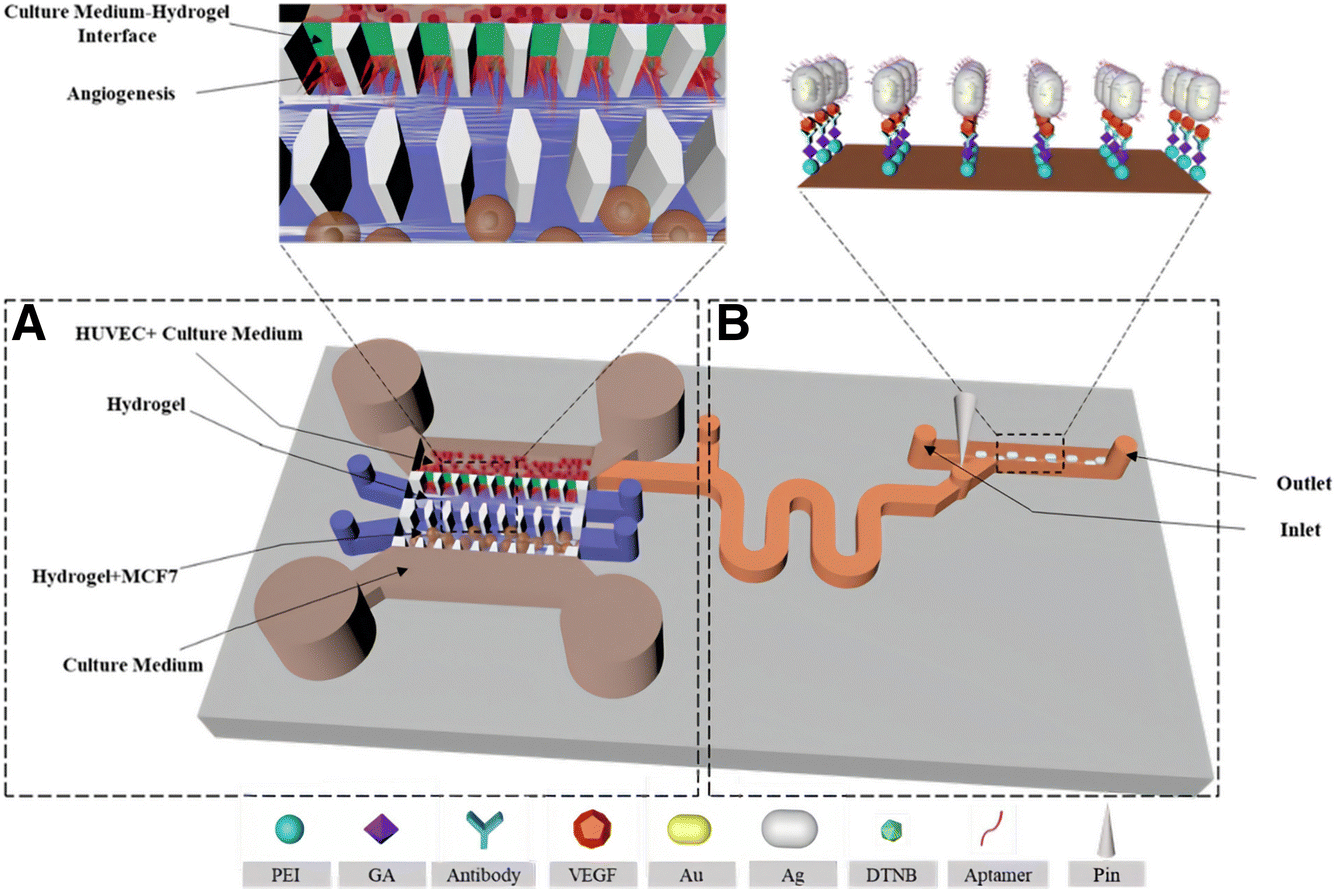 | ||
| Fig. 14 Schematic illustration of a microfluidic chip for tumor cell culture (A) and SERS sensing (B). [Adapted with permission from ref. 208 Copyright (2019) American Chemical Society] | ||
Surface receptors are commonly upregulated in tumoral cells to promote growth and ensure survival.209 In fact, any abnormality in surface receptor regulation allows cells to escape apoptosis, thus playing key roles in many tumoral processes (proliferation, invasiveness, chemoresistance, etc.), and being appealing targets for therapy development and cancer tissue classification.210 The cancer biomarker HER2 is upregulated in most breast cancers. Samanta et al. studied the expression of this receptor by cancer cells in mice bearing xenografts generated from SKBR-3 cells.211 SERS tags were prepared by conjugating AuNPs coated with lipoic acid-containing amine acetylated tricarbocyanines,which performed as RaRs, to two HER2-recognition motifs: anti-HER2 monoclonal antibody and single-chain fragment variable anti-HER2 antibody. The authors injected the NPs (tail-vein injection) into mice and obtained the SERS spectra of the tumor site through the skin, under NIR laser irradiation. The signal from the tricabocyanines on the tag was recorded only at the tumor site and no signal was detected at other anatomical locations, which indicates that SERS can specifically detect HER2 positive tumors in vivo.
Similar to multiplex bioimaging, whereby multiple RaRs are used, multiple biomarkers can be actively targeted via rational design of SERS tags. This approach can be beneficial for increasing diagnostic accuracy in vivo. For example, Dinish et al. reported the detection of three different cancer-related receptors (EGFR, CD44, and TGFβRII) in a subcutaneous MDA-MB-231 breast cancer xenograft mouse model (Fig. 15A).212 The authors used AuNPs bearing three different NIR-active RaRs, cyanine 5 (Cy5), MGITC, and rhodamine 6G (Rh6G), and three different antibodies against EGFR, a cell surface receptor for epidermal growth factor family members, CD44, a cell surface adhesion molecule related to tumor growth and metastasis, and TGF receptor II, which binds anti-proliferative TGFβ and is down-regulated during breast carcinogenesis. The efficacy of active targeting was supported by comparing SERS spectra in control mice that were injected with non-antibody conjugated tags to those injected with antibody-conjugated tags. In the former, the disappearance of the RaR spectra was observed after 1 h due to elimination of the NPs from the models (Fig. 15B). In the latter, the peaks related to the RaRs were still observed after 48 h due to selective binding of the antibodies with their receptors, causing a delay in elimination (observed after 72 h; Fig. 15C).
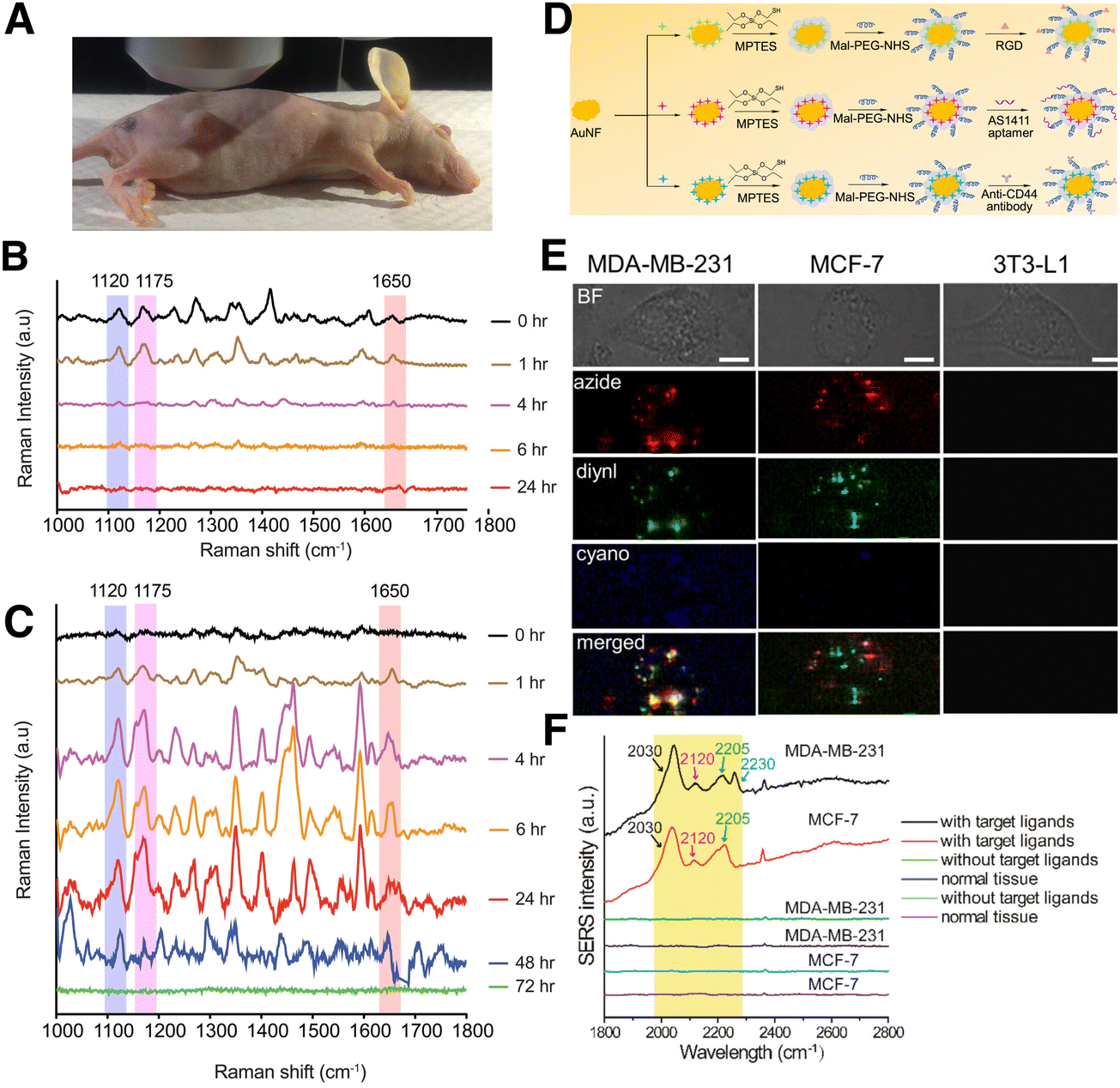 | ||
| Fig. 15 In vivo detection of cancer-related biomarkers in a xenograft tumor: (A) Setup for experiments in tumor bearing mice. (B) Control SERS spectra from tumor site showing the peaks at 1120, 1175, and 1650 cm−1 right after injection of non-antibody bearing SERS tags and quick clearance from the mouse body due to non-specific bonding. (C) Representative SERS spectra from the tumor site, showing the peaks at 1120, 1175, and 1650 cm−1 from the antibody conjugated SERS tags bound to TGFβRII, CD44, and EGFR biomarkers, respectively. Multiplex SERS spectra were recorded up to 48 hours, followed by tag clearance from the mouse body after ∼72 hours. [Adapted from ref. 212 with permissions from Springer Nature]. (D) Scheme describing the fabrication of bioorthogonal SERS tags: gold nanoflowers (AuNF) are functionalized with (3-mercaptopropyl)triethoxysilane (MPTES), which is then coupled to either a targeting peptide (RGD), aptamer (AS1411), or antibody (anti-CD44), via coupling with a maleimide and NHS ester functionalized polyethylene glycol (Mal-PEG-NHS; Mn = 5 kDa). (E) Multiplex SERS image of breast cancer (MDA-MB-231 and MCF7) and healthy cells (3T3-L1) after incubation with the SERS tags. The maps reveal the presence of azide (2120 cm−1, red), vinyl (2205 cm−1, green), and cyano (2230 cm−1, blue) groups, corresponding to the aptamer, RGD peptide, and antibody coated tags, respectively. (F) SERS spectra obtained from MDA-MB-231 and MCF-7 tumor-bearing mice after intravenous delivery of SERS tags, highlighting the intensities of the cyano, azide, and vinyl peaks. [Adapted with permission from ref. 138,139 Copyright (2019) American Chemical Society]. | ||
Beyond antibodies, aptamers serve as low-cost alternatives for multiplex detection that retain the affinity and specificity of antibodies while displaying lower immunogenicity and longer tissue penetration (like small molecules). In recent examples by Tang's group, breast cancer diagnostic capabilities of aptamer-functionalized SERS tags were explored. MCF7 cells are known to overexpress nucleolin (a Bcl-2 mRNA binding protein involved in apoptosis regulation) and Mucin 1 (a transmembrane glycoprotein that plays a role in metastasis). Bioorthogonal nanotags were functionalized by attaching dithiolane and diynl RaRs to AuNRs then adding lipoic acid labelled oligonucleotide aptamers A1411 and MUC1 for targeting nucleolin and Mucin 1, respectively.138,139 Both tag types showed significantly higher uptake in MCF7 cells compared to mouse fibroblasts in vitro. Following tail vein injection in MCF7 tumor-bearing mice, the particles accumulated in the liver, spleen, and tumor, but showed no pathological effects on the main organs, therefore exhibiting good biocompatibility and capacity for specific identification of MCF7 cells. Later, the same group used a combination of Arg-Gly-Asp (RGD) peptide, aptamers (A1411), and anti-CD44 coatings for their binding to breast cancer MCF7 and MDA-MB-231 cells both in vitro and in vivo (Fig. 15D). MDA-MB-231 overexpress CD44 compared to MCF7, therefore, there was comparatively high localization of the tags functionalized with anti-CD44 in MDA-MB-231 2D cell cultures and tumors (Fig. 15E and F). These results ultimately point towards possibilities for aptamer- and antibody-functionalized SERS tags for distinguishing between different cancers.
5.2. Sensing in cancer therapeutics
One of the most appealing applications of 3D cell models comprises testing of antitumoral drugs, which is not only expected to reduce the use of animal models, but also to be more efficient as a high-throughput platform. The incorporation of SERS may serve to monitor spectral changes of cellular metabolites (i.e., cholesterol, trypsin, guanine), as shown by Altunbek and colleagues, to test the chemotherapeutic response of HeLa cell spheroids to doxorubicin and paclitaxel.213 Similarly, Kapara et al. analyzed the drug response of estrogenic receptor alpha (Erα)-positive breast cancer spheroids.119 MCF7 breast cancer cells were targeted with SERS tags coated with 1,2-bis(4-pyridyl) ethylene (BPE) and anti-ERα, and the effect of the drug Fulvestrant on tag uptake was evaluated. Further than just tracking drug efficacy, 3D scaffolds containing AuNRs were employed by Plou et al. to uncover the diffusion profile of SERS-active drugs. More specifically, they studied the diffusion rate of methylene blue (MB, a photosensitizer used in photodynamic therapy of cancer) through a 3D MCF-7 culture,72 and correlated drug diffusion with cell death in a custom cell culture device (Fig. 16A). This study involved the design of a chip where the plasmonic scaffold was printed from a plasmonic bioink of label-free AuNRs and gelatin-alginate, ultimately yielding a suitable spatiotemporal resolution for mapping the diffusion of MB through MCF-7 cell-laden Matrigel (Fig. 16B and C). | ||
| Fig. 16 (A) Setup integrating nanocomposite scaffolds within a tumor cell environment and a reservoir for drug delivery. (B) Confocal 3D reconstruction of MCF-7 cells embedded in Matrigel within the supporting scaffold. (C) MB diffusion patterns along the X-axis in the scaffolds, scaffolds embedded in Matrigel, and Matrigel loaded with cells. [Adapted with permissions from ref. 72 Copyright (2021) American Chemical Society]. | ||
It is also worth mentioning that, when working with complex biological samples, the task of pinpointing distinctive SERS fingerprints for specific biomolecules is challenging, primarily due to the coexistence of numerous components within the same sample. Consequently, establishing a direct correlation between vibrational peaks and the presence of specific metabolites becomes a difficult task. In this context, incorporating techniques such as multivariate analysis and artificial intelligence, machine learning in particular, has become an interesting solution. Such techniques range from fundamental principal component analysis (PCA), which simplifies the dimensionality of SERS spectra to reveal data variations, to more advanced machine learning methods like deep learning, which extract pertinent information and utilize it for data classification.214
5.3. Theranostics
Although the primary focus of this review is on 3D SERS bioimaging and biosensing, we find it valuable to briefly discuss some recent developments in “theranostics”, that is, the simultaneous application of SERS tags for diagnostic and therapeutic purposes, a widely explored area often pursued in tandem with SERS biosensing/imaging. As mentioned in the beginning of Section 3, thermoplasmonic heating may be one of the main challenges when performing SERS measurements on biological materials. Even though it is usually desirable to minimize thermoplasmonic heating, the same effect can be exploited for therapeutic purposes. For example, Zeng et al. explored thermal ablation of breast cancer (MCF7) cells in vitro and in vivo in tumor-bearing mice.215 They used SERS to track the accumulation of the designed tags – Agcore@Aushell nanostars capped with the dye 3.3′-diethylthiatricarbocyanine iodide (RaR) – in the tumor. In this design, the gold shell assisted in shielding the Ag core, thereby improving biocompatibility. With 80% light-to-heat conversion at 808 nm irradiation, photothermal heating under 140 mW irradiation for 10 s reduced cell viability down to ∼5%.Active targeting ligands can also be used for specific binding to upregulated proteins and more efficient delivery of SERS tags to tumor sites in vivo, followed by combined detection, bioimaging, and PTT.138 In one example, Pal and colleagues designed multimodal SERS tags based on AuNRs selectively binding to folate receptor 1 (FOLR1) in breast cancer cells. In vivo and in vitro studies showed preferential uptake by MDA-MB-468 cells due to their overexpression of FOLR1, compared to MCF-7 and MDA-MB-231 cells. Following a mild photothermal treatment at 3 W cm−2 for 5 min, specific reduction in size of MDA-MB-468 tumors was achieved. Photothermal treatment may also require the combined use with other techniques, to ensure full tumor removal. Recently, Wen et al. demonstrated 100% tumor removal with silica-coated AuNSts tags, containing PNTP and a PEG stabilizing coating. Even though the light-to-heat conversion was lower than in the previous examples (44.7% at 808 nm), image-guided resection (see Section 4.3) proved highly effective toward removing the primary tumor and subsequently residual microtumors by thermoplasmonic heating.
Thermonanoplasmonic SERS tags can also carry chemotherapeutics, for combined photothermal therapy (PTT)-chemotherapy. Yin et al. developed silica-coated AuNSts tags, subsequently coated with gold sphere satellite structures, then finally functionalized with RGD and doxorubicin.216 This configuration led to high doxorubicin loading efficiency into HeLa cells, as well as >85% light-to-heat conversion. In vivo SERS measurements showed that nanocarriers functionalized with 4-MBA were efficiently retained at the injection site within tumor-bearing mice, for at least 2 h. Upon triggering localized heating by irradiation with NIR light, doxorubicin was selectively released. Ultimately, thermoplasmonic heating yielded five-fold higher nuclear loading of doxorubicin, compared to the negative control, and tumor size was shown to decrease irreversibly after an irradiation treatment of just 5 min.
5.4. pH sensing
Aside from biomarkers, pH is a key parameter of cell microenvironments because cellular activity is influenced or followed by pH changes. As a general example, intracellular pH (pHi) homeostasis is disrupted when cells undergo apoptosis.217 It is well known that cancer cells exhibit different pHi and extracellular pH (pHe) profiles compared to normal cells, due to the Warburg effect, which describes how cancer cells undergo aerobic glycolysis at higher rates than healthy cells (Fig. 17). Detection of pHi provides useful information regarding early cancer cell development, viability, invasion behaviors, and nanomaterial endocytic pathways, whereas pHe monitoring enables complementary information on viability, invasion, and proliferation, and has practical utility for identifying tumor boundaries (3D SERS image-guided tumor excision).218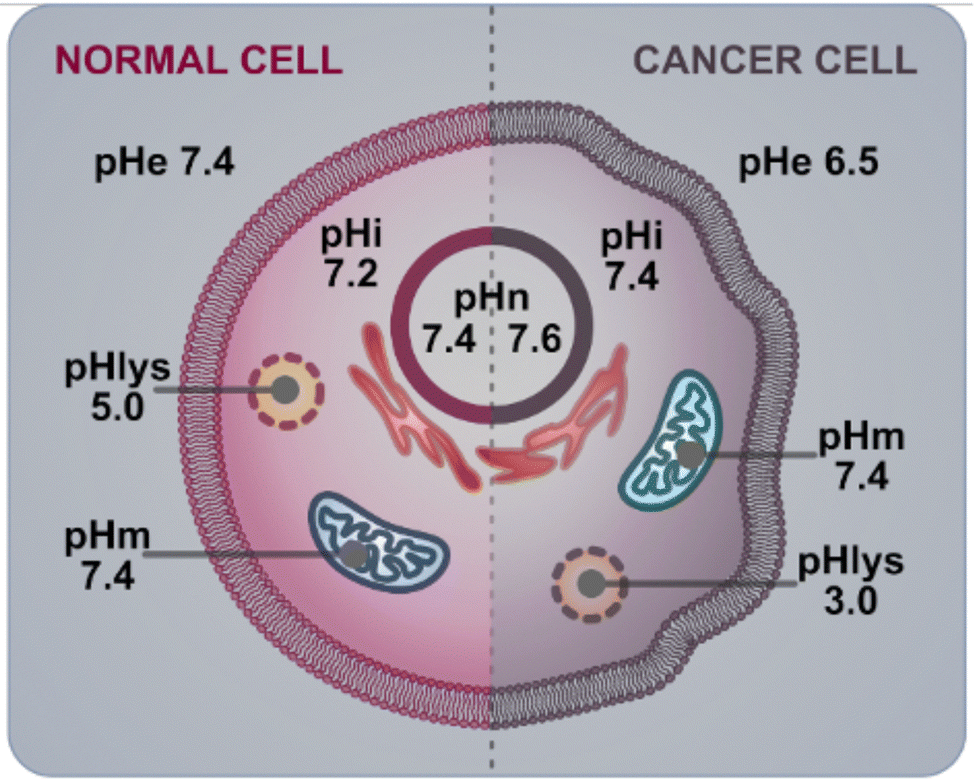 | ||
| Fig. 17 Diagram comparing the pH profile of a healthy and a cancerous cell. | ||
Ideally, a pH sensor should provide biologically appropriate precision and sensing range, achieve high/targeted intracellular delivery (in the case of pHi), and be effective and easy to implement in realistic 3D models. Thus far, various strategies for monitoring pH in 2D and 3D have been developed, including those that incorporate electrochemical measurements/nanoprobes, pH-sensitive fluorophores, metal oxide thin films, conductive polymers, and plasmonic nanomaterials.219–222 As emphasized in the earlier sections, compared to other approaches, SERS pH tags are attractive because they are exceptionally photostable, sensitive with a broad sensing range, minimally invasive, highly multiplexable, and capable of providing additional chemical information about the surrounding environment. As pH sensors, plasmonic NPs coated with pH-responsive ligands are capable of high sensitivity (down to ∼0.1 pH units) and may cover tunable sensing ranges (2–12 pH), depending on the chosen nanomaterial and ligand, for evaluation of either pHi or pHe. Two of the most common pH-responsive RaRs include 4-MPy and 4-MBA. In most cases, pH is determined using a the “ratiometric approach,” whereby the SERS spectra are recorded in buffer solutions with different pH values, then, the change in intensity or integrated peak area of one or more (summed) select pH-sensitive peaks is evaluated. Finally, the ratio between the pH-sensitive peaks and a non-pH sensitive base peak is evaluated, and the values are fit to a calibration curve following the Henderson–Hasselbalch equation. The curve is then used to calculate an unknown pH value from SERS spectra measured from the sample.
The SERS spectra of pH-sensitive molecules, as with other RaRs, depends on their concentration and orientation, which is influenced by their packing density and the particle's composition and crystal structure. For pH sensitive RaRs, the packing density can also influence the effective pKa, which in turn modulates the sensing range and the overall calibration curve for the sensor. In the work by Phan and Haes, Ag@Au@SiO2 NPs functionalized with 4-MBA were prepared with different incubation times.223 These authors monitored SERS peak shifts and changes in intensity while pH was raised from 4.5 to 7.5 in 1-unit increments (Fig. 18A and B). Ratiometric comparison of the intensity of pH sensitive peaks (indicated by black dashed lines Fig. 18A and B) to the base peak (the C–S stretching, νCS at 1080 cm−1) showed that longer incubation times (18 h) gave a ∼1 unit higher estimated pKa value (pKa = 7.7) compared to shorter incubation times (pKa = 6.9 at 30 min) (Fig. 18C). This difference was attributed to the increased ligand density and π–π stacking interactions. Increased π–π stacking resulted in a decrease of electron density in the rings and an increase of electron density at the carboxylic acid group, thereby increasing the bond strength of the COO–H group and reducing the acidity (Fig. 18D).223 Although this study was carried out using a 2D plasmonic substrate, the result is ultimately important for the accurate and effective implementation of pH sensors in 3D systems, and is important to consider when measuring pHi or pHe, which will be discussed using concrete examples in in the following sections.
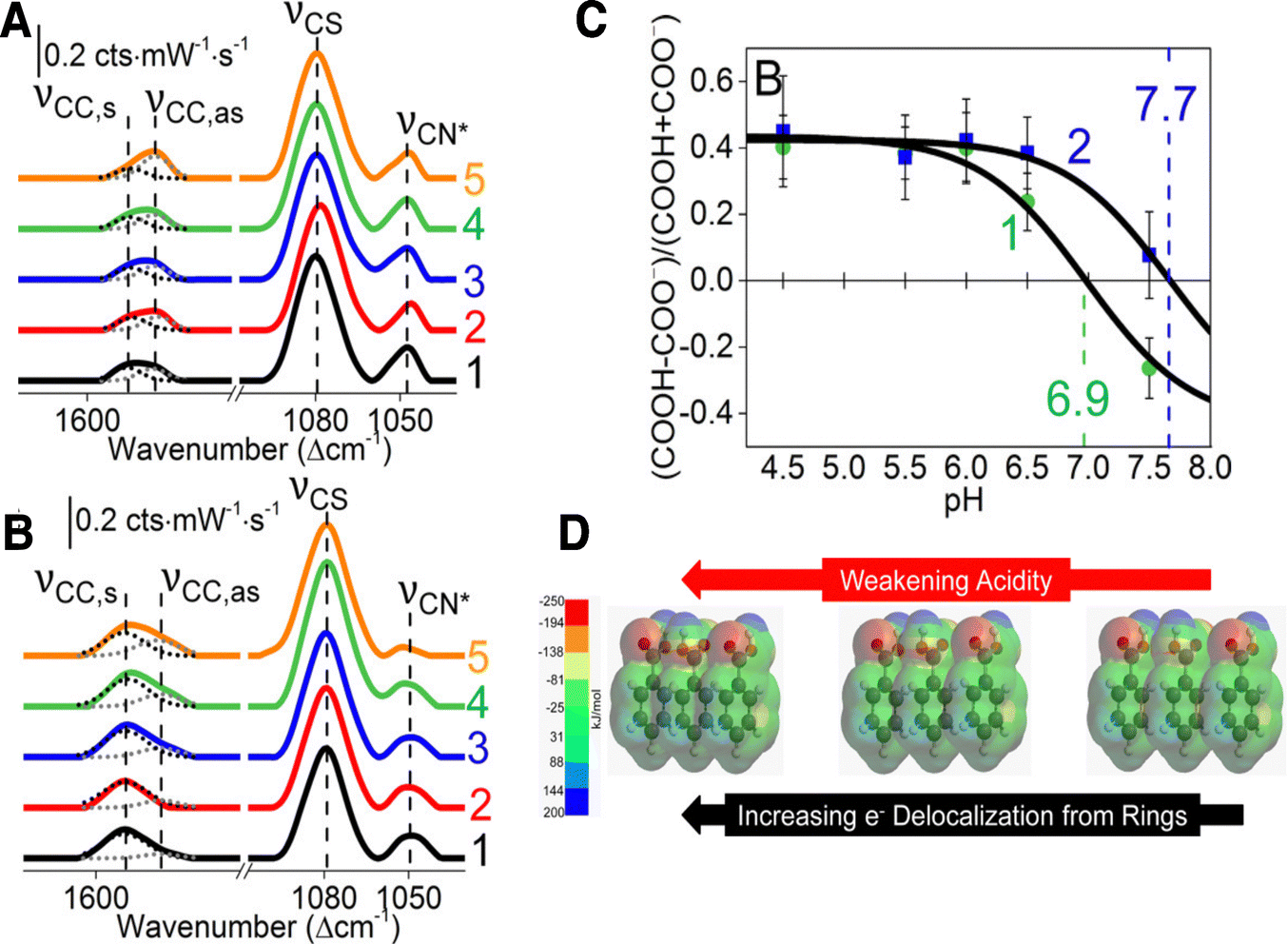 | ||
| Fig. 18 (A) and (B): SERS spectra of Ag@Au@SiO2 incubated with 4-MBA for (A) 30 min and (B) 18 h at different pH values: (1) 4.5, (2) 5.5, (3) 6.0, (4) 6.5, and (5) 7.5. (C) Comparison of ratiometrically prepared calibration curves for tags incubated for 30 min (green text, pKa 6.9) and 18 h (blue text, pKa 7.7). (D) Energy density simulations of 4-MBA ligands with increasing packing density (from left to right), showing that higher density is associated with stronger acidity. [Adapted with permission from ref. 223 Copyright (2018) American Chemical Society]. | ||
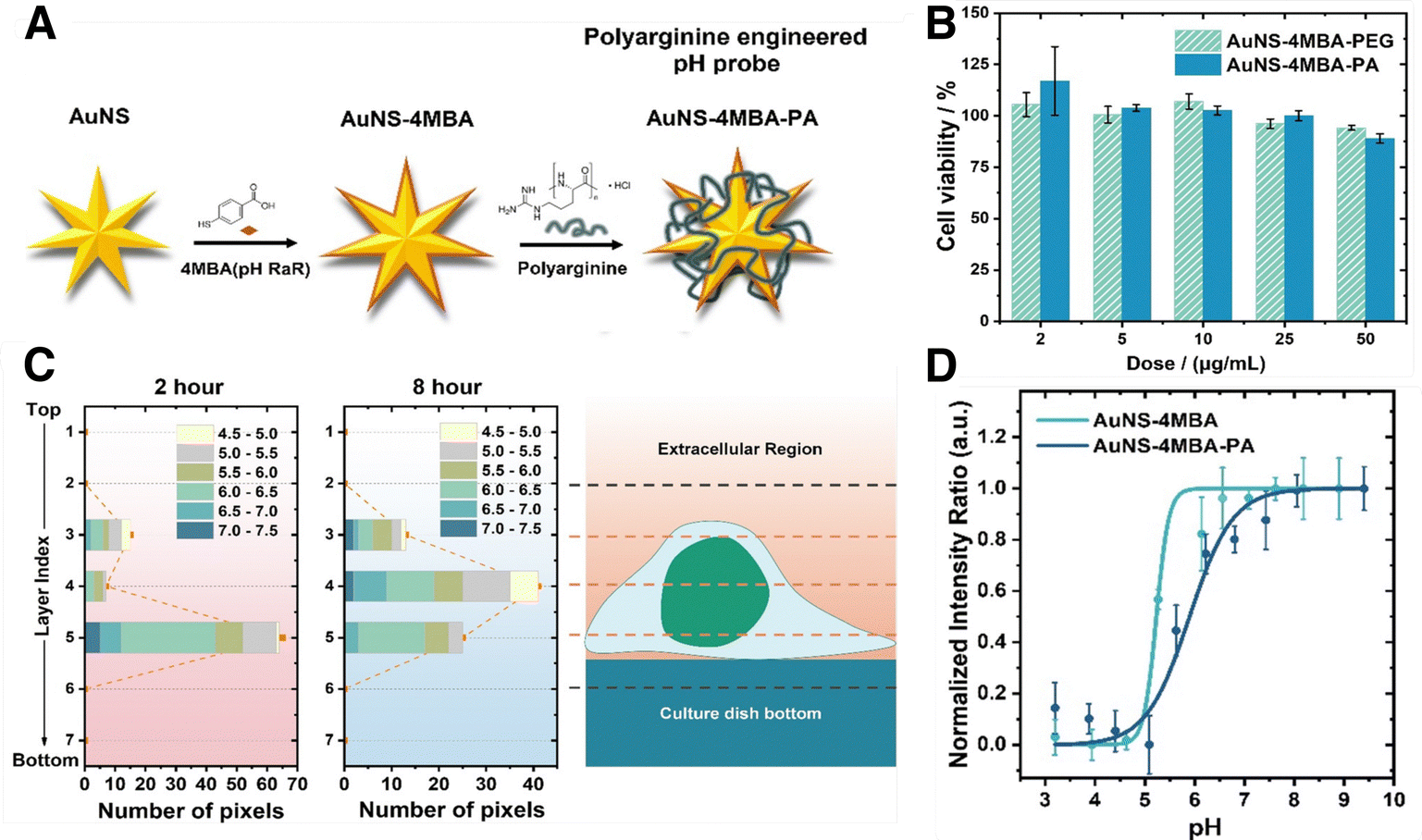 | ||
| Fig. 19 (A) Scheme illustrating the fabrication of pH SERS tags from AuNSts capped with pH-sensitive 4-MBA and PA. (B) Uptake of tags capped with PEG and positively charged poly-L-arginine by MCF7 breast cancer cells, evaluated with inductively coupled plasma mass spectrometry. (C) Analysis of particle distribution and sensed pH, in an individual cancer cell over time. (D) Comparison of pH sensing calibration curve from tags with and without PA stabilizing coating. [Adapted with permission from ref. 183 Copyright (2020) American Chemical Society]. | ||
Compared to 4-MBA, the other common pH sensitive RaR, 4-MPy, is primarily in its deprotonated neutral form at pH higher than ∼3, and therefore lacks sufficient Coulombic repulsion to remain stable in aqueous suspension or biological media. Zheng and co-workers demonstrated that the colloidal stability of 4-MPy-coated gold nanospheres could be improved by conjugation to different biomolecules.227 In their work, it was shown that subsequent functionalization with the protein bovine serum albumin (BSA) could stabilize the structures within a range of pH 5.5 to 9.0, compared to non-BSA functionalized tags, which exhibited aggregation. These AuNP@4-MBA@BSA tags were shown to maintain colloidal stability, even in high ionic strength solutions. Similar to the results of the 4-MBA study, the coated tags exhibited more reliable and reproducible pH sensing, as well as high cell uptake (CaSki; human cervical epidermoid carcinoma), compared to non-functionalized tags. In a later work from the same group, subsequent functionalization with a cell penetrating peptide (transactivator of transcription, TAT) led to further enhanced cell uptake after 2 h incubation, as evaluated by comparing the SERS intensities of the 1093 cm−1 characteristic peak of 4-MPy.140 Notably, the cells exhibited a uniform distribution of the tags throughout the cytoplasm, whereas without the TAT peptide coating the tags have difficulty escaping lysosomes, ultimately limiting detailed pH studies in the subcellular regime. Other important considerations for pHi measurements include the potential errors that may arise due to interference from the background, as demonstrated by Capocefalo and colleagues.228 Their study showed that pH estimation could be inaccurate by several tenths of a pH unit depending on overlap of cell-related signals and the pH-sensitive peaks. Thus, high-accuracy measurements from the ratiometric approach should consider the background SERS spectra or apply environments that exactly recapitulate the conditions of the sample. For pHi sensing, where there are unavoidable and non-uniform background signals from proteins and lipids, implementing either of these two approaches would be time-consuming and challenging. In addition, beyond absolute and integrated peak intensities, some works also suggest that monitoring peak shifts can further enhance the pH-sensitivity.229
So far, pHe sensing in 3D configurations has been demonstrated in microencapsulated cells, organoids, and tissues. In the work of Zhou, pHe sensing was performed for cells contained in semipermeable alginate membranes (Fig. 20A and B).225 One challenge for the real-world implementation of encapsulated cell therapies is the death of cells within such membranes. SERS sensing could be used to map acidification at the capsule periphery (Fig. 20C and D). In Fig. 20E, it can be observed that capsules containing dead cells had a significantly lower pH than those with live ones, which was supported by metabolic characterization (Fig. 20F). Skinner et al. developed pHe tags with high SERS signals via the assembly of dense gold nanosphere coatings on polystyrene microspheres, further functionalized with 4-MBA.232 These tags were applied to test the accumulation of metabolic acids in patient-derived airway organoid models, relevant for studies of cystic fibrosis. Notably, pH differences as low as 0.1 units could be identified, demonstrating that the 3D configuration contributes to the production of acidic metabolites that, in turn, provide appropriate conditioning of the microenvironment. Overall, the implementation of this design for other types of organoids could have far reaching impact toward the development of disease models incorporating SERS sensors. In a recent study by Choudhury and colleagues on ex vivo ovine lung models, accuracies of ±0.07 pH units could be achieved using an endoscopic optrode functionalized with gold nanoshells@4-MBA for alveolar pH sensing. Despite the advances in sensitivity of pH-responsive SERS nanotags, there are still very few examples of pHe sensors for ex vivo and in vivo applications.
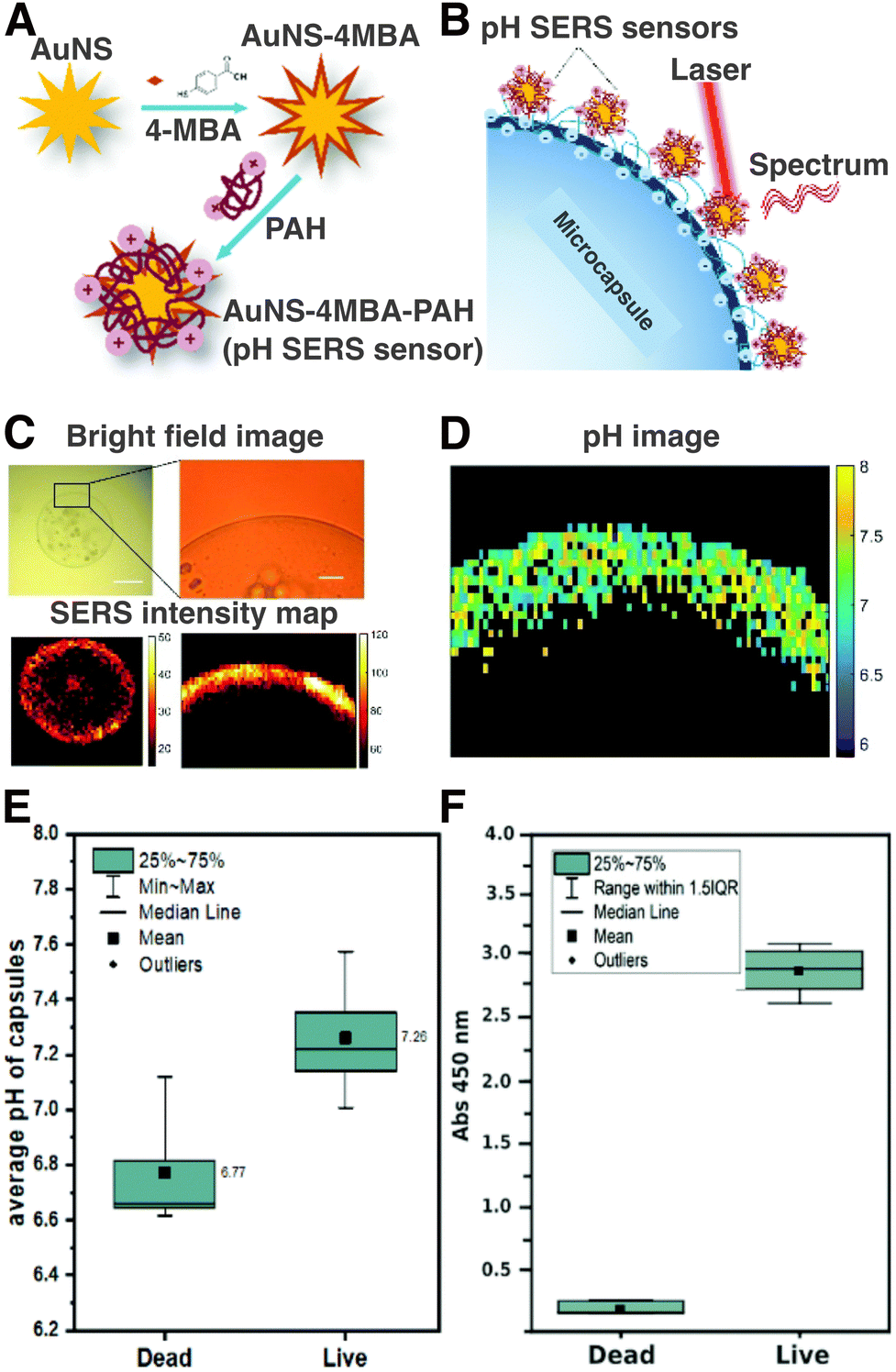 | ||
| Fig. 20 (A) Schematic fabrication process of pH-sensitive SERS tags capped with pH-responsive 4-MBA and stabilizing PAH. (B) Configuration of pH sensors on alginate microcapsules containing Murine C2C12 myoblasts. (C) Brightfield image (top) of microencapsulated cells and the corresponding SERS map (bottom). (D) Map of pH distribution at the microcapsules derived from SERS mapping. (E) and (F): graphs comparing the (E) pH and (F) metabolic activity of dead and live microencapsulated cells 24 h after labelling. [Adapted from ref. 225 with permission from the Royal Society of Chemistry]. | ||
Finally, similar limitations to the ratiometric approach also apply for pHe sensing, creating opportunities for the exploitation of more advanced data analysis methods like PCA and machine learning. While PCA alone does not solve the issue of variable contributions from the background, Williams et al. showed that pH changes can be captured with higher precision using PCA, with ∼70% lower error for each pH value compared to the ratiometric approach.233 For machine learning, a key study by Vikesland's group validated several multivariate regression models for pH prediction with 4-MPy-functionalized stacked gold nanodisk arrays, demonstrating successful pH determination across five different media (not utilized for model training).234 Overall, while machine learning analysis has progressed significantly for SERS sensing of biomolecules,235,236 the implementation of such techniques for both pHe and pHi in biological systems is still in its early stages, and can be a key element paving the way for more successful demonstrations of SERS pH sensing in complex 3D environments.
5.5. Practical considerations for SERS clinical application
SERS holds great promise for various clinical applications. However, practical implementation of this technology in clinical settings presents its own set of challenges. On one hand, for routine diagnostic implementation of SERS in vivo, it is imperative to navigate through various regulatory frameworks and conduct comprehensive safety evaluations regarding nanomaterials administration. Such considerations depend on national regulations and, therefore, require a region-specific approach. The challenges are many, for example: nanomaterials/nanomedicines lack a unified classification criterion; analytical procedures differ from one nanomaterial to another; current pre-clinical biodistribution and toxicological studies fail to mimic the real complexity of human body; etc.237 There are few examples of clinical implementation of nanomaterials, mainly encompassing polymeric micelles, liposomes, and lipid nanoparticles, whereas few examples of metallic nanomaterials have made it to the market and the clinic.238 In the case of AuNPs, several systems have reached various stages of clinical trials, but none has been clinically approved to date.239 Ultimately, despite these challenges, some strategies may be pursued to achieve clinical approval. For instance, the use of simple nanotag compositions (few components) can speed-up the approval process, and also benefit production scalability. Targeting high risk/mortal diseases with a need for novel therapeutics, i.e., cancer, should also increase the probability of clinical approval. Overall, it is important that any clinical risks (accumulation of gold in the body and other side effects) are outweighed by the benefit of using AuNP systems, meaning that they should result in a higher therapeutic efficacy.On the other hand, the ease of implementation of SERS measurements in the clinic also requires consideration. In many of the SERS platforms highlighted so far, the end goal is to develop devices compatible with in vitro cell cultures and ex vivo screening of patient samples (i.e., patient-derived 3D models, biopsies, microfluidic sensing, etc.). In this regard, easy-to-use, and simple instrumentation that can be readily integrated into existing medical and diagnostic workflows are needed. Conventional Raman spectrometers have an optical configuration in which lasers can be applied via flexible optical fibers to excite the sample and collect the scattered light. Therefore, Raman spectrometers are easily customizable. Actually, various designs for handheld Raman instruments compatible with clinical settings have been developed for in situ measurements, such as the SpectroPen described in Section 4.2, many of them being commercially available.240,241 Moreover, available spectral analysis software can assist in interpretation of the spectra for molecular identification, to facilitate clinical use. However, one of the primary challenges here is the cost of instrument components: laser light source(s), high-performance optical fibers, etc. An additional constraint arises from the limited penetration depth of the excitation laser into body tissues, which restricts its application to superficial or readily accessible tumors. A significant stride forward occurred in 2015, with the release of an endoscopic probe designed for SERS imaging.242 Nevertheless, it is crucial to address these obstacles, to fully harness the potential benefits of SERS in medical diagnostics and treatment monitoring.
6. Summary and outlook
In this review, we highlight the importance of constructing 3D biomimetic models, and give an overview of the fabrication techniques and common geometries applied in these systems. General design rules and strategies for incorporating SERS tags within 3D systems are discussed. It is noted that SERS tags should be biocompatible, stable in biologically relevant conditions, and strongly interact with the target system/analyte. Then, the latest developments in 3D SERS bioimaging and sensing are summarized. Some important considerations regarding SERS bioimaging include minimizing or making additional considerations to address the scattering background, using appropriate data processing tools, and multimodal bioimaging for enhancing contrast. Regarding biosensing in 3D systems, SERS substrates can be used to analyze secreted biomolecules/metabolites from 3D models, which has been explored using microfluidic platforms.208,235,243,244 SERS sensors can also be directly incorporated into the 3D model, and this configuration has been applied, both for differentiating between cell types and for tracking certain parameters of the microenvironment, including pH.Based on the research progress so far, we can identify various areas that require further exploration, namely: (i) fully leveraging the capabilities of SERS for chemical and biomolecule identification in 3D, (ii) improving multiplexing possibilities, (iii) refining signal processing tools, (iv) addressing limitations towards clinical and practical implementation of SERS, and (v) expanding 3D SERS bioimaging and biosensing beyond 3D cancer models. While SERS has been successfully implemented for the quantitative detection in 2D of a multitude of analytes, including therapeutics,245 neurotransmitters,246 metabolites/proteins,247–249 reactive oxygen species,250,251 DNA,252 pathogens,253 and more,200 still very few works take full advantage of the rich chemical information that SERS can provide in 3D systems. The implementation of label-free SERS in 3D models that recapitulate real biological environments or with ex/in vivo models, could provide key insights at the molecular level for early diagnosis and disease monitoring and/or for the development of novel therapies.
One of the advantages of SERS imaging is the possibility to customize SERS tags to target specific tumor microenvironments, which play a key role in the resulting efficacy of diagnostic and therapeutic approaches. SERS tags can be conjugated with a series of antibodies, aptamers, or biomolecules, which can be tailored to achieve a unique cocktail for simultaneous targeting of different cell types. Taking into account the diverse characteristics of different cancer types, SERS precision and sensitivity could be improved. Responsive and dynamic SERS probes could also be useful to adapt to cancer progression and recognize temporal changes within the tumor microenvironment. Responsiveness to changes in the composition of the cell membrane or external parameters, such as pH or temperature, could pave the way toward smart SERS tags that can dynamically adapt to the tumor microenvironment. By exploring the tailored design of SERS tags for specific tumor microenvironments, we have the potential to elevate the precision, sensitivity, and clinical applicability of SERS technologies for effective cancer therapies.
In terms of multimodal bioimaging, there are many examples combining SERS with fluorescence-based imaging, however, other imaging modes could also be coupled with SERS, which have been comparatively under-explored in 3D models, including magnetic, acoustic, and thermometric-based imaging. We finally provided some examples where complex SERS signals require processing via advanced data analysis techniques and machine learning. We propose that the continued development of user-friendly analytical tools is imperative, especially for the advancement of quantitative biosensing in complex environments. Although the specific focus of this review was mainly geared toward cancer applications, we note that there are still very few examples of 3D SERS bioimaging and biosensing for other diseases, such as for the study of Alzheimer's,254 Parkinson's,255 cystic fibrosis,232 immune disorders,256etc. Additionally, the emerging implementation of chiral plasmonic NPs, either functionalized into different substrate/host materials or in colloidal suspension, may add enantiomeric specificity for SERS sensing, which is another crucial aspect of drug development.257
Table of acronyms
| Acronym | Definition |
| AgNP | Silver nanoparticle |
| AgNR | Silver nanorod |
| AuNP | Gold nanoparticle |
| AuNR | Gold nanorod |
| AuNSt | Gold nanostar |
| ATP | 4-aminothiophenol |
| BSA | Bovine serum albumin |
| 4-BPT | 4-bisphenylthiol |
| BT | Benzenethiol |
| CT | Computerized tomography |
| Cy5 | Cyanine 5 |
| DTNB | 5,5’-dithiobis(2-nitrobenzoic acid) |
| dECM | Decellularized extracellular matrix |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| EpCAM | Epithelial cell adhesion molecule |
| HeLa | Henrietta Lacks cervical cancer cell line |
| HER2 | Human epidermal growth factor receptor 2 |
| HDF | Human dermal fibroblasts |
| ICP-MS | Ion coupled plasma- mass spectroscopy |
| J774 | Macrophage cell line |
| LSPR | Localized surface plasmon resonance |
| 4-MBA | 4-mercaptobenzoic acid |
| 4-MBT | 4-methylbenzenethiol |
| MCF7 | Michigan Cancer Foundation 7 cell line |
| MDA | M. D. Anderson epithelial human breast cancer cell line |
| MDA-MB-231 | Epithelial human breast cancer cell line |
| MGITC | Malachite green isothiocyanate |
| 4-MPy | 4-mercaptopyridine |
| MRI | Magnetic resonance imaging |
| MSOT | Multispectral optoacoustic tomography |
| 4-MSTP | 4-(methylsulfanyl)thiophenol |
| 1-NAT | 1-napthalenethiol |
| 2-NAT | 2-napthalenethiol |
| NBT | nitrobenzenethiol |
| NIR | Near-infrared |
| NP | Nanoparticle |
| 2-NPT | 2-napthalenethiol |
| 4-NTP | 4-nitrothiophenol |
| PA | Poly-L-arginine hydrochloride |
| PAH | Poly(allylamine hydrochloride) |
| PALM | Photo-activated localization microscopy |
| 4-ATP | 4-aminothiophenol |
| PCA | Principal component analysis |
| PC3 | Human prostatic carcinome cell line |
| PDT | Photodynamic therapy |
| PDX | Patient-derived xenograft |
| PEG | Poly(ethylene glycol) |
| PET | Positron emission tomography |
| PI-DETA | Poly(isoprene)-diethylenetriamine |
| PI-b-PEG | Poly(isoprene)-block-poly(ethylene glycol) |
| PLGA | Poly(lactic-co-glycolic acid) |
| PMA | Dodecylamine-modified polyisobutylene-alt-maleic polymer |
| 4-NTP | 4-nitrothiophenol |
| PVA | poly(vinyl alcohol) |
| PVP | Polyvinylpyrrolidone |
| RaR | Raman reporter molecule |
| Rh6G | Rhodamine 6G |
| Ru(bpy)3 | tris(2,2′-bipyridyl)ruthenium(II) chloride hexahydrate |
| SEM | Scanning electron microscopy |
| SEHRS | Surface-enhanced hyper Raman spectroscopy |
| (SE)SORS | (Surface-enhanced) spatial-offset Raman spectroscopy |
| SERS | Surface-enhanced Raman spectroscopy |
| SERRS | Surface-enhanced resonance Raman spectroscopy |
| STED | Stimulated emission depletion microscopy |
| STORM | Stochastic optical reconstruction microscopy |
| 4T1 | Human breast cancer cell line |
| TEM | Transmission electron microscopy |
| TERS | Tip-enhanced Raman spectroscopy |
| TGF-β | Transforming growth factor-β |
| TRS | Transmission Raman spectroscopy |
| UV | Ultraviolet |
| U87-MG | Human glioblastoma cell line |
| VEGF | Vascular endothelial growth factor |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the European Research Council (ERC-AdG-2017 #787510) and by the European Comission (EUSMI, Grant Number 731019). C.G.A. thanks the Spanish State Research Agency for a Juan de la Cierva Incorporación Fellowship (IJC2019-040827-I). L.T.A. thanks the Spanish Ministry of Universities for a Formación de Profesorado Universitario Fellowship (FPU-05867). Fig. 1, 2, 5 and 10 were created using BioRender.com.References
- J. J. Hornberg, F. J. Bruggeman, H. V. Westerhoff and J. Lankelma, BioSystems, 2006, 83, 81–90 CrossRef CAS PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- C. Sawyers, Nature, 2004, 432, 294–297 CrossRef CAS PubMed.
- Q. Gao, G. Zhou, S. Lin, R. Paus and Z. Yue, Exp. Dermatol., 2019, 28, 413–418 CrossRef PubMed.
- L. Hutchinson and R. Kirk, Nat. Rev. Clin. Oncol., 2011, 8, 189–190 CrossRef PubMed.
- N. M. Anderson and M. C. Simon, Curr. Biol., 2020, 30, R921–R925 CrossRef CAS PubMed.
- M. Timaner, O. Beyar-Katz and Y. Shaked, Curr. Protoc. Cell Biol., 2016, 70, 19.18.1–19.18.12 Search PubMed.
- R. C. Dutta and A. K. Dutta, Biotechnol. Adv., 2009, 27, 334–339 CrossRef CAS PubMed.
- S. R. Horman, C. Hogan, K. D. Reyes, F. Lo and C. Antczak, Future Med. Chem., 2015, 7, 513–525 CrossRef CAS PubMed.
- K. Kingwell, Nat. Rev. Drug Discovery, 2017, 16, 6–7 CrossRef CAS PubMed.
- R. Edmondson, J. J. Broglie, A. F. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS PubMed.
- R. Augustine, S. N. Kalva, R. Ahmad, A. A. Zahid, S. Hasan, A. Nayeem, L. McClements and A. Hasan, Transl. Oncol., 2021, 14, 101015 CrossRef CAS PubMed.
- E. C. Costa, A. F. Moreira, D. De Melo-Diogo, V. M. Gaspar, M. P. Carvalho and I. J. Correia, Biotechnol. Adv., 2016, 34, 1427–1441 CrossRef PubMed.
- J. W. Lichtman and J.-A. Conchello, Nat. Methods, 2005, 2, 910–919 CrossRef CAS PubMed.
- D. Davydova, A. de la Cadena, D. Akimov and B. Dietzek, Laser Photonics Rev., 2016, 10, 62–81 CrossRef CAS.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS PubMed.
- J. Choi, E. K. Lee, J. Choo, J. Yuh and J. W. Hong, Biotechnol. J., 2015, 10, 1682–1688 CrossRef CAS PubMed.
- A. R. Kherlopian, T. Song, Q. Duan, M. A. Neimark, M. J. Po, J. K. Gohagan and A. F. Laine, BMC Syst. Biol., 2008, 2, 74 CrossRef PubMed.
- A. Downes and A. Elfick, Sensors, 2010, 10, 1871–1889 CrossRef PubMed.
- D. I. Ellis, D. P. Cowcher, L. Ashton, S. O’Hagan and R. Goodacre, Analyst, 2013, 138, 3871–3884 RSC.
- E. Brauchle and K. Schenke-Layland, Biotechnol. J., 2013, 8, 288–297 CrossRef CAS PubMed.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- J. A. Creighton, Vibrational Spectroscopy of Adsorbates, Springer-Verlag, Berlin Heidelberg, New York, 1980, vol. 15, pp. 145–164 Search PubMed.
- F. W. King, R. P. Van Duyne and G. C. Schatz, J. Chem. Phys., 1978, 69, 4472–4481 CrossRef CAS.
- T. Itoh, M. Procházka, Z.-C. Dong, W. Ji, Y. S. Yamamoto, Y. Zhang and Y. Ozaki, Chem. Rev., 2023, 123, 1552–1634 CrossRef CAS PubMed.
- S. Schlücker, Surface Enhanced Raman Spectroscopy: Analytical, Biophysical and Life Science Applications, John Wiley & Sons, 2011 Search PubMed.
- S. M. Morton and L. Jensen, J. Am. Chem. Soc., 2009, 131, 4090–4098 CrossRef CAS PubMed.
- M. Prochazka, Surface-Enhanced Raman Spectroscopy, Springer International Publishing, Cham, 2016 Search PubMed.
- S. Schlücker, ChemPhysChem, 2009, 10, 1344–1354 CrossRef PubMed.
- S. A. Maier, Plasmonics: fundamentals and applications, Springer, 2007, vol. 1 Search PubMed.
- D. K. Gramotnev and S. I. Bozhevolnyi, Nat. Photonics, 2010, 4, 83–91 CrossRef CAS.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783–826 CrossRef CAS.
- R. Alvarez-Puebla, L. M. Liz-Marzán and F. J. García de Abajo, J. Phys. Chem. Lett., 2010, 1, 2428–2434 CrossRef CAS.
- P. Nordlander, C. Oubre, E. Prodan, K. Li and M. I. Stockman, Nano Lett., 2004, 4, 899–903 CrossRef CAS.
- F. Hao, C. L. Nehl, J. H. Hafner and P. Nordlander, Nano Lett., 2007, 7, 729–732 CrossRef CAS PubMed.
- N. Valley, N. Greeneltch, R. P. Van Duyne and G. C. Schatz, J. Phys. Chem. Lett., 2013, 4, 2599–2604 CrossRef CAS.
- Y. Fang, N.-H. Seong and D. D. Dlott, Science, 2008, 321, 388–392 CrossRef CAS PubMed.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- P. G. Etchegoin and E. C. Le Ru, Phys. Chem. Chem. Phys., 2008, 10, 6079–6089 RSC.
- J. Kneipp, ACS Nano, 2017, 11, 1136–1141 CrossRef CAS PubMed.
- A. Kamińska, E. Witkowska, K. Winkler, I. Dzięcielewski, J. L. Weyher and J. Waluk, Biosens. Bioelectron., 2015, 66, 461–467 CrossRef PubMed.
- H. Kearns, R. Goodacre, L. E. Jamieson, D. Graham and K. Faulds, Anal. Chem., 2017, 89, 12666–12673 CrossRef CAS PubMed.
- Z. Cheng, R. Wang, Y. Xing, L. Zhao, J. Choo and F. Yu, Analyst, 2019, 144, 6533–6540 RSC.
- Y. Zhou, J. Liu, T. Zheng and Y. Tian, Anal. Chem., 2020, 92, 5910–5920 CrossRef CAS PubMed.
- S. S. Panikar, G. Ramírez-García, S. Sidhik, T. Lopez-Luke, C. Rodriguez-Gonzalez, I. H. Ciapara, P. S. Castillo, T. Camacho-Villegas and E. De La Rosa, Anal. Chem., 2019, 91, 2100–2111 CrossRef CAS PubMed.
- H.-Y. Wu and B. T. Cunningham, Nanoscale, 2014, 6, 5162–5171 RSC.
- G. A. Vinnacombe-Willson, Surface Functionalization, Patterning, and in Situ Growth of Gold Nanoparticles, University of California, Los Angeles, 2022 Search PubMed.
- K. Białkowska, P. Komorowski, M. Bryszewska and K. Miłowska, Int. J. Mol. Sci., 2020, 21, 6225 CrossRef PubMed.
- A. Moscona and H. Moscona.
- F. Pampaloni, E. G. Reynaud and E. H. K. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839–845 CrossRef CAS PubMed.
- J. W. Haycock, 3D cell culture: a review of current approaches and techniques, Springer, 2011 Search PubMed.
- X. Cui, Y. Hartanto and H. Zhang, J. R. Soc., Interface, 2017, 14, 20160877 CrossRef PubMed.
- E. C. Costa, A. F. Moreira, D. De Melo-Diogo, V. M. Gaspar, M. P. Carvalho and I. J. Correia, Biotechnol. Adv., 2016, 34, 1427–1441 CrossRef PubMed.
- S. F. Abu-Absi, J. R. Friend, L. K. Hansen and W.-S. Hu, Exp. Cell Res., 2002, 274, 56–67 CrossRef CAS PubMed.
- S. J. Han, S. Kwon and K. S. Kim, Cancer Cell Int., 2021, 21, 152 CrossRef PubMed.
- D. Antoni, H. Burckel, E. Josset and G. Noel, Int. J. Mol. Sci., 2015, 16, 5517–5527 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- M. C. Cushing and K. S. Anseth, Science, 2007, 316, 1133–1134 CrossRef CAS PubMed.
- Y. C. Choi, J. S. Choi, B. S. Kim, J. D. Kim, H. I. Yoon and Y. W. Cho, Tissue Eng., Part C, 2012, 18, 866–876 CrossRef CAS PubMed.
- L. T. Saldin, M. C. Cramer, S. S. Velankar, L. J. White and S. F. Badylak, Acta Biomater., 2017, 49, 1–15 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- J. Thiele, Y. Ma, S. M. C. Bruekers, S. Ma and W. T. S. Huck, Adv. Mater., 2014, 26, 125–148 CrossRef CAS PubMed.
- J. Lou and D. J. Mooney, Nat. Rev. Chem., 2022, 6, 726–744 CrossRef CAS PubMed.
- C. Vila-Parrondo, C. García-Astrain and L. M. Liz-Marzán, Adv. Colloid Interface Sci., 2020, 283, 102237 CrossRef CAS PubMed.
- D.-W. Cho, B. S. Kim, J. Jang, G. Gao, W. Han and N. K. Singh, 3D Bioprinting: Modeling In Vitro Tissues and Organs Using Tissue-Specific Bioinks, Springer International Publishing, 2019 Search PubMed.
- S. Kyle, Z. M. Jessop, A. Al-Sabah and I. S. Whitaker, Adv. Healthcare Mater., 2017, 6, 1700264 CrossRef PubMed.
- S. V. Murphy and A. Atala, Nat. Biotechnol., 2014, 32, 773–785 CrossRef CAS PubMed.
- J. Rosendahl, A. Svanström, M. Berglin, S. Petronis, Y. Bogestål, P. Stenlund, S. Standoft, A. Ståhlberg, G. Landberg, G. Chinga-Carrasco and J. Håkansson, Bioengineering, 2021, 8, 97 CrossRef CAS PubMed.
- C. García-Astrain, E. Lenzi, D. Jimenez De Aberasturi, M. Henriksen-Lacey, M. R. Binelli and L. M. Liz-Marzán, Adv. Funct. Mater., 2020, 30, 2005407 CrossRef.
- J. Plou, B. Molina-Martínez, C. García-Astrain, J. Langer, I. García, A. Ercilla, G. Perumal, A. Carracedo and L. M. Liz-Marzán, Nano Lett., 2021, 21, 8785–8793 CrossRef CAS PubMed.
- D. L. Hughes, A. Hughes, Z. Soonawalla, S. Mukherjee and E. O’Neill, Cancers, 2021, 13, 2870 CrossRef CAS PubMed.
- C. Perleberg, A. Kind and A. Schnieke, Dis. Models Mech., 2018, 11, dmm030783 CrossRef PubMed.
- Y. Liu, W. Wu, C. Cai, H. Zhang, H. Shen and Y. Han, Signal Transduction Targeted Ther., 2023, 8, 160 CrossRef PubMed.
- L. G. Campana, M. Bullo, P. Di Barba, F. Dughiero, M. Forzan, M. E. Mognaschi, P. Sgarbossa, A. L. Tosi, A. Bernardis and E. Sieni, Technol. Cancer Res. Treat., 2018, 17, 1533033818789693 Search PubMed.
- J. Jung, Toxicol. Res., 2014, 30, 1–5 CrossRef PubMed.
- L. Pompili, M. Porru, C. Caruso, A. Biroccio and C. Leonetti, J. Exp. Clin. Cancer Res., 2016, 35, 1–8 CrossRef PubMed.
- J. J. Morton, G. Bird, Y. Refaeli and A. Jimeno, Cancer Res., 2016, 76, 6153–6158 CrossRef CAS PubMed.
- Z. Lei, X. Ren, S. Wang, X. Liang and Y. Tang, OncoTargets Ther., 2016, 545–555 CAS.
- K. Majumder, N. Arora, S. Modi, R. Chugh, A. Nomura, B. Giri, R. Dawra, S. Ramakrishnan, S. Banerjee and A. Saluja, J. Gastrointest. Surg., 2016, 20, 53–65 CrossRef PubMed.
- J. Jin, K. Yoshimura, M. Sewastjanow-Silva, S. Song and J. A. Ajani, Cancers, 2023, 15, 4352 CrossRef CAS PubMed.
- A. Khademhosseini and R. Langer, Nat. Protoc., 2016, 11, 1775–1781 CrossRef CAS PubMed.
- K. C. Bantz, A. F. Meyer, N. J. Wittenberg, H. Im, Ö. Kurtuluş, S. H. Lee, N. C. Lindquist, S.-H. Oh and C. L. Haynes, Phys. Chem. Chem. Phys., 2011, 13, 11551–11567 RSC.
- T. Köker, N. Tang, C. Tian, W. Zhang, X. Wang, R. Martel and F. Pinaud, Nat. Commun., 2018, 9, 607 CrossRef PubMed.
- S. Yu, Y. Yin, J. Chao, M. Shen and J. Liu, Environ. Sci. Technol., 2014, 48, 403–411 CrossRef CAS PubMed.
- J. Liu, K. G. Pennell and R. H. Hurt, Environ. Sci. Technol., 2011, 45, 7345–7353 CrossRef CAS PubMed.
- F. Wang, R. G. Widejko, Z. Yang, K. T. Nguyen, H. Chen, L. P. Fernando, K. A. Christensen and J. N. Anker, Anal. Chem., 2012, 84, 8013–8019 CrossRef CAS PubMed.
- R. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef CAS PubMed.
- X. Huang, S. Neretina and M. A. El-Sayed, Adv. Mater., 2009, 21, 4880–4910 CrossRef CAS PubMed.
- B. Andreiuk, F. Nicolson, L. M. Clark, S. R. Panikkanvalappil, M. Rashidian, S. Harmsen and M. F. Kircher, Nanotheranostics, 2022, 6, 10–30 CrossRef PubMed.
- J. Xie, Q. Zhang, J. Y. Lee and D. I. C. Wang, ACS Nano, 2008, 2, 2473–2480 CrossRef CAS PubMed.
- S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587–1595 CrossRef CAS PubMed.
- X. Zhuo, D. Vila-Liarte, S. Wang, D. Jimenez de Aberasturi and L. M. Liz-Marzán, Chem. Mater., 2023, 35, 5689–5698 CrossRef CAS.
- G. Baffou, F. Cichos and R. Quidant, Nat. Mater., 2020, 19, 946–958 CrossRef CAS PubMed.
- R. Rodríguez-Oliveros and J. A. Sánchez-Gil, Opt. Express, 2012, 20, 621–626 CrossRef PubMed.
- G. Baffou and R. Quidant, Laser Photonics Rev., 2013, 7, 171–187 CrossRef CAS.
- E. Lenzi, M. Henriksen-Lacey, B. Molina, J. Langer, C. D. L. de Albuquerque, D. Jimenez de Aberasturi and L. M. Liz-Marzán, ACS Sens., 2022, 7, 1747–1756 CrossRef CAS PubMed.
- G. A. Vinnacombe-Willson, N. Chiang, L. Scarabelli, Y. Hu, L. K. Heidenreich, X. Li, Y. Gong, D. T. Inouye, T. S. Fisher, P. S. Weiss and S. J. Jonas, ACS Cent. Sci., 2020, 6, 2105–2116 CrossRef CAS PubMed.
- Y.-T. Yang, I.-L. Hsu, T.-Y. Cheng, W.-J. Wu, C.-W. Lee, T.-J. Li, C. I. Cheung, Y.-C. Chin, H.-C. Chen, Y.-C. Chiu, C.-C. Huang and M.-Y. Liao, Anal. Chem., 2019, 91, 8213–8220 CrossRef CAS PubMed.
- E. Lázaro-Ibáñez, F. N. Faruqu, A. F. Saleh, A. M. Silva, J. Tzu-Wen Wang, J. Rak, K. T. Al-Jamal and N. Dekker, ACS Nano, 2021, 15, 3212–3227 CrossRef PubMed.
- E. Lenzi, D. Jimenez de Aberasturi and L. M. Liz-Marzán, ACS Sens., 2019, 4, 1126–1137 CrossRef CAS PubMed.
- X.-S. Zheng, P. Hu, Y. Cui, C. Zong, J.-M. Feng, X. Wang and B. Ren, Anal. Chem., 2014, 86, 12250–12257 CrossRef CAS PubMed.
- A. S. D. S. Indrasekara, B. J. Paladini, D. J. Naczynski, V. Starovoytov, P. V. Moghe and L. Fabris, Adv. Healthcare Mater., 2013, 2, 1370–1376 CrossRef CAS PubMed.
- H. Wang, H. Zhang, L. Zhang, S. Zhao, Y. Chen, Y. Wan, Q. Zhang and L. Xia, RSC Adv., 2021, 11, 37443–37448 RSC.
- S. Lee, H. Chon, J. Lee, J. Ko, B. H. Chung, D. W. Lim and J. Choo, Biosens. Bioelectron., 2014, 51, 238–243 CrossRef CAS PubMed.
- D. Jimenez de Aberasturi, M. Henriksen-Lacey, L. Litti, J. Langer and L. M. Liz-Marzán, Adv. Funct. Mater., 2020, 30, 1909655 CrossRef CAS.
- D. Jimenez de Aberasturi, A. B. Serrano-Montes, J. Langer, M. Henriksen-Lacey, W. J. Parak and L. M. Liz-Marzán, Chem. Mater., 2016, 28, 6779–6790 CrossRef CAS.
- M. Schumacher, D. Jimenez de Aberasturi, J.-P. Merkl, L. Scarabelli, E. Lenzi, M. Henriksen-Lacey, L. M. Liz-Marzán and H. Weller, Adv. Opt. Mater., 2022, 10, 2102635 CrossRef CAS.
- X. Zhuo, M. Henriksen-Lacey, D. Jimenez de Aberasturi, A. Sánchez-Iglesias and L. M. Liz-Marzán, Chem. Mater., 2020, 32, 5879–5889 CrossRef CAS PubMed.
- Z. A. Nima, M. Mahmood, Y. Xu, T. Mustafa, F. Watanabe, D. A. Nedosekin, M. A. Juratli, T. Fahmi, E. I. Galanzha, J. P. Nolan, A. G. Basnakian, V. P. Zharov and A. S. Biris, Sci. Rep., 2014, 4, 4752 CrossRef CAS PubMed.
- J. Yang, Z. Wang, S. Zong, C. Song, R. Zhang and Y. Cui, Anal. Bioanal. Chem., 2012, 402, 1093–1100 CrossRef CAS PubMed.
- S. Rodal-Cedeira, A. Vázquez-Arias, G. Bodelón, A. Skorikov, S. Núñez-Sánchez, A. Laporta, L. Polavarapu, S. Bals, L. M. Liz-Marzán, J. Pérez-Juste and I. Pastoriza-Santos, ACS Nano, 2020, 14, 14655–14664 CrossRef CAS PubMed.
- S. Barua and S. Mitragotri, Nano Today, 2014, 9, 223–243 CrossRef CAS PubMed.
- J. Yue, T. J. Feliciano, W. Li, A. Lee and T. W. Odom, Bioconjugate Chem., 2017, 28, 1791–1800 CrossRef CAS PubMed.
- M. Bhamidipati and L. Fabris, Bioconjugate Chem., 2017, 28, 449–460 CrossRef CAS PubMed.
- P. T. Sujai, M. M. Joseph, G. Saranya, J. B. Nair, V. P. Murali and K. K. Maiti, Nanoscale, 2020, 12, 6971–6975 RSC.
- S. M. McCabe, G. Q. Wallace, S. Sloan-Dennison, W. J. Tipping, N. C. Shand, D. Graham, M. Boyd and K. Faulds, Analyst, 2023, 148, 3247–3256 RSC.
- A. Kapara, K. A. Findlay Paterson, V. G. Brunton, D. Graham, M. Zagnoni and K. Faulds, Anal. Chem., 2021, 93, 5862–5871 CrossRef CAS PubMed.
- L. E. Jamieson, V. L. Camus, P. O. Bagnaninchi, K. M. Fisher, G. D. Stewart, W. H. Nailon, D. B. McLaren, D. J. Harrison and C. J. Campbell, Nanoscale, 2016, 8, 16710–16718 RSC.
- E. J. Zeman and G. C. Schatz, in Dynamics on Surfaces, ed. B. Pullman, J. Jortner, A. Nitzan and B. Gerber, Springer, Netherlands, Dordrecht, 1984, vol. 17, pp. 413–424 Search PubMed.
- M. Bhamidipati and L. Fabris, Bioconjugate Chem., 2017, 28, 449–460 CrossRef CAS PubMed.
- P. Lavrador, M. R. Esteves, V. M. Gaspar and J. F. Mano, Adv. Funct. Mater., 2021, 31, 2005941 CrossRef CAS.
- P. Thoniyot, M. J. Tan, A. A. Karim, D. J. Young and X. J. Loh, Adv. Sci., 2015, 2, 1400010 CrossRef PubMed.
- C. Wang, N. T. Flynn and R. Langer, Adv. Mater., 2004, 16, 1074–1079 CrossRef CAS.
- R. Contreras-Montoya, A. B. Bonhome-Espinosa, A. Orte, D. Miguel, J. M. Delgado-López, J. D. G. Duran, J. M. Cuerva, M. T. Lopez-Lopez and L. Álvarez De Cienfuegos, Mater. Chem. Front., 2018, 2, 686–699 RSC.
- S. E. Lehman, J. M. McCracken, L. A. Miller, S. Jayalath and R. G. Nuzzo, Adv. Healthcare Mater., 2021, 10, 2001040 CrossRef CAS PubMed.
- Y. Tsuge, T. Moriya, Y. Moriyama, Y. Tokura and S. Shiratori, ACS Appl. Mater. Interfaces, 2017, 9, 15122–15129 CrossRef CAS PubMed.
- H. Kang, S. Jeong, Y. Park, J. Yim, B. Jun, S. Kyeong, J. Yang, G. Kim, S. Hong, L. P. Lee, J. Kim, H. Lee, D. H. Jeong and Y. Lee, Adv. Funct. Mater., 2013, 23, 3719–3727 CrossRef CAS.
- K. Zhang, C. Hao, Y. Huo, B. Man, C. Zhang, C. Yang, M. Liu and C. Chen, Lasers Med. Sci., 2019, 34, 1849–1855 CrossRef PubMed.
- S. Mosca, P. Dey, T. A. Tabish, F. Palombo, N. Stone and P. Matousek, J. Biophotonics, 2020, 13, e201960092 CrossRef PubMed.
- Y. Zhang, R. Chen, F. Liu, P. Miao, L. Lin and J. Ye, Small Methods, 2023, 7, 2201334 CrossRef CAS PubMed.
- D. A. Stuart, J. M. Yuen, N. Shah, O. Lyandres, C. R. Yonzon, M. R. Glucksberg, J. T. Walsh and R. P. Van Duyne, Anal. Chem., 2006, 78, 7211–7215 CrossRef CAS PubMed.
- H. Kang, S. Jeong, Y. Park, J. Yim, B. Jun, S. Kyeong, J. Yang, G. Kim, S. Hong and L. P. Lee, Adv. Funct. Mater., 2013, 23, 3719–3727 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- R. M. Davis, B. Kiss, D. R. Trivedi, T. J. Metzner, J. C. Liao and S. S. Gambhir, ACS Nano, 2018, 12, 9669–9679 CrossRef CAS PubMed.
- H. Chen, W. Zhang, G. Zhu, J. Xie and X. Chen, Nat. Rev. Mater., 2017, 2, 17024 CrossRef CAS PubMed.
- J. Wang, D. Liang, Q. Jin, J. Feng and X. Tang, Bioconjugate Chem., 2020, 31, 182–193 CrossRef CAS PubMed.
- J. Wang, D. Liang, J. Feng and X. Tang, Anal. Chem., 2019, 91, 11045–11054 CrossRef CAS PubMed.
- A. M. Fales, H. Yuan and T. Vo-Dinh, Mol. Pharmaceutics, 2013, 10, 2291–2298 CrossRef CAS PubMed.
- S. McAughtrie, K. Lau, K. Faulds and D. Graham, Chem. Sci., 2013, 4, 3566 RSC.
- E. Lenzi, L. Litti, D. Jimenez de Aberasturi, M. Henriksen-Lacey and L. M. Liz-Marzán, J. Raman Spectrosc., 2021, 52, 355–365 CrossRef CAS.
- Y. Chen, X. Bai, L. Su, Z. Du, A. Shen, A. Materny and J. Hu, Sci. Rep., 2016, 6, 19173 CrossRef CAS PubMed.
- C. Liu, J. Dong, Z. Zhang, K. Fu, D. Wang, X. Mi, S. Yue, X. Tan and Y. Zhang, Anal. Chem., 2023, 95, 13880–13888 CrossRef CAS PubMed.
- L. A. Dykman and N. G. Khlebtsov, Chem. Rev., 2014, 114, 1258–1288 CrossRef CAS PubMed.
- B. D. Chithrani and W. C. W. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS PubMed.
- X.-T. Pan, X.-Y. Yang, T.-Q. Mao, K. Liu, Z.-Z. Chen, L.-N. Ji, D.-C. Jiang, K. Wang, Z.-Z. Gu and X.-H. Xia, Biosensors, 2022, 12, 875 CrossRef CAS PubMed.
- L. Fang, X.-T. Pan, K. Liu, D. Jiang, D. Ye, L.-N. Ji, K. Wang and X.-H. Xia, ACS Appl. Mater. Interfaces, 2023, 15, 20677–20685 CrossRef CAS PubMed.
- S. Vantasin, W. Ji, Y. Tanaka, Y. Kitahama, M. Wang, K. Wongravee, H. Gatemala, S. Ekgasit and Y. Ozaki, Angew. Chem., 2016, 128, 8531–8535 CrossRef.
- A. Chakraborty, A. Ghosh and A. Barui, J. Raman Spectrosc., 2020, 51, 7–36 CrossRef CAS.
- J. V. Jokerst, A. J. Cole, D. Van de Sompel and S. S. Gambhir, ACS Nano, 2012, 6, 10366–10377 CrossRef CAS PubMed.
- A. M. Mohs, M. C. Mancini, S. Singhal, J. M. Provenzale, B. Leyland-Jones, M. D. Wang and S. Nie, Anal. Chem., 2010, 82, 9058–9065 CrossRef CAS PubMed.
- L. Li, R. Jiang, B. Shan, Y. Lu, C. Zheng and M. Li, Nat. Commun., 2022, 13, 5249 CrossRef CAS PubMed.
- P. Wang, Y. Fan, L. Lu, L. Liu, L. Fan, M. Zhao, Y. Xie, C. Xu and F. Zhang, Nat. Commun., 2018, 9, 2898 CrossRef PubMed.
- H. Karabeber, R. Huang, P. Iacono, J. M. Samii, K. Pitter, E. C. Holland and M. F. Kircher, ACS Nano, 2014, 8, 9755–9766 CrossRef CAS PubMed.
- Y. Wen, V. X. Truong and M. Li, Nano Lett., 2021, 21, 3066–3074 CrossRef CAS PubMed.
- L. A. Lane, R. Xue and S. Nie, Curr. Opin. Chem. Biol., 2018, 45, 95–103 CrossRef CAS PubMed.
- H. He, Y. Zhang, S. Zhu, J. Ye and L. Lin, J. Phys. Chem. C, 2022, 126, 12575–12581 CrossRef CAS.
- S. Lin, Z. Cheng, Q. Li, R. Wang and F. Yu, ACS Sens., 2021, 6, 3912–3932 CrossRef CAS PubMed.
- Y. Liu, J. R. Ashton, E. J. Moding, H. Yuan, J. K. Register, A. M. Fales, J. Choi, M. J. Whitley, X. Zhao, Y. Qi, Y. Ma, G. Vaidyanathan, M. R. Zalutsky, D. G. Kirsch, C. T. Badea and T. Vo-Dinh, Theranostics, 2015, 5, 946–960 CrossRef CAS PubMed.
- C. De La Encarnación, D. Jimenez De Aberasturi and L. M. Liz-Marzán, Adv. Drug Delivery Rev., 2022, 189, 114484 CrossRef PubMed.
- M. Quintanilla, Y. Zhang and L. M. Liz-Marzán, Chem. Mater., 2018, 30, 2819–2828 CrossRef CAS.
- M. Quintanilla, M. Henriksen-Lacey, C. Renero-Lecuna and L. M. Liz-Marzán, Chem. Soc. Rev., 2022, 51, 4223–4242 RSC.
- M. Quintanilla, I. García, I. De Lázaro, R. García-Alvarez, M. Henriksen-Lacey, S. Vranic, K. Kostarelos and L. M. Liz-Marzán, Theranostics, 2019, 9, 7298–7312 Search PubMed.
- D.-E. Lee, H. Koo, I.-C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656–2672 RSC.
- Z. Wang, S. Zong, J. Yang, J. Li and Y. Cui, Biosens. Bioelectron., 2011, 26, 2883–2889 CrossRef CAS PubMed.
- S. Lee, H. Chon, S.-Y. Yoon, E. K. Lee, S.-I. Chang, D. W. Lim and J. Choo, Nanoscale, 2012, 4, 124–129 RSC.
- A. B. Serrano-Montes, J. Langer, M. Henriksen-Lacey, D. Jimenez de Aberasturi, D. M. Solís, J. M. Taboada, F. Obelleiro, K. Sentosun, S. Bals, A. Bekdemir, F. Stellacci and L. M. Liz-Marzán, J. Phys. Chem. C, 2016, 120, 20860–20868 CrossRef CAS.
- M. S. Strozyk, D. J. de Aberasturi, J. V. Gregory, M. Brust, J. Lahann and L. M. Liz-Marzán, Adv. Funct. Mater., 2017, 27, 1701626 CrossRef.
- E. Lenzi, D. Jimenez de Aberasturi, M. Henriksen-Lacey, P. Piñeiro, A. J. Muniz, J. Lahann and L. M. Liz-Marzán, ACS Appl. Mater. Interfaces, 2022, 14, 20708–20719 CrossRef CAS PubMed.
- J. Qian, L. Jiang, F. Cai, D. Wang and S. He, Biomaterials, 2011, 32, 1601–1610 CrossRef CAS PubMed.
- Y. Zhang, J. Qian, D. Wang, Y. Wang and S. He, Angew. Chem., Int. Ed., 2013, 52, 1148–1151 CrossRef CAS PubMed.
- Y. Zhang, H. Liu, J. Tang, Z. Li, X. Zhou, R. Zhang, L. Chen, Y. Mao and C. Li, ACS Appl. Mater. Interfaces, 2017, 9, 17769–17776 CrossRef CAS PubMed.
- A. A. Stacy and R. P. Van Duyne, Chem. Phys. Lett., 1983, 102, 365–370 CrossRef CAS.
- S. Harmsen, R. Huang, M. A. Wall, H. Karabeber, J. M. Samii, M. Spaliviero, J. R. White, S. Monette, R. O’Connor, K. L. Pitter, S. A. Sastra, M. Saborowski, E. C. Holland, S. Singer, K. P. Olive, S. W. Lowe, R. G. Blasberg and M. F. Kircher, Sci. Transl. Med., 2015, 7, 271ra7–271ra7 Search PubMed.
- V. Neuschmelting, S. Harmsen, N. Beziere, H. Lockau, H.-T. Hsu, R. Huang, D. Razansky, V. Ntziachristos and M. F. Kircher, Small, 2018, 14, 1800740 CrossRef PubMed.
- D. Graham, W. E. Smith, A. M. T. Linacre, C. H. Munro, N. D. Watson and P. C. White, Anal. Chem., 1997, 69, 4703–4707 CrossRef CAS.
- R. J. Stokes, A. Macaskill, J. A. Dougan, P. G. Hargreaves, H. M. Stanford, W. E. Smith, K. Faulds and D. Graham, Chem. Commun., 2007, 2811–2813 RSC.
- A. Ingram, L. Byers, K. Faulds, B. D. Moore and D. Graham, J. Am. Chem. Soc., 2008, 130, 11846–11847 CrossRef CAS PubMed.
- H. Cho, B. R. Baker, S. Wachsmann-Hogiu, C. V. Pagba, T. A. Laurence, S. M. Lane, L. P. Lee and J. B.-H. Tok, Nano Lett., 2008, 8, 4386–4390 CrossRef CAS PubMed.
- G. Sabatté, R. Keir, M. Lawlor, M. Black, D. Graham and W. E. Smith, Anal. Chem., 2008, 80, 2351–2356 Search PubMed.
- G. McNay, D. Eustace, W. E. Smith, K. Faulds and D. Graham, Appl. Spectrosc., 2011, 65, 825–837 CrossRef CAS PubMed.
- Y. Zhang, D. Jimenez De Aberasturi, M. Henriksen-Lacey, J. Langer and L. M. Liz-Marzán, ACS Sens., 2020, 5, 3194–3206 CrossRef CAS PubMed.
- R. A. Ando, N. P. W. Pieczonka, P. S. Santos and R. F. Aroca, Phys. Chem. Chem. Phys., 2009, 11, 7505 RSC.
- X. Zou, Y. Wang, W. Liu and L. Chen, Analyst, 2017, 142, 2333–2337 RSC.
- S. Kang, W. Nam, W. Zhou, I. Kim and P. J. Vikesland, ACS Appl. Nano Mater., 2021, 4, 5768–5777 CrossRef CAS.
- F. Madzharova, Z. Heiner and J. Kneipp, Chem. Soc. Rev., 2017, 46, 3980–3999 RSC.
- S. Mosca, P. Dey, M. Salimi, B. Gardner, F. Palombo, N. Stone and P. Matousek, Anal. Chem., 2021, 93, 6755–6762 CrossRef CAS PubMed.
- N. Stone, K. Faulds, D. Graham and P. Matousek, Anal. Chem., 2010, 82, 3969–3973 Search PubMed.
- S. N. K. M. L. GR, Chem. Sci., 2011, 2, 776–780 RSC.
- P. Matousek, M. D. Morris, N. Everall, I. P. Clark, M. Towrie, E. Draper, A. Goodship and A. W. Parker, Appl. Spectrosc., 2005, 59, 1485–1492 CrossRef CAS PubMed.
- S. M. Asiala, N. C. Shand, K. Faulds and D. Graham, ACS Appl. Mater. Interfaces, 2017, 9, 25488–25494 CrossRef CAS PubMed.
- B. Sharma, K. Ma, M. R. Glucksberg and R. P. Van Duyne, J. Am. Chem. Soc., 2013, 135, 17290–17293 CrossRef CAS PubMed.
- A. S. Moody, P. C. Baghernejad, K. R. Webb and B. Sharma, Anal. Chem., 2017, 89, 5688–5692 CrossRef CAS PubMed.
- R. A. Odion, P. Strobbia, B. M. Crawford and T. Vo-Dinh, J. Raman Spectrosc., 2018, 49, 1452–1460 CrossRef CAS.
- F. Nicolson, L. E. Jamieson, S. Mabbott, K. Plakas, N. C. Shand, M. R. Detty, D. Graham and K. Faulds, Chem. Commun., 2018, 54, 8530–8533 RSC.
- F. Nicolson, B. Andreiuk, C. Andreou, H.-T. Hsu, S. Rudder and M. F. Kircher, Theranostics, 2019, 9, 5899 CrossRef CAS PubMed.
- Y. Kitahama and Y. Ozaki, Analyst, 2016, 141, 5020–5036 RSC.
- S. Mosca, C. Conti, N. Stone and P. Matousek, Nat. Rev. Methods Primers, 2021, 1, 21 CrossRef CAS.
- S. Tanwar, K. K. Haldar and T. Sen, J. Am. Chem. Soc., 2017, 139, 17639–17648 CrossRef CAS PubMed.
- M. Bhamidipati, H.-Y. Cho, K.-B. Lee and L. Fabris, Bioconjugate Chem., 2018, 29, 2970–2981 CrossRef CAS PubMed.
- S. C.-H. Tsao, J. Wang, Y. Wang, A. Behren, J. Cebon and M. Trau, Nat. Commun., 2018, 9, 1482 CrossRef PubMed.
- S. Cervo, E. Mansutti, G. Del Mistro, R. Spizzo, A. Colombatti, A. Steffan, V. Sergo and A. Bonifacio, Anal. Bioanal. Chem., 2015, 407, 7503–7509 CrossRef CAS PubMed.
- A. Stefancu, V. Moisoiu, R. Couti, I. Andras, R. Rahota, D. Crisan, I. E. Pavel, C. Socaciu, N. Leopold and N. Crisan, Nanomedicine, 2018, 13, 2455–2467 CrossRef CAS PubMed.
- L. Shi, Y. Li and Z. Li, Light: Sci. Appl., 2023, 12, 234 CrossRef CAS PubMed.
- J. Hoarau-Véchot, A. Rafii, C. Touboul and J. Pasquier, Int. J. Mol. Sci., 2018, 19, 181 CrossRef PubMed.
- J. Folkman, Nat. Med., 1995, 1, 27–30 CrossRef CAS PubMed.
- Z. Qian, J. Fei, S. Zong, K. Yang, L. Li, R. Liu, Z. Wang and Y. Cui, ACS Sens., 2020, 5, 208–216 CrossRef CAS PubMed.
- A. Antignani, E. C. H. Ho, M. T. Bilotta, R. Qiu, R. Sarnvosky and D. J. FitzGerald, Biomolecules, 2020, 10, 1331 CrossRef CAS PubMed.
- M. Brisset, M. Grandin, A. Bernet, P. Mehlen and F. Hollande, EMBO Mol. Med., 2021, 13, e14495 CrossRef CAS PubMed.
- A. Samanta, K. K. Maiti, K. Soh, X. Liao, M. Vendrell, U. Dinish, S. Yun, R. Bhuvaneswari, H. Kim and S. Rautela, Angew. Chem., Int. Ed., 2011, 50, 6089 CrossRef CAS PubMed.
- U. S. Dinish, G. Balasundaram, Y.-T. Chang and M. Olivo, Sci. Rep., 2014, 4, 4075 CrossRef CAS PubMed.
- M. Altunbek, D. Çetin, Z. Suludere and M. Çulha, Talanta, 2019, 191, 390–399 CrossRef CAS PubMed.
- J. Plou, P. S. Valera, I. García, C. D. L. De Albuquerque, A. Carracedo and L. M. Liz-Marzán, ACS Photonics, 2022, 9, 333–350 CrossRef CAS PubMed.
- L. Zeng, Y. Pan, S. Wang, X. Wang, X. Zhao, W. Ren, G. Lu and A. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 16781–16791 CrossRef CAS PubMed.
- B. Yin, W. K. H. Ho, X. Xia, C. K. W. Chan, Q. Zhang, Y. M. Ng, C. Y. K. Lam, J. C. W. Cheung, J. Wang and M. Yang, Small, 2023, 19, 2206762 CrossRef CAS PubMed.
- D. Lagadic-Gossmann, L. Huc and V. Lecureur, Cell Death Differ., 2004, 11, 953–961 CrossRef CAS PubMed.
- S. Gilles, C. Kaulen, M. Pabst, U. Simon, A. Offenhäusser and D. Mayer, Nanotechnology, 2011, 22, 295301 CrossRef CAS PubMed.
- M. T. Ghoneim, A. Nguyen, N. Dereje, J. Huang, G. C. Moore, P. J. Murzynowski and C. Dagdeviren, Chem. Rev., 2019, 119, 5248–5297 CrossRef CAS PubMed.
- M. Aref, E. Ranjbari, J. J. García-Guzmán, K. Hu, A. Lork, G. A. Crespo, A. G. Ewing and M. Cuartero, Anal. Chem., 2021, 93, 15744–15751 CrossRef CAS PubMed.
- J. Guo, A. Sesena Rubfiaro, Y. Lai, J. Moscoso, F. Chen, Y. Liu, X. Wang and J. He, Analyst, 2020, 145, 4852–4859 RSC.
- B. P. Nadappuram, K. McKelvey, R. Al Botros, A. W. Colburn and P. R. Unwin, Anal. Chem., 2013, 85, 8070–8074 CrossRef CAS PubMed.
- H. T. Phan and A. J. Haes, J. Phys. Chem. C, 2018, 122, 14846–14856 CrossRef CAS.
- K. Sokołowska, S. Malola, M. Lahtinen, V. Saarnio, P. Permi, K. Koskinen, M. Jalasvuori, H. Häkkinen, L. Lehtovaara and T. Lahtinen, J. Phys. Chem. C, 2019, 123, 2602–2612 CrossRef.
- Y. Zhang, I. Gallego, J. Plou, J. L. Pedraz, L. M. Liz-Marzán, J. Ciriza and I. García, Nanoscale, 2021, 13, 14354–14362 RSC.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, Nano Lett., 2007, 7, 2819–2823 CrossRef CAS PubMed.
- X.-S. Zheng, P. Hu, Y. Cui, C. Zong, J.-M. Feng, X. Wang and B. Ren, Anal. Chem., 2014, 86, 12250–12257 CrossRef CAS PubMed.
- A. Capocefalo, D. Mammucari, F. Brasili, C. Fasolato, F. Bordi, P. Postorino and F. Domenici, Front. Chem., 2019, 7, 413 CrossRef CAS PubMed.
- Z. Zhang, K. Bando, K. Mochizuki, A. Taguchi, K. Fujita and S. Kawata, Anal. Chem., 2019, 91, 3254–3262 CrossRef CAS PubMed.
- M. Xu, X. Ma, T. Wei, Z.-X. Lu and B. Ren, Anal. Chem., 2018, 90, 13922–13928 CrossRef CAS PubMed.
- L. E. Jamieson, D. J. Harrison and C. J. Campbell, Analyst, 2015, 140, 3910–3920 RSC.
- W. H. Skinner, N. Robinson, G. R. Hardisty, H. Fleming, A. Geddis, M. Bradley, R. D. Gray and C. J. Campbell, Chem. Commun., 2023, 59, 3249–3252 RSC.
- A. Williams, K. J. Flynn, Z. Xia and P. R. Dunstan, J. Raman Spectrosc., 2016, 47, 819–827 CrossRef CAS.
- S. Kang, W. Nam, W. Zhou, I. Kim and P. J. Vikesland, ACS Appl. Nano Mater., 2021, 4, 5768–5777 CrossRef CAS.
- J. Plou, P. S. Valera, I. García, D. Vila-Liarte, C. Renero-Lecuna, J. Ruiz-Cabello, A. Carracedo and L. M. Liz-Marzán, Small, 2023, 2207658 CrossRef CAS PubMed.
- N. M. Ralbovsky and I. K. Lednev, Chem. Soc. Rev., 2020, 49, 7428–7453 RSC.
- S. Đorđević, M. M. Gonzalez, I. Conejos-Sánchez, B. Carreira, S. Pozzi, R. C. Acúrcio, R. Satchi-Fainaro, H. F. Florindo and M. J. Vicent, Drug Delivery Transl. Res., 2022, 12, 500–525 CrossRef PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- R. Zhang, F. Kiessling, T. Lammers and R. M. Pallares, Drug Delivery Transl. Res., 2023, 13, 378–385 CrossRef PubMed.
- J. Jehlička and A. Culka, Anal. Chim. Acta, 2022, 1209, 339027 CrossRef PubMed.
- I. A. Bratchenko, L. A. Bratchenko, A. A. Moryatov, Y. A. Khristoforova, D. N. Artemyev, O. O. Myakinin, A. E. Orlov, S. V. Kozlov and V. P. Zakharov, Exp. Dermatol., 2021, 30, 652–663 CrossRef CAS PubMed.
- E. Garai, S. Sensarn, C. L. Zavaleta, N. O. Loewke, S. Rogalla, M. J. Mandella, S. A. Felt, S. Friedland, J. T. C. Liu, S. S. Gambhir and C. H. Contag, PLoS One, 2015, 10, e0123185 CrossRef PubMed.
- A. Kamińska, E. Witkowska, A. Kowalska, A. Skoczyńska, P. Ronkiewicz, T. Szymborski and J. Waluk, Anal. Methods, 2016, 8, 4521–4529 RSC.
- X.-S. Zheng, I. J. Jahn, K. Weber, D. Cialla-May and J. Popp, Spectrochim. Acta, Part A, 2018, 197, 56–77 CrossRef CAS PubMed.
- X. Huang, B. Sheng, H. Tian, Q. Chen, Y. Yang, B. Bui, J. Pi, H. Cai, S. Chen, J. Zhang, W. Chen, H. Zhou and P. Sun, Acta Pharm. Sin. B, 2023, 13, 1303–1317 CrossRef CAS PubMed.
- P. Wang, M. Xia, O. Liang, K. Sun, A. F. Cipriano, T. Schroeder, H. Liu and Y.-H. Xie, Anal. Chem., 2015, 87, 10255–10261 CrossRef CAS PubMed.
- X. X. Han, B. Zhao and Y. Ozaki, Anal. Bioanal. Chem., 2009, 394, 1719–1727 CrossRef CAS PubMed.
- S. Laing, K. Gracie and K. Faulds, Chem. Soc. Rev., 2016, 45, 1901–1918 RSC.
- J. Plou, I. García, M. Charconnet, I. Astobiza, C. García-Astrain, C. Matricardi, A. Mihi, A. Carracedo and L. M. Liz-Marzán, Adv. Funct. Mater., 2020, 30, 1910335 CrossRef CAS.
- M. I. Anik, A. Pan, M. G. Jakaria, S. A. Meenach and G. D. Bothun, ACS Appl. Nano Mater., 2022, 5, 14356–14366 CrossRef CAS.
- S. Lin, H. Ze, X. Zhang, Y. Zhang, J. Song, H. Zhang, H. Zhong, Z. Yang, C. Yang and J. Li, Angew. Chem., Int. Ed., 2022, 61, e202203511 CrossRef CAS PubMed.
- T. Kang, S. M. Yoo, I. Yoon, S. Y. Lee and B. Kim, Nano Lett., 2010, 10, 1189–1193 CrossRef CAS PubMed.
- Y. Wang, S. Rauf, Y. S. Grewal, L. J. Spadafora, M. J. A. Shiddiky, G. A. Cangelosi, S. Schlücker and M. Trau, Anal. Chem., 2014, 86, 9930–9938 CrossRef CAS PubMed.
- Y. Zhan, R. Fei, Y. Lu, Y. Wan, X. Wu, J. Dong, D. Meng, Q. Ge and X. Zhao, Analyst, 2022, 147, 4124–4131 RSC.
- I. Badillo-Ramírez, B. Landeros-Rivera, J. M. Saniger, J. Popp and D. Cialla-May, Analyst, 2023, 148, 1848–1857 RSC.
- J. E. L. Villa, I. Garcia, D. Jimenez de Aberasturi, V. Pavlov, M. D. P. T. Sotomayor and L. M. Liz-Marzán, Biosens. Bioelectron., 2020, 165, 112418 CrossRef CAS PubMed.
- B. Tadgell and L. M. Liz-Marzán, Chem. – Eur. J., 2023, e202301691 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |