
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring pathological link between antimicrobial and amyloid peptides
Yijing
Tang†
a,
Yanxian
Zhang†
b,
Dong
Zhang
 c,
Yonglan
Liu
d,
Ruth
Nussinov
*ef and
Jie
Zheng
*a
c,
Yonglan
Liu
d,
Ruth
Nussinov
*ef and
Jie
Zheng
*a
aDepartment of Chemical, Biomolecular, and Corrosion Engineering, The University of Akron, Ohio 44325, USA. E-mail: zhengj@uakron.edu
bDivision of Endocrinology and Diabetes, Department of Pediatrics, School of Medicine, Stanford University, Palo Alto, CA 94304, USA
cThe Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, Georgia 30332, USA
dCancer Innovation Laboratory, National Cancer Institute, Frederick, MD 21702, USA
eComputational Structural Biology Section, Frederick National Laboratory for Cancer Research, Frederick, MD 21702, USA. E-mail: nussinor@mail.nih.gov
fDepartment of Human Molecular Genetics and Biochemistry Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel
First published on 23rd July 2024
Abstract
Amyloid peptides (AMYs) and antimicrobial peptides (AMPs) are considered as the two distinct families of peptides, characterized by their unique sequences, structures, biological functions, and specific pathological targets. However, accumulating evidence has revealed intriguing pathological connections between these peptide families in the context of microbial infection and neurodegenerative diseases. Some AMYs and AMPs share certain structural and functional characteristics, including the ability to self-assemble, the presence of β-sheet-rich structures, and membrane-disrupting mechanisms. These shared features enable AMYs to possess antimicrobial activity and AMPs to acquire amyloidogenic properties. Despite limited studies on AMYs–AMPs systems, the cross-seeding phenomenon between AMYs and AMPs has emerged as a crucial factor in the bidirectional communication between the pathogenesis of neurodegenerative diseases and host defense against microbial infections. In this review, we examine recent developments in the potential interplay between AMYs and AMPs, as well as their pathological implications for both infectious and neurodegenerative diseases. By discussing the current progress and challenges in this emerging field, this account aims to inspire further research and investments to enhance our understanding of the intricate molecular crosstalk between AMYs and AMPs. This knowledge holds great promise for the development of innovative therapies to combat both microbial infections and neurodegenerative disorders.
 Yijing Tang | Yijing Tang received her BE in Polymer Science and materials from Beijing University of Chemical Technology and her MS in Polymer Science from the University of Akron. After completing her PhD study in 2024 under the supervision of Prof. Jie Zheng at the University of Akron, she joined the group of Prof. Brian Bacskai and Prof. Steven Hou at the Massachusetts General Hospital and Harvard Medical School as a Postdoc research fellow. Since then, her research has primarily focused on developing in vivo fluorescent labeling techniques to monitor real-time progression and dynamics of neurodegeneration. |
 Yanxian Zhang | Dr Yanxian Zhang earned her PhD in Chemical, Biomolecular, and Corrosion Engineering from the University of Akron, focusing on amyloid peptide cross-seeding and inhibitor development. Her work revealed critical interactions between antimicrobial human α-defensins and amyloid peptides, notably inhibiting amyloid aggregation. Since 2021, she has been a postdoctoral researcher at Stanford University, developing biopharmaceuticals by merging molecular biology, chemical engineering, and protein engineering to enhance stability and optimize pharmacological profiles. |
 Dong Zhang | Dr Dong Zhang earned his PhD in Chemical, Biomolecular, and Corrosion Engineering from The University of Akron in 2022, under the supervision of Professor Jie Zheng. He is currently a postdoctoral fellow in Professor Younan Xia's group at the Georgia Institute of Technology. His research focuses on the rational design and synthesis of polymers and nanomaterials for applications in environmental, biological, and biomedical fields. |
 Yonglan Liu | Dr Yonglan Liu earned her PhD in Chemical Engineering from the University of Akron. She currently is a research scientist at the Cancer Innovation Laboratory, National Cancer Institute, National Institutes of Health. Her research centers on computational biology, biophysics, and chemistry, with specialized expertise in cancer biology, neurodegenerative diseases, neurodevelopmental disorders, structural biology, drug discovery, and materials science. Dr. Liu employs machine learning and molecular dynamics simulations to innovate in these fields of study, aiming to revolutionize our understanding and treatment of complex diseases. Her work is driven by a passion for bridging interdisciplinary approaches to tackle critical biomedical challenges. |
 Ruth Nussinov | Ruth Nussinov is at the Frederick National Laboratory of Cancer Research, NCI, an adjunct Professor at the Chemistry Department at the University of Maryland, and emeritus at the Medical school at Tel Aviv University. She authored over 750 scientific papers, is Editor-in-Chief of Current Opinion in Structural Biology, was Editor-in-Chief of PLoS Computational Biology, is Associate Editor and on Editorial Boards of multiple journals. She is a frequent speaker in Domestic and International meetings, symposia, and academic institutions, won multiple awards, elected fellow of several societies, and was twice a highly cited researcher. Her NCI website gives further details. https://ccr.cancer.gov/ruth-nussinov. |
 Jie Zheng | Prof. Zheng, a Full Professor of Dept. of Chemical, Biomolecular, and Corrosion Engineering at the University of Akron, earned his PhD from the University of Washington in 2005. His research spans disease-related proteins, biomaterials, and structural biology, incorporating experimental methods, biophysical analysis, molecular simulations, and machine-learning models. He has authored over 290 journal papers, achieving an h-index of 82 and accumulating over 23 |
1. Introduction
Amyloids and antimicrobial peptides are generally considered as two distinct families characterized by their diverse sequences, structures, biological functions, and targets. Amyloid peptides (AMYs) are widely recognized as causative agents in the development of many neurodegenerative diseases, including Alzheimer's disease (AD), type II diabetes (T2D), and Parkinson's disease (PD).1–4 The prevailing “amyloid cascade hypothesis” for the past 30 years postulates that the accumulation of misfolded amyloid aggregates, characterized by their highly ordered, β-sheet structures (namely amyloids),5 in human tissues is a key pathological feature of neurodegenerative diseases. Unlike the pathological characteristics of amyloid peptides, antimicrobial peptides (AMPs), particularly those broad-spectrum antibacterial ones, are aimed at combating diseases caused by the infection and inflammation of various pathogens (e.g., viruses, parasites, bacteria, fungi).6Despite the disparate functions of AMYs and AMPs, numerous studies revealed that certain amyloid and antimicrobial peptides unexpectedly exhibit additional functions typically associated with the other class.7 AMPs, such as protegrin-1,8 plantaricin A,9 uperin 3.5,10 magainin,11,12 dermaseptin S9,13 have demonstrated their ability to form amyloid-like fibrils enriched in β-sheet structures, resembling the characteristic morphology of classic amyloid fibrils. Similarly, several AMYs, such as Aβ, hIAPP, α-syn, and SAA,7,14–16 exhibited antibacterial activity against various bacterial strains (e.g., Candida albicans, Escherichia coli, Staphylococcus epidermidis, etc.)17–19 and antiviral activity against different viruses (e.g., influenza virus A, herpes simplex virus, H3N2, H1N1, etc).20,21 AMYs appear to act as a class of innate immune defense molecules, functioning by utilizing toxic amyloid aggregates to eliminate a broad spectrum of pathogens through the disruption of their cell membranes thus crucial functions. The shared functionality between AMYs and AMPs could be attributed to their common β-sheet-rich structures, enabling a strong binding affinity to cell membranes. Such strong membrane-activating interaction allows β-sheet-rich AMYs and AMPs to catalyze toxin-like channel formation and membrane depolarization as the primary mode-of-action of membrane disruption,22 ultimately leading to the elimination of host target cells and pathogens. Therefore, AMP amyloidogenicity and AMY antimicrobial activity are suggested to originate from their shared innate and conserved characteristics throughout evolution.6
More importantly, recent studies revealed intriguing pathological connections between amyloid and antimicrobial peptides, likely arising from their shared structural and functional characteristics. While this area is still relatively less explored, there is emerging evidence that certain pairs of AMYs and AMPs, such as hIAPP and aurein, Aβ and PG1, α-/β-defensin and Aβ/hIAPP/hCT,23,24 LL-37 and Aβ/hIAPP,25,26 α-syn and CsgA/CsgC/CsgE,27–29 can interact with each other, leading to the formation of amyloid fibrils that share similar conformations and pathological properties. This phenomenon, referred to as cross-seeding, highlights the intricate interplay between amyloid and antimicrobial peptides in pathological processes. As a result, the cross-seeding between AMYs and AMPs, likely facilitated by their similar β-sheet structures, often modulates amyloid aggregation through acceleration, inhibition, or modification of amyloid-induced cytotoxicity.30,31 This dynamic crosstalk between AMYs and AMPs highlights the potential for therapeutic interventions targeting amyloid-related diseases by harnessing the functional properties of antimicrobial peptides.
Pathologically, the cross-seeding between AMYs and AMPs gives rise to the “microbial infection hypothesis” and the “neuroinflammation hypothesis”. Both hypotheses offer insights into the underlying pathological mechanisms of neurodegenerative diseases. Since neuroinflammation is recognized as a significant risk factor in the development of pathological hallmarks of neurodegenerative diseases, both pathogens (including viruses, bacteria, and fungi) and amyloid aggregates (including Aβ, hIAPP, α-syn, and SAA) have been reported to induce persistent neuroinflammation by activating a long-lasting immune response. This chronic neuroinflammation, which arises as the consequence of both amyloid aggregation and microbial infection, contributes to the detrimental effects on neuronal health and leads to the eventual neurodegeneration.32,33
Emerging evidence from both experimental and clinical studies revealed a remarkable connection, and even a feedback loop, between amyloid formation and neuroinflammation, in addition to their individual contributions to the pathogenesis of neurodegenerative diseases. This connection is facilitated through bidirectional and continuous communication between amyloid proteins and gut microbiota.34,35 Certain amyloid proteins can influence the activity of gut microbiota, while the gut microbiota, in turn, can modulate amyloid aggregation and its associated toxicity. These findings shed light on the potential involvement of the brain-gut-microbiota axis in neurodegenerative mechanisms.33,36 The intricate interplay between amyloid formation, neuroinflammation, and the gut microbiota represents a complex and dynamic relationship that underlies the pathogenesis of these diseases, underscoring antimicrobial peptides as promising agents to regulate the detrimental functions of both.
Here, we present a historical timeline tracing the discovery of both native and alternative functions, as well as cross-seeding interactions, between AMPs and AMYs (Fig. 1). Overall, we aim to provide a comprehensive overview of the pathological relationship between AMYs and AMPs, with emphasis on their shared structural/functional characteristics, cross-seeding, and the biological roles of AMPs in the pathogenesis of neurodegeneration. Work on the pathological link between AMYs and AMPs is still at its early stage. We hope that our perspective, which outlines current challenges and future direction, will inspire efforts to exploit the potential of antimicrobial peptides in the development of AMP-based drugs in the treatment of neurodegenerative diseases.
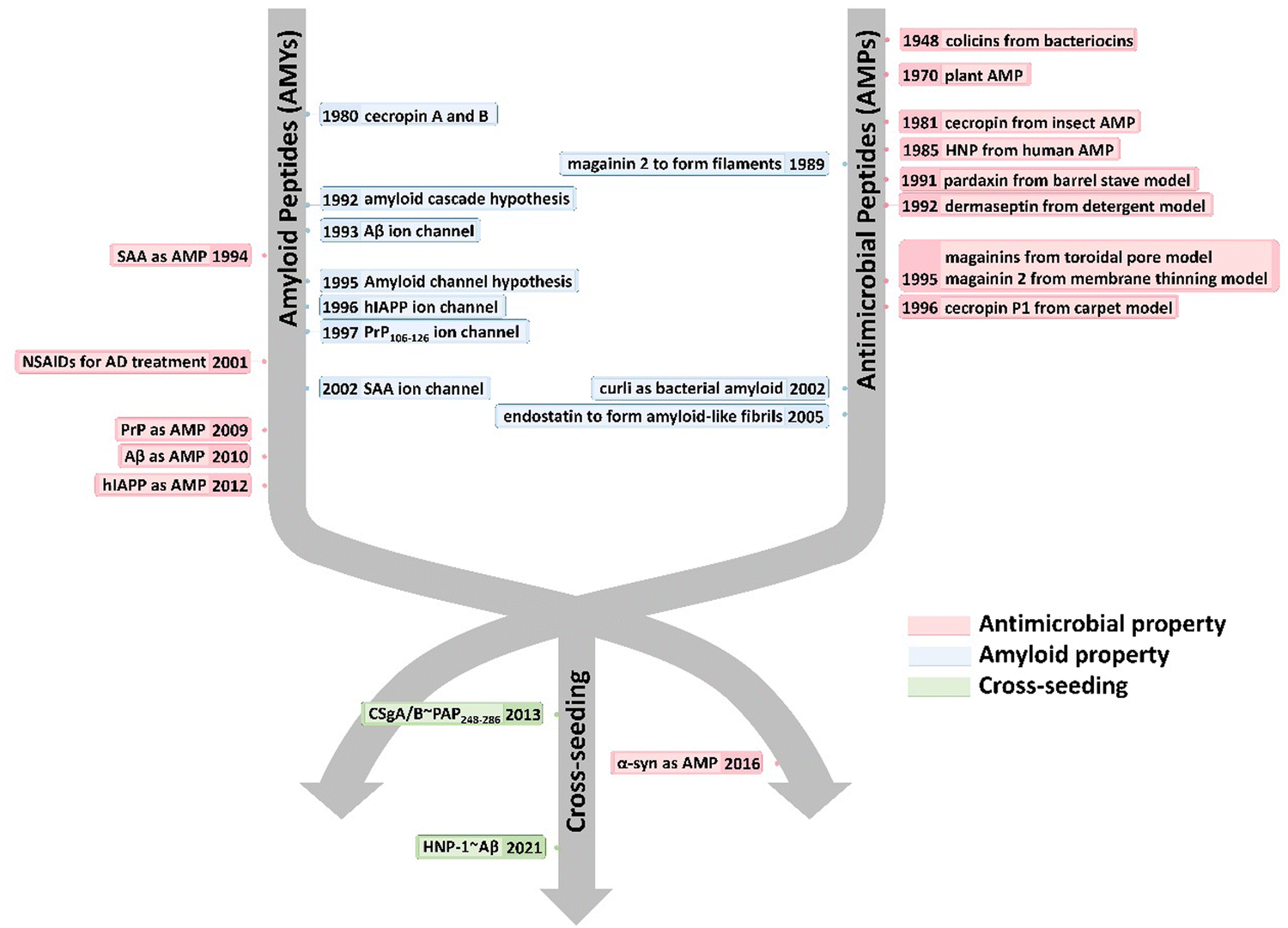 | ||
| Fig. 1 Historical timeline for discovering the native and alternative functions and cross-seeding of antimicrobial peptides (AMPs) and amyloid peptides (AMYs). | ||
2. Pathological links in sequence, structure, and function similarities between antimicrobial peptides and amyloid peptides
2.1. Sequence and structural similarities between AMPs and AMYs
AMYs and AMPs exhibit distinct sequence and structural characteristics related to their intrinsic antimicrobial activity and peptide aggregation property. As reported in antimicrobial peptides database (APD)37,38 and collection of anti-microbial peptides (CAMP)39 database, AMPs contain over 2400 sequences,40 while AMYs have a much smaller sequence pool, comprising approximately 30 sequences associated with 25 different neurodegenerative diseases. Typically, AMYs demonstrate amphiphilicity with a high hydrophobic residue content,4 while AMPs of typically 10–40 amino acids long are rich in cationic residues (lysine and arginine) and hydrophobic residues (leucine, valine, and isoleucine) to counterbalance positive charges.41 A notable commonality between the two peptide classes is the prevalence of hydrophobic residues, crucial for membrane insertion and stabilization through preferential hydrophobic interactions with lipid membranes.42–44 Both AMYs and AMPs frequently demonstrate a pronounced preference for hydrophobic residues such as Ile, Val, and aromatic amino acids.40,45 Sequence analysis from AMP databases reveals that the average hydrophobic content of AMPs is 42%. Within this content, Leu, Ala, Cys, Ile, Val, and Phe are frequently occurring residues in AMP sequences, while Met is rarely observed in AMPs (<1.2%).46 Differently, all AMYs inherently possess the capability to form β-structure-rich aggregates, suggesting that AMY sequences may exhibit a higher tendency for aggregation compared to other peptides. Computational analysis, exploring the aggregation propensity of both amyloidogenic and non-amyloidogenic sequences,47–49 along with experimental investigations,50 reveals that amino acids such as Val, Trp, Phe, Cys, Tyr, Ser, and Ile are more favorable for amyloid formation, while charged residues like Asp, Lys, Glu, and Arg are less favored. Aligned with the hydrophobicity indexes of amino acids, these residues play a dual role: strengthening the association force in peptide-cell membrane interactions and providing more robust stabilization of the peptide aggregates. The distinct hydrophobic residue composition among different AMYs and AMPs contributes to the diversity in their structures and functions, which is underscored by the linkage of the hydrophobicity to their hemolytic potential.While positive charge serves as a distinguishing feature between AMPs and AMYs, however both classes demonstrate stronger interactions with anionic lipid membranes compared to neutral zwitterionic lipid membranes. The prevalence of positively charged residues in AMPs with average net charge of +3.2e promotes selectivity, facilitating the initiation of contacts and the adoption of a surface position on anionic membranes. Typical Arg-rich AMPs (e.g., apidaecin, buforin II, indolicidin) can spontaneously translocate into bacterial cells without membrane perturbationn.51–54 Upon translocation, these Arg-rich AMPs interact with DNA, RNA, ribosomes, and other intracellular components via electrostatic interactions, ultimately leading to cell death. However, the presence of positively charged residues like Arg and Lys appears to discourage their occurrence in amyloid sequences. This may be attributed to electrostatic repulsion, as charge–charge stacking disfavors the self-aggregation of AMYs. On the other hand, the prevalence of negatively charged residues in AMYs enables complex formation with cations, facilitating the transport of cations across the membrane.
The amphiphilic nature of AMPs achieves a balance between polar and nonpolar components, notably featuring a lower prevalence of Gln and Asn in AMPs. Cys is notably abundant (>14%) in β-hairpin AMPs, underscoring the prevalence of disulfide bonds as common structural motifs essential for maintaining a stable amphipathic structure.40 In contrast, although strong hydrophobicity plays a crucial role in AMY aggregation, the mere bias toward hydrophobicity does not provide a sound explanation for amyloid structures and aggregation. Polar residues are equally essential to facilitate the formation of specific β-sheet organizations. Given that typical, parallel β-sheets in amyloid fibrils are predominantly stabilized by interchain hydrogen bonds between β-sheets, Gln and Asn are highly favored in AMYs. These residues contribute to the formation of a ladder structure crucial for stabilizing amyloid fibrils when Gln/Asn harboring peptides adopt an in-register, parallel β-sheet arrangement, as evidenced by GNNQQNY. While proline tends to discourage the β-structure in amyloid sequences, it does not exhibit a distinct preference in AMP sequences.
Overall, an amphipathic characteristic is a common sequence feature shared between AMPs and AMYs, although specific attributes may vary to achieve their unique structures and distinct native functions. AMPs commonly exhibit amphipathic structures with an abundance of hydrophobic and cationic residues but fewer polar residues. Key contributors to the positive charge, necessary for membrane interaction, are Arg and Lys, while cysteine residues may form disulfide bonds, enhancing stability. This amphipathicity enables selective interaction with microbial membranes. AMYs feature a propensity for hydrophobic amino acids, presence of aromatic residues, glycine-rich segments for flexibility, and occurrence of charged residues, and these sequence elements contribute to the adoption of β-structure-rich structure in their aggregates. The presence of various residue types contributes to peptide–membrane interactions in distinct ways. Charged residues are known for initiating contacts and membrane adsorption through electrostatic interactions, hydrophobic residues increase the likelihood of the adsorbed peptides to partition into the hydrophobic interior of the membranes, and polar residues achieve their functions by forming hydrogen bonding networks with lipid polar groups and adjusting secondary structures for membrane interactions. Moreover, charged and polar residues play a vital role in determining the correct orientation for peptide adsorption on and insertion into the membrane, with specific tilt angles.55 A strong correlation in Fig. 2 is evident for 80% of the amino acid residues found in both antimicrobial and amyloid-like regions. The probability of an individual residue being situated in either aggregation-prone or antimicrobial domains is well correlated, with the notable exception of positively charged residues that favors in antimicrobial region, but not aggregation-prone region.56 Illustratively, AMYs arrange hydrophobic, hydrophilic, and charged residues sequentially, as seen in patterns like CCCHHHPPPPPPPPPPPPPC, while AMPs adopt an alternative sequence, exemplified by PHCPHCPHCPHCPHCPHC (e.g., VKRWKKWRWKWKKWV).57 Statistical analysis of compositional preferences in naturally occurring AMPs and AMYs may identify specific sequence fragments for membrane binding, transmembrane insertion, and structural transition, which could serve as the basis for engineering specific sequences for AMPs and AMYs, thereby endowing them with new alternative functions.
 | ||
| Fig. 2 Functional residue propensity in both amyloid and antimicrobial sequence regions of antimicrobial peptides (AMPs) and amyloid peptides (AMYs). Reproduced with permission from ref. 56. | ||
Structurally, AMPs display four distinctive conformations—α-helical, β-sheet, extended, and disordered states, predominantly determined by solution NMR and X-ray methods. Among these, helical- and β-related structures are the most populated conformations. In many instances, AMPs exhibit distinct structures in solution compared to when they interact with membranes. Upon exposure to cell membranes, they undergo structural transitions or peptide aggregation to facilitate membrane adsorption or insertion. In contrast, AMYs lack defined structures in their native states, however, under pathological conditions, AMYs consistently adopt an in-register, β-sheet organization in their fibrillar aggregates, demonstrating a remarkable independence from both their individual sequences and the cellular environment. Commonly, fibrillar amyloid aggregates exhibit several structural motifs, including (i) the self-complementing polar or non-polar van der Waals (VDW) zippers between (anti)parallel sheets in sequences like GNNQQNY,58,59 Aβ,60,61 hIAPP62 and (ii) chemically-identical amino acid ladders such as Asn and Gln, and π–π stacking.63 These motifs provide structure-based forces to stabilize β-sheet conformations. Different from AMYs, the presence of cell membranes appears to accelerate amyloid formation, impacting the kinetics of β-sheet structures rather than their thermodynamics. β-structure motifs are present in both AMPs and AMYs (Fig. 3). In AMPs they are often stabilized by disulfide bridges between conserved Cys residues (e.g., protegrin I, tachyplesin, human β-defensin, gomesin)42,64–66 (Fig. 3a), while those of AMYs (e.g., hIAPP, Aβ, PrP,) are typically stabilized by salt bridges60,62 (Fig. 3b). Deletion of disulfide bonds in AMPs (e.g., PG-1, α-defensins) leads to loss of β-hairpin and membrane-activated antimicrobial activity,67–69 emphasizing the essential role of disulfide bonds in the structure and function of AMPs. AMPs without disulfide bonds, including ll37,70 melittin,71 magainin,72,73 cecropin A,74,75 and dermaseptins,76 initially adopt a random coil in solution and undergo a random-coil-to-helix transition upon interaction with the membrane. The lengths of these β-sheets typically span the membrane bilayer thickness, in which mismatches between the hydrophobic regions of the β-strands and the lipids significantly influence the peptides lateral and orientational movements within the membranes.
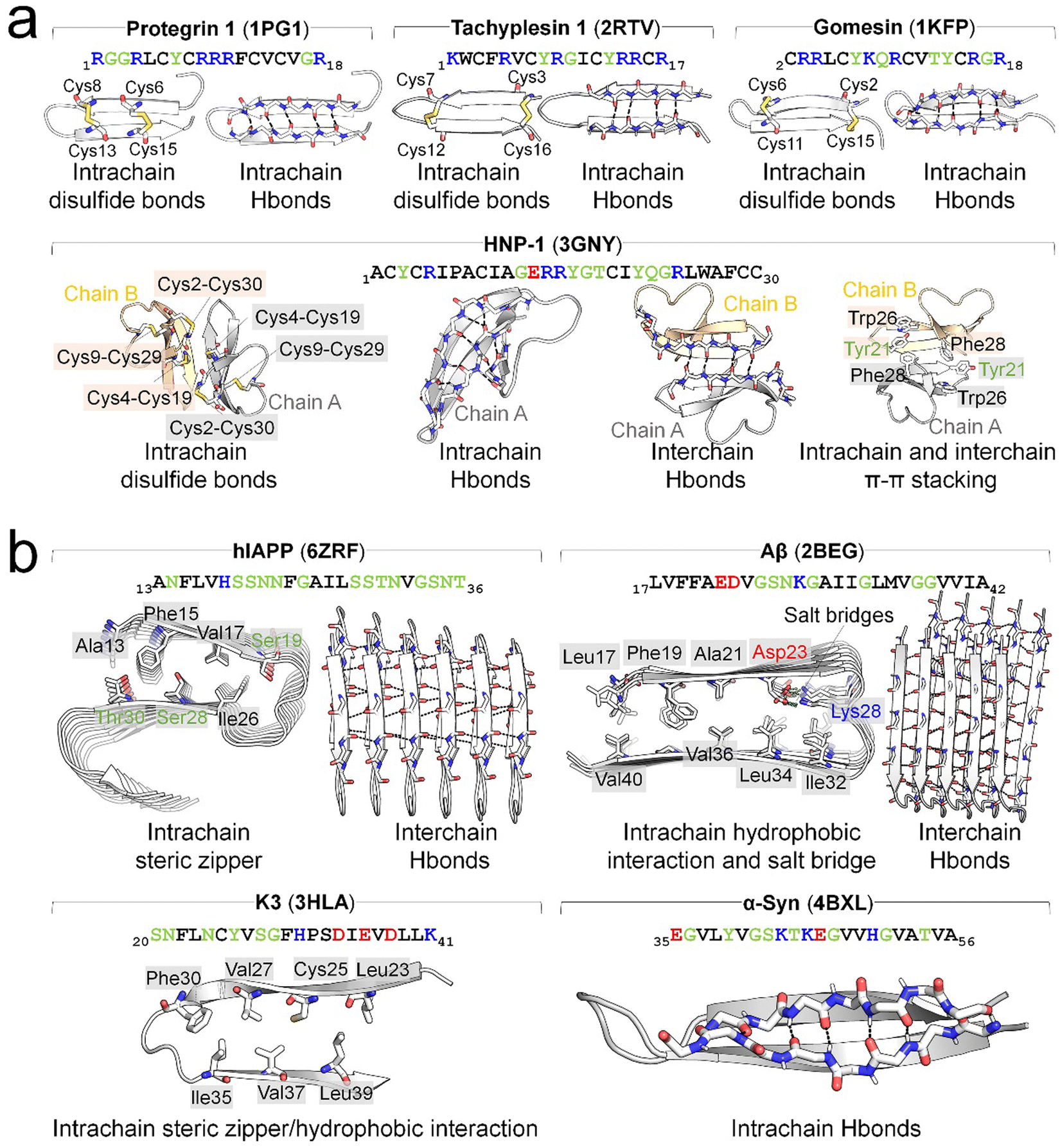 | ||
| Fig. 3 Overview of typical β-hairpin structure of (a) AMPs of protegrin 1 (PDB: 1PG1), tachyplesin 1 (PDB: 2RTV), gomesin (1KFP), and human β-defensin (HNP-1, PDB: 3GNY) and (b) AMYs of hIAPP (PDB: 6ZRF), Aβ (PDB: 2BEG), K3 of β2-microtubulin (PDB: 3HLA), and α-Syn (PDB: 4BXL). The β-hairpin structures are stabilized by different interactions, specifically, with disulfide bonds between Cys residues in AMPs, and steric zippers (e.g., hIAPP and K3), hydrophobic interactions (e.g., Aβ and K3), and salt bridges between positive and negative residues (e.g., Aβ) in AMYs. Intrachain backbone-backbone hydrogen bonds (H-bonds) are prevalent and crucial for stabilizing the β-hairpin structures of AMPs and are also observed in some AMYs, such as α-Syn. For dimerization or oligomerization of both AMPs and AMYs, interchain backbone-backbone H-bonds serve as the primary interactions. Additional interactions, though sporadic, contribute to maintaining β-hairpin structures. Examples include intrachain and interchain π–π stacking observed in HNP-1. H-bonds and salt bridges are denoted by black and lime dashes, respectively. The text of positively charged, negatively charged, hydrophobic, and hydrophilic residues are drafted in blue, red, black, and green, respectively. | ||
2.2. Common membrane disruption mechanisms of AMPs and AMYs
Antimicrobial peptides (AMPs) and amyloidogenic peptides (AMYs) disrupt cell membranes through both receptor-mediated and nonreceptor-mediated mechanisms. Receptor-mediated mechanisms involve the recognition of target cells via specific receptors such as sugars, lipids, or proteins, leading to cellular responses including cell necrosis and apoptosis through varied cellular processes.35,77,78 However, this review primarily addresses nonreceptor-mediated mechanisms, focusing on direct interactions of these peptides with the membrane that lead to increased membrane permeability through various modes (barrel-stave, toroidal-pore, carpet, and detergent models).79 Such direct membrane interaction process can be generalized into three sequential steps: initial membrane adsorption, deeper peptide insertion, and reorganization within the membrane (Fig. 4a). Although this model simplifies the dynamics, it underscores the critical factors influencing these interactions, including intrinsic physicochemical properties of peptides (size, sequence, secondary structure, net charge, charge distribution, hydrophobicity, and amphiphilic character), dynamic properties of peptides (aggregation, adsorption, orientation, and insertion), characteristics of cell membranes (composition, headgroup size, surface charge, hydrogen bonding capacity, and bilayer elastic properties), and environmental conditions (peptide concentration, temperature, ionic strength, pH).80,81 Membrane disruption by AMPs and AMYs occurs through two broad mechanisms:82 non-poration and poration (Fig. 4b). Non-poration does not involve crossing the membrane but can disrupt its integrity. For instance, in the carpet model, peptides disrupt the membrane without forming pores, extracting lipids to create micelles and causing local, transient defects. This mechanism is employed by peptides such as cecropin and melittin, which align parallel to the membrane, disrupting lipid organization and inducing thinning. In contrast, poration involves forming transmembrane pores or channels, typically occurring when peptides insert into the membrane and oligomerize to create structures that facilitate the passage of ions and molecules. The barrel-stave and toroidal models are common scenarios, where monomeric peptides either insert and subsequently form pores, or peptide oligomers directly insert to create pores.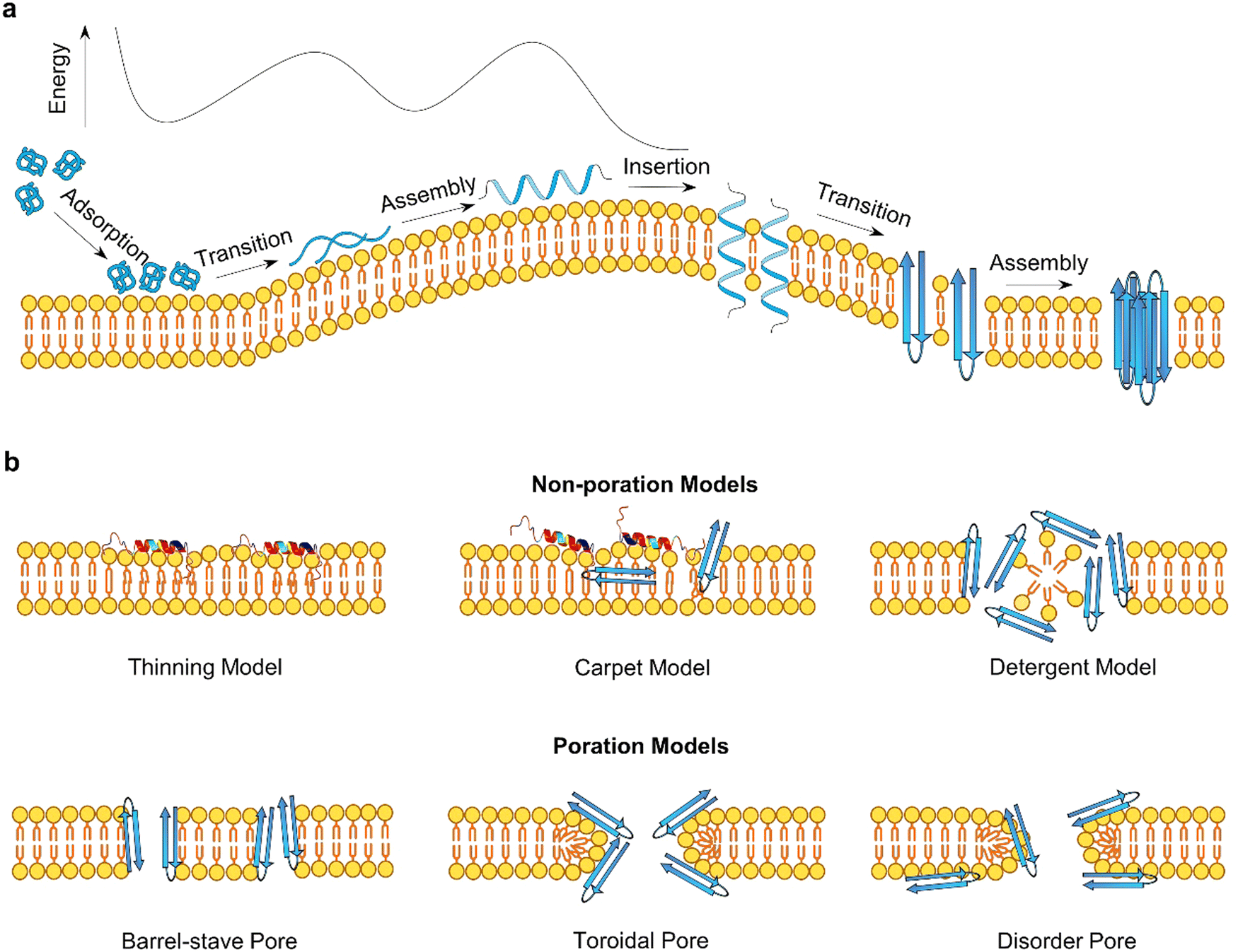 | ||
| Fig. 4 (a) A general peptide–membrane interaction process in a free energy landscape. It involves initial peptide adsorption, structural transition, self-assembly, insertion pathways, (b) illustrating distinct non-poration (carpet model, detergent model, and membrane thinning model) and poration (barrel-stave pore, toroidal pore, disorder pore) membrane disruption mechanisms of AMPs and AMYs. | ||
In these non-poration, membrane-disruption events (Fig. 4b), AMP and AMY peptides could function as “detergents”83,84 extracting specific lipids from membranes to form peptide/lipid micelles, anchoring themselves in the polar headgroup region to cause lateral membrane expansion and thinning, or triggering lipids clustering and segregation and subsequent lipid phase transitions.68,69,73,85,86 In the carpet or detergent models, both AMYs and AMPs align parallel to the membrane, with the hydrophilic side of the peptide facing the headgroups through electrostatic interactions, while the hydrophobic portion faces the lipid tails through hydrophobic interactions. These mechanisms have been reported for AMPs such as cecropin,83 buforin 2,87 aurein 1.2,88 Citropin,88 and melittin89 to achieve their antimicrobial activity at high peptide densities or peptide-to-lipid ratios, as well as for AMYs like Aβ,90 hIAPP,91 α-syn,92 and PrP93 to induce cell toxicity and neurodegeneration. Additional evidence for the non-poration models comes from the observation that certain peptides are too short to span the membrane and form a pore. Examples include Mastoparan (14 residues),94 KLLKLLLKLLLKLLK (15 residues),95 (Aib-Lys-Aib-Ala)n=1–5.96 These non-poration membrane destabilization events typically involve the membrane interactions of monomeric AMPs or AMYs, resulting in the creation of permeation and leakage pathways that facilitate the crossing of small molecules and ions through lipid bilayers.97
Distinct from non-poration actions, AMPs (e.g., PG-1) and AMYs (e.g., Aβ, hIAPP) can form trans-membrane pores or ion channels, generally through two main processes depending on peptide-to-lipid ratios.98–101 The first process involves monomeric peptides initially inserting into the membrane, followed by reorganization to create pores. In the second scenario, peptide oligomers directly insert and immediately form pores. Both scenarios require peptide oligomerization, occurring after and before membrane insertion, respectively. Essential to this process is the precise assembly of a specific number of peptides, achieving optimal hydrophobic alignment with lipid chains and reducing exposed hydrophobic and charged residues, crucial for functional transmembrane pore formation. Two primary pore topologies, “barrel-stave” and “toroidal”, derived from α-helices AMPs, are central to understanding peptide-induced pore formation. In barrel-stave pores, peptide chains align vertically and parallel to the membrane lipids, creating stability through hydrophobic interactions. Examples include AMPs such as alamethicin, ceratotoxins, and distinctin,55,102–104 and AMYs such as Aβ, hIAPP, and α-syn.105–107 In contrast, toroidal pores form when peptides bind to lipid headgroups, inducing a positive curvature that results in a torus-shaped opening, with peptides arranged circularly. Peptides known to form toroidal pores include magainins,73 mastoparan-X,48,49 viroporin, and PG-142 among AMPs, as well as Aβ,108,109 PrP(106–126),110 and hIAPP111 among AMYs.
Visualizing is comprehending. While AMP transmembrane pores are directly characterized by X-ray and NMR, amyloid pores – formed by Aβ,112–115 hIAPP,116,117 α-synuclein,118 and serum amyloid A,116,119,120 and K3 peptides derived from β2-microglobulin,114,121,122 ABri, and ADan – are typically analyzed using fluorescence leakage tests in giant vesicles, ionic conductance, confocal microscopy, electrophysiology, cell calcium imaging, and molecular dynamics (MD) simulations. Direct observation of these amyloid pores remains challenging due to difficulties in isolating pure amyloid oligomers for crystallization. Often, their existence is inferred from activities, such as channel conductance, calcium imaging, neuritic degeneration, mitochondrial damage.81 Alternatively, AFM/SEM images and molecular modeling enables the reconstruction of amyloid pores at low-to-medium resolutions (Fig. 5), showing structural similarities with AMP pores. Both amyloid and AMP pores are characterized by donut-shaped supramolecular arrangements of loosely connected oligomeric aggregates with β-sheet structures,113,123,124 reminiscent of pore-forming bacterial toxins. Despite variations in pore conformations, types, and subunit interactions with bilayers, these irregularly shaped and dynamic pores, with typical inner diameters of 1–3 nm and outer diameters of 6–10 nm,125 maintain an open state, allowing the uncontrolled passage of ions and molecules without specific ion selectivity. Unlike traditional gated ion channels, these non-gated ion pores lack mechanisms to regulate their opening and closure, indicating a fundamental difference in their functional properties.
 | ||
| Fig. 5 Comparative characterizations of (a) antimicrobial pores formed by melittin (Reproduced with permission from ref. 126 Copyright © 2015 American Chemical Society), amhelin (Reproduced with permission from ref. 127 Copyright 2013 National Academy of Science), perforin-2 (Reproduced with permission from ref. 128 Copyright © 2022 The Author(s)), perforin (Reproduced with permission from ref. 129 Copyright © 2021 The Royal of Chemistry) by AFM images (upper panel) and melittin (Reproduced with permission from ref. 130 Copyright © 2015 American Chemical Society), protegrin-1 (Reproduced with permission from ref. 131 Copyright © 2008 American Chemical Society), pleurocidin (Reproduced with permission from ref. 132 Copyright © 2021 American Chemical Society), kaempferol (Reproduced with permission from ref. 133 Copyright © 2022 American Chemical Society), mutacin 1140 (Reproduced with permission from ref. 134 Copyright © 2019 the Owner Societies), chrysophsin-3 (Reproduced with permission from ref. 135 Copyright © 2018 The Royal of Chemistry) by molecular simulations (lower panel); (b) amyloid pores formed by Aβ1–40 (Reproduced with permission from ref. 116 Copyright © 2005 National Academy of Sciences, U.S.A.), ovalbumin (Reproduced with permission from ref. 136 Copyright © 2013 American Chemical Society), α-synuclein,116 ABri,116 albebetin (Reproduced with permission from ref. 137 Copyright © 2004 American Chemical Society), hIAPP,116 serum amyloid A,116 ADan,116 K3 (Reproduced with permission from ref. 121 2009 American Chemical Society) by AFM images (upper panel) and Aβ (Reproduced with permission from ref. 138 Copyright © 2013 American Chemical Society), Medin (Reproduced with permission from ref. 139 Copyright © 2020 Biophysical Society), hIAPP (Reproduced with permission from ref. 106 Copyright © 2012 Elsevier B.V.) K3,121 FKFEFKFE (Reproduced with permission from ref. 140 Copyright © 2022 American Chemical Society), α-synuclein64–92 (Reproduced with permission from ref. 141 Copyright © 2023 American Chemical Society) by molecular simulations (lower panel). | ||
Computational models, utilizing NMR-resolved β-strand-turn-β-strand amyloid monomers, have been employed to reconstruct diverse oligomeric pore structures in amyloids like Aβ, hIAPP, serum amyloid A, β2-microtublin.60,142–144 These computational pores feature varied peptide counts (12 to 36 monomers), pore sizes (inner diameters of 2–4 nm and outer diameters of 7–12 nm), and topologies interacting with lipid bilayers.117 The U-shaped structures of these amyloid pores, consisting of multiple, loosely connected, mobile subunits, are consistent with AFM images and reveal dynamic behaviors,116 such as multiple conductance, weak cation selectivity, and voltage independence, and responses to inhibitors such as Congo red and zinc.119,122,145,146 Importantly, computationally constructed pores do not consistently maintain a stable, open pore-like structure, instead factors like lateral bilayer pressure, hydrophobic interactions, and thermal instability can destabilize these pores, leading to pore collapse and reflecting the complex role of amyloid pores in various disorders.
Taken together, AMPs and AMYs, despite their sequence and structural diversity, share common features such as a well-defined β-structure and a hydrophobic, amphipathic region. These attributes support their biological functions, with AMYs displaying antimicrobial activities and AMPs showing self-aggregation properties. Crucially, peptide oligomerization initiates and promotes interactions that lead to the formation of transmembrane pores and curved membranes, both capable of disrupting normal membrane permeability. This oligomerization not only transforms nonamyloidogenic peptides into pathogenic variants,147 but also enhances the antimicrobial efficacy of AMP monomers,77 highlighting its essential role in regulating membrane permeability. Moreover, the mechanisms of membrane disruption by AMPs and AMYs are multifaceted, involving various actions like transmembrane pore formation and membrane fusion. These processes are part of a broader phenomenon seen in diverse peptides, including cell-penetrating peptides, pore-forming toxins, glycopeptides, and lipopeptides. Such membrane-activating peptides play crucial roles across biological domains, not only enabling viruses and bacteria to attack host cells but also serving as a defense mechanism in both invertebrates and vertebrates through membrane-disruption strategies.
2.3. Pathological links between antimicrobial peptides and amyloid peptides
The pathological link between antimicrobial peptides (AMPs) and amyloid peptides (AMYs) can be traced back to their common association with microbial contexts. Mechanistically, in addition to the well-established role of amyloid aggregation, microbial infection has emerged as a significant contributor to the pathogenesis of neurodegenerative diseases.4,35 The “amyloid aggregation hypothesis” posits that the formation of toxic amyloid aggregates with β-rich structures is a requirement for the progression of neurodegenerative diseases, leading to cellular degeneration and eventual cell death.4 Conversely, the “microbial infection hypothesis” suggests that neuroinflammation induced by bacteria, viruses, and fungi contributes to the pathology of neurodegenerative diseases through a persistent immune response.32,33 While these two hypotheses propose distinct mechanisms for neurodegenerative diseases, emerging evidence from experimental and clinical studies has unveiled an intricate relationship between amyloid formation and microbial infection. Bidirectional and continuous communication between amyloid proteins and the gut microbiota (particularly bacterial amyloids) has emerged as a critical factor in the pathogenic connection between these two phenomena.34,35 This suggests a molecular crosstalk between the amyloid aggregation hypothesis and the microbial infection hypothesis.Interestingly, recent research has uncovered the convergence of certain features between AMYs and AMPs, adding another layer of complexity to their interplay. Some AMYs, typically associated with disease pathology, exhibit antimicrobial activity, while certain AMPs, known for their role in host defense, display amyloidogenic potentials. The dual functionality of some AMYs and AMPs provides new evidence for manipulating their sequences, structures, and activities to develop therapeutic intervention strategies for combating both microbial infections and neurodegenerative disorders. The discovery of dual functionality in certain AMYs and AMPs presents compelling evidence for the potential manipulation of these attributes as a basis for developing therapeutic intervention strategies combating both microbial infections and neurodegenerative disorders.
Several AMPs, including phenol-soluble modulin,148 plantaricin A,9 longipin,149 melittin,150 dermaseptin S9,76 magainin 2,12 temporins,151 aurein,152,153 uperin,10,154 LL-37,155 protegrin-1,156 defensins,23,24 and AMC-K9 conjugate,157 have been observed to exhibit self-assembly into amyloid-like fibrils with β-rich structures, either in buffer solutions or on lipid membranes. These self-assembled AMP fibrils bear similarity to pathological amyloid fibrils associated with human disease. The identification of AMPs as capable of forming amyloid-like structures highlights an intriguing aspect of their biological activity beyond their well-known antimicrobial properties. The resemblance between these self-assembled AMP fibrils and disease-related amyloid fibrils raises important questions regarding their functional implications and the underlying mechanisms governing their assembly. Considering that AMPs are implicated in the pathogenesis of neurodegenerative diseases associated with “microbial infection hypothesis”, further investigations focusing on cross-seeding interactions between these AMPs and different amyloid proteins may lead to the identification of dual-functional, multiple-target inhibitors. Such AMP inhibitors would have the potential to simultaneously impede pathology-associated amyloid aggregation and microbial infection pathways.
In parallel, recent studies have challenged the conventional perception of AMYs as exclusively pathogenic and useless substances. Instead, emerging evidence reveals the diverse biological functions of AMYs, encompassing their beneficial roles as reservoirs for certain polypeptide hormones storage,158 participation in cell adhesion,159 contribution to the development of functionalized biomaterials and nanomaterials,160,161 and demonstration of antibacterial activity.7,162 Unlike some AMPs that possess amyloidogenic properties, AMYs are increasingly recognized as ancient and highly conserved innate immune effectors involved in the prevention of microbial infections. Since the initial discovery in 2002 that serum amyloid A (SAA) can cause significant damage to various bacteria cells through the formation of voltage-independent and poorly selective ion channels,119 numerous other AMYs have been found to possess antimicrobial activity against common bacteria and fungi, including PrP23–231 and its truncated variants,163 Aβ,17 hIAPP,18 hCT,23,24 α-syn,15 and SAA.164 In some cases, the antimicrobial potency of these AMYs is equivalent to or even greater than that of LL-37 antimicrobial peptide. These findings provide compelling evidence that AMYs exert their antimicrobial and cytotoxic activities through a shared mechanism involving the formation of ion-permeable channels in the cell membranes of pathogens. Also, these findings not only expand our understanding of the biological roles of AMYs in relation to the “microbial infection hypothesis”, but also challenge the simplistic notion that they are solely deleterious, as commonly associated with the “amyloid aggregation hypothesis.”
Emerging but limited studies have revealed the occurrence of cross-seeding between bacterial amyloids and AMYs, as well as between AMPs and AMYs. This is likely due to the presence of these peptides in the blood circulation and cerebrospinal fluid, which raises the possibility of their co-aggregation or cross-seeding with other amyloidogenic proteins.165–167 The former cross-seeding between bacterial amyloids and AMYs seems to establish a pathogenic link, possibly creating a loop that contributes to disease propagation from microbial/virus infection to amyloid pathogenies through the brain-gut-microbiota.33–35 Unlike the cross-seeding between bacterial amyloids and AMYs, the cross-seeding observed in certain AMPs-AMYs pair systems, such as human β-defensin and Aβ/hIAPP/hCT,23 human α-defensin and Aβ/hIAPP/hCT,23 LL-37 and Aβ/hIAPP,25,26 α-syn and CsgA/CsgC/CsgE,27–29 presents a more intricate scenario. This cross-seeding enables AMPs to modulate the aggregation and misfolding of different AMYs. However, the changes in amyloid aggregation induced by AMPs do not necessarily lead to a reduction in amyloid-induced cell toxicity and the preservation of AMPs's antimicrobial activity, presumably due to the formation of AMP-AMY complexes and alterations in the distribution of AMY aggregates. On a positive note, recent studies have identified specific AMPs as dual-functional, multiple-target inhibitors capable of simultaneously blocking amyloid aggregation and microbial infection pathways. These AMPs show the potential to interrupt the interlinked pathological processes and bidirectional communication between amyloid aggregation and microbial infection. This discovery introduces an innovative approach to explore and repurpose a diverse range of antimicrobial peptides that inherently possess both bacterial-killing and amyloid inhibition functions, so as to effectively reconcile the “amyloid cascade hypothesis” and the “microbial infection hypothesis”. Additionally, the occurrence of amyloid cross-seeding, where various disease-related amyloid proteins form structurally similar amyloid fibrils, has been frequently observed.30,31,168 This phenomenon poses an additional risk factor for the initiation and progression of other neurodegenerative diseases. Surprisingly, seemingly unrelated amyloid proteins can trigger pathological events in different neurodegenerative diseases through amyloid cross-seeding. This further complicates the understanding of the amyloid aggregation hypothesis and its association with microbial infection.36
The COVID-19 pandemic has prompted researchers to investigate a potential connection between SARS-CoV-2 infection and amyloidosis. Preliminary studies have reported some interesting findings. It has been observed that the spike protein (S-protein) of the SARS-CoV-2 virus contains aggregation-prone heparin-binding sequences, and this characteristic enables the S-protein to aggregate into amyloid-like fibrils at a faster rate compared to Aβ and α-synuclein.169 In another study, S-protein does not affect α-synuclein aggregation, while the SARS-CoV-2 nucleocapsid protein (N-protein) significantly accelerates the aggregation process.170 Additionally, molecular dynamics simulations have indicated that the SK9 fragment from the E-protein of SARS-CoV-2 promotes the formation of amyloid structures in serum amyloid A.171 These findings highlight the potential involvement of SARS-CoV-2 viral proteins in amyloid formation but require further investigation to explore the cross-seeding interactions between specific SARS-CoV-2 proteins (e.g., S-protein, N-protein, E-protein) and different amyloid peptides (e.g., Aβ, α-synuclein, tau, and prions), as well as reveal the potential impact of these interactions on disease pathology.
These findings highlight the intricate and multifaceted nature of the pathological links between antimicrobial peptides and amyloid peptides. The bidirectional communication between amyloid proteins and the gut microbiota, coupled with the phenomenon of amyloid cross-seeding, provides new insights into the complex interplay between amyloid formation and microbial infection in the development and progression of neurodegenerative diseases. Further exploration of these pathological links holds promise for advancing our understanding of neurodegenerative diseases and may open avenues for the development of novel therapeutic strategies targeting both amyloid aggregation and microbial infection.
3. Amyloid property of antimicrobial peptides
Antimicrobial peptides (AMPs), also known as host defense peptides (HDPs), are abundant in the brain and other immune-privileged tissues and function as innate defense mechanisms against a variety of microorganisms and pathogens. AMPs, typically comprising 50–100 amino acids, show significant sequence and structural diversity, but mainly fall into two categories based on their secondary structures: α-helical and β-sheet peptides. These peptides disrupt bacterial cell walls, protein and nucleic acid synthesis, enzymatic activities, and membrane integrity, and possess antiviral, antifungal, antitumor, and immunomodulatory properties. The structural and functional similarities between AMPs and amyloids (AMYs) highlight fundamental biological connections, with emerging evidence of amyloidogenic properties in certain AMPs across various organisms, including animals, amphibians, insects, plants, and microbes, suggesting a more universally conserved role for AMPs linked to their amyloid properties.Mechanistically, the neuroinflammation seen in neurodegenerative diseases35 is closely linked to microbial infections caused by viruses (such as HSV-1,172 HIV,173 HHV-6A174), bacteria (such as gut bacteria,175 liver bacteria Helicobacter pylori,176Chlamydia pneumoniae177), fungi (such as Candida species, Cladosporium, Cryptococcus178), and SEVI.179 These infections can compromise the blood–brain barrier (BBB), trigger persistent immune responses, and ultimately contribute to neurodegeneration.32,33 Bacterial amyloid proteins, found in the gut microbiota, share structural similarities with amyloid proteins in the central nervous system (CNS). Both bacterial and amyloid aggregates are targeted by the immune system, leading to prolonged inflammation180,181 and activation of microglia,182 which exacerbate neurodegeneration. Exposure to bacterial amyloids can enhance immune responses against neuronal amyloids, potentially crossing the compromised BBB and disrupting brain function in individuals with infections. Below, Table 1 presents a range of antimicrobial peptides (AMPs), including natural AMPs from various sources and synthetic or engineered AMPs with amyloid-like properties.
| Category | Name | Source | Sub-category | Target microbes | Secondary structure | Amyloid-like fibrils | Other key findings | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Anti-Gram (+) | Anti-Gram (−) | Anti-fungal | ||||||||
| Mammal AMPs | Bovine lactoferricin | Bovine | N/A | Enterococcus sp, Staphylococcus sp | E. coli, P. aeruginosa, Shigella Jexneri | C. albicans | Distorted antiparallel β-sheet (2D NMR) | N/A | N/A | 183 |
| Indolicidin | Bovine | Cathelicidin | S. aureus | E. coli | Aspergillus fumigatus | N/A | Yes (Congo red) | Form amyloid-like fibers in the presence of phosphatidylserine containing liposomes | 184 | |
| PG-1 | Porcine | Cathelicidin | MRSA, vancomycin-resistant Enterococcus | E. coli, P. aeruginosa | C. albicans | β-hairpin (NMR) | Yes (ThT, AFM) | N/A | 8 | |
| LL-37 | Human | Cathelicidin | Enterococcus hirae, S. aureus | E. coli, P. aeruginosa | C. albicans | α-helix (NMR) | Yes (ThT, TEM, Cryo-EM) | Human LL-37 active core (residues 17–29) can form into a protein fibril of densely packed helices | 155 | |
| HD-6 | Human | Defensin | B. breve, L. acidophilus, B. adolescentis, B. longum | S. Typhimurium, Y. enterocolitica | C. albicans | Triple-stranded β-sheet (XRD) | Yes (SEM) | N/A | 185 | |
| Eosinophil cationic protein | Human | Ribonuclease A | S. aureus | E. coli | N/A | α + β folding (XRD) | Yes (Congo red, ThT, TEM) | N/A | 186 | |
| Amphibian AMPs | Magainin 2 | African clawed frog | Magainin | S. aureus | E. coli, A. calcoaceticus, P. vulgaris, H. pylori | T. basicola | α-helix (NMR) | Yes (TEM) | N/A | 11 |
| Dermaseptin S9 | South American hylid frog | Dermaseptin |
B.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) megaterium, L. monocytogenes megaterium, L. monocytogenes |
E. coli, S. typhimurium | N/A | β-sheet (CD, FTIR) | Yes (TEM, Congo red) | N/A | 13 and 76 | |
| Dermaseptin PD-3-7 | South American hylid frog | Dermaseptin | B. subtilis | E. coli | N/A | Amphipathic α-helix (CD) | Yes (Congo red, ThT, TEM) | Monomeric PD-3-7 displayed no antibiotic action, whereas amorphous aggregates of PD-3-7 release from the amyloid depot mediating a strong cytotoxic effect | 187 and 188 | |
| Uperin 3.5 | Australian toadlet | Uperin | M. luteus, S. aureus, S. hominis, S. epidermidis | N/A | N/A | Cross-α | Yes (ThT, CD, TEM, XRD, cryo-EM) | Antibacterial activity is induced by a chameleon cross-α/cross-β secondary structure switch of uperin 3.5 fibrils | 189 and 190 | |
| Arthropod AMPs | cupiennin-1 | Spider | N/A | M. luteus, S. aureus | K. pneumoniae | C. parapsilosis | α-helix/cross-β (XRD) | Yes (TEM) | N/A | 190 |
| Lasioglossin LL-I | Eusocial bee | N/A | M. luteus, S. aureus, B. subtilis | E. coli, P. aeruginosa | N/A | α-helix/cross-β (XRD) | Yes (TEM) | N/A | 190 | |
| Cecropin-C | Mosquitoes | Cecropin | M. luteus | P. carotovorum | M. rileyi | α-helix/cross-β (XRD) | Yes (TEM) | N/A | 190 | |
| Melittin | Honeybee | N/A | S. haemoliticus | K. pneumoniae, P. aeruginosa | Aspergillus, Botrytis, Candida, Colletotrichum, Fusarium, Malassezia, Neurospora, Penicillium, Saccharomyces, Trichoderma, Trichophyton, Trichosporon. | α-helix (NMR) | Yes (ThT, Congo red, TEM) | Amyloid-like aggregates formed by melittin in the presence of SDS | 150 | |
| Longipin | Opiliones | N/A | M. luteus | E. coli, S. marcescens | Candida | β-sheet (CD, FTIR) | Yes (ThT) | Fold into β-sheet structure and form into amyloid-like fibril in the presence of a lipid bilayer | 149 | |
| Microbe AMPs | Microcin B17 | Gram (−) E.coli | Ribosomally bacteriocins (microcins) | N/A | E. coli | N/A | N/A | Yes (TEM) | N/A | 191 |
| Microcin E492 | Gram (−) K. pneumoniae RYC492 | Ribosomally bacteriocins (microcins) | N/A | Escherichia, Klebsiella, Salmonella, Citrobacter, Enterobacter | N/A | β-sheet (CD) | Yes (Congo red, ThT, TEM) | Loss of toxicity after in vivo MccE492 amyloid formation. | 192 | |
| Plantaricin A | Gram (+) L. plantarum C11 | Ribosomally bacteriocins (class IIa) | S. aureus, L. Monocytogenes, Bacillus | E. coli, Salmonella | N/A | N/A | Yes (phase contrast microscopy) | The formation of amyloid-like fibrils is induced by PS-containing liposomes. | 9 | |
| Plantaricin J | Gram (+) L. plantarum C11 | Ribosomally bacteriocins (class IIb) | M. luteus | N/A | N/A | Cross-β (XRD) | Yes (TEM) | N/A | 190 | |
| Plantaricin K | Gram (+) L. plantarum C11 | M. luteus | N/A | N/A | Cross-β (XRD) | Yes (ThT, TEM) | N/A | |||
| cOB1 | Gram (+) E. faecalis | Sex pheromone | E. faecalis | N/A | N/A | β-sheet (CD) | Yes (Congo red, ThT, TEM) | Antibacterial ability of cOB1 is not affected by the formation of cOB1 aggregates | 193 | |
| sakacin P | Gram (+) L. sakei | Ribosomally bacteriocins (class IIa) | L. sake, L. coryneformis, E. faecalis, C. piscicola | N/A | N/A | S shaped antiparallel β-sheet (in silico) | N/A | N/A | 194 | |
| Gramicidin S | Gram (+) B. brevis | Non ribosomally | A. aureus, B. subtilis | E. coli | C. Albicans | β-helix (NMR) | N/A | N/A | 195 | |
| Tyrocidines | Gram (+) B. aneurinolyticus | Non ribosomally | L. monocytogenes | N/A | N/A | S shaped antiparallel β-sheet (CD, in silico) | N/A | Different dimer models are studied. | 196 | |
| Plant AMPs | RsAFP-19 | Radish seed | Defensin | N/A | N/A | Candida, Aspergillus, Fusarium, B. cinerea, N. crassa | Cross-β (XRD) | Yes (ThT, TEM, AFM) | loss of the antifungal ability after “gel-like” RsAFP-19 fibrils formation. | 197 |
| Cn-AMP2 | Cocos nucifera | Defensin | B. subtilis, S. aureus | E. coli, P. aeruginosa | N/A | N/A | Yes (Congo red, ThT, TEM) | N/A | 198 | |
| Synthetic or Engineered AMPs | C16-Gn(IIKK)nI-NH2 | N/A | N/A | S. mutans | N/A | N/A | β-sheet (CD) | Yes (ThT, CD, AFM, TEM) | β-sheet lipidated AMPs exhibit either comparable or diminished antimicrobial acitivity | 199 |
| FF8 | N/A | N/A | N/A | E. coli | N/A | β-sheet and β-turn (CD, FTIR) | Yes (AFM) | FF8 remains random coil structure under physiological conditions, but specifically triggered by the negatively charged lipid membrane and self-aggregate into nanofibrils | 200 | |
| HPRP-A1 isomers | Gram (+) H. pylori | Sequence shuffling | N/A | E. coli, P. aeruginosa, K. pneumoniae | N/A | α-helix, β-sheet, random coil (CD) | N/A | Reduction of antibacterial ability after the formation of HRPR-A1 amyloid aggregates antibacterial efficiency: α-helix > β-sheet > random coil | 201 | |
| (KIAGKIA)3-NH2 | PGLa derivative (α-helix) | Residues substitution | S. aureus | E. coli, P aeruginosa | N/A | β-sheet (CD, FTIR) | N/A | (KIGAKI)3-NH2 is highly selective to bacterial membranes as compared to mammalian membranes | 202 | |
| GL13K | Salivary protein BPIFA2 (α-helix) | Residues substitution | S. aureus | E. coli | N/A | β-sheets (CD, NMR) | No (AFM) | GL13K is highly selective for anionic bacterial model membranes (DOPG) compared to zwitterionic (neutral) eukaryotic model membranes (DOPC). | 203 and 204 | |
| Pardaxin diasteromer | Pardaxin fragment (α-helix) | Stereoisomer substitution | B. subtilis, B. megaterium | E. coli, A. calcoaceticus | N/A | β-sheet (FTIR) | N/A | The diastereomer retain potent antimicrobial activity, while the hemolytic activity was abolished. | 205 | |
| GS14K4 | GS14 (β-sheet) | Stereoisomer substitution | S.epidermidis, E. faecalis, C. xerosis | E. coli, P. aeruginosa, S.typhimurium | C. Albicans | Non β-sheets (CD, NMR) | N/A | GS14K4 shows high specificity for microbial cells than human cells. | 206 | |
| Temporin L analogue | Temporin L (α-helix) | Residues and stereoisomer substitution | S. aureus, S. epidermidis, B. megaterium | E. coli, P. aeruginosa, A. baumannii, K. pneumoniae | N/A | β-sheet (CD) | N/A | Temporin L analogue remians unstrauctured in water, while forms β-aggregates in liposome mimikcing bacterial membranes | 207 | |
| (IRIK)2-NH2 | N/A | N/A | S. epidermidis S. aureus | E. coli P. aeruginosa | C. Albicans | β-sheet (CD) | N/A | N/A | 208 | |
| (IRVK)3-NH2 | N/A | N/A | S. epidermidis S. aureus | E. coli P. aeruginosa | C. Albicans | β-sheet (CD) | N/A | N/A | 208 | |
| FSKRGY | N/A | N/A | S. aureus | N/A | N/A | β-sheet (CD) | N/A | N/A | 209 | |
3.1. Natural AMPs from mammals
Numerous studies were conducted to unravel the relationship between the self-association of AMPs and membrane disruption. Several models have been proposed, similar to those describing the membrane disruption of AMYs.83,210,211 The identification of a distorted antiparallel β-sheet structure in bovine lactoferricin B, a 25-residue AMP, suggests a potentially beneficial α-to-β secondary structure transition in antimicrobial activity. This observation implies a resemblance to the pathological amyloid formation (Fig. 6a).183 Subsequently, several AMPs from various animals have been recognized for their amyloid-like properties. | ||
| Fig. 6 Illustrations of natural AMPs from mammals with amyloid property. (a) Bovin lactoferricin characterized by a distorted antiparallel β-sheet structure in solution as elucidated by 2D NMR (Reproduced with permission from ref. 183 Copyright © 1998 American Chemical Society.) (PDB: 1LFC). (b) Bovine cathelicidin indolicidin capable of forming amyloid-like fibers induced by acidic phospholipids; a left image displays phase-contrast microscopy, while a right image exhibits a polarizing microscopy after Congo red staining. (Reproduced with permission from ref. 184 Copyright © 2005 American Chemical Society.) (c) Porcine cathelicidin PG-1, featuring a cysteine-rich β-hairpin structure (PDB: 1PG1), spontaneously forming amyloid fibrils within hours in the AFM image. (Reproduced with permission from ref. 8 Copyright © 2014 Elsevier Inc.) (d) Human cathelicidin LL-37 is an α-helical AMP (PDB: 2K6O). The active core of LL-37 (residues 17–29) mimics the ability of the full-length peptide to self-assemble into densely packed helical protein fibrils, as demonstrated by transmission electron microscopy (TEM, middle image) and the resolved crystal structure (right image). (Reproduced with permission from ref. 155 Copyright © 2020 Springer Nature Limited) (e) Human α-defensin HD-6 adopting a triple-stranded β-sheet structure (PDB: 1ZMQ). HD-6 self-assembles into elongated fibrils that entrap bacteria, as evidenced by SEM images of wild-type S. Typhimurium incubated with vehicle (upper image) and HD-6 (bottom images); scale bar is 5 μm. (Reproduced with permission from ref. 185 Copyright © 2024 American Association for the advancement of Science) (f) Human ribonuclease ECP is an AMP with an α + β folding topology (PDB: 1DYT). ECP demonstrates amyloid-like aggregation capacity as depicted in the TEM micrograph of ECP aggregates; scale bar is 1 μm. (Reproduced with permission from ref. 186 Copyright © 2010 American Chemical Society.) Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
Cathelicidins, along with defensins, constitute a significant category of cationic AMPs and represent a pivotal component of the immune system in diverse vertebrates, including humans and other animals. This cationic AMP family is featured by a highly conserved N-terminal cathelin domain and a diverse C-terminal antimicrobial domain exhibiting α-helical, β-hairpin, or proline/arginine-rich characteristics. Within the extensive family of over 30 cathelicidin members found in mammals, numerous AMPs have been identified as amyloidogenic peptides. For instance, the 13-residue AMP indolicidin, derived from bovine species, demonstrates the ability to form amyloid-like fibers in the presence of liposomes containing phosphatidylserine (Fig. 6b).184 The protegrin group of cathelicidin AMPs, isolated from porcine leukocytes, exhibits a distinctive cysteine-rich β-sheet structure.212 The unique conformation of protegrins, featuring a two-stranded antiparallel β-sheet stabilized by two cysteine bridges with strands connected by a β-turn, suggests potential self-interactions similar to those of AMYs. The amyloidogenic property of protegrins is evident, particularly in the case of PG-1, which exhibits rapid kinetics in forming amyloid fibrils (Fig. 6c).8 Moreover, LL-37, as the only human cathelicidin, has been extensively studied for its amyloid-like property. LL-37, an α-helical AMP essential in the first line of defense against local infections and systemic pathogen invasions,213 exhibits initial evidence suggesting that its antimicrobial effects arise from compromising the microbe's membrane barrier through the formation of cytotoxic amyloid-like fibers in the presence of acidic phospholipids.214 These amyloid-like structures have been studied for their role in immune responses and interactions with host cells. The fibrillation of LL-37 is critical for DNA binding and affects receptors in the immune system.215 Further studies have identified an active core of LL-37 (residues 17–29) that mimics the ability of full-length LL-37 to self-assemble into densely packed helices forming a protein fibril (Fig. 6d).155 Recent studies on LL-37 suggested a potent inhibitory effect on amyloid aggregation,25,26 suggesting the complex role that AMPs may play in response to external stimuli.
Defensins, characterized as small cysteine-rich cationic proteins, play a central role in the host defense mechanisms of granulocytic leukocytes, mucosal surfaces, skin, and other epithelia. Three defensin subfamilies—α-defensins, β-defensins, and θ-defensins—are expressed in animals. Typically spanning 18–45 amino acids, defensins feature three or four highly conserved disulfide bonds, and their tertiary structures are dominated by turn-linked β-strands, resulting in compact folded structures with favorable self-assembly properties. Human α-defensin 6 (HD-6) showcases a unique innate immune mechanism, wherein it self-assembles into elongated fibrils that effectively entrap bacteria, preventing microbial invasion (Fig. 6e).185,216 Notably, certain subgroups of defensins, with their unique β-strand-rich conformations, exhibit a potent inhibitory effect on amyloid peptides. This inhibitory effect has been observed in human α-defensin HNP-1, rabbit α-defensin NP-3A,24 HD-6, and human β-defensin HBD-1.23 These defensins can cross-seed with three amyloid peptides—Aβ, hIAPP, and hCT—hindering their aggregation into amyloid fibrils from both monomers and oligomers. These findings suggest a therapeutic potential for AMPs in the context of amyloid-related diseases. This raises an intriguing question about the potential bidirectional communication between microbial infection and amyloid formation and opening avenues for further exploration.
Beyond AMPs derived from the innate immune system, certain basic proteins exhibit potent toxicity against microbes and viruses. The eosinophil cationic protein (ECP), located in the eosinophil primary matrix, belongs to the Ribonuclease A superfamily. Apart from its involvement in tissue-remodeling processes, ECP serves as an AMP with a broad spectrum of action against bacteria, and at higher concentrations, displays cytotoxic activity to eukaryotic cells. Recently, the in vitro formation of amyloid-like aggregation of ECP has been reported (Fig. 6f). This discovery may offer new insights into the antimicrobial mechanism of the protein involving amyloid formation, and its potential toxicity to host tissues during inflammation processes.186 The amyloid-like fibril propensity of ECP suggests a connection to the presence of eosinophil infiltration in AD. These findings imply that amyloidosis in mammals could be a more general outcome potentiated by the immunomodulatory and infection control functions of AMPs.
3.2. Natural AMPs from amphibians
In 1989, the first documentation of an AMP with aggregation properties emerged, revealing the spontaneous polymerization of magainin 2.11 Magainin 2, a 23-residue AMP derived from the skin of the African clawed frog Xenopus laevis, possesses the ability to form filaments with a diameter of 13 nm with a periodic helical substructure (Fig. 7a). This observation suggests a crucial aspect of peptide–lipid interactions, implicating polymerization in membrane-disrupting antibiotic activities. Following the identification of magainin 2, numerous natural AMPs sourced from amphibians have been reported.217,218 | ||
| Fig. 7 Illustrations of natural AMPs from amphibians with amyloid property. (a) Magainin 2, a helical AMP derived from the African clawed frog's skin (PDB: 2LSA), can spontaneously polymerize into 13 nm filaments with a periodic helical substructure, revealed by TEM images; scale bar is 100 nm. (Reproduced with permission from ref. 11 Copyright © 1989 John Wiley & Sons, Inc.) (b) Dermaseptin S9, found in hylid frog skin secretions, exhibits a β-sheet-rich conformation with a high aggregation propensity in aqueous environments, as identified by CD spectrum (left image) and TEM (right image); scale bar is 50 nm. (Reproduced with permission from ref. 76 Copyright © 2008 John Wiley & Sons, Inc.) (c) Dermaseptin PD-3-7 with a unique negative net charge opposed to other dermaseptins forms amyloid-like fibrils at acidic pH as observed by TEM (left image; scale bar is 200 nm), while it exhibits reversibly amyloid-like aggregates in a pH-dependent manner as monitored by ThT fluorescence changes (right image). (Reproduced with permission from ref. 188 Copyright © 2009 John Wiley & Sons, Inc.) (d) Uperin 3.5, an α-helical peptide from the Australian toadlet's skin, can self-assemble into elongated amyloid fibrils, as confirmed by ThT (upper left panel) and TEM (upper right panel, scale bar is 300 nm). Crystal structures of uperin 3.5 in the presence of bacterial cells or membrane mimetics reveal a cross-α amyloid fibril architecture (middle panel, PDB: 6GS3), integral to antimicrobial activity. (Reproduced with permission from ref. 189 Copyright © 2021 National Academy of Science) In the absence of lipids, uperin 3.5 forms a 3-blade symmetrical propeller of nine peptides per fibril layer with tight β-sheet interfaces (bottom panel, PDB: 7QV5). (Reproduced with permission from ref. 152 Copyright © 2022 Springer Nature Limited) Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
Skin secretions of hylid frogs comprise a diverse array of genetically related AMPs, collectively termed dermaseptins. This peptide family forms a superfamily characterized by marked diversity, predominantly exhibiting a cationic nature with an amphipathic α-helical structure. Dermaseptin S9, a representative member of the dermaseptins superfamily, exhibits amyloidogenic properties (Fig. 7b).76 Mechanistic studies on dermaseptin S9 revealed that its largely hydrophobic middle segment serves as a structural foundation for the formation of β-strand, subsequently facilitating self-assembly into amyloid-like fibrils.13 The antimicrobial activity of Dermaseptin S9 is attributed to the same hydrophobic segment, which can adopt an α-helical conformation, supported by its cationic N- and C-termini when bound to anionic target membranes.219 The amyloid-like properties are not exclusive to dermaseptin S9 but extend to other dermaseptins as well. Dermaseptin PD-3-7, for instance, stands out as a unique example within the dermaseptin family, featuring a negative net charge at neutral pH due to the presence of three aspartic acid residues. The peptide can self-assemble into reversible amyloid fibrils in a pH-controlled manner (Fig. 7c). Through the transition from low pH to pH exceeding 5.0, metastable amorphous aggregates of PD-3-7 form and release from the amyloid depot, inducing a robust cytotoxic effect.187,188 This observation introduces a novel natural defense strategy involving amyloid deposits, wherein a temporary cytotoxic agent can be rapidly generated and released in response to microenvironmental factors such as pH. The dual functionality of dermaseptin peptides suggests a connection between amyloid and antimicrobial characteristics, prompting the exploration of potential associations between the two properties.
The connection between amyloid and antimicrobial activities gains further support with the discovery that amyloid-like self-assembly serves as a mechanism regulating AMPs. Uperins, representing a family of AMPs, are α-helical peptides consisting of 13 to 19 residues secreted on the skin of Uperoleia mjobergii (Australian toadlet). A wide variety of uperin peptides have shown their intriguing aggregation propensity, among which uperin 3.5 exhibits the highest propensity.220 A simulation study exploring helix-to-coil transitions in individual uperin 3.x (x = 4, 5, 6) peptides demonstrates an inverse relationship between the helical stability of peptides and their tendency to form structures rich in β-sheets. These findings underscore the significance of helical intermediates in the amyloidogenesis pathway for uparin AMPs.154 The crystal structure of uperin 3.5 presents a distinctive helical cross-α amyloid fibril formed on membranes, recapitulating properties of β-sheets and contributing to its antibacterial activity. However, in the absence of lipids, the same peptide primarily forms cross-β fibrils.189 Cryo-EM further elucidates the amyloid cross-β fibrils, revealing mated β-sheets at atomic resolution (Fig. 7d).152 These secondary structure transitions suggest a role as structural and functional cross-α/β chameleons. Building on these observations, a recent computational screen successfully identified new sequences of fibril-forming AMPs (ffAMPs) from living organisms, particularly in amphibians.190 Examples from amphibians, including cyanophlyctin secreted on the skin of the amphibian Euphlyctis cyanophlyctis, citropin-1.3 secreted from the granular dorsal and submental glands of the Blue Mountains tree frog Litoria citropa, brevinin-2SKb isolated from the stream brown frog Rana sakuraii, temporin-1CEa extracted from the skin of the Asiatic grass frog Rana chensinensis, bombinin H4 secreted on the skin of the yellow-bellied toad Bombina variegate, and aurein 3.3 secreted by Ranoidea raniformis (Southern bell frog), all exhibit cross-β and cross-α amyloid properties.190 These findings prompt hypotheses about the prevalent role of fibril secondary structure switching in regulating antimicrobial activities in AMPs, providing a new perspective on the amyloid-antimicrobial link.
3.3. Natural AMPs from arthropods
Insects have evolved a diverse array of AMPs to defend themselves against a broad spectrum of pathogens, often displaying a propensity to form large amyloid-like aggregates. The cross-α/β amyloid properties are notably prevalent in various natural AMPs found in insects, exemplified by cupiennin-1 from the spider venom of Cupiennius salei, lasioglossin LL-I isolated from the venom of the eusocial bee Lasioglossum laticeps, and cecropin-C produced by Anopheles gambiae mosquitoes (Fig. 8a).190 | ||
| Fig. 8 Illustrations of natural AMPs from arthropods with amyloid property. (a) Cupiennin-1 from the spider venom of Cupiennius salei, lasioglossin LL-I from the venom of the eusocial bee Lasioglossum laticeps, and cecropin-C from Anopheles gambiae mosquitoes can form amyloid-like fibrils, as visualized by TEM; scale bar is 200 nm. (Reproduced with permission from ref. 190 Copyright © 2022 American Chemical Society.) (b) Melittin from the honeybee (PDB: 6DST) adopts a helical conformation (CD spectrum, upper right panel) and forms large globular oligomers and some fibrillar species (AFM, bottom left panel) in the presence of SDS. Melittin aggregates exhibit ThT fluorescence (bottom right panel), indicating the presence of amyloid-like β-sheet structures. (Reproduced with permission from ref. 150 Copyright © 2015 Singh et al.) (c) Longipin, derived from the harvestman Acutisoma longipes, forms amyloid-like structures in the presence of lipid-vesicles, as evaluated by ThT. The inset shows representative emission spectra of longipin and POPG or POPC vesicles incubated for 1, 5, and 30 minutes. (Reproduced with permission from ref. 149 Copyright © 2016 Sayegh et al.) (d) Papiliocin (PDB: 2LA2), a cecropin originally found in the haemolymph of Hyalophora cecropia, shares high structural similarity with Aβ42. (Reproduced with permission from ref. 221 Copyright © 2020 Springer Nature Limited) All structures are depicted from left to right as a ribbon, a schematic secondary structure with helices shown as cylinders, and a surface representation highlighting the distribution of polar (green) and apolar (orange) residues. Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
Cecropins, initially discovered in the hemolymph of Hyalophora cecropia, are AMPs of 31–37 residues and constitute the essential part of the innate immune system of insects. Besides cecropin-C exhibiting cross-α/β amyloid properties, their aggregation tendencies have been demonstrated, closely associated with their antimicrobial activity. Early investigations into the aggregation of cecropin P1 in the presence of membrane proposed the widely cited “carpet model” of AMPs’ mechanism of action. In this model, cecropin P1 adheres extensively to the pathogen's membrane, causing membrane deformation and eventually destruction when the concentration of cecropin P1 exceeds a critical threshold.83 Similarly, cecropin A demonstrates concentration-dependent membrane activity, implying that its binding and state of aggregation determine its antimicrobial activity against bacterial membranes.222 Another study on cecropin AD and POPC/POPG vesicles reveals concentration-dependent positive cooperativity, indicating potential cecropin aggregates formation in the lipid phase.223 However, more data is required to firmly establish the amyloid-like properties of these cecropins.
Melittin, an extensively studied AMP, is a 26-residue C-terminal amidated peptide derived from the honeybee (Apis mellifera). While there is no direct evidence to support the ability of free melittin to form amyloid structures, the peptide has been reported to generate amyloid-like aggregates in the presence of sodium dodecyl sulfate (SDS). Melittin rapidly oligomerizes to form helix-rich oligomers in the presence of SDS, and further aggregation into fibrils has been demonstrated. The amyloid-like aggregates induce ThT fluorescence, indicating the presence of β-sheet structures (Fig. 8b).150 Additionally, melittin oligomers exhibit cytotoxic and hemolytic activity, likely due to the accumulation of helix-rich oligomers on the cell surface. Similarly, longipin, an unstructured AMP consisting of 18 residues derived from the harvestman Acutisoma longipes, has been shown to fold into a β-sheet structure and form amyloid-like fibril in the presence of a lipid bilayer (Fig. 8c).149
In addition to the observed amyloid formation properties of natural AMPs from arthropods, the connection between AMYs and AMPs is further underscored by structural similarities shared between representative amyloid peptide Aβ and natural AMPs from various organisms. Papiliocin, a cecropin-like peptide discovered in the Asian butterfly Papilio Xuthus, exhibits the highest structural resemblance to Aβ among the analyzed AMPs. In a comparative analysis between Aβ and papiliocin, it was observed that both peptides display a similar tilt angle between helices, and their surfaces, particularly in the C-terminus, exhibit a comparable distribution of apolar residues (Fig. 8d).221 Such structural and sequence similarity highlights an emerging connection between AMYs and AMPs based on shared structural characteristics.
3.4. Natural AMPs from microbes
Based on biosynthesis pathways, microbial AMPs can be categorized into ribosome- and nonribosome-synthesized AMPs,224 constituting a diverse group of antibacterial proteins found in various bacterial strains. Similar to their eukaryotic counterparts, microbial AMPs are small in size, cationic, and amphiphilic or hydrophobic. However, a significant distinction lies in their antibacterial efficiency; eukaryotic AMPs are typically active at micro-molar concentrations, while microbial AMPs are highly potent, acting at pico- to nano-molar concentrations.225 Despite their potency, bacterial AMPs often exhibit a relatively narrow killing spectrum, specifically affecting bacteria closely related to the producing strain.226 This limitation is attributed to their specific modes of action. Bacterial AMPs kill or inhibit the growth of closely related species by binding to specific cell surface receptors, inducing pore formation in the cytoplasmic membrane, DNA/RNA degradation, and inhibition of transcription, translation, or DNA replication.227Ribosome-synthesized AMPs, commonly known as bacteriocins, are prevalent in Gram-negative bacteria, particularly within the Enterobacteriaceae family (e.g., Escherichia and Klebsiella). These bacteriocins are commonly categorized into microcins (small peptides, <10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) or colicins (large proteins, 25–80000 Da). A representative example of microcin is Microcin B17 (MccB17), a 43-residue peptide produced by E. coli. MccB17 showcases antibacterial activity by inhibiting DNA replication and inducing the SOS response in susceptible bacteria.228 MccB17 was the first bacterial AMP discovered to exhibit amyloid-forming capabilities. This groundbreaking discovery, dating back to 1986, was captured by electron microscopy, revealing highly ordered and conspicuously long filaments reminiscent of amyloid fibrils191 (Fig. 9a). Microcin E492 (MccE492), a peptide naturally produced by Klebsiella pneumoniae RYC492, exhibits activity against various Enterobacteriaceae strains, including Escherichia, Klebsiella, Citrobacter, Salmonella, and Enterobacter.229 Unlike other bacteriocins, MccE492 employs a mechanism involving the formation of ion-permeable pores, leading to the depolarization and permeabilization of the bacterial cytoplasmic membrane, ultimately resulting in a lethal loss of membrane potential.230,231 Additionally, MccE492 demonstrates the capability to form amyloid-like fibrils in vitro, sharing structural, morphological, biochemical, and kinetic properties with disease-related AMYs.192 This aligns with the observation that Mcc E492 extends beyond antibacterial activity and has been found to induce eukaryotic cell apoptosis and necrosis.232 Computational analysis using different algorithms (such as AGGRESCAN, AmyloidMutants, etc.) identified the region MccE49254–63 (VNVPIPVLIG) as the pro-amyloidogenic stretch. Consistently, a mutant of MccE492 lacking residues 54–63 exhibited a significantly reduced tendency for intracellular aggregation and displayed slower kinetics in in vitro polymerization.233 However, similar to other amyloid-like AMPs, in vivo MccE492 amyloid-like aggregation is associated with the loss of antibacterial capability192 (Fig. 9b), supporting the emerging perspective that mature amyloid fibrils may not be inherently harmful; rather, they might serve as inert end products or play a protective role by isolating toxic intermediates.
000 Da) or colicins (large proteins, 25–80000 Da). A representative example of microcin is Microcin B17 (MccB17), a 43-residue peptide produced by E. coli. MccB17 showcases antibacterial activity by inhibiting DNA replication and inducing the SOS response in susceptible bacteria.228 MccB17 was the first bacterial AMP discovered to exhibit amyloid-forming capabilities. This groundbreaking discovery, dating back to 1986, was captured by electron microscopy, revealing highly ordered and conspicuously long filaments reminiscent of amyloid fibrils191 (Fig. 9a). Microcin E492 (MccE492), a peptide naturally produced by Klebsiella pneumoniae RYC492, exhibits activity against various Enterobacteriaceae strains, including Escherichia, Klebsiella, Citrobacter, Salmonella, and Enterobacter.229 Unlike other bacteriocins, MccE492 employs a mechanism involving the formation of ion-permeable pores, leading to the depolarization and permeabilization of the bacterial cytoplasmic membrane, ultimately resulting in a lethal loss of membrane potential.230,231 Additionally, MccE492 demonstrates the capability to form amyloid-like fibrils in vitro, sharing structural, morphological, biochemical, and kinetic properties with disease-related AMYs.192 This aligns with the observation that Mcc E492 extends beyond antibacterial activity and has been found to induce eukaryotic cell apoptosis and necrosis.232 Computational analysis using different algorithms (such as AGGRESCAN, AmyloidMutants, etc.) identified the region MccE49254–63 (VNVPIPVLIG) as the pro-amyloidogenic stretch. Consistently, a mutant of MccE492 lacking residues 54–63 exhibited a significantly reduced tendency for intracellular aggregation and displayed slower kinetics in in vitro polymerization.233 However, similar to other amyloid-like AMPs, in vivo MccE492 amyloid-like aggregation is associated with the loss of antibacterial capability192 (Fig. 9b), supporting the emerging perspective that mature amyloid fibrils may not be inherently harmful; rather, they might serve as inert end products or play a protective role by isolating toxic intermediates.
 | ||
| Fig. 9 Illustrations of natural AMPs from microbes with amyloid property. (a) MccB17 produced from E. coli can form amyloid-like fibrils, as visualized by EM. (Reproduced with permission from ref. 191 Copyright © 1986 John Wiley & Sons, Inc.) (b) MccE492 produced by Klebsiella pneumoniae RYC492 possesses the capability to form β-sheet-rich, amyloid-like fibrils, as evidenced by CD spectrum and TEM (left images). Scale bars are 200 nm. The occurrence of amyloid-like fibril formation, verified by the immunoblot in the right inset image, is correlated with the diminished antibacterial activity. (Reproduced with permission from ref. 192 Copyright © 2005 Elsevier Inc.) (c) Plantaricin A (plA) generated from L. plantarum C11 exhibits the ability to form amyloid-like fibrils in the presence of PS-containing liposomes, as demonstrated through phase-contrast microscopy (left image) and Congo red staining observed via fluorescent microscopy (right image). Scale bars are 10 μm (left) and 15 (right). (Reproduced with permission from ref. 9 Copyright © 2005 Elsevier B.V.) (d) Plantaricin J (plJ) and Plantaricin K (plK) derived from L. plantarum C11 exhibit the ability to form amyloid-like fibrils, as observed by TEM (left image) and ThT fluorescence (right image). Scale bars are 200 nm. (Reproduced with permission from ref. 190 Copyright © 2022 American Chemical Society.) (e) Tyrocidines (TyrA and TyrC) derived from B. aneurinolyticus adopt an antiparallel β-sheet confirmation, as indicated by CD spectrum. (Reproduced with permission from ref. 196 Copyright © 2013 American Chemical Society.) (f) cOB1 produced from E. faecalis shows amyloid-like properties, as demonstrated by the enhanced ThT fluorescence (left image), the presence of amyloid-like fibrils by TEM (left inset image), and β-sheet confirmation by CD spectrum (right image). (Reproduced with permission from ref. 193 Copyright © 2019 John Wiley & Sons, Inc.) Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
In Gram-positive bacteria, bacteriocins are classified into two main classes: lantibiotics (class I) and nonlantibiotics (class II).234 Class II bacteriocins can be further divided into four subclasses: pediocin-like bacteriocins (class IIa), two-component bacteriocins (class IIb), circular bacteriocins (class IIc), and unmodified, linear, non-pediocin-like bacteriocins (class IId).235 Notably, bacteriocins produced by lactic acid bacteria (LAB), covering diverse types from class I to class IIa-d, have garnered significant attention due to their unique attributes such as food-grade safety and heat stability. As a result, LAB-produced bacteriocins find widespread use in the food industry as natural biopreservatives. In comparison to bacteriocins from Gram-negative bacteria, those originating from LAB exhibit a broader spectrum of activity, extending their antibacterial efficacy beyond closely related species. These LAB-produced bacteriocins have demonstrated effectiveness against various food spoilage and pathogenic bacteria, including Gram-positive strains such as Listeria monocytogenes, Clostridium perfringens, Bacillus cereus, and Gram-negative strains like Salmonella enteritidis and Escherichia coli, contributing to the extension of the shelf life of food products.236
Plantaricin A (plA), a 26-residue peptide produced by Lactobacillus plantarum C11, a strain of LAB found in the human microbial flora, exhibits membrane-permeabilizing antimicrobial activity. The interaction of plA with membranes is highly dependent on the lipid composition of the membrane. Intriguingly, plA has been observed to form supramolecular protein-lipid amyloid-like fibrils upon binding to negatively charged phospholipid-containing membranes (Fig. 9c). This suggests a potential mechanistic link between fibril formation and the cytotoxicity of plA.9 Another bacteriocin produced by Lactobacillus plantarum C11, plantaricin J (plJ), and plantaricin K (plK), are two synergistic peptides with little sequence similarity, measuring 25 and 32 residues, respectively. Typically, they are used in equal amounts to achieve optimal antibacterial activity, potentially by forming an active complex. When considered separately, both plJ and plK can individually form amyloid-like fibrils, as observed by TEM. Distinctively, plJ displayed a typical cross-β pattern in fiber X-ray diffraction, whereas plK formed a mixed population with the major polymorph being of cross-β content, along with the ability to induce ThT fluorescence (Fig. 9d).190 However, the amyloid-forming capability of the mixture of plJ and plK remains unclear. The wild-type and G6A-substituted N-terminal domain of pediocin-like bacteriocins of sakacin P (class IIa), obtained from Lactobacillus sakei, have been identified as an S-shaped three-stranded antiparallel β-sheet-like domain.194 It remains to be investigated in future studies whether this β-sheet conformation is associated with an amyloid-like structure and whether it contributes to the antibacterial activity of sakacin P.
Unlike conventional ribosomal protein synthesis, nonribosome synthesized peptides are secondary metabolites produced by intricate enzymatic machinery, typically categorized by the linearity or cyclization of the molecule. Among these, a group of nonribosomal AMPs (e.g., gramicidin S (GS), tyrocidines (Tyr), loloatins, and laterocidin) originating from Gram-positive Bacillus strains share structural similarities, notably featuring a cyclic decapeptide. Structurally, GS demonstrates 50% identity with Tyr, specifically sharing the conserved pentapeptide unit of FPV(Orn)L. Additionally, GS195 and Tyr196 also share a similar secondary structure of β-sheets in solution. To delve into specifics, the β-sheet conformations of tyrocidines and gramicidin S are distinct, i.e., tyrocidines have an S-shaped, three-stranded antiparallel β-sheet-like domain (Fig. 9e), whereas gramicidin S adopts a β-helix structure. More recently, a new antimicrobial innate immune peptide called cOB1 has been identified. Originating from Enterococcus faecalis, cOB1 (VAVLVLGA) operates as a sex pheromone, exerting its natural antimicrobial effects to restrict the growth of multidrug-resistant Enterococcus faecalis in the gut at a picomolar concentration.237 Experimental evidence indicates that cOB1 forms amyloid-like structures, as supported by both in silico predictions and in vitro assays involving Congo red, ThT staining, CD, and TEM morphology (Fig. 9f).193 These structures are postulated to serve as the nucleating core, potentially facilitating enhanced biofilm formation.
3.5. Natural AMPs from plants
Plant AMPs exhibit several shared characteristics with those from animals, amphibians, and insects, such as (i) small peptides ranging from 10 to 54 amino acids in length; (ii) positive charges to interact with negatively charged microbial membranes; (iii) amphipathic structure for interacting with and disrupting the lipid membranes of microorganisms, all of which make them effective in broad-spectrum antibacterial, antifungal, and antiviral activities. However, plant AMPs are distinguished by their high cysteine content, playing a pivotal role in stabilizing, and maintaining the structural integrity of these peptides. The presence of cysteine residues enables the formation of 3–5 intramolecular disulfide bridges, contributing to the stabilization of the secondary structure, enhancing resistance to degradation by proteolytic enzymes and environmental factors, and thereby maintaining bioactivity. Plant AMPs are categorized into distinct subgroups, such as thionins, defensins, cyclotides, hevein, knottin, α-hairpinin, lipid transfer proteins, and snakins, based on their sequence similarity, number of cysteine residues, disulfide bond patterns, and tertiary structure.238,239While diverse plant AMPs have been extensively studied for their antimicrobial activities, the connection between these peptides and amyloidogenicity remains poorly understood. To date, only two peptides within the defensin category have been reported to exhibit a high propensity for amyloid fibril formation, suggesting that amyloidogenicity is not a generic feature of plant AMPs. One example is RsAFP-19, a 19 amino acid C-terminal fragment derived from the radish seed (Raphanus sativus) antifungal protein (RsAFP). RsAFP-19 has been demonstrated to possess amyloid fibril-forming properties by a synergistic approach combing computational methods (i.e., TANGO) and experimental techniques (i.e., fluorescent-binding, morphology, and secondary structures). Evidently, RsAFP-19 exhibits characteristics typical of classical amyloid fibrils, including ThT assay fluorophore-binding intensity, narrow protofilaments observed by AFM and TEM, and a cross-β structure from X-ray fiber diffraction analysis. In contrast, NaD1-19, a plant defensin peptide structurally and functionally related to RsAFP-19, derived from the ornamental tobacco plant Nicotiana alata, does not show any propensity for amyloid fibril formation,197 highlighting the specificity and diversity of amyloidogenic properties in plant AMPs. Interestingly, challenging the assumption that amyloid formation is intricately linked to antimicrobial properties, the “gel-like” RsAFP-19 amyloid fibrils formed after 1-month storage (freezing and thawing) completely lost their anti-fungal activity (Fig. 10a), suggesting the irrelevance of fibril formation to the biological functions of RsAFP.197 The other example is Cn-AMP2, a plant defensin derived from the liquid endosperm of coconut (Cocos nucifera), which is rich in hydrophobic residues (a characteristic shared with AMYs) and exhibits a natural tendency to form amyloid-like fibrillary structures comparable to Aβ (Fig. 10b). However, the antibacterial effect of fibrillar Cn-AMP2 remains unstudied, creating uncertainty about the link between the amyloidogenic structure and its biological functions.198
 | ||
| Fig. 10 Illustrations of natural AMPs from plants with amyloid property. (a) RsAFP-19 produced from Radish seed shows amyloid-like properties, as indicated by the enhanced ThT fluorescence and the presence of amyloid-like fibrils by TEM (left images). The scale bar is 200 nm. The occurrence of amyloid-like fibril formation correlates with the reduction in antibacterial activity (right image). (Reproduced with permission from ref. 197 Copyright © 2013 Elsevier B.V.) (b) Cn-AMP2 generated from Cocos nucifera exhibits amyloid-like properties, as evidenced by the enhanced ThT fluorescence (left image) and the presence of amyloid-like fibrils by TEM (right image). The scale bar is 200 nm. (Reproduced with permission from ref. 198 Copyright © 2016 John Wiley & Sons, Inc.) Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
3.6. Synthetic or engineered AMPs
Most naturally occurring antimicrobial peptides are characterized by cationic linear sequences that tend to fold into amphipathic α-helices. This structural feature plays a crucial role in inducing membrane leakage, contributing to their broad-spectrum antimicrobial activity and rapid action against microbial membranes. Despite these advantages, AMPs with amphipathic α-helices face certain limitations, including the requirement for high concentrations to effectively eliminate target organisms, susceptibility to proteolytic degradation by enzymes, high toxicity to host cells, and limited selectivity that may lead to unintended interactions with host cells. To address these challenges and enhance the antimicrobial efficacy of AMPs, rational design strategies to explore AMPs with stable β-sheet structures are essential. However, there is ongoing debate about the preference for the development of β-sheet species into amyloid-like aggregates.The interaction between amyloid aggregates and antimicrobial properties is complex and multifaceted. Although formation of amyloid-like fibrils can create pores or channels in microbial membranes, exhibiting potential antimicrobial capabilities, the prevailing consensus is that self-assembly is detrimental to antibacterial potency. On one hand, a simple and straightforward approach to increase the folding ability of short peptides is acylation with long-chain fatty acids (i.e., lipidation). In the design of lipidated peptides, palmitic acid was conjugated to the N-terminus of (IIKK)nI-NH2 (n = 2–3). This lipidation enhanced the hydrophobic interaction and induced the formation of β-sheet-rich nanofibrils resembling amyloid fibrils, as confirmed by ThT, CD, and TEM analyses199 (Fig. 11a). However, the antimicrobial activity of these lipidated AMPs was either comparable or diminished. This reduction was primarily attributed to stronger homo-interactions (i.e., self-aggregation) compared to hetero-interactions, resulting in limited interaction with bacteria.199 On the other hand, the delivery of such large, self-assembled AMPs to infectious locations poses challenges, resulting in reduced efficiency. The critical aspect lies in understanding when and where these amyloid-like fibrils form. As proof of concept, a designed peptide, KRRFFRRK (FF8), remains in a random coil structure under physiological conditions but is specifically triggered by the negatively charged lipid membrane to self-aggregate into nanofibrils (Fig. 11b). This structure exhibits enhanced antimicrobial capability compared to a control peptide, GG8 (KRRGGRRK), without the ability to self-assemble,200 implying that formation of amyloid-like fibrils occurs subsequent to FF8 being transported to the membrane, reducing the transportation pressure of self-assembling AMPs.
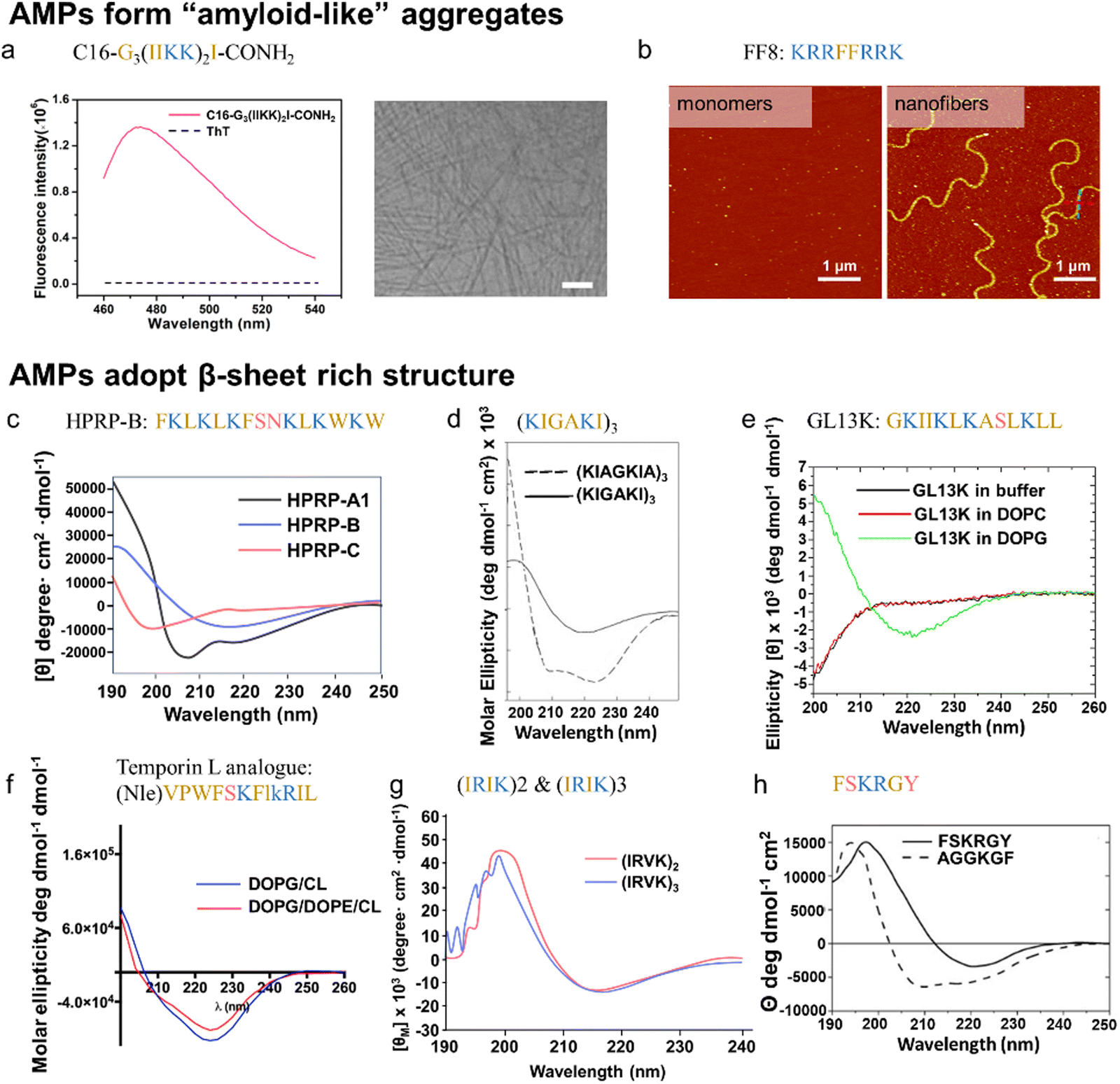 | ||
| Fig. 11 Illustrations of synthetic or engineered AMPs with amyloid-forming or β-sheet formation properties. (a) C16-G3(IIKK)2I-CONH2, an engineered AMPs via terminal alkylation, forms amyloid-like aggregates, as indicated by the enhanced ThT fluorescence (left image) and the presence of amyloid-like fibrils by cryo-TEM (right image). The scale bar is 100 nm. (Adapted with permission from ref. 199 Copyright © 2020 American Chemical Society). (b) FF8 (b) FF8 demonstrates the ability to form amyloid-like nanofibrils under pH 9.4, while maintaining a monomeric state at pH 7.4, as visualized by AFM. (Reproduced with permission from ref. 200 Copyright © 2020 The Royal Society of Chemistry.) (c) HPRP-A1 isomers are designed to adopt different conformations, where HPRPA1 assumes an α-helix, HPRPB adopts a β-sheet, and HPRPB adopts a random coil conformation, as confirmed by the CD spectrum. (Adapted with permission from ref. 201 Copyright © 2015 John Wiley & Sons, Inc.). (d) (KIGAKI)3, designed from α-helical (KIAGKIA)3, adopts β-sheet structure in the presence of POPG LUV, as indicated by CD spectrum. (Adapted with permission from ref. 202 Copyright © 2001 Elsevier Inc.). (e) GL13K folds into β-sheet structure in the presence of anionic DOPG liposomes, while retaining unstructured in the presence of neutral DOPC liposomes and PBS buffer, as confirmed by CD spectrum (Reproduced with permission from ref. 204 Copyright © 2013 Elsevier B.V.) (f) Temporin L analogue adopts a β-type conformation when exposed to liposomes mimicking bacterial membranes, as evidenced by CD spectrum. (Reproduced with permission from ref. 207 Copyright © 2022 MDPI (Basel, Switzerland).) (g) (IRIK)2 and (IRIK)3 demonstrate β-sheet folding in the presence of membrane-mimicking environment, specifically in a 25 mM SDS micelles solution. (Adapted with permission from ref. 208 Copyright © 2013 John Wiley & Sons, Inc.). (h) FSGRGY – a pore-forming peptide – adopts β-sheet structure, in contrast to non-pore-forming AGGKGF that exhibits some α-helical secondary structure, as depicted by CD spectrum. (Reproduced with permission from ref. 209 Copyright © 2005 National Academy of Science.) Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
To date, there has been only limited research exploring the self-assembly capabilities of newly designed AMPs into larger amyloid aggregates. Several studies suggested a tendency for these peptides to fold into β-sheets. Further investigation is necessary to explore the potential transformation of β-sheets into amyloid-like fibrils. To this end, a number of approaches aimed at re-engineering naturally occurring AMPs, such as sequence shuffling, residue or stereoisomer substitution, guided by the structure–activity relationship. These modifications aim to bring about changes in charge, conformation, sequence, length, amphipathicity, hydrophobicity, self-aggregation, and other key properties. These efforts are geared towards engineered AMPs to enhance their properties and overcome inherent limitations.
In this regard, a significant area of investigation involves transforming α-helical AMPs into β-sheet AMPs, representing a current focal point in the field. One illustrative instance is HPRP-A1, a 15-residue α-helical AMP derived from the N-terminus of the Helicobacter pylori ribosomal protein L1. HPRP-A1 served as the foundational peptide for designing isomers with distinct secondary structures (i.e., α-helical structure with different helicity, β-sheet structure, and random coil structure, Fig. 11c). This was achieved by reshuffling the peptide sequence while maintaining an identical amino acid composition to eliminate the influence of other properties, such as peptide length, charge, and hydrophobicity. A comparative analysis of HPRP-A1 isomers with different secondary structures revealed a hierarchy in antibacterial efficacy, with the order being α-helix > β-sheet > random coil.201 This aligns with the prevailing consensus indicating that a decline in antibacterial capability occurs following the formation of amyloid aggregates.
Inspired by a derivative of PGLa, (KIAGKIA)3-NH2, known for its amphipathic α-helix structure, (KIGAKI)3-NH2 was engineered to adopt an amphipathic β-sheet structure (Fig. 11d). A direct comparison reveals that the β-sheet variant, (KIGAKI)3-NH2, exhibits antimicrobial activity comparable to that of its α-helical counterpart, (KIAGKIA)3-NH2, but with higher selectivity toward bacterial membranes over mammalian membranes.202 Notably, (KIGAKI)3-NH2 exhibits higher selectivity in binding to and inducing leakage in membranes rich in phosphatidylethanolamine, a neutral phospholipid prevalent in bacterial plasma membranes, compared to those rich in phosphatidylcholine, a major neutral lipid in mammalian plasma membranes.240 Furthermore, a single substitution of Ile (I) with Trp (W) in (KIGAKI)3-NH2, while preserving the β-sheet structure, enhances its binding affinity to membranes containing acidic phospholipids (characteristic of bacterial membranes) over zwitterionic phospholipids (characteristic of mammalian membranes), underscoring the specificity of β-sheet (KIGAKI)3-NH2 and its analogs for targeting bacterial membranes.241 Similarly, GL13K, engineered from the salivary protein BPIFA2 by substituting charged residues with Lys (K), gained a newfound antibacterial capacity and a preference for folding into β-sheets in anionic DOPG membranes, as opposed to zwitterionic (neutral) eukaryotic DOPC membranes203,204 (Fig. 11e). However, it is crucial to note that the adoption of a β-sheet structure in GL13K does not necessarily lead to the formation of amyloid-like fibrils. Subsequent investigations have revealed that GL13K does not form fibrils under physiological conditions, effectively dispelling the possibility of pathological amyloid presence.242
Beyond residue substitution, the incorporation of stereoisomers, specifically substituting L-amino acids with their D-enantiomers, offers advantages in enhancing resistance to enzymatic degradation and improving stability. An illustrative example involves the substitution of D-amino acids into a cytolytic α-helical pardaxin fragment, inducing a structural transition to a β-sheet conformation. This modification not only leads to a structural shift but also results in a functional transition, transforming from high toxicity towards both bacteria and erythrocytes to specific cytolytic activity targeted at bacteria while sparing erythrocytes.205 Similarly, the gramicidin S analogue (GS14) with a β-sheet structure demonstrates broad-spectrum antimicrobial capability against both Gram-positive/negative bacteria and fungi, but it poses a toxicity risk to red blood cells. To address this concern, GS14 was modified by substituting D-amino acids with L-amino acids, disrupting the β-sheet structure and significantly reducing toxicity to red blood cells.206 Furthermore, the combination of residue and stereoisomer substitution/addition has been investigated in the modification of Temporin L, an antimicrobial peptide with the ability to form α-helical aggregates, aiming to enhance peptide stability and effectiveness. These modifications encompass the addition of a norleucine residue at the N-terminus, the substitution of Q3 to P3 and G10 to K10, as well as the replacement of L9 and K10 with l9 and k10. This tailored modification strategy ensures that the Temporin L analogue remains unstructured in an aqueous environment but adopts a β-type conformation when exposed to liposomes mimicking bacterial membranes (Fig. 11f), resulting in improved antibacterial efficacy.207 These findings underscore the promising advantage of β-sheet-forming AMPs in selectively targeting and disrupting bacterial membranes while minimizing the impact on host cell membranes.
While employing wild-type AMPs as templates for chemical modifications to design new AMPs has its drawbacks, such as the potential for increased immunogenicity due to extended sequences which may elevate manufacturing costs, and a high similarity to host defense AMPs that could trigger resistance, recent research by Novabiotics Ltd demonstrates promising advancements in this area. Utilizing HDP templates, they have developed new antimicrobial and immunomodulatory compounds, with several products currently undergoing clinical trials. This underscores the potential of such strategies, although optimizing efficacy and safety profiles remains a challenge.243 Hence, the strategic development of short synthetic peptides that bear minimal resemblance to naturally occurring AMP sequences is anticipated to be a promising approach for creating safe and effective AMPs for clinical applications. Pursuing this objective, a series of short synthetic β-sheet folding AMPs, consisting of short recurring (X1Y1X2Y2)n=2–3-NH2, have been designed. Here, X and Y represent hydrophobic (i.e., V, I, F, W) and cationic (R, K) residues, respectively, which facilitate interaction with microbial membranes. These designed β-sheet folding AMPs (confirmed by CD, Fig. 11g) demonstrate wide-ranging antimicrobial efficacy against Gram-positive bacteria such as S. epidermidis and S. aureus, Gram-negative bacteria like E. coli and P. aeruginosa, and the yeast C. albicans. Among these AMPs, (IRIK)2-NH2 and (IRVK)3-NH2 emerge as the most potent, inhibiting sessile biofilm bacteria growth and inducing biomass reduction.208
The D-amino acid-substituted β-sheet-forming peptides, (IRIK)2-NH2-all-D and (IRVK)3-NH2-all-D, demonstrate enhanced antimicrobial activities, extending to a series of clinically relevant antibiotic-resistant bacteria (i.e., methicillin-resistant S. aureus, vancomycin-resistant Enterococci, A. baumannii, M. tuberculosis, etc.). Additionally, these peptides exhibit improved protease stability, making them promising candidates for therapeutic applications in the fight against antibiotic resistance.244,245 However, the limitations in the number of rational designs for AMPs may stem from a poor understanding of the fundamental principles governing the correlation between self-assembly and action mechanisms, as well as the complex nature of diverse AMPs. In contrast to trial-and-error attempts, the utilization of rational combinatorial libraries offers a potent method for the selection and engineering of novel pore-forming sequences. Construction of the combinatorial library involved employing β-sheets as a foundational framework and subsequent screening to obtain good hits. One example is FSKRGY, a novel AMP that self-assembles into β-sheet pores in membranes to exhibit antimicrobial properties209 (Fig. 11h).
4. Antimicrobial activity of amyloid peptides
Amyloid aggregation involves the transition from unstructured soluble monomers to β-sheet-rich amyloid fibrils, hallmark features of protein misfolding diseases like AD, PD, and T2D.246,247 The abnormal accumulation of these fibrils leads to deposits in organs, causing cellular dysfunction and tissue damage. Recently, “functional amyloid fibrils” have gained recognition for their positive biological roles,248 such as curli fibrils in bacteria and silk proteins in insects, which aid in biofilm formation and structural support.249,250 Despite belonging to different peptide families, AMYs and AMPs share notable similarities. Both involve hydrophobic residues crucial for aggregation in AMYs and microbial membrane interactions in AMPs.22 As discussed in Section 3, many AMPs, like AMYs adopt β-sheet structures and can self-assemble into aggregates, particularly during microbial interactions. Both AMYs and AMPs disrupt lipid bilayers, causing cell or bacterial death.7,18 This section presents a range of AMYs (Fig. 12 and Table 2), such as Aβ (red, Alzheimer's disease), hIAPP (green, type 2 diabetes), α-syn (yellow, Parkinson's disease), PrP (blue, prion disease), and SAA (grey, systemic amyloid A), which exhibit antimicrobial properties. | ||
| Fig. 12 Summary of amyloid peptides with anti-bacterial, anti-fungal, and anti-viral activity in terms of minimal inhibitory concentrations (MIC) and the number of targeted stains. *MIC data are not available in the references. | ||
| AMD | Sequence | Targeted bacteria | Targeted fungi | Targeted virus | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # of strains | Anti-Gram (+) | Anti-Gram (−) | MIC (μg mL−1) | # of strains | Anti-fungal | MIC (μg mL−1) | # of strains | Anti-viral | MIC (μg mL−1) | |||
| Aβ | Aβ40 | 12 | S. epidermidis, S. aureus, S. pneumoniae, L. monocytogenes, E. faecalis, S. agalactiae, S. mitis, L. acidophilus | E. coli, P. gingivalis, A. actinomycetemcomitans, F. nucleatum | 1.56–50 | 1 | C. albicans | 0.78 | 3 | HSV-1, H3N2, H1N1 | 7.5–> 20 | 17 and 251–253 |
| Aβ42 | 10 | S. epidermidis, S. aureus, S. pneumoniae, L. monocytogenes, E. faecalis, S. agalactiae, L. acidophilus | E. coli, P. gingivalis, A. actinomycetemcomitans | 1.56–12.5 | 1 | C. albicans | 0.78 | 3 | HSV-1, H3N2, H1N1 | 1–>20 | 17, 23, 24, 153, 157 and 251–253 | |
| Aβ35–42 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 1 | H3N2 | 6 | 254 | |
| Aβ22–42 | 1 | N/A | E. coli | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 254 and 255 | |
| Aβ2–42 | 1 | E. faecalis | N/A | 6.25 | N/A | N/A | N/A | N/A | N/A | N/A | 254 and 255 | |
| Methylated-GNPs-Aβ32–37 | 5 | S. aureus, E. faecalis | E. coli, P. aeruginosa, K. pneumoniae | >500 | N/A | N/A | N/A | N/A | N/A | N/A | 256 | |
| KK | 1 | N/A | E. coli | 375 | N/A | N/A | N/A | N/A | N/A | N/A | 257 | |
| K3(FA)4K3 | 2 | N/A | Salmonella, E. coli | 32–63 | N/A | N/A | N/A | N/A | N/A | N/A | 258 | |
| K6(FA)4 | 2 | N/A | Salmonella, E. coli | 63–125 | N/A | N/A | N/A | N/A | N/A | N/A | 258 | |
| hIAPP | hIAPP37 | 3 | S. aureus S. epidermidis | E. coli | >20 | N/A | N/A | N/A | N/A | N/A | N/A | 14, 23, 24, 153 and 157 |
| α-syn | α-syn140 | 4 | S. aureus, S. epidermidis | E. coli, P. aeruginosa | 2.8 | 8 | C. albicans, C. tropicalis, F. neoformans, A. flavus, A. fumigatus, A. parasiticus, R. solani, T. harzianum | 5.6–44.8 | 2 | WNV, VEEV | N/A | 15 and 259 |
| α-syn1–60 | 4 | S. aureus, S. epidermidis | E. coli, P. aeruginosa | 9.6–19.2 | 4 | C. albicans, F. neoformans, A. flavus, R. solani | 38.4–76.8 | N/A | N/A | N/A | 15 | |
| α-syn1–95 | 4 | S. aureus, S. epidermidis | E. coli, P. aeruginosa | 7.6–15.2 | 4 | C. albicans, F. neoformans, A. flavus, R. solani | 15.2–30.4 | N/A | N/A | N/A | 15 | |
| PrP | PrP23–231 | 1 | N/A | E. coli | 35 | 1 | C. parapsilosis | 70 | N/A | N/A | N/A | 163 |
| PrP23–144 | 1 | N/A | E. coli | <12 | N/A | N/A | N/A | N/A | N/A | N/A | 163 | |
| PrP90–231 | 1 | N/A | E. coli | <15 | N/A | N/A | N/A | N/A | N/A | N/A | 163 | |
| SAA | SAA104 | 4 | S. aureus | E. coli, UTI89, F11 | 1–5 | 2 | C. albicans, C. dubliniensis | 40–>100 | 4 | HCV, Phil, PR-8 | <2–8 | 16, 164 and 260–264 |
4.1. Amyloid-β peptide (associated with Alzheimer disease)
Aβ, an inadvertent byproduct of amyloid protein precursor (APP) catabolism in the brain and peripheral tissues, is widely recognized as a crucial pathological hallmark of AD. Aβ is generated in both AD and healthy individuals through extracellular and intramembrane endoproteolytic cleavage of APP via β-secretase/γ-secretase pathways.265 The degradation of Aβ is facilitated by the Aβ-degrading enzyme neutral endopeptidase (NEP).266 Under normal circumstances, Aβ maintains a dynamic equilibrium between production and degradation, with a steady low level (approximately nanomolar concentration) of soluble Aβ.267 However, an imbalance in this equilibrium leads to elevated Aβ deposition in the brain, contributing to the onset and progression of AD. Despite extensive efforts to develop therapeutic strategies targeting Aβ inhibition, none have received approval due to limited efficacy or adverse side effects.268,269 For instance, clinical studies on Aβ immunotherapy with AN1792 showed that AD patients experienced a higher incidence of subacute meningoencephalitis—a brain inflammatory disease caused by infection—resulting in the termination of the trial.270,271 Similar challenges have been encountered in other Aβ-related inhibition therapies, such as AD02272 and CAD106.273 These observations suggest a potential physiological role of Aβ beyond its pathological implications, prompting the consideration of maintaining normal Aβ doses and forms rather than pursuing complete clearance.Recently, a novel model for AD amyloidogenesis, known as the “antimicrobial protection hypothesis” has emerged, complementing the traditional “amyloid cascade hypothesis”.274 According to this hypothesis, Aβ is not a functionless peptide released into the cell by accident or genetic predisposition. Instead, akin to AMPs, Aβ production is stimulated as part of an innate immune response to activate neuroinflammatory pathways (e.g., microglia and proinflammatory cytokines) and eliminate foreign threats. As a byproduct, Aβ subsequently plays a secondary role in AD pathology by inducing chronic activation of these pathways, leading to sustained inflammation and neurodegeneration.32 Notably, Aβ has been identified as an AMP in vitro, exhibiting activity against 8 of 16 common and clinically relevant pathogens (e.g., Gram-positive bacteria, Gram-negative bacteria, fungi), with antimicrobial capacity equivalent to or greater than LL-37, a human innate AMP17,253 (Fig. 13a). Recent findings from the same group indicate that the expression of Aβ is associated with an increased survival rate in different bacterial and fungal infection models, such as in vitro mammalian cells, in vivo nematodes, and mice. Conversely, APP knockout mice, exhibiting immunodeficiency associated with low Aβ expression, showed higher mortality than wild-type mice after infection, further highlighting the protective role of Aβ as an AMP in innate immunity.275 Brain tissues from AD patients exhibit higher antimicrobial activity than samples from age-matched non-AD individuals, correlating with Aβ levels in the brain.17
 | ||
| Fig. 13 Antimicrobial activity of Aβ. (a) Antibacterial efficacy of Aβ42 against E. faecalis growth, in comparison to untreated and LL-37-treated bacteria, through colony forming unit (CFU) counting and western blot analyses using mAβ 6E10 or anti-LL-37 antibodies to further evaluate the impact on bacterial cultures. (Reproduced with permission from ref. 17 Copyright © 2010 Soscia et al.) (b) Antiviral ability of Aβ42 in an HSV1 Encephalitis 5XFAD Mouse Model. 5XFAD mice exhibit an extended survival rate compared to wild-type mice after the injection of HSV1 into the hippocampal region of each brain hemisphere. (Reproduced with permission from ref. 251 Copyright © 2018 Elsevier Inc.) (c) Comparative analysis of the antifungal efficacy of monomeric Aβ42, amyloid-β-derived diffusible oligomeric ligands (ADDLs), and protofibrillar Aβ42 against C. albicans, with the descending order of anti-fungal efficiency: oligomers > monomers > protofibrils. (Reproduced with permission from ref. 275 Copyright © 2016 American Association for the Advancement of Science.) (d) Comparative analysis of the antifungal activity of cell-derived Aβ42, Aβ40, LL-37, and synthetic Aβ42 against C. albicans, with the descending order of anti-fungal efficiency: LL-37 > cell-derived Aβ42 > cell-derived Aβ40 > synthetic Aβ42. (Adapted with permission from ref. 275 Copyright © 2016 American Association for the Advancement of Science). (e) Comparative analysis of the antiviral ability (neutralization) of Aβ42 fragments lacking C-terminals (i.e., Aβx–40) versus those retaining C-terminals (i.e., Aβx–42) on Phil IAV H3N2-infected MDCK cells, emphasizing the crucial role of C-terminals in Aβ42 in antiviral ability. (Reproduced with permission from ref. 254 Copyright © 2018 White et al.) (f) Antibacterial efficacy of peptides KK (red) and KY (blue) against E. coli. (Reproduced with permission from ref. 257 Copyright © 2019 MDPI (Basel, Switzerland).) (g) Antibacterial efficacy of peptides K3(FA)4K3 and K6(FA)4 against E. coli. (Reproduced with permission from ref. 258 Copyright © 2020 American Chemical Society.) Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
In addition to microbial infection, there is growing evidence suggesting the potential involvement of viral infection in the development of AD.276,277 Members of the herpes virus family, specifically HSV-1, HSV-2, CMV, and HHV-6, are frequently identified in the brains of AD patients, where they colocalize within Aβ plaques.278,279 Intriguingly, direct interactions between Aβ and HSV-1 have been demonstrated both in vitro and in vivo. In these studies, Aβ was shown to bind to the surface glycoproteins of HSV-1, triggering a general protective viral entrapment response in various cell lines challenged with HSV-1. This response inhibited HSV-1 replication and upregulated miRNA-146.21,280 Similarly, 5xFAD mice (a transgenic AD mouse model overexpressing human Aβ) subjected to hippocampal HSV-1 inoculation exhibited an improved survival rate compared to wild-type counterparts251 (Fig. 13b). Beyond HSV-1, Aβ has demonstrated its ability to prevent infection of cultured cells by other viruses, including HHV-6,251 as well as seasonal and pandemic strains of H1N1 and H3N2 of the influenza virus.252
The inherent oligomerization and fibrillization behavior observed in AMP function (as discussed in Section 2.2) suggests that the form of Aβ oligomers may not be inherently abnormal in the context of AD pathophysiology. Instead, Aβ oligomerization may occur as an adaptive response to optimize antimicrobial activities, akin to established AMPs. As illustrated in Fig. 13c, synthetic Aβ oligomers (referred to as amyloid-β-derived diffusible oligomeric ligands, ADDLs) exhibit higher potency against Candida compared to non-oligomerized peptides (i.e., monomers and protofibrils).275 In a separate comparative analysis, cell-derived Aβ, encompassing diverse polymorphic molecular forms, demonstrates superior antimicrobial and antiviral activity compared to homogenous synthetic Aβ251,275 (Fig. 13d). Various hypotheses regarding the mechanism have been proposed, including (i) enhanced binding of soluble Aβ oligomers to microbial cell wall carbohydrates via a heparin-binding domain (VHHQKL);275 (ii) amyloidogenic-induced microbial agglutination, entrapping microbes in a network of β-amyloid;281,282 (iii) insertion of Aβ oligomers into the lipid bilayer cell membrane;283 and (iv) pore-forming ability of β-sheet-rich Aβ oligomers on the plasma membrane via toxic ion channels.7 Considering the intrinsic properties of oligomeric Aβ, they are relatively unstable and can exist in various aggregation states, morphologies, and sizes,284 thereby creating a diverse and polymorphic oligomer pool capable of targeting a broader spectrum of pathogens.
To date, over 40 distinct variants/isoforms of the Aβ peptide, featuring N- and C-terminal truncations, have been identified in secretory compartments and peripheral blood, forming a complex mixture with lengths ranging from 37 to 43 amino acids.285,286 Among these, Aβ1–40 is the most prevalent, while Aβ1–42 is recognized as the most amyloidogenic and neurotoxic form.287,288 Efforts to pinpoint the crucial antimicrobial domains of Aβ have involved extensive comparisons between full-length and truncated Aβx–40/Aβx–42. Regardless of the origin (synthetic or cell-derived), Aβ1–42 consistently exhibits more robust antibacterial and antiviral activity than Aβ1–40.17,275 This is evident in lower minimum inhibitory concentration (MIC) values against the same types of microorganisms (Fig. 13d), emphasizing the significance of Ile41 and Ala42 located at the C-terminus of Aβ1–42. The pivotal role of these domains is reinforced by observations that truncated Aβx–42 variants (e.g., Aβ22–42, Aβ35–42) retain antimicrobial activities, while fragments lacking C-terminals (e.g., Aβ1–34, Aβ1–28, Aβ22–40, Aβ33–40) lose such capabilities254,255 (Fig. 13e). These differences likely arise from (i) the higher amyloidogenicity of Aβx-42, inducing agglutination and mediating oligomerization through a loop between Met35 and Ala42;289 (ii) the heightened neurotoxicity of Aβx–42, directly inducing bacterial death;287 (iii) the increased hydrophobicity of Aβx–42, leading to higher binding to the surface of microorganisms; and (iv) the enhanced uptake of bacteria by neutrophils for Aβx–42. Studies suggest an increased tendency for aggregation in N-terminal truncated Aβx–42 variants.290,291 Accordingly, Aβ22–42 demonstrates a notable ability to enhance neutrophil uptake of IAV and E. coli, surpassing that of the full-length Aβ1–42.254 However, further truncation to Aβ35–42 decreases E. coli uptake, likely attributed to the absence of the β-turn-β confirmation formed by a salt bridge between Lys28 and Asp23.254
Given the robust anti-microorganism behavior of Aβ, numerous studies have explored variations inspired by its core motif. In one such investigation, two variables were introduced: N-methylation modification and the functionalization of gold nanoparticles (GNPs). These modifications were assessed for their anti-bacterial capabilities in comparison to fragments containing naked Aβ32–37 (i.e., CGGIGLMVG and CGGGGGIGLMVG). The results demonstrated a significant enhancement in antimicrobial efficacy with N-methylated peptides, further amplified when conjugated with GNPs.256 Additionally, specific Aβ segments, namely GAIIG (Aβ29–33) and KLVFFA (Aβ16–21), have been identified as self-assembling building blocks capable of spontaneously forming a β-sheet amyloid core.292,293 This intrinsic ability suggests potential fibrillization-mediated antimicrobial properties. Consequently, these segments were utilized as starting sequences for designing experimental functional scaffolds, including KYKGAIIGNIK, KYRSGAITIGY, K3(FA)4K3, and K6(FA)4. As anticipated, all these peptides demonstrated the capacity to self-assemble into amyloid fibrils, exhibiting a potent bactericidal effect against E. coli257,258 (Fig. 13f and g).
4.2. Human islet amyloid polypeptide (associated with type 2 diabetes)
Presently, it is clear that type 2 diabetes (T2D) arises from a combination of factors, encompassing inadequate response to insulin (insulin resistance) and impaired insulin secretion by the pancreatic islet β cells. This culminates the elevated blood glucose level.294,295 However, the precise pathological mechanisms underlying these processes remain elusive. On one hand, a pathological correlation exists between aggregated human islet amyloid polypeptide (hIAPP) and type 2 diabetes (T2D), evident in clinical observations where over 90% of T2D patients exhibit the presence of hIAPP deposits in the form of extracellular fibrillar aggregates within their pancreatic tissue.296 It was postulated that alterations in the local microenvironment, particularly changes in the hydrophobicity of hIAPP, play a pivotal role in amyloid formation.297 This, in turn, contributes to pancreatic β dysfunction, cell death, and ultimately triggers the onset of T2D.On the other hand, recent studies have investigated into the connection between T2D and microbial infection/inflammation. Similar to the speculation in AD, hIAPP might also be produced as a form of inflammatory response to high levels of blood glucose or external pathogens. Clinically, the onset of T2D often coincides with or follows a pathogen infection, especially pancreatitis.298 The microbiome plays a crucial role in both indirect and direct contributions to T2D development. Numerous studies have highlighted that gut microbial dysbiosis can indirectly contribute to the onset of T2D. When microbial dysbiosis occurs, it can lead to changes in the function and permeability of the intestinal barrier. This, in turn, has the potential to activate the innate immune system and modify signaling pathways, triggering low-grade inflammation, ultimately leading to insulin resistance and possibly T2D.299,300 Moreover, direct infections by viruses (hepatitis C, cytomegalovirus),301,302 bacteria (H. pylori, Lactobacillus, C. pneumoniae),303–305 and fungi (C. albicans)306 are also linked to an increased risk of developing T2D or worsening its symptoms. In this context, an elevated number of immune cells are detected in the pancreatic islets, accompanied by heightened levels of cytokines, chemokines, and IL-1.307,308 These seem to be part of an immune response aimed at eliminating foreign intruders. This observation prompts consideration of whether hIAPP production might be a consequence of this inflammatory response, akin to the antimicrobial role of Aβ in AD.
Structurally, both hIAPP and AMPs share a net positive charge and exhibit amphipathic characteristics, crucial features enabling their interactions with negatively charged lipid membranes. Notably, hIAPP demonstrates antimicrobial activity against clinically relevant bacteria such as S. aureus and E. coli (Fig. 14a and b). Certainly, hIAPP demonstrated a significant inhibition of bacteria growth during the incubation period, particularly evident from 5 hours for S. aureus and 2 hours for E. coli. However, these differences gradually diminished as the incubation progressed, nearly disappearing at the end, possibly due to peptide degradation. In comparison to the extensively discussed Aβ42 as a potent AMP (Section 4.1), hIAPP exhibited greater potency in inhibiting the growth of E. coli. Furthermore, the antimicrobial efficacy of hIAPP varied based on its amyloid states, with the ascending order of antimicrobial capacity being freshly-prepared hIAPP monomers > protofibrillar hIAPP > fibrillar hIAPP (Fig. 14c and d) at incubation timepoints of 4, 7.5, and 12 hours.14 Our series of studies342 further confirmed hIAPP's antimicrobial properties against S. aureus, S. epidermidis, and E. coli. Due to variations in batches and concentrations of hIAPP tailored for distinct projects, the antimicrobial efficacy exhibited variability, with growth inhibition ranging from 14% to 36% for S. aureus, 8% to 53% for S. epidermidis, and 26% to 32% for E. coli. Overall, it is essential to emphasize that research focused on the antimicrobial properties of hIAPP is limited and ongoing. The exact mechanisms linking hIAPP aggregation, microbial infection, and T2D are still under exploration.
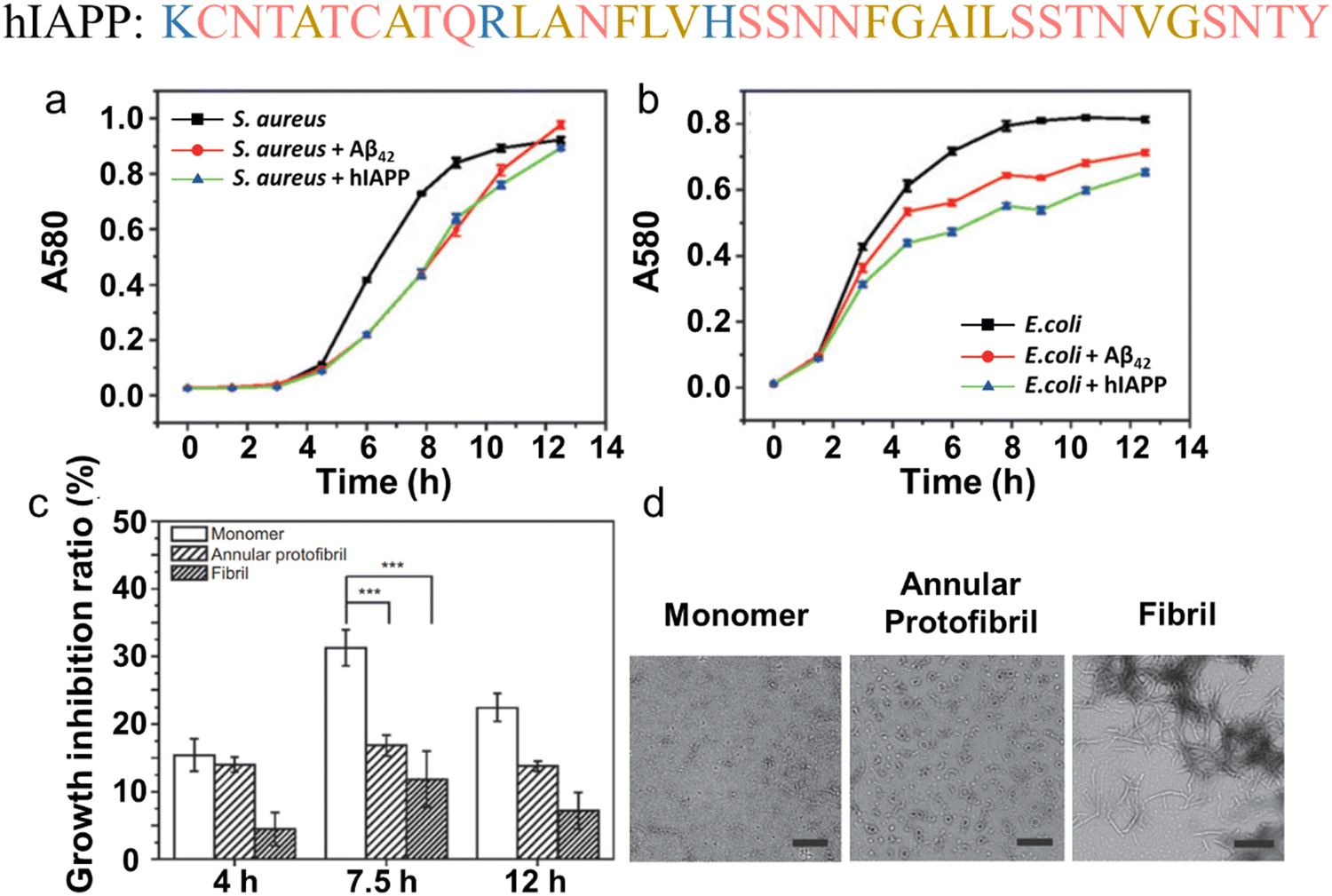 | ||
| Fig. 14 Antimicrobial activity of hIAPP. Antibacterial efficacy of hIAPP against (a) S. aureus and (b) E. coli, in comparison to untreated and Aβ42-treated bacteria, monitored by the turbidimetry method. A comparative analysis of (c) antibacterial activity and (d) morphology by TEM of monomeric, annular protofibrillar, and fibrillar hIAPP against S. aureus, with a decreasing order of antibacterial efficiency: monomers > annular protofibrils > fibrils. All scale bars represent lengths of 500 nm. (Reproduced with permission from ref. 14 Copyright © 2012 by Walter de Gruyter Berlin Boston.) Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
4.3. α-Synuclein (associated with Parkinson disease)
α-Synuclein (α-syn), a widespread protein prominently found in the brain, is linked to Parkinson's disease (PD) and other neurodegenerative conditions.309 It has been established that the characteristic proteinaceous deposits of α-syn constitute the primary component of Lewy bodies and Lewy neurites, hallmark pathologies in PD, cortical Lewy body dementia (LBD), and multiple system atrophy (MSA).310,311 Recently, α-syn and its fragment α-syn61–95 (also known as non-Aβ component, NAC) were identified in association with AD, accumulating with Aβ in senile plaques.312 Consequently, strategies targeting the production, aggregation, spread, and degradation of α-syn have been actively pursued to combat these diseases.313 Unfortunately, none of these approaches has successfully advanced to the clinical stage, reflecting the challenge of balancing the inhibition of α-syn pathological activities with the preservation of its physiological function. While the precise functions of α-syn remain elusive, compelling evidence suggests its role as a membrane protein in its physiological state. This is evident through (i) the localization of α-syn at pre-synaptic terminals of neurons, implying an association with membranes as synaptic vesicles are membrane-bound structures,314 and (ii) the binding interaction between α-syn and synaptic membranes, facilitating its various cellular functions such as neurotransmitter release, synaptic vesicle trafficking, and cellular membrane fusion.315–317 The affinity of α-syn for membranes can be attributed to favorable electrostatic interactions, particularly with the numerous positively charged lysine residues in the N-terminus of α-syn and the negatively charged phospholipid membranes. This binding mechanism closely resembles the working principle of AMPs, prompting further exploration of potential physiological functions of α-syn as an AMP through membrane-targeting models.318,319Clearly, clinical observations suggest that patients undergoing antiviral therapy for hepatitis C virus (HCV) are less prone to developing PD,320 indicating a potential link between pathogen exposure and PD risk. Similar implications arise regarding an elevated PD risk in patients with bacterial infections (e.g., Helicobacter pylori) or fungal infections (e.g., Malassezia).321 While epidemiological studies have yet to establish direct associations between microbial infections and PD risk, let alone determine their sequential occurrence, at the molecular level, it is evident that full length α-syn1–140 can directly inhibit the growth of bacteria (e.g., Staphylococcus aureus and E. coli, etc., Fig. 15a), yeast (e.g., Candida albicans, Candida troppicalis, etc., Fig. 15b),15 and mold (e.g., Aspergillus flavus, Aspergillus fumigatus, and Rhizoctonia solani, etc.) with extremely high efficiency, as indicated by low MIC values ranging from 0.2 to 3.2 μM. When comparing different α-syn fragments with the full-length α-syn, an antimicrobial activity ranks as follows: α-syn1–140 > α-syn1–95 (MIC = 0.8–3.2 μM) > α-syn1–60 (MIC = 1.6–12.8 μM) > α-syn61–140 (MIC > 25.6 μM) ≈ α-syn96–140 (MIC > 25.6 μM). This ranking highlights the crucial roles of highly conserved N-terminal region (1–65) compared to less conserved C-terminal region (96–140). Further investigation into the targeted sites of α-syn demonstrated that rhodamine-labeled α-syn accumulated on the cell surface of E. coli, while it accumulates in the cytoplasm of C. albicans. This suggests that α-syn interacts with bacterial membranes and fungal cytoplasmic compounds in microbial cells, leading to membrane leakage and inhibition of cell growth. Moreover, the endogenous neuronal expression of α-syn in a mice model has been demonstrated to inhibit the replication of viruses, such as West Nile virus (WNV) and Venezuelan equine encephalitis virus (VEEV).259 This inhibition reduces the likelihood of viral infection, injury, and disease in the central nervous system, as evidenced by lower WNV viral titers and loads, along with a higher survival rate among mice (Fig. 15c).
 | ||
| Fig. 15 Antimicrobial activity of α-synuclein. (a) Antibacterial efficacy of α-syn at 0.2–1.6 μM against S. aureus and E. coli, compared to untreated bacteria (control, C). (Reproduced with permission from ref. 15 Copyright © 2016 Elsevier Inc.) (b) Antifungal efficacy of α-syn at 0.2–1.6 μM against C. albicans, compared to untreated fungal (Ccntrol, C) (Adapted with permission from ref. 15 Copyright © 2016 Elsevier Inc.). (c) Antiviral efficacy of α-syn in inhibiting WNV growth, as indicated by the lower mortality rate in Snca+/+ mice (wild type) than Snca−/− mice (homozygous knockout lack α-syn). (Reproduced with permission from ref. 259 Copyright © 2016 American Society for Microbiology.) Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
A plausible deduction is that monomeric α-syn is produced as an AMP in response to immune activation, serving to protect and eliminate external threats. Once the threats are neutralized or eradicated, the body ideally returns to a state of equilibrium, and any remaining antimicrobial agents (i.e., α-syn) are degraded or recycled. However, during this process, α-syn monomers may undergo aggregation before complete degradation, a phenomenon triggered by bacteria.322 Consequently, all forms of α-syn, including monomeric, oligomeric, and aggregated forms, collectively contribute to inducing neuroinflammation. In this scenario, α-syn and immune responses appear to occur synchronously during infections, forming a self-reinforcing cycle. Any dysregulation of this coordination may transform the virtuous cycle of infection defense into a vicious cycle of neuroinflammation and neurodegeneration.
4.4. Prion protein (associated with prion disease)
Prion diseases, also referred to as transmissible spongiform encephalopathies (TSEs), constitute a spectrum of neurodegenerative disorders. This group includes Creutzfeldt–Jakob disease (CJD), fatal familial insomnia (FFI), kuru, Gerstmann-Sträussler-Scheinker disease (GSS), and variably protease-sensitive prionopathy (VPSPr) in humans.323 Similar to other amyloid-forming peptides, the soluble cellular prion-related protein (PrPc) undergoes a conformational change from α-helix to β-sheet during pathogenesis. This results in the formation of an insoluble protease-resistant isoform (PrPsc), which further aggregates in the brain, forming deposits that contribute to the onset of neurodegenerative diseases. Importantly, PrPsc can act as a seed or template, binding with PrPc and facilitating the PrPc-to-PrPsc conversion, thereby intensifying the progression of neurodegenerative diseases.324 Unlike other amyloid diseases with uncertain transmissibility and infections, prion diseases are well-established to be transmissible between individuals, inducing chronic infection and/or disease in different species—presenting an additional potential pathological route for disease induction. Prnp-knockout mice lacking PrP expression are not susceptible to prion infection, emphasizing the inflammatory role of PrP in prion pathogenesis.325 Increasing evidence suggests that the most infectious particles are PrP-folding intermediates, specifically small oligomers consisting of 12–24 monomers.326,327 This aligns with the widely accepted notion that small oligomers of the misfolded protein are primarily responsible for neurotoxicity.246In addition to the putative neurotoxicity attributed to misfolded amyloids, the loss of normal PrPC function is considered integral to neurodegenerative processes. PrP serves diverse physiological functions, including cellular differentiation, neuronal excitability, myelin maintenance, and metal ion homeostasis.328–331 Other studies have demonstrated an increase in PrP expression during bacterial infection332 and inflammatory wounding/diseases,163,333 suggesting that PrP may play a crucial role in inducing innate immune responses. PrP has been identified as an antimicrobial agent against both Gram-negative (e.g., E. coli, P. aeruginosa) and Gram-positive bacteria (B. subtilis, S. aureus) (Fig. 16a), as well as fungi (e.g., C. parapsilosis, Fig. 16b) under normal (pH = 7.4) and low pH (pH = 5.5) conditions.163 When comparing the antibacterial effects of PrP with its truncated variants, a descending antimicrobial ability is observed in the order of PrP23–231 > PrP23–144 > PrP90–231, highlighting the significance of the N-terminal region. A more detailed comparison between full-length and a series overlapping peptide sequences comprising 20 amino acids, as well as shorter variants, further emphasizes the importance of the N-terminal part of PrP, especially the unstructured N-terminal (KKRPK) region of the protein (Fig. 16c), which is mainly attributed to (i) the highly positive sequence of KKRPK interacting with negatively charged microbial membranes, (ii) the heparin-binding site enhancing the binding and disruption of microbial membranes, and (iii) the histidine-rich region coordinating interactions with metal ions (e.g., Zn2+ and Cu2+).110,163 Substantial evidence suggests that the antimicrobial activity primarily depends on the formation of unstable oligomers, rather than mature amyloid fibrils, to disrupt bacterial membranes via a carpet or detergent model.110 Despite significant efforts over the past two decades to investigate the pathological (infectious and neurotoxic) and physiological (antimicrobial) nature of prion proteins, a major gap persists in our understanding of the vicious loop involving PrPc–PrPsc-infections-PrPc. Here, we propose two potential scenarios, with the hope that they prove useful for future research and validation: (i) the conversion of PrPc to PrPsc induces inflammatory infection and upregulation of antimicrobial PrPc and (ii) external microbial threats induce PrPc upregulation, leading to PrPc–PrPsc conversion.
 | ||
| Fig. 16 Antimicrobial activity of prion protein. (a) Antibacterial efficacy of prion protein aginst E. coli in a concentration-dependent manner. (b) Antifungal efficacy of prion protein aginst C. parapsilosis in a concentration-dependent manner. (c) Antimicrobial efficacy of prion protein fragments, with and without KKRPK motif, agsinst E. coli and C. parapsilosis. (Reproduced with permission from ref. 163 Copyright © 2009 Pasupuleti et al.) Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
4.5. Serum amyloid A (associated with systemic amyloid A)
Serum amyloid A (SAA), a highly conserved acute-phase protein present in nearly all mammalian species, is primarily synthesized by hepatocytes in the liver.334 However, its production is not exclusive to hepatocytes, as various cells in extrahepatic tissues also contribute to its synthesis. The SAA family comprises four protein isoforms, each consisting of 104 amino acids. In humans, the acute-phase isoforms SAA1 and SAA2 (collectively referred to as SAA in this review) are prominently induced by inflammatory signals, particularly interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and lipopolysaccharide (LPS) during the acute-phase response (APR). SAA3 is a pseudogene in humans. Human SAA4, identified as a constitutive isoform, is presumed to play a housekeeping role rather than serving as a responsive element to inflammation, constituting the most abundant serum SAA form in healthy individuals. Similarly, in mice, SAA1 and SAA2 are the major forms of SAA proteins produced by hepatocytes, while SAA4 is constitutively expressed. A notable distinction between human and mouse SAA lies in the SAA3 isoform, which codes for a functional SAA protein and emerges as the primary form in mouse inflammatory tissues.335,336Originally identified as a major component of amyloid A (AA) fibrillar deposits associated with reactive systemic amyloidosis,337 SAA has since been recognized for its dual role. SAA primarily functions as an apolipoprotein of high-density lipoproteins (HDL), the principal carrier of SAA in the bloodstream, and acts as a secondary (reactive) precursor inducing pathological AA amyloidosis. In the absence of the acute-phase response (APR), SAA is constitutively produced at relatively low levels, contributing to the maintenance of lipid metabolism balance. APR process involves displacing apolipoprotein A1 (apoA1) via its N-terminal lipid binding sites (SAA1–15) to form acute-phase HDL. This modified HDL participates in various functions, including (i) regulating lipid metabolism and cholesterol efflux regulation;338 (ii) recruiting immune cells (e.g., monocytes and polymorphonuclear leukocytes) to sites of inflammation;339 (iii) promoting the expression of several proinflammatory cytokines, such as TNF-α, IL-6, IL-8, IL-1β, and the growth-promoting granulocyte-macrophage colony-stimulating factor;340 (iv) inducing directional migration of human mast cells;341 and (vi) serving as an immune opsonin for Gram-negative bacteria, including E. coli and P. aeruginosa.342 In normal cases, elevated SAA levels decline rapidly as inflammation resolves. However, any abnormal prolonged or excessive inflammation or infectious diseases can lead to pathological conditions, including secondary amyloidosis, where SAA contributes to the formation of amyloid deposits.
While a clear clinical association between SAA and inflammatory infections exists, the precise physiological role of SAA during microbial infection remains elusive. The bacteriotoxicity of SAA was initially observed in 1992 during an investigation utilizing E. coli to express recombinant SAA at high production levels. This expression led to a notable decrease in the volume of cells, reaching 70–80% of the initial volume within 2 hours, indicative of cell lysis260 (Fig. 17a). Subsequent studies confirmed that SAA toxicity is connected to the formation of ion channels in the bacterial membrane,119 resembling the behavior observed in other amyloid peptides.117,343,344 The formation of such channels is attributed to both host cell damage and antimicrobial activity. On one hand, it particularly affects cells in the kidney, liver, and spleen, which require electrically tight membranes for their ionic exchange functions. This is supported by clinical evidence that renal dysfunction is the most common symptom at the onset of AA amyloidosis.345 On the other hand, amyloid-forming SAA variants (e.g., human SAA1, mouse SAA2) can effectively induce bacterial lysis, a capability not shared by SAA4. This distinction highlights the essential requirements of a β-pleated sheet confirmation and the hydrophobicity of the N-terminus to interact with the hydrophobic component of the bacterial cell.119 This interaction occurs with high affinity (KD = 10−7–10−8 M) and in a rapid manner (∼15 minutes).164 In addition to E. coli, SAA exhibits binding affinity with a diverse array of Gram-negative bacteria, including S. typhimurium, S. flexneri, K. pneumoniae, V. cholerae, and P. aeruginosa, while showing no binding affinity for Gram-positive bacteria such as S. pneumoniae and S. aureus (Fig. 17b). This strain-specific binding capability is closely linked to the outer membrane protein A (OmpA)/OprF family conserved ligands, present in nearly all Gram-negative bacteria. These ligands act as opsonins, enhancing bacterial clearance by modulating macrophages and neutrophils.164,342 OmpA-deficient E. coli or OprF-deficient P. aeruginosa did not bind to SAA, underscoring the role of SAA as a pattern-recognition innate immune protein.164
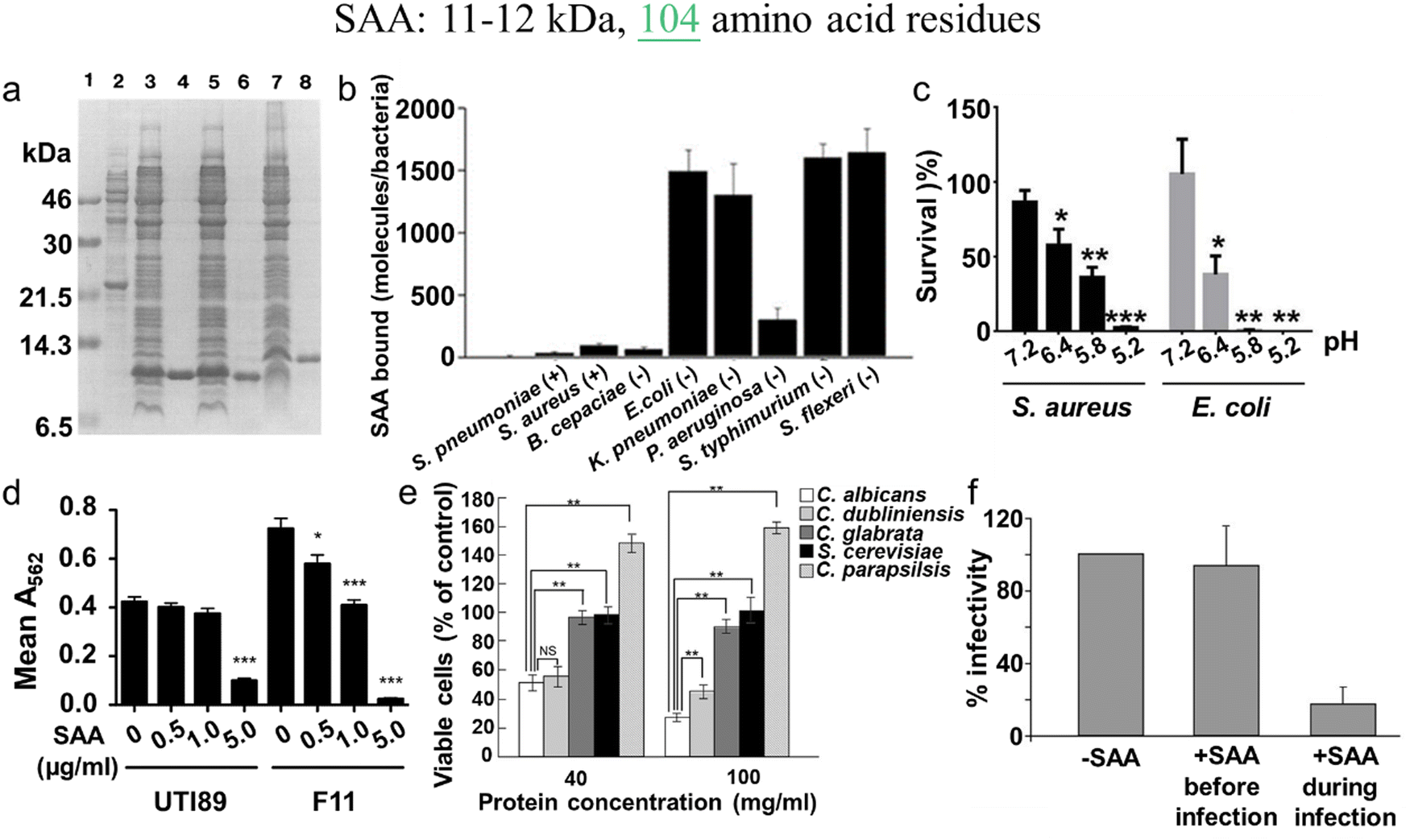 | ||
| Fig. 17 Antimicrobial activity of serum amyloid A (SAA). (a) Antibacterial efficacy of recombinant SAA against E. coli, as indicated by the presence of E. coli lysates subsequent to SAA expression in E. coli. Lane 1: molecular weight markers. Lane 2: lysates of non-induced E. coli. Lane 3,5,7: lysates of E. coli expressing SAA1, SAA2, SAA4. Lane 4,6,8: purified rSAA1, rSAA2, rSAA4. (Reproduced with permission from ref. 260 Copyright © 1994 Published by Elsevier B.V.) (b) Stain-selective antibacterial efficacy of SAA. Where SAA binds to Gram-negative bacteria while not interacting with Gram-positive bacteria. (Reproduced with permission from ref. 164 Copyright © 2005 Elsevier Inc.) (c) pH-dependent antibacterial efficacy of SAA against S. aureus and E. coli., highlighting its enhanced antibacterial activity under acidic conditions. (Reproduced with permission from ref. 16 Copyright © 2020 Elsevier Inc.) (d) Antibacterial efficacy of SAA against uropathogenic E. coli (UPEC, e.g., UTI89 and F11) through the inhibition of their biofilm formation. Biofilm levels were quantified by measuring A562. (Reproduced with permission from ref. 261 Copyright © 2012 Erman et al.) (e) Stain-selective antifungal efficacy of rhSAA, where rhSAA exhibits anti-fungal activity specifically against C. albicans or closely related species of C. dubliniensis, excluding other fungal species. (Reproduced with permission from ref. 262 Copyright © 2019 American Society for Microbiology.) (f) Antiviral efficacy of SAA against Huh-7 cells infected by HCVpp. (Reproduced with permission from ref. 263 and 264 Copyright © 2016 American Association for the Study of Liver Diseases.) Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
Clinically, investigations into the bactericidal activity of SAA have systematically explored different microenvironments and utilized distinct gene knockout mouse models to understand its efficacy in various infectious diseases. In comparison to physiological neutral conditions, SAA has demonstrated heightened bactericidal activity under acidic conditions, particularly at pH 5.2 (Fig. 17c). This suggests its potential as an effective antibacterial agent in addressing cutaneous infections, a characteristic environment of the skin surface. This effectiveness was further demonstrated using SAA1/2 double knockout mice (SAA1/2 DKO), which exhibited impaired clearance of S. aureus during cutaneous infections.16 Similarly, SAA1/2 DKO mice proved to be more susceptible to dextran sodium sulfate (DSS)-induced colitis, showcasing increased weight and blood loss, along with higher histological disease scores compared to wild-type controls.346 A robust mouse urinary tract infections (UTI) model utilized the introduction of uropathogenic E. coli (UPEC, e.g., UTI89 and F11) into the urinary tract to induce SAA expression. The results revealed heightened levels of SAA1/2 in response to UTI, demonstrating its role in blocking biofilm formation by uropathogens261 (Fig. 17d).
In addition to its antibacterial function, SAA also exhibits anti-fungal activity against Candida albicans. The potential anti-fungal properties of recombinant SAA were initially demonstrated in 1990, showcasing its role as a potent activator that enhances calcium mobilization, cell-surface antigen expression, lactoferrin secretion, phagocytosis, and the anti-Candida activity of polymorphonuclear cells.347 However, direct evidence of SAA's anti-fungal capabilities became clearer in 2019 when a systemic infection model was employed. In this model, mouse SAA1 was upregulated following the induction of C. albicans infection. During the infection, both human and mouse recombinantly expressed SAA1 exhibited species-specific anti-fungal activity by binding to the surface of C. albicans or closely related species of C. dubliniensis (but not C. glabrata, S. cerevisiae, and C. parapsilsis) cells (Fig. 17e). This binding disrupted the integrity of the fungal cell membrane, leading to cell death.262 Further investigation by the same research group delved into the molecular mechanisms of SAA1's anti-fungal activity and indicated that SAA1 mainly targets Als3 (agglutinin-like sequence 3), a cell wall adhesin of C. albicans, inducing rapid cell aggregation and subsequent death.348
Upregulated expression of SAA has been observed in various viral infections affecting both animals and humans. For instance, SAA levels were notably elevated in COVID-19 patients at the onset of hospitalization, even in cases with mild respiratory symptoms. This highlights SAA's potential as an effective predictive factor for severe COVID-19, demonstrating an accuracy of 89%.349,350 Additionally, SAA has been reported to exhibit antiviral activity against the hepatitis C virus (HCV) by inhibiting its entry into cells. However, this antiviral activity is limited to the timing of SAA addition and virus adsorption (i.e., SAA addition during infection > SAA addition before infection)263,264 (Fig. 17f). Another study demonstrates the antiviral effect of SAA by directly binding to the influenza A virus (IAV) and enhancing the uptake of the virus by neutrophils.351 SAA has demonstrated the capacity to block HIV-1 infection of host cells through CCR5 receptors. Notably, SAA emerges as one of the initial systemic antiviral responses to HIV-1, detected as early as 5–7 days before the first detection of plasma viral RNA and significantly earlier than other systemic cytokines.352
5. Cross-seeding between antimicrobial, bacterial-secreted, and amyloid peptides
The concept of cross-seeding between AMPs, bacterial-secreted peptides, and amyloid peptides is supported by several lines of indirect evidence. Firstly, the co-existence and often co-localization of these peptides in various tissues, blood vessels, and spinal fluids have led to conceptual frameworks proposing molecular interactions and cross-seeding among these diverse peptide aggregates. Secondly, additional evidence from previous sections has demonstrated the alternative antimicrobial activity of certain AMPs (AMPs) and the amyloid-like aggregation property of specific amyloids (AMYs), as well as some shared sequential and structural features in both AMPs and AMYs. Thirdly, some investigations into cross-seeding among amyloid peptides, particularly in cases where (i) multiple neuropathologies co-occur in patients with two or more forms of dementia and (ii) both amyloid aggregation and microbial infection are observed, confirm that such co-occurrences can be key pathological causes of neurodegenerative diseases.353 Building on the aforementioned findings and logical connections, the cross-seeding concept appears to offer a plausible explanation for both direct and indirect molecular crosstalk and spreading mechanisms between neurodegenerative diseases and microbial infections. These cross-seeding interactions involve the mutual modulation of peptide aggregations in a transmissible manner, both in vitro and in vivo. The identified cross-seeding systems between antimicrobial peptides, bacterial-secreted peptides, and amyloid peptides are still limited. However, antimicrobial peptides, owing to their intrinsic antimicrobial activity, represent a vast and native resource that has been relatively underexplored for repurposing as potential and effective amyloid inhibitors (Fig. 18). Hence, gaining a comprehensive understanding of the new functions of AMYs and AMPs, along with the molecular events that underlie their cross-seeding, spreading, and crosstalk in mixed pathologies, is of paramount importance for the development of new therapeutic strategies. | ||
| Fig. 18 Historical literature review of the first-discovered or important amyloid modulators including both non-AMPs and AMPs. The inclusion of a negative (−) or positive (+) sign in brackets denotes the amyloid inhibition or promotion effect by these modulators. | ||
5.1. Cross-seeding between antimicrobial peptides and amyloid peptides
Amyloid peptides (AMYs), traditionally associated with neurodegenerative diseases, have a strong tendency to aggregate into fibrillar formation, contributing to pathological processes. Conversely, antimicrobial peptides play a crucial role in the innate immune response, targeting and neutralizing microbial invaders. However, emerging research has unveiled unexpected intersections between these two peptide classes through a phenomenon known as cross-seeding. This cross-seeding interaction showcases how amyloid peptides can impact the behavior of antimicrobial peptides and vice versa. This underscores the intricate nature of peptide-peptide interactions, offering a rich avenue for exploring novel mechanisms, implications, and potential applications in the realms of health and diseases within the context of this cross-seeding phenomenon.:amyloid molar ratio from 0.005 to 1 resulted in a significant reduction in ThT signals for Aβ from 11 to 94%, hIAPP from 16 to 49%, and hCT from 35 to 93% (Fig. 19b).23 This HD-6-induced amyloid inhibition effect led to the formation of less-fibrillar, amorphous-like aggregates, while retaining the original disordered structures. In the HBD-1:amyloid systems across various molar ratios (1:0.005–1:1), HBD-1 notably decreased ThT fluorescence (a marker for amyloid fibril presence) by 44–93%, reduced the rate of aggregation (halving the aggregation time) by 71–96%, postponed the shift towards β-structures, lowered β-structure content by 4–28%, and broke down mature, larger, and thicker fibrils into smaller, thinner forms. Comprehensive evaluations using ThT, CD, AFM, and SPR methods across different amyloid–defensin systems highlight that the defensins amyloid aggregation inhibitory effects depend on both dosage and peptide sequence. Significant observations include: (i) HD-6 and HBD-1 exhibit sequence-specific effectiveness in inhibiting amyloid aggregation, ranked as Aβ > hCT > hIAPP for HBD-1 and in the order hCT > Aβ > hIAPP for HNP-1 and NP-3A. (ii) At lower concentrations, α-defensins more effectively slow the expansion of amyloids from smaller to larger aggregates, while at higher doses, they primarily prevent the formation of amyloid nuclei during the lag phase. These differences indicate the existence of interspecies energy barriers that influence the interactions between different α-defensins and amyloid proteins.
 | ||
| Fig. 19 Identified cross-seeding systems between antimicrobial peptides and amyloid peptides, including (a) HNP-1 and Aβ, hIAPP, hCT by ThT, (Reproduced with permission from ref. 24 Copyright © 2021 The Royal of Chemistry) (b) HD-6 and Aβ, hIAPP, hCT by ThT, (Reproduced with permission from ref. 23 Copyright © 2022 The Royal of Chemistry) where both HNP-1 and HD-6 exhibit a general inhibition property against the fibrillization of different amyloid peptides. (c) LL-37 and Aβ42 by SPR imaging (Reproduced with permission from ref. 25 Copyright © 2017 IOS Press) and LL-37 and hIAPP by ThT, (Reproduced with permission from ref. 26 Copyright © 2020 John Wiley & Sons, Inc.) where LL-37 acts as a nanomolar inhibitor to prevent the fibril formation of both Aβ and hIAPP and their associated cell toxicity; (d) LL-III and α-synuclein by phase-contrast light and fluorescence microscopy, (Reproduced with permission from ref. 354 Copyright © 2021 MDPI (Basel, Switzerland)) where the addition of LL-III to a monomeric α-synuclein solution facilitates the formation of LL-III-enriched droplet clusters, effectively preventing the conversion of α-synuclein into mature fibrils; (e) CAP37, CG, NE and Aβ by ThT, (Reproduced with permission from ref. 355 Copyright © 2021 Bentham Science Publishers) where CAP37 (circles), CG (inverted triangles), and NE (triangles) inhibit the kinetic aggregation of Aβ through different pathways, with varied inhibition efficiencies ranked in a decreasing order of NE > CAP37 > CG. (f) Magainin 2 and rIAPP by liposome leakage assay, (Reproduced with permission from ref. 18 Copyright © 2013 National Academy of Science) where magainin 2 and rIAPP exhibit full cross-cooperativity, leading to equilibrium membrane leakage that is 100 times greater than the simple sum of the activities of individual peptides. Amino acid residues of each AMP are color-coded to reflect their properties: polar uncharged residues in rose, polar charged residues in blue, and non-polar residues in yellow. | ||
The four defensins also exhibited a dose-dependent protective role in rescuing cells from amyloid-induced toxicity to varying extents. Specifically, in the presence of three amyloid peptides, HNP-1 improved cell viability by 23–28%, NP-3A by 12–35%, HD-6 by 7–55%, and HBD-1 by 32–51%, while reducing cell toxicity by 9–31%, 10–31%, 11–33%, and 16–25%, respectively. The aggregated cellular data indicate various potential mechanisms through which defensins could reduce amyloid-induced toxicity. Potent interactions between defensins and amyloid peptides may lessen the creation of harmful amyloid aggregates known to disrupt cell membranes, resulting in the formation of defensin–amyloid complexes that are either less toxic or non-toxic and relatively harmless to cell membranes. Additionally, defensins may competitively decrease the binding propensity of amyloids to cell membranes, collectively endowing defensins with an improved cell protection function. On the other hand, cross-seeds of defensins with the three different amyloid peptides generally demonstrated comparable or even higher antimicrobial efficiency than the corresponding pure amyloid peptides or defensins.
Considering that β-rich configurations are prevalent in amyloid aggregates independent of sequence, the ability of α-defensins to inhibit amyloids in a sequence-independent manner mainly stems from their interaction with amyloid proteins via β-structure engagements. This interaction is explained by the “conformational selection binding” mechanism, which suggests that defensins with β-structures, or any peptides capable of forming β-structures, are predisposed to engage with amyloids that share similar β-structural features. This mechanism competitively reduces amyloid–amyloid interactions, thereby preventing amyloid aggregation of Aβ, hIAPP, and hCT, along with the resultant amyloid-induced toxicity. Additionally, it contributes to antimicrobial protection.
Similarly, considering the presence of both LL-37 and hIAPP in the pancreas and their notable (42%) sequence similarity, LL-37 has been identified to interact with hIAPP with nanomolar affinity. This cross-seeding mechanism proves effective in preventing hIAPP aggregation and associated pancreatic β-cell damage and neuroinflammation in vitro.26 The inhibitory action of LL-37 is achieved by its attachment to (1) initial hIAPP species, encapsulating them into soluble, non-fibrillar mixed complexes, and (2) established hIAPP fibrils, transforming them into assemblies that cannot seed further aggregation (Fig. 19c). LL-37 emerges as having a significant connection to the pathogenesis of T2D through various pathways, e.g., LL-37 has been observed to suppress pancreatic β-cell inflammation in a mouse model, while concurrently promoting insulin and glucagon secretion, ultimately enhancing islet function.357 These studies suggest a potential protective role for LL-37, acting as a molecular inhibitor of both Aβ and hIAPP, in the pathogenesis of both AD and T2D. Particularly noteworthy is the resemblance of sporadic AD to what is often termed as ‘type 3 diabetes’ occurring in brain tissue.358
The limited studies on AMPs-AMYs systems indicate that cross-seeding can cooperatively induce membrane leakage and bacterial cell death through different mechanisms. In some cases, both AMPs and AMYs show strong antimicrobial activity. The discovery of antimicrobial peptides with β-structure, which can simultaneously inhibit microbial infection and amyloid aggregation, expands their potential as multi-target amyloid inhibitors. These AMPs, while existing in limited quantities, provide a molecular foundation for further engineering and designing new variants. These variants could combine antimicrobial, anti-amyloid, and immunomodulatory functions, making them promising candidates for multifunctional drugs in combating both microbial infections and amyloid-related issues. These multiple-functional AMPs introduce a novel concept, resembling “killing two birds with one stone.” This concept successfully integrates the “amyloid cascade hypothesis” with the “microbial infection hypothesis”. The functional and structural links between antimicrobial peptides and amyloid proteins illustrate their natural abilities in both bacterial elimination and amyloid suppression functions.
5.2. Cross-seeding between bacterial-secreted peptides and amyloid peptides
The gut-brain axis, involving bidirectional communication between the gut and the brain, has garnered significant interest due to its potential relevance in human disease and health. Throughout this axis, the gut and the brain are interconnected systems that can influence each other's functioning, thus representing a possible connection between the gut microbiome, their metabolites, and neurological disorders. Recent research presented compelling evidence supporting the notion that symptoms of Alzheimer can be transferred to a healthy young organism through the gut microbiota, establishing a causative role that gut microbiota may have in the disease.359 A growing body of symptomatic, physiological, and pathological data suggest that cross-seeding occurs between microbial amyloid curli presented in the gut and amyloid peptides found in the brain.27,360–362 Curli fibrils are assembled through bacterial secretion of unfolded amyloid proteins (e.g., CsgA, CsgC, CsgE, FapCS) (Fig. 20a). Interestingly, these curli fibrils exhibit some similarities to amyloid fibrils, including self-aggregation into β-rich fibrils, surface adhesion to epithelial cells, and participation in bacteriophage defense mechanisms. Biochemical studies have provided compelling evidence that the native bacterial chaperones of curli possess the ability to interact with diverse human amyloid proteins, thereby influencing their aggregation pathways in vitro and in vivo, including αSyn,27–29,322 Aβ,29 Tau,363 hIAPP,364,365 and cellular prion protein (PrPC).366 Specifically, In the in vivo study of curli-producing Escherichia coli (CsgA) in mice27 and in worm,367 a promotion of αSyn aggregation and inflammation was observed in both the gut and the brain, leading to a worsening of motor impairment and gastrointestinal (GI) dysfunction. However, in the two cases of variants of CsgA that were incapable of forming amyloids and the introduction of an amyloid inhibitor that prevented CsgA expression in the gut, they both did not affect αSyn aggregation in the brain of mice. Different from the cross-β amyloid conformation characteristic of curli fibrils, phenol-soluble modulins (PSM) are amyloidogenic proteins originating from Staphylococcus aureus that adopt a cross-α fibrillar structure.368 Both CsgE and PSMα have been observed to accelerate αSyn amyloidogenesis in vitro28,369 (Fig. 20b). The higher cross-α content observed in PSMα3 has been associated with an augmented promotional effect on the aggregation of Aβ40,370 implying a highly diverse conformational interplay in cross-seeding interaction (Fig. 20c). Remarkably, pre-formed CsgA seeds have also been identified as accelerators of Aβ aggregation, and it is noteworthy that fibrillation inhibitors are shared between CsgA and Aβ.371 Similarly, Aβ displayed favorable binding with FapC amyloid fragments (FapCS) of Pseudomonas aeruginosa, leading to the acceleration of cross-seeding fibrils (Fig. 20c).34 FapCS acted as a catalyst and propagated its structural characteristics for promoting Aβ amyloidogenesis in vitro, in silico, and in a zebrafish model of AD. The robust seeding capacity for Aβ by FapCS induced multiple pathological indicators, including behavior, cognitive memory function, cerebral Aβ burden, synaptic health, production of reactive oxygen species (ROS), and cell degeneration in both neuronal cells and zebrafish. Both CsgA and CsgB can cross-seed with the prostatic acid phosphatase fragment of PAP248–286 (Fig. 20d). Acting as catalytic agents, they have a moderate impact on the nucleation rate and a significant effect on enhancing fiber growth from existing nuclei.361 This suggests a more intricate scenario in which cross-seeding partially bypasses the nucleation step but has limited influence on fiber elongation.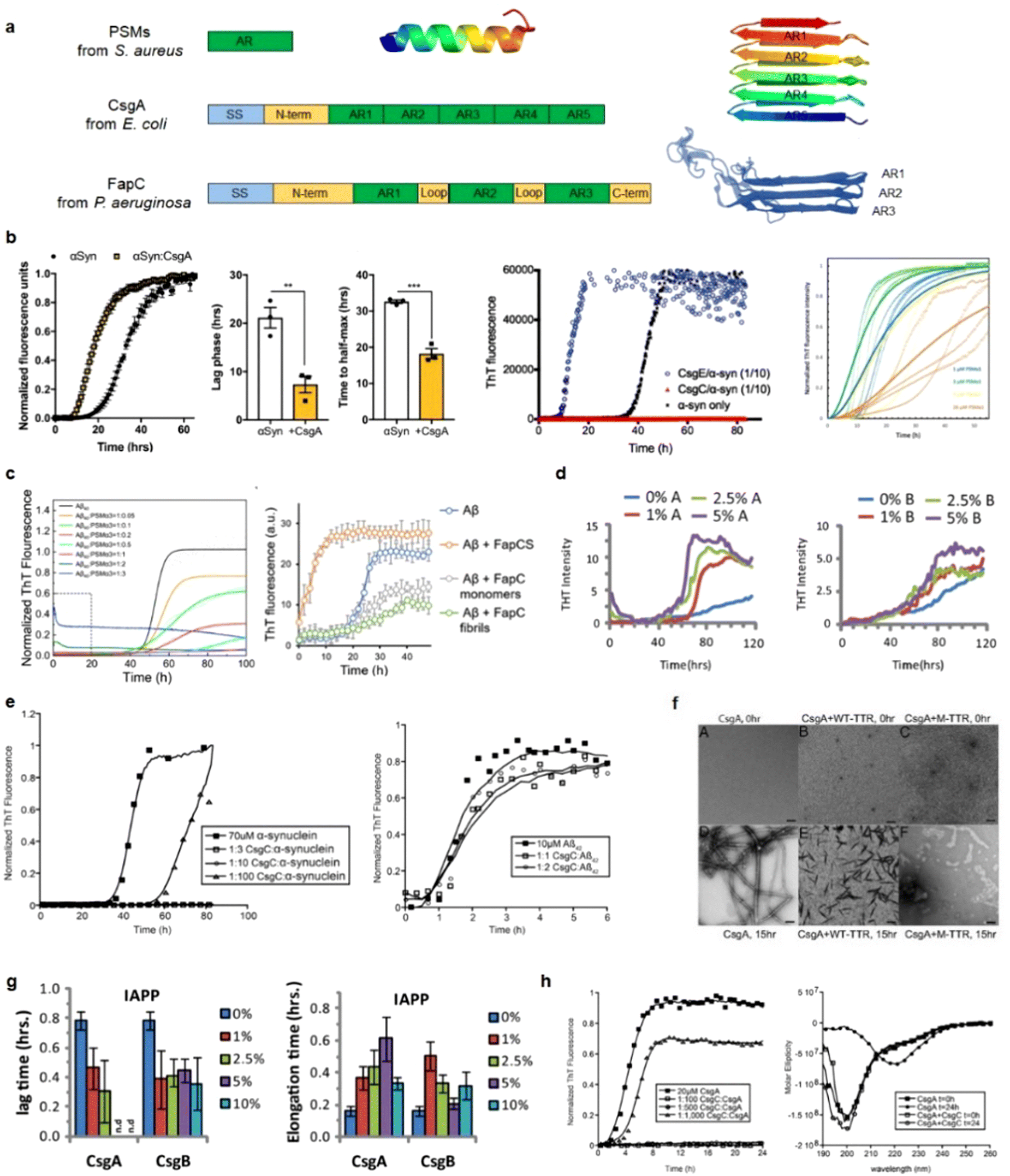 | ||
| Fig. 20 Cross-seeding interactions between bacterial-secreted peptides and amyloid peptides. (a) Schematic representation of distinct bacterial biofilm-forming functional amyloids. Signal sequences (SS) are depicted in blue, amyloid regions (AR) in green, and loop/linker and terminus regions in yellow. PSMs are short proteins adopting a cross-α fibrillar structure (PDB: 5I55). CsgA forms a β-helix with five imperfect AR repeats stacking on top of each other (PDB: 8ENQ), while FapC also forms a β-helix with three ARs stacking on top of each other (FapC model modified from ref. 372 Copyright © 2018 The Authors). (b) CsgA (Reproduced with permission from ref. 27 Copyright © 2020 eLife Science Publications Ltd.) CsgE (Reproduced with permission from ref. 28 Copyright © 2015 Chorell et al.) and PSMα1 (Reproduced with permission from ref. 369 Copyright © 2021 MDPI (Basel, Switzerland)) have demonstrated the ability to accelerate αSyn amyloidogenesis. (c) PSMα3 exhibits a promotional effect on the aggregation of Aβ40. (Reproduced with permission from ref. 370 Copyright © 2023 MDPI (Basel, Switzerland)). Similarly, FapCS plays a catalytic role in promoting Aβ amyloidogenesis. (Reproduced with permission from ref. 34 Copyright © 2023 John Wiley & Sons, Inc.) (d) Both CsgA and CsgB can cross-seed with the prostatic acid phosphatase fragment of PAP248-286, facilitating its conversion into the amyloid SEVI. (Reproduced with permission from ref. 361 Copyright © 2013 PeerJ. Inc.) (e) CsgC inhibits the amyloid assembly of αSyn, while demonstrating no inhibitory effect on Aβ42 aggregation. (Reproduced with permission from ref. 29 Copyright © 2015 Elsevier Inc.) (f) TTR and its mutants selectively inhibit the conversion of CsgA into amyloid-like fibrils. (Reproduced with permission from ref. 373 Copyright © 2017 National Academy of Sciences) (g) Cross-seeding between CsgA/CsgB and hIAPP results in a reduction in the lag-time but a significant inhibition of hIAPP elongation.361 (h) CsgC can inhibit CsgA amyloid formation at sub-stoichiometric concentrations and maintain CsgA in a non-β-sheet-rich conformation.29 | ||
In contrast to findings indicating that bacterial amyloid acts as a trigger, promoting amyloid aggregation through cross-seeding, some studies observed an opposite aspect: bacterial curli possesses a potent and selective inhibitor of amyloid formation. CsgC inhibited amyloid assembly of αSyn, while having no inhibition effect on Aβ42 aggregation in vitro (Fig. 20e).29 Human wild-type tetrameric transthyretin (TTR, associated with AD) and its mutants were found to exert a selective inhibitory effect on the conversion of CsgA into amyloid-like fibrils in vitro (Fig. 20f).373 Notably, this effect was observed specifically with CsgA and not CsgB, indicating that the inhibitory mechanism is likely linked to its cross-seeding interaction with CsgA. Furthermore, transthyretin exhibited a comparable inhibitory effect on amyloid formation and subsequent toxicity induced by Aβ,374,375 hIAPP,376 and HypF-N377 in both in vitro experiments and mouse models.
Adding to the complexity, the cross-seeding of Csg with various amyloid proteins led to intricate outcomes, which were dependent on the specific amyloid sequences and concentrations at play. When CsgA and CsgB were cross-seeded with hIAPP, they caused a reduction in the lag-time of hIAPP amyloid formation but significantly inhibited hIAPP elongation (Fig. 20g).361 On the other hand, the impact of CsgA and CsgB on the fibrillization rate of Aβ40 was more nuanced. The fibrillization rate demonstrated a modest decrease at lower concentrations of both CsgA and CsgB, while it slightly increased at higher concentrations.361 These findings suggest that microbial amyloid proteins may possess broad cross-seeding activity, but they could serve as both an inhibitor and enhancer of amyloid fibrillization, influenced by the specific amyloid protein involved and the concentrations of the interacting components. Importantly, under exceptional instances, microbial amyloid proteins possess the capability to cross-seed with each other. CsgC was found to inhibit CsgA amyloid formation at sub-stoichiometric concentrations, effectively maintaining CsgA in a non-β-sheet-rich conformation (Fig. 20h).29 These findings not only provide additional evidence of the intricate interactions between microbial amyloid proteins and their potential modulatory effects (either acceleration, inhibition, or both at different aggregation stages) on amyloid formation pathways, relevant to a conformational relationship underlying their cross-seeding interactions.
These findings along the gut-brain axis highlight the potential interplay and multifaceted nature between microbial amyloids and human amyloid proteins within the gut environment, with complex pathological consequences between amyloid aggregation and bacterial-induced inflammation.27 Such interplay leads to an interesting hypothesis that exposure to microbial amyloids in the gastrointestinal tract might play a crucial role in accelerating amyloid aggregation and disease progression in both the gut and the brain. The presence of human and microbial amyloids in the circulation and cerebrospinal fluid opens possibilities for multiple cross-seedings between different pairs of amyloidogenic proteins. This includes cross-seeding between α-syn and hIAPP,378 Aβ and hIAPP,379 Aβ and α-syn,380 Aβ and tau,381 Aβ and transthyretin,375 hIAPP and insulin.382 Additionally, cross-seeding can also occur between human and microbial amyloid proteins, such as Natural silk from the silkworm Bombyx mori, the prion protein Sup35 from the yeast Saccharomyces cerevisiae, and the curli protein CsgA from the bacterium Escherichia coli, all of which have been shown to enhance the amyloidosis of amyloid protein A (AA) in mice.36 The cross-seedings between various amyloidogenic proteins, originating from both human and microbial sources, adds another layer of complexity to the gut-brain axis and its association with amyloid-related diseases, microbial-related pathologies, and other neurological disorders such as autism spectrum disorder, depression, and anxiety. This growing understanding of the intricate connections between the gut microbiome, amyloid proteins, and various neurological conditions offers insights into potential therapeutic approaches and preventive measures in the realm of neurodegenerative and neuropsychiatric disorders.
6. Computational understanding of antimicrobial peptides and amyloid peptides
While numerous studies have explored the native antimicrobial activity of AMPs and the misfolding and self-aggregation properties of AMYs, less efforts have been devoted to computational modeling and simulations of the alternative functions of self-aggregation into amyloid-like fibrils of AMPs, the antimicrobial activity of AMPs, and the cross-seeding between them. This exploration is attributed to the absence of atomic structures for these peptides and fewer identified systems involving alternative functions and cross-seeding interactions between AMPs and AMYs.6.1. Alternative function of antimicrobial peptides and amyloid peptides
Current computational strategies for studying alternative function of AMPs and AMYs predominantly focus on the membrane disruption mode of AMYs and the stable β-structure organization of AMPs.100 For AMPs, the self-association and multimerization into β-structures play a pivotal role in their activities, with β-structures being widely recognized for their ability to disrupt cell membrane potential. In Fig. 21, PG-1 assembles into fibrils rich in β-structures, featuring both antiparallel and parallel β-sheets. These twisted fibrils exhibit significant structural similarities to classical amyloids. The β-forming PG-1 peptides have the capability to permeate and self-organize into oligomeric pores within various lipid bilayers, leading to non-specific ion leakage and membrane disruption akin to the membrane pores induced by AMPs.8,114,383 When comparing β-rich AMPs and amyloids like Aβ, besides their shared cytotoxicity and amyloidogenicity, they also possess a common structural motif, contributing to their ability to form channels. Microsecond time-scale MD simulations of uperin-3.5 unveiled two crucial factors of peptide concentrations and helical intermediates that contribute to the formation of β-sheet-rich amyloid-like structures.154,384 Driven by strong hydrophobic interactions, uperin-3.5 rapidly assembled into polymorphic aggregates initially devoid of any β-structures. Subsequently, a gradual structural transition occurred and progressed from random coils to α-helices and ultimately to β-sheets. Although a complete structural transition and formation of β-sheet aggregates were not observed within the typical timescale, a noticeable increase in β-sheet content at the expense of α-helices is evident, suggesting that partial helical conformations may offer a more accessible energetic pathway to the stable formation of amyloid aggregates. Replica Exchange Molecular Dynamics (REMD) simulations of two tyrocidines from Bacillus aneurinolyticus demonstrated their propensity to readily aggregate into distinct dimers with varying orientations (parallel vs. antiparallel between two monomers), each adopting a classical β-turn structure.196 Despite structural similarities among the dimers, the underlying forces of stabilization differ, with some primarily associated by hydrogen bonds and others by hydrophobic interactions. The diverse orientations and organizations of tyrocidine dimers suggest a polymorphic nature in peptide aggregation similar to amyloid peptides.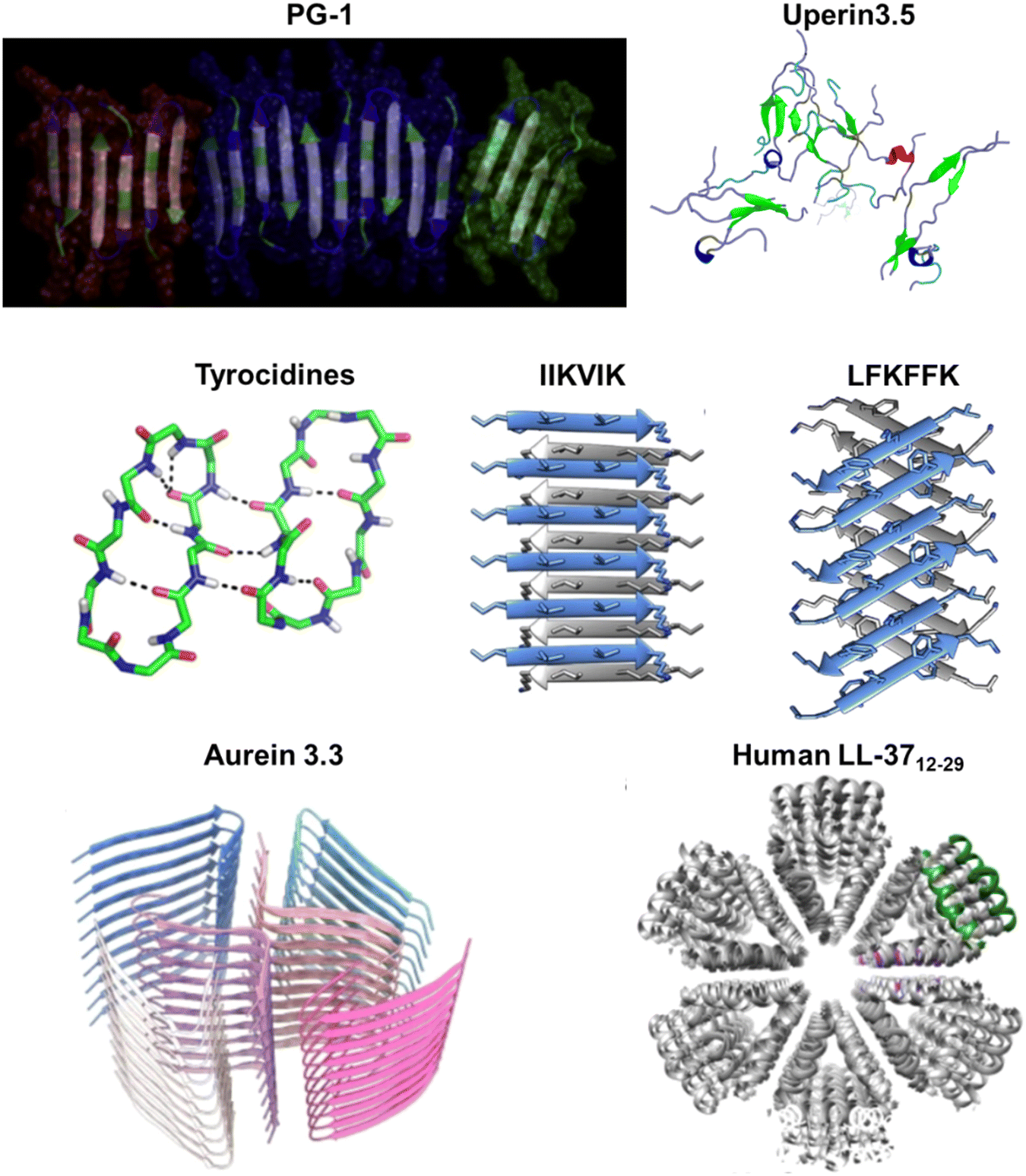 | ||
| Fig. 21 Computational investigation of self-aggregation propensity of antimicrobial peptides to form amyloid-like fibrils with distinct secondary structures and peptide organizations (cross-β sheets, cross-α sheets, α-helices). Examined peptides include PG-1 (Reproduced with permission from ref. 8 Copyright © 2011 Elsevier Inc.), uperin 3.5 (Reproduced with permission from ref. 384 Copyright © 2023 The Owner Societies), tyrocidines (Reproduced with permission from ref. 196 Copyright © 2013 American Chemistry Society), IIKVIK from PSMα1 and LFKFFK from PSMα3 (Reproduced with permission from ref. 385 Copyright © 2018 Springer Nature Limited), aurein 3.3 (Reproduced with permission from ref. 152 Copyright © 2022 Springer Nature Limited), human LL-3717–29 (Reproduced with permission from ref. 155 Copyright © 2020 Springer Nature Limited). | ||
A combination of computational modeling and x-ray microcrystallography has elucidated atomic structures of short segments from the Staphylococcus aureus phenol-soluble modulin (PSM) peptide family.148,385 PSM peptides demonstrated the capacity to form amyloid-like fibrils, but adopted distinct secondary structures, resulting in diverse cell toxicities. IIKVIK from PSMα1 and IIKIIK from PSMα4 formed classical canonical cross-β amyloid fibrils, where pairs of β-sheets tightly interlocked through a dry interface, creating a steric zipper. In contrast, LFKFFK from PSMα3 self-assembled into novel cross-α fibrils, with α-helices stacking perpendicularly to the fibril axis. The structural disparities among PSMs correlate with distinct functionalities: PSMα3-formed α-helical fibrils exhibited the highest toxicity to human cells, while PSMα1- and PSMα4-formed β-structure fibrils conveyed minimal toxicity but enhanced biofilm formation. The new discovery of cross-α fibrillation introduces an innovative grasp of the polymorphic structure of amyloid-like fibrils and its influence on cell toxicity. Similarly, a collaboration between cryo-electron microscopy (cryo-EM) and computational modeling has yielded the atomic structure of cross-β fibrils of aurein 3.3. This structure reveals six kinked β-sheet chains arranged in an unconventional in-plane layer.152 The six-chain organization comprises two inner chains forming an overall S-shape, with two β-strand conformations within a single chain located in an inner area exhibiting C21 symmetry and four outer chains adopting a V-shape conformation wrapped around this inner cross in C41 symmetry. Such organization requires strong interlocking β-sheet interactions and precise geometric matching to create tightly staggered β-sheets in both lateral and fibrillar directions. Lastly, diverging from the exclusively β-sheet-rich amyloid-like fibrils formed by AMPs and the cross-α amyloid fibrils of PSMα3, human LL-3717–29 adopted a distinct supra-helical fibril structure with unique secondary structure features.155 LL-3717–29 fibrils are formed through the assembly of densely packed helices. In the lateral direction, the cross-section reveals six helices forming a hexameric structure with a central, hydrophobic pore. These helical fibrillar structures prevent LL-3717–29 from stacking on top of each other perpendicular to the fibril axis, and as a result, they do not bind to the amyloid indicator dye Thioflavin T. Nevertheless, LL-3717–29 fibrils exhibit remarkable thermal stability, retaining their structural integrity without disassembling even after exposure to an 80 °C heat shock.
Computational investigations into the antimicrobial activities of AMYs predominantly focus on their membrane disruption mechanisms. These mechanisms have been discussed in preceding sections and will not be reiterated here. These computational studies highlight the structure–function relationship between amyloid aggregation and antimicrobial activity for AMPs at the structural level. Various AMPs demonstrate the capability to self-assemble into amyloid fibrillar structures, each exhibiting distinct secondary structures and peptide organizations (cross-β sheets, cross-α sheets, α-helices), with specific functional roles in the eradication of bacterial cells.
6.2. Cross-seeding between antimicrobial peptides and amyloid peptides
Fundamentally, the tightly packed β-sheet configuration acts as a structural base and a template for interaction during amyloid cross-seeding. This high degree of structural resemblance helps diminish barriers across species, enhancing the binding and recognition between various species and thus encouraging the formation of heterogeneous amyloid assemblies. The only recent computational study married molecular docking and MD simulations to investigate the binding structures between the HNP-1 dimer and amyloid pentamers formed by Aβ and hIAPP.24 HNP-1 showed strong affinity for both the β-sheet and U-turn regions of Aβ and hIAPP pentamers. This dual binding mode indicates possible pathways through which HNP-1 inhibits amyloid formation. By binding to these regions, HNP-1 not only interferes with the lateral associations and extension processes of amyloid aggregation but also alters the primary and secondary structures of Aβ or hIAPP, enhancing its ability to inhibit amyloid formation. The interaction of HNP-1 with amyloid aggregates also highlighted specific interfacial residues and their preferences for binding. In the Aβ pentamer, HNP-1 strongly interacts with Leu17, Val18, Phe19, and Phe20 from the N-terminal β-sheet, and Ile32, Gly33, Leu34, Met35, Val36, Gly37, Gly38, and Val40 from the C-terminal β-sheet (Fig. 22a). In contrast, HNP-1 showed a preference for binding to Leu27, Ser29, and Asn31 from the C-terminal β-sheet of hIAPP (Fig. 22b). | ||
| Fig. 22 Computational exploration of cross-seeding between (a) HNP-1 and Aβ and (b) HNP-1 and hIAPP by MD simulations. HNP-1 demonstrates favorable bindings to both the β-sheet and U-turn regions of Aβ and hIAPP oligomers but exhibiting distinct residue binding preferences in each case. | ||
In contrast to the growing body of experimental findings revealing new alternative functions (e.g., plantaricin A, longipin, melittin, dermaseptin S9, magainin 2, temporin B, Indolicidin) and identifying AMP-AMY cross-seeding systems, there has been a limited number of computational simulations that follow or parallel these experimental investigations at nanoscale or atomic scale. The scarcity of computational studies may trace back to the fundamental challenges associated with protein (mis)folding and structural transitions during the aggregation process, whether involving the same or different peptides. Overcoming high energy barriers within long timescales, on the order of microseconds and beyond, is a crucial aspect that needs to be addressed in exploring these intricate phenomena by current computational approaches. Nonetheless, there is substantial potential for further computational studies in this area.
7. Conclusions and perspectives
AMYs and AMPs belong to distinct families in terms of sequence identity, structural characteristics, gene information, biological function, and pathological implications. However, recent studies revealed intriguing overlaps and interconnections between these two peptide families, hinting at a deeper pathological relationship. Despite their apparent differences, increasing evidence suggests that both AMYs and AMPs share certain structural and functional features, likely acquired through natural evolution. Both AMYs and AMPs exhibit a propensity to adopt β-sheet-rich structures upon aggregation and possess common membrane-disruption mechanisms. These shared characteristics, including peptide self-assembly, oligomerization, the presence of β-rich structures, and common modes of membrane interaction—such as the creation of membrane pores and membrane thinning—observed in both AMPs and AMYs, not only open the door for AMYs to acquire antimicrobial activity and AMPs to exhibit amyloidogenic properties, but also hold potential implications for other membrane-activating peptides and proteins. These common structure-function traits provide possible targets for designing universal defense strategies against viral and amyloid-related diseases. Although challenging to design, approaches such as stabilizing membranes, inhibiting pore formation through small drugs or peptides, and preventing peptide oligomerization or structural transitions toward β-structures could represent therapeutic strategies applicable across a spectrum of diseases, including emerging threats like COVID-19.More importantly, the cross-seeding phenomenon between AMYs and AMPs is found to play a significant role in the pathogenesis of neurodegenerative diseases and host defense against microbial infections. Cross-seeding involves mutual induction and propagation of peptide aggregation, leading to the formation of mixed amyloid complexes. This interplay is associated with the progression and exacerbation of neurodegenerative disorders, such as Alzheimer's, Parkinson's, and amyotrophic lateral sclerosis. Cross-seeding highlights the remarkable adaptive nature of AMYs and AMPs, allowing them to contribute to the innate immune response and combat microbial pathogens. Understanding the intricate connection between AMYs and AMPs through cross-seeding mechanisms will help uncover their roles in disease pathology and potential therapeutic interventions.
With only a few studies to date, exploration of the AMY-AMP connection may start with comparative studies that assess the antimicrobial activity of AMYs and the amyloidogenic properties of AMPs. From a bioinformatic perspective, several amyloid datasets, such as TANGO,47 ZipperDB,58 Waltz-DB2.0,386 Zyggregator,387 and PASTA2,388 have been developed for the identification of amyloid-like aggregation-prone regions in protein or peptide sequences. These datasets, primarily derived from amyloid sequences, offer valuable predictive information for the propensity of specific segments in amyloid proteins and the identification of hotspots within these sequences. However, the predictive accuracy and reliability of these datasets may be compromised when applied to non-amyloid proteins or sequences that do not originate from amyloidogenic regions. Given the increasingly complex pathological interplay between amyloid peptide aggregation and antimicrobial peptide activity, conducting comparative sequence analysis between membrane-activating peptides (e.g., cell penetrating peptides, glycopeptides, lipopeptides) and AMYs and AMPs would facilitate de novo peptide design approaches. This involves employing techniques such as high-throughput combinatorial library screening, structure–activity relationship modeling, and predictive algorithms, AI/ML models, with the integration of non-coded modifications, to advance both peptide chemistry and understanding of intricate mechanisms in protein-membrane interactions.
Despite the recent rapid advancements in data-driven artificial intelligence (AI) and machine learning (ML), there is a lack of comprehensive computational studies aimed at screening large antimicrobial datasets to identify AMPs with amyloidogenic properties, as well as amyloid datasets to identify AMYs with antimicrobial activity. Developing innovative data/model-driven deep learning algorithms will serve to facilitate the rational design of peptides or the repurposing of existing AMPs and AMYs with dual antimicrobial and amyloidogenic properties. Utilizing large datasets of AMPs and smaller datasets of AMYs, AI/ML-driven models would permit extracting valuable structure–property features, enabling the discovery and design of peptides with specific characteristics such as β-structure self-assembly sequences, membrane-disruption actions, and antimicrobial activity. The design outcomes, successful or failed, can be used as feedback to refine and optimize the AI/ML models through iterative training processes. This iterative approach ultimately enhances the efficiency of peptide design, leading to the development of highly effective and novel peptide candidates.
In addition to AI/ML modeling, the investigation of AMP-AMY cross-seeding remains crucial for understanding both host defense mechanisms and amyloid aggregation processes. Molecular simulations, including molecular docking and molecular dynamics, are powerful tools for identifying and exploring the binding modes between AMPs and AMYs at both atomic and coarse-grained levels. Simulations can provide useful information such as binding residues, binding sites, structural characteristics, affinity, and specificity of the peptide interactions. Computational mutagenesis techniques can further validate the key binding sequences, quaternary structures, and hotspot residues involved in these peptide interactions. Advancements in hardware technologies (e.g., GPU-based simulations) and innovative algorithms (e.g., graph theory) hold promise for mapping out the complex interaction patterns within specific AMP-AMY systems. By leveraging these computational approaches, researchers can gain a deeper understanding of the intricate interplay between AMPs and AMYs and shed light on their potential roles in host defense and amyloid-related diseases.
In addition to computational efforts, it is vital to conduct in vitro and in vivo experimental research to further explore the complementary functions of AMPs and AMYs. A key aspect to investigate is the self-assembly properties of both peptide families for determining the effective states of peptide monomers, oligomers, or fibrils for antimicrobial activity in the case of AMYs and for neurotoxicity towards neuron cells in the case of AMPs. These experimental studies will contribute to expanding the pool of datasets for AMPs and AMYs with dual anti-amyloid and antibacterial activities. This iterative process, which combine experimental validation with computationally informed design, would accelerate the discovery, development, and optimization of new self-assembled peptides, which could be further translated into sustainable and cost-effective therapeutic interventions.
Cross-seeding between these peptide families has significant biological importance and scientific interest. Aggregation through both acceleration and inhibition demonstrated the ability to reduce amyloid-induced cytotoxicity. The pathological implications of this relationship extend beyond the mere formation of amyloid fibrils. Mechanistically, cross-seeding between AMYs and AMPs contributes to the “microbial infection hypothesis” and the “neuroinflammation hypothesis”. The bidirectional communication between amyloid proteins and gut microbiota highlights the critical role of the brain-gut-microbiota axis in the pathogenesis of neurodegenerative diseases. In a broader perspective, it is important to investigate the potential cross-seeding interactions between specific pathogen-related proteins (e.g., viral capsid proteins, bacterial surface proteins, or fungal amyloid proteins) and amyloid peptides (e.g., Aβ, tau, hIAPP, α-synuclein, and prions), including their direct assembly, aggregation, and fibrillation. Unraveling the interplay between pathogen-related proteins and amyloid peptides holds the key to understanding the mechanisms underlying amyloid formation in infectious diseases and neurodegenerative disorders. Furthermore, by exploring the potential impact of these cross-seeding interactions on disease pathology, novel therapeutic targets and strategies for intervention may be uncovered, paving the way for future advancements in combating these debilitating conditions.
As briefly summarized from the above, AMPs and AMYs appear to be two sides of the same coin. Despite some progress, the connection between AMPs and AMYs is still unclear. Firstly, there is a lack of studies that explore the underlying mechanisms and interactions between these two peptide families. The precise molecular crosstalk and interplay between AMPs and AMYs are not yet fully understood. Secondly, the existing literature primarily focuses on individual aspects of either AMPs or AMYs, with limited emphasis on their overlapping properties and functional connections. This hinders a comprehensive understanding of the complex relationship between these peptides. Additionally, the available datasets for AMPs and AMYs are often limited in size and diversity, which restricts the accuracy and generalizability of predictive models. Further research could unravel this intricate connection. Endeavors will not only deepen our fundamental understanding of the intricate workings of the innate immune system and the pathogenesis of neurodegenerative diseases, but also hold great potentials for advancing therapeutic interventions and innovative approaches in both antimicrobial strategies and neurodegenerative disease treatments.
Declarations
Ethics approval and consent to participate: not applicable; consent for publication: yes; availability of data and materials: all data are available in the main text or the supplementary materials.Author contributions
The manuscript was written with contributions from all authors, who have all approved the final version.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There is no competing interests.Acknowledgements
We thank financial support from NSF-CBET-2107619. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and the Intramural Research Program of the NIH Clinical Center.References
- C. Soto and S. Pritzkow, Nat. Neurosci., 2018, 21, 1332–1340 CrossRef CAS PubMed.
- J. Gandhi, A. C. Antonelli, A. Afridi, S. Vatsia, G. Joshi, V. Romanov, I. V. J. Murray and S. A. Khan, Rev. Neurosci., 2019, 30, 339–358 CrossRef.
- A. Gamez, P. Yuste-Checa, S. Brasil, A. Briso-Montiano, L. R. Desviat, M. Ugarte, C. Perez-Cerda and B. Perez, Clin. Genet., 2018, 93, 450–458 CrossRef CAS.
- P. H. Nguyen, A. Ramamoorthy, B. R. Sahoo, J. Zheng, P. Faller, J. E. Straub, L. Dominguez, J. E. Shea, N. V. Dokholyan, A. De Simone, B. Ma, R. Nussinov, S. Najafi, S. T. Ngo, A. Loquet, M. Chiricotto, P. Ganguly, J. McCarty, M. S. Li, C. Hall, Y. Wang, Y. Miller, S. Melchionna, B. Habenstein, S. Timr, J. Chen, B. Hnath, B. Strodel, R. Kayed, S. Lesne, G. Wei, F. Sterpone, A. J. Doig and P. Derreumaux, Chem. Rev., 2021, 121, 2545–2647 CrossRef CAS.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184–185 CrossRef CAS PubMed.
- V. Baltutis, P. D. O'Leary and L. L. Martin, Chempluschem, 2022, 87, e202200240 CrossRef CAS PubMed.
- B. L. Kagan, H. Jang, R. Capone, F. Teran Arce, S. Ramachandran, R. Lal and R. Nussinov, Mol. Pharm., 2012, 9, 708–717 CrossRef CAS.
- H. Jang, F. T. Arce, M. Mustata, S. Ramachandran, R. Capone, R. Nussinov and R. Lal, Biophys. J., 2011, 100, 1775–1783 CrossRef CAS PubMed.
- H. Zhao, R. Sood, A. Jutila, S. Bose, G. Fimland, J. Nissen-Meyer and P. K. Kinnunen, Biochim. Biophys. Acta, 2006, 1758, 1461–1474 CrossRef CAS PubMed.
- A. N. Calabrese, Y. Liu, T. Wang, I. F. Musgrave, T. L. Pukala, R. F. Tabor, L. L. Martin, J. A. Carver and J. H. Bowie, Chembiochem, 2016, 17, 239–246 CrossRef CAS PubMed.
- R. Urrutia, R. A. Cruciani, J. L. Barker and B. Kachar, FEBS Lett., 1989, 247, 17–21 CrossRef CAS.
- D. W. Juhl, E. Glattard, M. Lointier, P. Bampilis and B. Bechinger, Front. Cell Infect. Microbiol., 2020, 10, 526459 CrossRef CAS PubMed.
- L. Caillon, J. A. Killian, O. Lequin and L. Khemtemourian, PLoS One, 2013, 8, e75528 CrossRef CAS.
- L. Wang, Q. Liu, J.-C. Chen, Y.-X. Cui, B. Zhou, Y.-X. Chen, Y.-F. Zhao and Y.-M. J. B. C. Li, Biol. Chem., 2012, 393, 641–646 CrossRef CAS.
- S. C. Park, J. C. Moon, S. Y. Shin, H. Son, Y. J. Jung, N. H. Kim, Y. M. Kim, M. K. Jang and J. R. Lee, Biochem. Biophys. Res. Commun., 2016, 478, 924–928 CrossRef CAS.
- H. Zheng, H. Li, J. Zhang, H. Fan, L. Jia, W. Ma, S. Ma, S. Wang, H. You, Z. Yin and X. Li, J. Biol. Chem., 2020, 295, 2570–2581 CrossRef CAS PubMed.
- S. J. Soscia, J. E. Kirby, K. J. Washicosky, S. M. Tucker, M. Ingelsson, B. Hyman, M. A. Burton, L. E. Goldstein, S. Duong and R. E. J. P. O. Tanzi, PloS one, 2010, 5, e9505 CrossRef PubMed.
- N. B. Last and A. D. Miranker, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6382–6387 CrossRef CAS.
- M. L. Gosztyla, H. M. Brothers and S. R. Robinson, J. Alzheimers Dis., 2018, 62, 1495–1506 CAS.
- U. H. Iqbal, E. Zeng and G. M. Pasinetti, Int. J. Mol. Sci., 2020, 21, 4920 CrossRef CAS.
- K. Bourgade, H. Garneau, G. Giroux, A. Y. Le Page, C. Bocti, G. Dupuis, E. H. Frost and T. Fulop, Jr., Biogerontology, 2015, 16, 85–98 CrossRef CAS.
- M. Zhang, J. Zhao and J. Zheng, Soft Matter, 2014, 10, 7425–7451 RSC.
- Y. Tang, D. Zhang, X. Gong and J. Zheng, Chem. Sci., 2022, 13, 7143–7156 RSC.
- Y. Zhang, Y. Liu, Y. Tang, D. Zhang, H. He, J. Wu and J. Zheng, Chem. Sci., 2021, 12, 9124–9139 RSC.
- E. De Lorenzi, M. Chiari, R. Colombo, M. Cretich, L. Sola, R. Vanna, P. Gagni, F. Bisceglia, C. Morasso and J. S. Lin, J. Alzheimer's Dis., 2017, 59, 1213–1226 CAS.
- V. Armiento, K. Hille, D. Naltsas, J. S. Lin, A. E. Barron and A. Kapurniotu, Angew. Chem., Int. Ed., 2020, 59, 12837–12841 Search PubMed.
- T. R. Sampson, C. Challis, N. Jain, A. Moiseyenko, M. S. Ladinsky, G. G. Shastri, T. Thron, B. D. Needham, I. Horvath, J. W. Debelius, S. Janssen, R. Knight, P. Wittung-Stafshede, V. Gradinaru, M. Chapman and S. K. Mazmanian, Elife, 2020, 9, e53111 CrossRef CAS.
- E. Chorell, E. Andersson, M. L. Evans, N. Jain, A. Gotheson, J. Aden, M. R. Chapman, F. Almqvist and P. Wittung-Stafshede, PLoS One, 2015, 10, e0140194 CrossRef PubMed.
- M. L. Evans, E. Chorell, J. D. Taylor, J. Aden, A. Gotheson, F. Li, M. Koch, L. Sefer, S. J. Matthews, P. Wittung-Stafshede, F. Almqvist and M. R. Chapman, Mol. Cell, 2015, 57, 445–455 CrossRef CAS.
- B. Ma and R. Nussinov, J. Mol. Biol., 2012, 421, 172–184 CrossRef CAS.
- B. Ren, Y. Zhang, M. Zhang, Y. Liu, D. Zhang, X. Gong, Z. Feng, J. Tang, Y. Chang and J. Zheng, J. Mater. Chem. B, 2019, 7, 7267–7282 RSC.
- E. A. Newcombe, J. Camats-Perna, M. L. Silva, N. Valmas, T. J. Huat and R. Medeiros, J. Neuroinflammation, 2018, 15, 1–26 CrossRef.
- K. Kowalski and A. Mulak, J. Neurogastroenterol. Motil., 2019, 25, 48–60 CrossRef.
- I. Javed, Z. Zhang, J. Adamcik, N. Andrikopoulos, Y. Li, D. E. Otzen, S. Lin, R. Mezzenga, T. P. Davis, F. Ding and P. C. Ke, Adv. Sci., 2020, 7, 2001299 CrossRef CAS.
- Y. Tang, D. Zhang, X. Gong and J. Zheng, Biophys. Chem., 2022, 281, 106735 CrossRef CAS PubMed.
- K. Lundmark, G. T. Westermark, A. Olsén and P. Westermark, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6098–6102 CrossRef CAS.
- Z. Wang and G. Wang, Nucleic Acids Res., 2004, 32, D590–592 CrossRef CAS PubMed.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2009, 37, D933–937 CrossRef CAS PubMed.
- S. Thomas, S. Karnik, R. S. Barai, V. K. Jayaraman and S. Idicula-Thomas, Nucleic Acids Res., 2010, 38, D774–780 CrossRef CAS.
- P. G. A. Aronica, L. M. Reid, N. Desai, J. Li, S. J. Fox, S. Yadahalli, J. W. Essex and C. S. Verma, J. Chem. Inf. Model., 2021, 61, 3172–3196 CrossRef CAS.
- L. T. Nguyen, E. F. Haney and H. J. Vogel, Trends Biotechnol., 2011, 29, 464–472 CrossRef CAS PubMed.
- R. Mani, S. D. Cady, M. Tang, A. J. Waring, R. I. Lehrer and M. Hong, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16242–16247 CrossRef CAS.
- R. I. Lehrer and T. Ganz, Curr. Opin. Immunol., 1999, 11, 23–27 CrossRef CAS PubMed.
- M. Stefani, FEBS J, 2010, 277, 4602–4613 CrossRef CAS PubMed.
- H. H. Tsai, K. Gunasekaran and R. Nussinov, Structure, 2006, 14, 1059–1072 CrossRef CAS PubMed.
- B. H. Gan, J. Gaynord, S. M. Rowe, T. Deingruber and D. R. Spring, Chem. Soc. Rev., 2021, 50, 7820–7880 RSC.
- A. M. Fernandez-Escamilla, F. Rousseau, J. Schymkowitz and L. Serrano, Nat. Biotechnol., 2004, 22, 1302–1306 CrossRef CAS PubMed.
- K. Matsuzaki, S. Yoneyama, O. Murase and K. Miyajima, Biochemistry, 1996, 35, 8450–8456 CrossRef CAS.
- M. P. Cabrera, D. S. Alvares, N. B. Leite, B. M. de Souza, M. S. Palma, K. A. Riske and J. R. Neto, Langmuir, 2011, 27, 10805–10813 CrossRef.
- M. I. Ivanova, M. J. Thompson and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4079–4082 CrossRef CAS.
- M. Castle, A. Nazarian, S. S. Yi and P. Tempst, J. Biol. Chem., 1999, 274, 32555–32564 CrossRef CAS.
- J. Portelinha, S. S. Duay, S. I. Yu, K. Heilemann, M. D. J. Libardo, S. A. Juliano, J. L. Klassen and A. M. Angeles-Boza, Chem. Rev., 2021, 121, 2648–2712 CrossRef CAS PubMed.
- C. B. Park, H. S. Kim and S. C. Kim, Biochem. Biophys. Res. Commun., 1998, 244, 253–257 CrossRef CAS.
- T. J. Falla, D. N. Karunaratne and R. E. Hancock, J. Biol. Chem., 1996, 271, 19298–19303 CrossRef CAS.
- M. B. Ulmschneider, M. S. Sansom and A. D. Nola, Biophys. J., 2006, 90, 1650–1660 CrossRef CAS PubMed.
- M. Torrent, J. Valle, M. V. Nogues, E. Boix and D. Andreu, Angew. Chem., Int. Ed., 2011, 50, 10686–10689 CrossRef CAS.
- L. Jin, X. Bai, N. Luan, H. Yao, Z. Zhang, W. Liu, Y. Chen, X. Yan, M. Rong, R. Lai and Q. Lu, J. Med. Chem., 2016, 59, 1791–1799 CrossRef CAS PubMed.
- M. J. Thompson, S. A. Sievers, J. Karanicolas, M. I. Ivanova, D. Baker and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4074–4078 CrossRef CAS.
- J. Zheng, B. Ma, C. J. Tsai and R. Nussinov, Biophys. J., 2006, 91, 824–833 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS.
- A. T. Petkova, R. D. Leapman, Z. Guo, W. M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262–265 CrossRef CAS.
- N. F. Dupuis, C. Wu, J. E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2009, 131, 18283–18292 CrossRef CAS.
- M. Sunde, L. C. Serpell, M. Bartlam, P. E. Fraser, M. B. Pepys and C. C. Blake, J. Mol. Biol., 1997, 273, 729–739 CrossRef CAS.
- K. T. Nguyen, J. T. King and Z. Chen, J. Phys. Chem. B, 2010, 114, 8291–8300 CrossRef CAS.
- S. H. White, W. C. Wimley and M. E. Selsted, Curr. Opin. Struct. Biol., 1995, 5, 521–527 CrossRef CAS PubMed.
- S. Troeira Henriques, N. Lawrence, S. Chaousis, A. S. Ravipati, O. Cheneval, A. H. Benfield, A. G. Elliott, A. M. Kavanagh, M. A. Cooper, L. Y. Chan, Y. H. Huang and D. J. Craik, ACS Chem. Biol., 2017, 12, 2324–2334 CrossRef CAS PubMed.
- C. P. Hill, J. Yee, M. E. Selsted and D. Eisenberg, Science, 1991, 251, 1481–1485 CrossRef CAS PubMed.
- G. Guigas and M. Weiss, Biophys. J., 2006, 91, 2393–2398 CrossRef CAS PubMed.
- A. Zemel, A. Ben-Shaul and S. May, Biophys. J., 2004, 86, 3607–3619 CrossRef CAS PubMed.
- G. Wang, J. Biol. Chem., 2008, 283, 32637–32643 CrossRef CAS.
- A. S. Ladokhin and S. H. White, J. Mol. Biol., 1999, 285, 1363–1369 CrossRef CAS PubMed.
- K. Matsuzaki, K. Sugishita, M. Harada, N. Fujii and K. Miyajima, Biochim. Biophys. Acta, 1997, 1327, 119–130 CrossRef CAS PubMed.
- S. Ludtke, K. He and H. Huang, Biochemistry, 1995, 34, 16764–16769 CrossRef CAS.
- S. M. Gregory, A. Cavenaugh, V. Journigan, A. Pokorny and P. F. Almeida, Biophys. J., 2008, 94, 1667–1680 CrossRef CAS PubMed.
- D. Andreu, R. B. Merrifield, H. Steiner and H. G. Boman, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 6475–6479 CrossRef CAS.
- C. Auvynet, C. El Amri, C. Lacombe, F. Bruston, J. Bourdais, P. Nicolas and Y. Rosenstein, FEBS J, 2008, 275, 4134–4151 CrossRef CAS PubMed.
- S. Malekkhaiat Häffner and M. Malmsten, Curr. Opin. Colloid Interface Sci., 2018, 38, 56–79 CrossRef.
- Y. Tang, D. Zhang, Y. Zhang, Y. Liu, X. Gong, Y. Chang, B. Ren and J. Zheng, ACS Appl. Bio Mater., 2020, 3, 8286–8308 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Chem. Rev., 2010, 110, 4820–4838 CrossRef CAS.
- A. A. Strömstedt, L. Ringstad, A. Schmidtchen and M. Malmsten, Curr. Opin. Colloid Interface Sci., 2010, 15, 467–478 CrossRef.
- S. M. Butterfield and H. A. Lashuel, Angew. Chem., Int. Ed., 2010, 49, 5628–5654 CrossRef CAS PubMed.
- M. Zhang, B. Ren, H. Chen, Y. Sun, J. Ma, B. Jiang and J. Zheng, Israel J. Chem., 2017, 57, 586–601 CrossRef CAS.
- E. Gazit, I. R. Miller, P. C. Biggin, M. S. Sansom and Y. Shai, J. Mol. Biol., 1996, 258, 860–870 CrossRef CAS.
- L. Chen, X. Li, L. Gao and W. Fang, J. Phys. Chem. B, 2015, 119, 850–860 CrossRef CAS.
- H. Bouvrais, P. Meleard, T. Pott, K. J. Jensen, J. Brask and J. H. Ipsen, Biophys. Chem., 2008, 137, 7–12 CrossRef CAS PubMed.
- Y. Pouny, D. Rapaport, A. Mor, P. Nicolas and Y. Shai, Biochemistry, 1992, 31, 12416–12423 CrossRef CAS PubMed.
- S. Kobayashi, K. Takeshima, C. B. Park, S. C. Kim and K. Matsuzaki, Biochemistry, 2000, 39, 8648–8654 CrossRef CAS.
- E. E. Ambroggio, F. Separovic, J. H. Bowie, G. D. Fidelio and L. A. Bagatolli, Biophys. J., 2005, 89, 1874–1881 CrossRef CAS.
- M. Monette and M. Lafleur, Biophys. J., 1995, 68, 187–195 CrossRef CAS PubMed.
- A. B. Reiss, H. A. Arain, M. M. Stecker, N. M. Siegart and L. J. Kasselman, Rev. Neurosci., 2018, 29, 613–627 CAS.
- A. K. Bishoyi, P. H. Roham, K. Rachineni, S. Save, M. A. Hazari, S. Sharma and A. Kumar, Biol. Chem., 2021, 402, 133–153 CAS.
- Y. C. Wong and D. Krainc, Nat. Med., 2017, 23, 1–13 CrossRef CAS PubMed.
- A. Aguzzi and J. Falsig, Nat. Neurosci., 2012, 15, 936–939 CrossRef CAS.
- I. R. Mellor and M. S. Sansom, Proc. R. Soc. London, Ser. B, 1990, 239, 383–400 CAS.
- L. Beven, S. Castano, J. Dufourcq, A. Wieslander and H. Wroblewski, Eur. J. Biochem., 2003, 270, 2207–2217 CrossRef CAS.
- Y. Higashimoto, H. Kodama, M. Jelokhani-Niaraki, F. Kato and M. Kondo, J. Biochem., 1999, 125, 705–712 CrossRef CAS.
- R. G. Panchal, M. L. Smart, D. N. Bowser, D. A. Williams and S. Petrou, Curr. Pharm. Biotechnol., 2002, 3, 99–115 CAS.
- R. Capone, F. G. Quiroz, P. Prangkio, I. Saluja, A. M. Sauer, M. R. Bautista, R. S. Turner, J. Yang and M. Mayer, Neurotox Res, 2009, 16, 1–13 CrossRef CAS PubMed.
- R. Lal, H. Lin and A. P. Quist, Biochim. Biophys. Acta, 2007, 1768, 1966–1975 CrossRef CAS.
- H. Jang, J. Zheng, R. Lal and R. Nussinov, Trends Biochem. Sci., 2008, 33, 91–100 CrossRef CAS.
- N. Arispe, J. C. Diaz and O. Simakova, Biochim. Biophys. Acta, 2007, 1768, 1952–1965 CrossRef CAS PubMed.
- B. Leitgeb, A. Szekeres, L. Manczinger, C. Vagvolgyi and L. Kredics, Chem. Biodiversity, 2007, 4, 1027–1051 CrossRef CAS.
- Y. Bessin, N. Saint, L. Marri, D. Marchini and G. Molle, Biochim. Biophys. Acta, 2004, 1667, 148–156 CrossRef CAS.
- D. Raimondo, G. Andreotti, N. Saint, P. Amodeo, G. Renzone, M. Sanseverino, I. Zocchi, G. Molle, A. Motta and A. Scaloni, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6309–6314 CrossRef CAS.
- H. Jang, F. T. Arce, S. Ramachandran, R. Capone, R. Lal and R. Nussinov, J. Mol. Biol., 2010, 404, 917–934 CrossRef CAS PubMed.
- J. Zhao, Y. Luo, H. Jang, X. Yu, G. Wei, R. Nussinov and J. Zheng, Biochim. Biophys. Acta, 2012, 1818, 3121–3130 CrossRef CAS.
- L. Y. Tan, K. H. Tang, L. Y. Y. Lim, J. X. Ong, H. Park and S. Jung, Biomolecules, 2022, 12, 507 CrossRef CAS PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS.
- N. Arispe, H. B. Pollard and E. Rojas, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10573–10577 CrossRef CAS.
- P. Walsh, G. Vanderlee, J. Yau, J. Campeau, V. L. Sim, C. M. Yip and S. Sharpe, J. Biol. Chem., 2014, 289, 10419–10430 CrossRef CAS PubMed.
- X. Li, M. Wan, L. Gao and W. Fang, Sci. Rep., 2016, 6, 21614 CrossRef CAS.
- H. Jang, J. Zheng and R. Nussinov, Biophys. J., 2007, 93, 1938–1949 CrossRef CAS PubMed.
- H. Jang, F. T. Arce, R. Capone, S. Ramachandran, R. Lal and R. Nussinov, Biophys. J., 2009, 97, 3029–3037 CrossRef CAS PubMed.
- H. Jang, F. Teran Arce, S. Ramachandran, R. Capone, R. Lal and R. Nussinov, J. Phys. Chem. B, 2010, 114, 9445–9451 CrossRef CAS.
- L. Connelly, H. Jang, F. T. Arce, R. Capone, S. A. Kotler, S. Ramachandran, B. L. Kagan, R. Nussinov and R. Lal, J. Phys. Chem. B, 2012, 116, 1728–1735 CrossRef CAS PubMed.
- A. Quist, I. Doudevski, H. Lin, R. Azimova, D. Ng, B. Frangione, B. Kagan, J. Ghiso and R. Lal, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10427–10432 CrossRef CAS.
- T. A. Mirzabekov, M. C. Lin and B. L. Kagan, J. Biol. Chem., 1996, 271, 1988–1992 CrossRef CAS.
- H. A. Lashuel, B. M. Petre, J. Wall, M. Simon, R. J. Nowak, T. Walz and J. P. T. Lansbury, J. Mol. Biol., 2002, 322, 1089–1102 CrossRef CAS PubMed.
- Y. Hirakura, I. Carreras, J. D. Sipe and B. L. Kagan, Amyloid, 2002, 9, 13–23 CrossRef CAS.
- L. Wang, H. A. Lashuel, T. Walz and W. Colon, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15947–15952 CrossRef CAS.
- M. Mustata, R. Capone, H. Jang, F. T. Arce, S. Ramachandran, R. Lal and R. Nussinov, J. Am. Chem. Soc., 2009, 131, 14938–14945 CrossRef CAS PubMed.
- Y. Hirakura and B. L. Kagan, Amyloid, 2001, 8, 94–100 CrossRef CAS.
- H. Jang, B. Ma, R. Lal and R. Nussinov, Biophys. J., 2008, 95, 4631–4642 CrossRef CAS PubMed.
- R. Capone, M. Mustata, H. Jang, F. T. Arce, R. Nussinov and R. Lal, Biophys. J., 2010, 98, 2644–2652 CrossRef CAS.
- J. H. Viles, Angew. Chem., Int. Ed., 2023, 62, e202215785 CrossRef CAS.
- D. Gimenez, O. L. Sanchez-Munoz and J. Salgado, Langmuir, 2015, 31, 3146–3158 CrossRef CAS.
- P. D. Rakowska, H. Jiang, S. Ray, A. Pyne, B. Lamarre, M. Carr, P. J. Judge, J. Ravi, U. I. Gerling, B. Koksch, G. J. Martyna, B. W. Hoogenboom, A. Watts, J. Crain, C. R. Grovenor and M. G. Ryadnov, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8918–8923 CrossRef CAS.
- F. Jiao, F. Dehez, T. Ni, X. Yu, J. S. Dittman, R. Gilbert, C. Chipot and S. Scheuring, Nat. Commun., 2022, 13, 5039 CrossRef CAS.
- A. W. Hodel, J. A. Rudd-Schmidt, J. A. Trapani, I. Voskoboinik and B. W. Hoogenboom, Faraday Discuss., 2021, 232, 236–255 RSC.
- D. Sun, J. Forsman and C. E. Woodward, Langmuir, 2015, 31, 752–761 CrossRef CAS.
- A. A. Langham, A. S. Ahmad and Y. N. Kaznessis, J. Am. Chem. Soc., 2008, 130, 4338–4346 CrossRef CAS PubMed.
- R. Talandashti, F. Mehrnejad, K. Rostamipour, F. Doustdar and A. Lavasanifar, J. Phys. Chem. B, 2021, 125, 7163–7176 CrossRef CAS PubMed.
- F. A. Daison, N. Kumar, S. Balakrishnan, K. Venugopal, S. Elango and P. Sokkar, J. Chem. Inf. Model, 2022, 62, 40–48 CrossRef CAS.
- R. Pokhrel, N. Bhattarai, P. Baral, B. S. Gerstman, J. H. Park, M. Handfield and P. P. Chapagain, Phys. Chem. Chem. Phys., 2019, 21, 12530–12539 RSC.
- A. Catte, M. R. Wilson, M. Walker and V. S. Oganesyan, Soft Matter, 2018, 14, 2796–2807 RSC.
- M. Bhattacharya, N. Jain, P. Dogra, S. Samai and S. Mukhopadhyay, J. Phys. Chem. Lett., 2013, 4, 480–485 CrossRef CAS.
- L. A. Morozova-Roche, V. Zamotin, M. Malisauskas, A. Ohman, R. Chertkova, M. A. Lavrikova, I. A. Kostanyan, D. A. Dolgikh and M. P. Kirpichnikov, Biochemistry, 2004, 43, 9610–9619 CrossRef CAS PubMed.
- H. Jang, F. T. Arce, S. Ramachandran, B. L. Kagan, R. Lal and R. Nussinov, J. Phys. Chem. B, 2013, 117, 11518–11529 CrossRef CAS PubMed.
- S. Younger, H. Jang, H. A. Davies, M. J. Niemiec, J. G. N. Garcia, R. Nussinov, R. Q. Migrino, J. Madine and F. T. Arce, Biophys. J., 2020, 118, 2769–2782 CrossRef CAS PubMed.
- Y. Yang, H. Distaffen, S. Jalali, A. J. Nieuwkoop, B. L. Nilsson and C. L. Dias, ACS Chem. Neurosci., 2022, 13, 2766–2777 CrossRef CAS.
- M. Maurer and T. Lazaridis, J. Chem. Inf. Model, 2023, 63, 7171–7179 CrossRef CAS PubMed.
- S. Luca, W. M. Yau, R. Leapman and R. Tycko, Biochemistry, 2007, 46, 13505–13522 CrossRef CAS PubMed.
- T. Luhrs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef CAS.
- K. Iwata, T. Fujiwara, Y. Matsuki, H. Akutsu, S. Takahashi, H. Naiki and Y. Goto, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18119–18124 CrossRef CAS.
- N. Arispe, H. B. Pollard and E. Rojas, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1710–1715 CrossRef CAS PubMed.
- Y. Hirakura, W. W. Yiu, A. Yamamoto and B. L. Kagan, Amyloid, 2000, 7, 194–199 CrossRef CAS.
- H. Jang, F. T. Arce, S. Ramachandran, R. Capone, R. Azimova, B. L. Kagan, R. Nussinov and R. Lal, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6538–6543 CrossRef CAS.
- E. Tayeb-Fligelman, N. Salinas, O. Tabachnikov and M. Landau, Structure, 2020, 28(301–313), e306 Search PubMed.
- R. S. Sayegh, I. F. Batista, R. L. Melo, K. A. Riske, S. Daffre, G. Montich and P. I. da Silva Jr, PLoS One, 2016, 11, e0167953 CrossRef PubMed.
- P. K. Singh, D. Ghosh, D. Tewari, G. M. Mohite, E. Carvalho, N. N. Jha, R. S. Jacob, S. Sahay, R. Banerjee, A. K. Bera and S. K. Maji, PLoS One, 2015, 10, e0120346 CrossRef PubMed.
- R. Sood, Y. Domanov and P. K. Kinnunen, J. Fluoresc., 2007, 17, 223–234 CrossRef CAS PubMed.
- R. Bucker, C. Seuring, C. Cazey, K. Veith, M. Garcia-Alai, K. Grunewald and M. Landau, Nat. Commun., 2022, 13, 4356 CrossRef PubMed.
- Y. Tang, D. Zhang, X. Gong and J. Zheng, J. Mater. Chem. B, 2023, 11, 7920–7932 RSC.
- S. Ray, S. Holden, A. K. Prasad, L. L. Martin and A. S. Panwar, J. Phys. Chem. B, 2020, 124, 11659–11670 CrossRef CAS PubMed.
- Y. Engelberg and M. Landau, Nat. Commun., 2020, 11, 3894 CrossRef CAS PubMed.
- Y. Tang, D. Zhang and J. Zheng, ACS Chem. Neurosci., 2023, 14, 3143–3155 CrossRef CAS.
- Y. Zhang, Y. Tang, Y. Liu, D. Zhang and J. Zheng, Adv. Funct. Mater., 2021, 31, 2102978 CrossRef CAS.
- S. K. Maji, M. H. Perrin, M. R. Sawaya, S. Jessberger, K. Vadodaria, R. A. Rissman, P. S. Singru, K. P. Nilsson, R. Simon, D. Schubert, D. Eisenberg, J. Rivier, P. Sawchenko, W. Vale and R. Riek, Science, 2009, 325, 328–332 CrossRef CAS PubMed.
- S. L. Gras, A. K. Tickler, A. M. Squires, G. L. Devlin, M. A. Horton, C. M. Dobson and C. E. MacPhee, Biomaterials, 2008, 29, 1553–1562 CrossRef CAS PubMed.
- H. Sakai, K. Watanabe, Y. Asanomi, Y. Kobayashi, Y. Chuman, L. Shi, T. Masuda, T. Wyttenbach, M. T. Bowers, K. Uosaki and K. Sakaguchi, Adv. Funct. Mater., 2013, 23, 4881–4887 CrossRef CAS.
- D. Li, E. M. Jones, M. R. Sawaya, H. Furukawa, F. Luo, M. Ivanova, S. A. Sievers, W. Wang, O. M. Yaghi, C. Liu and D. S. Eisenberg, J. Am. Chem. Soc., 2014, 136, 18044–18051 CrossRef CAS PubMed.
- D. Chen, X. Liu, Y. Chen and H. Lin, Appl. Microbiol. Biotechnol., 2022, 106, 7711–7720 CrossRef CAS.
- M. Pasupuleti, M. Roupe, V. Rydengard, K. Surewicz, W. K. Surewicz, A. Chalupka, M. Malmsten, O. E. Sorensen and A. Schmidtchen, PLoS One, 2009, 4, e7358 CrossRef PubMed.
- R. Hari-Dass, C. Shah, D. J. Meyer and J. G. Raynes, J. Biol. Chem., 2005, 280, 18562–18567 CrossRef CAS PubMed.
- X. Zhan, B. Stamova, L. W. Jin, C. DeCarli, B. Phinney and F. R. Sharp, Neurology, 2016, 87, 2324–2332 CrossRef CAS.
- D. C. Emery, D. K. Shoemark, T. E. Batstone, C. M. Waterfall, J. A. Coghill, T. L. Cerajewska, M. Davies, N. X. West and S. J. Allen, Front. Aging Neurosci., 2017, 9, 195 CrossRef PubMed.
- E. Y. Lee, Y. Srinivasan, J. de Anda, L. K. Nicastro, C. Tukel and G. C. L. Wong, Front. Immunol., 2020, 11, 1629 CrossRef CAS PubMed.
- M. I. Ivanova, Y. Lin, Y. H. Lee, J. Zheng and A. Ramamoorthy, Biophys. Chem., 2021, 269, 106507 CrossRef CAS.
- O. Tavassoly, F. Safavi and I. Tavassoly, ACS Chem. Neurosci., 2020, 11, 3704–3706 CrossRef CAS PubMed.
- S. A. Semerdzhiev, M. A. A. Fakhree, I. Segers-Nolten, C. Blum and M. Claessens, ACS Chem. Neurosci., 2022, 13, 143–150 CrossRef CAS PubMed.
- A. K. Jana, A. B. Greenwood and U. H. E. Hansmann, J. Phys. Chem. B, 2021, 125, 9155–9167 CrossRef CAS PubMed.
- D. D. Li Puma, R. Piacentini, L. Leone, K. Gironi, M. E. Marcocci, G. De Chiara, A. T. Palamara and C. Grassi, Stem Cells, 2019, 37, 1467–1480 CrossRef CAS PubMed.
- M. Ortega and B. M. Ances, J. Neuroimmune Pharmacol., 2014, 9, 483–491 CrossRef.
- D. Bortolotti, V. Gentili, A. Rotola, E. Caselli and R. Rizzo, Alzheimers Res Ther, 2019, 11, 104 CrossRef CAS PubMed.
- F. Pistollato, S. Sumalla Cano, I. Elio, M. Masias Vergara, F. Giampieri and M. Battino, Nutr. Rev., 2016, 74, 624–634 CrossRef PubMed.
- C. Roubaud-Baudron, P. Krolak-Salmon, I. Quadrio, F. Megraud and N. Salles, Neurobiol. Aging, 2012, 33(1009), e1011–1009 Search PubMed.
- C. J. Hammond, L. R. Hallock, R. J. Howanski, D. M. Appelt, C. S. Little and B. J. Balin, BMC Neurosci., 2010, 11, 121 CrossRef.
- D. Vigasova, M. Nemergut, B. Liskova and J. Damborsky, Microb. Cell Fact., 2021, 20, 25 CrossRef CAS.
- Y. Tang, D. Zhang, Y. Zhang, Y. Liu, Y. Miller, K. Gong and J. Zheng, Commun. Biol., 2022, 5, 417 CrossRef CAS.
- I. Blasko, R. Veerhuis, M. Stampfer-Kountchev, M. Saurwein-Teissl, P. Eikelenboom and B. Grubeck-Loebenstein, Neurobiol. Dis., 2000, 7, 682–689 CrossRef CAS PubMed.
- J. W. Kinney, S. M. Bemiller, A. S. Murtishaw, A. M. Leisgang, A. M. Salazar and B. T. Lamb, Alzheimer's Dementia: Transl. Res. Clin. Interventions, 2018, 4, 575–590 CrossRef.
- Z. Wang, P. Vilekar, J. Huang and D. F. Weaver, ACS Chem. Neurosci., 2020, 11, 4152–4168 CrossRef CAS PubMed.
- P. M. Hwang, N. Zhou, X. Shan, C. H. Arrowsmith and H. J. Vogel, Biochemistry, 1998, 37, 4288–4298 CrossRef CAS.
- H. Zhao, A. Jutila, T. Nurminen, S. A. Wickstrom, J. Keski-Oja and P. K. Kinnunen, Biochemistry, 2005, 44, 2857–2863 CrossRef CAS PubMed.
- H. Chu, M. Pazgier, G. Jung, S.-P. Nuccio, P. A. Castillo, M. F. de Jong, M. G. Winter, S. E. Winter, J. Wehkamp, B. Shen, N. H. Salzman, M. A. Underwood, R. M. Tsolis, G. M. Young, W. Lu, R. I. Lehrer, A. J. Bäumler and C. L. Bevins, Science, 2012, 337, 477–481 CrossRef CAS PubMed.
- M. Torrent, F. Odorizzi, M. V. Nogues and E. Boix, Biomacromolecules, 2010, 11, 1983–1990 CrossRef CAS.
- R. Gossler-Schofberger, G. Hesser, M. M. Reif, J. Friedmann, B. Duscher, J. L. Toca-Herrera, C. Oostenbrink and A. Jilek, Arch. Biochem. Biophys., 2012, 522, 100–106 CrossRef.
- R. Gossler-Schofberger, G. Hesser, M. Muik, C. Wechselberger and A. Jilek, FEBS J, 2009, 276, 5849–5859 CrossRef.
- N. Salinas, E. Tayeb-Fligelman, M. D. Sammito, D. Bloch, R. Jelinek, D. Noy, I. Usón and M. Landau, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2014442118 CrossRef CAS.
- P. Ragonis-Bachar, B. Rayan, E. Barnea, Y. Engelberg, A. Upcher and M. Landau, Biomacromolecules, 2022, 23, 3713–3727 CrossRef CAS PubMed.
- J. Davagnino, M. Herrero, D. Furlong, F. Moreno and R. Kolter, Proteins: Struct., Funct., Bioinf., 1986, 1, 230–238 CrossRef CAS.
- S. Bieler, L. Estrada, R. Lagos, M. Baeza, J. Castilla and C. Soto, J. Biol. Chem., 2005, 280, 26880–26885 CrossRef CAS PubMed.
- S. Gour, V. Kumar, M. Rana and J. K. Yadav, J. Pept. Sci., 2019, 25, e3178 CrossRef.
- G. Fimland, J. Pirneskoski, J. Kaewsrichan, A. Jutila, P. E. Kristiansen, P. K. Kinnunen and J. Nissen-Meyer, Biochim. Biophys. Acta, 2006, 1764, 1132–1140 CrossRef CAS PubMed.
- A. Stern, W. A. Gibbons and L. C. Craig, Proc. Natl. Acad. Sci. U. S. A., 1968, 61, 734–741 CrossRef CAS.
- G. Munyuki, G. E. Jackson, G. A. Venter, K. E. Kover, L. Szilagyi, M. Rautenbach, B. M. Spathelf, B. Bhattacharya and D. van der Spoel, Biochemistry, 2013, 52, 7798–7806 CrossRef CAS.
- M. Garvey, S. Meehan, S. L. Gras, H. J. Schirra, D. J. Craik, N. L. Van der Weerden, M. A. Anderson, J. A. Gerrard and J. A. Carver, Biochim. Biophys. Acta, 2013, 1834, 1615–1623 CrossRef CAS.
- S. Gour, V. Kaushik, V. Kumar, P. Bhat, S. C. Yadav and J. K. Yadav, J. Pept. Sci., 2016, 22, 201–207 CrossRef CAS.
- K. Liu, L. Yang, X. Peng, J. Wang, J. R. Lu and H. Xu, Langmuir, 2020, 36, 1737–1744 CrossRef CAS PubMed.
- Z. Shen, Z. Guo, L. Zhou, Y. Wang, J. Zhang, J. Hu and Y. Zhang, Biomater. Sci., 2020, 8, 2031–2039 RSC.
- X. T. Mai, J. Huang, J. Tan, Y. Huang and Y. Chen, J. Pept. Sci., 2015, 21, 561–568 CrossRef CAS PubMed.
- J. Blazyk, R. Wiegand, J. Klein, J. Hammer, R. M. Epand, R. F. Epand, W. L. Maloy and U. P. Kari, J. Biol. Chem., 2001, 276, 27899–27906 CrossRef CAS.
- N. Harmouche, C. Aisenbrey, F. Porcelli, Y. Xia, S. E. D. Nelson, X. Chen, J. Raya, L. Vermeer, C. Aparicio, G. Veglia, S. U. Gorr and B. Bechinger, Biochemistry, 2017, 56, 4269–4278 CrossRef CAS PubMed.
- V. Balhara, R. Schmidt, S. U. Gorr and C. Dewolf, Biochim. Biophys. Acta, 2013, 1828, 2193–2203 CrossRef CAS.
- Z. Oren, J. Hong and Y. Shai, Eur. J. Biochem., 1999, 259, 360–369 CrossRef CAS.
- D. L. Lee and R. S. Hodges, Biopolymers, 2003, 71, 28–48 CrossRef CAS.
- R. Bellavita, E. Buommino, B. Casciaro, F. Merlino, F. Cappiello, N. Marigliano, A. Saviano, F. Maione, R. Santangelo, M. L. Mangoni, S. Galdiero, P. Grieco and A. Falanga, Antibiotics, 2022, 11, 1285 CrossRef CAS PubMed.
- Z. Y. Ong, S. J. Gao and Y. Y. Yang, Adv. Funct. Mater., 2013, 23, 3682–3692 CrossRef CAS.
- J. M. Rausch, J. R. Marks and W. C. Wimley, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10511–10515 CrossRef CAS.
- D. Rapaport and Y. Shai, J. Biol. Chem., 1991, 266, 23769–23775 CrossRef CAS.
- K. Matsuzaki, O. Murase, N. Fujii and K. Miyajima, Biochemistry, 1995, 34, 6521–6526 CrossRef CAS PubMed.
- V. N. Kokryakov, S. S. L. Harwig, E. A. Panyutich, A. A. Shevchenko, G. M. Aleshina, O. V. Shamova, H. A. Korneva and R. I. Lehrer, FEBS lett., 1993, 327, 231–236 CrossRef CAS.
- M. Frohm, B. Agerberth, G. Ahangari, M. Ståhle-Bäckdahl, S. Lidén, H. Wigzell and G. H. Gudmundsson, J. Biol. Chem., 1997, 272, 15258–15263 CrossRef CAS.
- R. Sood, Y. Domanov, M. Pietiäinen, V. P. Kontinen and P. K. J. Kinnunen, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 983–996 CrossRef CAS PubMed.
- E. Y. Lee, C. Zhang, J. Di Domizio, F. Jin, W. Connell, M. Hung, N. Malkoff, V. Veksler, M. Gilliet and P. Ren, Nat. Commun., 2019, 10, 1012 CrossRef PubMed.
- P. Chairatana and E. M. Nolan, J. Am. Chem. Soc., 2014, 136, 13267–13276 CrossRef CAS PubMed.
- E. Soravia, G. Martini and M. Zasloff, FEBS Lett., 1988, 228, 337–340 CrossRef CAS.
- G. Diamond, M. Zasloff, H. Eck, M. Brasseur, W. L. Maloy and C. L. Bevins, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3952–3956 CrossRef CAS.
- O. Lequin, A. Ladram, L. Chabbert, F. Bruston, O. Convert, D. Vanhoye, G. Chassaing, P. Nicolas and M. Amiche, Biochemistry, 2006, 45, 468–480 CrossRef CAS PubMed.
- J. H. Bowie, F. Separovic and M. J. Tyler, Peptides, 2012, 37, 174–188 CrossRef CAS.
- A. Pastore, F. Raimondi, L. Rajendran and P. A. Temussi, Commun. Biol., 2020, 3, 135 CrossRef CAS PubMed.
- L. Silvestro, K. Gupta, J. N. Weiser and P. H. Axelsen, Biochemistry, 1997, 36, 11452–11460 CrossRef CAS.
- H. S. McHaourab, J. S. Hyde and J. B. Feix, Biochemistry, 1994, 33, 6691–6699 CrossRef CAS PubMed.
- S. L. Puan, P. Erriah, M. M. A.-A. Baharudin, N. M. Yahaya, W. N. I. W. A. Kamil, M. S. M. Ali, S. A. Ahmad, S. N. Oslan, S. Lim and S. Sabri, Appl. Microbiol. Biotechnol., 2023, 107, 5569–5593 CrossRef CAS.
- J. Nissen-Meyer and I. F. Nes, Arch. Microbiol., 1997, 167, 67–77 CrossRef CAS PubMed.
- M. A. Riley and J. E. Wertz, Annu. Rev. Microbiol., 2002, 56, 117–137 CrossRef CAS PubMed.
- M. A. Riley, Annu. Rev. Genet., 1998, 32, 255–278 CrossRef CAS.
- M. Herrero and F. Moreno, J. Gen. Microbiol., 1986, 132, 393–402 CAS.
- V. D. Lorenzo, Arch. Microbiol., 1984, 139, 72–75 CrossRef PubMed.
- D. Destoumieux-Garzon, X. Thomas, M. Santamaria, C. Goulard, M. Barthelemy, B. Boscher, Y. Bessin, G. Molle, A. M. Pons, L. Letellier, J. Peduzzi and S. Rebuffat, Mol. Microbiol., 2003, 49, 1031–1041 CrossRef CAS.
- V. De Lorenzo and A. Pugsley, Antimicrob. Agents Chemother., 1985, 27, 666–669 CrossRef CAS PubMed.
- C. Hetz, M. R. Bono, L. F. Barros and R. Lagos, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2696–2701 CrossRef CAS.
- P. Aguilera, A. Marcoleta, P. Lobos-Ruiz, R. Arranz, J. M. Valpuesta, O. Monasterio and R. Lagos, Front. Microbiol., 2016, 7, 35 Search PubMed.
- R. H. Perez, T. Zendo and K. Sonomoto, Microb. Cell Fact., 2014, 13, 1–13 CrossRef.
- P. D. Cotter, C. Hill and R. P. Ross, Nat. Rev. Microbiol., 2005, 3, 777–788 CrossRef CAS PubMed.
- A. Thakur, Master thesis, University of Horticulture & Forestry Nauni (Solan), India, 2017.
- J. Nakayama, Y. Abe, Y. Ono, A. Isogai and A. Suzuki, Biosci. Biotechnol. Biochem., 1995, 59, 703–705 CrossRef CAS PubMed.
- J. P. Tam, S. Wang, K. H. Wong and W. L. Tan, Pharmaceuticals, 2015, 8, 711–757 CrossRef CAS.
- W. F. Broekaert, B. P. Cammue, M. F. De Bolle, K. Thevissen, G. W. De Samblanx, R. W. Osborn and K. Nielson, CRC Crit. Rev. Plant Sci., 1997, 16, 297–323 CrossRef CAS.
- Y. Jin, J. Hammer, M. Pate, Y. Zhang, F. Zhu, E. Zmuda and J. Blazyk, Antimicrob. Agents Chemother., 2005, 49, 4957–4964 CrossRef CAS PubMed.
- H. Kamimori, J. Blazyk and M. I. Aguilar, Biol. Pharm. Bull., 2005, 28, 148–150 CrossRef CAS.
- H. Youssef and C. E. DeWolf, Langmuir, 2019, 36, 660–665 CrossRef.
- G. Bell and P. H. Gouyon, Microbiology, 2003, 149, 1367–1375 CrossRef CAS.
- Z. Y. Ong, J. Cheng, Y. Huang, K. Xu, Z. Ji, W. Fan and Y. Y. Yang, Biomaterials, 2014, 35, 1315–1325 CrossRef CAS PubMed.
- G. Zhong, J. Cheng, Z. C. Liang, L. Xu, W. Lou, C. Bao, Z. Y. Ong, H. Dong, Y. Y. Yang and W. Fan, Adv. Healthcare Mater., 2017, 6, 1601134 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS.
- M. G. Iadanza, M. P. Jackson, E. W. Hewitt, N. A. Ranson and S. E. Radford, Nat. Rev. Mol. Cell Biol., 2018, 19, 755–773 CrossRef CAS.
- D. Otzen and R. Riek, Cold Spring Harbor Perspect. Biol., 2019, 11, a033860 CrossRef CAS.
- N. Van Gerven, R. D. Klein, S. J. Hultgren and H. Remaut, Trends Biotechnol., 2015, 23, 693–706 CAS.
- B. Dai, C. J. Sargent, X. Gui, C. Liu and F. Zhang, Biomacromolecules, 2019, 20, 2015–2023 CrossRef CAS.
- W. A. Eimer, D. K. V. Kumar, N. K. N. Shanmugam, A. S. Rodriguez, T. Mitchell, K. J. Washicosky, B. György, X. O. Breakefield, R. E. Tanzi and R. D. J. N. Moir, Neuron, 2018, 99(56–63), e53 Search PubMed.
- M. R. White, R. Kandel, S. Tripathi, D. Condon, L. Qi, J. Taubenberger and K. L. Hartshorn, PloS one, 2014, 9, e101364 CrossRef.
- Y. Liao, H. Chen, C. Qiu, H. Shen, Z. He, Z. Song and W. Zhou, Research Square, 2023 DOI:10.21203/rs.3.rs-3300215/v1.
- M. R. White, R. Kandel, I. N. Hsieh, X. De Luna and K. L. Hartshorn, PLoS One, 2018, 13, e0194001 CrossRef PubMed.
- P. Spitzer, M. Condic, M. Herrmann, T. J. Oberstein, M. Scharin-Mehlmann, D. F. Gilbert, O. Friedrich, T. Gromer, J. Kornhuber, R. Lang and J. M. Maler, Sci. Rep., 2016, 6, 32228 CrossRef CAS PubMed.
- N. M. Hemmaragala, H. Abrahamse and B. P. George, IET Nanobiotechnol., 2017, 11, 377–382 CrossRef.
- C. Kokotidou, S. V. R. Jonnalagadda, A. A. Orr, G. Vrentzos, A. Kretsovali, P. Tamamis and A. Mitraki, Biomolecules, 2019, 10, 7 CrossRef PubMed.
- X. Sha, P. Li, Y. Feng, D. Xia, X. Tian, Z. Wang, Y. Yang, X. Mao and L. Liu, ACS Appl. Bio Mater., 2020, 3, 3648–3655 CrossRef CAS.
- E. L. Beatman, A. Massey, K. D. Shives, K. S. Burrack, M. Chamanian, T. E. Morrison and J. D. Beckham, J. Virol., 2016, 90, 2767–2782 CrossRef CAS.
- T. Yamada, B. Kluve-Beckerman, J. J. Liepnieks and M. D. Benson, Biochim. Biophys. Acta, 1994, 1226, 323–329 CrossRef CAS.
- A. Erman, K. Lakota, K. Mrak-Poljsak, M. G. Blango, V. Krizan-Hergouth, M. A. Mulvey, S. Sodin-Semrl and P. Veranic, PLoS One, 2012, 7, e32933 CrossRef CAS.
- J. Gong, J. Wu, M. Ikeh, L. Tao, Y. Zhang, J. Bing, C. J. Nobile and G. Huang, Antimicrob. Agents Chemother., 2019, 64, e01975-19 CrossRef PubMed.
- M. Lavie, C. Voisset, N. Vu-Dac, V. Zurawski, G. Duverlie, C. Wychowski and J. Dubuisson, Hepatology, 2006, 44, 1626–1634 CrossRef CAS.
- Z. Cai, L. Cai, J. Jiang, K.-S. Chang, D. R. van der Westhuyzen and G. Luo, J. Virol., 2007, 81, 6128–6133 CrossRef CAS.
- T. Sato, P. Kienlen-Campard, M. Ahmed, W. Liu, H. Li, J. I. Elliott, S. Aimoto, S. N. Constantinescu, J. N. Octave and S. O. Smith, Biochemistry, 2006, 45, 5503–5516 CrossRef CAS.
- N. Nalivaeva, N. Belyaev, I. Zhuravin and A. Turner, Int. J. Alzheimers Dis, 2012, 2012, 383796 CAS.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS.
- R. Penninkilampi, H. M. Brothers and G. D. Eslick, J. Neuroimmune Pharmacol., 2017, 12, 194–203 CrossRef PubMed.
- K. G. Yiannopoulou and S. G. Papageorgiou, J. Cent. Nerv. Syst. Dis., 2020, 12, 1179573520907397 Search PubMed.
- S. Gilman, M. Koller, R. S. Black, L. Jenkins, S. G. Griffith, N. C. Fox, L. Eisner, L. Kirby, M. B. Rovira, F. Forette, J. M. Orgogozo and A. N. S. Team, Neurology, 2005, 64, 1553–1562 CrossRef CAS.
- J. M. Orgogozo, S. Gilman, J. F. Dartigues, B. Laurent, M. Puel, L. C. Kirby, P. Jouanny, B. Dubois, L. Eisner, S. Flitman, B. F. Michel, M. Boada, A. Frank and C. Hock, Neurology, 2003, 61, 46–54 CrossRef CAS PubMed.
- A. Schneeberger, S. Hendrix, M. Mandler, N. Ellison, V. Burger, M. Brunner, L. Frolich, N. Mimica, J. Hort, M. Rainer, D. Imarhiagbe, A. Kurz, O. Peters, H. J. Gertz, L. Tierney, F. Mattner, W. Schmidt and B. Dubois, J. Prev. Alzheimers Dis., 2015, 2, 103–114 CAS.
- R. Vandenberghe, M. E. Riviere, A. Caputo, J. Sovago, R. P. Maguire, M. Farlow, G. Marotta, R. Sanchez-Valle, P. Scheltens, J. M. Ryan and A. Graf, Alzheimers Dement, 2017, 3, 10–22 Search PubMed.
- R. D. Moir, R. Lathe and R. E. Tanzi, Alzheimers Dement, 2018, 14, 1602–1614 CrossRef PubMed.
- D. K. Kumar, S. H. Choi, K. J. Washicosky, W. A. Eimer, S. Tucker, J. Ghofrani, A. Lefkowitz, G. McColl, L. E. Goldstein, R. E. Tanzi and R. D. Moir, Sci. Transl. Med., 2016, 8, 340ra372 Search PubMed.
- L. De Vlieger, R. E. Vandenbroucke and L. Van Hoecke, Drug Discovery Today, 2022, 27, 103340 CrossRef CAS PubMed.
- M. Sochocka, K. Zwolinska and J. Leszek, Curr. Neuropharmacol., 2017, 15, 996–1009 CAS.
- W. R. Lin, M. A. Wozniak, R. J. Cooper, G. K. Wilcock and R. F. Itzhaki, J. Pathol., 2002, 197, 395–402 CrossRef CAS.
- R. Piacentini, G. De Chiara, D. D. Li Puma, C. Ripoli, M. E. Marcocci, E. Garaci, A. T. Palamara and C. Grassi, Front. Pharmacol., 2014, 5, 97 Search PubMed.
- K. Bourgade, A. Le Page, C. Bocti, J. M. Witkowski, G. Dupuis, E. H. Frost and T. Fulop, Jr., J Alzheimers Dis., 2016, 50, 1227–1241 CAS.
- D. K. Kumar, W. A. Eimer, R. E. Tanzi and R. D. Moir, Neurodegener Dis Manag, 2016, 6, 345–348 CrossRef.
- M. Torrent, D. Pulido, M. V. Nogues and E. Boix, PLoS Pathog, 2012, 8, e1003005 CrossRef CAS PubMed.
- H. Jang, L. Connelly, F. T. Arce, S. Ramachandran, B. L. Kagan, R. Lal and R. Nussinov, J. Chem. Theory Comput., 2013, 9, 822–833 CrossRef CAS PubMed.
- Y. R. Huang and R. T. Liu, Int. J. Mol. Sci., 2020, 21, 4477 CrossRef CAS PubMed.
- E. Portelius, G. Brinkmalm, A. J. Tran, H. Zetterberg, A. Westman-Brinkmalm and K. Blennow, Neurodegener. Dis., 2009, 6, 87–94 CrossRef CAS PubMed.
- A. Guntert, H. Dobeli and B. Bohrmann, Neuroscience, 2006, 143, 461–475 CrossRef CAS.
- J. C. J. A. C. N. Phillips, ACS Chem. Neurosci., 2019, 10, 2843–2847 CrossRef CAS.
- L. Gu and Z. Guo, Biochem. Biophys. Res. Commun., 2021, 534, 292–296 CrossRef CAS.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CrossRef CAS PubMed.
- C. J. Pike, M. J. Overman and C. W. Cotman, J. Biol. Chem., 1995, 270, 23895–23898 CrossRef CAS PubMed.
- D. Meral and B. Urbanc, Biophys. J., 2013, 104, 389a CrossRef.
- J. P. Colletier, A. Laganowsky, M. Landau, M. Zhao, A. B. Soriaga, L. Goldschmidt, D. Flot, D. Cascio, M. R. Sawaya and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16938–16943 CrossRef CAS.
- C. Kokotidou, S. V. R. Jonnalagadda, A. A. Orr, M. Seoane-Blanco, C. P. Apostolidou, M. J. van Raaij, M. Kotzabasaki, A. Chatzoudis, J. M. Jakubowski, E. Mossou, V. T. Forsyth, E. P. Mitchell, M. W. Bowler, A. L. Llamas-Saiz, P. Tamamis and A. Mitraki, FEBS Lett., 2018, 592, 1777–1788 CrossRef CAS PubMed.
- R. A. DeFronzo, E. Ferrannini, L. Groop, R. R. Henry, W. H. Herman, J. J. Holst, F. B. Hu, C. R. Kahn, I. Raz, G. I. Shulman, D. C. Simonson, M. A. Testa and R. Weiss, Nat. Rev. Dis. Primers, 2015, 1, 15019 CrossRef PubMed.
- S. Chatterjee, K. Khunti and M. J. Davies, Lancet, 2017, 389, 2239–2251 CrossRef CAS.
- P. Westermark and E. J. D. Wilander, Diabetologia, 1983, 24, 342–346 CrossRef CAS PubMed.
- S. Chakraborty, B. Chatterjee and S. J. B. C. Basu, Biophys. Chem., 2012, 168, 1–9 CrossRef PubMed.
- R. A. Noel, D. K. Braun, R. E. Patterson and G. L. Bloomgren, Diabetes Care, 2009, 32, 834–838 CrossRef.
- S. Sharma and P. Tripathi, J. Nutr. Biochem., 2019, 63, 101–108 CrossRef CAS PubMed.
- A. K. Sikalidis and A. Maykish, Biomedicines, 2020, 8, 8 CrossRef CAS PubMed.
- C. Naing, J. W. Mak, S. I. Ahmed and M. Maung, World J. Gastroenterol., 2012, 18, 1642 CrossRef PubMed.
- S. G. Yoo, K. D. Han, K. H. Lee, Y. La, D. E. Kwon and S. H. Han, Diabetes Metab. J., 2019, 43, 815–829 CrossRef PubMed.
- J. Sato, A. Kanazawa, F. Ikeda, T. Yoshihara, H. Goto, H. Abe, K. Komiya, M. Kawaguchi, T. Shimizu and T. Ogihara, Diabetes Care, 2014, 37, 2343–2350 CrossRef CAS PubMed.
- A. Bener, R. Micallef, M. Afifi, M. Derbala, H. M. Al-Mulla and M. A. J. T. J. G. Usmani, Turk J. Gastroentero., 2007, 18, 225–229 Search PubMed.
- C. Wang, D. Gao and B. Kaltenboeck, J. Infect. Dis., 2009, 200, 279–287 CrossRef CAS PubMed.
- S. Al Mubarak, A. A. Robert, J. K. Baskaradoss, K. Al-Zoman, A. Al Sohail, A. Alsuwyed and S. Ciancio, J. Infect. Public Health, 2013, 6, 296–301 CrossRef PubMed.
- M. Y. Donath, M. Boni-Schnetzler, H. Ellingsgaard and J. A. Ehses, Physiology, 2009, 24, 325–331 CrossRef CAS PubMed.
- M. Boni-Schnetzler, J. Thorne, G. Parnaud, L. Marselli, J. A. Ehses, J. Kerr-Conte, F. Pattou, P. A. Halban, G. C. Weir and M. Y. Donath, J. Clin. Endocrinol. Metab., 2008, 93, 4065–4074 CrossRef.
- S. Kim, J. H. Seo and Y. H. Suh, Parkinsonism Relat. Disord., 2004, 10(Suppl 1), S9–S13 CrossRef.
- L. Breydo, J. W. Wu and V. N. Uversky, Biochim. Biophys. Acta, 2012, 1822, 261–285 CrossRef CAS PubMed.
- M. C. Bennett, Pharmacol. Ther., 2005, 105, 311–331 CrossRef CAS.
- O. EI-Agnaf and G. J. B. S. T. Irvine, Biochem. Soc. Trans., 2002, 30, 559–565 CrossRef.
- C. R. Fields, N. Bengoa-Vergniory and R. Wade-Martins, Front. Mol. Neurosci., 2019, 12, 299 CrossRef CAS PubMed.
- D. L. Fortin, M. D. Troyer, K. Nakamura, S. Kubo, M. D. Anthony and R. H. Edwards, J. Neurosci., 2004, 24, 6715–6723 CrossRef CAS PubMed.
- D. Sulzer and R. H. Edwards, J. Neurochem., 2019, 150, 475–486 CrossRef CAS.
- C. R. Pacheco, C. N. Morales, A. E. Ramirez, F. J. Munoz, S. S. Gallegos, P. A. Caviedes, L. G. Aguayo and C. M. Opazo, J. Neurochem., 2015, 132, 731–741 CrossRef CAS.
- K. J. Vargas, S. Makani, T. Davis, C. H. Westphal, P. E. Castillo and S. S. Chandra, J. Neurosci., 2014, 34, 9364–9376 CrossRef.
- F. Savini, M. R. Loffredo, C. Troiano, S. Bobone, N. Malanovic, T. O. Eichmann, L. Caprio, V. C. Canale, Y. Park, M. L. Mangoni and L. Stella, Biochim. Biophys. Acta Biomembr., 2020, 1862, 183291 CrossRef CAS PubMed.
- Y. Luo and Y. Song, Int. J. Mol. Sci., 2021, 22, 11401 CrossRef CAS PubMed.
- W. Y. Lin, M. S. Lin, Y. H. Weng, T. H. Yeh, Y. S. Lin, P. Y. Fong, Y. R. Wu, C. S. Lu, R. S. Chen and Y. Z. Huang, JAMA Neurol., 2019, 76, 1019–1027 CrossRef PubMed.
- H. Wang, X. Liu, C. Tan, W. Zhou, J. Jiang, W. Peng, X. Zhou, L. Mo and L. Chen, Brain Behav., 2020, 10, e01549 CrossRef.
- S. G. Chen, V. Stribinskis, M. J. Rane, D. R. Demuth, E. Gozal, A. M. Roberts, R. Jagadapillai, R. Liu, K. Choe, B. Shivakumar, F. Son, S. Jin, R. Kerber, A. Adame, E. Masliah and R. P. Friedland, Sci. Rep., 2016, 6, 34477 CrossRef CAS PubMed.
- I. Poggiolini, D. Saverioni and P. Parchi, Int. J. Cell Biol., 2013, 2013, 910314 Search PubMed.
- R. A. Moore, L. M. Taubner and S. A. Priola, Curr. Opin. Struct. Biol., 2009, 19, 14–22 CrossRef CAS PubMed.
- H. Bueler, A. Aguzzi, A. Sailer, R. A. Greiner, P. Autenried, M. Aguet and C. Weissmann, Cell, 1993, 73, 1339–1347 CrossRef CAS PubMed.
- J. R. Silveira, G. J. Raymond, A. G. Hughson, R. E. Race, V. L. Sim, S. F. Hayes and B. J. N. Caughey, Nature, 2005, 437, 257–261 CrossRef CAS PubMed.
- P. Tixador, L. Herzog, F. Reine, E. Jaumain, J. M. Chapuis, A. L. Dur, H. Laude and V. Beringue, PLoS Pathog, 2010, 6, e1000859 CrossRef PubMed.
- A. R. Castle and A. C. Gill, Front. Mol. Biosci., 2017, 4, 19 Search PubMed.
- R. Linden, V. R. Martins, M. A. Prado, M. Cammarota, I. Izquierdo and R. R. Brentani, Phys. Rev., 2008, 88, 673–728 CAS.
- F. Prestori, P. Rossi, B. Bearzatto, J. Laine, D. Necchi, S. Diwakar, S. N. Schiffmann, H. Axelrad and E. D’Angelo, J. Neurosci., 2008, 28, 7091–7103 CrossRef CAS PubMed.
- J. Bremer, F. Baumann, C. Tiberi, C. Wessig, H. Fischer, P. Schwarz, A. D. Steele, K. V. Toyka, K.-A. Nave and J. Weis, Nat. Neurosci., 2010, 13, 310–318 CrossRef CAS.
- P. C. Konturek, K. Bazela, V. Kukharskyy, M. Bauer, E. G. Hahn and D. Schuppan, World J. Gastroenterol., 2005, 11, 7651–7656 CrossRef CAS PubMed.
- J. Pammer, W. Weninger and E. Tschachler, Am. J. Pathol., 1998, 153, 1353–1358 CrossRef CAS PubMed.
- M. D. Beer, T. Yuan, M. Kindy, B. Asztalos, P. Roheim and F. D. Beer, J. Lipid Res., 1995, 36, 526–534 CrossRef.
- A. Butler and A. S. Whitehead, Immunogenetics, 1996, 44, 468–474 CrossRef CAS PubMed.
- R. L. Meek and E. P. Benditt, J. Exp. Med., 1986, 164, 2006–2017 CrossRef CAS PubMed.
- E. P. Benditt and N. Eriksen, Am. J. Pathol., 1971, 65, 231–252 CAS.
- C. L. Banka, T. Yuan, M. C. de Beer, M. Kindy, L. K. Curtiss and F. C. de Beer, J. Lipid Res., 1995, 36, 1058–1065 CrossRef CAS.
- R. Badolato, J. M. Wang, W. J. Murphy, A. R. Lloyd, D. F. Michiel, L. L. Bausserman, D. J. Kelvin and J. J. Oppenheim, J. Exp. Med., 1994, 180, 203–209 CrossRef CAS PubMed.
- C. Song, K. Hsu, E. Yamen, W. Yan, J. Fock, P. K. Witting, C. L. Geczy and S. B. Freedman, Atherosclerosis, 2009, 207, 374–383 CrossRef CAS PubMed.
- N. Olsson, A. Siegbahn and G. Nilsson, Biochem. Biophys. Res. Commun., 1999, 254, 143–146 CrossRef CAS PubMed.
- C. Shah, R. Hari-Dass and J. G. Raynes, Blood, 2006, 108, 1751–1757 CrossRef CAS PubMed.
- M. C. Lin, T. Mirzabekov and B. L. Kagan, J. Biol. Chem., 1997, 272, 44–47 CrossRef CAS PubMed.
- N. Arispe, E. Rojas and H. B. Pollard, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 567–571 CrossRef CAS.
- L. Obici and G. Merlini, Swiss Med. Wkly., 2012, 142, w13580 Search PubMed.
- E. R. Eckhardt, J. Witta, J. Zhong, R. Arsenescu, V. Arsenescu, Y. Wang, S. Ghoshal, M. C. de Beer, F. C. de Beer and W. J. de Villiers, BMC Gastroenterol., 2010, 10, 133 CrossRef.
- R. Badolato, J. M. Wang, S. L. Stornello, A. N. Ponzi, M. Duse and T. Musso, J. Leukoc. Biol., 2000, 67, 381–386 CrossRef CAS.
- J. Gong, J. Bing, G. Guan, C. J. Nobile and G. Huang, Antimicrob. Agents Chemother., 2020, 64, e00024-20 CrossRef.
- X. N. Mo, Z. Q. Su, C. L. Lei, D. F. Chen, H. Peng, R. C. Chen, L. Sang, H. K. Wu and S. Y. Li, Respirology, 2020, 764 CrossRef.
- H. Li, X. Xiang, H. Ren, L. Xu, L. Zhao, X. Chen, H. Long, Q. Wang and Q. Wu, J. Infect., 2020, 80, 646–655 CrossRef CAS PubMed.
- M. R. White, I. N. Hsieh, X. De Luna and K. L. Hartshorn, J. Leukocyte Biol., 2021, 110, 155–166 CrossRef CAS.
- H. B. Kramer, K. J. Lavender, L. Qin, A. R. Stacey, M. K. Liu, K. D. Gleria, A. Simmons, N. Gasper-Smith, B. F. Haynes, A. J. McMichael, P. Borrow and B. M. Kessler, PLoS Pathog, 2010, 6, e1000893 CrossRef PubMed.
- S. Makin, Nature, 2018, 559, S4–S7 CrossRef CAS.
- R. Oliva, S. K. Mukherjee, L. Ostermeier, L. A. Pazurek, S. Kriegler, V. Bader, D. Prumbaum, S. Raunser, K. F. Winklhofer, J. Tatzelt and R. Winter, Chemistry, 2021, 27, 11845–11851 CrossRef CAS PubMed.
- A. Kasus-Jacobi, J. L. Washburn, C. A. Land and H. A. Pereira, Curr. Alzheimer Res, 2021, 18, 414–427 CrossRef CAS PubMed.
- A. F. Gombart, N. Borregaard and H. P. Koeffler, FASEB J, 2005, 19, 1067–1077 CrossRef CAS PubMed.
- L. D. Pound, C. Patrick, C. E. Eberhard, W. Mottawea, G. S. Wang, T. Abujamel, R. Vandenbeek, A. Stintzi and F. W. Scott, Diabetes, 2015, 64, 4135–4147 CrossRef CAS.
- J. Sun, L. Furio, R. Mecheri, A. M. van der Does, E. Lundeberg, L. Saveanu, Y. Chen, P. van Endert, B. Agerberth and J. Diana, Immunity, 2015, 43, 304–317 CrossRef CAS PubMed.
- S. Grabrucker, M. Marizzoni, E. Silajdzic, N. Lopizzo, E. Mombelli, S. Nicolas, S. Dohm-Hansen, C. Scassellati, D. V. Moretti, M. Rosa, K. Hoffmann, J. F. Cryan, O. F. O'Leary, J. A. English, A. Lavelle, C. O'Neill, S. Thuret, A. Cattaneo and Y. M. Nolan, Brain, 2023, 146, 4916–4934 CrossRef PubMed.
- T. Harach, N. Marungruang, N. Duthilleul, V. Cheatham, K. D. Mc Coy, G. Frisoni, J. J. Neher, F. Fåk, M. Jucker and T. Lasser, Sci. Rep., 2017, 7, 41802 CrossRef CAS PubMed.
- K. Hartman, J. R. Brender, K. Monde, A. Ono, M. L. Evans, N. Popovych, M. R. Chapman and A. Ramamoorthy, PeerJ, 2013, 1, e5 CrossRef.
- A. Cattaneo, N. Cattane, S. Galluzzi, S. Provasi, N. Lopizzo, C. Festari, C. Ferrari, U. P. Guerra, B. Paghera, C. Muscio, A. Bianchetti, G. D. Volta, M. Turla, M. S. Cotelli, M. Gennuso, A. Prelle, O. Zanetti, G. Lussignoli, D. Mirabile, D. Bellandi, S. Gentile, G. Belotti, D. Villani, T. Harach, T. Bolmont, A. Padovani, M. Boccardi, G. B. Frisoni and I.-F. Group, Neurobiol. Aging, 2017, 49, 60–68 CrossRef CAS.
- M. Sanchez-Tapia, A. Mimenza-Alvarado, L. Granados-Dominguez, A. Flores-Lopez, A. Lopez-Barradas, V. Ortiz, C. Perez-Cruz, H. Sanchez-Vidal, J. Hernandez-Acosta, J. A. Avila-Funes, M. Guevara-Cruz, A. R. Tovar and N. Torres, Nutrients, 2023, 15, 932 CrossRef CAS.
- O. Meza-Barajas, I. Aranda, A. Binmahfooz, A. Newell and S. Jayasinghe, bioRxiv, 2019, preprint DOI:10.1101/772392.
- N. Khan, M. Arlantico, E. Feliciano, H. Nguyen and S. Jayasinghe, FASEB J, 2018, 32, lb59 Search PubMed.
- I. Cherny, L. Rockah, O. Levy-Nissenbaum, U. Gophna, E. Z. Ron and E. Gazit, J. Mol. Biol., 2005, 352, 245–252 CrossRef CAS PubMed.
- C. Wang, C. Y. Lau, F. Ma and C. Zheng, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2106504118 CrossRef CAS.
- E. Tayeb-Fligelman, O. Tabachnikov, A. Moshe, O. Goldshmidt-Tran, M. R. Sawaya, N. Coquelle, J. P. Colletier and M. Landau, Science, 2017, 355, 831–833 CrossRef PubMed.
- C. Haikal, L. Ortigosa-Pascual, Z. Najarzadeh, K. Bernfur, A. Svanbergsson, D. E. Otzen, S. Linse and J.-Y. Li, Int. J. Mol. Sci., 2021, 22, 11594 CrossRef CAS PubMed.
- B. Peng, S. Xu, Y. Liang, X. Dong and Y. Sun, Biomimetics, 2023, 8, 459 CrossRef CAS PubMed.
- S. Perov, O. Lidor, N. Salinas, N. Golan, E. Tayeb-Fligelman, M. Deshmukh, D. Willbold and M. Landau, PLoS Pathog, 2019, 15, e1007978 CrossRef CAS.
- S. L. Rouse, S. J. Matthews and M. S. Dueholm, J. Mol. Biol., 2018, 430, 3685–3695 CrossRef CAS PubMed.
- N. Jain, J. Aden, K. Nagamatsu, M. L. Evans, X. Li, B. McMichael, M. I. Ivanova, F. Almqvist, J. N. Buxbaum and M. R. Chapman, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12184–12189 CrossRef CAS.
- J. N. Buxbaum, Z. Ye, N. Reixach, L. Friske, C. Levy, P. Das, T. Golde, E. Masliah, A. R. Roberts and T. Bartfai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2681–2686 CrossRef CAS PubMed.
- X. Li, X. Zhang, A. R. Ladiwala, D. Du, J. K. Yadav, P. M. Tessier, P. E. Wright, J. W. Kelly and J. N. Buxbaum, J. Neurosci., 2013, 33, 19423–19433 CrossRef CAS PubMed.
- S. Wasana Jayaweera, S. Surano, N. Pettersson, E. Oskarsson, L. Lettius, A. L. Gharibyan, I. Anan and A. Olofsson, Biomolecules, 2021, 11, 411 CrossRef.
- R. Cascella, S. Conti, B. Mannini, X. Li, J. N. Buxbaum, B. Tiribilli, F. Chiti and C. Cecchi, Biochim. Biophys. Acta, 2013, 1832, 2302–2314 CrossRef CAS PubMed.
- I. Horvath and P. Wittung-Stafshede, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12473–12477 CrossRef CAS.
- R. Hu, M. Zhang, H. Chen, B. Jiang and J. Zheng, ACS Chem. Neurosci., 2015, 6, 1759–1768 CrossRef CAS.
- P. K. Mandal, J. W. Pettegrew, E. Masliah, R. L. Hamilton and R. Mandal, Neurochem. Res., 2006, 31, 1153–1162 CrossRef CAS.
- I. C. Stancu, B. Vasconcelos, D. Terwel and I. Dewachter, Mol Neurodegener., 2014, 9, 51 CrossRef PubMed.
- P. Liu, S. Zhang, M. S. Chen, Q. Liu, C. Wang, C. Wang, Y. M. Li, F. Besenbacher and M. Dong, Chem. Commun., 2012, 48, 191–193 RSC.
- H. Jang, B. Ma, T. B. Woolf and R. Nussinov, Biophys. J., 2006, 91, 2848–2859 CrossRef CAS.
- A. K. Prasad, L. L. Martin and A. S. Panwar, Phys. Chem. Chem. Phys., 2023, 25, 12134–12147 RSC.
- N. Salinas, J. P. Colletier, A. Moshe and M. Landau, Nat. Commun., 2018, 9, 3512 CrossRef PubMed.
- N. Louros, K. Konstantoulea, M. De Vleeschouwer, M. Ramakers, J. Schymkowitz and F. Rousseau, Nucleic Acids Res., 2020, 48, D389–D393 CrossRef CAS.
- G. G. Tartaglia and M. Vendruscolo, Chem. Soc. Rev., 2008, 37, 1395–1401 RSC.
- I. Walsh, F. Seno, S. C. Tosatto and A. Trovato, Nucleic Acids Res., 2014, 42, W301–307 CrossRef CAS PubMed.
Footnote |
| † Authors contribute equally to this paper. |
| This journal is © The Royal Society of Chemistry 2024 |