
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strategies to address key challenges of metallacycle/metallacage-based supramolecular coordination complexes in biomedical applications
Dongdong
Xu
a,
Yang
Li
*a,
Shouchun
Yin
 *a and
Feihe
Huang
*bc
*a and
Feihe
Huang
*bc
aKey Laboratory of Organosilicon Chemistry and Materials Technology of Ministry of Education, College of Materials, Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou 311121, P. R. China. E-mail: liyang@hznu.edu.cn; yinsc@hznu.edu.cn
bStoddart Institute of Molecular Science, Department of Chemistry, Zhejiang University, Hangzhou 310058, P. R. China. E-mail: fhuang@zju.edu.cn
cZhejiang-Israel Joint Laboratory of Self-Assembling Functional Materials, ZJU-Hangzhou Global Scientific and Technological Innovation Center, Zhejiang University, Hangzhou, 311215, P. R. China
First published on 22nd February 2024
Abstract
Owing to their capacity for dynamically linking two or more functional molecules, supramolecular coordination complexes (SCCs), exemplified by two-dimensional (2D) metallacycles and three-dimensional (3D) metallacages, have gained increasing significance in biomedical applications. However, their inherent hydrophobicity and self-assembly driven by heavy metal ions present common challenges in their applications. These challenges can be overcome by enhancing the aqueous solubility and in vivo circulation stability of SCCs, alongside minimizing their side effects during treatment. Addressing these challenges is crucial for advancing the fundamental research of SCCs and their subsequent clinical translation. In this review, drawing on extensive contemporary research, we offer a thorough and systematic analysis of the strategies employed by SCCs to surmount these prevalent yet pivotal obstacles. Additionally, we explore further potential challenges and prospects for the broader application of SCCs in the biomedical field.
 Dongdong Xu | Dongdong Xu was born in Henan, China, in 1991. He received his PhD degree from Max Planck Institute for Polymer Research in 2020 under the direction of Prof. Katharina Landfester and Prof. Seraphine Wegner. He is currently a lecturer at Hangzhou Normal University. His research interests are focused on the construction of functional supramolecular coordination complexes and protein modified biomaterials. |
 Yang Li | Yang Li was born in Hubei, China, in 1989. She received her PhD degree from Zhejiang University for Chemistry Research in 2018 under the direction of Prof. Guping Tang. She is currently an Associate Professor at Hangzhou Normal University. Her research interests are mainly in the synthesis of functional supramolecular coordination complexes and exploration of their applications in bio-imaging and cancer therapy. |
 Shouchun Yin | Shouchun Yin received his PhD degree from the University of Science and Technology of China under the guidance of Prof. Wenfang Shi and Prof. Hongyao Xu in 2005. He worked in Prof. Xi Zhang's group at the University of Tsinghua from 2006 to 2007, Prof. Win Dehaen's group at Katholieke Universiteit Leuven from 2008 to 2010, and Prof. Peter J. Stang's group at the University of Utah from 2016 to 2017, as a postdoctor, respectively. He is currently a Professor at Hangzhou Normal University. His research interests are supramolecular coordination complexes, supramolecular polymers, opto-electric functional materials, fluorescent probes, and dyes. |
 Feihe Huang | Feihe Huang is a Changjiang Scholar Chair Professor of Zhejiang University. His current research is focused on supramolecular polymers and nonporous adaptive crystals (NACs). Awards and honours he has received include the Chinese Chemical Society AkzoNobel Chemical Sciences Award, The Cram Lehn Pedersen Prize in Supramolecular Chemistry, the Royal Society of Chemistry Polymer Chemistry Lectureship Award, and the Bruno Werdelmann Lectureship Award. His publications have been cited more than 32 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 476 times. His h-index is 98. He sits/sat on the Advisory Boards of JACS, Chem. Soc. Rev., ChemCommun, Macromolecules, ACS Macro Lett., and Polym. Chem. He is an Editorial Board Member of Mater. Chem. Front.
476 times. His h-index is 98. He sits/sat on the Advisory Boards of JACS, Chem. Soc. Rev., ChemCommun, Macromolecules, ACS Macro Lett., and Polym. Chem. He is an Editorial Board Member of Mater. Chem. Front.1. Introduction
Synthesizing therapeutic systems that involve strong covalent bonds is complex and cumbersome, often leading to challenges in achieving precise drug release and preserving drug activity due to their highly stable structures. These issues also complicate clinical translation.1–5 Consequently, the simple and flexible supramolecular self-assembly approach, leveraging interactions such as van der Waals,6 π–π stacking,7 hydrophobic,8 electrostatic,9 metal-coordination,10 hydrogen bonding,11 and host–guest interactions,12 has garnered significant interest in the biomedical field. Specifically, supramolecular systems assembled through weak non-covalent bonds (such as hydrophobic, hydrogen, and electrostatic interactions) often exhibit unstable structures. This instability can lead to disassembly in the complex in vivo microenvironments, resulting in diminished drug efficacy and potential harm to normal cells.13 Conversely, supramolecular self-assembled systems driven by metal-coordination bonds, possessing a strength intermediate between strong covalent and weak non-covalent bonds, offer relative stability and dynamic flexibility.14,15 Supramolecular coordination complexes (SCCs) developed by Verkade,16 Fujita,17,18 Stang,19,20 Nitschke,21–23 Therrien,24 Newkome,25 Raymond,26,27 and Mirkin28,29 have been intensively researched in the fields of functional polymers, molecular imaging, catalysis, biomedicine, sensing, and more.30–37SCCs typically comprise two-dimensional (2D) metallacycles and three-dimensional (3D) metallacages. These structures can exhibit customized shapes, sizes, topologies, and physicochemical properties by adjusting the chemical structures of the metal-based acceptors and organic donors, alongside the coordination angles.38–42 Over the past two decades, the chemotherapeutic function of metal ions in the coordination center, such as platinum (Pt),43 ruthenium (Ru),44 palladium (Pd),45 and iridium (Ir),46 combined with advancements in functional ligand design, has positioned SCCs as pivotal in linking multiple distinct functional molecules through metal–nitrogen (N) coordination bonds (e.g., Pt(II)–N and Ru(II)–N). This capability plays a significant role in enhancing therapeutic modalities, overcoming the limitations of traditional therapeutic methods, and advancing theranostics, among other biomedical applications.30–32 For instance, to enhance the generation of reactive oxygen species (ROS), SCCs can be assembled using Pt(II)–N or Ru(II)–N coordination bonds. These bonds leverage the heavy atom effect to facilitate intersystem crossing relaxation, increasing the yield of ROS in photodynamic therapy (PDT).47 Additionally, to address the challenges of poor PDT efficacy in the hypoxic microenvironments of tumor tissues and the inhibition of photothermal therapy (PTT) due to heat shock protein expression in tumor cells, SCCs can be employed to develop a synergistic PDT/PTT model.48 To tackle the limitations of short excitation wavelength and low tissue penetration depth in traditional phototherapy, BODIPY (fluoroboron dipyrrole) derivatives, featuring high quantum yields and stable chemical/photophysical properties,49–51 or organic ligands with a donor–acceptor–donor (D–A–D) structure52 can be employed for incorporation into SCCs. This approach extends the π-conjugation planes and enhances the push–pull electronic effect across the molecules, red-shifting the emission wavelength of the ligand and enabling phototherapy in the second near-infrared (NIR-II) region. Furthermore, to meet the dual requirements of diagnosis and treatment and to overcome the aggregation-induced quenching (ACQ) of fluorophores and the heavy atom effect on fluorescence imaging, aggregation-induced emission (AIE) units like tetraphenylethylene (TPE),53–57 triphenylamine,58,59 and other molecules can be integrated into the SCC system. This integration aids in constructing a theranostic platform.60 These strategic approaches address specific challenges across various domains, significantly broadening and deepening the biomedical applications of SCCs. Numerous reviews have focused on aspects such as molecular structure design, PDT/PTT applications, and theranostics in the near-infrared/infrared region.30,42,47,48,52 However, the path to clinical application of SCCs remains extensive. Beyond the aforementioned specific issues, SCCs also encounter several general but critical obstacles, including water solubility, biosafety, biocompatibility, potential toxicity, and biodistribution, which must be overcome before their real-world implementation. Despite considerable progress in these areas, comprehensive and up-to-date reviews addressing these advancements are lacking.
We emphasize the significant strategies employed by SCCs to address key challenges in biomedical applications. Our focus includes methods to impart appropriate water solubility to hydrophobic SCCs, enhancing their biocompatibility, strategies to improve cellular selectivity and reduce potential toxicity for increased biosafety, and techniques for precise drug distribution within cells to analyze active sites and mechanisms (Fig. 1). Finally, we summarize the current state-of-the-art research, discuss additional challenges for future applications, and propose potential research directions to tackle these issues.
 | ||
| Fig. 1 Schematic representation of general strategies employed by supramolecular coordination complexes (SCCs) to enhance water solubility and biostability and reduce potential toxicity and side effects for biomedical applications. | ||
2. Strategies to improve the water solubility and biostability of SCCs
The preparation process of SCCs typically progresses from synthesizing simple small-molecule components to assembling metallacycles/metallacages, predominantly conducted in organic solvents. Most chemotherapeutic drugs (such as doxorubicin (DOX) and paclitaxel (PTX)), phototherapeutic agents (like BODIPY), and AIE molecules, including TPE, possess hydrophobic conjugate groups. Consequently, the multifunctional SCCs constructed in this manner are typically hydrophobic and rigid structures. The hydrophobic nature of SCCs facilitates their rapid and firm binding to the lipophilic cell membrane surfaces,61 enhancing their cellular uptake efficiency. However, this hydrophobicity leads to their rapid aggregation and precipitation in the aqueous environment of living organisms, posing challenges in maintaining their in vivo stability and biocompatibility. Consequently, enhancing water solubility is crucial for the further biomedical application of SCCs. Presently, two primary strategies are employed to improve their water solubility (Fig. 2). One involves encapsulating hydrophobic SCCs within nanocarriers featuring hydrophilic surfaces.62–64 The other strategy is to chemically modify SCCs with hydrophilic groups or long chains, conferring a degree of water solubility (carrier-free).65–67 | ||
| Fig. 2 Schematic representation of general strategies of supramolecular coordination complexes (SCCs) to enhance water solubility and biostability for biomedical applications. | ||
2.1. Encapsulate SCCs with nanocarriers
Nanocarrier encapsulation strategies typically involve creating nanoparticles or vesicles that encapsulate SCCs within a hydrophilic exterior. The therapeutic efficacy of these nanocarriers is influenced by factors such as their surface charge, size, and chemical composition.68 Given that cell membranes are usually negatively charged, positively charged nanocarriers can facilitate enhanced cell adhesion and promote rapid endocytosis. However, a positive surface charge on nanocarriers may result in increased nonspecific protein adsorption in the bloodstream and heightened nonspecific phagocytosis by cells of the reticuloendothelial system.69 Therefore, nanocarriers with negatively or neutrally charged surfaces are also favored to mitigate these issues.70 Regarding their size, while the diameter range of drug delivery systems (DDS) is defined as 1–1000 nm,71 practical applications typically employ nanocarriers with diameters of 10–200 nm. This size range prevents early elimination by glomerular filtration due to the small carrier size (below 10 nm)72 and mitigates phagocytosis and destruction by the reticuloendothelial system for large-sized carriers. Consequently, this size range facilitates selective extravasation from the blood vessels and passive targeting to tumor tissues, leveraging the enhanced permeability and retention (EPR) effect.73–75 Data suggest that an appropriate nanocarrier size can maximize the benefits of the EPR effect, increasing drug concentration in tumor tissue by up to 10–100 times compared to healthy tissue.76 This can lead to more effective therapies at lower doses. However, there is still no definitive consensus on the precise size that ensures nanocarriers effectively overcome physiological barriers, while simultaneously exhibiting potent anticancer activity and minimal side effects.77 Indeed, the nanocarrier size is not static during blood circulation but typically decreases over time. Therefore, our subsequent discussion will consider both nanocarrier size and structure. In this context, liposomes prepared using DSPE–PEG (distearoyl phosphatidylethanolamine–polyethylene glycol) and block copolymers, exemplified by Pluronics, are two commonly utilized materials in nanocarrier fabrication. The key characteristics of this strategy are outlined in Table 1.| Topological structures | Nanocarriers used | Particle size (nm) | Biostability test | Aqueous solution | Load efficiency | Ref. |
|---|---|---|---|---|---|---|
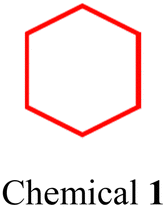
|
DSPE–mPEG2000 | 145 | 2 days | PBS | — | 83 |

|
DSPE–PEG2000–MAL | 49 | 7 days | H2O | — | 84 |

|
DSPE–mPEG5000 | 32 | 20 h | PBS + 10% FBS | — | 85 |

|
DSPE–PEG5000 | 220 | 7 days | PBS/PBS + 10% FBS/blood | — | 86 |

|
DSPE–mPEG5000 | 240 | 7 days | PBS + 10% FBS | — | 87 |

|
F127 | 150 | 21 days | H2O | 10% | 101 |

|
F127 | 130 | 4 days | H2O | 4.98% | 103 |
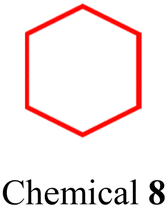
|
F127 | 110 | 7 days | PBS/FBS/H2O | — | 104 |

|
mPEG-b-PLGA | 65 | 7 days | PBS/H2O | — | 105 |

|
mPEG-b-PBLG | 134 ± 13.7 | — | — | 6.1% | 106 |

|
Heavy chain ferritin | 13.32 | 1 day | PBS/PBS + 10% FBS | — | 107 |

|
PEG/melanin dots | 100 | 7 days | PBS + 10% FBS | — | 108 |
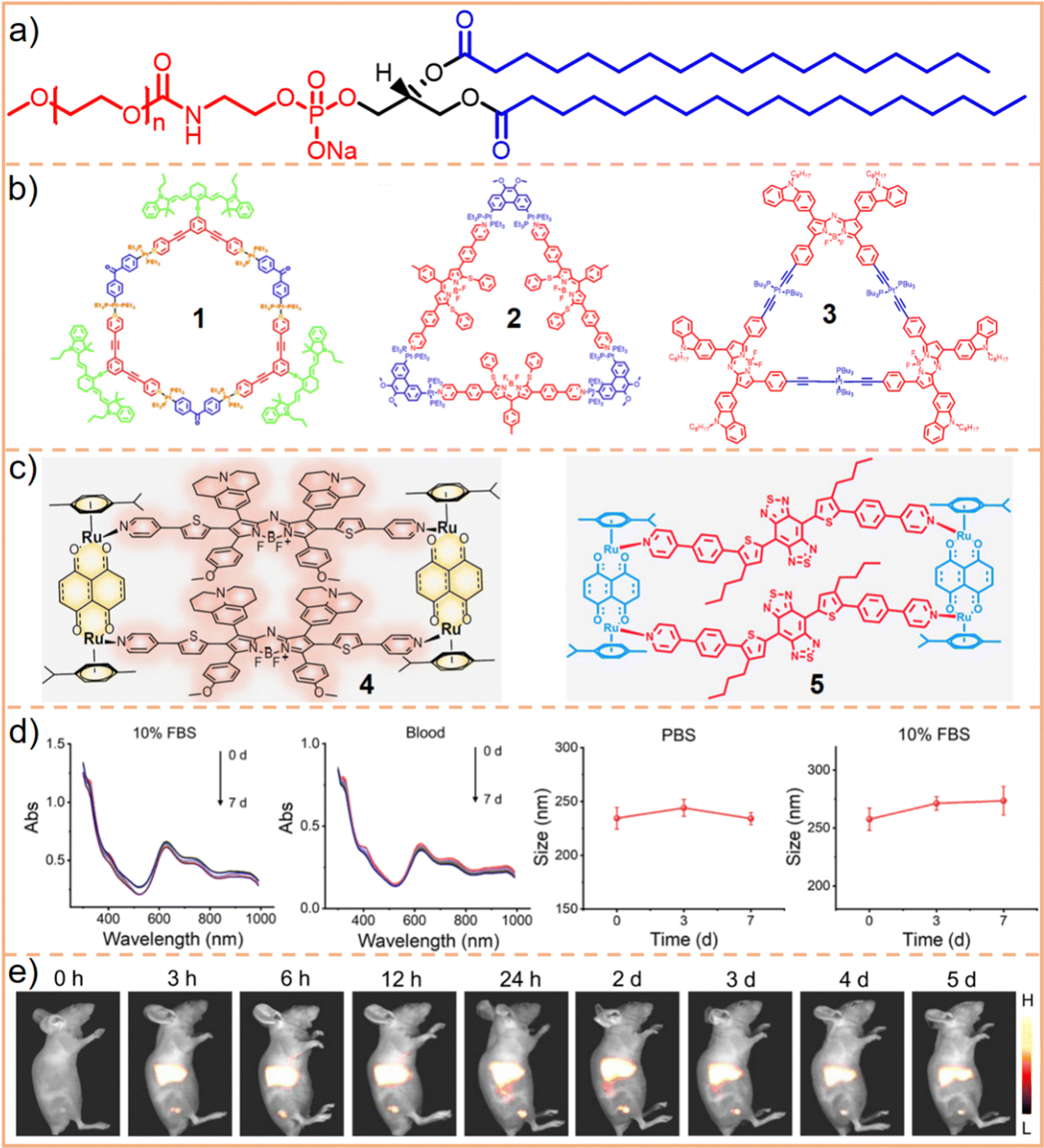 | ||
| Fig. 3 (a) Chemical structure of DSPE–PEG, where the hydrophobic chains are depicted in blue and the hydrophilic motif in red. Typically, n represents the chain length or molecular weight of PEG, such as DSPE–PEG2000 (n = 45) and DSPE–PEG5000 (n = 113). (b) Chemical structures of SCCs 1, 2, and 3, and (c) SCCs 4 and 5. (d) SCC 4 nanoparticles (encapsulated by DSPE–PEG5000), showing their hydrodynamic diameter when incubated in 10% FBS and whole blood and changes after incubation with PBS and 10% FBS over time. (e) Fluorescence images of tumor models post-injection with SCC 4 nanoparticles. This figure is adapted from ref. 86 with permission from Springer Nature, Copyright 2022. | ||
As illustrated in Fig. 3b, hexagonal83 (chemical 1) and triangular84,85 (chemicals 2 and 3) metallacycles were encapsulated using DSPE–PEG derivatives (DSPE–mPEG: DSPE-methoxy PEG; DSPE–PEG–MAL: DSPE-maleimide PEG), forming water-soluble nanoparticles. Our findings and those of our colleagues demonstrate that the size of these nanoparticles is influenced by the molecular structure and morphology of metallacycles. For instance, hexagonal metallacycles (chemical 1) possess a larger spatial structure compared to triangular metallacycles (chemical 2), leading to the formation of larger nanoparticles (145 nm vs. 49 nm). Furthermore, the final nanoparticle size is influenced by factors, including the type and molecular weight of PEG, the operational method, the size of the filter used, the ratio of SCCs to DSPE–PEG, and the stacking arrangement of nanoparticles. Consequently, predicting the size of nanoparticles is challenging, even when metallacycles of similar structures are synthesized using the same nanoprecipitation technique. However, these nanoparticles generally exhibit robust stability in aqueous solutions or physiological environments (such as phosphate-buffered saline (PBS) and fetal bovine serum (FBS)), maintaining consistent particle size and spectroscopic properties for durations ranging from 20 h to one week. After DSPE–PEG encapsulated nanoparticles are internalized by cells through fusion with the cell membrane phospholipid bilayer or endocytosis, the acidic lysosomal environment cleaves the Pt(II)–N coordination bond. This process releases the smaller components of SCCs, facilitating a synergistic chemotherapeutic effect with the heavy metals. Consequently, DSPE–PEG encapsulation ensures SCC stability during transport and preserves its therapeutic efficacy. This dual functionality substantially enhances the potential of hydrophobic SCCs to be utilized in physiological environments.
Compared to those formed through Pt(II)–N coordination, SCCs created using Ru(II)–N coordination exhibit slightly higher water solubility and can be directly utilized in PBS or FBS for in vitro experiments. Sun et al.86 synthesized a tetragonal metallacycle based on Ru-pyridine coordination (Fig. 3c, chemical 4), demonstrating effective PDT and PTT in the NIR-II region. These metallacycles show potential as antitumor phototherapeutic agents. However, for in vivo applications, encapsulating SCCs with DSPE–PEG remains essential to ensure prolonged circulation time and passive tumor targeting. The nanoparticles resulting from this encapsulation exhibit stable size and optical properties across various environments (PBS, FBS, whole blood) for up to one week (Fig. 3d). Moreover, a gradual increase in fluorescence intensity at the tumor site was observed, peaking 24 h post-injection, with the signal persisting for up to 5 d and exhibiting a high signal-to-background ratio (Fig. 3e). This suggests that DSPE–PEG encapsulated nanoparticles possess effective tumor accumulation capabilities. Additionally, they engineered and assembled different tetragonal Ru metallacycles87 utilizing a D–A–D structure (Fig. 3c, chemical 5). The nanoparticles produced following encapsulation with DSPE–mPEG5000 demonstrated efficacy in treating bacteria-infected wounds. These outcomes further underscore that DSPE–PEG encapsulation significantly enhances the applicability of SCCs in biomedical contexts.
 | ||
| Fig. 4 (a) Chemical structure of Pluronic F127, with the hydrophobic chain (PPO) depicted in blue and the hydrophilic motif (PEO) represented in red. Typically, a = 101 and b = 56 for F127. Chemical structures of SCCs 6 (b), 7 (c), and 8 (d). Chemical structures of SCC 9 (e), SCC 10 (f), mPEG-b-PLGA (g), and mPEG-b-PBLG (h). (i) Chemical structure of SCC 11 and the following assembly process of nanoparticles. Figure adapted from ref. 107 with permission from the Royal Society of Chemistry, Copyright 2023. (j) Chemical structure of SCC 12 and the following assembly process of nanoparticles. Figure adapted from ref. 108 with permission from Science China Press and Springer-Verlag, Copyright 2021. | ||
Compared to the hydrophobicity conferred by DSPE–PEG, which comprises only two alkyl chains with 18 saturated carbon atoms each, the hydrophobicity imparted by F127 with its PPO component of up to 3600 g mol−1 is markedly stronger. Consequently, F127 is frequently utilized for encapsulating certain highly hydrophobic SCCs or SCCs combined with hydrophobic small molecule drugs. Accordingly, we selected F127 for encapsulating BODIPY-based rhomboidal metallacycles (Fig. 4b, chemical 6)101 and various polydentate complexes.102 Both cases resulted in the formation of stable nanoparticles. Additionally, F127 has been utilized to encapsulate rhomboidal metallacycles composed of the highly hydrophobic AIE moiety triphenylamine and the dicyano group (Fig. 4c, chemical 7).103 Conversely, Kim et al.104 employed F127 to encapsulate hexagonal metal macrocycles (Fig. 4d, chemical 8) and hydrophobic NIR-II fluorescent molecules, achieving a stable particle size for up to one week in FBS, PBS, and water. These examples underscore the vital role of F127 in enhancing the compatibility of SCCs for various applications.
Beyond F127, various block copolymers have been explored for encapsulating SCCs. These copolymers typically feature mPEG as the hydrophilic group, while the hydrophobic chain's structure is modified to achieve specific encapsulation effects. Principally, for the purpose of improving the encapsulation efficiency, more flexible chains can be designed for the ligands or acceptors of SCCs, which facilitate the encapsulation of SCCs by polymers such as DSPE–PEG, F127, or other polymers through the entanglement with the flexible chains. However, if SCCs themselves have a large π-plane that does not contain any flexible chains, amphiphilic block polymers containing aromatic rings are a better choice to encapsulate metallacycles/metallacages by planar stacking between the carriers and SCCs. For instance, Wang et al.105 synthesized an amphiphilic polymer, mPEG-b-PLGA, by covalently linking hydrophobic polylactic acid (PLGA) with mPEG. This polymer successfully encapsulated a 3D metallacage (Fig. 4e, chemical 9) constructed via Pt(II)–N coordination, maintaining stability for one week in PBS and water. We106 substituted PLGA with poly(γ-benzyl-L-glutamate) (PBLG) to encapsulate a metallacage composed of anthracene, porphyrin, and a Pt(II) acceptor (Fig. 4f, chemical 10). The resulting nanoparticles enhanced the water solubility of the metallacage, and the increased hydrophobicity provided by PBLG helped maintain its integrity. In summary, compared to F127, mPEG-b-PLGA (Fig. 4g) possesses two modifiable hydrophobic chains, while mPEG-b-PBLG (Fig. 4h) incorporates an aromatic ring, rendering it more suitable for encapsulating compounds with highly hydrophobic and large rigid structures. Therefore, given that SCC 10 not only features a larger conjugation structure but lacks a flexible chain, unlike SCC 9, conventional amphiphilic block polymers (F127, mPEG-b-PLGA) may not yield effective encapsulation. Contrastingly, using amphiphilic block polymers containing aromatic rings facilitates metallacage encapsulation by leveraging the stacking interactions between the benzene rings and the rigid planes of SCCs. This tailored design of block copolymers efficiently addresses the requirements for adaptability and biostability in the complex SCC systems within in vivo environments.
2.2. Carrier-free SCCs prepared by functional motif modification
For a DDS, drug loading capacity (DL = m1/(m1 + m2), where m1 = mass of the drug and m2 = mass of the carrier) is a crucial metric that directly influences the dosage required for clinical application. Typically, a higher DL is preferable as it more readily meets clinical requirements. However, approaches utilizing nanocarriers to encapsulate SCCs often lead to a low DL,109,110 indicating that a substantial proportion of the material introduced into the body serves no therapeutic function but merely acts as a transport medium. This necessitates additional in vivo degradation, potentially impacting liver function and biliary excretion, posing short- or long-term toxicity risks to the human body.111,112 Consequently, despite the significant role that carriers play in DDS research, only a few, such as Pluronics and PEG, have received approval from the FDA. Moreover, using materials like Pluronics, which are relatively non-toxic in animal models, is subject to stringent limitations. For instance, the maximum permissible daily exposure for intramuscular injection of the inactive ingredient Pluronic F68 is limited to 4 mg;88 an intradermal injection of 137.5 mg kg−1 Pluronic F127 can lead to significant increases in cholesterol and triglycerides. Similarly, intraperitoneal injection of Pluronic F127 at dosages of 0.5–1 g kg−1 induces a hyperlipidemic effect.113,114 While nanocarriers can be employed to encapsulate SCCs to enhance their water solubility and biological stability, challenges such as low DL capacity and the excessive use of carrier materials must also be considered. Presently, the typical encapsulation techniques for SCCs, including nanoprecipitation,115–117 nanoemulsification,118–120 and thin-film hydration,121–123 face difficulties in addressing these specific challenges. In this context, a carrier-free self-delivery system can be developed by modifying SCCs to have an amphiphilic structure. In this system, SCCs themselves are an integral part of the carrier, eliminating the need for additional materials. This strategy effectively mitigates the toxicity issues associated with the degradation of excess carriers. Simultaneously, it ensures a degree of water solubility for SCCs, effectively resulting in a DL capacity of 100%.124 Unlike direct amphiphilic macromolecule encapsulation, this approach typically involves modifying hydrophobic donors or acceptors with hydrophilic small molecules125,126 or polymer chains.66,127 These modified components are then assembled into SCCs, which subsequently self-assemble into nanoparticles or vesicles due to their amphiphilic nature. These hydrophilic segments can reduce the adhesion of proteins and cells to the material and shield the hydrophobic components of SCCs from hydrolysis and enzymatic degradation. This enhances the circulation stability of SCCs in the bloodstream and facilitates the long-term sustained release of the materials. However, this carrier-free approach might require using small amounts of organic solvents, such as methanol, for nanoparticle formation. Additionally, maintaining long-term size stability under physiological conditions often presents challenges compared to the first method, highlighting an area that needs further exploration and improvement. Overall, developing carrier-free SCCs remains a promising strategy to enhance their water solubility. The characteristics of this approach are detailed in Table 2.| Topological structures | Hydrophilic units used | Particle size (nm) | Biostability test | Aqueous solution | Ref. |
|---|---|---|---|---|---|

|
Glycosyl group | 80 | — | H2O + 5% CH3OH | 128 |
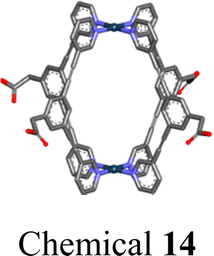
|
Peptide-(PEG)2 | — | 1 day | H2O | 129 |
|
Glycosyl group | 145 (chemical 15) | — | H2O + 50% CH3OH | 130 |

|
210 (chemical 16) | ||||

|
1008 (chemical 17) | ||||

|
(PEG)6 | 78 (chemical 18), 77 (chemical 19), 69 (chemical 20) | — | H2O + 6% CH3OH | 131 |
|
(PEG)3 | 110 | 1 day | PBS + 10% FBS | 65 |
|
PBEMA-b-POEGMA on SCCs | 80–120 | 2 days | PBS + 10% FBS | 133 |

|
POEGMA on SCCs | 130 | — | H2O | 134 |
We engineered a hydrophilic glycosyl group onto the 120° pyridine ligand and combined it with a 60° Pt(II) acceptor to construct a rhomboidal supramolecular metallacycle (chemical 13, Fig. 5a) exhibiting amphiphilic characteristics.128 At low concentrations, these rhomboidal SCCs can self-assemble into 80 nm nanovesicles in an aqueous environment (Fig. 5b). These nanovesicles are capable of acting as carriers for the hydrophobic anticancer drug DOX (Fig. 5c). The encapsulated DOX demonstrates prolonged release under neutral conditions and accelerated release in acidic environments, where the increased protonation of DOX enhances its solubility and leads to SCC disassembly. This release behavior is facilitated by the stabilizing effect of the glycosyl groups in an aqueous environment and the encapsulation within the amphiphilic SCCs (Fig. 5d). These findings demonstrate that amphiphilic SCCs can effectively serve as carriers in DDSs. Casini et al.129 modified a pyridine ligand with PepH3, a peptide that can translocate across the blood–brain barrier. This modification resulted in the formation of a water-soluble homoleptic metallacage upon the addition of coordination Pd2+ ions. Furthermore, this metallacage (chemical 14, Fig. 5e) was utilized as a targeted delivery system to encapsulate a radiotherapy agent. Chen et al.130 compared the structures of amphiphilic metallacycles, assembled in various ratios using different angular Pt(II) receptors on 120° pyridine ligands modified with galactoside. They also examined the sizes of nanoparticles or vesicles that are formed by their aggregation. Transmission electron microscopy (TEM) analysis revealed the diverse morphologies of nanoparticles and vesicles formed through the self-assembly of a galactoside-modified pyridine ligand with different acceptors. Specifically, after [2 + 2] self-assembly with the acceptor, resulting in a rhomboidal metallacycle (chemical 15), nanoparticles with an average size of 145 nm were observed (Fig. 6a). In the case of [3 + 3] self-assembly, which formed a hexagonal metallacycle (chemical 16), the structures manifested as 210 nm nanovesicles (Fig. 6b). Furthermore, [6 + 6] self-assembly, forming a larger hexagonal metallacycle (chemical 17), produced micrometer-sized microvesicles of 1.08 μm (Fig. 6c). These findings suggest a linear correlation between the size of the metallacycle and the resultant nanoparticle size for a given structural framework. The micrometer-sized vesicles formed by the [6 + 6] self-assembly exhibited potent biofilm inhibition properties against Staphylococcus aureus. Furthermore, they were effective in alleviating pneumonia in mice caused by this bacterium, demonstrating their potential therapeutic applications.
 | ||
| Fig. 5 (a) Chemical structure of the amphiphilic rhomboidal metallacycle 13 and schematic illustration of the formation of nanomicelles, and applications in the encapsulation of DOX. Dynamic light scattering (DLS) spectrum of nanomicelles before (b) and after (c) encapsulation of DOX. (d) Drug release curves of the metallacycle within DOX at pH = 7.5 and 5.0. Figure adapted from ref. 128 with permission from American Chemical Society, Copyright 2020. (e) PepH3 functionalized pyridine ligand and the formed metallacage 14. Figure adapted from ref. 129 with permission from American Chemical Society, Copyright 2021. | ||
 | ||
| Fig. 6 Schematic representation of the formation and hierarchical self-assembly morphologies of amphiphilic metallacycles and transmission electron microscopy (TEM) images of (a) [2 + 2] chemical 15, (b) [3 + 3] chemical 16, and (c) [6 + 6] chemical 17. Figure adapted from ref. 130 with permission from American Chemical Society, Copyright 2020. | ||
 | ||
| Fig. 7 Structures of metallacycles 18 (a), 19 (b), 20 (c), and 21 (d). | ||
 | ||
| Fig. 8 (a) Schematic representation of the preparation of a metallacycle with a supramolecular block copolymer (chemical 22) and (b) the chemical structure of PBEMA-b-POEGMA. Figure adapted from ref. 133 with permission from American Chemical Society, Copyright 2020. (c) Chemical structure of metallacycle 23. | ||
3. Strategies to reduce the potential toxicity and side effects of SCCs
As with any therapeutic intervention, potential toxicities and side effects must be thoroughly considered when administering drugs. Some of these adverse effects arise from the inherent chemical activity of the drug. For instance, the ROS generated by the interaction between DOX and iron can inflict damage on the myocardium,135,136 leading to myofibrillar loss and cytoplasmic vesiculation. Others are related to the method of administration. For example, the FDA-approved drug DOXIL (DOX encapsulated in PEG liposomes) exhibits a high tendency to aggregate at the skin due to its PEG coating. During administration, minute quantities of the drug may seep out of capillaries into the palms and soles, causing redness, swelling, tenderness, discomfort and even pain due to skin peeling. This condition is known as palmar-plantar erythrodysesthesia or hand-foot syndrome.137–139 Additionally, certain side effects may result from the treatment modality, such as chemotherapy-induced hepatitis B.140–142 As SCCs progress toward clinical trials, these medical challenges must be addressed to align with the principles of precision medicine. As illustrated in Fig. 9, critical issues for SCCs at the cellular level include: (1) cell selectivity: the ability to differentiate and target diseased cells while sparing normal cells is essential for minimizing collateral damage and enhancing treatment efficacy; (2) heavy metals: the risk of heavy metal leakage from SCCs during transport poses a significant safety concern. Ensuring the containment and stability of these complexes is crucial to prevent unintended systemic exposure. On the other hand, the therapeutic index can be optimized by using dual metal coordination centers; and (3) intracellular distribution: understanding the specific action sites and mechanisms of SCCs within cells is vital for predicting their therapeutic impact and potential side effects. Each of these factors plays a fundamental role in the successful development and application of SCCs in clinical settings. Addressing these challenges is paramount to harnessing the full potential of SCCs in precision medicine.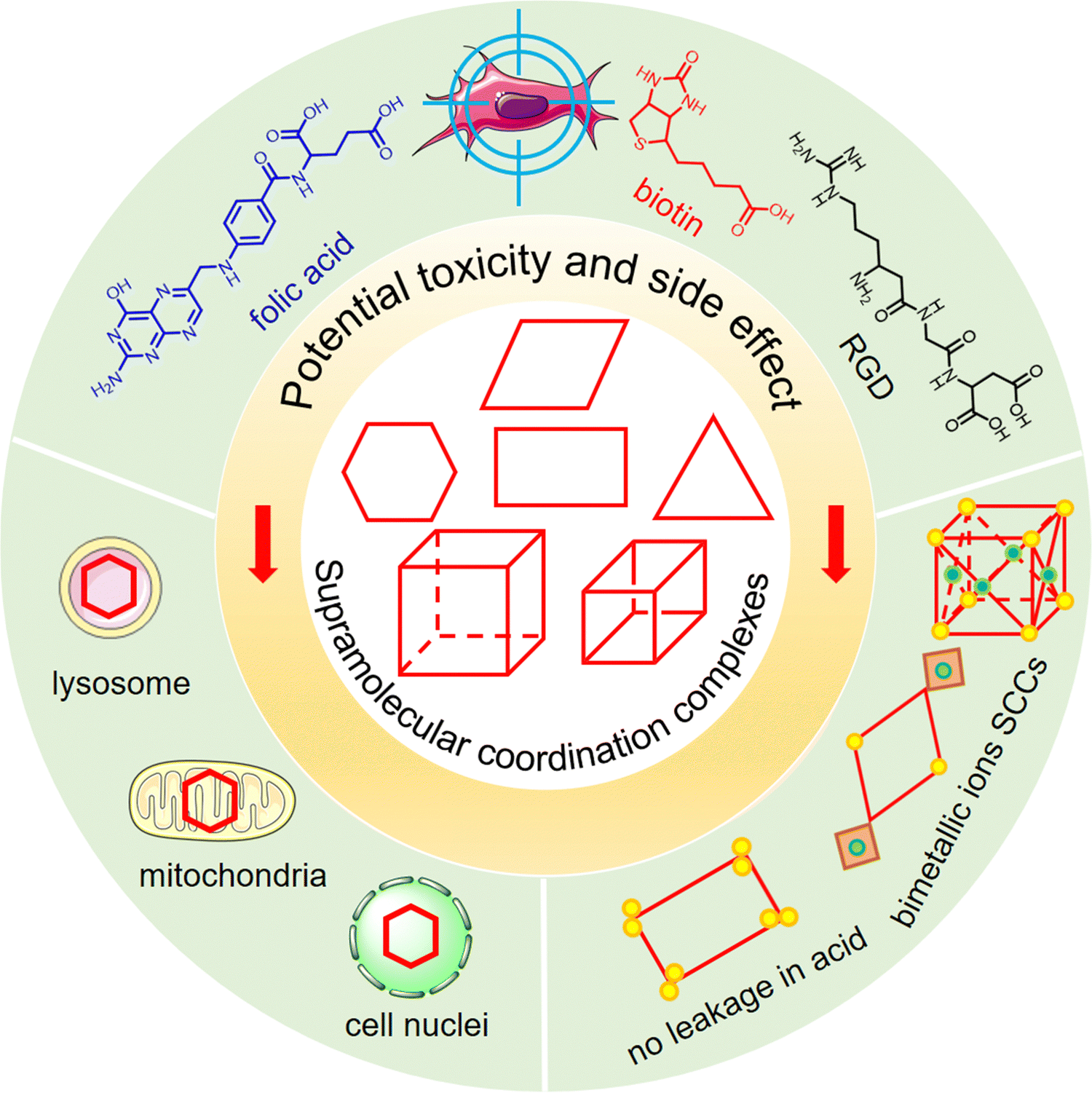 | ||
| Fig. 9 Schematic representation of general strategies of SCCs to reduce the potential toxicity and side effects for biomedical applications. | ||
3.1. Reduce the potential toxicity and side effects of SCCs by active cell selectivity
The non-selective drug action on diseased and healthy cells in vivo is a primary contributor to its toxic side effects. Enhancing the cell selectivity of SCCs to effectively distinguish between these two cell states is critical for minimizing these adverse effects. Although SCCs, as currently assembled, generally demonstrate improved cell uptake and, to a certain extent, better selectivity between diseased and healthy cells compared to their small-molecule counterparts, their performance in this regard is not yet optimal. This relative selectivity may be attributed to SCCs possessing multiple positive charges, a certain degree of lipophilicity, and unique ring tension.42,143–149 However, further improvements are necessary to achieve satisfactory selectivity for clinical applications. Various strategies have been employed to encapsulate SCCs into nanoparticles or vesicles to enhance their water solubility and biostability while conferring a passive EPR effect. This effect is intended to augment SCC accumulation in tumor tissues. However, the effectiveness of the EPR effect is subject to ongoing scrutiny due to several factors:150–161 (1) heterogeneity of the vascular system: there is considerable variability in the vascular systems across species and tumor types, which can significantly impact the distribution and effectiveness of nanomedicines; (2) tumor microenvironment variability: the diverse characteristics of tumor microenvironments, such as pH, interstitial pressure, and extracellular matrix composition, can influence the action and efficacy of nanomedicines; and (3) low delivery efficiency to solid tumors: studies indicate that only 0.7% of systematically administered nanomedicines reach solid tumors. The significant attrition rate in the efficacy of nanomedicines, primarily due to insufficient accumulation of nanoparticles in tumors and suboptimal pharmacokinetics, has led to skepticism about their effectiveness. This situation suggests that an excessive emphasis on the EPR effect might be misleading in the design of nanomedicines.162,163 In this scenario, modifying SCCs with tumor-targeting moieties, such as folic acid (FA),164 biotin,165 Arg–Gly–Asp (RGD)166–168 sequence, and other peptides,169 offers a promising strategy. This approach aims to selectively concentrate the drugs in tumor tissues and cells through active recognition and binding of these targeting moieties to specific molecules and proteins present at tumor sites. Consequently, this endows SCCs with active cellular selectivity. For a more comprehensive understanding, the general pathway of SCCs equipped with targeting units is depicted in Fig. 10. Additionally, the characteristics of SCCs with active cell selectivity are outlined in Table 3.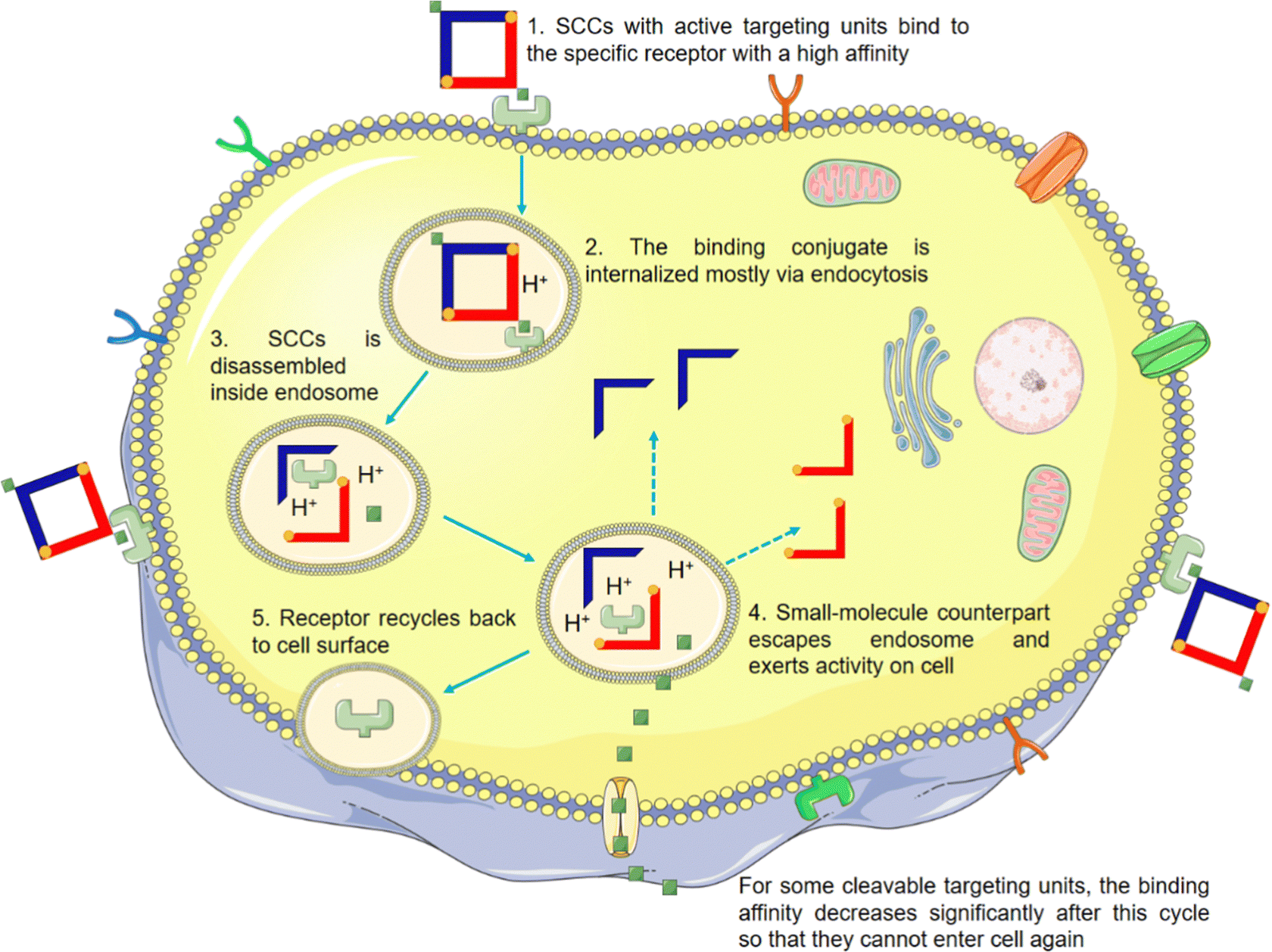 | ||
| Fig. 10 Schematic representation of tumor cellular uptake of SCCs modified with targeting motifs (e.g., FA, biotin, cRGDfk, etc.). Typically, a receptor binds to these modified SCCs with high affinity, facilitating endocytosis and internalization. Inside the lysosome/endosome, SCCs disassemble, allowing the small-molecule counterparts to escape and exert their activity within the cell. Subsequently, the vesicle, with receptors, returns to the cell plasma membrane. For certain cleavable targeting units, the binding affinity significantly decreases after this cycle, preventing further cellular entry. | ||
| Target units used | Targeting receptors | Cell lines | Targeting outcomes | Therapy outcomes | Ref. |
|---|---|---|---|---|---|
| FA | FR | Higher intracellular Pt concentration | Lower IC50 | 164 | |
| DU145 | 1.6-fold | 49%; with 1.4-fold higher level of apoptosis and necrosis | |||
| HeLa | 2.3-fold | 20% | |||
| A549 | 1.9-fold | 56% | |||
| HCT116 | 2.8-fold | 40% | |||
| HEK293 (FR-negative) | 1.1-fold | 84% | |||
| Biotin | BR | Fluorescence image | Lower IC50 | 165 | |
| 4T1 | Brighter | 20% | |||
| HepG2 | Brighter | 19% | |||
| HCT116 (BR-negative) | No difference | 92% | |||
| cRGDfk | αvβ3 integrin | U87MG | 2.5-fold higher fluorescence intensity; 2.1-fold higher intracellular Pt concentration | Pretreatment with free cRGDfK resulted in diminished cytotoxicity | 207 |
| cRGDfk | αvβ3 integrin | A2780 | Pretreatment with free cRGDfK resulted in diminished internalization | 3.8-fold higher IC50 after pretreatment with free cRGDfK | 210 |
| (Tyr3)-octreotate peptide | sst2R | CHOsst2 | — | — | 169 |
FA and its receptor, the folate receptor (FR), a glycosylated phosphatidylinositol-linked membrane glycoprotein, exhibit a very high affinity under physiological conditions. When FA is conjugated with a carrier, this complex can enter cells expressing the FR via the same pathway as free FA.176–178 Utilizing this characteristic, Stang et al.164 employed an FA-modified PEG-PLGA block copolymer (Fig. 11a) to encapsulate a square metallacycle. This metallacycle (3-NF-FA) was composed of a camptothecin-modified pyridine ligand and a combretastatin A-4-modified Pt(II) acceptor. Cell uptake analysis revealed that 3-NF-FA achieved a higher intracellular Pt concentration compared to its non-modified counterpart, 3-NF, in FR-positive HeLa cell lines (Fig. 11b). This increased uptake was not observed in FR-negative HEK293 cells (Fig. 11c). Further experiments showed that when FR-positive cells were pre-treated with free FA, 3-NF-FA uptake significantly decreased. Conversely, pre-treating HEK293 cells, which lack FR, with free FA had negligible effects on the endocytosis of 3-NF-FA, highlighting its potential for selective delivery in cells overexpressing FR. Consequently, 3-NF-FA demonstrated a greater cytotoxicity than 3-NF (Fig. 11d). Moreover, apoptosis analysis revealed that treatment with 3-NF-FA induced a higher level of apoptosis and necrosis, totaling 50.14%, whereas a lower proportion of apoptotic and necrotic cells was observed in cells treated with 3-NF (34.93%). These results confirmed that the introduction of FA enhances cellular uptake in cancer cells through FR-mediated endocytosis. This leads to higher drug concentrations within the cancer cells, effectively achieving targeted cell selectivity.
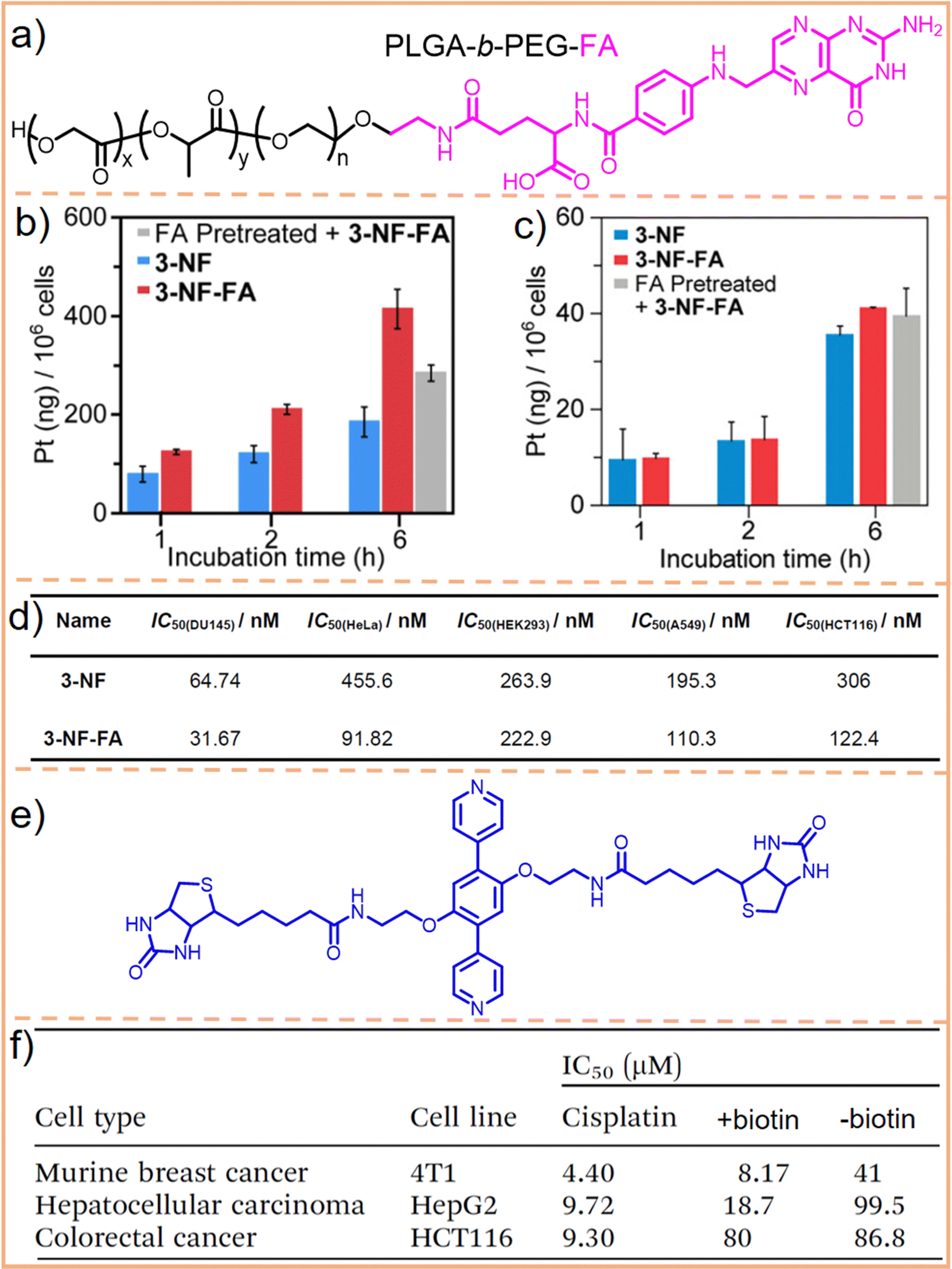 | ||
| Fig. 11 (a) Chemical structure of PLGA-b-PEG-FA. Cell uptake analysis of 3-NF-FA and 3-NF in FR-positive HeLa cells (b), and FR-negative HEK293 cells (c), with or without preincubation with 1 mM free FA for 2 h. (d) Cytotoxicity analysis of 3-NF and 3-NF-FA on various cell lines. Figure adapted from ref. 164 with permission from American Chemical Society, Copyright 2022. (e) Chemical structure of the biotin-modified pyridine ligand and (f) IC50 values of the metallacage with or without biotin modification and cisplatin towards HepG2, 4T1, and HCT116 cells. Figure adapted from ref. 165 with permission from the Royal Society of Chemistry, Copyright 2020. | ||
Compared to FA, biotin exhibits a relatively low molecular weight and simple chemical structure. It also has low toxicity (biotin is one kind of B vitamin, also known as vitamin H, vitamin B7, and coenzyme R, which is indispensable for the normal metabolism of fats and proteins) and a highly specific tumor-targeting capability due to its interaction with the biotin receptor (BR, a glycoprotein overexpressed in tumor cells, exhibits a high affinity for biotin and its derivatives).179–181 We utilized biotin to modify DSPE–PEG macromolecules and encapsulated 3D metallacages within them.182 This modification endowed metallacages with excellent biostability and conferred them with targeted delivery capabilities through receptor-mediated endocytosis. Recently, Zhang et al.165 utilized biotin to modify a pyridine ligand (Fig. 11e), assembling it into a 3D supramolecular metallacage. This modification aimed at differentiating between BR-positive hepatocellular carcinoma cells (HepG2) and murine breast cancer cells (4T1), and BR-negative colorectal cancer cells (HCT116). The findings revealed that HepG2 and 4T1 cells incubated with the biotin-modified metallacages exhibited high fluorescence brightness. Additionally, the half maximal inhibitory concentration (IC50) of metallacages for 4T1 and HepG2 cells was merely 20% and 19%, respectively, of that observed for metallacages without biotin modification. Conversely, IC50 of metallacages for HCT116 cells did not significantly differ (Fig. 11f). These results demonstrate the efficacy of biotin in enhancing the tumor-targeting capabilities of SCCs.
In our previous work, we utilized cRGDfk to modify the amphiphilic block polymer PEG-b-PEBP (poly(2-ethylbutoxy phospholane)) (Fig. 12a). This modified polymer was then employed to encapsulate 3D metallacages, assembled from porphyrin, organoplatinum(II), and disodium terephthalate.207 The effectiveness of this approach was evaluated using confocal laser scanning microscopy (CLSM) and inductively coupled plasma mass spectrometry (ICP-MS). The results from these analyses demonstrated that integrin αvβ3 receptor-mediated endocytosis significantly enhanced the tumor cell targeting capability of metallacages. As depicted in Fig. 12b, the fluorescence intensity of cRGDfk-modified nanoparticles significantly increased over time. After 4 h of incubation, the fluorescence intensity of these cRGDfk-modified nanoparticles was 2.5 times higher than that of nanoparticles without cRGDfk modification. This observation indicates enhanced cellular uptake due to the specific interaction of the cRGDfk modification with integrin receptors. Furthermore, when cells were pre-treated with free cRGDfk before introducing nanoparticles, the fluorescence intensity considerably decreased. This reduction suggested that the presence of free cRGDfk competes with the cRGDfk-modified nanoparticles for integrin binding, inhibiting their internalization. Likewise, pre-treatment with cRGDfk resulted in a reduction in the intracellular Pt content (Fig. 12c). These experiments proved that the binding of cRGDfk to integrin receptors enhances the cellular uptake and internalization of metallacages. Chen et al.208 and Zheng et al.209 employed a similar strategy to encapsulate 3D metallacages with varying structures. This methodology effectively endowed SCCs with the capability of actively recognizing tumor cells, leveraging the targeting potential of the cRGDfk peptides. Building upon this concept, Mao et al.210 further leveraged the integrin-mediated endocytosis and the elevated concentration of reduced glutathione (GSH) found in tumor cells. They accomplished this by modifying poly(ethylene glycol) methacrylate (POEGMA)–poly(2-azidomethyl benzoyl glycerol methacrylate) (PAZMB) with cRGDfk (Fig. 12d). These modified copolymers were then used to encapsulate hexagonal metallacycles. The hydrophilic nature of POEGMA ensures prolonged circulation time of nanoparticles in the bloodstream. PAZMB enhances the loading capacity of metallacycles through π–π stacking and enables GSH-responsive drug release. This responsive release mechanism is triggered by a reduction-induced cascade elimination reaction of the azidomethyl benzoyl group. Incorporating cRGDfk into the system enhanced the active selectivity of metallacycles toward tumor cells. This was demonstrated by the marked decrease in the fluorescence intensity of metallacycles in tumor cells, along with a significant increase in their IC50 value against tumor cells (from 1.88 ± 0.14 to 7.16 ± 0.54 μM) following the addition of free cRGDfk. This finding suggests that the presence of free cRGDfk competes with the cRGDfk-modified metallacycles for binding to integrin receptors, reducing the uptake and effectiveness of metallacycles in tumor cells. Beyond the cRGDfk peptide, various other types of polypeptides have been explored as tumor cell targeting moieties. Casini et al.169 developed the Pd2L2 metallacycle (where L = 3,5-bis(3-ethynylpyridine)phenyl) and modified it with an octapeptide on the SCCs, targeting the somatostatin-2 receptor (sst2R), which is pathologically overexpressed in various neuroendocrine tumors. This specific targeting approach aimed to achieve selective binding to tumor cells (Fig. 12e). Using peptides to modify metallacycles/metallacages is a strategic response to the unique endogenous characteristics of tumor cells. Such targeted designs are crucial for enhancing the active selectivity of SCCs towards tumor cells, which contributes to reducing their toxic side effects on healthy cells.
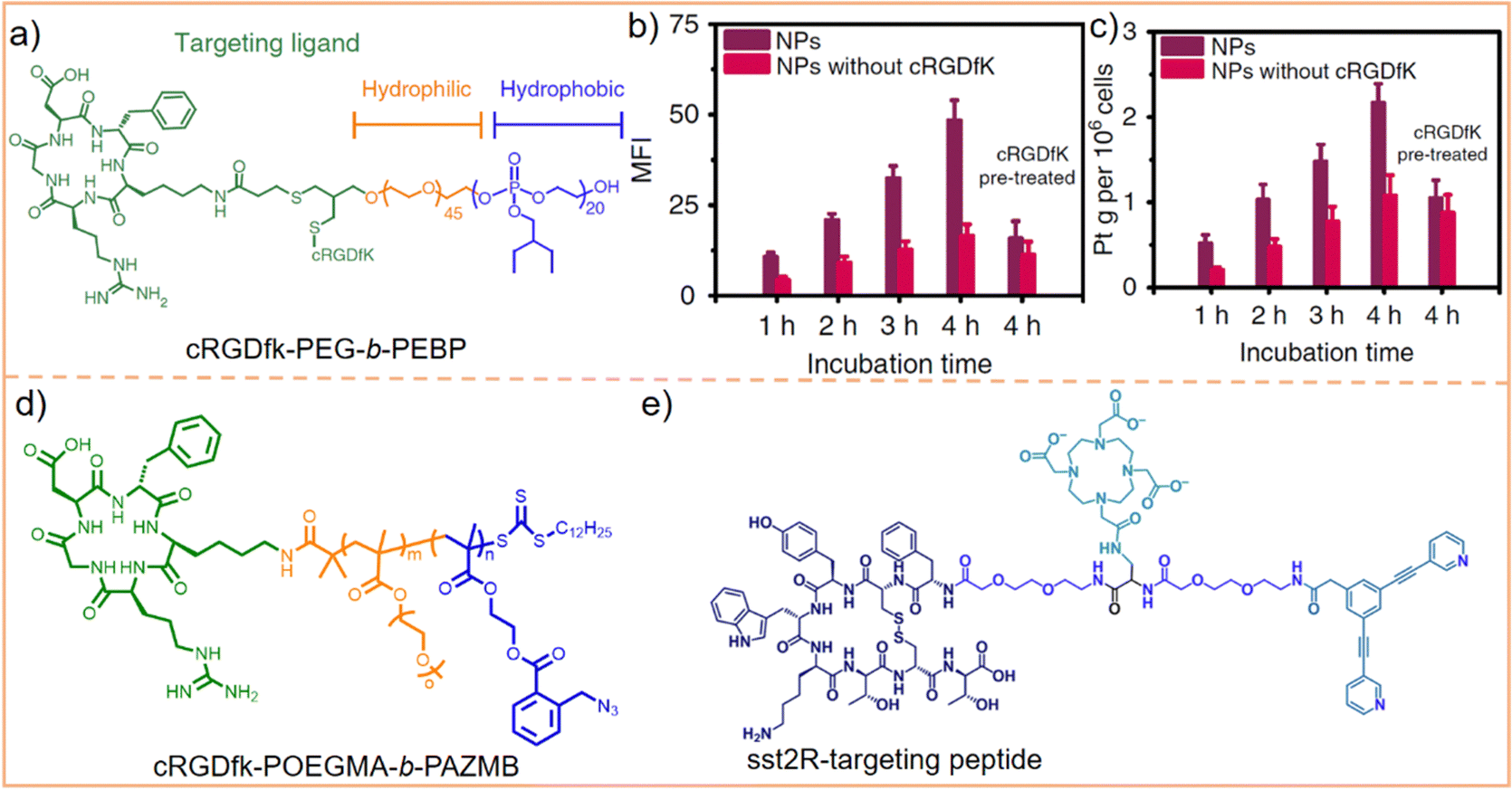 | ||
| Fig. 12 (a) Chemical structure of cRGD-PEG-b-PEBP. (b) Mean fluorescence intensity (MFI) and (c) intracellular Pt amounts of tumor cells after treatment with nanoparticles with or without cRGDfK for different incubation times. Figure adapted from ref. 207 with permission from Springer Nature, Copyright 2018. (d) Chemical structure of cRGD-POEGMA-b-PAZMB and (e) chemical structure of the sst2R-targeting peptide functionalized pyridine ligand. | ||
3.2. Reduce the potential toxicity and side effects of SCCs by changing the metal coordination centers
While various metals can be utilized to form coordination bonds, the assembly of SCCs predominantly employs heavy metals with strong coordination capabilities, such as Pt, Ru, Pd, and Ir. These heavy metals serve dual roles in assembled SCCs. They can act as chemotherapeutic agents, effectively killing tumor cells or bacteria. However, the excessive use of heavy metals poses significant health risks. They can potentially lead to DNA (deoxyribonucleic acid) mutations, autoimmune diseases, respiratory and hearing problems, and damage to vital organs, including the intestines, kidneys, and bone marrow.211–213 The issue of heavy metal misuse due to drug resistance is a significant concern. For instance, organoplatinum, commonly used as a coordination center in the assembly of SCCs, exhibits effects similar to those of cisplatin. However, congenital and acquired resistance to platinum-based chemotherapy has been observed in patients over long-term clinical use,214–218 significantly limiting the effectiveness of these drugs. Additionally, heavy metal misuse can stem from the instability of the assembled SCCs. If these complexes are not sufficiently stable, they may disassemble during transport within the body, leading to the unintended release of metal ions into non-diseased tissues. Altering the metal ion species at the coordination center is a viable option to reduce the potential toxicity of SCCs in biomedical applications. Opting for lighter metals that are essential to human health as coordination centers could be an ideal solution. However, the weaker coordination capabilities of these lighter metals compared to their heavy metal counterparts have led to their limited application in the SCC research in the biomedical field. For instance, Chan et al.219 synthesized diverse heteroleptic metalla-supramolecules using cadmium-N coordination bonds. However, their work was primarily focused on the synthesis aspect, with no subsequent exploration of biomedical applications. Current research focuses on enhancing the efficacy of SCCs in killing cancer cells or bacteria and improving their stability to mitigate the potential toxicity and side effects. To provide a clearer understanding, the general characteristics of SCCs achieved by altering the metal coordination centers are summarized in Table 4.| Topological structures | Metal ions | Physicochemical properties outcomes | Therapy outcomes | Ref. |
|---|---|---|---|---|
|
Ir(III)–Pd(II) | 25.3-fold higher TPA; 1.3-fold higher 1O2 quantum yield; brighter fluorescence image | Lower IC50; 5-fold higher PDI; 28% tumor volume at 20 days of post-treatment | 220 |
|
Ru(II)–Pt(II) | 16.5-fold higher TPA; 1.1-fold higher 1O2 quantum yield; brighter fluorescence image | Lower IC50; 5–33-fold higher PDI for different cell lines | 221 |
|
Ru(II)–Pt(II) | 33.9-fold higher TPA; 1.3-fold higher 1O2 quantum yield; brighter fluorescence image | 59% lower IC50; 11.2-fold higher PDI | 222 |

|
Pd(II) | Less than 8% change in the intensity of absorption spectra at pH = 7.4, 6.8, and 5.3 | Imaging-guided PDT; no in vivo experiment | 227 |

|
Ru(II) | Imidazole–Ru(II) coordination; no significant changes in the absorbance spectra from pH 4.5 to 8.0 | Multimodal therapy; no in vivo experiment | 228 |

|
Pt(II) | Maintained for two days at pH 5.5 without obvious changes | Synergistic photochemotherapy; no in vivo experiment | 229 |
 | ||
| Fig. 13 Scheme of bimetallic SCCs assembled (a) by the donor and the acceptor with two species of metal ions and (b) by the donor with one metal ion and the acceptor with another metal ion. (c) Chemical structures of the Ir(III)–Pd(II) metallacage and the Ru(II)–Pt(II) metallacycle (d) and the metallacage (e). | ||
Utilizing this concept, Su et al.220 successfully assembled eight Ir(III)-based photosensitizers with four Pd(II)-based receptors into a Pd4Ir8 metallacage (Fig. 13c). This structure demonstrated a maximum two-photon absorption (TPA) cross-section of 783 GM, while that of the Ir(III) ligand under the same experimental conditions peaked at 31 GM. This enhanced two-photon activation ability of the Pd4Ir8 metallacage facilitates two-photon PDT in tumor treatments and effectively increases the treatable depth of photosensitizers. Moreover, owing to an enlarged conjugation plane and efficient energy transfer facilitated by its highly ordered cubic cage structure, the singlet oxygen (1O2) quantum yield of the metallacage was higher than that of the Ir(III) ligand (0.84 vs. 0.67). Consequently, the bimetallic Pd4Ir8 metallacage demonstrated superior cell-killing ability and a higher PDI (the ratio of photocytotoxicities to dark cytotoxicities) compared to the monometallic Ir(III) ligand. Stang et al. designed heterometallic Ru(II)–Pt(II) metallacycles221 and metallacages222 (Fig. 13d and e, respectively). The metallacage exhibited the highest TPA and enhanced 1O2 quantum yield compared to both the metallacycle and the Ru(II) ligand. This superior performance resulted in better therapeutic efficiency and a higher phototoxicity index against tumor cells. These experimental results strongly suggest that incorporating bimetallic ions is a viable strategy for augmenting the therapeutic capabilities of SCCs.
Moreover, imaging agents used for cellular imaging require structural stability in complex biological systems. This stability is crucial for maintaining the consistency and sustainability of signal intensity, making stable SCCs particularly advantageous for imaging-guided therapies. Chen et al.227 designed a square metallacycle based on Pd–N coordination, named Pd8L4 (where L refers to N,N-3,5-dimethylpyrazole substituted perylene tetracarboxylic acid diimides) (Fig. 14a). This metallacycle demonstrated exceptional stability, with less than an 8% change in the intensity of absorption spectra under normal physiological conditions (pH = 7.4), in the slightly acidic microenvironment of tumor tissues (pH = 6.8, Fig. 14b) and in the more acidic environment of lysosomes (pH = 5.3, Fig. 14c). This robust stability makes Pd8L4 an ideal candidate for imaging-guided PDT of tumors. Sun et al.228 designed imidazole ligands and coordinated them with Ru(II) to synthesize various tetragonal metallacycles (Fig. 14d). These metallacycles exhibited exceptional stability, with no significant changes observed in their absorbance spectra across a pH range from 4.5 to 8.0. Song et al.229 developed a triangular metallacycle (Fig. 14e), demonstrating remarkable stability over two days in a slightly acidic environment at pH 5.5 (Fig. 14f). These studies provide valuable insights into the development of acid- and alkali-resistant SCCs, setting a precedent for future research aimed at creating SCCs with even greater stability and safety.
 | ||
| Fig. 14 (a) Construction of the Pd8L4 metallacycle and changes in the absorbance spectra of the metallacycle versus time in PBS buffer at (b) pH = 6.8 and (c) pH = 5.3. Figure adapted from ref. 227 with permission from the Royal Society of Chemistry, Copyright 2020. (d) The design and chemical structures of various Ru metallacycles. (e) Schematic representation of synthesis of the metallacycle and (f) absorption profiles of the metallacycle at pH = 5.5 at different times. Figure adapted from ref. 229 with permission from American Chemical Society, Copyright 2021. | ||
3.3. Reduce the potential toxicity and side effects of SCCs by monitoring their subcellular distribution for precision medicine
The subcellular distribution of drugs within eukaryotic cells plays a crucial role in determining their efficacy and toxicity. Studying this distribution is vital for drug targeting strategies and for minimizing toxic side effects, aligning with the fundamental principles of precision medicine.230 Eukaryotic cells comprise various organelles, each with distinct biological characteristics and functions. These include the nucleus, mitochondria, lysosomes, endoplasmic reticulum, and Golgi apparatus, among others. Investigating how drugs are distributed across these different organelles can offer valuable insights into the specific sites and mechanisms of action of these drugs. For instance, mitochondria can release cytochrome c into the cytoplasm, which activates caspase 3, a key enzyme in the execution phase of apoptosis.231–233 Drugs that target mitochondria and influence this pathway can effectively induce apoptosis in diseased cells, such as cancer cells. The Golgi plays a role in protein processing and trafficking, and its dysfunction can trigger cell death. Particularly, oxidative stress within the Golgi apparatus can initiate an apoptotic pathway.234 Finally, DNA damage within the nucleus can directly lead to cell death. Currently, leveraging insights from studies on the chemical structure–localization relationship and organelle feature–localization relationship of small molecules,235,236 the research into the subcellular distribution of SCCs is advancing. Inside the cell, SCCs typically accumulate in lysosomes and mitochondria,237–242 except for certain metallacycles/metallacages that enter the nucleus.243 For a more comprehensive understanding, the general features of SCCs with different subcellular locations are summarized in Table 5.| Subcellular locations | Internalization pathways | Outcomes of distribution study | Outcomes of accumulation | Ref. |
|---|---|---|---|---|
| Lysosomes | — | Lysosome correlation coefficients were in the range of 0.73–0.89 for different cells | Lysosome-targeted cell imaging | 239 |
| Lysosomes | Energy-dependent clathrin-mediated and caveolae-mediated pathways | Lysosome correlation coefficient = 0.75; 78.5% of SCCs in lysosomes, 17.6% in mitochondria | Lysosomal damage; mitochondrial dysfunction; cell apoptosis | 240 |
| Lysosomes | Energy-dependent caveolae-mediated endocytic pathway | Lysosome correlation coefficient = 0.7; Ru content was 1.73-fold higher in lysosomes than that in mitochondria | Lysosomal and mitochondrial damage and dysfunction; cell cycle arrest at the G2/M phase; cell apoptosis | 241 |
| Mitochondria | — | 62.5 ± 4.3% of Ru in the mitochondria | Mitochondrial membrane depolarization and superoxide generation; mitochondrial dysfunction; cell apoptosis | 289 |
| Mitochondria/lysosomes | Energy-dependent endocytosis | 55% 27 was in cytosolic fraction, and less in the mitochondria (37%); 58% 28 was in mitochondria, followed by the cytosolic (29%) fraction | Disruption of lysosomal integrity, mitochondrial membrane depolarization, perturbation of autophagy, necrotic cell death and cell apoptosis | 238 |
| Mitochondrial DNA | — | Mitochondrial correlation coefficient = 0.88 (30); higher mitochondrial DNA binding capacity with binding to the minor groove of DNA | The most efficient PDT efficacy with PDI > 33.3 and 97.5% total apoptotic ratio; cell apoptosis | 237 |
| Cell nucleus | — | Metallacycle target genes were located in chromosomal regions enriched with G4 structures and exhibited bright fluorescence in cell nucleus | Metallacycles interfere with the G4 epitope recognition by the quadruplex specific antibody BG4, replacing the latter to bind to G4 and kill tumor cells | 243 |
| Cell nucleus | Clathrin-mediated endocytosis pathway | Cytotoxic protein and genome-editing proteins were delivered into cell nucleus | Tumor cell death and specific protein expression | 312 |
| Cell nucleus | Caveolae/lipid raft-mediated endocytosis pathway | Higher protein delivery efficiency than commercial reagents; a majority of protein was released into cytosol and accumulated within nucleus after 18 h of delivery | Molecular chaperone was delivered into cell nucleus for neurodegenerative disease treatment | 313 |
Given the significance of lysosomes in mediating cell death, investigating the localization of SCCs within lysosomes is crucial and necessary. Sun et al.240 assembled an Ru-based metallacycle (chemical 24, Fig. 15a) for colocalization experiments. After 6 h of incubation, predominant metallacycle accumulation in lysosomes was observed (correlation coefficient = 0.75), while smaller amounts of these complexes were found in the mitochondria. Further analysis using laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) revealed that 78.5% of metallacycles appeared in lysosomes, 17.6% in mitochondria, and almost none in the nucleus (Fig. 15b). On this basis, the internalization of metallacycles was confirmed to occur through the energy-dependent clathrin-mediated and caveolae-mediated pathways. Building on these findings, Sun et al. developed a π-expansive Ru receptor and a ligand with higher ROS yield. This was achieved using extended non-planar π-conjugation 3,4-ethylenedioxythiophene, which replaced the phenyl group to facilitate the intersystem crossing process.241 The newly assembled metallacycle (chemical 25, Fig. 15c) demonstrated a high localization coefficient (= 0.7) and high Ru content in lysosomes (Fig. 15d), indicating that it was primarily distributed within lysosomes. Moreover, an acridine orange (AO, a lysosomotropic dye that emits red fluorescence when localized within the acidic environment of lysosomes but shows green fluorescence in the cytosol) staining experiment was performed. This experiment revealed that, upon laser irradiation, the accumulation of 25 in lysosomes generated ROS, leading to the loss of lysosomal membrane integrity and lysosome dysfunction (Fig. 15e), eventually causing cell apoptosis. Fan et al.239 utilized an assembled metallacycle encapsulating five kinds of dyes and conducted colocalization experiments in three cell types (with correlation coefficients ranging from 0.73 to 0.89), confirming that the metallacycle predominantly accumulates in lysosomes. These results corroborate that SCCs first accumulate in lysosomes and contribute to cell death through lysosomal damage.
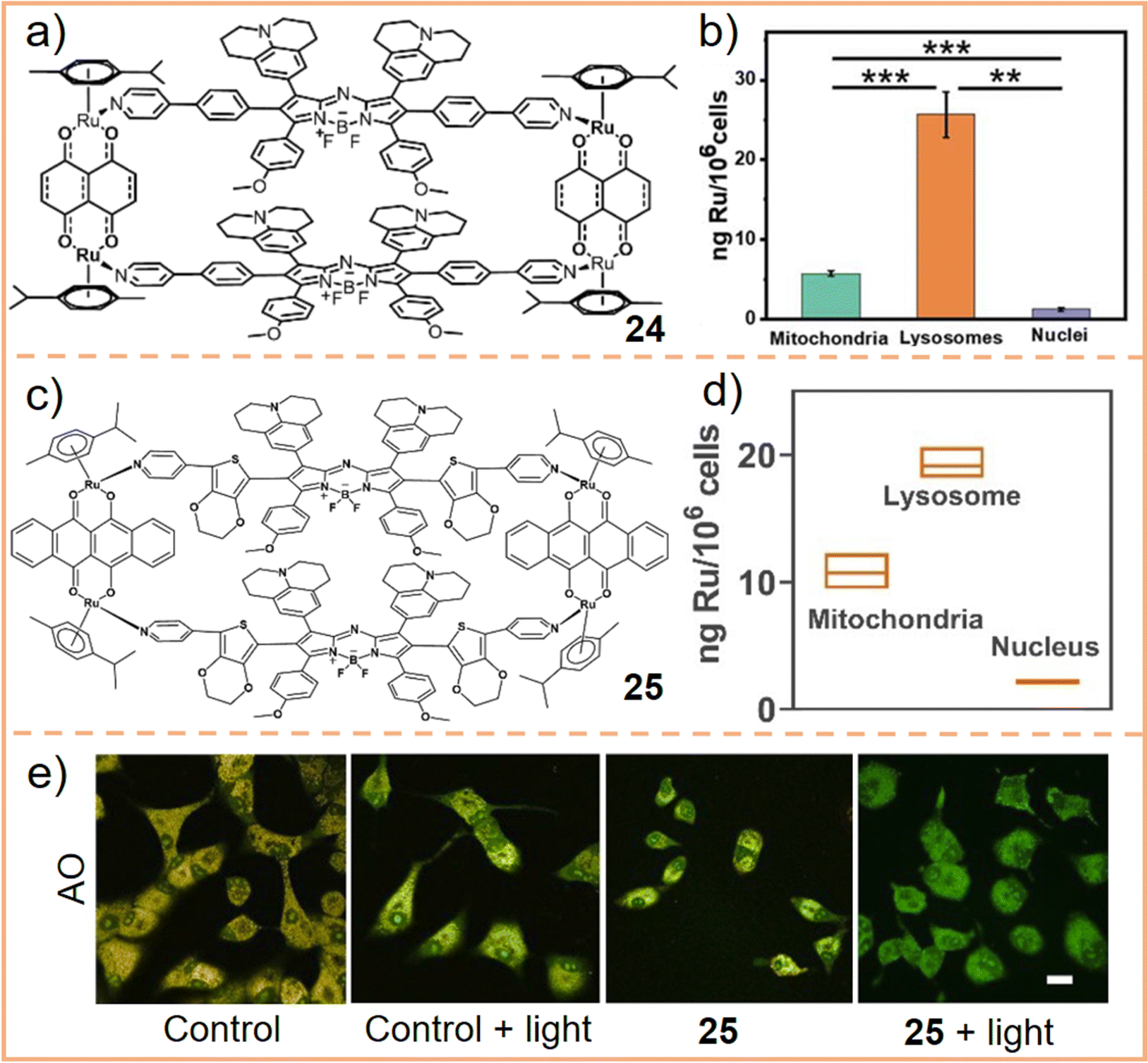 | ||
| Fig. 15 (a) Chemical structure of an Ru(II)-based metallacycle 24. (b) Subcellular distribution and quantification of Ru in A549 cells as inferred from ICP-MS studies. Figure adapted from ref. 240 with permission from the Royal Society of Chemistry, Copyright 2022. (c) Chemical structure of an Ru(II)-based metallacycle 25 and (d) Ru distribution in different organs with the incubation of the metallacycle for 6 h. (e) AO after incubation with the metallacycle or medium (control), followed by irradiation with or without laser illumination. Scale bar: 25 μm. Figure adapted from ref. 241 with permission from Wiley-VCH, Copyright 2023. | ||
The consistent observation of SCCs localizing primarily in lysosomes across different studies underscores the potential of targeting lysosomes in the development of SCC-based therapies. By exploiting the lysosome's role in cell metabolism and death, researchers can design SCCs that selectively activate cell death mechanisms in diseased cells, such as cancer cells, offering a promising strategy for targeted therapeutic interventions.
Gou et al.289 developed arene–Ru(II) supramolecular complexes (chemical 26, Fig. 16a), capable of self-assembling into supramolecular vesicles through non-covalent interactions in water. Subcellular localization studies revealed that upon entering a cell, Ru predominantly accumulated in the mitochondria, accounting for 62.5 ± 4.3%. Conversely, only 3.5% appeared in the nucleus, with the rest 31.7% dispersed in other cellular fractions (Fig. 16b). Staining experiments using JC-1 and MitoSOX Red, which assess mitochondrial membrane potential and superoxide generation capacities respectively, demonstrated that chemical 26 causes mitochondrial membrane polarization and superoxide production when exposed to laser irradiation. This led to mitochondrial dysfunction, which was further linked to inducing apoptosis in tumor cells. These findings underscore that mitochondria are the primary targets of the arene–Ru(II) complex.
 | ||
| Fig. 16 (a) Chemical structure of the arene–Ru(II) complex (chemical 26). (b) Ru uptake in the different cellular compartments of MDA-MB-231 cells treated with 26. Figure adapted from ref. 289 with permission from Springer Nature, Copyright 2022; (c) schematic drawing of the proposed anti-cancer mechanisms of action of 27 and 28. Figure adapted from ref. 238 with permission from the Royal Society of Chemistry, Copyright 2021. | ||
Building on the established connection between mitochondria and lysosomes, Che et al.238 explored the physiological process of SCCs from lysosomal localization to mitochondrial release and their role in cell death. They created a planar Pt(II) terpyridyl scaffold linked to a glucose moiety (27), which undergoes cleavage by β-glucosidase, forming compound 28. Their results demonstrated that 27 predominantly accumulated in lysosomes, leading to necrotic cell death, whereas 28 was more prevalent in mitochondria, causing late apoptosis. Necrosis resulted from lysosomal integrity disruption and autophagy perturbation. TEM analysis of cells treated with 27 revealed a significant increase in double-membrane cytosolic vesicles, indicative of autophagosomes. Based on these distribution patterns, they suggested an anticancer mechanism: Pt(II)-containing supramolecular structures accumulate in lysosomes and mitochondria, resulting in lysosomal integrity disruption, mitochondrial membrane depolarization, autophagy perturbation, and ultimately, cancer cell death (Fig. 16c).
Furthermore, given the distinct characteristics of mitochondrial DNA, several researchers have concentrated on strategies for localizing SCCs within mitochondrial DNA. Metallohelical supramolecular systems are particularly favored in this context due to their protein-like α-helical structure. This structural mimicry inherently enhances their DNA-targeting properties. However, Duan et al.237 discovered that mesocate helical supramolecular systems exhibit a higher DNA-binding ability compared to traditional α-helices. They synthesized two mesocate supramolecules, labeled 29 and 30 (Fig. 17a), demonstrating impressive mitochondrial localization (with correlation coefficients of 0.91 and 0.88, respectively, Fig. 17b). Additionally, their DNA-binding capabilities were investigated through an ethidium bromide (EB) displacement assay using natural calf-thymus DNA (ct-DNA). Fig. 17c and d illustrate that the fluorescence of EB-bound ct-DNA was effectively quenched following the addition of the mesocate complexes. The binding constants (Kapp) were calculated at 0.67 × 107 M−1 for 29 and 1.12 × 107 M−1 for 30. These results indicated that their inherent DNA-targeting ability, formed through noncovalent interactions, can be enhanced by extending the length of the diamine linkers (from 29 to 30). Compared to metal ions in α-helical structures, exhibiting Kapp values between 0.39 × 107 M−1 and 0.66 × 107 M−1, mesocate supramolecular complexes demonstrate a considerably higher binding capacity. Molecular docking studies revealed distinct binding preferences: 29 predominantly binds in the major groove of DNA (Fig. 17e), with an average binding energy of −6.35 kcal mol−1. Conversely, 30 favors the minor groove of DNA (Fig. 17f), indicated by a lower binding energy of −7.82 kcal mol−1 in the minor groove compared to −6.18 kcal mol−1 in the major groove. These findings suggest that the mesocate structure is critical for enhancing the interactions of supramolecular complexes with DNA. Consequently, 29 and 30 induced a higher rate of apoptosis in tumor cells, with rates of 88.9% and 97.5%, respectively. Overall, these studies significantly contribute to our understanding of the distribution of SCCs in cells, advancing research into the mechanisms of cell death caused by SCCs and aiding the development of precision medicine.
 | ||
| Fig. 17 (a) Chemical structures of 29 and 30. (b) Determination of intercellular localization of 29 and 30, and fluorescence quenching of the EB bound ct-DNA upon addition of 29 (c) and 30 (d). Scale bar: 20 μm. Molecular modeling for the interaction of 29 (e) and 30 (f) with DNA. Figure adapted from ref. 237 with permission from Wiley-VCH, Copyright 2020. | ||
SCC accumulation in the cell nucleus has been intensively studied by Tereni et al.243 They developed a hexagonal Pt(II) metallacycle (chemical 31, Fig. 18a) and co-incubated it with cisplatin to test cell viability. The results indicated a slight antagonistic effect, implying that both might target nucleic acids but with different action modes. Gene set enrichment analysis revealed that 31 preferentially targets genes in chromosomal regions rich in G4 structures. Consequently, the metallacycle accumulates in the nucleus, exhibiting bright fluorescence upon entering the cell. However, when cells were also treated with pyridostatin, a known G4 stabilizer, the fluorescence intensity of 31 diminished. This suggests that the accumulation of 31 is linked to the presence of G4 structures. However, when cells were incubated with both 31 and cisplatin, the fluorescence intensity remained unaltered, indicating distinct DNA binding mechanisms for the two compounds. Further investigation through photobleaching experiments and immunofluorescence studies showed that 31 interferes with G4 epitope recognition by the quadruplex-specific antibody BG4. After entering the cell nucleus, 31 replaces BG4 to bind to G4, thereby killing tumor cells. While in-depth studies on the localization of SCCs in cell nuclei are less common compared to analyses of their presence in lysosomes and mitochondria, some innovative designs have been developed. For example, Wang et al. created discrete organometallic cages assembled into supramolecular nanoparticles, which were then fused with proteins carrying nuclear localization signals. These signals direct the intracellularly released protein to the nucleus. Through the clathrin-mediated endocytosis312 or caveolae/lipid raft-mediated endocytosis pathway,313 various proteins (e.g., GFP, green fluorescence protein) and protein-based drugs are delivered into the nucleus (Fig. 18b). Such efforts significantly contribute to the advancement of precision medicine by elucidating the subcellular distribution of SCCs, whether through exploring their DNA binding mechanisms or by achieving targeted release of cargoes within SCCs.
 | ||
| Fig. 18 (a) Chemical structure of a hexagonal Pt(II) metallacycle 31. (b) Images of HeLa cells transfected with GFP alone and in GFP-SCC nanoparticles. Nuclei were stained with Hoechst 33342 (blue). Scale bar: 20 μm. Figure adapted from ref. 312 with permission from Wiley-VCH, Copyright 2021. | ||
4. Conclusion and perspectives
In the biomedical realm, numerous treatment modalities, including theranostics,314–316 imaging-guided therapy,317–319 synergistic therapy,320–322 and precision medicine, necessitate the involvement of various functional molecules. These molecules operate independently but synergistically contribute to achieving the ultimate therapeutic goal. During these treatments, it is essential to precisely control the dosage and location of different molecules to ensure targeted efficacy, a task challenging to accomplish through simple mixing of different components. In this context, the capability of SCCs to dynamically connect two or more molecules at a fixed ratio and angle offers them distinct advantages in these applications. To address the prevalent yet critical challenges associated with the use of SCCs, such as water solubility, biostability, and potential toxicity, significant progress has been made in recent years. This includes state-of-the-art research involving the use of DSPE–PEG-like liposomes and block copolymers like F127 as nanocarriers. These enhance the aqueous solubility and in vivo circulation stability of SCCs. Notably, the lengths and molecular weights of hydrophilic and hydrophobic segments in F127 block copolymers can be easily adjusted. Moreover, direct modification of hydrophilic small molecules, polypeptides, or PEG macromolecules on metallacycles/metallacages enables SCCs to self-assemble into carrier-free nanoparticles or vesicles. This approach improves water solubility and offers higher drug loading capacity. Additionally, targeted molecular modification allows SCCs to distinguish between diseased and normal cells. Altering the metal coordination centers can enhance the cancer/bacterial killing efficiency or prevent leakage of heavy metal ions from SCCs. Furthermore, colocalization experiments are being conducted to study the intracellular distribution of SCCs, aiming to elucidate their mechanism of action. These strategies are pivotal in reducing SCCs’ side effects and focusing on precision medicine. Despite considerable progress, SCCs still face numerous challenges before clinical application, and many common issues across different systems must be addressed and resolved.(1) SCCs are commonly administered intravenously, a method where the drugs must navigate several challenges before effectively targeting cells: circulation, accumulation, penetration, internalization, and drug release. Forming nanoparticles or vesicles with hydrophilic surfaces aids in maintaining the stability of SCCs in blood circulation, prolonging their presence. However, this hydrophilicity hinders their ability to cross the lipophilic outer cell membrane, posing challenges in cellular uptake and internalization. Conversely, injecting hydrophobic metallacycles/metallacages without hydrophilic chain modifications enhances cellular entry but compromises circulation stability under physiological conditions, risking premature disassembly before reaching the target. Hence, a delicate balance between circulation stability and cellular uptake efficiency of SCCs is essential, but studies in this area are scarce. One potential approach is investigating the lipophilic/hydrophilic properties of various SCCs (before and after nanoparticle or vesicle formation). By comparing circulation stability and cellular uptake, a reasonable balance might be found. Although this endeavor demands extensive time and involves rigorous basic and comparative research, which can be tedious, the potential for yielding valuable, straightforward results makes it a promising avenue to explore.
(2) PEG has long been a cornerstone in DDSs, used extensively for encapsulating liposomes or block copolymers and modifying hydrophilic chains. As a versatile and enduring material in DDSs, PEG enhances drug stability, prevents adhesion by proteins in the bloodstream, and is highly biocompatible. However, research has shown that PEG can provoke the production of anti-PEG antibodies (being recognized by B cells as a foreign substance, leading to the generation of anti-PEG IgM and IgG),323–325 which results in rapid clearance of PEG-containing drugs and compromises their therapeutic efficacy. Consequently, there is a pressing need to develop new materials. Replacing PEG with biological small molecules, peptides, or even proteins to provide a degree of hydrophilicity to SCCs could be a viable approach to mitigate the production of anti-PEG antibodies. The current challenge with these alternatives, compared to PEG, lies in their relatively weak hydrophilicity and flexibility. Adjusting their molecular weight, chain length, and hydrophilic force is challenging but represents a crucial area for future research and development. Addressing these issues could lead to more effective and less immunogenic drug delivery systems.
(3) Utilizing heavy metal ions in the self-assembly of SCCs presents both advantages and challenges. While the toxicity of these ions can be harnessed to kill targeted cells or bacteria, they can also harm normal cells, leading to severe side effects. Although constructing SCCs via light metal coordination-driven self-assembly is a known strategy, related research remains scarce. Current efforts are more focused on enhancing the targeting capability and individual treatment efficacy of SCCs, such as increasing the ROS yield for PDT, boosting photothermal conversion efficiency for PTT, and designing SCCs with dual-drug combinations or heteronuclear bimetallic ions. The goal is to achieve better therapeutic outcomes with lower doses of SCCs, reducing their toxic side effects. However, these design strategies often involve more complex donor/receptor structures or additional functional molecules, potentially increasing side effects while improving efficacy. Therefore, finding a balance between enhancing treatment effectiveness and minimizing side effects in SCC design is critical. This requires a multifaceted approach combining theoretical, experimental, simulation, and computational research to identify and assess key factors. These include the geometric structure, composition, and defects of SCCs, their pharmacokinetics, internalization mechanisms, and active sites of action. Such comprehensive evaluation can determine the overall performance of SCCs and indicate potential side effects, guiding the development of safer and more effective therapeutic solutions.
(4) Compared to healthy cells, tumor cells exhibit abnormalities in histology (angiogenesis,326–328 lymphangiogenesis,329–331 axonogenesis and neurogenesis332–335), cell types (fibroblast activation,336–338 pericytes,339–341 and adipocyte change342–344), biophysical status (hypoxia345–347 and low pH348–350), and biochemical indices (up-regulation of the concentration of small molecules, such as glucose,351–353 glutathione,354–356 adenosine-5′-triphosphate,357–359 guanosine-5′-triphosphate,360–362 and large molecules, such as aminopeptidase N,363–365 alkaline phosphatase,366–368 cathepsin,369–371 and matrix metallopeptidases372–374). Targeting these abnormalities can enhance treatment efficacy and reduce toxic side effects. However, the design of stimuli-responsive small molecule drugs has outpaced the development of SCCs. The requirement for angular and structural symmetry in the assembly of SCCs, typically from symmetrical ligands or acceptors, limits the variety of small molecular counterparts that can be used. Consequently, most SCCs currently target tumor cells through modifications with molecules like FA, biotin, and RGD or utilize the passive EPR effect. This approach falls short of meeting diverse clinical needs. A significant challenge for SCCs is broadening their stimulus responsiveness. Beyond synthesizing and structurally modifying ligands or acceptors, encapsulation post-assembly can impart additional functions. For instance, traditional amphiphilic polymer encapsulation of SCCs can be enhanced by coating them with various cell membranes, such as erythrocytes,375–377 macrophages,378–380 homologous tumor cells,381–384 or hybrid membranes.385–387 This method can improve blood circulation ability, reduce immunogenicity, and enhance internalization and invasion capabilities in tumor tissues. Such strategies can significantly expand the applications of SCCs in the biomedical field.
(5) In biomedical applications, designing treatments tailored to the specific characteristics of different tumor cells or bacteria is crucial. Numerous types of tumor cells exhibit consistent abnormalities in certain physiological parameters or biochemical indices. A common strategy involves exploiting these abnormalities, such as the high expression of FR and biotin receptors, to modify SCCs and enhance their targeting capabilities. However, the expression levels of these receptors can vary significantly among different types of cancer cells. For instance, certain lung, renal, colon, and breast cancer cell lines388,389 overexpress the BR at higher levels than the FR. Therefore, carefully selecting targeting motifs is vital for achieving better targeting efficiency and therapeutic outcomes. Currently, most research on SCCs does not delve deeply into the issue of selectively targeting tumor cell characteristics. Instead, these studies often focus on demonstrating the therapeutic effects of SCCs on tumor cells, comparing the efficacy before and after modification with targeting motifs. To address this gap, future studies could involve a parallel comparison of different targeting motifs within the same cell line when examining the targeting efficiency of SCCs with a specific structure. Although this approach is not overly complex, it holds significant value in providing more comprehensive data for future designs.
(6) The acid cleavability of SCCs is advantageous as it ensures their disassembly at the tumor site, allowing for the independent release of ligands and receptors. This feature enables the assembly of two molecules with distinct functions into a single entity without altering their pharmacological activity and pharmacokinetics. However, to enhance the adaptability of SCCs to the complex in vivo physiological environment and maintain their stability during transportation, certain SCCs are designed to resist both acidic and alkaline conditions. This resistance prevents the leakage of small-molecule counterparts and metal ions, reducing potential toxicity and side effects. However, this acid–alkali resistance also means that these SCCs may lose their ability to disassemble at the tumor site or within lysosomes. This limitation adds to the research complexity, as it eliminates the possibility of independent ligand or receptor release. Consequently, researchers must consider SCCs as intact molecules and assess how changes in their molecular structure could impact their pharmacological activity and bioactivity. Balancing the stability of SCCs to prevent leakage of small-molecule counterparts or metal ions while ensuring their acid-responsive release at the tumor site is a critical aspect of their design. However, the relationship between the structure of SCCs and the leakage of metal ions, along with their potential toxic effects, has not been thoroughly and systematically explored. Studies on stability in acidic or alkaline environments have primarily been incidental, often using absorption or emission spectroscopy, without specific focus on the effects of SCC leakage or the comparative impacts of acid-cleavable versus non-cleavable SCCs on cellular/bacterial growth or systemic toxicity in vivo. This lack of targeted research means that the acid–alkali resistance of SCCs is more often a chance discovery rather than the result of intentional design. Given the known properties of some SCCs, follow-up research need not be overly complicated. Routine in vitro and in vivo experiments, such as ICP-MS, CLSM, flow cytometry, and MTT assays, can provide valuable insights. Based on these findings, researchers can undertake targeted designs starting from the molecular structure of SCCs. This approach will help clarify the optimal balance between stability and acid cleavability of SCCs in biomedical applications.
(7) Understanding the subcellular distribution of SCCs is crucial for deciphering their anticancer and antibacterial mechanisms. This knowledge facilitates the design of specific drug molecules that can minimize the side effects of SCCs, aligning with the core principles of precision medicine. Research has confirmed that SCCs predominantly localize within lysosomes, mitochondria, and nuclei. However, establishing a clear chemical structure–localization relationship and organelle feature–localization relationship akin to those for small molecule drugs remains a distant goal. To progress in this area, more data supporting the localization results of SCCs with varied structures are necessary. Additionally, exploring organelle-targeting motifs for localization in other organelles, such as the Golgi apparatus and endoplasmic reticulum, could provide more active sites and action mechanisms for SCCs. This would significantly enrich the study of their subcellular distribution. Given these challenges, while the potential of SCCs in the biomedical field is promising, there is still considerable work to be done.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the Natural Science Foundation of Zhejiang Province (grants LZ23B040001, and LY23E030003), the National Natural Science Foundation of China (grants 21971049, and 51903070), and “Ten-thousand Talents Plan” of Zhejiang Province (grant number 2019R52040), and the “General scientific research project” of Zhejiang Provincial Department of Education (grant Y202249956).Notes and references
- X. Z. Ai, C. J. H. Ho, J. Aw, A. B. E. Attia, J. Mu, Y. Wang, X. Y. Wang, Y. Wang, X. G. Liu, H. B. Chen, M. Y. Gao, X. Y. Chen, E. K. L. Yeow, G. Liu, M. Olivo and B. G. Xing, Nat. Commun., 2016, 7, 10432 CrossRef CAS PubMed.
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- Q. Guan, G. B. Wang, L. L. Zhou, W. Y. Li and Y. B. Dong, Nanoscale Adv., 2020, 2, 3656–3733 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- S. M. Li, J. Zou, L. F. Tan, Z. B. Huang, P. Liang and X. W. Meng, Chem. Eng. J., 2022, 446, 137148 CrossRef CAS.
- X. Zhang, X. Fan, G. Q. Zhu, Y. T. Wang, H. X. Ding, H. P. Lin, Y. Y. Li, Q. Li, J. Z. Gao, M. H. Pan and Q. M. Guo, J. Phys. Chem. C, 2020, 124, 12589–12595 CrossRef CAS.
- A. S. Y. Law, L. C. C. Lee, K. K. W. Lo and V. W. W. Yam, J. Am. Chem. Soc., 2021, 143, 5396–5405 CrossRef CAS PubMed.
- K. D. M. Rao, M. Hossain, A. Roy, A. Ghosh, G. S. Kumar, P. Moitra, T. Kamitya, S. Acharya and S. Bhattacharya, Nanoscale, 2020, 12, 11986–11996 RSC.
- G. Tang, Y. Y. Tian, Y. H. Gao, Z. Y. Zhou, X. Chen, Y. Li, X. Y. Yu, H. C. Wang, X. Li and Y. S. Cao, ACS Nano, 2022, 16, 4892–4904 CrossRef CAS PubMed.
- B. Chen, C. C. Chu, E. Ren, H. R. Lin, Y. Zhang, P. Y. Wang, H. Yao, A. L. Liu, G. Liu and X. H. Lin, Front. Chem., 2022, 10, 870769 CrossRef CAS PubMed.
- M. Dergham, S. M. Lin and J. Geng, Angew. Chem., Int. Ed., 2022, 61, e202114267 CrossRef CAS PubMed.
- S. Loescher and A. Walther, Angew. Chem., Int. Ed., 2020, 59, 5515–5520 CrossRef CAS PubMed.
- J. T. Xu, J. Wang, J. Ye, J. Jiao, Z. G. Liu, C. J. Zhao, B. Li and Y. J. Fu, Adv. Sci., 2021, 8, 21011101 Search PubMed.
- E. N. Salgado, R. J. Radford and F. A. Tezcan, Acc. Chem. Res., 2010, 43, 661–672 CrossRef CAS PubMed.
- C. B. He, D. M. Liu and W. B. Lin, Chem. Rev., 2015, 115, 11079–11108 CrossRef CAS PubMed.
- P. Stricklen and J. Verkade, J. Am. Chem. Soc., 1983, 105, 2494–2495 CrossRef CAS.
- P. Ballester, M. Fujita and J. Rebek, Chem. Soc. Rev., 2015, 44, 392–393 RSC.
- M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645–5647 CrossRef CAS.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed.
- S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC.
- M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161–171 RSC.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS PubMed.
- A. M. Lifschitz, M. S. Rosen, C. M. McGuirk and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 7252–7261 CrossRef CAS PubMed.
- X. Li, X. Zhao, W. Wang, Z. Shi, Y. Zhang, Q. Tian, Y. Yao, C. He and C. Duan, Coord. Chem. Rev., 2023, 495, 215366 CrossRef CAS.
- G. Yu, M. Jiang, F. Huang and X. Chen, Curr. Opin. Chem. Biol., 2021, 61, 19–31 CrossRef CAS PubMed.
- J. Zhou, L. Rao, G. Yu, T. R. Cook, X. Chen and F. Huang, Chem. Soc. Rev., 2021, 50, 2839–2891 RSC.
- Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889–3919 RSC.
- G. T. Williams, C. J. E. Haynes, M. Fares, C. Caltagirone, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2021, 50, 2737–2763 RSC.
- H. Wang, S. Mumtaz, H. Li, J. Liu and F. Yang, Future Gener. Comput. Syst., 2021, 117, 145–154 CrossRef.
- W. X. Gao, H. N. Zhang and G. X. Jin, Coord. Chem. Rev., 2019, 386, 69–84 CrossRef CAS.
- C. Guo, A. C. Sedgwick, T. Hirao and J. L. Sessler, Coord. Chem. Rev., 2021, 427, 213560 CrossRef CAS PubMed.
- H. Y. Lin, Y. T. Wang, X. Shi, H. B. Yang and L. Xu, Chem. Soc. Rev., 2023, 52, 1129–1154 RSC.
- Y. Sun and P. J. Stang, Aggregate, 2021, 2, e94 CrossRef CAS.
- C. Yin, J. Du, B. Olenyuk, P. J. Stang and Y. Sun, Inorganics, 2023, 11, 54 CrossRef CAS.
- B. Li, T. He, Y. Fan, X. Yuan, H. Qiu and S. Yin, Chem. Commun., 2019, 55, 8036–8059 RSC.
- Y. Xu, Y. Dou, Q. Li, H. Ye, Y. Li, S. Qiu, X. Xiong, J. Li and Y. Sun, Coord. Chem. Rev., 2023, 493, 215320 CrossRef CAS.
- W. Jogadi and Y. R. Zheng, Curr. Opin. Chem. Biol., 2023, 73, 102276 CrossRef CAS PubMed.
- V. Balzani, G. Bergamini, F. Marchioni and P. Ceroni, Coord. Chem. Rev., 2006, 250, 1254–1266 CrossRef CAS.
- I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834–1895 CrossRef CAS PubMed.
- D. Rota Martir and E. Zysman-Colman, Chem. Commun., 2019, 55, 139–158 RSC.
- J. Wang, Q. Gong, L. Jiao and E. Hao, Coord. Chem. Rev., 2023, 496, 215367 CrossRef CAS.
- N. Kwon, H. Kim, X. Li and J. Yoon, Chem. Sci., 2021, 12, 7248–7268 RSC.
- Q. Wu, Y. Zhu, X. Fang, X. Hao, L. Jiao, E. Hao and W. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 47208–47219 CrossRef CAS PubMed.
- S. Bian, X. Zheng, W. Liu, J. Li, Z. Gao, H. Ren, W. Zhang, C. S. Lee and P. Wang, Biomaterials, 2023, 298, 122130 CrossRef CAS PubMed.
- Y. Tian, H. Zhou, Q. Cheng, H. Dang, H. Qian, C. Teng, K. Xie and L. Yan, J. Mater. Chem. B, 2022, 10, 707–716 RSC.
- Y. Liu, Y. Li, S. Koo, Y. Sun, Y. Liu, X. Liu, Y. Pan, Z. Zhang, M. Du, S. Lu, X. Qiao, J. Gao, X. Wang, Z. Deng, X. Meng, Y. Xiao, J. S. Kim and X. Hong, Chem. Rev., 2022, 122, 209–268 CrossRef CAS PubMed.
- Z. Guo, J. Zhao, Y. Liu, G. Li, H. Wang, Y. Hou, M. Zhang, X. Li and X. Yan, Chin. Chem. Lett., 2021, 32, 1691–1695 CrossRef CAS.
- Y. Liu, Z. Guo, Y. Guo, G. Li, S. Yang, X. Yan, Y. Shen and J. Wang, Chin. Chem. Lett., 2023, 34, 108531 CrossRef CAS.
- C. Mu, Z. Zhang, Y. Hou, H. Liu, L. Ma, X. Li, S. Ling, G. He and M. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12293–12297 CrossRef CAS PubMed.
- Y. Yin, Z. Chen, R.-H. Li, F. Yi, X. C. Liang, S. Q. Cheng, K. Wang, Y. Sun and Y. Liu, Inorg. Chem., 2022, 61, 2883–2891 CrossRef CAS PubMed.
- X. Yan, M. Wang, T. R. Cook, M. Zhang, M. L. Saha, Z. Zhou, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 4580–4588 CrossRef CAS PubMed.
- Z. Yin, X. Chang, J. Zang, S. Lin, Z. Zhou, T. Liu, L. Ding, H. Peng and Y. Fang, J. Mater. Chem. C, 2022, 10, 10429–10438 RSC.
- Y. Li, S. S. Rajasree, G. Y. Lee, J. Yu, J.-H. Tang, R. Ni, G. Li, K. N. Houk, P. Deria and P. J. Stang, J. Am. Chem. Soc., 2021, 143, 2908–2919 CrossRef CAS PubMed.
- H. Shen, C. Xu, F. Sun, M. Zhao, Q. Wu, J. Zhang, S. Li, J. Zhang, J. W. Y. Lam and B. Z. Tang, ChemMedChem, 2022, 17, e202100578 CrossRef CAS PubMed.
- R. S. Gamage, J. L. Chasteen and B. D. Smith, Bioconjugate Chem., 2023, 34, 961–971 CrossRef CAS PubMed.
- Y. Sun, F. Ding, Z. Zhou, C. Li, M. Pu, Y. Xu, Y. Zhan, X. Lu, H. Li, G. Yang, Y. Sun and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1968–1973 CrossRef CAS PubMed.
- C. Huang, T. Shi, J. Zhang, Y. Sun, T. Ma, W. Li, Y. Li, H. Qiu and S. Yin, Dyes Pigm., 2023, 210, 110932 CrossRef CAS.
- F. Chen, Y. Li, X. Lin, H. Qiu and S. Yin, Polymers, 2021, 13, 370 CrossRef CAS PubMed.
- Y. Sun, F. Ding, Z. Chen, R. Zhang, C. Li, Y. Xu, Y. Zhang, R. Ni, X. Li, G. Yang, Y. Sun and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16729–16735 CrossRef CAS PubMed.
- J. Zhang, M. Jiang, Y. Li, J. Yu, H. Qiu, M. Gu, Y. Li and S. Yin, Dyes Pigm., 2022, 206, 110679 CrossRef CAS.
- W. Chen, X. Li, C. Liu, J. He, M. Qi, Y. Sun, B. Shi, H. Sepehrpour, H. Li, W. Tian and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30942–30948 CrossRef CAS PubMed.
- E. Villemin, Y. C. Ong, C. M. Thomas and G. Gasser, Nat. Rev. Chem., 2019, 3, 261–282 CrossRef CAS.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2012, 64, 302–315 CrossRef.
- S. Svenson, Eur. J. Pharm. Biopharm., 2009, 71, 445–462 CrossRef CAS PubMed.
- P. Huang, X. Wang, X. Liang, J. Yang, C. Zhang, D. Kong and W. Wang, Acta Biomater., 2019, 85, 1–26 CrossRef CAS PubMed.
- S. D. Li and L. Huang, Mol. Pharmaceutics, 2008, 5, 496–504 CrossRef CAS PubMed.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- R. Duncan, S. Dimitrijevic and E. Evagorou, STP Pharma Sci., 1996, 6, 237–263 Search PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. Chan, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed.
- K. K. Gill, A. Kaddoumi and S. Nazzal, J. Drug Targeting, 2015, 23, 222–231 CrossRef CAS PubMed.
- N. Wauthoz and K. Amighi, Eur. J. Lipid Sci. Technol., 2014, 116, 1114–1128 CrossRef CAS.
- G. Khuller, M. Kapur and S. Sharma, Curr. Pharm. Des., 2004, 10, 3263–3274 CrossRef CAS PubMed.
- T. McPherson, A. Kidane, I. Szleifer and K. Park, Langmuir, 1998, 14, 176–186 CrossRef CAS.
- A. Hucknall, S. Rangarajan and A. Chilkoti, Adv. Mater., 2009, 21, 2441–2446 CrossRef CAS.
- B. Huang, X. Liu, G. Yang, J. Tian, Z. Liu, Y. Zhu, X. Li, G. Yin, W. Zheng and L. Xu, CCS Chem., 2022, 4, 2090–2101 CrossRef CAS.
- G. Li, X. Zhang, W. Zhao, W. Zhao, F. Li, K. Xiao, Q. Yu, S. Liu and Q. Zhao, ACS Appl. Mater. Interfaces, 2020, 12, 20180–20190 CrossRef CAS PubMed.
- X. Lin, F. Chen, X. Yu, H. Wang, H. Qiu, Y. Li, S. Yin and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203994119 CrossRef CAS PubMed.
- Y. Xu, C. Li, S. Lu, Z. Wang, S. Liu, X. Yu, X. Li and Y. Sun, Nat. Commun., 2022, 13, 2009 CrossRef CAS PubMed.
- Y. Xu, C. Li, X. Ma, W. Tuo, L. Tu, X. Li, Y. Sun, P. J. Stang and Y. Sun, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2209904119 CrossRef CAS PubMed.
- K. C. de Castro, J. C. Coco, É. M. Dos Santos, J. A. Ataide, R. M. Martinez, M. H. M. do Nascimento, J. Prata, P. R. M. L. da Fonte, P. Severino and P. G. Mazzola, J. Controlled Release, 2023, 353, 802–822 CrossRef CAS PubMed.
- L. Li, L. Mu and V. Torchilin, J. Drug Delivery Sci. Technol., 2007, 17, 389–392 CrossRef CAS.
- J. Yu, H. Qiu, S. Yin, H. Wang and Y. Li, Molecules, 2021, 26, 3610 CrossRef CAS PubMed.
- M. Wang, B. Wu, J. D. Tucker, P. Lu and Q. Lu, Drug Delivery, 2016, 23, 3224–3233 CrossRef CAS PubMed.
- P. Singla, O. Singh, S. Sharma, K. Betlem, V. K. Aswal, M. Peeters and R. K. Mahajan, ACS Omega, 2019, 4, 11251–11262 CrossRef CAS PubMed.
- A. Pitto-Barry and N. P. Barry, Polym. Chem., 2014, 5, 3291–3297 RSC.
- R. J. P J, O. S. Oluwafemi, S. Thomas and A. O. Oyedeji, J. Drug Delivery Sci. Technol., 2022, 72, 103390 CrossRef.
- B. Shriky, A. Kelly, M. Isreb, M. Babenko, N. Mahmoudi, S. Rogers, O. Shebanova, T. Snow and T. Gough, J. Colloid Interface Sci., 2020, 565, 119–130 CrossRef CAS PubMed.
- G. Dumortier, J. L. Grossiord, F. Agnely and J. C. Chaumeil, Pharm. Res., 2006, 23, 2709–2728 CrossRef CAS PubMed.
- M. S. H. Akash and K. Rehman, J. Controlled Release, 2015, 209, 120–138 CrossRef CAS PubMed.
- M. Kurahashi, K. Kanamori, K. Takeda, H. Kaji and K. Nakanishi, RSC Adv., 2012, 2, 7166–7173 RSC.
- N. U. Khaliq, J. Lee, S. Kim, D. Sung and H. Kim, Pharmaceutics, 2023, 15, 2102 CrossRef CAS PubMed.
- K. S. Patil, A. A. Hajare, A. S. Manjappa, H. N. More and J. I. Disouza, J. Drug Delivery Sci. Technol., 2021, 65, 102685 CrossRef CAS.
- J. Zhang, J. Yu, W. Li, Y. Fan, Y. Li, Y. Sun, S. Yin and P. J. Stang, Inorganics, 2022, 10, 80 CrossRef CAS.
- J. Zhang, Y. Li, M. Jiang, H. Qiu, Y. Li, M. Gu and S. Yin, ACS Biomater. Sci. Eng., 2023, 9, 821–830 CrossRef CAS PubMed.
- H. Jia, T. Shi, T. He, Y. Li and S. Yin, Dalton Trans., 2023, 52, 4296–4302 RSC.
- F. Ding, Z. Chen, W. Y. Kim, A. Sharma, C. Li, Q. Ouyang, H. Zhu, G. Yang, Y. Sun and J. S. Kim, Chem. Sci., 2019, 10, 7023–7028 RSC.
- Y. Qin, X. Chen, Y. Gui, H. Wang, B. Z. Tang and D. Wang, J. Am. Chem. Soc., 2022, 144, 12825–12833 CrossRef CAS PubMed.
- H. Zhu, Q. Li, B. Shi, F. Ge, Y. Liu, Z. Mao, H. Zhu, S. Wang, G. Yu, F. Huang and P. J. Stang, Angew. Chem., Int. Ed., 2020, 59, 20208–20214 CrossRef CAS PubMed.
- X. Min, M. Li, W. Zhang, R. H. Li, Z. Zhang, P. Wang, W. Su, F. Li, Y. Sun and Y. Liu, J. Mater. Chem. B, 2023, 11, 1090–1099 RSC.
- C. Li, P. P. Jia, Y. L. Xu, F. Ding, W. C. Yang, Y. Sun, X. P. Li, G. Q. Yin, L. Xu and G. F. Yang, Sci. China: Chem., 2021, 64, 134–142 CrossRef CAS.
- K. Cai, X. He, Z. Song, Q. Yin, Y. Zhang, F. M. Uckun, C. Jiang and J. Cheng, J. Am. Chem. Soc., 2015, 137, 3458–3461 CrossRef CAS PubMed.
- J. Zhang, S. Li, F. F. An, J. Liu, S. Jin, J. C. Zhang, P. C. Wang, X. Zhang, C. S. Lee and X. J. Liang, Nanoscale, 2015, 7, 13503–13510 RSC.
- D. Landesman-Milo and D. Peer, J. Controlled Release, 2012, 161, 600–608 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed.
- J. M. Blonder, L. Baird, J. C. Fulfs and G. J. Rosenthal, Life Sci., 1999, 65, 261–266 CrossRef PubMed.
- T. P. Johnston, L. B. Nguyen, W. A. Chu and S. Shefer, Int. J. Pharm., 2001, 229, 75–86 CrossRef CAS PubMed.
- E. Lepeltier, C. Bourgaux and P. Couvreur, Adv. Drug Delivery Rev., 2014, 71, 86–97 CrossRef CAS PubMed.
- W. S. Saad and R. K. Prud’homme, Nano Today, 2016, 11, 212–227 CrossRef CAS.
- C. J. M. Rivas, M. Tarhini, W. Badri, K. Miladi, H. Greige-Gerges, Q. A. Nazari, S. A. G. Rodríguez, R. Á. Román, H. Fessi and A. Elaissari, Int. J. Pharm., 2017, 532, 66–81 CrossRef PubMed.
- A. A. Date, N. Desai, R. Dixit and M. Nagarsenker, Nanomedicine, 2010, 5, 1595–1616 CrossRef CAS PubMed.
- R. J. Wilson, Y. Li, G. Yang and C. X. Zhao, Particuology, 2022, 64, 85–97 CrossRef CAS.
- K. B. Sutradhar and M. L. Amin, Eur. J. Nanomed., 2013, 5, 97–110 Search PubMed.
- H. Zhang, Liposomes: Methods Protoc., 2017, pp. 17–22 Search PubMed.
- V. Ravalika and A. K. Sailaja, Nano Biomed. Eng., 2017, 9, 242–248 CAS.
- S. Karki, H. Kim, S.-J. Na, D. Shin, K. Jo and J. Lee, Asian J. Pharm. Sci., 2016, 11, 559–574 CrossRef.
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS PubMed.
- W. Chen, J. He, H. Li, X. Li and W. Tian, Supramol. Chem., 2020, 32, 597–604 CrossRef CAS.
- A. G. Cheetham, P. Zhang, Y. A. Lin, L. L. Lock and H. Cui, J. Am. Chem. Soc., 2013, 135, 2907–2910 CrossRef CAS PubMed.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840–853 CrossRef CAS PubMed.
- Y. Li, X. Yuan, J. Yu, Y. Fan, T. He, S. Lu, X. Li, H. Qiu and S. Yin, ACS Appl. Bio Mater., 2020, 3, 8061–8068 CrossRef CAS PubMed.
- B. Woods, R. D. Silva, C. Schmidt, D. Wragg, M. Cavaco, V. Neves, V. F. Ferreira, L. Gano, T. S. Morais and F. Mendes, Bioconjugate Chem., 2021, 32, 1399–1408 CrossRef CAS PubMed.
- G. Tao, T. Ji, N. Wang, G. Yang, X. Lei, W. Zheng, R. Liu, X. Xu, L. Yang and G. Q. Yin, ACS Macro Lett., 2019, 9, 61–69 CrossRef PubMed.
- Y. Fan, J. Zhang, Y. Li, Q. Chen, Z. Ni, H. Zhou, J. Yu, H. Qiu and S. Yin, Mater. Chem. Front., 2022, 6, 633–643 RSC.
- Z. Wang, L. He, B. Liu, L. P. Zhou, L. X. Cai, S. J. Hu, X. Z. Li, Z. Li, T. Chen, X. Li and Q. F. Sun, J. Am. Chem. Soc., 2020, 142, 16409–16419 CrossRef CAS PubMed.
- J. Zhou, G. Yu, J. Yang, B. Shi, B. Ye, M. Wang, F. Huang and P. J. Stang, Chem. Mater., 2020, 32, 4564–4573 CrossRef CAS.
- J. Tian, B. Huang, S. Weng, W. Zheng and W. Zhang, Mater. Today Adv., 2022, 14, 100229 CrossRef CAS.
- E. Gammella, S. Recalcati and G. Cairo, Oxid. Med. Cell. Longevity, 2016, 2016, 8629024 Search PubMed.
- Y. Qin, T. Guo, Z. Wang and Y. Zhao, J. Mater. Chem. B, 2021, 9, 4793–4803 RSC.
- A. Gabizon, H. Shmeeda and Y. Barenholz, Clin. Pharmacokinet., 2003, 42, 419–436 CrossRef CAS PubMed.
- D. Lorusso, A. Di Stefano, V. Carone, A. Fagotti, S. Pisconti and G. Scambia, Ann. Oncol., 2007, 18, 1159–1164 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- R. Loomba, A. Rowley, R. Wesley, T. J. Liang, J. H. Hoofnagle, F. Pucino and G. Csako, Ann. Intern. Med., 2008, 148, 519–528 CrossRef PubMed.
- R. Dillon, G. M. Hirschfield, M. E. D. Allison and K. P. Rege, BMJ, 2008, 337, a423 CrossRef PubMed.
- W. Yeo, K. C. Lam, B. Zee, P. S. K. Chan, F. K. F. Mo, W. M. Ho, W. L. Wong, T. W. T. Leung, A. T. C. Chan, B. Ma, T. S. K. Mok and P. J. Johnson, Ann. Oncol., 2004, 15, 1661–1666 CrossRef CAS PubMed.
- S. Gao, X. Yan, G. Xie, M. Zhu, X. Ju, P. J. Stang, Y. Tian and Z. Niu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23437–23443 CrossRef CAS PubMed.
- P. Jeyakkumar, Y. Liang, M. Guo, S. Lu, D. Xu, X. Li, B. Guo, G. He, D. Chu and M. Zhang, Angew. Chem., Int. Ed., 2020, 59, 15199–15203 CrossRef CAS PubMed.
- G. Y. Wu, X. Shi, H. Phan, H. Qu, Y. X. Hu, G. Q. Yin, X. L. Zhao, X. Li, L. Xu, Q. Yu and H. B. Yang, Nat. Commun., 2020, 11, 3178 CrossRef CAS PubMed.
- Z. Zhang, H. Ye, F. Cai and Y. Sun, Dalton Trans., 2023, 52, 15193–15202 RSC.
- Y. Wang, C. Wang, R. Long, Y. Cao, D. Fan, M. Cen, L. Cao, Y. Chen and Y. Yao, Chem. Commun., 2019, 55, 5167–5170 RSC.
- S. Datta, M. L. Saha and P. J. Stang, Acc. Chem. Res., 2018, 51, 2047–2063 CrossRef CAS PubMed.
- Y. Tian, X. Yan, M. L. Saha, Z. Niu and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 12033–12036 CrossRef CAS PubMed.
- S. M. Kim, P. H. Faix and J. E. Schnitzer, J. Controlled Release, 2017, 267, 15–30 CrossRef CAS PubMed.
- Y. Nakamura, A. Mochida, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2016, 27, 2225–2238 CrossRef CAS PubMed.
- A. Nel, E. Ruoslahti and H. Meng, ACS Nano, 2017, 11, 9567–9569 CrossRef CAS PubMed.
- E. Huynh and G. Zheng, Nanomedicine, 2015, 10, 1993–1995 CrossRef CAS PubMed.
- H. Nakamura, F. Jun and H. Maeda, Expert Opin. Drug Delivery, 2015, 12, 53–64 CrossRef CAS PubMed.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- W. C. W. Chan, Acc. Chem. Res., 2017, 50, 627–632 CrossRef CAS PubMed.
- F. Danhier, J. Controlled Release, 2016, 244, 108–121 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- J. W. Nichols and Y. H. Bae, J. Controlled Release, 2014, 190, 451–464 CrossRef CAS PubMed.
- Q. Wang, Q. Liang, J. Dou, H. Zhou, C. Zeng, H. Pan, Y. Shen, Q. Li, Y. Liu, D. T. Leong, W. Jiang and Y. Wang, Nat. Nanotechnol., 2024, 19, 95–105 CrossRef CAS PubMed.
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575 CrossRef CAS PubMed.
- I. de Lázaro and D. J. Mooney, Nat. Mater., 2020, 19, 486–487 CrossRef PubMed.
- Anonymous, Nat. Mater., 2020, 19, 477 CrossRef PubMed.
- P. Zhang, Z. Zhou, W. Long, Y. Yan, Y. Li, T. Fu, Y. Liu, Z. Zhao, W. Tan and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202255119 CrossRef CAS PubMed.
- X. Wang, Q. Su, Z. Zhang, J. Yang, Y. Zhang and M. Zhang, Chem. Commun., 2020, 56, 8460–8463 RSC.
- G. Yu, B. Zhu, L. Shao, J. Zhou, M. L. Saha, B. Shi, Z. Zhang, T. Hong, S. Li and X. Chen, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6618–6623 CrossRef CAS PubMed.
- X. Jiang, Z. Zhou, H. Yang, C. Shan, H. Yu, L. Wojtas, M. Zhang, Z. Mao, M. Wang and P. J. Stang, Inorg. Chem., 2020, 59, 7380–7388 CrossRef CAS PubMed.
- G. Yu, S. Yu, M. L. Saha, J. Zhou, T. R. Cook, B. C. Yung, J. Chen, Z. Mao, F. Zhang and Z. Zhou, Nat. Commun., 2018, 9, 4335 CrossRef PubMed.
- S. Deiser, M. Drexler, G. Moreno-Alcántar, M. Irl, C. Schmidt, T. Günther and A. Casini, Inorg. Chem., 2023, 62, 20710–20720 CrossRef CAS PubMed.
- A. C. Marques, P. J. Costa, S. Velho and M. H. Amaral, J. Controlled Release, 2020, 320, 180–200 CrossRef CAS PubMed.
- S. Maiti and P. Paira, Eur. J. Med. Chem., 2018, 145, 206–223 CrossRef CAS PubMed.
- R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670–6683 RSC.
- P. S. Low and S. A. Kularatne, Curr. Opin. Chem. Biol., 2009, 13, 256–262 CrossRef CAS PubMed.
- P. Tagde, G. T. Kulkarni, D. K. Mishra and P. Kesharwani, J. Drug Delivery Sci. Technol., 2020, 56, 101613 CrossRef CAS.
- P. S. Low, W. A. Henne and D. D. Doorneweerd, Acc. Chem. Res., 2008, 41, 120–129 CrossRef CAS PubMed.
- E. J. Roy, U. Gawlick, B. A. Orr and D. M. Kranz, Adv. Drug Delivery Rev., 2004, 56, 1219–1231 CrossRef CAS PubMed.
- C. M. Paulos, M. J. Turk, G. J. Breur and P. S. Low, Adv. Drug Delivery Rev., 2004, 56, 1205–1217 CrossRef CAS PubMed.
- X. B. Zhao and R. J. Lee, Adv. Drug Delivery Rev., 2004, 56, 1193–1204 CrossRef CAS PubMed.
- D. N. Heo, D. H. Yang, H. J. Moon, J. B. Lee, M. S. Bae, S. C. Lee, W. J. Lee, I. C. Sun and I. K. Kwon, Biomaterials, 2012, 33, 856–866 CrossRef CAS PubMed.
- H. Li, G. Bruce, N. Childerhouse, G. Keegan, G. Mantovani and S. Stolnik, J. Controlled Release, 2023, 357, 333–341 CrossRef CAS PubMed.
- N. U. Deshpande and M. Jayakannan, Biomacromolecules, 2018, 19, 3572–3585 CrossRef CAS PubMed.
- G. Yu, T. R. Cook, Y. Li, X. Yan, D. Wu, L. Shao, J. Shen, G. Tang, F. Huang, X. Chen and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13720–13725 CrossRef CAS PubMed.
- X. Sun, Y. Li, T. Liu, Z. Li, X. Zhang and X. Chen, Adv. Drug Delivery Rev., 2017, 110–111, 38–51 CrossRef CAS PubMed.
- S. H. Wang and J. Yu, Biomaterials, 2018, 156, 1–15 CrossRef CAS PubMed.
- B. M. Cooper, J. Iegre, D. H. O'Donovan, M. Ölwegård Halvarsson and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1480–1494 RSC.
- K. Kurrikoff, D. Aphkhazava and Ü. Langel, Curr. Opin. Pharmacol., 2019, 47, 27–32 CrossRef CAS PubMed.
- F. Araste, K. Abnous, M. Hashemi, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Controlled Release, 2018, 292, 141–162 CrossRef CAS PubMed.
- O. Larsson, A. Girnita and L. Girnita, Br. J. Cancer, 2005, 92, 2097–2101 CrossRef CAS PubMed.
- S. Favelyukis, J. H. Till, S. R. Hubbard and W. T. Miller, Nat. Struct. Biol., 2001, 8, 1058–1063 CrossRef CAS PubMed.
- J. Zha and M. R. Lackner, Clin. Cancer Res., 2010, 16, 2512–2517 CrossRef CAS PubMed.
- H. Hamidi, M. Pietilä and J. Ivaska, Br. J. Cancer, 2016, 115, 1017–1023 CrossRef CAS PubMed.
- P. H. Wu, A. E. Opadele, Y. Onodera and J. M. Nam, Cancers, 2019, 11, 1783 CrossRef CAS PubMed.
- K. Ahmad, E. J. Lee, S. Shaikh, A. Kumar, K. M. Rao, S. Y. Park, J. O. Jin, S. S. Han and I. Choi, Semin. Cancer Biol., 2021, 69, 325–336 CrossRef CAS PubMed.
- S. L. Goodman and M. Picard, Trends Pharmacol. Sci., 2012, 33, 405–412 CrossRef CAS PubMed.
- K. Chen and X. Chen, Theranostics, 2011, 1, 189–200 CrossRef CAS PubMed.
- F. Ciardiello and G. Tortora, Clin. Cancer Res., 2001, 7, 2958–2970 CAS.
- J. Mendelsohn and J. Baselga, Semin. Oncol., 2006, 33, 369–385 CrossRef CAS PubMed.
- C. Yewale, D. Baradia, I. Vhora, S. Patil and A. Misra, Biomaterials, 2013, 34, 8690–8707 CrossRef CAS PubMed.
- B. F. El-Rayes and P. M. LoRusso, Br. J. Cancer, 2004, 91, 418–424 CrossRef CAS PubMed.
- F. Danhier, A. Le Breton and V. Préat, Mol. Pharmaceutics, 2012, 9, 2961–2973 CrossRef CAS PubMed.
- L. Ge, X. You, K. Huang, Y. Kang, Y. Chen, Y. Zhu, Y. Ren, Y. Zhang, J. Wu and H. Qian, Biomater. Sci., 2018, 6, 125–135 RSC.
- M. Nieberler, U. Reuning, F. Reichart, J. Notni, H. J. Wester, M. Schwaiger, M. Weinmüller, A. Räder, K. Steiger and H. Kessler, Cancers, 2017, 9, 116 CrossRef PubMed.
- S. Liu, Mol. Pharmaceutics, 2006, 3, 472–487 CrossRef CAS PubMed.
- S. Liu, Bioconjugate Chem., 2009, 20, 2199–2213 CrossRef CAS PubMed.
- Z. Liu, F. Wang and X. Chen, Drug Dev. Res., 2008, 69, 329–339 CrossRef CAS PubMed.
- C. Adessi and C. Soto, Curr. Med. Chem., 2002, 9, 963–978 CrossRef CAS PubMed.
- G. Yu, S. Yu, M. L. Saha, J. Zhou, T. R. Cook, B. C. Yung, J. Chen, Z. Mao, F. Zhang, Z. Zhou, Y. Liu, L. Shao, S. Wang, C. Gao, F. Huang, P. J. Stang and X. Chen, Nat. Commun., 2018, 9, 4335 CrossRef PubMed.
- G. Yu, B. Zhu, L. Shao, J. Zhou, M. L. Saha, B. Shi, Z. Zhang, T. Hong, S. Li, X. Chen and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6618–6623 CrossRef CAS PubMed.
- B. Yang, H. Liu, H. Yang, W. Chen, J. Wu, X. Feng, R. Tong, H. Yu, Y. Chen, Z. Lv, W. Sun, B. He, J. Wu, G. Yu, Z. Mao and S. Zheng, J. Mater. Chem. B, 2019, 7, 6476–6487 RSC.
- Y. Ding, Z. Tong, L. Jin, B. Ye, J. Zhou, Z. Sun, H. Yang, L. Hong, F. Huang, W. Wang and Z. Mao, Adv. Mater., 2022, 34, 2106388 CrossRef CAS PubMed.
- A. J. Danford, D. Wang, Q. Wang, T. D. Tullius and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12311–12316 CrossRef CAS PubMed.
- E. Rudolph, S. Hann, G. Stingeder and C. Reiter, Anal. Bioanal. Chem., 2005, 382, 1500–1506 CrossRef CAS PubMed.
- J. H. Schiller and G. Bittner, Clin. Cancer Res., 1999, 5, 4287–4294 CAS.
- B. Köberle, M. T. Tomicic, S. Usanova and B. Kaina, Biochim. Biophys. Acta, Rev. Cancer, 1806, 2010, 172–182 Search PubMed.
- A.-M. Florea and D. Büsselberg, Cancers, 2011, 3, 1351–1371 CrossRef CAS PubMed.
- L. Amable, Pharmacol. Res., 2016, 106, 27–36 CrossRef CAS PubMed.
- L. Galluzzi, I. Vitale, J. Michels, C. Brenner, G. Szabadkai, A. Harel-Bellan, M. Castedo and G. Kroemer, Cell Death Dis., 2014, 5, e1257 CrossRef CAS PubMed.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869–1883 CrossRef CAS PubMed.
- S. C. Wang, K. Y. Cheng, J. H. Fu, Y. C. Cheng and Y. T. Chan, J. Am. Chem. Soc., 2020, 142, 16661–16667 CrossRef CAS PubMed.
- Y. Y. Liu, H. J. Yu, Y. P. Wang, C. J. Li, X. F. Wang, C. G. Ye, H. L. Yao, M. Pan and C. Y. Su, Mater. Chem. Front., 2022, 6, 948–955 RSC.
- Z. Zhou, J. Liu, T. W. Rees, H. Wang, X. Li, H. Chao and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5664–5669 CrossRef CAS PubMed.
- Z. Zhou, J. Liu, J. Huang, T. W. Rees, Y. Wang, H. Wang, X. Li, H. Chao and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20296–20302 CrossRef CAS PubMed.
- E. L. Eskelinen, Y. Tanaka and P. Saftig, Trends Cell Biol., 2003, 13, 137–145 CrossRef CAS PubMed.
- J. A. Mindell, Annu. Rev. Physiol., 2012, 74, 69–86 CrossRef CAS PubMed.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS.
- R. A. Gatenby, E. T. Gawlinski, A. F. Gmitro, B. Kaylor and R. J. Gillies, Cancer Res., 2006, 66, 5216–5223 CrossRef CAS PubMed.
- L. He, L. X. Cai, M. H. Li, G. L. Zhang, L. P. Zhou, T. Chen, M. J. Lin and Q. F. Sun, Chem. Sci., 2020, 11, 7940–7949 RSC.
- L. Tu, C. Li, C. Liu, S. Bai, J. Yang, X. Zhang, L. Xu, X. Xiong and Y. Sun, Chem. Commun., 2022, 58, 9068–9071 RSC.
- S. Lv, Y. Miao, D. Zheng, X. Li, D. Liu and F. Song, Mol. Pharmaceutics, 2021, 18, 1229–1237 CrossRef CAS PubMed.
- K. Qiu, Y. Chen, T. W. Rees, L. Ji and H. Chao, Coord. Chem. Rev., 2019, 378, 66–86 CrossRef CAS.
- F. J. Bock and S. W. G. Tait, Nat. Rev. Mol. Cell Biol., 2020, 21, 85–100 CrossRef CAS PubMed.
- B. A. Carneiro and W. S. El-Deiry, Nat. Rev. Clin. Oncol., 2020, 17, 395–417 CrossRef PubMed.
- S. Fulda and D. Vucic, Nat. Rev. Drug Discovery, 2012, 11, 109–124 CrossRef CAS PubMed.
- Z. Jiang, Z. Hu, L. Zeng, W. Lu, H. Zhang, T. Li and H. Xiao, Free Radical Biol. Med., 2011, 50, 907–917 CrossRef CAS PubMed.
- Q. Li, T. Zhou, F. Wu, N. Li, R. Wang, Q. Zhao, Y.-M. Ma, J.-Q. Zhang and B.-L. Ma, Drug Metab. Rev., 2018, 50, 430–447 CrossRef CAS PubMed.
- N. Zheng, H. N. Tsai, X. Zhang, K. Shedden and G. R. Rosania, Mol. Pharmaceutics, 2011, 8, 1611–1618 CrossRef CAS PubMed.
- X. Li, J. Wu, L. Wang, C. He, L. Chen, Y. Jiao and C. Duan, Angew. Chem., Int. Ed., 2020, 59, 6420–6427 CrossRef CAS PubMed.
- K.-C. Tong, P.-K. Wan, C.-N. Lok and C.-M. Che, Chem. Sci., 2021, 12, 15229–15238 RSC.
- Q. Feng, T. Yang, L. Ma, X. Li, H. Yuan, M. Zhang, Y. Zhang and L. Fan, ACS Appl. Mater. Interfaces, 2022, 14, 38594–38603 CrossRef CAS PubMed.
- C. Li, Y. Xu, L. Tu, M. Choi, Y. Fan, X. Chen, J. L. Sessler, J. S. Kim and Y. Sun, Chem. Sci., 2022, 13, 6541–6549 RSC.
- L. Tu, C. Li, X. Xiong, J. Hyeon Kim, Q. Li, L. Mei, J. Li, S. Liu, J. Seung Kim and Y. Sun, Angew. Chem., Int. Ed., 2023, 62, e202301560 CrossRef CAS PubMed.
- Y. Xu, C. Li, S. Lu, Z. Wang, S. Liu, X. Yu, X. Li and Y. Sun, Nat. Commun., 2022, 13, 2009 CrossRef CAS PubMed.
- O. Domarco, C. Kieler, C. Pirker, C. Dinhof, B. Englinger, J. M. Reisecker, G. Timelthaler, M. D. García, C. Peinador, B. K. Keppler, W. Berger and A. Terenzi, Angew. Chem., Int. Ed., 2019, 58, 8007–8012 CrossRef CAS PubMed.
- C. Settembre, A. Fraldi, D. L. Medina and A. Ballabio, Nat. Rev. Mol. Cell Biol., 2013, 14, 283–296 CrossRef CAS PubMed.
- W. W. Y. Yim and N. Mizushima, Cell Discovery, 2020, 6, 6 CrossRef CAS PubMed.
- J. P. Luzio, B. A. Rous, N. A. Bright, P. R. Pryor, B. M. Mullock and R. C. Piper, J. Cell Sci., 2000, 113, 1515–1524 CrossRef CAS PubMed.
- R. E. Lawrence and R. Zoncu, Nat. Cell Biol., 2019, 21, 133–142 CrossRef CAS PubMed.
- J. P. Luzio, M. D. J. Parkinson, S. R. Gray and N. A. Bright, Biochem. Soc. Trans., 2009, 37, 1019–1021 CrossRef CAS PubMed.
- P. Saftig and J. Klumperman, Nat. Rev. Mol. Cell Biol., 2009, 10, 623–635 CrossRef CAS PubMed.
- S. R. Bonam, F. Wang and S. Muller, Nat. Rev. Drug Discovery, 2019, 18, 923–948 CrossRef CAS PubMed.
- A. Serrano-Puebla and P. Boya, Ann. N. Y. Acad. Sci., 2016, 1371, 30–44 CrossRef PubMed.
- Ana M. Villamil Giraldo, H. Appelqvist, T. Ederth and K. Öllinger, Biochem. Soc. Trans., 2014, 42, 1460–1464 CrossRef CAS PubMed.
- Y. Chen, Z. Yang, S. Wang, Q. Ma, L. Li, X. Wu, Q. Guo, L. Tao and X. Shen, Adv. Healthcare Mater., 2023, 12, 2202150 CrossRef CAS PubMed.
- P. Boya and G. Kroemer, Oncogene, 2008, 27, 6434–6451 CrossRef CAS PubMed.
- A. Serrano-Puebla and P. Boya, Biochem. Soc. Trans., 2018, 46, 207–215 CrossRef CAS PubMed.
- C. Oberle, J. Huai, T. Reinheckel, M. Tacke, M. Rassner, P. G. Ekert, J. Buellesbach and C. Borner, Cell Death Differ., 2010, 17, 1167–1178 CrossRef CAS PubMed.
- H. Erdal, M. Berndtsson, J. Castro, U. Brunk, M. C. Shoshan and S. Linder, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 192–197 CrossRef CAS PubMed.
- F. Wang, R. Gómez-Sintes and P. Boya, Traffic, 2018, 19, 918–931 CrossRef CAS PubMed.
- U. Repnik, M. Hafner Česen and B. Turk, Mitochondrion, 2014, 19, 49–57 CrossRef CAS PubMed.
- M. F. Chung, K. J. Chen, H. F. Liang, Z. X. Liao, W. T. Chia, Y. Xia and H.-W. Sung, Angew. Chem., Int. Ed., 2012, 51, 10089–10093 CrossRef CAS PubMed.
- Z. Liu, Y. Xiao, W. Chen, Y. Wang, B. Wang, G. Wang, X. Xu and R. Tang, J. Mater. Chem. B, 2014, 2, 3480–3489 RSC.
- H. Lima Jr, L. Jacobson, M. Goldberg, K. Chandran, F. Diaz-Griffero, M. P. Lisanti and J. Brojatsch, Cell Cycle, 2013, 12, 1868–1878 CrossRef PubMed.
- T. Yamashima and S. Oikawa, Prog. Neurobiol., 2009, 89, 343–358 CrossRef CAS PubMed.
- E. Gulbins, S. Dreschers and J. Bock, Exp. Physiol., 2003, 88, 85–90 CrossRef CAS PubMed.
- S. Desagher and J.-C. Martinou, Trends Cell Biol., 2000, 10, 369–377 CrossRef CAS PubMed.
- D. R. Green and J. C. Reed, Science, 1998, 281, 1309–1312 CrossRef CAS PubMed.
- C. Wang and R. J. Youle, Annu. Rev. Genet., 2009, 43, 95–118 CrossRef CAS PubMed.
- H. Kong and N. S. Chandel, J. Biol. Chem., 2018, 293, 7499–7507 CrossRef CAS PubMed.
- S. M. Richard, G. Bailliet, G. L. Páez, M. S. Bianchi, P. I. Peltomäki and N. S. O. Bianchi, Cancer Res., 2000, 60, 4231–4237 CAS.
- G. D. Dakubo, R. L. Parr, L. C. Costello, R. B. Franklin and R. E. Thayer, J. Clin. Pathol., 2006, 59, 10–16 CrossRef CAS PubMed.
- N. O. Bianchi, M. S. Bianchi and S. M. Richard, Mutat. Res., Rev. Mutat. Res., 2001, 488, 9–23 CrossRef CAS PubMed.
- F. Ciccarese and V. Ciminale, Front. Oncol., 2017, 7, 117 CrossRef PubMed.
- S. Mani, G. Swargiary and S. J. Ralph, Mitochondrion, 2022, 62, 50–73 CrossRef CAS PubMed.
- C.-H. Ni, C.-S. Yu, H.-F. Lu, J.-S. Yang, H.-Y. Huang, P.-Y. Chen, S.-H. Wu, S.-W. Ip, S.-Y. Chiang, J.-G. Lin and J.-G. Chung, Environ. Toxicol., 2014, 29, 740–749 CrossRef CAS PubMed.
- B. G. Heerdt, M. A. Houston and L. H. Augenlicht, Cancer Res., 2005, 65, 9861–9867 CrossRef CAS PubMed.
- L. D. Zorova, V. A. Popkov, E. Y. Plotnikov, D. N. Silachev, I. B. Pevzner, S. S. Jankauskas, V. A. Babenko, S. D. Zorov, A. V. Balakireva, M. Juhaszova, S. J. Sollott and D. B. Zorov, Anal. Biochem., 2018, 552, 50–59 CrossRef CAS PubMed.
- E. N. Maldonado, J. Patnaik, M. R. Mullins and J. J. Lemasters, Cancer Res., 2010, 70, 10192–10201 CrossRef CAS PubMed.
- N. J. Haradhvala, P. Polak, P. Stojanov, K. R. Covington, E. Shinbrot, J. M. Hess, E. Rheinbay, J. Kim, Y. E. Maruvka, L. Z. Braunstein, A. Kamburov, P. C. Hanawalt, D. A. Wheeler, A. Koren, M. S. Lawrence and G. Getz, Cell, 2016, 164, 538–549 CrossRef CAS PubMed.
- M. Ljungman, Chem. Rev., 2009, 109, 2929–2950 CrossRef CAS PubMed.
- A. Torgovnick and B. Schumacher, Front. Genet., 2015, 6, 157 Search PubMed.
- A. Marnef and G. Legube, Curr. Opin. Cell Biol., 2017, 46, 1–8 CrossRef CAS PubMed.
- N. Chatterjee and G. C. Walker, Environ. Mol. Mutagen., 2017, 58, 235–263 CrossRef CAS PubMed.
- M. Saki and A. Prakash, Free Radical Biol. Med., 2017, 107, 216–227 CrossRef CAS PubMed.
- K. V. Ferry, T. C. Hamilton and S. W. Johnson, Biochem. Pharmacol., 2000, 60, 1305–1313 CrossRef CAS PubMed.
- U. Repnik and B. Turk, Mitochondrion, 2010, 10, 662–669 CrossRef CAS PubMed.
- F. Guerra, G. Girolimetti, R. Beli, M. Mitruccio, C. Pacelli, A. Ferretta, G. Gasparre, T. Cocco and C. Bucci, Cells, 2019, 8, 452 CrossRef CAS PubMed.
- L. F. Burbulla, P. Song, J. R. Mazzulli, E. Zampese, Y. C. Wong, S. Jeon, D. P. Santos, J. Blanz, C. D. Obermaier, C. Strojny, J. N. Savas, E. Kiskinis, X. Zhuang, R. Krüger, D. J. Surmeier and D. Krainc, Science, 2017, 357, 1255–1261 CrossRef CAS PubMed.
- Y. C. Wong, D. Ysselstein and D. Krainc, Nature, 2018, 554, 382–386 CrossRef CAS PubMed.
- G. Xu, C. Li, C. Chi, L. Wu, Y. Sun, J. Zhao, X. H. Xia and S. Gou, Nat. Commun., 2022, 13, 3064 CrossRef CAS PubMed.
- D. S. Goodsell, Stem Cells, 2006, 24, 514–515 CrossRef PubMed.
- J. Raber, C. Zhu and L. A. Eriksson, J. Phys. Chem. B, 2005, 109, 11006–11015 CrossRef CAS PubMed.
- I. L. Zilberberg, V. I. Avdeev and G. M. Zhidomirov, THEOCHEM, 1997, 418, 73–81 CrossRef CAS.
- D. S. Eric, M. B. James and B. H. Stephen, Mol. Pharmacol., 1999, 56, 633 CrossRef.
- M.-H. Baik, R. A. Friesner and S. J. Lippard, J. Am. Chem. Soc., 2003, 125, 14082–14092 CrossRef CAS PubMed.
- J. R. Mitchell, J. H. J. Hoeijmakers and L. J. Niedernhofer, Curr. Opin. Cell Biol., 2003, 15, 232–240 CrossRef CAS PubMed.
- E. C. Friedberg, Nat. Rev. Cancer, 2001, 1, 22–33 CrossRef CAS PubMed.
- J. A. Marteijn, H. Lans, W. Vermeulen and J. H. J. Hoeijmakers, Nat. Rev. Mol. Cell Biol., 2014, 15, 465–481 CrossRef CAS PubMed.
- M. Baretti and D. T. Le, Pharmacol. Ther., 2018, 189, 45–62 CrossRef CAS PubMed.
- D. C. Chung and A. K. Rustgi, Gastroenterology, 1995, 109, 1685–1699 CrossRef CAS PubMed.
- P. Modrich, Science, 1994, 266, 1959–1960 CrossRef CAS PubMed.
- T. Aparicio, R. Baer and J. Gautier, DNA Repair, 2014, 19, 169–175 CrossRef CAS PubMed.
- T. Helleday, J. Lo, D. C. van Gent and B. P. Engelward, DNA Repair, 2007, 6, 923–935 CrossRef CAS PubMed.
- K. K. Khanna and S. P. Jackson, Nat. Genet., 2001, 27, 247–254 CrossRef CAS PubMed.
- M. Nechay, D. Wang and R. E. Kleiner, Cell Chem. Biol., 2023, 30, 906–919 CrossRef CAS PubMed.
- H. Hu, B. Li, J. Wang, Y. Tan, M. Xu, W. Xu and H. Lu, Biomed. Pharmacother., 2023, 163, 114778 CrossRef CAS PubMed.
- Q. Cao, Y. Li, E. Freisinger, P. Z. Qin, R. K. O. Sigel and Z.-W. Mao, Inorg. Chem. Front., 2017, 4, 10–32 RSC.
- W. Liu, Y. F. Zhong, L. Y. Liu, C. T. Shen, W. Zeng, F. Wang, D. Yang and Z. W. Mao, Nat. Commun., 2018, 9, 3496 CrossRef PubMed.
- E. Palma, J. Carvalho, C. Cruz and A. Paulo, Pharmaceuticals, 2021, 14, 605 CrossRef CAS PubMed.
- I. Alessandrini, M. Recagni, N. Zaffaroni and M. Folini, Int. J. Mol. Sci., 2021, 22, 5947 CrossRef CAS PubMed.
- R. Hänsel-Hertsch, A. Simeone, A. Shea, W. W. I. Hui, K. G. Zyner, G. Marsico, O. M. Rueda, A. Bruna, A. Martin, X. Zhang, S. Adhikari, D. Tannahill, C. Caldas and S. Balasubramanian, Nat. Genet., 2020, 52, 878–883 CrossRef PubMed.
- X. H. Zheng, Y. F. Zhong, C. P. Tan, L. N. Ji and Z. W. Mao, Dalton Trans., 2012, 41, 11807–11812 RSC.
- J. Liu, T. Luo, Y. Xue, L. Mao, P. J. Stang and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 5429–5435 CrossRef CAS PubMed.
- J. Liu, J. Sheng, L. Shao, Q. Zheng, W. Li, X. Chen, L. Mao and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 26740–26746 CrossRef CAS PubMed.
- S. Jeelani, R. J. Reddy, T. Maheswaran, G. Asokan, A. Dany and B. Anand, J. Pharm. BioAllied Sci., 2014, 6, S6 CrossRef CAS PubMed.
- E.-K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS PubMed.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- L. Xing, B. Thorndyke, E. Schreibmann, Y. Yang, T. F. Li, G. Y. Kim, G. Luxton and A. Koong, Med. Dosim., 2006, 31, 91–112 CrossRef PubMed.
- H. Y. Yoon, S. Jeon, D. G. You, J. H. Park, I. C. Kwon, H. Koo and K. Kim, Bioconjugate Chem., 2017, 28, 124–134 CrossRef CAS PubMed.
- Y. Huang, S. He, W. Cao, K. Cai and X.-J. Liang, Nanoscale, 2012, 4, 6135–6149 RSC.
- J. Huo, Q. Jia, H. Huang, J. Zhang, P. Li, X. Dong and W. Huang, Chem. Soc. Rev., 2021, 50, 8762–8789 RSC.
- C. He, Z. Tang, H. Tian and X. Chen, Adv. Drug Delivery Rev., 2016, 98, 64–76 CrossRef CAS PubMed.
- W. Fan, B. Yung, P. Huang and X. Chen, Chem. Rev., 2017, 117, 13566–13638 CrossRef CAS PubMed.
- J. J. Verhoef, J. F. Carpenter, T. J. Anchordoquy and H. Schellekens, Drug Discovery Today, 2014, 19, 1945–1952 CrossRef CAS PubMed.
- G. T. Kozma, T. Shimizu, T. Ishida and J. Szebeni, Adv. Drug Delivery Rev., 2020, 154, 163–175 CrossRef PubMed.
- P. Zhang, F. Sun, S. Liu and S. Jiang, J. Controlled Release, 2016, 244, 184–193 CrossRef CAS PubMed.
- D. Huang, H. Lan, F. Liu, S. Wang, X. Chen, K. Jin and X. Mou, Int. J. Clin. Exp. Med., 2015, 8, 8369–8376 Search PubMed.
- P. Carmeliet and R. K. Jain, Nat. Rev. Drug Discovery, 2011, 10, 417–427 CrossRef CAS PubMed.
- R. K. Jain, Science, 2005, 307, 58–62 CrossRef CAS PubMed.
- J. Hagendoorn, R. Tong, D. Fukumura, Q. Lin, J. Lobo, T. P. Padera, L. Xu, R. Kucherlapati and R. K. Jain, Cancer Res., 2006, 66, 3360–3364 CrossRef CAS PubMed.
- A. Alitalo and M. Detmar, Oncogene, 2012, 31, 4499–4508 CrossRef CAS PubMed.
- R. P. Kataru, C. L. Ly, J. Shin, H. J. Park, J. E. Baik, S. Rehal, S. Ortega, D. Lyden and B. J. Mehrara, Cancer Immunol. Res., 2019, 7, 1345–1358 CrossRef CAS PubMed.
- M. Madeo, P. L. Colbert, D. W. Vermeer, C. T. Lucido, J. T. Cain, E. G. Vichaya, A. J. Grossberg, D. Muirhead, A. P. Rickel, Z. Hong, J. Zhao, J. M. Weimer, W. C. Spanos, J. H. Lee, R. Dantzer and P. D. Vermeer, Nat. Commun., 2018, 9, 4284 CrossRef PubMed.
- G. E. Ayala, H. Dai, M. Powell, R. Li, Y. Ding, T. M. Wheeler, D. Shine, D. Kadmon, T. Thompson, B. J. Miles, M. M. Ittmann and D. Rowley, Clin. Cancer Res., 2008, 14, 7593–7603 CrossRef CAS PubMed.
- D. Hanahan and M. Monje, Cancer Cell, 2023, 41, 573–580 CrossRef CAS PubMed.
- F. Winkler, H. S. Venkatesh, M. Amit, T. Batchelor, I. E. Demir, B. Deneen, D. H. Gutmann, S. Hervey-Jumper, T. Kuner, D. Mabbott, M. Platten, A. Rolls, E. K. Sloan, T. C. Wang, W. Wick, V. Venkataramani and M. Monje, Cell, 2023, 186, 1689–1707 CrossRef CAS PubMed.
- G. Biffi and D. A. Tuveson, Physiol. Rev., 2021, 101, 147–176 CrossRef CAS PubMed.
- N. I. Nissen, M. Karsdal and N. Willumsen, J. Exp. Clin. Cancer Res., 2019, 38, 115 CrossRef PubMed.
- A. Arina, C. Idel, E. M. Hyjek, M.-L. Alegre, Y. Wang, V. P. Bindokas, R. R. Weichselbaum and H. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7551–7556 CrossRef CAS PubMed.
- G. Bergers, S. Song, N. Meyer-Morse, E. Bergsland and D. Hanahan, J. Clin. Invest., 2003, 111, 1287–1295 CrossRef CAS PubMed.
- V. G. Cooke, V. S. LeBleu, D. Keskin, Z. Khan, J. T. O'Connell, Y. Teng, M. B. Duncan, L. Xie, G. Maeda, S. Vong, H. Sugimoto, R. M. Rocha, A. Damascena, R. R. Brentani and R. Kalluri, Cancer Cell, 2012, 21, 66–81 CrossRef CAS PubMed.
- A. L. Ribeiro and O. K. Okamoto, Stem Cells Int., 2015, 2015, 868475 Search PubMed.
- K. M. Nieman, I. L. Romero, B. Van Houten and E. Lengyel, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1831, 2013, 1533–1541 Search PubMed.
- Q. Wu, B. Li, J. Li, S. Sun, J. Yuan and S. Sun, Biomark. Res., 2021, 9, 1–21 CrossRef PubMed.
- Q. Wu, B. Li, Z. Li, J. Li, S. Sun and S. Sun, J. Hematol. Oncol., 2019, 12, 95 CrossRef PubMed.
- M. S. Nakazawa, B. Keith and M. C. Simon, Nat. Rev. Cancer, 2016, 16, 663–673 CrossRef CAS PubMed.
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437–447 CrossRef CAS PubMed.
- S. R. McKeown, Br. J. Radiol., 2014, 87, 20130676 CrossRef CAS PubMed.
- L. E. Gerweck and K. Seetharaman, Cancer Res., 1996, 56, 1194–1198 CAS.
- M. Stubbs, P. M. J. McSheehy, J. R. Griffiths and C. L. Bashford, Mol. Med. Today, 2000, 6, 15–19 CrossRef CAS PubMed.
- A. J. Thistlethwaite, D. B. Leeper, D. J. Moylan, III and R. E. Nerlinger, Int. J. Radiat. Oncol., Biol., Phys., 1985, 11, 1647–1652 CrossRef CAS PubMed.
- L. Szablewski, Biochim. Biophys. Acta, Rev. Cancer, 2013, 1835, 164–169 CrossRef CAS PubMed.
- R. A. S. Nascimento, R. E. Özel, W. H. Mak, M. Mulato, B. Singaram and N. Pourmand, Nano Lett., 2016, 16, 1194–1200 CrossRef CAS PubMed.
- N. Hay, Nat. Rev. Cancer, 2016, 16, 635–649 CrossRef CAS PubMed.
- B. J. Altman, Z. E. Stine and C. V. Dang, Nat. Rev. Cancer, 2016, 16, 619–634 CrossRef CAS PubMed.
- D. R. Wise and C. B. Thompson, Trends Biochem. Sci., 2010, 35, 427–433 CrossRef CAS PubMed.
- A. A. Cluntun, M. J. Lukey, R. A. Cerione and J. W. Locasale, Trends Cancer, 2017, 3, 169–180 CrossRef CAS PubMed.
- S. Trabanelli, D. Očadlíková, S. Gulinelli, A. Curti, V. Salvestrini, R. de Paula Vieira, M. Idzko, F. Di Virgilio, D. Ferrari and R. M. Lemoli, J. Immunol., 2012, 189, 1303–1310 CrossRef CAS PubMed.
- P. Pellegatti, L. Raffaghello, G. Bianchi, F. Piccardi, V. Pistoia and F. Di Virgilio, PLoS One, 2008, 3, e2599 CrossRef PubMed.
- V. Vultaggio-Poma, A. C. Sarti and F. Di Virgilio, Cells, 2020, 9, 2496 CrossRef CAS PubMed.
- P. A. Smethurst and M. Griffin, Biochem. J., 1996, 313, 803–808 CrossRef CAS PubMed.
- T. Inaoka and K. Ochi, J. Bacteriol., 2002, 184, 3923–3930 CrossRef CAS PubMed.
- A. Meshkini, Cell Biochem. Biophys., 2014, 70, 27–32 CrossRef CAS PubMed.
- T. Tokuhara, N. Hattori, H. Ishida, T. Hirai, M. Higashiyama, K. Kodama and M. Miyake, Clin. Cancer Res., 2006, 12, 3971–3978 CrossRef CAS PubMed.
- R. Pasqualini, E. Koivunen, R. Kain, J. Lahdenranta, M. Sakamoto, A. Stryhn, R. A. Ashmun, L. H. Shapiro, W. Arap and E. Ruoslahti, Cancer Res., 2000, 60, 722–727 CAS.
- A. Kehlen, U. Lendeckel, H. Dralle, J. Langner and C. Hoang-Vu, Cancer Res., 2003, 63, 8500–8506 CAS.
- J. Zhou, X. Du and B. Xu, Angew. Chem., Int. Ed., 2016, 55, 5770–5775 CrossRef CAS PubMed.
- M. Uhlén, L. Fagerberg, B. M. Hallström, C. Lindskog, P. Oksvold, A. Mardinoglu, Å. Sivertsson, C. Kampf, E. Sjöstedt, A. Asplund, I. Olsson, K. Edlund, E. Lundberg, S. Navani, C. A.-K. Szigyarto, J. Odeberg, D. Djureinovic, J. O. Takanen, S. Hober, T. Alm, P.-H. Edqvist, H. Berling, H. Tegel, J. Mulder, J. Rockberg, P. Nilsson, J. M. Schwenk, M. Hamsten, K. von Feilitzen, M. Forsberg, L. Persson, F. Johansson, M. Zwahlen, G. von Heijne, J. Nielsen and F. Pontén, Science, 2015, 347, 1260419 CrossRef PubMed.
- F. J. Benham, J. Fogh and H. Harris, Int. J. Cancer, 1981, 27, 637–644 CrossRef CAS PubMed.
- I. Podgorski and B. F. Sloane, Biochem. Soc. Symp., 2003, 70, 263–276 CrossRef CAS PubMed.
- M. H. Abdulla, M. A. Valli Mohammed, K. Al Khayal, A. Al Shkieh, A. Zubaidi, R. Ahmad, K. Al Saleh, O. Al Obeed and J. McKerrow, Oncol. Rep., 2017, 37, 3175–3180 CrossRef CAS PubMed.
- C. S. Gondi and J. S. Rao, Expert Opin. Ther. Targets, 2013, 17, 281–291 CrossRef CAS PubMed.
- L. Zhang, J. Shi, J. Feng, H. Klocker, C. Lee and J. Zhang, Prostate Cancer Prostatic Dis., 2004, 7, 327–332 CrossRef CAS PubMed.
- L. M. Coussens, B. Fingleton and L. M. Matrisian, Science, 2002, 295, 2387–2392 CrossRef CAS PubMed.
- R. E. Vandenbroucke and C. Libert, Nat. Rev. Drug Discovery, 2014, 13, 904–927 CrossRef CAS PubMed.
- L. Rao, Q. F. Meng, L. L. Bu, B. Cai, Q. Huang, Z. J. Sun, W. F. Zhang, A. Li, S. S. Guo, W. Liu, T. H. Wang and X. Z. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 2159–2168 CrossRef CAS PubMed.
- M. Gao, C. Liang, X. Song, Q. Chen, Q. Jin, C. Wang and Z. Liu, Adv. Mater., 2017, 29, 1701429 CrossRef PubMed.
- L. Rao, B. Cai, L. L. Bu, Q. Q. Liao, S. S. Guo, X. Z. Zhao, W. F. Dong and W. Liu, ACS Nano, 2017, 11, 3496–3505 CrossRef CAS PubMed.
- Y. Xia, L. Rao, H. Yao, Z. Wang, P. Ning and X. Chen, Adv. Mater., 2020, 32, 2002054 CrossRef CAS PubMed.
- Y. Zhang, K. Cai, C. Li, Q. Guo, Q. Chen, X. He, L. Liu, Y. Zhang, Y. Lu, X. Chen, T. Sun, Y. Huang, J. Cheng and C. Jiang, Nano Lett., 2018, 18, 1908–1915 CrossRef CAS PubMed.
- H. Cao, Z. Dan, X. He, Z. Zhang, H. Yu, Q. Yin and Y. Li, ACS Nano, 2016, 10, 7738–7748 CrossRef CAS PubMed.
- Z. Chen, P. Zhao, Z. Luo, M. Zheng, H. Tian, P. Gong, G. Gao, H. Pan, L. Liu, A. Ma, H. Cui, Y. Ma and L. Cai, ACS Nano, 2016, 10, 10049–10057 CrossRef CAS PubMed.
- J. Y. Zhu, D. W. Zheng, M. K. Zhang, W. Y. Yu, W. X. Qiu, J. J. Hu, J. Feng and X. Z. Zhang, Nano Lett., 2016, 16, 5895–5901 CrossRef CAS PubMed.
- Q. Hu, W. Sun, J. Wang, H. Ruan, X. Zhang, Y. Ye, S. Shen, C. Wang, W. Lu, K. Cheng, G. Dotti, J. F. Zeidner, J. Wang and Z. Gu, Nat. Biomed. Eng., 2018, 2, 831–840 CrossRef CAS PubMed.
- H. Sun, J. Su, Q. Meng, Q. Yin, L. Chen, W. Gu, P. Zhang, Z. Zhang, H. Yu, S. Wang and Y. Li, Adv. Mater., 2016, 28, 9581–9588 CrossRef CAS PubMed.
- Q. Jiang, Y. Liu, R. Guo, X. Yao, S. Sung, Z. Pang and W. Yang, Biomaterials, 2019, 192, 292–308 CrossRef CAS PubMed.
- S. Rezaei, R. F. de Araújo Júnior, I. L. G. da Silva, T. Schomann, C. Eich and L. J. Cruz, Biomater. Adv., 2023, 151, 213456 CrossRef CAS PubMed.
- H. Y. Chen, J. Deng, Y. Wang, C. Q. Wu, X. Li and H.-W. Dai, Acta Biomater., 2020, 112, 1–13 CrossRef CAS PubMed.
- G. Russell-Jones, K. McTavish, J. McEwan, J. Rice and D. Nowotnik, J. Inorg. Biochem., 2004, 98, 1625–1633 CrossRef CAS PubMed.
- A. Raza, A. Singh, S. Amin, J. E. Spallholz and A. K. Sharma, Chem. – Biol. Interact., 2022, 365, 110071 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |